Изобретение касается новых азольных противогрибковых средств широкого спектра действия и их получения; оно дополнительно относится к их составу, так же как к их применению в качестве лекарственного средства.
Системные грибковые инфекции человека относительно редки в странах умеренного климата, и многие из грибков, которые могут становиться патогенными, обычно сосуществуют в организме человека или являются обычными для окружающей среды. Однако в последние десятилетия появились доказательства увеличивающихся случаев распространения по всему миру многочисленных угрожающих жизни системных грибковых инфекций, и они сейчас представляют наибольшую угрозу для многих восприимчивых больных, особенно тех, которые уже госпитализированы. Большая часть этого увеличения связана с улучшением жизнеспособности людей с пониженным иммунитетом и с постоянным применением противомикробных препаратов.
Более того, типичная флора для многих обычных грибковых инфекций также изменяется, что представляет собой эпидемиологический сдвиг, имеющий все возрастающее значение. Больные с наибольшим риском включают тех, которые имеют нарушения функций иммунной системы, возникающие либо первично в результате иммуносупрессии, вызываемой цитотоксическими лекарствами или ВИЧ-инфекцией, либо вторично по отношению к другим ослабляющим болезням, таким как рак, острая лейкемия, инвазивные хирургические вмешательства или продолжительное применение антимикробных препаратов.
Наиболее обычными системными грибковыми инфекциями у человека являются кандидоз, аспергиллез, гистоплазмоз, кокцидоидомикоз, паракокцидоидомикоз, бластомикоз и криптококкоз.
Для лечения и профилактики системных грибковых инфекций у больных с ослабленным иммунитетом применяют такие противогрибковые средства, как кетоканазол, итраконазол и флуконазол. Однако растет беспокойство по поводу устойчивости грибков к некоторым из этих агентов, особенно тех из них, которые обладают более узким спектром действия, например флуконазолу. Хуже того, в медицинском мире известно, что около 40% людей, страдающих от тяжелых системных грибковых инфекций, могут с трудом или совсем не могут применять лекарства перорально. Эта неспособность обусловлена тем фактом, что такие больные находятся в коме или страдают от тяжелого гастропареза. Следовательно, применение нерастворимых или малорастворимых антигрибковых средств, таких как итраконазол, которые трудно ввести внутривенно, крайне затруднено у указанной группы больных.
Следовательно, существует потребность в новых антигрибковых препаратах, предпочтительно широкого спектра противогрибкового действия, против которых не существует устойчивости и которые можно вводить внутривенно. Предпочтительно противогрибковые препараты должны также быть пригодны для фармацевтических композиций, которые подходят для перорального введения. Это позволяет врачу продолжать лечение тем же лекарством после того, как больной выйдет из состояния, при котором требуется внутривенное введение указанного лекарства.
В US-4267179 раскрыты гетероциклические производные (4-фенилпиперазин-1-ил-арилокси-метил-1,3-диоксолан-2-ил)-метил-1Н-имидазолов и 1Н-1,2,4-триазолов, применимые в качестве противогрибковых средств. Указанный патент охватывает итраконазол, который сейчас является широко распространенным по всему миру противогрибковым средством широкого спектра действия.
US-4916134 указывает на 4-[4-[4-[(2,4-дифторофенил)-2-(1Н-азолилметил)-1,3-диоксолан-4-ил] метокси] фенил] -1-пиперазинил] фенил] триазолоны, имеющие улучшенные антимикробные свойства.
В US-4791111 раскрыты производные [[4-[4-(4-фенил-1-пиперазинил)феноксиметил] -1,3-диоксолан-2-ил] -1Н-имидазолов и 1Н-1,2,4-триазолов, структурно близких к некоторым соединениям настоящего изобретения, где указывается на то, что они имеют улучшенные антимикробные свойства. Особенно соединение, которое представляет собой здесь цис-4-[4-[4-[4-[[2-(2,4-дифторофенил)-2-(1Н-1,2,4-триазол-1-илметил)-1,3-диоксолан-4-ил] -метокси] -фенил] -1-пиперазинил] фенил] -2,4-дигидро-2-(2-гидрокси-1-метилпропил)-3Н-1,2,4-триазолон, причем указанное соединение является смесью стереоизомеров всех возможных энантиомеров и диастереомеров, имеющих цисконфигурацию у 1,3-диоксоланового кольца.
В WO 93/19061 раскрыты [2R-[2α, 4α, 4(R*)]], [2R-[2α, 4α, 4(S*)]], [2S-[2α, 4α, 4(S*)] ] и [2S-[2α, 4α, 4(R*)]] стереоспецифические изомеры итраконазола, которые, как указано, имеют большую растворимость в воде, чем соответствующие их диастереомерные смеси.
В WO 95/19983 раскрыты производные [[4-[4-(4-фенил-1-пиперазинил)феноксиметил]1,3-диоксолан-2-ил]метил]-1Н-имидазолы и 1Н-1,2,4-триазолы, структурно сходные с некоторыми соединениями настоящего изобретения, которые, как указывается, являются водорастворимыми антимикробными соединениями.
В WO 95/17407 раскрыты тетрагидрофурановые противогрибковые средства, так же как в WO 96/38443 и в WO 97/00255. В последних двух публикациях раскрыты тетрагидрофурановые противогрибковые средства, которые, как указывается, являются растворимыми и/или суспендируемыми в водной среде и пригодными для в/в введения и содержат группы-заместители, которые легко превращаются in vivo в гидроксильные группы.
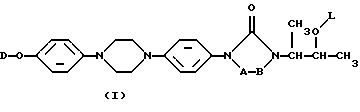
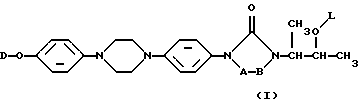
Настоящее изобретение касается новых соединений формулы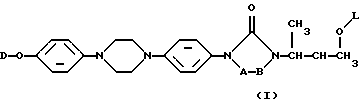
их N-оксидных форм, их фармацевтически приемлемых аддитивных солей и их стереохимически изомерных форм, где -А-В- образуют двухвалентный радикал формулы
-N=СН- (а);
-CH=N- (b);
-СН=СН- (с),
где один атом водорода в радикалах (а) и (b) может быть замещен C1-6алкильным радикалом и до двух атомов водорода в радикале (с) может быть замещено C1-6алкильным радикалом;
L представляет собой ацильную часть аминокислоты и, таким образом, -O-L является группой сложного эфира аминокислоты;
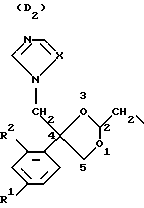
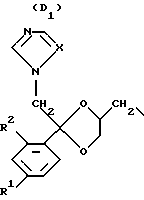
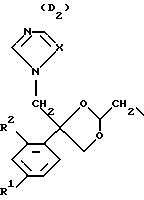
D является радикалом формулы (D1) или (D2)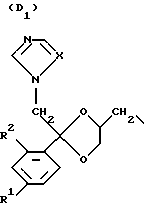
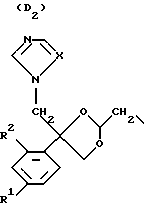
где X является N или СН;
R1 представляет собой галоген;
R2 является водородом или галогеном;
В определениях, представленных выше и ниже, термин галоген представляет собой фтор, хлор, бром и иод; C1-6алкил характерен для углеводородов с прямой или разветвленной цепью, имеющих от 1 до 6 атомов углерода, таких как, например, метил, этил, пропил, бутил, пентил или гексил, и их возможных разветвленных изомеров.
В определении L термин "аминокислота" обозначает, включая, но не ограничиваясь этим
- 20α-аминокислоты, обычно присутствующие в белках, такие как глицин, аланин, валин, лейцин, изолейцин, метионин, пролин, фенилаланин, триптофан, серии, треонин, цистеин, тирозин, аспарагин, глутамин, аспарагиновая кислота, глутаминовая кислота, лизин, аргинин и гистидин; и
- относительно редко встречающиеся аминокислоты, которые были идентифицированы в определенных типах белков, такие как, например, 4-гидроксипролин, гидроксилизин, десмозин и изодесмозин; и
- более 150 других аминокислот, присутствующих в биологическом материале в свободной или комбинированной форме, но никогда не присутствующих в белках, являются ли они α-, β-, γ- или δ- аминокислотами, или они имеют L- или D-конфигурацию, такие как, например, β-аланин, гомоцистеин и гомосерин, цитруллин, орнитин, γ-аминомасляная кислота, D-глутаминовая кислота и D-аланин; и
- синтетические аналоги аминокислот, такие как, например, фенилглицин, п-фторофенилаланин, тионин, норлейцин и тому подобное.
В определении L термин "аминокислота" также подразумевает включение тех аминокислот, в которых аминная часть является одно- или двузамещенной; в таких соединениях L может быть представлен -L'-NRXRY. Примеры RX и RY включают водород, C1-6алкил и известные специалистам в этой области защитные группы для аминной части, например трет-бутилоксикарбонильная, бензоилоксикарбонильная, трифторометоксикарбонильная или те защитные группы, которые упоминаются в главе 7 "Protective Groups in Organic Synthesis" by T.Greene и P.Wuyts (John Wiley & Sons, Inc.1991). RX и RY могут также образовывать совместно с атомом азота аминной части аминокислоты кольцо, такое, например, как пирролидиновое, пиперидиновое, морфолиновое, пиперазиновое или замещенное пиперазиновое кольцо, причем указанный замещенный пиперазин представляет собой пиперазиновое кольцо, замещенное в 4-м положении пиперазинового кольца, например, C1-6алкилом, гидроксиС1-6алкилом, аминоС1-6алкилом, моно- или ди(C1-6алкил)аминоС1-6алкилом.
Например, в случае если L представляет собой ацильную часть N,N-диэтилглицина, то L' представляет собой -С(=O)-CH2- и -NRXRY представляет собой -N(СН2СН3)2.
Многие аминокислоты коммерчески доступны и перечислены в Novabiochem's 1997/1998 Catalog & Peptide Synthesis Handbook (Calbiochem-Novabiochem AG, Laufelfingen, Switzerland). Подразумевается, что эти коммерчески доступные аминокислоты также включаются в термин "аминокислота", как это применяется в определении L.
Подразумевается, что фармацевтически приемлемые аддитивные соли, как упоминалось здесь выше, включают терапевтически активные нетоксичные кислые формы аддитивных солей, которые способны образовывать соединения формулы (I). Последние удобно получать путем обработки основной формы такими подходящими кислотами, как неорганические кислоты, например галогенводородные кислоты, например хлористоводородная, бромистоводородная и т.п.; серная кислота; азотная кислота; фосфорная кислота и т.п.; или органические кислоты, например уксусная, пропионовая, гликолевая, 2-гидроксипропионовая, 2-оксопропионовая, щавелевая, малоновая, фумаровая, яблочная, винная, 2-гидрокси-1,2,3-пропантрикарбоновая, метансульфоновая, этансульфоновая, бензолсульфоновая, 4-метилбензолсульфоновая, циклогексансульфаминовая, 2-гидроксибензойная, 4-амино-2-гидроксибензойная и подобные этим кислоты. И, наоборот, солевая форма может быть превращена в свободную основную форму путем обработки щелочью.
Соединения формулы (I), содержащие кислотные протоны, могут быть превращены в их терапевтически активные нетоксичные металло- или аминоформы аддитивных солей путем обработки подходящими органическими и неорганическими основаниями. Подходящие основные формы солей включают, например, соли аммония, соли щелочных и щелочноземельных металлов, например соли лития, натрия, калия, магния, кальция и т.п., соли с органическими основаниями, например бензатин, N-метил-D-глюкамин, 2-амино-2-(гидроксиметил)-1,3-пропандиол, соли гидрабамина и соли с аминокислотами, такими как, например, аргинин, лизин и тому подобное. И наоборот, солевая форма может быть превращена в свободную кислую форму путем обработки кислотой.
Термин аддитивная соль также включает гидраты и сольвентные аддитивные формы, которые способны образовывать соединения формулы (I). Примерами таких форм являются, например, гидраты, алкоголяты и тому подобное.
Подходящие солевые формы соединений настоящего изобретения включают фумаровую, янтарную, L-яблочную, щавелевую, малеиновую, L-винную и гидрохлоридную кислую форму соли, так же как гидратированные формы.
Термин "стереохимически изомерные формы", употребляемый здесь ранее, обозначает все возможные стереоизомерные формы, в которых могут существовать соединения формулы (I), включая также все энантиомеры, энантиомерные смеси и диастереомерные смеси. Если специально не упоминается или не указывается, химическое обозначение соединений подразумевает смесь всех возможных стереохимически изомерных форм, причем указанные смеси содержат все диастереомеры и энантиомеры основной молекулярной структуры. То же самое применимо к описываемым здесь промежуточным продуктам, используемым для получения конечных продуктов формулы (I).
Энантиомерно чистые формы соединений и промежуточных продуктов, как здесь указывается, определяются как по существу свободные от других энантиомерных или диастереомерных форм указанных соединений или промежуточных продуктов той же основной молекулярной структуры.
Асимметрические центры могут иметь R- или S-конфигурацию. Термины цис- и транс- применяются здесь в соответствии с номенклатурой Chemical Abstracts и относятся к положению заместителей в кольцевой части, более конкретно в диоксолановом кольце соединений формулы (I). В последнем случае, когда устанавливают цис- или транс- конфигурацию, рассматриваются заместитель с наибольшим старшинством у углеродного атома во 2-м положении диоксоланового кольца и заместитель с наибольшим старшинством у углеродного атома в 4-м положении диоксоланового кольца (причем старшинство заместителя определяется в соответствии с правилами последовательности Кан-Ингольд-Прелог). Когда два указанных заместителя с наибольшим старшинством находятся по одну и ту же сторону кольца, то конфигурацию обозначают как цис, если это не так, конфигурацию обозначают как транс.
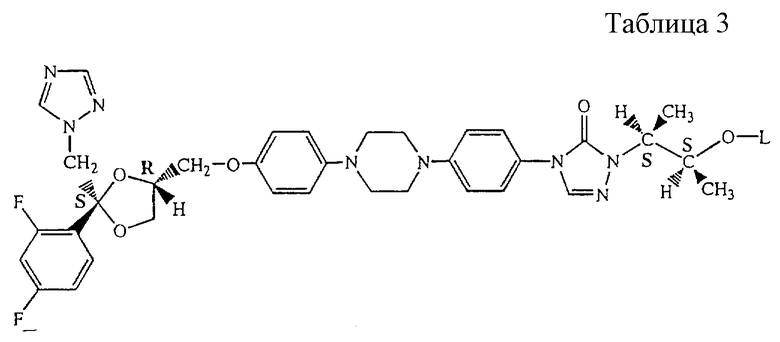
Все соединения формулы (I) содержат по меньшей мере 4 асимметрических центра. Употребляемые здесь стереохимические обозначения, обозначающие стереохимическую конфигурацию каждого из 4-х или более асимметрических центров, даны также в соответствии с номенклатурой Chemical Abstracts. Например, абсолютная конфигурация асимметричных углеродных атомов соединения 23, как описано здесь далее в примере В.2, то есть [2S-[2α, 4 [(R*, R*)]]]-2-[4-[4-[4-[4-[[2-(2,4-дифторофенил)-2-(1Н-1,2,4-триазол-1-илметил)-1,3-диоксолан-4-ил] метокси] -фенил] -1-пиперазинил] -фенил]-4,5-дигидро-5-оксо-1Н-1,2,4-триазол-1-ил] -1-метилпропил L-фенилаланина изображена здесь ниже. Диоксолановое кольцо в этом соединении имеет цис-конфигурацию.
[(R*, R*)]]]-2-[4-[4-[4-[4-[[2-(2,4-дифторофенил)-2-(1Н-1,2,4-триазол-1-илметил)-1,3-диоксолан-4-ил] метокси] -фенил] -1-пиперазинил] -фенил]-4,5-дигидро-5-оксо-1Н-1,2,4-триазол-1-ил] -1-метилпропил L-фенилаланина изображена здесь ниже. Диоксолановое кольцо в этом соединении имеет цис-конфигурацию.
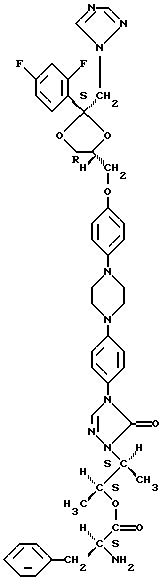
Далее в соответствии с номенклатурой Chemical Absracts за названием радикала следует название аминокислоты, с которой образуется эфир, в котором эта аминокислота представляет ацильную группу. Например, в соединении 23 L-фенилаланин этерифицируется с указанной замещенной 1-метилпропильной группой.
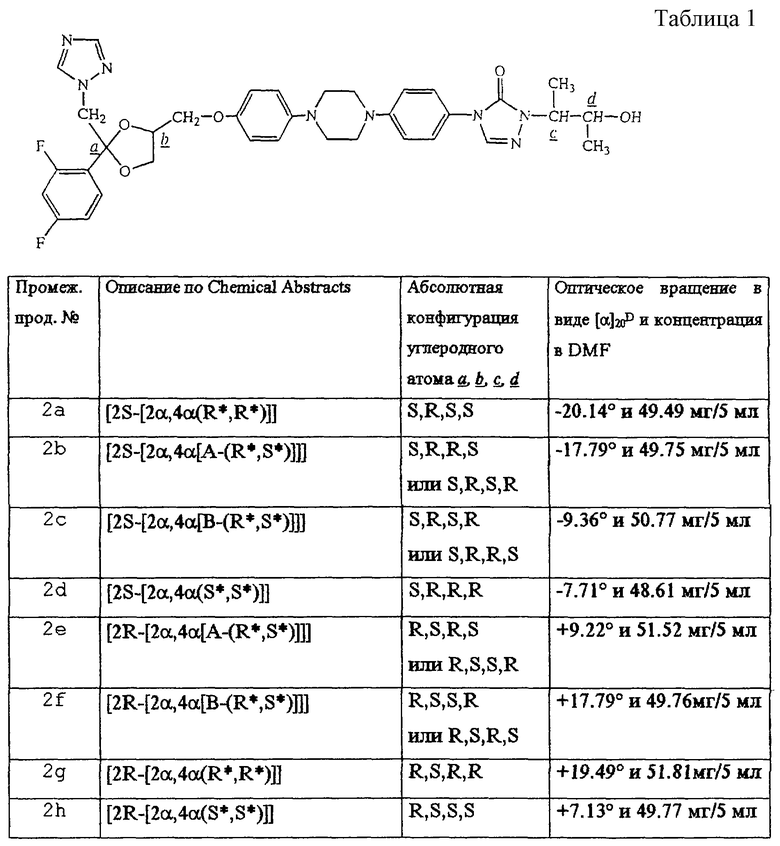
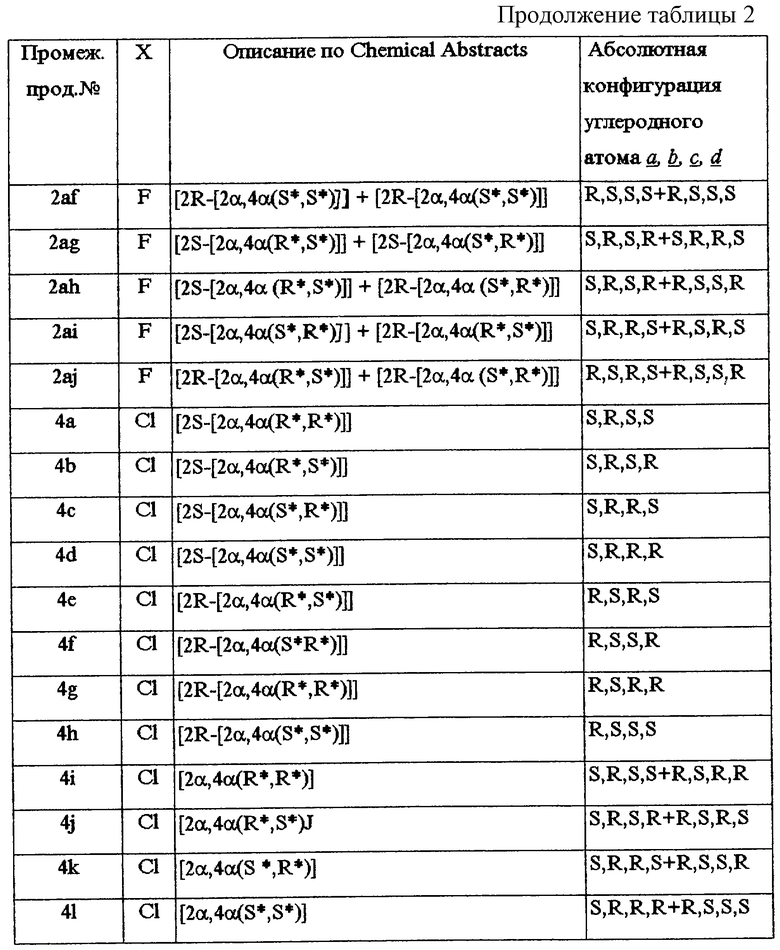
Та же номенклатура Chemical Absracts употребляется для обозначения смесей энантиомеров. Например, обозначение промежуточного продукта 2i, то есть [2α, 4α(R*, R*)] , указывает на то, что промежуточный продукт 2i представляет собой смесь двух энантиомеров, описываемых стереохимически как [2S-[2α, 4α(R*, R*)]] и [2R-[2α, 4α(R*, R*)]] соответственно.
Нумерация атомов в диоксолановом кольце в соответствии с номенклатурой Chemical Absracts дана ниже для радикалов D1 и D2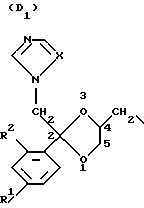
У некоторых соединений формулы (I) и промежуточных продуктов, применяемых при их получении, не определяли экспериментально абсолютную стереохимическую конфигурацию. В этих случаях стереохимически изомерная форма, которая была выделена в первую очередь, обозначалась как "А", а во вторую очередь как "В", без дальнейшего указания их действительной стереохимической конфигурации. Однако указанные изомерные формы "A" и "В" могут недвусмысленно характеризоваться по, например, их оптическому вращению, которое в случае "А" и "В" имеет энантиомерное отношение. Специалисты в этой области в состоянии определить абсолютную конфигурацию таких соединений, применяя для этого хорошо известные способы, такие как, например, дифракция рентгеновских лучей.
Например, промежуточный продукт 2b, описываемый стереохимически как [2S-[2α, 4α[А-(R*, S*)] ] ] , обозначает энантиомер, имеющий либо [2S-[2α, 4α[(R*, S*)]]], либо [2S-[2α, 4α[(S*, R*)]]] конфигурацию, и недвусмысленно характеризуется по его оптическому вращению, составляющему [α]
Подразумевается, что N-оксидные формы соединений настоящего изобретения включают соединения формулы (I), в которых один или несколько атомов азота окислены до так называемого N-оксида.
Применяемый здесь далее термин "соединения формулы (I)" обозначает также то, что в него включаются N-оксидные формы, их фармацевтически приемлемые аддитивные соли и их стереохимически изомерные формы.
В плане настоящего изобретения -А-В- представляет собой подходящий радикал формулы (b).
D является подходящим радикалом формулы D1.
Х является подходящим N.
R1 и R2 соответственно являются идентичными, предпочтительно хлор- или фтор-. В частности, оба R1 и R2 представляют собой фтор.
Интересной группой соединений настоящего изобретения являются те соединения формулы (I), в которых L представляет собой радикал формулы (а)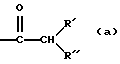
где R' представляет собой амино; моно- или ди(C1-6алкил)амино; амино(C1-6алкил); C1-6алкилоксикарбониламино; бензилоксикарбониламино; трифторометоксикарбониламино; 1-пирролидинил; 1-пиперидинил; 4-морфолинил; 1-пиперазинил или 1-пиперазинил, замещенный C1-6алкилом, гидроксиС1-6алкилом, aминоC1-6алкилом или С1-6алкиламиноС1-6алкилом;
R'' представляет собой водород; C1-6алкил; арил; C1-6алкил, замещенный арилом, С1-6алкилтио, индолилом, амино, гидрокси, меркапто, аминокарбонилом, карбоксилом, гуанидинилом, имидазолилом;
или
R' и R'' совместно образуют -CH2-CH2-CH2-NH-;
арил является фенилом или фенилом, замещенным гидрокси или галогеном.
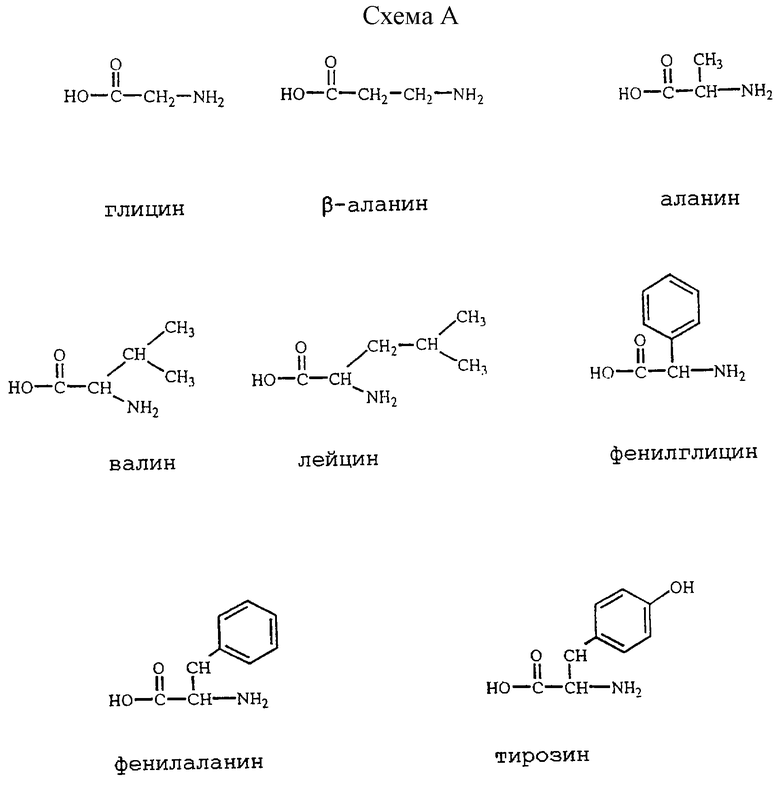
Более интересная группа содержит те соединения формулы (I), в которых L представляет собой ацильную часть одной из следующих аминокислот (см. схему A в конце описания).
или тех их производных, в которых аминная часть является одно- или двузамещенной C1-6алкилом или однозамещенной трет-бутилоксикарбонилом.
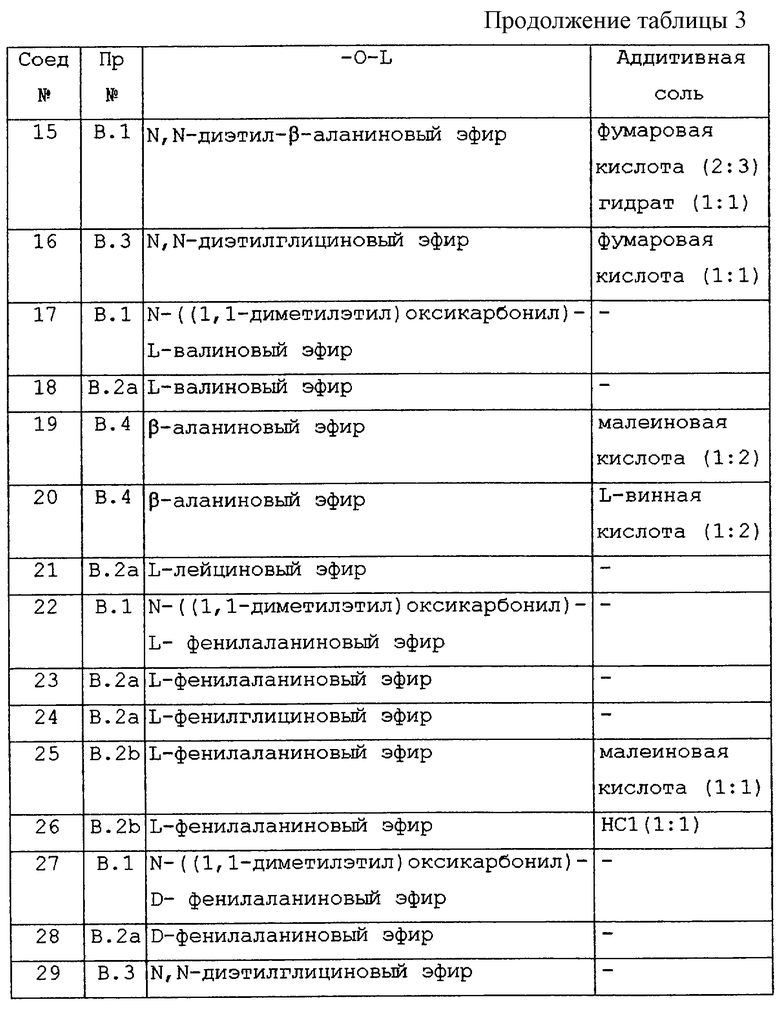
Особенно интересными ацильными частями являются те из них, которые происходят из аланина, β-аланина, глицина, лейцина, валина, фенилглицина, фенилаланина и их N-трет-бутилоксикарбонильного производного и N,N-диэтилглицина и N, N-диэтил-β-аланина; особенно глицина, β-аланина, L-аланина, L-валина, 1-лейцина, L-фенилглицина, L-фенилаланина, D-фенилаланина, N-((1,1-диметилэтил)оксикарбонил)-β-аланина, N-((1,1-диметилэтил)оксикарбонил)-глицина, N, N-диэтилглицина, N,N-диэтил-β-аланина, N-((1,1-диметилэтил)оксикарбонил)-L-аланина, N-((1,1-диметилэтил)оксикарбонил)-L-лейцина, N-((1,1-диметилэтил)оксикарбонил)-L-фенилглицина, N-((1,1-диметилэтил) оксикарбонил)-L-валина, N-((1,1-диметилэтил)оксикарбонил)-L-фенилаланина, N-((1,1-диметилэтил)оксикарбонил)-D-фенилаланина.
Особыми соединениями являются те соединения формулы (I), в которых D является радикалом формулы D1, где Х является N, а R1 и R2, оба представляют собой фтор; и -А-В- представляет собой радикал формулы (b); a L представляет собой ацильную часть лейцина, валина, фенилглицина, фенилаланина и их трет-бутилкарбонил производных; или L представляет собой ацильную часть N, N-диэтилглицина.
Другими особыми соединениями являются те соединения формулы (I), в которых D, является ли он D1 или D2, имеет цис- конфигурацию.
Предпочтительными соединениями являются те соединения, в которых D является радикалом формулы D1, в котором заместители в диоксолановом кольце имеют цис-конфигурацию и атом углерода номер 2 диоксоланового кольца имеет абсолютную S- конфигурацию, как изображено здесь ниже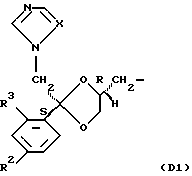
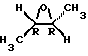
Другими предпочтительными соединениями являются те соединения, в которых 1-метилпропильная часть имеет трео- конфигурацию, то есть два хиральных углерода 1-метилпропильной части (оба хиральных атома углерода на фигуре, представленной ниже помечены звездочкой) имеют идентичные абсолютные конфигурации, например, оба они имеют R- конфигурацию или оба они имеют S-конфигурацию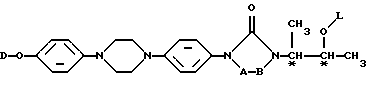
Более предпочтительными соединениями являются соединения формулы (I) в их энантиомерно чистых формах, в частности те соединения формулы (I), в которых два хиральных углерода 1-метилпропильной части оба имеют S-конфигурацию, а D является радикалом формулы D1, в котором заместители в диоксолановом кольце имеют цис-конфигурацию и атом углерода номер 2 диоксоланового кольца имеет абсолютную S-конфигурацию, которая соответствует тем соединениям формулы (I), в которых D является радикалом формулы D1, имеющим [2S-[2α, 4α[(R*, R*)]]] конфигурацию.
Наиболее предпочтительными являются соединения:
2-[4-[4-[4-[4-[[2-(2,4-дифторфенил)-2-(1H-1,2,4-триазол-1-илметил)-1,3-диоксолан-4-ил] метокси]фенил]-1-пиперазинил]фенил]-4,5-дигидро-5-оксо-1H-1,2,4-триазол-1-ил]-1-метилпропил N,N-диэтилглицин;
2-[4-[4-[4-[4-[[2-(2,4-дифторфенил)-2-(1Н-1,2,4-триазол-1-илметил)-1,3-диоксолан-4-ил] метокси] -фенил]-1-пиперазинил]-фенил]-4,5-дигидро-5-оксо-1H-1,2,4-триазол-1-ил]-1-метилпропил L-фенилаланин;
2-[4-[4-[4-[4-[[2-(2,4-дифторфенил)-2-(1H-1,2,4-триазол-1-илметил)-1,3-диоксолан-4-ил] метокси] -фенил]-1-пиперазинил]-фенил]-4,5-дигидро-5-оксо-1H-1,2,4-триазол-1-ил]-1-метилпропил L-лейцин;
2-[4-[4-[4-[4-[[2-(2,4-дифторфенил)-2-(1H-1,2,4-триазол-1-илметил)-1,3-диоксолан-4-ил] метокси] -фенил]-1-пиперазинил]-фенил]-4,5-дигидро-5-оксо-1H-1,2,4-триазол-1-ил]-1-метилпропил L-валин;
2-[4-[4-[4-[4-[[2-(2,4-дифторфенил)-2-(1H-1,2,4-триазол-1-илметил)-1,3-диоксолан-4-ил] метокси] -фенил]-1-пиперазинил]-фенил]-4,5-дигидро-5-оксо-1H-1,2,4-триазол-1-ил] -1-метилпропил L-фенилглицин; их N-оксидные формы, их фармацевтически приемлемые аддитивные соли и их стереохимические изомерные формы, особенно их [2S-[2α, 4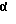 [(R*, R*)]]] форма.
[(R*, R*)]]] форма.
Обозначения переменных в том виде, в котором они применяются в следующей последовательности реакций, являются теми же, что и указанные выше, если это специально не оговорено.
Соединения настоящего изобретения могут быть получены с применением известных науке методов этерификации, например, тех из них, которые описаны в "Principles of Peptide Synthesis", M.Bodanszky, Springer-Verlag Berlin Heidelberg, 1984. Особые последовательности реакций описаны здесь ниже.
Соединения формулы (I) в общем виде могут быть получены путем О-ацилирования промежуточного спирта формулы (II) с помощью ацилирующего агента формулы (III), где W1 является реагирующей удаляемой группой, такой как галоген, азидо, или активированной кислотной функцией, например, галогенфениловым эфиром, таким как пентахлор- или пентафторфениловым эфиром, и связан с ацильной частью L. Указанная реакция может быть проведена с помощью известных науке процедур ацилирования, например, путем перемешивания реагентов в реакционно-инертном растворителе, необязательно с дополнительным присутствием основания для нейтрализации кислоты, которая образуется в процессе реакции. В противоположном варианте О-ацилирование осуществляют путем применения подходящего сопрягающего реагента, такого как дициклогексилкарбодиимид или его функциональное производное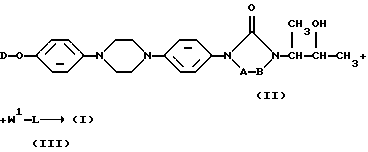
В этом и последующих препаратах продукты реакции могут быть отделены от реакционной среды и, если необходимо, дополнительно очищены в соответствии с обычными методологиями, известными науке, такими как, например, экстракция, кристаллизация, растирание и хроматография.
Соединения формулы (I) могут быть также получены путем О-алкилирования фенола формулы (IV) алкилирующим реагентом формулы (V), где W2 является реагирующей удаляемой группой, такой как галоген или сульфонилоксигруппа. Указанная реакция может быть проведена путем перемешивания реагентов в реакционно-инертном растворителе, необязательно с дополнительным присутствием подходящего основания для нейтрализации кислоты, которая образуется в процессе реакции. В соединениях и промежуточных продуктах, упоминаемых здесь далее, заместители представляют собой указанные выше, за исключением специально оговоренных случаев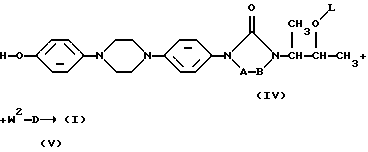
Получение промежуточных продуктов формулы (V), где D является радикалом формулы D1, раскрыто в патенте США 4267179.
Как указывалось здесь выше, переменный L может быть также представлен как L'-NRXRY, две части которого, то есть L'- и -N-NRXRY, применяют в следующей схеме реакций: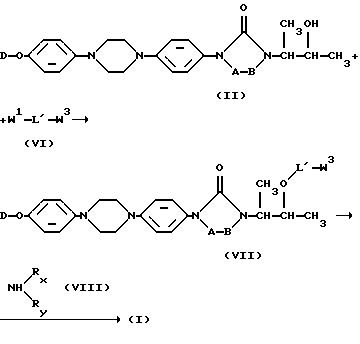
Указанная выше схема реакций отражает получение соединений формулы (I) путем О-ацилирования промежуточного продукта формулы (II) с помощью реагента формулы (VI), где W3 является реагирующей удаляемой группой, такой как галоген, и W1 является указанной выше и связан с ацильной частью L'; и последующего взаимодействия полученного таким образом промежуточного продукта формулы (VII) с амином формулы (VIII).
Соединения формулы (I) могут также преобразовываться друг в друга, следуя известным в данной области превращениям. Например, соединения формулы (I), в которых L содержит защищенную аминную часть, может превращаться в соединения формулы (I), в которых указанная аминная часть является незамещенной, при использовании известных в данной области способов удаления защитных групп, например, путем взаимодействия с трифторуксусной кислотой в соответствующем растворителе, например дихлорметане.
Соединения формулы (I) могут также превращаться в соответствующие N-оксидные формы, следуя известным в данной области способам превращения трехвалентного азота в его N-оксидную форму. Указанная реакция N-окисления может обычно проводиться путем взаимодействия исходных веществ формулы (I) с подходящей органической или неорганической перекисью. Подходящие неорганические перекиси включают, например, перекись водорода, перекиси щелочных или щелочноземельных металлов, например перекись натрия, перекись калия; подходящие органические перекиси могут включать надкислоты, такие как, например, надбензойная кислота или галогензамещенная надбензойная кислота, например 3-хлорнадбензойная кислота, алкановые надкислоты, например надуксусная кислота, алкилгидропероксиды, например трет-бутилгидропероксид. Подходящими растворителями являются, например, вода, низшие алканолы, например этанол и тому подобное, углеводороды, например толуол, кетоны, например 2-бутанон, галогенированные углеводороды, например дихлорметан, и смеси таких растворителей.
Некоторые промежуточные продукты и исходные материалы, применяемые в указанных выше последовательностях реакции, коммерчески доступны или могут быть синтезированы в соответствии с процедурами, описанными в других патентах, например в US-4791111, US-4931444 и US-4267179. Некоторые способы получения промежуточных продуктов настоящего изобретения описаны здесь ниже.
Например, промежуточные продукты формулы (II) могут быть получены путем O-алкилирования реагента формулы (IX) алкилирующим реагентом формулы (V) с последующими процедурами О-алкилирования, описанными здесь выше, для получения соединений формулы (I)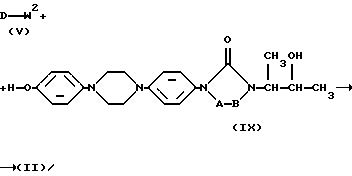
Промежуточные продукты формулы (II) могут быть также получены путем О-алкилирования реагента формулы (X) алкилирующим реагентом формулы (V) с последующими процедурами О-алкилирования, описанными здесь выше, для получения соединений формулы (I), и последующим восстановлением образованного таким образом промежуточного соединения формулы (XI). Указанное восстановление можно выполнять путем перемешивания промежуточного соединения формулы (XI) с восстанавливающим реагентом, таким, например, как борогидрид натрия в реакционно-инертном растворителе, например, таком как дихлорметан, метанол или их смесь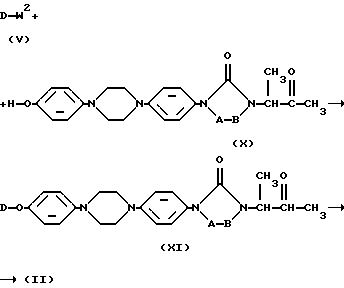
Получение промежуточного соединения формулы (X) раскрыто в US-4931444.
Промежуточные продукты формулы (XI) могут быть также получены путем N-алкилирования промежуточного соединения формулы (XII) с последующими известными в данной области процедурами N-алкилирования с помощью алкилирующего реагента формулы (XIII), в которой W4 является подходящей удаляемой группой, например галогеном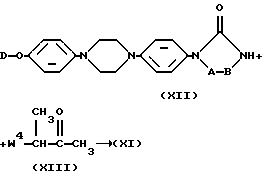
Стереохимически чистые изомерные формы соединений и промежуточных продуктов настоящего изобретения можно получать с помощью применения известных в данной области методов. Диастереомеры можно разделять с помощью методов физического разделения, таких как избирательная кристаллизация и хроматографическое разделение, например жидкостная хроматография. Энантиомеры можно отделять друг от друга путем избирательной кристаллизации их диастереомерных солей с оптически активными кислотами. В противоположном варианте энантиомеры могут быть разделены с помощью хроматографической техники с применением хиральных стационарных фаз. Указанные чистые стереохимически изомерные формы могут также происходить из соответствующих чистых стереохимически изомерных форм соответствующих исходных материалов с условием, что реакция протекает стереоизбирательно или стереоспецифично. Если желателен специфический стереомер, предпочтительно чтобы указанное соединение синтезировалось путем стереоизбирательного или стереоспецифического методов получения. В этих методах обычно выгодно применять чистые в отношении энантиомеров исходные материалы. Стереохимически изомерные формы соединений формулы (I) предназначены, очевидно, для включения в объем настоящего изобретения.
Как установлено здесь выше, энантиомерно чистые формы соединений формулы (I) образуют предпочтительную группу соединений. Из этого следует, что энантиомерно чистые формы промежуточных продуктов формулы (II), их N-оксидные формы и их аддитивные солевые формы особенно применимы при получении энантиомерно чистых соединений формулы (I). Для получения соединений формулы (I) с соответствующей конфигурацией также пригодны энантиомерные смеси и диастереомерные смеси промежуточных продуктов формулы (II). Указанные энантиомерно чистые формы, а также энантиомерные и диастереомерные смеси промежуточных продуктов формулы (II) считаются новыми.
Особенно предпочтительными промежуточными продуктами формулы (II) являются 4-[4-[4-[4-[[2-(2,4-дифторофенил)-2-(1H-1,2,4-триазол-1-илметил)-1,3-диоксолан-4-ил] метокси] фенил] -1-пиперазинил]фенил]-2,4-дигидро-2-(2-гидрокси-1-метилпропил)-3H-1,2,4-триазол-3-он в его [2S-[2α, 4α[(R*, R*)]]] в его энантиомерно чистой форме и соответствующий 2,4-дихлорофенильный аналог.
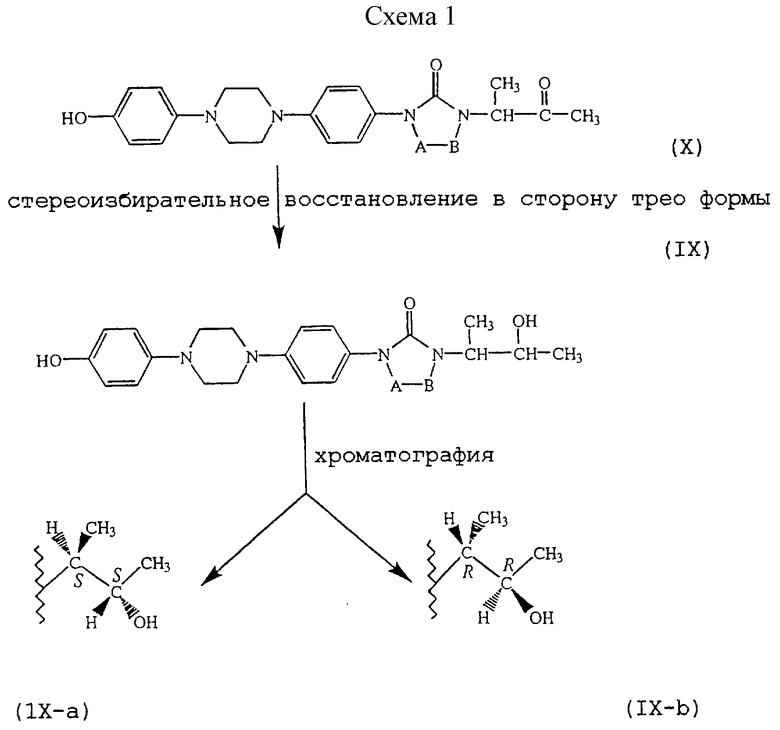
В частности, [2S-[2α, 4α[(R*, R*)]]] энантиомерно чистая форма промежуточных продуктов формулы (II) может быть получена с помощью реакции соответствующей энантиомерно чистой формы промежуточного продукта (IX), то есть [S-(R*, R*)] формы, представленной формулой (IХ-а), с соответствующей энантиомерно чистой формой промежуточного продукта (V), то есть [2S-(2α, 4α)] формой, предсталенной (V-а), согласно последовательности реакций, как описано выше.
Стереоизбирательный синтез промежуточного продукта (IХ-а), начиная с промежуточного продукта (X), может проводиться, как описано на схеме 1 (см. в конце описания).
Подходящие условия стереоизбирательного восстановления включают применение К-селектрида в подходящем растворителе, таком как, например, диметилацетамид или тетрагидрофуран; применение борогидрида натрия необязательно в комбинации с СеСl3•7Н2О, ZnCl2 или CaCl2•2H2O в подходящем растворителе, таком как, например, диметилацетамид, диметилформамид, метанол или тетрагидрофуран. Указанные условия восстановления благоприятствуют образованию трео формы 2-гидрокси-1-метилпропильной части, то есть формы, в которой два асимметричных атома углерода имеют идентичную абсолютную конфигурацию. Перекристаллизация полученной смеси после стереоизбирательного восстановления может даже дополнительно улучшить соотношение трео/эритро в сторону трео формы. Желаемая [S-(R*, R*)] форма может затем быть выделена хроматографически с применением хиральной стационарной фазы, такой как, например, Chiralpak AD (амилоза 3,5 диметилфенилкарбомат), полученный от Daicel Chemical Industries, Ltd, Japan.
Алкоксифенильные производные промежуточных соединений формулы (IХ-а) могут быть получены в соответствии с той же последовательностью реакций, что и на схеме 1.
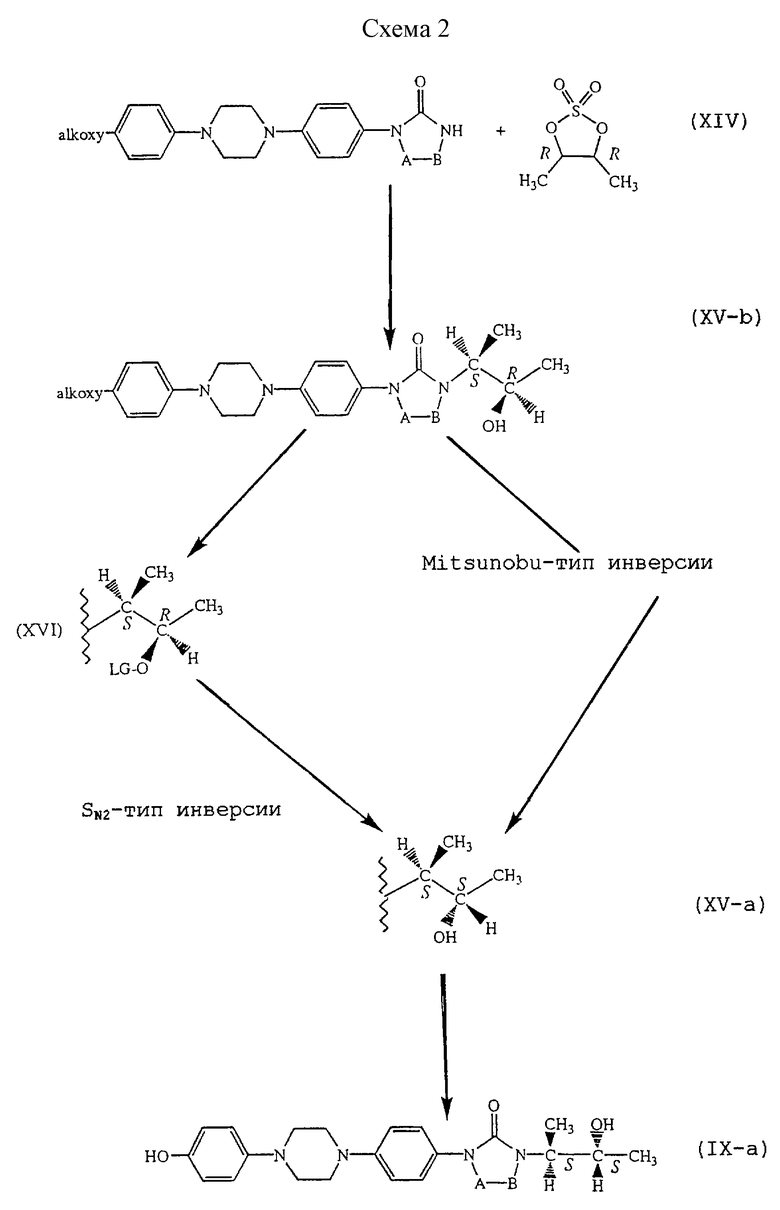
Противоположный путь получения промежуточных соединений формулы (IХ-а) или их алкоксифенильных аналогов отражен на схеме 2 (см. в конце описания).
Реакция промежуточного соединения формулы (XIV) с (4R-транс)-4,5-диметил-2,2-диоксид-1,3,2-диоксатиоланом может быть выполнена в подходящем растворителе, предпочтительно в полярном апротонном растворителе, таком как, например, диметилацетамид или N,N-диметилформамид, и в присутствии основания, такого как, например, трет-бутанолат калия, гидроокись калия или гидрид калия. Впоследствии к реакционной смеси может быть добавлена кислота, такая как серная кислота, с получением в результате этого промежуточного продукта формулы (XV-b), в котором 2-гидрокси-1-метилпропильная часть имеет эритро форму. Затем атом углерода, несущий спиртовую функцию указанной 2-гидрокси-1-метилпропильной части, эпимеризуют, предпочтительно со 100% инверсией, получая таким образом промежуточный продукт (XV-a), в котором 2-гидрокси-1-метилпропильная часть имеет трео форму. Оба пути являются удобными.
Первый путь включает трансформацию спиртовой функции в подходящую удаляемую группу O-LG путем, например, превращения гидроксильной группы с помощью органической кислоты, такой как, например, карбоновая кислота, например уксусная кислота или 4-нитробензойная кислота; или сульфокислота, например п-толуолсульфокислота или метансульфокислота; получая таким образом промежуточный продукт формулы (XVI). Атом углерода, несущий уходящую группу в указанном промежуточном продукте (XVI), может впоследствии быть эпимеризован, предпочтительно со 100% инверсией, путем реакции SN2-типа с подходящим нуклеофильным реагентом, таким как, например, алкоголят, например бензилоксигруппа; гидроокисная соль (гидроокись) щелочного металла, например гидроокись натрия или гидроокись калия; ацетат, например ацетат натрия. Указанную реакцию проводят в подходящем растворителе, предпочтительно в полярном апротонном растворителе, таком как, например, диметилацетамид, N-метилпирролидинон, диметилимидазолидинон или сульфолан. В том случае, если в реакции SN2-типа применяют алкоголят или ацетат, у полученного таким образом промежуточного продукта может быть удалена защитная группа с помощью известной науке техники удаления защитных групп, получая в результате этого спиртовой промежуточный продукт формулы (XV-а).
Альтернативным путем инверсии стереохимии атома углерода, несущего спиртовую функцию, является применение реакции Mitsunobu. Спиртовая функция промежуточного продукта формулы (XV-b) активируется с помощью диизопропилазодикарбоксилата или его функционального производного, такого как диэтилазодикарбоксилат, в присутствии трифенилфосфина и полярного апротонного растворителя, такого как, например, 4-нитробензойная кислота, уксусная кислота, монохлоруксусная кислота. Полученный таким образом эфир, в котором 2-гидрокси-1-метилпропильная часть трансформирована в трео форму, может впоследствии быть гидролизован с применением известной науке техники гидролиза с получением в результате этого промежуточного продукта формулы (XV-a).
Наконец, алкоксифенильная часть промежуточных продуктов формулы (XV-a) может быть трансформирована в фенольную часть с применением, например, бромноватой кислоты в уксусной кислоте в присутствии тиосульфата натрия, с получением таким образом промежуточного продукта формулы (IХ-а).
Подходящей альтернативой (4R-транс)-4,5-диметил-2,2-диоксид-1,3,2-диоксатиолану являются следующие энантиомерно чистые промежуточные соединения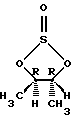
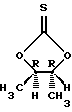
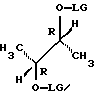
где LG является удаляемой группой, такой как, например, п-толуолсульфонил.
Промежуточные продукты формулы (IX-b), где 2-гидрокси-1-метилпропиловая часть находится в [R-(R*, R*)] форме, могут быть получены с применением той же последовательности реакций, что и изображенная на схеме 2, но с заменой (4R-транс)-4,5-диметил-2,2-диоксид-1,3,2-диоксатиолана его энантиомером (4S-транс)-4,5-диметил-2,2-диоксид-1,3,2-диоксатиоланом.
В альтернативном варианте последовательности реакций схемы 2 промежуточное соединение формулы (XIV) может быть непосредственно присоединено к энантиомерно чистому промежуточному продукту, такому как [R-(R*, S*)]-3-бром-2-бутанол-4-нитробензоат, или его функциональному производному, при этом таким образом непосредственно получают промежуточное соединение формулы (XV-а).
Интересно, что особенно энантиомерно чистые формы промежуточных продуктов формулы (IV) могут быть синтезированы с применением типа реакции Mitsunobu в схеме 2, в которой карбоновая кислота, применяемая в реакции с активированным спиртом формулы (XV-b), заменяется защищенной аминокислотой. Защитная группа аминокислоты необязательно может быть удалена с помощью известных науке способов.
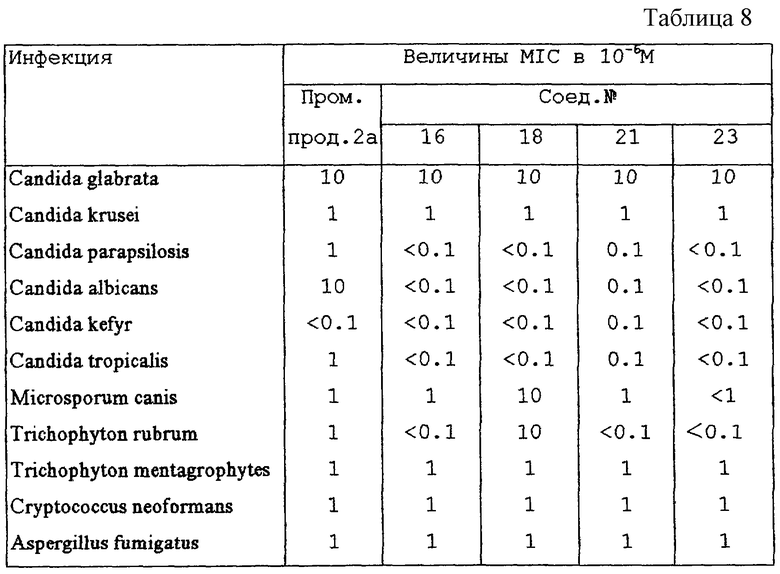
Соединения формулы (I), их фармацевтически приемлемые аддитивные соли и их стереохимически изомерные формы являются полезными средствами для борьбы с грибками in vivo. Более того, профиль растворимости в водных растворах соединений формулы (I) делает их пригодными для внутривенного введения. Настоящие соединения, как обнаружено, активны против широкого спектра грибков, таких как Candida spp., например Candida albicans, Candida glabrata, Candida crusei, Candida parapsilosis, Candida kefyr, Candida tropicalis; Aspergillus spp., например Aspergillus fumigatus, Aspergillus niger, Aspergillus flavus; Crytococcus neoformans; Sporothrix schenckii; Fonsecaea spp.; Epidermophyton floccosum; Microsporum canis; Trichophyton spp.; Fusarium spp.; и некоторые кожные гифомицеты.
Чистые энантиомеры, энантиомерные смеси и диастереомерные смеси промежуточных продуктов формулы (II) также являются антигрибковыми средствами, характеризующимися благоприятным фармакологическим профилем в отношении антигрибковой активности и наличия побочных эффектов.
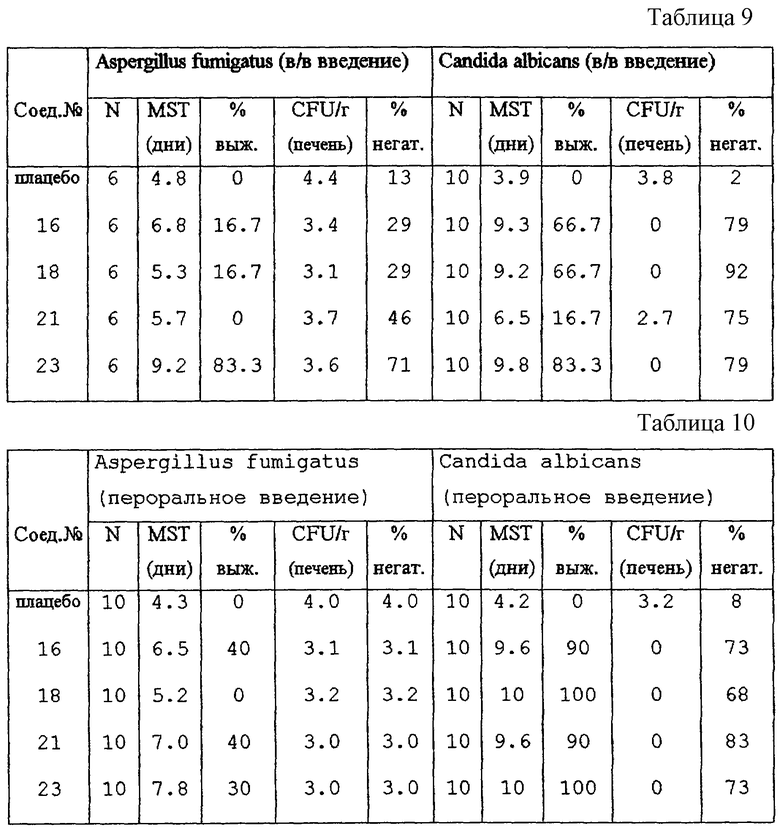
Химическую стабильность некоторых соединений формулы (I) определяли, как показано здесь ниже в экспериментальной части. Эксперименты показывают, что метаболическое разрушение настоящих соединений до промежуточных продуктов формулы (II) является органоспецифичным и происходит с трудом. Далее, эксперименты in vitro показывают, что соединения формулы (I) обладают улучшенной внутренней способностью тормозить рост грибков, например Candida albicans, при сравнении с промежуточными соединениями формулы (II), противогрибковая активность которых указана в US-4791111. Указанные эксперименты in vitro включают восприимчивость грибков к настоящим соединениям, как описано в фармакологическом примере здесь ниже. Другие эксперименты in vitro, такие как определение эффектов настоящих соединений на синтез стеролов, например, у Candida albicans, показывают их противогрибковую эффективность. Эксперименты in vivo на нескольких моделях на мышах, морских свинках и крысах показывают также, что после как перорального, так и внутривенного введения настоящие соединения являются сильными противогрибковыми средствами.
Соединения настоящего изобретения имеют также хорошую усвояемость при пероральном поступлении.
С точки зрения полезного применения соединений формулы (I) предложен способ лечения теплокровных животных, включая человека, страдающих от грибковых инфекций. Указанный метод включает системное или местное введение эффективного количества соединения формулы (I), его N-оксидной формы, его фармацевтически приемлемой аддитивной соли или его возможной стереоизомерной формы теплокровным животным, включая человека. Следовательно, соединения формулы (I) предлагаются для применения в качестве лекарственного средства, в особенности предлагается применение соединения формулы (I) в производстве лекарства, полезного для лечения грибковых инфекций.
Настоящее изобретение предлагает также композиции для лечения или предотвращения грибковых инфекций, включающие терапевтически эффективное количество соединения формулы (I) и фармацевтически приемлемый носитель или разбавитель.
С точки зрения их полезных фармакологических свойств соединения, являющиеся целью изобретения, могут быть включены в различные фармацевтические формы с целью введения. Для получения фармацевтических композиций настоящего изобретения терапевтически эффективное количество конкретного соединения в основной или солевой форме в качестве активного ингредиента объединяют с тщательно примешиваемым фармацевтически приемлемым носителем, который может иметь широкий спектр форм в зависимости от формы препарата для желаемого введения. Эти фармацевтические композиции желательно представлять в единицах лекарственной формы, подходящей предпочтительно для перорального, ректального, местного, чрескожного или парентерального введения. Например, при получении композиций в виде лекарственной формы для перорального введения может быть применена любая обычная фармацевтическая среда, такая как, например, вода, гликоли, масла, спирты и тому подобное, в случае перорального применения жидких препаратов, таких как суспензии, сиропы, эликсиры и растворы; или твердые носители, такие как крахмалы, сахара, каолин, смазывающие вещества, связующие соединения, дезинтегрирующие агенты и тому подобное в случае порошков, пилюль, капсул и таблеток. Из-за простоты их введения таблетки и капсулы представляют собой наиболее выгодную лекарственную форму для перорального введения, в случае которого, очевидно, применяют твердые фармацевтические носители. В качестве подходящих композиций для местного нанесения здесь могут быть процитированы все композиции, обычно применяемые для местного введения лекарств, например кремы, желе, повязки, шампуни, настойки, пасты, мази, лечебные мази, порошки и тому подобное. Для парентеральных композиций носитель обычно представляет собой стерильную воду, по крайней мере чаще всего, хотя могут быть включены и другие ингредиенты, например циклодекстрины для достижения растворимости. Могут быть получены растворы для инъекций, например, в которых носитель включает солевой раствор, раствор глюкозы или смесь солевого раствора и раствора глюкозы. Могут быть также получены суспензии для инъекций, в случае которых могут быть применены подходящие жидкие носители, суспендирующие агенты и тому подобное. В композициях, подходящих для чрескожного введения, носитель необязательно включает агент, повышающий проницаемость и/или подходящий увлажняющий агент, необязательно соединенный с подходящими добавками любой природы в небольших количествах, причем эти добавки не вызывают значительного вредного воздействия на кожу. Указанные добавки могут ускорять введение в кожу и/или могут быть полезными при получении желаемых композиций. Эти композиции могут вводиться разными путями, например, в виде пластыря для чрескожного введения, в виде нанесенного пятна, в виде мази. Для парентеральных композиций носитель обычно включает стерильную воду, по крайней мере чаще всего. Могут быть получены растворы для инъекций, например, в случае которых подходящие жидкие носители включают солевой раствор, раствор глюкозы или смесь солевого раствора и раствора глюкозы. Могут быть также получены суспензии для инъекций, в случае которых могут быть применены подходящие жидкие носители, суспендирующие агенты и тому подобное. Для парентеральных композиций могут быть включены и другие ингредиенты, например, для достижения растворимости, например циклодекстрины. Подходящие циклодекстрины представляют собой α-, β-, γ-циклодекстрины или их эфиры и их смешанные эфиры, где одна или более гидроксильных групп ангидроглюкозных единиц циклодекстрина замещены C1-6алкилом, в частности метилом, этилом или изопропилом, например, произвольно метилированным β-CD; гидроксиС1-6алкилом, в частности гидроксиэтилом, гидроксипропилом или гидроксибутилом; карбоксиС1-6алкилом, в частности карбоксиметилом или карбоксиэтилом; С1-6алкилкарбонилом, в частности ацетилом. Особенно заслуживают внимания в качестве комплексообразующих соединений и/или солюбилизаторов β-CD, произвольно метилированный β-CD, 2,6-диметил-β-CD, 2-гидроксиэтил-β-CD, 2-гидроксиэтил-γ-СО, 2-гидроксипропил-γ-СО и (2-карбокси-метокси)пропил-β-CD и в особенности 2-гидроксипропил-β-CD (2-HP-β-CD).
Термин смешанный эфир обозначает производные циклодекстрина, в которых по меньшей мере две гидроксильные группы циклодекстрина этерифицированы разными группами, такими как, например, гидроксипропильная или гидроксиэтильная.
Среднее молярное замещение (M. S.) применяют в качестве меры среднего количества молей алкоксильных единиц ангидроглюкозы. Степень среднего замещения (D.S.) обозначает среднее количество замещенных гидроксилов на единицу ангидроглюкозы. M.S. и D.S. величины могут быть определены с помощью различных аналитических методов, таких как ядерный магнитный резонанс (ЯМР), масс-спектрометрия (МС) и инфракрасная спектроскопия (ИК). В зависимости от используемого метода могут быть получены слегка различающиеся величины для данного циклодекстринового производного. Предпочтительно, чтобы измеренные с помощью масс-спектрометрии величины M.S. лежали в диапазоне от 0,125 до 10 и величины D.S. находились в диапазоне от 0,125 до 3.
Особенно выгодно составлять упомянутые выше фармацевтические композиции в единицах лекарственной формы для простоты введения и единообразия дозировки. Единица лекарственной формы, применяемая здесь в описании и формуле изобретения, относится к физически дискретным единицам, подходящим в качестве единых единиц лекарственной формы, при этом каждая единица содержит предварительно определенное количество активного ингредиента, рассчитанное на индукцию желаемого терапевтического эффекта, в комплексе с требуемым фармацевтическим носителем. Примерами таких единиц лекарственных форм служат таблетки (включая или таблетки с насечкой, или таблетки, покрытые оболочкой), капсулы, пилюли, пакетики порошков, облатки, растворы или суспензии для введения, дозировки в виде чайной ложки, дозировки в виде столовой ложки и тому подобное и их кратные части.
Специалисты в области лечения теплокровных животных, страдающих от заболеваний, вызываемых грибками, могут легко определить терапевтически эффективную суточную дозу из представленных здесь результатов тестов. В целом ожидается, что терапевтически эффективная суточная доза должна составлять от 0,05 мг/кг до 20 мг/кг веса тела.
Экспериментальная часть
Далее здесь "DMF" обозначает N,N-диметилформамид, "MIK" обозначает метилизобутилкетон, "DIPE" обозначает диизопропиловый эфир.
А. Получение промежуточных продуктов
Пример А-1
Смесь (±)-2,4-дигидро-[4-[4-[4-(4-гидроксифенил)-1-пиперазинил] -фенил] -2-(1-метил-2-оксопропил)-3H-1,2,4-триазол-3-она (0,06 моля) в DMF (500 мл) охлаждали до -10oС и затем перемешивали под током N2. По каплям добавляли 1 М раствор три-сек-бутилборогидрида калия в тетрагидрофуране (150 мл). Смеси давали медленно нагреться до комнатной температуры и затем выливали в воду. Осадок отфильтровывали, промывали СН3ОН и кристаллизовали из СН3ОН. Осадок отфильтровывали и высушивали. Остаток очищали с помощью HPLC [высокоэффективной жидкостной хроматографии] на CHIRALPAC AD (элюент:этанол). Были собраны две чистые фракции, из которых были выпарены растворители. Каждый из остатков растирали в СН3ОН. Осадок отфильтровывали и высушивали, что дало 7,3 г [S-(R*, R*)]-2,4-дигидро-2-(2-гидрокси-1-метилпропил)-4-[4-[4-(4-гидроксифенил)-1-пиперазинил] -фенил] -3H-1,2,4-триазол-3-она (промеж. прод. 1a) [α]
Сходным образом получали:
[A-(R*, S*)] -2,4-дигидро-2-(2-гидрокси-1-метилпропил)-4-[4-[4-(4-гидроксифенил)-1-пиперазинил] -фенил] -3Н-1,2,4-триазол-3-он (промеж. прод. 1b) [α]
[В-(R*, S*)] -2,4-дигидро-2-(2-гидрокси-1-метилпропил)-4-[4-[4-(4-гидроксифенил)-1-пиперазинил] -фенил] -3H-1,2,4-триазол-3-он (промеж. прод. 1с) [α]
[R-(R*, R*)] -2,4-дигидро-2-(2-гидрокси-1-метилпропил)-4-[4-[4-(4-гидроксифенил)-1-пиперазинил] -фенил] -3H-1,2,4-триазол-3-он (промеж. прод. 1d) [α]
(R*, S*)-2,4-дигидро-2-(2-гидрокси-1-метилпропил)-4-[4-[4-(4-гидроксифенил)-1-пиперазинил]-фенил]-3H-1,2,4-триазол-3-он (промеж. прод. 1е).
Сходным образом получали:
(R*, R*)-2,4-дигидро-2-(2-гидрокси-1-метилпропил)-4-[4-[4-(4-гидроксифенил)-1-пиперазинил]-фенил]-3Н-1,2,4-триазол-3-он (промеж. прод. 1f);
[R-(R*, R*)+R-(R*, S*)]-2,4-дигидро-2-(2-гидрокси-1-метилпропил)-4-[4-[4-(4-гидроксифенил)-1-пиперазинил] -фенил] -3Н-1,2,4-триазол-3-он (промеж. прод. 1g);
[R-(R*, R*)+S-(R*, S*)]-2,4-дигидро-2-(2-гидрокси-1-метилпропил)-4-[4-[4-(4-гидроксифенил)-1-пиперазинил] -фенил] -3Н-1,2,4-триазол-3-он (промеж. прод. 1h) ;
[S-(R*, R*)+R-(R*, S*)]-2,4-дигидро-2-(2-гидрокси-1-метилпропил)-4-[4-[4-(4-гидроксифенил)-1-пиперазинил] -фенил] -3Н-1,2,4-триазол-3-он (промеж. прод. 1i);
[S-(R*, R*)+S-(R*, S*)]-2,4-дигидро-2-(2-гидрокси-1-метилпропил)-4-[4-[4-(4-гидроксифенил)-1-пиперазинил] -фенил] -3Н-1,2,4-триазол-3-он (промеж. прод. 1j);
Пример А-2
Смесь цис-(2S)-4-метилбензолсульфоната 2-(2,4-дифторфенил)-2-(1H-1,2,4-триазол-1-илметил)-1,3-диоксолан-4-метанола (эфира) (0,0134 моля), промежуточного продукта (1а) (0,0122 моля) и NaOH (0,013 моля) в DMF (200 мл) перемешивали при 60oС под струей азота в течение ночи. Смесь охлаждали и выливали в воду. Осадок отфильтровывали и высушивали. Остаток очищали колоночной хроматографией на силикагеле (элюент: CH2Cl2/CH3OH от 94/6 до 0/100). Собирали чистые фракции и растворитель упаривали. Остаток растирали в MIK. Осадок отфильтровывали и сушили, что дало 4.7 г (56%) [2S-[2α, 4α[(R*, R*] ] ]-4-[4-[4-[4-[[2-(2,4-дифторофенил)-2-(1H-1,2,4-триазол-1-илметил)-1,3-диоксолан-4-ил] метокси] -фенил]-1-пиперазинил]-фенил]-2,4-дигидро-2-(2-гидрокси-1-метилпропил)-3Н-1,2,4-триазол-3-она (промеж. прод. 2а) [α]
В таблице 1 перечислены промежуточные продукты, полученные аналогично примеру А. 2. Асимметричные углеродные атомы отмечены  их абсолютная конфигурация и оптическое вращение также показаны в таблице 1.
их абсолютная конфигурация и оптическое вращение также показаны в таблице 1.
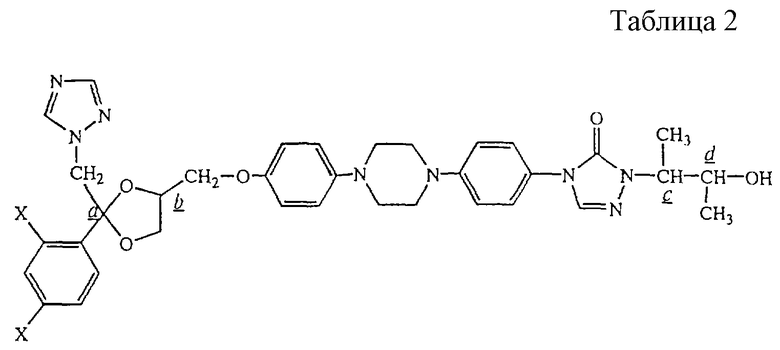
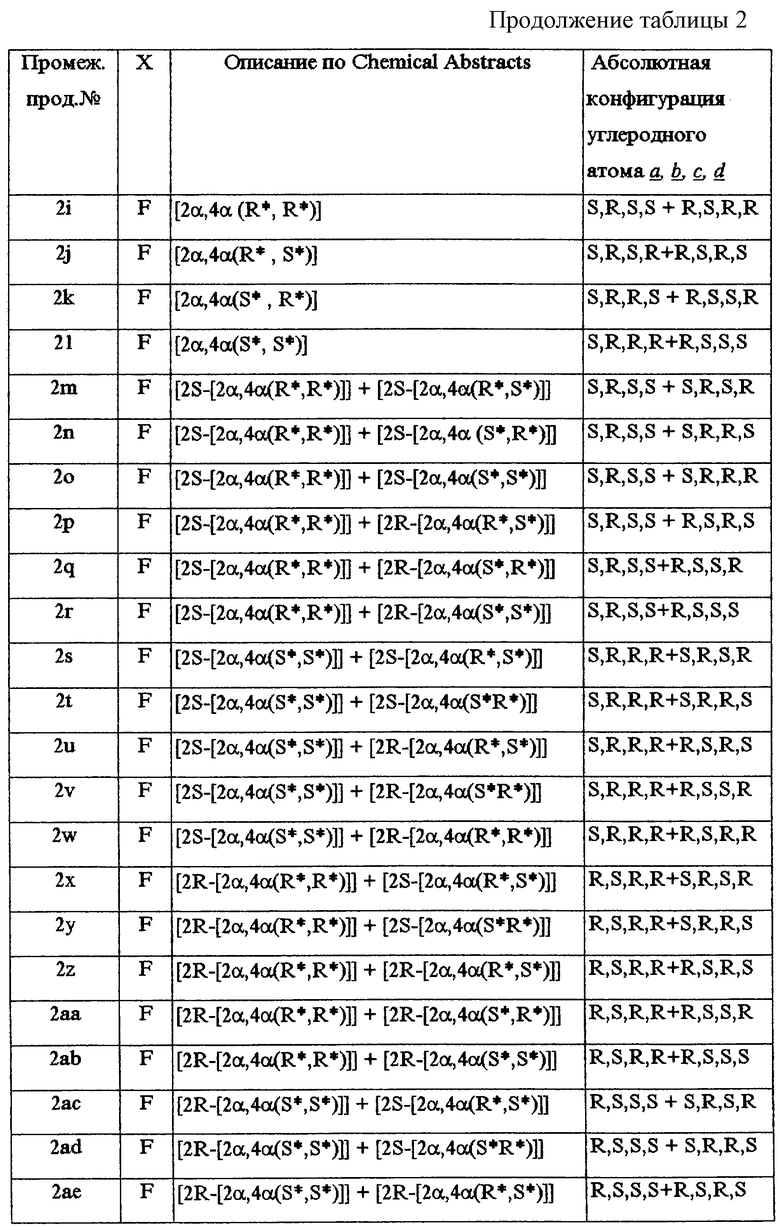
В таблице 2 перечислены промежуточные продукты, полученные аналогично примеру А.2.
Пример А-3
Смесь промежуточного продукта 2а (0,01 моля) и хлорангидрида хлоруксусной кислоты (0,0115 моля) в CH2Cl2 перемешивали при комнатной температуре. Добавляли пиридин (0,02 моля) и смесь перемешивали в течение 2 часов, промывали водой, сушили, фильтровали и растворитель упаривали. Остаток кристаллизовали из MIK/DIPE. Осадок фильтровали и высушивали, что дало 6,7 г (87%) [2S-[2α, 4α[(R*, R*)]]]-2-[4-[4-[4-[4-[[2-(2,4-дифторфенил)-2-(1H-1,2,4-триазол-1-илметил)-1,3-диоксолан-4-ил] метокси]-фенил]-1-пиперазинил]-фенил] -4,5-дигидро-5-оксо-1H-1,2,4-триазол-1-ил] -1-метилпропил-хлорацетата (промеж. прод. 3).
Пример А.4
а) 2,4-дигидро-4-[4-[4-(4-метоксифенил)-1-пиперазинил] фенил] -3Н-1,2,4-триазол-3-он (0,15 моля), полученный, как описано в WO 94/18978, перемешивали в диэтилацетамиде (500 мл) при 60oС. Добавляли трет-бутилат калия (0,165 моля). Смесь перемешивали при 100oС под током N2 в течение 1 часа и затем охлаждали до 50oС. По каплям добавляли (4R-транс)-4,5-диметил-2,2-диоксид-1,3,2-диоксатиолан (0,165 моля). Смесь перемешивали при 50-60oС в течение 2 часов. По каплям добавляли раствор концентрированной H2SO4 (20 мл). Смесь перемешивали при 60oС в течение 2 часов. Добавляли Н2О (20 мл). Смесь перемешивали при 60oС в течение 20 часов, охлаждали, выливали в H2O (1000 мл), защелачивали 50% NaOH и перемешивали. Осадок отфильтровывали, промывали H2O и высушивали. Остаток растворяли в CH2Cl2/CH3OH. Смесь фильтровали и растворитель выпаривали. Остаток растирали в 2-пропаноле, отфильтровывали и высушивали. Остаток очищали на силикагеле на стеклянном фильтре (элюент: CH2Cl2/CH3OH 99/1). Собирали чистые фракции и растворитель упаривали. Остаток растирали в CH2Cl2 (150 мл), отфильтровывали и высушивали при 110oС, что дало 0,37 г [S-(R*, S*)]-2,4-дигидро-2-(2-гидрокси-1-метилпропил)-4-[4-[4-(4-метоксифенил)-1-пиперазинил] -фенил] -3Н-1,2,4-триазол-3-она (промеж.прод. 5а). [α]
b) К 2, 4-дигидро-4-[4-[4-(4-метоксифенил)-1-пиперазинил]-фенил]-3Н-1,2,4-триазол-3-ону (0,0925 моля), полученному, как описано в WO 94/18978, добавляли 1-метокси-2-пропанол (700 мл), воду (700 мл) и NaOH (50%; 4,8 мл). Полученную смесь нагревали до 45oС и добавляли при перемешивании при 45oС транс-2,3-диметил-оксиран (0,1387 моля). Реакционную смесь перемешивали в течение 68 часов при 45oС и в течение 60 часов при 60oС, после чего охлаждали до 20oС. Дополнительно добавляли NaOH (50%; 4,8 мл). Реакционную смесь перемешивали в течение 64 часов при 50oС, в течение 18 часов при 100oС, после чего охлаждали на ледяной бане. Смесь разделяли фильтрованием на осадок (1) и фильтрат (2). Осадок (1) высушивали, перерастворяли в CH2Cl2 (100 мл) и отфильтровывали. Соответствующий фильтрат выпаривали и остаток высушивали, что дало 2,2 г (R*, S*)-2,4-дигидро-2-(2-гидрокси-1-метилпропил)-4-[4-[4-(4-метоксифенил)-1-пиперазинил] -фенил]-3Н-1,2,4-триазол-3-она (промеж.прод. 5b). Фильтрат (2) упаривали. Остаток перемешивали в CH2Cl2 (150 мл) и отфильтровывали. Соответствующий фильтрат упаривали и остаток высушивали, что дало 7,4 г (R*, S*)-2,4-дигидро-2-(2-гидрокси-1-метилпропил)-4-[4-[4-(4-метоксифенил)-1-пиперазинил] -фенил] -3Н-1,2,4-триазол-3-она (промеж. прод. 5b). Две фракции промежуточного продукта 5b объединяли и далее очищали с помощью активированного угля, колоночной хроматографии и перекристаллизации, что дало 1,5 г (общий выход 3,9%) (R*, S*)-2,4-дигидро-2-(2-гидрокси-1-метилпропил)-4-[4-[4-(4-метоксифенил)-1-пиперазинил] -фенил] -3Н-1,2,4-триазол-3-она (промеж.прод. 5b).
Пример А-5
Смесь промежуточного продукта 5а (0,00327 моля), трифенилфосфина (0,00806 моля) и п-нитробензойной кислоты (0,00717 моля) в тетрагидрофуран/диметилацетамиде 3/2 (50 мл) нагревали до полного растворения. Затем по каплям добавляли диэтилазодикарбоксилат (0,00806 моля). Смесь перемешивали при комнатной температуре в течение 90 минут и при 50oС в течение 1 часа. При 50oС добавляли раствор NaOH (1N; 90 мл). Смесь выливали в воду (100 мл) и NaOH (1N; 90 мл) и затем перемешивали. Осадок отфильтровывали и перекристаллизовывали из 2-пропанола (60 мл). Смесь перемешивали в течение 48 часов. Преципитат отфильтровывали и высушивали, что дало 0,98 г (71%) [S-(R*, R*)] -2,4-дигидро-2-(2-гидрокси-1-метилпропил)-4-[4-[4-(4-метоксифенил)-1-пиперазинил]-фенил]-3Н-1,2,4-триазол-3-она (промеж.прод. 5с).
Пример А-6
а) N, N-диметил-4-пиридинамин (0,01062 моля) и промежуточный продукт 5а (0,00708 молей) суспендировали в CH2Cl2 (50 мл). По каплям при комнатной температуре добавляли раствор метансульфонилхлорида (0,01062 моля) в CH2Cl2 (30 мл). Смесь перемешивали при комнатной температуре в течение 2-х дней. Повторно добавляли N,N-диметил-4-пиридинамин (0,00352 моля) и метансульфонилхлорид (0,00358 молей). Смесь перемешивали в течение ночи, промывали водой (2 х 100 мл), высушивали, фильтровали через декалит и растворитель выпаривали. Осадок растворяли в MIК (150 мл). Добавляли активированный уголь (0,5 г). Смесь кипятили, фильтровали теплой и перемешивали в течение 2 часов. Преципитат отфильтровывали и высушивали, что дало 1,7 г (50%) [S-(R*, S*)] -2,4-дигидро-2-(2-метансульфонилокси-1-метилпропил)-4-[4-[4-(4-метоксифенил)-1-пиперазинил]фенил]-3Н-1,2,4-триазол-3-она (промеж.прод. 5d).
b) В соответствии с процедурой, описанной в Nakamura et al., (J.A.C.S. 1985, 107 р2138), промежуточный продукт 5d (0,001 моль) добавляли к раствору КОН (0,03 г) в СН3ОН (7 мл) и тетрагидрофурану (3 мл). Смесь перемешивали при 100oС в течение 4 часов, что дало [S-(R*, R*)]-2,4-дигидро-2-(2-гидрокси-1-метилпропил)-4-[4-[4-(4-метоксифенил)-1-пиперазинил] фенил] -3Н-1,2,4-триазол-3-он (промеж. прод. 5с).
В. Получение конечных соединении
Пример В-1
Смесь N-[(1,1-диметилэтокси)карбонил-L-фенилаланина (0,023 моля), промежуточного продукта (2а) (0,01 моля), дициклогексилкарбодиимида (0,046 моля) и N,N-диметил-4-пиридинамина (0,046 моля) в СН2Сl2 (200 мл) перемешивали при комнатной температуре в течение ночи. Добавляли воду (200 мл) и смесь перемешивали в течение 1 часа и экстрагировали CH2Cl2. Органическую фазу отделяли, промывали водой, высушивали, фильтровали и растворитель выпаривали. Остаток очищали колоночной хроматографией на силикагеле (элюент: СН2Сl2/СН3ОН 99/1). Чистые фракции собирали и растворитель выпаривали, что дало 10,8 г (86,7%) [2S-[2α, 4α[(R*, R*)]]]-2-[4-[4-[4-[4-[[2-(2,4-дифторофенил)-2-(1H-1,2,4-триазол-1-илметил)-1,3-диоксолан-4-ил] метокси] -фенил] -1-пиперазинил] фенил] -4,5-дигидро-5-оксо-1H-1,2,4-триазол-1-ил] -1-метилпропил N-[(1,1-диметилэтокси) (соединение 22).
Пример В-2
a) Смесь соединения 22 (0,0075 моля) в трифторуксусной кислоте (15 мл) и CH2Cl2 (150 мл) перемешивали в течение ночи. Смесь вливали в раствор NaНСО3, перемешивали в течение 30 минут и экстрагировали CH2Cl2. Органическую фазу отделяли, промывали, высушивали, фильтровали и растворитель выпаривали. Остаток очищали хроматографией на флэш-колонке с силикагелем (элюент: СН2С2/СН3ОН 96/4). Чистые фракции собирали и растворитель выпаривали. Осадок растирали в порошок в DIPE. Преципитат отфильтровывали и высушивали, что дало 3,6 г [2S-[2α, 4α[(R*, R*)]]]-2-[4-[4-[4-[4-[[2-(2,4-дифторфенил)-2-(1H-1,2,4-триазол-1-илметил)-1,3-диоксолан-4-ил] метокси] -фенил] -1-пиперазинил]-фенил]-4,5-дигидро-5-оксо-1Н-1,2,4-триазол-1-ил]-1-метилпропил L-фенилаланин (соединение 23).
b) Соединение 23 (0,00359 моля) растворяли в 2-пропаноне (25 мл). Добавляли раствор (Z)-2-бутендикислоты (0,00359 моля) в 2-пропаноне (5 мл). Смесь перемешивали в течение 16 часов. Преципитат отфильтровывали, промывали 2-пропаноном (2,5 мл) и высушивали, что дало 3,12 г [2S-[2α, 4α[(R*, R*)]]]-2-[4-[4-[4-[4-[[2-(2,4-дифторфенил)-2-(1H-1,2,4-триазол-1-илметил)-1,3-диоксолан-4-ил] метокси] -фенил]-1-пиперазинил]-фенил]-4,5-дигидро-5-оксо-1H-1,2,4-триазол-1-ил] -1-метилпропил L-фенилаланин (Z)-2-бутендиоата (1: 1) (соединение 25).
Пример В-3
Смесь промежуточного продукта (3) (0,0081 моля) и N,N-диэтиламина (0,027 моля) в DMF (50 мл) перемешивали при комнатной температуре в течение 8 часов. Смеси давали отстаиваться в течение 5 дней, затем вливали ее в воду и экстрагировали CH2Cl2. Органическую фазу отделяли, промывали водой, высушивали, фильтровали и растворитель выпаривали. Остаток очищали колоночной хроматографией на силикагеле (элюент: CH2Cl2/CH3OH 98/2). Чистые фракции собирали и растворитель выпаривали. Осадок растворяли в СН3СN (200 мл) и превращали в кислую соль (Е)-2-бутендикислоты (1:1). Преципитат отфильтровывали и высушивали, что дало 5 г (67%) [2S-[2α, 4α-[(R*, R*]]]-2-[4-[4-[4-[4-[[2-(2,4-дифторфенил)-2-(1H-1,2,4-триазол-1-илметил)-1,3-диоксолан-4-ил] метокси] фенил]-1-пиперазинил]фенил]-4,5-дигидро-5-оксо-1H-1,2,4-триазол-1-ил]-1-метилпропил N,N-диэтилглицин (Е)-2-бутендиоата (соединение 16).
Пример В-4
[2S-[2α, 4α-[(R*, R*)]]]-2-[4-[4-[4-[4-[[2-(2,4-дифтор-фенил)-2-(1H-1,2,4-триазол-1-илметил)-1,3-диоксолан-4-ил] метокси] фенил] -1-пиперазинил] фенил] -4,5-дигидро-5-оксо-1H-1,2,4-триазол-1-ил] -1-метилпропил β-аланин (0,0028 моля) растворяли в теплом этаноле (25 мл). К смеси добавляли (-)-(S)-гидроксибутандикислоту (0,0061 моль) и смесь кипятили до полного растворения. Полученному чистому раствору давали остыть до комнатной температуры и растворитель выпаривали. Остаток перемешивали в 2-пропаноне, отфильтровывали, затем высушивали, что дало 1,53 г (53%) [2S-[2α, 4α-[(R*, R*)]]]-2-[4-[4-[4-[4-[[2-(2,4-дифторфенил)-2-(1H-1,2,4-триазол-1-илметил)-1,3-диоксолан-4-ил] метокси] фенил] -1-пиперазинил] фенил]-4,5-дигидро-5-оксо-1H-1,2,4-триазол-1-ил] -1-метилпропил β-аланин (S)-гидроксибутандиоат (1: 2) моногидрата (соединение 12).
В таблице 3 перечислены соединения формулы (I), которые были получены в соответствии с одним из указанных выше примеров, обозначенных в колонке "Пр. ".
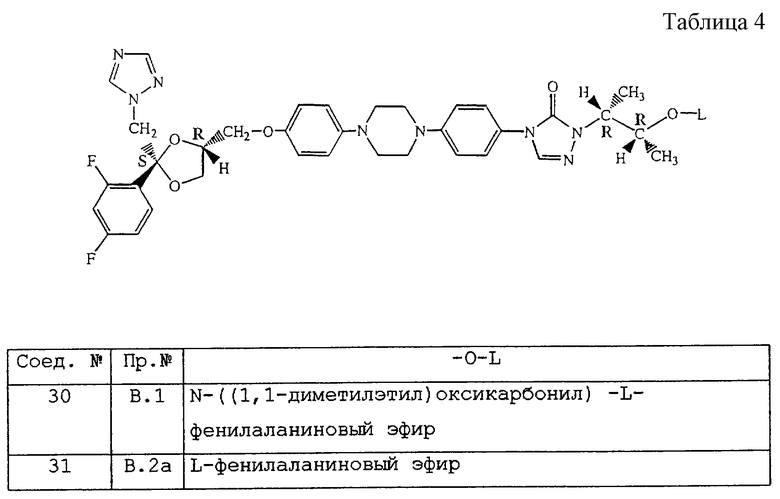
Таблица 4 (см. в конце описания).
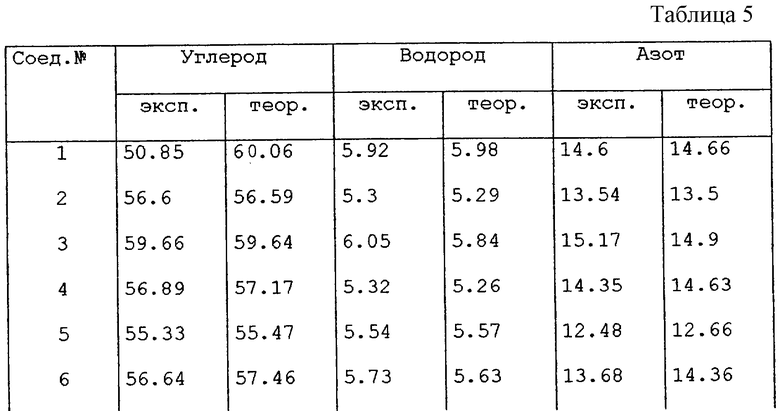
В таблице 5 представлены результаты элементного анализа, как экспериментального (заголовок колонки "эксп."), так и теоритического (заголовок колонки "теор."), для величин содержания углерода, водорода и азота в соединениях, полученных здесь выше в экспериментальной части.
С. Физикохимический пример
Пример С-1: Растворимость
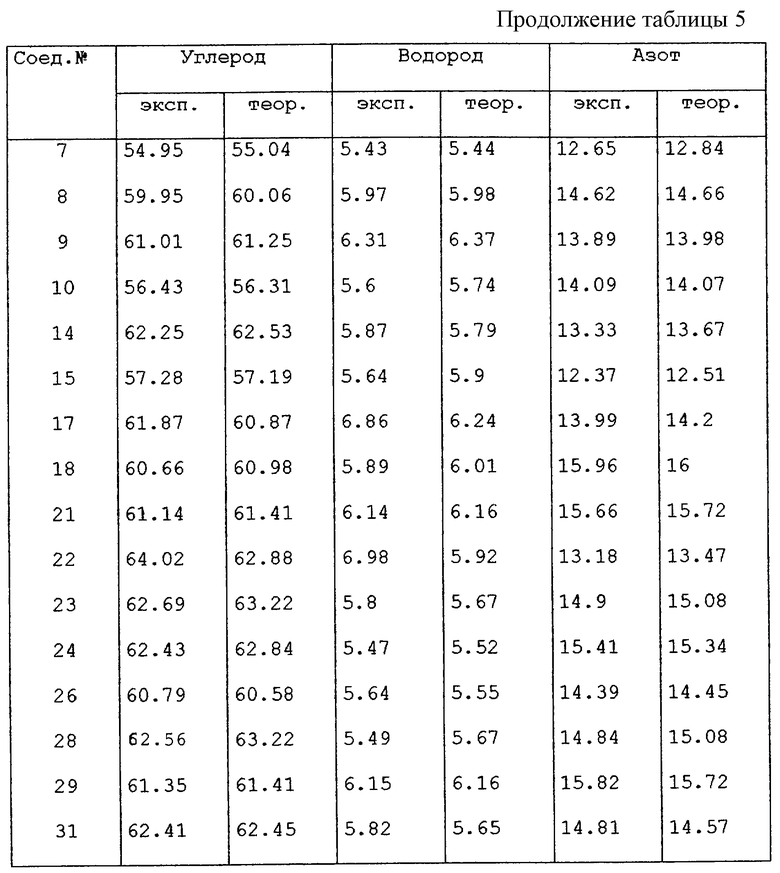
К растворителю добавляли избыток соединения (тип растворителя указан в таблице 6). Смесь встряхивали в течение 1 дня при комнатной температуре. Осадок отфильтровывали. Измеряли рН оставшегося растворителя, как показано в таблице 6. Концентрацию соединения, указанную в колонке "Растворимость", измеряли с помощью ВЭЖХ.
Таблица 7 (см. в конце описания).
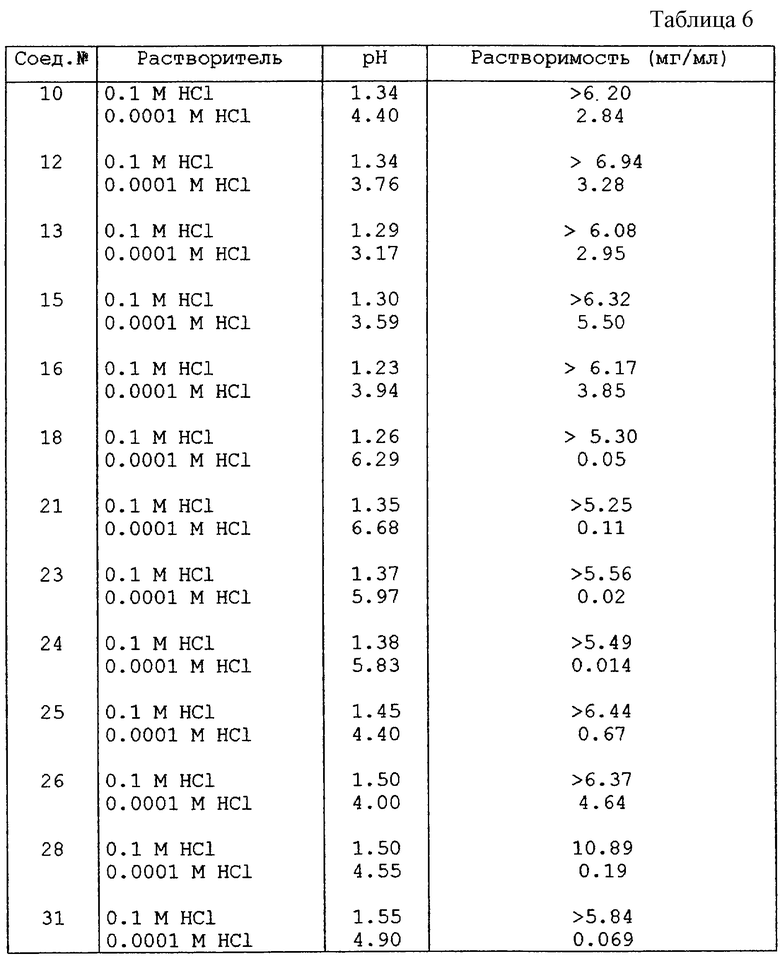
Пример С-2: Химическая стабильность
50 мг тестируемого соединения помещали в открытый стеклянный сосуд при 40oС и относительной влажности 75%. Через одну неделю определяли количество оставшегося тестируемого соединения.
D. Фармакологические примеры
Пример D-1: Определение чувствительности грибков
Для оценки активности тестируемых соединений in vitro применяли панель изолятов Candida и одиночные изоляты дерматофитов Microsporum canis, Trichophyton rubrum и Т. mentagrophytes; Aspergillus fumigates и Cryptococcus neoformans. Инокуляты готовили в виде бульонных культур (дрожжи) или как суспензии грибкового материала, сделанные из культур на скошенном агаре (плесневые грибы). Тестируемые соединения переносили пипеткой из маточного раствора DMSO в воду для получения серий 10-кратных разведении. Грибковые инокуляты суспендировали в среде для выращивания CYG (F.C. Odds, Journal of Clinical Microbiology, 29, (2735-2740, 1991) до приблизительно 50,000 колоний-формирующих единиц (CFU) на мл и добавляли к водному раствору тестируемых лекарств.
Культуры помещали в 96-луночные пластиковые планшеты для микротитрования и инкубировали их в течение 2 дней при 37oС (Candida spp.) или в течение 5 дней при 37oС (другие грибки). Рост микрокультур измеряли по их оптической плотности (OD), измеренной при длине волны 405 нм. OD для культур с тестируемыми соединениями рассчитывали как процент от контроля, OD для культур без лекарств. Торможение роста до 35% от контроля или ниже считали значительным торможением.
Минимальная ингибиторная концентрация (MICs; в 10-6 M) промежуточного продукта 2 как главного метаболита и некоторых из соединений формулы (I) для Candida glabrata, Candida krusei, Candida parapsilosis, Candida albicans, Candida kefyr, Candida tropicalis, Microsporum canis, Trichophyton rubrum, Trichophyton mentagrophytes, Cryptococcus neoformans, Aspergillus fumigatus представлены в таблице 8.
Пример D-2: Диссеминированный аспергилез и канадидоз у морских свинок
Во всех экспериментах применяли специальных, свободных от патогенных микроорганизмов (SPF) морских свинок (весом 400-500 г). Животным, которых лечили путем внутривенной инфузии, в левую яремную вену вводили катетер. Животным вводили Aspergillus fumigatus (4,000 CFU/г веса тела) или Candida albicans (40,000 CFU/г веса тела) либо через латеральную вену пениса, либо через имплантированный катетер. Лечение с помощью внутривенных инъекций (5 мг/кг/день) начинали через 1 час после инфицирования. Тестируемые составы вводили затем в последующие дни в виде двух одночасовых инфузий, разделенных пятичасовым периодом, ежедневно, суммарно 19 инфузий в течение 9,5 дней. Лечение тестируемыми соединениями перорально (5 мг/кг/день) начинали через 1 час после инфицирования и повторяли два раза в день до 10-го дня после инфицирования (всего 19 приемов препаратов). Для каждой группы тестируемых животных (число тестируемых животных в группе дано в колонке "N") записывали среднее время жизни (MST) в днях, а также % выживаемости (% выж.). Животных каждой группы, тех которые умирали в течение эксперимента и тех, которые пережили эксперимент и были забиты, исследовали post mortem на количество Aspergillus fumigatus и Candida albicans во внутренних органах (печени, селезенке, почках, легких и мозге). Оставшееся количество CFU/г в печени, позитивной по отношению к культурам, подсчитывали и представляли в таблице 9 (после внутривенной обработки) и в таблице 10 (после перорального введения) в виде среднего log10CFU/г. Колонки "% негат." в таблицах 9 и 10 отражают общий процент негативных в отношении культур внутренних органов после обработки. Следовательно, более эффективные тестируемые соединения характеризуются более высокими значениями в колонках "MST", "% выж." и "% негат." и более низкими в колонках "CFU/г".
Е. Пример составления композиции
Пример E.I: Раствор для инъекции.
1,8 грамм метил-1,4-гидроксибензоата и 0,2 грамма едкого натра растворяли в приблизительно 0,5 л кипящей воды для инъекций. После охлаждения до приблизительно 50oС туда добавляли при перемешивании 0,05 грамма пропиленгликоля и 4 грамма активного ингредиента. Раствор охлаждали до комнатной температуры и доводили водой для инъекций до 1 л, что давало раствор, включающий 4 мг/мл активного ингредиента. Раствор стерилизовали при фильтрации и заполняли им стерильные контейнеры.

Изобретение относится к области органической химии. Описывается сложный эфир общей формулы (I), его N-оксидная форма, фармацевтически приемлемая аддитивная соль или его стехиометрически изомерная форма, где -А-В- образуют двухвалентный радикал формулы: -N=CH- (a), -CH=N- (b), где один атом водорода в радикалах (а) и (b) может быть замещен C1-6 алкильным радикалом; L представляет собой ацильную часть аминокислоты; D является радикалом формулы (D1) или (D3), где Х является N; R1 представляет собой галоген; R2 является водородом или галогеном. Также описываются способы получения соединений формулы (I), энантиомерно чистая форма промежуточного соединения, энантиомерная смесь промежуточных продуктов, фармацевтическая композиция, обладающая противогрибковой активностью широкого спектра действия, и способ получения фармацевтической композиции. Технический результат - получены новые соединения, обладающие полезными биологическими свойствами. 7 с. и 11 з.п. ф-лы, 10 табл.
N-оксидная форма, фармацевтически приемлемая аддитивная соль или его стехиометрически изомерная форма,
где -А-В- образуют двухвалентный радикал формулы:
-N= CH- (a),
-CH= N- (b),
где один атом водорода в радикалах (а) и (b) может быть замещен C1-6 алкильным радикалом;
L представляет собой ацильную часть аминокислоты;
D является радикалом формулы
(D1)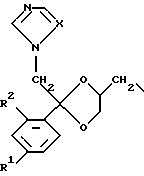
или (D2)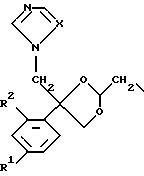
где X является N;
R1 представляет собой галоген;
R2 является водородом или галогеном.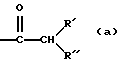
где R' представляет собой амино, моно- или ди(С1-6алкил)амино, моно- или ди(С1-6алкил)аминоС1-6алкил, амино(С1-С6)алкил или С1-6алкилоксикарбониламино;
R'' представляет собой водород, C1-6алкил; арил, С1-6алкил, замещенный арилом или амино;
арил является фенилом или фенилом, замещенным пщрокси или галогеном.
или тех их производных, в которых аминная часть является моно- или дизамещенной C1-6алкилом или однозамещенной трет-бутилоксикарбонилом.
2-[4-[4-[4-[4-[[2-(2,4-дифторфенил)-2-(1H-1,2,4-триазол-1-илметил)-1,3-диоксолан-4-ил] метокси] фенил] -1-пиперазинил] -фенил] -4,5-дигидро-5-оксо-1H-1,2, 4-триазол-1-ил] -1-метил-пропил N, N-диэтилглицин;
2-[4-[4-[4-[4-[[2-(2,4-дифторфенил)-2-(1H-1,2,4-триазол-1-илметил)-1,3-диоксолан-4-ил] метокси] -фенил] -1-пиперазинил] -фенил] -4,5-дигидро-5-оксо-1H-1,2,4-триазол-1-ил] -1-метил-пропил L-фенилаланин;
2-[4-[4-[4-[4-[[2-(2,4-дифторфенил)-2-(1H-1,2,4-триазол-1-илметил)-1,3-диоксолан-4-ил] метокси] -фенил] -1-пиперазинил] -фенил] -4,5-дигидро-5-оксо-1H-1,2,4-триазол-1-ил] -1-метил-пропил L-лейцин;
2-[4-[4-[4-[4-[[2-(2,4-дифторфенил)-2-(1H-1,2,4-триазол-1-илметил)-1,3-диоксолан-4-ил] метокси] -фенил] -1-пиперазинил] -фенил] -4,5-дигидро-5-оксо-1H-1,2,4-триазол-1-ил] -1-метил-пропил L-валин;
2-[4-[4-[4-[4-[[2-(2,4-дифторфенил)-2-(1H-1,2,4-триазол-1-илметил)-1,3-диоксолан-4-ил] метокси] -фенил] -1-пиперазинил] -фенил] -4,5-дигидро-5-оксо-1H-1,2,4-триазол-1-ил] -1-метил-пропил L-фенилглицин; его N-оксидную форму, его фармацевтически приемлемую аддитивную соль и его стереохимически изомерную форму.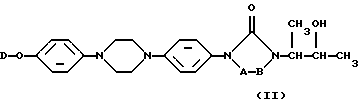
его N-оксида или его аддитивной соли, где D и -А-В-, как указано в п. 1.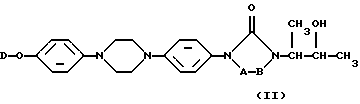
их N-оксидов или их аддитивных солей, где D и -А-В-, как определено в п. 1.
[2α, 4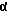 [(R*, R*)] ] -4-[4-[4-[4-[[2-[2,4-дифторфенил)-2-(1H-1,2,4-триазол-1-илметил)-1,3-диоксолан-4-ил] метокси] фенил] -1-пиперазинил] фенил] -2,4-дигидро-2-(2-гидрокси-1-метилпропил)-3H-1,2,4-триазол-3-он.
[(R*, R*)] ] -4-[4-[4-[4-[[2-[2,4-дифторфенил)-2-(1H-1,2,4-триазол-1-илметил)-1,3-диоксолан-4-ил] метокси] фенил] -1-пиперазинил] фенил] -2,4-дигидро-2-(2-гидрокси-1-метилпропил)-3H-1,2,4-триазол-3-он.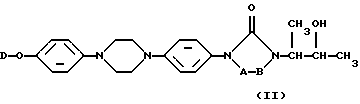
где D и -А-В- такие, как определены в п. 1,
с помощью ацилирующего реагента формулы (III)
W1-L, (III),
где W1 является реакционноспособной удаляемой группой, связанной с ацильной частью L, и L такое, как определено в п. 1, проводят путем перемешивания реагентов в реакционно-инертном растворителе, с необязательным добавлением в смесь основания для нейтрализации кислоты, которая образуется в процессе реакции;
и, если это желательно, преобразование соединений формулы (I) друг в друга с помощью известных в данной области превращений; и далее, если это желательно, превращение соединений формулы (I) в терапевтически активную нетоксичную кислую аддитивную соль путем обработки кислотой, или наоборот, превращение кислой аддитивной солевой формы в свободное основание путем обработки щелочью; и, если это желательно, получение их стехиометрически изомерных форм или их N-оксидных форм.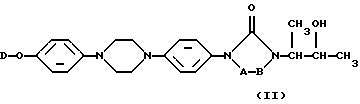
подвергают O-ацилированию с помощью реагента формулы (VI)
W1-L'-W3, (VI)
с последующим взаимодействием полученного таким образом промежуточного продукта формулы (VII)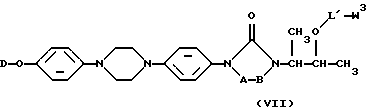
с амином формулы (VIII)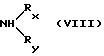
где W3 является реакционноспособной удаляемой группой; D и -А-В- такие, как определены в п. 1, причем NRxRy является необязательно одно- или дизамещенной аминогруппой аминокислоты, как определено в отношении L в п. 1, причем L' идентичен L, как указано в п. 1, исключая необязательно одно- или дизамещенную аминогруппу;
и, если это желательно, преобразование соединений формулы (I) друг в друга с помощью известных в данной области превращений; и далее, если это желательно, преобразование соединений формулы (I) в терапевтически активную нетоксичную кислотно-аддитивную соль путем обработки кислотой, или наоборот, преобразование кислотно-аддитивной солевой формы в свободное основание путем обработки щелочью; и, если это желательно, получение их стехиометрически изомерных форм или их N-оксидных форм.
Приоритет по пунктам:
11.02.1997 - по пп. 1-9 и 14-19;
15.10.1997 - по пп. 10-13.
| Прибор для очистки паром от сажи дымогарных трубок в паровозных котлах | 1913 |
|
SU95A1 |
| Приспособление в пере для письма с целью увеличения на нем запаса чернил и уменьшения скорости их высыхания | 1917 |
|
SU96A1 |
| Бесколесный шариковый ход для железнодорожных вагонов | 1917 |
|
SU97A1 |
| Прибор для очистки паром от сажи дымогарных трубок в паровозных котлах | 1913 |
|
SU95A1 |
| RU 94045798 A1, 20.07.1996 | |||
| RU 2056421 C1, 20.03.1996. | |||
