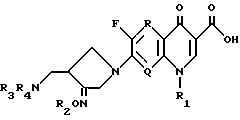
Настоящее изобретение относится к новому производному хинолин (нафтиридин) карбоновой кислоты, проявляющему превосходную антибактериальную активность. Более конкретно настоящее изобретение касается нового производного хинолин (нафтиридин) карбоновой кислоты, представленного следующей ниже формулой (I), которое имеет 4-аминометил-3-оксимпирролидиновый заместитель в положении 7 хинолонового ядра и проявляет превосходную антибактериальную активность в противоположность известным хинолоновым антибактериальным средствам, проявляющим слабую активность против штаммов грамположительных бактерий, а также имеет широкий антибактериальный спектр и сильно улучшенные фармакокинетические свойства: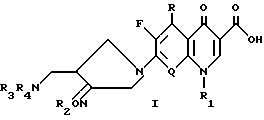
и его фармацевтически приемлемой нетоксичной соли, физиологически гидролизуемого сложного эфира, сольвата и изомера,
где
R представляет водород, метил или амино;
Q представляет C-H, C-F, C-Cl, C-OH, C-CH3, C-O-CH3 или N;
R1 представляет циклопропил, этил или фенил, который замещен одним или несколькими атомом(ами) фтора;
R2 представляет одно из следующих a)-e):
a) водород, неразветвленный или разветвленный C1-C4 аклил, циклопропил, циклопропилметил, C3-C6 алкинил, 2 - галогенэтил, метоксиметил, метоксикарбонилметил, арил или аллил,
b) группу следующей формулы (I)
где
X представляет водород, 2, 3 или 4- фтор, циан, нитро, метокси, C1-C4 алкил или 2,4 - дифтор,
c) группу следующей формулы (2)
d) гетероарилметил следующей формулы (3)
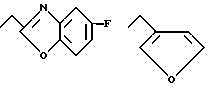
e) группу следующей формулы (4)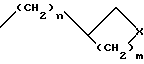
где
n=0 или 1;
m = 0,1 или 2;
X- представляет метилен, O или N;
R3 и R4 независимо друг от друга представляет водород или C1-C3 - алкил или R3 и R4 вместе с атомом азота, к которому они присоединены, могут образовывать кольцо.
Настоящее изобретение касается также способа получения соединения формулы (I), определение которого дано выше, и антибактериальной композиции, содержащей соединение формулы (I) и в качестве активного компонента.
С тех пор как впервые в 1962 г. была введена в употребление в качестве средства для лечения инфекции мочевого тракта налидиксовая кислота (см. G.I. Zesher, et al/, J. Med. Chem., 5, 1063 - 1065, (1962)), было разработано много разных антибактериальных средств из хинолинкарбоновой кислоты, включая оксолиновую кислоту, розоксацин, пипемидиновую кислоту и т.д. Но эти ранее разработанные антибактериальные средства обладают низкой активностью против штаммов грамположительных бактерий и потому были использованы только против грамотрицательных штаммов.
Недавно был создан норфлоксацин, являющийся хинолоновым соединением, имеющим фтор в положении 6 (см. H. Koda, et al., J. Med. Chem., 23, 1358 - 1363 (1980)), после чего были проведены широкие исследования по разработке различных хинолоновых антибактериальных соединений. Но, поскольку норфлоксацин проявляет низкую антибактериальную активность против грамположительных штаммов бактерий и плохо распределяется и всасывается в живом организме, его использовали только для лечений болезней, таких, как инфекционные заболевания мочевого тракта и желудочно-кишечного тракта, венерические заболевания и т.п. Затем были созданы ципрофлоксацин (см.R.Wise, et al., Antimicrob. Agents Chemother, 23, 559 (1983)), офлоксацин (см. K. Sata, et al, Antimicrob. Agents Chemother, 22, 548, (1982)) и т.п. Эти антибактериальные средства проявляют высокую и широкую антибактериальную активность по сравнению с ранее разработанными бактериальными соединениями и потому нашли широкое практическое применение для лечения болезней в клинических условиях.
Применяемые или проходящие клинические испытания соединения в основном являются производными, имеющими пиперазиновый заместитель в положении 7 хинолонового ядра, как у ципрофлоксацина или офлоксацина. Однако в результате исследования по разработке хинолоновых соединений, имеющих более высокую и широкую антибактериальную активность, было обнаружено, что соединение, имеющее 3- амино- или 3- аминометилпирролидиновую группу, введенную в 7- положение, проявляет более высокую активность против грамположительных штаммов по сравнению с соединениями, имеющими 7- пиперазиновую группу, с сохранением при этом высокой активности против грамотрицательных штаммов. Но, к сожалению, соединения, имеющие пирролидиновый заместитель, имеют низкую растворимость в воде по сравнению с соединениями, имеющими пиперазиновый заместитель, и потому их антибактериальная активность in vivo не так, высока, как активность in vitro. Поэтому были проведены дальнейшие многочисленные исследования по устранению указанного недостатка соединений, имеющих пирролидиновый заместитель, то есть по повышению растворимости в воде и улучшению фармакокинетических свойств.
В результате было сделано много сообщений о таких исследованиях. Например, было описано, что производные (( 2S, 4S)-4-амино-2-метилпирролидинил) нафтиридина (см. Rosen, T.Chu, D.T.W. и т.д., J. Med. Chem, 1988, 31, 1598 - 1611) или производные (транс -3- амино-4-метилпирролидинил) нафтиридина (см. Matsumoto J. , et al., Proceedings of the 14 th International Congress of Chemоtheropy; Ishigami I., Ed.; Uniwersity of Tokyo Press: Tokyo, 1985, стр 1519-1520) показывают 20-40- кратное повышение растворимости в воде, повышенную биологическую доступность и улучшенные фармакокинетические свойства по сравнению с соединениями, не имеющими метильной группы, при одинаковой антибактериальной активности in vitro.
Кроме того, делались попытки уменьшить недостатки ранее разработанных хинолоновых соединений, такие, как относительно низкая антибактериальная активность против штаммов грамположительных бактерий, низкая растворимость в воде и плохие фармакокинетические свойства, путем введения различных функциональных групп (вместо аминогруппы) в пирролидиновый или пиперазиновый фрагмент. Как об одной из таких попыток сообщалось о некоторых соединениях, имеющих оксимгруппу, введенную в 7- аминовый фрагмент хинолоновых соединений. Например, исследователи ф. "Аббот" сообщили в научном журнале (J. Med. Chem. , 1992, 35, 1392-1398), что хинолоновое соединение, имеющее представленную ниже общую формулу [А], в котором в качестве заместителя в положении 7 хинолонового ядра является 3- оксим (или метилоксим) пирролидиновая группа или 4- оксим (или метилоксим) - пиперидиновая группа, проявляет хорошую антибактериальную активность против грамположительных штаммов: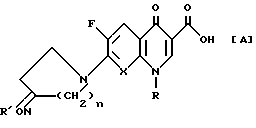
где
R представляет циклопропил или 2,4-дифторфенил;
R' представляет водород или метил;
X представляет C-H, C-F или N; и
n 1 или 2.
Соединение [А] имеет некоторые недостатки, состоящие в том, что оно проявляет достаточно высокую антибактериальную активность против грамположительных штаммов, но относительно низкую активность против грамотрицательных штаммов, а также проявляет относительную низкую антибактериальную активность при испытании in vivo.
Кроме того, в японской выложенной публикации патента N (Hei) 01-100165 (1989) описано соединение, имеющее следующую общую формулу [В]: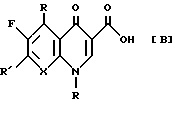
где
R представляет циклопропил, 2,4-дифторфенил или 4-гидроксифенил;
X представляет C-H, C-F или C-Cl; и
R' представляет заместитель, производный от оксим - или гидроксиаминопирролидина.
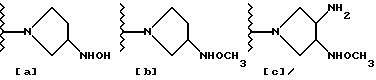
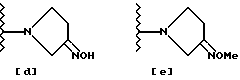
В частности, в упомянутой японской выложенной публикации в качестве заместителя R' очень широко раскрыты группы, производные от оксим- или гидроксиаминопирролидина. Но конкретно в качестве примера указаны лишь 3- гидроксиаминопирролидиновая [представленная ниже формула (а)], 3-метоксиаминопирролидиновая [представленная ниже формула (b)]; 3-амино-4-метоксиаминопирролидиновая [представленная ниже формула (c)], 3- оксимпирролидиновая [представленная ниже формула (d)] и 3- метилоксимпирролидиновая [представленная ниже формула (e)] группы, а пирролидиновый заместитель, имеющий и 3-оксим- и 4 аминометильную группу, нигде конкретно не упомянут.
Далее, в публикации более раннего европейского патента N 0541086 раскрыто хинолоновое соединение следующей общей формулы [C]: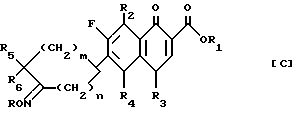
где
R и R1 независимо друг от друга представляют водород или C1 - C5-алкил;
R2 - водород, амино, фтор или гидрокси;
R3 - C3 - C7-циклоалкил;
R4 - метокси или фтор;
R5 и R6 могут быть одинаковыми или отличающимися друг от друга и независимо друг от друга представляют водород или алкил или R5 и R6 вместе могут образовывать C3 - С5-циклоалкил;
m = 0 или 1;
n= 1 - 3, целое число.
У соединений [C] , раскрытых в упомянутой публикации европейского патента, типичным заместителем в положении 7 хинолонового ядра является группа, имеющая следующую структуру:
Однако соединение формулы [C] не включает соединения, имеющего как оксимгруппу, так и аминометильную группу в положении 7 и, следовательно, отличается от соединения в соответствии с настоящим изобретением.
Общим отличительным признаком известных оксим- или гидроксиамин-производных соединений, как упомянуто выше, является то, что они проявляют относительно высокую активность против грамположительных штаммов, включающих штаммы MR SA (Methicillin Resistant Staphyloccus aureus), в сравнении с равнее разработанными хинолоновыми соединениями, но проявляют низкую активность против грамотрицательных штаммов в сравнении с антибактериальными средствами, включающими офлоксацин или ципрофлоксацин. Поэтому можно сказать, что их антибактериальный спектр может быть более узким, чем антибактериальный спектр известного антибактериального соединения офлоксацина или ципрофлоксацина.
Поэтому, основываясь на описанном выше известном уровне техники, данные изобретатели провели обширные исследования по разработке нового оксим-антиметильного соединения, проявляющего высокую антибактериальную активность против широкого спектра патогенных штаммов, включая резистентные штаммы, а также улучшенные фармакокинетические свойства и высокую всасываемость в живом организме, путем введения различных замещенных пирролидиновых групп в 7-положение хинолинового ядра и определения фармакологических активностей получающихся соединений. В результате было обнаружено, что хинолоновые соединения приведенной выше общей формулы (I) с введенной в 7-положение хинолинового ядра 4-аминометил-3- (необязательно замещенный) оксим-пиррилидиновой группой могут удовлетворять указанным требованиям и потому составлять настоящее изобретение.
Таким образом, задачей настоящего изобретения является создание нового производного хинолин (нафтиридин) карбоновой кислоты приведенной выше формулы (I), проявляющего высокую антибактериальную активность против широкого спектра патогенных штаммов, включая как грамположительные, так и грамотрицательные штаммы бактерий, а также имеет хорошие фармакологические свойства.
Другой задачей настоящего изобретения является создание способа получения нового производного хинолин (нафтиридин) - карбоновой кислоты, имеющего формулы (I).
И еще одной задачей настоящего изобретения является создание антибактериальной композиции, содержащей новое производное хинолин (нафтиридин) карбоновой кислоты формулы (I) в качестве активного компонента.
Выше указаны некоторые из более существенных задач настоящего изобретения. Указанные задачи следует рассматривать как просто иллюстрирующие некоторые из более существенных признаков и применений настоящего изобретения. Применяя настоящее изобретение иным образом или модифицируя его в пределах его раскрытого объема, можно получить другие полезные результаты. Поэтому другие задачи и более полное понимание настоящего изобретения будут очевидны из приведенного ниже подробного описания в дополнение к объему изобретения, определенному в формуле изобретения.
Во-первых, в соответствии с настоящим изобретением предлагаются новое производное хинолин(нафтиридин)карбоновой кислоты, имеющее следующую формулу I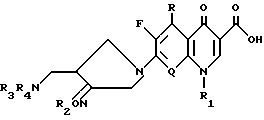
и его фармацевтически приемлемая нетоксичная соль, физиологически гидролизуемый сложный эфир, сольват и изомер,
где
R - водород, метил или амино;
Q - C-H, C-F, C-Cl, C-OH, C-CH3, C-O-CH3 или N;
R1 - циклопропил, этил или фенил, замещенный одним или несколькими атомом(ами) фтора;
R2 - одно из следующих a) - e):
a) водород, неразветвленный или разветвленный C1 - C4 алкил, циклопропил, циклопропилметил, C3 - C6-алкил, 2-галогенэтил, метоксиметил, метоксикрабонилметил, арил или аллил,
b) группу следующей формулы (1)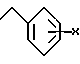
где X - водород, 2,3 или 4-фтор, циано, нитро, метокси, C1 - C4-алкил или 2,4-дифтор, c) группу следующей формулы (2)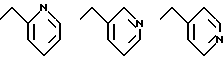
d) гетероарилметил следующей формулы (3)
e) группу следующей формулы (4)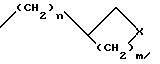
где n = 0 или 1; m = 0, 1 или 2 и X представляет метилен, O или N;
R3 и R4 независимо друг от друга представляют водород или C1 - C3-алкил или R3 и R4 вместе с атомом азота, к которому они присоединены, могут образовывать кольцо.
Из соединений приведенной выше формулы (I), имеющих превосходную антибактериальную активность, широкий антибактериальный спектр и отличные фармакокинетические свойства, предпочтительными являются те, у которых Q представляет C-H, C-F, C-Cl, C-OMe или N, R представляют водород или амино группу, R1 представляет циклопропил или 2,4-дифторфенил, R2 представляет водород, метил, этил, изопропил, трет-бутил, фенил, пропаргил, гомопропаргил, 2-фторэтил, бензил, 2-фторбензил или 2-цианбензил и R3 и R4 представляет водород.
Более предпочтительные соединения формулы (I) включает те, у которых Q представляет C-H, C-Cl, C-F или N, R представляет водород или амин, R1 представляет циклопропил, R2 представляет метил, трет-бутил, гомопропаргил, 2-фторэтил, бензил или 2-фторбензил и R3 и R4 представляют водород.
В пирролидиновой части соединения формулы (I) 4-углеродный атом, у которого замещена аминометильная группа, является асимметрическим углеродным атомом и потому соединение может быть в виде R- или S- или смеси R- и S-изомеров. Кроме того, в результате наличия (необязательно замещенной) оксимгруппы в положении 3 пирролидинового фрагмента соединение формулы (I) может быть в форме син- или анти-изомеров, в зависимости от их геометрической структуры. Таким образом, настоящее изобретение охватывает также все эти геометрические изомеры и их смеси.
Соединение формулы (I) в соответствии с настоящим изобретением может образовывать фармацевтически приемлемую нетоксичную соль. Такой солью является соль с неорганическими кислотами, такими, как хлористоводородная, бромистоводородная, фосфорная, серная и т.д., соль с органическими карбоновыми кислотами, такими, как уксусная, трифторуксусная, лимонная, малеиновая, щавелевая, янтарная, бензойная, винная, фумаровая, миндальная, аскорбиновая или яблочная, или с сульфокислотами, такими, как метансульфокислота, паратолуолсульфокислота и т.д., и соль с другими кислотами, которые как правило известны и традиционно используются при получении соединений на основе хинолина. Эти соли присоединения кислоты могут быть получены в соответствии с традиционным способом превращения.
Во-вторых, в соответствии с настоящим изобретением предлагается способ получения нового соединения формулы (I).
В соответствии с настоящим изобретением соединение формулы (I) может быть получено путем химического взаимодействия соединения формулы (II) с соединением формулы (III) или его солью, как показано в следующей ниже схеме 1 реакции:
В приведенной выше схеме R, R1, R2, R3, R4 и Q - такие, как указанные выше, и X представляют атом галогена, предпочтительно хлор, бром или фтор.
Согласно приведенной выше схеме 1 реакции соединения формулы (I) в соответствии с настоящим изобретением может быть получено путем перемещения соединения формулы (II) и соединения формулы (III) в присутствии растворителя в течение 1-20 часов при температуре между комнатной температурой и 200oC с добавлением подходящего основания. В этой реакции соединение формулы (III) может быть использовано в виде свободного соединения или соли с кислотой, такой, как хлористоводородная, бромистоводородная или трифтоуксусная.
В качестве растворителя для описанной выше реакции может быть использован любой растворитель, который не оказывает вредного влияния на реакцию. Предпочтительно можно использовать ацетонитрил, диметилформамид (DMF - ДМФ), диметилсульфоксид (DMSO - ДМСО), пиридин или гексаметилфосфорамид (HMPA - ГМФА).
Эту реакцию обычно проводят в присутствии акцептора кислоты. В этом случае, чтобы повысить выход реакции на относительно дорогой исходный материал (II), реагент (III) используют в избыточном количестве, например, эквимолекулярном количестве до 10-кратного молярного количества, предпочтительно эквимолекулярном количестве до 5-кратного молярного количества, по отношению к исходному материалу (II). При использовании регента (III) в избыточном количестве непрореагировавшее соединение формулы (III), которое остается после реакции, может быть извлечено и вновь использовано в другой реакции. Акцептор кислоты, который предпочтительно может быть использован в этой реакции, включает неорганические основания, такие, как гидрокарбонат натрия, карбонат калия и т. д., и органические основания, такие, как триэтиламин, диизопропилэтиламин, пиридин, N,N-диметиланилин, N,N-диметиламинопиридин, 1,8-диазабицикло[5.4.0]унден-7-ен (DBU), 1,4-диазабицикло[2.2.2]октан (DABCO) и т.д.
Соединение формулы (I) в соответствии с настоящим изобретением может быть также получено способом, показанным в следующей ниже схеме 2 реакции, при котором вводят защитную группу P в один из радикалов R3 и R4 соединения формулы (III), где R3 и R4 - водород, для получения соединения формулы (III'), в котором аминогруппа защищена группой P, защищенное соединение формулы (III') вводят во взаимодействие с соединением формулы (II) при таких же условиях, как в схеме I реакции, и затем полученное соединение формулы (I') освобождают от защиты, удаляя защитную группу P, для образования требуемого соединения формулы (I).
Схема 2 реакции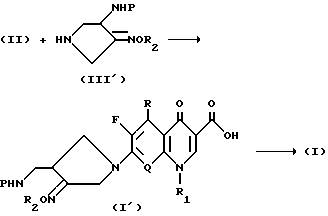
В приведенной выше схеме R, R1, R2 и Q такие, как указанные выше; и P представляет аминозащитную группу.
В реакции согласно приведенной выше схеме 2 соединение формулы (III') может быть использовано в виде свободного соединения или соли с хлористоводородной, бромистоводородной или трифторуксусной кислотой, как соединение формулы (III), используемое в схеме 1 реакции.
В качестве подходящей амино-защитной группы P в соединении формулы (III') может быть использована любая защитная группа, традиционно используемая в органической химии, которая может быть легко удалена после реакции без разрушения структуры целевого соединения. Конкретные примеры защитных групп, которые могут быть использованы для этой цели, включают формил, ацетил, трифторацетил, бензоил, пара-толуолсульфонил, метоксикарбонил, этоксикарбонил, трет-бутоксикарбонил, бензилоксикарбонил, параметооксибензилоксикарбонил, трихлорэтоксикарбонил, бета-иодэтоксикарбонил, бензил, пара-метоксибензил, тритил, тетрагидропиранил и т.д.
После окончания реакции амино-защитная группа в полученном соединении формулы (I') может быть удалена путем гидролиза, сольволиза или восстановления в зависимости от свойства использованной защитной группы. Например, соединение формулы (I') обрабатывают в растворителе в присутствии или отсутствии кислоты или основания при температуре 0-130oC, чтобы удалить защитную группу. В качестве кислоты, которая может быть использована для этой цели, можно назвать неорганическую кислоту, такую, как хлористоводородная, бромистоводородная, серная, фосфорная и т.д., органическую кислоту, такую, как уксусная, трифторуксусная, муравьиная, толуолсульфокислота и т.д., и кислоту Льюиса, такую, как трибромид бора, алюминийхлорид и т.д. В качестве основания для этой цели можно использовать гидроксид щелочного или щелочноземельного металла, такой, как гидроксид натрия, гидроксид бария и т.д., карбонат щелочного металла, такой, как карбонат натрия, карбонат кальция и т.д., алкоксид щелочного металла, такой, как метоксид натрия, этоксид натрия и т. д. , или ацетат натрия и т.п. Реакция может быть осуществлена в присутствии растворителя, например воды или органического растворителя, такого, как этанол, тетрагидрофуран, диоксан, этиленгликоль, уксусная кислота и т.д. , или смеси такого органического растворителя и воды. При необходимости эта реакция может быть также осуществлена в отсутствии любого растворителя.
Кроме того, когда защитными группами являются пара-толуолсульфонил, бензил, тритил, пара-метоксибензил, бензилоксикарбонил, пара-метоксибензилоксиркарбонил, трихлорэтоксикарбонил, бета-иодэтоксикарбонил и т. п. , эти группы могут быть эффективно удалены путем восстановления. Хотя условия реакции восстановления для удаления защитной группы могут быть изменены в зависимости от свойств использованной защитной группы, обычно восстановление может быть осуществлено струей газообразного водорода в инертном растворителе в присутствии катализатора, такого, как платина, палладий, никель Ренея и т. д. , при температуре 10-100oC или металлическим натрием или металлическим литиев в аммиаке, при температуре от -50 до -10oC.
Соединение формулы (I), используемое в качестве исходного материала в настоящем изобретении, является известным соединением и может быть легко получено известным способом, описанным в более ранней публикации (см. J.M. Domogala et al. , J. Med. Chem., 34, 1142 (1991); J.M. Domogala et al., J. Med. Chem. 31, 991 (1988); D. Bougard et al., J. Med. Chem. 35, 518 (1992)).
Соединение формулы (III), используемое в настоящем изобретении в качестве другого исходного материала, может быть легко получено в соответствии со способом, представленным в следующих ниже схемах 3, 4 и 5 реакций.
Схема 3 реакции.
Реакционная схема 3.
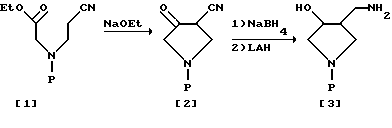

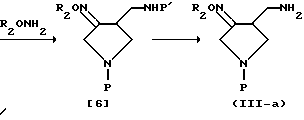
Схема 4 реакции.
В приведенных выше схемах 3 и 4 защитные группы P и P' независимо друг от друга представляют такую же амино-защитную группу, как указанные для P в связи с соединением формулы (III'), и могут быть одинаковыми или отличающимися друг от друга; и Py представляет пиридин.
Ниже дано конкретное описание способа, представленного в схемах 3 и 4.
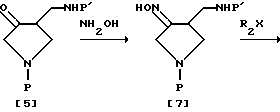
В соответствии со схемой 3 сначала цианоэфир [1], имеющий защищенную аминогруппу, может быть введен во взаимодействие с этоксидом натрия в растворителе (таким, как этанол) для получения 3-кето-4-цианпирролидина [2]. Полученный цианпирролидин [2] восстанавливают газообразным водородом и в присутствии платинового катализатора для получения аминоспирта [3]. В этом случае цианпирролидин [2] может быть восстановлен посредством другого восстановителя для получения аминоспирта [3]. Например, кетоновая и цианогруппы могут быть восстановлены алюмогидридом лития (LAH), комплексом борогидрид натрия - хлорид кобальта (NaBH4 - CoCl3) или борогидридом лития (LiBH4). В соответствии с еще одним вариантом аминоспирт [3] может быть синтезирован путем восстановления сначала кетоновой группы в гидроксильную группу посредством борогидрида натрия (NaBH4) и затем восстановления цианогруппы алюмогидридом лития (LAH). Затем аминогруппу полученного таким образом аминоспирта [3] избирательно защищают, чтобы получить защищенный амин [4], который после этого обрабатывают смесью триоксид серы (SO3) - пиридин в диметилсульфоксидном растворителе (см. Parikh J.R. and Doering W.U.E., J. Am. Chem. Soc. , 1967, 89, 5505) или окисляют другим окислителем, чтобы получить кетоновое соединение [5] . Полученное кетоновое соединение [5] вводят затем во взаимодействие с O-замещенным гидроксиамином формулы R2ONH2 для получения целевого замещенного оксимсоединения [6], с которого может быть снята защита подходящим способом, выбранным в зависимости от вида защитной группы, для получения целевого оксимсоединения (III), в котором R3 и R4 - водород, т.е. соединения формулы (III-a).
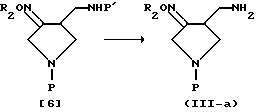
В соответствии с другим вариантом способом, представленным на схеме 4, кетоновое соединение [5] вводят во взаимодействие с гидроксиамином, чтобы получить целевое оксимсоединение [7] , и соединение [7] вводят во взаимодействие с электрофильным соединением формулы R2X, которое может вводить требуемую R2 группу, в присутствии основания, чтобы получить оксимпроизводное формулы [6], которое затем освобождают от защиты подходящим способом, выбранным в зависимости от вида защитной группы, так же, как в схеме 3, чтобы получить целевое оксимсоединение (III-a).
Соединение формулы (III), где R3 и R4 аминометильной группы в положении 4 пирролидина - иные, чем водород, т.е. соединение формулы (III-b) может быть получено по следующей ниже схеме 5 реакции.
Схема 5 реакции.


В приведенной выше схеме R'3 и R'4 - такие же, как указанные для R3 и R4 в связи с соединением формулы (I), при условии, что они не могут быть водородом.
В соответствии со способом по схеме 5 сначала аминовое соединение [3] обрабатывают C1 - C3-альдегидом и затем восстанавливают, чтобы получить замещенное аминовое соединение [8], и полученное аминовое соединение [8] обрабатывают смесью триоксид серы (SO3) - пиридин в диметилсульфоксидном растворителе или окисляют другим окислителем, чтобы получить кетоновое соединение [9]. Полученное кетоновое соединение [9] может быть обработано так же, как в способе обработки кетонового соединения [5] в схемах 3 и 4, чтобы синтезировать целевое соединение формулы (III-b).
Описанные выше способы синтеза будут более конкретно разъяснены в следующих примерах получения.
В соответствии с настоящим изобретением предлагается также антибактериальная композиция, содержащая новое соединение формулы (I), указанное выше, или его фармацевтически приемлемую соль в качестве активного компонента и фармацевтически приемлемый носитель. При использовании такой антибактериальной композиции для клинического назначения она может быть приготовлена в форме твердых, полутвердых или жидких фармацевтических препаратов для перорального, парентерального или местного введения путем объединения соединения формулы (I) с фармацевтически приемлемым инертным носителем. Фармацевтически приемлемый инертный носитель, который может быть использован для этой цели, может быть твердым или жидким. Для приготовления твердого или полутвердого фармацевтического препарата в форме порошков, таблеток, диспергирующихся порошков, желатиновых капсул, крахмальных капсул (облаток), суппозиториев и мазей обычно используют твердые носители. Используемый твердый носитель предпочтительно представляет собой одно или несколько веществ, выбранных из группы, состоящей из разбавителей, вкусовых и ароматизирующих веществ, солюбилизирующих, смазывающих, суспендирующих, связывающих, разрыхляющих веществ и т.д., или вещества, заключенные в капсулу. В случае порошкового препарата он содержит тонкоизмельченный активный компонент в количестве от 5 - 10 до 80% в носителе. Конкретные примеры подходящего твердого носителя включают карбонат магния, стеарат магния, тальк, сахар, лактозу, пектин, декстрин, крахмал, желатин, трагакант, метилцеллюлозу, натрийкарбоксиметилцеллюлозу, низкокипящий воск, масло какао и т.д. Благодаря легкости их введения таблетки, порошки и крахмальные и желатиновые капсулы являются наиболее целесообразными твердыми лекарственными формами для перорального введения.
Жидкие лекарственные формы включают растворы, суспензии и эмульсии. Например, инъецируемая лекарственная форма для парентерального введения может быть изготовлена в виде водного или водно-пропиленгликолевого раствора, изотоничность, pH и т.п. можно регулировать в соответствии с физиологическим состоянием живого организма. Жидкий препарат может быть также изготовлен в виде раствора в водном полиэтиленгликолевом растворе. Водный раствор для перорального введения может быть изготовлен путем растворения активного компонента в воде и добавления к нему подходящих красящего, вкусового, стабилизирующего и сгущающего вещества. Водная суспензия, пригодная для перорального введения, может быть изготовлена диспергированием тонкоизмельченного активного компонента в вязких веществах, таких, как природная или синтетическая камедь, метилцеллюлоза, натрийкарбоксиметилцеллюлоза и другие известные суспендирующие средства.
Особенно целесообразно изготавливать вышеупомянутые фармацевтические препараты в виде унифицированно дозированной лекарственной формы для легкого введения и равномерной дозировки. Унифицированно дозированные формы препарата - это физически раздельные элементы, пригодные для использования в качестве унифицированной (единичной) дозы и содержащие каждый заданное количество активного компонента, рассчитанное для получения требуемого терапевтического эффекта. Такая лекарственная форма на один прием может быть изготовлена в упакованном виде, например в виде таблетки, капсулы или порошка, насыпанного в пузырек или ампулу, или мази, геля или пасты в тюбике или склянке.
Хотя количество активного компонента, содержащегося в унифицированно дозированной лекарственной форме, может изменяться, его обычно можно регулировать в пределах от 1 до 100 мг в зависимости от эффективности выбранного активного компонента.
При использовании активного соединения формулы (I) в соответствии с настоящим изобретением в качестве лекарственного средства для лечения бактериальных инфекционных заболеваний его предпочтительно вводят в количестве примерно 6 - 14 мг на килограмм массы тела на первой стадии. Однако вводимая доза может быть изменена в соответствии с потребностью конкретного больного, тяжестью подлежащего лечению инфекционного заболевания, выбранного соединения и т.п.
Предпочтительная доза, подходящая для некоторого состояния, может быть определена специалистом в данной области традиционным образом. Обычно терапевтическое лечение начинают с количества меньшего, чем оптимальная доза активного соединения, а затем вводимую дозу понемногу увеличивают, пока не получат оптимальный лечебный эффект. Для удобства суммарная суточная доза может быть разделена на несколько порций и введена за несколько раз.
Как было упомянуто выше, соединение в соответствии с настоящим изобретением проявляет высокую бактериальную активность широкого спектра против самых разных патогенных организмов, включая штаммы грамположительных и грамотрицательных бактерий. Антибактериальная активность предлагаемого соединения против грамотрицательных штаммов сравнима или выше, чем антибактериальная активность известных антибактериальных средств (например, ципрофлоксацина), и, в частности, антибактериальная активность предлагаемого соединения против грамположительных штаммов намного выше, чем у известных антибактериальных средств. Кроме того, предлагаемое соединение также проявляет очень высокую антибактериальную активность против штаммов, стойких к известным хинолоновым соединениям.
Что касается фармакокинетических свойств, то соединение в соответствии с настоящим изобретением имеет высокую растворимость в воде и потому может хорошо всасываться в живом организме (по сравнению с известными хинолоновыми соединениями), показывая очень высокую биологическую доступность. Биологический полупериод существования предлагаемого соединения в организме намного длиннее, чем у известных хинолоновых соединений, и потому предлагаемое соединение можно вводить один раз в сутки, что будет достаточным для использования его в качестве антибактериального средства.
Кроме того, поскольку соединение в соответствии с настоящим изобретением менее токсично, оно может быть эффективно использовано для профилактики и лечения болезней, вызываемых бактериальным заражением, у теплокровных животных, включая человека.
Настоящее изобретение более конкретно разъяснено на следующих ниже примерах. Однако следует понимать, что описанные ниже получения и примеры предназначены иллюстрировать настоящее изобретение, а не каким-либо образом ограничивать настоящее изобретение.
Получение 1.
Синтез этилового эфира (2-цианэтиламино)уксусной кислоты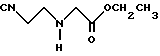
В 80 мл дистиллированной воды растворяли 139,6 г (1 моль) гидрохлорида этилового эфира глицина, и к полученному раствору добавляли 230 мл водного раствора 67,3 г (1,2 моль экв. ) гидроксида калия. Затем к реакционному раствору добавляли 106,2 г (2 моль экв.) акрилонитрила с одновременным перемешиванием и нагреванием при 50 - 60oC. Реакционную смесь перемешивали 5 часов с нагреванием и затем отделяли органический слой. Водный слой экстрагировали диэтиловым эфиром и экстракт объединяли с упомянутым выше отделенным органическим слоем. Объединенный органический слой осушали над безводным сульфатом магния и фильтровали. Фильтрат концентрировали под пониженным давлением для удаления растворителя. Остаток перегоняли под пониженным давлением (100 - 150oC/ 10,25 мм рт. ст.), и в результате получили 65,6 г (выход: 48%) указанного в заголовке соединения.
1H ЯМР (CDCl3 м.д.): δ 4,20 (2H, кв), 3,48 (2H, с), 2,96 (2H, т), 2,54 (2H, т), 1,30 (3H, т) MS (FAB, m/e): 157 (M + H)
Получение 2.
Синтез 4-циано-1-(N-трет-бутоксикарбонил)-пирролидин-3-она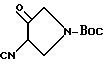
В вышеприведенной формуле и далее Boc представляет трет-бутоксикарбонил. 29 г (0,186 моль) соединения, полученного в получении 1, растворяли в 200 мл хлороформа и полученный раствор вливали в колбу объемом 1 л. Затем туда добавляли 45 г (1,1 моль экв.) ди-трет-бутоксикарбонилдикарбоната, и реакционную смесь перемешивали в течение 17 часов при комнатной температуре. Реакционный раствор концентрировали, и остаток разводили 250 мл абсолютного этанола. Полученный раствор добавляли к раствору этоксида натрия (Na OEt), полученному путем добавления 6 г токарной стружки металлического натрия (Na) к 220 мл абсолютного этанола, при нагреве с обратным холодильником. Реакцию непрерывно проводили в течение еще одного часа при нагреве с обратным холодильником. Реакционный раствор концентрировали под пониженным давлением, и остаток разводили водой, а затем промывали метиленхлоридом. Доводили pH водного слоя до 4 с помощью 1 н. HCl и экстрагировали этилацетатом. Экстракт сушили над безводным сульфатом магния и затем фильтровали. Фильтрат концентрировали, получив в результате стехиометрическое количество указанного в заголовке соединения в неочищенном состоянии.
1H ЯМР (CDCl3 м.д.): δ 4,5 - 3,5 (5H, м), 1,5 (9H, с), MC (FAB, m/e): 211 (M + H)
Получение 3.
Синтез гидрохлорида 4-аминометил-1-(N-трет-бутоксикарбонил)- пирролидин-3-ола
В смеси 357 мл абсолютного этанола и 7 мл хлороформа растворяли 3 г (14 ммоль) соединения, полученного в Получении 2, и полученный раствор вливали в колбу. Затем туда добавляли каталитическое количество оксида платины (Pt O2). После удаления воздуха из реакционной колбы под пониженным давлением реакционную смесь перемешивали 72 часов при комнатной температуре с продувкой газообразным водородом из баллона, заполненного газообразным водородом. Реакционный раствор фильтровали и фильтрат концентрировали, получив в результате стехиометрическое количество указанного в заголовке соединения.
1H ЯМР (CDCl3 м.д.): δ 8,0 (2H, шир. с), 3,5 - 2,0 (7H, м), 3,3 (2H, с), 1,38 (9H, с), MC (FAB, m/e): 217 (M + H)
Получение 4.
Синтез 4-(N-трет-бутоксикарбонил)аминометил-1- (N-бутоксикарбонил)пирролидин-3-ола
Способ A.
В смеси 456 мл диоксана и 268 мл дистиллированной воды растворяли 20 г (0,094 моль) соединения, полученного в Получении 3, и полученный раствор регулировали 1 н. водным раствором гидроксида натрия до pH 9. Затем к раствору добавляли 30,9 г (1,5 моль экв.) ди-трет-бутоксикарбонилдикарбоната, и реакционную смесь перемешивали в течение 30 минут при комнатной температуре и концентрировали под пониженным давлением. Остаток разводили метиленхлоридом. После добавления к реакционному раствору воды отделяли органический слой, а водный слой подкисляли до pH 4 и затем экстрагировали метиленхлоридом. Экстракт объединяли с органическим слоем, отделенным ранее, и объединенный раствор осушали над безводным сульфатом магния и концентрировали. Остаток очищали путем колоночной хроматографии, получив в результате 17 г (выход: 57%) указанного в заголовке соединения.
1H ЯМР (CDCl3 м.д.): δ 4,95 (1H, м), 4,1 (1H, м), 3,5 (2H, м) 3,3 - 3,0 (4H, м), 2,1 (1H, M), 1,45 (18H, C);
MC (FAB, m/e): 317 (M + H)
Способ B.
В колбу емкостью 1 л вносили 10 г (0,047 моль) соединения, полученного в Получении 2, и затем растворяли его путем добавления 500 мл сухого тетрагидрофурана. Этот раствор охлаждали до -3oC над баней со льдом и хлоридом натрия и затем добавляли к нему порциями в течение 20 минут 3,8 г (0,094 моль) алюмогидрида лития (LAH). После окончания добавления реакционную смесь перемешивали в течение часа под водяной баней со льдом. После окончания реакции в реакционной смеси осторожно добавляли последовательно 4 мл воды, 4 мл 15%-ного водного раствора гидроксида натрия и 12 мл воды. Всю смесь интенсивно перемешивали в течение 3 часов при комнатной температуре и добавляли к ней 10 г безводного сульфата магния. Эту смесь перемешивали и затем фильтровали, и фильтрат концентрировали для стехиометрического получения продукта. Полученный продукт разводили 200 мл смеси (2:1 по объем) диоксана и воды, и добавляли к нему при комнатной температуре 12,3 г (0,056 моль) ди-трет-бутоксикарбонилдикарбоната. Реакционный раствор перемешивали в течение часа при комнатной температуре до окончания реакции и затем концентрировали. Остаток разводили опять этилацетатом, промывали насыщенным водным раствором хлорида натрия, осушали над безводным сульфатом магния и фильтровали. Фильтрат концентрировали и затем остаток очищали путем колоночной хроматографии с использованием в качестве элюента смеси (2:1 по объему) гексана и этилацетата, что дало в результате 8,2 г (выход: 55%) указанного в заголовке соединения.
Способ C.
В 4 л метанола растворяли 210 г (1 моль) соединения, полученного в Получении 2, и этот раствор вливали в снабженный термометром реакционный сосуд емкостью 6 л. Внутреннюю температуру реакционного сосуда понижали до 10oC под баней с сухим льдом и ацетоном. В сосуд добавляли порциями 76 г (2 моль) борогидрида натрия (NaBH4), поддерживая при этом внутреннюю температуру в сосуде на уровне 10-13oC. После окончания добавления реакционную смесь перемешивали еще 30 минут при той же самой температуре, чтобы весь кетон мог быть восстановлен в спирт. Затем к реакционной смеси добавляли в течение 10 минут 243 г (1 моль) гидрата хлорида кобальта. После окончания реакции полученный твердый комплекс растворяли в 4 л аммиачной воды и этот раствор разбавляли 8 л воды и затем экстрагировали этилацетатом. Органический слой промывали насыщенным рассолом, осушали над безводным сульфатом магния и фильтровали. Фильтрат концентрировали и смешивали со смесью 1,5 л диоксана и 0,5 л дистиллированной воды. К смеси добавляли 212 г ди-трет-бутоксикарбонилдикарбоната, и всю смесь перемешивали 2 часа при комнатной температуре. После окончания реакции реакционную смесь концентрировали под пониженным давлением, разводили опять дихлорметаном, промывали водой, осушали над безводным сульфатом магния и затем фильтровали. Фильтрат концентрировали и затем очищали путем хроматографии на колонках с силикагелем (в качестве элюента использовали смесь гексана и этилацетата с отношением 2:1), что дало в результате 202 г (выход: 64%) указанного в заголовке соединения.
Способ D.
В колбу емкостью 1 л вносили 10 г (0,047 моль) соединения, полученного в Получении 2, и растворяли его добавлением 500 мл метанола. Этот раствор охлаждали под ледяной баней и добавляли к нему порциями в течение 20 минут 3,6 г (0,094 моль) боргидрида натрия. Реакционную смесь перемешивали еще 30 мин, чтобы завершить реакцию, и затем концентрировали под пониженным давлением, разводили этилацетатом, промывали водой, осушали над безводным сульфатом магния и затем фильтровали. Фильтрат концентрировали, получив в результате соединение с требуемой кетоновой группой, восстановленной в спирт. 10,1 г (0,047 моль) полученного спиртового соединения растворяли в 200 мл сухого тетрагидрофурана, и этот раствор охлаждали до -5oC под баней со льдом и солью. К раствору добавляли в течение 20 минут 2,6 г (0,066 моль) алюмогидрида лития. Реакционную смесь перемешивали еще 30 минут при той же самой температуре для завершения реакции, и затем к смеси добавляли по порядку 2,6 мл воды, 2,6 мл 15%-ного раствора гидроксида натрия и 7,8 мл воды. Эту смесь перемешивали в течение часа при комнатной температуре. После добавления 6 г безводного сульфата магния смесь перемешивали еще 30 минут и фильтровали. Полученный продукт разводили 200 мл смеси диоксан-вода (2:1 по объему) и добавляли к нему порциями 12,3 г (0,056 моль) ди-трет-бутоксикарбонилдикарбоната. Смесь перемешивали 30 минут, чтобы завершить реакцию, и затем концентрировали, разводили этилацетатом, промывали насыщенным рассолом, осушали над безводным сульфатом магния и затем фильтровали. Фильтрат концентрировали и остаток очищали путем колоночной хроматографии, получив в результате 12,3 г (выход: 83%) указанного в заголовке соединения.
Получение 5.
Синтез 4-(N-трет-бутоксикарбонил)аминометил-1-(N-трет- бутоксикарбонил)пирролидин-3-она.

В 64 мл диметилсульфоксида растворяли 14 г (0,044 моль) соединения, полученного в Получении 4, и к полученному раствору добавляли 18,5 мл (3 моль экв.) триэтиламина. Эту смесь охлаждали под ледяной баней. Когда стенка реакционной колбы начинала замораживаться, к смеси добавляли порциями 12,7 г (1,8 моль экв. ) окислителя пиридин-триоксид серы (Py-SO3). По окончании добавления удаляли ледяную баню и реакционный раствор перемешивали в течение 3 часов при комнатной температуре, разбавляли водой и затем экстрагировали этилацетатом. Экстракт осушали над безводным сульфатом магния и концентрировали до получения стехиометрического количества указанного в заголовке соединения в неочищенном виде.
1H-ЯМР (CDCl3, м.д.): δ 4,95 (1H, шир. с), 4,15 - 2,7 (6H, м), 2,8 (1H, шир.), 1,45 (9H, с), 1,40 (9H, с).
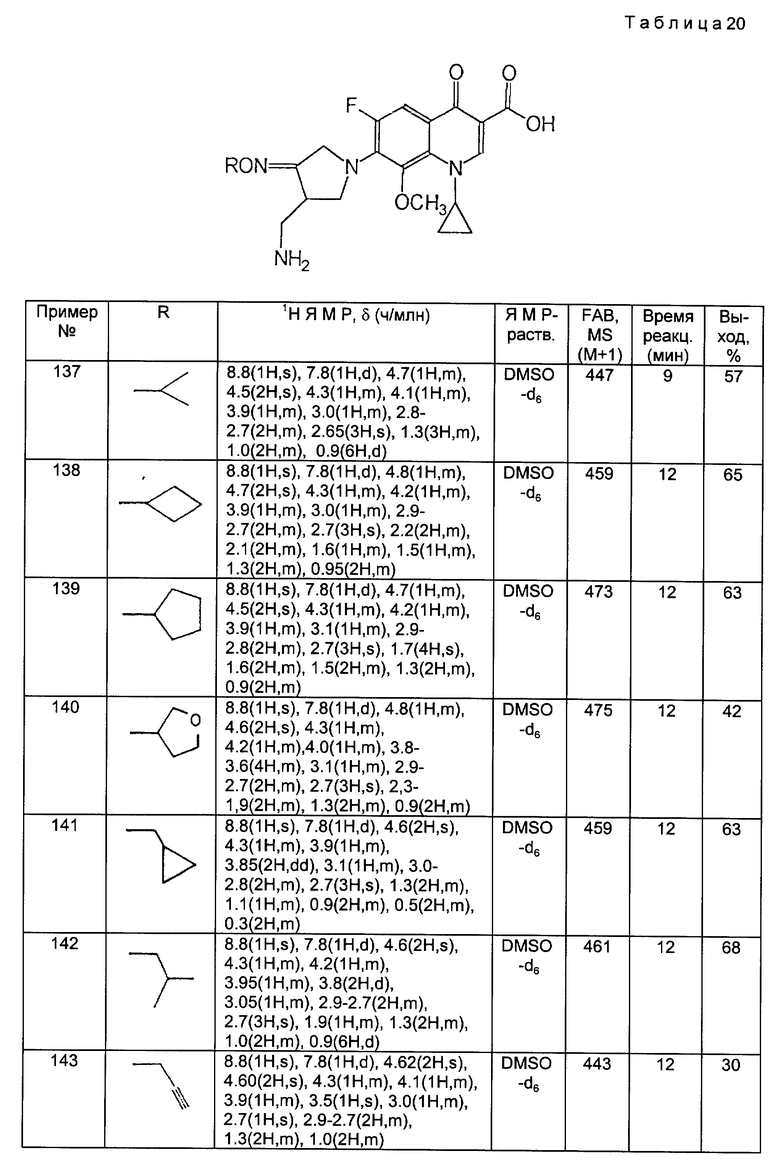
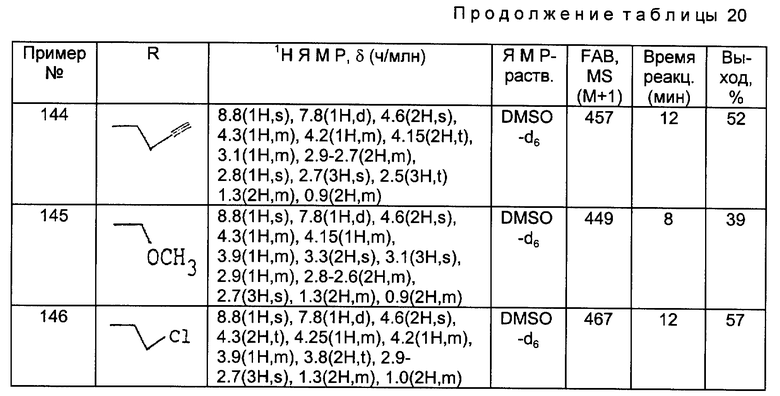
MC (FAB, m/e): 315 (M+H)
Получение 6.
Синтез 1-(N-трет-бутоксикарбонил)-4-(N-трет-бутоксикарбонил)- аминометилпирролидин-3-оноксима.
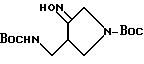
В смеси 6 мл 95%-ного этанола и 3 мл тетрагидрофурана (ТГФ) растворяли 300 мг соединения, полученного в Получении 5, и этот раствор вливали в реакционный сосуд емкостью 30 мл. Туда добавляли 232 мг (3,5 моль экв.) гидроксиамингидрохлорида (NH2OH•HCl), после чего добавляли 281 мг (3,5 моль экв. ) гидрокарбоната натрия, растворенного в 1,5 мл дистиллированной воды. Реакционную смесь перемешивали 40 минут при 40oC под масляной баней, чтобы завершить реакцию, охлаждали и затем концентрировали под пониженным давлением. Остаток разводили метиленхлоридом, промывали насыщенным водным раствором хлорида натрия, осушали над безводным сульфатом магния и фильтровали. Фильтрат концентрировали, и фильтрат подвергали колоночной хроматографии на силикагеле, элюируя смесью (1:1 по объему) гексан-этилацетат, в результате чего получили 230 мг (выход 73%) указанного в заголовке соединения.
1H ЯМР (CDCl3, м.д.): δ 9,70 (1H, шир. с), 5,05 (1H, шир. с), 4,2 (2H, шир.); 3,83 (1H, м), 3,5 - 3,2 (3H, м), 3,0 (1H, м); 1,42 (18H, с).
МС (FAB, m/e): 330 (M+H).
Получение 7.
Синтез 1-(N-трет-бутоксикарбонил)-4-(N-трет-бутоксикарбонил)- аминометилпирролидин-3-он-бензилоксима.

К 15 мл дихлорметана добавляли 659 мг соединения, полученного в Получении 6, 193 мг тетра-н-бутиламмонийбромида и 855 мг бензилбромида, после чего к полученной смеси добавляли 5 мл 15%-ного водного раствора гидроксида натрия. Реакционную смесь перемешивали в течение 30 минут при комнатной температуре. Органический слой отделяли, осушали над безводным сульфатом магния и фильтровали. Фильтрат перегоняли под пониженным давлением, и остаток очищали путем хроматографии на стеклянных колонках, получив в результате 776 мг (выход: 92%) указанного в заголовке соединения.
1H ЯМР (CDCl3, м.д.): δ 7,38 (5H, м), 5,13 (2H, с), 4,92 (1H, м), 4,13 (2H, м), 3,76 (1H, м), 3,41 (1H, м), 3,25 (2H, м), 3,02 (1H, м), 1,50 (9H, с), 1,49 (9H, с).
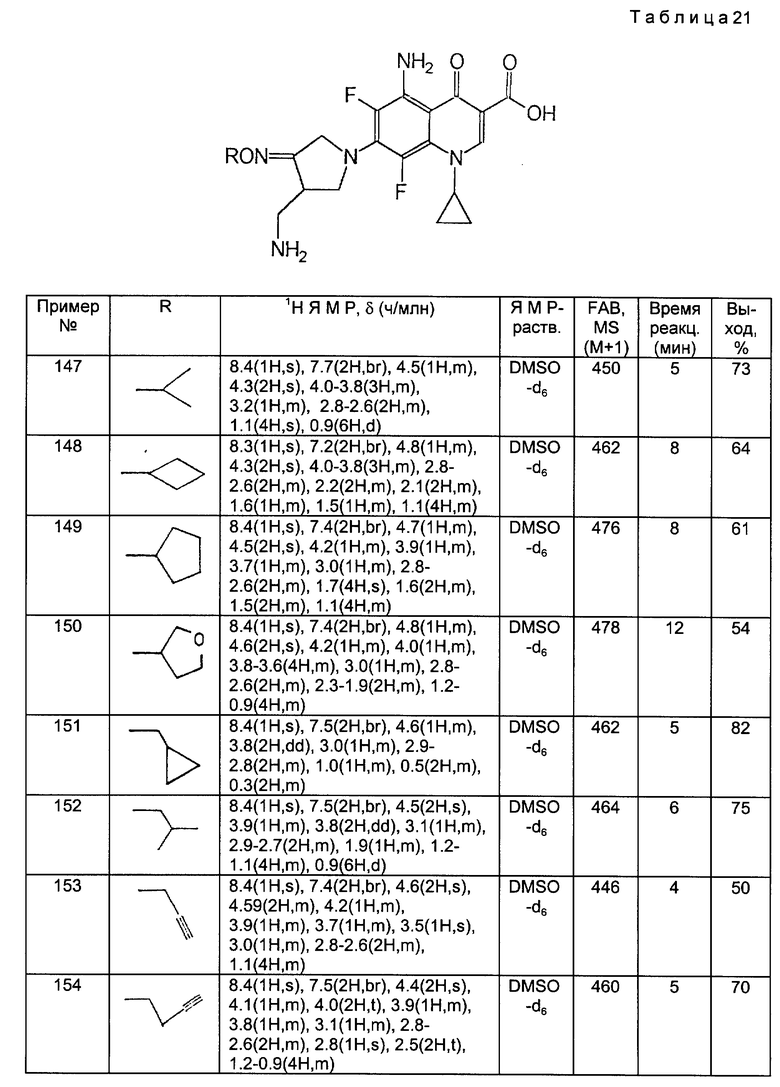
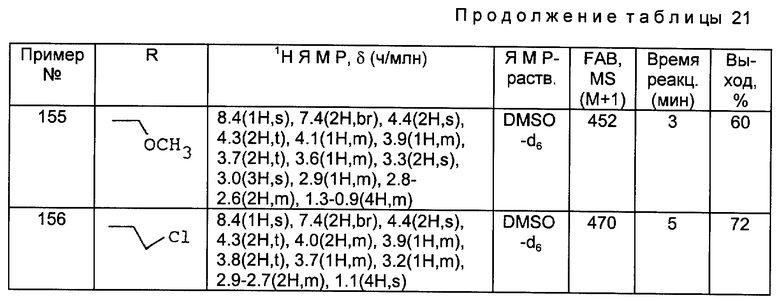
МС (FAB, m/e): 420 (M+H)
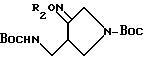
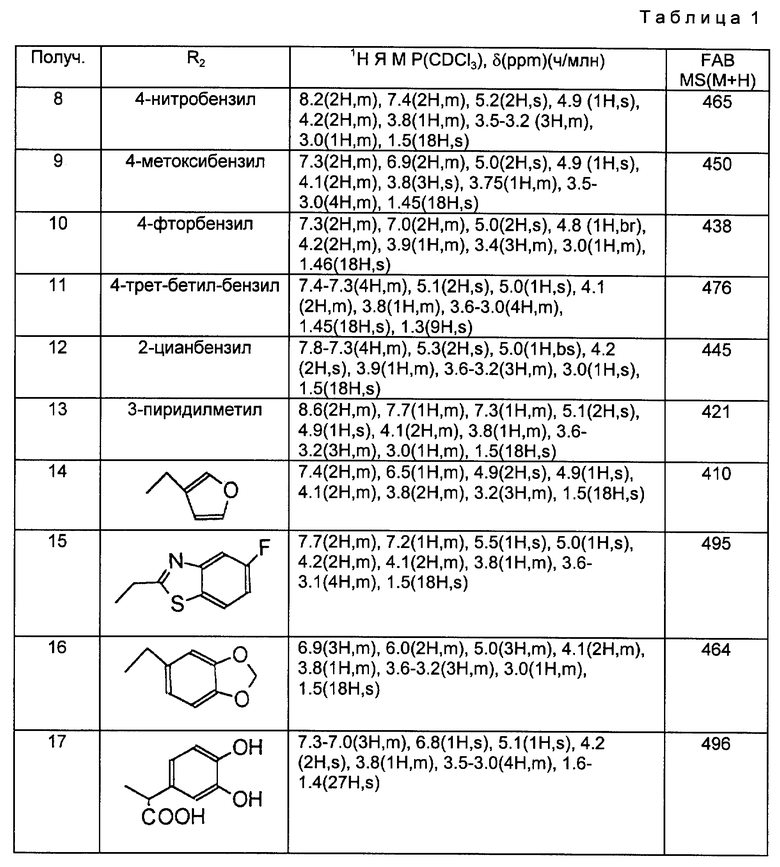
Получения 8 - 17.
Аминосоединения, перечисленные в следующей ниже таблице 1, были получены в соответствии с такой же процедурой, как в Получении 7, за исключением того, что вместо бензилбромида использовали соответствующие производные бензилбромида, имеющие такую структуру R2, какая представлена в таблице 1.
Используемые в данных далее таблицах условные обозначения и сокращения имеют следующие значения:
Prep. Получение
NMR ЯМР (Ядерно-магнитный резонанс)
MS МС (Масс-спектр)
ppm м.д. (миллионные доли)
s (с. (синглет)
d д. (дуплет)
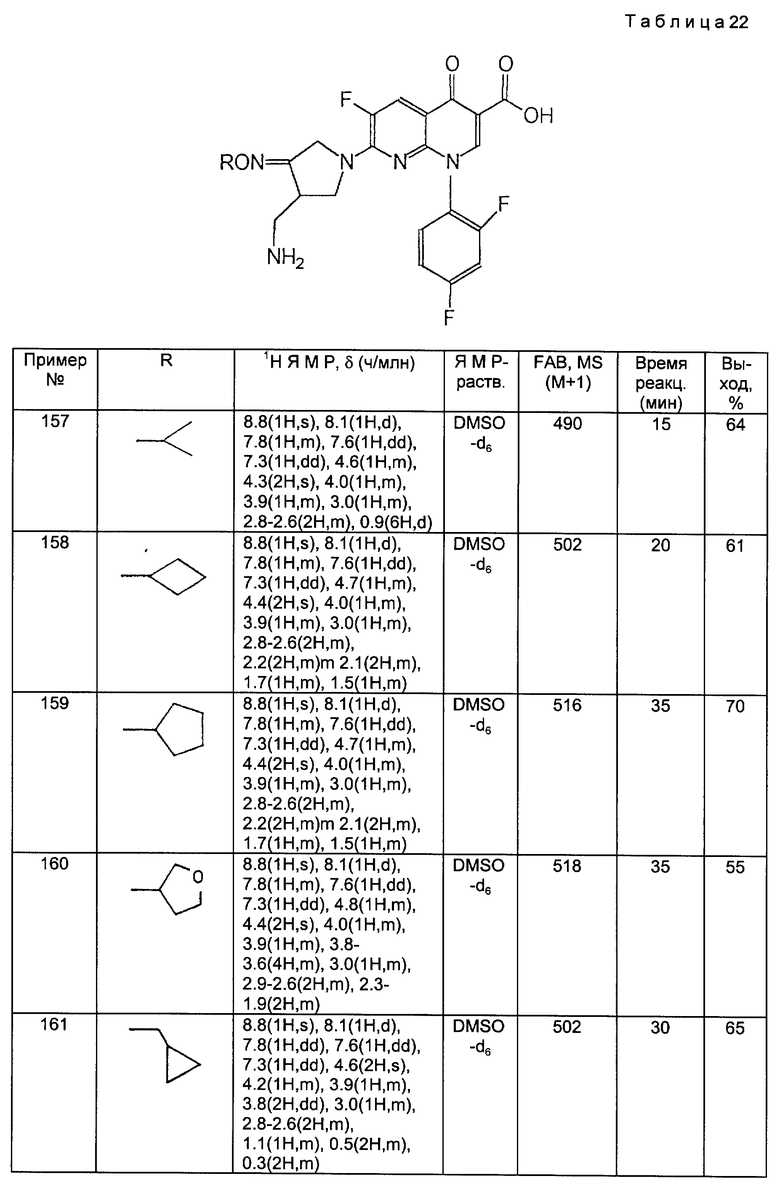
t т. (триплет)
q кв. (квартет)
m м. (мультиплет)
dd дд. (дуплет дуплетов)
b, br широкий
Ex., Examp. Пример
NMR solv. Растворитель для ЯМР
Reac. time (min, hr) Время реакции (мин, ч)
Yield Выход
DMSO ДМСО (диметилсульфоксид)
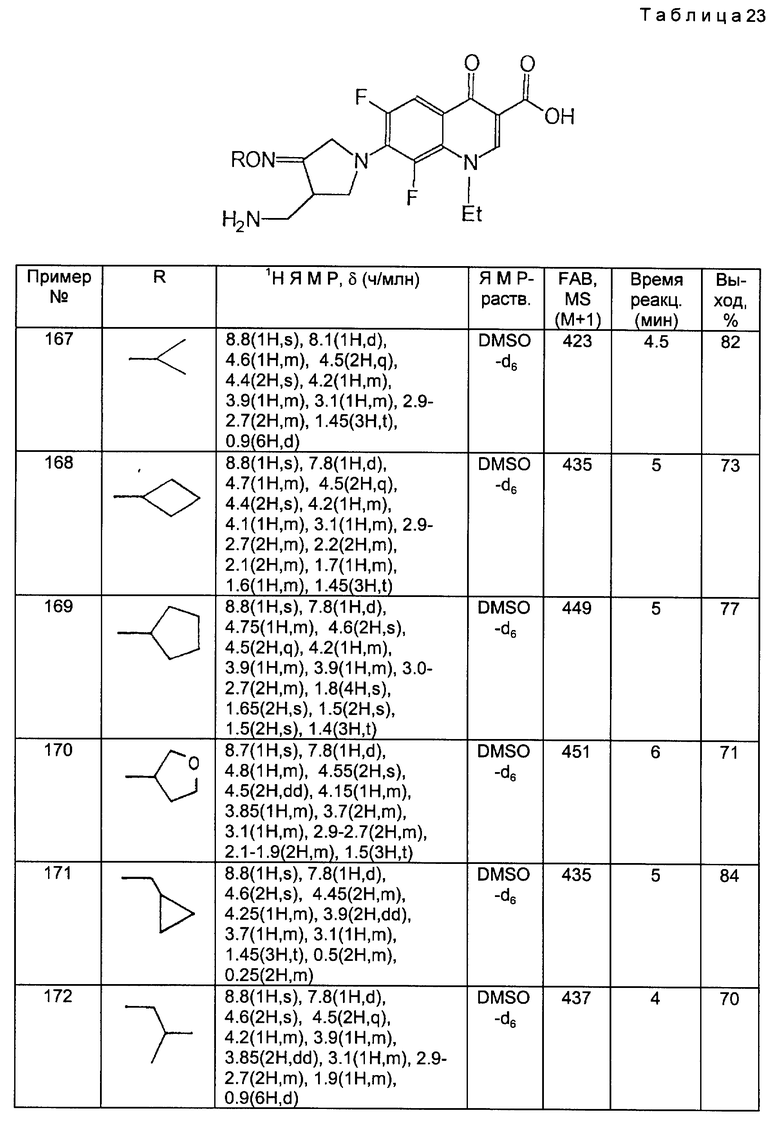
Et этил
Ph фенил
t-Bu трет-бутил
Получение 18
Синтез дигидрохлорида 4-аминометилпирролидин-3-он-бензилоксима,
20 мл метанола охлаждали до 5oC, после чего к нему медленно добавляли 10 мл ацетилхлорида. Эту смесь перемешивали в течение 30 минут, и к ней добавляли 990 мг полученного в получении 7 соединения, растворенного в 10 мл метанола. Реакционную смесь перемешивали 50 минут при комнатной температуре и концентрировали при пониженном давлении. Остаток промывали этилацетатом и высушивали, получив в результате 648 мг (выход: 94%) указанного в заголовке соединения в виде желтого твердого вещества.
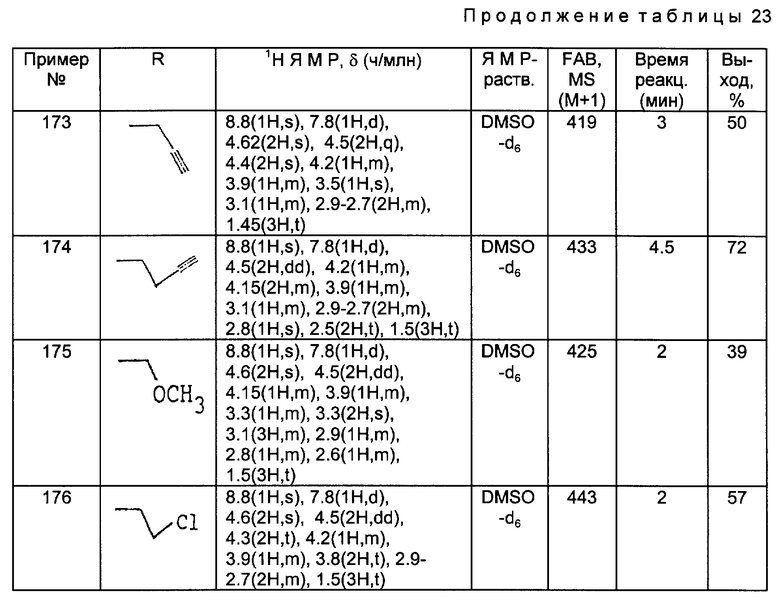
1H ЯМР (DMCO - d6, м.д.): δ 10,0 (1H, м) 8,35 (2H, м), 7,40 (5H, м), 5,18 (2H, с), 4,00 (2H, с) 3,69 (1H, м), X 3,40 (2H, м), 3,12 (2H, с)
MC (FAB,m/ e): 220 (M+H)
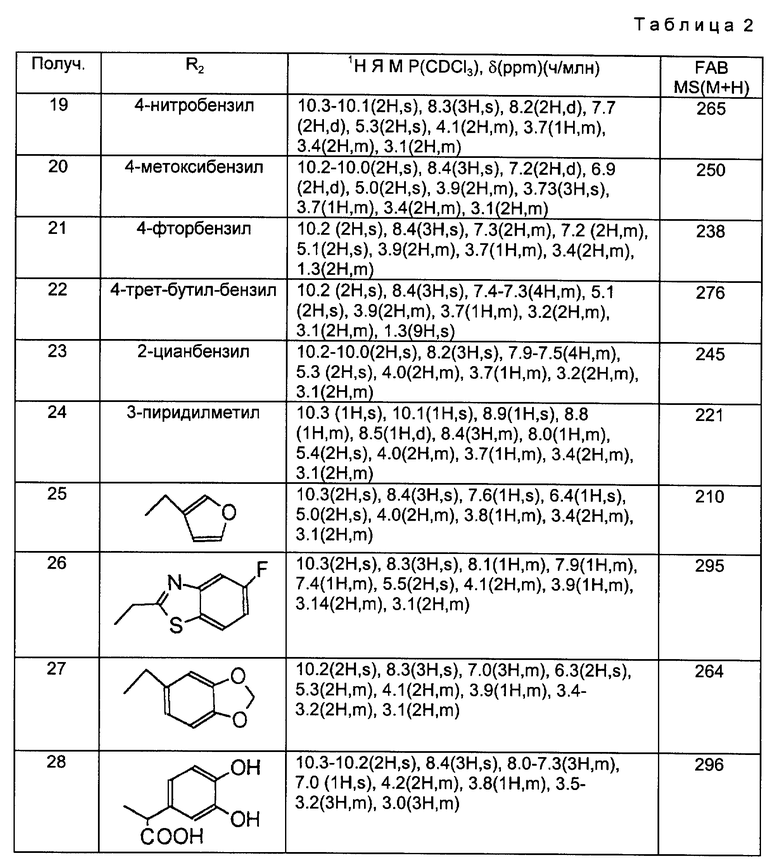
Получения 19 - 28.
Соединения, полученные в следующей ниже таблице 2, были получены из аминосоединений, полученных в Получениях 8 - 17, в соответствии с такой же процедурой, как в Получении 18.
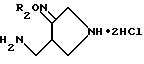
Получение 29.
Синтез 1-(N-трет-бутоксикарбонил)-4-(N-трет-бутоксикарбонил)- аминометилпирролидин-3-он-трет-бутилоксима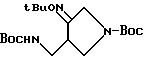
300 мг соединения, полученного в Получении 5, растворяли в смеси 6 мл 95%-ного этанола и 3 мл тетрагидрофурана (ТГФ), и этот раствор вливали в реакционный сосуд емкостью 30 мл. Туда добавляли 487 мг (3,5 моль экв.) о-трет-бутилгидроксиамингидрохлорида и затем добавляли 281 мг (3,5 моль экв) гидрокарбоната натрия, растворенного в 1,5 мл дистиллированной воды. Реакционную смесь перемешивали в течение 40 минут при 40oC под масляной баней, чтобы завершить реакцию, и затем охлаждали, концентрировали под пониженным давлением, разводили метиленхлоридом, промывали насыщенным водным раствором хлорида натрия, осушали над безводным сульфатом магния и затем фильтровали. Фильтрат концентрировали и остаток подвергали колоночной хроматографии на силикагеле, элюируя смесью (1:1 по объему) гексана и этилацетата, и в результате получили 285 мг (выход: 80%) указанного в заголовке соединения.
1H ЯМР (CDCI3, м.д.): δ 5,10 (1H, шир. с), 4,05 (2H, с), 3,71 (1H, дд), 3,43 (1H, шир.) 3,2 (2H, т) 3,0 (1H, м) 1,42 (18H, с), 1,30 (9H, с)
MC (FAB, m/e): 386 (M+H)
Получение 30.
Синтез 1-(N-трет-бутоксикарбонил)-4-(N-трет-бутоксикарбонил)-аминометилпирролидин -3-он-3-бутинилоксима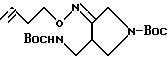
А. Синтез 3-бутинилгидроксиламина.
В 15 мл сухого тетрагидрофурана растворяли 0,35 г (5 ммоль) 3-бутинола, 0,86 г (5,25 ммоль) N-гидроксифталимида и 1,44 г (5,5 ммоль) трифенилфосфина, и в полученный раствор добавляли затем в течение 30 минут 1,05 г (6 ммоль) диэтилазодикарбоксилата. Смесь перемешивали в течение 10 минут при комнатной температуре, после чего перегоняли под пониженным давлением для удаления растворителя. К остатку добавляли 50 мл смеси (1:1%) этилацетата и гексана. Осажденный твердый материал отфильтровывали, и фильтрат концентрировали. Остаток очищали путем колоночной хроматографии (гексан-этилацетат, 9: 1 о/о). Полученное белое твердое вещество [0,54 г, выход 50%, 1H ЯМР (CDCI3, м.д.): δ 7,85 (2H, м), 7,75 (2H, м), 4,2 (2H, т), 2,8 (2H, дд), 2,5 (2H, дд), 2,1 (1H, с), FAB MC (POS): [M+H]+=216] растворяли в 12 мл метиленхлорида и к полученному раствору добавляли по каплям 0,25 г (5 ммоль) гидрата гидразина, разведенного 4 мл метанола. Осадок твердого вещества отфильтровывали и фильтрат концентрировали при низкой температуре под пониженным давлением, получив в результате 0,2 г (выход 93%) указанного в заголовке соединения.
1H ЯМР (CDCI3, м. д.): δ 9,5 (2H, шир.), 4,5 (2H, т), 2,8 (2H, м) 2,4 (2H, м), 2,05 (1H, с)
MC (FAB, m/e): 86 (M+H)+
B. Синтез соединения, указанного в заголовке.
В 5 мл метанола растворяли 0,45 г (1,43 ммоль) соединения, полученного в Получении 5 и 0,2 г (2,35 ммоль) 3-бутинилгидроксиамина, и проводили реакцию в течение 12 часов при 60oC. Реакционный раствор концентрировали под пониженным давлением и остаток подвергали колоночной хроматографии (этилацетат - гексан, 1:4 о/о), в результате чего получили 0,59 г (стехиометрическое количество) указанного в заголовке соединения.
1H ЯМР (CDCI3, м.д.): 5,0 (1H, м.), 4,15 (2H, т), 4,0 (2H, с), 3,75 (1H, м), 3,6 - 3,2 (3H, м) 3,0 (1H, м), 2,5 (2H, м), 2,0 (1H, с), 1,45 (18H, с)
FAB MC (POS): 382 (M+H)+
Получения 31 - 36.
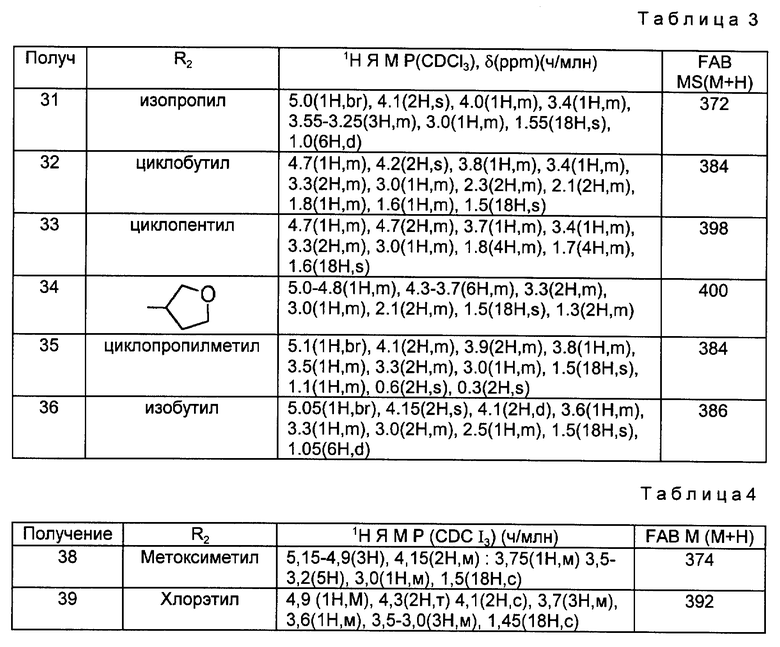
Аминосоединения, перечисленные в следующей ниже таблице 3, были получены в соответствии с такой же процедурой, как в Получении 30, за исключением того, что вместо 3-бутинола использовали соответствующие производные спирта, имеющие такую структуру R2, какая представлена в таблице 3.
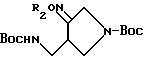 - Таблица 3
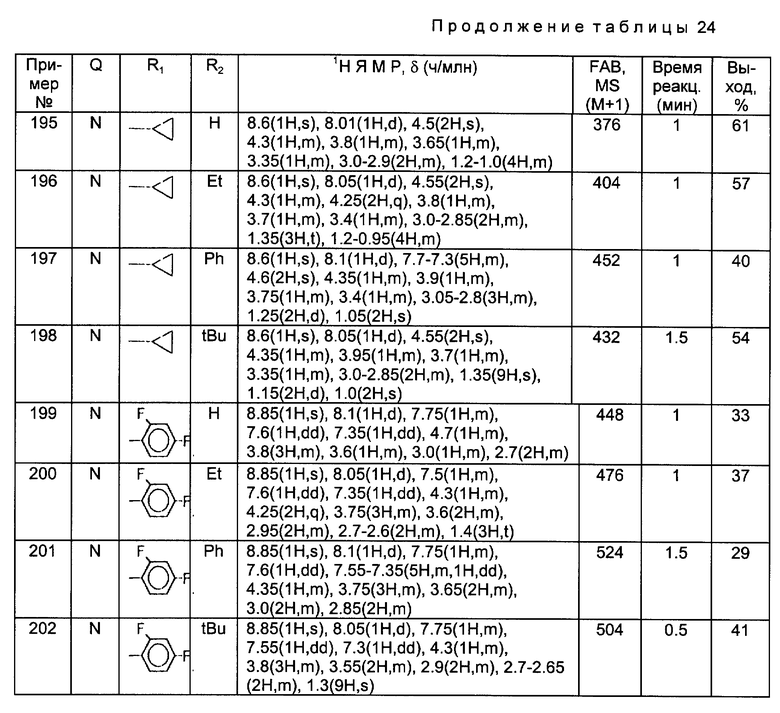
- Таблица 3
Получение 37.
Синтез 1-(N-трет-бутоксикарбонил)-4-(N-трет-бутоксикарбонил)- аминометилпирролидин-3-он-пропаргилоксима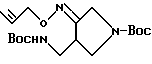
К 15 мл дихлорметана добавляли 659 мг соединения, полученного в Получении 6, 193 мг тетра-н-бутиламмонийбромида и 855 мг пропаргилбромида и к полученной смеси добавляли 5 мл 15%-ного водного раствора гидроксида натрия. Эту смесь перемешивали в течение 30 минут при комнатной температуре. Органический слой отделяли, осушали над безводным сульфатом магния и затем фильтровали. Фильтрат перегоняли под пониженным давлением, и остаток очищали путем хроматографии на стеклянной колонке, получив в результате 776 мг (выход 92%) указанного в заголовке соединения.
1H ЯМР (CDCI3, м.д.): δ 4,92 (1H, м), 4,13 (2H, м) 3,76 (1H, м), 3,41 (1H, м), 3,25 (2H, м), 3,02 (1H, м), 1,50 (9H, с), 1,49 (9H, с)
MC (FAB, m/e): 368 (M+H)
Получения 38 и 39.
Аминосоединения, перечисленные в следующей ниже таблице 4, были получены в соответствии с такой же процедурой, как в Положении 37, за исключением того, что вместо пропаргила использовали соответствующие производные алкила, имеющие представленную в таблице 4 структуру R2.

Получение 40.
Синтез 4-аминометилпирролидин-3-он-трет-бутилоксимдигидрохлорида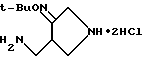
5 мл метанола охлаждали до 0oC и медленно добавляли к нему 3 мл ацетилхлорида. Эту смесь перемешивали в течение 10 минут и добавляли к ней 640 мг полученного в Получении 29 соединения, растворенного в 10 мл метанола. Реакционную смесь перемешивали 20 минут при комнатной температуре и концентрировали при пониженном давлении. Остаток отфильтровывали и промывали диэтиловым эфиром и высушивали, получив в результате 390 мг (выход 91%) указанного в заголовке соединения в виде белого твердого вещества.
1Н ЯМР (DMCO -d6, м.д.): δ 10,0 - 9,6 (2H, шир, с х 2), 8,20 (3H, шир.) 3,90 (2H, дд), 3,61 (1H.с.), 3,40 (2H, шир. с), 3,12 (2H шир. с.), 1,25 (9H, с)
МС (FAB, m/e) : 186 (M + H)
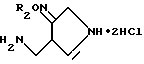
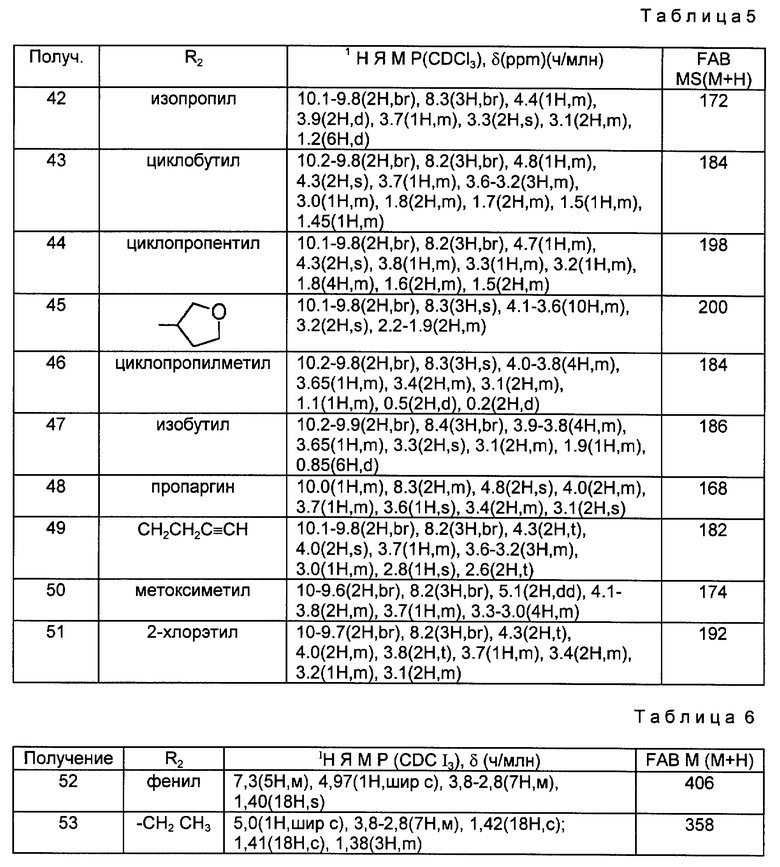
Получения 41 - 50.
Соединения Получений 41-50, перечисленные в следующей ниже таблице 5, были получены из соединений, полученных в Получениях 30-40 в соответствии с такой же процедурой, как в Получении 40.
Получение 51.
Синтез 4-(N-трет-бутоксикарбонил)аминометил-1- (N-трет-бутоксикарбонил)пирролидин-3-он-O-метилоксима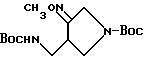
260 мг (8,28 • 10-4 моль) соединения, полученного в Получении 5, растворяли в смеси 5 мл 95%-ного этанола и 2,5 мл тетрагидрофурана, и полученный раствор вливали в реакционный сосуд. К раствору добавляли 256 мг (3,7 моль экв.) метоксиамингидрохлорида, а также 257 мг (3,7 моль экв.) гидрокарбоната натрия (NaHCO3), растворенного в 2,5 мл дистиллированной воды. Реакционную смесь перемешивали в течение 1 часа при 40oC на масляной бане, концентрировали под пониженным давлением, промывали последовательно водным раствором аммоний-хлорида и водным раствором хлорида натрия, осушали над безводным сульфатом магния и затем фильтровали. Фильтрат концентрировали, получив в результате 250 мг (выход 88%) указанного в заголовке соединения.
1H ЯМР (CDCl3, м.д.) : δ 4,98 (1H, шир.с.), 3,81 (3H, с), 3,75 - 2,80 (7H, м), 1,40 (18H, с)
МС (FAB, m/e) : 344 (M+H)
Получения 52 и 53
Соединения, перечисленные в следующей ниже таблице 6, были получены в соответствии с такой же процедурой, как в Получении 51, за исключением того, что вместо метоксиамингидрохлорида использовали феноксиамингидрохлорид или этоксиамингидрохлорида.

Получение 54.
Синтез 4-аминометилпирролидин-3-он-O-метилоксимдитрифторацетата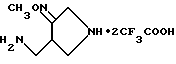
К 250 мг соединения, полученного в Получении 51, добавляли 5 мл трифторуксусной кислоты, и полученную смесь перемешивали в течение 20 минут при комнатной температуре. Реакционную смесь концентрировали под пониженным давление, растворяли в наименьшем количестве ацетонитрила и затем приводили в твердое состояние диэтиловым эфиром, получив в результате 220 мг (выход 84%) указанного в заголовке соединения в очищенном состоянии.
1H ЯМР (CD3OD, м.д.) : δ 4,1 (2H, с.), 3,96 (3H, с), 3,83 (1H, дд), 3,7 - 3,2 (6H, м)
МС (FAB, m/e) : 144 (M+H)
Получения 55 - 57
Соответствующие соединения Получений 55-57 были получены из соединений, полученных в Получениях 6, 52, 53 (соответственно) в соответствии с такой же процедурой, как в Получении 54.
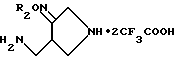
Пример 1.
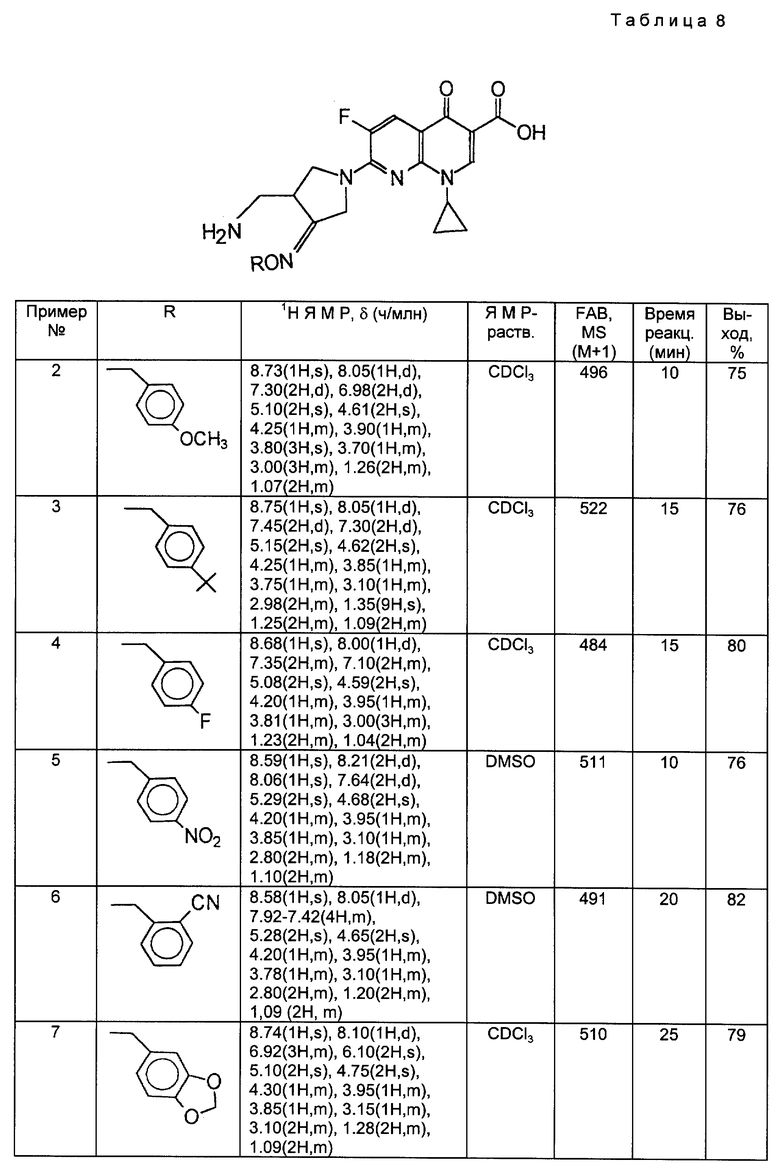
Синтез 7-(4-аминометил-3-бензилоксииминопирролидин-1-ил) -1-циклопропил-6-фтор-1,4-дигидро-4-оксо-1,8-нафтиридин-3-карбоновой кислоты.
622 мг 7-хлор-1-циклопропил-6-фтор-1,4-дигидро-4-оксо-1,8- нафтиридин-3-карбоновой кислоты и 643 мг соединения, полученного в Получении 18, суспендировали в 15 мл ацетонитрила. Эту суспензию охлаждали под водяной баней со льдом, и затем к ней медленно добавляли 1,0 мл 1,8 - диазабицикло[5.4.0]-ундец-7-ена (DBU). Реакционную смесь перемешивали в течение 1,5 часов при комнатной температуре и после добавления 15 мл воды концентрировали. Концентрированную суспензию фильтровали. Отфильтрованный твердый продукт промывали водой и этанолом, и получили в результате 584 мг (выход 57%) указанного в заголовке соединения.
1H ЯМР (DMCO -d6, м.д.) : δ 8,59 (1H, с), 8,03 (1H, д), 7,40 (5H, м), 5,14 (2H, с), 4,75 (2H,с), 4,18 (1H, м), 3,94 (1H, м), 3,83 (1H, м), 3,35 (2H, м), 3,05 (1H, м), 2,81 (1H, м), 2,73 (1H, м), 1,25 - 1,05 (4H, м)
МС (FAB, m/e) : 466 (M+H)
Примеры 2 - 11.
Тот же самый исходный материал, что и в Примере 1, вводили во взаимодействие с каждым из соединений, полученных в Получениях 19 - 28, в соответствии с такой же, как в Примере 1, процедурой и получили соответственные соединения, перечисленные в следующей ниже таблице 8.
Пример 12.
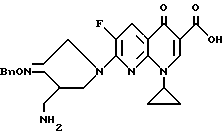
Синтез 7-(4-аминометил-3-бензилоксииминопирролидин-1-ил) -1-циклопропил-6-фтор-1,4-дигидро-4-оксохинолин-3-карбоновой кислоты.
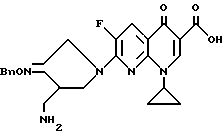
530 мг 1-циклопропил-6,7-дифтор-1,4-гидиро-4-оксохинолин-3-карбоновой кислоты и 584 мг соединения, полученного в Получении 8, суспендировали в 15 мл ацетонитрила. Эту суспензию охлаждали под водяной баней со льдом, и затем медленно добавляли к ней 913 мг 1,8-диизабицикло [5.4.0] ундец-7-ена (DBU). Реакционную смесь перемешивали в течение 2 часов при 80oC и затем, после добавления 15 мл воды, концентрировали. Концентрированную суспензию фильтровали. Отфильтрованный твердый продукт промывали водой и этанолом, получив в результате 631 мг (выход 68%) указанного в заголовке соединения.
1H ЯМР (ДМСО - d6, м.д.) : δ 8,60 (1H, с), 7,92 (1H, д), 7,38 (5H, м), 5,10 (2H, с), 4,87 (2H, с), 4,10 (1H, м), 3,94 (1H, м), 3,86 (1H, м), 3,37 (2H, м), 3,02 (1H, м), 2,38 (1H, м), 2,73 (1H, м), 1,25 - 1,05 (4H, м)
MC (FAB, m/e) : 465 (M+H)
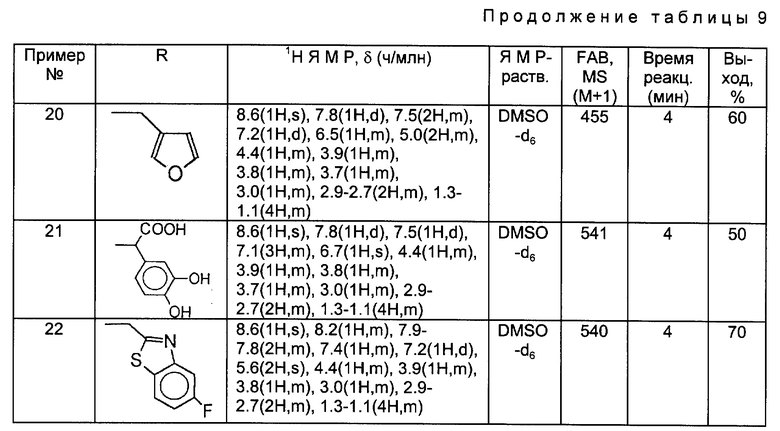
Примеры 13-22.
Тот же самый исходный материал, что и в Примере 12, вводили во взаимодействие с каждым из соединений, полученных в Получениях 19-28, в соответствии с такой же, как в Примере 12, процедурой и получили соответственные соединения, перечисленные в следующей ниже таблице 9.
Примеры 23.
Синтез 7-(4-аминометил-3-бензилоксииминопирролидин-1-ил)циклопропил-6,8- дифтор-1,4-дигидро-4-оксохинолин-3-карбоновой кислоты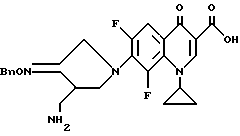
566 мг 1-циклопропил-6,7,8-трифтор-1,4-дигидро-4-оксохинолин-3-карбоновой кислоты и 584 мг соединения, полученного в Получении 8, суспендировали в 15 мл ацетонитрила. Полученную суспензию охлаждали под водяной баней со льдом и затем медленно добавляли к ней 913 мг 1,8-диазобицикло [5.4.0] ундец-7-ена (DBU). Реакционную смесь перемешивали в течение 2 часов при 80oC и затем после добавления 10 мл воды концентрировали. Концентрированную суспензию фильтровали. Отфильтрованный твердый продукт промывали водой и этанолом, получив в результате 704 мг (выход 73%) указанного в заголовке соединения. 1H ЯМР (ДМСО-d6 м.д.) : δ 8,64 (1H, с), 7,99 (1H, д), 7,41 (5H, м), 5,10 (2H, с), 4,73 (2H, с), 4,18 (1H, м), 3,92 (1H, м), 3,86 (1H, м), 3,37 (2H, м), 3,02 (1H, м), 2,83 (1H, м), 2,73 (1H, м), 1,25 - 1,05 (4H, м)
MC (FAB, m/n) : 483 (M+H)
Примеры 24 - 33.
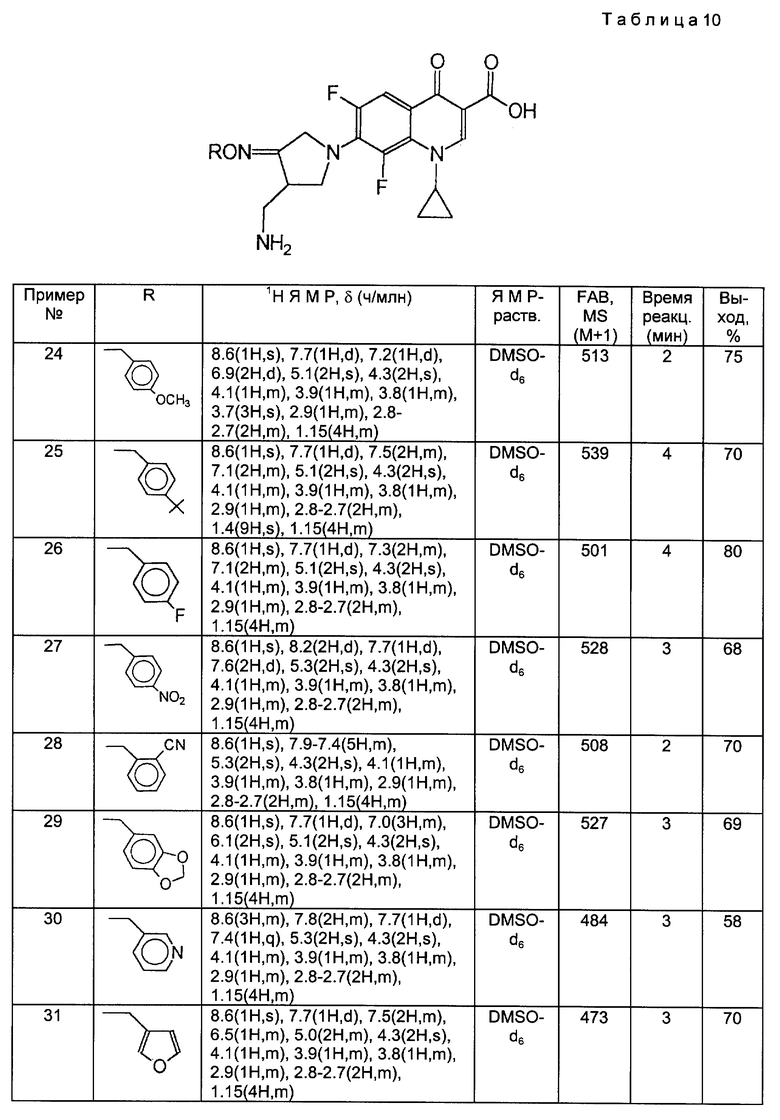
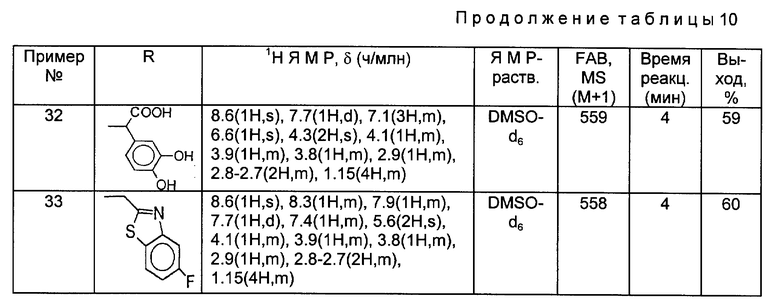
Такой же самый исходный материал, как в Примере 23, вводили во взаимодействие с каждым из соединений, полученны[ в Получениях 19-28, в соответствии с такой же, как в Примере 23, процедурой и получили соответственные соединения, перечисленные в следующей ниже таблице 10.
Примеры 34
Синтез 7-(4-аминометил-3-бензилоксииминопирролидин-1-ил)-8-хлор-1-циклопропил- 6-фтор-1,4-дигидро-4-оксохинолин-3-карбоновой кислоты.
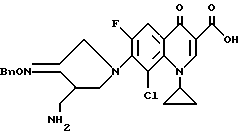
598 мг 8-хлор-1-циклопропил-6,7-дифтор-1,4-дигидро-4-оксохинолин-3-карбоновой кислоты и 584 мг соединения, полученного в Получении 8, суспендировали в 15 мл ацетонитрила, и затем к полученной суспензии медленно добавляли 913 мг 1,8-диазобицикло [5.4.0]ундец-7-ена (DBU). Реакционную смесь перемешивали 3 часа при 80oC и после добавления 15 мл воды концентрировали. Концентрированную суспензию фильтровали. Отфильтрованный твердый продукт промывали водой и диэтиловым эфиром, получив в результате 510 мг (выход 52%) указанного в заголовке соединения.
1H ЯМР (ДМСО - d6 м.д.) : δ/ 8,78 (1H, с), 7,91 (1H, д), 7,41 (5H, м), 5,16 (2H, с), 4,74 (2H, с), 4,16 (1H, м), 3,90 (1H, м), 3,85 (1H, м), 3,35 (2H, м), 3,02 (1H, м), 2,82 (1H, м), 2,75 (1H, м), 1,30 - 1,10 (4H, м)
MC (FAB, m/e) : 499 (M+H)
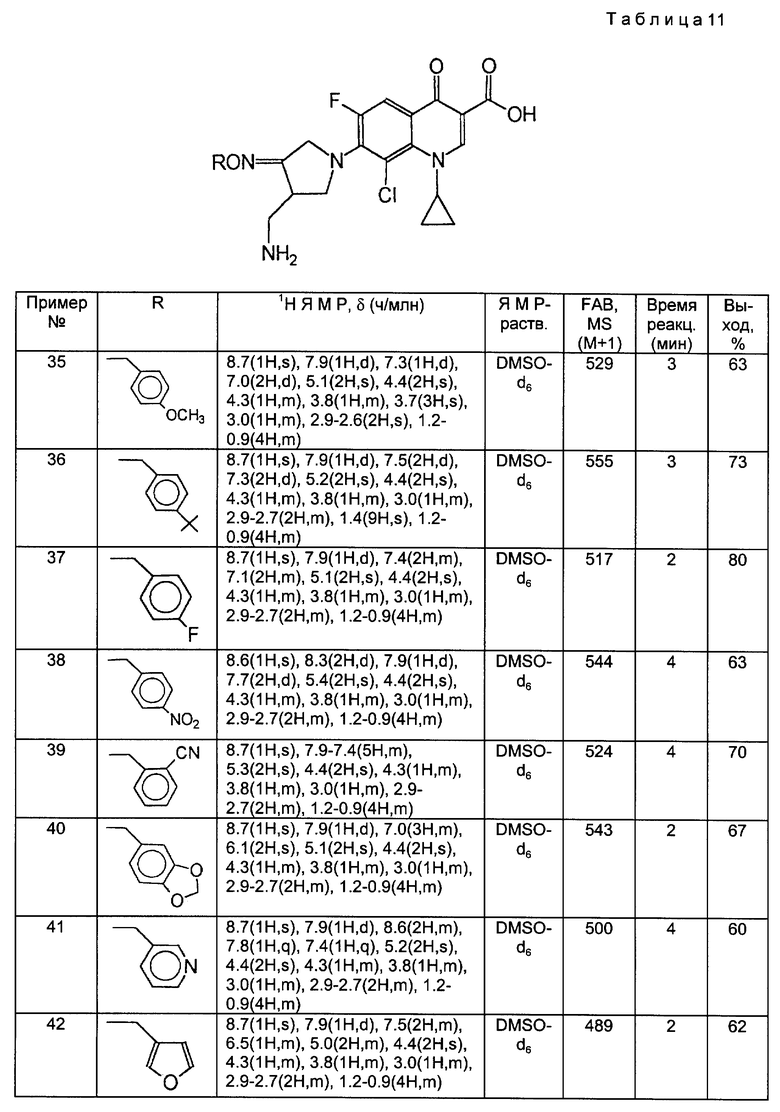
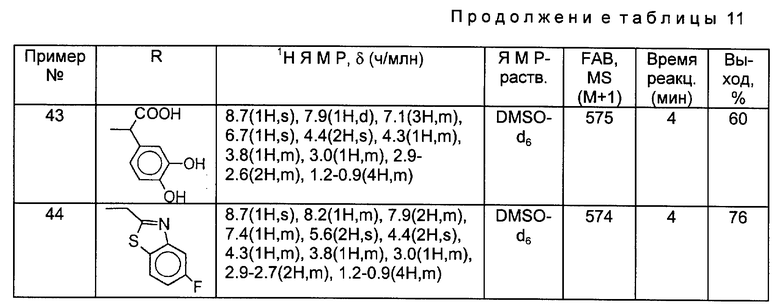
Примеры 35 - 44.
Такой же самый исходный материал, как в Примере 34, вводили во взаимодействие с каждым из соединений, полученных в Получениях 19-28, в соответствии с такой же, как в Примере 34, процедурой и получили соответственные соединения, перечисленные в следующей ниже таблице 11.
Пример 45.
Синтез 7-(4-аминометил-3-бензилоксииминопирролидин-1-ил)-1-циклопропил-6- фтор-8-метокси-1,4-дигидро-4-оксохинолин-3-карбоновой кислоты.

590 мг 1-циклопропил-6,7-дифтор-8-метокси-1,4-дигидро-4-оксохинолин-3-карбоновой кислоты и 584 мг соединения, полученного в Получении 8, суспендировали в 15 мл ацетонитрила, и к полученной суспензии медленно добавляли 913 мг 1,8-диазабицикло [5.4.0] ундец-7-ена (DBU). Реакционную смесь перемешивали в течение 2 часов при 80oC и затем, после добавления 15 мл воды, перемешивали еще 30 минут при комнатной температуре и фильтровали. Отфильтрованный твердый продукт промывали водой и диэтиловым эфиром и получили в результате 465 мг (выход 47%) указанного в заголовке соединения.
1H ЯМР (ДМСО-d6, м. д.) : δ 8,61 (1H, с), 7,99 (1H, д), 7,40 (5H, м), 5,15 (2H, с), 4,74 (2H, с), 4,17 (1H, м), 3,95 (1H, м), 3,83 (1H, м), 3,60 (3H, с), 3,35 (2H, м), 3,02 (1H, м),2,80 (1H, м), 2,71 (1H, м), 1,30 - 1,10 (4H, м)
MC (FAB, m/e) : 495 (M+H)
Примеры 46-55.
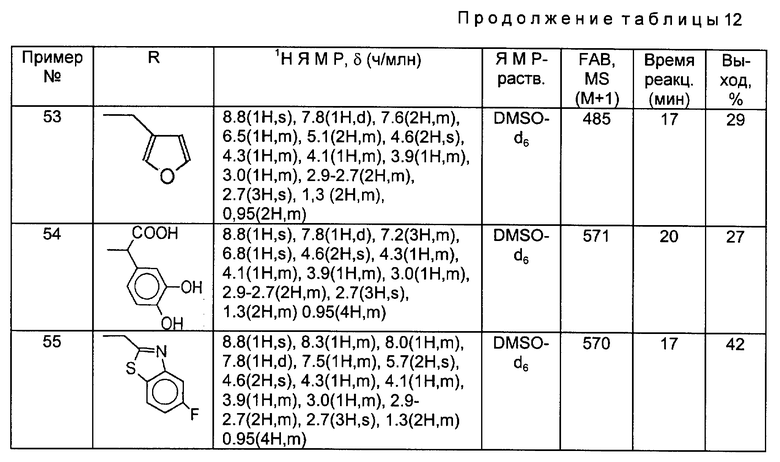
Такой же самый исходный материал, как в Примере 45, вводили во взаимодействие с каждым из соединений, полученных в Получениях 19-28, в соответствии с такой же, как в Примере 45, процедурой и получили соответственные соединения, перечисленные в следующей ниже таблице 12.
Пример 56
Синтез 5-амино-7-(4-аминометил-3-бензилоксииминопирролидин-1-ил)-1-циклопропил-6,8-дифтор-1,4-дигидро-4-оксохинолин-3-карбоновой кислоты
448 мг 5-амино-1-циклопропил-6,7,8-трифтор-1,4-дигидро-4-оксохинолин-3-карбоновой кислоты и 438 мг соединения, полученного в Получении 8, суспендировали в 15 мг ацетонитрила, и затем к полученной суспензии медленно добавляли 585 мг 1,8-диазобицикло[5.4.0]ундец-7-ена (DBU). Реакционную смесь нагревали в течение 6 часов при 80oC и добавляли к ней 10 мл воды. Полученную суспензию фильтровали. Отфильтрованный твердый продукт промывали водой, ацетонитрилом и диэтиловым эфиром, получив в результате 395 мг (выход 53%) указанного в заголовке соединения.
1H ЯМР (ДМСО-d6, м. д.) : δ 8,62 (1H, с), 7,92 (1H, д), 7,40 (5H, м), 6,10 (2H, шир.с.), 5,13 (2H, с), 4,73 (2H, с), 4,15 (1H, м), 3,95 (1H, м), 3,82 (1H, м), 3,35 (2H, м), 3,01 (1H, м), 2,80 (1H, м), 2,73 (1H, м), 1,25 - 1,05 (4H, м)
MC (FAB, m/e) : 498 (M+H)
Примеры 57-66.
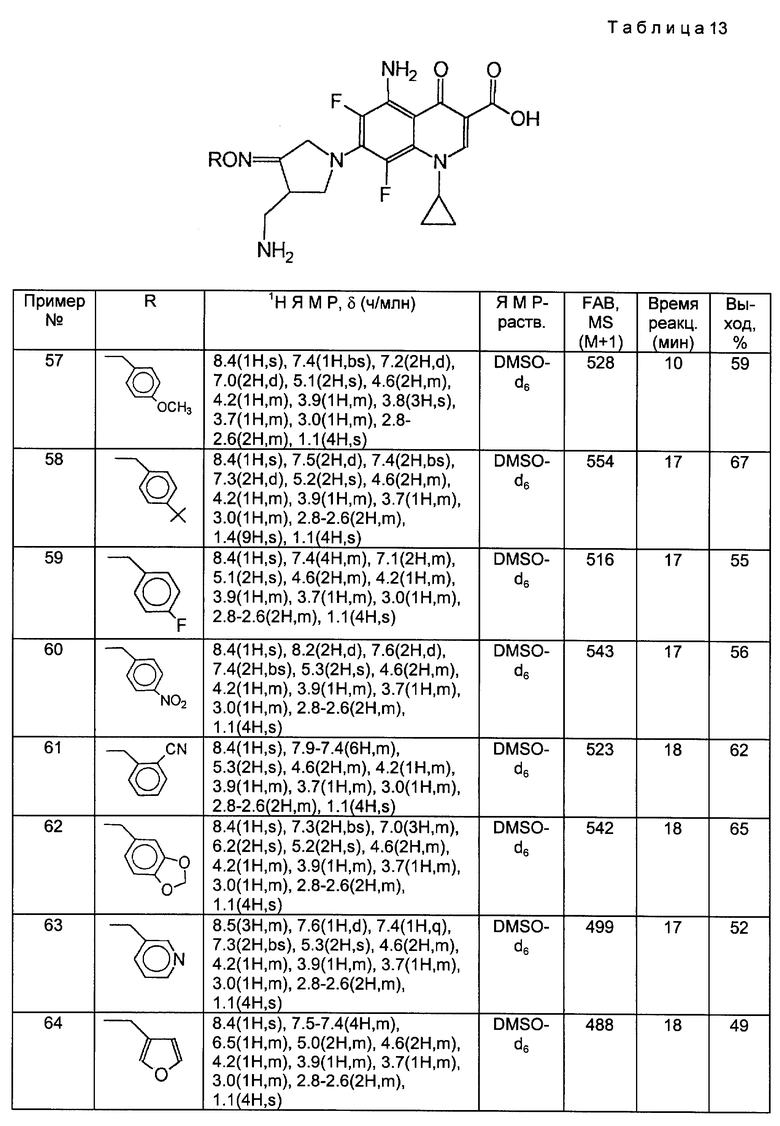
Такой же самый исходный материал, как в Примере 56, вводили во взаимодействие с каждым из соединений, полученных в Получениях 19 - 28, в соответствии с такой же, как в Примере 56, процедурой и получили соответственные соединения, перечисленные в следующей ниже таблице 13.
Пример 67.
Синтез-7-(4-аминометил-3-бензилоксииминопирролидин-1-ил) -1-(2,4-дифторфенил)-6-фтор-1,4-дигидро-4-оксо-1,8-нафтиридин-3-карбоновой кислоты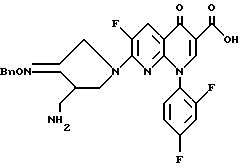
806 мг 7-хлор-1-(2,4-дифторфенил)-6-фтор-1,4-дигидро-4-оксо-1,8-нафтиридин-3- карбоновой кислоты и 438 мг соединения, полученного в Получении 8, суспендировали в 15 мл ацетонитрила, и к полученной суспензии медленно добавляли 913 мг 1,8-диазабицикло[5.4.0]ундец-7-ена (DBU). Реакционную смесь перемешивали в течение часа при комнатной температуре и после добавления 15 мл воды перемешивали еще 30 минут и фильтровали. Отфильтрованный твердый продукт промывали водой и ацетонитрилом, получив в результате 524 мг (выход 65%) указанного в заголовке соединения.
1H ЯМР (ДМСО-d6, м. д.) : δ 8,82 (1H, с), 8,21 (1H, д), 7,85 (1H, м), 7,56 (1H, м), 7,40 (6H, м), 5,16 (2H, с), 4,76 (2H, c), 4,18 (1H, м), 3,94 (1H, м), 3,81 (1H, м), 3,34 (2H, м), 3,04 (1H, м), 2,82 (1H, м) 2,73 (1H, м), 1,30 - 1,00 (4H, м)
MC (FAB, m/e) : 538 (M+H)
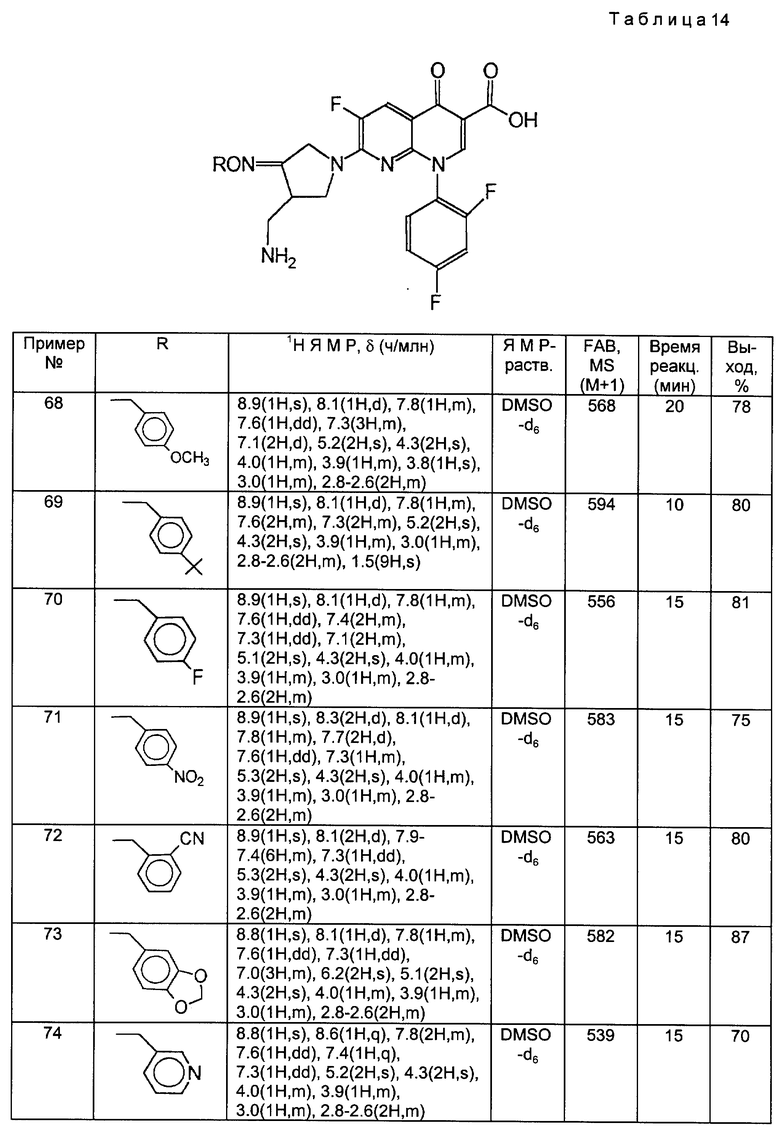
Примеры 68-77.
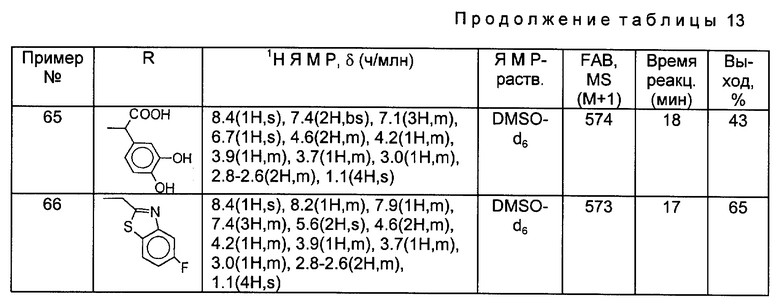
Такой же самый исходный материал, как в примере 67, вводили во взаимодействие с каждым из соединений, полученных в Получениях 19-28, в соответствии с такой же, как в примере 67, процедурой и получили соответственные соединения, перечисленные в следующей ниже таблице 14.
Пример 78.
Синтез 7-(4-аминометил-3-бензилоксииминопирролидин-1-ил)-1-этил -6,8-дифтор-1,4-дигидро-4-оксохинолин-3-карбоновой кислоты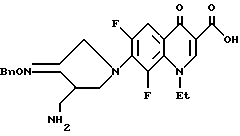
353 мг 1-этил-6,7,8-трифтор-1,4-дигидро-4-оксохинолин-3-карбоновой кислоты и 380 мг соединения, полученного в Получении 8, суспендировали в 15 мл ацетонитрила, и затем к полученной суспензии медленно добавляли 593 мг 1,8-диазабицикло[5.4.0]ундец-7-ена (DBU). Реакционную смесь перемешивали 2,5 часа при 80oC, а затем после добавления 15 мл воды перемешивали еще 30 минут под баней с холодной водой и фильтровали. Отфильтрованный твердый продукт промывали водой, ацетонитрилом и диэтиловым эфиром, получив в результате 391 мг (выход 64%) указанного в заголовке соединения.
1H ЯМР (ДМСО -d6, м.д.) : δ 8,8 (1H, с), 7,8 (1H, д), 7,40 (5H, м), 5,10 (2H, с), 4,6 (2H, кв), 4,4 (2H, дд.); 4,0 (1H, м), 3,1 (1H, м), 3,7 (1H, м), 2,8 (2H, ддд); 1,46 (3H, т.)
MC (FAB, m/e) : 471 (M+H)
Примеры 79-88.
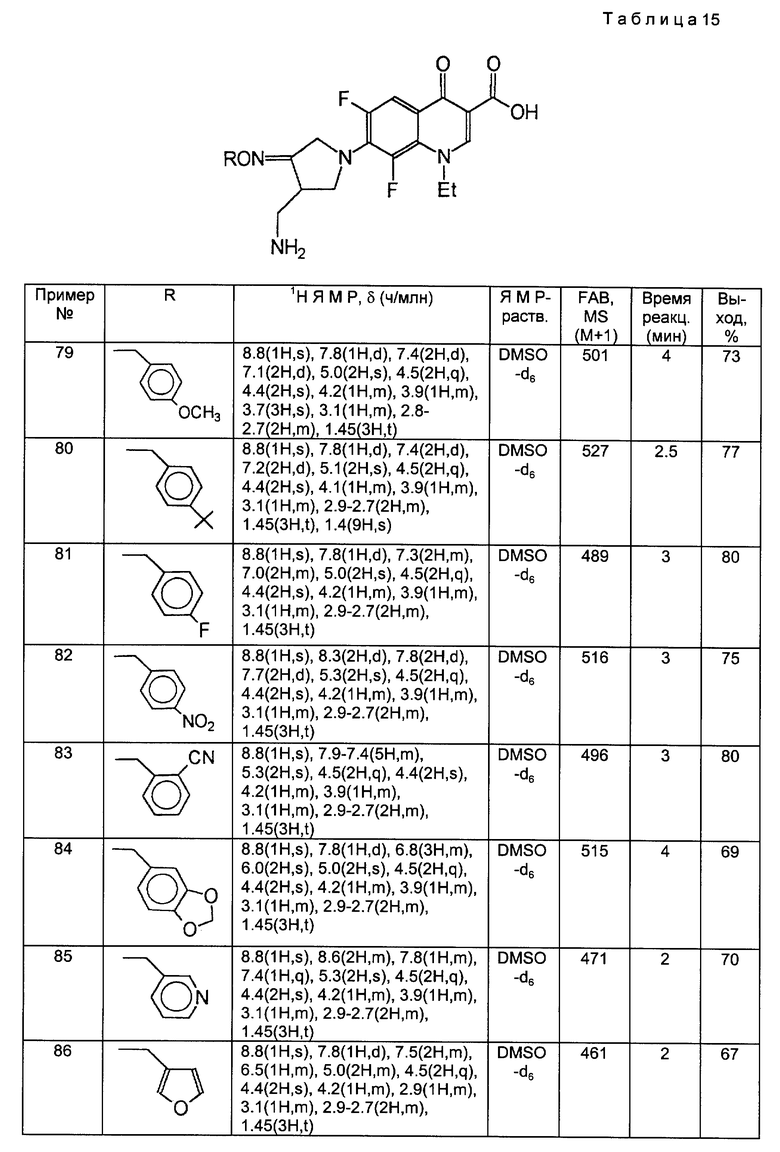
Такой же самый исходный материал, как в Примере 78, вводили во взаимодействие с каждым из соединений, полученных в Получениях 19-28, в соответствии с такой же, как в Примере 78, процедурой и получили соответственные соединения, приведенные в следующей ниже таблице 15.
Пример 89.
Синтез 7-(4-аминометил-3-трет-бутилоксииминопирролидин-1-ил)-1-циклопропил- 6-фтор-1,4-дигидро-4-оксо-1,8-нафтиридин-3-карбоновой кислоты
141 мг (0,5 ммоль) 7-хлор-1-циклопропил-6-фтор-4-оксо-1.4-дигидро-[1,8] нафтиридил-3-карбоновой кислоты и 143 мг (0,55 ммоль) 4-аминометилпирролидин-3-он-трет-бутилоксимдигидрохлорида тщательно суспендировали в 2,5 мл ацетонитрила. Затем к полученной суспензии медленно добавляли по каплям 230 мг 1,8-диазабицикло [5.4.0] унденц-7-ена. Реакционную смесь перемешивали в течение 30 минут при комнатной температуре и после добавления 1 мл воды интенсивно перемешивали еще 10 минут и фильтровали. Отфильтрованный твердый продукт промывали последовательно смесью (4:1 по объему, 2 мл) ацетонитрила и воды и ацетонитрилом (2 мл х 2), а затем диэтиловым эфиром и высушивали, получив в результате 132 мг (выход 61%) указанного в заголовке соединения.
1H ЯМР (ДМСО -d6, м.д.) : δ/ 8,6 (1H, с), 8,1 (1H, д), 4,6 (2H, с), 4,2 (1H, дд), 3,9 (1H, дд), 3,7 (1H, м), 3,3 (1H, дд), 2,9 - 2,7 (2H, ддд), 1,3 (9H, с), 1,2 (2H, м), 1,1 (2H, м)
FAB MC (POS) : 432 (M+H)+
Пример 90.
Синтез 7-(3-аминометил-4-трет-бутилоксииминопирролидин-1-ил)-1-циклопропил-6,8- дифтор-4-оксо-1,4-дигидрохинолин-3-карбоновой кислоты
141 мг (0,5 ммоль) 1-циклопропил-6,7,8-трифтор-4-оксо-1,4-дигидрохинолин-3-карбоновой кислоты и 143 мг (0,55 ммоль) 3-аминометил-4-трет-бутилоксииминопирролидиндигидрохлорида нагревали с обратным холодильником так же, как в Примере 89, в течение 2,5 часов и охлаждали до комнатной температуры. Затем полученный продукт отделяли и очищали путем препаративной ВЭЖХ, получив в результате 151 мг (выход 67%) указанного в заголовке соединения.
1Н ЯМР (DMCO-d6, м. д.): δ 8,6 (1H, с), 7,7 (1H, д), 4,3 (2H, с), 4,1 (1H, м), 3,9 (1H, м), 3,8 (1H, м), 2,9 (1H, м), 2,8 - 2,7 (2H, м), 1,3 (9H, м), 1,15 (4H, с)
FAB MC (POS) :[M + H]+ = 449.
Пример 91.
Синтез 8-хлор-1-циклопропил-6-фтор-[7-(3-аминометил-4-третбутилоксииминопирролидин- 1-ил)]-4-оксо-1,4-дигидрохинолин-3-карбоновой кислоты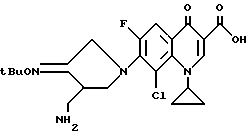
150 мг (0,5 ммоль) 8-хлор-1-циклопропил-6,7-дифтор-4-оксо-1,4-дигидрохинолин-3- карбоновой кислоты вводили в реакцию так же, как в примере 90. Затем реакционный раствор концентрировали и остаток очищали путем препаративной ВЭЖХ, что дало в результате 148 мг (выход 64%) указанного в заголовке соединения.
1H ЯМР (DMCO-d6, м.д.) : δ 8,7 (1H, с), 7,9 (1H, д), 4,4 (2H, с), 4,3 (1H, м), 3,8 (1H, м) 3,7 (1H, м), 3,0 (1H, м), 2,9 - 2,7 (2H, м), 1,3 (9Н, с), 1,2 - 0,9 (4H, м)
FAB MC (POS) : [M + H]+ = 465
Пример 92.
Синтез 7-(3-аминометил-3-трет-бутилоксииминопирролидин-1-ил)- 1-циклопропил-6-фтор-4-оксо-1,4-дигидрохинолин-3-карбоновой кислоты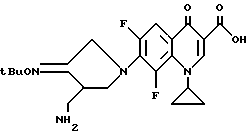
132 мг (0,5 ммоль) 1-циклопропил-6,7-дифтор-4-оксо-1,4-дигидрохинолин-3-карбоновой кислоты нагревали с обратным холодильником, как в Примере 89, в течение 3,5 часов. Затем полученный остаток подвергали препаративной ВЭЖХ и в результате получили 129 мг (выход 60%) указанного в заголовке соединения.
1Н ЯМР (DMCO-d6, м.д.) : δ 8,6 (1H, с), 7,8 (1H, д), 7,2 (1H, д), 4,4 (2H, с), 3,9 (1H, м), 3,8 (1H, м), 3,7 (1H, м) 3,0 (1H, м), 2,9 - 2,7 (2H, м), 1,4 (9H, с), 1,3 - 1,1 (4H, м)
FAB MC (POS) : [M + H]+ = 431
Пример 93.
Синтез 5-амино-7-(3-аминометил-4-трет-бутилоксииминопирролидин-1-ил)-1-циклопропил- 4-оксо-1,4-дигидрохинолин-3-карбоновой кислоты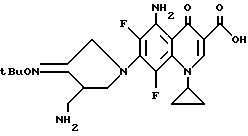
148 мг (0,5 ммоль) 5-амино-1-циклопропил-6.7,8-трифтор-4-оксо-1,4-дигидрохинолин-3-карбоновой кислоты нагревали с обратным холодильником, как в Примере 89, в течение 8 часов. Затем полученный остаток очищали путем препаративной ВЭЖХ и в результате получили 151 мг (выход 65%) указанного в заголовке соединения.
1Н ЯМР (DMCO-d6, м. д.) : δ 8,6 (1H, с), 7,5 (2H, шир.), 4,3 (2H, с), 4,0-3,8 (3H, м), 3,2 (1H, м), 2,8 - 2,6 (2H, м), 1,3 (9H, с), 1,1 (4H, м)
FAB MC (POS) : [M + H]+ = 464
Пример 94.
Синтез 7-(3-аминометил-4-трет-бутилоксиимино-пирролидин-1-ил)-1- циклопропил-6-фтор-8-метокси-4-оксо-1,4-дигидрохинолин-3-карбоновой кислоты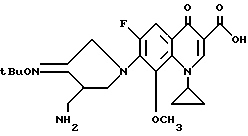
148 мг (0,5 ммоль) 1-циклопропил-6,7-дифтор-8-метокси-4-оксо-1,4-дигидрохинолин-3-карбоновой кислоты нагревали с обратным холодильником, как в Примере 89, в течение 10 часов. Затем полученный остаток очищали путем препаративной ВЭЖХ и в результате получили 92 мг (выход 40%) указанного в заголовке соединения.
1Н ЯМР (DMCO-d6, м.д.) : δ/ 8,9 (1H, с), 7,8 (1H, д), 4,5 (2H, с), 4,3 (1H, м), 4,1 (1H, м), 3,9 (1H, м), 3,0 (1H, м), 2,8 - 2,7 (2H, м), 2,7 (3H, с), 1,3 (9H, с), 1,25 (2H, м), 0,9 (2H, с)
FAB MC (POS) : [M + H]+=461
Пример 95.
Синтез 7-(3-аминометил-4-трет-бутилоксииминопирролидин-1-ил)-1-(2,4-дифторфенил)-6- фтор-4-оксо-1,4-дигидро-1,8-нафтиридин-3-карбоновой кислоты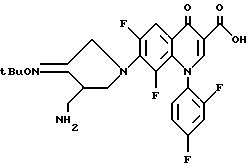
168 мг (0,5 ммоль) 6,7-дифтор-1-(2,4-дифторфенил)-4-оксо-1,4-дигидронафтиридин-3-карбоновой кислоты и 143 мг (0,55 ммоль) 3-аминометил-4-трет-бутилокси-иминопирролидиндигидрохлорида суспендировали в 3 мл сухого ацетонитрила. К полученной суспензии добавляли 230 мг (1,5 ммоль) 1.8-диазабицикло [5.4.0] ундец-7-ена, и реакционную смесь перемешивали в течение 15 минут при комнатной температуре и затем обрабатывали так, как в примере 89, получив в результате 203 мг (выход 81%) указанного в заголовке соединения.
1Н ЯМР (DMCO-d6, м. д.): δ 8,9 (1H, с), 8,1 (1H, д), 7,8 (1H, м), 7,7 (1H, дд), 7,3 (1H, дд), 4,3 (2H, с), 4,0 (1H, м), 3,9 (1H, м), 3,0 (1H, м), 2,8 - 2,6 (2H, м), 1,3 (9H, с)
FAB MC (POS) : [M + H]+ = 504
Пример 96.
Синтез 7-(3-аминометил-4-трет-бутилоксииминопирролидин-1-ил)- 6,8-дифтор-1-этил-4-оксо-1,4-дигидрохинолин-3-карбоновой кислоты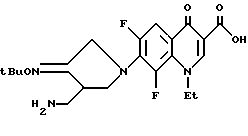
136 мг (0,5 ммоль) 1-этил-6,7,8-трифтор-4-оксо-1,4-дигидрохинолин-3-карбоновой кислоты нагревали с обратным холодильником, как в примере 89, в течение 5 часов. Затем полученный остаток очищали путем препаративной ВЭЖХ и в результате получили 170 мг (выход 78%) указанного в заголовке соединения.
1H ЯМР (DMCO-d6, м.д.) : δ 8,8 (1H, с), 7,8 (1H, д), 4,5 (2H, кв.), 4,4 (2H, с), 4,2 (1H, м), 3,9 (1H, м), 3,1 (1H, м), 2,9 - 2,7 (2H, м), 1,45 (3H, т.), 1,3 (9H, с)
FAB MC (POS) : [M + H]+ = 437
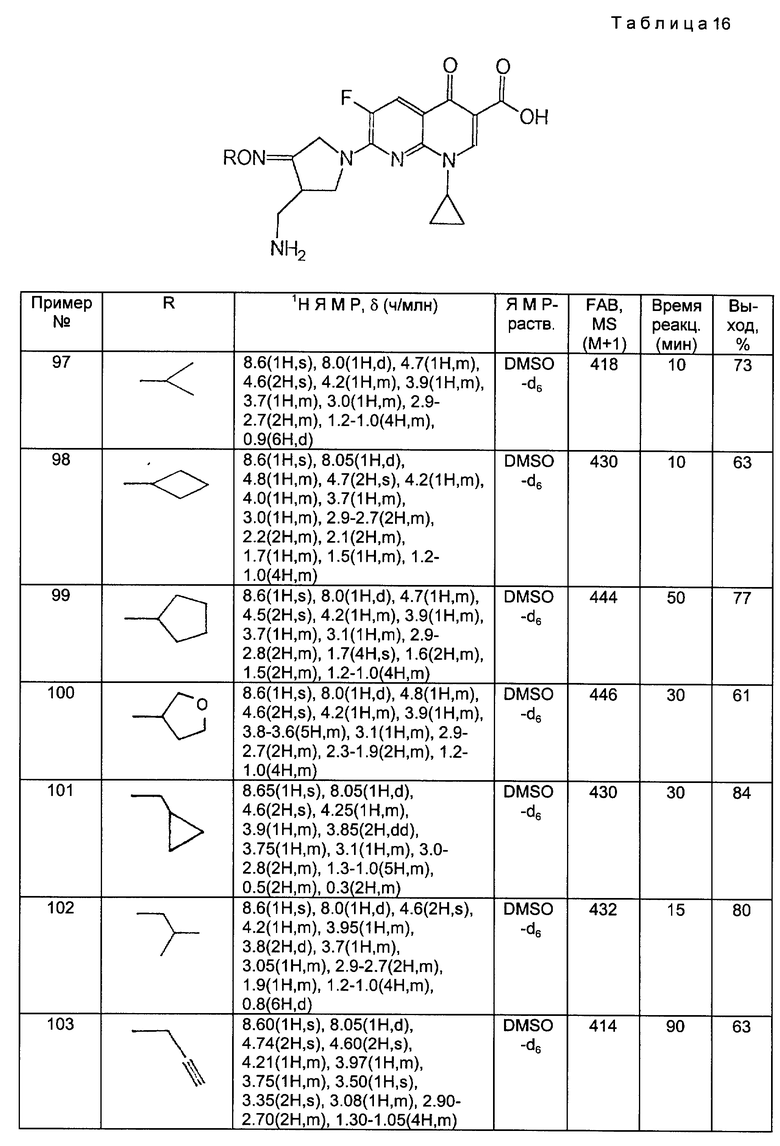
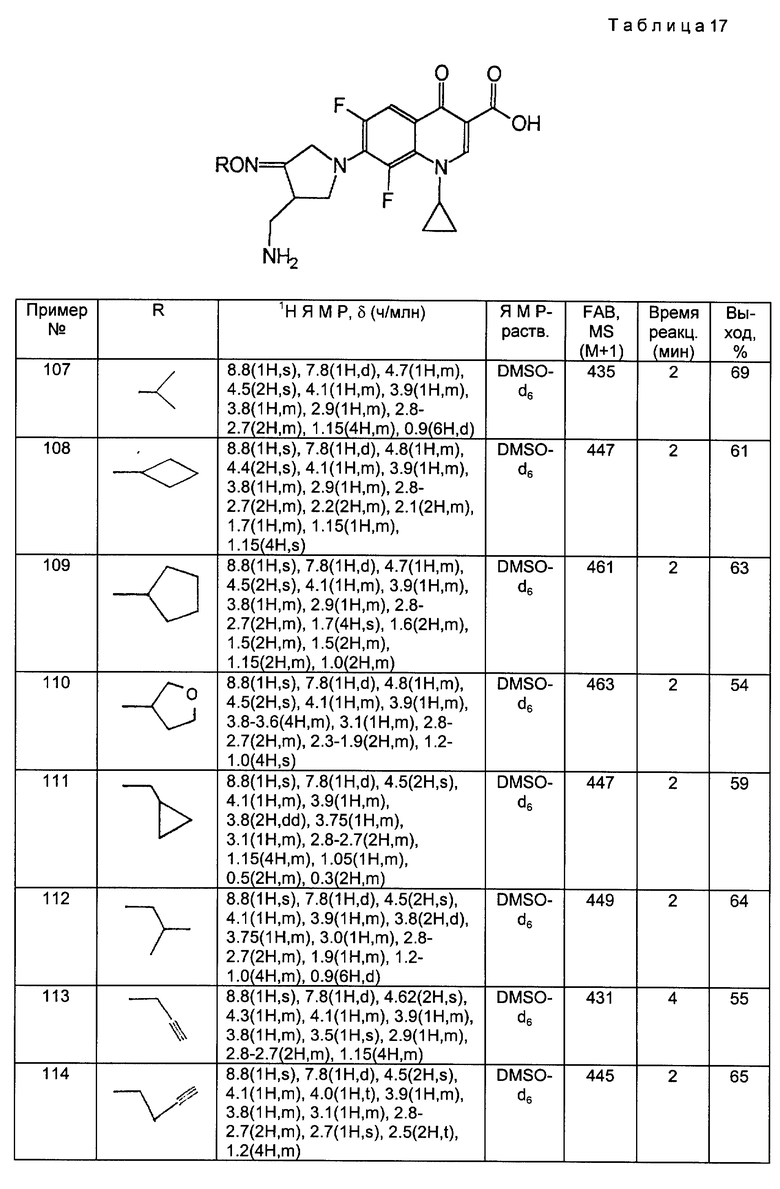
Примеры 97 - 176.
Аминосоединения, полученные в Получениях 41 - 50, обрабатывали в соответствии с такой же, как в Примерах 89 - 96, процедурой и получили соответственные соединения 97 - 176, для которых данных ЯМР и МС приведены в следующих ниже таблицах 16 - 23.
Пример 177.
Синтез 7-(4-амино-3-метоксииминопирролидин-1-ил)-1-циклопропил- 6,8-дифтор-4-оксо-1,4-дигидрохинолин-3-карбоновой кислоты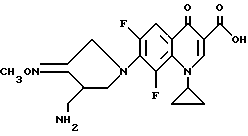
К 23 мл сухого ацетонитрила добавляли 2,83 г (10 ммоль) 1-циклопропил-6,7,8-трифтор-4-оксо-1,4-дигидрохинолин-3-карбоновой кислоты и 4,27 г (11,5 ммоль) - 4-аминометилпирролидин-3-он-O-метилоксимдитрифторацетата. Затем к полученной смеси добавляли 4,6 г (30 ммоль) 1,8-диазабицикло[5.4.0]ундец-7-ена, и смесь нагревали с обратным холодильником в течение 1,5 часов и затем охлаждали до комнатной температуры. К реакционному раствору добавляли 15 мл дистиллированной воды. Осажденный твердый продукт отделяли и высушивали, получив в результате 2,24 г (выход 55%) указанного в заголовке соединения.
1Н ЯМР (DMCO-d6, м.д.): δ 8,6 (1H, с), 7,75 (1H, д), 4,35 (2H, с), 4,1 - 3,9 (2H, м), 3,8 (3H, с), 3,7 (1H, м), 3,35 (1H, м), 2,9 - 2,6 (2H, м), 1,25 (2H, д), 0,95 (2H, с)
FAB MC (POS) : [M + H] = 407
Пример 178.
Синтез 7-(4-аминометил-3-метоксииминопирролидин-1-ил)-8-хлор-1- циклопропил-6-фтор-4-оксо-1,4-дигидрохинолин-3-карбоновой кислоты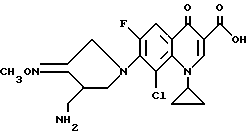
141 мг (0,5 ммоль) 1-циклопропил-8-хлор-6,7-дифтор-4-оксо-1,4- дигидрохинолин-3-карбоновой кислоты и 205 мг (0,55 ммоль) 4-аминометилпирролидин-3-он-O-метилоксимдитрифторацетата вводили в реакцию на один час таким же образом, как в Примере 177. Затем реакционный раствор концентрировали, и остаток очищали путем препаративной ВЭЖХ, получив в результате 88 мг (выход 42%) указанного в заголовке соединения.
1Н ЯМР (DMCO-d6, м.д.): δ 8,7 (1H, с), 7,85 (1H, д), 4,4 (1H, м), 3,75 (3H, с), 3,7 (3H, м), 3,4 (2H, м), 3,0 - 2,7 (2H, м), 1,25 (2H, д), 1,0 (2H, с)
FAB MC (POS) : [M + H] = 423
Пример 179.
Синтез 7-(4-аминометил-3-метоксиимипирролидин-1-ил)-1- циклопропил-6-фтор-4-оксо-1,4-дигидрохинолин-3-карбоновой кислоты
132 мг (0,5 ммоль) 1-циклопропил-6,7-дифтор-4-оксо-1,4- дигидрохинолин-3-карбоновой кислоты и 205 мг (0,55 ммоль) 4-аминометилпирролидин-3-он-O-метилоксимдитрифторацетата вводили в реакцию на 3 часа таким же образом, как в Примере 177. Затем реакционный раствор концентрировали и остаток очищали путем препаративной ВЭЖХ, получив в результате 73 мг (выход 37%) указанного в заголовке соединения.
1Н ЯМР (DMCO-d6, м.д.): δ/ 8,6 (1H, С), 7,86 (1H, д), 7,2 (1H, д), 4,4 (2H, д), 3,9 (1H, м), 3,85 (3H, с), 3,8 - 3,65 (2H, м), 3,0 (1H, м), 2,9 - 2,7 (2H, м), 1,3 (2H, м), 1,1 (2H, м)
FAB MC (POS) : [M + H] = 389
Пример 180.
Синтез 7-(4-аминометил-3-метоксииминопирролидин-1-ил)-1- циклопропил-6-фтор-4-оксо-1,4-дигидро[1,8]нафтиридин-3-карбоновой кислоты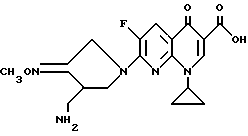
141 мг (0,5 ммоль) 1-циклопропил-7-хлор-6-фтор-4-оксо-1,4-дигидро[1,8] нафтиридин-3-карбоновой кислоты и 205 мг (0,5 ммоль) 4-аминометилпирролидин-3-он-O- метилоксимдитрифторацетата вводили в реакцию на 0,5 часа таким же образом, как в Примере 177, и в результате получили 167 мг (выход 85%) указанного в заголовке соединения.
1Н ЯМР (DMCO-d6, м.д.): δ 8,6 (1H, с), 8,05 (1H, д), 4,55 (2H, с), 4,3 (1H, м), 3,85 (3H, с) (1H, м), 3,7 (1H, м), 3,1 - 3,0 (2H, м), 1,2 - 1,0 (4H, м)
FAB MC (POS) : [M + H] = 390
Пример 181
Синтез 7-(4-аминометил-3-метоксииминопирролидин-1-ил)-1-(2,4- дифторфенил)-6-фтор-4-оксо-1,4-дигидро-[1,8]нафтиридин-3-карбоновой кислоты
177 мг (0,5 ммоль) 1-(2,4-дифторфенил)-7-хлор-6-фтор- 4-оксо-1,4-дигидро-[1,8] нафтиридин-3-карбоновой кислоты и 205 мг (0,55 ммоль) 4-аминометилпирролидин-3-он-О-метилоксимдитрифторацетата вводили в реакцию на 0,5 часа так, как в примере 177, и в результате получили 59 мг (выход 25%) указанного в заголовке соединения.
1H ЯМР (DMCO -d6, м.д.) : δ 8,85 (1H, с), 8,05 (1H, д), 7,75 (1H, дд), 7,6 (1H, дд), 7,35 (1H, дд), 4,3 (2H, м), 3,8 (3H, с, 1H, м) 3,6 (1H, м), 3,0 (1H, м), 2,7 (2H, м)
FAB MC (POS) : [M+H] = 462
Пример 182.
Синтез 1-циклопропил-5-амино-6,8-дифтор-7- (4-аминометил-3-метилоксииминопирролидин-1-ил)- 4-оксо-1,4-дигидрохинолин-3-карбоновой кислоты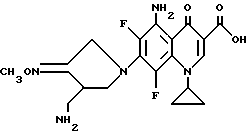
148 мг (0,5 ммоль) 1-циклопропил-5-амино-6,7,8-трифтор- 4-оксо-1,4-дигидрохинолин-3-карбоновой кислоты и 205 мг (0,55 ммоль)-4-аминометилпирролидин-3-он-О-метилоксимдитрифторацетата нагревали с обратным холодильником, как в примере 177, в течение 4 часов. Затем реакционный раствор концентрировали, и остаток очищали путем препаративной ВЭЖХ, получив в результате 84 мг (выход 40%) указанного в заголовке соединения.
1H ЯМР (DMCO -d6, м.д.) : δ 8,49 (1H, с), 7,28 (2H, шир. с), 4,3 (2H, с), 3,9 (2H, м), 3,8 (3H, с), 3,7 (1H, м), 2,6 - 2,8 (3H, м), 1,05 (4H, м)
FAB МС (POS) : [M+H]+ = 422
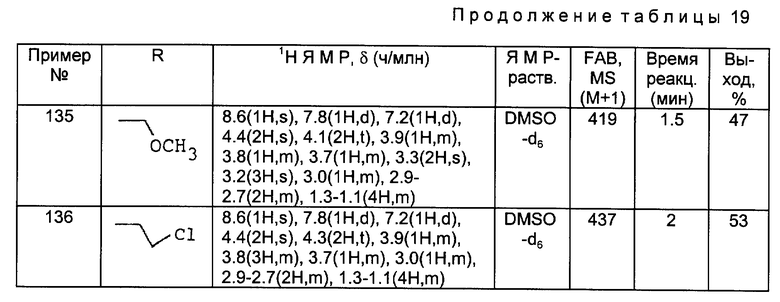
Примеры 183 - 202.
Соединения, полученные в Получениях 40 и 55-57, обрабатывали в соответствии с такой же, как в примерах 177-182, процедурой и получили соответственные соединения 183-202, данные ЯМР и МС для которых приведены в следующей ниже таблице 24.
Биологический пример 1
Испытание на антибактериальную активность in vitro.
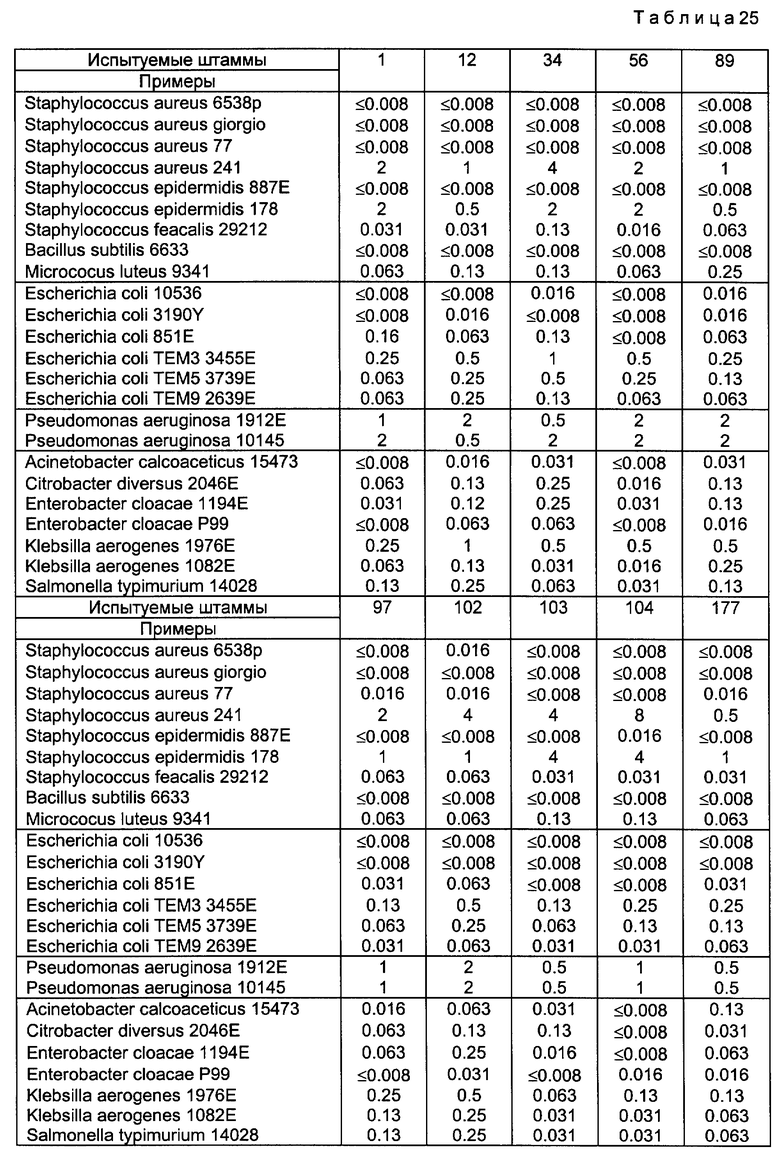
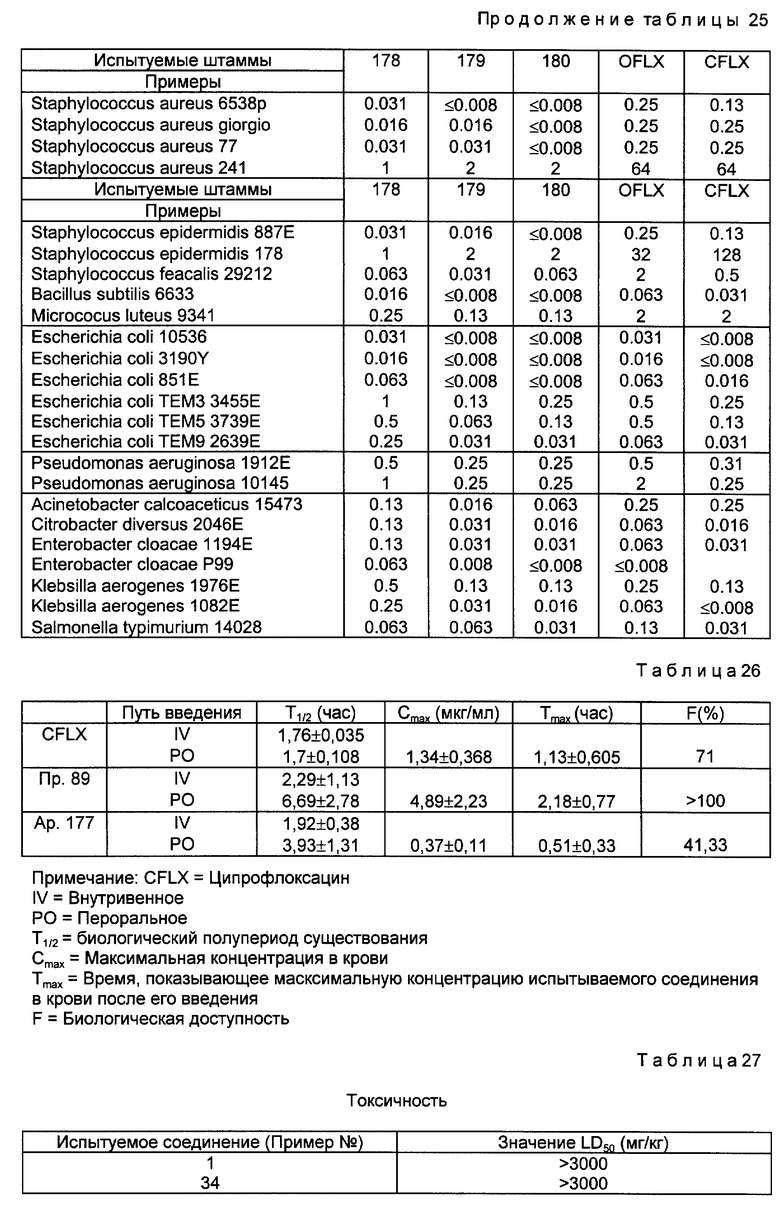
Антибактериальную активность соединений в соответствии с настоящим изобретением определяли путем измерения их минимальной ингибирующей концентрации (MIC, мкг/мл) против стандартных штаммов, клинически изолированных штаммов и штаммов, стойких к некоторым антибактериальным средствам. В этом испытании в качестве средства для сравнения использовали известные антибактериальные соединения (офлоксацин и ципрофлоксацин). Минимальную ингибирующую концентрацию можно было определить путем разведения соединений для испытания в соответствии с методом двукратного разведения, диспергирования разведенных испытуемых соединений в агаровой среде Мюллера-Хинтона и затем засева 5 мкл стандартного штамма, имеющего 107 CFU (колониеобразующих единиц) на мл, на среде, который затем выращивали в течение 18 часов при 37oC. Измеренные результаты представлены в таблице 25.
Минимальная ингибирующая концентрация испытуемых соединений (мкг/мл) приведена в табл. 25.
Примечание к табл. 25: ОFLX - офлоксацин, СFLX - ципрофлоксацин.
Биологический пример 2.
Фармакокинетическое испытание.
Параметры фармакокинетических свойств соединений в соответствии с настоящим изобретением определяли с использованием крыс SD (самцов) весом примерно 230 ± 10 г. В частности, испытываемые соединения в соответствии с настоящим изобретением вводили в количестве 20 мг/кг массы тела в испытуемых крыс через бедренные вены. Затем с определенными интервалами после введения испытываемых соединений брали кровь из бедренной вены и анализировали методом агаровых ячеек, чтобы измерить концентрацию испытываемых соединений в крови, по которой вычисляли фармакокинетические параметры : полупериод существования лекарственного вещества в организме (T1/2) и AUC (площадь под кривой). Полученные фармакокинетические параметры представлены в табл. 26
Биологический пример 3.
Испытание на острую пероральную токсичность
Для определения острой пероральной токсичности соединений, полученных в примерах 1 и 34, перорально вводили испытуемый раствор, содержащий соединения в различных концентрациях, в организм мужских особей мышей ICR в количестве 10 мл на кг массы тела. В течение 7 дней после введения наблюдали летальность и состояние испытуемых мышей, и по этим наблюдениям было вычислено значение LD50 (мг/кг). Полученные результаты представлены в табл. 27.
Хотя настоящее изобретение описано на предпочтительном конкретном варианте его осуществления, для специалистов в данной области понятно, что описание на предпочтительном варианте выполнено лишь для примера и что могут быть внесены различные изменения в детали структуры, сочетание и расположение частей в пределах существа и объема настоящего изобретения.













Настоящее изобретение относится к новому хинолоновому соединению, проявляющему высокую антибактериальную активность. Более конкретно настоящее изобретение касается нового производного хинолин (нафтиридин) карбоновой кислоты, представленного приведенной ниже формулой, которое имеет 4-аминометил- 3-оксимпирролидиновый заместитель в положении 7 хинолонового ядра и проявляет высокую антибактериальную активность и противоположность известным хинолоновым антибактериальным средствам, имеющим низкую активность против штаммов грам-положительных бактерий, а также имеет широкий антибактериальный спектр и сильно улучшенные фармакокинематические свойства: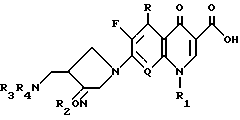
где R, R1, R2, R3, R4 и Q - такие же, как указанные в описании. 3 с. и 8 з.п. ф-лы, 27 табл.
где R представляет водород или аминогруппу;
Q представляет C-H, C-F, C-Cl, C-O-CH3 или N;
R1 представляет циклопропил или фенил, замещенный одним или несколькими атомом(ами) фтора;
R2 представляет водород, неразветвленный или разветвленный C1 - C4-алкил, циклопропилметил, C3 - C6-алкинил, 2-галогенэтил, метоксиметил, бензил, аллил, фенил, циклоалкил C4 - C6 или группу формулы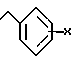
где X представляет водород, 2, 3 или 4-фтор, циано, нитрометокси, C1 - C4-алкил или 2,4-дифтор,
или группу формулы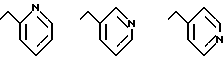
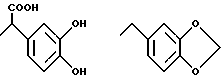

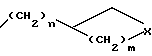
где n = 0 или 1;
m = 0, 1 или 2;
X представляет метилен, O или N;
R3 и R4 - водород.
где R представляет водород или аминогруппу;
Q - C-H, C-F, C-Cl, C-O-CH3 или N;
R1 - циклопропил или фенил, замещенный одним или несколькими атомом(ами) фтора;
R2 представляет водород, неразветвленный или разветвленный C1-C4-алкил, циклопропилметил, C3-C6-алкинил, 2-галогенэтил, метоксиметил, бензил, алкил, фенил, циклоалкил C4 - C6 или группу формулы
где X - водород, 2,3 или 4-фтор, циано, нитро, метокси, C1 - C4-алкил или 2,4-дифтор,
или группу формулы

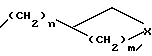
где n = 0 или 1;
m = 0, 1 или 2;
X - метилен, O или N;
R3 и R4 - водород,
отличающийся тем, что осуществляют реакцию соединения формулы II
где Q, R и R1 - такие же, как указанные выше;
X - галоген,
с соединением формулы III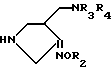
где R2, R3 и R4 - такие же, как указанные выше,
в растворителе в присутствии акцептора кислоты.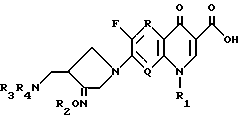
где R представляет водород или аминогруппу;
Q - C-H, C-F, C-Cl, C-O-CH3 или N;
R1 - циклопропил или фенил, замещенный одним или несколькими атомом(ами) фтора;
R2 - водород, неразветвленный или разветвленный C1 - C4-алкил, циклопропилметил, C3 - C6-алкинил, 2-галогенэтил, метоксиметил, бензил, аллил, фенил, циклоалкил C4 - C6, или группу формулы
где X - водород, 2,3 или 4-фтор, циано, нитро, метокси, C1 - C4-алкил или 2,4-дифтор;
или группу формулы
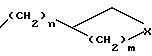
где n = 0 или 1;
m = 0, 1 или 2;
X - метилен, O или N;
R3 и R4 - водород,
отличающийся тем, что осуществляют реакцию соединения формулы II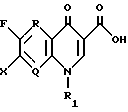
где Q, R и R1 - такие же, как указанные выше;
X - галоген,
с соединением формулы III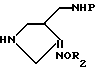
где R2 - такой же, как указанный выше;
P - аминозащитная группа,
в присутствии основания и затем у полученного соединения удаляют аминозащитную группу P.
| EP, 0541086, C 07 D 401/04, 1992. |

