Изобретение относится к способу получения новых химических веществ, обладающих ценными фармацевтическими свойствами, и касается производных N-(3-гидрокси-4-пиперидинил)-(дигидробензо- фуран, дигидро-2Н-бензопиран или дигидробензодиоксин)карбоксамида, обладающих активностью по стимулированию желудочно-кишечной перистальтики.
Известны производные (3-гидрокси-4-пиперидинил)бензамида, обладающие стимулирующей активностью желудочно-кишечной перистальтики.
Производные N-(3-гидрокси-4-пиперидинил)-(дигидробензофуран, дигидро-2Н-бензопиран или дигидробензодиоксин)карбо- ксамида по изобретению превосходят указанные известные соединения по фармацевтической активности указанного вида.
Соединения по изобретению имеют формулу I
L-N


 R2 (I) где А является радикалом формулы -СН2-СН2- (а-1) -СН2-СН2-СН2- (а-2) или -СН2-СН2-О- (а-3) причем один или два атома водорода в указанном радикале (а-1) могут быть замещены С1-4-алкильным радикалом
R2 (I) где А является радикалом формулы -СН2-СН2- (а-1) -СН2-СН2-СН2- (а-2) или -СН2-СН2-О- (а-3) причем один или два атома водорода в указанном радикале (а-1) могут быть замещены С1-4-алкильным радикалом
R1 является галоидом,
R2 амино-группой,
R3 водородом или С1-4-алкилом,
L С3-6-циклоалкилом, С3-6-алкенилом или L является радикалом формулы -AIk -R5 (b-1) -AIk -X-R6 (b-2) -AIk -Y-C/=O/-R8 (b-3) или -AIk -Y-C/=O/ -NR10R11 (b-4) где каждый AIk является С1-6-алкандиилом, и
R5 является водородом, циано, С3-6-циклоалкилом, фенилом, необязательно замещенным галоидом или Het,
R6 является водородом, С1-6-алкилом, С3-6-циклоалкилом, галоидфенилом, необязательно замещенным С1-4-алкилкарбонилом, 3-циано-2-пиридинилом, 2-метил-5-пиридилом, 4-гидрокси-2-пиримидинилом, 2-метил-3-пиразинилом или 3,4-дигидро-4-оксо-2-хиназолинилом;
Х является О или NH,
R8 является водородом, С1-6-алкилом, 2,4,6-триметоксифенилом, 3,4,5-триметоксифенилом, 2,6-дихлорфенилом или С1-6-алкокси;
Y является NR9 или простой связью; указанный R9 является водородом, С1-4-алкилом или фенилом;
R10 и R11 каждый независимо является С1-6-алкилом или R10 и R11, взятые вместе с атомом азота, к которому они присоединены, могут образовать пирролидинильное кольцо;
Het является системой простого циклического эфира, выбранной в группе, состоящей из ;
; 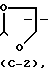 R12
R12
 ; или
; или 
 где R12 является водородом или С1-4-алкилом; или
где R12 является водородом или С1-4-алкилом; или
Het является гетероциклической системой, выбранной в группе, состоящей из пиридинила или бензимидазолила, замещенного С1-6-алкилом, или
Het является моноциклической амидной системой, выбранной в группе, состоящей из



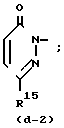 где R14 является водородом или С1-6-алкилом,
где R14 является водородом или С1-6-алкилом,
R15 является галоидом, С1-6-алкилом или фенилом,
G1 является -СН2-СН2-, СН=СН- или -С(=О)-СН2- или
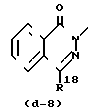
Het является бициклической амидной системой, выбранной в группе, состоящей из
 ;
;

 N
N  ;
;  или
или 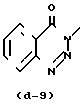 где R16 является С1-6-алкилом или фенилметилом,
где R16 является С1-6-алкилом или фенилметилом,
R17 является С1-6-алкилом и
R18 является водородом или галоидом,
G3 является -S-(CH2)2- или -S-CH=CH-,
G4 является -CH=CH-CH=CH-, -CH=CCI-CH=CH-, -CH=N-CH=CH- или -N=CH-N=CH-.
В объем изобретения входят также соли и стереоизомеры указанных соединений.
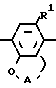
Объектом изобретения является также способ получения указанных соединений, согласно которому осуществляют N-алкилирование пиперидина формулы II
H-N

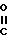
 R2 (II) где R1, R2, R3 и А имеют значения, указанные в формуле (I), промежуточным продуктом формулы L-W (III), где L имеет значения, указанные в формуле (I), а W является галоидом или сульфонилоксигруппой; или альдегидом формулы L' 0 (IV),
R2 (II) где R1, R2, R3 и А имеют значения, указанные в формуле (I), промежуточным продуктом формулы L-W (III), где L имеет значения, указанные в формуле (I), а W является галоидом или сульфонилоксигруппой; или альдегидом формулы L' 0 (IV),
L' 0 является соединением формулы L-H, у которого два соседних атома водорода в С1-6-алкандииле или С3-6-циклоалкандииле замененыО; или алкеном формулы NC-CH= CH2 (V), в реакционно инертном растворителе, необязательно в присутствии основания, иодидной соли или восстанавливающего агента; и необязательно, при желании, восстанавливают соединение формулы
NC Alk N


 R2 (I-c)
R2 (I-c)
где R1, R2, R3, A и AIk определены в формуле (I), в реакционно инертном растворителе в присутствии катализатора и в атмосфере водорода, в результате получают соединение формулы
H2N Alk N


 R2 (I-d) и затем вводят указанное соединение формулы (I-d) в реакцию с реагентом формулы R-W, где R является R6, -С/=O/-R8 или -C/=O/R10R11 и
R2 (I-d) и затем вводят указанное соединение формулы (I-d) в реакцию с реагентом формулы R-W, где R является R6, -С/=O/-R8 или -C/=O/R10R11 и
W является галоидом или метилтиогруппой, необязательно в реакционно инертном растворителе, необязательно в присутствии основания, при этом получают соединение формулы
R  - Alk N
- Alk N

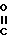
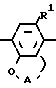 R2 (I-e) где R1, R2, R3, R9 AIk и А определены в формуле (I), или, при желании, проводят деацетализацию соединения формулы
R2 (I-e) где R1, R2, R3, R9 AIk и А определены в формуле (I), или, при желании, проводят деацетализацию соединения формулы
R Alk N
Alk N

 R2 (I-f) где R1, R2, R3, R8 AIk и А определены в формуле (I), в реакционно инертном растворителе в присутствии кислоты, при этом получают соединение формулы
R2 (I-f) где R1, R2, R3, R8 AIk и А определены в формуле (I), в реакционно инертном растворителе в присутствии кислоты, при этом получают соединение формулы
R Alk
Alk  N
N


 R2 (I-g) или, при желании, превращают соединение формулы
R2 (I-g) или, при желании, превращают соединение формулы
C1-6 алкил O  __
__  - Alk N
- Alk N


 R2 (I-h) где R1, R2, R3, R6, AIk и А определены в формуле (I), в реакционно инертном растворителе в присутствии кислоты, при этом получают соединение формулы
R2 (I-h) где R1, R2, R3, R6, AIk и А определены в формуле (I), в реакционно инертном растворителе в присутствии кислоты, при этом получают соединение формулы
R NH Alk N
NH Alk N

 R2 (I-i) и, при желании превращают соединение формулы (I) в его терапевтически активную нетоксичную соль присоединения при обработке кислотой, или наоборот, превращают кислую соль в свободное основание при обработке щелочью, и/или получают стереохимически изомерную форму.
R2 (I-i) и, при желании превращают соединение формулы (I) в его терапевтически активную нетоксичную соль присоединения при обработке кислотой, или наоборот, превращают кислую соль в свободное основание при обработке щелочью, и/или получают стереохимически изомерную форму.
В используемых здесь определениях "галоид" является общим для фтора, хлора, брома и иода; "алкил С1-С6" определяет насыщенные углеводородные радикалы с прямой или разветвленной цепью, имеющие от 1 до 6 атомов углерода, такие как, например, метил, этил, пропил, бутил, гексил, 1-метилэтил, 2-метилпропил и аналогичные; "циклоалкил С3-С6" является родовым определением для циклопропила, циклобутила, циклопентила и циклогексила; "циклоалканон С5-С6" является родовым для циклопентанона и циклогексанона; "алкенил С3-С6" определяет углеводородные радикалы с прямой или разветвленной цепью, содержащие одну двойную связь и имеющие от 3 до 6 углеводородных атомов, такие как, например, 2-пропенил, 3-бутенил, 2-бутенил, 2-пентенил, 3-пентенил, 3-метил-2-бутенил и аналогичные; и, когда алкенил С3-С6 замещен у гетероатома, тогда атом углерода указанного алкенила С3-С6, связанный с указанным гетероатомом, является предпочтительно насыщенным; "алкандиил С1-С6" определяет двухвалентные углеводородные радикалы с прямой или разветвленной цепью, содержащие от 1 до 6 атомов углерода, такие как, например, 1,2-этандиил, 1,3-пропандиил, 1,4-бутандиил, 1,5-пентандиил, 1,6-гександиил и их разветвленные изомеры.
Имеется в виду, что упомянутые соли включают терапевтически активные нетоксичные аддитивные солевые формы, которые способны образовывать соединения формулы (I). Последние могут удобным образом получаться с помощью обработки формы основания такими соответствующими кислотами, как неорганические кислоты, например галоидуглеводородные кислоты, например хлористоводородная, бромистоводородная и аналогичные, серная кислота, азотная кислота, фосфорная кислота и аналогичные; или органические кислоты, например уксусная, пропановая, гидроксиуксусная, 2-гидроксипропановая, 2-оксопропановая, этандионовая, пропандионовая, бутандионовая, (Z)-2-бутандионовая, (Е)-2-бутандионовая, 2-гидроксибутандионовая, 2,3-дигидроксибутандио- новая, 2-гидрокси-1,2,3-пропантрикарбоновая, метансульфоновая, этаносульфоновая, бензолсульфоновая, 4-метилбензолсульфоновая, циклогексансульфаминовая, 2-гидроксибензойная, 4-амино-2-гидроксибензойная и аналогичные кислоты. Наоборот, форма соли может с помощью обработки щелочью превращаться в форму свободного основания.
Соединения формулы (I), содержащие кислотные протоны, могут также превращаться в их терапевтически активные нетоксичные формы солей с металлами или амином с помощью обработки соответствующими органическими или неорганическими основаниями.
Термин "аддитивная соль" также включает гидраты и аддитивные формы с растворителем, которые соединения формулы (I) способны образовывать. Примерами таких форм являются, например, гидраты, алкоголяты и аналогичные.
Соединения формулы (I) имеют, по крайней мере, два асимметричных атома углерода в своей структуре, а именно атомы, расположенные в 3- и 4-положении пиперидинового ядра. Стереохимически изомерные формы соединений формулы (I) подпадают под объем изобретения. Кроме того, соединения изобретения могут образовывать цис/транс изомеры, более конкретно, заместители в указанных 3- и 4-положениях пиперидинового ядра могут иметь или транс- или цис-конфигурацию; такие цис/транс изомеры также подпадают под объем изобретения.
Реакция N-алкилирования соединения (II) соединением (III) удобным образом проводится в реакционно инертном растворителе, таком как, например, вода, ароматический углеводород, например бензол, метилбензол, диметилбензол, хлорбензол, метоксибензол и аналогичные, алканол, например метанол, этанол, 1-бутанол и аналогичные, галоидированные углеводороды, например дихлорметан, трихлорметан и аналогичные, сложный эфир, например этилацетат, γ-бутиролактон и аналогичные, кетоны, например 2-пропанон, 4-метил-2-пентанон и аналогичные; простой эфир, например 1,4-диоксан-1,1'-оксибисэтан, тетрагидрофуран и аналогичные, в полярном апротонном растворителе, например N, N-диметилформамиде, N,N-диметилацетамиде, диметилсульфоксиде, гексаметилфосфортриамиде, 1,3-диметил- 3,4,5,6-тетрагидро-2(1Н)-пиримидиноне, 1,3-диметил-2-имидазолидиноне, 1,1,3,3-тетраметилмочевине, нитробензоле, 1-метил-2-пирро- лидиноне и аналогичных, или в смеси таких растворителей.
Для улавливания кислоты, которая выделяется в течение хода реакции, может использоваться добавление соответствующего основания, такого как, например, карбонат, бикарбонат, карбоксилат, амид, окись, гидроокись или алкоголят щелочного или щелочно-земельного металла, например карбонат натрия, бикарбонат натрия, карбонат калия, окись кальция, ацетат натрия, амид натрия, гидроокись натрия, метилат натрия и аналогичные или органического основания, такого как, например, амин, например, N,N-диметил-4-пиридинамин, N, N-диэтилэтанамин, N-(1-метилэтил)-2-пропанамин, 1,4-диазабицикло-(2,2,2)-октан, 4-этилморфолин и аналогичные. В некоторых случаях может быть подходящим добавление иодидной соли, предпочтительно иодида щелочного металла, или кроун эфира, например 1,4,7,10,13,16-гексаоксациклооктадекана и аналогичных. Перемешивание и несколько повышенные температуры могут усиливать скорость реакции. Дополнительно, может быть благоприятным проведение указанного N-алкилирования в инертной атмосфере, такой как, например, свободный от кислорода аргон или азотный газ. Альтернативно указанное N-алкилирование может осуществляться при применении известных в данной области техники условий реакций с катализом фазового переноса. Такие условия включают перемешивание реагентов с подходящим основанием и необязательно в инертной атмосфере, как описана выше, в присутствии подходящего катализатора фазового переноса, такого как, например, галогенид, гидроокись, кислый сульфат триалкилфенилметиламмония, тетраалкиламмония, тетраалкилфосфония, тетраарилфосфония, и аналогичные катализаторы. Для усиления скорости реакции могут быть подходящими несколько повышенные температуры.
На данном и следующих этапах получения реакционные продукты могут выделяться из реакционной смеси и, если необходимо, дополнительно очищаться в соответствии с методиками, обычно известными в технике, такими как, например, экстракция, перегонка, кристаллизация, тритурирование или хроматография.
Соединения формулы (I) могут также превращаться друг в друга с использованием известных в технике приемов превращения функциональных групп. Некоторые примеры таких процедур будут приведены ниже.
Соединения формулы (I), содержащие гидрокси-функцию, могут О-алкилироваться в соответствии с известными в технике приемами О-алкилирования, например с помощью перемешивания с соответствующим алкилирующим агентом, если необходимо, в присутствии гидрида натрия.
Соединения формулы (I), содержащие защитное диоксолановое кольцо, могут деацетализироваться, давая соответствующие оксосоединения. Такая деацетализация может проводиться в соответствии с процедурами, широко известными в данной области техники, например с помощью реакции исходных веществ в водно-кислотной среде.
Соединения формулы (I), содержащие циано-заместитель, могут превращаться в соответствующие амины с помощью перемешивания и, если необходимо, нагревания исходных циано-соединений в среде, содержащей водород, в присутствии соответствующего катализатора, такого как, например, платина на угле, никель Ренея и аналогичные катализаторы, и необязательно в присутствии основания, такого как, например, амин, например, N,N-диэтилэтанамин и аналогичные, или гидроокись, например гидроокись натрия и аналогичные. Подходящими растворителями являются, например, алканолы, например метанол, этанол и аналогичные; простые эфиры, например тетрагидрофуран и аналогичные, или смесь таких растворителей.
Чистые стереохимически изомерные формы соединений формулы (I) и промежуточных соединений формулы (II) могут быть получены с использованием известных в технике приемов. Диастереоизомеры могут разделяться с помощью физических методов разделения, таких как, селективная кристаллизация и приемы хроматографии, например распределение в противотоке, и энантиомеры могут отделяться друг от друга с помощью селективной кристаллизации их диастереомерных солей с оптически активными кислотами и их оптически активированными производными.
Цис- и транс-диастереомерные рацематы могут далее расщепляться на их оптические изомеры, цис (+), цис (-), транс (+) и транс (-) с применением известных методик.
Чистые стереохимически изомерные формы могут также получаться из соответствующих чистых стереохимически изомерных форм соответствующих исходных материалов при условии, что реакция происходит стереоспецифическим образом.
Соединения формулы (I), содержащие алкеновый фрагмент, могут присутствовать в "Е" или "Z" форме, причем указанные Е- и Z-обозначения имеют значения, описанные в J. Org. Chem. 35, 2849-2868 (1970).
Соединения формулы (I), их фармацевтически приемлемые соли и возможные стереоизомерные формы обладают благоприятными свойствами стимулирования желудочно-кишечной перистальтики. В частности, настоящие соединения обнаруживают значительное действие по усилению перистальтики на ободочную (толстую) кишку. О последнем свойстве четко свидетельствуют результаты, полученные в описанном ниже испытании по "сокращениям, вызванным восхождением ободочной кишки".
Стимулирующее действие предлагаемых соединений формулы (I) на перистальтику (двигательную активность) желудочно-кишечной системы может дополнительно подтверждаться, например, различными испытаниями, описанными в The Journal Pharmacology and Experimental Therapeutics, 234, 775-783 (1985) и в Drug Development Research, 8. 243-250 (1986). Испытание "опустошение из желудка жидкой пищи у крыс", описанное в последней статье, и испытание "опустошение из желудка бескалорийной пищи у собак после назначения лидамидина" дополнительно обнаружили, что характерные представители ряда соединений также значительно ускоряют опустошение желудка.
В дополнение к сказанному, соединения формулы (I), их фармацевтически приемлемые кислотно-аддитивные соли и возможные стереоизомерные формы имеют особенный рецептор-связывающий профиль. Некоторые группы соединений изобретения, особенно соединения, в которых радикал А не замещен алкилом С1-С6, имеют плохую 5НТ3 антагонистическую активность, индуцированную высокими дозами серотонина на подвздошной кишке морской свинки. Большинство соединений изобретения не показывают какого-либо явного заметного сходства рецептор-связывания с серетонэргическим 5НТ1 и серотонэргическим 5НТ2 рецепторами и не имеют почти или вообще никакой допаминэргической антагонистической активности.
Ввиду своих полезных свойств усиления желудочно-кишечной перистальтики на основе указанных соединений могут быть приготовлены разнообразные формы для целей приема.
Для приготовления фармацевтических композиций изобретения эффективное количество конкретного соединения, в форме основания для кислотно-аддитивной соли, в качестве активного ингредиента, тщательно смешивается с фармацевтически приемлемым носителем, который может иметь разнообразные формы в зависимости от формы препарата, предлагаемого к назначению. Эти фармацевтические композиции представлены в форме единичных доз, предпочтительно для назначения орально, ректально или с помощью парентеральных инъекций. Например, при получении композиций в виде оральных дозировок могут применяться любые из обычных фармацевтических сред, такие как, например, вода, гликоли, масла, спирты и аналогичные, в случае оральных жидких препаратов, таких как суспензии, сиропы, эликсиры и растворы, или твердые носители, такие, как крахмалы, сахара, каолин, смазочные агенты, дезинтегрирующие агенты и аналогичные, в случае порошков, пилюль, капсул и таблеток. Вследствие простоты их приема таблетки и капсулы представляют наиболее благоприятные формы оральных дозированных единиц, и в этом случае очевидно применяются твердые фармацевтические носители. Для парентеральных композиций носитель обычно включает стерильную воду, по крайней мере, большую часть, хотя могут включаться другие ингредиенты, например, для того, чтобы способствовать растворимости. Инъецируемые растворы, например, могут приготавливаться, в которых носитель включает физиологический раствор, раствор глюкозы или смесь физиологического раствора и раствора глюкозы. Могут также приготавливаться инъецируемые суспензии, в этом случае могут применяться соответствующие жидкие носители, суспендирующие агенты и аналогичные. В композициях, подходящих для назначения через кожу, носитель необязательно включает агент, усиливающий проникновение, и/или подходящий смачивающий агент, необязательно в сочетании с небольшими количествами подходящих добавок любой природы, которые не оказывают значительного вредного воздействия на кожу. Указанные добавки могут облегчать применение к коже и/или могут быть полезными для получения желаемых композиций. Эти композиции могут назначаться различными путями, например трансдермальным путем, локально, в виде мази. Кислотно-аддитивные соли соединений (I) вследствие их повышенной растворимости в воде по сравнению с соответствующими формами основания являются очевидно более подходящими для приготовления водных композиций.
Особенно благоприятно изготавливать упомянутые фармацевтические композиции в форме дозированных единиц для легкости их назначения и равномерности доз. Форма дозированных единиц относится к физически дискретным единицам, подходящим в виде единичных доз, каждая единица содержит заданное количество активного ингредиента, рассчитанное на получение нужного терапевтического эффекта, в сочетании с требуемым фармацевтическим носителем. Примерами таких единичных дозированных форм являются таблетки (включающие таблетки в виде ядра или с нанесенным покрытием), капсулы, пилюли, порошки, вафли, инъецируемые растворы или суспензии, количества, составляющие полную чайную ложку, столовую ложку, и аналогичные, и сегрегированные множественные сочетания их.
Ввиду их способности стимулировать перистальтику желудочно-кишечной системы, в частности их способности усиливать двигательную активность толстой кишки, описываемые соединения полезны для приведения в норму или для улучшения опорожнения желудка и кишечника у субъектов, страдающих расстроенной перистальтикой, например, пониженной перистальтикой желудка и/или тонких, и/или толстых кишок.
Ввиду полезности предлагаемых соединений их предлагают для лечения теплокровных животных, страдающих расстройствами двигательной активности желудочно-кишечной системы, такими, как, например, гастропарез, диспепсия, сопровождающаяся метеоризмом, безъязвенная диспепсия, псевдонепроходимость, и особенно нарушенное прохождение содержимого через толстую кишку. При этом предусматривается общее назначение эффективного для стимуляции двигательной активности желудочно-кишечного тракта количества соединения формулы (I), N-окиси, фармацевтически приемлемой кислотно-аддитивной соли или его возможной стереоизомерной формы теплокровным животным. Некоторые конкретные соединения изобретения также обладают терапевтической ценностью при лечении двигательной активности верхнего пищеварительного тракта и расстройства гастроэзофагеального рефлюкса.
Соединения по изобретению являются малотоксичными. Специалисты в данной области техники могли бы легко определить эффективное количество, стимулирующее двигательную активность, по результатам испытаний, представленным ниже.
В общем считается, что эффективная доза составляет от 0,001 до 10 мг/кг массы тела, и более предпочтительно от 0,01 до 1 мг/кг массы тела. Примеры предназначены для иллюстрации, но не для ограничения изобретения во всех его аспектах.
Экспериментальная часть.
А. Получение промежуточных продуктов.
П р и м е р 1.
а). К раствору 8,1 мас.ч. 4-амино-5-хлор-2,3-дигидро-2,2-диметил-7-бензофуранкар- боновой кислоты в 218 мас.ч. трихлорметана и 3,43 мас.ч. N, N-диэтилэтанамина по каплям добавлялось 3,63 мас.ч. этилхлороформата, при поддержании температуры ниже 10оС. После перемешивания в течение 1/2 ч при 10оС все добавлялось к раствору 6,26 мас.ч. этил-4-амино-3-метокси-1-пиперидинкарбокси- лата в 145 мас. ч. трихлорметана при 10оС. Перемешивание продолжалось в течение 1/2 ч при комнатной температуре. Реакционная смесь промывалась водой, 5% гидроокисью натрия и водой, затем сушилась, фильтровалась и выпаривалась. Остаток суспендировался в 2,2'-оксибиспропане. Продукт отфильтровывался и сушился с получением 12,3 мас.ч. (93,2%) этил-цис-4-[(4-амино-5-хлор-2,3-дигидро-2,2-диметил-7-бен- зофуранил)карбониламино]-3-метокси-1- пиперидинкарбоксилата (промежуточный продукт 1).
b). Смесь 12,3 мас.ч. промежуточного продукта 1, 15,9 мас.ч. гидроокиси калия и 156 мас.ч. 2-пропанола перемешивалась в течение 12 ч при температуре обратного потока. Реакционная смесь выпаривалась и к остатку добавлялась вода. Все выпаривалось снова и остаток разбавлялся водой. Продукт экстрагировался дихлорметаном (2 раза) и объединенные экстракты сушились, фильтровались и выпаривались. Остаток очищался с помощью хроматографии на колонке (силикагель; СН2Cl2/CH3OH-/NH3) 90 10). Элюент желаемой фракции выпаривался и остаток суспендировался в 2,2'-оксибиспропане. Продукт отфильтровывался и сушился с получением 7,24 мас.ч. (71,0%) цис-4-амино-5-хлор-2,3-дигидро-N-(3-мето- кси-4-пиперидинил)-2,2-диметил-7-бензо- фуранкарбоксамида, т.пл. 179,3оС (промежуточный продукт 5).
Аналогичным образом были также получены промежуточные продукты, перечисленные в табл. 1.
П р и м е р 2.
а). Раствор 9,1 мас.ч. 5-хлор-2,3-дигидро-4-бензофуранамина [описан в J. Het. Chem. 17(6) 1333 (1980)] 9,6 мас.ч. N-бромсукцинимида и 130,5 мас.ч. бензола перемешивался в течение 1 ч при температуре обратного потока. Растворитель выпаривался и остаток растворялся в 387,4 мас.ч. трихлорметана. Раствор промывался водой (2 х 200 мас.ч.). Органический слой сушился, фильтровался и выпаривался. Остаток очищался с помощью хроматографии на колонке (силикагель; С6Н14/СН2Cl2 50 50). Элюент желаемой фракции выпаривался с получением 11,8 мас.ч. (87,9%) 7-бром-5-хлор-2,3-дигидро-4-бензофуранамина (промежуточный продукт 8).
b). К охлаждаемой (-70оС) и перемешиваемой смеси 15,6 мас.ч. раствора н. бутиллития в гексане 2,5 молярности и 44,5 мас.ч. тетрагидрофурана по каплям добавлялся раствор 4 мас.ч. промежуточного продукта 8 в 26,7 мас.ч. тетрагидрофурана в потоке азота. Реакционная смесь перемешивалась в течение 1 ч при приблизительно -60оС и вливалась в насыщенную суспензию двуокиси углерода (лед) в 44,5 мас.ч. тетрагидрофурана. Массе давалась возможность нагреться до комнатной температуры с одновременным перемешиванием и добавлялось 80 мас. ч. воды. Водный слой нейтрализовался соляной кислотой, и образовавшийся осадок отфильтровывался и сушился в вакууме при 60оС с получением 1,1 мас.ч. (32,2%) 4-амино-5-хлор- 2,3-дигидро-7-бензофуранкарбоновой кислоты;
Т.пл. 258,4оС (промежуточный продукт 9).
Аналогичным образом была получена 8-амино-7-хлор-2,3-дигидро-1,4-бензодиок- син-5-карбоновая кислота (промежуточный продукт 10).
П р и м е р 3.
а). Смесь 40 мас. ч. метил-4-(ацетиламино)-5-хлор-2-(2-пропинокси)бензоата и 172 мас.ч. феноксибензола перемешивалась в течение 45 мин при 230оС. После охлаждения реакционная смесь вливалась в петролейный эфир. Органический слой отделялся, сушился, фильтровался и выпаривался. Остаток очищался с помощью хроматографии на колонке (силикагель; CH2Cl2/CH3OH 97 3). Элюент желаемых фракций выпаривался и остаток кристаллизовался из ацетонитрила с получением 11,9 мас.ч. метил 5-(ацетиламино)-6-хлор-2Н-1-бензопиран-8-карбокси- лата (промежуточный продукт 11).
b). Смесь 31,3 мас.ч. промежуточного продукта 11,31 мас.ч. N,N-диэтилэтанамина и 395 мас.ч. метанола гидрировалась при нормальном давлении при комнатной температуре с 4 мас.ч. 10% катализатора палладия на угле. После того как рассчитанное количество водорода поглощалось, катализатор отфильтровывался и фильтрат выпаривался. Остаток суспендировался в воде, и продукт экстрагировался дихлорметаном (2 раза). Объединенные экстракты промывались водой, сушились, фильтровались и выпаривались. Остаток очищался дважды с помощью хроматографии на колонке (силикагель; CH2Cl2/CH3OH 97,5 2,5). Элюент желаемой фракции выпаривался, и остаток кристаллизовался из ацетонитрила. Продукт отфильтровывался и сушился с получением 19,1 мас.ч. (69,7%) метил-5-(ацетиламино)-3,4-дигидро-2Н-1-бензопиран-8-карбоксилата.
Т.пл. 175,1оС (промежуточный продукт 12).
с). Смесь 19,1 мас.ч. промежуточного продукта 12, 10,22 мас.ч. N-хлорсукцинимида и 237 мас.ч. ацетонитрила перемешивалось в течение 1 ч при температуре обратного потока. После охлаждения реакционная смесь вливалась в 300 мас.ч. воды. Продукт экстрагировался дихлорметаном (2 раза) и объединенные экстракты промывались водой, сушились, фильтровались и выпаривались. Остаток суспендировался в 2,2'-оксибиспропане. Продукт отфильтровывался и сушился с получением 17,8 мас.ч. (81,5%) метил-5-(ацетиламино)-6-хлор-3,4-дигидро-2Н-1-бензопиран-8-карбоксилата. Т.пл. 184,2оС (промежуточный продукт 13).
d). Смесь 1,34 мас.ч. промежуточного продукта 13, 2,62 мас.ч. гидроокиси калия и 20 мас.ч. воды перемешивалась в течение 3 ч при температуре обратного потока. После охлаждения реакционная смесь подкислялась до рН 4 концентрированной соляной кислотой. Осадок отфильтровывался и сушился с получением 0,65 мас. ч. (60,7%) 5-амино-6-хлор-3,4-дигидро-2Н-1-бензопир- ан-8-карбоновой кислоты. Т.пл. 225,9оС (промежуточный продукт 14).
П р и м е р 4.
а). К раствору 104,6 мас.ч. метил-2-гидроокси-4-(ацетиламино)бензоата в 470 мас. ч. N, N-диметилформамида порциями добавлялось 24 мас.ч. дисперсии гидрида натрия в минеральном масле (50%) в атмосфере азота. После перемешивания в течение 1 ч при комнатной температуре к смеси добавлялся раствор 55,2 мас.ч. 3-хлор-2-метил-1-пропена в 47 мас.ч. N,N-диметилформамида. Перемешивание продолжалось в течение 3 дней при 50оС. Реакционная смесь выпаривалась, и остаток растворялся в дихлорметане. Данный раствор промывался водой, 10% гидроокисью натрия и водой и затем сушился, фильтровался и выпаривался. Остаток кристаллизовался из 2,2'-оксибиспропана. Продукт отфильтровывался и сушился с получением 65,8 мас.ч. (50%) метил-4-(ацетиламина)-2-[(2-метил-2-пропенил)окси]бензоата (промежуточный продукт 15).
b). Смесь 72 мас.ч. промежуточного продукта 15 и 226 мас.ч. 1-метил-2-пирролидинона перемешивалась в течение 1,5 ч при температуре обратного потока. После охлаждения реакционная смесь вливалась в ледяную воду. Продукт экстрагировался дихлорметаном (2 раза), и объединенные экстракты промывались водой, сушились, фильтровались и выпаривались. Остаток кристаллизовался из 2,2'-оксибиспропана. Продукт отфильтровывался и сушился с получением 35,4 мас.ч. (49,8%) метил-4-(ацетиламина-2-гидрокси-3-2-метил-2-пропенил)бензоата. Маточная жидкость выпаривалась, и остаток последовательно суспендировался в воде и перекристаллизовывался из 2,2'-оксибиспропана с получением дополнительно 17,6 мас.ч. (24,8%) метил-4-(ацетиламина-2-гидрокси-3-2-метил-2-про-пенил)бензоата. Общий выход: 53,0 мас.ч. 74,6% (промежуточный продукт 16).
с). Смесь 126 мас.ч. промежуточного продукта 16 и 1220 мас.ч. муравьиной кислоты перемешивалась в течение 20 ч при температуре обратного потока. После охлаждения реакционная смесь вливалась в смесь лед-вода и все экстрагировалось дихлорметаном (2 раза). Объединенные экстракты промывались 10% гидроокисью натрия и водой и затем сушились, фильтровались и выпаривались. Остаток суспендировался в 2,2'-оксибиспропане. Продукт отфильтровывался и сушился с получением 105,5 мас.ч. (83,8%) метил-4-(ацетиламина)-2,3-дигидро-2,2-диметил-7-бензофуранкар- боксилата (промежуточный продукт 17).
d). Смесь 10,5 мас.ч. промежуточного продукта 17, 5,87 мас.ч. N-хлорсукцинимида и 158 мас.ч. ацетонитрила перемешивалась в течение 1 ч при температуре обратного потока. После охлаждения реакционная смесь вливалась в ледяную воду. Продукт экстрагировался дихлорметаном (2 раза), и объединенные экстракты сушились, фильтровались и выпаривались. Остаток суспендировался в 2,2'-оксибиспропане. Продукт отфильтровывался и сушился с получением 11,9 мас. ч. (99,9%) метил-4-(ацетиламина)-5-хлор-2,3-дигидро-2,2-диметил-7-бензофуранкарбоксилат а (промежуточный продукт 18).
е). Смесь 11,9 мас.ч. промежуточного продукта 18, 22,4 мас.ч. гидроокиси калия и 200 мас. ч. воды перемешивалась в течение 3 ч при температуре обратного потока. После охлаждения реакционная смесь подкислялась до рН 4-5. Осадок отфильтровывался и сушился с получением 8,1 мас.ч. (83,8%) 4-амина-5-хлор-2,3-дигидро-2,2-диметил-7-бензофуранкарбоновой кислоты (промежуточный продукт 19).
В. Получение конечных соединений.
П р и м е р 5. Смесь 3,9 мас.ч. промежуточного продукта 2, 2,54 мас.ч. карбоната натрия, 1 кристаллик йодистого калия и 144 мас.ч. 4-метил-2-пентанона перемешивалась в течение 1 ч при температуре обратного потока с использованием водного сепаратора. После добавления 3,2 мас.ч. 1-(2-хлорэтил)-3-этил-2,3-дигидро-1Н-бензи- мидазол-2-она, перемешивание продолжалось в течение ночи при температуре обратного потока. Реакционная смесь промывалась водой. Органический слой сушился, фильтровался и выпаривался. Остаток очищался с помощью хроматографии на колонке (силикагель; CH2Cl2/CH3OH 96 4). Элюент желаемой фракции выпаривался и остаток кристаллизовался из 2,2'-оксибиспропана. Продукт сушился в вакууме при 70оС с получением 2,30 мас.ч. (37,3% ) цис-4-амино-5-хлор-N-[1-(2-(3-этил-2,3-дигидро-2-оксо-1Н-бензимидазол-1-ил)э тил)си-4-пиперидинил] -2,3-дигидро-7-бензофу- ранкарбоксамидо. Т.пл. 173,7оС (соединение 1).
П р и м е р 6. Смесь 4,2 мас.ч. монобромгидрата 3-(2-бромэтил)-2-метил-4Н-хиназолин-4-она, 3,3 мас. ч. промежуточного продукта 2, 4,24 мас.ч. карбоната натрия, 160 мас.ч. 4-метил-2-пентанона и нескольких кристалликов йодистого калия перемешивалась в течение 20 ч при температуре обратного потока. Растворитель выпаривался и остаток распределялся между трихлорметанолом и водой. Органический слой промывался водой, сушился, фильтровался и выпаривался. Остаток очищался дважды с помощью хроматографии на колонке (силикагель; CHCl3/CH3OH 97 3; HPLC; силикагель; C6H5-CH3) изо-С3Н7OH 80 20). Элюент желаемой фракции выпаривался и остаток кристаллизовался из ацетонитрила. Продукт отфильтровывался и сушился в вакууме при 60оС с получением 3,10 мас.ч. (60,5%) цис-4-амино-5-хлор-2,3-дигидро-N-[3-мето- кси-1-(2-(2-метил-4-оксо-3(4Н)-хиназолин) этил)-4-пиперидинил] -7-бензофуранкарбок- самида.
Т.пл. 274,9оС (соединение 30).
П р и м е р 7. Смесь 4,07 мас.ч. промежуточного продукта 7, 3,82 мас.ч. карбоната натрия и 200 мас.ч. 4-метил-2-пентанона перемешивалась при нагревании с обратным холодильником (с водным сепаратором) в течение 1 ч. Затем добавлялось 2,7 мас. ч. 6-хлор-2-(3-хлорпропил)-2Н-пиридазин-3-она и перемешивание при температуре обратного потока продолжалось в течение ночи. Реакционная смесь выпаривалась и остаток брался в дихлорметан. Данный раствор промывался водой, сушился, фильтровался и выпаривался. Остаток очищался с помощью хроматографии на колонке (силикагель; CH2Cl2/CH3OH/NH3/ 95 5). Элюент желаемой фракции выпаривался и остаток отверждался в 2,2'-оксибиспропане. Продукт отфильтровывался и сушился с получением 3,9 мас.ч. (63,7%) цис-5-амино-6-хлор-N-[1-(3-(3-хлор-1,6-дигидро-6-оксо-1-пиридазинил)пропил)- 3-мединил]-3,4-дигидро-2Н-бензопиран-8-кар- боксамида.
Т.пл. 149,5оС (соединение 136).
П р и м е р 8. Смесь 3,4 мас.ч. промежуточного продукта 7, 3,16 мас.ч. тетрагидро-2-фуранметанолметансульфоната (сложный эфир), 80 мас.ч. 4-метил-2-пентанона и 1,58 мас.ч. карбоната натрия перемешивалась при нагревании с обратным холодильником (с водным сепаратором) в течение 30 ч. Реакционная смесь выпаривалась и остаток разбавлялся водой. Продукт экстрагировался дихлорметаном (2 раза) и объединенные экстракты промывались водой, сушились, фильтровались и выпаривались. Остаток очищался с помощью хроматографии на колонке (силикагель; CH2Cl2/CH3OH 95 5). Элюент желаемой фракции выпаривался и остаток суспендировался в 2,2'-оксибиспропане. Продукт отфильтровывался и сушился с получением 2,44 мас.ч. (57,6%) цис-5-амино-6-хлор-3,4-дигидро-N-(3-метокси-1-)(тет- рагидро-2-фуранил)метил(-4-пиперидинил)-2Н-1-бензопиран-8-карбоксамида. Т.пл. 158,1оС (соединение 76).
П р и м е р 9. Смесь 3,53 мас.ч. промежуточного продукта 5, 2,1 мас.ч. 1-(3-хлорпропил)-3-этил-2-имидазолидинона, 94 мас.ч. N,N-диметилформамида и 1,58 мас. ч. карбоната натрия перемешивалась в течение 20 ч при 70оС. Реакционная смесь выпаривалась и остаток разбавлялся водой. Продукт экстрагировался дихлорметаном (2 раза) и объединенные экстракты промывались водой, сушились, фильтровались и выпаривались. Остаток очищался с помощью хроматографии на колонке (силикагель; CH2Cl2/CH3OH/NH3/ 96 4). Элюент желаемой фракции выпаривался и остаток превращался в соль этандиоата в 2-пропаноле. Продукт отфильтровывался и сушился с получением 4,18 мас.ч. (70,0%) этандиоата цис-4-амино-5-хлор-N-[1(3-(3-этил-2-оксо-1-ими- дазолидинил)пропил)-3-метокси-4-пипери- динил] -2,3-дигидро-2,2-диметил-7-бензофу- ранкарбоксамида (1 1).
Т.пл. 208,0оС (соединение 121).
П р и м е р 10. Смесь 2,6 мас.ч. 2-йодметил-1,3-диоксолана, 3,3 мас.ч. промежуточного продукта 2, 2,12 мас.ч. карбоната натрия и 47 мас.ч. N,N-диметилформамида перемешивалась в течение 3 дней при 70оС. После охлаждения реакционная смесь выпаривалась. Остаток распределялся между дихлорметаном и водой. Органический слой отделялся, промывался водой, сушился, фильтровался и выпаривался. Остаток очищался с помощью хроматографии на колонке (силикагель; CH2Cl2/CH3OH 95 5). Элюент желаемой фракции выпаривался и остаток кристаллизовался из ацетонитрила (к которому добавлялось несколько капель воды). Продукт отфильтровывался при 0оС и сушился в вакууме при 40оС с получением 2,3 мас.ч. (55,8%) цис-4-амино-5-хлор-N-[1(1,3-диоксолан-2-ил-метил)-3-метокси-4-пиперидинил]-23-дкарбоксамида.
Т.пл. 149,1оС (соединение 83).
П р и м е р 11. Смесь 2,78 мас.ч. 1-(3-хлорпропил)-2-метил-1Н-бензимидазола, 3,3 мас.ч. промежуточного продукта 2, 2,04 мас.ч. N,N-диэтилэтанамина и 94 мас. ч. N,N-диметилформамида перемешивалась в течение 20 ч при 70оС. Реакционная смесь выпаривалась и к остатку добавлялась вода. Продукт экстрагировался дихлорметаном (2 раза) и объединенные экстракты промывались водой, сушились, фильтровались и выпаривались. Остаток кристаллизовался из ацетонитрила (к которому добавлялось несколько капель воды) с получением 2,30 мас.ч. (44,6%) моногидрата цис-4-амино-5-хлор-2,3-дигидро-N-[3-метокси-1-(3-(2-мет- ил-1Н-бензимидазол-1-ил)пропил)-4-пипе- ридинил]-7-бензофуранкарбоксамида. Т.пл. 151,5оС (соединение 27).
П р и м е р 12. Смесь 3,3 мас.ч. промежуточного продукта 2, 4,4 мас.ч. этил N-(2-оксоэтил)-N-фенилкарбамата, 2 мас.ч. раствора тиофена в 4% метаноле гидрировалась при нормальном давлении и при 50оС с 2 мас.ч. 5% катализатора платина на угле. После того как рассчитанное количество водорода поглощалось, катализатор отфильтровывался и фильтрат выпаривался. Остаток разбавлялся водой и продукт экстрагировался дихлорметаном (2 раза). Объединенные экстракты сушились, фильтровались и выпаривались. Остаток очищался с помощью хроматографии на колонке (силикагель; CH2Cl2/CH3OH 95 5). Элюент желаемой фракции выпаривался и остаток суспендировался в 2,2'-оксибиспропане. Продукт отфильтровывался и сушился с получением 3,08 мас.ч. (58,6%) полугидрата этил цис-N-[2-[4-[[(4-амино-5-хлор-2,3-дигидро-7-бензофуранил)-карбон- ил] амино] -3-метокси-1-пиперидинил] этил] N-фенилкарбамата. Т.пл. 116,4оС (соединение 57).
П р и м е р 13. К перемешиваемой смеси 3,4 мас.ч. промежуточного продукта 7, 2 мас.ч. тетрагидрофурана, 2 мас.ч. раствора тиофена в 4% метаноле и 119 мас.ч. метанола по каплям добавлялась смесь 11 мл раствора ацетальдегида в 10% тетрагидрофуране и 8,9 мас.ч. татрагидрофурана в течение гидрирования. После завершения гидрирования катализатор отфильтровывался и фильтрат выпаривался. Остаток растворялся в дихлорметане и этот раствор промывался водой (2 раза), сушился, фильтровался и выпаривался. Остаток перекристаллизовывался из ацетонитрила. Продукт отфильтровывался и сушился с получением 2,66 мас.ч. (72,3%) цис-5-амино-6-хлор-N-(1-этил-3-метокси-4-пиперидинил)-3,4-дигидро-2Н-1-бензо пи- ран-8-карбоксамида. Т. пл. 153,8оС (соединение 81).
П р и м е р 14. Смесь 3 мас.ч. 1-гексаналя, 3,7 мас.ч. промежуточного продукта 3, 1 мас.ч. раствора тиофена в 4% метанола и 242,5 мас.ч. 2-метоксиэтанола гидрировалась при нормальном давлении и при 50оС с 2 мас.ч. 5% катализатора платина на угле. После того как рассчитанное количество водорода поглощалось, катализатор отфильтровывался и фильтрат выпаривался. Остаток очищался с помощью хроматографии на колонке (силикагель; CH2Cl2/CH3OH/NH3/ 98 2). Элюент желаемой фракции выпаривался и остаток кристаллизовался из 2,2'-оксибиспропана. Продукт сушился в вакууме при 70оС с получением 3,20 мас. ч. (68,5% ) цис-4-амино-5-хлор-N-(1-гексил-3-гидрокси-4-пиперидинил)-2,3-дигидро-7-бензо фуран- карбоксамида.
Т.пл. 13,4оС (соединение 8).
П р и м е р 15. Смесь 4,5 мас.ч. (1,1-диметилэтил)(2-оксоэтил)метилкарбамата, 5,5 мас.ч. промежуточного продукта 2, 1 мас.ч. раствора тиофена в 4% метаноле, 198 мас.ч. метанола и 2 мас.ч. ацетата калия гидрировалась при нормальном давлении и при комнатной температуре с 2 мас.ч. 10% катализатора палладий на угле. После того как рассчитанное количество водорода поглощалось, катализатор отфильтровывался и фильтрат выпаривался. Остаток распределялся между трихлорметаном и водой. Органический слой отделялся, промывался водой, сушился, фильтровался и выпаривался. Остаток отверждался в 2,2'-оксибиспропане (к которому добавлялось несколько капель воды). Продукт отфильтровывался при 0оС и сушился в вакууме при 40оС с получением 6,3 мас. ч. (76,6% ) (1,1-диметилэтил)-цис-[2-[4-[(4-амино-5-хлор-2,3 -дигидро-7-бензофуранил)карбониламино] 3-метокси-1-пиперидинил]этил]метилкарбама- та (соединение 41).
П р и м е р 16. К нагреваемому с обратным холодильником раствору 17,4 мас. ч. промежуточного продукта 2 в 195 мас.ч. 2-пропанола добавлялось 4,03 мас. ч. 2-пропаннитрила. Перемешивание при температуре дефлегмации продолжалось в течение 18 ч. Реакционная смесь выпаривалась и остаток кристаллизовался из 2-пропанола. Продукт отфильтровывался и сушился в вакууме при 60оС с получением 14,8 мас.ч. (73,7%) цис-4-амино-5-хлор-N-[1-(2-цианоэтил)-3-метокси-4-пиперидинил]-2, 3-дигидро-7-бензофуранкарбоксамида.
Т.пл. 190,7оС (соединение 97).
П р и м е р 17. Раствор 15,7 мас.ч. цис-4-амино-5-хлор-N-[1-(цианометил)-3-ме- токси-4-пиперидинил] -2,3-дигидро-7-бензо- фуранкарбоксамида в 178 мас.ч. тетрагидрофурана и 158 мас.ч. метанола гидрировался при нормальном давлении и при комнатной температуре с 6 мас.ч. никеля Ранея. После того как рассчитанное количество водорода поглощалось, катализатор отфильтровывался и фильтрат выпаривался. Остаток очищался с помощью хроматографии на колонке (силикагель; CH2Cl2/CH3OH(NH3) 93 7). Элюент желаемой фракции выпаривался и остаток кристаллизовался из ацетонитрила (к которому добавлялось несколько капель воды). Продукт отфильтровывался при 0оС и сушился в вакууме при 40оС с получением 8,5 мас.ч. (53,6%) цис-4-амино-N-[1-(2-аминоэтил)-3-метокси-4-пиперидинил]-5-хлор-2,3- дигидро-7-бензофуранкарбоксамида (соединение 35).
П р и м е р 18. К охлаждаемой (ледяная баня) смеси 3,8 мас.ч. моногидрата цис-4-амино-5-хлор-2,3-дигидро-N-[3-метокси-1-[2- (метиламино)этил)-4-пиперидинил] -7-бензо- фуранкарбоксамида в 104,3 мас.ч. трихлорметана добавлялось 1,3 мас.ч. 1-пирролидинкарбонил хлорида. После перемешивания в течение 15 мин при 0оС по каплям добавлялось 1,31 мас.ч. N,N-диэтилэтанамина с поддержанием температуры до 10оС. Перемешивание продолжалось в течение 20 ч при комнатной температуре. Реакционная смесь промывалась водой, сушилась, фильтровалась и выпаривалась. Остаток кристаллизовался из ацетонитрила (к которому добавлялось немного воды). Продукт отфильтровывался при 0оС и сушился в вакууме при 40оС с получением 3,3 мас.ч. (73,6%) моногидрата цис-4-амино-5-хлор-2,3-дигидро-N-[3-метокси-1-[2-[метил(1-пир- ролидинилкарбонил)амино]этил]-4-пипери- динил]-7-бензофуранкарбоксамида.
Т.пл. 112,0оС (соединение 43).
П р и м е р 19. Смесь 1,4 мас.ч. 2-хлор-3-пиридинкарбонитрила, 3,2 мас. ч. цис-4-амино-N-[1-(4-аминобутил)-3-метокси-4-пи- перидинил]-5-хлор-2,3-дигидро-7-бензофу- ранкарбоксамида, 65,8 мас. ч. N,N-диметилформамида и 1,3 мас.ч. карбоната натрия перемешивалась в течение 20 ч при 70оС. Растворитель выпаривался и остаток растворялся в трихлорметане. Органический слой промывался водой, сушился, фильтровался и выпаривался. Остаток очищался с помощью хроматографии на колонке (силикагель; CHCl3/CH3OH(NH3) 98 2). Элюент желаемой фракции выпаривался и остаток кристаллизовался из 2,2'-оксибиспропана. Продукт сушился в вакууме при 60оС с получением 1,44 мас.ч. (35,4%) полугидрата цис-4-амино-5-хлор-N-[1-[4-[(3-циано-2-пи- ридинил)амино]бутил] -3-метокси-4-пипери- динил]-2,3-дигидро-7-бензофуранкарбокса- мида.
Т.пл. 129,7оС (соединение 6).
П р и м е р 20. Смесь 1,18 мас.ч. 2-хлор-4(3Н)-хиназолина, 2,40 мас.ч. соединения 35 и минимального количества N,N-диметилформамида перемешивалась в течение 3 ч при 120оС. После охлаждения реакционная смесь распределялась между дихлорметаном и метанолом. Органический слой отделялся, сушился, фильтровался и выпаривался. Остаток очищался с помощью хроматографии на колонке (силикагель; CH2Cl2/CH3OH 90 10). Элюент желаемой фракции выпаривался и остаток кристаллизовался из ацетонитрила (к которому добавлялось немного воды). Продукт отфильтровывался при 0оС и сушился с получением 0,95 мас. ч. (37,5% ) полуторного гидрата цис-4-амино-5-хлор-2,3-дигидро-N-[1-[2-[(3,4-дигидро-4-оксо-2-хиназолинил)ам и- но]этил]-3-метокси-4-пиперидинил] -7-бензо- фуранкарбоксамида. Т.пл. 191,8оС (соединение 88).
П р и м е р 21. Смесь 4,69 мас.ч. дигидрохлорида цис-4-амино-N-[1-(2-аминоэтил)-3-метокси-4-пиперидинил] -5-хлор-2,3-дигид- ро-2,2-диметил-7-бензофуранкарбоксамида, 1,54 мас.ч. 2-хлор-3-метилпиридазина и 1,68 мас.ч. окиси кальция перемешивалась в течение 20 ч при 120оС. После охлаждения реакционная смесь разбавлялась водой и продукт экстрагировался дихлорметаном (3 раза). Объединенные экстракты сушились, фильтровались и выпаривались. Остаток очищался с помощью хроматографии на колонке (силикагель; CH2Cl2/CH3OH(NH3) 95 5). Элюент желаемой фракции выпаривался и остаток преобразовывался в соль этандиоата в 2-пропаноле. Продукт отфильтровывался и сушился с получением 1,38 мас.ч. (23,1%) моногидрата этандиоата (1 1) цис-4-амино-5-хлор-2,3-дигидро-N-[3-метокси-1-[2-[(3-метил-2-пиразинил)амино этиперидинил] -2,2-диметил-7-бензофуранкар- боксамида. Т. пл. 117,1оС (соединение 170).
П р и м е р 22. Смесь 5 мас.ч. цис-5-амино-N-[1-(3-аминопропил)-3-метокси-4-пипери- динил] -6-хлор-3,4-дигидро- 2Н-1-бензопиран-8-карбоксамида, 3,2 мас.ч. 2-метилтио-4-пиримидинола и 79 мас.ч. ацетонитрила перемешивалась в течение 2 дней (конец недели) при температуре дефлегмации. Реакционная смесь выпаривалась и остаток распределялся между дихлорметаном и аммиаком (водным). Водный слой отделялся и реэкстрагировался дихлорметаном (2 раза). Объединенные дихлорметановые слои сушились, фильтровались и выпаривались. Остаток очищался с помощью хроматографии на колонке (силикагель; CH2Cl2/CH3OH(NH3) 95 5). Элюент двух желаемых фракций выпаривался и остатки отдельно кристаллизовались из ацетонитрила. Продукт отфильтровывался и сушился в вакууме при 70оС с получением первой фракции из 2,22 мас.ч. (35,2%) полугидрата цис-5-амино-6-хлор-3,4-дигидро-N-[1-[3-[(4-гидрокси-2- пиримидинил) амино] пропил]-3-метокси-4-пиперидинил] 2H-1- бензопиран-8-карбоксамида, т. пл. 142,6оС и второй фракции 1,00 мас.ч. (15,9%) полугидрата цис-5-амино-6-хлор-3,4-дигидро-N-[1-[3-[(4-гидрокси-2- пиримидинил)амино]пропил] -3-метокси-4-пипе- ридинил]-2Н-1- бензопиран-8-карбоксамида.
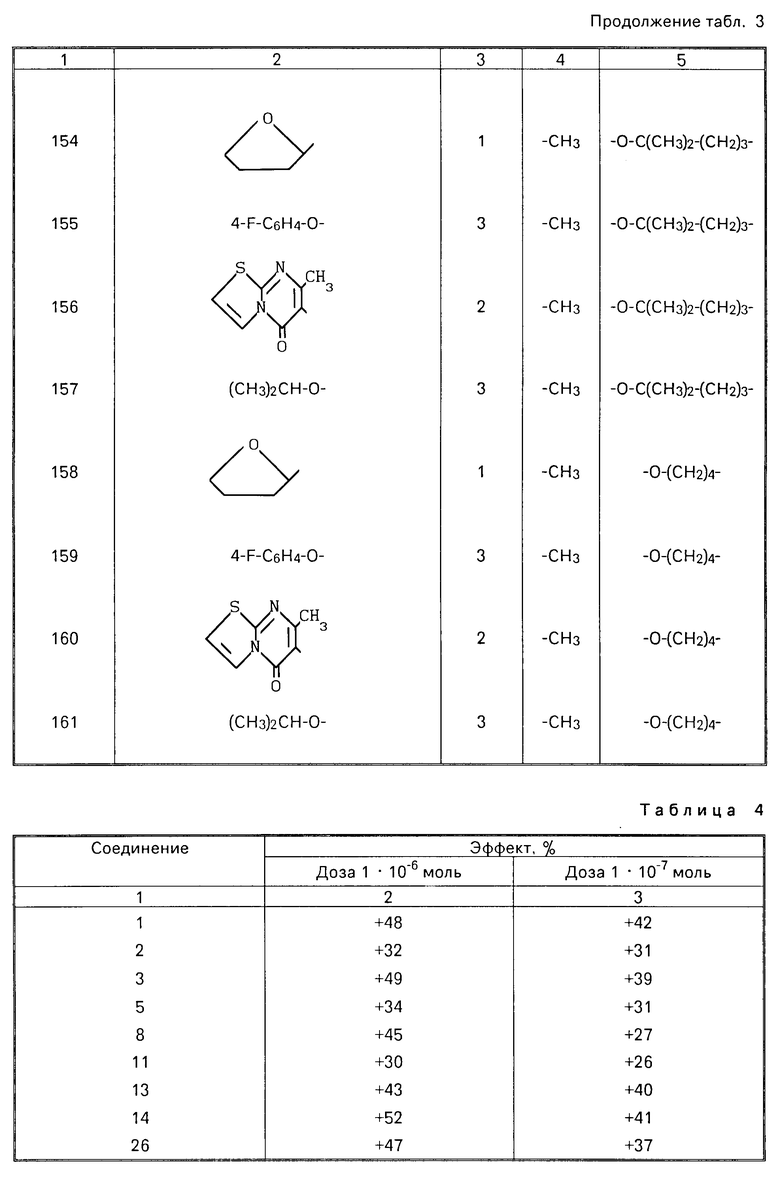
Т. пл. 143,5оС. Общий выход: 3,22 мас.ч. (51,1%) продукта (соединение 128).
П р и м е р 23. Смесь 5,4 мас.ч. цис-4-амино-5-хлор-2,3-дигидро-N-[3-метокси-1- [3-(2-метил-1,3-диоксолан-2-ил)пропил] -4-пиперидинил] -2,2-диметил-7-бензофур ан- карбоксамида и 85 мл водного 1% раствора серной кислоты перемешивалась в течение 2 ч при температуре дефлегмации. После охлаждения реакционная смесь подщелачивалась аммиаком и экстрагировалась дихлорметаном (2 раза). Объединенные экстракты сушились, фильтровались и выпаривались. Остаток очищался с помощью хроматографии на колонке (силикагель; СН2Cl2/CH3OH 95 5). Элюент желаемой фракции выпаривался и остаток суспендировался в 2,2'-оксибиспропане. Продукт отфильтровывался и сушился с получением 2,4 мас.ч. (51,6%) полугидрата цис-4-амино-5-хлор-2,3-дигидро-N-[3-метокси-1-(4-оксопе- нтил)-4-пиперидинил]-2,2-диметил-7-бензо- фуранкарбоксамида. Т.пл. 137,7оС (соединение 112).
П р и м е р 24. Смесь 6,3 мас.ч. соединения 41, 23,4 мас.ч. 2-пропанола, насыщенного соляной кислотой и 198 мас.ч. метанола перемешивалась в течение 15 мин при температуре обратного потока. После охлаждения реакционная смесь выпаривалась. Остаток поглощался водой и все подщелачивалось аммиаком. Продукт экстрагировался трихлорметаном и экстракт сушился, фильтровался и выпаривался. Остаток кристаллизовался из ацетонитрила (к которому добавлялось несколько капель воды). Продукт отфильтровывался при 0оС и сушился в вакууме при 40оС с получением 3,8 мас.ч. (72,9%) моногидрата цис-4-амино-5-хлор-2,3-дигидро-N-[3-метокси-1-[2-(метиламино)этил] -4-пипери- динил]-7-бензофуранкарбоксамида (соединение 42).
П р и м е р 25. К охлажденному на ледяной бане раствору 2,4 мас.ч. цис-4-амино-N-[1-[2-аминоэтил] -3-метокси-4-пи- перидинил] -5-хлор-2,3-дигидро-7-бензо- фуранкарбоксамида в 74,5 мас.ч. трихлорметана добавляют 0,86 мас.ч. N,N-диэтилэтанамина, а затем по каплям прибавляют раствор 0,77 мас.ч. этилхлорформата в 29,8 мас.ч. трихлорметана. После 0,5 ч перемешивания при комнатной температуре прибавляют воду. Отделяют органический слой, сушат, фильтруют и выпаривают. Остаток очищают на хроматографической колонке (силикагель, СH2Cl2/CH3OH(NH3) 98 2). Элюент целевой фракции выпаривают и кристаллизуют остаток из ацетонитрила (к которому прибавлено несколько капель воды). Продукт фильтруют при 0оС и сушат в вакууме при 40оС, получают 0,95 мас. ч. (32,5%) полу-гидрата этил-цис-[2-[4[(4-амино-5-хлор-2,3-дигидро-7-бензофуранил)карбонилами- но]-3-метокси-1-пиперидинил]этил]карба- мата (соединение 36).
Т.пл. 145,2оС.
Соединения, приведенные в табл. 2, приготавливались согласно аналогичным способам, которые описаны в любом из предыдущих примеров 5-25.
П р и м е р 26. Соединения, приведенные в табл. 3, приготовлялись согласно аналогичным способам, которые описаны в любом из представленных примеров.
С. Фармакологические примеры.
Полезные свойства предлагаемых соединений стимулировать желудочно-кишечную перистальтику, в частности их способность усиливать сократительную способность толстой кишки могут быть продемонстрированы с помощью следующего испытания.
П р и м е р 27. Сокращения, вызываемые восхождением толстой (ободочной) кишки.
Эксперимент проводился согласно следующей методике. Сегменты толстой кишки длиной 4,5 см вертикально суспендировались с предварительной нагрузкой 2 г в 100 мл раствора Де Jalon (KCl 5,6 ммоль; CaCl2 x x 2H2O 0,54 ммоль; NaHCO3 5,9 ммоль; NaCl 154,1 ммоль; глюкоза 2,8 ммоль) при температуре 37,5оС и насыщении газовой смесью 95% O2 и 5% CO2. Сокращения измерялись изотонически с помощью контрольной установки датчика смещения НР 7 ДСДТ-1000, JSID.
После периода стабилизации около 20 мин давалось 3,4 х 10-6 моль метахолина с интервалом времени в 15 мин. Когда получались воспроизводимые сокращения, в промывочный раствор вводилось испытываемое соединение. Эффект соединения исследовался в течение 10 мин и выражался относительно максимальных концентраций, вызываемых метахолином в количестве 3,4 х 10-6 моль. Эффект для характерных представителей ряда соединений формулы (I) показан в табл. 4 и 5.
Соединения A, B, C, E и F описаны в [1] соединение D в [2]
Д. Примеры композиций.
П р и м е р 28. Капли для орального приема.
500 мас. ч. активного ингредиента растворялось в 0,5 л 2-гидроксипропановой кислоты и 1,5 л полиэтиленгликоля при 60-80оС. После охлаждения до 30-40оС добавлялось 35 л полиэтиленгликоля и смесь хорошо перемешивалась. Затем добавлялся раствор 1750 мас.ч. сахариннатрия в 2,5 л очищенной воды и при перемешивании добавлялось 2,5 л ароматизирующего вещества какао и сколько нужно полиэтиленгликоля до объема 50 л, давая раствор оральных капель, содержащий 10 мг/мл активного ингредиента. Получающийся раствор заполнялся в подходящие контейнеры.
П р и м е р 29. Раствор для орального приема.
9 мас. ч. метил-4-гидроксибензоата и 1 мас.ч. пропил-4-гидроксибензоата растворялись в 4 л кипящей очищенной воды. В 3 л данного раствора растворялись сначала 10 мас.ч. 2,3-дигидроксибутандионовой кислоты, после 20 мас.ч. активного ингредиента. Этот раствор объединялся с оставшейся частью первого раствора и добавлялось 12 л 1,2,3-пропантриола и 3 л 70% раствора сорбита. 40 мас.ч. сахариннатрия растворялось в 0,5 л воды и добавлялось 2 мл малиновой и 2 мл крыжовниковой эссенции. Последний раствор объединялся с первым, добавлялась вода в нужном количестве до объема 20 л, давая оральный раствор, содержащий 5 мг активного ингредиента на полную чайную ложку (5 мл). Получающийся раствор заполнялся в подходящие контейнеры.
П р и м е р 30. Капсулы.
20 мас.ч. активного ингредиента, 6 мас.ч. лаурилсульфата натрия, 56 мас. ч. крахмала, 56 мас.ч. лактозы, 0,8 мас.ч. коллоидной двуокиси кремния и 1,2 мас. ч. стеарата магния энергично перемешивались вместе. Получающаяся смесь впоследствии заполнялась в 1000 подходящих затвердевших желатиновых капсул, содержащих 20 мг активного ингредиента каждая.
П р и м е р 31. Таблетки, покрытые пленкой.
Получение ядра таблеток. Смесь 100 мас.ч. активного ингредиента, 570 мас. ч. лактозы и 200 мас.ч. крахмала хорошо перемешивалась, после этого увлажнялась раствором 5 мас.ч. додецилсульфата натрия и 10 мас.ч. поливинилпирролидона (Kollidon К 90®) в примерно 200 мл воды. Смесь влажного порошка просеивалась, сушилась и просеивалась снова. Затем к ней добавлялось 100 мас. ч. микрокристаллической целлюлозы (Avicel) и 15 мас.ч. гидрированного растительного масла (Sterotex). Все хорошо смешивалось и прессовалось в таблетки, давая 10000 таблеток, содержащих 10 мг активного ингредиента каждая.
Покрытие. К раствору 10 мас.ч. метилцеллюлозы (Methocel 60 HG) в 75 мл денатурированного этанола добавлялся раствор 5 мас.ч. этилцеллюлозы (Ethocel 22 cps) в 150 мл дихлорметана. Затем к нему добавлялось 75 мл дихлорметана и 2,5 мл 1,2,3-пропантриола. 10 мас.ч. полиэтиленгликоля расплавлялось и растворялось в 75 мл дихлорметана. Последний раствор добавлялся к первому, затем добавлялось 2,5 мас.ч. октадеканоата магния, 5 мас.ч. поливинилпирролидона и 30 мл концентрированной суспензии (Opasprax K-1-2109) и все гомогенизировалось. Ядра таблеток покрывались полученной смесью в покрывающем устройстве.
П р и м е р 32. Инъецируемый раствор.
1,8 мас. ч. метил-4-гидроксибензоата и 0,2 мас.ч. пропил-4-гидроксибензоата растворялись примерно в 0,5 л кипящей воды для инъекций. После охлаждения до примерно 50оС при перемешивании добавлялось 4 мас.ч. молочной кислоты, 0,05 мас. ч. пропиленгликоля и 4 мас.ч. активного ингредиента. Раствор охлаждался до комнатной температуры и в него добавлялась вода для инъекций в необходимом количестве до 1 л, давая раствор, содержащий 4 мг/мл активного ингредиента. Раствор стерилизовался фильтрацией (U.S.P. XVII стр. 811) и заполнялся в стерильные контейнеры.
П р и м е р 33. Медицинские свечи или суппозитории.
3 мас. ч. активного ингредиента растворялось в растворе 3 мас.ч. 2,3-дигидроксибутандионовой кислоты в 25 мл полиэтиленгликоля 400. 12 мас.ч. поверхностно-активного вещества (SPAN) и триглицериды (Witepsol 555) в необходимом до 300 мас.ч. количестве расплавлялись вместе. Последняя смесь хорошо перемешивалась с прежним раствором. Полученная таким образом смесь выливалась в формы при температуре 37-38оС для формирования 100 суппозирориев, содержащих 30 мг/мл активного ингредиента каждый.
П р и м е р 34. Инъецируемый раствор.
60 мас. ч. активного ингредиента и 12 мас.ч. бензилового спирта хорошо перемешивались и добавлялось кунжутное масло в количестве необходимом для доведения смеси до 1 л, давая раствор, содержащий 60 мг/мл активного ингредиента. Раствор стерилизовался и заполнялся в стерильные контейнеры.













Использование: в медицине, в качестве стимулятора желудочно-кишечной перистальтики. Сущность изобретения продуктопроизводные N-(3-гидрокси-4-пиперидинил)-(дигидробензофуран, дигидро-2Н-бензопиран или дигидробензодиоксин) карбоксамид ф-лы I, где R1, R2, R3, L и A имеют соответсвующие значения. Реагент 1: соединение ф-лы II, где R1, R2, R3, L и A имеют соответсвующие значения. Реагент 2: L - W, L - имеют соответствующие значения. W - атом галогена или сульфонилоксигруппа; или альдегид L′=0, где L′=0 являются соединением ф-лы L - H, у которого два соседних атома водорода в C1-C6 алкандииле или C3-C6 циклоалкандииле заменены = 0, или алкен ф-лы NC-CH=CH2. Процесс проводят в среде реакционно инертного растворителя, необязательно в присутствии основания, иодидной соли или восстанавливающего агента с последующими превращениями одного целевого продукта в другой по желанию и выделением целевого продукта в свободном виде или в виде терапевтически активной нетоксичной соли, или в виде стереохимически изомерной формы. Структура ф-л I и II (см. чертеж). 1 ил., 5 табл.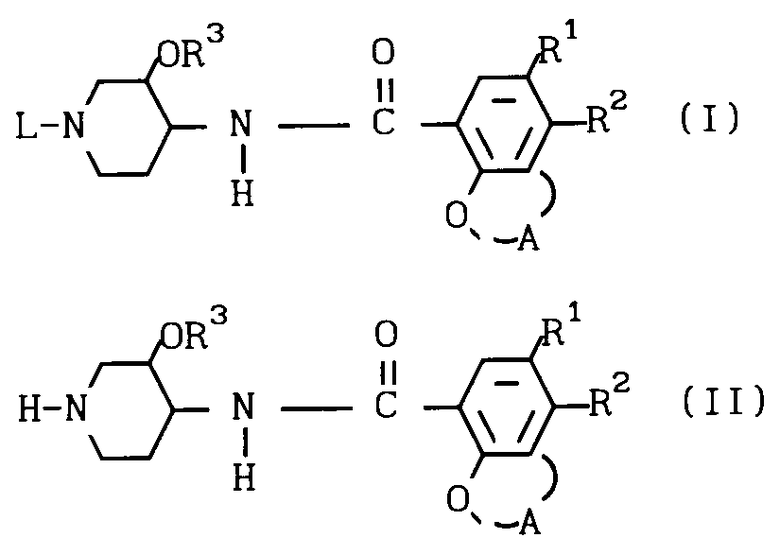
Способ получения производных N-(3-гидрокси-4-пиперидинил)-(дигидробензофуран, дигидро-2Н-бензопиран или дигидробензодиоксин)-карбоксамида общей формулы I
их солей или стереохимически изомерной формы, где А радикал формулы
-СН2 СН2- (а 1);
-СН2 СН2 СН2- (а 2);
или
-СН2 СН2 О- (а 3)
причем один или два атома водорода в указанном радикале (а 1) могут быть замещены С1 С4-алкильным радикалом;
R1 галоген;
R2 аминогруппа;
R3 водород или С1 С4-алкил;
L С3 С6-циклоалкил, С3 С6-алкенил или L - радикал формулы
-Alk R5 (в 1);
-Alk X R6 (в 2);
-Alk Y C/=O/ R8 (в 3)
или
-Alk Y C/=O/ NR10R11 (в 4)
где каждый Alk С1 С6-алкандиил;
R5 водород, циано С3 С6-циклоалкил, фенил, необязательно замещенный атомом галогена или Het;
R6 водород, С1 С6-алкил, С3 - С6-циклоакил, галоидфенил, необязательно замещенный С1 - С4-алкилкарбонилом, 3-циано-2-пипиридинилом, 2-метил-5-пиридилом, 4-гидрокси-2-пиримидинилом, 2-метил-3-пиразинилом или 3,4-дигидро-4-оксо-2-хиназолинилом;
Х О или NH;
R8 водород, С1 С6-алкил, 2,4,6-триметоксифенил, 3,4,5-триметоксифенил, 2,6-дихлорфенил или С1 С6-алкокси,
Y NR9 или простая связь;
R9 водород, С1 С4-алкил или фенил;
R10 и R11 каждый независимо С1 - С6-алкил
или R10 и R11, взятые вместе с атомом азота, к которому они присоединены, могут образовывать пирролидинильное кольцо;
Het система простого циклического эфира, выбранная в группе, состоящей из
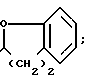

где R12 водород или С1 С4-алкил,
или Het гетероциклическая система, выбранная в группе, состоящей из пиридинила или бензимидазолила, замещенного C1 C6-алкилом,
или Het моноциклическая амидная система, выбранная в группе, состоящей из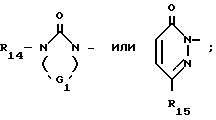
где R14 водород или С1 С6-алкил;
R15 галоген, С1 С6-алкил или фенил;
G1 -СН2 СН2-, СН СН-, или -С(=О)-СН2-;
или Het бициклическая амидная система, выбранная в группе, состоящей из



где R16 С1 С6-алкил или фенилметил;
R17 С1 С6-алкил;
R18 водород или галоген;
G3 -S (CH2)2- или -S CH CH-;
G4 -CH CH CH CH-, -CH CCl CH CH-, -CH N CH CH- или -N CH N CH-;
или их солей, или их стереохимически изомерной формы, отличающийся тем, что осуществляют N-алкилирование пиперидина формулы II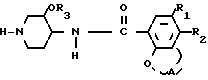
где R1, R2, R3 и А имеют указанные значения,
промежуточным продуктом формулы III
L W,
где L имеет указанные значения;
W галоген или сульфонилоксигруппа,
или альдегидом формулы IV
L' 0,
где L' 0 является соединением формулы L H, у которого два соседних атома водорода в С1 С6-алкандииле или С3 - С6-циклоалкандииле замененыО,
или алкеном формулы V NC CH CH2, в реакционно-инертном растворителе, необязательно в присутствии основания, иодидной соли или восстанавливающего агента
и необязательно, при желании, восстанавливают соединение формулы I-с
где R1, R2, R3, А и Alk имеют указанные значения,
в реакционно-инертном растворителе в присутствии катализатора и в атмосфере водорода, в результате получают соединение формулы I-d
которое подвергают взаимодействию с соединением формулы VI
RW,
где R R6 C(= O) R8 или -C(=O) R10, R11, где R6, R8, R10 и R11 имеют указанные значения;
W галоген или метилтиогруппа,
необязательно в реакционно-инертном растворителе, необязательно в присутствии основания, при этом получают соединение формулы I-е
где R, R1, R2, R3, R9, Alk и А имеют указанные значения,
или, при желании, проводят деацетализацию соединения формулы 1-f
где R1, R2, R3, R8, Alk и А имеют указанные значения,
в реакционно-инертном растворителе в присутствии кислоты, при этом получают соединение формулы 1-g
или, при желании, превращают соединение формулы 1-h
где R1, R2, R3, R6, Alk и А имеют указанные значения,
в реакционно-инертном растворителе в присутствии кислоты, при этом получают соединение формулы 1-i
и, при желании, превращают соединение формулы I в его терапевтически активную нетоксичную соль при обработке кислотой или, наоборот, превращают кислую соль в свободное основание обработкой щелочью и/или получают стереохимически изомерную форму.
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| СПОСОБ ЭЛЕКТРОЛИТИЧЕСКОГО ОСАЖДЕНИЯ СПЛАВА КАДМИЙ—ОЛОВО | 0 |
|
SU299566A1 |
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
| Механизм для сообщения поршню рабочего цилиндра возвратно-поступательного движения | 1918 |
|
SU1989A1 |

