Изобретение относится к области органической химии и медицины. В EP N 0389037, 1990, описаны производные N-(3-гидрокси-4-пиперидинил) (дигидробензофуран, дигидро-2H-бензопиран или дигидробензодиоксин)карбоксамида и в EP N 0445862, 1991, описаны производные N-(4-пиперидинил) (дигидробензофуран- или дигидробензо-2H-бензопиран)карбоксамида. В обеих заявках описана способность этих соединений стимулировать желудочно-кишечную перистальтику. Производные диметилдигидробензофурана и диметилдигидро-2H-бензопирана изобретения проявляют 5-HT3-антагонизм (антагонизм к 5-гидрокситриптамину у 5-HT3-рецепторов).
Изобретение относится к способу лечения теплокровных животных, страдающих 5-HT3-медиированными нарушениями, например тревожными состояниями, психозами, депрессивными состояниями, шизофренией, нарушениями познавательной способности, злоупотреблениями лекарственными средствами, мигренью, рвотой, синдромом раздраженной толстой кишки и аналогичными нарушениями, системным введением этому теплокровному животному эффективного 5-HT3-антагонистического количества соединения формулы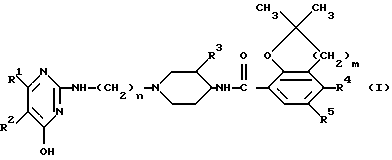
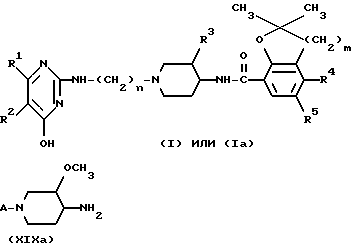
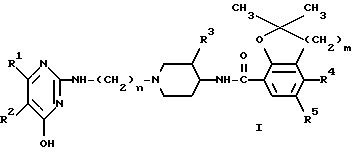
или фармацевтически пригодной соли с кислотой или стереохимического изомера этого соединения
где
R1 и R2 - водород или R1 и R2 вместе образуют двухвалентный радикал формулы (a), (b) или (c):
-CH=CH-CH=CH- (a),
-CH=C(Cl)-CH=CH- (b),
-CH=CH-C(Cl)=CH- (c);
n = 2,3 или 4;
R3 - водород или метоксигруппа;
m = 1 или 2;
R4 - водород, аминогруппа или C1-C3- алкилкарбониламиногруппа;
R5 - водород или галоген.
Изобретение относится также к применению соединений формулы I и их фармацевтически пригодных солей с кислотами и стереохимических изомеров для получения лекарственного препарата, предназначенного для лечения 5-HT3-медиированных нарушений, например тревожных состояний, психоза, депрессивных состояний, шизофрении, нарушений познавательной способности, злоупотребления лекарственными средствами, мигрени, рвоты, синдрома раздраженной толстой кишки и аналогичных нарушений.
В указанных выше и последующих определениях термин "галоген" обозначает фтор, хлор, бром и иод, предпочтительно хлор; C1-C4-алкил обозначает содержащие от 1 до 4 атомов углерода насыщенные углеводородные радикалы с нормальной или разветвленной цепью, например метил, этил, пропил, 1-метилэтил, бутил, 1-метилпропил, 2-метилпропил или 1,1-диметилэтил, предпочтительно метил. C1-C6-Алкил обозначает C1-C4-алкил и более высшие гомологи его, например пентил и гексил. C1-C3-Алкилкарбонил обозначает ацилы нормального или разветвленного строения, например метилкарбонил, этилкарбонил, пропилкарбонил, предпочтительно метилкарбонил.
Термин "фармацевтически пригодная соль" с кислотой обозначает нетоксичные, терапевтически активные соли с кислотами, которые могут образовать соединения формулы (I). Соединения формулы (I), обладающие основными свойствами, можно превратить в соответствующие терапевтически активные, нетоксичные соли с кислотами обработкой свободного основания подходящим количеством соответствующей кислоты обычными методиками. Примерами подходящих кислот являются неорганические кислоты, такие как галогеноводородные кислоты, например соляная, бромистоводородная и подобные кислоты, серная кислота, азотная кислота, фосфорная кислота и подобные кислоты, или органические кислоты, например уксусная, пропионовая, гидроксиуксусная, 2-гидроксипропионовая, 2-оксопропионовая, пропандионовая, бутандионовая, (Z)-2-бутендионовая, (E)-2-бутендионовая, 2-гидроксибутандионовая, 2,3-дигидроксибутандионовая, 2-гидрокси-1,2,3-пропантрикарбоновая, циклогексансульфаминовая, 2-гидроксибензойная, 4-амино-2-гидроксибензойная и подобные кислоты. Термин "фармацевтические пригодные соли" включает также сольваты, которые соединения формулы (I) могут образовать, например алкоголяты, в частности гидраты.
Соединения формулы (I) могут также находиться в виде их таутомерных форм. Предполагается, что такие формы, хотя они подробно не указываются выше, включены в объем изобретения.
Термин "стереохимические изомеры", обозначает различные изомерные формы, которые могут иметь соединения формулы (I). Если не оговорено особо или не указано иначе, по химическому определению соединения представляют собой смесь всех возможных стереохимических изомерных форм, причем эти смеси содержат все диастереомеры и/или энантиомеры основной молекулярной структуры. Предполагается, что все стереохимически изомерные формы соединений формулы (I) как в чистом виде, так и в смеси друг с другом включены в объем изобретения.
Термин "энантиомерно чистый" обозначает соединения, имеющие содержание энантиомера от по меньшей мере 94% (т.е. минимальное содержание одного энантиомера 97% и максимальное содержание другого энантиомера 3%) до 100% (т.е. содержание одного энантиомера 100% и отсутствие другого), в частности соединения, имеющие содержание энантиомера от 96% до 100%, более конкретно соединения, имеющие содержание энантиомера от 98% до 100%. Термин "энантиомерно обогащенный" обозначает соединения, имеющие содержание энантиомера от более чем 0% до около 94%. Термины "диастереомерно обогащенные" и "диастереомерно чистые", которые будут приведены ниже, следует понимать аналогичным образом, но для содержания диастереомера в указанной смеси.
Для применения в качестве 5-HT3-антагонистов интересны соединения формулы (I), у которых R5 представляет собой галоген, предпочтительно хлор.
Для применения в качестве 5-HT3-антагонистов интересны также соединения формулы (I), у которых R4 представляет собой водород или аминогруппу.
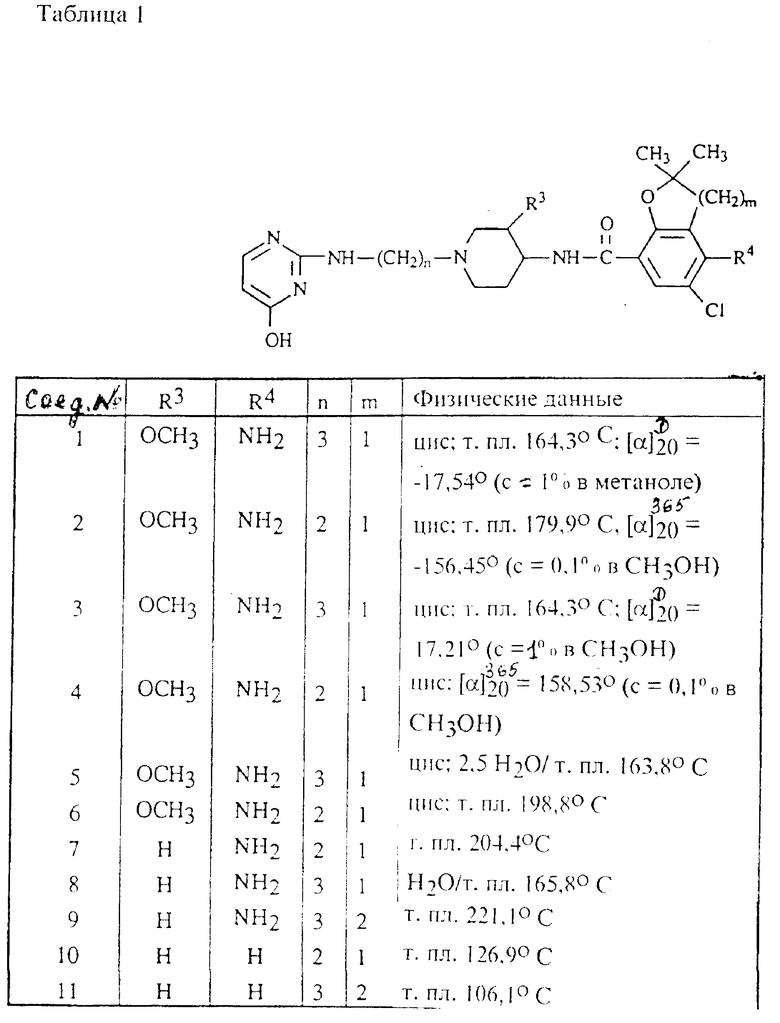
Для применения в качестве 5-HT3-антагонистов наиболее интересны соединения формулы (I), у которых R1 и R2 - водород; n = 1 или 2; R3 - метоксигруппа и имеет цис-конфигурацию; m = I; R4 - аминогруппа и R5 - галоген.
В частности, интересными для применения в качестве 5-HT3-антагонистов являются соединения формулы (I), в которой R3 представляет собой метоксигруппу и имеет цис-конфигурацию, которые являются левовращающими.
Предпочтительными соединениями являются (-)-цис-4-амино-5-хлор-2,3-дигидро-N-[1-[3-[(3,4-дигидро-4-оксо-2 -пиримидинил)амино]пропил]-3-метокси-4-пиперидинил] -2,2- деметил-7-бензофуранкарбоксамид и (-)-цис-4-амино-5-хлор-N-[1-[2-[(3,4-дигидро-4-оксо-2 -пиримидинил)амино] этил]-2,3-дигидро-3-метокси-4-пиперидинил]-2,2-диметил-7- бензофуран карбоксамид и их фармацевтически пригодные соли с кислотами.
Соединения формулы (I), у которых R3 представляет собой метоксигруппу и имеет цис-конфигурацию, представлены формулой (I-a). Указанные далее промежуточные продукты, у которых R3 представляет собой метоксигруппу и возможно имеет цис-конфигурацию, будут обозначены путем добавления буквы -a к их номерам.
Дополнительным признаком изобретения является тот факт, что левовращающие энантиомеры соединений формулы (I), у которых R3 представляет собой метоксигруппу и имеет цис-конфигурацию, т.е. левовращающие энантиомеры соединений формулы (I-a), считаются новыми соединениями.
Соединения формулы (I) можно обычно получать известными способами, например описанными в EP N 389037, и другими известными способами. Некоторые промежуточные продукты формул (II), (III), (IV), (V), (VI), (VII), (IX), (X) и (XIII) описаны в EP N 0389037, EP N 0445862 и EP N 0076350. Некоторые способы получения соединений формулы (I), особенно соединений формулы (I-a) и новых промежуточных продуктов будут описаны ниже.
В указанных способах получения продукты реакции можно выделить из реакционной смеси и, если необходимо, далее очищать методиками, обычно известными в данной области, например экстракцией, перегонкой, кристаллизацией, растиранием и хроматографией.
Для упрощения структурного изображения соединений формулы (I) и некоторых исходных соединений и промежуточных продуктов их радикал формулы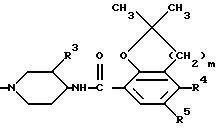
далее будет обозначаться символом D, и радикал формулы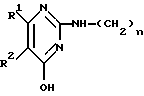
далее будет обозначаться символом L.
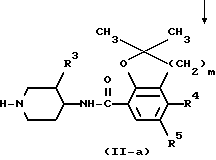
Соединения формулы (I) можно получить N-алкилированием пиперидина формулы (II) промежуточным продуктом формулы (III)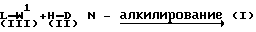
W1, приведенный в схеме реакции соединения (III) с (II) и последующих реакционных схемах, обозначает соответствующую удаляемую группу, такую, как галоген, например хлор, бром или иод, или сульфонилоксигруппа, например метансульфонилоксигруппа, 4-метилбензолсульфонилоксигруппа и подобные удаляемые группы. Реакцию N-алкилирования соединения (II) соединением (III) обычно проводят известными способами алкилирования.
Соединения формулы (I) можно также получить N-ацилированием амина формулы (IV) карбоновой кислотой формулы (V) или ее функциональным производным, например ацилгалогенидом, симметричным или смешанным ангидридом или эфиром, предпочтительно активированным эфиром, известными методами.
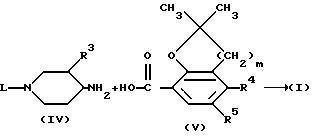
Может быть целесообразно защитить амино- или гидроксигруппы в процессе проведения реакции, чтобы избежать нежелательные побочные реакции. Подходящие защитные группы представляют собой легко удаляемые группы, например C1-C4-алкилкарбонил, C1 - C4-алкилоксикарбонил, фенилметил, трет-бутил и подобные защитные группы.
Соединения формулы (I) можно также получить N-алкилированием промежуточного продукта формулы (VII) алкилирующим реагентом формулы (VI), в которой R6 представляет собой водород или C1-C6-алкил и W2 представляет собой соответствующую удаляемую группу, такую как галоген, например хлор, бром или иод; сульфонилоксигруппа, например метансульфонилоксигруппа, 4-метилбензолсульфонилоксигруппа; C1-C6-алкилоксигруппа, например метоксигруппа, этоксигруппа; C1-C6-алкилтиогруппа, например метилтиогруппа, этилтиогруппа. Когда R6 представляет собой C1-C6-алкил, образуется промежуточный продукт формулы (VIII), который можно затем превратить в конечное соединение путем отщепления защитной простой эфирной группы. Такое отщепление можно осуществить обработкой промежуточного продукта формулы (VIII) кислотой, такой как галогеноводородная кислота, например соляная кислота.
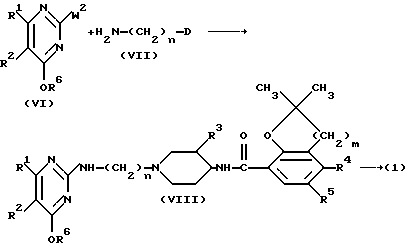
Соединения формулы (I) можно альтернативно получить N-алкилированием 2-аминопиридина формулы (IX) промежуточным продуктом формулы (X).
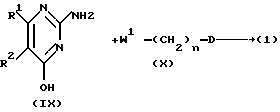
Реакция алкилирования соединений формулы (VI) соединениями (VII) и соединений формулы (IX) соединениями (X) можно проводить известными способами, например перемешиванием и иногда нагреванием реагентов без растворителя или в смеси с инертным органическим растворителем, таким как спирт, например 2-пропанол, бутанол, диполярный апротонный растворитель, например ацетонитрил, возможно в присутствии подходящего основания, например карбоната калия.
Соединения формулы (I) можно также превратить друг в друга известными реакциями превращений.
Аминогруппы можно превратить в C1 - C3-алкилкарбониламиногруппы известными реакциями N-ацилирования и, наоборот, C1 - C3-алкилкарбониламиногруппы можно превратить в аминогруппы известными реакциями гидролиза.
Соединения формулы (I), у которых R5 представляет собой водород, можно превратить в соответствующие соединения, у которых R5 представляет собой галоген, при помощи известных методик галогенирования.
Промежуточные продукты формулы (VII) можно получить N-алкилированием промежуточного продукта формулы (II) реагентом формулы (XI) с последующим удалением защитной группы P у полученного таким образом промежуточного продукта (XIII) по известным методикам реакций.
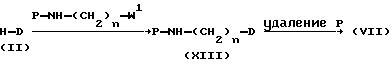
В соединениях формул (XI) и (XIII) и других промежуточных продуктах, содержащих группу P, приведенных схем P представляет собой подходящую защитную группу, легко удаляемую, например, гидрогенолизом или гидролизом. Предпочтительными защитными группами являются, например, C1 - C4-алкилкарбонилы, например метилкарбонил, этилкарбонил; C1 - C4-алкоксикарбонилы, например этоксикарбонил, 1,1'-диметилэтилоксикарбонил; тригалогенметилкарбонилы, например трифторметилкарбонил; дифенилметил; трифенилметил или арилметил, где арил представляет собой фенил, возможно имеющий до двух заместителей, выбранных из C1 - C4-алкоксигрупп и галогенов.
Промежуточные продукты формулы (II) можно получить по известным методикам образования амидов реакций соответственно замещенного пиперидина формулы (XIV) с промежуточной кислотой формулы (V) или ее функциональным производным с последующим удалением защитной группы P1 известными методами. P1 представляет собой легко удаляемую защитную группу и имеет такое же значение, как указанная выше группа P.
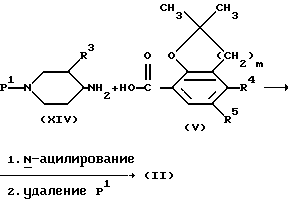
Промежуточные продукты формулы (XIV), у которых R3 представляет собой метоксигруппу и имеет цис-конфигурацию, т.е. 3-метокси-4-аминопиперидины формулы (XIV-a), можно получить, например, каталитическим гидрированием имина формулы (XVI-a) с последующим превращением вторичного амина формулы (XV-a) в 3-метокси-4-аминопиперидины формулы (XIV-a) гидрогенолизом. Имины формулы (XVI-a) можно получить известными методами образования иминов из 3-метокси-4-оксопиперидина формулы (XVII-a) и имина формулы (XVIII).
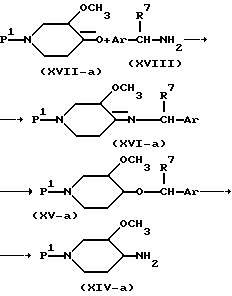
В промежуточных продуктах формул (XVIII), (XVI-а) и (XV-а) R7 представляет собой водород, C1-C6-алкил или гидрокси-C1-C6-алкил и Ar представляет собой фенил, возможно замещенный галогеном, C1-C6-алкилом, C1-C6-алкилоксигруппой, или нафтил, возможно замещенный галогеном, C1-C6-алкилом, C1-C6-алкилоксигруппой.
Последовательность реакций от промежуточного продукта формулы (XVII-а) до образования промежуточного продукта формулы (XIV-а) можно также проводить по методике в одной емкости.
Энантиомерно обогащенные или энантиомерно чистые промежуточные продукты формулы (XV-а) и (XIV-а) можно получить одним из следующих способов.
Исходный рацемический 3-метокси-4-оксопиперидин формулы (XVII-а) или соответствующий кеталь, такой как ди-C1-C6- алкилкеталь, например 4,4-диэтокси-3-метоксипиперидин, можно разделить на его энантиомеры и затем превратить в энантиомерно чистый цис-3-метокси-4-аминопиперидин формулы (XIV-а), как описано выше. Такое разделение на энантиомеры можно проводить, например, колоночной хроматографией с применением хиральной неподвижной фазы, например Chiracell OD.
Альтернативно промежуточный имин формулы (XVI-а) можно получить, применяя один из энантиомеров хирального амина формулы (XVIII), в которой R7 имеет указанные выше значения, кроме водорода, причем эти амины представлены формулой (XVIII-b), например (-)-(R)- α -аминобензолэтанолом или (+)-S - α -аминобензолэтанол, которые после гидрирования превращаются в диастереомерные амины формулы (XV-а), которые можно, как обычно, разделить методами физического разделения, например методиками селективной кристаллизации или хроматографическими методами. Удалением арилметила формулы Ar-CH(R7) у соответствующих диастереомерных аминов формулы (XV-а) гидрогенолизом получают соответствующие энантиомерные 3-метокси-4-аминопиперидины формулы (XIV-а).
Однако в процессе оптимизации указанной выше реакционной последовательности был найден другой путь получения энантиомерно чистых 3-метокси-4-аминопиперидинов формулы (XIV-а). При проведении реакции рацемического кетона, например 3-метокси-4-оксопиперидина формулы (XVII-а) с энантиомерно чистым хиральным амином формулы (XVIII-b), например (-)-(S)- α -метилбензиламином, и последующего гидрирования образованного таким образом имина формулы (XVI-а) ожидаемое соотношение диастереомерных аминов формулы (XV-а) составляет около 1: 1. Однако было найдено, что после проведения указанной выше реакционной последовательности полученное диастереомерное соотношение значительно отличается от соотношения 1:1. Другими словами, амины формулы (XV-а) были диастереомерно обогащенными или даже диастереомерно чистыми. Следовательно, в процессе реакционной последовательности один диастереомер превращается в другой конфигурационным обращением стереоцентра, имеющего метоксигруппу.
Следовательно, найден новый и изобретательский способ получения новых энантиомерно обогащенных или энантиомерно чистых 3-метокси-4-аминопиперидинов формулы (XIV-а) и в более общем виде промежуточных продуктов формулы (XIX-а) по методике, описанной далее более подробно.

В формулах (XIX-а), (XX-а), (XXI-а) и (XXII-а) радикал A представляет собой водород, -(CH2)n - NH2, -(CH2)n - NH-P, P1 или L, где n, P, P1 и L имеют указанные выше значения. Реакцией рецемической смеси 3-метокси-4-оксопиперидина формулы (XXII-а) с одним энантиомером хирального амина формулы (XXIII), у которого R8 представляет собой C1-C6-алкил или гидрокси-C1-C6-алкил, Ar представляет собой фенил, возможно замещенный галогеном, C1-C6-алкилом, C1-C6-алкилоксигруппой; или нафтил, возможно замещенный галогеном, C1-C6-алкилом, C1-C6-алкилоксигруппой, получают диастереомерную смесь промежуточного имина формулы (XXI-а). Эту реакцию можно проводить с применением известных методик образования иминов, например перемешиванием реагентов при температуре кипения в инертном в реакционных условиях растворителе, таком как ароматический углеводород, например метилбензол, с применением прибора Дина-Старка.
Имин формулы (XXI-а) можно выделить и, если необходимо, очистить, например, колоночной хроматографией, перегонкой или кристаллизацией. Затем имин можно гидрировать перемешиванием его в атмосфере водорода в пригодном растворителе, таком как спирт, например метанол или этанол; простой эфир, например тетрагидрофуран или 2,2'-оксибиспропан; сложный эфир, например этилацетат; ароматический углеводород, например метилбензол, в присутствии подходящих катализаторов, например палладия на угле, платины на угле, родия на угле и подобных катализаторов, получая диастереомерно обогащенный или диастереомерно чистый амин формулы (XX-а).
В альтернативном варианте промежуточный имин формулы (XXI-а) не выделяют. В этом случае рацемическую смесь 3-метокси-4-оксопиперидина формулы (XXII-а) обрабатывают одним из энантиомеров хирального амина формулы (XXIII) в условиях гидрирования, получая диастереомерно обогащенные или диастереомерно чистые промежуточные амины формулы (XX-а). Эту реакцию проводят в условиях, аналогичных описанным выше. Однако в этом случае реакцию предпочтительно проводят в смеси с кислотой, например уксусной кислотой, щавелевой кислотой, хлоруксусной кислотой, 2-гидрокси-1,2,3-пропантрикарбоновой кислотой и, в частности, (-)-[S-(R*, R*)]-2,3-дигидроксибутандионовой кислотой, особенно когда растворителем является спирт.
В аминах формулы (XXIII) R8 представляет собой гидроксиметил, метил или этил, особенно метил, и Ar предпочтительно представляет собой незамещенный фенил или нафтил, особенно фенил. Предпочтительными аминами формулы (XXIII) являются энантиомеры α -метилбензиламина, т.е. (-)-(S)- α -метилбензиламина или (+)-(R)- α -метилбензиламин.
Иногда в процессе реакции гидрирования может образоваться небольшое количество транс-3-метокси-4-аминосоединения, которое можно удалить кристаллизацией или хроматографией.
Предпочтительный путь получения диастереомерно обогащенного или чистого амина формулы (XX-а) предусматривает получение сначала имина формулы (XXI-а) с применением энантиомера α -метилбензиламина и последующее гидрирование имина формулы (XXI-а) перемешиванием его в метилбензоле в атмосфере водорода с применением родиевого катализатора.
Чтобы избежать нежелательное дальнейшее гидрирование некоторых функциональных групп у реагентов и продуктов реакции, полезно добавлять в реакционную среду яд для катализатора, например тиофен, хинолин-серу и т.д. Повышенные давления и/или температуры могут повышать скорость реакции.
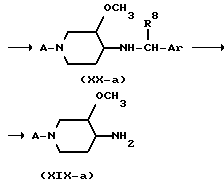
Полученный промежуточный продукт формулы (XX-а) имеет соотношение диастереомеров, которое очень сильно отличается от соотношения 1:1. Другими словами, промежуточное соединение формулы (XX-а) диастереомерно обогащенное или диастереомерно чистое. Соответствующие диастереомерные формы можно затем, если нужно, разделять с применением обычных физических методов, например хроматографией или фракционной кристаллизацией, возможно после образования соли. Полученные таким образом диастереомерно чистые амины формулы (XX-а) можно затем гидрогенолизом для удаления хиральной дополнительной группы Ar-CH(R8) превратить в энантиомерно чистые 3-метокси-4-аминопиперидины формулы (XIX-а).
Заслуживает внимания замечание, что конфигурация стереоцентра, имеющего метоксигруппу, определяется конфигурацией применяемого исходного энантиомерно чистого амина формулы (XVIII). Следовательно, любую конфигурацию этого стереоцентра можно получить отбором одного или другого энантиомера амина формулы (XXIII). Можно далее отметить, что выбор кислоты, применяемой в процессе гидрирования имина, можно также влиять в определенной степени на соотношении диастереомеров аминов формулы (XIX-a). Выбор катализатора также может оказывать влияние в определенной степени на количество образованного транс-4-амино-3-метоксисоединения.
Диастереомерно обогащенные или диастереомерно чистые промежуточные продукты формулы (XX-a) и энантиомерно обогащенные или энантиомерно чистые промежуточные продукты формулы (XX-a) и их фармацевтически пригодные соли с кислотами считаются новыми соединениями. Новыми соединениями считаются также энантиомерно обогащенные или энантиомерно чистые промежуточные продукты формул (II-a), (IV-a), (VII-a), (X-a), (XIII-a), (XIV-a) и их фармацевтически пригодные соли с кислотами. Эти промежуточные продукты можно получить, как описано выше, из энантиомерно обогащенных или энантиомерно чистых промежуточных продуктов формулы (XIV-a).
С применением такой методики и описанных выше энантиомерно обогащенных или энантиомерно чистых промежуточных продуктов предложен новый и изобретательский способ получения энантиомерно обогащенных или энантиомерно чистых соединений формулы (I-a), особенно левовращающих энантиомеров соединений формулы (I-a).
Очевидно, что диастереомерные рацематы цис- и трансизомеров соединений формул (I), (I-a) или любых других промежуточных продуктов можно также расщепить на их оптические изомеры, цис-(+)-, цис-(-)-, транс-(+)- и транс-(-)-изомеры при помощи известных методик. Диастереомеры можно разделить физическими способами разделения, например селективной кристаллизацией и хроматографией, например противоточным распределением. Энантиомеры можно разделить друг от друга селективной кристаллизацией их диастереомерных солей с энантиомерно чистыми кислотами или их энантиомерно чистыми производными.
Соединения формулы (I) и их фармацевтически пригодные соли и стереоизомеры являются антагонистами 5-HT3-рецепторов, что доказывается тем фактом, что они активны в проявлении, например, антагонизма химиорефлексу von Bejold-Jarish, индуцированному серотонином (5-HT) у крыс (Pharmacology and Toxicology, 70, 11, 17-22 (1992)). Этот тест описан ниже как пример 10.
Соединения формулы (I), особенно соединения формулы (I-a), активны в течение долгого периода времени. Кроме того, соединения формулы (I), особенно соединения формулы (I-a), проявляют высокую степень безвредности для сердечно-сосудистой системы.
Ввиду проявления 5-HT3-антагонизма соединения изобретения можно выпускать в различных фармацевтических формах для целей введения. Для получения этих фармацевтических препаратов эффективное количество данного соединения в виде свободного основания или соли с кислотой, применяемого в качестве активного компонента, тесно смешивают с фармацевтически пригодным носителем. В зависимости от формы препарата, требуемого для введения, применяют различные формы носителя. Эти фармацевтические препараты желательно выпускать в виде унифицированных доз, пригодных предпочтительно для введения перорально, ректально или перентеральной инъекцией. Например, для получения препаратов в пероральной дозированной форме в качестве обычной фармацевтической среды можно применять, например, воду, гликоли, масла, спирты и подобные вещества в случае пероральных жидких препаратов, например суспензий, сиропов, эликсиров и растворов, или твердые носители, например крахмалы, сахара, каолин, смазывающие вещества, связующие, дезинтегрирующие средства и подобные вещества в случае порошков, пилюль, капсул и таблеток. По причине легкости введения таблетки и капсулы являются наиболее благоприятной пероральной дозированной формой. Ясно, что в этом случае применяют твердые фармацевтические носители. Для парентеральных препаратов носитель обычно представляет собой стерильную воду, по меньшей мере в большой части, хотя можно включать другие компоненты, например компоненты для повышения растворимости. Можно получить, например, инъецируемые растворы, в которых носитель представляет собой соляной раствор, раствор глюкозы или смесь соляного раствора и раствора глюкозы. Можно также получить инъецируемые суспензии, в этом случае можно применять соответствующие жидкие носители, суспендирующие средства и подобные средства. В препаратах, пригодных для чрескожного введения, носитель возможно содержит средство, усиливающее проникновение активного компонента, и/или подходящее смачивающее средство, возможно в комбинации с подходящими добавками любой природы в небольших количествах. Но эти добавки не должны оказывать значительное вредное действие на кожу. Эти добавки могут облегчать введение активного компонента в кожу и/или могут быть полезны для приготовления требуемых препаратов. Эти препараты можно вводить различными методами, например в виде чрескожных повязок, нанесением пятен на кожу или в виде мазей. Соли соединений формулы (I) с кислотами благодаря повышенной растворимости по сравнению с соответствующим основанием явно более пригодны для приготовления водных препаратов.
Особенно предпочтительно готовить указанные выше препараты в форме дозы на один прием для облегчения введения их и постоянства дозировки. Формой дозы на один прием в описании изобретения называют физически дискретные единицы, пригодные в качестве унифицированных доз, причем каждая единица содержит заданное количество активного компонента, рассчитанное для индуцирования желаемого терапевтического действия, в сочетании с требуемым фармацевтическим носителем. Примерами таких дозированных форм на один прием являются таблетки (включая таблетки с метками и оболочками), капсулы, пилюли, пакеты с порошком, облатки инъецируемые растворы или суспензии, препараты в количестве чайных или столовых ложек и т.д., и такие формы в виде кратных этой дозе частей.
Благодаря 5-HT3-антагонистической активности соединения формулы (I) и особенно новые соединения формулы (I-a) пригодны для лечения 5-HT3-медиированных нарушений, например тревожных состояний, психоза, депрессивного состояния (Arzneim, Forsch. , 42(1), 239-246 (1992)), шизофрении, нарушений познавательной способности, например недостаточной памяти (Arzneim, Forsch. , 42(1) 246-249 (1992)), злоупотребления лекарственными средствами, мигрени, рвоты, например индуцированной приемом цитотоксичных лекарственных средств и радиацией (Drugs 42(4), 551-568 (1991)), синдрома раздраженной толстой кишки, особенно диарреядоминирующего синдрома раздраженной толстой кишки, и аналогичных нарушений. Следовательно, в изобретении предлагается способ лечения теплокровных животных, страдающих 5-HT3-медиированными заболеваниями, например лечения тревожных состояний, психоза, депрессивных состояний, шизофрении, нарушений познавательной памяти, злоупотребления лекарственными средствами, мигрени, рвоты, например, индуцированной цитотоксичными лекарственными средствами или радиацией, синдрома раздраженной толстой кишки, особенно диарреядоминирующего синдрома раздраженной толстой кишки, и аналогичных нарушений. Этот способ предусматривает системное введение теплокровным животным эффективного 5-HT3-антагонистического количества соединения формулы (I), его фармацевтически пригодной соли с кислотой или стереоизомера.
Соединения формулы (I) пригодны для приготовления лекарственного препарата, предназначенного для лечения 5-HT3-медиированных нарушений. Новые соединения формулы (I-a) пригодны в качестве лекарственного средства.
В общем предполагается, что эффективное количество должно быть от около 0,001 мг/кг до около 50 мг/кг массы тела, предпочтительно от около 0,02 мг/кг до около 5 мг/кг массы тела. Способ лечения может включать также введение активного компонента по схеме, предусматривающей прием его 2 - 4 раза в день.
Экспериментальная часть
A. Получение промежуточных продуктов
Пример 1
a) 3,4,4-Триметокси-1-(фенилметил)пиперидин (0,676 моль) очищали колоночной хроматографией на силикагеле (элюент: CH2Cl2/CH3OH, 98:2). Чистые фракции собирали и растворитель выпаривали. Этот остаток (смесь энантиомеров) разделяли на энантиомеры колоночной хроматографией на колонке с Chiracell OD (элюент: гексаны/2-пропанол, 98,5:1,5). Отбирали фракцию, соответствующую первому хроматографическому пику, и из нее выпаривали растворитель. Образец очищали перегонкой (т.кип. 120oC при 0,5 мм рт.ст.), получая 56 г (-)-3,4,4-триметокси-1-(фенилметил)пиперидина с [α]
Отбирали фракцию, соответствующую второму хроматографическому пику, и из нее выпаривали растворитель. Образец очищали перегонкой (т.кип. 120oC при 0,5 мм рт.ст.), получая 64 г (+)-3,4,4-трирметокси-1-(фенилметил)пиперидин с [α]
b) Смесь промежуточного продукта 1 (0,21 моль) в метаноле (600 мл) гидрировали при 50o с применением в качестве катализатора 10% (3 г) палладия на угле. После поглощения H2 (1 экв) катализатор отделяли фильтрованием. В фильтрат добавляли оксид кальция (0,63 моль). Реакционную смесь перемешивали при комнатной температуре. По каплям добавляли этилхлорформиат (0,63 моль) и реакционную смесь перемешивали 2 ч при 50oC, затем при комнатной температуре в течение ночи. Растворитель удаляли выпариванием. К остатку добавляли метилбензол. Суспензию фильтровали и фильтрат выпаривали. Остаток очищали перегонкой, получая 32,6 г (63%) (-)-этил-3,4,4-триметокси-1-пиперидинкарбоксилата с [α]
c) Смесь промежуточного продукта 3 (0,132 моль), 4-метилбензолсульфокислоты (0,6 г) в 2-пропаноле (180 мл) и воде (30 мл) перемешивали и кипятили с обратным холодильником 18 ч. Реакционную смесь охлаждали и добавляли N, N-диэтилэтанамин (0,6 мл). Растворитель удаляли выпариванием (температуру поддерживали ниже 40oC). Остаток растворяли в CH2Cl2. Остаток промывали два раза насыщенным раствором NaCl. Органический слой отделяли, сушили (MgSO4), фильтровали и выпаривали для удаления растворителя. Остаток очищали перегонкой, получая 19,2 г (-)-этил-3-метокси-4-оксо-1-пиперидинкарбоксилата (72,3%) с [α]
d) Смесь промежуточного продукта 4 (0,095 моль) и бензолметанамина (0,11 моль) в метаноле (200 мл) гидрировали в атмосферных условиях с применением в качестве катализатора 10% (2 г) палладия на активированном угле в присутствии 4%-ного раствора тиофена в 2',2-оксибиспропане (2 мл). После поглощения водорода катализатор отделяли фильтрованием и фильтрат выпаривали. Остаток растворяли в метаноле (250 мл) и полученную смесь гидрировали при 50oC с применением в качестве катализатора 10% (2 г) палладия на активированном угле. После поглощения водорода (1 экв) катализатор отделяли фильтрованием и фильтрат выпаривали. Остаток очищали перегонкой (т. кип. 85oC при 0,1 мм рт. ст.), получая 13,4 г (70%) этил-(-)-цис-4-амино-3-метокси-1-пиперидинкарбоксилата с [α]
Аналогичным образом, но из промежуточного продукта 2 получали также (+)-этил-цис-4-амино-3-метокси-1-пиперидинкарбоксилат с [α]
Пример 2
a) Смесь этил-3-метокси-4-оксо-1-пиперидинкарбоксилата (0,5 моль), (-)-(S)- α -метилбензолметанамина (0,53 моль), моногидрата 4-метилбензолсульфокислоты (1,25 г) и метилбензола (625 мл) перемешивали и кипятили с обратным холодильником с применением прибора Дина-Старка в течение 3 ч. Реакционную смесь выпаривали и перегоняли, получая 121 г (79,5%) (-)-этил-[цис(S)]-3-метокси-4-[(1-фенилэтил)имино]-1- пиперидинкарбоксилата (промежуточный продукт 7).
b) Смесь промежуточного продукта 7 (0,4 моль) и метилбензола (750 мл) гидрировали при комнатной температуре и атмосферном давлении с применением в качестве катализатора родия на угле (5 г). После поглощения водорода (1 экв) катализатор отделяли фильтрованием и фильтрат выпаривали. Остаток растворяли в 4-метил-2-пентаноне и превращали в соль с 4-метилбензолсульфокислотой (1 : 1), применяя моногидрат 4-метилбензолсульфокислоты (1 экв). Соль фильтровали и сушили. Эту фракцию перекристаллизовывали два раза из смеси 2,2'-оксибиспропан/метанол (250 мл/180 мл). Осажденный продукт фильтровали и сушили, получая 61,7 г (32,5%) 4-метилбензолсульфоната (-)-этил-[цис(S)]-3-метокси-4-[(1-фенилэтил)амино] -1-пиперидинкарбоксилата (1 : 1) с [α]
Аналогичным образом, но применяя (+)-(R)- α -метилбензолметанамин, получали 4-метилбензолсульфонат (+)-этил-[цис(R)] -3-метокси-4-[(1-фенилэтил)амино] -1- пиперидинкарбоксилата (1 : 1) с [α]
Пример 3
a) Смесь этил-3-метокси-4-оксо-1-пиперидинкарбоксилата (0,2 моль), (-)-(S)- α -метилбензолметанамина (0,4 моль) и (-)-[S-(R*,R*)]-2,3-дигидроксибутандионовой кислоты (0,2 моль) в метаноле (500 мл) гидрировали при комнатной температуре и атмосферном давлении с применением в качестве катализатора 10% (2 г) палладия на активированном угле в присутствии 4%-ного раствора тиофена в 2,2'-оксибиспропане (2 мл). После поглощения H2 (1 экв) катализатор отделяли фильтрованием и фильтрат выпаривали. Остаток распределяли между метилбензолом и H2O/NH4OH. Органический слой отделяли, сушили (MgSO4), фильтровали и выпаривали для удаления растворителя. Остаток растворяли в 4-метил-2-пентаноне и превращали в соль с 4-метилбензолсульфокислотой (1 : 1), применяя моногидрат 4-метилбензолсульфокислоты (1 экв). Соль фильтровали и сушили. Эту фракцию перекристаллизовывали из смеси 2,2'-оксибиспропан/-CH3OH (500 мл/100 мл). Смесь перемешивали 24 ч. Осадок отделяли фильтрованием и сушили (вакуум, 50oC), получая 32 г 4-метилбензолсульфоната (-)-этил-[цис(S)]-3-метокси-4-[(1-фенилэтил)амино]-1- пиперидинкарбоксилата (1 : 1) с [α]
Аналогичным образом, но с применением (+)-(R)- α -метилбензолметанамина также получали 4-метилбензолсульфонат (+)-этил-[цис(R)]-3-метокси-4-[(1-фенилэтил)амино] -1- пиперидинкарбоксилата (1 : 1) (промежуточный продукт 10).
b) Промежуточный продукт 8 (0,067 моль) превращали в свободное основание при помощи водного аммиака. Эту смесь экстрагировали метилбензолом. Органический слой отделяли, сушили (MgSO4), фильтровали и выпаривали для удаления растворителя. Оставшееся свободное основание растворяли в метаноле (250 мл) и гидрировали при комнатной температуре и атмосферном давлении с применением в качестве катализатора 10% (2 г) палладия на активированном угле. После поглощения H2 (1 экв) катализатор отделяли фильтрованием и фильтрат выпаривали. Остаток очищали перегонкой (т. кип. 85oC при 0,1 мм рт. ст. ), получая 9,9 г (79,2%) этил-(-)-цис-4-амино-3-метокси-1-пиперидинкарбоксилата (промежуточный продукт 5).
Аналогичным образом, но из промежуточного продукта 9 также получали (+)-этил-цис-4-амино-3-метокси-1-пиперидинкарбоксилат (промежуточный продукт 6).
Пример 4
Смесь 53,3 г этил-3-метокси-4-оксо-1-пиперидинкарбоксилата (описан в EP-патенте N 76350), 33 г (-)-(R)- α -аминобензолэтанола и 700 мл этанола кипятили с обратным холодильником в течение ночи. После охлаждения реакционную смесь выпаривали и остаток перегоняли, получая 59,1 г (92%) этил-(R)-4-[(2-гидрокси-1-фенилэтил)имино] - 3-метокси-1-пиперидинкарбоксилата с т. кип. 180oC (давление 3,75 • 10-4 Pa) (промежуточный продукт 6).
b) Раствор 59,1 г промежуточного продукта 10 в 500 мл этанола гидрировали при нормальном давлении и при комнатой температуре с применением в качестве катализатора 2 г платины на угле. После поглощения рассчитанного количества водорода катализатор отделяли фильтрованием и фильтрат выпаривали. Остаток очищали на NH2-силикагеле (элюент: смесь хлористый метилен/циклогексан/метанол, 60 : 40 : 0,5). Чистые фракции собирали, из них выпаривали элюент, получая 18 г (30%) этил-(-)-[4(R)цис]-4-[(2-гидрокси-1-фенилэтил)амино] -3-метокси-1- пиперидинкарбоксилата в виде остатка. [α]
c) Раствор 18 г промежуточного продукта 11 в 250 мл метанола гидрировали при нормальном давлении и комнатной температуре с применением в качестве катализатора 2 г палладия на угле (10%). После поглощения рассчитанного количества водорода катализатор отделяли фильтрованием и фильтрат выпаривали. Остаток перегоняли, получая 6,2 г (55%) этил-(-)-цис-4-амино-3-метокси-1-пиперидинкарбоксилата (промежуточный продукт 5).
Пример 5
a) 4-Амино-5-хлор-2,3-дигидро-2,2-диметил-7-бензофуранкарбоновую кислоту (описана в EP N 0389037) (0,05 моль) растворяли в смеси N,N-диэтилэтанамина (7 мл) и трихлорметана (250 мл). Этилкарбонохлоридат (0,05 моль) добавляли по каплям при температуре ниже 10oC. Реакционную смесь перемешивали 30 мин при температуре ниже 10oC. Смесь добавляли в раствор промежуточного продукт 5 (0,047 моль) в трихлорметане (250 мл), перемешивали при 10oC. Реакционную смесь перемешивали 30 мин при комнатной температуре, затем промывали водой, 5%-ным NaOH и снова водой. Органический слой отделяли, сушили (MgSO4), фильтровали и растворитель выпаривали. Остаток очищали колоночной хроматографией на силикагеле (элюент: CH2Cl2/CH3OH, 98:2). Чистые фракции собирали и растворитель выпаривали, получая 19 г (94%) (+)-этил-цис-4-[(4-амино- 5-хлор-2,3-дигидро-2,2-диметил-7-бензофуранил)карбониламино] - 3-метокси-1-пиперидинкарбоксилата (промежуточный продукт 12).
b) Смесь промежуточного продукта 12 (0,045 моль) и едкого кали (0,45 моль) в 2-пропаноле (300 мл) перемешивали и кипятили с обратным холодильником в течение 12 ч. Реакционную смесь охлаждали и растворитель выпаривали. К остатку добавляли воду (100 мл). Растворитель выпаривали. Остаток распределяли между дихлорметаном и водой. Органический слой отделяли, промывали водой, сушили (MgSO4), фильтровали и растворитель выпаривали. Остаток очищали колончатой хроматографией на силикагеле (элюент: CH2Cl2/(CH3OH/NH3), 97:3). Чистые фракции собирали и растворитель выпаривали. Остаток сушили (вакуум 50oC), получая 12,5 г (+)-цис-4-амино-5-хлор-2,3-дигидро-N-(3-метокси-4-пиперидинил)-2,2- диметил-7-бензофуранкарбоксамида (77,2%) с [α]
Пример 6
а) Смесь промежуточного продукта 13 (0,017 моль), этил-(2-хлорэтил)карбамата (0,02 моль) и N,N-диэтилэтанамина (0,022 моль) в N,N-диметилформамиде (150 мл) перемешивали 72 ч при 70oC. Реакционную смесь охлаждали и растворитель выпаривали. Остаток распределяли между дихлорметаном и водой. Органический слой отделяли, сушили (MgSO4), фильтровали и выпаривали для удаления растворителя. Остаток очищали пропусканием через силикагель на стеклянном фильтре (элюент: CH2Cl2/CH3OH, 97:3). Чистые фракции собирали и выпаривали растворитель, получая 5 г (+)-этил-цис-[2-[4-[[(4-амино-5-хлор-2,3-дигидро-2,2-диметил -7-бензофуранил)карбонил]амино]-3-метокси-1-пиперидинил] этил] карбамата (63%) с [α]
Пример 7
a) Смесь промежуточного продукта 13 (0,023 моль) и 2-пропенонитрила (0,028 моль в 2-пропаноле (150 мл) перемешивали и кипятили с обратным холодильником 16 ч. Реакционную смесь охлаждали и растворитель выпаривали, получая 8 г (85,5%) (-)-цис-4-амино-5-хлор- N -[1-(2-цианоэтил)-3-метокси-4-пиперидинил] -2,3-дигидро-2, 2-диметил-7-бензофуранкарбоксамида с [α]
b) Смесь промежуточного продукта 16 (0,02 моль) в метаноле (250 мл) и тетрагидрофуране (100 мл) гидрировали при атмосферных условиях с применением в качестве катализатора никеля Ренея (3 г). После поглощения водорода (2 экв) катализатор отделяли фильтрованием и фильтрат выпаривали, получая 7 г (85,2%) (-)-цис-4-амино- N -[1-(3-аминопропил)-3-метокси-4-пиперидинил]-5-хлор -2,3-дигидро-2,2- диметил-7-бензофуранкарбоксамида (промежуточный продукт 17).
Пример 8
a) Промежуточный продукт 17 (0,769 моль) растворяли в 1-бутаноле (2310 мл) (требуется нагревание до 50oC). При 30oC добавляли (гетерогенно) карбонат калия (1,538 моль). Добавляли затем 2-хлор-4-метоксипиримидин (0,960 моль) и реакционную смесь нагревали до температуры кипения (104oC). Реакционную смесь перемешивали и кипятили с обратным холодильником 11 ч. Смесь оставляли для охлаждения до 20oC и затем в нее добавляли воду (769 мл) и смесь перемешивали 15 мин. Слои разделяли. Органический слой выпаривали (60oC при 1,66 мм Hg),получая 458,9 г (92,1%) (±)-цис-4-амино-5-хлор-2,3-дигидро- N -[3-метокси-1-[3-[(4-метокси-2-пиримидинил)амино]пропил]-4- пиперидинил]-2,2-диметил-7-бензофуранкарбоксамида (промежуточный продукт 18).
b) Соляную кислоту в 2-пропаноле (434 мл) добавляли по каплям в течение 15 мин в раствор промежуточного продукта 18 (0,769 моль) в 4-метил-2-пентаноне (3845 мл), смесь перемешивали при 15 - 20oC (если было нужно, охлаждали ледяной баней). Реакционную смесь перемешивали 1 ч при 15oC. Осадок отделяли фильтрованием, промывали 4-метил-2-пентаноном (769 мл) и сушили (50oC, вакуум), получая 425,9 г (93,6%) дигидрохлорида (±)-цис-4-амино-5- хлор-2,3-дигидро-N-[3-метокси-1-[3-[(4-метокси-2- пиримидинил)амино]пропил]-4-пиперидинил]-2,2 -диметил-7-бензофуранкарбоксамида (промежуточный продукт 19).
B. Получение конечных соединений
Пример 9. Смесь промежуточного продукта 17 (0,017 моль) и 2-метилтио-4-пиримидинона (0,022 моль) в ацетонитриле (300 мл) перемешивали и кипятили с обратным холодильником в течение 16 ч. Добавляли дополнительное количество 2-метилтио-4-пиримидинона (2 г) и реакционную смесь перемешивали и кипятили с обратным холодильником 16 ч. Реакционную смесь охлаждали, растворитель выпаривали. Остаток очищали пропусканием через силикагель на стеклянном фильтре (элюент: CH2Cl2/CH3OH/(CH3OH/NH3), 90:9:1). Чистые фракции собирали и растворитель выпаривали. Остаток растирали в 2,2'-оксибиспропане. Твердую часть отделяли фильтрованием и сушили (комнатная температура, вакуум), получая 2,6 г (29,7%) (-)-цис-4- амино-5-хлор-2,3-дигидро- N -[1-[3-[(3,4-дигидро-4-оксо-2-пиримидинил)амино] пропил] -3- метокси-4-пиперидинил] -2,2-диметил-7-бензофуранкарбоксамида с т.пл. 164,3oC и [α]
Аналогичным образом были получены соединения, представленные в табл.1.
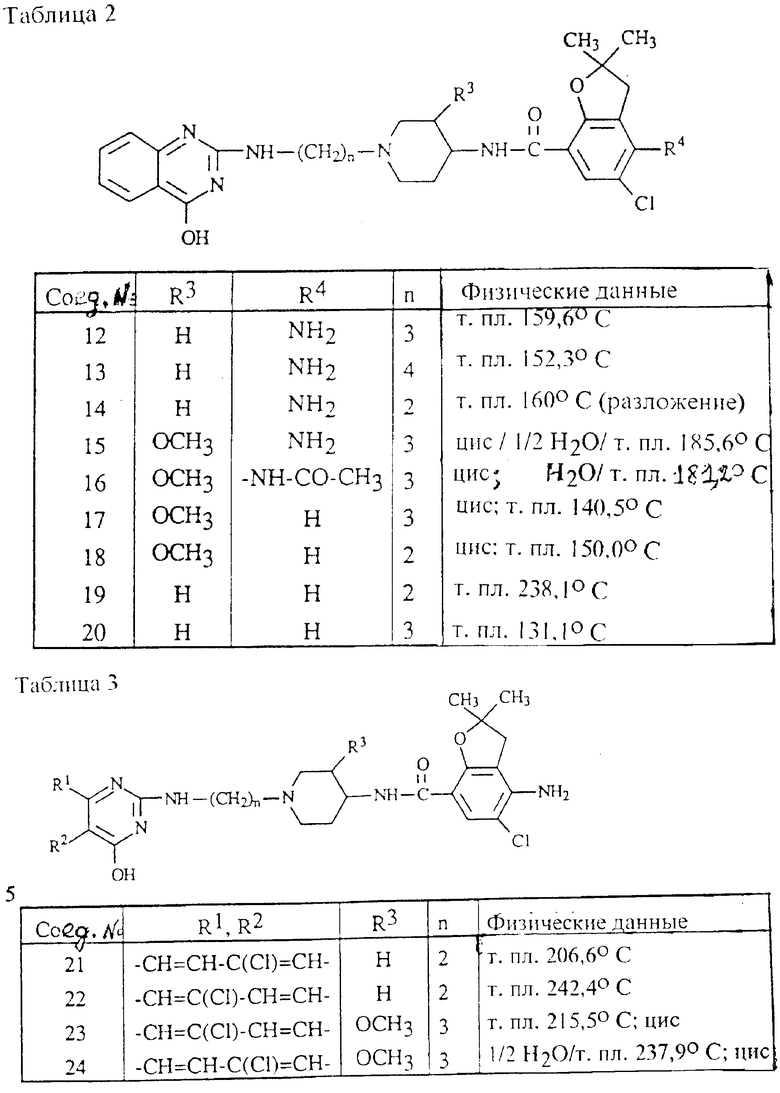
Пример 10. Смесь 4,15 г 2-хлор-4-гидроксихиназолина, 4,57 г 4-амино-N-[1-(3-аминопропил)-4-пиперидинил] -5-хлор-2,3-дигидро- 2,2-диметил-7-бензофуранкарбоксамида (описан в EP N 0445862) и 0,80 г оксида кальция перемешивали в течение 1 ч при 140oC. Реакционную смесь растворяли в смеси дихлорметана и метанола. Весь раствор промывали водой, сушили, фильтровали и выпаривали. Остаток очищали два раза колоночной хроматографией (силикагель; CH2Cl2/CH3OH(NH3), 90:10; CH2Cl2/CH3OH(NH3), 88:12). Элюент целевой фракции выпаривали и остаток кипятили в 2,2'-оксибиспропане. Продукт фильтровали и сушили, получая 3,2 г (50,8%) 4-амино-5-хлор-2,3-дигидро-N-[1-[3-[4-гидрокси-2-хиназолинил)амино] пропил] - 4-пиперидинил] -2,2-диметил-7-бензофуранкарбоксамида с т.пл. 159,6oC (соединение 12).
Аналогичным образом получали соединения, представленные в табл. 2.
Пример 11
Смесь 2,6 г 2,6-дихлор-4-хинаолинола (описан в J. Med. Chem., 1968, p. 130), 3,7 г 4-амино-N-[1-(2-аминоэтил)-4-пиперидинил]-5-хлор- 2,3-дигидро-2,2-диметил-7-бензофуранкаробксамида (описан в EP N 0445862), 0,8 г оксида кальция и 5,64 г N, N-диметилацетамида перемешивали 3 ч при 140oC. После охлаждения реакционную смесь выпаривали и остаток растворяли в смеси дихлорметана и метанола. Целый продукт промывали водой. Частично осажденный продукт отделяли фильтрованием (первая фракция). Органический слой декантировали, сушили, фильтровали и выпаривали (вторая фракция). Объединенную фракцию очищали два раза колоночной хроматографией (силикагель; CH2Cl2/CH3OH(NH3), 95 : 5; CH2Cl2/CH3OH, 92 : 8). Элюент целевой фракции выпаривали и остаток кристаллизовали из ацетонитрила. Продукт фильтровали при 0oC и сушили в вакууме при 60oC, получая 1 г (18,3%) 4-амино-5-хлор-N-[1-[2-[(6-хлор-4-гидрокси-2- хиназолинил)амино] этил] -4-пиперидинил]-2,3-дигидро-2,2-диметил-7- бензофуранкарбоксамида с т.пл. 206,6oC (соединение 21).
Аналогичным образом были получены соединения, представленные в табл.3.
Пример 12. Воду (2880 мл) добавляли в промежуточный продукт 19 (0,72 моль) для полного его растворения, затем по каплям добавляли соляную кислоту (193 мл). Реакционную смесь нагревали до температуры кипения (95oC) и затем перемешивали и кипятили с обратным холодильником 24 ч. При кипячении добавляли дополнительно соляную кислоту (128,6 мл). Реакционную смесь перемешивали и кипятили с обратным холодильником 2,5 ч. Нагревание прекращали и добавляли дихлорметан (360 мл). Слои разделяли и в водную фазу добавляли дихлорметан (1080 мл). Двухфазную смесь подщелачивали при 20 - 25oC добавлением гидроксида аммония (433 мл) до pH выше 10 в течение 30 мин, причем требовалось наружное охлаждение. Смесь сначала представляла собой гомогенную систему, затем при pH 6 - 7 образовался осадок, который растворялся при более высоких значениях pH. Слои разделяли. Водный слой экстрагировали дихлорметаном (360 мл). Органические экстракты объединяли, сушили и выпаривали (40oC, вакуум). Остаток сушили (40oC, вакуум), получая 321,2 г (88,3%) (-)-цис-4-амино-5-хлор N-[1-[3-[(3,4-дигидро-4-оксо-2- пиримидинил] амино] пропил] -3-метокси-4-пиперидинил] -2,3-дигидро-2,2- диметил-7-бензофуранкарбоксамида (соединение 1).
C. Фармакологический пример
Пример 13. Определение по методике von Bezold-Jorish
Самопроизвольно гипертензивные самцы крыс (± 6 мес) анестезировали ингаляцией эфиром и бедренную вену и артерию рассекали и канюлировали полиэтиленовым катетером. Для индуцирования местной анестезии в рану вокруг канюлей вводили лидокаин (20%).
Животных помещали в клетки Bollman и артериальный катетер соединяли с датчиком тензиометрического измерения кровяного давления и анализировали систолическое давление. Когда животные полностью просыпались, им вводили инъекцией контрольный серотонин (0,04 мг/кг) через катетер бедренной вены. Ответная реакция систолического кровяного давления на внутривенную инъекцию серотонина обычно развивается в три фазы: 1) короткое и острое понижение (рефлекс von Bezold-Jorish ), 2) повышение, и 3) долго длящееся повышение систолического кровяного давления. Ингибирование первого острого понижения кровяного давления (рефлекс von Bezold-Jorish ) рассматривается как критерий 5-HT3-антагонизма. Через некоторое время после контрольной инъекции серотонина внутрибрюшинной инъекцией вводили испытуемое соединение. Через 30 мин серотонин инъецировали снова внутривенно и регистрировали наличие или отсутствие первого короткого и острого понижения давления. Ту же процедуру повторяли через 60 мин. Соединения испытывали при различных дозах.
Наименьшую активную дозу (LAD), которая приведена в табл. 4, можно определить как дозу (в мг/кг массы тела), при которой по меньшей мере половина испытуемых животных проявляет ингибирование рефлекса von Bezold-Jorish.
D. Примеры препаратов
Примерами типичных фармацевтических препаратов в дозах на один прием, пригодных для системного или местного введения теплокровным животным в соответствии с изобретением служат следующие составы.
Термин "активный компонент" (А.I.), применяемый во всех этих примерах, относится к соединению формулы (I), его фармацевтически пригодной соли с кислотой или стереохимическому изомеру.
Пример 14. Пероральные растворы
9 г метил-4-гидроксибензоата и 1 г прпил-4-гидроксибензоата растворят в 4 л кипящей очищенной волы. В 3 л этого раствора растворяют сначала 10 г 2,3-дигидроксибутандионовой кислоты и затем 20 г А.I. Последний раствор объединяют с остальной частью первого раствора и в объединенную часть добавляют 12 л 1,2,3-пропантриола и 3 л 70%-ного раствора сорбита. В 0,5 л воды растворяют 40 г натриевой соли сахарина и добавляют 2 мл малиновой и 2 мл крыжовниковой эссенции. Последний раствор объединяют с первым, добавляют необходимое количество воды для достижения общего объема 20 л, получая пероральный раствор, содержащий 5 мг A.I. в одной чайной ложке (5 мл). Полученный раствор помещают в подходящие емкости.
Пример 15. Капсулы
Смесь 20 г А.I., 6 г лаурилсульфата натрия, 56 г крахмала, 56 г лактозы, 0,8 г коллоидного диоксида кремния и 1,2 г стеарината магния энергично перемешивают. Полученной смесью затем наполняют 1000 подходящих отвержденных желатиновых капсул, каждая полученная капсула содержит 20 мг А.I.
Пример 16. Таблетки с оболочкой
Приготовление сердцевины таблеток. Смесь 100 г A.I., 570 г лактозы и 200 г крахмала хорошо перемешивают и затем увлажняют раствором 5 г додецилсульфата натрия и 10 г поливинилпирролидона в около 200 мл воды. Влажную порошкообразную смесь просеивают, сушат и просеивают снова. Затем добавляют 100 г микрокристаллической целлюлозы и 15 г гидрогенизированного растительного масла. Всю массу хорошо перемешивают и прессуют в таблетки, получая 10 000 таблеток, каждая из которых содержит 10 мг активного компонента.
Покрытие оболочкой. В раствор 10 г метилцеллюлозы в 75 мг денатурированного этилового спирта добавляют раствор 5 г этилцеллюлозы в 150 мл дихлорметана. Затем добавляют 75 мл дихлорметана и 2,5 мл 1,2,3-пропантриола. Расплавляют 10 г полиэтиленгликоля и добавляют в 75 мл дихлорметана. Последний раствор добавляют в первый и затем добавляют 2,5 г октадеканоата магния, 5 г поливинилпирролидона и 30 мл концентрированной суспензии красящего средства и всю массу гомогенизируют. Сердцевины таблеток покрывают полученной таким образом смесью в устройстве для образования оболочек.
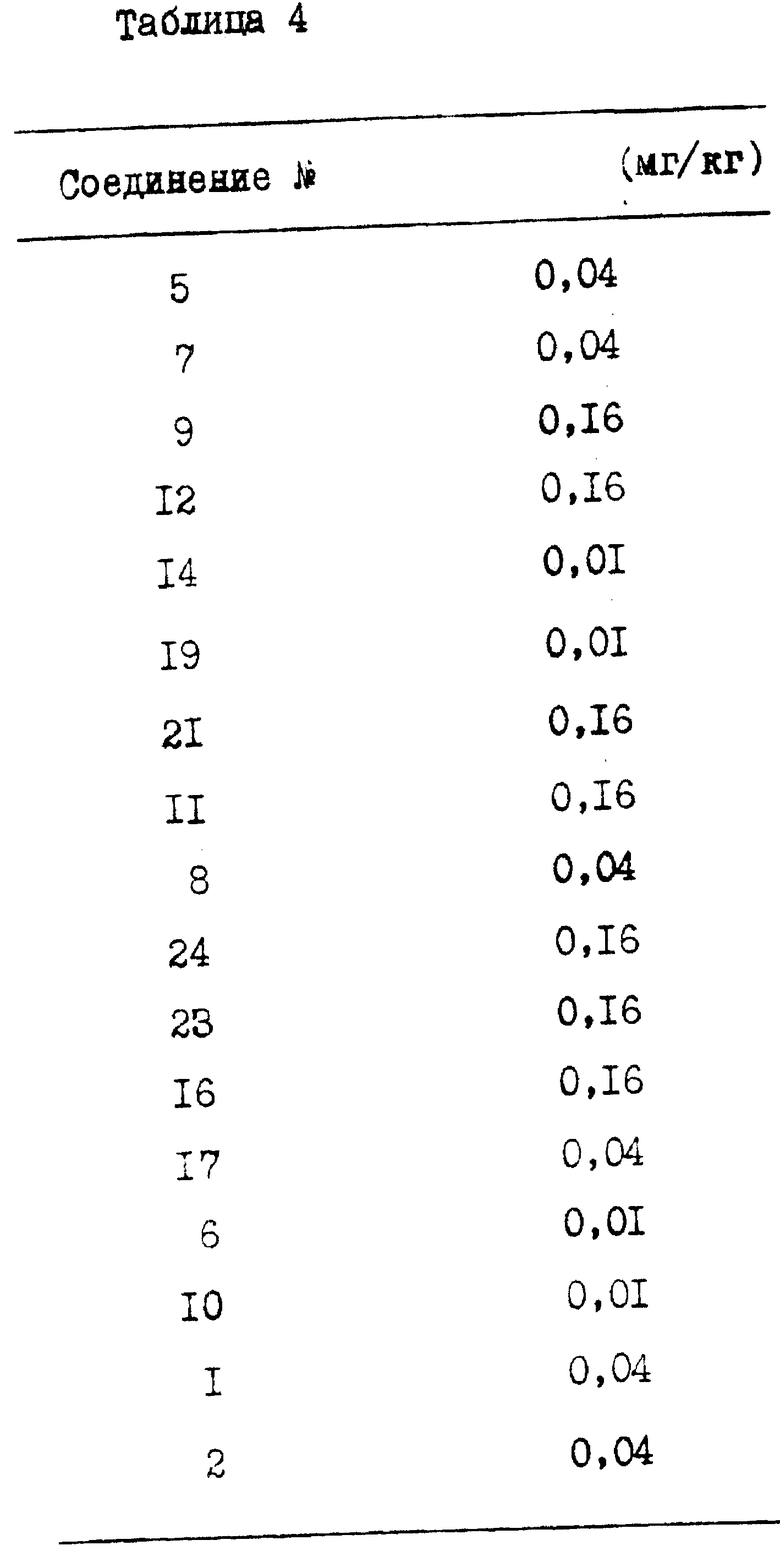
\ \ 1 Изобретение относится к области органической химии и медицины. Предложены новые соединения общей формулы (I), где R1 и R2 представляют собой водород или R1 и R2 вместе образуют двухвалентный радикал (a) -CH = CH-CH= С-; (b) -CH = C(Cl)-CH = CH- или (c) -CH = CH-C(Cl) = CH-; n - число 2, 3 или 4; R3 - водород или метоксигруппа; m - число 1 или 2; R4 - водород, аминогруппа или C1-C3-алкилкарбониламиногруппа; R5 - водород или галоген, их стереохимические изомеры, и промежуточные продукты синтеза формулы XIXa, и левовращающийся изомер формулы Ia (R3 = OCH3). Предложены способы получения новых соединений. Предложены фармацевтическая композиция и средство на основе соединений общей формулы I для лечения 5-HT3-медиированных нарушений, 6 с. и 4 з.п. ф-лы, 4 табл.
 \
\
или его фармацевтически пригодная аддитивная соль кислоты или стереохимический изомер этого соединения,
где R1 и R2 - атом водорода или R1 и R2 вместе образуют двухвалентный радикал формулы
-CH=CH-CH=CH-, (a),
-CH=C(Cl)-CH=CH-, (b),
-CH=CH-C(Cl)=CH-, (c);
n = 2, 3 или 4;
R3 - атом водорода или метоксигруппа;
m = 1 или 2;
R4 - атом водорода, аминогруппа или C1 - C3-алкилкарбониламиногруппа;
R5 - атом водорода или галогена.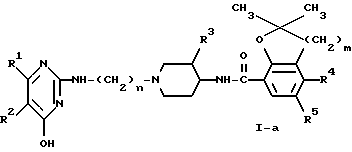
или его фармацевтически пригодная аддитивная соль кислоты,
где R1 и R2 - атом водорода или R1 и R2 вместе образуют двухвалентный радикал формулы
-CH=CH-CH=CH-, (a),
-CH=C(Cl)-CH=CH-, (b), или
-CH=CH-C(Cl)=CH-, (c);
n - целое число, равное 2, 3 или 4;
R3 представляет собой метоксигруппу и имеет цис-конфигурацию;
m - целое число, равное 1 или 2;
R4 - атом водорода, аминогруппа или C1 - C3-алкилкарбониламиногруппа;
R5 - атом водорода или галогена.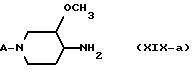
или его фармацевтически пригодная аддитивная соль кислоты,
где A - P1, где P1 - C1 - C4-алкилкарбонил, C1 - C4-алкилоксикарбонил, тригалогенметилкарбонил или фенилметил.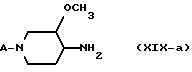
где A - P1, где P1 - C1 - C4-алкилкарбонил, C1 - C4-алкилоксикарбонил, тригалогенметилкарбонил или фенилметил,
отличающийся тем, что осуществляют реакцию рецемического 3-метокси-4-оксо-пиперидина формулы XXII-а, где A имеет указанные значения, с одним энантиомером хирального амина формулы XXIII, где R8 - C1 - C6-алкил или гидрокси-C1 - C6-алкил и Ar - фенил, возможно замещенный галогеном, C1 - C6-алкилом или C1 - C6-алкилоксигруппой, или нафтил, возможно замещенный галогеном, C1 - C6-алкилом или C1 - C6-алкилоксигруппой, с образованием промежуточного продукта формулы XXI-а, где A, Ar и R8 имеют указанные значения, которое гидрируют в присутствии катализатора, например палладия на угле, платины на угле или родия на угле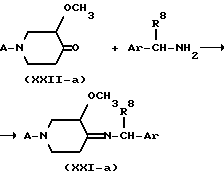
с образованием диастереомерно обогащенного или диастереомерно чистого промежуточного продукта формулы XX-а, где A, Ar и R8 имеют указанные значения, и последующим удалением хиральной вспомогательной группы Ar-CH(R8)-, и, если нужно, превращают промежуточный продукт формулы XIX-а в его кислую аддитивную соль обработкой кислотой или, наоборот, превращают аддитивную соль кислоты в свободное основание обработкой щелочью.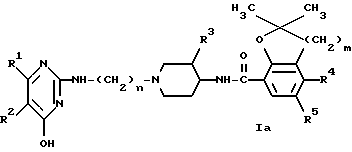
где R3, R4 и R5 имеют указанные значения,
отличающийся тем, что осуществляют следующие стадии: а) проводят реакцию энантиомерно обогащенного или энантиомерно чистого промежуточного продукта формулы XIV-а, где P1 - C1 - C4-алкилкарбонил, C1 - C4-алкилоксикарбонил, тригалогенметилкарбонил или фенилметил, с кислотой формулы V или ее функциональным производным с последующим удалением защитной группы P1 и получают энантиомерно обогащенный или энантиомерно чистый промежуточный продукт формулы II-а, где R3, R4 и R5 и m имеют указанные значения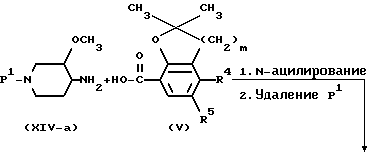
где R3, R4 и R5 имеют указанные значения: b) N-алкилируют энантиомерно обогащенный или энантиомерно чистый промежуточный продукт формулы II-а реагентом формулы XI, где P - подходящая защитная группа, легкоудаляемая, например, гидрогенолизом или гидролизом, W1 - удаляемая группа, такая как галоген или метансульфонилоксигруппа, с последующим удалением защитной группы P, получая энантиомерно обогащенный или энантиомерно чистый промежуточный продукт формулы VII-а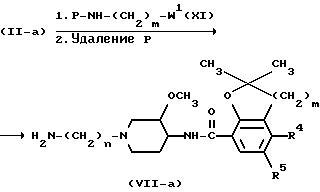
с) реакцию энантиомерно обогащенного или энантиомерно чистого промежуточного продукта формулы VII-а, где R4, R5, m и n имеют указанные значения, с реагентом формулы VI, где R6 - водород или C1 - C6-алкил, и W2 представляет собой соответствующую удаляемую группу, такую как, например, галоген, например хлор, бром или иод, сульфонилоксигруппу, например метансульфонилоксигруппу, метилбензолсульфонилоксигруппу, C1 - C6-алкилоксигруппу, например метокси-, этокси-, C1 - C6-алкилтиогруппу, например метилтио-, этилтио-, а R2 имеет указанные значения, и, если необходимо, отщепляют защитную эфирную группу для получения энантиомерно обогащенного или энантиомерно чистого соединения формулы I-а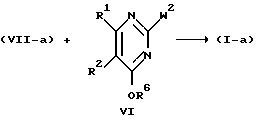
и, если нужно осуществляют дальнейшую очистку энантиомерно обогащенного соединения формулы I-а для получения энантиомерно чистого соединения формулу I-а, и, если далее нужно, превращают соединение формулы I-а в фармацевтически приемлемую аддитивную кислую соль обработкой кислотой или, наоборот, превращают аддитивную кислую соль в свободное основание обработкой щелочью.
| EP, 0242973, A, C 07 D 403/06, 1987 | |||
| EP,0397365, A1, C 07 D 417/14, 1990 | |||
| EP, 0309043, A1, C 07 D 211/58, 1989 | |||
| EP, 0445862, A1, C 07D 405/12, 1991 . |

