Настоящее изобретение относится к гетероцикло-циклическим аминным производным формулы I и к их фармацевтически приемлемым солям. Соединения формулы I являются ингибиторами холинэстеразы и могут быть использованы для улучшения памяти у пациентов, страдающих деменцией и болезнью Альцгеймера.
Болезнь Альцгеймера обусловлена перерождением холинергических нейронов в базальном переднем мозге, которые играют фундаментальную роль в познавательных мозгах, включая память. См., Becker и др., Drug Development Research, 12, 163-195 (1988). В результате такой дегенерации, у пациентов, страдающих этим заболеванием, обнаруживается заметное снижение синтеза ацетилхолина, холинацетилтрансферазной активности, ацетилхолинэстеразной активности и поглощения холина.
Известно, ингибиторы ацетилхолинэстеразы являются эффективными средствами для повышения холинергической активности и могут быть использованы для улучшения памяти у пациентов, страдающих болезнью Альцгеймера. Посредством ингибирования ацетилхолинэстеразы эти соединения способствуют повышению уровня нейротрансмиттера ацетилхолина в мозге и тем самым способствуют улучшению памяти. В вышеупомянутой работе Becker и др. указывается, что поведенческие изменения, имеющие место после ингибирования холинэстеразы, очевидно, соответствуют предсказанным пиковым уровням ацетилхолина в мозге. В этой работе также обсуждается эффективность трех известных ингибиторов ацетилхолинэстеразы, а именно физостигнина, метрифоната и тетрагидроаминоакридина.
Европейская патентная заявка ЕР A 0229391 относится к пиперидиновым производным формулы
R1-X-A-R2.
Европейская патентная заявка ЕР A 296560 относится к циклическому аминовому соединению формулы
Краткое описание изобретения
Настоящее изобретение относится к соединениям формулы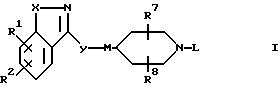
где
R1 и R2 независимо выбирают из водорода, (C1-C6)-алкокси, бензилокси, фенокси, гидрокси, фенила, бензила, галогена, нитро, циано, COR5, -COOR5, -CONHR5, -NR5COR6, -OCONR5R6, -NR5R6, -NHCOOR5, (C1-C6)-алкила, необязательно замещенного 1-3 атомами фтора; SOpCH2-фенила или SOp(C1-C6)-алкила, где p=0, 1 или 2; пиридилметилокси или тиенилметилокси; 2-оксазолила, 2-тиазолила и бензолсульфонамида; где фенильные части указанных фенокси-, бензилокси-, фенильной, бензильной и бензосульфонамидной групп; пиридильные и тиенильные части указанных пиридилметилокси- или тиенилметилокси-групп; и оксазолильные и тиазольные части указанных 2-оксазолильной и 2-тиазолильной группы могут быть, но необязательно, замещенными 1 или 2 заместителями, независимо выбранными из галогено-, (C1-C4)-алкила, трифторметила, (C1-C4)-алкокси, циано, нитро и гидрокси;
либо R1 и R2, если они связаны с соседними атомами углерода и если X является кислородом, серой или NR4, где R4 является водородом или (C1-C4)-алкилом, могут образовывать вместе с атомами углерода, с которыми они связаны, группы формулы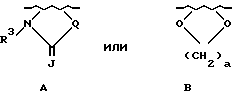
где
J - является кислородом, серой или NR4;
a = 1 или 2, R3 является водородом или (C1-C6)-алкилом;
Q является кислородом, серой, NH, CHCH3, C(CH3)2, -CH=CH- или (CH2)1, где L - является целым числом от 1 до 3;
X представляет собой кислород, серу, CH=CH-, -CH=N-, -N=N- или NR4, где R4 является водородом или (C1-C4)-алкилом;
Y представляет собой -(CH2)m-, -CH=CH(CH2)n-, -NR4(CH2)m- или -O(CH2)m-, где R4 определен выше, n является целым числом от 0 до 3, а m является целым числом от 1 до 3;
R5 и R6 независимо выбирают из водорода, (C1-C6)-алкила, фенила или бензила, где фенильные части указанных фенильной и бензильной групп могут быть, но необязательно, замещенными одними или двумя заместителями, независимо выбранными из фторо-, хлоро-, бромо-, иодо-, (C1-C4)-алкила, трифторметила, (C1-C4)-алкокси, циано и гидрокси; либо NR5R6 вместе образуют кольцо с 4-6 членами, в котором один атом является азотом, а другие атомы являются атомами углерода, кислорода или азота (например, пирролидинил, пиперидинил, морфолино, пиперазинил или N-метилпиперазинил), либо NR5COR6 вместе образуют циклическое лактамовое кольцо с 4-8 членами;
M представляет собой -CH- или азот;
L представляет собой фенил, фенил-(C1-C6)-алкил, циннамил или пиридилметил, где фенильные части указанных фенильной и фенил-(C1-C6)-алкильной групп могут быть, но необязательно, замещенными 1-3 заместителями, независимо выбранными из (C1-C6)-алкила, (C1-C6)-алкокси, (C1-C4)-алкоксикарбонила, (C1-C4)-алкилкарбонила, - ОCONR5R6, -NHCOOR5 или галогеногруппы; либо L представляет собой группу формулы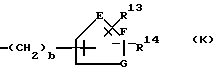
где
b является целым числом от 1 до 4;
R13 и R14 независимо выбирают из водорода, (C1-C4)-алкила, галогеногруппы и фенила;
E и F независимо выбирают из -CH- и азота;
G представляет собой кислород, серу или NR4, где R4 определен выше, при условии, что если E и F оба являются азотом, то один из R13 и R14 отсутствует;
R7 и R8 независимо выбирают из водорода, (C1-C6)-алкила, (C1-C6)-алкоксикарбонила, (C1-C6)-алкилкарбонила и (C1-C6)-(C1-C6)-алкокси, при условии, что указанная (C1-C6)-алкоксигруппа не связана с углеродом, который является атомом, соседним с атомом азота.
Настоящее изобретение также относится к фармацевтически приемлемым кислым аддитивным солям соединений формулы I. Примерами являются соли кислоты, п-толуолсульфоновой кислоты, малеиновой кислоты, фумаровой кислоты, лимонной кислоты, янтарной кислоты, салициловой кислоты, щавелевой кислоты, бромистоводородной кислоты, фосфорной кислоты, метансульфоновой кислоты, винной кислоты, ди-п-толуолвинной кислоты и миндальной кислоты.
Настоящее изобретение также относится к фармацевтической композиции, предназначенной для ингибирования холинэстеразы и содержащей соединениt формулы I или его фармацевтически приемлемую кислую аддитивную соль и фармацевтически приемлемый носитель.
Настоящее изобретение также относится к способу ингибирования холинэстеразы у млекопитающих, заключающемуся в том, что млекопитающему вводят определенное количество соединения формулы I или его фармацевтически приемлемой кислой аддитивной соли, являющееся эффективным для ингибирования холинэстеразы.
Настоящее изобретение также относится к способу улучшения памяти либо лечения или предупреждения болезни Альцгеймера у млекопитающих, заключающемуся в том, что млекопитающему вводят определенное количество соединения формулы I или его фармацевтически приемлемой кислой аддитивной соли, являющееся эффективным для улучшения памяти, или для лечения, или предупреждения болезни Альцгеймера.
Настоящее изобретение также относится к соединениям формулы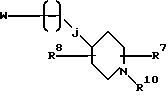
где
W является уходящей группой; j равно целому числу от 0 до 2; R10 является азот-блокирующей группой; а R7 и R8 независимо выбирают из водорода, (C1-C6)-алкила, (C1-C6)-алкоксикарбонила, (C1-C6)-алкилкарбонила и (C1-C6)-алкокси, при условии, что указанная (C1-C6)-алкокси-группа не связана с атомом углерода, соседним с атомом углерода, который является соседним с атомом азота. Эти соединения могут быть использованы в качестве промежуточных соединений в синтезе соединений формулы I.
Настоящее изобретение также относится к соединениям формулы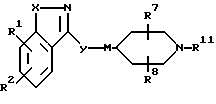
где
R1, R2, R7, R8, X, и Y и M определены выше, а R11 является водородом или азотной защитной группой. Эти соединения могут быть использованы в качестве промежуточных соединений в синтезе соединений формулы I.
Настоящее изобретение также относится к соединениям формулы: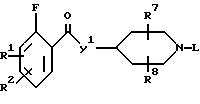
где
R1, R2, R7, R8 и L определены выше;
Y' является -CH=CH-(CH2)n- или -(CH2)m-.
Эти соединения могут быть использованы в качестве промежуточных соединений в синтезе соединений формулы I.
Используемый в настоящем описании термин "млекопитающее" включает в себя человека.
Используемый в настоящем изобретении термин "галогено" означает хлоро, бромо, иодо или фторо.
Термин "алкил", используемый в настоящем описании, означает, если это не оговорено особо, насыщенные моновалентные углеводородные радикалы, имеющие разветвленные, прямые или циклические части или их комбинации.
Термин "(C1-C4)-алкилкарбонил" относится к заместителю формулы
где
R15 является (C1-C4-алкилом).
Термин "C1-C4-алкоксикарбонил" относится к заместителю формулы V, указанной выше, где R15 является (C1-C4)-алкоксигруппой.
Термин "(C1-C6)-алкоксикарбонил" относится к заместителю формулы V, указанной выше, где R15 является (C1-C6)-алкоксигруппой.
Термин "(C1-C6)-алкилкарбонил" относится к заместителю формулы V, указанной выше, где R15 является (C1-C6)-алкилом.
Предпочтительными соединениями настоящего изобретения являются соединения формулы I, в которых X является кислородом или серой, Y является -CH2-, -CH2-CH2-; M является -CH, является бензилом; R1 и R2 являются (C1-C6)-алкилом; (C1-C6)-алкокси; NR5R6 или NR5COR6; R3 является водородом или (C1-C6)-алкилом; J является кислородом или серой; а Q является CH(CH3), CH(CH3)2, -CH=CH или (CH2)1, и их фармацевтически приемлемые соли:
3-[2-[1-(фенилметил)-4-пиперидинил]этил]-1,2-бензизоксазол;
5-метил-3-[2-[1-(фенилметил)-4-пиперидинил]этил]-1,2- бензизоксазол;
5,6-диметил-3-[2-[1-(фенилметил)-4-пиперидинил]этил]-1,2- бензизоксазол;
5-метокси-3-[2-[1-(фенилметил)-4-пиперидинил]этил]-1,2- бензизоксазол;
6-метокси-3-[2-[1-(фенилметил)-4-пиперидинил]этил]-1,2- бензизоксазол;
7-метокси-3-[2-[1-(фенилметил)-4-пиперидинил]этил]-1,2- бензизоксазол;
6-ацетамидо-3-[2-[1-(фенилметил)-4-пиперидинил]этил]-1,2- бензизоксазол;
6-амино-3-[2-[1-(фенилметил)-4-пиперидинил]этил]-1,2- бензизоксазол;
6-бензамид-3-[2-[1-(фенилметил)-4-пиперидинил]этил]-1,2- бензизоксазол;
6-бензолсульфонамид-3-[2-[1-(фенилметил)-4-пиперидинил] этил] -1,2- бензизоксазол;
6-(4-морфолинил)-3-[2-[1-(фенилметил)-4-пиперидинил] -этил] -1,2- бензизоксазол;
5,7-дигидро-3-[2-[1-(фенилметил)-4-пиперидинил] этил]-6H-пирроло [4,5-f] -1,2-бензизоксазол-6-он;
1-[2-[1-(фенилметил)-4-пиперидинил]этил]изохинолин;
3-[2-[1-(фенилметил)-4-пиперидинил]этил]-1,2-бензизотиазол;
4-[2-[1-(фенилметил)-4-пиперидинил]этил]-1,3-хиназолин;
6-гидрокси-3-[2-[1-(фенилметил)-4-пиперидил]этил]-1,2-бензизоксазол;
6-бромо-3-[2-[1-(фенилметил)-4-пиперидил]этил]-1,2-бензизоксазол;
6-циано-3-[2-[1-(фенилметил)-4-пиперидил]этил]-1,2-бензизоксазол;
6-карбоксамид-3-[2-[1-(фенилметил)-4-пиперидил]этил]-1,2-бензизоксазол;
3-[(1-фенилметил-4-пиперидил)метокси]-1,2-бензизоксазол;
3-[1-фенилметил-4-пиперидил)метиламино]-1,2-бензизоксазол;
3-[2-[1-фенилметил)-4-пиперидил]этиламино]-1,2-бензизоксазол;
3-[3-[1-(фенилметил)-4-пиперидил]пропил]-1,2-бензизоксазол;
транс-3-[2-[1-(фенилметил)-4-пиперидил]этенил]-1,2-бензизоксазол;
3-[2-[1-(фенилметил)-4-пиперазинил]этил]-1,2-бензизоксазол;
5,7-дигидро-7-метил-3-[2-[1-(фенилметил)-4-пиперадинил] -этил] -6H- пирроло[4,5-f]-1,2-бензизоксазол-6-он;
5,7-дигидро-7-этил-3-[2-[1-(фенилметил)-4-пиперидинил] этил] -6H-пирроло [4,5-f]-1,2-бензизоксазол-6-он;
5,7-дигидро-3-[2-[1-(1-(2-хлоро-5-тиофенметил)-4-пиперидинил] этил] -6H- пирроло-[4,5-f]-1,2-бензизоксазол-6-он;
5,7-дигидро-3-[2-[1-(2-метил-4-тиазолметил)-4-пиперидинил] этил] -6H-пирроло-[4,5-f]-1,2-бензизоксазол-6-он;
3-[2-[1-(3-бромофенилметил)-4-пиперидинил]5,7-дигидро-6H- пирроло[4,5-f] -1,2-бензизоксазол-6-он;
3-[2-[1-(4-бромофенилметил)-4-пиперидинил] этил] -5,7-дигидро-6H- пирроло[4,5-f]-1,2-бензизоксазол-6-он;
5,7-дигидро-3-[3-[1-(фенилметил)-4-пиперидинил] пропил] -6H- пирроло[4,5-f]-1,2-бензизоксазол-6-он;
3-[2-[1-(фенилметил)-4-пиперидинил] этил] -5,6,8-тригидро-7H- изоксазоло[4,5-q]хинолин-7-он;
6,8-дигидро-3-[2-[1-(фенилметил)-4-пиперидинил] этил]-7H- пирроло[5,4-q] -1,2-бензизоксазол-7-он;
5,7-дигидро-3-[2-[1-(фенилметил)-4-пиперидинил] этилен] -6H- пирроло[5,4-f]-1,2-бензизоксазол-6-он;
3-[2-[1-(фенилметил)-4-пиперидинил]этил]-1H-индазол
и фармацевтически приемлемые соли указанных соединений.
Примерами других соединений формулы I являются
6-фениламино-3-[2-[1-(фенилметил)-4-пиперидинил] этил] -1,2- бензизоксазол;
6-(2-тиазолил)-3-[2-[1-(фенилметил)-4-пиперидинил]этил]-1,2- бензизоксазол;
6-(2-оксазолил)-3-[2-[1-(фенилметил)-4-пиперидинил] этил]-1,2- бензизоксазол;
6-пирролидинил-3-[2-[1-(фенилметил)-4-пиперидинил]этил]-1,2- бензизоксазолил;
6-пиперидинил-3-[2-[1-(фенилметил)-4-пиперидинил] этил]-1,2- бензизоксазол;
5,7-дигидро-5,5-диметил-3-[2-[1-(фенилметил)-4-пиперидинил] этил]-6H-пирроло-[4,5-f]-1,2- бензизоксазол-6-он;
5,7-дигидро-3-[2-[1-(фенилметил)-4-пиперидинил] этил] -7- н-пропил-6H-пирроло-[4,5-f]-1,2-бензизоксазол-6-он;
5,7-дигидро-3-[2-[1-(фенилметил)-4-пиперидинил] этил] -7- пропил-6H-пирроло-[4,5-f]-1,2-бензизоксазол-6-он;
5,6-дигидро-3-[2-[1-(фенилметил)-4-пиперидинил]этил]-6H- пирроло-[4,5-f] -1,2-бензизоксазол-6-он;
3-[2-[1-(фенилметил)-4-пиперидинил] этил] -6-фенилметилсульфон- 1,2-бензизоксазол;
1-метил-3-[2-[1-(фенилметил)-4-пиперидинил]этил]-1H-индазол и
3-[1-фенилметил-4-пиперидинил)метил]-1,2бензизоксазол.
Соединения формулы I могут иметь оптические центры, а поэтому могут существовать в виде различных изомерных форм. В объем настоящего изобретения входят все стереоизомеры соединений формулы I, включая их смеси.
Подробное описание изобретения
Получение соединений формулы I и некоторых исходных материалов, используемых для синтеза этих соединений, проиллюстрировано в нижеприведенных реакционных схемах. В этих реакционных схемах и в последующем их обсуждении R1, R2, R3, R4, R5, R6, R7, R8, R13, R14, E, G, X, Y, M, L, a, b, I, m, n, p и структуры I, A, B и K являются такими, как они были определены выше, если это не оговорено особо.
Все статьи, монографии, патенты и патентные заявки, цитируемые в нижеприведенном обсуждении, вводятся в настоящее описание посредством ссылки. Схемы 1 - 6 приведены в конце текста.
Получение соединений формулы I, в которых Y является -(CH2)m и M является -CH-, проиллюстрировано на схеме 1. Эти соединения изображена на схеме 1 и обозначены как соединения формулы I-A (где L представляет собой фенил-(C1-C6)-алкил, пиридилметил или группу формулы K) и как соединения формулы I-B (где представляет собой фенил или циннамил).
В схеме 1 соединения формулы I-A могут быть получены путем депротонирования соединения формулы II с использованием основания в присутствии или с последующим добавлением алкилирующего агента формулы III, где R10 представляет собой азотзащитную группу, а W представляет собой уходящую группу. Если R10 является азотзащищающей группой, то в результате этой реакции образуется промежуточное соединение формулы IV. Это промежуточное соединение затем подвергают разблокированию с получением вторичного пиперидина формулы VI в виде свободного основания или соли свободного основания, после чего указанное свободное основание или его соль алкилируют с использованием соединения формулы WL, где W определен выше, а L представляет собой фенил(C1-C6)-алкил, пиридилметил или группу формулы K.
Примерами подходящих уходящих групп (W) являются мезилат, тозилат, хлорид, иодид или бромид. Примерами подходящих азотзащитных групп (R10) являются амиды, такие как N-формил и N-ацетил, и карбоматы, такие как т-бутоксикарбомат (ВОС). Предпочтительными азотзащитными группами являются ВОС. Подходящими основаниями, используемыми для получения соединений формулы IV, являются сильные основания, такие как диизопропиламид лития (LDA), н-бутиллитий, s-бутиллитий, и гексаметилдисилазид лития или натрия, или калия (LiHMDS, NaHMDS или KHMDS). Предпочтительными являются LDA и s-бутиллитий.
Реакцию соединения формулы II с соединением формулы III в основном, осуществляют в полярном апротонном растворителе, таком как диэтиловый эфир, 1,2-диметоксиэтан или тетрагидрофуран (ТГФ). Температура реакции может варьироваться от около -70 до около 30oC. Предпочтительно, если указанную реакцию проводят в ТГФ при около -70oC.
В основном соединения формулы II подвергают депротонированию в присутствии соединения формулы III. Однако в случае если соединение формулы II имеет более одного протона кислоты, то предпочтительно сначала осуществлять стадию депротонирования с последующим немедленным и быстрым добавлением алкилирующего агента формулы III.
Защитная группа (R10) может быть удалена из соединений формулы IV с образованием соответствующих соединений формулы VI традиционными способами, хорошо известными специалистам. Например, если R10 является ВОС или другим карбоматом, то эта группа может быть удалена с использованием кислоты, такой как бромоводород (газообразный или жидкий), хлороводород (газообразный или жидкий) или трифторуксусная кислота. В случае использования трифторуксусной кислоты может быть добавлен акцептор т-бутил-катионов, например, такой, как тиоанизол. Если в качестве разблокирующего агента используется кислота, то в результате будет продуцироваться не свободное основание соединения формулы VI, а его кислая аддитивная соль. Подходящими растворителями являются неполярные растворители, такие как метиленхлорид, а также полярные растворители, такие как диэтиловый эфир, этилацетат, диоксан, спирты, например, метанол или этанол, и вода. Температуры могут варьироваться в пределах от около -20oC и приб. до температуры перегонки. Предпочтительно использовать трифторуксусную кислоту в метиленхлориде в отсутствие или в присутствии тиоанизола при около 0oC.
Альтернативно, если R10 является ВОС, то она может быть удалена с использованием триалкилсилилтрифторометансульфонатного производного, такого как триметилсилил-, триэтилсилил- или т-бутилдиметилсилил-трифторметансульфонат, в присутствии ароматического или третичного аминового основания, такого как 2,6-лутидин или триэтиламин. Подходящими растворителями для данной реакции являются неполярные растворители, такие как метиленхлорид, и полярные растворители, такие как ТГФ, диэтиловый эфир или ДМФ. Температуры могут варьироваться в пределах от около -20oC до комнатной температуры. Для осуществления данной реакции предпочтительно использовать триметилсилилтрифторметан-сульфонат и 2,6-лутидин в метиленхлориде при температуре от около 0oC и примерно до комнатной температуры.
Промежуточный вторичный пиперидин формулы VI, полученный в виде свободного основания или соли, как описано выше, подвергают реакции с 2-10 эквивалентами основания, а затем с алкилирующими агентом формулы WL, где W является группой, определенной выше, а L является фенил-(C1-C6)-алкилом, пиридилметилом или группой формулы K. Подходящими основаниями являются третичные амины, такие как триэтиламин и диизопропилэтиламин; ароматические амины, такие как пиридин и диметиламинопиридин; и карбонаты металлов, такие как бикарбонат натрия, или карбонат натрия, калия или цезия. Если W является хлоридом, то в качестве катализатора может быть добавлен иодид (иодид калия или иодид тетра-н-бутиламмония). Подходящими растворителями являются неполярные растворители, такие как метиленхлорид, и полярные растворители, такие как диметилформамид, ТГФ, ацетонитрил, ацетон, диоксан, и спирты, такие как метанол или этанол. Реакцию алкилирования предпочтительно проводить в присутствии триэтиламина в метиленхлориде при комнатной температуре или в присутствии карбоната натрия в диметил формамиде при комнатной температуре.
Альтернативно, промежуточный вторичный пиперидин формулы VI, в случае, если он был получен в виде соли после удаления защитной группы, может быть подвергнут депротонированию с получением свободного амина путем его растворения или суспендирования в соответствующем растворителе (например, в метиленхлориде или этиленацетате), смешивания с водным бикарбонатом натрия или водным гидроксидом натрия или калия и выделения свободного амина из органического слоя с использованием стандартной техники экстрагирования. Полученный свободный амин может быть затем подвергнут алкилированию с использованием соответствующего алкилирующего агента формулы WL в условиях, описанных выше, и с использованием 1-2 эквивалентов соответствующего основания.
Исходные материалы формулы II могут быть получены способами, хорошо известными специалистам. Если X является кислородом, то исходные 3-метил-1,2-бензоизоксазолы могут быть получены способами, аналогичными описанным Wunsch и др., Adv. Heterocycl. Chem., 1967, 8, 277; Smalley P.K., Adv. Heterocycl. Chem. 1981, 29, 2 и Thakar и др., Indian J. Chem. 1977, 15B, 1058. Соответствующие b-гидроксиацетофеноны могут быть превращены в соответствующие оксимы с помощью реакции с гидрохлоридом гидроксиламина в присутствии соответствующего основания, такого как гидроксид калия или натрия, ацетат натрия или пиридин, предпочтительно водный гидроксид калия или водный ацетат натрия, в полярном растворителе, таком как метанол, этанол или вода, а предпочтительно в этаноле, при температуре примерно от комнатной температуры до около 120oC. Затем оксим превращают в соответствующий оксимацетат путем ацетилирования с использованием подходящего ацетилирующего агента, такого как уксусный ангидрид. Эта реакция может быть проведена при температурах примерно от комнатной температуры до температуры перегонки растворителя. Предпочтительными являются температуры 80-130oC.
Реакция замыкания кольца с образованием бензизоксазолового кольца может быть проведена путем нагревания чистого оксимацетата при температуре около 125 - 200oC при атмосферном давлении или пониженном давлении, например от около 0,01 мм рт.ст. (1,33 • 10-5 бар) до около 760 мм рт.ст. (1,01 бар). Реакцию замыкания кольца предпочтительно проводить путем нагревания оксимацетата при температуре перегонки в соответствующем основании, таком как пиридин, или путем нагревания оксимацетата при температуре около 130oC в полярном растворителе, таком как ДМФ или ДМСО (диметилсульфоксид) в присутствии нескольких эквивалентом соответствующего основания, такого как пиридин или 2,6-лутидин.
Альтернативно, замыкание кольца может быть осуществлено непосредственно из оксима с помощью его реакции с ацил- или сульфонилхлоридом, таким как оксалил- или тионилхлорид, в присутствии ароматического амина, такого как пиридин (см. , Kalkote и др., Aust, J. Chem., 1977, 30, 1847). Подходящими растворителями являются полярные растворители, такие как диэтиловый эфир или ТГФ. Температуры могут варьироваться в пределах от около 0oC и примерно до комнатной температуры. Другим способом замыкания кольца является обработка оксима одним или менее эквивалентами основания, такого как гидроксид калия, в полярном растворителе, таком как метанол, при температурах в пределах примерно от комнатной температуры до около 100oC (Crabbe и др., J. Chem. Soc. Perkin. Trans., 1, 1973, 2220).
Если X является серой, то исходные 3-метил-1,2-бензизотиазолы могут быть получены из о-метилтиоацетофенонов в соответствии с процедурами, описанными выше для бензизоксазолов (см., McLinnon и др., Can. J. Chem., 1988, 66, 1405 и работы, цитированные в настоящем описании). b-Метилтиоацетофеноны превращают в соответствующие оксимы и замыкание кольца осуществляют непосредственно путем реакции с соответствующим ацилирующим агентом, таким как уксусный ангидрид в основании, таком как пиридин. Температура реакции может быть примерно от комнатной температуры до около 130oC, а предпочтительно около 120oC.
Если X является NR4, где R4 является водородом, то исходные 3-метил-1H-индазолы могут быть получены в соответствии с методами, описанными Behr и др. "Pyrazoles, Pyrazolines, Pyrazolidines, Indazoles, and Condensed Ring", Heterocyclic Compounds, R. H. Wiley, Ed., 1967, 289; Barch и др., J. Heterocycl. Chem., 1984, 21, 1063; Hannig и др., Pharmazie 1976, 31, 534; Barton и др., J. Chem. Soc. Chem. Comm., 1982, 450; Ruechardtn и др., Liebigs Ann. Chem. , 1980, 908 и Rees и др., J. Chem. Soc. D, 1971, 827. N-алкилирование 3-метил-1H-индазолов (X= NR4, R4 = (C1-C4)-алкил) может быть осуществлено в соответствии с описанием Behr и др., "Pyrazoles, Pyrazolines, Pyrazolidines, Indazoles, and Condensed Rings", Heterocyclic Compounds, R.H. Wiley, Ed., 1967, 309; Palmer и др., J. Chem. Soc. Perkin Trans. II, 1975, 1695 и Claramunt и др., Heterocycles 1985, 23, 2895.
Если X является -CH=CH-, то исходные 1-метилизохинолины могут быть получены методами Bischler-Napieralksi или Pictet spengler (см., "Organic Reactions", vol, VI, главы 2 м 3, с., 74 - 190, John Wiley & Sons, Нью-Йорк, 1951).
Если -N=CH-, то исходные 4-метилхиназолины могут быть получены методами, описанными Byford и др., Indian J. Chem., 1988, 27B, 396; Higashino T., Chem Pharm. Bull. , 1962, 10, 1043; и Uff и др., J. Chem. Soc., Perkin Trans I, 1986, 2295.
Если X является -CH=N-, то исходные 1-метилфталазины могут быть получены методами, описанными Kant и др., J. Heterocycl. Chem., 1985, 22, 1065 и работы, цитированные в настоящем описании; Acheson. и др., J. Chem. Soc. C, 1966, 2218 и Gabriel и др., Chem. Ber. 1987, 30, 3022.
Если X является -N=N-, то исходные 4-метил-1,2,3-бензотриазины могут быть получены методами, описанными Adger и др., J. Chem. Soc. Perkin Trans I, 1975, 31; Boulton и др., Ibid, 1988, 1509 и Rees и др., J. Chem. Soc. D, 1971, 828.
Если R1 и R2 оба являются NH2, то исходный материал формулы II может быть получен из соответствующего NHAc-предшественника (Ac=ацетил) с помощью кислотного гидролиза. Кислотный гидролиз может быть осуществлен с использованием соляной кислоты при температурах от около 50 до около 120oC. Однако предпочтительным является нагревание до температуры перегонки (около 120oC) в 1 н. HCl. Соответствующие соединения NHBZ (BZ=бензоил) или NHSO2C6H5 могут быть получены из соответствующих амино-производных с помощью реакции с подходящим бензоилом или бензосульфонилхлоридом в присутствии основания, такого как триэтиламин, пиридин или диметиламинопиридин. Подходящими растворителями являются метиленхлорид, ТГФ, диэтиловый эфир или диметилформамид. Температуры реакции могут составлять от около -20 до около 80oC. Если один или оба R1 и R2 являются NHBZ, то предпочтительно использовать триэтиламин/диметиламинопиридин в метиленхлориде при комнатной температуре. Если один или оба R1 и R2 являются NHSO2Ph, то предпочтительно использовать пиридин в метиленхлориде при 0oC.
Циклические диалкиламино-соединения формулы II (т.е. соединения, в которых один или оба R1 и R2 являются NR5R6, где NR5R6 вместе образуют кольцо) могут быть также получены из соответствующего амино-производного путем алкилирования с использованием соответствующего бис-галогенидного реагента формулы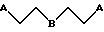
где каждый A независимо является бромидом или хлоридом;
B является кислородом или (CH2)q, где q = 0, 1 или 2,
в присутствии соответствующего основания, такого как триэтиламин или диизопропилэтиламин (основание Хьюнига), в подходящем неполярном растворителе, таком как толуол или ксилол. Алкилирование обычно осуществляют при температуре примерно от комнатной температуры до около 150oC. Предпочтительно, если реакция протекает в присутствии основания Хьюнига в толуоле при около 120oC (температура перегонки) (см. Verboom и др., J. Org. Chem. 1984, 49, 269).
Альтернативно, эти циклические диалкиламино-производные могут быть получены путем нуклеофильного замещения ароматического фторида соответствующим циклическим амином. Подходящими для этой реакции растворителями являются полярные апротонные растворители, такие как диметилсульфоксид (ДМСО), диметилформамид (ДМФ), ацетонитрил, пиридин и гексаметилфосфорамид. Предпочтительными являются ацетонитрил и пиридин. Реакция может протекать в присутствии основания, такого как третичные или ароматические амины (например, триэтиламин, диизопропилэтиламин, пиридин или диметиламинопиридин), а предпочтительно пиридин или триэтиламин. Реакция может быть осуществлена при температуре примерно от комнатной и до около 160oC, а предпочтительно от около 80 до около 160oC.
Если R1 и R2 вместе с атомами углерода, с которыми они связаны, образуют группу формулы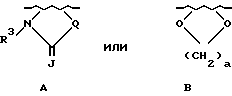
а
X является кислородом или серой, то исходное соединение формулы II может быть получено с помощью процедуры, показанной ниже и проиллюстрированной лишь для случаев, когда R1 и R2 образуют группу формулы A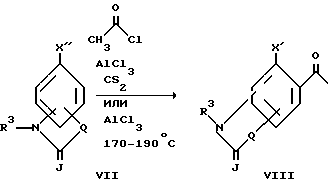
Сначала получают соединение формулы VIII, где X является гидрокси, тиолом или метилсульфидом, путем ацилирования Фриделя-Крафта, соответствующего соединения формулы VII, где X является метокси или метилсульфидом, с помощью ацилирующего агента, такого как ацетилхлорид или уксусный ангидрид, а предпочтительно ацетилхлорид, в присутствии кислоты Льюиса, такой как хлорид алюминия, тетрахлорид титана или трифторидэтеарат бора, а предпочтительно хлорид алюминия. Подходящими растворителями являются сероуглерод, 1,2-дихлорэтан и нитробензол. Предпочтительными являются сероуглерод и 1,2-дихлорэтан. В основном эту реакцию осуществляют при температуре примерно от комнатной температуры до около 200oC, а предпочтительно от около 50 до около 100oC.
Альтернативно, соединение формулы VIII, где X является гидроксигруппой, может быть также получено путем перегруппировки Фриса, которой подвергают соответствующее соединение формулы VII, где XII является ацетилоксигруппой. Смесь VII и кислоты Льюиса, такой как хлорид алюминия, трифторидэтеарат бора или тетрахлорид титана, нагревают при температуре от около 80 до около 200oC в отсутствие или в присутствии растворителя, такого как нитробензол или 1,2-дихлорэтан. Предпочтительно, если перегруппировку Фриса проводят без растворителя с использованием хлорида алюминия при 170-190oC.
Соединения формулы VIII, полученные вышеуказанным способом, могут быть превращены в соответствующие исходные соединения формулы II с помощью процедуры, описанной выше для 1,2-бензизоксазолов и 1,2-бензизотиазолов.
В схеме 1 соединения формулы III могут быть получены из соответствующих соединений, в которых W является гидроксигруппой, стандартными способами. Например, соединения формулы III, где W является иодидом, могут быть получены с помощью реакции гидрокси-противочасти с иодидом и трифенилфосфином в присутствии основания, такого как пиридин или имидазол в неполярном растворителе, таком как бензол или толуол, при температуре примерно от комнатной до около 130o. Предпочтительно, если реакция протекает в бензоле в присутствии пиридина при около 90oC (температура перегонки).
Соединения формулы I-B могут быть получены путем депротонирования соединения формулы II с использованием основания в присутствии или с последующим добавлением алкилирующего агента формулы III, где R10 является фенилом или циннамилом, а W является таким, как он был определен выше. Подходящие и предпочтительные основания, растворители и условия являются такими же, как были описаны выше для получения соединений формулы IV.
На схеме 2 проиллюстрировано получение соединений формулы I, где Y является -CH= CH(CH2)n-, путем альдольной конденсации. Эти соединения обозначены на схеме 2 как соединения формулы I-C. Согласно этой схеме соединение формулы IX, где R9 является водородом, подвергают депротонированию с использованием основания и с последующим немедленным и быстрым добавлением альдегида X. Подходящими основаниями и растворителями являются основания и растворители, описанные выше для первой реакции, проиллюстрированной на схеме 1. Реакция может протекать при температуре от около -78oC и примерно до комнатной температуры. Предпочтительно, если данную реакцию проводят с использованием диизопропиламида лития в ТГФ при около -78oC с последующим нагреванием до комнатной температуры.
Если в процессе реакции образуется промежуточный спирт, то он может быть дегидрирован с получением олефина в стандартных кислотных условиях при помощи кислоты, такой как разбавленная соляная кислота, п-толуолсульфоновая кислота, или п-толуолсульфонат пиридиния, а предпочтительно при помощи п-толуолсульфоновой кислоты в растворителе, таком как бензол, толуол, ТГФ или метиленхлорид, при температуре от около 0 до около 130oC. Предпочтительно, если гидрирование проводят в бензоле при около 80oC (температура перегонки) с азетропным удалением воды. Дегидрирование может быть также осуществлено посредством обработки реагентом Берджесса (Et
Альтернативно, промежуточный спирт может быть превращен в хорошую уходящую группу, такую как мезилат или тозилат, с последующим элиминированием с помощью соответствующего основания. Мезилат или тозилат могут быть получены в стандартных условиях путем осуществления реакции между спиртом и метилсульфонилхлоридом или п-толуолсульфонилхлоридом в присутствии основания, такого как триэтиламин, диизопропиламин или пиридин. Подходящими растворителями являются метиленхлорид и ТГФ, причем из них предпочтительным является метиленхлорид. Реакция может быть проведена при температуре от около 0 до около 60oC, а предпочтительно от около 0oC и примерно до комнатной температуры. Затем может быть осуществлено элиминирование с образованием олефина, которое проводят с использованием основания, такого как диазабициклоундекан или диазабициклононан, в соответствующем растворителе, таком как бензол, метиленхлорид или ТГФ, а предпочтительно в бензоле или метиленхлориде, при температуре от около 0 до около 100oC, а предпочтительно примерно от комнатной температуры и до около 100oC.
Соединения формулы I-C могут быть также получены с помощью реакции Виттига из соединений формулы IX, где R9 является бромом, хлором или йодом. Согласно этой процедуре соединение формулы IX превращают в его фосфониевую соль путем обработки трифенилфосфином в неполярном растворителе, таком как бензол, толуол или ксилол, а предпочтительно в толуоле, при температуре примерно от комнатной температуры до около 150oC, а предпочтительно от около 80 до около 120oC. Фосфониевая соль может быть затем подвергнута депротонированию с использованием сильного основания, такого как гидрид натрия, т-бутоксид калия, гидрид калия или н-бутиллитий, в подходящем растворителе, таком как диэтиловый эфир или ТГФ, при температуре от около 0 до около 80oC. Предпочтительно, если депротонирование протекает в присутствии гидрида натрия в ТГФ примерно при комнатной температуре.
Схема 2 иллюстрирует альтернативное получение соединений формулы I, где X является кислородом или NR4, Y является (CH2)m или -CH=CH(CH2)n, а M является углеродом (т. е. -CH-). На схеме 2 эти соединения обозначены как соединения формулы I-F (т.е. соединения в которых Y является -CH=CH(CH2)n, а n' представляет собой целое число от 0 до 3), и как соединения формулы I-G (т. е. соединения, в которых Y является (CH2)m, а n' представляет собой целое число от 0 до 1). В схеме 2 соединения формул I-F могут быть получены путем депротонирования соединения формулы XV с использованием соответствующего основания и с последующим добавлением альдегида формулы XVI, и в результате этой реакции образуется промежуточное соединение формулы XVII. Это промежуточное соединение затем превращают в соединения формулы I-F с помощью реакции с соответствующим амином.
Подходящими основаниями для использования в получении соединений формулы XVII являются диизопропиламид лития, гексаметилдисилазид лития, натрия или калия или н-бутиллитий, а предпочтительными основаниями являются диизопропиламид лития или гексаметилдисилазид лития. Реакцию соединения формулы XV с соединением формулы XVI в основном осуществляют в полярном апротонном растворителе, таком как диэтиловый эфир, 1,2-диметоксиэтан или тетрагидрофуран. Температуры реакции могут составлять от -78 до 80oC. Предпочтительно, если данная реакция протекает в ТГФ при -78oC с последующими нагреванием до комнатной температуры.
Соединение формулы I-F затем получают из промежуточного соединения формулы XVII при помощи реакции с амином, таким как гидразин или гидроксиламин, в присутствии основания, такого как гидроксид натрия или калия, карбонат натрия или калия или алкоксид натрия или калия (метоксид или этоксид), а предпочтительно гидроксида натрия или калия. В некоторых случаях, если амином является гидразин, добавление основания не является обязательным. Подходящими для этой реакции растворителями являются метанол, этанол, i-пропанол, вода, либо если амин является гидразином, то растворителем может быть сам гидразин. Температурный диапазон составляет от около 50 до 120oC. Однако предпочтительно, чтобы соединение XVII подвергалось реакции с гидразином при 120oC (температура перегонки) либо с гидроксиламином и гидроксидом калия в EtOH/воде при 100oC (тем-ра перегонки). После реакции с гидроксиламином полученный промежуточный оксим может быть выделен, а затем циклизован с получением соединения формулы I-F в соответствии с подходящими и предпочтительными условиями, описанными выше для получения исходных соединений 3-метил-1,2-бензизоксазолов.
Соединения формулы I-G могут быть получены путем восстановления промежуточного соединения формулы XVII с получением соединения формулы XVIII. Затем промежуточное соединение формулы XVII может быть подвергнуто реакции с амином с получением соединения формулы I-G. Промежуточное соединение формулы XVII восстанавливают до промежуточного соединения формулы XVIII с использованием газообразного водорода в присутствии катализатора, такого как палладированный уголь, оксид платины или уголь, покрытый родием, а предпочтительно в присутствии окиси платины в полярном растворителе, таком как этилацетат, тетрагидрофуран, этанол или уксусная кислота, а предпочтительно в EtOH. Давление может варьироваться от атмосферного до 50 фунт/кв.дюйм (3,45 бар), а предпочтительно 40-50 фунт/кв.дюйм (2,76-3,45 бар), а температура может варьироваться в пределах от комнатной температуры до 80oC, а предпочтительной температурой является комнатная.
Соединения формулы I-G могут быть затем получены из промежуточного соединения формулы XVIII при подходящих и предпочтительных условиях, описанных выше, для получения соединения формулы I-F из промежуточного соединения формулы XVII.
На схеме 3 проиллюстрировано получение соединений формулы I, в которых Y является -O(CH2)m- или -NR4(CH2)m. Эти соединения в схеме 3 обозначены формулой I-D. В схеме 3 соединения формулы I-D могут быть получены с помощью реакции соединения формулы XI, где R12 является хлоро- или бромогруппой, с нуклеофилом формулы XII, где Z является -NHR4 или -OH. Если Y является NR4(CH2)m (т. е. Z является -NHR4), то амин формулы XII обычно подвергают реакции с соответствующим соединением формулы XI либо в отсутствие растворителя, либо в полярном растворителе, таком как диметилформамид (ДМФ), диметилсульфоксид (ДМСО), ТГФ или пиридин. Предпочтительными растворителями являются ДМСО и ДМФ. Может быть также добавлен акцептор кислоты, такой как диазабициклоундекан, пиридин, лутидин, триэтиламин, или карбонаты металлов, такие как карбонат калия, натрия или цезия. Предпочтительными являются карбонаты металлов, например карбонат цезия. Реакция может быть осуществлена при температуре примерно от комнатной до около 160oC, а предпочтительно от около 100 до около 160oC.
Если Y является O(CH2)m (т.е. Z является -OH), то образуется анион алкоксида, который реагирует с соединением формулы XI. Согласно этой процедуре спирт (XIII) подвергают депротонированию с использованием соответствующего основания, после чего добавляют соответствующее соединение формулы XI и полученную смесь нагревают. Примерами подходящих оснований является натрий, гидрид натрия и гидрид калия, причем предпочтительным является гидрид натрия. Подходящими растворителями являются ТГФ, ДМФ и ДМСО, при этом предпочтительными являются ТГФ и ДМФ. Эту реакцию в основном проводят при температуре от около 40 до около 160oC. Предпочтительными температурами являются от около 60 до около 160oC.
Альтернативно, соединения формулы I-D могут быть получены с помощью реакции соединение формулы XI с нуклеофилом формулы XIX, где Z и R10 определены выше. В результате этой реакции образуется промежуточное соединение формулы XXX, которое затем подвергают разблокированию и получают вторичный пиперидин формулы XXI в виде свободного основания или соли свободного основания, после чего это свободное основание или его соль алкилируют с использованием соединения формулы WL, где W является таким, как он был определен выше, а L является фенил-(C1-C6)-алкилом, пиридилметилом или группой формулы K.
Подходящими и предпочтительными основаниями, растворителями и условиями для реакции соединения формулы XI с нуклеофилом формулы XIX являются основания, растворители и условия, аналогичные тем, которые были описаны для реакции соединения формулы XI с нуклеофилом формулы XII для получения соединений формулы I-D. Подходящими и предпочтительными основаниями, растворителями и условиями для превращения соединений формул XX и XXI в соединения формул I-D являются основания, растворители и условия, аналогичные тем, которые были описаны в схеме 1 для реакции соединений формул IV и VI с получением соединений формулы I-A.
В схеме 4 проиллюстрировано получение соединений формулы I, где Y является -(CH2)m- и M является азотом. Эти соединения в схеме 4 и далее обозначаются как соединения формулы I-E. В этой схеме 4 соединение формулы XIII, где R12 является хлоро-, бромо- или иодогруппой, подвергают реакции с соединением формулы XIV. Эта реакция может быть осуществлена в присутствии акцептора кислоты, такого как пиридин, 2,6-лутидин или карбонат металла (например, бикарбонат натрия или карбонат натрия и калия). Если R12 является хлоро- или бромогруппой, то может быть добавлено каталитическое количество активатора замещения. Примерами подходящих активаторов замещения являются иодид натрия, иодид калия или иодид тетра-н-бутиламмония. В основном эту реакцию проводят в неполярном растворителе, таком как толуол или ксилол, или в полярном растворителе, таком как ТГФ, ДМФ или ДМСО, а предпочтительно в ксилоле или ДМФ, при температуре примерно от комнатной до около 160oC, а предпочтительно от около 90 до около 160oC.
На схеме 5 проиллюстрировано получение соединений формулы I, где X является -N=CH-, а M является углеродом (т.е. -CH). На схеме 5 и далее эти соединения обозначены как соединения формулы I-H. На этой схеме соединения формулы II могут быть депротонированы с использованием одного эквивалента основания и с последующим добавлением одного эквивалента основания и с последующим добавлением силилирующего депротонирования с использованием второго эквивалента того же основания и последующего добавления алкилирующего агента формулы III с соответствующей обработкой получают промежуточное соединение формулы IV. Это промежуточное соединение затем подвергают разблокированию, как описано в схеме 1, и получают вторичный пиперидин формулы VI в виде свободного основания или его соли, после чего это свободное основание или его соль подвергают алкилированию с использованием соединения формулы WL, где W определен выше, а L представляет собой фенил (C1-C6)-алкил, пиридилметил или группу формулы K.
Подходящими основаниями, растворителями и температурами для депротонирования соединения формулы II являются основания, растворители и температуры, аналогичные тем, которые были описаны выше для первой реакции схемы 1, а предпочтительно LDA в ТГФ при температуре от 0oC до комнатной температуры. После добавления первого эквивалента основания добавляют силилирующий агент, такой как триметилсилил или триэтилсилилхлорид, а предпочтительно триметилсилилхлорид. Затем добавляют второй эквивалент того же самого основания с последующим добавлением алкилирующего агента формулы III. Триметилсилильную группу затем удаляют в кислотных условиях путем размешивания сырой реакционной смеси с разбавленной соляной кислотой при комнатной температуре в течение 30-60 мин. После этого сырую реакционную смесь подщелачивают водным карбонатом натрия или водным гидроксидом натрия или калия, а предпочтительно водным гидроксидом натрия, и промежуточное соединение формулы IV экстрагируют органическим растворителем с помощью стандартной техники экстрагирования. Подходящими и предпочтительными условиями для трансформации промежуточных соединений формулы IV в соединения формулы I-H являются условия, аналогичные тем, которые были описаны в схеме 1 для получения соединений формулы I-A.
Схема 6 иллюстрирует получение соединений формулы I, где Y является (CH2)m, M является углеродом (т. е.-CH-), J является серой, а Q является CHCH3, C(CH3)2, -CH=CH или (CH2)1. На схеме 6 эти соединения обозначены как соединения формулы I-I. В схеме 6 соединение формулы I-I может быть получено из соответствующего соединения формулы IV'', где J является кислородом, при помощи реакции этого соединения с сульфидом фосфора, в результате которой образуется промежуточное соединение формулы XXII. Затем это промежуточное соединение подвергают разблокированию, как описано в схеме 1, и получают вторичный пиперидин формулы VI'' в виде свободного основания или соли свободного основания, после чего указанное свободное основание или его соль подвергают алкилированию с использованием соединения формулы WL, где W определен выше, а L представляет собой фенил (C1-C6)-алкил, пиридилиетил, или группу формулы K.
Превращение соединения формулы IV'' в промежуточное соединение формулы XXII осуществляют с использованием сульфида фосфора, такого как пентасульфид фосфора (P2S10) или реагент Lawesson [2,4-бис(4-метоксифенил)-1,3-дитиа-2,4-дифосфенат-2,4-дисульфид] , в неполярном растворителе, таком как бензол, толуол или ксилол. Температуры реакции могут составлять 50-160oC. Предпочтительно, если реакция протекает в присутствии реагента Lawesson в толуоле при 80oC. Подходящими и предпочтительными условиями для трансформации промежуточных соединений формулы XXII в соединения формулы I-I являются условия, аналогичные тем, которые были описаны в схеме 1 для получения соединений формулы I-A.
Схема 7 иллюстрирует получение соединений формулы I, где Y является (CH2)m, M является углеродом (т.е. -CH-), а R3 является (C1-C6)-алкилом. На схеме 7 эти соединения обозначены как соединения формулы I-J. На этой схеме соединение формулы I-J может быть получено из соответствующих соединений формулы IV''', где R3 является водородом, путем депротонирования с помощью основания и с последующим добавлением соответствующего алкилирующего агента (предпочтительно (C1-C6)-алкилхлорида, бромида или иодида), в результате чего получают промежуточное соединение формулы XXIII. Это промежуточное соединение затем подвергают разблокированию, как описано в схеме 1, и получают вторичный пиперидин формулы VI в виде свободного основания или соли свободного основания, после чего это основание или соль подвергают алкилированию с помощью соединения формулы WL, где W определен выше, а L представляет собой фенил-(C1-C6)-алкил, пиридилметил или группу формулы K.
Подходящими основаниями для превращения соединения формулы IV в соединение формулы XXIII являются гидрид натрия, гидрид калия, диизопропиламид лития или н-бутиллитий, а предпочтительным основанием является гидрид натрия. Обычно реакцию осуществляют в полярном растворителе, таком как тетрагидрофуран, диметилформамид или 1,2-диметоксиэтан, при температуре в пределах -78 - 80oC. Предпочтительно, если эта реакция протекает в диметилформамиде при комнатной температуре. Подходящими и предпочтительными условиями для превращения промежуточных соединений формулы XXIII в соединения формулы I-J являются условия, аналогичные тем, которые были описаны в схеме 1 для получения соединений формулы I-A.
Альтернативно, соединение формулы I-J может быть получено непосредственно из соответствующего соединения формулы I-A, где L представляет собой фенил-(C1-C6)-алкил, пиридилметил или группу формулы K, а R3 является водородом или I-B', где L является фенилом или циннамилом, а R3 является водородом. Соединения формулы I-A' и I-B' получают способами, описанными в схеме 1, для получения соединений I-A и I-B. Подходящими и предпочтительными соединениями, растворителями и условиями для трансформации соединений формулы I-A и I-B в соединения формулы I-J являются основания, растворители и условия, аналогичные тем, которые описаны выше для получения соединений формулы XXIII.
Если один или оба R1 и R2 являются OH, то соединения формулы I могут быть получены из соответствующего -OMe-предшественника путем деалкилирования с использованием кислоты Льюиса, такой как трихлорид алюминия, трихлорид бора, трибромид бора, или протонной кислоты, такой как водная хлористоводородная или бромистоводородная кислота. Для реакции с использованием кислот Льюиса подходящими растворителями являются неполярные растворители, такие как бензол, толуол, дихлорметан, или 1,2-дихлорэтан. Температуры могут составлять от -78 до 120oC. Предпочтительно, если реакция протекает в присутствии водной бромистоводородной кислоты (48%) при 100-120oC (тем-ра перегонки).
Если один или оба R1 и R2 являются NH2, то соединения формулы I могут быть получены из соответствующего NHAc-предшественника (Ac=ацетил) путем кислого гидролиза в подходящих и предпочтительных условиях, описанных выше, для получения исходных материалов формулы II, в которых один или оба R1 и R2 являются NH2. Соответствующие нитриловые (-CN) соединения могут быть получены из соответствующих аминосоединений посредством образования диазониевой соли с помощью реакции аминосоединения с азотистой кислотой (полученной из водной соляной кислоты и нитрита натрия) с последующей нейтрализацией и добавлением к CuCN. Подходящими растворителями являются полярные протонные растворители, такие как вода или двухфазные смеси с неполярными растворителями, такими как бензол, толуол или ксилол. Нейтрализация может быть осуществлена путем добавления основания, такого как карбонат натрия, карбонат калия, гидроксид натрия или гидроксид калия, до тех пор, пока pH не будет равен 7. Температуры реакции могут составлять от -20 до 60oC. При этом, предпочтительно, чтобы образование диазониевой соли протекало в воде при 0oC, с последующей нейтрализацией при помощи карбоната натрия и добавлением полученной диазониевой соли к двухфазной смеси водного Cu CN и толуола при 0oC и последующим нагреванием до 50oC.
Если один или оба R1 и R2 являются карбоксамидом (-CONH2), то соединения формулы I могут быть получены из соответствующего нитрилового (-CN)-предшественника при помощи его реакции с основанием, таким как гидроксид натрия, гидроксид калия или гидроксид тетраметиламмония, в полярном растворителе, таком как вода, метанол, этанол или т-бутанол. Реакция может протекать при температурах от комнатной до 120oC. Предпочтительно, если реакцию осуществляют с гидроксидом калия в т-бутаноле при 85-100oC.
Соединения формулы I, не относящиеся к формулам I-A-I, могут быть получены способами, непосредственно вытекающими из вышеописанных процедур, или другими хорошо известными способами.
В каждой из вышеописанных реакций давление не является критическим параметром. Обычно, давление составляет в пределах от около 0,5-3 атм (0,5-3 бар), и для удобства предпочтительно использовать давление окружающей среды (т. е. около 1 атм). Кроме того, для тех реакций, где предпочтительная температура колеблется в зависимости от конкретных реагентов, нельзя указать какую-либо предпочтительную температуру. Для таких реакций предпочтительные температуры для конкретных реагентов могут быть определены путем контролирования реакции с помощью тонкослойной хроматографии.
Соединения формулы I и их фармацевтически приемлемые соли (называемые далее "активными соединениями настоящего изобретения") могут быть введены пациенту различными способами, например перорально в виде капсул или таблеток; парентеральной в виде стерильных растворов или суспензий и в некоторых случаях внутривенно в виде раствора. Соединения настоящего изобретения в виде свободных оснований могут быть введены в виде их фармацевтически приемлемых кислых аддитивных солей.
Суточная доза активных соединений настоящего изобретения обычно варьируется от около 1 до 300 мг/день для среднего взрослого человека и может быть введена в виде разовой или разделенной дозы.
При парентеральном введении в виде раствора или суспензии активные соединения настоящего изобретения присутствуют в концентрации по крайней мере 1 мас.%, а предпочтительно от около 4 до 70 мас.% (по полной массе стандартной формы). Парентеральная стандартная форма обычно содержит от около 5 до 100 мг активного соединения (или соединений).
Активные соединения настоящего изобретения могут быть введены перорально в сочетании с инертным разбавителем или съедобным носителем, либо они могут быть заключены в желатиновые капсулы или спрессованы в таблетки. Такие препараты должны содержать по крайней мере 0,5% активного соединения, но эта концентрация может варьироваться в зависимости от конкретной формы и может составлять от 4 до 70 мас.% (по полной массе стандартной формы). Пероральная стандартная форма обычно содержит 1,0-300 мг активного носителя соединения.
Способность активных соединений настоящего изобретения ингибировать холинэстеразу может быть определена рядом стандартных биологических или фармацевтических тестов. Один из таких методов определения ингибирования холинэстеразы описан Ellman и др., "A New and Rapid Colorimetric Determination of Acetycholinesterase Activity", Biochem, Pharm., 1, 88 (1961).
Настоящее изобретение проиллюстрировано примерами, приведенными ниже. При этом следует указать, что эти примеры не ограничивают изобретения до конкретных деталей. Приведенные точки плавления не скорректированы. Спектры протонного магнитного резонанса (1H-ЯМР) и спектры C13-ядерного магнитного резонанса (C13-ЯМР) измеряли для растворов в дейтерохлороформе (CDCl3), если только это не указано особо, а пиковые положения выражали в миллионных долях (м.д.). Формы пиков обозначали следующим образом: с (синглет); д (дублет); т (триплет); кв. (квартет); м (мультиплет); шир. (широкий). Все частоты выражали в Гц. 1М-растворы диизопропиламида лития были свежеприготовлены путем добавления н-бутиллития (1,6 - 2,5 М в гексанах) к раствору диизопропиламина в терагидрофуране при 0oC.
Пример 1.
3-[2-[1-(Фенилметил)-4-пиперидинил]этил]-1,2-бензизоксазолмалеат
a) 1,4-Пиперидиндикарбоновая кислота, 1-(1,1-диметилэтил)сложный эфир, 4-этиловый сложный эфир
Раствор этилизонипекотата (20,0 г, 0,127 М) и триэтиламина (17,8 мл, 0,127 М) в 1:1 -смеси диоксана и H2O (1,2 л) охлаждали до 0oC. Через 15 мин добавляли т-ВОС-ангидрид (35,2 г, 0,161 М) и полученную смесь оставляли на ночь для нагревания до комнатной температуры. Затем смесь экстрагировали этилацетатом (4 раза) и объединенные органические слои промывали 1 н. соляной кислотой, водой и солевым раствором, а затем осушали сульфатом магния, фильтровали и концентрировали, в результате чего получали светло-оранжевое маслообразное вещество, а после дистилляции Kugelrohr (0,5 торр (6,67 бар), 80 - 90oC) получали целевой карбамат (30,69 г, 94%) в виде бесцветного маслообразного продукта.
1Н-ЯМР (CDCl3) δ : 4,11 (кв. 2H, J = 7,2 Гц); 3,97 - 4,05 (м, 2H); 2,80 (шир. , т, 2H, J = 11,6 Гц); 2,40 (тт, 1H, J = 22,0 Гц, J = 3,9 Гц); 1,81 - 1,86 (м, 2H); 1,52 - 1,66 (м, 2H); 1,43 (с, 9H); 1,23 (т, 3H, J = 7,2 Гц).
b) 4-Гидроксиметил-1-пиперидинкарбоновая кислота, 1-(1,1-диметилэтил)сложный эфир
Алюмогидрид лития (4,3 г, 0,114 М) добавляли к холодному (0oC) раствору карбомата, полученного в стадии а) (26,57 г, 0,103 М) в тетрагидрофуране (ТГФ) (1 л). Через 30 мин ледяную баню удаляли и реакционную смесь размешивали в течение ночи при комнатной температуре. Затем к смеси осторожно добавляли сульфатдекагидрат натрия до тех пор, пока не прекратится выделение газа. После 1-часового размешивания, смесь фильтровали через слой ЦелитаТМ, а фильтрат концентрировали. После перекристаллизации (из этилацетата/гексана) получали целевой спирт (20,67 г, 93%) в виде белого твердого продукта.
1Н-ЯМР (CDCl3) δ : 4,04 - 4,26 (м, 2H); 3,49 (д, 2H, J = 6,4 Гц); 2,70 (шир. с, 2H, J = 12,0 Гц); 1,6 - 1,73 (м, 3H); 1,47 (с, 9H); 1,15 (ддд, 2H, J = 23,2 Гц, J = 12,0 Гц, J = 4,3 Гц).
c) 4-Иодометил-1-пиперидинкарбоновая кислота, 1-(1,1-диметилэтил)сложный эфир
Трифенилфосфин (31,0 г, 0,119 М) добавляли к смеси иода (29,0 г, 0,114 М) в бензоле (1 л). Через 5 мин добавляли пиридин (18,5 мл, 0,228 М), а затем спирт, полученный в стадии (b) (20,5 г, 0,095 М). Полученную смесь нагревали с обратным холодильником в течение 1,5 ч. Затем смесь охлаждали, фильтровали, а фильтрат промывали насыщенным тиосульфатом натрия (Na2S2O3) и солевым раствором, осушали сульфатом магния, фильтровали и концентрировали. После очистки с помощью хроматографии на силикагеле (10% ___→ 20% этилацетата/гексана) получали целевой иодид (28,5 г, 92%) в виде прозрачного маслообразного продукта. Этот продукт охлаждали и получали белое твердое вещество. Т. пл. 58 - 59oC.
1Н-ЯМР (CDCl3) δ 4,09 (шир. д, 2H, J = 13,1 Гц); 3,08 (д, 2H, J = 6,5 Гц); 2,66 (шир. т; 2H, J = 13,1 Гц); 1,80 (шир. д., 2H, J = 12,9 Гц); 1,52 - 1,64 (м, 1H); 1,43 (с, 9H); 1,11 (ддд, 2H, J = 24,7 Гц); J = 12,7 Гц, J = 4,3 Гц).
d) 4-[2-[1,2-Бензизоксазол-3-ил] этил] -1-пиперидинкарбоновая кислота, 1-(1,1-диметилэтил)сложный эфир
Смесь 3-метил-1,2-бензизоксазола (0,410 г, 3,08 мМ) и иодида, полученного в стадии (c) (1,05 г, 3,23 мМ), в сухом ТГФ (3,2 мл), охлаждали до -78oC. Затем по капле добавляли свежеприготовленный 1М диизопропиламид лития (LDA) (3,1 мл, 3,1 мМ) и полученный желто-оранжевый раствор размешивали 25 мин при -78oC. После этого добавляли насыщенный хлорид аммония и смесь экстрагировали 3 раза этилацетатом. Объединенный органический слой промывали солевым раствором, а затем осушали сульфатом магния, фильтровали и концентрировали. После очистки с помощью флеш-хроматографии на силикагеле (10% ___→ 20% этилацетат/гексан) получали целевое соединение (0,430 г, 42%) в виде бесцветного маслообразного вещества.
1Н-ЯМР (CDCl3) δ 7,62 (д, 1H, J = 8,0 Гц); 7,49 - 7,55 (м, 2H); 7,25 - 7,31 (м, 1H); 4,09 (м, 2H); 3,00 (т, 2H, J = 7,8 Гц); 2,66 (шир. т, 2H, J = 13,0 Гц); 1,71 - 1,84 (м, 4H); 1,47 - 1,53 (м, 1H); 1,43 (с, 9H); 1,14 (ддд, 2H, J = 24,5 Гц, J = 12,1 Гц, J = 4,1 Гц).
e) 3-[2-[1-(Фенилметил)-4-пиперидинил]этил]-1,2-бензизоксазол-матеат
Трифторуксусную кислоту (ТФК) (7 мл) по капле добавляли к холодному (0oC) раствору пиперидина, полученного в стадии (d) (0,50 г, 1,51 мМ) в метиленхлориде (7 мл). Полученный раствор размешивали 30 мин при 0oC. Летучие вещества удаляли при пониженном давлении, а избыток ТФЕ удаляли путем концентрирования (2 раза) из толуола. Сырой продукт снова растворяли в метиленхлориде (10 мл), а затем добавляли триэтиламин (0,42 мл, 3,01 мМ) и бензилбромид (0,18 мл, 1,51 мМ). Полученную смесь размешивали в течение ночи (15 ч) при комнатной температуре. Смесь промывали водой и солевым раствором, а затем осушали сульфатом магния, фильтровали и концентрировали. После флеш-хроматографии на силикагеле (50% этилацетат/гексан) получали целевое соединение - свободное основание (0,350 г, 73%) в виде бесцветного маслообразного продукта.
Малеат (соль) получали путем добавления раствора малеиновой кислоты (0,108 г, 0,930 мМ) в этиловом эфире (20 мл) к раствору свободного основания (0,297 г, 0,926 мМ) в этиловом эфире (20 мл). Полученное твердое белое вещество собирали и промывали этиловым эфиром. Выход: 0,35 г, 87%. Т. пл. 146,4 - 147,6oC
EIMS (эмиссионная масс-спектроскопия) (не исходный) 319,1; 303,1; 185,2; 172,1; 91,1.
1Н-ЯМР (CDCl3) δ 7,60 (д, J = 8, 1H); 7,51 - 7,52 (м, 2H); 7,37-7,49 (м, 5H); 7,25-7,32 (м, 1H); 6,30 (с, 2H); 4,16 (с, 2H); 3,45-3,51 (м, 2H); 2,98 (т, J=7,4); (2H); 2,60-2,70 (м, 2H); 1,84-1,95 (м, 4H); 1,60-1,82 (м, 3H).
13С-ЯМР δ 169,5; 163,0; 157,7; 135,8; 131,1; 130,1; 130,0; 129,3; 128,5; 123,5; 121,3; 121,1; 110,0; 60,6; 52,1; 32,8; 28,8; 22,1.
ИК (KBr): 2944, 2927, 2921, 2499-2518 (шир.), 2329-2388 (шир.), 1583, 1517, 1473, 1458, 1445, 1438, 1383, 1360, 782 см-1
Анализ для C21H24N2O • C4H4O4:
Вычислено: C 68,79; H 6,47; N 6,42.
Найдено: C 68,80; H 6,35; N 6,27.
Пример 2.
5-Метил-3-[2-[1-(фенилметил)-4-пиперидинил]этил]1,2-бензизоксазолмалеат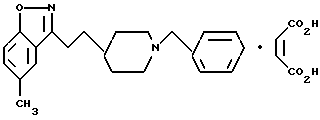
4-[2-[-5--Метил-1,2-бензизоксазол-3-ил] этил] -2-пиперидинкарбоновая кислота, 1-(1,1-диметилэтил)сложный эфир
Повторяли процедуру, описанную в примере 1d, с использованием 3,5-диметил-1,2-бензизоксазола (0,500 г, 3,40 мМ), 4-иодометил-1-пиперидинкарбоновой кислоты, 1-(1,1-диметилэтил)-сложного эфира (1,20 г, 3,74 мМ) и 1M диизопропиламида лития (LDA) (3,74 мл, 3,74 мМ) в сухом ТГФ (5 мл). После очистки получали целевое соединение (0,910 г, 78%) в виде прозрачного маслообразного продукта.
1H-ЯМР (CDCl3) δ 7,28-7,40 (м, 3H); 4,04-4,11 (м, 2H); 2,94 (т, 2H, J= 7,8 Гц); 2,64 (шир.т, 2H, J=12,3 Гц); 2,43 (с, 3H); 1,70-1,99 (м, 4H); 1,42 (с, 9H); 1,41-1,55 (м, 1H); 1,13 (ддд, 2H, J=24,4 Гц); J=12,0 Гц); J= 4,1 Гц).
о) 5-Метил-3-[2-[1-(фенилметил)-4-пиперидинил]этил]-1,2- бензизоксазолмалеат
Повторяли процедуру, описанную в примере Ie, с использованием пиперидина, полученного в стадии (а) (0,910 г, 2,64 мМ), и ТФК (13 мл) в метиленхлориде (CH2Cl2) (13 мл), и триетиламина (3,7 мл, 26,4 мМ), и бензилбромида (0,32 мл, 2,69 мМ) в метиленхлориде (CH2Cl2) (20 мл). После очистки получали целевое соединение - свободное основание в виде прозрачного маслообразного продукта (0,56 г, 63%).
Малеат (соль) получали путем добавления раствора малеиновой кислоты (0,20 г, 1,72 мМ) в этиловом эфире (Et2O) (10 мл) к раствору свободного основания (0,56 г, 1,67 мМ) в Et2O (40 мл). Образовавшееся белое твердое вещество собирали и промывали этиловым эфиром. Выход: 0,70 г, 93%. Т.пл. 149-151oC.
1H-ЯМР (CDCl3) δ 7,27-7,43 (м, H8); 6,32 (с, 2H); 4,17 (с, 2H); 3,51 (шир.д, J=11,6 Гц, 2H); 2,96 (т, J=7,3 Гц, 2H); 2,66 (шир.т, J=10,8 Гц, 2H); 2,45 (с, 3H); 1,60-1,97 (м, 7H).
13C-ЯМР δ : 169,4; 161,6; 157,3; 135,7; 133,2; 131,6; 131,0; 130,1; 129,3; 128,4; 121,4; 120,2; 60,6; 52,0; 32,8; 32,7; 28,7; 22,0; 21,1.
ИК (KBr) 2934; 2848; 2499; 2362; 1701; 1617; 1572; 1487; 1454; 1357 см-1.
EIMS (не исходный) 333,1; 317,2; 185,1; 172,1; 91,1 (основание).
Анализ для C22H26N2O • C4H4O4:
Вычислено: C 69,32; H 6,71; N 6,22.
Найдено: C 69,18; H 6,48; N 6,08.
Пример 3.
5,6-Диметил-3-[2-[1-(фенилметил)-4-пиперидинил] этил] -1,2- бензизоксазолмалеат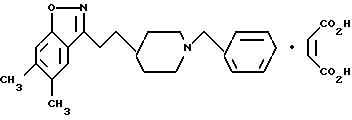
а) 4-[2-[5,6-Диметил-1,2-бензизоксазол-3-ил]этил]-1-пиперидинкарбоновая кислота, 1-(1,1-диметилэтил)сложный эфир
Повторяли процедуру, описанную в примере 1d, с использованием 3,5,6-триметил-1,2-бензизоксазола (0,600 г, 3,73 мМ), 4-иодометил-1-пиперидинкарбоновой кислоты, 1-(1,1-диметилэтил) сложного эфира (1,30, 4,10 мМ)) и 1M LDA (4,10 мл, 4,10 мМ) в сухом ТГФ (10 мл). После очистки получали целевое соединено (1,04 г, 78%) в виде прозрачного маслообразного вещества.
1H-ЯМР (CDCl3) δ 7,32 (с, 1H); 7,27 (с, 1H); 4,04-4,10 (м, 2H); 2,93 (т. , 2H, J= 7,8 Гц); 2,64 (шир.т. 2H, J=11,8 Гц); 2,35 (с, 3H); 2,32 (с, 3H); 1,70-1,80 (м, 4H); 1,43 (с, 9H); 1,43-1,51 (м, 1H); 1,13 (ддд, 2H, J= 24,3 Гц); J=12,3 Гц; J= 4,2 Гц).
о) 5,6-Диметил-3-[2-[1-(фенилметил)-4-пиперидинил] этил]-1,2- бензизоксазолмалеат
Повторяли процедуру, описанную в примере 1e, с использованием пиперидина, полученного в стадии (A) (1,04 г, 2,90 мМ), и ТФК (16 мл) в CH2Cl2 (16 мл), и триэтиламина (4,2 мл, 29,0 мМ), и бензилбромида (0,36 мл, 3,03 мМ) в CH2Cl2 (20 мл). После очистки получали целевое соединение (свободное основание) (0,53 г, 52%) в виде прозрачного маслообразного продукта.
Малеат (соль) получали путем добавления раствора малеиновой кислоты (0,18 г, 1,55 мМ) в Et2O (10 мл) к раствору свободного основания (0,53 г, 1,52 мМ) в Et2O (25 мл). Образовавшееся белое твердое вещество собирали и промывали этиловым эфиром.
Выход: 0,65 г, 92%. Т.пл. 182-183,5oC.
1H-ЯМР (CDCl3) δ 7,30-7,41 (м, 7H); 6,32 (с, 2H); 4,17 (с, 2H); 3,51 (шир. д, J=11,8 Гц; 2H); 2,95 (т, J=7,2, 2H); 2,65 (шир. т, J= 11,7 Гц, 2H); 2,38 (с, 3H); 2,34 (с, 3H); 1,59 - 1,96 (м, 7H).
13С-ЯМР δ 169,4; 162,3; 157,1; 140,2; 135,7; 132,6; 131,1; 130,1; 129,3; 128,3; 120,3; 119,2; 110,0; 60,6; 52,0; 32,7; 28,7; 22,0; 20,9; 29,9.
EIMS 347,2; 331,1; 185,1; 172,1; 91,1 (основание)
ИК (KBr) 2949; 2914; 2512; 2420; 1580; 1476; 1456; 1449; 1358 см-1.
Анализ для C23H28N2O • C4H4O4 • 1/4H2O:
Вычислено: C 69,14; H 6,93; N 5,97.
Найдено: C 69,27; H 6,83; N 5,91.
Пример 4.
5-Метокси-3-[2-[1-(фенилметил)-4-пиперидинил] этил] -1,2- бензизоксазолмалеат
а) 4-[2-[5-Метокси-1,2-бензизоксазол-3-ил] этил] -1-пиперидинкарбоновая кислота. 1-(1,1-диметилэтил)сложный эфир
Повторяли процедуру, описанную в примере 1d, с использованием 5-метокси-3-метил-1,2-бензизоксазола (0,32 г, 1,96 мМ) 4-иодометил-1-пиперидинкарбоновой кислоты, 1-(1,1-диметилэтил)сложного эфира (0,70 г, 2,15 мМ) и IM LDA (2,0 мл, 2,0 мМ) в сухом ТГФ (2 мл). После очистки получали целевое соединение (0,62 г, 87%) в виде прозрачного маслообразного продукта.
1Н-ЯМР (CDCl3) δ 7,46 (д, 1H, J = 9,1 Гц); 7,17 (дд, 1H, J = 9,1 Гц, J = 2,5 Гц); 6,96 (д, 1H, J = 2,4 Гц); 4,09-4,16 (м, 2H), 3,87 (c, 3H); 2,99 (т, 2H, J = 7,8 Гц); 2,68 (шир. т, 2H, J = 12,3 Гц); 1,74-1,85 (м, 4H); 1,46-1,64 (м, 1H); 1,46 (c, 9H); 1,17 (ддд, 2H, J = 22,3 Гц, J = 12,2 Гц; J = 4,2 Гц).
о) 5-Метокси-3-[2-[1-(фенилметил)-4-пиперидинил] этил]-1,2- бензизоксазолмалеат
Повторяли процедуру, описанную в примере Ie, с использованием пиперидина, полученного в стадии (а) (0,58 г, 1,61 мМ), и ТФК (7 мл) в CH2Cl2 (7 мл), и триэтиламина (0,50 мл, 3,6 мМ), и бензилбромида (0,195 мл, 1,64 мМ) в CH2Cl2 (10 мл). После очистки получали целевое соединение (свободное основание) (0,27 г, 48%) в виде прозрачного маслообразного продукта.
Малеат (соль) получали путем добавления раствора малеиновой кислоты (0,080 г, 0,69 мМ) в Et2O (10 мл) к раствору свободного основания (0,24 г, 0,68 мМ) в Et2O (20 мл). Образовавшееся белое твердое вещество собирали и промывали этиловым эфиром. Выход: 0,29 г, 91%, т.пл. 143,5-145oC.
1Н-ЯМР (CDCl3) δ 7,35-7,42 (м, 6H); 7,13 (дд, J1 = 9,1, J2 = 2,5, 1H); 6,92 (д, J = 2,4, 2H); 6,30 (c, 2H); 4,17 (c, 2H); 3,83 (c, 3H); 3,46-3,51 (м, 2H); 2,94 (т, J = 7,3, 2H); 2,60-2,80 (м, 2H); 1,60-1,96 (м, 7H).
13С-ЯМР δ 169,4; 158,4; 157,6; 156,3; 135,8; 131,0; 130,0; 129,2; 128,4; 121,5; 120,3; 110,6; 101,1; 60,5; 56,0; 52,0; 32,7; 32,5; 28,7; 22,0.
ИК (KBr) 2942, 2927, 2916, 2518, 2366, 1616, 1572, 1544, 1521, 1480, 1454, 1443, 1384, 1357, 1220 см-1.
EIMS: 349,2; 333,2; 318,1; 259,1; 185,1; 172,1; 91,1 (основание).
Анализ для C22H26N2O2 • C4H4O4:
Вычислено: C 66,94; H 6,48; N 6,00.
Найдено: C 67,21; H 6,52; N 5,94.
Пример 5.
6-Метокси-3-[2-[1-(фенилметил)-4-пиперидинил]этил]-1,2-бензизоксазол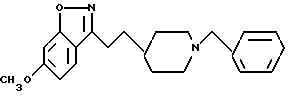
а) 4-[2-[6-Метокси-1,2-бензизоксазол-3-ил] этил] -1-пиперидинкарбоновая кислота, 1-(1,1-диметилэтил)сложный эфир
Повторяли процедуру, описанную в примере 1d, используя 6-метокси-3-метил-1,2-бензизоксазол (0,32 г, 1,96 мМ), 4-иодометил-1-пиперидинкарбоновой кислоты 1-(1,1-диметилэтил)сложного эфира (0,70 г, 2,15 мМ) и IM LDA (2,0 мл, 2,0 мМ) в сухом ТГФ (3 мл). После очистки получали целевое соединение (0,57 г, 80%) в виде белого твердого продукта. Т.пл. 95-96oC.
1Н-ЯМР (CDCl3) δ 7,47 (д, 1H, J = 8,7 Гц); 6,99 (д, 1H, J = 2,1 Гц); 6,91 (дд, 1H, J = 8,6 Гц, J = 2,1 Гц); 4,08-4,11 (м, 2H); 3,89 (c, 3H); 2,97 (т, 2H, J = 7,8 Гц); 2,68 (шир.т., 2H, J = 12,7 Гц); 1,72-1,84 (м, 4H); 1,46-1,60 (м, 1H); 1,46 (c, 9H); 1,16 (ддд, 2H, J = 24,6 Гц, J = 12,3 Гц; J = 4,3 Гц).
b) 6-Метокси-3-[2-[1-(фенилметил)-4-пиперидинил]этил]-1,2-бензизоксазол
Повторяли процедуру, описанную в примере 1e, используя пиперидин, полученный в стадии (а) (0,49 г, 1,36 мМ), и ТФК (7 мл) в CH2Cl2 (7 мл), и триэтиламин (0,85 мл, 6,1 мМ), и бензилбромид (0,165 мл, 1,39 мМ) в CH2Cl2 (8 мл). После очистки получали целевое соединение (0,265 г, 55%) в виде белого твердого вещества. Т.пл. 90,5-91,5oC.
1Н-ЯМР (CDCl3) δ 7,47 (д, J = 8,7; 1H); 7,21-7,33 (м, 5H); 6,98 (д, J = 1,8, 1H); 6,90 (дд, J1 = 8,7, J2 = 2,0, 1H); 3,88 (c, 3H); 3,50 (c, 2H); 2,88-2,98 (м, 4H); 1,96 (шир.т, J = 10,6, 2H); 1,74-1,83 (м, 4H); 1,27-1,34 (м, 3H).
13-ЯМР δ 164,8; 162,0; 158,3; 138,4; 129,3; 128,1; 126,9; 121,4; 115,0; 113,8; 92,6; 63,4; 55,7; 53,7; 35,3; 34,3; 32,6; 22,6.
ИК (KBr) 2924, 2913, 2797, 2758, 1625, 1608, 1276, 1196, 1154, 734 см-1.
EIMS 349,2; 333,6; 259,1; 185,1; 172,1; 91,0 (основание).
Анализ для C22H26N2O2:
Вычислено: C 75,40; H 7,8; N 7,99.
Найдено: C 75,52; H 7,63; N 7,94.
Пример 6.
7-Метокси-3-[2-[1-(фенилметил)-4-пиперидинил] этил] -1,2- бензизоксазолфумарат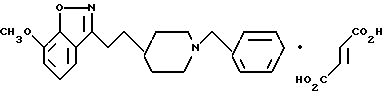
а) 4-[2-[7-Метокси-1,2-бензизоксазол-3-ил] этил-1-пиперидинкарбоновая кислота. 1-(1,1-диметилэтил)сложный эфир
Повторяли процедуру, описанную в примере Id, используя 7-метокси-3-метил-1,2-бензизиксазол (0,30 г, 1,84 мМ), 4-иодометил-1-пиперидинкарбоновой кислоты 1-(1,1-диметилэтил)сложный эфир (0,60 г, 1,85 мМ) и 1M LDA (1,9 мл, 1,9 мМ) в сухом ТГФ (2 мл). После очистки получали целевое соединение (0,41 г, 62%) в виде бледно-желтого маслообразного вещества.
1H-ЯМР (CDCl3) δ 7,12-7,10 (м, 2H); 6,91(дд, 1H, J=6,5 Гц, J=2,2 Гц); 3,98-4,07 (м, 2H); 3,98 (с, 3H); 2,95 (т, 2H, J=7,8 Гц); 2,62 (шир.т, 2H, J= 12,2 Гц); 1,67-1,78 (м, 4H); 1,40-1,48 (м, 1H); 1,40 (с, 9H); 1,10 (ддд, 2H, J=24,5 Гц, J=12,5 Гц, J=4,3 Гц).
b) 7-Метокси-3-[2-[1-(фенилметил)-4-пиперидинил]этил]-1,2- бензизоксазолфумарат
Повторяли процедуру, описанную в примере 1e, используя пиперидин, полученный в стадии (a) (0,40 г, 1,11 мМ), и ТФК (6 мл) в CH2Cl2 (6 мл), и триэтиламин (0,34 мл, 2,44 мМ), и бензилхлорид (0,14 мл, 1,18 мМ) в CH2Cl2 (140 мл). После очистки получали целевое соединение (свободное основание) (0,080 г, 21%) в виде прозрачного маслообразного продукта.
Фумаратную соль получали путем добавления раствора фумаровой кислоты (0,025 г, 0,213 мМ) в этаноле EtOH (2 мл) к раствору свободного основания (0,071 г, 0,203 мМ) в этиловом эфире (10 мл). После концентрирования до получения объема 4-5 мл образовывался беловато-розовый осадок. Этот твердый осадок собирали и промывали этиловым эфиром. Выход: 0,065 г, 69%.
Т.пл. 138-139oC.
1H-ЯМР (CDCl3) δ 7,27-7,41 (м, 7H); 7,19 (д, J=7,7, 1H); 6,59 (с, 2H); 3,97 (с, 3H); 3,65 (с. 2H); 2,90-3,01 (м, 4H); 2,18 (шир.т, J=10,8, 2H); 1,67-1,77 (м, 4H); 1,26-1,32 (м, 3H).
13 C-ЯМР δ 166,6; 158,8; 152,4; 144,0; 136,5; 134,4; 129,4; 128,3; 127,5; 124,9; 122,9; 113,2; 111,4; 61,5; 56,2; 52,6; 34,3; 33,4; 30,7; 22,0.
ВРМС: вычислено (свободное основание) 350,1992. Найдено: 350,1984.
ИК (KBr) 1705, 1531, 1266, 756, 642 см-1.
Анализ для C22H26N2 • C4H4O4:
Вычислено: C 66,94; H 6,48; N 6,00.
Найдено: C 66,76; H 6,31; N 5,61.
Пример 7.
6-Ацетамида-3-[2-[1-(фенилметил)-4-пиперидинил] этил]-1,2- бензизоксазол-гемифумарат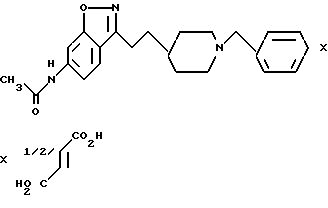
а) 4-[2-[6-Ацетамидо-1,2-бензизоксазол-3-ил] этил] пиперидикарбоновая кислота, 1-(1,1-диметилэтил) сложный эфир
Свежеприготовленный 1M LDA (11,0 мл, 11,0 мМ) по капле и быстро добавляли к охлажденному (-78oC) раствору 6-N-ацетил-3-метилбензизоксазола (1,0 г, 5,26 М) в ТГФ (50 мл). Сразу после завершения добавления одной порцией добавляли раствор 1-(1,1-диметилэтилового) сложного эфира 4-иодометил-1-пиперидинкарбоновой кислоты (1,71 г, 5,26 мМ) в ТГФ (8 мл). Затем добавляли насыщенный хлорид аммония (NH4Cl) и полученную смесь экстрагировали этилацетатом (EtOAc) (3 раза). Объединенный органический слой промывали солевым раствором, осушали сульфатом магния, фильтровали и концентрировали.
После очистки с помощью флеш-хроматографии на силикагеле (20% ___→ 50% EtOAc/CH2Cl2) получали целевое соединение (1,56 г, 76%) в виде белого твердого продукта. Т.пл. 142-143oC.
1H-ЯМР (CDCl3) (CH2Cl2 ____→ 10% 8,76 (с, 1H); 8,05 (с, 1H); 7,48 (д, 1H) J= 8,5 Гц); 7,32 (дд, 1H, J=8,6 Гц; J=1,5 Гц); 4,06 (шир. д, 2H, J=11,5 Гц); 2,94 (т, 2H, J= 7,8 Гц); 2,66 (шир.т, 2H, J=11,8 Гц); 2,20 (с, 3H); 1,69-1,80 (м, 4H); 1,41-1,47 (м, 1H); 1,44 (c, 9H); 1,12 (ддд, 2H, J=23,8 Гц; J=12,0 Гц, J=3,9 Гц).
b) 6-Ацетамидо-3-[2-[1-(фенилметил)-4-пиперидинил] этил]- 1,2-бензизоксазол-фумарат
Трифторуксусную кислоту (ТФК) (4 мл) по капле добавляли к холодному (0oC) раствору пиперидина, полученного в стадии (а) (0,40 г, 1,03 мМ) в CH2Cl2 (8 мл). Полученный раствор размешивали 30 мин при 0oC. Летучие вещества удаляли при пониженном давлении, а избыток ТФК удаляли путем концентрирования (2 раза) из толуола. Неочищенный продукт снова растворяли в метиленхлориде (10 мл) и добавляли триэтиламин (1,44 мл, 10,3 мМ) и бензилбромид (0,184 мл, 1,55 мМ). Полученную смесь размещали 6 ч при комнатной температуре. Затем смесь промывали водой и солевым раствором и осушали сульфатом магния, фильтровали и концентрировали. После очистки с помощью флеш-хроматографии на силикагеле δ MeOH/CH2Cl2) получали целевое соединение (свободное основание) (0,270 г, 69%) в виде белого твердого вещества.
Фумаратную соль получали путем добавления раствора фумаровой кислоты (0,091 г, 0,788 мМ) в этаноле (EtOH) (5 мл) к раствору свободного основания (0,270 г, 0,716 мМ) в CH2Cl2 (20 мл). После концентрирования полученный твердый продукт перекристаллизовывали из этанола и получали белые игольчатые кристаллы.
Выход: 0,17 г, 48%. Т.пл. 225-226oC.
1H-ЯМР (DМCO-d6) 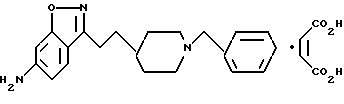 10,37 (с, 1H); 8,13 (c, 1H); 7,76 (д, 1H, J=8,5); 7,25-7,36 (м, 6H); 6,59 (c, 2H); 3,54 (c, 2H); 2,83-2,96 (м, 4H); 2,10 (c, 3H); 2,01 (шир.т, 2H, J=11,1); 1,69-1,73 (м, 4H); 1,20-1,28 (м, 3H).
10,37 (с, 1H); 8,13 (c, 1H); 7,76 (д, 1H, J=8,5); 7,25-7,36 (м, 6H); 6,59 (c, 2H); 3,54 (c, 2H); 2,83-2,96 (м, 4H); 2,10 (c, 3H); 2,01 (шир.т, 2H, J=11,1); 1,69-1,73 (м, 4H); 1,20-1,28 (м, 3H).
Анализ для C23H27N3O2 • 1/2 C4H4O4 • 1/4H2O:
Вычислено: C 68,24; H 6,76; N 9,55.
Найдено: C 68,35; H 6,63; N 9,35.
Пример 8.
6-Амино-3-[2-[1-(фенилметил)-4-пиперидинил] этил] -1,2-бензизоксазол- малеат
δ
Смесь 6-ацетато-3-[2-[1-(фенилметил)-4-пиперидинил] этил] - 1,2-бензизоксазола (0,30 г, 0,79 мМ) в 1н. HCl (10 мл) нагревали с обратным холодильником в течение 30 мин. Охлажденную реакционную смесь подщелачивали путем добавления 10% NaOH и экстрагировали 2 раза этилацетатом. Объединенный органический слой промывали солевым раствором и осушали сульфатом магния, затем фильтровали, концентрировали и получали целевое соединение - свободное основание (0,259 г, 98%) в виде маслообразного вещества.
Мономалеатную соль получали путем добавления раствора малеиновой кислоты (0,099 г, 0,85 мМ) в EtOH (5 мл) к раствору свободного основания (0,26 г, 0,77 мМ) в CH2Cl2 (3 мл). После концентрирования остаток растирали с этиловым эфиром и получали белый порошок. Выход: 0,29 г, 64%. Т.пл. 173,0-173,5oC
1H-ЯМР (DMCO-d6) 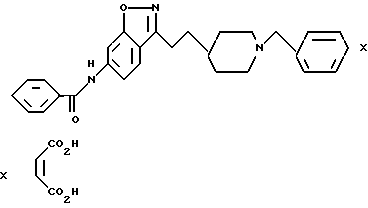 7,41-7,47 (м. 6H); 6,58-6,63 (м, 2H); 6,06 (c, 2H); 5,87 (шир.с, 2H); 4,26 (c, 2H); 3,29-3,38 (м, 2H); 2,80-2,95 (м, 4H); 1,90 (шир.д, J=12,5, 2H); 1,25-1,80 (м, 5H).
7,41-7,47 (м. 6H); 6,58-6,63 (м, 2H); 6,06 (c, 2H); 5,87 (шир.с, 2H); 4,26 (c, 2H); 3,29-3,38 (м, 2H); 2,80-2,95 (м, 4H); 1,90 (шир.д, J=12,5, 2H); 1,25-1,80 (м, 5H).
13C-ЯМР (DMCO-d6) 167,4; 164,8; 157,4; 151,9; 136,0; 131,2; 130,1; 129,5; 128,9; 121,9; 112,8; 110,7; 90,9; 59,3; 51,5; 32,6; 32,6; 28,6; 21,6.
EIMS (не исходн.) 289, 268, 218, 190 (основание).
ИК (KBr) 3483, 3384, 2929, 2526, 1633, 1619, 1582, 1515, 1474, 1459, 1438, 1389, 1379, 1359, 977, 702 см-1.
Анализ для C21H25N2O • C4H4O4:
Вычислено: C 66,50; H 6,47; N 9,31.
Найдено: C 66,49; H 6,43; N 9,22.
Пример 9.
6-Бензамид-3-[2-[1-(фенилметил)-4-пиперидинил] этил] -1,2 -бензизоксазол-малеат
δ
6-Бензамид-3-метил-1,2-бензизоксазол
Бензоилхлорид (0,56 мл, 4,82 мМ) добавляли к раствору 6-амино-3-[2-[1-(фенилметил)-4-пиперидинил]этил]-1,2-бензизоксазола (0,70 г, 4,72 мМ, триэтиламина (1,35, 9,69 мМ) и 4-диметиламинопиридина (0,07 г, 0,57 мМ) в CH2Cl2 (30 мл). Полученную смесь размешивали при комнатной температуре в течение ночи. Эту гетерогенную смесь концентрировали, а полученный твердый остаток собирали, промывали водой и эфиром и осушали воздухом, в результате чего получали целевое соединение (1,02 г, 86%) в виде беловатого твердого вещества. Небольшой образец очищали путем перекристаллизации из этанола и получали чистый хлопьевидный продукт.
Т.п. 213-214oC.
1H-ЯМР (DMCO-d6) (30% ___→ 50% 10,6 (с, 1H); 8,30 (с, 1H); 7,98 (д, 2H, J= 6,9 Гц); 7,80 (д, 1H, J= 8,6 Гц); 7,68 (д, 1H, J=8,8 Гц); 7,52-7,63 (м, 3H); 2,53 (с, 3H).
b) 4-[2-[6-Бензамид-1,2-бензизоксазол-3-ил]этил]-1 -пиперидинкарбоновая кислота. 1-(1,1-диметилэтил)сложный эфир
Повторяли процедуру, описанную в примере 7а, используя бензамид, полученный в стадии (a) (1,0 г, 3,96 мМ), IM LDA (7,95 мл, 7,95 мМ) и 4-иодометил-1-пиперидинкарбоновой кислоты 1-(1,1-диметилэтил)сложный эфир (1,30 г, 4,00 мМ) в сухом ТГФ (50 мл), за исключением того, что после добавления реагентов смесь размешивали при -78oC в течение 1,5 ч. После очистки путем хроматографии δ EtOAc/гексан) получали целевое соединение (1,54 г, 87%) в виде бледно-желтого твердого вещества. Небольшой образец очищали путем перекристаллизации (CH2Cl2/Et2O) и получали белое твердое вещество. Т.п. 177-178,5oC.
1H-ЯМР (CDCl3) δ 8,61 (c, 1H); 8,15 (c, 1H); 7,86 (д, 2H, J=7,4 Гц); 7,39-7,53 (м, 5H); 4,03 (шир.д, 2H, J=12,7 Гц); 2,92 (т, 2H, J= 8,0 Гц); 2,50-2,73 (м, 2H); 1,60-1,80 (м, 4H); 1,40-1,45 (м, 1H); 1,41 (c, 9H); 1,13 (ддд, 2H, J=24,0 Гц); J=12,2 Гц; J=3,8 Гц).
c) 6- Бензамид-3-[2-[1-(фенилметил)-4-пиперидинил]этил] -1,2-бензизоксазолмалеат \\\ Трифторуксусную кислоту (ТФК) (8,4 мл) по капле добавляли к холодному раствору (0oC) пиперидина, полученного в стадии (b) (0,70 г, 1,56 мМ) в CH2Cl2 (10 мл). Полученный раствор размешивали 30 мин при 0oC. Летучие вещества удаляли при пониженном давлении, а избытое ТФК удаляли путем концентрирования (2 раза) из толуола. Сырой продукт растворяли в ТГФ 10 мл и добавляли триэтиламин (2,1 мл, 15,1 мМ), а затем бензилбромид (0,21 мл, 1,77 мМ). Полученную смесь разбавляли этилацетатом и размешивали при комнатной температуре в течение ночи. Затем смесь промывали водой и солевым раствором, осушали сульфатом магния, фильтровали и концентрировали. После очистки с помощью флеш-хроматографии на силикагеле (30% EtOAc/CH2Cl2 100% EtOAc) получали целевое соединение - свободное основание (0,280 г, 41%) в виде бледно-желтого твердого вещества.
Малеат (соль) получали путем добавления раствора малеиновой кислоты (0,081 г, 0,702 мМ) в EtOH (5 мл) к раствору свободного основания (0,280 г, 0,638 мМ) в CH2Cl2 (20 мл). После концентрирования полученный твердый продукт перекристаллизовывали из EtOH/CH2Cl2 и получали белое твердое вещество. Выход: 0,208 г, 59%. Т.пл. 181,5-183,0oC.
1H-ЯМР (DMCO-d6) δ 10,65 (с, 1H); 8,31 (с, 1H); 7,98 (д, J=7,2, 2H); 7,85 (д, J= 8,6, 1H); 7,48-7,71 (м, 9H); 6,03 (c, 2H); 4,25 (шир.с, 2H); 3,20-3,60 (м, 4H); 2,89-3,02 (т, при 2,99 J=7,5 и м, 4H); 1,40-1,97 (м, 7H).
13C-ЯМР (DMCO-d6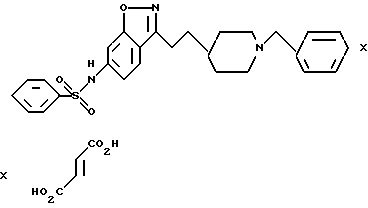 167,2; 166,2; 163,0; 158,1; 141,5; 135,9; 134,6; 132,0; 131,2; 129,5; 128,9; 128,5; 127,8; 121,9; 117,01; 116,8; 99,9; 59,5; 51,7; 32,8; 32,5; 28,8; 21,7.
167,2; 166,2; 163,0; 158,1; 141,5; 135,9; 134,6; 132,0; 131,2; 129,5; 128,9; 128,5; 127,8; 121,9; 117,01; 116,8; 99,9; 59,5; 51,7; 32,8; 32,5; 28,8; 21,7.
EIMS 439,2; 422,2; 100; 383; 348; 293; 185.
ИК (KBr): 2934, 2919, 1657, 1610, 1579, 1536, 1499, 1491, 1462, 1453, 1352 см-1.
Анализ для C28H29N3O2 • C4H4O4:
Вычислено: C 69,17; H 5,99; N 7,56.
Найдено: C 68,81; H 5,90; N 7,49.
Пример 10.
6-Бензолсульфонамид-3-[2-[1-(фенилметил)-4-пиперидинил] этил] - 1,2-бензизоксазол-фумарат
δ
a) 6-Бензолсульфонамид-3-метил-1,2-бензизоксазол
Бензолсульфонилхлорид (0,528 мл, 4,14 мМ) добавляли к холодному (0oC) раствору 6-амино-3-[2-[1-(фенилметил)-4-пиперидинил] этил] -1,2-бензизоксазола (0,613 г, 4,14 мМ) и пиридина (0,670 мл, 8,28 мМ) в CH2Cl2 (30 мл). Через 1,3 ч добавляли насыщенный бикарбонат натрия (NaHCO3), полученную смесь размешивали при комнатной температуре в течение ночи. Органический слой отделяли и промывали водой и солевым раствором, осушали сульфатом магния, фильтровали и концентрировали. После очистки флеш-хроматографии на силикагеле (5% EtOAc/CH2Cl2) получали целевое соединение (0,867 г, 83%) в виде белого твердого вещества. Т.пл. 183-184oC.
1H-ЯМР (CDCl3) (20% ___→ 40% 10,9 (шир.с, 1H); 7,84 (д, 2H, J=6,7 Гц); 7,68(д, 1H, J=8,5 Гц); 7,52-7,63 (м, 3H); 7,34 (д, 1H, J=1,5 Гц); 7,11 (дд, 1H, J=8,5 Гц, J=1,7 Гц); 2,52 (с, 3H).
(b) 4-2-6-Бензолсульфонамид-1,2-бензизоксазол-3-ил-этил-1- пиперидикарбоновая кислота, 1-(1,1-диметилэтил)сложный эфир
Повторяли процедуру, описанную в примере 7a, используя бензолсульфонамид, полученный в стадии (a) (0,60 г, 2,08 мМ), IM LDA (4,58 мл, 4,58 мМ) и 1-(1-диметилэтил) сложный эфир 4-иодометил-1-пиперидинкарбоновой кислоты (0,813 г, 2,50 мМ) в сухом ТГФ (70 мл), за исключением того, что после добавления реагентов смесь размешивали при -78oC в течение 10-15 мин. После очистки с помощью хроматографии δ EtOAc/гексаны) получали целевое соединение (0,997 г, 99%) в виде белого пенистого вещества. Т.пл. 66-67oC.
1H-ЯМР (CDCl3) (CH2Cl2 ____→ 5% 7,85 (дд, 2H, J=8,3 Гц, J=1,6 Гц); 7,35 (-7,57 (м, 6H); 7,02 (дд, 1H, J=8,5 Гц, J=1,6 Гц); 4,11 (шир.д, 2H, J=13,2 Гц); 2,94 (т, 2H, J=7,4 Гц); 2,68 (шир.т, 2H, J=12,8 Гц); 1,71-1,77 (м, 4H); 1,46 (c, 9H); 1,46-1,55 (м, 1H); 1,15 (ддд, 2H, J=23,6 Гц, J=11,7 Гц, J=3,9 Гц).
c) 6-Бензолсульфонамид-3-[2-[1-(фенилметил)-4-пиперидинил]этил] -1,2-бензизоксазол-фумарат
Триметилсилилтрифторометансульфонат (1,30 мл, 6,76 мМ) по капле добавляли к холодному (0oC) раствору пиперидина, полученного в стадии (b) (0,819 г, 1,69 мМ), и 2,6-лутидина (0,590 мл, 5,07 мМ) в CH2Cl2 (17 мл). Через полтора часа добавляли насыщенный бикарбонат натрия (NaHCO3) и полученную смесь размешивали 15 мин при комнатной температуре. Образовавшийся белый осадок собирали путем фильтрации и снова растворяли в воде при pH 2. Этот кислый водный слой экстрагировали 2 раза метиленхлоридом и 1 раз этилацетатом. Все органические слои объединяли, осушали сульфатом магния, фильтровали и концентрировали. Полученный белый остаток суспендировали в ТГФ (30 мл) и ДФМ (50 мл) и добавляли триэтиламин (0,40 мл, 2,86 мМ) и бензилбромид (0,22 мл, 1,86 мМ). Полученную гетерогенную смесь размешивали при комнатной температуре в течение 24 ч (время получения более гомогенной смеси). Эту смесь концентрировали и к остатку добавляли CH2Cl2. Органический слой промывали водой и солевым раствором, осушали сульфатом магния, фильтровали и концентрировали. После очистки с помощью флеш-хроматографии на силикагеле δ MeOH/CH2Cl2) получали целевое соединение (свободное соединение) (0,334 г, 49%) в виде белого пенистого вещества.
Фумарат (соль) получали путем добавления раствора фумаровой кислоты (0,040 г, 0,345 мМ) в EtOH ( 3 мл) к раствору свободного основания (0,150 г, 0,315 мМ) в CH2Cl2 (6 мл). После концентрирования остаток растирали с этиловым эфиром и получали белое твердое вещество. Выход: 0,151 г, 81%.
1H-ЯМР (DMCO-d6) 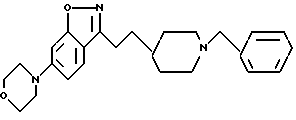 7,82 (д, 1H, J= 7,0); 7,70 (д, 1H, J = 8,6); 7,51 - 7,60 (м, 3H); 7,27 - 7,32 (м, 6H); 7,07 (дд, 1H, J= 8,6, J=1,6); 6,59 (с, 2H); 5,55 (с, 2H); 2,82 - 2,90 (м, 4H); 20,3 (шир, т. 2H, J=11,5); 1,61 - 1,70 (м, 4H); 1,20 - 1,25 (м, 3H).
7,82 (д, 1H, J= 7,0); 7,70 (д, 1H, J = 8,6); 7,51 - 7,60 (м, 3H); 7,27 - 7,32 (м, 6H); 7,07 (дд, 1H, J= 8,6, J=1,6); 6,59 (с, 2H); 5,55 (с, 2H); 2,82 - 2,90 (м, 4H); 20,3 (шир, т. 2H, J=11,5); 1,61 - 1,70 (м, 4H); 1,20 - 1,25 (м, 3H).
Пример 11.
6-(4-морфолинил)-3-[2-[1-(фенилметил)-4-пиперидинил] этил] -1,2-бензизоксазол
,β,β′
a) 3-Метил-6-(4-морфолинил)-1,2-бензизоксазол
Смесь 6-амино-3-[2-[1-(фенилметил)-4-пиперидинил]этил]1,2-бензизоксазола (0,230 г, 1,55 мМ) δ дибромодиэтилового эфира (0,397 г, 171 мМ) и диизопропилэтиламина (основания Хюнига, 0,648 мл, 3,71 мМ) в толуоле (2,5 мл) нагревали 15 ч при 120oC. Охлажденную смесь разбавляли этилацетатом и промывали водой м солевым раствором, затем осушали сульфатом магния, фильтровали и концентрировали. После этого осуществляли таким же способом еще две отдельные реакции с использованием 6-амино-3-[2-[1-(фенилметил)-4-пиперидинил] этил]-1,2- бензизоксазола (0,050 г, 0,34 мМ и 0,150 г, 1,01 мМ). Неочищенный продукт, полученный из трех реакций, объединяли и очищали с помощью флеш-хроматоографии на силикагеле (1% MeOH/CH2Cl2), в результате чего получали целевое соединение (0,499 г, 79%) полный выход в виде бледно-желтого твердого вещества. Затем очищали небольшой образец путем перекристаллизации (EtOAc/гексаны) и получали белое твердое вещество. Т.пл. 138,5 - 139,5oC.
1H-ЯМР (CDCl3) (5% ___→ 40% 7,46 (д, 1H, J=8,7 Гц); 6,95 (дд, 1H, J=8,7 Гц, J=2,0 Гц); 6,90 (д, 1H, J=1,9 Гц); 3,88 (т. 4H, J=4,9 Гц); 3,27 (т, 4H, J=4,8 Гц); 2,52 (с, 3H).
b) 4[2-[6-(4-Морфолинил)-1,2-бензизоксазол-3-ил] этил]-1- пиперидинкарбоновая кислота. 1-(1,1-диметилэтил) сложный эфир
Повторяли процедуру, описанную в примере 7a, используя морфолино-производное, полученное в стадии (a) (0369 г, 1,69 мМ), 1M LDA (1,86 мл, 1,86 мМ), и 4-иодометил-1-пиперидинкарбоновой кислоты 1-(1,1-диметилэтил) сложный эфир (0,605 г, 1,86 мМ) в сухом ТГФ (8 мл). Осуществляли также еще одну реакцию с вышеуказанным морфолино-производным (0,100 г, 0,46 мМ). Сырой продукт, полученный из этих двух реакций, объединяли и после очистки путем хроматографии EtOAc/гексаны) получали целевое соединение (0,477 г, 53%) в виде белого твердого продукта. Затем небольшой образец очищали путем перекристаллизации (EtOAc/гексаны) и получали белое твердое вещество.
Т.пл. 164 - 165oC.
1H-ЯМР (CDCl3) δ 7,46 (д, 1H, J=8,6 Гц); 6,92 - 6,97 (м, 2H); 4,02 - 4,15 (м, 1H); 3,89 (т, 4H, J=4,9 Гц); 3,27 (т, 4H, J=4,9 Гц); 2,95 (т, 2H, J=7,8 Гц); 2,7 (шир. т, 2H, J=12,1 Гц); 1,74 - 1,80 (м, 4H); 1,46 - 1,56 (м, 1H); 1,46 (с, 9H); 1,10 - 1,22 (м, 2H).
c) 6-(4-Морфолинил)-3-[2-[1-(фенилметил)-4-пиперидинил]этил -1,2-бензизоксазол
Трифторуксусную кислоту (ТФК) (5 мл) добавляли к холодному (0oC) раствору пиперидина, полученного в стадии (b) (0,40 г, 0,96 мМ), и тианизола (1,13 мл, 9,60 мМ) в CH2Cl2 (10 мл). Полученную смесь размешивали 30 мин при 0oC. Летучие вещества удаляли при пониженном давлении и к остатку добавляли 1н. NaOH. Водный слой экстрагировали этилацетатом ( 2 раза) и объединенный органический слой промывали водой и солевым раствором, осушали сульфатом магния, фильтровали и концентрировали. Полученное желтое маслообразное вещество снова растворяли в метиленхлориде (10 мл) и к раствору добавляли триэтиламин (0,267 мл, 1,92 мМ) и бензилбромид (0,148 мл, 11,25 мМ). Полученную смесь размешивали при комнатной температуре в течение ночи. После этого смесь промывали водой и солевым раствором, осушали сульфатом магния, фильтровали и концентрировали. После очистки с помощью флеш-хроматографии на силикагеле (CH2Cl2 ____→ 10% MeOH/CH2Cl2), получали целевое соединение (0,285 г, 73%) в виде белого твердого продукта. Затем небольшой образец очищали путем перекристаллизации из этанола и получали белое твердое вещество. Т.пл. 129 - 130oC.
1H-ЯМР (CDCl3) δ 7,44 (д, J=8,7, 1H); 7,23 - 7,30 (м, 5H); 6,89 - 6,93 (м, 2H); 3,87 (т, J=4,8, 4H); 3,47 (с, 2H); 3,25 (т, J=4,8, 4H); 2,85 - 2,94 (м, 4H); 1,92 (шир. т. J=10,8, 2H); 1,72 - 1,79 (м, 4H); 1,21 - 14,31 (м, 3H).
13C-ЯМР: 165, 158, 153,4; 138,8; 129,2; 128,1; 126,9; 121,3; 113,3; 94,5; 66,7; 63,5; 53,7; 49,1; 35,3; 34,4; 32,1; 22,7.
ИК (KBr): 3020, 2920, 1950, 1895, 1815, 1722, 1620, 1450, 1250, 1122, 738 см-1.
Анализ для C25H31N3O2:
Вычислено: C 74,04; H 7,70; N 10,36.
Найдено: C 73,75; H 7,69; N 10,36.
Пример 12.
5,7-Дигидро-3-[2-[1-(фенилметил)-4-пиперидинил] этил] -6H- пирроодо[4-, 5-f]-1,2-бензизоксазол-6-он-малеат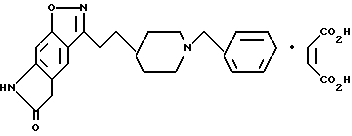 a) 5-Ацетил-1,3-дигидро-6-гидрокси-2H-индол-2-он
a) 5-Ацетил-1,3-дигидро-6-гидрокси-2H-индол-2-он
Ацетилхлорид (4,09 мл, 0,0575 М) добавляли к суспензии трихлорида алюминия (AlCl3) (35,36 г, 0,265 М) в сероуглероде (CS2) (250 мл). Через 2 - 3 мин добавляли 6-метоксиоксиндол (7,22 г, 0,0442 М). Полученную смесь нагревали с обратным холодильником в течение 2,5 ч (со временем смесь становилась в виде черного дегтя). Избыток растворителя декантировали, и к остатку осторожно добавляли ледяную воду. Полученную смесь размешивали в течение ночи. Полученное твердое бледно-желтое вещество собирали, промывали водой, осушали в высоком вакууме и получали целевое соединение (7,32 г, 87%).
1H-ЯМР (DMCO-d6) δ 13,0 (с, 1H); 10,8 (с, 1H); 7,70 (с, 1H); 6,30 (с, 1H); 3,40 (с, 2H); 2,54 (с, 3H).
b) 5-Ацетил-1,3-дигидро-6-гидрокси-2H-индол-2-он, 5-оксим
Водный раствор гидроксиламина-гидрохлорида (8,26 г, 0,119 М) и тригидрат ацетата натрия (16,9 г, 0,124 М) добавляли к смеси кетона, полученного в стадии (a) (9,88 г, 0,0517 M) в EtOH(600 мл). Полученную смесь нагревали с обратным холодильником в течение 20 ч. Горячую реакционную смесь фильтровали, твердый остаток собирали и промывали этанолом. После сушки получали целевое соединение (10,11 г, 95%) в виде бледно-желтого вещества.
1H-ЯМР (DMCO-d6) δ 12,0 (с, 1H); 11,4 (с, 1H); 10,5 (с, 1H); 7,29 (с, 1H); 6,35 (с, 1H); 3,38 (с, 2H); 2,20 (с, 3H).
c) 5-Ацетил-1,3-дигидро-6-гидрокси-2-индол-2-он, 5-оксимацетат
Гетерогенную смесь оксима, полученного в стадии (b) (7,15 г, 34,7 мМ), в уксусной ангидриде (55 мл) нагревали в течение 2 ч при 80oC. Охлажденную реакционную смесь фильтровали, а собранное твердое вещество промывали водой. После осушки получали целевое соединение (4,67 г, 54%) в виде бледно-желтого твердого вещества.
1H-ЯМР (DMCO-d6) δ 11,3 (С, 1H); 10,6 (с, 1H); 7,35 (с, 1H); 6,44 (с, 1H); 3,41 (с, 2H); 2,37 (с, 3H), 2,21 (с, 3H).
d) 5,7-Дигидро-3-метил-6H-пирроло[4,5-f]-1,2-бензизоксазол-6-он
Смесь оксимацетата, полученного в стадии с (4,48 г, 18,0 мМ), и пиридина (14,6 мл, 180 мМ) в диметилформамиде (ДМФ) (660 мл) нагревали 4 ч при 125 - 130oC. Охлажденную реакционную смесь выливали на воду и экстрагировали 4 раза этилацетатом. Объединенный органический слой промывали водой и солевым раствором, осушали сульфатом магния, фильтровали и концентрировали. После очистки с помощью флеш-хроматографии на силикагеле (50% EtOAc/ гексаны ____→ 100% EtOAc) получали целевое соединение (2,20 г, 65%) в виде бледно-желто-оранжевого твердого продукта. Т.пл. EtOAc 264 - 265oC. (разл.).
1H-ЯМР (DMCO-d6) δ 10,8 (с, 1H); 7,60 (с, 1H); 6,98 (с, 1H); 3,57 (с, 2H); 2,47 (с, 3H).
e) 4-[2-[5,7-Дигидро-6H-пирроло[4,5-f] -1,2-бензизоксазол-6-он-3-ил] этил]-1-пиперидинкарбоновая кислота. 1-(1,1-диметил)этил сложный эфир
Повторяли процедуру, описанную в примере 7a, используя бензизоксазол, полученный в стадии d (2,33 г, 12,4 мМ), IM LDA (40,9 мл, 40,9 мМ), и 4-иодометил-1-пиперидин-карбоновой кислоты 1-(1,1-диметилэтил) сложный эфир (4,42 г, 13,6 мМ) в сухом ТГФ (500 мл), за исключением того, что после добавления реагентов смесь размешивали 4 ч при -78oC. После очистки с помощью хроматографии ( 20% ____→ 30% EtOAc/CH2Cl2) получали исходный материал (0,210 г, 9%) и целевое соединение (2,75 г, 58%) в виде беловатого твердого вещества.
1H-ЯМР (CDCl3) δ 8,48 (с, 1H); 7,44 (с, 1H); 7,03 (с, 1H); 4,08-4,14 (м, 2H); 3,63 (с, 2H); 2,97 (т, 2H, J=7,8 Гц); 2,69 (шир.т, 2H, J=12,8 Гц); 1,74-1,84 (м, 4H); 1,46-1,55 (м, 1H); 1,46 (с, 9H); 1,18 (ддд, 2H, J=24,4 Гц; J=12,1 Гц, J=4,3 Гц).
f) 5,7-Дигидро-3-[2-[1-(фенилметил)-4-пиперидинил] этил] - 6H-пирроло[4,5-f]-1,2-бензизоксазол-6-он-малеат
Трифторуксусную кислоту (ТФК) (3,3 мл) по капле добавляли к холодному раствору (0oC) пиперидина, полученного в стадии (e) (0,50 г, 1,30 мМ) в CH2Cl2 (13 мл). Через 30 мин смесь концентрировали и избыток ТФК удаляли путем концентрирования из толуола (2-3 раза). Неочищенный остаток растворяли в ДМФ (12,5 мл) и добавляли карбонат натрия (Na2CO3) (0,689 г, 6,50 мМ) и бензилхлорид (0,186 мл, 1,56 мМ). Полученную смесь размешивали 4 ч при комнатной температуре. Реакционную смесь фильтровали и фильтрат концентрировали в вакууме. Остаток растворяли в метиленхлориде, промывали солевым раствором, осушали сульфатом магния, фильтровали и концентрировали. После очистки с помощью хроматографии на силикагеле (CH2Cl2 10% MeOH/CH2Cl2) получали целевое соединение (свободное основание) (0,343 г, 70%) в виде белого твердого вещества.
Малеат (соль) получали путем добавления раствора малеиновой кислоты (0,061, 0,528 мМ) в этаноле (EtOH) (1 мл) к раствору свободного основания (0,180 г, 0,48 мМ) в CH2Cl2 (10 мл). После концентрирования соль очищали путем перекристаллизации из изопропанола, в результате чего получали беловатое твердое вещество. Выход: 0,173 г, 73% Т.пл. 194-195oC.
1H-ЯМР (DMCO-d6) δ 10,82 (с, 1H); 7,65 (с, 1H); 7,48 (с, 5H); 7,00 (с, 1H); 6,03 (с, 1H); 4,24 (шир.с, 2H); 3,58 (с, 2H); 3,25-3,38 (м, 2H); 2,94 (т, 2H, J=7,6 Гц); 2,82-2,97 (м, 2H); 1,86-1,96 (м, 2H); 1,62=1,76 (м, 2H); 1,30-1,60 (м, 3H).
Анализ для C23H25N3O2• C4H4O4:
Вычислено: C 65,97; H 5,95; N 8,55.
Найдено: C 65,98; H 6,04; N 8,54.
Пример 13.
1-[2-[1-(фенилметил)-4-пиперидинил]изохинолинмалеат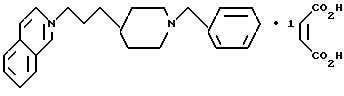
a) 4-[2-[1-Изонинолин] этил] -1-пиперидинкарбоновая кислота. 1-(1,1-диметилэтил)сложный эфир
Повторяли процедуру, описанную в примере 7a, используя 1-метилизохинолин (0,50 г, 3,49 мМ), 1М LDA (4,2 мл, 4,2 мМ) и 1-(1,1-диметилэтил)сложный эфир 4-иодометил-1-пиперидинкарбоновой кислоты (1,2 г, 3,84 мМ) в сухом ТГФ (45 мл), за исключением того, что после добавления реагентов смесь размешивали 1,75 ч при -78oC. После очистки с помощью хроматографии ( 30% ____→ EtOAc/толуол) получали целевое соединение (0,784 г, 66%) в виде желтого маслообразного вещества.
1H-ЯМР (CDCl3) δ 8,38 (д, 1H, J=5,8 Гц); 8,08 (д, 1H, J=8,3 Гц); 7,77 (д, 1H, J=8,0 Гц); 7,62 (т, 1H, J=7,1 Гц); 7,54 (т, 1H, J=7,1 Гц); 7,46 (д, 1H, J=5,7 Гц); 4,08 (шир.с, 2H); 3,25-3,30 (м, 2H); 2,67 (шир.т, 2H, J=12,3 Гц); 1,73-1,81 (м, 4H); 1,49-1,63 (м, 1H); 1,42 (с, 9H); 1,17 (ддд, 2H, J= 24,6 Гц, J=12,1 Гц, J=3,8 Гц).
b) 1-[2-[1-(фенилметил)-4-пиперидинил]этил]изохинолинмалеат
Повторяли процедуру, описанную в примере 1e, используя пиперидин, полученный в стадии (a) (0,713 г, 2,10 мМ), и ТФК (7 мл) в CH2Cl2 (14 мл), и триэтиламин (2,9 мл, 21,0 мМ), и бензилбромид (0,275 мл, 2,31 мМ) в CH2Cl2 (60 мл). После очистки ( CH2Cl2 _____→ 5% MeOH/CH2Cl2) получали целевое соединение (свободное основание) (0,26 г, 38%) в виде бледно-желтого маслообразного вещества.
Малеат (соль) получали путем добавления раствора малеиновой кислоты (0,10 г, 0,867 мМ) в EtOH (3 мл) к раствору свободного основания (0,26 г, 0,788 мМ) в CH2Cl2 (7 мл). После концентрирования соль очищали путем перекристаллизации из холодного (0oC) этилацетата и получали беловатое твердое вещество.
Выход: 0,195 г, 56%.
1H-ЯМР (DMCO-d6) δ 8,39 (д, 1H, J=5,7 Гц); 8,26 (д, 1H, J=8,3 Гц); 7,97 (д, 1H, J=8,1 Гц); 7,77 (т, 1H, J=7,4 Гц); 7,66-7,71 (м, 2H); 7,49 (с, 5H); 6,05 (с, 2H); 4,28 (шир. с, 2H); 3,27-3,32 (м, 2H); 2,87-2,90 (м, 2H); 1,76-2,03 (м, 6H), 1,55-1,69 (м, 1H); 1,33-1,46 (м, 2H).
Пример 14.
3-[2-[1-(Фенилметил)-4-пиперидинил]этил]- 1,2-бензизотиазолмалеат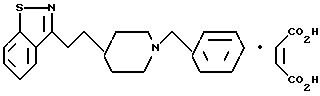
a) 4-[2-[1,2-Бензизотиазол-3-ил] этил] -1-пиперидинкарбоновая кислота. 1-(1,1-диметилэтил) сложный эфир
Повторяли процедуру, описанную в примере 1o, используя 3-метил-1,2-бензизотиазол (0,50 г, 3,35 мМ), 1-(1,1-диметилэтил) сложный эфир 4-иодометил-2-пиперидинкарбоновой кислоты (1,20 г, 3,69 мМ) и 1М диизопропиламид лития (LDA) (3,35 мл, 3,35 мМ) в сухом ТГФ (100 мл). После очистки получали целевое соединение (0,582 г, 50%) в виде желтоватого маслообразного вещества.
1H-ЯМР (CDCl3) δ 7,91-7,96 (м, 2H); 7,43-7,52 (м, 2H); 4,05-4,14 (м, 2H); 3,15 (т, 2H, J=7,9 Гц); 2,69 (шир.т, 2H, J=12,1 Гц); 1,74-1,88 (м, 4H); 1,46-1,60 (м, 1H); 1,46 (с, 9H); 1,29-1,10 (м, 2H).
b) 3-[2-[1-(Фенилметил)-4-пиперидинил]этил]-1,2-бензизотиазол-малеат
Повторяли процедуру, описанную в примере 1e, используя пиперидин, полученный в стадии (a) (0,102 г, 0,29 мМ), и трифторуксусную кислоту (ТФК) (0,75 мл) в метиленхлориде (3 мл), и триэтиламин (0,202 мг, 1,45 мМ), и бензилбромид (0,038 мл, 0,32 мМ) в метиленхлориде (3 мл). После очистки получали целевое соединение (свободное основание) (0,058 г, 62%) в виде прозрачного маслообразного вещества.
Малеат (соль) получали путем добавления раствора малеиновой кислоты (0,020 г, 0,17 мМ) в этаноле (3 мл) к раствору свободного основания (0,058 г, 0,17 мМ) в метиленхлориде (3 мл). Полученную смесь концентрировали и остаток растирали с Et2O. Полученное белое твердое вещество фильтровали и фильтрат собирали, в результате чего получали целевое соединение (0,077 г, 96%). Т.пл. 175-176oC.
1H-ЯМР (CDCl3) δ 8,14-8,21 (м, 2H); 7,63 (т, J=7,4, 1H); 7,50-7,55 (м, 6H); 6,04 (с, 2H); 4,26 (шир.с, 2H); 3,35 (шир.с, 2H); 3,15 (т, J=7,6, 2H); 2,80-2,92 (м, 2H); 1,92-2,00 (м, 2H); 1,78-1,88 (м, 2H); 1,54-1,65 (м, 1H); 1,35-1,35 (и, 2H);
13C-ЯМР (DMCO-d6) δ 167,3; 166,2; 151,6; 136,0; 134,2; 131,3; 130,1; 129,5; 128,9; 127,9; 124,9; 123,6; 120,6; 59,4; 51,6; 32,5; 33,5; 28,9; 27,8.
ИК (KBr) 3030, 2910, 2350, 1700, 1575, 1445, 1350 см-1.
HIMS: 336,2 (M+, свободное основание); 319,1; 245,1; 185,1; 172,1; 91,0 (основание).
Анализ для C21H24N2S• C4H4O4:
Вычислено: C 66,35; H 6,24; N 6,19.
Найдено: C 66,21; H 5,93; N 6,03.
Пример 15.
4-[2-[1-(Фенилметил)-4-пиперидил]этил]-1,3-хиназолинмалеат
а) 4-[2-[1,3-Хиназол-4-ил]этил]-1-пиперидинкарбоновая кислота, 1-(1,1-диметилэтил) сложный эфир
Свежеприготовленный 1M LDA (4,2 мл, 4,2 мМ) добавляли к раствору 4-метил-1,3-хиназолина (0,60 г, 4,2 мМ) в ТГФ (35 мл) при 0oC. К полученному желтому раствору добавляли чистый триметилсилилхлорид (0,53 мл, 4,2 мМ). Затем ледяную баню удаляли и реакционную смесь размешивали 3 мин. После этого смесь снова охлаждали до 0oC и добавляли второй эквивалент 1М LDA (4,2 мл, 4,2 мМ). Затем добавляли раствор 1-(1,1-диметилэтил)сложный эфир 4-иодометил-1-пиперидинкарбоновой кислоты (1,49 г, 4,6 мМ) в ТГФ (10 мл) и размешивали в течение 1 ч при 0oC. После этого добавляли разбавленную HCl (1 н.) и смесь размешивали при комнатной температуре 30 мин. Затем реакционную смесь подщелачивали путем добавления 1 н. NaOH и экстрагировали этилалцетатом. Органический слой промывали солевым раствором, осушали сульфатом магния, фильтровали и концентрировали. После очистки с помощью флеш-хроматографии на силикагеле (20% ____→ 50% EtOAc-гексаны) получали целевое соединение (0,466 г, 33%) в виде маслообразного вещества.
1H-ЯМР (CDCl3) δ 9,21 (с, 1H); 8,12 (д, 1H, J = 7,7 Гц); 8,05 (д, 1H, J = 8,4 Гц); 7,90 (т, 1H, J = 7,8 Гц); 7,65 (т, 1H, J = 7,7 Гц); 4,08 - 4,17 (м, 2H); 3,32 (т, 2H, J = 8,3 Гц); 2,72 (шир.т, 2H, J = 12,1 Гц); 1,78 - 1,90 (м, 4H); 1,57 - 1,59 (м, 1H); 1,47 (с, 9H); 1,18 - 1,29 (м, 2H).
б) 4-[2-[1-(Фенилметил)-4-пиперидил]этил]-1,3- хиназолинмалеат
Повторяли процедуру, описанную в примере 1e, используя пиперидин, полученный в стадии (а) (0,429 г, 1,26 мМ), и ТФК (3,5 мл) в CH2Cl2 (13 мл), и триэтиламин (0,88 мл, 6,3 мМ), и бензилбромид (0,17 мл, 1,39 мМ) в CH2Cl2 (22 мл). После очистки с помощью хроматографии ( CH2Cl2 ____→ 10% MeOH - CH2Cl2) получали целевое соединение (свободное основание) (0,179 г, 13%) в виде бесцветного маслообразного продукта.
Малеат (соль) получали путем добавления раствора малеиновой кислоты (0,052 г, 0,45 мМ) в этиловом эфире (10 мл) к раствору свободного основания (0,135 г, 0,41 мМ) в Et2O (200 мл). Полученное белое твердое вещество собирали путем фильтрации, в результате чего получали целевое соединение (0,103 г, 56%). Т.пл. 121 - 122oC.
1H-ЯМР (DMCO-d6) δ 9,16 (с, 1H); 8,33 (д, 1H, J = 8,4 Гц); 7,98 - 8,01 (м, 2H); 7,72 - 7,78 (м, 1H); 7,43 (с, 5H); 6,02 (с, 2H); 4,08 (шир.с, 2H); 3,32 (т, 2H, J = 7,7 Гц); 3,15 - 3,35 (м, 2H); 2,60 - 2,80 (м, 2H); 1,89 - 1,94 (м, 2H); 1,76 - 1,79 (м, 2H); 1,30 - 1,60 (м, 3H).
ИК (KBr): 3037, 2923, 1705, 1571, 1386 см-1.
EIMS 331 (M+, свободное основание); 318, 248, 143, 77 (основание).
Анализ для C22H25N3 • C4H4O4:
Вычислено: C 69,78; H 6,53; N 9,39.
Найдено: C 69,46; H 6,61; N 9,26.
Пример 16.
6-Гидрокси-3-[2-[1-(фенилметил)-4-пиперидил]этил]-1,2- бензизоксазол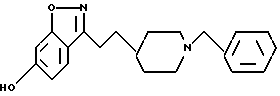
Смесь 6-метокси-3-[2-[1-(фенилметил)-4-пиперидинил] этил] - 1,2-бензизоксазол (0,11 г, 0,288 мМ) в 48%-ном водном HBr (10 мл) нагревали 16 ч при 110oC. Затем смесь подщелачивали путем добавления насыщенного NaHCO3 и экстрагировали метиленхлоридом. Органический слой осушали сульфатом магния, фильтровали и концентрировали. После очистки с помощью флэш-хроматографии на силикагеле ( (3 ____→ 6% MeOH - CH2Cl2) получали целевое соединение (0,055 г, 57%) в виде белого твердого вещества. Т. пл.148 - 149oC.
1H-ЯМР (CDCl3) δ 7,70 (шир.с, 1H); 7,27 - 7,37 (м, 6H); 6,74 - 6,80 (м, 2H); 3,63 (с, 2H); 3,04 (шир.д, 2H, J = 10,8 Гц); 2,88 (т, 2H, J = 7,7 Гц); 2,05 - 2,20 (м, 2H); 1,65 - 1,95 (м, 4H); 1,30 - 1,60 (м, 3H).
ИК (KBr): 3080, 3040, 2945, 1624, 1437, 1384 см-1.
EIMS: 336,2 (M+), 319,2; 255,0; 185,1; 91,1 (основание).
ВРМС (высоко разрез. масс-спектрометрия) для C21H24N2O2:
Вычислено: 336,18382.
Найдено: 336,18187.
Пример 17.
6-Бромо-3-[2-[1-(фенилметил)-4-пиперидил]этил]-1,2- бензизоксазолмалеат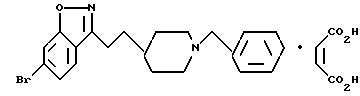
а) 4-[2-[6-Бромо-1,2-бензизоксазол-3-ил] этил] - пиперидинкарбоновая кислота, 1-(1,1-диметилэтил)сложный эфир
Повторяли процедуру, описанную в примере 1d, используя 6-бромо-3-метил-1,2-бензизоксазол (1,02 г, 4,81 мМ), 4-иодометил-1-пиперидинкарбоновой кислоты 1-(1,1-диметилэтил)сложный эфир (1,72 г, 5,29 мМ) и 1М LDA (5,30 мл, 5,30 мМ) в сухом ТГФ (40 мл), за исключением того, что после добавления реагентов смесь размешивали полтора часа при -78oC. После очистки получали целевое соединение (0,697 г, 35%) в виде бледно-желтого твердого вещества.
1H-ЯМР (CDCl3) δ 7,77 (с, 2H); 7,52 (д, 1H, J = 7,2 Гц); 7,45 (д, 1H, J = 7,2 Гц); 4,11 (шир.д, 2H, J = 14,3 Гц); 3,02 (т, 2H, J = 7,2 Гц); 4,11 (шир. д, 2H, J = 14,3 Гц); 3,02 (т, 2H, J = 7,2 Гц); 2,70 (дт, 2H, J = 12,9 Гц, J = 3,6 Гц); 1,7 - 1,85 (м, 4H); 1,49 (с, 9H); 1,45 - 1,55 (м, 1H); 1,09 - 1,29 (и, 2H).
b) 6-Бромо-3-[2-[1-(фенилметил)-4-пиперидинил] этил] -1,2- бензизоксазолмалеат
Повторяли процедуру, описанную в примере 1e, используя пиперидин, полученный в стадии (a) (0,398 г, 0,972 мМ), и ТФК (4 мл) с CH2Cl2 (26 мл), и триэтиламин (1,6 мл, 11,8 мМ), и бензилбромид (0,155 мл, 1,3 мМ) в CH2Cl2 (12 мл). Осуществляли также другую реакцию с вышеуказанным пиперидином (0,102 г, 0,249 мМ). Сырой продукт, полученный от этих двух реакций, объединяли и после очистки получали целевое соединение (свободное основание) (0,021 г, 4%) в виде твердого желтовато-коричневатого продукта.
Малеат (соль) получали путем добавления раствора малеиновой кислоты (0,023 г, 0,198 мМ) в этаноле (5 мл) к раствору свободного основания (0,072 г, 0,18 мМ) в метиленхлориде (6 мл). После концентрации остаток перекристаллизовывали из этанола и получали целевое соединение (0,035 г, 38%) в виде белого твердого продукта. Т.пл. 156,8-157,5oC.
1H-ЯМР (DMCO-d6) δ 8,08 (с, 1H); 7,87 (д, 1H, J=8,4 Гц); 7,58 (д, 1H, J= 8,5 Гц); 7,47 (с, 5H); 6,03 (с, 2H); 4,23 (шир. с, 2H); 3,25-3,40 (м, 2H); 3,02 (т, 2H, J=7,5 Гц); 2,80-2,95 (м, 2H); 1,88-2,00 (м, 2H); 1,70-1,80 (м, 2H); 1,30-1,60 (м, 3H).
13C-ЯМР (DMCO-d6) δ 167,2; 162,8; 158,6; 136,0; 131,1; 129,5; 128,9; 127,0; 124,0; 123,7; 120,6; 113,1; 59,6; 51,9; 32,7; 28,9; 21,6;
EIMS: 398 (M+), 381, 309, 252, 200, 185 (основание), 172, 91.
BPMC для C21H23BrN2O:
Вычислено: 398,0994.
Найдено: 398,0941.
Пример 18.
6-Циано-3-[2-[1-(фенилметил)-4-пиперидил]этил]-1,2- бензизоксазол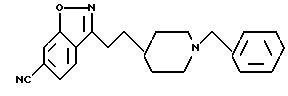
Раствор NaNO2 (0,112 г, 1,62 мМ) в воде (4 мл) добавляют к раствору 6-амино-3-[2-[1-(фенилметил)-4-пиперидинил]этил]-1,2- бензизоксазола (0,534 г, 1,59 Мм) в 28% HCl (20 мл), выдерживавшемуся при 0oC. Полученную смесь нейтрализовали до pH 7 путем осторожного добавления твердого Na2CO3. Затем нейтральную смесь порциями добавляли к хорошо размешанной смеси толуола (75 мл), льда и свежеприготовленного раствора CuCN (Organic Synthesis, Coll. Vol. 1, с. 514; CuSO4: 0,318 г 1,99 мМ). Полученную смесь выдерживали 30 мин при 0oC, а затем 2 ч при комнатной температуре, а затем нагревали 5 мин при 50oC. После этого смесь экстрагировали этилацетатом, а органическую фазу промывали водой, солевым раствором, осушали сульфатом магния, фильтровали и концентрировали. После очистки с помощью флеш-хроматографии на силикагеле (3% MeOH-CH2Cl2) получали целевое соединение (0,236 г, 43%) в виде бледно-оранжевого твердого продукта. После перекристаллизации небольшого образца (этанол-гексан) получали целевое соединение в виде беловатого твердого вещества. Т.пл. 113-144,5oC.
1H-ЯМР (CDCl3) δ 7,89 (с, 1H); 7,77 (д, 1H, J = 8,4 Гц); 7,57 (д, 1H, J = 8,7 Гц); 7,24-7,33 (м, 5H); 3,51 (с, 2H); 3,04 (т, 2H, J = 7,9 Гц); 2,92 (шир. д, 2H, J = 11,2 Гц); 1,97 (шир.т, 2H, J = 10,7 Гц); 1,74-1,85 (м, 4H); 1,26-1,40 (м, 3H).
1C-ЯМР (CDCl3) δ 161,7; 158,8; 129,2; 128,2; 127,0; 126,3; 125,1; 122,5; 118,1; 114,5; 113,2; 63,4; 53,6; 35,2; 34,0; 32,0; 22,6.
ИК (CHCl3): 2830, 2720, 2160, 1600, 1425, 1385 см-1.
FAB-MC (масс-спектроскопия путем бомбардировки быстрыми атомами): 346 (M+ + 1), 309, 275, 239, 155, 119 (основание).
ВРМС для C22H23N3O:
Вычислено 345, 1842.
Найдено: 345,1836.
Пример 19.
6-Карбоксамид-3-[2-[1-(фенилметил)-4-пиперидил]этил]1,2- бензизоксазол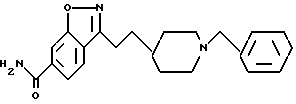
Измельченный в порошок гидроксида калия (KOH) (0,150 г, 2,68 мМ) добавляли к смеси 6-циано-3-[2-[1-(фенилметил)-4-пиперидил]этил]-1,2- бензизоксазола (0,250 г, 0,724 мМ) в т-BuOH (10 мл). Полученную смесь нагревали 20 мин при 85oC. Затем охалжденную реакционную смесь выливали поверх солевого раствора и экстрагировали метиленхлоридом. Органическую фазу промывали 10% NaOH, солевым раствором, осушали сульфатом магния, фильтровали и концентрировали. Сырой продукт очищали путем перекристаллизации (из EtOAc-гексана) и получали целевое соединение (0,114 г, 43%) в виде белого твердого вещества.
Т.пл. 181-182oC.
1H-ЯМР (DMCO-d6) δ 8,20 (шир.с, 1H, -NH); 8,15 (с, 1H); 7,97 (д, 1H, J = 8,4 Гц); 7,87 (д, 1H, J=8,1 Гц); 7,64 (шир.с, 1H-NH), 7,22-7,31 (м, 5H); 3,42 (с, 2H); 3,02 (т, 2H, J = 7,8 Гц); 2,78 (шир.д, 2H, J = 11,5 Гц); 1,87 (шир.т, 2H, J = 10,9 Гц); 1,69-1,73 (м, 4H); 1,22-1,26 (м, 3H).
13C-ЯМР (DMCO-d6) δ 167,1; 162,0; 158,9; 138,9; 136,3; 128,7; 126,8; 126,8; 123,3; 122,0; 108,7; 62,5; 53,2; 34,9; 33,7; 31,6; 22,0.
FAB-MC: 346 (M+ + 1), (основание), 321, 272, 185, 172.
Пример 20.
3-[(1-Фенилметил-4-пиперидил)метокси)-1,2-бензизоксазолфумарат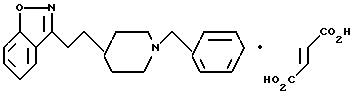
а) 4-[(1,2-Бензизоксазол-3-ил)оксиметил] -1-пиперидил- карбоновая кислота, 1-(1,1-диметилэтил)сложный эфир
NaH (60% дисперсия в минеральном масле, 0,941 г, 23,53 мМ) добавляли порциями к раствору 1-(1,1диметилэтил)сложного эфира 4-гидроксиметил-1-пиперидинкарбоновой кислоты (4,82 г, 22,41 мМ) в ДМФ (220 мл) при 0oC. Через 10 мин реакционную смесь нагревали до комнатной температуры и добавляли 3-хлоро-1,2-бензизоксазол (3,44 г, 22,41 мМ). Полученную смесь нагревали 16 ч при 115oC. Затем реакционную смесь разбавляли этилацетатом и промывали водой (4х), солевым раствором, осушали сульфатом магния, фильтровали и концентрировали. После очистки с помощью флеш-хроматографии на силикагеле (10% EtOAc-гексан) получали целевое соединение (4,16 г, 56%) в виде белого твердого вещества. Т.пл. 103-104,5oC.
1H-ЯМР (CDCl3) δ 7,61 (д, 1H, J=7,0 Гц); 7,48-7,54 (м, 1H); 7,41 (д, 1H, J= 8,5 Гц); 7,22-7,27 (м, 1H); 4,28 (д, 2H, J=6,5 Гц); 4,16 (шир.д, 2H, J= 13,3 Гц); 2,75 (дт, 2H, J=13,2 Гц); J= 2,6 Гц); 2,04-2,13 (м, 1H); 1,83 (шир. д, 2H, J=13,7 Гц); 1,45 (с, 9H); 1,30 (ддд, 2H, J=24,9 Гц, J=12,5 Гц, J=4,2 Гц).
b) 3-[(1-Фенилметил-4-пиперидил)метокси]-1,2-бензизоксазол) -фурамат
Трифторуксусную кислоту (ТФК) (10 мл) по капле добавляли к раствору пиперидина, полученного в стадии (a) (0,827 г, 2,49 мМ) в CH2Cl2 (25 мл) при 0oC. Полученную смесь размешивали 20 мин при 0oC. Затем смесь концентрировали, а избыток ТФК удаляли путем концентрирования из толуола. Остаток распределяли между метиленхлоридом и насыщенным MaHCO3. Органический слой осушали сульфатом магния, фильтровали и концентрирорвали. Сырой продукт (0,192 г, 0,827 мМ) растворяли этиленхлориде (8 мл) и добавляли триэтиламин (0,58 мл, 4,13 мМ), а затем бензилбромид (0,128 мл, 1,07 мМ). Полученную смесь размешивали в течение ночи (18 ч) при комнатной температуре. Затем реакционную смесь промывали водой, солевым раствором, осушали сульфатом магния, фильтровали и концентрировали. После очистки с помощью флеш-хроматографии на силикагеле (5% MeOHCH2Cl2) получали целевое соединение (свободное основание) (0,199 г, 25%) в виде беловатого твердого вещества.
Фумарат (соль) получали путем добавления фумаровой кислоты (0,074 г, 0,633 мМ), растворенной в минимальном количестве EtOH, к раствору свободного основания (0,186 г, 0,58 мМ) в метиленхлориде (6 мл). После концентрации сырую соль очищали путем перекристаллизации (EtOH-Et2O), получали целевое соединение (0,134 г, 53%) в виде беловатого твердого вещества. Т.пл. 163-164oC.
1H (DMCO-d6) δ 7,74 (д, 1H, J=7,8 Гц); 7,61 - 7,69 (м, 2H); 7,26 - 7,41 (м, 6H); 6,60 (с, 2H); 4,28 (д, 2H, J=6,4 Гц); 3,65 (с, 2H); 2,07 (шир. д, 2H, J= 11,4 Гц); 2,20 (шир. т, 2H, J=11,7 Гц); 1,90 - 2,05 (м, 1H); 1,81 (шир. д, 2H, J=12,8 Гц); 1,38 - 1,49 (м, 2H).
13C-ЯМР (DMCO-d6) δ 166,5; 166,1; 163,3; 137,0; 134,3; 131,1; 129,3; 128,3; 127,3; 123,6; 120,8; 113,5; 110,3; 74,2; 61,7; 52,2; 34,7; 27,6.
ИК (KBr) 2980, 2550, 1706, 1650, 1616, 1573, 1447, 1374 cv-1
EIMC: 305, 185, 172, 90 (основание).
BPMC для C20H22N2O2 (свободное основание):
Вычислено: 322,1682.
Найдено: 322,1719.
Анализ для C20H22N2O2• C4H4O4•0,75H2O:
Вычислено: C 63,78; H 6,13; N 6,20.
Найдено: C 63,88; H 5,85; N 6,14.
Пример 21.
3-[(1-Фенилметил-4-пиперидил)метиламино]-1,2бензизоксазол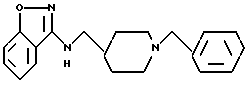
Смесь 3-хлоро-1,5-бензизаксазола (0,238 г, 1,55 мМ), 4-аминометил-1-фенилметилпиперидина (0,316 г, 1,55 мМ) и K2CO3 (0,214 г, 1,55 мМ) в DMCO (10 мл) нагревали при 150oС в течение 20 ч. Охлажденную реакционную смесь разбавляли этилацетатом (75 мл) и выливали в воду (200 мл). Органическую фазу разделяли и промывали солевым раствором, осушали сульфатом магния, фильтровали и концентрировали. Полученный коричневый маслообразный продукт очищали с помощью флеш-хроматографии на силикагеле (4% MeOH-CH2Cl2) и получали целевое соединение (0,084 г, 17%) светло-желтого маслообразного вещества.
1H-ЯМР (CDCl3): δ 7,44 - 7,50 (м, 2H); 7,38 (д, 1H, J=8,8 Гц); 7,16 - 7,31 (м, 6H); 4,35 - 4,42 (м, 1H, -NH-); 3,51(с, 2H); 3,33 (т, 2H, J=6,2 Гц); 2,92 (шир. д, 2H, J=11,5 Гц); 1,99 (т, 2H, J=11,6 Гц); 1,77 (шир. д, 2H, J=12,7 Гц); 1,75 - 1,80 (м, 1H); 1,33 - 1,45 (м, 2H).
13C-ЯМР (CDCl3) δ 162,8; 158,6; 137,8; 129,8; 128,2; 127,2; 122,1; 119,7; 116,2; 110,1; 63,2; 53,3; 49,4; 35,2; 29,9; 29,7.
ИК (KBr) 3290, 2924, 2852, 1614, 1564, 1450, 1365 см-1.
EIMS: 321 (M+), 230, 201, 185, 172, 91 (основание).
BPMC для C20H23N3O.
Вычислено: 321,1842.
Найдено: 321,1825.
Пример 22.
3-[2-[(Фенилметил)-4-пиперидил)этиламин]-1,2бензизоксазол
Повторяли процедуру, описанную в примере 21, используя 3-хлоро-1,2-бензизоксазол (0,682 г, 4,44 мМ), 4-аминоэтил-1- фенилметилпиперидин (0,970 г, 4,44 мМ) и K2CO3 (0,614 г, 4,44 мМ) в DМCO (30 мл). После очистки получали целевое соединение (0,231 г, 15%) в виде бледно-желтого маслообразного продукта.
1H-ЯМР (CDCl3) δ 7,45 - 7,54 (м, 2H); 7,24 - 7,40 (м, 6H); 7,19 (т, 1H, J= 7,8 Гц); 4,40 (шир. т, 1H, J=5,5 Гц); 3,52 (с, 2H); 3,45 (дт, 2H, J=7,4 Гц, J= 6,0 Гц); 2,91 (шир. д, 2H, J=11,8 Гц); 1,99 (шир. т, 2H, J=11,2 Гц) 1,62 - 1,73 (м, 4H); 1,37 - 1,52 (м, 3H).
13C-ЯМР (CDCl3) δ 162,7; 158,6; 137,8; 129,8; 129,4; 128,2; 127,1; 122,1; 119,8; 116,3; 110,0; 63,3; 53,6; 41,5; 36,1; 33,4; 32,0.
EIMC: 335 (M+), 244, 199, 186, 172, 91 (основание).
BPMC для C21H25N3O:
Вычислено 335,1998.
Найдено: 335,1909.
Пример 23.
3-[3-[1-(Фенилметил)-4 пиперидил]пропил]-1,2бензизоксазол-малеат
a) 4-[2-Этокси-2-оксоэтилидин] -1-пиперидинкарбоновая кислота, 1-(1,1-диметилэтил)сложный эфир
Раствор триэтилфосфоноацетата (3,1 мл, 15,69 мМ) в свежедистиллированном 1,2-диметоксиэтана (ДМЭ, 12,5 мл) добавляли к суспензии NaH (60% дисперсии в минеральном масле, 0,75 г, 18,18 мМ) в ДМЭ (6,5 мл). Полученную смесь размешивали в течение 1 ч при комнатной температуре и добавляли раствор 1-(1,1-диметилэтил)сложного эфира 4-кето-1-пиперидинкарбоновой кислоты (2,5 г, 12,55 мМ) в ДМЭ (12,5 мл). После размешивания в течение ночи (15 ч) смесь концентрировали. Остаток очищали с помощью флеш-хроматографии на силикагеле (5 ____→ 20% EtOAc-гексаны) и получали целевое соединение (3,06 г, 91%) в виде белого твердого вещества.
1H-ЯМР (CDCl3) δ 5,71 (с, 1H); 4,16 (кв., 2H, J= 7,1 Гц); 3,45 - 3,53 (м, 4H); 2,94 (т, 2H, J=5,7 Гц); 1,28 (т, 2H, J=5,6 Гц); 1,48 (с, 9H); 1,28 (т, 3H, J=7,1 Гц).
b) 4-Этоксикарбонилметил-1-пиперидинкарбоновая кислота, 1-(1,1-диметилэтил)сложный эфир
Смесь олефина, полученного в стадии (a) (3,05 г, 11,3 мМ), и 10% Pd/C (1,2 г, 1,13 мМ) в EtOH (50 мл) гидрировали в шейкере Парра при давлении 50 фунт/кв. дюйм (3,45 бар) в течение полутора часов. Полученную смесь фильтровали через прокладку из ЦелитаТМ, а фильтрат концентрировали, в результате чего получали целевое соединение (3,07 г, количественный выход) в виде бесцветного маслообразного продукта.
1H-ЯМР (CDCl3) δ 4,14 (кв., 2H, J=7,0 Гц); 4,05 - 4,12 (м, 2H); 2,72 (шир. дт, 2H, J=12,1 Гц, J=2,8 Гц); 2,23 (д, 2H, J=7,2 Гц); 1,85 - 1,95 (м, 1H); 1,61 - 1,66 (м, 2H); 1,45 (с, 9H); 1,26 (т, 3H, J=7,0 Гц); 1,05 - 1,23 (м, 2H).
c) 4-Гидроксиэтил-1-пиперидинкарбоновая кислота. 1-(1,1-диметилэтил)сложный эфир
Повторяли процедуру, описанную в примере 1b, используя сложный эфир, полученный в стадии (b) (3,06 г, 11,3 мМ), и алюмогидрид лития (0,47 г, 12,4 мМ) в ТГФ (105 мл). После очистки с помощью флеш-хроматографии на силикагеле (50% EtOAc-гексаны) получали непрореагировавший исходный материал (0,73 г, 24%) и целевое соединение (1,74 г, 68%) в виде бесцветного маслообразного продукта.
1H-ЯМР (CDCl3) δ 4,07 (шир.д. 2H, J=14,1 Гц); 3,71 (т, 2H, J=6,5 Гц); 2,69 (шир.т. 2H, J=12,5 Гц); 1,50-1,70 (м, 6H); 1,45 (с, 9H); 1,=5-1,15 (м, 2H).
d) 4-Иодоэтил-1-пиперидинкарбоновая кислота, 1-(1-1-диметилэтил)сложный эфир
Повторяли процедуру, описанную в примере 1c, используя спирт, полученный в стадии (c) (1,74 г, 7,59 мМ), трифенилфосфин (2,49 г, 9,49 мМ), йод (2,31 г, 9,11 мМ) и пиридин (1,5 мл, 18,2 мМ) в бензоле (50 мл). После очистки получали целевое соединение (2,27 г, 98%) в виде бесцветного маслообразного продукта.
1H-ЯМР (CDCl3) δ 4,09 (шир.д, 2H, J=11,4 Гц); 3,22 (т, 2H, J=7,3 Гц); 2,70 (шир. т, 2H, J=12,5 Гц); 1,78 (кв., 2H, J=6,9 Гц); 1,47-1,68 (м, 3H); 1,46 (с, 9H); 1,12 (ддд, 2H, J=24,2 Гц); J=12,9 Гц, J=4,3 Гц).
e) 4-[3-[1,2-Бензизоксазол-3-ил)пропил] -1-пиперидинкарбоновая кислота, 1-(1,1-диметилэтил)сложный эфир
Повторяли процедуру, описанную в примере 1d, используя 3-метил-1,2-бензизоксазол (0,412 г, 3,09 мМ), иодид, полученный в стадии о (1,15 г, 3,4 мМ), и 1M LDA (3,4 мл, 3,4 мМ) в ТГФ (8 мл). После очистки получали целевое соединение (0,694 г, 65%) в виде бледно-желтого маслообразного продукта.
1H-ЯМР (CDCl3) δ 7,65 (д. 1H, J=8,0 Гц); 7,54-7,57 (м, 2H); 7,27-7,34 (м, 1H); 4,08 (шир.д, 2H, J=12,5 Гц); 2,99 (т, 2H, J= 7,6 Гц); 2,66 (шир.т, 2H, J= 12,0 Гц); 1,86-1,92 (м, 2H); 1,63-1,68 (м, 4H); 1,45 (с, 9H); 1,36-1,45 (м, 1H); 1,06-1,12 (м, 2H).
f) 3-[3-[1-(Фенилметил)-4-пиперидил]пропил]-1,2-бензизоксазол-малеат
Повторяли процедуру, описанную в примере 1e, используя пиперидин, полученный в стадии (e) (0,544 г, 1,58 мМ), и ТФК (4 мл) в метиленхлориде (16 мл), и триэтиламин (1,1 мл, 7,9 мМ), и бензилбромид (0,21 мл, 1,74 мМ) в метиленхлориде (10 мл). После очистки с помощью хроматографии (2 -____→ 5% MeOH-CH2Cl2) получали целевое соединение (свободное основание) (0,285 г, 54%) в виде бледно-желтого маслообразного продукта.
Малеат (соль) получали путем добавления малеиновой кислоты (0,109 г, 0,94 мМ), растворенной в минимальном количестве этанола, к раствору свободного основания (0,285 г, 0,85 мМ) в метиленхлориде (10 мл). После концентрирования остаток очищали путем перекристаллизации (EtOFc) и получали целевое соединение (0,292 г, 76%) в виде беловатого твердого вещества.
Т.пл. 134,5-135,5oC.
1H-ЯМР (DMCO-d6) δ 7,90 (д. 1H, J=7,9 Гц); 7,61-7,72 (м, 2H); 7,47 (с, 5H); 7,38 (т, 1H, J=7,3 Гц); 6,04 (с, 2H); 4,26 (шир.с, 2H); 3,22/3,45 (м, 2H); 2,99 (т, 2H, J=7,4 Гц); 2,70-2,96 (м, 2H); 1,70-1,90 (м, 4H); 1,40-1,65 (м, 1H); 1,20-1,40 (м, 4H).
13C-ЯМР (DMCO-d6) δ 167,2; 162,2; 158,4; 135,8; 131,2; 130,3; 129,5; 128,9; 123,5; 122,1; 121,2; 109,7; 59,3; 51,7; 34,8; 32,6; 28,9; 24,5; 24,1.
ИК (КBr): 2942, 1705, 1581, 1460, 1359 см-1..
EIMS: 334 (M+, свободное основание), 243, 202, 173 (основание), 91.
Анализ для C22H26N2O • C4H4O4:
Вычислено: C 69,31; H 6,71; N 6,22.
Найдено: C 69,11; H 6,64; N 6,46.
Пример 24.
Транс-3-[2-[-1-(фенилметил-4-пиперидил]этенил]-1,2-бензизоксазолмалеат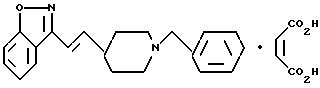
NaH (60% дисперсия в минеральном масле, 0,10 г, 2,51 мМ) добавляли к смеси 3-трифенилфосфонийметил-1,2-бензизоксазолбромида (1,19 г, 2,51 мМ) в ТГФ (10 мл). После 1-часового размешивания при комнатной температуре добавляли раствор 4-карбоксальдегид-1-фенилметилпиперидина (0,51 г, 2,51 мМ) в ТГФ (2 мл). Полученную смесь размешивали 4 ч и фильтровали. Фильтрат концентрировали, а остаток распределяли между диэтиловым эфиром и водой. Отделенный органический слой осушали сульфатом магния, фильтровали и концентрировали. После очистки с помощью флеш-хроматографии на силикагеле (40% EtOAc-гексан) получали целевое соединение (свободное основание) (0,483 г, 60%) в виде бесцветного маслообразного продукта.
Малеат (соль) получали путем добавления малеиновой кислоты (0,194 г, 1,67 мМ), растворенной в минимальном количестве этанола, к раствору свободного основания (0,483 г, 1,52 мМ) в Et2O (25 мл). Полученное белое твердое вещество собирали путем фильтрации и получали целевое соединение (0,581 г, 88%).
Т.пл. 174-175oC
1H-ЯМР (DMCO-d6) δ 8,16 (д. 1H, J=7,8 Гц); 7,75 (д, 1H, J=8,4 Гц); 7,67 (т. 1H, J= 8,2 Гц); 7,41-7,55 (м, 6H); 6,96 (шир.дд, 1H, J=16,5 Гц); J=5,6 Гц); 6,78 (д, 1H, J=16,5 Гц); 6,08 (с, 2H); 4,33 (с, 2H); 3,32-3,39 (м, 2H); 2,95-3,15 (м, 2H); 2,50-2,70 (м, 1H); 2,06 (шир.д, 2H, J=12,6 Гц); 1,60-1,90 (м, 2H).
13C-ЯМР (DMCO-d6) δ: 167,3; 162,7; 154,9; 142,7; 135,9; 131,2; 130,4; 130,2; 129,5; 128,9; 124,2; 122,7; 119,4; 116,5; 109,9; 59,2; 51,0; 36,2; 27,8.
ИК (KBr) 3035, 2944, 1708, 1588, 1472, 1360 см-1.
EIMS : 3,18 (M+, свободное основание), 201, 227, 172, 91 (основание).
Анализ для C21H22N2O • C4H4O4:
Вычислено: C 69,11; H 6,03; N 6,45.
Найдено: C 69,04; H 6,27; N 6,38.
Пример 25.
3-[2-[1-(фенилметил)-4-пиперазинил] этил] -1,2-бензизоксазол, дигидрохлоридная соль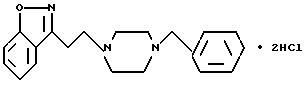
Смесь 3-[2-хлорэтил] -1,2-бензизоксазола (0,55 г, 3,03 мМ) и N-бензилпиперазина (1,06 мл, 6,06 мМ) в ксилоле (4 мл) нагревали в течение 4,75 ч при 150oC. Охлажденную смесь разбавляли этиленхлоридом и промывали водой. Органический слой сушили сульфатом магния, фильтровали и концентрировали. После очистки с помощью флеш-хроматографии на силикагеле (50% EtOAc-гексан) (100% EtOAc) получали целевое соединение (свободное основание) (0,337 г, 35%) В виде бледно-желтого маслообразного продукта.
Дигидрохлоридную соль получали путем барботирования избытка хлороводорода через раствор свободного основания (0,337 г, 1,05 мМ) в этиловом эфире (50 мл). Образовавшееся белое твердое вещество собирали путем фильтрации и получали целевое соединение (0,259 г, 63%). Т. пл. 233-234oC.
H1-ЯМР (D2O) δ: 7,79 (д, 1H, J=8,0 Гц); 7,53-7,69 (м, 2H); 7,50 (с, 5H); 7,40 (ддд, 1H, J=7,9 Гц, J=6,6 Гц, J=1,3 Гц) 4,47 (с, 2H); 3,82 (т, 2H, J= 7,5 Гц); 3,60-3,80 (м, 8H); 3,55 (т, 2H, J=7,5 Гц).
13C-ЯМР (D2O) δ 165,5; 158,0; 134,2; 133,6; 132,4; 130,3; 127,1; 124,4; 123,2; 112,8; 63,4; 56,6; 51,8; 51,0; 23,2.
ИК (KBr) 2989, 1608, 1436, 1375 см-1.
EIMS : 312 (М+, свободное основание), 256, 189, 91 (основание).
Анализ для C20H23N3O•2HCl:
Вычислено: C 60,92; H 6,39; N 10,66.
Найдено: C 60,64; H 6,57; N 10,42.
Пример 26.
5,7-Дигидро-7-метил-3-[2-[1-(фенилметил)-4-пиперидинил] этил] -6H-пирроло[4,5-f]-1,2-бензизоксазол-6-он-мезилат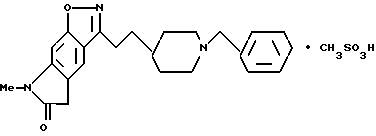
NaH (60% дисперсия в минеральном масле, 0,048 г, 1,2 мМ) добавляли к раствору 5,7-дигидро-3-[2-[1-(фенилметил)-4-пиперидинил] этил] -6H-пирроло[4,5-f] -1,2-бензизоксазол-6-она (0,374 г, 1,0 мМ) с ДМФ (10 мл) при комнатной температуре. После прекращения выделения газообразного водорода добавляли метилиодид (0,093 мл, 1,5 мМ) и полученную смесь размешивали в течение 5,5 ч. Затем добавляли насыщенный NH4Cl и воду (> 50 мл). Реакционную смесь экстрагировали метиленхлоридом, а органический слой осушали сульфатом магния, фильтровали и концентрировали. После очистки с помощью флаш-хроматографии на силикагеле ( CH2Cl2 ____→ 3% MeOH-CH2Cl2) получали целевое соединение (свободное основание( (0,056 г, 14%) в виде беловатого пенистого продукта.
Мезилат (соль) получали путем добавления метансульфоновой кислоты (0,009 мл, 0,144 мМ) к раствору свободного основания (0,056 г, 0,044 мМ) в метиленхлориде (5 мл). После концентрирования остаток растирали с этиловым эфиром и получали целевое соединение (0,049 г, 70%) в виде беловатого твердого вещества. Т.пл. 164-165oC (разлож.).
1H-ЯМР (DMCO-d6) 7,69 (с, 1H); 7,49 (с, 5H); 7,32 (с, 1H); 4,28 (с, 2H); 3,64 (с, 2H); 3,35 (шир.д, 2H. J=11,6 Гц); 3,18 (с, 3H); 2,85-2,99 (м, 4H); 2,30 (с, 3H); 1,95 (шир.д, 2H, J=12,9 Гц); 1,66-1,80 (м, 2H); 1,45-1,60 (м, 1H); 1,35-1,44 (м, 2H).
EIMS : 389 (М+, свободное основание), 298, 217, 200, 185 (основание), 172.
BPMC для C24H27N3O2•CH3 SO3H:
Вычислено 389, 2104.
Найдено: 389,2075.
Пример 27.
5,7-Дигидро-7-этил-3-[2-[1-(фенилметил)-4-пиперидинил] этил] -6H- пирроло[4,5-f]-1,2-бензизоксазол-6-он мезилат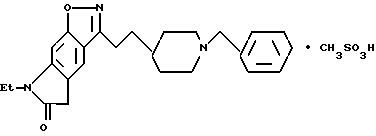
Повторяли процедуру, описанную в примере 26, используя 5,7-дигидро-3-[2-[1-(фенилметил)-4-пиперидинил] этил] -6H-пирроло[4,5-f] -1,2-бензизоксазол-6-он (0,374 г, 1,0 мМ), NaH (0,068 г, 1,7 мМ) и этилиодид (0,16 мл, 2,0 мМ) в ДМФ (10 мл). После очистки получали целевое соединение (свободное основание) (0,076 г, 19%) в виде бледно-желтого маслообразного продукта.
Мезилат (соль) получали путем добавления метансульфоновой кислоты (0,007 мл, 0,112 мМ) к раствору свободного основания (0,045 г, 0,112 мМ) в CH2Cl2 (5 мл). После концентрирования остаток растирали с Et2O и получали целевое соединение (0,042 г, 75%) в виде беловатого твердого продукта (гигроскопичного). Т. пл. ≈162oC (разл. при >60oC).
1H-ЯМР (DMCO-d6) δ 7,69 (с, 1H); 7,48 (с, 5H); 7,39 (с, 1H); 4,27 (c, 2H); 3,77 (шир. кв., 2H, J=7,1 Гц); 3,64 (c, 2H); 3,34-3,39 (м, 2H); 2,92-2,98 (м, 4H); 2,30 (с, 3H); 1,94 (шир.д, 2H, J=12,8); 1,66-1,78 (м, 2H); 1,30-1,60 (м, 3H); 1,16 (т, 3H, J=7,1 Гц).
13C-ЯМР (DMCO-d6) 174,5; 163,2; 158,1; 147,0; 131,4; 131,0; 129,8; 129,6; 128,9; 121,2; 117,0; 115,2; 90,0; 59,3; 51,6; 34,5; 34,4; 33,0; 32,6; 28,5; 21,6; 12,3.
ИК (KBr) 2934, 1716, 1630, 1605, 1466, 1330 см-1.
EIMS 403 (М+, свободное основание), 386, 312, 185 (основание), 172.
BPMC для C25H29N3O2 • CH3SO3H:
Вычислено: 403,22605.
Найдено: 403,22761.
Пример 28.
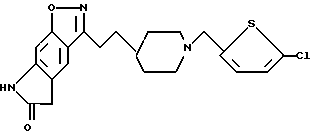
5,7-Дигидро-3-[2-[1-(2-хлоро-5-тиофенметил)-4-пиперидинил] этил] -6H-пирроло[4,5-f]-1,2-бензизоксазол-6-он
Повторяли процедуру, описанную в примере 12f, используя 4-[2-[5,7-дигидро-6H-пирроло[4,5, -f] -1,2-бензизоксазол-6-он-3-ил] этил] - 1-пиперидинкарбоновой кислоты 1-(1; 1-диметилэтил) сложный эфир (0,328 г, 0,851 мм) и ТФК (2 мл) в CH2Cl2(8 мл). В стадии алкилирования использовали лишь часть неочищенной соли: Na2CO3 (0,175 г, 1,66 мМ) и 2-хлоро-5-хлорометилтиофен (0,048 мл, 0,40 мМ) в ДМФ (3 мл). После очистки (2-4% MeOH-CH2Cl2) получали целевое соединение (0,0514 г, 43%) в виде белого твердого вещества. Т.пл. 202-204oC (разлож).
1H-ЯМР (DMCO-d6) δ 10,8 (с, 1H); 7,62 (с, 1H); 6,79 (с, 1H); 6,92 (д, 1H, J=3,7 Гц); 6,80 (д, 1H, J=3,7 Гц); 3,57 (с, 2H); 3,55 (с, 2H); 2,81-2,93 (м, 4H); 1,91 (шир.т, 2H, J=10,7 Гц); 1,61-1,71 (м, 4H); 1,11-1,23 (м, 3H).
13C-ЯРМ (DMCO-d6) δ: : 176,7; 162,9; 158,3; 146,5; 142,8; 127,0; 126,1; 125,1; 123,2; 117,1; 115,1; 90,1; 57,0; 53,0; 35,0; 34,8; 33,9; 31,6; 21,9.
ИК (KBr) 3174, 2950, 1702, 1631, 1453, 1330 см-1.
EIMS: 398, 382, 350, 322, 236, 172, 91, 81 (основание).
BPMC для C21H22ClN3O2S:
Вычислено: 415,1122.
Найдено: 415,1085.
Анализ для C21H22ClN3O2S • 0,5H2O:
Вычислено: C 59,36; H 5,46; N 9,89.
Найдено: C 59,21; H 5,12; N 9,65.
Пример 29.
5,7-Дигидро-3-[2-[1-(2-метил-4-тиазолометил)-4-пиперидинил] этил]-6H-пирроло[4,5-f]-1,2-бензизоксазол-6-он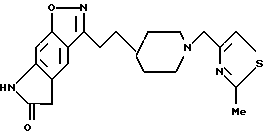
Повторяли процедуру, описанную в примере 12f, используя 4-[2-[5,7-дигидро-6H-пирроло[4,5-f] -1,2-бензизоксазол-6-он-3-ил] этил]-1- пиперидинкарбоновой кислоты 1-(1,1-диметилэтил)сложный эфир (0,367 г, 0,952 мМ) и ТФК (2,5 мл) в метиленхлориде (10 мл), и Na2CO3(1,01 г, 9,52 мМ), и 4-хлорметил-2-метилтиазола гидрохлоридную соль (0,210 г, 1,142 мМ) в ДМФ (10 мл). После очистки с помощью хроматографии (4% MeOH-EtOAc) с последующей перекристаллизацией (EtOAc-гексан) получали целевое соединение (0,074 г, 20%) в виде белого твердого вещества. Т.пл.172-173oC (разлож.).
1H-ЯМР (CDCl3) δ 9,40 (шир.с, 1H); 7,41 (с, 1H); 7,05 (с, 1H); 7,02 (с, 1H); 3,72 (с, 2H); 3,61 (с, 2H); 3,07 (шир.д, 2H, J=11,3 Гц); 2,93 (т, 2H, J= 7,8 Гц); 2,69 (с, 3H); 2,15 (шир.т, 2H, J=10,8 Гц); 1,76-1,85 (и, 4H); 1,42-1,55 (м, 3H).
13C-ЯРМ (CDCl3) δ: : 177,4; 166,0; 163,6; 158,3; 151,7; 145,1; 122,2; 116,8; 116,3; 91,5; 57,9; 53,4; 35,4; 34,8; 34,0; 31,2; 22,5; 19,2.
ИК (KBr) 3101, 3015, 2938, 2924, 1713, 1633, 1456, 1328 см-1.
EIMS: 396 (M+), 379, 284, 267, 206 (основание).
BPMC для C21H24N4O2S:
Вычислено 396,1621.
Найдено: 396,1631.
Пример 30.
3-[2-[1-(3-Бромфенилметил)-4-пиперидинил] этил] -5,7-дигидро-6H-пирроло [4,5-f]-1,2-бензизоксазол-6-он
Повторяли процедуру, описанную в примере 12f, используя 1-[1,1-диметилэтил)сложный эфир 4-[2-[5,7-дигидро-6H-пирроло-[4,5-f] - 1,2-бензизоксазол-6-он-3-ил]этил]-1-пиперидинкарбоновой кислоты (0,50 г, 1,30 мМ) и ТФК (3 мл) в метиленхлориде (12 мл), и Na2CO3 (0,689 г, 6,5 мМ) и 3-бромбензилбромид (0,46 г, 1,84 мМ) в ДМФ (20 мл). После очистки с помощью хроматографии (CH2Cl2 ____→ 5% MeOE-CH2Cl2) получали целевое соединение (0,314 г, 53%) в виде бледно-желтого твердого вещества. После перекристаллизации (EtOAc, два раза) получали белый твердый продукт (0,076 г, 13%). Т.пл.173-174oC.
1H-ЯРМ (CDCl3) δ 8,77 (с, 1H); 7,47 (с, 1H); 7,41 (с, 1H); 7,36 (д, 1H, J-7,7 Гц); 7,23 (д, 1H, J=7,9 Гц); 7,16 (т, 1H), J=7,7 Гц); 7,00 (с, 1H); 3,61 (с, 2H); 3,46 (с, 2H); 2,86-2,96 (м, 4H); 1,90-2,02 (м, 2H); 1,70-1,85 (м, 4H); 1,30-1,42 (м, 3H).
13C-ЯРМ (CDCl3) δ : 177,1; 163,5; 158,4; 144,7; 132,0; 130,1; 129,8; 127,8; 122,3; 122,0; 116,5; 91,5; 62,6; 53,6; 35,3; 35,2; 34,3; 31,9; 22,6.
Пример 31.
3-[2-[1-(4-Бромфенилметил)-4-пиперидинил] этил] -5,7-дигидро-6H-пирроло [4,5-f]-1,2-бензизоксазол-6-он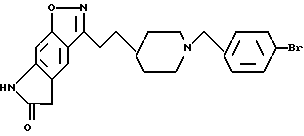
Повторяли процедуру, описанную в примере 12f, используя 4-[2-[5,7-дигидро-6H-пирроло[4,5-f] -1,2-бензизоксазол-6-он-3-ил] этил]- 1-пиперидинкарбоновой кислоты 1-(1,1-диметилэтил)сложный эфир (0,50 г, 1,3 мМ) и ТФК (3 мл) в CH2Cl2 (12 мл), и Na2CO3 (0,689 г, 6,5 мМ), и 4 -бромбензилбромид (0,39 г, 1,56 мМ) в ДМФ (15 мл). После очистки с помощью хроматографии (CH2Cl2 ____→ 5% MeOH-CH2Cl2) получали целевое соединение (0,415 г, 70%) в виде беловатого твердого вещества.
Т.пл.177-178oC.
1H-ЯМР (CDCl3) δ 9,98 (шир.с, 1H); 7,37-7,40 (м, 3H); 7,16 (д, 2H, J = 8,2 Гц); 6,99 (с, 1H); 3,60 (с, 2H); 3,42 (с, 2H); 2,87-2,94 (м, 4H); 1,94 (шир.т, 2H, J = 10,5 Гц); 1,65-1,80 (м, 4H); 1,20-1,35 (м, 3H).
13C-ЯМР (CDCl3) δ : 178,1; 163,5; 158,4; 145,2; 137,3; 131,2; 130,8; 122,2; 120,7; 116,7; 116,3; 91,5; 62,5; 53,5; 35,5; 35,2; 34,2; 31,9; 22,5.
Пример 32.
5,7-Дигидро-3-[3-[1-(фенилметил)-4-пиперидинил] пропил] - 6H-пирроло[4,5-f]-1,2-бензизоксазол-6-он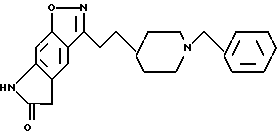
а) 4-[3-[5-, 7-Дигидро-6H-пирроло[4,5-f] -1,2-бензизоксазол- 6-он-3-ил] пропил]-1-пиперидинкарбоновая кислота, 1-(1,1-диметилэтил)сложный эфир
Повторяли процедуру, описанную в примере 7а, используя 5,7-дигидро-3-метил-6H-пирроло[4,5-f] -1,2-бензизоксазол-6-он (0,13 г, 0,69 мМ), IM LDA (2,8 мл, 2,8 мМ) и 4-одоэтил-1-пиперидинкарбоновой кислоты 1-(1,1-диметилэтил)сложный эфир (0,233 г, 0,69 мМ) в сухом ТГФ (14 мл), за исключением того, что после добавления реагентов смесь размешивали при -78oC в течение 4 ч. После очистки с помощью хроматографии (10% _____→ 50% EtOAc-CH2Cl2) получали восстановленный исходный материал (0,031 г, 24%) и целевое соединение (0,129 г, 47%) в виде бесцветного маслообразного продукта.
1H-ЯМР (CDCl3) δ: 9,50 (с, 1H); 7,42 (с, 1H); 7,04 (с, 1H); 4,06 (шир.д, 2H, J = 14,5 Гц); 3,62 (с, 2H); 2,91 (т, 2H, J = 7,5 Гц); 2,66 (дт, 2H, J = 13,0 Гц, J = 2,0 Гц); 1,81-1,87 (м, 2H); 1,65 (шир.д, 2H, J = 12,3 Гц); 1,44 (с, 9H); 1,34-1,43 (м, 3H); 1,09-1,20 (М, 2H).
о) 5,7-Дигидро-3-[3-[3-[1-(фенилметил)-4-пиперидил] пропил] - 6H-пирроло[4,5-f]-1,2-бензизоксазол-6-он
Повторяли процедуру, описанную в примере 12f, используя пиперидин, полученный в стадии (а) (0,114 г, 0,29 мМ), и ТФК (1,5 мл) в CH2Cl2 (6 мл), и Na2CO3 (0,154 г, 1,45 мМ), и бензилбромид (0,042 мл, 0,35 мМ) в ДМФ (5 мл). После очистки с помощью хроматографии (CH2Cl2 ____→ 5% MeOH-CH2Cl2) получали целевое соединение (0,70 г, 62%) в виде белого пенистого твердого продукта. Этот продукт перекристаллизовывали (CH2Cl2) и получали белое твердое вещество (0,054 г, 48%).
Т.пл. 164-166oC.
1H-ЯМР (CDCl3) δ: 9,73 (шир.с, 1H); 7,41 (с, 1H); 7,24-7,34 (м, 5H); 6,99 (с, 1H); 3,61 (с, 2H); 3,56 (с, 2H); 2,86-2,97 (м, 4H); 1,97-1,05 (м, 2H); 1,76-1,88 (м, 2H); 1,65-1,70 (м, 2H); 1,26-1,38 (м, 5H).
13C-ЯМР (CDCl3) 177,7; 163,6; 158,3; 145,2; 129,5; 128,3; 127,3; 122,2; 116,8; 116,4; 91,6; 63,2; 53,6; 36,1; 35,5; 35,3; 31,9; 25,4; 25,0.
ИК (KBr) 3150, 3096, 2930, 1705, 1634, 1495, 1345 см-1.
EIMS: 389 (M+), 372, 298, 202, 172, 108, 91 (основание).
BPMC для C24H27N3O:
Вычислено: 389,2104.
Найдено: 389,2107.
Пример 33.
3-[2-[1-(Фенилметил)-4-пиперидин] этил] -5,6,8-тригидро-7H- изоксазоло[4,5-g]хинолин-7-он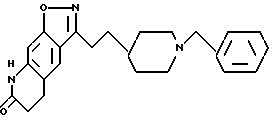
а) 6-Ацетил-3,4-дигидро-7-гидрокси-2H-хинолин-2-он
Ацетилхлорид (2,0 мл, 28,1 мл) добавляли к смеси 3,4-дигидро- 7-метокси-2H-хинолин-2-она (1,99 г, 11,2 мМ) в 1,2-дихлорэтане (30 мл). Полученную смесь охлаждали до 0oC и порциями добавляли AlCl3 (6,0 г, 44,98 мМ). Реакционную смесь нагревали с обратным холодильником в течение 2 ч. После этого реакционную смесь осторожно выливали на воду со льдом, размешивали как минимум в течение 1 ч (а как максимум в течение ночи) и экстрагировали метиленхлоридом. Органический слой промывали солевым раствором, осушали сульфатом магния, фильтровали и концентрировали, в результате чего получали целевое соединение (1,89 г, 82%) в виде беловатого твердого продукта.
1H-ЯМР (DMCO-d6) δ: : 12,4 (С, 1H); 10,4 (с, 1H); 7,74 (с, 1H); 6,38 (с, 1H); 2,86 (т, 2H, J = 7,4 Гц); 2,56 (с, 3H); 2,46-2,56 (м, 1H).
о) 6-Ацетил-3,4-дигидро-7-гидрокси-2H-хинолин-2-он, 6-оксим
Водный раствор гидрохлорида гидроксиамина (1,47 г, 21,2 мМ) и тригидрата ацетата натрия (3,0 г, 22,1 мМ) добавляли к смеси кетона, полученного в стадии (A) (1,89 г, 9,2 мМ) в EtOH (100 мл). Полученную смесь нагревали с обратным холодильником в течение 4 ч. Затем реакционную смесь концентрировали, а остаток размешивали с водой. Полученное твердое вещество собирали путем фильтрации и промывали этанолом и этиловым эфиром, в результате чего получали целевое соединение (1,67 г, 82%) в виде беловатого твердого вещества.
Т.пл. 286,5-287,7oC (разлож.).
1H-ЯМР (DMCO-d6) δ 11,7 (С, 1H); 11,3 (с, 1H); 10,1 (с, 1H); 7,28 (с, 1H); 6,37 (с, 1H); 2,81 (т, 2H, J = 7,5 Гц); 2,43 (т, 2H, J = 7,5 Гц); 2,21 (с, 3H).
c) 6-Ацетил-3,4-дигидро-7-гидрокси-2H-хинолин-2-он, 6-оксимацетат
Гетерогенную смесь оксима, полученного в стадии b (1,67 г, 7,57 мМ) в уксусном ангидриде (13 мл), нагревали при 80oC в течение полутора часов. Реакционную смесь концентрировали, а избыток уксусного ангидрида удаляли путем концентрирования из толуола. После осушки получали целевое соединение (1,7 г, 86%) в виде беловатого твердого продукта.
1H-ЯМР (DMCO-d6) δ 11,0 (с, 1H); 10,2 (с, 1H); 7,36 (с, 1H); 6,44 (с, 1H); 2,82 (т, 2H, J=7,5 Гц); 2,45 (т, 2H, J=7,5 Гц); 2,38 (с, 3H); 2,22 (с, 3H).
o) 5,6,8-Тригидро-7H-изоксазоло-[4,5-g]хинолин-7-он
Смесь оксимацетата, полученного в стадии (c) (1,58 г, 6,02 мМ), и пиридина (4,8 мл, 60,2 мМ) в ДМФ (75 мл) нагревали при 125-130oC в течение 2 ч. Реакционную смесь концентрировали в вакууме, а остаток очищали путем перекристаллизации EtOAc, в результате чего получали целевое соединение (0,80 г, 66%) в виде бледно-желтого твердого продукта.
Т.пл. 309-311oC (разлож.).
1H-ЯМР (DMCO-d6) δ 10,4 (с, 1H); 7,62 (с, 1H); 7,02 (с, 1H); 2,99 (т, 2H, J=7,4 Гц); 2,49-2,53 (м, 2H); 2,47 (с, 3H).
e) 4-[2-[5,6,8-Тригидро-7H-изоксазоло[4,5-g] хинолин-7-он-3-ил- этил]-1-пиперидинкарбоновая кислота, 1-(1,1-диметилэтил)сложный эфир
Повторяли процедуру, описанную в примере 7a, используя бензизоксазол, полученный в стадии d (0,47 г, 2,3 мМ), 1М LDA (8,1 мл, 8,1 мМ) и 1-(1,1-диметилэтил)сложный эфир 4-иодометил-1-пиперидинкарбоновой кислоты (0,75 г, 2,3 мМ) в сухом ТГФ (150 мл), за исключением того, что после добавления реагентов смесь размешивали 3,5 ч при -78oC. Затем проводили еще одну реакцию, которую осуществляли аналогичным образом с использованием бензизоксазола, полученного в стадии d (0,206 г, 1,02 мМ). Неочищенный продукт из этих двух реакций объединяли и очищали с помощью хроматографии (EtOAc), получали целевое соединение (0,72 г, 54%) в виде белого твердого вещества.
1H-ЯМР (DMCO-d6) δ: 10,4 (с, 1H); 7,67 (с, 1H); 7,02 (с, 1H); 3,93 (шир. д, 2H, J=13,4 Гц); 2,89-3,31 (м, 4H); 2,57-2,75 (шир.м, 2H); 2,49-2,53 (м, 2H); 1,64-1,72 (м, 4H); 1,38 (с, 9H); 1,36-1,50 (м, 1H); 1,01-1,16 (м, 2H).
f) 3-[2-[1-(Фенилметил)-4-пиперидинил]этил]-5,6,8-тригидро-7H- изоксазоло[4,5-g]хинолин-7-он
Повторяли процедуру, описанную в примере 12f, используя пиперидин, полученный в стадии (e) (0,55 г, 13,37 мМ), и ТФК (3,5 мл) в CH2Cl2 (14 мл), и Na2CO3 (0,758 г, 7,14 мМ), и бензилбромид (0,23 мл, 1,93 мМ) в ДМФ (14 мл). После очистки с помощью хроматографии (5 --> 30% MeOH - CH2Cl2) получали целевое соединение в виде белого твердого продукта. Образец перекристаллизовывали из EtOAc-MeOH.
Т.пл.: 164,4-165,9oC.
1H-ЯМР (CDCl3) δ 9,37 (c, 1H); 7,38 (c, 1H); 7,22-7,36 (м, 5H); 7,01 (с, 1H); 3,55 (с, 2H); 3,09 (т, 2H, J=7,4 Гц); 2,91-2,97 (м, 4H); 2,70 (т, 2H, J=7,4 Гц); 2,01 (шир.т, 2H, J=10,3); 1,75-1,80 (м, 4H); 1,39-1,50 (м, 3H).
1C-ЯМР (CDCl3) δ 170,6; 162,0; 158,2; 141,1; 140,9; 128,7; 128,2; 126,8; 121,2; 120,3; 115,7; 94,6; 62,5; 53,2; 34,9; 33,8; 31,6; 30,2; 24,8; 21,9.
ИК (KBr) 3172, 3088, 2821, 1694, 1631, 1447, 1381 см-1.
EIMS 389 (M+), 388, 372, 298, 185, 172, 91 (основание).
BPMC для C24H27N3O2:
Вычислено: 389, 2104.
Найдено: 389, 2102.
Пример 34.
6,8-Дигидро-3-[2-[1-(фенилметил)-4-пиперидинил] этил]-7H- пирроло[5,4-g] -1,2-бензизоксазол-7-он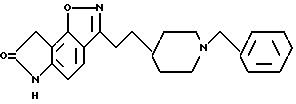
a) 5-Ацетил-1,3-дигидро-4-гидрокси-2H-индол-2-он
Хорошо размешанную смесь 4-ацетилокси-1,3-дигидро-2H-индол-2-она (0,876 г, 4,58 мМ) и AlCl3 (1,83 г, 13,7 мМ) помещали в колбу, имеющую форму слезы, и эту колбу погружали в масляную баню, предварительно нагретую до 190oC, а затем нагревали в течение 1 ч. Затем в охлажденную реакционную смесь осторожно добавляли воду со льдом и размешивали в течение 1,5 ч. Затем добавляли конц. HCl и смесь экстрагировали этилацетатом. Органический слой осушали сульфатом магния, фильтровали и концентрировали. После очистки с помощью флеш-хроматографии на силикагеле (I ___→ 3% MeOH-CH2Cl2) получали целевое соединение (0,441 г, 50%) в виде беловатого твердого продукта.
1H-ЯМР (DMCO-d6) δ: 12,6 (с, 1H); 10,8 (с, 1H); 7,85 (д, 1H, J = 8,4 Гц); 6,49 (д, 1H, J=8,4 Гц); 3,41 (с, 2H); 2,57 (с, 3H).
o) 5-Ацетил-1,3-дигидро-4-гидрокси-2H-индол-2-он, 5-оксим)
Повторяли процедуру, описанную в примере 33b, используя кетон, полученный в стадии (a), (0,40 г, 2,09 мМ), водный раствор гидрохлорида NH2OH (0,29 г, 4,18 мМ) и тригидрат NaOAc (0,569 г, 4,18 мМ) в EtOH (32 мл). Через 3 ч смесь разбавляли водой и экстрагировали этилацетатом. Органический слой осушали сульфатом магния, фильтровали и концентрировали, в результате с чего получали целевое соединение (0,436 г, с количественным выходом) в виде беловатого твердого вещества.
1H-ЯМР (DMCO-d6) δ: 12,0 (с, 1H); 11,4 (с, 1H); 10,4 (с, 1H); 7,35 (д, 1H, J=8,2 Гц); 6,41 (9д, 1H, J=8,3 Гц); 3,34 (с, 2H); 2,22 (с, 3H).
c) 5-Ацетил-1,3-дигидро-4-гидрокси-2H-индол-2-он, 5-оксимацетат
Повторяли процедуру, описанную в примере 33c, используя оксим, полученный в стадии (b) (0,392 г, 1,9 мМ) в Ac2O (10 мл). Полученное твердое вещество восстанавливали из любых неорганических солей, полученных в предыдущей стадии, путем размешивания в H2O. После фильтрации и осушки получали целевое соединение (0,417 г, 88%) в виде красновато-розоватого твердого вещества.
1H-ЯМР (DMCO-d6) δ 11,45 (с, 1H); 10,6 (с, 1H); 7,50 (д, 1H, J=8,2 Гц); 6,49 (д, 1H, J=8,2 Гц); 3,40 (с, 2H); 2,41 (с, 3H), 2,23 (с, 3H).
d) 6,8-Дигидро-3-метил-7H-пирроло[5,4-g]-1,2-бензизоксазол-7-он
Повторяли процедуру, описанную в примере 33b, используя оксимацетат, полученный в стадии (c) (0,334 г, 1,35 мМ), и пиридин (0,55 мл, 6,75 мМ) в ДМФ (25 мл). После обработки остаток очищали с помощью флеш-хроматографии на силикагеле (50 ___→ 75% EtOAc-гексаны) и получали целевое соединение (0,086 г, 34%) в виде белого твердого продукта.
Т.пл. 259-260oC (разлож.).
1H-ЯМР (DMCO-d6) δ: 10,79 (с, 1H); 7,67 (д, 1H, J=8,5 Гц); 6,92 (д, 1H, J=8,3 Гц); 3,73 (с, 2H); 2,49 (с, 3H).
e) 4-[2-[6,8-Дигидро-7H-пирроло-[5,4-g] -1,2-бензизоксазол- 7-он-3-ил] этил]-1-пиперидинкарбоновая кислота, 1-(1,1-диметилэтил)сложный эфир
Повторяли процедуру, описанную в примере 7a, используя бензизоксазол, полученный в стадии (d) (0,040 г, 0,213 мМ), 1М LDA (0,85 мл, 0,85 мМ) и 1-(1,1-диметилэтил)сложный эфир 4-иодометил-1-пиперидинкарбоновой кислоты (0,078 г, 1,234 мМ) в сухом ТГФ (20 мл), за исключением того, что после добавления реагентов смесь размешивали в течение 4 ч при -78oC. После очистки с помощью хроматографии (25 ___→ 45% EtOAc-CH2Cl2) получали целевое соединение (0,042 г, 51%) в виде бледно-желтого твердого продукта.
1H-ЯМР (CDCl3) δ: 8,85 (с, 1H); 7,53 (д, 1H, J= 8,1 Гц); 6,95 (д, 1H, J= 8,3 Гц); 4,08-4,14 (м, 2H); 3,78 (с, 2H); 2,99 (т, 2H, J=7,8 Гц); 2,68 (шир. т, 2H, J=12,1 Гц); 1,73-1,84 (м, 4H); 1,46-1,60 (м, 1H); 1,46 (с, 9H); 1,17 (ддд, 2H, J=23,2 Гц; J=12,1 Гц; J=4,3 Гц).
f) 6,8-Дигидро-3-[2-[1-(фенилметил-4-пиперидинил] этил] - 7H-пирроло[5,4-g]-1,2-бензизоксазол-7-он
Повторяли процедуру, описанную в примере 12f, используя пиперидин, полученный в стадии (e) (0,042 г, 0,109 мМ), ТФК (1,5 мл) в CH2Cl2 (6 мл), Na2CO3 (0,058 г, 0,545 мМ) и бензилбромид (0,016 мл, 0,131 мМ) в ДМФ (6 мл). После очистки с помощью хроматографии (I ___→ 10% MeOH-CH2Cl2) получали целевое соединение (0,018 г, 44%) в виде беловатого твердого продукта. Т.пл. 188-189oC.
1H-ЯМР (CDCl3) δ 9,59 (с, 1H); 7,51 (д, 1H, J= 8,1 Гц); 7,23-7,33 (м, 5H); 6,94 (д, 1H, J=8,2 Гц); 3,77 (с, 2H); 3,56 (с, 2H); 2,93-2,99 (м, 2H); 2,02 (шир.т, 2H, J=10,7 Гц); 1,75-1,79 (м, 4H); 1,26-1,39 (м, 3H).
13C-ЯМР (CDCl3) δ: 177,2; 158,9; 158,5; 145,0; 129,4; 128,3; 127,2; 121,5; 117,9; 107,1; 105,1; 63,2; 53,5; 35,1; 34,1; 33,8; 31,7; 29,7; 22,5.
Пример 35.
5,7-Дигидро-3-[2-[1-(фенилметил)-4-пиперидинил] этил- 6H-пирроло-[5,4-f] -1,2-бензизоксазол-6-он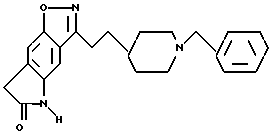
a) 6-Ацетил-1,3-дигидро-5-гидрокси-2H-индол-2-он
Повторяли процедуру, описанную в примере 33a, используя 1,3-дигидро-5-метокси-2H-индол-2-он (4,4 г, 26,96 мМ), ацетилхлорид (4,8 мл, 67,41 мМ) и AlCl3 (14,4 г, 107,8 мМ) в 1,2-дихлорэтане (210 мл) в течение 6 ч. После обработки водой и размешивания в течение ночи получали осадок. Этот желтый твердый осадок собирали путем фильтрации и осушали, в результате чего получали целевое соединение (2,7 г, 52%).
1H-ЯМР (DMCO-d6) δ: 12,0 (с, 1H); 10,4 (с, 1H); 7,12 (с, 1H); 6,89 (с, 1H); 3,54 (с, 2H); 2,61 (с, 3H).
о) 6-Ацетил-1,3-дигидро-5-гидрокси-2H-индол-2-он, 6-оксим
Повторяли процедуру, описанную в примере 33b, используя кетон, полученный в стадии (a) (2,7 г, 14,4 мМ), водный раствор NH2OH-гидрохлорида (2,26 г, 32,5 мМ) и NaOAc-тригидрат (4,6 г, 33,9 мМ) в EtOH (155 мл), и получали целевое соединение (2,7 г, 93%) в виде беловатого твердого вещества.
1H-ЯМР (LMCO-d6) δ: 11,5 (с, 1H); 11,3 (с, 1H); 10,2 (с, 1H); 6,81 (с, 1H); 6,78 (с, 1H); 43,44 (с, 2H); 2,22 (с, 3H).
c) 6-Ацетил-1,3-дигидро-5-гидрокси-2H-индол-2-он, 6-оксимацетат
Повторяли процедуру, описанную в примере 33c, используя оксим, полученный в стадии (d) (2,7 г, 13,1 мМ) в Ac2O (65 мл), в течение 3 ч, и получали целевое соединение (2,93 г, 90%) в виде беловатого твердого продукта.
1H-ЯМР (DMCO-d6) δ: 10,4 (с, 1H); 10,2 (с, 1H); 6,86 (с, 1H); 6,81 (с, 1H); 3,48 (с, 2H); 2,37 (с, 3H); 2,22 (с, 3H).
d) 5,7-Дигидро-3-метил-6H-пирроло[5,4-f]-1,2-бензизоксазол-6-он
Повторяли процедуру, описанную в примере 33d, используя оксимацетат, полученный в стадии (c) (2,75 г, 10,97 мМ), и пиридин (8,9 мл, 109,7 мМ) в ДМФ (110 мл). После концентрирования в вакууме остаток очищали с помощью флэш-хроматографии на силикагеле (2% MeOH-CH2Cl2) и получали (0,245 г, 12%) в виде светло-желтого твердого продукта.
1H-ЯМР (DMCO-d6) δ 10,6 (с, 1H); 7,58 (с, 1H); 7,03 (с, 21H); 3,63 (с, 2H); 2,50 (с, 3H).
e) 4-[2-[5,7-Дигидро-6H-пирроло[5,4-f] -1,2-бензизоксазол- 6-он-3-ил] этил]-1-пиперидинкарбоновая кислота, 1-(1,1-диметилэтил)сложный эфир
Повторяли процедуру, описанную в примере 7a, используя бензизоксазол, полученный в стадии (d) (0,152 г, 0,808 мМ), 1М LDA (3,2 мл, 3,2 мМ) и 1-(1,1-диметилэтил)сложный эфир 4-иодометил-1-пиперидинкарбоновой кислоты (0,135 г, 0,970 мМ) в сухом ТГФ (30 мл), за исключением того, что после добавления реагентов смесь размешивали в течение 4,5 ч при -78oC. После очистки с помощью хроматографии (50% ___→ EtOAc-CH2Cl2) получали неразделимую смесь (0,153 г, 1,6:1) исходного материала и целевого соединения соответственно в виде бледно-желтого мягкого твердого вещества. В другом эксперименте, проведенном отдельно, получали лучшее отношение исходного материала к целевому соединению, а именно 1:3.
1H-ЯМР (CDCl3) δ: 9,84 (с, 1H); 7,45 (с, 1H); 7,05 (с, 1H); 4,05-4,15 (м, 2H); 3,69 (с, 2H); 2,96 (т, 2H, J=7,8 Гц); 2,68 (шир.т, 2H, J=11,8 Гц); 1,72-1,82 (м, 4H); 1,45 (с, 9H); 1,43-1,53 (м, 1H); 1,15 (ддд, 2H, J=23,6 Гц, J=12,1 Гц, J=4,0 Гц).
f) 5,7-Дигидро-3-[2-[1-(фенилметил)-4-пиперидинил] этил] -6H- пирроло[5,4-f]-1,1-бензизоксазол-6-он
Повторяли процедуру, описанную в примере 12f, используя смесь, полученную в стадии (a) (0,153), и ТФК (1,5 мл) в метиленхлориде (6 мл). В стадии алкилирования использовали лишь часть неочищенной солевой смеси (0,081 г): Na2CO3 (0,041 г, 0,388 мМ) и бензилбромида (0,012 мл, 0,101 мМ) в ДМФ (6 мл). Внимание: для стадии алкилирования растворитель (ДМФ) тщательно дегазировали с использованием серебра, а обработку насыщенным NaHCO3 не проводили (вместо этого был использован солевой раствор). После очистки (2 ____→ 6% MeOH-CH2Cl2) получали целевое соединение (0,018 г, 60%) по отношению к нужному исходному материалу в виде светло-желтого твердого продукта.
Т.пл. 204,5-205,5oC (разлож.).
1H-ЯМР (CDCl3) δ: 8,09 (с, 1H); 7,46 (с, 1H); 7,27-7,33 (м, 5H); 6,99 (с, 1H); 3,68 (с, 2H); 3,50-3,52 (м, 2H); 2,90-2,99 (м, 4H); 1,92-2,05 (м, 2H); 1,70-1,80 (м, 4H); 1,30-1,40 (м, 4H).
Пример 36.
3-[2-[1-(Фенилметил)-4-пиперидинил]этил]-1H-индазолмалеат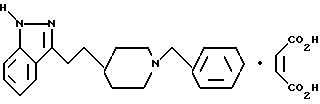
a) 1-[2-Фторфенил)-3-[1-(фенилметил)-4-пиперидинил]пропанон
Гексаметилдисилазид лития (1М, 14,5 мл, 14,5 мМ) добавляли к раствору о-фтороацетофенона (2,0 г, 14,5 мМ) в ТГФ (100 мл) при -78oC. Полученную смесь оставляли нагреваться до -20oC в течение 30-60 мин, а затем снова охлаждали до -78oC. Затем добавляли раствор 1-(1,1-диметилэтил)сложного эфира 4-карбоксальдегид-1-пиперидинкарбоновой кислоты (2,94 г, 14,5 мМ) в ТФГ (20 мл). Смесь выдерживали 30 мин при -78oC, а затем оставляли нагреваться до комнатной температуры и размешивали в течение 40 мин. После этого добавляли NH4Cl и реакционную смесь экстрагировали этилацетатом. Органический слой осушали сульфатом магния, фильтровали и концентрировали. После очистки с помощью флеш-хроматографии на силикагеле (30 ___→ 50% EtOAc/-CH2Cl2) получали желтый маслообразный продукт 32,14 г). 1H-ЯМР-анализ обнаружил смесь соединений (два главных компонента). Дальнейшую очистку не проводили. И в таком виде смесь использовали в следующей стадии.
Часть смеси, полученной выше (0,661 г), и PtO2 (0,070 г, 0,31 мМ) в EtOH подвергали гидрогенизации в шейкере Парра при давлении 48 фунт/кв. дюйм (3,31 бар) в течение 2 ч. Затем смесь фильтровали через прокладку из Целита и фильтрат концентрировали. Затем осуществляли еще одну реакцию с использованием вышеуказанной смеси (1,00 г). Неочищенные продукты из этих реакций объединяли и очищали с помощью флеш-хроматографии (I ___→ 3% MeOH-CH2Cl2), а затем 30% EtOAc-гексаны), в результате чего получали целевое соединение (0,192 г, 5,3%) (полный) в виде бесцветного маслообразного продукта.
1H-ЯМР (CDCl3) δ 7,85 (дт, 1H, J=7,6 Гц, J=1,8 Гц); 7,47-7,56 (м, 1H); 7,09-7,33 (м, 7H); 43,49 (с, 2H); 2,95-3,03 (м, 2H); 2,89 (шир.д, 2H, J=11,4 Гц); 1,94 (шир.т, 2H, J=11,1 Гц); 1,63-1,77 (м, 4H); 1,24-1,39 (м, 3H).
EIMS 325 (M+), 202, 188, 172, 91, 66 (основание).
b) 3-[2-[1-Фенилметил)-4-пиперидинил]этил]-1H-индазолмалеат
Смесь кетона, полученного в стадии (b) (0,178 г, 0,55 мМ) в безводном гидразине (10 мл), нагревали с обратным холодильником в течение 3 ч. Затем реакционную смесь охлаждали, добавляли воду, а смесь экстрагировали этиленхлоридом. Органический слой осушали сульфатом магния, фильтровали и концентрировали. После очистки с помощью радиальной хроматографии на силикагеле (CH2Cl2 ___→ 10% MeOH-CH2Cl2) получали целевое соединение - свободное основание (0,047 г, 27%) в виде бесцветного маслообразного продукта.
Малеат (соль) получали путем добавления раствора малеиновой кислоты (0,010 г, 0,085 мМ) в Et2O (5 мл) к раствору свободного основания (0,027 г, 0,085 мМ) в Et2O (75 мл). Полученное твердое белое вещество собирали путем фильтрации и получали целевое соединение (0,014 г, 38%).
Т.пл. 151,5-152,5oC.
1H-ЯМР (DMCO-d6) δ: 12,64 (с, 1H); 7,72 (д, 1H, J=8,0 Гц); 7,44-7,47 (м, 6H); 7,31 (т, 1H, J= 7,3 Гц); 7,06 (т, 1H, J=7,4 Гц); 6,02 (с, 2H); 4,24 (шир. с, 2H); 2,93 (т, 2H, J=7,6 Гц); 1,9-2,00 (м, 2H); 1,60-1,80 (м, 3H); 1,30-1,50 (м, 4H).
В отношении биологических данных.
Представленные в таблице данные свидетельствуют о способности соединений ингибировать ацетилхолинэстеразу. Значения IC50 были получены методом, упомянутым в описании и описанным в статье Ellman et al., Biochem. Pharm., 1, 88 (1961).
Пример 37.
Препарат 1 (таблетки)
(1) Соединение примера 4 (малеат) - 50 г
(2) Лактоза - 100 г
(3) Кукурузный крахмал - 15 г
(4) Кальций карбоксиметилцеллюлоза - 44 г
(5) Стеарат магния - 1 г
1000 таблеток - 210 г
Компоненты (1), (2) и (3) и 30 г компонента (4) смешивали с водой, сушили в вакууме и гранулировали. Полученный гранулированный порошок смешивали с 14 г компонента (4) и 1 г компонента (5) и смесь загружали в таблетирующую машину. Получали 1000 таблеток, содержащих 50 мг компонента (1) в каждой таблетке.
Пример 38.
Препарат 2
(1) Соединение примера 9 (малеат) - 10 г
(2) Лактоза - 4,5 г
(3) Кукурузный крахмал - 4,5 г
(4) Стеарат магния - 1 г
100 капсул - 20 г
Все компоненты тщательно перемешивали и смесью наполняли желатиновые капсулы. Получали 100 капсул, содержащих 100 мг компонента (1) в каждой капсуле.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ АНТРАНИЛОВОЙ КИСЛОТЫ ИЛИ ИХ ФАРМАКОЛОГИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ, ПРОМЕЖУТОЧНЫЕ ПРОДУКТЫ ДЛЯ ИХ ПОЛУЧЕНИЯ И ЛЕКАРСТВЕННЫЙ ПРЕПАРАТ НА ИХ ОСНОВЕ | 1994 |
|
RU2128644C1 |
| СОЕДИНЕНИЯ АЗОЛА, СПОСОБЫ ИХ ПОЛУЧЕНИЯ, ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ, СПОСОБЫ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ОБЛАДАЮЩАЯ ПРОТИВОГРИБКОВОЙ АКТИВНОСТЬЮ | 1995 |
|
RU2142947C1 |
| ПРОИЗВОДНЫЕ КОНДЕНСИРОВАННЫХ ПОЛИЦИКЛИЧЕСКИХ ГЕТЕРОЦИКЛИЧЕСКИХ СОЕДИНЕНИЙ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1996 |
|
RU2167877C2 |
| СОЕДИНЕНИЯ С КОНДЕНСИРОВАННЫМ 1,3-ДИГИДРОИМИДАЗОЛЬНЫМ ЦИКЛОМ | 2003 |
|
RU2328498C9 |
| СОЕДИНЕНИЕ, ЕГО ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМАЯ СОЛЬ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1992 |
|
RU2095366C1 |
| ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМАЯ СОЛЬ, ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ | 1994 |
|
RU2176640C2 |
| НОВЫЕ КОНДЕНСИРОВАННЫЕ ПРОИЗВОДНЫЕ ИМИДАЗОЛА, ИНГИБИТОР ДИПЕПТИДИЛПЕПТИДАЗЫ IV, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СПОСОБ ЛЕЧЕНИЯ И ПРИМЕНЕНИЕ НА ИХ ОСНОВЕ | 2003 |
|
RU2297418C9 |
| ХИНОЛОНОВОЕ СОЕДИНЕНИЕ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2009 |
|
RU2544530C2 |
| ПРОИЗВОДНЫЕ ЭТАНОЛАМИНА, СПОСОБ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБ ЛЕЧЕНИЯ | 1993 |
|
RU2125983C1 |
| КОНДЕНСИРОВАННЫЙ ПИРИДАЗИН ИЛИ ЕГО ФАРМАКОЛОГИЧЕСКИ ПРИЕМЛЕМАЯ СОЛЬ, СРЕДСТВО, ПРОЯВЛЯЮЩЕЕ ИНГИБИРУЮЩУЮ АКТИВНОСТЬ В ОТНОШЕНИИ ЦИКЛИЧЕСКОЙ ГМФ- ФОСФОДИЭСТЕРАЗЫ | 1995 |
|
RU2128175C1 |
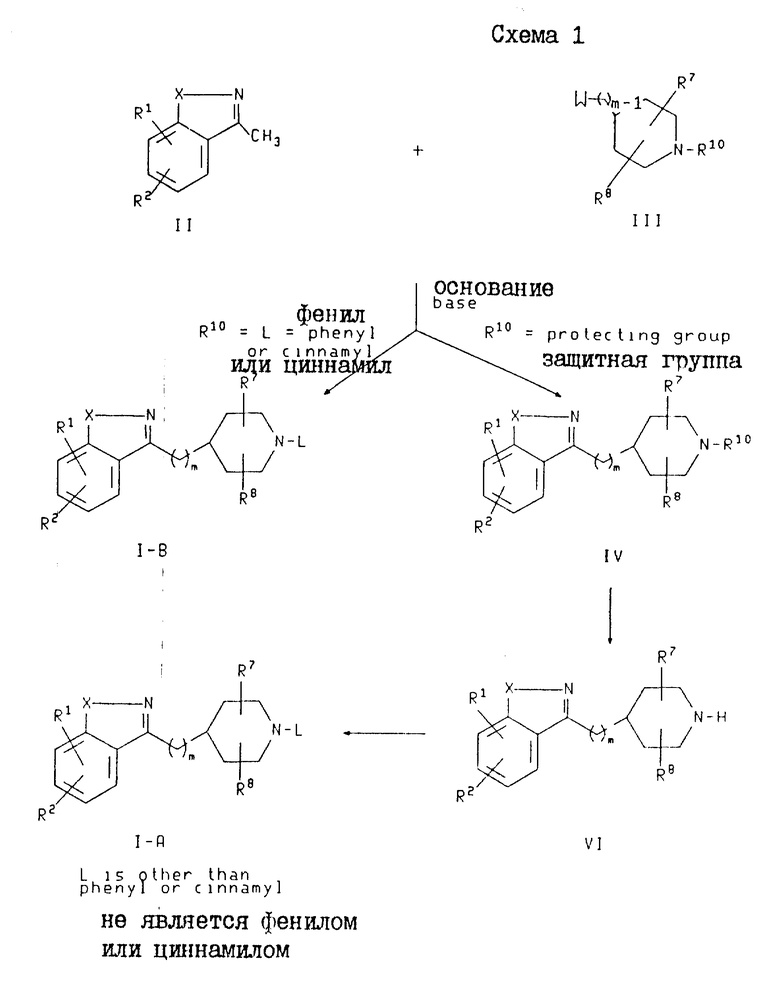
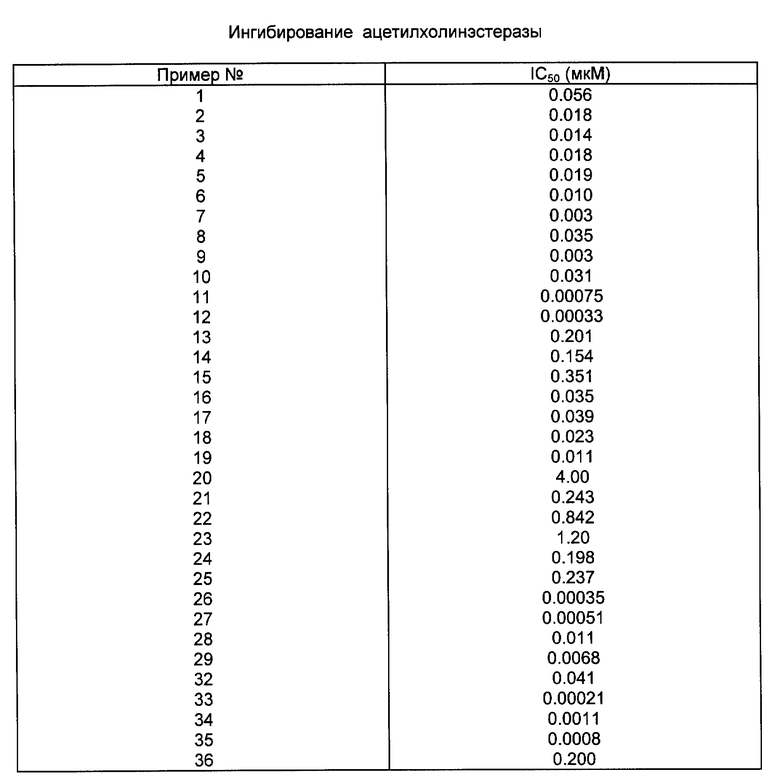


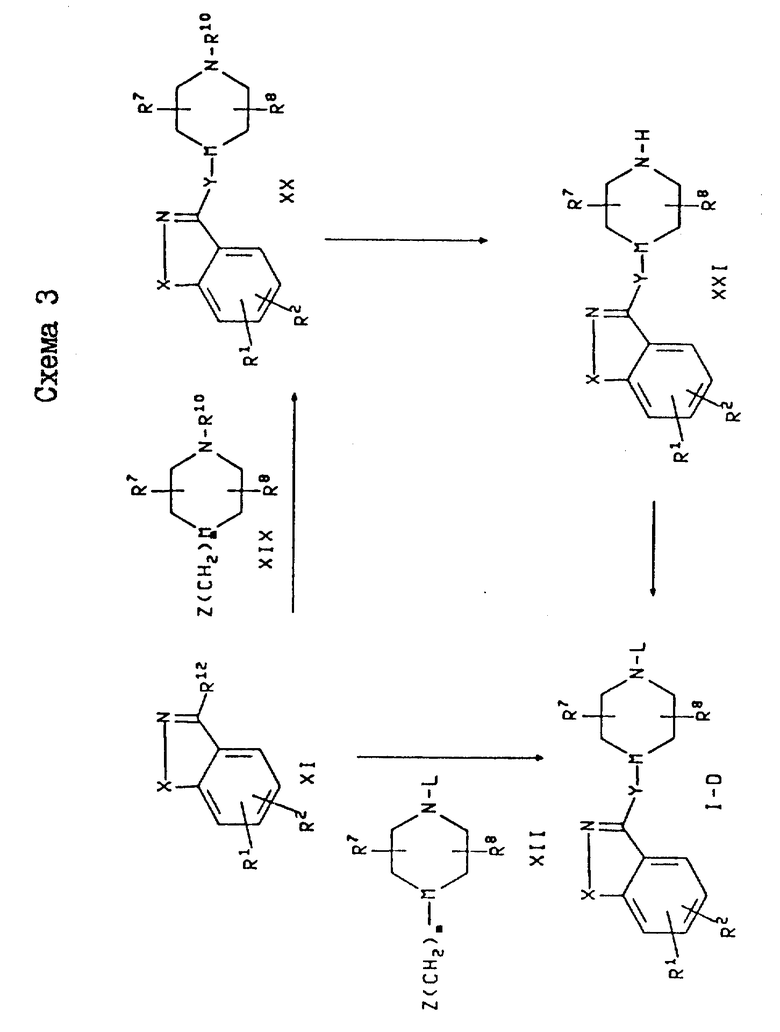
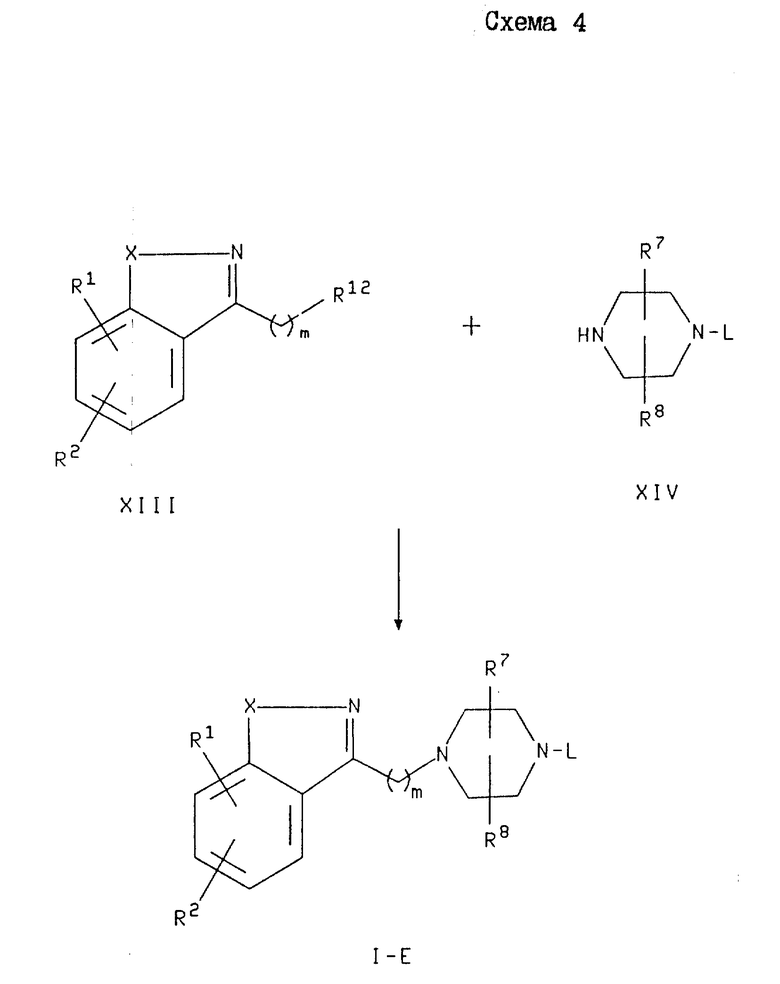
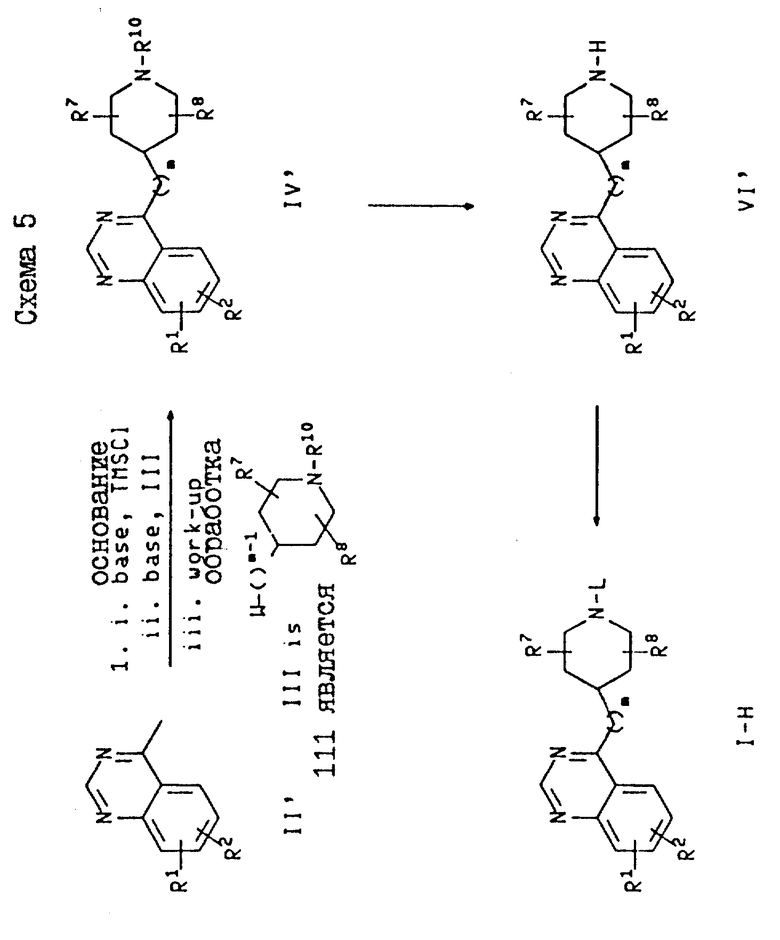
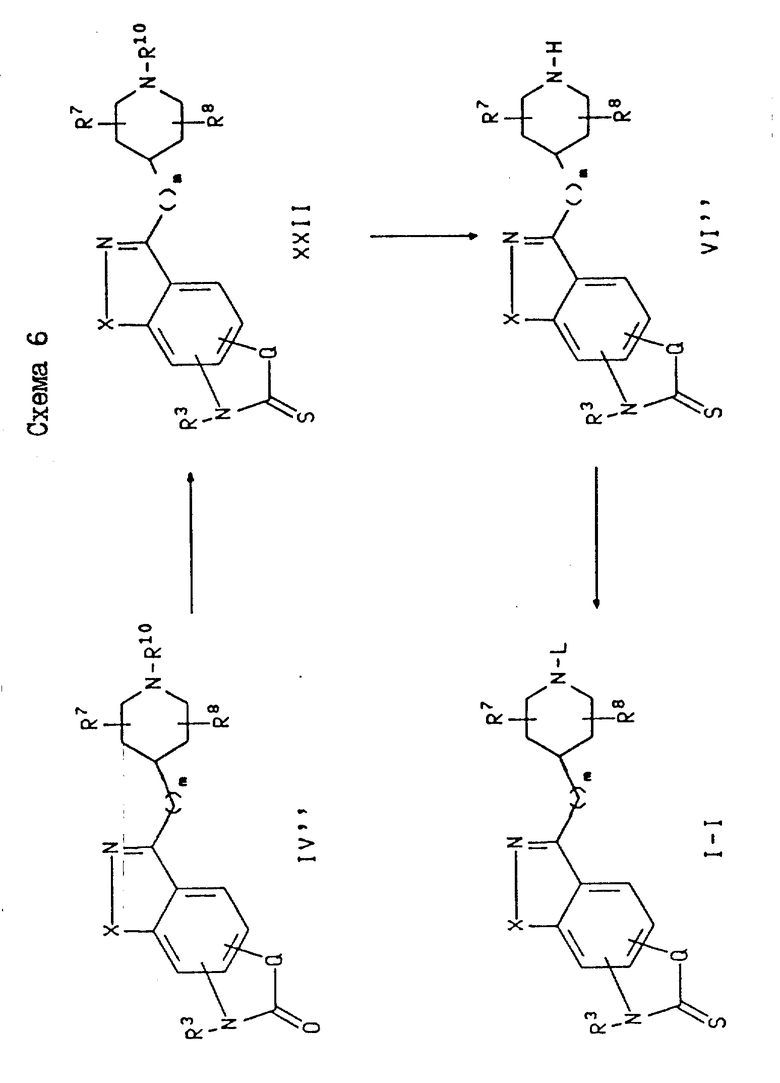
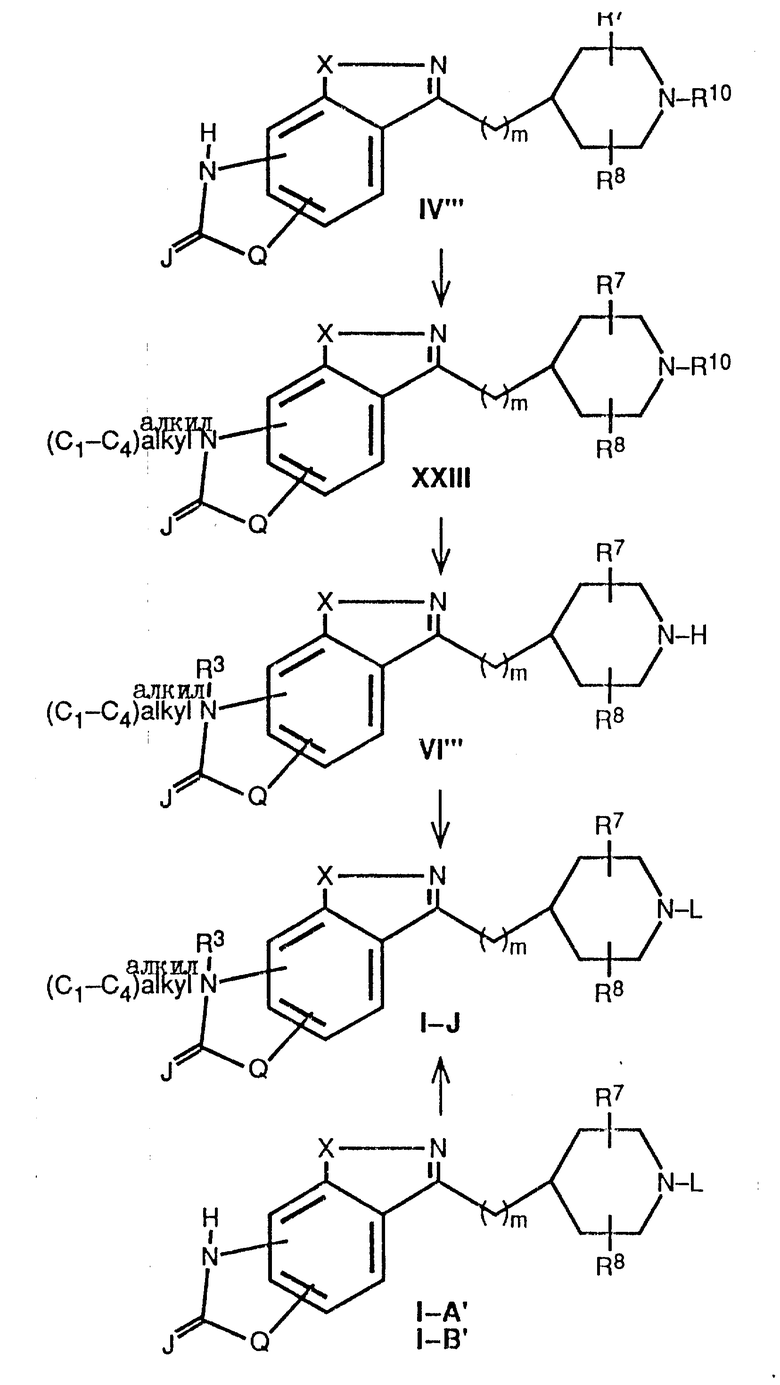
Использование в химии гетероциклических соединений, обладающих ингибирующей холинэстеразу активностью. Раскрыты гетероцикло-циклические производные аминов формулы I, где Х, M, L, R1, R2, R7, R8 представлены на с. 121-124 описания, или их соли, промежуточные соединения, фармацевтическая композиция и способ ингибирования холинэстеразы на основе соединений формулы I, способ получения этих соединений. 6 с. и 4 з.п. ф-лы, 1 табл. 
где R1 и R2 независимо выбирают из водорода, (C1-C6)-алкила, (C1-C6)-алкокси, циано-, амино-, амидо-, гидроксигруппы, галогена, ацетамидо-, бензамидо-, бензолсульфонамидо-, морфолиногруппы, R1 и R2 взятые вместе могут образовать пирролидон, который может быть замещен (C1-C6)-алкилом, а также пиперидиновое кольцо и хинолингруппу;
R7 и R8 - водород;
X представляет собой кислород, серу, -NH, -CH=CH-, -CH-N-группу;
Y представляет собой (C1-C6)-алкил, (C1-C6)-алкокси, (C2-C6)-алкилен, (C1-C6)- алкиламиногруппу;
M представляет собой азот или -CH-группу;
L представляет собой фенил-(C1-C6)-алкил, фенильная группа которого может быть замещена, но не обязательно, галогеном, или галогентиенил-(С1-С6)-алкил, или (С1-С6)-алкилтиазо-(C1-C6)-алкил,
или их фармацевтически приемлемые соли.
3-[2-[1-(фенилметил)-4-пиперидинил]этил]-1,2-бензизоксазол;
5-метил-3-[2-[1-(фенилметил)-4-пиперинидил]этил]- 1,2-бензизоксазол;
5,5-диметил-3-[2-[1-(фенилметил)-4-пиперинидинил] этил] -1,2-бензизоксазол;
5-метокси-3-[2-[1-фенилметил)-4-пиперинидил]этил]-1,2-бензизоксазол;
6-метокси-3-[2-[1-(фенилметил)-4-пиперинидил]этил]-1,2-бензизоксазол;
7-метокси-3-[2-[1-(фенилметил) -4-пиперинидил]этил]-1,2-бензизоксазол;
6-ацетамидо-3-[2-[1- (фенилметил)-4-пиперинидил]этил]-1,2-бензизоксазол;
6-амино-3-[2-1-(фенилметил)-4-пиперидинил]этил]-1,2-бензизоксазол;
6-бензамид-3-[2-[1-(фенилметил)-4-пиперидинил]этил]-1,2-бензизоксазол;
6-бензолсульфонамид-3-[2-[1-(фенилметил)-4-пиперинидил]этил] 1,2-бензизоксазол;
6-(4-морфолинил)-3-[2-[1-(фенилметил)-4-пиперинидил] этил]-1,2-бензизоксазол;
5,7-дигидро-3-[2-[1-(фенилметил)-4-пиперинидил] этил]- 6Н-пирроло[4,5-f] -1,2-бензизоксазол-6-он;
3-[2-[1-(фенилметил)-4-пиперинидил]этил]-1,2-бензизоксазол;
6-гидрокси-3-[2-[1-(фенилметил) -4-пиперидил]этил]-1,2-бензизоксазол;
6-бромо-3-[2-[1-(фенилметил)-4-пиперидил]этил]-1,2-бензизоксазол;
6-циано-3-[2-[1- (фенилметил)-4-пиперидил]этил]-1,2-бензизоксазол;
6-карбоксамид-3- [2-[1-(фенилметил)-4-пиперидил]этил]-1,2-бензизоксазол;
3-[(1-фенилметил-4-пиперидил)метокси]-1,2-бензизоксазол;
3-[(1-фенилметил-4-пиперидил)метиламино]-1,2-бензизоксазол;
3-[1-[(1-фенилметил)-4-пиперидил]этиламино]-1,2-бензизоксазол;
3-[3-[(1-(фенилметил)-4-пиперидил]пропил]-1,2-бензизоксазол;
транс-3-[2-[1-(фенилметил)-4-пиперидил]этенил]-1,2-бензизоксазол;
3-[2-[1-(фенилметил)-4-пиперазинил]этил]-1,2-бензизоксазол;
5,7-дигидро-7-метил-3-[2-[1-(фенилметил) -4-пиперидинил]этил]-6Н-пирроло[4,5-f]-1,2-бензизоксазол-6-он;
5,7-дигидро-7-этил-3-[2-[1-(фенилметил)-4-пиперидинил] этил]-6Н-пирроло[4,5-f]-1,2-бензизоксазол-6-он;
5,7-дигидро-3-[2-[1-(2-хлоро-5-тиофенметил)-4-пиперинил] этил]-6Н-пирроло[4,5-f]-1,2-бензизоксазол-6-он;
5,7-дигидро-3-[2-[1-(2-метил-4-тиазолметил)-4-пиперидинил] этил] -6Н- пирроло[4,5-f]-1,2-бензизоксазол-6-он;
3-[2-[1-(3-бромофенилметил)-4-пиперидинил] этил] -5,7- дигидро-6Н-пирроло[4,5-f]-1,2-бензизоксазол-6-он;
3-[2-[1-(4- бромофенилметил)-4-пиперидинил] этил]-5,7-дигидро-6Н-пирроло[4,5-f] -16-бензизоксазол-6-он;
5,7-дигидро-3-[3-[1-(фенилметил)-4- пиперидинил] пропил] -6Н-пирроло[4,5-f]-1,2-бензизоксазол-6-он;
3-[2-[1-(фенилметил)-4-пиперидинил] этил] -5,6,8-тригидро-7Н- изоксазоло[4,5-g]хинолин-7-он;
6,8-дигидро-3-[2-[1- (фенилметил)-4-пиперидинил]этил]-7Н-пирроло[5,4-g] -1,2-бензизоксазол-7-он;
5,7-дигидро-3-[2-[1-(фенилметил)-4-пиперидинил] этил]-6Н-пирроло[5,4 -f] -1,2-бензизоксазол-6-он;
3-[2-[1-(фенилметил) -4-пиперидинил]этил]-1Н-индазол,
или фармацевтически приемлемые соли таких соединений.
3-[2-[1-(фенилметил)-4-пиперидинил]этил]-1,2-бензизоксазол алеат;
5,6-диметил-3-[2-[1-(фенилметил)-4- пиперидинил] этил] -1,2-бензизоксазолмалеат;
5-метокси-3- [2-[1-(фенилметил)-4-пиперидинил] этил]-1,2-бензизоксазолмалеат;
7-метокси-3-[2-[1-(фенилметил)-4-пиперидинил]этил]-1,2- бензизоксазолфумарат;
6-бензамид-3-[2-[1-(фенилметил)-4- пиперинидил] этил] -1,2-бензизоксазолмалеат; и
6-бензолсульфонамид-3- [2-[1-(фенилметил)-4-пиперидинил]этил]-1,2-бензизоксазолфумарат.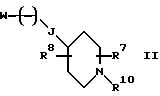
где R7 и R8 - водород;
R10 имеет значения, определенные для L в п. 1;
W - уходящая группа;
j является целым числом от 1 до 2.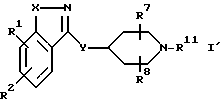
где R1, R2, R7, R8, X, Y, M определены в п. 1;
R11 имеет значения, определенные для L в п. 1.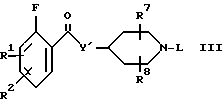
где R1, R2, R7, R8 и L определены в п. 1;
Y' представляет собой -CH=CH-(CH2)n или -(CH2)m, где n = 1 - 3, m = 1 - 3.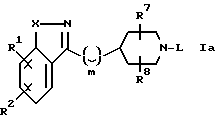
где R1, R2, R7, R8, X, L имеют значения, определенные в п. 1;
m = 1 - 3,
или их фармацевтически приемлемых солей, отличающийся тем, что соединение формулы VI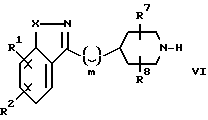
где R1, R2, R7, R8, X, m имеют указанные значения,
подвергают взаимодействию с соединением формулы
WL,
где L имеет указанные значения;
W - отщепляемая группа,
с последующим выделением целевого продукта в свободном виде или в виде фармацевтически приемлемых солей.
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| EP, 229391, кл | |||
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| EP, 296560, кл | |||
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
| EP, 428437, кл | |||
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |

