Область техники.
Настоящее изобретение относится к новым избирательно действующим ингибиторам тромбина, имеющим следующую формулу (I):
в которой
R1 представляет собой ацетил, замещенный арилом или арилокси, или представляет собой сульфонил, замещенный замещенным или незамещенным арилом или N-содержащей гетероциклической группой,
X представляет собой группу формулы 
R2 и R3 независимо друг от друга представляют собой водород; циклоалкил, замещенный или незамещенный карбоксилом или алкоксикарбонилом; арилалкилокси; гидрокси; или низший алкил, замещенный или незамещенный карбоксилом, алкоксикарбонилом или гидрокси, или
R2 и R3 вместе с атомом азота, к которому они присоединены, могут образовывать пиперидиновую группу, замещенную карбоксилом или алкоксикарбонилом,
R4 представляет собой водород, низший алкил или низший алкокси,
R5 представляет собой алкансульфонил; алкоксикарбонил; алкилкарбонил; формил; низший алкил; арил, замещенный или незамещенный алкокси или галогеналкилом; или гидроксизамещенный низший алкил, и
R6 и R7 независимо друг от друга представляют собой водород, низший алкил или амино.
Некоторые соединения формулы (I) могут проявлять высокую ингибирующую активность по отношению к тромбину даже при пероральном применении и, таким образом, представляют большую ценность.
Настоящее изобретение также относится к способу получения соединения формулы (I) и к фармацевтической композиции для подавления активности тромбина, которая содержит соединение формулы (I) в качестве активного ингредиента.
ПРЕДПОСЫЛКИ.
Широко известно, что процесс свертывания крови включает в себя множество сложных ферментативных реакций, заключительная стадия которых включает в себя реакцию превращения протромбина в тромбин. Тромбин, полученный в результате заключительной стадии процесса свертывания крови, активирует тромбоциты и переводит фибриноген в фибрин, который затем преобразуется в высокомолекулярное вещество путем полимеризации и который приобретает поперечные сшивки благодаря действию активированного фактора крови XIII с образованием нерастворимого кровяного сгустка. Соответственно, тромбин играет важную роль в процессе свертывания крови. Тромбин также активирует факторы крови V и VIII, которые, в свою очередь, ускоряют свертывание крови по механизму обратной связи.
Таким образом, поскольку ингибиторы тромбина действуют как эффективные антикоагулянты и, в то же время, могут подавлять активацию тромбоцитов и продукцию и стабилизацию фибрина, длительное время предпринималось множество попыток найти способ предотвращения свертывания крови и лечения тромбозов различного рода, используя новое соединение, которое способно подавлять активность тромбина.
Однако соединение, способное подавлять лишь активность тромбина, ограничено в применении в качестве эффективного противосвертывающего и тромболитического средства. Причиной является тот факт, что, поскольку тромбин представляет собой одну из сериновых протеаз, а в организме человека, особенно в крови, присутствует большое количество сериновых протеаз, схожих с трипсином, обычно плазмина, то эффективный ингибитор тромбина, как правило, обладает высокой ингибирующей активностью по отношению к таким сериновым протеазам. В соответствии с такой характерной чертой тромбина, при разработке ингибиторов тромбина очень важным является, чтобы соединение-ингибитор обладало меньшей ингибирующей активностью по отношению к прототипу сериновых протеаз, такому как трипсин, чем по отношению к тромбину.
При таких условиях было проведено большое количество исследований с целью разработки избирательно действующих ингибиторов тромбина, который мог бы эффективно подавлять активность тромбина и, в то же время, обладал бы низкой ингибирующей активностью по отношению к трипсину. В результате был разработан Аргатробан (Argatroban), имеющий следующую формулу (A), производного арилсульфониларгинина (патенты США 4258192 и 4201863).
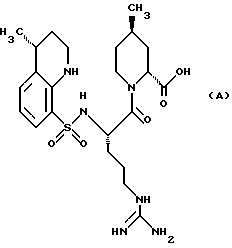
Аргатробан проявляет высокую ингибирующую активность по отношению к тромбину, которая в 250 раз превышает активность по отношению к трипсину (Biochemistry 1984, 23, с. 85-90). Однако он может быть получен только путем сложного способа синтеза. Он был выпущен в продажу в Японии в 1990 году.
В дополнение к этому, был также разработан NAPAP, имеющий следующую формулу (B), арилсульфонильного производного бензамидина.
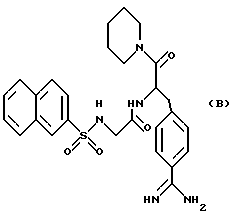
Данное соединение может быть легко синтезировано и обладает высокой ингибирующей активностью по отношению к тромбину. Однако оно имеет недостаток в том, что ингибирующая активность по отношению к тромбину лишь в 50 раз превышает активность по отношению к трипсину (J. Biol. Chem. 1991, 266, с. 20085- 20093).
Далее, сообщалось о Ro 46-6240, имеющем следующую формулу (C), как о соединении, обладающем улучшенной избирательностью по отношению к тромбину по сравнению с трипсином. Данное соединение указывает на возможность разработки внутривенно вводимого состава благодаря его короткому периоду полужизни в крови, но не дает возможности перорального введения (J. Med. Chem. 1994, 37, 3889-3901).

В дополнение к этому, сообщалось о недавно разработанном соединении на основе пиперазида как о дающем, в некоторой степени, возможность перорального введения у крыс, но обладающем низкой избирательностью по отношению к тромбину (WO 94/18185). Таким образом, не ожидается, что такие соединения в данной области будут перспективны.
Таким образом, авторы настоящего изобретения провели всесторонние исследования в целях разработки определенного соединения, которое может быть легко синтезировано, проявляет более высокую ингибирующую активность по отношению к тромбину, чем по отношению к трипсину, и также может вводиться перорально. В результате заявители обнаружили, ингибитор тромбина формулы (I) по настоящему изобретению отвечает таким требованиям и таким образом создали настоящее изобретение.
ОПИСАНИЕ ИЗОБРЕТЕНИЯ.
Соответственно, объектом настоящего изобретения является новый ингибитор тромбина формулы (I), как определен выше, который может быть введен перорально и который обладает высокой избирательностью по отношению к тромбину.
Другим объектом целью настоящего изобретения является способ получения ингибитора тромбина формулы (I).
Далее, еще одним объектом настоящего изобретения является фармацевтическая композиция для предотвращения свертывания крови и лечения тромбозов различного рода, которая содержит ингибитор тромбина формулы (I) в качестве активного ингредиента.
ПРЕДПОЧТИТЕЛЬНЫЙ СПОСОБ ОСУЩЕСТВЛЕНИЯ ИЗОБРЕТЕНИЯ.
С одной стороны, настоящее изобретение относится к новому соединению, представленному следующей формулой (I):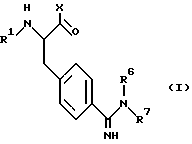
его фармацевтически приемлемой соли, гидрату, сольвату и изомеру, в которых
R1 представляет собой ацетил, замещенный арилом или арилокси, или представляет собой сульфонил, замещенный замещенным или незамещенным арилом или N-содержащей гетероциклической группой,
X представляет собой группу формулы 
R2 и R3 независимо друг от друга представляют собой водород; циклоалкил, замещенный или незамещенный карбоксилом или алкоксикарбонилом; арилалкилокси; гидрокси; или низший алкил, замещенный или незамещенный карбоксилом, алкоксикарбонилом или гидрокси, или
R2 и R3 вместе с атомом азота, к которому они присоединены, могут образовывать пиперидиновую группу, замещенную карбоксилом или алкоксикарбонилом,
R4 представляет собой водород, низший алкил или низший алкокси,
R5 представляет собой алкансульфонил; алкоксикарбонил; алкилкарбонил; формил; низший алкил; арил, замещенный или незамещенный алкокси или галогеналкилом; или гидроксизамещенный низший алкил,
и
R6 и R7 независимо друг от друга представляют собой водород, низший алкил или амино.
В определении каждого заместителя соединения формулы (I) по настоящему изобретению термин "низший алкил" означает насыщенный неразветвленный или разветвленный углеводородный радикал, содержащий от 1 до 4 атомов углерода, такой как метил, этил, изопропил, изобутил, трет-бутил и т.д.; термин "аралкилокси" означает алкоксигруппу, замещенную ароматическим кольцом, таким как бензилокси и т.д.; и термин "циклоалкил" означает циклическую алкильную группу, содержащую от 3 до 8 атомов углерода, такую как циклопентил.
Среди соединений формулы (I), приведенной выше, предпочтительным является соединение, в котором
R1 представляет собой ацетил, замещенный нафтилом или нафтилокси, или представляет собой сульфонил, замещенный нафтилом или фенилом, которые могут быть замещены или не замещены от одного до четырех заместителями, выбранными из группы, включающей низший алкил, низший алкокси и диалкиламино,
X представляет собой группу формулы 
R2 и R3 независимо друг от друга представляют собой C3-6-циклоалкил, замещенный или незамещенный карбоксилом или метоксикарбонилом; бензилокси; низший алкил, замещенный или незамещенный карбоксилом, метоксикарбонилом или гидрокси; или гидрокси, или
R2 и R3 вместе с атомом азота, к которому они присоединены, может образовывать пиперидиновую группу, замещенную карбоксилом или метоксикарбонилом,
R4 представляет собой водород,
R5 представляет собой метансульфонил, этоксикарбонил, формил, этил, фенил, метилкарбонил, гидроксиэтил или фенил, который может быть замещен или не замещен трифторметилом или этокси, и
R6 и R7 независимо друг от друга представляют собой водород, метил или амино.
Типичный пример соединения формулы (I) по настоящему изобретению включает следующее:
(S)-N-циклопентил-N-метил-3-(4-амидразонофенил)-2-(2-нафтилсульфониламино)пропионамид,
(S)-N-бутил-N-метил-3-(4-амидразонофенил)-2-(2- нафтилсульфониламино)пропионамид,
(S)-N-циклопентил-N-пропил-3-(4-амидразонофенил)-2-(2-нафтилсульфониламино)пропионамид,
(S)-N-циклопентил-N-(2-бензилоксиэтил)-3-(4-амидразонофенил)-2-(2-нафтилсульфониламино)пропионамид,
(S)-N-циклопентил-N-бутил-3-(4-амидразонофенил)-2-(2-нафтилсульфониламино)пропионамид,
(S)-N-циклопентил-N-этил-3-(4-амидразонофенил)-2-(2-нафтилсульфониламино)пропионамид,
(S)-N-циклопентил-N-метил-3-[4-(метиламидино)фенил] -2-(2-нафтилсульфониламино)пропионамид,
(S)-N-циклопентил-N-метил-3-[4-(1,1-диметиламидино)фенил] -2-(2-нафтилсульфониламино)пропионамид,
(S)-N-циклопентил-N-метил-3-(4-амидразонофенил)-2-[(4-метокси-2,3,6-триметилбензол)сульфониламино]пропионамид,
(S)-N-циклопентил-N-гидрокси-3-(4-амидразонофенил)-2-(2-нафтилсульфониламино)пропионамид,
(S)-N-циклопентил-N-(2-гидроксиэтил)-3-(4-амидразоно-фенил)-2-(2-нафтилсульфониламино)пропионамид,
(S)-N-циклопентил-N-метил-3-[4-(метиламидино)фенил] -2-[(4-метокси-2,3,6-триметилбензол)сульфониламино]пропионамид,
(S)-N,N-диметил-3-(4-амидразонофенил]-2-(2-нафтилсульфониламино)пропионамид,
(S)-N, N-диметил-3-[4-(1-метиламидино)фенил] -2-(2-нафтилсульфониламино)пропионамид,
(S)-N-циклогексил-N-метил-3-(4-амидразонофенил)-2-(2-нафтилсульфониламино)пропионамид,
(S)-N-циклопропил-N-метил-3-(4-амидразонофенил)-2-(2-нафтилсульфониламино)пропионамид,
(S)-3-[4-(амидразоно)фенил]-N-циклопентил-N-метил-2-(2-нафталин-1-илацетиламино)пропионамид,
(S)-3-[4-(амидразоно)фенил] -N-циклопентил-N-метил-2-(5-диметиламинонафталин-1-сульфониламино)пропионамид,
(S)-3-[4-(амидразоно)фенил] -N-циклопентил-N-метил-2-(5-метоксинафталин-1-сульфониламино)пропионамид,
(S)-3-[4-(амидразоно)фенил]-N-циклопентил-N-метил-2-(6,7-диметоксинафталин-2-сульфониламино)пропионамид,
(S)-3-[4-(метиламидино)фенил]-N-циклопентил-N-метил-2-(5-диметиламинонафталин-1-сульфониламино)пропионамид,
(S)-3-[4-(амидразоно)фенил] -N-циклопентил-N-метил-2-(нафталин-2-сульфониламино)пропионамид,
(S)-3-[4-(амидразоно)фенил] -N-циклопентил-N-метил-2-[2-(нафталин-1-илокси)ацетиламино)пропионамид,
(S)-3-[4-(амидразоно)фенил] -N-циклопентил-N-метил-2-[2-(нафталин-2-илокси)ацетиламино)пропионамид,
метиловый эфир {[3-(4-(амидразонофенил)-(S)-2-(нафталин-2- сульфониламино)пропионил]метиламино}уксусной кислоты,
{ [3-(4-(амидразонофенил)-(S)-2-(нафталин-2-сульфониламино)пропионил] метиламино}уксусная кислота,
метиловый эфир (S)-2-{[3-(4-амидразонофенил)-(S)-2-(нафталин-2-сульфониламино)пропионил]метиламино}пропионовой кислоты,
(S)-2-{[3-(4-амидразонофенил)-(S)-2-(нафталин-2-сульфониламино) пропионил]метиламино}пропионовая кислота,
метиловый эфир (R)-2-{[3-(4-амидразонофенил)-(S)-2-(нафталин-2-сульфониламино)пропионил]метиламино}пропионовой кислоты,
(R)-2-{[3-(4-амидразонофенил)-(S)-2-(нафталин-2-сульфониламино)пропионил]метиламино}пропионовая кислота,
метиловый эфир (R)-2-{[3-(4-амидразонофенил)-(S)-2-(нафталин-2-сульфониламино)пропионил]метиламино}-3-метилмасляной кислоты,
(R)-2-{[3-(4-амидразонофенил)-(S)-2-(нафталин-2- сульфониламино)пропионил]метиламино}-3-метилмасляная кислота,
метиловый эфир 3-{[3-(4-амидразонофенил)-(S)-2-(нафталин-2-сульфониламино)пропионил]метиламино}пропионовой кислоты,
3-{ [3-(4-амидразонофенил)-(S)-2-(нафталин-2-сульфониламино)пропионил] метиламино}пропионовая кислота,
метиловый эфир 4-{[3-(4-амидразонофенил)-(S)-2-(нафталин-2-сульфониламино)пропионил]метиламино}масляной кислоты,
4-{ [3-(4-амидразонофенил)-(S)-2-(нафталин-2-сульфониламино)пропионил] метиламино}масляная кислота,
метиловый эфир {[3-(4-амидразонофенил)-(S)-2-(нафталин-2-сульфониламино)пропионил]циклопропиламино}уксусной кислоты,
{ [3-(4-амидразонофенил)-(S)-2-(нафталин-2-сульфониламино)пропионил] циклопропиламино}уксусная кислота,
метиловый эфир {[3-(4-амидразонофенил)-(S)-2-(нафталин-2-сульфониламино)пропионил]бутиламино}уксусной кислоты,
{ [3-(4-амидразонофенил)-(S)-2-(нафталин-2-сульфониламино)пропионил] бутиламино}уксусная кислота,
метиловый эфир {[3-(4-амидразонофенил)-(S)-2-(нафталин-2-сульфониламино)пропионил]циклопентиламино}уксусной кислоты,
{ [3-(4-амидразонофенил)-(S)-2-(нафталин-2-сульфониламино)пропионил] циклопентиламино}уксусная кислота,
метиловый эфир 1-{[3-(4-амидразонофенил)-(S)-2-(нафталин-2-сульфониламино)пропионил]метиламино}циклопентанкарбоновой кислоты,
1-{ [3-(4-амидразонофенил)-(S)-2-(нафталин-2-сульфониламино)пропионил] метиламино}циклопентанкарбоновая кислота,
метиловый эфир 1-{[3-(4-амидразонофенил)-(S)-2-(нафталин-2-сульфониламино)пропионил]метиламино}циклопентанкарбоновой кислоты,
1-{ [3-(4-амидразонофенил)-(S)-2-(нафталин-2-сульфониламино)пропионил] метиламино}циклопентанкарбоновая кислота,
этиловый эфир 2-{ [3-(4-амидразонофенил)-(S)-2-(нафталин-2-сульфониламино)пропионил]метиламино}циклопентанкарбоновой кислоты,
2-{ [3-(4-амидразонофенил)-(S)-2-(нафталин-2-сульфониламино)пропионил] метиламино}циклопентанкарбоновая кислота,
метиловый эфир (S)-2-{[3-(4-амидразонофенил)-(S)-2-(нафталин-2-сульфониламино)пропионил]метиламино}-3-метилмасляной кислоты,
метиловый эфир 1-[3-(4-амидразонофенил)-(S)-2-(нафталин-2-сульфониламино)пропионил]пиперидин-(R)-2-карбоновой кислоты,
1-[3-(4-амидразонофенил)-(S)-2-(нафталин-2-сульфониламино)пропионил] пиперидин-(R)-2-карбоновая кислота,
[1-(4-амидразоно)бензил-2-оксо-2-(4-метилсульфонилпиперазинил)этил] амид (S)-нафталин-2-сульфоновой кислоты,
[1-(4-амидразоно)бензил-2-(4-формилпиперазинил)-2-оксоэтил] амид (S)-нафталин-2-сульфоновой кислоты,
[1-(4-амидразоно)бензил-2-(4-этилпиперазинил)-2-оксоэтил]амид (S)-нафталин-2-сульфоновой кислоты,
[1-(4-амидразоно)бензил-2-оксо-2-(4-фенилпиперазинил)этил] амид (S)-нафталин-2-сульфоновой кислоты,
[1-(4-амидразоно)бензил-2-оксо-2-[4-(3-трифторметилфенил)пиперазинил)этил]амид (S)-нафталин-2-сульфоновой кислоты,
[2-(4-ацетилпиперазинил)-1-(4-амидразоно)бензил-2-оксоэтил]амид (S)-нафталин-2-сульфоновой кислоты,
[1-(4-амидразоно)бензил-2-оксо-2-[4-(2-гидроксиэтил)пиперазинил] этил] амид (S)-нафталин-2-сульфоновой кислоты,
и
[1-(4-амидразоно)бензил-2-оксо-2-[4-(2-этоксифенил)пиперазинил]этил]амид (S)-нафталин-2-сульфоновой кислоты.
Соединение формулы (I) по настоящему изобретению также может образовывать фармацевтически приемлемую соль. Подходящие фармацевтически приемлемые соли соединения (I) могут включать соли добавления кислот, образованные кислотами, которые могут образовывать нетоксичную соль добавления кислоты, содержащую фармацевтически приемлемый анион, например неорганических кислот, таких как хлористоводородная кислота, серная кислота, азотная кислота, фосфорная кислота, бромистоводородная кислота, иодистоводородная кислота и т.д., органических карбоновых кислот, таких как винная кислота, муравьиная кислота, лимонная кислота, уксусная кислота, трихлоруксусная кислота, глюконовая кислота, бензойная кислота, молочная кислота, фумаровая кислота, малеиновая кислота и т.д., сульфоновых кислот, таких как метансульфоновая кислота, бензолсульфоновая кислота, п-толуолсульфоновая кислота, нафталинсульфоновая кислота и т.д., и тому подобных кислот.
В дополнение к этому, поскольку соединение формулы (I) по настоящему изобретению может включать в свой состав асимметричный атом углерода, оно может находиться в виде рацемата, смеси диастереомеров и отдельного диастереомера. Все такие изомеры включены в объем настоящего изобретения. То есть, изомеры соединения формулы (I) могут быть представлены следующим образом:
в котором R6 представляет метил или амино.
С другой точки зрения, настоящее изобретение также относится к способу получения соединения формулы (I), как определено выше.
В соответствии с настоящим изобретением, соединение формулы (I) может быть получено путем взаимодействия соединения формулы (II) с соединением формулы (III), как показано на следующей схеме реакции 1.
Схема реакции 1.
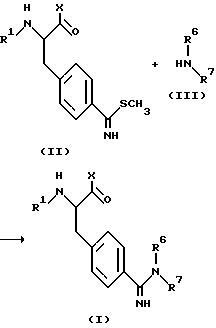
В приведенной выше схеме реакции X, R1, R6 и R7 определены, как описано выше.
Как изображено на приведенной выше схеме реакции 1, соединение формулы (I) по настоящему изобретению может быть получено путем взаимодействия метилмеркаптопроизводного формулы (II) с производным амина формулы (III) в качестве нуклеофильного вещества. Данная реакция может быть предпочтительно проведена в присутствии растворителя. Хотя в данной реакции может быть применен любой органический растворитель, который не влияет отрицательно на ход реакции, для данной цели применяют, главным образом, спиртовой растворитель, такой как метанол, этанол, пропанол и т.д.
В приведенной выше реакции реакционные условия, включая количество реагентов, температуру реакции, время реакции и т.д., могут быть определены в зависимости от вида конкретного реагента, применяемого квалифицированным специалистом в данной области. Хотя температура реакции может варьироваться в пределах существенного интервала, как правило, особенно предпочтительным является проведение реакции при от 0oC до 50oC. В дополнение к этому, реакция обычно занимает от 0,5 до 5 часов и предпочтительно может быть проведена за от 1 до 2 часов.
После того, как данная реакция завершается, продукт реакции может быть отделен и очищен в соответствии с обычными методиками переработки, например хроматографией, перекристаллизацией и т.д.
Метилмеркаптопроизводные формулы (II), применяемое в качестве промежуточного продукта для получения соединения формулы (I) в схеме реакции 1, может быть получено в соответствии со схемой реакции 2 или 3, как описано ниже.
Схема реакции 2: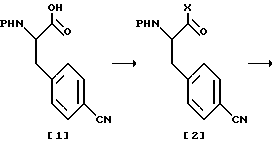
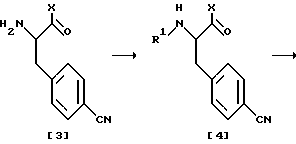
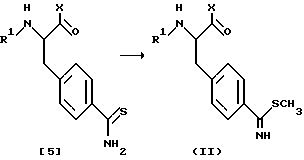
Схема реакции 3:


В приведенных выше схемах реакций
X, R1, R6 и R7 определены, как описано выше, и P представляет собой аминозащитную группу.
В дальнейшем, схемы реакций 2 и 3 пояснены конкретно.
В схеме реакции 2 сначала к C-концу соединения [1] присоединяют аминогруппу X с получением соединения [2], амино-защитную группу с N-конца которого удаляют с получением соединения [3]. Затем на лишенный защиты N-конец соединения [3] вводят группу R1 с получением соединения [4]. Альтернативно, в соответствии со схемой реакции 3, сначала на N-конец соединения [7] вводят группу R1, а затем к C-концу присоединяют аминогруппу X с получением соединения [4].
Конкретно, в соответствии со схемой реакции 2, к соединению формулы [1] присоединяют аминпроизводное, соответствующее заместителю X с получением соединения формулы [2] , с N-конца которого удаляют аминозащитную группу с получением соединения формулы [3]. Затем на N-конец соединения формулы [3] вводят группу R1 с получением нитрильного производного формулы [4], которое затем насыщают сероводородом в присутствии пиридина и триэтиламина с получением тиоамидного соединения формулы [5]. Данное тиоамидное соединение затем метилируют с помощью метилирующего агента, такого как иодметан, диметилсульфат, метилтрифлат и т.д., с получением желаемого метилмеркаптосоединения формулы (II).
В соответствии со схемой реакции 3, сначала на N-конец соединения формулы [7] вводят группу R1 путем взаимодействия соединения [7] с соединением формулы [6] и затем на C-конец полученного соединения формулы [8] вводят аминогруппу путем присоединения к соединению [8] аминпроизводного соединения, соответствующего заместителю X, с получением соединения формулы [4], которое затем подвергают той же процедуре, что и в схеме реакции 2, с получением желаемого метилмеркаптопроизводного формулы (II).
Сшивающий агент, который может быть применен для процедуры присоединения в схемах реакций 2 и 3, включает одно или несколько веществ, выбранных из группы, включающей дициклогексилкарбодиимид (ДЦК), 3-этил-3'-(диметиламино)пропилкарбодиимид (ЭДК), хлорангидрид бис-(2-оксо-3-оксазолидинил)фосфиновой кислоты (БОФ-С1) и дифенилфосфорилазид (ДФФА), но не ограничивается ими.
Хотя соединения - карбоновые кислоты [1] и [7], применяемые в схемах реакций 2 и 3, могут применяться в виде свободных кислот, предпочтительно они могут применяться в виде своих реакционноспособных производных, например галогенангидридного производного или другого активированного сложноэфирного производного в целях облегчения протекания реакции. Для реакции присоединения аминового соединения с образованием амидной связи или реакции присоединения спирта с образованием сложноэфирной связи особенно необходимо активированное сложноэфирное производное карбоновой кислоты. Такие реакционноспособные производные включают обычные производные, которые могут быть получены в соответствии с методикой, обычно применяемой в данной области техники. Например включено галогенангидридное производное хлорангидрид; и активированное сложноэфирное производное включает ангидрид карбоновой кислоты, являющийся производным алкоксикарбонилгалогенида, такого как метоксикарбонилхлорид, изобутилоксикарбонилхлорид и т.д., и сшивающий агент, сложноэфирное производное N-гидроксифталимида, сложноэфирное производное N-гидроксисукцинимида, сложноэфирное производное N-гидрокси-5-норбонен-2',3'-дикарбоксиимида, сложноэфирное производное 2,4,5-трихлорфенола и т.д., но не ограничивается ими.
Эффект ингибирования тромбином соединения формулы (I) по настоящему изобретению может быть обнаружен путем определения константы диссоциации Ki, представленной следующим уравнением, в соответствии с известной методикой, описанной в литературе (Methods in Enzymology, т. 80, с. 341-361; Biochemistry 27, с. 2144-2151 (1988)).
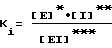
* [E] - концентрация свободного фермента;
** [I] - концентрация несвязанного ингибитора;
*** [EI] - концентрация комплекса фермент-ингибитор.
Константа диссоциации Ki обозначает степень диссоциации комплекса фермент-ингибитор тромбина. Соответственно, низкое значение константы диссоциации говорит о высокой связующей способности ингибитора тромбина по отношению к ферменту и таким образом устанавливается, что ингибитор тромбина обладает высокой ингибирующей активностью по отношению к тромбину. Такая константа диссоциации может быть определена путем взаимодействия тромбина с определенным субстратом, который дает окраску, будучи гидролизованным под действием тромбина, и последующего измерения степени развития окраски в зависимости от времени по методу спектрофотометрии.
В настоящем изобретении в качестве субстратного вещества для тромбина применяют Хромозим ТН (Chromozym ТН) (Gly-Pro-Arg-4-нитроанилидацетат), который дает окраску при действии тромбина. Хромозим ТН гидролизуется тромбином с получением желтого пара-нитроанилина. Соответственно, количество полученного таким образом желтого пара-нитроанилина может быть измерено как изменение поглощения с течением времени для определения ингибирующей активности по отношению к тромбину соединения по настоящему изобретению. Активность фермента может быть определена исходя из скорости изменения поглощения и затем может быть непосредственно связана со способностью ингибитора тромбина подавлять активность фермента (Methods in Enzymology, т. 80, с. 341-361; Biochemistry 27, с. 2144-2151, 1988).
Для определения избирательности соединения по настоящему изобретению по отношению к тромбину по сравнению с трипсином измеряют ингибирующую активность соединения формулы (I) по отношению к трипсину как значение Ki в соответствии с той же методикой, что и приведенная выше методика для определения ингибирующей активности по отношению к тромбину, а затем рассчитывают отношение активности по отношению к трипсину к активности по отношению к тромбину. В данном случае, процедура определения ингибирующей активности по отношению к трипсину существенно идентична таковой для тромбина за исключением того, что в качестве субстрата применяют N-бензоил-Val-Gly-Arg-пара-нитроанилидгидрохлорид.
Как результат определения ингибирующей активности по отношению к тромбину и трипсину соединения формулы (I) по настоящему изобретению может быть установлено, что соединение по настоящему изобретению проявляет отличную ингибирующую активность по отношению к тромбину и, более того, обладает высокой избирательностью по отношению к тромбину по сравнению с трипсином. В частности, значения избирательности соединений из Примеров 1 и 7 по отношению к тромбину по сравнению с трипсином составляют приблизительно 2900 раз и 26304 раз соответственно, тогда как значения избирательности известных ингибиторов тромбина, аргатробана (A) и NAPAP (B), составляют лишь 250 раз и 50 раз соответственно. Соответственно, может быть замечено, что соединение (I) по настоящему изобретению значительно усовершенствовано в плане избирательности по отношению к тромбину по сравнению с трипсином.
Как было указано выше, поскольку новое соединение формулы (I) по настоящему изобретению является ингибитором тромбина, который проявляет высокую ингибирующую активность по отношению к тромбину даже при пероральном введении и также обладает высокой избирательностью по отношению к тромбину по сравнению с трипсином, оно может использоваться для профилактики свертывания крови и лечения тромбозов различного рода.
Соответственно, третьим объектом настоящего изобретения является фармацевтическая композиция для профилактики свертывания крови и лечения тромбоза, которая содержит соединение формулы (I) или его фармацевтически приемлемую соль в качестве активного ингредиента.
Когда соединение формулы (I) по настоящему изобретению вводят субъекту в клинических целях, суточная доза соединения (I) может варьироваться, предпочтительно, в пределах от 0,001 мг до 10 мг на 1 кг массы тела. Однако при лечении определенного пациента может быть назначена особая доза сверх указанного выше интервала доз в зависимости от конкретного применяемого соединения, массы, пола и состояния здоровья данного пациента, диеты, времени и способа введения, скорости выведения соединения, других активных компонентов, применяемых в сочетании с ним и тяжести заболеваний, подвергающихся лечению.
Соединение по настоящему изобретению может вводиться в форме препарата для инъекций или препарата для перорального применения в соответствии с поставленной целью.
Препарат для инъекций, например стерилизованные водные или масляные суспензии для инъекций, могут быть приготовлены с применением подходящего диспергирующего агента, увлажняющего агента или суспендирующего агента в соответствии с известной методикой, обычно применяемой в области приготовления препаратов для инъекций. В качестве водного растворителя, подходящего для данной цели, могут быть применены вода, раствор Рингера или изотонический раствор NaCI. В дополнение к этому, хотя стерилизованное нелетучее масло может быть применено в качестве растворителя или суспендирующей среды, любое не вызывающее раздражение нелетучее масло, включая моно- и диглицериды, может быть применено в тех же целях. Более того, в препарат для инъекций может быть добавлена жирная кислота, такая как олеиновая кислота.
Твердый препарат для перорального применения может находиться в форме капсул, таблеток, пилюль, порошков и гранул, причем состав в виде капсул или таблеток является наиболее предпочтительным. Таблетки и пилюли могут быть приготовлены, предпочтительно, в виде состава с энтеросолюбильным покрытием. При составлении твердого препарата активное соединение формулы (I) по настоящему изобретению может быть объединено с фармацевтически приемлемым носителем, например с одним или более инертных разбавителей, таких как сахароза, лактоза, крахмал и т.д., смазывающими веществами, такими как силикат магния, дезинтеграторами, связывающими веществами и тому подобным.
Одной из главных характерных черт соединения формулы (I) по настоящему изобретению является то, что соединение формулы (I) проявляет хороший фармакологический эффект, даже когда его приготавливают в форме препарата для перорального применения и затем вводят перорально. Это можно продемонстрировать исходя из результатов фармакокинетических экспериментов на крысах и собаках в качестве экспериментального животного. В таких экспериментах может быть установлено, что активное соединение по настоящему изобретению сохраняется в крови в течение продолжительного промежутка времени, будучи введено перорально. Соответственно, соединение формулы (I) по настоящему изобретению является более применимым в свете того факта, что оно может эффективно применяться в форме препарата для перорального применения.
Далее, исходя из фармакокинетических экспериментов может быть также установлено, что активное соединение по настоящему изобретению может позволять достигать желаемой цели без острой токсичности у млекопитающих, включая крысу и собаку.
Когда ингибитор тромбина по настоящему изобретению вводят с целью получить противосвертывающий и тромболитический эффекты, он может быть введен в сочетании с одним или более веществом(ами), выбранными из группы, включающей тромболитические средства и средства для подавления активности тромбоцитов. В качестве тромболитического средства, которое может быть применено для данной цели, могут быть упомянуты t-PA, урокиназа, стрептокиназа; и аспирин, тиклопидин (ticlopidin), клопидрогель (clopidrogel), моноклональное тело 7Е3 и т. д. могут быть применены в качестве средства для подавления активности тромбоцитов.
Однако следует учитывать, что препарат, содержащий активное соединение по настоящему изобретению для лечения и профилактики тромбоза, не ограничивается таковыми, описанными выше, и может включать любой препарат, применимый для данной цели.
В дальнейшем, настоящее изобретение будет пояснено более конкретно с помощью рабочих примеров. Однако следует учитывать, что примеры приведены лишь в целях пояснения настоящего изобретения и не предназначены для ограничения настоящего изобретения каким-либо образом.
Получение 1.
Получение циклопентилметиламина. К раствору циклопентанона (10 мл, 113 ммоль) в метаноле (50 мл) и воде (50 мл) добавляли метиламингидрохлорид (7,6 г, 113 ммоль) и цианоборгидрид натрия (NaBH3CN) (7,1 г, 113 ммоль). Смесь нагревали с обратным холодильником в течение 12 ч при pH 6. Метанол выпаривали в условиях пониженного давления и остаток охлаждали до 0oC, доводили до pH 2, применяя 3н. соляную кислоту, и затем трижды промывали диэтиловым эфиром. Водный слой вновь охлаждали до 0oC и затем доводили до pH 11, применяя 6н. раствор гидроксида натрия. К данной смеси добавляли ангидрид третбутилоксикарбонила (24,5 г, 113 ммоль) в диоксане (50 мл). Раствор перемешивали в течение 3 часов при комнатной температуре и концентрировали в условиях пониженного давления до приблизительно 30 мл. Остаток экстрагировали добавлением этилацетата и промывали водным раствором 0,5н. соляной кислоты и насыщенным водным раствором бикарбоната натрия. Органический слой сушили над безводным сульфатом магния, фильтровали и концентрировали в условиях пониженного давления с получением белого твердого вещества, которое затем очищали по методу колоночной хроматографии (элюент = этилацетат: гексан = 7:3 (по объему)). По завершении очистки полученный твердый продукт растворяли в растворе 4н. HCl-диоксана (60 мл) и полученный раствор перемешивали в течение 30 минут при комнатной температуре. Растворитель выпаривали в вакууме с получением указанного в заголовке соединения (13,7 г. Выход: 90,5%).
1H ЯМР (CD3OD, миллионные доли) δ : 3,50 (м, 1Н), 2,68 (с, 3H), 2,10 (м, 2Н), 1,86-1,50 (м, 6Н).
Получение 2.
Получение (S)-N-циклопентил-N-метил-3-(4-цианофенил)-2-(бутилоксикарбониламино)пропионамида.
К раствору (S)-3-(4-цианофенил)-2-(бутилоксикарбониламино)пропионовой кислоты (0,7 г, 2,14 ммоль) в диметилформамиде (ДМФ, 6 мл) добавляли 1-(3-диметиламинопропил)-3-этилкарбодиимидгидрохлорид (ЭДК, 0,7 г) и 1-гидроксибензо-триазолгидрат (ГОБТ, 0,4 г) при 0oC. Смесь перемешивали до тех пор, пока они полностью не растворялись. К данной реакционной смеси добавляли соединение (0,4 г, 2,96 ммоль), полученное в Получении 1, и N-метилморфолин (1,0 мл) и затем температуру реакции медленно повышали до комнатной температуры. Реакционный раствор перемешивали в течение 3,5 часов. После завершения реакции реакционный раствор концентрировали в условиях пониженного давления для удаления летучих веществ и полученный остаток разбавляли этилацетатом, тщательно промывали водным насыщенным раствором гидрокарбоната натрия, разбавленной соляной кислотой и солевым раствором, сушили над безводным сульфатом натрия, фильтровали и затем концентрировали. Остаток очищали методом колоночной хроматографии (элюент = этилацетат:гексан = 7:3 (по объему)) с получением очищенного соединения указанного в заголовке (0,65 г. Выход: 73,0%).
1H ЯМР (CD3OD, миллионные доли) δ : 7,61 (м, 2Н), 7,32 (м, 2Н), 5,48, 5,01-4,86, 4,12 (3м, 3H), 2,75, 262 (2c, 3H); 2,90-1,20 (м, 17H).
Масс (FAB, m/e): 372 (М++1).
Получение 3.
Получение (S)-N-циклопентил-N-метил-3-(4-цианофенил)-2-(2-нафтилсульфониламино)пропионамида.
Соединение (0,65 г, 1,75 ммоль), полученное в Получении 2, растворяли в дихлорметане (3 мл) и затем охлаждали до -10oC и к нему добавляли трифторуксусную кислоту (ТФУ, 1 мл). Реакционную смесь перемешивали в течение 5 минут, медленно нагревали до комнатной температуры, перемешивали в течение 30 минут и затем концентрировали в условиях пониженного давления для удаления летучих веществ. Остаток сушили с помощью вакуумного насоса и затем к нему добавляли 6 мл ДМФ. Смесь охлаждали до 0oC и добавляли N,N-диизопропилэтиламин (1 мл). Реакционную смесь нагревали до комнатной температуры и перемешивали в течение приблизительно 5 минут. После добавления 2-нафталинсульфонилхлорида (0,47 г, 2,07 ммоль) реакционную смесь перемешивали в течение одного часа для завершения реакции и затем концентрировали в условиях пониженного давления для удаления летучих веществ. Остаток разбавляли этилацетатом, промывали насыщенным раствором гидрокарбоната натрия и солевым раствором, сушили над безводным сульфатом магния, фильтровали и концентрировали. Остаток очищали по методу колоночной хроматографии (элюент = этилацетат:гексан = 7:3 (по объему)) с получением указанного в заголовке соединения (0,65 г. Выход: 80,2%).
1H ЯМР (CDCl3, миллионные доли) δ : 8,28 (м, 1Н), 7,87 (м, 3H), 7,73 (м, 3H), 7,49 (м, 2Н), 5,92 (м, 1Н), 4,50, 4,32, 3,76 (м, м, м, 2Н), 2,95 (м, 2Н), 2,36, 2,22 (с, с, 3H), 1,60-1,20 (м, 6Н), 0,98, 0,80, 0,47 (м, м, м, 2Н).
Масс (FAB, m/e): 462 (М++1).
Пример 1.
Получение (S)-N-циклопентил-N-метил-3-(4-амидразонофенил)-2-(2-нафтилсульфониламино)пропионамида.
Соединение (0,65 г, 1,41 ммоль), полученное в Получении 3, растворяли в пиридине (10 мл) и полученный раствор помещали в колбу со штуцерами, в которую добавляли триэтиламин (0,45 мл). Реакционную колбу оборудовали таким образом, что газообразный сероводород (H2S) мог вводиться через один штуцер колбы, а выводиться через другой штуцер. Реакционный раствор насыщали газообразным сероводородом при перемешивании в течение приблизительно 10 минут, при чем бесцветный раствор приобретал зеленую окраску и затем постепенно - темно-коричневую. Колбу закрывали резиновой пробкой и оставляли стоять в течение 3 суток при комнатной температуре для завершения реакции. Затем реакционный раствор перегоняли в условиях пониженного давления для удаления летучих веществ и сушили с помощью вакуумного насоса. К полученному желтому твердому веществу добавляли вместе ацетон (15 мл) и иодметан (CH3I, 0,65 мл) и данную смесь нагревают с обратным холодильником в течение 30 минут. Данную реакционную смесь вновь перегоняли в условиях пониженного давления для удаления летучих веществ и сушили с помощью вакуумного насоса. Остаток растворяли в абсолютном метаноле (8 мл) и затем перемешивали. К данной смеси порциями трижды с интервалом 10 минут добавляли гидрат гидразина (H2NNH2•H2O, 0,12 мл, 1,98 ммоль). После того, как реакция была завершена, реакционный раствор концентрировали и очищали по методу ЖХВР с получением указанного в заголовке соединения (0,63 г. Выход: 73,0%).
Условия ЖХВР:
элюент = метанол: вода (75:25 по объему), оба содержат CF3COOH в концентрации 0,1%
длина волны = 215 нм
скорость элюирования = 20 мл/мин
колонка = Delta РАК C 18 100  (30 х 300 мм)
(30 х 300 мм)
1H ЯМР (CD3OD, миллионные доли) δ : 8,28 (д, 1Н), 7,92 (м, 3H), 7,70-7,50 (м, 5Н), 7,35 (дд, 2Н), 4,60, 4,45 (т, т, 1Н), 4,12, 3,99 (м, м, 1Н), 3,00, 2,87 (м, м, 2Н), 2,49, 2,26 (с, с, 3H), 1,60-1,00, 0,73-0,52 (м, м, 8Н).
Масс (FAB, m/e): 494 (М++1).
Получение 4.
Получение трифторуксуснокислой соли бутилметиламина.
N-бутилоксикарбонилбутиламин (140 мг, 0,80 ммоль) растворяли в ДМФ (8 мл) и к нему добавляли гидрид натрия (NaH, 20 мг, 1 экв. масса) и иодметан (0,10 мл, 2 экв. масса). Реакционную смесь перемешивали в течение 30 минут при комнатной температуре и фильтровали через слой целита и затем концентрировали в условиях пониженного давления для удаления растворителя. Остаток разбавляли этилацетатом, промывали 0,5н водным раствором соляной кислоты, сушили над безводным сульфатом магния и фильтровали. Органический слой концентрировали в условиях пониженного давления и сушили с помощью вакуумного насоса с получением белого твердого вещества, которое затем растворяли в дихлорметане и охлаждали до 0oC. К данной смеси добавляли 1 мл трифторуксусной кислоты (ТФУ). Реакционный раствор перемешивали в течение 30 минут при комнатной температуре, концентрировали в условиях пониженного давления и затем сушили с помощью вакуумного насоса с получением указанного в заголовке соединения (0,16 г) в количественном выходе.
1H ЯМР (CDCl3, миллионные доли) δ : 1,02 (т, 3H), 1,30-1,80 (м, 4Н), 3,04 (с, 3Н), 3,52 (т, 2Н), 8,20 (с, 2Н).
Пример 2.
Получение (S)-N-бутил-N-метил-3-(4-амидразонофенил)-2-(2-нафтилсульфониламино)пропионамида.
Проводили реакцию соединения, полученного в Получении 4, в соответствии с той же методикой, что и в Получениях 2 и 3, с получением промежуточного продукта (S)-N-бутил-N-метил-3-(4-цианофенил)-2-(2-нафтилсульфониламино)пропионамида (0,23 г), который применяли в качестве исходного продукта. Данный исходный продукт обрабатывали в соответствии с той же методикой, что и в Примере 1, с получением очищенного соединения указанного в заголовке (0,1 г. Выход: 40,0%).
1H ЯМР (CD3OD, миллионные доли) δ : 8,30 (д, 1Н), 7,98 (м, 3H), 7,81-7,30 (м, 7Н), 4,50 (м, 1Н), 3,25-2,55 (м, 4Н), 2,78, 2,45 (2 с, 3H), 1,40-0,50 (м, 7Н).
Масс (FAB, m/e): 482 (М++1).
Пример 3.
Получение (S)-N-циклопентил-N-пропил-3-(4-амидразонофенил)-2-(2-нафтилсульфониламино)пропионамида.
Использовали ту же методику, что и в Получении 1, за исключением того, что вместо метиламина применяли пропиламин, с получением циклопентилпропиламингидрохлорида, который затем обрабатывали в соответствии с той же методикой, что и в Получениях 2 и 3, с получением промежуточного продукта (S)-N-циклопентил-N-пропил-3-(4-цианофенил)-2-(2-нафтилсульфониламино)пропионамида (0,15 г). Данное промежуточное соединение применяли в качестве исходного продукта и обрабатывали в соответствии с той же методикой, что и в Примере 1, с получением очищенного соединения, указанного в заголовке (0,089 г. Выход: 55,1%).
1H ЯМР (CD3OD, миллионные доли) δ : 8,35-7,35 (м, 11Н), 4,62-4,32 (м, м, 1Н), 3,90, 3,70 (м, м, 1Н), 3,10-2,50 (м, 4Н), 1,70-0,50 (м, 13Н).
Масс (FAB, m/e): 522 (М++1).
Пример 4.
Получение (S)-N-циклопентил-N-(2-бензилоксиэтил)-3-(4-амидразонофенил)-2-(2-нафтилсульфониламино)пропионамида.
Использовали ту же методику, что и в Получении 1, за исключением того, что вместо метиламина применяли 2-бензилоксиэтиламин, с получением циклопентил-(2-бензилоксиэтил)амингидрохлорида, который затем обрабатывали в соответствии с той же методикой, что и в Получениях 2 и 3, с получением промежуточного продукта (S)-N-циклопентил-N-(2-бензилоксиэтил)-3-(4-цианофенил)-2-(2-нафтилсульфониламино)пропионамида (0,15 г). Данное промежуточное соединение применяли в качестве исходного продукта и обрабатывали в соответствии с той же методикой, что и в Примере 1, с получением очищенного соединения, указанного в заголовке (0,15 г. Выход: 93,8%).
1H ЯМР (CD3OD, миллионные доли) δ : 8,30-7,15 (м, 16Н), 4,64 (м, 1Н), 4,48, 4,39 (с, с, 1Н), 4,14, 4,00, 3,75, 3,45 (м, м, м, м, 3H), 3,10-2,70 (м, 5Н), 1,62-1,00 (м, 8Н).
Масс (FAB, m/e): 614 (М++1).
Пример 5.
Получение (S)-N-циклопентил-N-бутил-3-(4-амидразонофенил)-2-(2-нафтилсульфониламино)пропионамида.
Использовали ту же методику, что и в Получении 1, за исключением того, что вместо метиламина применяли бутиламин, с получением бутилциклопентиламингидрохлорида, который затем обрабатывали в соответствии с той же методикой, что и в Получениях 2 и 3, с получением промежуточного продукта (S)-N-циклопентил-N-бутил-3-(4-цианофенил)-2-(2-нафтилсульфониламино)пропионамида (0,28 г). Данное промежуточное соединение применяли в качестве исходного продукта и обрабатывали в соответствии с той же методикой, что и в Примере 1, с получением очищенного соединения, указанного в заголовке (0,18 г. Выход: 60%).
1H ЯМР (CD3OD, миллионные доли) δ : 8,32 (д, 1Н), 7,96 (м, 3Н), 7,78-7,55 (м, 5Н), 7,42 (дд, 2Н), 4,62, 4,30 (м, м, 1Н), 4,02, 3,90 (м, м, 1Н), 3,10-2,75, 2,55 (м, м, 4Н), 1,65-0,80 (м, 12Н), 0,65 (т, 3Н).
Масс (FAB, m/e): 536 (M++1).
Пример 6.
Получение (S)-N-циклопентил-N-этил-3-(4-амидразонофенил)-2-(2-нафтилсульфониламино)пропионамида.
Использовали ту же методику, что и в Получении 1, за исключением того, что вместо метиламина применяли этиламин, с получением циклопентилэтиламингидрохлорида, который затем обрабатывали в соответствии с той же методикой, что и в Получениях 2 и 3, с получением промежуточного продукта (S)-N-циклопентил-N-этил-3-(4-цианофенил)-2-(2-нафтилсульфониламино)пропионамида (0,21 г). Данное промежуточное соединение применяли в качестве исходного продукта и обрабатывали в соответствии с той же методикой, что и в Примере 1, с получением очищенного соединения, указанного в заголовке (0,11 г. Выход: 50,0%).
1H ЯМР (CD3OD, миллионные доли) δ : 8,32 (д, 1Н), 7,97 (м, 3Н), 7,75-7,55 (м, 5Н), 7,42 (м, 2Н), 4,60, 4,38 (м, м, 1Н), 3,98, 3,87 (м, м, 1Н), 3,20-2,70 (м, 4Н), 1,65-1,00 (м, 8Н), 0,95, 0,68 (т, т, 3Н).
Масс (FAB, m/e): 508 (M++1).
Пример 7.
Получение (S)-N-циклопентил-N-метил-3-[4-(метиламидино)фенил]-2-(2-нафтилсульфониламино)пропионамида.
Промежуточное соединение, полученное в Получении 3, применяли в качестве исходного продукта и обрабатывали в соответствии с той же методикой, что и в Примере 1, за исключением того, что метиламин в отличие от гидразина добавляли трижды с интервалом в один час, с получением очищенного соединения, указанного в заголовке (0,064 г. Выход: 8%).
1H ЯМР (CD3OD, миллионные доли) δ : 0,50-1,60 (м, 8Н), 2,00-2,49 (2 с, 3Н), 2,84 (м, 1Н), 2,96 (м, 1Н), 3,00 (с, 3Н), 4,05 (м, 1Н), 4,50 (м, 1Н), 7,20-8,30 (м, 11Н).
Масс (FAB, m/e): 493 (М++1).
Пример 8.
Получение (S)-N-циклопентил-N-метил-3-[4-(1,1-диметиламидино)фенил]-2-(2-нафтилсульфониламино)пропионамида.
Промежуточное соединение, полученное в Получении 3, применяли в качестве исходного продукта и обрабатывали в соответствии с той же методикой, что и в Примере 1, за исключением того, что диметиламин в отличие от гидразина добавляли трижды с интервалом в один час, с получением очищенного соединения, указанного в заголовке (0,18 г. Выход: 22%).
1H ЯМР (CD3OD, миллионные доли) δ : 0,60-1,60 (м, 8Н), 2,29, 2,54 (2 с, 3Н), 2,95 (м, 1Н), 3,06 (м, 1Н), 3,09 (с, 3Н), 3,31 (с, 3Н), 4,16 (м, 1Н), 4,60 (м, 1Н), 7,20-8,30 (м, 11Н).
Масс (FAB, m/e): 507 (М++1).
Пример 9.
Получение (S)-N-циклопентил-N-метил-3-(4-амидразонофенил)-2-(4-метокси-2,3,6-триметилбензолсульфониламино)пропионамида.
Соединение, полученное в Получении 2, обрабатывали в соответствии с той же методикой, что и в Получении 3, за исключением того, что вместо 2-нафталинсульфонилхлорида применяли 4-метокси-2,3,6-триметилбензолсульфонилхлорид, с получением промежуточного продукта (S)-N-циклопентил-N-метил-3-(4-цианофенил)-2-(4-метокси-2,3,6-триметилбензолсульфониламино)пропионамида (0,27 г). Данное промежуточное соединение применяли в качестве исходного продукта и обрабатывали в соответствии с той же методикой, что и в Примере 1, с получением очищенного соединения, указанного в заголовке (0,16 г. Выход: 57,1%).
1H ЯМР (CD3OD, миллионные доли) δ : 7,62 (д, 2Н), 7,40 (дд, 2Н), 6,70 (д, 1Н), 4,35, 3,90 (м, м, 2Н), 3,83 (д, 3H), 3,00 (м, 2Н), 2,50 (м, 9Н), 2,1 (с, 3H), 1,75-0,90 (м, 8Н).
Масс (FAB, m/e): 516 (М++1).
Получение 5.
Получение циклопентилгидроксиламина.
Гидроксиламингидрохлорид (H2NOH•HCl, 5,0 г, 71,95 ммоль) растворяли в воде (14 мл) и к нему добавляли метанол (30 мл) и циклопентанон (5,1 мл, 57,66 ммоль). Смесь перемешивали, охлаждали до 0oC, и значение pH затем доводили до 8 путем добавления 6н. водного раствора гидроксида натрия. К смеси добавляли цианоборгидрид натрия (NaBH3CN, 1,9 г, 30,24 ммоль). Смесь нагревали до комнатной температуры и затем перемешивали. Значение pH реакционной смеси поддерживали на уровне 4 путем порционного добавления смешанного раствора 6н. HCl (20 мл) и метанола (30 мл) по ходу реакции. Через 5 часов значение pH реакционной смеси устанавливали на уровне 7 и перегоняли в условиях пониженного давления для удаления метанола. Оставшийся реакционный раствор охлаждали до 0oC, значение pH вновь доводили до 11, насыщали хлоридом натрия и затем четыре раза экстрагировали добавлением хлороформа. Экстракт сушили над безводным сульфатом магния, фильтровали и концентрировали с получением указанного в заголовке соединения (3,4 г. Выход: 58,3%).
1H ЯМР (CDCl3, миллионные доли) δ : 7,20-5,00 (ушир. с, 1Н), 3,56 (м, 1Н), 1,90-1,45 (м, 9Н).
Пример 10.
Получение (S)-N-циклопентил-N-гидрокси-3-(4-амидразонофенил)-2-(2-нафтилсульфониламино)пропионамида.
Соединение, полученное в Получении 5, обрабатывали в соответствии с той же методикой, что и в Получениях 2 и 3, с получением промежуточного продукта (S)-N-циклопентил-N-гидрокси-3-(4-цианофенил)-2-(2-афтилсульфониламино)пропионамида (0,1 г). Данное промежуточное соединение применяли в качестве исходного продукта и обрабатывали в соответствии с той же методикой, что и в Примере 1, с получением очищенного соединения, указанного в заголовке (0,07 г. Выход: 63,6%).
1H ЯМР (CD3OD, миллионные доли) δ : 8,25-7,30 (м, 11H), 4,75 (м, 1Н), 4,30 (м, 1Н), 3,10, 2,75 (м, м, 2Н), 1,70-1,00 (м, 8Н).
Масс (FAB, m/e): 496 (М++1).
Пример 11.
Получение (S)-N-циклопентил-N-(2-гидроксиэтил)-3-(4-амидразонофенил)-2-(2-нафтилсульфониламино)пропионамида.
Соединение (0,15 г, 0,24 ммоль), полученное в Примере 4, растворяли в метаноле (10 мл) и затем к нему добавляли гидроксид палладия (0,02 мл). К реакционному сосуду присоединяли баллон с водородом. После перемешивания в течение 2 суток реакционную смесь фильтровали через слой целита и концентрировали. Остаток очищали по методу ЖХВР с получением указанного в заголовке соединения (0,08 г. Выход: 63,7%). Условия ЖХВР были идентичны таковым, примененным в Примере 1.
1H ЯМР (CD3OD, миллионные доли) δ : 7,80-7,00 (м, 11H), 4,47 (м, 1Н), 4,00 (м, 1Н), 3,60 (м, 2Н), 3,10-2,70 (м, 4Н), 1,70-1,10 (м, 8Н).
Масс (FAB, m/e): 524 (M++1).
Пример 12.
Получение (S)-N-циклопентил-N-метил-3-[4-(1,1-диметиламидино)фенил]-2-(4-метокси-2,3,6-триметилбензолсульфониламино)пропионамида.
Использовали ту же методику, что и в Примере 9, за исключением того, что вместо гидразина трижды с интервалом в один час добавляли метиламин, с получением очищенного соединения, указанного в заголовке (0,08 г. Выход: 28,3%).
1H ЯМР (CD3OD, миллионные доли) δ : 7,65 (д, 2Н), 7,40 (дд, 2Н), 6,70 (д, 1Н), 4,40, 3,90 (м, м, 2Н), 3,85 (д, 3Н), 3,20-2,90 (м, 5Н), 2,55 (м, 9Н), 2,10 (с, 3Н), 1,75-0,90 (м, 8Н).
Масс (FAB, m/e): 515 (M++1).
Пример 13.
Получение (S)-N, N-диметил-3-(4-амидразонофенил)-2-(2-нафтилсульфониламино)пропионамида.
Использовали ту же методику, что и в Получениях 2 и 3, за исключением того, что применяли диметиламин, с получением промежуточного продукта (S)-N, N-диметил-3-(4-цианофенил)-2-(2-нафтилсульфониламино)пропионамида (0,11 г). Данное промежуточное соединение применяли в качестве исходного продукта и обрабатывали в соответствии с той же методикой, что и в Примере 1, с получением очищенного соединения, указанного в заголовке (0,080 г. Выход: 53%).
1H ЯМР (CD3OD, миллионные доли) δ : 2,19 (с, 3Н), 2,62 (с, 3Н), 2,70-2,95 (м, 2Н), 4,41 (м, 1Н), 7,20-8,30 (м, 11Н).
Масс (FAB, m/e): 440 (М++1).
Пример. 14.
Получение (S)-N, N-диметил-3-[4-(1,1-диметиламидино)фенил]-2-(2-нафтилсульфониламино)пропионамида.
Использовали ту же методику, что и в Примере 13, за исключением того, что вместо гидразина из методики Примера 1 трижды с интервалом в один час добавляли метиламин, с получением очищенного соединения, указанного в заголовке (0,10 г. Выход: 48%).
1H ЯМР (CD3OD, миллионные доли) δ : 2,31 (с, 3Н), 2,75 (с, 3Н), 2,80-3,05 (м, 2Н), 3,07 (с, 3Н), 4,54 (м, 1Н), 7,30-8,40 (м, 11H).
Масс (FAB, m/e): 439 (M++1).
Пример 15.
Получение (S)-N-циклогексил-N-метил-3-(4-амидразонофенил)-2-(2-нафтилсульфониламино)пропионамида.
Использовали ту же методику, что и в Получении 1, за исключением того, что вместо циклопентанона применяли циклогексанон, с получением циклогексилметиламингидрохлорида, который затем обрабатывали в соответствии с той же методикой, что и в Получениях 2 и 3, с получением промежуточного продукта (S)-N-циклогексил-N-метил-3-(4-цианофенил)-2-(2-нафтилсульфониламино)пропионамида (0,21 г). Данное промежуточное соединение применяли в качестве исходного продукта и обрабатывали в соответствии с той же методикой, что и в Примере 1, с получением очищенного соединения, указанного в заголовке (0,17 г. Выход: 73,9%).
1H ЯМР (CD3OD, миллионные доли) δ : 8,32 (д, 1Н), 7,95 (м, 3Н), 7,75-7,53 (м, 5Н), 7,40 (дд, 2Н), 4,50, 4,21, 3,62 (м, м, м, 2Н), 3,05 (м, 1Н), 2,90 (м, 1Н), 2,55-2,40 (с, с, 3Н), 1,80-0,62 (м, 10Н).
Масс (FAB, m/e): 508 (М++1).
Пример 16.
Получение (S)-N-циклопропил-N-метил-3-(4-амидразонофенил)-2-(2-нафтилсульфониламино)пропионамида.
Использовали ту же методику, что и в Получении 1, за исключением того, что вместо циклопентанона применяли циклопропанон, с получением циклопропилметиламингидрохлорида, который затем обрабатывали в соответствии с той же методикой, что и в Получениях 2 и 3, с получением промежуточного продукта (S)-N-циклопропил-N-метил-3-(4-цианофенил)-2-(2-нафтилсульфониламино)пропионамида (0,21 г). Данное промежуточное соединение применяли в качестве исходного продукта и обрабатывали в соответствии с той же методикой, что и в Примере 1, с получением очищенного соединения, указанного в заголовке (0,06 г. Выход: 12%).
1H ЯМР (CD3OD, миллионные доли) δ : 0,40-0,90 (м, 4Н), 2,40 (с, 3Н), 2,75 (м, 1Н), 2,95 (м, 1Н), 7,20-8,30 (м, 11H).
Масс (FAB, m/e): 466 (M++1).
Получение 6.
Получение (S)-3-(4-цианофенил)-N-циклопентил-N-метил-2-(2-нафталин-1-ил-aцeтилaминo)пропионамида.
Соединение (0,51 г, 1,34 ммоль), полученное в Получении 2, растворяли в дихлорметане (3 мл) и затем охлаждали до -10oC и к нему добавляли трифторуксусную кислоту (ТФУ, 3 мл). Реакционную смесь перемешивали в течение 5 минут, медленно нагревали до комнатной температуры, вновь перемешивали в течение 30 минут и затем перегоняли в условиях пониженного давления для удаления летучих веществ. Остаток сушили с помощью вакуумного насоса и затем к нему добавляли ДМФ (10 мл). Данный раствор охлаждали до -10oC и к нему добавляли 1-(3-диметиламинопропил)-3-этилкарбодиимидгидрохлорид (ЭДК, 0,4 г) и 1-гидроксибензотриазол (ГОБТ, 0,2 г) и затем перемешивали до тех пор, пока они полностью не растворялись. К полученному раствору добавляли 1-нафталинуксусную кислоту (0,26 г, 1,4 ммоль) и N,N-диизопропилэтиламин (1,2 мл). Реакционную смесь медленно нагревали до комнатной температуры и перемешивали в течение 3 часов. По завершении реакции реакционный раствор перегоняли в условиях пониженного давления для удаления летучих веществ. Остаток разбавляли этилацетатом, тщательно промывали насыщенным раствором гидрокарбоната натрия, разбавленной соляной кислотой и солевым раствором, сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали и остаток очищали по методу колоночной хроматографии (метанол: хлороформ = 1:99 (по объему)) с получением указанного в заголовке соединения (0,42 г. Выход: 68%).
1H ЯМР (CDCl3, миллионные доли) δ : 8,0-6,8 (м, 11H), 6,5 (м, 1Н), 5,1-5,3 (м, 1Н), 4,8-4,2 (м, 1Н), 4,1-3,8 (м, 2Н), 3,0-2,6 (м, 5Н), 1,8-1,3 (м, 8Н).
Пример 17.
Получение (S)-3-[4-(амидразоно)фенил] -N-циклопентил-N-метил-2-(2-нафталин-1-ил-ацетиламино)пропионамида.
Соединение (0,21 г, 0,48 ммоль), полученное в Получении 6, растворяли в пиридине (3 мл) и полученный раствор помещали в колбу со штуцерами, в которую добавляли триэтиламин (0,2 мл). Реакционную колбу оборудовали таким образом, что газообразный сероводород (H2S) мог вводиться через один штуцер колбы, а выводиться через другой штуцер. Реакционный раствор насыщали газообразным сероводородом при перемешивании в течение приблизительно 10 минут, причем бесцветный раствор приобретал зеленую окраску и затем постепенно - темно-коричневую. Колбу закрывали резиновой пробкой и оставляли стоять в течение 3 суток при комнатной температуре для завершения реакции. Затем реакционный раствор перегоняли в условиях пониженного давления для удаления летучих веществ и сушили с помощью вакуумного насоса. К полученному желтому твердому веществу добавляли вместе ацетон (10 мл) и иодметан (CH3I, 0,3 мл) и смесь нагревали с обратным холодильником в течение 30 минут. Данную реакционную смесь вновь перегоняли в условиях пониженного давления для удаления летучих веществ и сушили с помощью вакуумного насоса. Остаток растворяли в абсолютном метаноле (5 мл) и затем перемешивали. К данной смеси порциями трижды с интервалом 10 минут добавляли гидрат гидразина (H2NNH2•H2O, 0,04 мл, 0,72 ммоль). После того, как реакция была завершена, реакционный раствор концентрировали и очищали по методу ЖХВР с получением указанного в заголовке соединения (0,16 г. Выход: 70%).
1H ЯМР (CD3OD, миллионные доли) δ : 8,0-7,3 (м, 11Н), 5,3-5,1 (м, 1Н), 4,8-4,2 (м, 1Н), 3,9 (с, 2Н), 3,2-2,9 (м, 2Н), 2,78, 2,72 (2 с, 3Н), 1,8-1,3 (м, 8Н).
Масс (FAB, m/e): 472 (M++1).
Пример 18.
Получение (S)-3-[4-(амидразоно)фенил] -N-циклопентил-N-метил-2-(5-диметиламинонафталин-1-сульфониламино)пропионамида.
Использовали ту же методику, что и в Получении 3, за исключением того, что вместо 2-нафталинсульфонилхлорида применяли 5-(N,N-диметиламино)-1-нафталинсульфонилхлорид, с получением промежуточного продукта (S)-3-(4-цианофенил)-N-циклопентил-N-метил-2-(5-диметиламинонафталин-1-сульфониламино)пропионамида (0,55 г, 1,09 ммоль). Данное промежуточное соединение затем обрабатывали в соответствии с той же методикой, что и в Примере 1, с получением очищенного соединения, указанного в заголовке (0,35 г. Выход: 60%).
1H ЯМР (CD3OD, миллионные доли) δ : 8,6-7,3 (м, 10Н), 4,9-3,9 (м, 2Н), 3,11 (с, 3Н), 3,0 (с, 3Н), 3,05-2,8 (м, 2Н), 2,5, 2,4 (2 с, 3Н), 1,7-0,9 (м, 10Н).
Масс (FAB, m/e): 537 (M++1).
Пример 19.
Получение (S)-3-[4-(амидразоно)фенил] -N-циклопентил-N-метил-2-(5-метоксинафталин-1-сульфониламино)пропионамида.
Использовали ту же методику, что и в Получении 3, за исключением того, что вместо 2-нафталинсульфонилхлорида применяли 5-метокси-1-нафталинсульфонилхлорид, с получением промежуточного продукта (S)-3-(4-цианофенил)-N-циклопентил-N-метил-2-(5-метоксинафталин-1-сульфониламино)пропионамида (0,18 г, 0,37 ммоль). Данное промежуточное соединение затем обрабатывали в соответствии с той же методикой, что и в Примере 1, с получением очищенного соединения, указанного в заголовке (0,12 г. Выход: 65%).
1H ЯМР (CD3OD, миллионные доли) δ : 8,5-7,0 (м, 10Н), 4,6-4,0 (м, 2Н), 4,2 (с, 3Н), 3,0-2,8 (м, 2Н), 2,48-2,45 (2 с, 3Н), 1,7-1,0 (м, 8Н).
Масс (FAB, m/e): 523 (M++1).
Пример 20.
Получение (S)-3-[4-(амидразоно)фенил] -N-циклопентил-N-метил-2-(6,7-диметоксинафталин-2-сульфониламино)пропионамида.
Использовали ту же методику, что и в Получении 3, за исключением того, что вместо 2-нафталинсульфонилхлорида применяли 6,7-диметокси-2-нафталинсульфонилхлорид, с получением промежуточного продукта (S)-3-(4-цианофенил)-N-циклопентил-N-метил-2-(6,7-диметоксинафталин-2-сульфониламино)пропионамида (2,2 г, 2,2 ммоль). Данное промежуточное соединение затем обрабатывали в соответствии с той же методикой, что и в Примере 1, с получением очищенного соединения, указанного в заголовке (1,55 г. Выход: 67%).
1H ЯМР (CD3OD, миллионные доли) δ : 8,4-7,3 (м, 9Н), 4,7-4,0 (м, 2Н), 4,2-4,0 (2 с, 6Н), 3,2-2,8 (м, 2Н), 2,6-2,2 (2 с, 3Н), 1,7-1,0 (м, 8Н).
Масс (FAB, m/e): 554 (M++1).
Пример 21.
Получение (S)-3-[4-(метиламидино)фенил] -N-циклопентил-N-метил-2-(5-диметиламинонафталин-1-сульфониламино)пропионамида.
Использовали ту же методику, что и в Получении 3, за исключением того, что вместо 2-нафталинсульфонилхлорида при меняли 5-(N,N-диметиламино)-1-нафталинсульфонилхлорид, с получением промежуточного продукта (S)-3-(4-цианофенил)-N-циклопентил-N-метил-2-(5-диметиламинонафталин-1-сульфониламино)пропионамида (0,3 г, 0,6 ммоль). Данное промежуточное соединение затем обрабатывали в соответствии с той же методикой, что и в Примере 1, за исключением того, что вместо 80% гидрата гидразина применяли метиламин, с получением очищенного соединения, указанного в заголовке (0,08 г. Выход: 25%).
1H ЯМР (CD3OD, миллионные доли) δ : 8,52-7,25 (м, 10Н), 4,55-4,42 (м, 2Н), 4,23-3,98 (м, 2Н), 3,09 (с, 3Н), 3,06-2,80 (м, 2Н), 2,90 (м, 6Н), 2,45 (с, 3Н), 2,38 (с, 3Н), 1,76-0,90 (м, 8Н).
Масс (FAB, m/e): 536 (M++1).
Пример 22.
Получение (S)-3-[4-(амидразоно)фенил] -N-циклопентил-N-метил-2-(нафталин-2-сульфониламино)пропионамида.
Использовали ту же методику, что и в Получении 3, за исключением того, что вместо 2-нафталинсульфонилхлорида применяли 1-нафталинсульфонилхлорид, с получением промежуточного продукта (S)-3-(4-цианофенил)-N-циклопентил-N-метил-2-(нафталин-2-сульфониламино)пропионамида (0,5 г, 1 ммоль). Данное промежуточное соединение затем обрабатывали в соответствии с той же методикой, что и в Примере 1, с получением очищенного соединения, указанного в заголовке (0,2 г. Выход: 40%).
1H ЯМР (CD3OD, миллионные доли) δ : 8,61-7,28 (м, 11Н), 4,55-4,41 (м, 2Н), 4,23-4,00 (м, 2Н), 3,00-2,84 (м, 2Н), 2,49, 2,41 (2 с, 3Н), 1,70-1,00 (м, 8Н).
Масс (FAB, m/e): 494 (M++1).
Пример 23.
Получение (S)-3-[4-(амидразоно)фенил] -N-циклопентил-N-метил-2-[2-(нафталин-1-илокси)ацетиламино]пропионамида.
Использовали ту же методику, что и в Получении 6, за исключением того, что вместо 1-нафталинуксусной кислоты применяли (1-нафтокси)уксусную кислоту, с получением промежуточного продукта (S)-3-(4-цианофенил)-N-циклопентил-N-метил-2-[2-(нафталин-1-илокси)ацетиламино]пропионамида (0,59 г, 1,3 ммоль). Данное промежуточное соединение затем обрабатывали в соответствии с той же методикой, что и в Примере 17, с получением очищенного соединения, указанного в заголовке (0,38 г. Выход: 60%).
1H ЯМР (CD3OD, миллионные доли) δ : 8,25-6,8 (м, 11Н), 5,38-5,22 (м, 2Н), 4,77-4,39 (м, 2Н), 4,70 (с, 2Н), 3,21-3,05 (м, 2Н), 2,88-2,77 (2 с, 3Н), 1,92-1,40 (м, 8Н).
Масс (FAB, m/e): 488 (M++1).
Пример 24.
Получение (S)-3-[4-(амидразоно)фенил] -N-циклопентил-N-метил-2-[2-(нафталин-2-илокси)ацетиламино]пропионамида.
Использовали ту же методику, что и в Получении 6, за исключением того, что вместо 1-нафталинуксусной кислоты применяли (2-нафтокси)уксусную кислоту, с получением промежуточного продукта (S)-3-[4-(аминогидразоно)фенил] -N-циклопентил-N-метил-2-[2-(нафталин-2-илокси)ацетиламино]пропионамида (0,7 г, 1,54 ммоль). Данное промежуточное соединение затем обрабатывали в соответствии с той же методикой, что и в Примере 17, с получением очищенного соединения, указанного в заголовке (0,59 г. Выход: 63%).
1H ЯМР (CD3OD, миллионные доли) δ : 7,80-7,14 (м, 11Н), 5,35-5,18 (м, 1Н), 4,76-4,35 (м, 1Н), 4,61 (с, 2Н), 3,18-3,05 (м, 2Н), 2,85-2,75 (2 с, 3Н), 1,85-1,25 (м, 8Н).
Масс (FAB, m/e): 488 (M++1).
Получение 7.
Получение метилового эфира N-трет-бутоксикарбонил-N-метиламиноуксусной кислоты.
Гидрохлорид метилового эфира глицина (1,0 г, 8,2 ммоль) растворяли в воде (12 мл) и 1н водном растворе гидроксида натрия (8,2 мл) и затем к нему добавляли 1,4-диоксан. К данной смеси добавляли ди-трет-бутилдикарбонат (2,2 г, 9,8 ммоль) при 0oC и смесь нагревали до комнатной температуры и перемешивали в течение 2 часов. Летучие вещества удаляли из реакционной смеси в условиях пониженного давления и остаток разбавляли этилацетатом, тщательно промывали водным насыщенным раствором гидрокарбоната натрия, разбавленной соляной кислотой и насыщенным солевым раствором, сушили над безводным сульфатом натрия, фильтровали и затем концентрировали. Полученный продукт растворяли в диметилформамиде (ДМФ, 10 мл). К данному раствору медленно добавляли гидрид натрия (NaH, 0,25 г, 6,4 ммоль) и затем добавляли по каплям иодметан (CH3I, 1,1 мл). Смесь медленно нагревали до комнатной температуры и перемешивали в течение 3 часов при той же температуре. Смесь фильтровали через слой целита и концентрировали в условиях пониженного давления. Остаток разбавляли этилацетатом, тщательно промывали водным насыщенным раствором гидрокарбоната натрия, разбавленной соляной кислотой и солевым раствором, сушили над безводным сульфатом натрия, фильтровали и затем концентрировали. Остаток очищали по методу колоночной хроматографии, применяя этилацетат/гексан в качестве элюента с получением очищенного соединения, указанного в заголовке (1,0 г. Выход: 65%).
1H ЯМР (CDCl3, миллионные доли) δ : 1,45 (д, 9Н), 2,95 (с, 3Н), 3,78 (с, 3Н), 3,92 (с, 1Н), 4,00 (с, 1Н).
Масс (FAB, m/e): 204 (М+1).
Получение 8.
Получение метилового эфира {[(S)-2-(трет-бутоксикарбониламино)-3- (4-цианофенил)пропионил]метиламино}уксусной кислоты.
(S)-2-(трет-Бутоксикарбониламино)-3-(4-цианофенил)пропионовую кислоту (0,5 г, 1,72 ммоль) растворяли в диметилформамиде (ДМФ). Полученный раствор охлаждали до 0oC и затем к нему добавляли 1-(3-диметиламинопропил)-3-этилкарбодиимидгидрохлорид (ЭДК, 0,39 г) и 1-гидроксибензотриазол (ГОБТ, 0,28 г) и затем перемешивали до тех пор, пока они полностью не растворялись. Отдельно от них, соединение (0,35 г, 1,72 ммоль), полученное в Получении 7, растворяли в дихлорметане (2 мл) и охлаждали до -10oC. К ним добавляли трифторуксусную кислоту (2 мл) и смесь перемешивали в течение 5 минут, медленно нагревали до комнатной температуры, вновь перемешивали в течение 30 минут и затем перегоняли в условиях пониженного давления для удаления летучих веществ. Полученное таким образом соединение и N-метилморфолин (1 мл) добавляли к раствору, как было достигнуто выше, и затем реакционный раствор медленно нагревали до комнатной температуры и перемешивали в течение 3,5 часов. По завершении реакции реакционный раствор перегоняли в условиях пониженного давления для удаления летучих веществ. Остаток разбавляли этилацетатом, тщательно промывали водным насыщенным раствором гидрокарбоната натрия, разбавленной соляной кислотой и солевым раствором, сушили над безводным сульфатом натрия, фильтровали и затем концентрирорвали, Остаток очищали по методу колоночной хроматографии, применяя этилацетат/гексан (3/7, по объему) в качестве элюента, с получением указанного в заголовке соединения 0,58 г. Выход: 90%).
1H ЯМР (CDCl3, миллионные доли) δ : 1,40 (м, 9Н), 3,08 (с, 3Н), 2,95-3,25 (м, 2Н), 3,78 (с, 3Н), 3,89-4,35 (м, 2Н), 4,95 (м, 1Н), 5,52 (д, 1Н), 7,35 (м, 2Н), 7,60 (м, 2Н).
Масс (FAB, m/e): 376 (М+1).
Получение 9.
Получение метилового эфира 1-{ [3-(4-цианофенил)-(S)-2-(нафталин-2-сульфониламино)пропионил]метиламино}уксусной кислоты.
Соединение (0,57 г, 1,52 ммоль), полученное в Получении 8, растворяли в дихлорметане (2 мл) и затем охлаждали до -10oC и к нему добавляли трифторуксусную кислоту (ТФУ, 2 мл). Реакционную смесь перемешивали в течение 5 минут, медленно нагревали до комнатной температуры, перемешивали в течение 30 минут и затем концентрировали в условиях пониженного давления для удаления летучих веществ. Остаток сушили с помощью вакуумного насоса и затем к нему добавляли ДМФ (10 мл). Данный раствор охлаждали до -10oC и к нему добавляли N, N-диизопропилэтиламин (1 мл). Данный реакционный раствор нагревали до комнатной температуры и перемешивали в течение приблизительно 5 минут и затем туда добавляли 2-нафталинсульфонилхлорид (0,41 г, 1,82 ммоль). Реакционную смесь перемешивали в течение одного часа для завершения реакции и затем перегоняли в условиях пониженного давления для удаления летучих веществ. Остаток разбавляли этилацетатом, дважды промывали водой, сушили над безводным сульфатом магния и затем фильтровали. Фильтрат концентрировали и остаток очищали по методу колоночной хроматографии, применяя этилацетат/гексан (1/1, по объему) в качестве элюента, с получением очищенного соединения, указанного в заголовке (0,55 г. Выход: 78%).
1H ЯМР (CDCl3, миллионные доли) δ : 2,88 (с, 3Н), 2,80-3,20 (м, 2Н), 3,80 (д, 3Н), 4,12 (д, 2Н), 4,58 (м, 1Н), 6,40 (д, 1Н), 7,20-8,40 (м, 11Н).
Масс (FAB, m/e): 466 (М+1).
Пример 25.
Получение метилового эфира {[3-(4-амидразонофенил)-(S)-2-(нафталин-2-сульфониламино)пропионил]метиламино}уксусной кислоты.
Соединение (0,55 г, 1,18 ммоль), полученное в Получении 9, растворяли в пиридине (10 мл) и полученный раствор помещали в колбу со штуцерами, в которую добавляли триэтиламин (0,45 мл). Реакционную колбу оборудовали таким образом, что газообразный сероводород (H2S) мог вводиться через один штуцер колбы, а выводиться через другой штуцер. Реакционный раствор насыщали газообразным сероводородом при перемешивании в течение приблизительно 10 минут, причем бесцветный раствор приобретал зеленую окраску и затем постепенно - темно-коричневую. Колбу закрывали резиновой пробкой и оставляли стоять в течение 3 суток при комнатной температуре. По завершении реакции реакционный раствор перегоняли в условиях пониженного давления для удаления летучих веществ и сушили с помощью вакуумного насоса. К полученному желтому твердому веществу добавляли вместе ацетон (10 мл) и иодметан (CH3I, 0,55 мл) и данную смесь нагревают с обратным холодильником в течение 30 минут. Данную реакционную смесь вновь перегоняли в условиях пониженного давления для удаления летучих веществ и сушили с помощью вакуумного насоса. Остаток растворяли в абсолютном метаноле (5 мл) и затем перемешивали. К данному раствору порциями трижды с интервалом 10 минут добавляли 80% гидрат гидразина (H2NNH2•H2O, 0,11 мл, 1,77 ммоль). После того, как реакция была завершена, реакционный раствор концентрировали и очищали по методу ЖХВР с получением указанного в заголовке соединения (0,25 г. Выход: 43%).
1H ЯМР (CD3OD, миллионные доли) δ : 2,95 (с, 3Н), 2,70-3,20 (м, 2Н), 3,54 (с, 3Н), 3,80 (д, 2Н), 4,55 (м, 1Н), 7,20-8,30 (м, 11Н).
Масс (FAB, m/e): 498 (М+1).
Пример 26.
Получение { [3-(4-амидразонофенил)-(S)-2-(нафталин-2-сульфониламино)пропионил]метиламино}уксусной кислоты.
Соединение (160 мг, 0,32 ммоль), полученное в Примере 25, растворяли в смешанном растворителе (4 мл) метанола и воды (3:1). К данному раствору медленно добавляли гидрат гидроксида лития (LiOH•H2O, 0,016 г, 0,38 ммоль) при 0oC и смесь перемешивали в течение 2 часов при комнатной температуре. По завершении реакции реакционный раствор концентрировали и очищали по методу ЖХВР с получением указанного в заголовке соединения (50 мг. Выход: 32%).
1H ЯМР (CD3OD, миллионные доли) δ : 2,20-2,60 (м, 2Н), 2,48 (с, 3Н), 2,78 (с, 3Н), 2,32 (м, 2Н), 4,12 (м, 1H), 6,80-7,80 (м, 11Н).
Масс (FAB, m/e): 484 (М+1).
Пример 27.
Получение метилового эфира (S)-2-{ [3-(4-амидразонофенил)-(S)-2-(нафталин-2-сульфониламино)пропионил]метиламино}пропионовой кислоты.
Использовали ту же методику, что и в Получении 7, за исключением того, что вместо метилового эфира глицина применяли метиловый эфир (L)-аланина, с получением метилового эфира (L)-(N-трет-бутоксикарбонил-N-метил)аланина, который затем обрабатывали в соответствии с той же методикой, что и в Получениях 8 и 9, с получением промежуточного продукта метилового эфира (S)-2-{ [3-(4-цианофенил)-(S)-2-(нафталин-2-сульфониламино]пропионил]метиламино} пропионовой кислоты (1,43 г). Данное промежуточное соединение обрабатывали в соответствии с той же методикой, что и в Примере 25, с получением очищенного соединения, указанного в заголовке (0,64 г. Выход: 48%).
1H ЯМР (CD3OD, миллионные доли) δ : 0,69, 0,88 (д, д, 3Н), 2,79, 2,95 (с, с, 3Н), 2,80, 3,06 (м, м, 2Н), 3,48, 3,57 (с, с, 3Н), 4,29 (м, 1Н), 4,55 (м, 1Н), 7,30-8,30 (м, 11Н).
Масс (FAB, m/e): 512 (М+1).
Пример 28.
Получение (S)-2-{ [3-(4-амидразонофенил)-(S)-2-(нафталин-2-сульфониламино)пропионил]метиламино}пропионовой кислоты.
Соединение, полученное в Примере 27, обрабатывали в соответствии с той же методикой, что и в Примере 26, с получением очищенного соединения, указанного в заголовке (0,06 г. Выход: 41%).
1H ЯМР (CD3OD, миллионные доли) δ : 0,64, 0,95 (д, д, 3Н), 2,76, 2,92 (с, с, 3Н), 2,83, 3,09 (м, м, 2Н), 4,37 (м, 1Н), 4,54 (м, 1Н), 7,30-8,40 (м, 11Н).
Масс (FAB, m/e): 498 (М+1).
Пример 29.
Получение метилового эфира (R)-2-{ [3-(4-амидразонофенил)-(S)-2-(нафталин-2-сульфониламино)пропионил]метиламино}пропионовой кислоты.
Использовали ту же методику, что и в Получении 7, за исключением того, что вместо метилового эфира глицина применяли метиловый эфир (D)-аланина, с получением метилового эфира (D)-(N-трет-бутоксикарбонил-N-метил)аланина, который затем обрабатывали в соответствии с той же методикой, что и в Получениях 8 и 9, с получением промежуточного продукта метилового эфира (R)-2-{ [3-(4-цианофенил)-(S)-2-(нафталин-2-сульфониламино]пропионил]метиламино} пропионовой кислоты (0,78 г). Данное промежуточное соединение обрабатывали в соответствии с той же методикой, что и в Примере 25, с получением очищенного соединения, указанного в заголовке (0,58 г. Выход: 70%).
1H ЯМР (CD3OD, миллионные доли) δ : 0,89, 1,21 (д, д, 3Н), 2,46, 2,94 (с, с, 3Н), 2,80, 3,08 (м, м, 2Н), 3,49, 3,78 (с, с, 3Н), 4,29 (м, 1Н), 4,59 (м, 1Н), 7,30-8,40 (м, 11Н).
Масс (FAB, m/e): 512 (М+1).
Пример 30.
Получение (R)-2-{ [3-(4-амидразонофенил)-(S)-2-(нафталин-2-сульфониламино)пропионил]метиламино}пропионовой кислоты.
Соединение (0,03 г, 0,059 ммоль), полученное в Примере 29, обрабатывали в соответствии с той же методикой, что и в Примере 26, с получением очищенного соединения, указанного в заголовке (0,01 г. Выход: 33%).
1H ЯМР (CD3OD, миллионные доли) δ: 0,90, 1,18 (д, д, 3Н), 2,44, 2,92 (с, с, 3Н), 2,82, 3,08 (м, м, 2Н), 4,33 (м, 1Н), 4,62 (м, 1Н), 7,30-8,40 (м, 11Н).
Масс (FAB, m/e): 498 (М+1).
Пример 31.
Получение метилового эфира (R)-2-{ [3-(4-амидразонофенил)-(S)-2-(нафталин-2-сульфониламино)пропионил]метиламино}-3-метилмасляной кислоты.
Использовали ту же методику, что и в Получении 7, за исключением того, что вместо метилового эфира глицина применяли метиловый эфир (D)-валина, с получением метилового эфира (D)-(N-трет-бутоксикарбонил-N-метил)валина, который затем обрабатывали в соответствии с той же методикой, что и в Получениях 8 и 9 с получением промежуточного продукта метилового эфира (R)-2-{ [3-(4-цианофенил)-(S)-2-(нафталин-2-сульфониламино] пропионил] метиламино}-3-метилмасляной кислоты (0,19 г). Данное промежуточное соединение обрабатывали в соответствии с той же методикой, что и в Примере 25, с получением очищенного соединения, указанного в заголовке (0,11 г. Выход: 55%).
1H ЯМР (CD3OD, миллионные доли) δ : 0,59, 0,70 (д, д, 3Н), 0,89, 0,98 (д, д, 3Н), 2,09, 2,21 (м, м, 1Н), 2,75, 3,06 (с, с, 3Н), 3,40, 3,68 (с, с, 3Н), 4,34, 4,38 (д, д, 1Н), 4,63, 4,70 (м, м, 1Н), 7,20-8,40 (м, 11Н).
Масс (FAB, m/e): 540 (M++1).
Пример 32.
Получение (R)-2-{ [3-(4-амидразонофенил)-(S)-2-(нафталин-2-сульфониламино)пропионил]метиламино}-3-метилмасляной кислоты.
Соединение, полученное в Примере 31, обрабатывали в соответствии с той же методикой, что и в Примере 26, с получением очищенного соединения, указанного в заголовке (0,04 г. Выход: 40%).
1H ЯМР (CD3OD, миллионные доли) δ : 0,57, 0,63 (д, д, 3Н), 0,92, 0,99 (д, д, 3Н), 2,09, 2,18 (м, м, 1Н), 2,74, 3,08 (с, с, 3Н), 4,18, 4,36 (д, д, 1Н), 4,64, 4,70 (м, м, 1Н), 7,20-8,40 (м, 11Н).
Масс (FAB, m/e): 526 (М+1).
Пример 33.
Получение метилового эфира 3-{[3-(4-амидразонофенил)-(S)-2-(нафталин-2-сульфониламино)пропионил]метиламино}пропионовой кислоты.
Использовали ту же методику, что и в Получении 7, за исключением того, что вместо метилового эфира глицина применяли метиловый эфир 3-аминопропионовой кислоты, с получением метилового эфира 3-(N-трет-бутоксикарбонил-N-метил)аминопропионовой кислоты, который затем обрабатывали в соответствии с той же методикой, что и в Получениях 8 и 9, с получением промежуточного продукта метилового эфира 3-{[3-(4-цианофенил)-(S)-2-(нафталин-2-сульфониламино]пропионил]метиламино}пропионовой кислоты (0,69 г). Данное промежуточное соединение обрабатывали в соответствии с той же методикой, что и в Примере 25, с получением очищенного соединения, указанного в заголовке (0,55 г. Выход: 74%).
1H ЯМР (CD3OD, миллионные доли) δ : 8,31, 7,97, 7,68, 7,48 (д, м, м, м, 11Н), 4,62, 4,51 (м, м, 1Н), 3,62, 3,55 (с, с, 3H), 3,05, 2,85 (м, м, 4Н), 2,80, 2,45 (с, с, 3H), 2,38, 1,91 (м, м, 2Н).
Масс (FAB, m/e): 512 (М++1).
Пример 34.
Получение 3-{ [3-(4-амидразонофенил)-(S)-2-(нафталин-2-сульфониламино)пропионил]метиламино}пропионовой кислоты.
Соединение, полученное в Примере 33, обрабатывали в соответствии с той же методикой, что и в Примере 26, с получением очищенного соединения, указанного в заголовке (0,17 г. Выход: 32%).
1H ЯМР (CD3OD, миллионные доли) δ : 8,31, 7,98, 7,78-7,37 (д, м, м, 11H), 4,65, 4,52 (м, м, 1H), 3,20-2,85 (м, 4Н), 2,80, 2,45 (с, с, 3H), 2,38, 1,91 (м, м, 2Н).
Масс (FAB, m.e): 498 (M++1).
Пример 35.
Получение метилового эфира 4-{[3-(4-амидразонофенил)-(S)-2-(нафталин-2-сульфониламино)пропионил]метиламино}масляной кислоты.
Использовали ту же методику, что и в Получении 7, за исключением того, что вместо метилового эфира глицина применяли метиловый эфир 4-аминомасляной кислоты, с получением метилового эфира (D)-(N-трет-бутоксикарбонил-N-метил)валина, который затем обрабатывали в соответствии с той же методикой, что и в Получениях 8 и 9, с получением промежуточного продукта метилового эфира 4-{ [3-(4-цианофенил)-(S)-2-(нафталин-2-сульфониламино] пропионил]метиламино}аминомасляной кислоты (0,51 г). Данное промежуточное соединение обрабатывали в соответствии с той же методикой, что и в Примере 25, с получением очищенного соединения, указанного в заголовке (0,40 г. Выход: 74%).
1H ЯМР (CD3OD, миллионные доли) δ : 8,32 (с, 1Н), 7,98 (м, 3Н), 7,78-7,36 (м, 7Н), 4,55 (м, 1Н), 3,72, 3,60 (с, с, 3Н), 3,10, 2,81 (м, м, 4Н), 2,79, 2,55 (с, с, 3Н), 2,22 (м, 1Н), 1,89 (м, 1Н), 1,63, 1,42 (м, м, 1Н), 1,18 (м, 1Н).
Масс (FAB, m/e): 526 (M++1).
Пример 36.
Получение 4-{ [3-(4-амидразонофенил)-(S)-2-(нафталин-2-сульфониламино)пропионил]метиламино}масляной кислоты.
Соединение, полученное в Примере 35, обрабатывали в соответствии с той же методикой, что и в Примере 26, с получением очищенного соединения, указанного в заголовке (0,12 г. Выход: 32%).
1H ЯМР (CD3OD, миллионные доли) δ : 8,32 (м, 1Н), 7,98 (м, 3Н), 7,78-7,35 (м, 7Н), 4,55 (м, 1Н), 3,05, 2,81 (м, м, 4Н), 2,79, 2,50 (с, с, 3Н), 2,18 (м, 1Н), 1,89 (м, 1Н), 1,35 (м, 1Н), 1,16 (м, 1Н).
Масс (FAB, m/e): 512 (M++1).
Получение 10.
Получение метилового эфира N-трет-бутоксикарбонил-N-циклопропилуксусной кислоты.
Циклопропиламин (1,34 г, 23,49 ммоль) смешивали с ДМФ (15 мл) и триэтиламином (3 мл) и смесь помещали в реакционный сосуд. В капельную воронку помещали метилбромацетат (2,2 мл, 23,49 ммоль) и ДМФ (5 мл). Реакционный сосуд охлаждали до 0oC и затем раствор, содержащийся в капельной воронке, добавляли по каплям в реакционный сосуд. По окончании добавления реакционную смесь нагревали до комнатной температуры и позволяли реакции протекать в течение 3,5 часов. После того, как реакция была завершена, к ней добавляли воду (10 мл) и 3 н. гидроксид натрия. К реакционной смеси добавляли 1,4-диоксан (10 мл) с последующим добавлением ангидрида бутилоксикарбонила (6,1 г, 27,95 ммоль). Реакционную смесь оставляли для протекания реакции на 3 часа при комнатной температуре и перегоняли в условиях пониженного давления для удаления летучих веществ. Остаток разбавляли этилацетатом, тщательно промывали насыщенным раствором гидрокарбоната натрия, разбавленной соляной кислотой и солевым раствором. Органический слой отделяли, сушили над безводным сульфатом магния и фильтровали. Растворитель удаляли из фильтрата в условиях пониженного давления. Остаток очищали по методу колоночной хроматографии (элюент: этилацетат/гексан (6/4, по объему)) с получением очищенного соединения, указанного в заголовке (2,3 г. Выход: 43%).
1H ЯМР (CDCl3, миллионные доли) δ : 3,95 (м, 2Н), 3,72 (м, 3Н), 2,75, 2,52 (ушир. с, ушир. с, 1Н), 1,45, 1,47 (с, с, 9Н), 0,80-0,45 (м, 4Н).
Масс (FAB, m/e): 230 (M++1).
Пример 37.
Получение метилового эфира {[3-(4-амидразонофенил)-(S)-2-(нафталин-2-сульфониламино)пропионил]циклопропиламино}уксусной кислоты.
Соединение, полученное в Получении 10, обрабатывали в соответствии с той же методикой, что и в Получениях 8 и 9, с получением промежуточного продукта метилового эфира { [3-(4-цианофенил)-(S)-2-(нафталин-2-сульфониламино]пропионил]циклопропиламино}уксусной кислоты (0,30 г). Данное промежуточное соединение обрабатывали в соответствии с той же методикой, что и в Примере 25, с получением очищенного соединения, указанного в заголовке (0,25 г. Выход: 77%).
1H ЯМР (CD3OD, миллионные доли) δ : 8,24 (с, 1Н), 7,93 (м, 3Н), 7,65 (м, 3Н), 7,42 (д, 2Н), 7,35 (д, 2Н), 5,02 (м, 1Н), 3,92 (д, 1Н), 3,64 (д, 1Н), 3,60 (с, 3Н), 3,19 (дд, 1Н), 2,80 (м, 2Н), 0,95, 0,85, 0,61 (м, м, м, 4Н).
Масс (FAB, m/e): 524 (M++1).
Пример 38.
Получение { [3-(4-амидразонофенил)-(S)-2-(нафталин-2-сульфониламино)пропионил]циклопропиламино}уксусной кислоты.
Соединение, полученное в Примере 37, обрабатывали в соответствии с той же методикой, что и в Примере 26, с получением очищенного соединения, указанного в заголовке (0,07 г. Выход: 29%).
1H ЯМР (CD3OD, миллионные доли) δ : 8,24 (с, 1Н), 7,93 (м, 3Н), 7,42 (д, 2Н), 7,35 (д, 2Н), 5,02 (м, 1Н), 3,95 (д, 1Н), 3,54 (д, 1Н), 3,20 (дд, 1Н), 2,80 (м, 2Н), 0,95, 0,85, 0,61 (м, м, м, 4Н).
Масс (FAB, m/e): 510 (M++1).
Пример 39.
Получение метилового эфира {[3-(4-амидразонофенил)-(S)-2-(нафталин-2-сульфониламино)пропионил]бутиламино}уксусной кислоты.
Использовали ту же методику, что и в Получении 10, за исключением того, что вместо циклопропиламина применяли бутиламин, с получением метилового эфира N-трет-бутоксикарбонил-N-бутиламиноуксусной кислоты, который затем обрабатывали в соответствии с той же методикой, что и в Получениях 8 и 9, с получением промежуточного продукта метилового эфира {[3- (4-цианофенил)-(S)-2-(нафталин-2-сульфониламино]пропионил]бутиламино}уксусной кислоты (0,31 г). Данное промежуточное соединение обрабатывали в соответствии с той же методикой, что и в Примере 25, с получением очищенного соединения, указанного в заголовке (0,19 г. Выход: 58%).
1H ЯМР (CD3OD, миллионные доли) δ: 8,30-7,32 (м, 11Н), 4,32-4,09 (м, 3Н), 3,55 (с, 3Н), 3,57-2,50 (м, 4Н), 1,26-0,50 (м, 7Н).
Масс (FAB, m/e): 540 (M++1).
Пример 40.
Получение { [3-(4-амидразонофенил)-(S)-2-(нафталин-2-сульфониламино)пропионил]бутиламино}уксусной кислоты.
Соединение, полученное в Примере 39, обрабатывали в соответствии с той же методикой, что и в Примере 26, с получением очищенного соединения, указанного в заголовке (0,12 г. Выход: 67%).
1H ЯМР (CD3OD, миллионные доли) δ : 8,30-7,10 (м, 11Н), 4,31-4,10 (м, 3Н), 3,52-2,55 (м, 4Н), 1,25-0,50 (м, 7Н).
Масс (FAB, m/e): 526 (М+1).
Пример 41.
Получение метилового эфира {[3-(4-амидразонофенил)-(S)-2-(нафталин-2-сульфониламино)пропионил]циклопентиламино}уксусной кислоты.
Использовали ту же методику, что и в Получении 10, за исключением того, что вместо циклопропиламина применяли циклопентиламин, с получением метилового эфира N-трет-бутоксикарбонил-N-циклопентиламиноуксусной кислоты, который затем обрабатывали в соответствии с той же методикой, что и в Получениях 8 и 9, с получением промежуточного продукта метилового эфира {[3-(4-цианофенил)-(S)-2-(нафталин-2-сульфониламино] пропионил] циклопентиламино} уксусной кислоты (0,23 г). Данное промежуточное соединение обрабатывали в соответствии с той же методикой, что и в Примере 25, с получением очищенного соединения, указанного в заголовке (0,12 г. Выход: 50%).
1H ЯМР (CD3OD, миллионные доли) δ : 8,35-7,35 (м, 11Н), 4,66, 4,33 (м, м, 1Н), 4,15 (м, 1Н), 3,75, 3,53 (м, м, 1Н), 3,61 (с, 3Н), 3,40-2,80 (м, 3Н), 1,90-0,60 (м, 8Н).
Масс (FAB, m/e): 552 (M++1).
Пример 42.
Получение { [3-(4-амидразонофенил)-(S)-2-(нафталин-2-сульфониламино)пропионил]циклопентиламино}уксусной кислоты.
Соединение, полученное в Примере 41, обрабатывали в соответствии с той же методикой, что и в Примере 26, с получением очищенного соединения, указанного в заголовке (0,4 г. Выход: 33%).
1H ЯМР (CD3OD, миллионные доли) δ : 8,35-7,35 (м, 1Н), 4,65, 4,32 (м, м, 1Н), 4,15 (м, 1Н), 3,75, 3,52 (м, м, 1Н), 3,41, 3,28-3,10, 2,80 (м, м, м, 3Н), 1,90-0,60 (м, 8Н).
Масс (FAB, m/e): 538 (M++1).
Получение 11.
Получение метилового эфира 1-(N-трет-бутоксикарбонил-N-метиламино)циклопентанкарбоновой кислоты.
Циклолейцин (3 г, 23,2 ммоль) растворяли в 1н. водном растворе гидроксида натрия (23,2 мл) и дистиллированной воде (7 мл) и затем к нему добавляли 1,4-диоксан (30 мл). К данной смеси добавляли ди-трет-бутилдикарбонат (6,1 г, 27,8 ммоль) при 0oC, смесь нагревали до комнатной температуры и перемешивали в течение 2 часов. Летучие вещества удаляли из реакционной смеси в условиях пониженного давления и остаток разбавляли этилацетатом, тщательно промывали водным насыщенным раствором гидрокарбоната натрия, разбавленной соляной кислотой и солевым раствором, сушили над безводным сульфатом натрия, фильтровали и затем концентрировали. Полученный белый твердый продукт растворяли в диметилформамиде (ДМФ, 30 мл). К данному раствору добавляли карбонат калия (4,8 г, 34,8 ммоль) и затем добавляли по каплям иодметан (CH3I, 14,4 мл, 232 ммоль). Смесь перемешивали в течение 2 часов при комнатной температуре и перегоняли в условиях пониженного давления для удаления летучих веществ. Оставшийся раствор разбавляли этилацетатом, тщательно промывали водным насыщенным раствором гидрокарбоната натрия, разбавленной соляной кислотой и насыщенным солевым раствором, сушили над безводным сульфатом натрия, фильтровали и затем концентрировали. Полученный продукт растворяли в диметилформамиде (ДМФ, 20 мл). К данному раствору медленно добавляли 60% гидрид натрия (NaH, 0,46 г, 11,4 ммоль) при 0oC и затем добавляли по каплям иодметан (CH3I, 1,8 мл, 28,4 ммоль). Смесь медленно нагревали до комнатной температуры и перемешивали в течение 3 часов при той же температуре. В реакционную смесь добавляли воду в целях удаления оставшийся гидрид натрия. Смесь фильтровали и концентрировали в условиях пониженного давления. Остаток разбавляли этилацетатом, тщательно промывали водным насыщенным раствором гидрокарбоната натрия, разбавленной соляной кислотой и насыщенным солевым раствором, сушили над безводным сульфатом натрия, фильтровали и затем концентрировали. Остаток очищали по методу колоночной хроматографии, применяя этилацетат/гексан (3/7, по объему) в качестве элюента с получением очищенного соединения, указанного в заголовке (2,0 г. Выход: 34%).
1H ЯМР (CDCl3, миллионные доли) δ : 1,35 (с, 9Н), 1,62 (м, 4Н), 1,78 (м, 2Н), 2,18 (м, 2Н), 2,90 (с, 3Н), 3,62 (с, 3Н).
Масс (FAB, m/e): 258 (М+1).
Пример 43.
Получение метилового эфира 1-{[3-(4-амидразонофенил)-(S)-2-(нафталин-2-сульфониламино)пропионил]метиламино}циклопентанкарбоновой кислоты.
Соединение, полученное в Получении 11, обрабатывали в соответствии с той же методикой, что и в Получениях 8 и 9, с получением промежуточного продукта метилового эфира 1-{ [3-(4-цианофенил)-(S)-2-(нафталин-2-сульфониламино] пропионил] циклопропиламино} циклопентанкарбоновой кислоты (0,28 г). Данное промежуточное соединение обрабатывали в соответствии с той же методикой, что и в Примере 25, с получением очищенного соединения, указанного в заголовке (0,56 г. Выход: 53%).
1H ЯМР (CD3OD, миллионные доли) δ : 0,52 (м, 1Н), 0,89 (м, 2Н), 1,29 (м, 2Н), 1,52 (м, 1Н), 1,76 (м, 2Н), 2,05 (м, 1Н), 2,75, 3,00 (м, м, 2Н), 2,88 (с, 3Н), 3,50 (с, 3Н), 4,48 (м, 1Н), 6,38 (м, 1Н), 7,30-8,40 (м, 11Н).
Масс (FAB, m/e): 552 (М+1).
Пример 44.
Получение 1-{ [3-(4-амидразонофенил)-(S)-2-(нафталин-2-сульфониламино)пропионил]метиламино}циклопентанкарбоновой кислоты.
Соединение, полученное в Примере 43, обрабатывали в соответствии с той же методикой, что и в Примере 26, с получением очищенного соединения, указанного в заголовке (0,01 г. Выход: 17%).
1H ЯМР (CD3OD, миллионные доли) δ : 0,42 (м, 1Н), 0,74 (м, 1Н), 1,25 (м, 2Н), 1,50 (м, 1Н), 1,78 (м, 2Н), 2,05 (м, 1Н), 2,75, 3,08 (м, м, 2Н), 2,98 (с, 3Н), 4,58 (м, 1Н), 7,40-8,40 (м, 11Н).
Масс (FAB, m/e): 538 (М+1).
Получение 12.
Получение этилового эфира 2-(N-трет-бутоксикарбонил-N-метил)аминоциклопентанкарбоновой кислоты.
В реакционный сосуд помещали этил-2-оксициклопентанкарбоксилат (10 мл, 67,49 ммоль) вместе с этанолом (100 мл). Затем для растворения реактивов туда добавляли метиламингидрохлорид (4,69 г, 68,13 ммоль) и воду (10 мл). В реакционный сосуд добавляли цианоборгидрид натрия (4,3 г, 68,43 ммоль) и значение pH смеси доводили до 6 и затем оставляли ее для протекания реакции на 12 часов или более при от 30 до 40oC. Реакционную смесь затем концентрировали в условиях пониженного давления, охлаждали до 0oC, доводили значение pH до 2, применяя 6 н. соляную кислоту и трижды промывали диэтиловым эфиром. Значение pH водного слоя вновь доводили до 10 и к нему добавляли то же количество диоксана. К данной смеси добавляли 1 эквивалентную массу ангидрида бутилоксикарбонила. Данную реакционную смесь оставляли для протекания реакции на 3 часа при комнатной температуре. После того, как реакция была завершена, реакционный раствор перегоняли в условиях пониженного давления для удаления летучих веществ, разбавляли этилацетатом и тщательно промывали насыщенным раствором гидрокарбоната натрия, разбавленной соляной кислотой и насыщенным солевым раствором. Органический слой сушили над безводным сульфатом магния, фильтровали и перегоняли в условиях пониженного давления для удаления растворителя. Остаток очищали по методу колоночной хроматографии (элюент = этилацетат:гексан = 1:1 (по объему)) с получением очищенного соединения, указанного в заголовке (5,82 г. Выход: 32%).
1H ЯМР (CDCl3, миллионные доли) δ : 4,55 (м, 1Н), 4,10 (м, 2Н), 2,79 (с, 3Н), 2,73 (с, 1Н), 2,00-1,40 (м, 6Н), 1,45 (с, 9Н), 1,24 (т, 3Н).
Масс (FAB, m/e): 272 (M++1).
Пример 45.
Получение этилового эфира 2-{[3-(4-амидразонофенил)-(S)-2-(нафталин-2-сульфониламино)пропионил]метиламино}циклопентанкарбоновой кислоты.
Соединение, полученное в Получении 12, обрабатывали в соответствии с той же методикой, что и в Получениях 8 и 9, с получением промежуточного продукта метилового эфира 2-{ [3-(4-цианофенил)-(S)-2-(нафталин-2-сульфониламино] пропионил] циклопропиламино} циклопентанкарбоновой кислоты (0,48 г). Данное промежуточное соединение затем обрабатывали в соответствии с той же методикой, что и в Примере 25, с получением очищенного соединения, указанного в заголовке (0,36 г. Выход: 71%).
1H ЯМР (CD3OD, миллионные доли) δ : 8,39-7,25 (м, 11Н), 4,78-4,40 (м, 2Н), 4,05 (м, 2Н), 3,05 (м, 1Н), 2,90-2,65 (м, 3Н), 2,50-2,40 (м, 2Н), 2,05, 1,90-1,30, 0,85 (м, м, м, 6Н), 1,28-1,15 (м, 3Н).
Масс (FAB, m/e): 566 (M++1).
Пример 46.
Получение 2-{ [3-(4-амидразонофенил)-(S)-2-(нафталин-2-сульфониламино)пропионил]метиламино}циклопентанкарбоновой кислоты.
Соединение, полученное в Примере 45, обрабатывали в соответствии с той же методикой, что и в Примере 26, с получением очищенного соединения, указанного в заголовке (0,086 г. Выход: 25%).
1H ЯМР (CD3OD, миллионные доли) δ : 8,39-7,20 (м, 11Н), 4,78-4,50 (м, м, 2Н), 3,05 (м, 1Н), 2,90-2,40 (м, 5Н), 2,10, 1,90-1,20, 0,75 (м, м, м, 6Н).
Масс (FAB, m/e): 538 (M++1).
Пример 47.
Получение метилового эфира (S)-2-{ [3-(4-амидразонофенил)-(S)-2-(нафталин-2-сульфониламино)пропионил]метиламино}-3-метилмасляной кислоты.
Проводили ту же процедуру, что и в Получении 7, за исключением того, что вместо метилового эфира глицина применяли метиловый эфир (L)-валина, с получением метилового эфира (L)-(N-трет-бутоксикарбонил-N-метил)валина, который затем обрабатывали в соответствии с той же методикой, что и в Получениях 8 и 9, с получением промежуточного продукта метилового эфира (S)-2-{ [3-(4-цианофенил)-(S)-2-(нафталин-2-сульфониламино] пропионил] метиламино}-3-метилмасляной кислоты (0,13 г). Данное промежуточное соединение обрабатывали в соответствии с той же методикой, что и в Примере 25, с получением очищенного соединения, указанного в заголовке (0,09 г. Выход: 69%).
1H ЯМР (CD3OD, миллионные доли) δ : 8,30, 7,95, 7,75-7,20 (м, м, м, 11H), 4,75-4,25 (м, 2H), 4,65, 3,39 (м, м, 3H), 3,15-2,65 (м, 4Н), 2,22, 2,15 (с, с, 1Н), 2,08, 1,90 (м, м, 1H), 1,00-0,55, 0,20 (м, м, 6H).
Масс (FAB, m/e): 540 (M++1).
Пример 48.
Получение метилового эфира 1-([3-(4-амидразонофенил)-(S)-2-(нафталин-2-сульфониламино)пропионил]пиперидин-(R)-2-карбоновой кислоты.
Использовали ту же методику, что и в Получении 7, за исключением того, что вместо метилового эфира глицина применяли метиловый эфир (D)-пипеколиновой кислоты, и полученный продукт затем обрабатывали в соответствии с той же методикой, что и в Получениях 8 и 9, с получением промежуточного продукта метилового эфира 1-{[3-(4-цианофенил)-(S)-2-(нафталин-2-сульфониламино]пропионил] пиперидин-(R)-карбоновой кислоты (0,18 г). Данное промежуточное соединение обрабатывали в соответствии с той же методикой, что и в Примере 25, с получением очищенного соединения, указанного в заголовке (0,16 г. Выход: 84%).
1H ЯМР (CD3OD, миллионные доли) δ : 8,32, 7,95, 7,78-7,35 (м, м, м, 11H), 4,71, 4,52 (м, м, 1H), 3,97, 3,80 (д, д, 1H), 3,73, 3,43 (c, c, 3H), 3,10, 2,83, 2,39 (м, м, м, 3H), 1,65-1,00, 0,30 (м, м, 6H).
Масс (FAB, m/e): 538 (M++1).
Пример 49.
Получение 1-[3-(4-амидразонофенил)-(S)-2-(нафталин-2-сульфониламино)пропионил]пиперидин-(R)-2-карбоновой кислоты.
Соединение, полученное в Примере 48, обрабатывали в соответствии с той же методикой, что и в Примере 26, с получением очищенного соединения, указанного в заголовке (0,03 г. Выход: 19%).
1H ЯМР (CD3OD, миллионные доли) δ : 8,35, 8,00, 7,75-7,30 (м, м, м, 11Н), 4,50, 4,20 (м, м, 1H), 3,89 (м, 1H), 3,10, 2,82, 2,45 (м, м, м, 3Н), 2,10 (м, 1Н), 1,65-1,00, 0,25 (м, м, 6Н).
Масс (FAB, m/e): 524 (M++1).
Получение 13.
Получение (S)-4-[2-(бутилоксикарбониламино)-3-(4-метилсульфонилпиперазинил)-3-оксопропил]бензонитрил.
(S)-3-(4-Цианофенил)-2-(бутилоксикарбониламино)пропионовую кислоту (0,5 г, 1,7 ммоль) растворяли в диметилформамиде (ДМФ, 20 мл) и затем охлаждали до 0oC. Затем к данному раствору добавляли 1-(3-диметиламинопропил)-3-этилкарбодиимидгидрохлорид (ЭДК, 0,5 г) и 1-гидроксибензотриазолгидрат (ГОБТ, 0,3 г) и перемешивали до тех пор, пока они полностью не растворялись там. К данной реакционной смеси добавляли 1-метансульфонилпиперазин (0,3 г) и N-метилморфолин (0,2 мл) и затем температуру реакции медленно поднимали до комнатной температуры. Реакционный раствор перемешивали в течение 3,5 часов. После завершения реакции реакционный раствор перегоняли в условиях пониженного давления для удаления летучих веществ и оставшийся раствор разбавляли этилацетатом, тщательно промывали водным насыщенным раствором гидрокарбоната натрия, разбавленной соляной кислотой и насыщенным солевым раствором, сушили над безводным сульфатом натрия, фильтровали и затем концентрировали. Остаток очищали по методу колоночной хроматографии, применяя этилацетат/гексан (6/4, по объему) в качестве элюента, с получением очищенного соединения, указанного в заголовке (0,7 г. Выход: 93%).
1H ЯМР (CDCl3, миллионные доли) δ : 7,7-7,3 (м, 4Н), 5,3 (м, 1Н), 4,8 (м, 1Н), 3,9-3,55 (м, 2Н), 3,55-2,8 (м, 8Н), 2,7 (с, 3Н), 1,5 (с, 9Н).
Получение 14.
Получение [1-(4-цианобензил)-2-(4-метилсульфонилпиперазинил)-2-оксоэтил] амида (S)-нафталин-2-сульфоновой кислоты.
Соединение (0,7 г, 1,6 ммоль), полученное в Получении 13, растворяли в дихлорметане (3 мл) и затем охлаждали до -10oC и к нему добавляли трифторуксусную кислоту (ТФУ, 3 мл). Реакционную смесь перемешивали в течение 5 минут, медленно нагревали до комнатной температуры, перемешивали в течение 3 минут и затем перегоняли в условиях пониженного давления для удаления летучих веществ. Оставшийся раствор сушили с помощью вакуумного насоса и затем к нему добавляли 20 мл ДМФ. Смесь охлаждали до 0oC и в нее добавляли 2-нафталинсульфонилхлорид (0,5 г) и диизопропилэтиламин (0,9 мл). Данную смесь перемешивали до тех пор, пока все реагенты полностью не растворялись. Реакционную смесь медленно нагревали до комнатной температуры и перемешивали в течение 3 часов. Реакционный раствор перегоняли в условиях пониженного давления для удаления летучих веществ. Оставшийся раствор разбавляли хлороформом, тщательно промывали водным насыщенным раствором гидрокарбоната натрия, разбавленной соляной кислотой и солевым раствором, сушили над безводным сульфатом натрия, фильтровали и концентрировали. Остаток очищали по методу колоночной хроматографии, применяя хлороформ/метанол (95/5, по объему) в качестве элюента, с получением очищенного соединения, указанного в заголовке (0,8 г. Выход: 95%).
1H ЯМР (CDCl3,, миллионные доли) δ : 8,3-7,2 (м, 11Н), 5,8 (м, 1Н), 4,5 (м, 1Н), 3,5-3,2 (м, 2Н), 2,45 (с, 3Н), 3,1-2,3 (м, 8Н).
Пример 50.
Получение [1-(4-амидразоно)бензил] -2-(4-метилсульфонилпиперазинил)-2-оксоэтил]амида (S)-нафталин-2-сульфоновой кислоты.
Соединение (0,4 г, 0,76 ммоль), полученное в Получении 14, растворяли в пиридине (5 мл) и полученный раствор помещали в колбу со штуцерами, в которую добавляли триэтиламин (0,3 мл). Реакционную колбу оборудовали таким образом, что газообразный сероводород (H2S) мог медленно вводиться через один штуцер колбы, а выводиться через другой штуцер. Реакционный раствор насыщали газообразным сероводородом при перемешивании в течение приблизительно 10 минут, причем бесцветный раствор приобретал зеленую окраску и затем постепенно - темно-коричневую. Колбу закрывали резиновой пробкой и оставляли стоять в течение 3 суток при комнатной температуре. Затем реакционный раствор перегоняли в условиях пониженного давления для удаления летучих веществ и сушили с помощью вакуумного насоса. К полученному желтому твердому веществу добавляли вместе ацетон (10 мл) и иодметан (0,5 мл) и данную смесь нагревают с обратным холодильником в течение 30 минут. Данную реакционную смесь вновь перегоняли в условиях пониженного давления для удаления летучих веществ и сушили с помощью вакуумного насоса. Остаток растворяли в абсолютном метаноле (5 мл) и затем перемешивали. К данному раствору порциями трижды с интервалом 10 минут добавляли 80% гидрат гидразина (0,06 мл). После того, как реакция была завершена, реакционный раствор концентрировали и затем очищали по методу ЖХВР с получением указанного в заголовке соединения (0,3 г. Выход: 65%).
1H ЯМР (CD3OD, миллионные доли) δ : 8,4-7,4 (м, 11Н), 4,6 (м, 1Н), 3,5-3,2 (м, 2Н), 2,55 (с, 3Н), 3,1-2,2 (м, 8Н).
Масс (FAB, m/e): 559 (M++1).
Пример 51.
Получение [1-(4-амидразоно)бензил-2-оксо-2-(4-этоксикарбонилпиперазинил)этил]амида (S)-нафталин-2-сульфоновой кислоты.
Использовали ту же методику, что и в Получении 13, за исключением того, что вместо 1-метансульфонилпиперазина применяли 1-пиперазинэтилкарбоксилат, и полученный продукт затем обрабатывали в соответствии с той же методикой, что и в Получении 14, с получением промежуточного продукта [1-(4-цианобензил)-2-оксо-2-(4-этоксикарбонилпиперазинил)этил] амида (S)-нафталин-2-сульфоновой кислоты (1 г, 1,9 ммоль). Данное промежуточное соединение обрабатывали в соответствии с той же методикой, что и в Примере 50, с получением очищенного соединения, указанного в заголовке (0,6 г. Выход: 56%).
1H ЯМР (CD3OD, миллионные доли) δ : 8,5-7,3 (м, 11Н), 4,55 (м, 1Н), 4,05 (м, 2Н), 3,2-2,4 (м, 10H), 1,2 (м, 3Н).
Масс (FAB, m/e): 553 (M++1).
Пример 52.
Получение [1-(4-амидразоно)бензил-2-(4-формилпиперазинил)-2-оксоэтил] амида (S)-нафталин-2-сульфоновой кислоты.
Использовали ту же методику, что и в Получении 13, за исключением того, что вместо 1-метансульфонилпиперазина применяли 1-пиперазинэтилкарбоксальдегид, и полученный продукт затем обрабатывали в соответствии с той же методикой, что и в Получении 14, с получением промежуточного продукта [1-(4-цианобензил)-2-(4-формилпиперазинил)-2-оксоэтил] амида (S)-нафталин-2-сульфоновой кислоты (7 г, 1,6 ммоль). Данное промежуточное соединение обрабатывали в соответствии с той же методикой, что и в Примере 50, с получением очищенного соединения, указанного в заголовке (0,5 г. Выход: 62%).
1H ЯМР (CD3OD, миллионные доли) δ : 8,36-7,44 (м, 11Н), 4,60 (м, 1Н), 3,46-2,80 (м, 9Н), 2,55 (м, 1Н).
Масс (FAB, m/e): 509 (M++1).
Пример 53.
Получение [1-(4-амидразоно)бензил-2-(4-этилпиперазинил)-2-оксоэтил]амида (S)-нафталин-2-сульфоновой кислоты.
Использовали ту же методику, что и в Получении 13, за исключением того, что вместо 1-метансульфонилпиперазина применяли 1-этилпиперазин, и полученный продукт затем обрабатывали в соответствии с той же методикой, что и в Получении 14, с получением промежуточного продукта [1-(4-цианобензил)-2-(4-этилпиперазинил)-2-оксоэтил]амида (S)-нафталин-2-сульфоновой кислоты (0,3 г, 0,6 ммоль). Данное промежуточное соединение обрабатывали в соответствии с той же методикой, что и в Примере 50, с получением очищенного соединения, указанного в заголовке (0,2 г. Выход: 56%).
1H ЯМР (CD3OD, миллионные доли) δ : 8,5-7,3 (м, 11Н), 4,6 (м, 1Н), 3,5-2,7 (м, 10Н), 2,2 (с, 2Н), 1,4-1,2 (м, 3Н).
Масс (FAB, m/e): 509 (M++1).
Пример 54.
Получение [1-(4-амидразоно)бензил-2-оксо-2-(4-фенилпиперазинил)этил] амида (S)-нафталин-2-сульфоновой кислоты.
Использовали ту же методику, что и в Получении 13, за исключением того, что вместо 1-метансульфонилпиперазина применяли 1-фенилпиперазин, и полученный продукт затем обрабатывали в соответствии с той же методикой, что и в Получении 14, с получением промежуточного продукта [1-(4-цианобензил)-2-оксо-2-(4-фенилпиперазинил)этил] амида (S)-нафталин-2-сульфоновой кислоты (0,5 г, 0,97 ммоль). Данное промежуточное соединение обрабатывали в соответствии с той же методикой, что и в Примере 50, с получением очищенного соединения, указанного в заголовке (0,3 г. Выход: 57%).
1H ЯМР (CD3OD, миллионные доли) δ: 8,35-7,54 (м, 11Н), 7,20 (м, 2Н), 6,86 (м, 1Н), 6,67 (м, 2Н), 4,56 (м, 1Н), 3,45 (м, 1Н), 3,25-2,92 (м, 5Н), 3,72 (м, 2Н), 2,42 (м, 1Н), 2,05 (м, 1Н).
Масс (FAB, m/e): 557 (M++1).
Пример 55.
Получение [1-(4-амидразоно)бензил-2-оксо-2-[4-(3-трифторметилфенил)пиперазинил]этил]амида (S)-нафталин-2-сульфоновой кислоты.
Использовали ту же методику, что и в Получении 13, за исключением того, что вместо 1-метансульфонилпиперазина применяли 1-(α,α,α--трифтор-м-толил)пиперазин, и полученный продукт затем обрабатывали в соответствии с той же методикой, что и в Получении 14, с получением промежуточного продукта [1-(4-цианобензил)-2-оксо-2-[4-(3-трифторметилфенил)пиперазинил)этил] амида (S)-нафталин-2-сульфоновой кислоты (0,5 г, 0,8 ммоль). Данное промежуточное соединение обрабатывали в соответствии с той же методикой, что и в Примере 50, с получением очищенного соединения, указанного в заголовке (0,3 г. Выход: 55%).
1H ЯМР (CD3OD, миллионные доли) δ : 8,35-6,7 (м, 15Н), 3,47 (м, 1Н), 3,3-3,0 (м, 5Н), 3,76 (м, 2Н), 2,45 (м, 1Н), 2,03 (м, 1Н).
Масс (FAB, m/e): 625 (M++1).
Пример 56.
Получение [2-(4-ацетилпиперазинил)-1-(4-амидразоно)бензил-2-оксоэтил]амида (S)-нафталин-2-сульфоновой кислоты.
Использовали ту же методику, что и в Получении 13, за исключением того, что вместо 1-метансульфонилпиперазина применяли 1-ацетилпиперазин, и полученный продукт затем обрабатывали в соответствии с той же методикой, что и в Получении 14, с получением промежуточного продукта [2-(4-ацетилпиперазинил)-1-(4-цианобензил)-2-оксоэтил] амида (S)-нафталин-2-сульфоновой кислоты (1,25 г, 2,55 ммоль). Данное промежуточное соединение обрабатывали в соответствии с той же методикой, что и в Примере 50, с получением очищенного соединения, указанного в заголовке (0,7 г. Выход: 53%).
1H ЯМР (CD3OD, миллионные доли) δ : 8,4-7,4 (м, 11Н), 4,5 (м, 1Н), 3,5-3,2 (м, 2Н), 3,1-2,2 (м, 8Н), 2,0 (с, 3Н).
Масс (FAB, m/e): 523 (M++1).
Пример 57.
Получение [1-(4-амидразоно)бензил-2-оксо-2-[4-(2-гидрокси)этилпиперазинил]этил]амида (S)-нафталин-2-сульфоновой кислоты.
Использовали ту же методику, что и в Получении 13, за исключением того, что вместо 1-метансульфонилпиперазина применяли 1-пиперазинэтан, и полученный продукт затем обрабатывали в соответствии с той же методикой, что и в Получении 14, с получением промежуточного продукта [1-(4-цианобензил)-2-оксо-2-[4-(2-гидроксиэтил)пиперазинил] этил]амида (S)-нафталин-2-сульфоновой кислоты (0,07 г, 0,14 ммоль). Данное промежуточное соединение обрабатывали в соответствии с той же методикой, что и в Примере 50, с получением очищенного соединения, указанного в заголовке (0,05 г. Выход: 68,5%).
1H ЯМР (CD3OD, миллионные доли) δ : 8,35-7,30 (м, 11Н), 4,60 (м, 1Н), 3,96-3,7 (м, 4Н), 3,70-2,80 (м, 10Н).
Масс (FAB, m/e): 525 (M++1).
Пример 58.
Получение [1-(4-амидразоно)бензил-2-оксо-2-[4-(2-этоксифенил)этилпиперазинил]этил]амида (S)-нафталин-2-сульфоновой кислоты.
Использовали ту же методику, что и в Получении 13, за исключением того, что вместо 1-метансульфонилпиперазина применяли 1-(2-этоксифенил)пиперазин, и полученный продукт затем обрабатывали в соответствии с той же методикой, что и в Получении 14, с получением промежуточного продукта [1-(4-цианобензил)-2-оксо-2-[4-(2-этоксифенил)пиперазинил] этил] амида (S)-нафталин-2-сульфоновой кислоты (0,4 г, 0,7 ммоль). Данное промежуточное соединение обрабатывали в соответствии с той же методикой, что и в Примере 50, с получением очищенного соединения, указанного в заголовке (0,26 г. Выход: 62%).
1H ЯМР (CD3OD, миллионные доли) δ : 8,36-6,54 (м, 15Н), 4,6 (м, 1Н), 4,01 (м, 2Н), 3,48 (м, 1Н), 3,28-3,08 (м, 5Н), 2,69 (м, 2Н), 2,38 (м, 1Н), 2,08 (м, 1Н), 1,35 (м, 3Н).
Масс (FAB, m/e): 601 (M++1).
Тест 1: ингибирующая активность по отношению к тромбину.
Способность соединения по настоящему изобретению подавлять активность тромбина определяли, как описано в дальнейшем.
В 1,5 мл кювету добавляли 1160 мкл 0,1М трис-буферного раствора (pH 7,8), содержащего 150 мМ NaCl и 0.1% ПЭГ 8000 (полиэтиленгликоль, молекулярная масса около 8000). Хромозим ТН растворяли в диметилсульфоксиде (ДМСО) в концентрации 10 мМ и полученный раствор разбавляли указанным трис-буферным раствором до концентрации 0,1 мМ, который затем применяли в качестве раствора субстрата. В кювету добавляли 225 мкл 0,1 мМ приготовленного таким образом раствора субстрата. Раствор ингибитора готовили путем растворения соединения-ингибитора тромбина по настоящему изобретению в диметилсульфоксиде в концентрации 10 мг/мл и последующего разбавления полученного раствора указанным трис-буферным раствором до концентрации 0,1 мг/мл, 0,01 мг/мл, 0,001 мг/мл и 0,0001 мг/мл. Полученный раствор ингибитора брали в количестве, соответствующем величине от 0 до 10 мкг ингибитора и затем разбавляли трис-буферным раствором до общего объема 100 мкл, который затем добавляли в кювету.
15 мкл раствора бычьего тромбина, растворенного в указанном выше трис-буферном растворе в концентрации 0,1 мг/мл, добавляли в кювету для инициации реакции ферментативного гидролиза. Количество пара-нитроанилина, образовавшегося в течение 2 минут с момента внесения фермента, отслеживали путем измерения поглощения на 381 нм. Был получен непрерывный спектр поглощения по времени реакции. Подобные эксперименты проводили с различными концентрациями ингибитора, получая при этом непрерывный спектр.
Для каждого спектра, исходя из значения угла наклона в первые 30 секунд реакционного времени, получали значение начальной скорости Vi и затем строили график зависимости обратного значения начальной скорости (1/Vi) от концентрации ингибитора. Исходя из графика, получали первичное уравнение, отвечающее точкам, изображенным на графике, и затем рассчитывали значение Ki, исходя из отрезка, отсекаемого на оси x по первичному уравнению, применяя уравнение ферментативной реакции. Значение Km, примененное для данного расчета, составляло 8,3 мкМ, что было получено путем изменения концентрации субстрата при постоянной концентрации фермента.
Значение константы скорости Ks получали, применяя тот же раствор в той же концентрации, что и в определении значения Ki, но с применением следующей методики эксперимента.
А именно, в 1,5 мл кювету добавляли 1160 мкл буферного раствора и туда добавляли 15 мкл раствора бычьего тромбина, имеющего концентрацию 0,1 мг/мл, и 100 мкл раствора ингибитора. Смесь выдерживали при комнатной температуре в течение 15 минут. Затем при добавлении 225 мкл 0,1 мМ раствора субстрата отслеживали изменение поглощения с течением времени в течение 2 минут. Из полученного непрерывного спектра определяли угол наклона линейного участка и представляли его в виде значения Vs. Подобный эксперимент проводили с различными концентрациями ингибитора, получая при этом значение Vs для каждой концентрации ингибитора, по которым строили график зависимости 1/Vs от концентрации ингибитора. Исходя из графика, получали первичное уравнение, отвечающее точкам, изображенным на графике, и затем рассчитывали значение Ki, исходя из отрезка, отсекаемого на оси x по первичному уравнению, применяя уравнение ферментативной реакции.
Независимо от этого, таким же образом, что и при определении ингибирующей активности по отношению к тромбину, как описано выше, определяли ингибирующую активность соединения по настоящему изобретению по отношению к трипсину.
В качестве субстрата применяли 20 мкМ раствор N-бензоил-Val-Gly-Arg-п-нитроанилид, а ингибитор применяли в различных концентрациях в интервале от 0 до 120 мкг. В дополнение к этому, в 0,1 н. HCl растворяли трипсин, доводили до концентрации 45 мкг/мл указанным буферным раствором непосредственно перед экспериментом и затем применяли в количестве 40 мкл. Как и в эксперименте с тромбином, общий объем реакционного раствора составлял 1,5 мл и остальные процедуры проводили таким же образом. Значение Km, применявшееся в расчете значения Ki, определяли в соответствии с той же методикой, что и в эксперименте, описанном выше, и оно составило 20,2 мкМ.
Ингибирующую активность соединения по настоящему изобретению по отношению к активности любого фермента, которую определяли в соответствии с указанной выше методикой, представляли в виде значений Ki и Ks, а избирательность по отношению к тромбину была представлена значением отношения ингибирующая активность по отношению к трипсину/ингибирующая активность по отношению к тромбину. Полученный таким образом результат описан в табл. 1.
Тест 2: фармакокинетический тест.
Методика проведения теста.
Самцов крыс и собак подвергали голоданию в течение 24 часов и использовали в качестве экспериментальных животных. Готовили 1% раствор (10 мг/мл) соединения из Примера 1, применяя физиологический раствор и затем вводили его экспериментальным животным внутривенно и перорально. Через заданные промежутки времени у животных забирали образцы крови и немедленно смешивали их с метанолом и сульфатом цинка. В итоге, верхний слой смеси подвергали количественному анализу в ультрафиолетовой области при длине волны 231 нм по методу ЖХВР для измерения концентрации лекарственного средства в крови.
Результаты теста.
Концентрация лекарственного средства и фармакокинетические параметры соединения из Примера 1 после внутривенного и перорального введения приведены в табл. 2 - 7. Когда соединение из Примера 1 вводили внутривенно, оно быстро распределялось и затем долго исчезало как в случае крыс, так и в случае собак, тогда как время полувыведения соединения у собак было вдвое или более выше, чем у крыс. Более того, время полувыведения соединения из Примера 1 было вдвое или более выше, чем таковое продажного аргатробана (40 минут) у человека (Osamu et al. Pharmacology and Therapy, том 14, прилож. 5, 1986). Между тем, также может быть определено, что биологическая доступность соединения из Примера 1 составляла 15% для крыс и 61% для собак при пероральном введении. Однако сообщалось, что аргатробан не всасывается в организм животных и человека, будучи введен перорально.
Как видно из приведенных выше результатов, соединение из Примера 1 проявляло лучшие фармакокинетические характеристики, чем аргатробан, с позиций всасывания при пероральном применении и времени полувыведения.
Хотя данное изобретение описано в его предпочтительной форме с определенной степенью конкретности, специалисты в данной области принимают во внимание тот факт, что настоящее описание предпочтительной формы приведено лишь в качестве примера и что можно прибегать к многочисленным изменениям в деталях построения, сочетания и расположения частей без отступления от сущности и сферы изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ 2-АМИНОТИАЗОЛКАРБОКСАМИДА | 1999 |
|
RU2193034C2 |
| ПРОИЗВОДНЫЕ ИМИДАЗОЛА, ОБЛАДАЮЩИЕ ИНГИБИРУЮЩЕЙ АКТИВНОСТЬЮ В ОТНОШЕНИИ ФАРНЕЗИЛТРАНСФЕРАЗЫ, СПОСОБ ИХ ПОЛУЧЕНИЯ, ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1998 |
|
RU2179975C1 |
| АКТИВАТОРЫ ГЛЮКОКИНАЗЫ И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ, СОДЕРЖАЩИЕ ИХ В КАЧЕСТВЕ АКТИВНОГО ИНГРЕДИЕНТА | 2008 |
|
RU2450001C2 |
| НОВЫЙ СПОСОБ ПОЛУЧЕНИЯ ИОПРОМИДА | 2009 |
|
RU2451667C1 |
| ПРОИЗВОДНЫЕ ХИНАЗОЛИНА ДЛЯ ТОРМОЖЕНИЯ РОСТА РАКОВЫХ КЛЕТОК И СПОСОБ ИХ ПОЛУЧЕНИЯ | 2005 |
|
RU2362773C2 |
| АГОНИСТ СФИНГОЗИН-1-ФОСФАТНЫХ РЕЦЕПТОРОВ, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СОДЕРЖАЩАЯ ЕГО В КАЧЕСТВЕ АКТИВНОГО ИНГРЕДИЕНТА | 2020 |
|
RU2798605C1 |
| НОВЫЕ АЦИКЛИЧЕСКИЕ НУКЛЕОЗИДФОСФОНАТНЫЕ ПРОИЗВОДНЫЕ, ИХ СОЛИ И СПОСОБ ПОЛУЧЕНИЯ ЭТИХ СОЕДИНЕНИЙ | 2002 |
|
RU2266294C2 |
| ПРОИЗВОДНЫЕ ХИНОЛИН- ИЛИ НАФТИРИДИНКАРБОНОВОЙ КИСЛОТЫ И СПОСОБЫ ИХ ПОЛУЧЕНИЯ | 1995 |
|
RU2120940C1 |
| 2-ЦИАНОПИРИМИДИН-4-ИЛКАРБАМАТ, ИЛИ ПРОИЗВОДНОЕ МОЧЕВИНЫ, ИЛИ ЕГО СОЛЬ И СОДЕРЖАЩАЯ ИХ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2019 |
|
RU2788740C2 |
| ПРИМЕНЕНИЕ ТАЛИПОРФИНА ИЛИ ЕГО ПРОИЗВОДНЫХ В ЛЕЧЕНИИ ЗАБОЛЕВАНИЙ СЕРДЦА, А ТАКЖЕ ИХ ПОЛУЧЕНИЕ | 2001 |
|
RU2350326C2 |
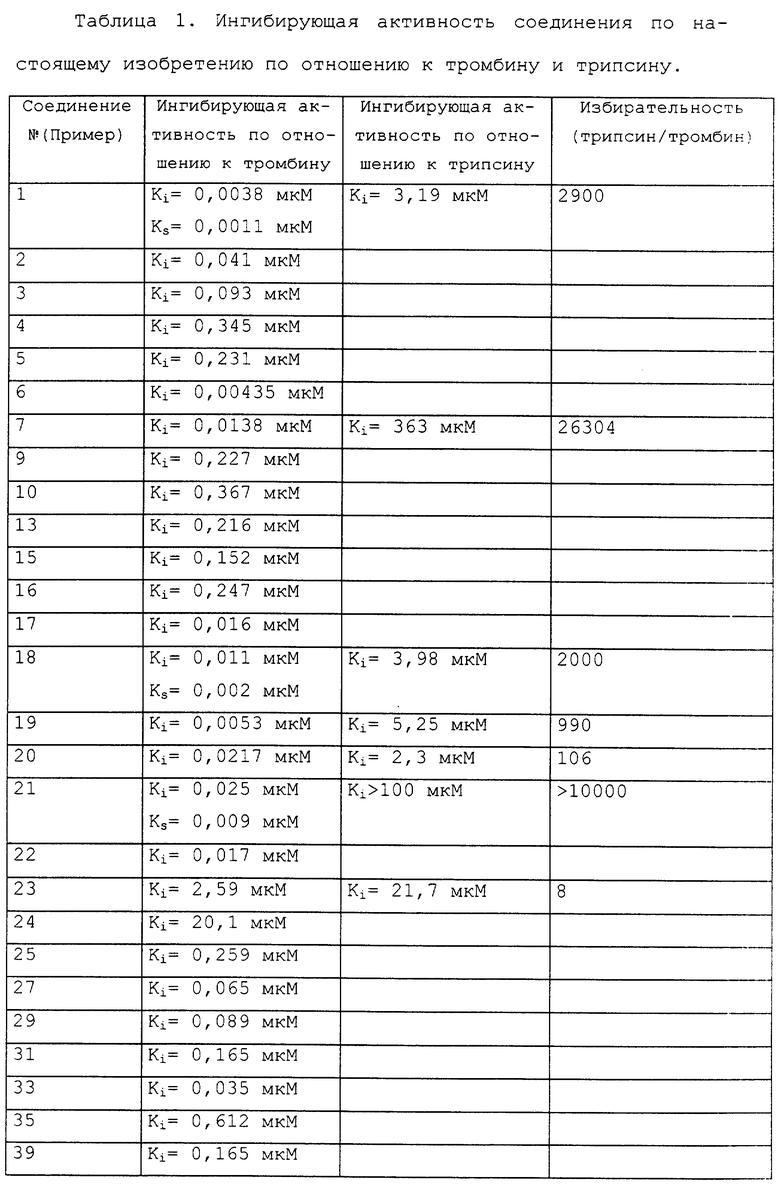




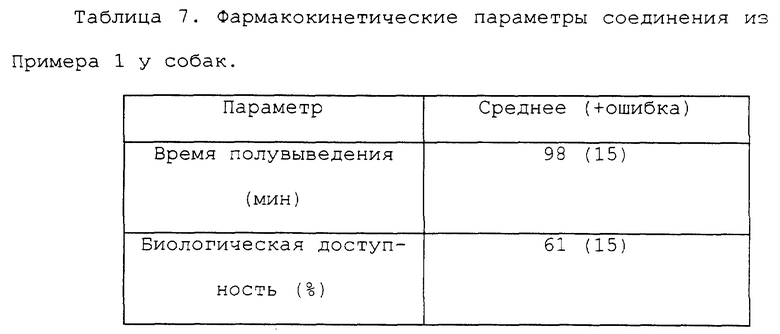
Описываются новые избирательно действующие ингибиторы тромбина, имеющие следующую формулу I
которые также эффективны при пероральном применении. Значения R1, R6, R7, X указаны в п.1 формулы изобретения. Описывается также фармацевтическая композиция на основе соединений формулы II для подавления активности тромбина. 2 с. и 2 з.п.ф-лы, 7 табл.
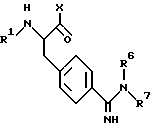
и их изомеры,
где R1 представляет собой ацетил, замещенный нафтилом или нафтилокси, или сульфонил, замещенный нафтилом, который, в свою очередь, может быть замещен низшей алкокси группой, диалкиламиногруппой; или замещенный фенилом, который, в свою очередь, замещен низшим алкилом, низшей алкоксигруппой;
X представляет собой группу формулы  или
или 
R2 и R3 независимо друг от друга представляют собой водород; C3 - C6-циклоалкил, который может быть замещен карбоксилом; гидрокси; низший алкил, незамещенный или замещенный метоксикарбонилом, гидрокси, карбоксилом; или R2 и R3 вместе с атомом азота, к которому они присоединены, могут образовывать пиперидиновую группу, замещенную карбоксилом;
R4 представляет собой водород;
R5 представляет собой метанолсульфонил, формил, алкоксикарбонил, фенил, незамещенный или замещенный трифторметилом или алкоксигруппой,
R6 представляет собой водород;
R7 представляет собой низший алкил, амино; при условии, что когда R1 - нафтил, X не может быть группой 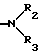
в которой R2 - водород, R3 - циклоалкил, низший алкил. или
или 
R2 и R3 независимо друг от друга представляют собой C3-6 циклоалкил, который может быть замещен карбоксилом; низший алкил, замещенный или незамещенный карбоксилом, метоксикарбонилом или гидрокси; или гидрокси, или R2 и R3 вместе с атомом азота, к которому они присоединены, могут образовывать пиперидиновую группу, замещенную карбоксилом, R4 представляет собой водород, R5 представляет собой метансульфонил, этоксикарбонил, формил, или фенил, который может быть замещен трифторметилом или этокси, R6 представляет собой водород и R7 представляет собой метил или амино.
| Экономайзер | 0 |
|
SU94A1 |
| Способ получения фармацевтически приемлемых солей производных бензамидина | 1984 |
|
SU1319784A3 |
| EP 0502536 A1, 09.09.92 | |||
| Устройство для закрепления деталей при копировальной обработке | 1975 |
|
SU560730A1 |

