Изобретение относится к каталитическим системам и более конкретно к форполимеризованным металлоцен/алюмоксановым каталитическим системам на носителе, которые могут быть использованы для полимеризации олефинов или смесей олефинов до полиолефинов. Они прежде всего пригодны для получения гранулированных изотактических полимеров пропилена большой молекулярной массы и с высокой температурой плавления.
Каталитические системы на носителе хорошо известны.
Техника нанесения каталитической системы на носитель весьма разнообразна. Так, например, техника применения гидратированных носителей или подложек представлена в заявках на Европейские патенты 367503 и 336593 и в описании к патенту США 4912075; способы применения инертных носителей и металлоценов различных типов представлены в патенте США 4659685. В патентах США 4808561 и 4701432 представлены способы, в которых носитель кальцинируют и в дальнейшем вводят в контакт с металлоценом и активатором или сокаталитическим компонентом.
В данной области техники известны способы получения полимерного продукта, характеризующегося однородными компактными сферическими частицами, узким распределением частиц по размерам и/или высокой объемной плотностью, с использованием катализаторов на носителе. В заявках на Европейские патенты 302424 и 354893 предлагается предварительная активация или предварительное введение в контакт металлоцена и активатора; в заявке на Европейский патент 426646 и в патенте США 4871705 предлагается форполимеризация каталитической системы на носителе в присутствии по меньшей мере одного олефина; в заявке на Европейский патент 279586 для получения порошкообразного полимера предлагается использовать тонкоизмельченный алюмоксан; а в заявке на Европейский патент 314797 предлагается использование нанесенной на носитель каталитической системы и наполнителей в этой системе, обладающих сродством к полиолефинам. В заявке на Европейский патент 518092 предлагается нанесение каталитической системы на тонкодисперсные частицы полимеризата.
Осуществление ранее известной технологии нанесения на носитель обычно позволяет получать каталитическую систему, каталитические характеристики которой хуже, чем у ее аналога без носителя. Активность получаемого катализатора на носителе ниже активности каталитической системы, не нанесенной на носитель. Получаемый полимер обычно обладает уменьшенной молекулярной массой и/или пониженной температурой плавления. Любые преимущества, достигаемые в отношении полимерной морфологии с использованием ранее известного катализатора на носителе, сводятся на нет ухудшающими эффектами вследствие засорения, образования мелочи, наслоения и комкования, которые обычно наблюдаются в полимеризационном реакторе при проведении полимеризации в газовой фазе, массе или суспензии.
Хорошо известно, что пониженная температура реакции полимеризации дала бы возможность увеличить температуру плавления и молекулярную массу получаемого полимера, но снижение температуры реакции полимеризации не всегда практично. Для промышленности привлекательны каталитические системы на носителе, которые способны работать при высоких реакционных температурах, т.е. при таких, которые превышают 60oC, поскольку они обычно позволяют повысить каталитическую активность и позволяют обходиться без создания особых условий охлаждения для отвода теплоты реакции, которая высвобождается во время полимеризации. При применении катализаторов на носителе необходимо принимать во внимание такие факторы, как каталитическая активность и условия реакции полимеризации, а также молекулярная масса и температура плавления получаемого полимера.
Для получения стереорегулярных полимеров с большой молекулярной массой, высокой температурой плавления и узким молекулярно-массовым распределением известно применение гомогенных или не нанесенных на носители металлоценовых каталитических систем. Так, например, в заявке на Европейский патент 0485822 сказано, что замещенные бис- инденилцирконоцены и алюмоксановые активаторы представляют собой каталитические системы, приемлемые для получения изотактического полимера с высокой температурой плавления, большой молекулярной массой и узким молекулярно-массовым распределением. Было установлено, что некоторые из таких каталитических систем являются высокоактивными каталитическими системами. Однако, как указано выше, после нанесения на инертные носители с использованием ранее известной технологии такие каталитические системы проявляют нежелательную тенденцию к потере активности и способности образовывать полимер большой молекулярной массы и/или с высокой температурой плавления.
В качестве ближайшего аналога выбрана заявка на ЕР 0485822, описывающая каталитическую систему на основе металлоцена и алюмоксана, нанесенных на неорганический пористый носитель, которая может быть форполимеризована в присутствии олефинового мономера(ов).
Суммируя предпосылки создания изобретения, можно сказать, что существуют металлоценовые каталитические системы, которые проявляют высокую активность и обеспечивают получение стереорегулярного полимера большой молекулярной массы, с высокой температурой плавления и узким молекулярно-массовым распределением. Однако при применении ранее известной техники нанесения в отношении ранее известных каталитических систем, проявляющих высокую активность при получении стереорегулярных полимеров, образуются каталитические системы на носителе, которые демонстрируют пониженную каталитическую активность в сравнении с их аналогами без носителей. Ранее известные каталитические системы обычно обеспечивают получение полимера с более низкими температурой плавления и молекулярной массой, чем у полимера, который получают с помощью аналогичной каталитической системы без носителя.
Задачей настоящего изобретения является создание каталитической системы на носителе, которая позволяет получать гранулированный высокоизо-тактический полиолефин, обладающий высокой температурой плавления, достигающей приблизительно 140oC или более, молекулярной массой примерно 100000 или более и узким молекулярно-массовым распределением. В данном описании молекулярную массу определяют как средневесовую молекулярную массу, а молекулярно-массовое распределение (ММР) определяют как соотношение между средневесовой молекулярной массой и среднечисленной молекулярной массой (Мw/Мn). Чтобы быть привлекательной с коммерческой точки зрения, такая каталитическая система не должна вызывать заметного или какого-либо вообще засорения реактора или образования наслоений и должна сохранять свою стабильность в течение достаточно продолжительного периода времени, позволяющего, если это необходимо, хранить и транспортировать катализатор на носителе.
Задача решается каталитической системой на основе металлоцена и алюмоксана, нанесенных на неорганический пористый носитель, полученной способом, включающим:
(а) контактирование металлоценового компонента (ов) с алюмоксаном в приемлемом растворителе с получением реакционного продукта, причем металлоцен отвечает общей формуле, приведенной ниже,
(б) нанесение на неорганический носитель полученного реакционного продукта, и
(в) необязательную форполимеризацию полученной при этом каталитической системы на носителе в присутствии олефинового мономера (ов).
Задача решается также каталитической системой на основе металлоцена и алюмоксана, нанесенных на неорганический пористый носитель, форполимеризованной в присутствии олефинового мономера(ов), полученной способом, включающим:
(а) контактирование металлоценового компонента (ов) с алюмоксаном в приемлемом растворителе с получением реакционного продукта, причем металлоцен отвечает общей формуле, приведенной ниже,
(б) нанесение на неорганический пористый носитель полученного реакционного продукта, и
(в) форполимеризацию полученной при этом каталитической системы на носителе в присутствии олефинового мономера (ов).
Задача также решается способом получения гомо- или сополимера пропилена, молекулярная масса которого составляет примерно 100000 г/моль или более, температура плавления равна примерно 140oC или выше, объемная плотность равна примерно 0,30 г/см3 или более, полимеризацией пропилена или сополимеризацией смесей пропилена с одним или несколькими другими олефинами при температуре полимеризационной реакции приблизительно 45oC или выше, под полимеризационным давлением 0,5 - 100 бар в газовой фазе, массе или суспензии в присутствии каталитической системы на основе металлоцена и алюмоксана, нанесенных на неорганический пористый носитель, необязательно форполимеризованной в присутствии олефинового мономера(ов), причем используют каталитическую систему, полученную способом, включающим:
(а) контактирование металлоценового компонента(ов) с алюмоксаном в приемлемом растворителе с получением реакционного продукта, причем металлоцен отвечает общей формуле, приведенной ниже,
(б) нанесение на неорганический пористый носитель полученного реакционного продукта, и
(в) необязательную форполимеризацию полученной при этом каталитической системы на носителе в присутствии олефинового мономера (ов).
Преимущественно пропилен полимеризуют до изотактического полипропилена.
Смеси пропилена с другими способными полимеризоваться олефинами преимущественно полимеризуют до сополимеров полипропилена.
Преимущественно каталитическую систему форполимеризуют с получением пропиленового полимера или его сополимеров, характеризующихся молекулярной массой примерно 100000 или более, температурой плавления примерно 140oC или выше и объемной плотностью примерно 0,30 г/см3 или более.
Задача решается также способом получения каталитической системы с использованием металлоцена и алюмоксана, включающим стадии:
а) контактирования металлоценового компонента (ов) с алюмоксаном в приемлемом растворителе с получением реакционного продукта, причем металлоценовый компонент определен ниже;
б) контактирование полученного реакционного продукта в приемлемом растворителе с пористым носителем;
в) удаление из образовавшейся суспензии практически всего растворителя;
г) получение каталитической системы на носителе; и
д) необязательную форполимеризацию полученной каталитической системы на носителе в присутствии олефинового мономера(ов).
Предлагаемый по изобретению катализатор на носителе можно применять в процессах полимеризации в газовой фазе, массе, растворе или суспензии при реакционной температуре, превышающей примерно 45oC.
Было установлено, что каталитические системы, нанесенные по способу изобретения, сохраняют каталитические рабочие характеристики или свойства подобно их аналогам, не нанесенным на носитель. К таким рабочим характеристикам относятся активность, способность обеспечить получение полимера с температурой плавления, молекулярной массой и морфологией, по которым он идентичен продукту, полученному с использованием их аналогов, но без носителя. Кроме того, было установлено, что такие каталитические системы на носителе в реакторе образуют незначительные или не образуют вообще засоров, мелочи или комков.
Эта каталитическая система на носителе особенно пригодна при получении полимеров пропилена, в частности гранулированного изотактического полипропилена большой молекулярной массы, с высокой температурой плавления и объемной плотностью в интервале от примерно 0,30 до примерно 0,45 г/см3 или более и средним размером частиц от примерно 500 до 1000 мкм и более. Получаемый изотактический полипропилен представляет собой легкосыпучий материал, простой в хранении и при транспортировке. Полученные полимеры предназначены для изготовления формованных изделий, пленок или волокон.
Используемые в описании термины "носитель" и "подложка" являются взаимозаменяющими и применительны для ссылки на любую структуру, которая может служить в качестве опоры для каталитического компонента или каталитической системы. Термином "металлоцен" обозначают производное такого π -связывающего остатка, как циклопентадиенильный (Cp) остаток или его производное, или вспомогательный лиганд, который координационно связан с атомом переходного металла. Используемый переходный металл представляет собой металл группы 4, 5 или 6, предпочтительно группы 4, а наиболее предпочтителен цирконий (Zr). Упомянутая Периодическая таблица элементов аналогична описанной системе обозначения New IUPAC notation in Hawiey's Condensed Chemical Dictionary, издание 11-е, переработанное, Sax and Lewis, Van Nostrand Reinhoid, New York, 1987 г.
А. Металлоценовый компонент
Металлоценовые компоненты подробно описаны в патенте США 5017714 и в заявке на Европейский патент 129368. Металлоценовые компоненты, используемые по настоящему изобретению, включают в себя атом переходного металла группы 4, 5 или 6, дициклопентадиенильные производные, предпочтительно бис-инденилметаллоценовые компоненты, отвечающие нижеследующей общей структурной формуле:
где М1 обозначает металл группы IV, V или VI Периодической таблицы, например титан, цирконий, гафний, ванадий, ниобий, тантал, хром, молибден и вольфрам, предпочтительно цирконий, гафний и титан, и наиболее предпочтительно цирконий;
R1 и R2 одинаковые или различные и обозначают атом водорода, C1-C10-алкильную группу, C1-C10-алкоксигруппу, C6-C10- арильную группу, C6-C10-арилоксигруппу, C2-C10-алкенильную группу, C7-C40-арилалкильную группу, C7-C40-алкиларильную группу, C8-C40-арилалкенильную группу или атом галогена;
R3 и R4 обозначают атом водорода;
R5 и R6 одинаковые или различные, предпочтительно одинаковые, и обозначают атом галогена, C1-C10-алкильную группу, которая может быть галоидированной, C6-C10-арильную группу, которая может быть галоидированной, C2-C10-алкенильную группу, C7-C40-арилалкильную группу, C7-C40-алкиларильную группу, C8-C40-арилалкенильную группу, радикал -NR
R7 обозначает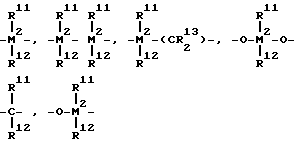
= BR11, =AIR11, -Ge-, -Sn-, -0-, -S-, =SO, =SO2, =NR11, =CO, PR11 или = P(O)R11;
где R11, R12 и R13 одинаковые или различные и обозначают атом водорода или атом галогена, C1-C20-алкильную группу, C1-C20-фторалкильную группу, C6-C30-арильную группу, C6-C30фторарильную группу, C1- C20-алкоксигруппу, C2-C20-алкенильную группу, C7-C40-арилалкильную группу, C8-C40-арилалкенильную группу, C7-C40-алкиларильную группу, или же R11 и R12 или R11 и R13 совместно со связывающими их атомами могут образовывать циклические системы;
М2 обозначает атом кремния, германия или олова, предпочтительно кремния или германия, наиболее предпочтительно кремния;
R8 и R9 одинаковые или различные и имеют те же значения, что и R11;
m и n одинаковые или различные и обозначает 0, 1 или 2, сумма m и n равна 0,1 или 2; а
радикалы R10 одинаковые или различные и имеют те же значения, что и R11, R12 и R13, где два смежных радикала R10 могут быть связаны с образованием кольцевой системы, предпочтительно кольцевой системы, содержащей приблизительно 4 - 6 углеродных атомов.
R1 и R2 предпочтительно обозначают C1-C3-алкильную группу, C1-C3-aлкоксигруппу, C6-C8-арильную группу, C6-C8-арилоксигруппу, C2-C4-алкенильную группу, C7-C10-арилалкильную группу, C7-C12-алкиларильную группу, C8-C12-арилалкенильную группу или хлор;
R5 и R6 предпочтительно одинаковые и предпочтительно обозначают атом фтора, хлора или брома, предпочтительно C1-C4-алкильную группу, которая может быть галоидирована, C6-C8-арильную группу, C2-C4-алкенильную группу, C7-C10арилалкильную группу, C7-C12алкиларильную группу, C8-C12-apилалкенильную группу.
Предпочтительно R15 обозначает атом хлора, C1-C3-алкильную группу или C6-C9-арильную группу.
Предпочтительно R11, RR12 и R13обозначают C1-C10-алкильную группу, C1-C10-фторалкильную группу, C6-C20-арильную группу, C6-C20-фторарильную группу, C1-C10-алкоксигруппу, C2-C10-алкенильную группу, C7-C20-арилалкильную группу, C8-C22-арилалкенильную группу или C7-C20-алкиларильную группу.
Термин "алкил" применителен в отношении линейных или разветвленных заместителей. Галогеном (при галоидированном состоянии) служит атом фтора, хлора, брома или иода, предпочтительно фтора или хлора.
Особенно предпочтительными металлоценами являются соединения формул
где M1обозначает Zr или Hf, R1 и R2 обозначают метил или хлор, а значения R5, R6, R8, R9, R10, R11 и R12 определены выше.
Хиральные металлоцены в виде рацематов используют для получения высших изотактических поли-1-олефинов.
Можно также использовать чистую R- или S-форму. С применением этих чистых стереоизомерных форм можно получать оптически активный полимер. Чтобы центр (т. е. атом металла) обеспечивал возможность стереорегулярной полимеризации, мезоформу металлоценов предпочтительно удалять.
Разделение стереоизомеров можно производить по известной в литературе технологии. Для особых продуктов возможно также использование рацемато/мезосмесей.
Обычно металлоцены получают с помощью многостадийного способа, в котором предусмотрены повторяющиеся депротонирование/металлирование ароматических лигандов и введение мостика и центрального атома в виде их галоидированных производных. Этот общий технический прием иллюстрирует нижеследующая реакционная схема:




X=Cl, Br, I или O-тозил,
Процессы получения описанных металлоценов проиллюстрированы в приведенных в описании пояснительных примерах, в Journal of Organometallic Chem.. 288, (1958), 63 - 67, и в заявке на Европейский патент 320762. JOCS и заявка на Европейский патент '762 включены в настоящее описание исключительно в качестве ссылок.
Иллюстрирующие, но не ограничивающие примеры металлоценов включают в себя:
диметилсиландиилбис(2-метил-4-фенил-1-инденил)ZrCl2;
димeтилсилaндиилбис(2-мeтил-4,5-бeнзoиндeнил)ZrCl2;
диметилсиландиилбис-(2-метил-4,6-диизопропилинденил)ZrCl2);
диметилсиландиилбис(2-этил-4-фенил-1-инденил)ZrCl2;
диметилсиландиилбис(4-нафтил-1-инденил)ZrCl2;
фенил(метил)силандиилбис(2-метил-4-фенил-1-инденил)ZrCl2;
диметилсиландиилбис(2-метил-4-(1-нафтил)-1-инденил)ZrCl2;
диметилсиландиилбис(2-метил-4-(2-нафтил)-1-инденил) ZrCl2;
диметилсиландиилбис(инденил)ZrCl2;
диметилсиландиилбис(2-метил-4,5-диизопропил-1-инденил)ZrCl2;
диметилсиландиилбис(2,4,6-триметил-1-инденил)ZrCl2;
фенил(метил)силандиилбис(2-метил-4,6-диизопропил-1- инденил)ZrCl2;
1,2-этaндиилбиc(2-мeтил-4,6-диизoпpoпил-1-индeнил)ZrCl2;
1,2-бутaндиилбиc(2-мeтил-4,6-диизoпpoпил-1-индeнил)ZrCl2;
димeтилcилaндиилбиc(2-мeтил-4-этил-1-индeнил)ZrCl2;
диметилсиландиилбис(2-метил-4-изопропил-1-инденил)ZrCl2;
диметилсиландиилбис(2-метил-4-трет.-бутил-1-инденил)ZrCl2;
фенил(метил)силандиилбис(2-метил-4-изопропил-1-инденил)ZrCl2;
димeтилcилaндиилбиc(2-этил-4-мeтил-1-индeнил)ZrCl2;
диметилсиландиилбис(2,4-диметил-1-инденил)ZrCl2;
диметилсиландиилбис(2-метил-4-этил-1-инденил)ZrCl2;
диметилсиландиилбис(2-метил- α -аценафт-1-инденил)ZrCl2;
фeнил(мeтил)cилaндиилбиc(2-мeтил-4,5-бeнзo-1-индeнил)ZrCl2;
фенил(метил)силандиилбис(2-метил-4,5-(метилбензо)-1-инденил) ZrCl2;
фенил(метил)силандиилбис (2-метил-4,5-(тетраметилбензо)-1-инденил)ZrCl2;
фенил(метил)силандиилбис(2-метил- α -аценафт-1-инденил)ZrCl2;
1,2-этандиилбис(2-метил-4,5-бензо-1-инденил)ZrCl2;
1,2- бутандиилбис(2-метил-4,5-бензо-1-инденил)ZrCl2;
диметилсиландиилбис(2-метил-4,5-бензо-1-инденил)ZrCl2;
1,2-этaндиилбиc(2,4,7-тpимeтил-1-индeнил)ZrCl2;
диметилсиландиилбис(2-метил-1-инденил)ZrCl2;
1,2-этандиилбис(2-метил-1-инденил)ZrCl2;
фенил(метил)силандиилбис(2-метил-1-инденил)ZrCl2;
дифенилсиландиилбис(2-метил-1-инденил)ZrCl2;
1,2-бутандиилбис(2-метил-1-инденил)ZrCl2;
диметилсиландиилбис(2-этил-1-инденил)ZrCl2;
диметилсиландиилбис(2-метил-5-изобутил-1-инденил)ZrCl2;
фенил(метил)силандиилбис(2-метил-5-изобутил-1-инденил)ZrCl2;
диметилсиландиилбис(2-метил-5-трет.-бутил-1-инденил)ZrCl2;
димeтилcилaндиилбиc(2,5,6-тpимeтил-1-индeнил)ZrCl2 и т.п.
Можно составить аналогичный список иллюстративных бис-инденильных вариантов, содержащих различные другие металлы, но поскольку такой список оказался бы почти идентичным тому, что уже приведен для подробного описания, этот список, по-видимому, существенного значения не имеет. Любому специалисту в данной области техники очевидно, что в композициях по настоящему изобретению могли бы быть использованы другие циклопентадиенильные производные и переходные металлы, отличные от циркония.
Б. Активаторный компонент
Активаторный или сокаталитический компонент по настоящему изобретению представляет собой алюмоксан, отвечающий общей формуле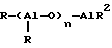
для олигомерного линейного алюмоксана и
для олигомерного, циклического алюмоксана, где значение каждого из n и m равно 1 - 40, наиболее предпочтительно 3 - 20, а R обозначает C1-C8-алкильную группу или R обозначает C6-C18арильную группу или водород, предпочтительно метильную группу, или же R может обозначать смеси алкильных и арильных заместителей.
Точное строение алюмоксана неизвестно. Алюмоксан или метилалюмоксан может быть получен различными известными способами. Независимо от способа, применяемого для получения алюмоксана, обычным для всех алюмоксановых растворов является варьирование содержания не подвергнутого конверсии исходного алюминиевого соединения в свободной форме или в форме аддукта.
В. Среда носителя
Обычно носителями могут служить любые органические или неорганические инертные твердые, в частности пористые, носители, такие, как тальк, неорганические окислы и смолистые материалы носителя, такие, как полиолефин, или такие материалы, которые могут быть использованы в качестве носителей, представленные в заявке на Европейский патент 519236.
Подходящие неорганические окисные материалы, которые желательно использовать, охватывают окислы элементов, относящиеся к группам 2, 3, 4, 5, 13, 14, 15, 16 Периодической таблицы элементов. Примеры окислов включают в себя двуокись кремния, окись алюминия, кремнийдиоксид-алюминийоксид и их смеси. Среди других неорганических окислов, которые можно применять либо индивидуально, либо в сочетании с двуокисью кремния, окисью алюминия или смесями кремнийдиоксид-алюминийоксид, следует упомянуть окись магния, двуокись титана, двуокись циркония и т.п. Можно применять другие подходящие материалы носителя, в частности тонкодисперсные полиолефины, например полиэтилен.
Если в естественных условиях носитель обладает низким влагосодержанием или остаточным содержанием растворителя, дегидратация или сушка перед его применением может оказаться необязательной. Но если это не так, например при использовании кремнийдиоксидного носителя, предусматривают дегидратацию или сушку. В предпочтительном варианте содержание воды или влаги в используемом носителе, определяемое по потерям при сжигании (ППС), составляет приблизительно 1% или меньше. Для достижения предпочтительных данных ППС можно проводить термическую дегидратацию или обработку для сушки носителя в вакууме или с обдувом сухим инертным газом, таким, как азот, при температуре от примерно 100oC до примерно 1000oC, предпочтительно от примерно 300oC до примерно 800oC. Параметры давления решающего значения не имеют. Однако обычно поддерживают нормальные условия. Продолжительность термической обработки может составлять от примерно 1 до примерно 24 часов. Однако эта операция может быть менее или более продолжительной при условии установления равновесия с поверхностными гидроксильными группами; обычно она занимает 4 - 8 часов.
Дегидратацию или сушку носителя можно проводить, подвергая носитель химической обработке для удаления воды и снижения концентрации поверхностных гидроксильных групп. Химическая обработка подвергает конверсии всю воду и гидроксильные группы в поверхностном слое окислов в инертные продукты. Пригодными химическими агентами являются, например, хлорсиланы, такие, как SiCl4, триметилхлорсилан, диметиламинотриметилсилан и т.п. Химическую дегидратацию проводят суспендированием неорганического материала в форме частиц, в частности двуокиси кремния, в инертном низкокипящем углеводороде, таком, как гексан. При химической дегидратационной обработке двуокись кремния необходимо содержать в атмосфере, свободной от влаги и кислорода. Затем в суспензию двуокиси кремния добавляют раствор в низкокипящем инертном углеводороде химического дегидратирующего агента, такого, как дихлордиметилсилан. Этот раствор медленно добавляют в суспензию. Во время реакции химической дегидратации температура находится в интервале от примерно 25oC до примерно 120oC, однако эту реакцию можно проводить при более высокой и более низкой температуре. Предпочтительная температура составляет от примерно 50oC до примерно 70oC. Процедуру химической дегидратации следует осуществлять до полного удаления влаги из порошкообразного материала носителя, на что указывает прекращение выделения газа. Обычно реакцию химической дегидратации проводят в течение от примерно 30 мин до примерно 16 ч, предпочтительно от 1 до 5 ч. По завершении химической дегидратации твердый порошкообразный материал отфильтровывают в атмосфере азота и промывают один или несколько раз сухим, не содержащим кислорода инертным растворителем. Для промывки в качестве растворителей, а также разбавителей, используемых при приготовлении суспензии и раствора химического дегидратирующего агента, можно применять любой подходящий инертный углеводород. Примерами таких углеводородов служат гептан, гексан, толуол, изопентан и т.п.
К характеристикам, которые определяют количество носителя, используемого для приготовления каталитических композиций, а также влияют на свойства получаемых полимеров, относятся конкретный размер частиц, удельные площадь поверхности, объем пор и число гидроксильных групп. Предпочтительные носители включают в себя двуокись кремния или другие материалы, которые характеризуются площадью поверхности приблизительно 10 - 500 м2 или пористостью примерно 0,2 - 3,5 см3 объема пор/грамм носителя (см3/г).
Можно применять органические носители, такие, как полистирол; при этом также предполагается, что они содержат минимальное количество или вообще не содержит влаги, остаточного растворителя процесса обработки или примесей, которые способны повлиять на рабочие свойства готовых каталитических систем.
Г. Нанесение каталитической системы на среду носителя
Обычно применяемая техника нанесения включает в себя контактирование металлоценового компонента, описанного выше, в соответствующем растворителе с алюмоксаном или метилалюмоксаном с получением растворимого реакционного продукта. Далее этот растворимый реакционный продукт вводят в контакт с дегидратированной подложкой или носителем, удаляют растворитель и полученную каталитическую систему на носителе сушат для обеспечения удаления практически всего или большей части остаточного растворителя из пор носителя. Получают легкосыпучую каталитическую систему.
Таким образом, способ получения легкосыпучей, предварительно полимеризованной каталитической системы на носителе включает в себя стадии
а) приготовления в подходящем растворителе металлоцен/алюмоксановой смеси, содержащийся в которой металлоцен описан выше;
б) контактирования смеси (а) с пористым, обычно неорганическим дегидратированным носителем;
в) удаления практически всего растворителя из образовавшейся суспензии;
г) получения каталитической системы на носителе и
д) необязательной форполимеризации указанной каталитической системы на носителе совместно с одним или несколькими олефиновыми мономерами с получением таким образом форполимеризованной каталитической системы на носителе для получения полимеров пропилена или его сополимеров, молекулярная масса которых составляет приблизительно 100000 или более, предпочтительно 150000 или более, температура плавления равна примерно 140oC или более, предпочтительно равна примерно 145oC или более, и объемная плотность равна приблизительно 0,30 г/см3 или более. Средний размер частиц полученного гранулированного полимера составляет от примерно 500 до примерно 1000 мкм или более.
Во всех случаях индивидуальные компоненты, а также выделенный каталитический компонент на носителе защищают от кислорода и влаги. Таким образом, реакции следует проводить в несодержащей кислород и влагу атмосфере, и выделенный катализатор на носителе содержат и хранят в атмосфере, которая не содержит влагу и кислород. Такие реакции предпочтительно проводить в присутствии сухого инертного газа, например, такого, как азот.
К предпочтительным растворителям для введения металлоцена в контакт с активатором относятся нефтепродукты и различные углеводороды, которые при реакционной температуре находятся в жидком состоянии и в которых по предпочтительному варианту растворимы индивидуальные компоненты. Тем не менее, растворимость индивидуальных компонентов необязательна при условии, что в выбранном растворителе растворим продукт реакции металлоцена с активатором. Иллюстрирующими примерами полезных или приемлемых растворителей служат алканы, в частности пентан, изопентан, гексан, гептан, октан и нонан, циклоалканы, в частности циклопентан и циклогексан, и ароматические продукты, такие, как бензол, толуол, этилбензол и диэтилбензол.
Количества алюмоксана и металлоцена, используемые при приготовлении каталитической системы на носителе, могут варьироваться в широком интервале. Однако молярное соотношение между алюминием в активаторе и переходным металлом в металлоцене составляет от примерно 12:1 до примерно 1000:1, более предпочтительно от примерно 50:1 до примерно 500:1.
Обычно в таких исследованиях использовали 30 вес.% MAO в толуоле, однако допустимы 10 вес.% в толуоле.
В одном варианте выполнения настоящего изобретения готовят толуольный раствор, содержащий металлоцен, в который добавляют толуольный раствор MAO (30 вес. % в толуоле). Количество растворителя должно быть достаточным для растворения реакционного продукта, адекватного отвода тепла от каталитических компонентов во время реакции и достижения хорошего перемешивания. В другом варианте для растворения металлоцена и MAO можно использовать один и тот же инертный растворитель или разные растворители. Металлоценовый раствор готовят и добавляют в приготовленный раствор MAO, проводя реакцию при комнатной температуре. Реакция между MAO и металлоценом протекает быстро, и желательно, чтобы продолжительность контактирования составляла приблизительно от одной минуты до одного часа, предпочтительно примерно десять минут. О ходе реакции между этими двумя компонентами можно судить благодаря ее экзотермической природе и изменению окраски. Тем не менее, экзотермия или изменения окраски необязательны: для определения окончания реакции можно пользоваться другой аналитической техникой.
Растворимый продукт реакции металлоцена с активатором в дальнейшем вводят в контакт с инертным носителем, обычно с двуокисью кремния, в сухом виде или в виде суспензии, которую готовят в том же самом или другом инертном растворителе. В предпочтительном варианте двуокись кремния добавляют в сухом виде. Компоненты можно добавлять в реакционный сосуд быстро или медленно. При контактировании образуется суспензия, включающая в себя металлоцен, алюмоксан и носитель, которую предпочтительно выдерживать при комнатной температуре в течение приблизительно от одной минуты до одного часа, наиболее предпочтительно примерно пять минут. Во время введения раствора металлоцена/MAO в контакт с носителем поддерживаемую температуру можно варьировать в широком диапазоне, например от 0oC до 100oC. Тем не менее, можно использовать и температуру, которая превышает или ниже интервала 0 - 100oC. Суспензию или смесь можно перемешивать с использованием тепла или без него и удалять из катализатора на носителе почти весь или большую часть растворителя. Предпочтительно удалять как растворитель, который обнаруживается визуально, так и практически весь растворитель, содержащийся в порах носителя. Удаление растворителя из смеси можно производить обычным путем, в частности, с помощью техники выпаривания в вакууме, при пониженном давлении, или техники мгновенного испарения. Нагрев можно производить до удаления свободного растворителя, обычно в течение от одного до трех часов, поддерживая температуру от примерно 30oC до примерно 60oC. Свободным растворителем считают тот, наличие которого можно визуально определить в реакционной смеси. Остаточным растворителем является тот, что захватывается порами носителя.
Другим вариантом удаления практически всего растворителя является сушка катализатора на носителе до "шламоподобного" состояния, когда удалена практически вся свободная жидкость, с последующей промывкой каталитического продукта низкокипящим углеводородом, таким, как пентан или гексан. После этого каталитическую систему на носителе можно подвергать дальнейшей обработке или использовать в полимеризационном реакторе.
Хотя этот катализатор на носителе можно применять как таковой, его предпочтительно предварительно полимеризовать с использованием одного или нескольких олефиновых мономеров, каждый из которых содержит 2 - 20 углеродных атомов, предпочтительно 2 - 8 углеродных атомов.
Д. Форполимеризация катализатора на носителе
Форполимеризация катализатора на носителе позволяет ослабить тенденцию катализатора вызывать засорение реактора и, как было установлено, улучшить регулирование морфологии частиц получаемого готового полимера. Хотя можно использовать олефиновый мономер, содержащий 2 - 20 углеродных атомов, катализатор на носителе предпочтительно предварительно полимеризовать с помощью этиленового или пропиленового мономера, наиболее предпочтительно этилена. По другому варианту катализатор на носителе можно форполимеризовать с использованием по меньшей мере двух различных мономеров, содержащих по 2 - 8 углеродных атома.
В одном из вариантов для форполимеризации катализатора этот катализатор на носителе суспендируют в изопентане или другом углеводороде, в котором не растворяется металлоцен или алюмоксан. В суспендированную смесь медленно добавляют этилен. Скорость добавления зависит от размеров применяемого реактора. Обычно, когда использовали реакторы лабораторного масштаба, расход вводимого потока при перемешивании составлял приблизительно 0,03 - 0,06 нкф/мин (нкф - кубический фут при нормальных условиях). По истечении приемлемого периода времени полимеризации этиленового мономера на катализаторе на носителе форполимеризованный катализатор фильтруют, пропуская через сито, декантируют жидкость с суспендированной смеси, промывают изопентаном, сушат в вакууме и выделяют форполимеризованную каталитическую систему. Форполимеризацию обычно проводят при температуре от -15oC до 60oC, предпочтительно ниже 25oC, в течение от примерно 10 мин до примерно 10 ч. Количество форполимера можно варьировать, оно, как правило, составляет от примерно 10% до примерно 1000%, обычно от примерно 100% до примерно 300%, от веса нефорполимеризованного катализатора на носителе.
Обычно получали форполимеризованный катализатор с размерами каталитических частиц от примерно 50 до примерно 100 мкм. Форполимеризованный катализатор позволял получать полимер с гранулированной морфологией, который обычно характеризовался размерами частиц примерно 500 - 1000 мкм и объемной плотностью в интервале от примерно 0,30 до примерно 0,50 г/см3 или более.
Как правило, чем активнее каталитическая система, тем труднее получать каталитическую систему на носителе, которая сохраняет свои рабочие характеристики в сравнении с ее не нанесенным на носитель аналогом. Активность и способность обеспечивать полимеризацию у таких связанных мостиками бис-индениловых производных металлоценов на носителе оказалась неожиданной, принимая во внимание их высокую активность без носителя.
Е. Полимеризация
Все растворители приобретали у поставщиков промышленной продукции, азот очищали и сушили над активированными молекулярными ситами. Алюминийалкильные реагенты (MAO) приобретали в виде растворов концентрации 10 - 30 вес.% в толуоле у фирмы Witco Corporation, PO Box 1227, Dublin, Ohio, 43017. В качестве двуокиси кремния использовали продукт Davison 948, который приобретали у фирмы W.R.Grace, Davison Chemical Division, 5603 Chemical Rd., Baltimore, Md. 21226, США, дегидратированный при температуре 800oC в токе азота в течение приблизительно восьми часов.
Полученный форполимеризованный, нанесенный на носитель каталитический продукт можно использовать в качестве единственного каталитического компонента для полимеризации олефинов, содержащих приблизительно от 2 до 20 углеродных атомов, или предпочтительно его можно применять совместно с алюминийалкиловой или алюмоксановой добавкой (или очистителем). Добавку предпочтительно использовать во время полимеризации, поскольку при этом обычно наблюдаются повышение каталитической активности и уменьшение засоров в реакторе. Количество добавки в реакторе связано с чистотой исходного мономера.
В общем процедуру полимеризации осуществляли следующим образом. ТЭАЛ, используемый в качестве добавки, загружали в чистый сухой двухлитровый автоклав, который предварительно продували азотом. Реактор закрывали и вводили в него 1000 мл жидкого пропилена. Реактор нагревали до целевой температуры, обычно составлявшей примерно 65oC, и вдували в него посредством трубки для добавления с помощью примерно 250 мл пропилена форполимеризованный катализатор на носителе, суспендированный приблизительно в 2 мл гексана. По истечении желаемого реакционного времени, обычно около 60 мин, реактор охлаждали и сбрасывали в атмосферу избыток пропилена. Осмотр реактора показывал, что он оказывался чистым и свободным от засоров, мелочи или полимерных комков. Удаляли и сушили гранулированный легкосыпучий полимер.
Ж. Изотактический полипропиленовый продукт
С помощью форполимеризованной каталитической системы на носителе, приведенной в описании, получали гранулированный легкосыпучий изотактический полипропилен, который характеризовался (а) молекулярной массой приблизительно 100000 г/моль или более, обычно свыше 150000, (б) температурой плавления примерно 140oC или более, обычно свыше 145oC, (в) объемной плотностью около 0,30 г/см3 или более, (г) узким молекулярно-массовым распределением, составляющим приблизительно 2,5 или менее, и (д) размерами частиц примерно 500 - 1000 мкм. Гомо- и сополимеры пропилена, к которым относятся изотактический полипропилен или смеси изотактического полипропилена и один или несколько других олефинов, могут быть получены проведением процессов полимеризации в газовой фазе, в массе, растворе или суспензии с использованием описанной каталитической системы на носителе при температуре реакции 45oC или более, предпочтительно при приблизительно 60oC или более. Полученный гомо- и сополимер пропилена можно использовать для получения формованных изделий, пленок и волокон.
Каталитическая система на носителе в соответствии с настоящим изобретением служит средством получения исключительно привлекательного полимерного продукта. Полученные полимеры явились неожиданностью, если принять во внимание те проблемы, с которыми связаны нанесенные на носитель высокоактивные металлоцены.
Отличительной особенностью каталитической системы на носителе, которая представляет особый интерес для производителей, является способность сводить к минимуму или устранить образование комков, наслоений, мелочи в реакторе или его засорение во время полимеризации. Кроме того, результатом ее использования является гранулированный полимерный продукт. Эти свойства особенно привлекательны для производителей, поскольку сводят к минимуму или устраняют необходимость остановки реакционной системы для очистки или частых профилактических работ и в дополнительном процессе обработки полученного полимера.
В таблице 1 отражены условия получения катализатора и рабочие характеристики катализатора при полимеризации с получением изотактического полипропилена. Проиллюстрированные каталитические системы форполимеризовали с этиленовым мономером при температуре приблизительно 25 - 60oC. Молекулярная масса полученного полимера превышала 100000, а температура плавления составляла приблизительно или превышала 146oC. Средняя объемная плотность полученного полимера равнялась примерно 0,45 г/см3, а размеры частиц превышали 500 мкм.
Свойства полимера определяли по таким методикам ASTM, как DSC - дифференциальная сканирующая калориметрия, GPC - гельпроникающая хроматография и элементарный анализ. Температуру плавления определяли с помощью обычного прибора для дифференциальной сканирующей калориметрии и указывали как вторую точку плавления.
Преимущества настоящего изобретения представляют исключительную ценность с практической и промышленной точек зрения. Оно продемонстрировало возможность нанесения металлоцена на носитель таким образом, что каталитическая система сохраняет многие рабочие характеристики своего аналога, не нанесенного на носитель, что обуславливает слабое или отсутствие засорения реактора во время полимеризации.
Хотя настоящее изобретение можно выполнить во многих различных вариантах, в описании подробно приведены некоторые конкретные варианты выполнения изобретения. Настоящее описание, включая примеры, иллюстрирует принципы изобретения и не ограничивает рамки изобретения конкретно представленными вариантами его выполнения.
Примеры
Представленные в описании пояснительные примеры демонстрируют использование металлоценового катализатора на носителе при получении изотактического полипропилена.
А. Синтез рац-диметилсиландиилбис(2-метил-4,5-бензоинаденил) цирконийдихлорида
Диэтилметил(2-нафтилметил)малонат (1)
5,15 г (224 ммоля) натрия растворяли с одновременным подогревом в 150 мл абсолютного этанола и при комнатной температуре добавляли 37,3 мл (217 ммоля) диэтилметилмалоната. При 0oC медленно по каплям добавляли раствор 50 г (217 ммоля) 2-бромметилнафталина (96%-ной чистоты) в 270 мл этанола и смесь кипятили с обратным холодильником дополнительно в течение от 4 до 5 ч. Ее выливали в смесь воды со льдом и экстрагировали этилацетатом. Объединенные органические фазы сушили над сульфатом натрия и упаривали. После сушки в вакууме, создаваемом масляным вакуумным насосом, полученный маслянистый остаток перемешивали с гексаном при 0oC, после чего кристаллизовали 55 г (81%) соединения 1.
Синтез 2-метил-З-нафтилпропионовой кислоты (2)
К 33,2 г (105 ммоля) соединения 1 в 70 мл этанола добавляли раствор 23,7 г (422 ммоля) гидроокиси калия в 50 мл воды и смесь кипятили с обратным холодильником 4 ч. После отпарки растворителя твердый остаток растворяли в этилацетате, добавляли воду и добавлением соляной кислоты величину pH доводили до 1. Водную фазу несколько раз экстрагировали этилацетатом. После сушки над сульфатом магния объединенные органические фазы полностью упаривали. Остаток перемешивали с гексаном для кристаллизации. Для декарбоксилирования окрашенное в бежевый цвет твердое вещество выдерживали при 175oC до завершения выделения газа. В виде окрашенного в бежевый цвет твердого вещества получали 21 г (94%) продукта 2.
Синтез 2-метил-6,7-бензоиндан-1-она (3)
22 мл хлористого тионила добавляли к 21 г (98 ммоля) соединения 2 в условиях отсутствия влаги и смесь кипятили с обратным холодильником 30 мин. Затем отгоняли избыток хлористого тионила. Остаток быстро освобождали от летучих соединений в вакууме, создаваемом масляным вакуумным насосом, и затем растворяли в 25 мл хлористого метилена в токе аргона в качестве инертного газа. Раствор медленно по каплям добавляли в суспензию 26 г (196 ммоля) трихлорида алюминия в 60 мл хлористого метилена и смесь дополнительно кипятили с обратным холодильником в течение 30 мин. Ее сливали на лед и экстрагировали хлористым метиленом. Объединенные органические фазы сушили над сульфатом натрия и выпаривали. Темный маслянистый остаток хроматографировали с использованием 600 г силикагеля 60. С использованием в качестве подвижной фазы смеси гексан/этилацет (9:3) элюировали 8,6 г (45%) соединения 3 (желтоватое твердое вещество).
Синтез 2-метил-4,5-бензоиндена (4)
2,2 г (59,5 ммоля) боргидрида натрия при комнатной температуре добавляли отдельными порциями в раствор 7,8 г (39,7 ммоля) инданона, соединения 3 в 400 мл смеси тетрагидрофурана с метанолом (2:1) и смесь перемешивали 14 ч. Раствор сливали на подкисленный HCl лед и экстрагировали эфиром. Объединенные органические фазы несколько раз промывали водой и сушили над сульфатом натрия. Окрашенный в оранжевый цвет маслоподобный продукт, который оставался после отпарки растворителя, растворяли в 240 мл толуола и раствор выдерживали при 80oC совместно с 570 мг (3,15 ммоля) п-толуолсульфокислоты в течение 15 мин. Его несколько раз промывали водой при комнатной температуре, сушили над сульфатом натрия и упаривали. Остаток хроматографировали с помощью 300 г силикагеля 60. 4,7 г (65%) индена 4 можно было элюировать (бесцветный маслоподобный продукт) с использованием в качестве подвижной фазы смеси гексана с диизопропиловым эфиром (20:1).
1H-ЯМР-спектрограмма (360 МГц, CDCl3): 8,02 (1, d), 7,84 (1, m), 7,59 (1, d), 7,52 (1, d), 7,38-7,48 (2, m), 7,06 (1, m), 3,42 (2, s), 2,25 (3, d).
Синтез диметилбис(2-метил-4,5-бензоинденил)силана (5)
10,2 мл (25,5 ммоля) 2,5М раствора бутиллития в гексане при комнатной температуре добавляли в раствор 4,6 г (25,5 ммоля) соединения 4 в 50 мл тетрагидрофурана и смесь кипятили с обратным холодильником 1 ч. Затем красный раствор при комнатной температуре по каплям добавляли в раствор 1,55 г (12 ммоля)диметилдихлорсилана в 10 мл тетрагидрофурана и смесь кипятили с обратным холодильником в течение от 5 до 6 ч. Реакционный раствор сливали в смесь воды со льдом и несколько раз экстрагировали эфиром. Объединенные органические фазы сушили над сульфатом натрия и упаривали, а остаток сушили в вакууме, создаваемом масляным вакуумным насосом. Его хроматографировали с помощью 300 г силикагеля 60. 500 мг непрореагировавшего исходного соединения 4 вначале можно было элюировать с использованием в качестве подвижной фазы смеси гексана/3% этилацетата. Затем той же подвижной фазой обрабатывали лигандную систему, соединение 5. После отпарки растворителя эту лигандную систему кристаллизовали (изомеры) из гексана. Выход составлял 1,7 г (34% или 44% в пересчете на инден, прореагировавшее соединение 4).
Синтез рац-диметилсиландиилбис(2-метил-4,5-бензоинденил)цирконий-дихлорида (6)
4,0 мл (10,2 ммоля) 2,5М раствора бутиллития в гексане при комнатной температуре в атмосфере аргона в качестве инертного газа добавляли в раствор 1,7 г (4,1 ммоля) соединения 5 в 20 мл тетрагидрофурана и эту смесь перемешивали при комнатной температуре 14 ч. Остаток, который получали после отпарки растворителя, сушили в вакууме, создаваемом масляным вакуумным насосом, и промывали гексаном. Полученный бледно-коричневый порошок сушили в вакууме, создаваемом масляным вакуумным насосом, при температуре от 40 до 50oC в течение нескольких часов и добавляли в суспензию 1,0 г (4,0 ммоля) четыреххлористого циркония в 25 мл хлористого метилена при -78oC. После подогрева смеси до комнатной температуры растворитель отгоняли и остаток экстрагировали 20 мл толуола для удаления мезоформы металлоцена, соединения 6. Остаток толуольного экстракта затем экстрагировали 40 мл хлористого метилена. Раствор концентрировали до небольшого объема и оставляли кристаллизоваться при -35oC. В общей сложности в нескольких фракциях в виде чистого рацемата выделяли 970 мг (42%) цирконоцена, соединения 6.
1H-ЯМР-спектрограмма рацемата (300 МГц, CDCl3): 7,96 (2, m), 7,78 (2, m), 7,60 (2, d), 7,48-7,56 (4, m), 7,36 (2, d), 7,27 (2, s, b-инд.-H), 2,37 (6, s, инд.-CH), 1,36 (6, s, Si-CH3). Масс-спектрограмма: 574 М+, точное расщепление, точная изотопная картина.
Нанесение на носитель каталитического соединения (6)
В 8-литровый сосуд, снабженный охлаждающей рубашкой и проходящей через верхнюю крышку эффективной мешалкой, вводили 925 мл 30%-ного по весу метилалюмоксана в толуоле. В токе азота с перемешиванием через иглу с раздвоенным концом добавляли суспензию 5,0 г соединения 6 в 700 мл толуола. После перемешивания в течение 10 мин в раствор в течение 20 мин добавляли 200 г дегидратированной двуокиси кремния (Davison 948, высушенной при 800oC). Суспензию перемешивали 10 мин, а затем, подключив верхнюю часть сосуда к вакуумной системе, через днище подавали слабый ток азота. Смесь выдерживали при 70oC с одновременным выпариванием растворителя в течение 9 ч. Сухой твердый остаток охлаждали до комнатной температуры в течение ночи. Для суспендирования твердых частиц добавляли 5 л изопентана и смесь охлаждали до 0oC. В перемешиваемую смесь по погруженной трубке вводили этилен с расходом 0,03 - 0,06 нкф/мин до тех пор, пока общее количество добавленного этилена не достигало 491 л. Перемешивание прекращали и твердым частицам давали осесть. Жидкость декантировали с твердых частиц, которые промывали дважды, используя каждый раз по 1,5 л изопентана. Мокрые твердые частицы переносили в сушильный шкаф в токе азота и отфильтровывали, пропуская через сито N 14 (размеры ячеек - 1,41 мм). Тонкодисперсные частицы отфильтровывали, промывали 4 л пентана и сушили в вакууме. Выход: 326 г.
Полимеризация с помощью соединения 6 на носителе
199 мг образца каталитического соединения 6 на носителе, суспендированного в 2 мл гексана, вводили с помощью струи 250 мл пропилена в 2-литровый автоклав, который предварительно продували азотом и нагревали до 65oC, содержавший 0,5 мл 1М раствора триэтилалюминия в гексане и 1000 мл пропилена. Реакцию проводили 1 ч. Автоклав охлаждали, вентилировали и открывали. Осмотр внутреннего пространства реактора показал, что он был чистым и свободным от засоров. Полимерный продукт сушили в вакууме. Выделяли 305 г легкосыпучего, гранулированного изотактического полипропиленового продукта. Мw составляла 273000, объемная плотность 0,43 г/см3, средний размер частиц 750 мкм.
Б. Синтез рац-диметилсиландиилбис(2-метилинденил)цирконийдихлорида
Синтез 2-Ме-индена (7)
110,45 г (0,836 моля) 2-инданона растворяли в 500 см диэтилового эфира и по каплям добавляли 290 см 3H (0,87 моля) эфирного раствора реактива Гриньяра (метиловый радикал) с такой скоростью, при которой обеспечивалось слабое кипение смеси. После осторожного кипячения смеси с обратным холодильником в течение 2 ч ее переносили в смесь льда с соляной кислотой и с использованием хлорида аммония величину pH доводили до 2 - 3. Органическую фазу отделяли, промывали NaHCO3 и раствором хлорида натрия и сушили, получая 98 г сырого продукта [2- гидрокси-2-метилиндана (соединения 7)], который в дальнейшем не очищали.
Затем соединение 7 растворяли в 500 см3 толуола, добавляли 3 г п-толуолсульфокислоты и смесь нагревали на сепараторе воды до полного удаления воды и упаривали. Остаток растворяли в дихлорметане и образовавшийся раствор фильтровали через силикагель. Фильтрат перегоняли в вакууме (80oC/10 мбар). Выход: 28,49 г (0,22 моля/26%).
Синтез соединения (7) описан также C.F.Koelsch, P.R. Johnson, J. Am. Chem. Soc., 65 (1943) 567-573.
Синтез (2-Ме-инден)2SiMe2 (8)
13 г (100 ммолей) 2-Ме-индена (соединения 7) растворяли в 400 см3 диэтилового эфира и по каплям, в течение 1 ч с охлаждением льдом добавляли 62,5 см3 1,6H (100 ммолей) раствора н-бутиллития в н-гексане. Смесь перемешивали при примерно 35oC еще 1 ч.
В 50 см3 Et2O вводили 6,1 см3 (50 ммолей) диметилдихлорсилана и по каплям в течение примерно 5 ч при 0oC добавляли раствор литиевой соли, смесь перемешивали в течение ночи при комнатной температуре и оставляли стоять с вечера пятницы до утра понедельника. Выпавший твердый осадок отфильтровывали и фильтрат упаривали досуха. Продукт экстрагировали с помощью небольших порций н-гексана и экстракты фильтровали и выпаривали, получая 5,7 г (18,00 ммолей) белых кристаллов. Маточный раствор упаривали и затем остаток очищали в хроматографической колонке (н-гексан/H2CCl2 в объемном соотношении 9:1), получая дополнительно 2,5 г (7,9 ммоля/52%) продукта (в виде изомерной смеси).
(SiO2; н-гексан/H2CCl2 в объемном соотношении 9:1) = 0,37.
Rf(SiO2; н-гексан/H2CCl2 в объемном соотношении 9:1) = 0,37.
1H-ЯМР- спектрограмма демонстрировала сигналы, которые ожидались для изомерной смеси, принимая во внимание сдвиг и степень интеграции.
Синтез Me2Si(2-Ме-Инд.)2ZrCl2 (9)
1,68 г (5,31 ммоля) хелатного лигандного диметилсилил-(2- метилинден) (соединения 8) вводили в 50 см3 ТГФ и при комнатной температуре по каплям в течение приблизительно 0,5 ч добавляли 6,63 см3 1,6H (10,61 ммоля) раствора н-бутиллития в н-гексане. Смесь перемешивали в течение примерно 2 ч при температуре около 35oC, растворитель отпаривали в вакууме, а остаток перемешивали с н-пентаном, фильтровали и сушили. Полученную дилитиевую соль при -78oC добавляли в суспензию 1,24 г (5,32 ммоля) ZrCl4 в 50 см3 CH2Cl2 и смесь перемешивали при этой температуре 3 ч. Затем смесь нагревали до комнатной температуры в течение ночи и упаривали. 1H-ЯМР-спектрограмма, помимо присутствия некоторого количества ZrClH4(ТГФ)2, показывала наличие рац/мезо-смеси. После перемешивания с н-пентаном и сушки твердый желтый остаток суспендировали в ТГФ, фильтровали и исследовали ЯМР-спектроскопией. После неоднократного повтора этих трех стадий получали 0,35 г (0,73 ммоля/14%) продукта, в котором обогащенность рацемической формой по данным 1H-ЯМР-спектрограммы превышала 17:1. Соединение (9) подтверждало точность элементарного анализа и нижеследующих ЯМР-сигналов (CDCl3, 100 МГц): d = 1,25 (s, 6H, Si-Me); 2,18 (s, 6H, 2-Ме); 6,8 (s, 2H, 3-H-Инд.); 6,92-7,75 (m, 8H, 4-7-H-Инд.).
Нанесение на носитель каталитического соединения (9)
В 8-литровый сосуд, снабженный охлаждающей рубашкой и эффективной мешалкой, проходившей через верхнюю крышку, добавляли 925 мл раствора метилалюмоксана в толуоле концентрацией 30 вес.%. При перемешивании в токе азота посредством иглы с раздвоенным концом добавляли суспензию 10 г соединения 9 в 700 мл толуола. После перемешивания в течение 10 мин в раствор в течение 20 мин добавляли 200 г дегидратированной двуокиси кремния (Davison 948, высушенной при 800oC). Эту суспензию перемешивали 10 мин, а затем, подключив верхнюю часть сосуда к вакуумной системе, через днище подавали слабый ток азота. Смесь выдерживали при 70oC с одновременным упариванием растворителя в течение 5 ч. Сухой твердый остаток охлаждали до комнатной температуры в течение ночи. Для суспендирования твердых частиц добавляли 6,4 л изопентана и смесь охлаждали до 0oC. В перемешиваемую смесь по погруженной трубке вводили этилен с расходом 2,5 - 3,4 л/мин до тех пор, пока общее количество добавленного этилена не достигало 392 л. Перемешивание прекращали и твердым частицам давали осесть. Жидкость декантировали с твердых частиц, которые промывали дважды, используя каждый раз по 3 л изопентана. Мокрые твердые частицы переносили в сушильный шкаф в токе азота и отфильтровывали через сито 14 (размеры ячеек - 1,41 мм). Образец отфильтровывали, промывали 4 л пентана и сушили в вакууме. Выход: 721 г.
Полимеризация с помощью соединения 9 на носителе
Осуществляли процедуру полимеризации соединения 6 с использованием 199 мг нанесенного на носитель соединения 9. Осмотр внутреннего пространства реактора показал, что он был чистым и свободным от засоров. Выход изотактического полипропилена был равным 143 г. Мw составляла 172000, объемная плотность 0,36 г/см3, а средний размер частиц 802 мкм. Температура плавления равна 147,9oC.
В. Синтез рац-диметилсиландиилбис (2-метил-4,6-диизопропилинденил)цирконийдихлорида
Синтез 2-метил-5,7-диизопропил-1-инданона (10) и 2-метил-4,6-диизопропил-1-инданона (11)
174 г (1300 ммолей) AlCl3 при комнатной температуре через воронку для дозирования твердых частиц осторожно добавляли в раствор 84,8 г (523 ммоля) 1,3-дизопропилбензола и 120 г (523 ммоля) 2-бромизобутирилбромида (98%-ной чистоты) в 600 мл хлористого метилена сорта для анализов. Смесь кипятили с обратным холодильником в течение последующих 20 ч и затем обрабатывали аналогично изложенному в примере А. Сырой продукт обрабатывали хроматографией на 3 кг силикагеля 60. Инданоны 10 и 11 можно было раздельно элюировать подвижной фазой в виде смеси гексан/15% этилацетат. С использованием той же самой подвижной фазы в качестве побочного продукта в последующую зону переводили соединение 2-метил-5-изопропил-1-инданон. Однако разделение обоих изомеров для последующих реакций оказывалось необязательным. Общий выход составлял 93 г (78%). 1H-ЯМР-спектрограмма (360 МГц, CDCl3): изомерная смесь (3: 2) 7,49 (d), 7,36 (d), 7,13 (s), 7,10 (s), 4,15 (септет), 3,25-3,40 (m), 3,10 (септет), 2,90-3,00 (m), 2,60-2,73 (m), 1,22-1,30 (m). Масс-спектрограмма: 230 М+, точная картина расщепления.
Синтез 2-метил-4,6-диизопропилидена (12) и 2-метил-5,7-диизопропилидена (13)
19,3 г (511 ммолей) NaBH4 при комнатной температуре вводили в раствор 78,5 г (341 ммолей) смеси изомеров 10 и 11 в 700 мл смеси растворителей тетрагидрофуран/метанол сорта для анализов (2:1). После перемешивания смеси при комнатной температуре в течение 2 ч добавляли 120-130 мл полуконцентрированной HCl и смесь экстрагировали диэтиловым эфиром. Объединенные органические фазы сушили над Na2SO4. Остаток, который получали после отпарки растворителя, растворяли в 500 мл хлористого метилена и смесь кипятили с обратным холодильником 15 мин совместно с 6,5 г (34 ммоля) п-толуолсульфокислоты. Остаток, который получали после отпарки растворителя, обрабатывали хроматографией на 1,5 кг силикагеля 60. 56 г смеси изомеров 12 и 13 можно было выделять в виде бесцветного маслоподобного продукта с помощью подвижной фазы в виде смеси гексан/диизопропиловый эфир в соотношении 20:1. Общий выход составлял 86%.
1H-ЯМР-спектрограмма (100 МГц, CDCl3): изомеры с двойными связями (1:1) 7,1 (m), 6,95 (m), 6,60 (m), 6,43, 3,25 (широк.), 2,75-3,20 (m), 2,12 (d), 1,28 (d), 1,25 (d).
Масс-спектрограмма: 214 M+, точная картина расщепления.
Синтез диметилбис(2-метил-4,6-диизопропилинденил)силана (14)
9,2 мл (22,8 ммоля) 2,5М раствора бутиллития в гексане при 0oC в токе аргона в качестве инертного газа вводили в раствор 4,9 г (22,8 ммоля) смеси изомеров 12 и 13 в 25 мл тетрагидрофурана и смесь кипятили с обратным холодильником в течение последующего часа. Затем раствор красного цвета при комнатной температуре по каплям добавляли в раствор 1,5г (11,4 мл) диметилдихлорсилана в 10 мл тетрагидрофурана и смесь кипятили с обратным холодильником 8 ч. Массу сливали в смесь воды со льдом и экстрагировали эфиром. Эфирную фазу сушили над сульфатом магния и выпаривали под пониженным давлением. Желтоватый маслоподобный продукт, который оставался, далее обрабатывали хроматографией на 500 г силикагеля 60. 1,4 г смеси инденовых изомеров 12/13 можно было вначале элюировать подвижной фазой в виде смеси гексан/5% хлористый метилен. Затем следовала лигандная система (гексан/8 % хлористый метилен). Вязкий маслоподобный продукт, который оставался после отпарки подвижной фазы, можно было кристаллизовать перемешиванием с метанолом на ледяной бане. Получали 3,1 г желтоватого твердого продукта. Выход составлял 56% или 84% в пересчете на прореагировавший инден.
1H-ЯМР-спектрограмма рацемата (100 МГц, CDCl3): изомеры с двойными связями (3: 1) 6,82-7,32 (m), 6,70 (m), 6,62 (m), 6,52 (m), 3,75 (s, широк.), 3,65 (s, широк. ), 3,35(s), 2,70-3,30 (m), 2,05-2,25 (m), 1,10-1,45 (m), 0,10-0,22 (m), от -0,15 до -0,32 (m).
Масс-спектрограмма: 484 M+, точное расщепление.
Синтез рац-диметилсиландиилбис(2-метил-4,6- диизопропилинденил)цирконийдихлорида (15)
6,3 мл (16,2 ммоля) 2,5М раствора бутиллития в гексане при комнатной температуре в токе аргона в качестве инертного газа вводили в раствор 3,1 г (6,5 ммоля) лигандной системы 14 в 25 мл диэтилового эфира и смесь перемешивали в течение ночи. После добавления 10 мл гексана окрашенную в бежевый цвет суспензию фильтровали и остаток промывали 20 мл гексана. Дилитиевую соль сушили в вакууме, создаваемом масляным вакуумным насосом, длительное время и затем при -78oC добавляли в суспензию 1,6 г (6,8 ммоля) ZrCl4 в 30 мл хлористого метилена. Смесь нагревали до комнатной температуры в течение 1 ч и перемешивали при этой температуре в течение последующих 30 мин. После отпарки растворителя оранжево-коричневый остаток экстрагировали 50 мл гексана. После отпарки растворителя в форме желтого порошка получали 2,6 г (60%) комплекса 6. Соотношение между рац- и мезоформами составляло 3:1. Перекристаллизацией из гексана в виде желтого кристаллического порошка в качестве чистого рацемата получали 1,3 г (30%) комплекса 15.
1H-ЯМР-спектрограмма (100 МГц, CDCl3): 7,27 (2, s, ароматический H), 7,05 (2, s, ароматический H), 6,80 (2, s, b-Инд.-H), 2,6-3,2 (4, m, изо-Pr-CH), 2,22 (6, s, Инд. -CH3), 1,15-1,40 (30, m, изо-Pr-CH3, Si-CH3). Масс-спектрограмма: 642 M+ (в отношении 90Zr), точная изотопная картина, точное расщепление.
Нанесение на носитель каталитического соединения 15
Раствор 102 мг комплекса 15, растворенного в 7 мл толуола, по каплям добавляли в 10 мл 30%-ного раствора метилалюмоксана в толуоле. После перемешивания раствора 10 мин при комнатной температуре в течение 5 мин добавляли 2,0 г двуокиси кремния Davison 948 (высушенной при 800oC). Для смыва боковых стенок реакционной колбы добавляли дополнительно 5 мл толуола. По истечении 15 мин растворитель упаривали в вакууме с одновременным подогревом до 90oC. Выход катализатора составлял 4,44 г.
Полимеризация с помощью соединения 15 на носителе
44 мг нанесенного на носитель соединения 15, суспендированного в 2 мл гексана, с помощью струи 250 мл пропилена вводили в 2-литровый автоклав, который предварительно продували азотом, подогретый до 65oC и содержавший 0,5 мл 1М раствора триэтилалюминия в гексане и 1000 мл пропилена. Реакцию проводили 1 ч. Автоклав охлаждали и вентилировали, а полимерный продукт сушили в вакууме. Выход пропилена составлял 75 г. Величина соотношения вязкостей расплава этого материала была равной 0,6, средневесовая молекулярная масса 438000, а молекулярно-массовое распределение 2,17.
Г. Синтез рац-диметилсиландиилбис(2-метил-4-фенилинденил) цирконийдихлорида
Синтез (+)-2-(2-фенилбензил)-пропионовой кислоты (16)
К 6,5 г (0,285 моля) натрия в 160 мл сухого этанола при комнатной температуре добавляли 48,6 г (0,279 моля) диэтилметилмалоната. В перемешиваемую реакционную смесь добавляли 70,4 г (0,285 моля) 2-фенилбензилбромида с такой скоростью, при которой теплота реакции поддерживала слабое кипение системы. Перемешивание при кипячении с обратным холодильником продолжали 3 ч. Добавляли 200 мл воды и 56 г (1 моль) гидроокиси калия и реакционную смесь кипятили с обратным холодильником 4 ч. Отгоняли растворители, добавляли воду в количестве, необходимом для полного растворения остатка, и для доведения величины pH до 1 добавляли концентрированную HCl. Осадок собирали на фильтре, сушили и нагревали в колбе до 130oC. Далее кислоту использовали как таковую. Выход: 58 г (85%).
1H-ЯМР-спектрограмма (CDCl3, 100 МГц): 11,7 (s, 1H, COOH), 7,1-7,5 (m, 9H, ароматич. H), 2,3-3,2 (m, 3H, CH и CH2), 0,9 (d, 3H, CH3).
Синтез (+)-2-метил-4-фенил-1-инданона (17)
Раствор 58 г (0,242 моля) соединения 16 в 60 мл (0,83 моля) хлористого тионила перемешивали 18 ч при комнатной температуре. Отгоняли избыток хлористого тионила, трижды добавляли по 100 мл толуола и удаляли в вакууме.
При 10oC в перемешиваемую суспензию 48 г (0,363 моля) трихлорида алюминия в 400 мл сухого толуола в течение 1 ч добавляли раствор хлорангидрида кислоты в 150 мл сухого толуола. После нагревания до 80oC в течение 3 ч смесь сливали на 500 г льда, подкисляли концентрированной HCl до величины pH 1 и трижды экстрагировали 200 мл диэтилового эфира. Объединенные органические слои промывали насыщенным водным раствором NaHCO3 и солевым раствором. В вакууме удаляли растворители. Инданон 17 использовали без дальнейшей очистки. Выход: 50 г (93%).
1H-ЯМР-спектрограмма (CDCl3 100 МГц): 7,2-7,8 (m, 8H, ароматич. H), 3,3 (dd, 1H, H-C(З)), 2,5-2,9 (m, 2H, H'-C(3) и H-C(2)), 1,3 (dd, 3H, CH3).
Синтез 2-метил-7-фенилиндена (18)
50 г (0,226 моля) соединения 17 в 450 мл ТГФ/метанола (2:1) обрабатывали при 0oC 12,8 г (0,34 моля) боргидрида натрия в виде нескольких порций. Реакционную смесь перемешивали при комнатной температуре 16 ч. Смесь сливали на 500 г льда, подкисляли концентрированной HCl до величины pH 1 и трижды экстрагировали 200 мл диэтилового эфира. Объединенные органические слои промывали солевым раствором и сушили над MgSO4. После удаления растворителя остаток кипятили с обратным холодильником 2 ч в 1000 мл толуола, содержавшего 2 г моногидрата п-толуолсульфокислоты. Для удаления кислого катализатора смесь промывали 200 мл насыщенного водного раствора NaHCO3. После удаления растворителя и хроматографической обработки на силикагеле (гексан/CH2Cl2, 9: 1)в виде бесцветного маслоподобного продукта получали инден 18. Выход: 42 г (90%).
1H-ЯМРспектрограмма (CDCl3, 100 МГц): 7,0-7,6 (m, 8H, ароматич. H), 6,5 (m, 1H, H-C(З)), 3,4 (s, 2H, CH2), 2,1 (sa, 3H, CH3).
Синтез диметилбис(2-метилфенилинденил)силана (19)
В раствор 15 г (73 ммоля) соединения 18 в 200 мл сухого толуола и 10 мл сухого ТГФ в атмосфере аргона при комнатной температуре вводили 29 мл (73 ммоля) 2,5М раствора бутиллития в гексане. После перемешивания при 80oC в течение 1 ч реакционную смесь охлаждали до 0oC и добавляли 4,7 г (36,5 ммоля) диметилсиландихлорида. Смесь выдерживали при 80oC 1 ч и после завершения реакции добавляли 100 мл воды. Органический слой отделяли, в вакууме удаляли растворитель и остаток очищали хроматографией на силикагеле (гексан/CH2Cl2, 9:1). Выход составлял 12 г (70%).
1H-ЯМР-спектрограмма (CDCl3, 100 МГц): 7,1-7,7 (m, 16H, ароматич. H), 6,8 (m, 2H, H-C(З)), 3,8 (s, 2H, H-C(1)), 2,2 (s, 6H, CH3), -0,2 (m, 6H, CH3Si).
Синтез рац-диметилсиландиилбис(2-метил-4-фенилинденил) цирконийдихлорида (20)
В раствор 6 г (13 ммолей) соединения 19 в 100 мл сухого толуола в атмосфере аргона при комнатной температуре вводили 10,6 мл (26 ммолей) 2,5М раствора бутиллития в гексане. После перемешивания при кипячении с обратным холодильником 3 ч реакционную смесь охлаждали до -25oC и добавляли 3,2 г (13,6 ммоля) четыреххлористого циркония. Реакционной смеси давали нагреться до комнатной температуры в течение 2 ч. Суспензию фильтровали через фритту Шленка G3 и остаток промывали 50 мл сухого толуола. В вакууме из объединенных экстрактов удаляли растворитель. В виде смеси рац- и мезо-форм 1:1 получали 9,0 г комплекса. После фракционной кристаллизации смеси 1:1 из сухого CH2CH2 получали 2,74 г (33%) чистого рацемата. 1H-ЯМР-спектрограмма (CDCl3, 100 МГц): 7,0-7,7 (m, 16H, ароматич. H), 6,9 (s, 2H, H-C(З)), 2,2 (s, 6H, CH3), 1,3 (m, 6H, CH3Si). Масс-спектрограмма (ЭИ) 626 (90Zr35Cl2).
Нанесение на носитель каталитического соединения 20
В профильтрованный раствор 0,05 г соединения 20 в 75 мл толуола при перемешивании водили 5,3 мл суспензии MAO (30% MAO в толуоле). По истечении пяти минут раствор становился красно-оранжевым и слегка мутнел. В него добавляли 6,0 г обычной дегидратированной двуокиси кремния (Davison 948, дегидратированной при 800oC) и перемешивали пятнадцать минут. Образовавшуюся суспензию выпаривали в роторном испарителе при 44oC в течение двадцати пяти минут, непосредственно после чего суспензия достигала "шламоподобного" состояния. По истечении еще 35 мин сушки при 46oC в виде светло-оранжевого твердого продукта выделяли 7,2 г твердых частиц.
Полимеризация с помощью соединения 20
В чистый и сухой двухлитровый автоклав, который продували парами пропилена, вводили ТЭАЛ (0,8 мл, 1,5М в гептане) и затем реактор закрывали и заполняли 750 мл жидкого пропилена. При температуре реактора 30oC по трубке для ввода с помощью 250 мл пропилена в реактор смывали катализатор 20 на носителе (в виде масляного шлама вес.%). Реактор быстро нагревали до 65oC. По истечении тридцати минут реактор охлаждали и сбрасывали в атмосферу избыток пропилена. Осмотр внутреннего пространства реактора показал, что он был чистым и свободным от засоров. Выделяли и сушили изотактический полипропилен. Каталитическая активность составляла 0,37 кг/г/ч, температура плавления, по данным дифференциальной сканирующей калориметрии, равнялась 150oC, средневесовая молекулярная масса Mw 748000 г/моль, MMP 2,2.
Полимерный продукт, полученный полимеризацией пропилена в присутствии каталитического соединения 6 на носителе по описанной выше методике, используют для изготовления волокна и нетканого материала.
Пример 1
Волокна были изготовлены в виде матовых (без блеска или с небольшим блеском) пучков пряжи путем механического вытягивания из экструдируемого расплава. Процесс был осуществлен на специальной технологической линии, поставленной J.J.Jenkins, Inc. (Stallings, NC).
Волокна вытягивали из расплава, имеющего температуру 232oC (450 o F), с использованием ненагретого прядильного диска, вращающегося в осевом направлении со скоростью 1000, 1500, 2000, 2500 и 3000 об/мин. Волокна формовали из расплава полипропилена с показателем текучести расплава (ПТР) 40, 51 и 68.
На разрывной машине "Инстрон" (модель 1122), снабженной компьютером с программным обеспечением Sintech Sima (Testworks II~), проводили измерение прочности на разрыв (г/денье) изготовленного пучка волокон. Образец толщиной 2,5 см с предварительной нагрузкой 0,1 г растягивали до разрыва со скоростью 500 мм/мин. Полученные результаты испытаний представлены в таблице 2.
Пример 2
Два образца полипропилена, полученного полимеризацией в присутствии каталитического соединения 6 на носителе по описанной выше методике, использовали для изготовления дутьем из расплава образцов нетканого материала (с весовой характеристикой 34 г/м), которые испытывали на воздухопроницаемость.
Воздухопроницаемость определяли по стандартной методике испытаний INDA 1ST 70/0-70(R82), соответствующей методике ASTM D737-75 и методике FTM'S 191 5450-1-1970. Измерения проводили на установке для испытаний на проницаемость от Frazier Precision Instrument Co., 210 Oakmont Ave., Gaitherburg, M.D. Полученные результаты представлены в таблице 3.
Этим завершается описание предпочтительных и других вариантов выполнения изобретения. Специалистам в данной области техники очевидна возможность эквивалентов описанных конкретных вариантов, которые охватываются рамками прилагаемой формулы изобретения.


Изобретение относится к металлоценовым каталитическим системам на носителе, необязательно форполимеризованным с использованием олефинового мономера, предпочтительно этилена или пропилена, которые можно применять при полимеризации пропилена до изотактического полипропилена. Обычно такой полученный изотактический полипропилен характеризуется мол.м. примерно 100000 г/моль или более, температурой плавления примерно 140oС или выше и гранулированной морфологией. Каталитическая система на носителе обычно обладает высокой каталитической активностью и при применении в полимеризационном реакторе обеспечивает протекание процесса с минимальным или без какого-либо образования в реакторе засоров, мелочи или комков. 5 с. и 16 з.п. ф-лы, 3 табл.

где M1 обозначает металл группы 4,5 или 6 Периодической таблицы;
R1 и R2 одинаковые или различные и обозначают атом водорода, C1 - C10 алкильную группу, C1 - C10 алкоксигруппу, C6 - C10 арильную группу, C6 - C10 арилоксигруппу, C2 - C10 алкенильную группу, C7 - C40 арилалкильную группу, C7 - C40 алкиларильную группу, C8 - C40 арилалкенильную группу или атом галогена;
R3 и R4 обозначают атом водорода;
R5 и R6 одинаковые или различные, предпочтительно одинаковые, и обозначают атом галогена, C1 - C10 алкильную группу, которая может быть галоидированной, C6 - C10 арильную группу, которая может быть галоидированной, C2 - C10 алкенильную группу, C7 - C40 арилалкильную группу, C7 - C40 алкиларильную группу, C8 - C40 арилалкенильную группу, радикал
-NR
-OSiR
где R15 обозначает атом галогена, C1 - C10 алкильную группу или C6 - C10 арильную группу;
R7 обозначает
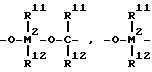
= BR11, =AlR11, -Ge-, -Sn-, -S-, -O-, =SO, =SO2, =NR11, =CO, PR11 или = P(O)R11,
где R11, R12 и R13 одинаковые или различные и обозначают атом водорода или атом галогена, C1 - C20 алкильную группу, C1 - C20 фторалкильную группу, C6 - C30 арильную группу, C6 - C30 фторарильную группу, C1 - C20 алкоксигруппу, C2 - C20 алкенильную группу, C7 - C40 арилалкильную группу, C8 - C40 арилалкенильную группу, C7 - C40 алкиларильную группу,
или же R11 и R12 или R11 и R13 совместно со связывающими их атомами могут образовывать циклические системы;
M2 обозначает атом кремния, германия или олова;
R8 и R9 одинаковые или различные и имеют те же значения, что и R11;
m и n одинаковые или различные и обозначают 0,1 или 2,
сумма m и n равна 0,1 или 2; а
радикалы R10 одинаковые или различные и имеют те же значения, что и R11, R12 и R13, где два смежных радикала R10 могут быть связаны с образованием кольцевой системы,
(б) нанесение на неорганический пористый носитель полученного реакционного продукта
и (с) необязательную форполимеризацию полученной при этом каталитической системы на носителе в присутствии олефинового мономера (ов).
где M1 обозначает металл группы 4,5 и 6 Периодической таблицы;
R1 и R2 одинаковые или различные и обозначают атом водорода, C1 - C10 алкильную группу, C1 - C10 алкоксигруппу, C6 - C10 арильную группу, C6 - C10 арилоксигруппу, C2 - C10 алкенильную группу, C7 - C40 арилалкильную группу, C7 - C40 алкиларильную группу, C8 - C40 арилалкенильную группу или атом галогена;
R3 и R4 обозначают атом водорода;
R5 и R6 одинаковые или различные, предпочтительно одинаковые, и обозначают атом галогена, C1 - C10 алкильную группу, которая может быть галоидированной, C6 - C10 арильную группу, которая может быть галоидированной, C2 - C10 алкенильную группу, C7 - C40 арилалкильную группу, C7 - C40 алкиларильную группу, C8 - C40 арилалкенильную группу, радикал
-NR
-PR
где R15 обозначает атом галогена, C1 - C10 алкильную группу или C6 - C10 арильную группу;
R7 обозначает

= BR11, =ALR11, -Ge-, -Sn-, -O-, -S-, =SO, =SO2, =NR11, =CO, PR11 или = P(O)R11,
где R11, R12 и R13 одинаковые или различные и обозначают атом водорода или атом галогена, C1 - C20 алкильную группу, C1 - C20 фторалкильную группу, C6 - C30 арильную группу, C6 - C30 фторарильную группу, C1 - C20 алкоксигруппу, C2 - C20 алкенильную группу, C7 - C40 арилалкильную группу, C8 - C40 арилалкенильную группу, C7 - C40 алкиларильную группу, или же R11 и R12 или R11 и R13 совместно со связывающими их атомами могут образовывать циклические системы;
M2 обозначает атом кремния, германия или олова;
R8 и R9 одинаковые или различные и имеют те же значения, что и R11;
m и n одинаковые или различные и обозначают 0,1 или 2,
сумма m и n равна 0,1 или 2;
радикалы R10 одинаковые или различные и имеют те же значения, что и R11, R12 и R13, где два смежных радикала R10 могут быть связаны с образованием кольцевой системы;
(б) нанесение на неорганический пористый носитель полученного реакционного продукта и (с) форполимеризацию полученной при этом каталитической системы на носителе в присутствии олефинового мономера (ов).
где M1 обозначает металл группы 4,5 и 6 Периодической таблицы;
R1 и R2 одинаковые или различные и обозначают атом водорода, C1 - C10 алкильную группу, C1 - C10 алкоксигруппу, C6 - C10 арильную группу, C6 - C10 арилоксигруппу, C2 - C10 алкенильную группу, C7 - C40 арилалкильную группу, C7 - C40 алкиларильную группу, C8 - C40 арилалкенильную группу или атом галогена;
R3 и R4 обозначают атом водорода;
R5 и R6 одинаковые или различные, предпочтительно одинаковые, и обозначают атом галогена, C1 - C10 алкильную группу, которая может быть галоидированной, C6 - C10 арильную группу, которая может быть галоидированной, C2 - C10 алкенильную группу, C7 - C40 арилалкильную группу, C7 - C40 алкиларильную группу, C8 - C40 арилалкенильную группу, радикал
-NR
-PR
где R15 обозначает атом галогена, C1 - C10 алкильную группу или C6 - C10 арильную группу;
R7 обозначает

= BR11, =ALR11, -Ge-, -Sn-, -O-, -S-, =SO, =SO2, =NR11, =CO, PR11 или = P(O)R11,
где R11, R12 и R13 одинаковые или различные и обозначают атом водорода или атом галогена, C1 - C20 алкильную группу, C1 - C20 фторалкильную группу, C6 - C30 арильную группу, C6 - C30 фторарильную группу, C1 - C20 алкоксигруппу, C2 - C20 алкенильную группу, C7 - C40 арилалкильную группу, C8 - C40 арилалкенильную группу, C7 - C40 алкиларильную группу, или же R11 и R12 или R11 и R13 совместно со связывающими их атомами могут образовывать циклические системы;
M2 обозначает атом кремния, германия или олова;
R8 и R9 одинаковые или различные и имеют те же значения, что и R11;
m и n одинаковые или различные и обозначают 0,1 или 2,
сумма m и n равна 0,1 или 2;
радикалы R10 одинаковые или различные и имеют те же значения, что и R11, R12 и R13, где два смежных радикала R10 могут быть связаны с образованием кольцевой системы;
(б) нанесение на неорганический пористый носитель полученного реакционного продукта и (с) необязательную форполимеризацию полученной при этом каталитической системы на носителе в присутствии олефинового мономера (ов).
| Литейная стопочная форма | 1974 |
|
SU485822A1 |
| СПОСОБ ПОЛУЧЕНИЯ КАРБОЦЕПНЫХ ЛИНЕЙНЫХ ПОЛИМЕРОВ | 1972 |
|
SU427022A1 |
| RU 94019418, 20.12.95 | |||
| Способ получения 3-диалкоксифосфонометил-1-глицидилгидантионов | 1973 |
|
SU530647A3 |
| СПОСОБ РЕГУЛИРОВАНИЯ ВЕЛИЧИНЫ ПОДАЧИ | 0 |
|
SU399347A1 |
| ВСЕСОЮЗНАЯПйТЕНтно-!?хн;:-;Е:кд1 | 0 |
|
SU363029A1 |
| ФЛЮС ДЛЯ СВАРКИ И ПАЙКИВ П т БU т^тт | 1973 |
|
SU433990A1 |
