Настоящее изобретение относится к способу получения этиленового сополимера. Более конкретно, оно относится к способу суспензионной полимеризации с получением эластомерного сополимера на этиленовой основе.
Среди эластомерных сополимеров на этиленовой основе вплоть до создания настоящего изобретения в промышленном масштабе производили только эластомеры на основе двойного сополимера этилена с пропиленом (EPM) и тройного этилен-пропиленового каучука с диеновым сомономером (EPDM).
В настоящее время процесс промышленного получения эластомеров EPM и EPDM проводят в присутствии катализатора Циглера-Натта на ванадиевой основе по методу полимеризации в растворе или суспензионной полимеризации.
В ходе проведения процессов в растворе сомономеры растворяют в растворителе, обычно в гексане, в котором растворяется также образующийся полимер. В суспензионных процессах реакционная среда формируется преимущественно за счет избытка жидкого пропилена, который выполняет функции полимеризационного разбавителя, а полимер образуется в виде твердого осадка, суспендированного в жидкой фазе.
Суспензионный процесс обладает рядом преимуществ перед процессом в растворе, а именно:
отсутствием проблем вязкости при перемешивании;
очень однородной реакционной средой;
более упрощенным удалением выделяющегося в результате реакции тепла;
повышенной производительностью реактора благодаря более высокой концентрации полимера в реакционной среде;
повышенным выходом процесса полимеризации;
возможностью получения полимеров с очень высокой средневесовой молекулярной массой;
экономией энергетических затрат на выделение полимера;
уменьшенными капитальными и производственными расходами.
Однако основная проблема в ходе проведения суспензионного процесса возникает из-за адгезивных свойств эластичного материала. Дело в том, что твердые частицы полимера проявляют тенденцию к слипанию между собой или прилипанию к поверхности стенки и перемешивающего элемента реактора. Это в значительной степени ухудшает диффузию этилена в реакционную среду и, что еще серьезнее, вызывает интенсивное загрязнение реактора, вследствие чего получение полимера оказывается сопряженным с очень большими затруднениями технологического порядка.
С целью избежать проблем в реакционную среду можно добавлять растворитель, в частности толуол или циклогексанон, который выполняет функции как средства, препятствующего накоплению загрязнений, так и носителя для каталитической системы. Было предложено также применять такой низкокипящий разбавитель, как пропан. Однако в результате резко ослабляются вышеуказанные достоинства суспензионного процесса.
Другое техническое решение, которое было предложено с целью обеспечить возможность проведения процесса в массе, состоит в добавлении в полимеризационный реактор антистатических агентов. Тем не менее такой технический прием оказывается не совсем удовлетворительным и, кроме того, обладает тем недостатком, что в конечный продукт попадают нежелательные соединения.
Недавно были описаны способы получения эластомерных этиленовых сополимеров в присутствии металлоцен/алюмоксановых катализаторов.
В описании к заявке на европейский патент N 347128 предлагается способ получения этилен/альфа-олефинового эластомера суспензионной полимеризацией с использованием цирконоцен-/алюмоксанового катализатора, нанесенного на силикагелевый носитель. В примерах представлено получение этилен/пропиленовых сополимеров в жидком пропилене. Указано, что если не происходит предварительной полимеризации находящегося на носителе катализатора с этиленом или другим альфа-олефином до его использования в процессе суспензионной полимеризации, неизменно происходит весьма интенсивное загрязнение реактора.
В описании к заявке на европейский патент N 535230 предлагается способ суспензионной полимеризации с получением сополимера на этиленовой основе, осуществление которого позволяет избежать возможности загрязнения. Такой способ осуществляют в присутствии как полисилоксановой добавки, так и нанесенного на силикагель цирконоцен/метилплюмоксанового катализатора. Все приведенные примеры относятся к этилен-пропиленовым эластомерам. В экспериментах сравнительных примеров, когда не предусматривалось использование никакой полисилоксановой добавки, наблюдались закупорка и заторы.
Совершенно неожиданно заявителями к настоящему времени было установлено, что процесс получения этилен-альфа-олефинового или этилен-альфа-полиенового эластомерного сополимера можно с успехом проводить по методу суспензионной полимеризации с использованием в качестве реакционной среды альфа-олефина, в присутствии катализатора на металлоценовой основе, без необходимости прибегать к обработке катализатора нанесением или предварительной полимеризацией или к использованию добавок, когда альфа-олефиновым мономером является 1-бутен.
Таким образом, целью настоящего изобретения является способ получения эластомерного сополимера на этиленовой основе, в соответствии с которым предусмотрена реакция суспензионной полимеризации смеси, включающей в себя этилен, 1-бутен и, возможно, небольшое количество полиона, в полимеризационной среде, которая состоит в основном из жидкого 1-бутена совместно с растворенным этиленовым газом, в присутствии каталитических количеств не подвергнутого предварительной полимеризации катализатора на основе металлоценового соединения титана, циркония или гафния.
Катализаторы, приемлемые для использования при осуществлении способа настоящего изобретения, могут быть получены, например, путем контактирования:
(А) металлоценового соединения формулы (I)
(C5R
возможно, после предварительной реакции с металлоорганическим алюминиевым соединением формулы (II)
AlR
где
M - атом металла, выбираемого из класса, который охватывает титан, цирконий и гафний; группы C5R NR1 или PR1, где заместители R1, идентичные или отличные друг от друга, определены по вышеизложенному, заместители 0, идентичные или отличные друг от друга, представляют собой атомы водорода или галогенов, группы OH, SH, R1, OR1, SR1, NR
NR1 или PR1, где заместители R1, идентичные или отличные друг от друга, определены по вышеизложенному, заместители 0, идентичные или отличные друг от друга, представляют собой атомы водорода или галогенов, группы OH, SH, R1, OR1, SR1, NR
(В) алюмоксановым соединением, возможно, в смеси с алюминиевым металлоорганическим соединением формулы (II)
AlR
где значения символа z и заместителей R3 определены выше, или по меньшей мере с одним соединением, способным вступать в реакцию с металлоценовым соединением с образованием катионоактивного алкилметаллоцена.
Величина молярного соотношения между алюминием и металлом металлоценового соединения обычно находится приблизительно от 100 до 10000, предпочтительнее от 300 до 5000, более предпочтительно приблизительно от 500 до 2000.
Предпочтительными металлоценовыми соединениями, приемлемыми для осуществления способа в соответствии с настоящим изобретением, оказываются такие соединения формулы (I), у которых металлом M является цирконий, то есть цирконоцены, а заместителями Q служат атомы хлора или гидрокарбильные группы, каждая из которых содержит от 1 до 7 углеродных атомов, предпочтительнее метильные группы.
Для того чтобы иметь возможность вводить 1-бутеновые звенья в полимерную цепь, в предпочтительном варианте не все заместители R1 одного и того же циклопентадиенильного кольца должны быть громоздкими радикалами. Так, например, подходящими металлоценами являются такие, у которых по меньшей мере один заместитель R1 (предпочтительнее по меньшей мере два заместителя R1) одного и того же циклопентадиенильного кольца должен представлять собой водородный атом. По другому варианту приемлемыми являются также металлоцены, у которых все заместители R1 образуют ароматические кольца.
Неограничивающими примерами металлоценовых соединений формулы (I) служат нижеследующие:
(Cp)2ZrCl2
(Ind)2ZrCl2
Me2Si(MeCp)2ZrCl2
C2H4(H4Ind)2ZrCl2
Ph2Si(Ind)2ZrCl2
(MeCp)2ZrCl2
(H4Ind)2ZrCl2
Me2Si(Ind)2ZrCl2
Me2Si(Ind)2ZrCl2
Me2SiCH2(Ind)2ZrCl2
C2H4(4,7-Me2Ind)2ZrCl2
C2H4(2,4,7-Me3Ind)2ZrCl2
C2H4(2,-MeH4Ind)2ZrCl2
C2H4(2,4,7-Me3H4Ind)2ZrCl2
Me2Si(3-MeInd)2ZrCl2
Me3Si(5,6-Me2Ind)2ZrCl2
Me2Si(3,4,7-Me3Ind)2ZrCl2
Me2Si(4,7-Me2H4Ind)2ZrCl2
Me2Si(Flu)2ZrCl2
Me2C(Flu)(Cp)2ZrCl2
C2H4(2-MeInd)2ZrCl2
(Me2Cp)2ZrCl2
Me2Si(Cp)2ZrCl2
C2H4(Ind)2ZrCl2
Ph(Me)Si(Ind)2ZrCl2
C2Me4(Ind)2ZrCl2
C2H4(3-MeInd)2ZrCl2
C2H4(5,6-Me2Ind)2ZrCl2
C2H4(3,4,7-Me3Ind)2ZrCl2
C2H4(4,7-Me2H4Ind)2ZrCl2
Me2Si(2-MeInd)2ZrCl2
Me2Si(4,7-Me2Ind)2ZrCl2
Me3Si(2,4,7-Me3Ind)2ZrCl2
Me2Si(2,-MeH4Ind)2ZrCl2
Me2Si(2,4,7-Me3H4Ind)2ZrCl2
C2H4(Flu)2ZrCl2
где Me - метил, Cp - циклопентадиенил, Ind - инденил, Flu - флуоренил, Ph - фенил, H4Ind-4,5,6,7-тетрагидроинденил.
Алюмоксановое соединение, приемлемое для осуществления способа в соответствии с настоящим изобретением, является линейным, разветвленным или циклическим соединением, молекула которого содержит по меньшей мере одну группу формулы (III)
где каждым из заместителей R4, которые идентичны или отличны друг от друга, может быть обозначена группа -O-Al(R4)2 или заместитель R1, где значения символа R1 определены выше, и, возможно, некоторые заместители R4 могут представлять собой атомы галогена.
Так, в частности, алюмоксановые соединения, которые могут быть использованы при осуществлении способа настоящего изобретения, представляет собой линейные алюмоксаны, отвечающие формуле (IV):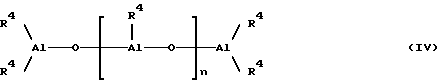
где
n - 0 или целое число от 1 до 40, и циклические алюмоксаны, отвечающие формуле (V):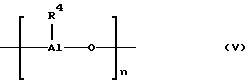
где
n - целое число от 2 до 40.
В формулах (IV) и (V) значения символа R4 определены выше, причем предпочтительным значением является углеводородная группа C1-C4, более предпочтительна метильная группа или изобутильная группа. Неограничивающими примерами алюмоксановых соединений, которые могут быть использованы при осуществлении способа настоящего изобретения, являются метилалюмоксан (МАО) и тетраизобутилдиалюмоксан (ТИБАО).
Неограничивающие примеры металлоорганических алюминиевых соединений формулы (II) охватывают нижеследующие продукты: Al(Me)3, Al(Et)3, AlH(Et)2, Al(iBu)3, AlH(iBu)2, Al(iHex)3, Al(C6H5)3, Al(CH2C6H5)3, Al(CH2CMе3)3, Al(CH2SiMe3)3, Al(Me2)iBu, Al(Me)2Et, AlMe(Et)2, AlMe(iBu)2, Al(Me)3iBu, Al(Me)2Cl, Al(Et)2Cl, AlEtCl2, Al2(Et)3Cl3, где Me - метил, Et - этил, iBu - изобутил, Hex - изогексил. К предпочтительным из них относятся (ТМА) и триизобутилалюминий (ТИБАЛ).
Неограничивающими примерами соединений, способных вступать в реакцию с металлоценовым соединением с образованием катионоактивного алкилметаллоцена, служат такие, которые отвечают формуле Y+Z-, где Y+ - кислота Бренстеда, которая способна отдавать протон и вступать в необратимую реакцию с заместителем Q соединения формулы (I), а Z--некоординационный совместимый анион, который способен стабилизировать активнодействующие каталитические компоненты и достаточно подвижен для замещения олефиновой основой. Соединения такого типа представлены, например, в описании к опубликованной заявке на международный патент WO 92/00333, которая в данном подробном описании упомянута, что совершенно очевидно, в качестве ссылки.
Способ суспензионной полимеризации по настоящему изобретению можно осуществлять в ходе проведения как периодического процесса, так и непрерывного процесса.
Полимеризационная температура обычно находится в пределах приблизительно от 0 до 200oC, в частности примерно от 20 до 100oC.
В соответствии с особенно выгодным вариантом настоящего изобретения после завершения реакции суспензионной полимеризации непрореагировавший газообразный этилен следует отпарировать из суспензии полимера в реакционной среде, которую отводят из реактора. Затем температуру суспензии повышают до образования раствора полимера в реакционной среде, которая после отпаривания этилена состоит практически из жидкого бутена. Такой раствор можно обрабатывать в смесителе, после чего полученный полимер выделяют выпариванием бутенового растворителя. Эту стадию предпочтительнее осуществлять в таком оборудовании, как экструдер для удаления летучих продуктов, благодаря чему продукт получают непосредственно в той форме, в которой он готов для переработки, то есть в форме шариков, таблеток и тому подобного.
Молекулы сополимеров, получаемых по способу настоящего изобретения, обычно содержат от 35 до 90 мол.%, предпочтительнее от 50 до 85 мол.%, этиленовых звеньев, от 5 до 65 мол.%, предпочтительнее от 15 до 50 мол.% звеньев, являющихся производными 1-бутена, и от 0 до 5 мол.%, предпочтительнее от 0 до 3 мол.% звеньев, являющихся производными полиена.
Сополимеры, молекулы которых включают в себя приблизительно до 80 мол.% этиленовых звеньев, преимущественно аморфны. Кристалличность сополимеров свидетельствует о том, что содержание этиленовых звеньев превышает приблизительно 80%, а теплота плавления (дельта Hf), когда количество этиленовых звеньев приближается к верхнему пределу в 90 мол.%, возрастает.
Полиены, которые могут быть использованы при осуществлении способа настоящего изобретения, представляют собой:
полиены, способные образовывать ненасыщенное звено, например такие, как:
несопряженные прямолинейные диены, в частности транс-1,4-гексадиен, цис-1,4-гексадиен, 6-метил-1, 5-гептадиен, 3,7-диметил-1,6-октадиен, 11-метил-1, 10-додекадиен;
моноциклические диолефины, в частности цис-1,5-циклооктадиен и 5-метил-1,5-циклооктадиен;
бициклические диолефины, в частности 4,5,8,9-тетрагидроинден и 6- и/или 7-метил-4,5,8,9-тетрагидродиен;
алкенил- или алкилиденнорборнены, в частности 5-этилиден-2-норборнен, 5-изопропилиден-2-норборнен, экзо-5-изопропенил-2-норборнен;
полициклические диолефины, в частности дициклопентадиен, трицикло-/6.2.1.02; 7/-4,9-ундекадиен и его 4-метилпроизводное;
несопряженные диолефины, способные к циклополимеризации, в частности 1,5-гексадиен, 1,6-гептадиен, 2-метил-1,5-гексадиен;
сопряженные диолефины, в частности бутадиен или изопрен.
Эластомерные сополимеры, получаемые при осуществлении способа настоящего изобретения, можно вулканизировать в соответствии с методами, которые известны для переработки эластомеров EPM и EPDM, например проведением процесса в присутствии перекисей или серы. Полученные продукты обладают ценными эластомерными свойствами, благодаря которым их можно использовать в тех областях, которые типичны для альфа-олефиновых эластомеров, в частности эластомеров EPM и EPDM.
Таким образом, осуществление способа настоящего изобретения дает возможность этилен/1-бутеновый или этилен/1-бутен/ полиеновый эластомерный сополимер получать в ходе проведения суспензионной реакции в среде жидкого сомономера, избегая загрязнения и необходимости использовать добавки или применять металлоценовый катализатор на носителе и в форме предварительно полимеризованного продукта.
Другое достоинство способа настоящего изобретения состоит в том, что такой полимер можно выделять без необходимости использования водяного пара в качестве инертного агента, содействующего отпарке.
Для чисто иллюстративных, а не ограничивающих рамки целей представлены нижеследующие примеры.
Характеристики
Дифференциальный сканирующий калориметрический анализ проводили с помощью прибора Перкина Элмера DSC7 при температуре от -25 до 180oC и скорости сканирования 10o/мин. Содержание 1-бутеновых звеньев в молекуле сополимера определяли 13C-ЯМР- спектрограммным анализом, который проводили с помощью прибора BRUKER AC200 при температуре 120oC. Образцы готовили растворением приблизительно 300 мг полимера в 2,5 мг смеси трихлорбензола с C2D2Cl4 в соотношении 3:1. Спектр фиксировали с использованием нижеследующих параметров:
задержка реакции - 12 с;
число импульсов - 2000 - 2500.
Характеристическую вязкость (ХВ) измеряли в тетрагидронафталине при температуре 135oC.
Молекулярно-массовое распределение (Mw/Mn) определяли с помощью гельпроникающей хроматографии, осуществляемой с использованием прибора WATERS 150 в орто-дихлорбензоле при температуре 135oC.
Для определения физико-механических характеристик полимеров с помощью каландра готовили смеси, которые обладали нижеследующим составом:
100 г сополимера;
30 г газовой сажи 550;
5 г окиси цинка;
1 г стеариновой кислоты;
1 г продукта Sartomer 206, промышленного продукта фирмы "Акномер";
4,5 г продукта Peroximon F40, промышленного продукта фирмы "Атокем".
Приготовленные смеси подвергали прямому прессованию с помощью 35-тонного пресса при удельном давлении 200 кг/см2 и температуре 165oC в течение 30-минутного периода времени. Из полученных образцов (200 х 120 х 2 мм) изготовили "гантели" для определения остаточного удлинения (200%, 1 мин, 23oC) и построения графика зависимости "напряжение-деформация". Скорость растяжения составляла 500 мм/мин.
Приготовление катализатора. - диметилсиландиил-бис(флуоренил)-цирконийдихлорид.
a) Получение диметил-бис(флуоренил)-силана.
120 мл (300 ммоль) 2,5 М раствора н-бутиллития в гексане по каплям добавляли в перемешиваемый раствор 50 г (300 ммоль) флуорена, растворенного в 400 мл тетрагидрофурана (ТГФ), поддерживая температуру раствора в ходе всей операции добавления на уровне 0oC. После завершения этой операции добавления раствор подогрели до комнатной температуры и перемешивание продолжали в течение 5 ч после прекращения газовыделения. Затем образовавшийся на этой стадии флуореновый анион по каплям добавляли в перемешиваемый раствор 0,15 моль диметилдихлорсилан, растворенного в 100 мл ТГФ, поддерживая в ходе операции добавления температуру на уровне 0oC. После завершения этой операции добавления раствор подогрели до комнатной температуры и перемешивание продолжали в течение 17 ч. Реакцию резко прекратили добавлением 150 мл воды и органический слой высушили над сульфатом магния. Растворители удалили в вакууме и собранный твердый материал перекристаллизовали из гексана, получив 37,8 диметилбисфлуоренилсилана (Me2SiFlu2), структуру которого и химическую чистоту подтвердили газовой хроматографией - масс-спектрометрией и 1Н-ЯМР-спектрометрией.
b) Получение диметилсиландиил-бис(флуоренил)-цирконийдихлорида.
8,5 г (0,0219 моль) лиганда Me2SiFlu2, полученного по вышеизложенному, растворили в 75 мл диэтилового эфира (Et2O). По каплям в этот раствор добавляли 31,25 мл метиллития (1,4 М раствор в диэтиловом эфире), поддерживая его температуру в ходе операции на уровне 0oC. После завершения операции добавления шлам подогрели до комнатной температуры и перемешивание продолжали в течение 5 ч после прекращения газовыделения. Далее фильтрованием удалили растворители и ярко-желтый порошок, который получили, промыли диэтиловым эфиром и пентаном, удалив все непрореагировавшие метиллитий и лиганд. Затем полученный таким образом лигандный дианион суспендировали в 100 мл диэтилового эфира и по каплям добавили в интенсивно перемешиваемую суспензию 5,1 г (0,0219 моль) четыреххлористого циркония в пентане, поддерживая в процессе добавления температуру на уровне - 78oC. После завершения этой операции добавления шламу дали нагреться до комнатной температуры и перемешивание продолжали в течение 17 ч. Далее шлам профильтровали ярко-красный твердый материал, который собрали, промыли диэтиловым эфиром и пентаном с последующей сушкой в вакууме при комнатной температуре. Выход продукта составил 13,56 г. Этот продукт использовали без дополнительной очистки в экспериментах нижеследующих примеров.
Этилен-бис(4,5,6,7-тетрагидроинденил)-цирконийдихлорид
а) Получение 1,2-бис(инденил)-этана.
Провели процесс получения, описанный в "Ewen. J., J. Am. Chem. Soc. 1987, 109, 6544, Suppl. mat.".
В 2-литровой 2-горлой круглодонной колбе в инертной атмосфере в 500 мл тетрагидрофурана растворили 50,8 г идена (437 ммоль) и раствор охладили до температуры -78oC. Затем осторожно, по каплям, в течение 1 ч добавили 175 мл н-бутиллития (2,5 М в гексане, 437,5 ммоль). Смеси дали нагреться до комнатной температуры и перемешивание продолжали в течение 4 ч.
Далее эту смесь охладили до температуры -78oC и по каплям в течение 20 мин добавили 40,42 г 1,2-дибромэтана (215 ммоль), растворенного в 100 мл тетрагидрофурана. В конце этой операции добавления температуру смеси повысили до 50oC и после перемешивания в течение 12 ч охладили до комнатной температуры и добавили в нее 20 мл воды.
Органическую фазу высушили и остаток подвергли экстракционной обработке пентаном.
Выпариванием в вакууме получили 28,65 г продукта. А выход составил 51,6%.
b) Получение этилен-бис(инденил)-цирконийдихлорида.
В 250-миллилитровую двугорлую круглодонную колбу, снабженную холодильником, загрузили 8 г (31 ммоль) 1,2-бисинденилэтана и 100 мл безводного тетрагидрофурана, приготовив таким образом желтый раствор. После охлаждения до температуры н-бутиллития в гексане (64 ммоль), в результате чего образовался осадок, который при нагревании вновь растворился с образованием красновато-желтого раствора.
В 250-миллилитровую четырехгорлую круглодонную колбу, снабженную холодильником, ввели 8,67 г четыреххлористого циркония (37,2 ммоль). После охлаждения до температуры -196oC в ней сконденсировали 50 мл тетрагидрофурана (очень резкоэкзотермическая реакция). Далее реакционной смеси дали нагреться до комнатной температуры и в течение 40 мин ее кипятили с обратным холодильником.
При комнатной температуре и при одновременном перемешивании раствор литиевой соли бисинденилэтана добавили в раствор аддукта тетрахлорид циркония/ТГФ и смесь перемешивали в темном месте в течение 20 ч.
При температуре 0oC пропускали пузырьки газообразного хлористого водорода, в результате чего образовался желтый раствор совместно с осадком того же цвета. Этот раствор сконцентрировали в вакууме выпариванием части растворителя, его охладили до температуры -20oC и профильтровали.
Далее осадок очистили экстракционной обработкой дихлорметаном, получив таким образом 2,3 г (выход - 14,7%) продукта.
c) Получение этилен-бис(4,5,6,7-тетрагидроинденил)- цирконийдихлорида.
Осуществили метод получения, описанный в "F.R.W.P. Wild, M. Wasiucionek. G. Hutter and H.H.Brintzinger, J. Organomet. Chem. 288, 1985, 63".
Суспензию 1 г этилен-бис(инденил)цирконийдихлорида (2,4 ммоль) и 80 г двуокиси платины в 25 мл дихлорметана гидрогенизовали в автоклаве под давлением водорода 100 бар в течение получаса при комнатной температуре. Реакционную смесь разбавили 500 мл дихлорметана, профильтровали и в вакууме выпарили растворитель.
Остаток после промывки пентаном перекристаллизовали из горячего толуола. Таким образом получили 640 мг (65%-ный выход) продукта.
Диметилсиландиил-бис(4,5,6,7-тетрагидроинденил)-цирконийхлорид
a) Получение бис-(инденил)-диметилсилана.
В 1-литровую 3-горлую круглодонную колбу, снабженную воронкой и патрубком для подачи азота, ввели 30 мл индена (257 ммоль) и 300 мл безводного тетрагидрофурана. Смесь охладили до температуры -80oC и осторожно, по каплям добавили в нее 170 мл 1,6 M раствора н-бутиллития в гексане (272 ммоль). Смеси дали вновь нагреться до комнатной температуры, выдержали с перемешиванием в течение 3 ч и добавили в раствор 15,5 мл (129 ммоль) дихлорметилсилана в 200 мл тетрагидрофурана.
После реакции в течение ночи смесь обработали 20 мл воды. Дали фазам разделиться, в вакууме выпарили растворитель и остаток обработали гексаном и высушили над безводным сульфатом натрия. После выпаривания гексана получили 38,5 г красного маслоподобного продукта, который очистили хроматографической обработкой на силикагеле (элюент-гексан). Выход продукта составил 18,8 г (51%).
b) Получение диметилсиландиил-бис(инденил)цирконийдихлорида
Осуществили процедуру, описанную в работе "W.A.Hermann et. al., Angew Chem. Int. Ed. Ebgl. 1989, 28, 1511".
9,4 г бис(инденил)-диметилсилана (32,59 ммоль), растворенного в 70 мл безводного тетрагидрофурана, обработали при температуре -78oC осторожным добавлением по каплям 40,7 мл 1,6 М раствора н-бутиллития в гексане (65,2 ммоль), в результате чего получили зеленый раствор. Этому раствору дали вновь нагреться до комнатной температуры, продолжая перемешивание в течение 1 ч.
Этот раствор, окраска которого сменилась на красную, по каплям в течение приблизительно 1 ч при комнатной температуре добавили в суспензию 12,4 г ZrCl4. 2ТГФ (32,9 ммоль) в 70 мл безводного тетрагидрофурана и оставили перемешиваться в течение 18 ч. В результате образовался оранжево-желтый осадок.
Объем реакционной смеси уменьшили до половины первоначального выпариванием в вакууме растворителя, выпавший осадок собрали фильтрованием и промыли в начале небольшим количеством тетрагидрофурана при температуре -20oC, а затем некоторым количеством диэтилового эфира. Выход продукта составил 4,97 г (34%).
c) Получение диметилсиландиил-бис(4,5,6,7-тетрагидроинденил)- цирконийдихлорида.
В 250-миллилитровую пробирку в инертной атмосфере добавили 2,856 г диметилсиландиил-бис(инденил)-цирконийдихлорида и 150 мл дихлорметана. По истечении 15 мин перемешивания приготовили оранжевый раствор. В этот раствор добавили 127,5 мг двуокиси платины и затем массу перенесли в 250-миллилитровый стеклянный автоклав, где ее выдержали под давлением водорода 2 ата в течение 1 ч, а затем под давлением водорода 4 ата в течение дополнительных 3 ч. Далее реакционную смесь профильтровали, остаток обработали 90 мл толуола и вновь профильтровали. После промывки пентаном твердый материал высушили в вакууме. В результате получили 1,092 г продукта.
Этилен-бис(4,7-диметил-1-инденил)-цирконийдихлорид
a) Получение 4,7-диметилиндена.
Синтез провели в соответствии с методом, который описан в "Organometallies, 1990, 9, 3098" (выход-п-ксилола - 54%).
b) Получение 1,2-бис(4,7-диметил-3-инденил)-этана.
38,2 г (265 ммоль) 4,7-диметилиндена растворили в 350 мл тетрагидрофурана и полученный раствор охладили до температуры 0oC. Затем по каплям в течение 2,5 ч добавили 165 мл 1,6 M раствора н-бутиллития в гексане (264 ммоль). После нагревания до комнатной температуры и перемешивания в течение 4 ч образовался пурпурный раствор 4,7-диметилиндениллития. Этот раствор охладили до температуры -70oC и обработали добавлением в течение 35 мин по каплям 25,3 г 1,2-дихлорметана (135 ммоль) в 15 мл тетрагидрофурана. После нагревания до комнатной температуры образовался бледно-желтый раствор; затем добавили воды. Органическую фазу собрали и высушили над сульфатом натрия. Растворитель удалили вакуумным выпариванием, получив 20 г сырого продукта (48%-ный выход).
c) Получение рацемического этилен-бис(4,7-диметил-1-инденил)-цирконийдихлорида.
Суспензию 10 г 1,2-бис(4,7-диметил-3-инденил)-этана (31,8 ммоль) в 80 мл тетрагидрофурана через полую иглу добавили в перемешиваемую суспензию 2,82 г KH (70,3 ммоль) в 160 мл тетрагидрофурана.
После затухания процесса выделения водорода образовавшийся коричневатый раствор отделили от избытка KH. Как этот раствор, так и раствор 12 г ZrСl4 (ТГФ)2 (31,8 ммоль) в 250 мл тетрагидрофурана по каплям через полую иглу в течение 3 ч добавили в колбу, содержавшую 50 мл интенсивно перемешиваемого тетрагидрофурана.
Образовались желтые раствор и осадок. После удаления в вакууме растворителя оранжево-желтый остаток (смесь рацемических и мезо-изомеров в соотношении 2,33:1 по 1H-ЯМР-спектрометрии) экстрагировали дихлорметаном до полного растворения всего оранжевого продукта. 1,7 г полученного желтого твердого продукта представляли собой единственный стереоизомер, а именно, мезо-изомер (выход - 11,3%).
В результате выпаривания дихлорметана из оранжевого раствора получили 4,9 г оранжевого твердого продукта, который соответствовал смеси 93,7% рацемических и 6,3% мезо-изомеров (выход - 32,5%). Далее этот твердый продукт перекристаллизовали из толуола при температуре - 20oC.
Бис(инденил)-цирконийдихлорид
Все операции проводили в инертной атмосфере. 0,7 мл индена (60 ммоль) растворили в 20 мл безводного тетрагидрофурана, раствор охладили до температуры -78oC и обработали 40,0 мл 1:5 M раствора н-бутиллития в гексане (60 ммоль). Эту смесь нагревали до комнатной температуры, в результате чего получили раствор, окрашенный в красный цвет.
В 100-миллилитровой круглодонной колбе, снабженной холодильником, 7 г тетрахлорида циркония (30 ммоль) охладили до температуры -78oC и обработали 30 мл тетрагидрофурана (экзотермическая реакция). После этого всю массу прокипятили с обратным холодильником в течение 30 мин, до тех пор, пока не образовался окрашенный в коричневый цвет раствор.
В раствор аддукта тетрахлорид циркония/ТГФ при комнатной температуре добавили раствор индениллития. При перемешивании массу выдержали в течение 2 ч (образовалась желтая суспензия), после чего полностью выпарили растворитель.
Остаток суспендировали в диэтиловом эфире, отфильтровали, повторно промыли диэтиловым эфиром и подвергли экстракционной обработке дихлорметаном. Раствор высушили и продукт промыли диэтиловым эфиром, а затем пентаном. Таким образом получили 4,35 г бис-инденилцирконийдихлорида (выход - 36,8%).
Метилалюмоксан
Метилалюмоксан (МАО) использовали в форме легкосыпучего белого порошка, полученного из технического 30%-ного по весу раствора в толуоле (фирма "Шеринг", средневесовая молекулярная масса 1400) путем удаления в вакууме летучих веществ (4 ч, 40oC, 0,1 мм рт.ст.)
Тетраизобутилдиалюмоксан
Тетраизобутилдиалюмоксан (ТИБАО) представлял собой технический продукт (30%-ный по весу раствор в циклогексане, выпускаемый фирмой "Шеринг АГ"), который был использован в поставляемом на рынок виде).
Примеры полимеризации
Примеры 1-4. В автоклав емкостью 2,6 л из нержавеющей стали, оборудованный магнитной мешалкой, манометром, датчиком температуры, системой для загрузки катализатора и для подачи мономеров и термостатируемой греющей рубашкой, предварительно продутый этиленом, при температуре 80oC ввели определенные количества воды, 1-бутена, которые указаны в табл.1. Параллельно этому раствор сокатализатора в толуоле (0,2 г/мл) добавили в диметилсиландиил-бис-(флуоренил)-цирконийдихлорид (2 мл раствора/мг циркония). Полученный раствор выдержали с перемешиванием в течение 5 мин при температуре 20oC, а затем требуемое количество ввели под давлением этилена в автоклав. После этого подавали смесь этилена с 1-бутеном в таком соотношении, чтобы поддерживать на постоянном уровне относительную концентрацию этилена и 1-бутена в растворе. Далее температуру быстро повысили до полимеризационного уровня. По истечении указанного в табл.1 промежутка времени реакцию полимеризации прекратили введением моноокиси углерода. После удаления продувкой непрореагировавших мономеров твердый продукт высушили в вакууме.
Условия проведения полимеризации и выход продукта представлены в табл. 1. Характеристики полученных полимеров приведены в табл. 2. В реакторе не наблюдалось никакого накопления загрязнений.
Примеры 5-8. Процедуру, описанную в примерах 1-4, повторяли полностью за исключением того, что в данном случае применяли 4,0-литровый автоклав из нержавеющей стали и что вместо диметилсиландиил-бис(флуоренил)-цирконийдихлорида использовали цирконоцены, указанные в табл. 1. В том случае, когда в качестве алюмоксанового соединения использовали МАО, перед добавлением катализатора использовали 50% его количества, которое вводили в автоклав.
Данные об условиях полимеризации и выходе продукта сведены в табл. 1. Характеристики полученных полимеров представлены в табл. 2. В реакторе не наблюдалось никакого накопления загрязнений.
Пример 9. В 1-литровый автоклав из нержавеющей стали ввели 255 г бутена. Температуру повысили до 50oC и ввели в него раствор, приготовленный смешением 0,16 мл 4,34 • 10-3 М толуольного раствора этилен-бис(4,5,6,7-тетрагидроинденил)- цирконийдихлорида с 2,45 мл 0,4 М толуольного раствора триизобутилалюминия в 7,5 мл толуола, содержащего 1,12 ммоль воды, и предварительным введением этих двух растворов в контакт между собой в течение 5 мин. После этого этилен подавали до тех пор, пока не было достигнуто избыточное давление в 4 атм, которое затем поддерживали на постоянном уровне с перемешиванием в течение 1 ч при температуре 50oC. После удаления непрореагировавшего мономера и сушки получили 7,90 г аморфного полимера (характеристическая вязкость - 2,96). Содержание бутеновых звеньев, определенное 13C-ЯМР- спектральным анализом, составляло 29,1 мол.%. В реакторе не наблюдалось никакого накопления загрязнений.
| название | год | авторы | номер документа |
|---|---|---|---|
| ЭЛАСТОМЕРНЫЕ СОПОЛИМЕРЫ ЭТИЛЕНА И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1994 |
|
RU2143441C1 |
| КАТАЛИЗАТОРЫ ДЛЯ (СО)ПОЛИМЕРИЗАЦИИ ОЛЕФИНОВ, СПОСОБ ПОЛУЧЕНИЯ ГОМО- ИЛИ СОПОЛИМЕРОВ ОЛЕФИНОВ | 1993 |
|
RU2132229C1 |
| КАТАЛИЗАТОРЫ И СПОСОБЫ ПОЛИМЕРИЗАЦИИ ОЛЕФИНОВ | 1995 |
|
RU2155774C2 |
| СПОСОБ ПОЛУЧЕНИЯ МОСТИКОВОГО МЕТАЛЛОЦЕНОВОГО СОЕДИНЕНИЯ, МОСТИКОВЫЙ БИС-ЦИКЛОПЕНТАДИЕНИЛЬНЫЙ ЛИГАНД | 1996 |
|
RU2168515C2 |
| КОМПОНЕНТЫ И КАТАЛИЗАТОРЫ ДЛЯ ПОЛИМЕРИЗАЦИИ ОЛЕФИНОВ | 1995 |
|
RU2161161C2 |
| СОПОЛИМЕР ЭТИЛЕНА С ОЛЕФИНОВЫМИ МОНОМЕРАМИ, СПОСОБ ЕГО ПОЛУЧЕНИЯ И КАТАЛИЗАТОР | 1993 |
|
RU2140930C1 |
| СОПОЛИМЕР ЭТИЛЕНА С ПРОПИЛЕНОМ, ЭЛАСТОМЕРНЫЙ СОПОЛИМЕР, ФОРМОВАННОЕ ИЗДЕЛИЕ, СПОСОБ ПОЛУЧЕНИЯ СОПОЛИМЕРА | 1994 |
|
RU2140426C1 |
| КАТАЛИЗАТОР НА НОСИТЕЛЕ ДЛЯ ПОЛИМЕРИЗАЦИИ ОЛЕФИНОВ (ВАРИАНТЫ), СПОСОБ ЕГО ПОЛУЧЕНИЯ (ВАРИАНТЫ), СПОСОБ ПОЛИМЕРИЗАЦИИ ОЛЕФИНОВ | 1994 |
|
RU2116316C1 |
| СОПОЛИМЕРЫ ЭТИЛЕНА И СПОСОБ ПОЛУЧЕНИЯ ПОЛИМЕРОВ НА ОСНОВЕ ЭТИЛЕНА | 1995 |
|
RU2155776C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПОЛИМЕРОВ ЭТИЛЕНА И ПРОДУКТЫ, ПОЛУЧЕННЫЕ ИЗ НИХ | 1994 |
|
RU2161627C2 |

Способ получения этилен/1-бутенового или этилен/1-бутен/диенового эластомера суспензионной полимеризацией мономеров в полимеризационной среде, состоящей в основном из избытка 1-бутена, который удерживают в жидкой форме, осуществляемый в присутствии катализатора на основе металлоценового соединения титана, циркония или гафния. Этот способ свободен от возможности загрязнения реактора, а его осуществление позволает выделять полимер без использования водяного пара в качестве инертного агента, содействующего отпарке. 12 з.п. ф-лы, 2 табл.
(А) металлоценового соединения формулы I
(C5R
где M - атом металла, выбираемого из класса, который охватывает титан, цирконий и гафний;
группы C5R1 5-m, которые могут быть как идентичными, так и различными, представляют собой одинаково или различно замещенные циклопентадиенильные кольца;
заместители R1, которые идентичны или отличны друг от друга, представляют собой водородные атомы, алкильные, алкенильные, арильные, алкиларильные или арилалкильные радикалы, каждый из которых содержит 1 - 20 углеродных атомов и которые могут также содержать атомы кремния или германия, или группы Si(CH3)3, или же два или четыре заместителя R1 одного и того же циклопентадиенильного кольца могут образовывать один или два кольца, каждое из которых содержит 4 - 6 углеродных атомов;
R2 - мостиковая группа, которая соединяет два циклопентадиенильных кольца и которую выбирают из групп формулы CR2 1, C2R4 1, SiR2 1, Si2R4 1, GeR2 1, Ge2R4 1, R2 1SiCR2 1, NR1 или PR1, где заместители R1, идентичные или отличные друг от друга, определены по вышеизложенному;
заместители Q, идентичные или отличные друг от друга, представляют собой атомы водорода или галогенов, группы OH, SH, R1, OR1, SR1, NR2 1 или PR2 1, где заместители R1, идентичные или отличные друг от друга, определены по вышеизложенному;
m - 0 или 1,
с (B) алюмоксановым соединением или по меньшей мере с одним соединением, способным вступать в реакцию с металлоценовым соединением с образованием катионоактивного алкилметаллоцена.
AlR
где заместители R3, которые идентичны или отличны друг от друга, представляют собой алкильные, алкенильные или алкиларильные радикалы, каждый из которых содержит 1 - 10 углеродных атомов и которые могут также содержать атомы кремния или германия;
z - 0 или 1.
AlR
где значения z и заместителей R3 определены выше.
где заместители R4 могут быть идентичными или отличными друг от друга и представляют собой группы -O-Al(R4)2 или заместители R1, где значения R1 определены выше, или атомы галогена при условии, что не все заместители R4 могут представлять собой атомы галогена.
| EP, 347128, кл | |||
| Топка с несколькими решетками для твердого топлива | 1918 |
|
SU8A1 |
| EP, 535230, кл | |||
| Топка с несколькими решетками для твердого топлива | 1918 |
|
SU8A1 |
| EP, 336593, кл | |||
| Топка с несколькими решетками для твердого топлива | 1918 |
|
SU8A1 |
