Область изобретения
Данное изобретение относится к синтетическому хинолоновому антибактериальному средству, которое эффективно в качестве лекарственных средств, ветеринарных препаратов, лекарств, применяемых в рыболовстве, или в качестве антибактериальных консервантов.
Уровень техники
С момента открытия норфлоксацина синтетические хинолоновые антибактериальные агенты были усовершенствованы с точки зрения антибактериальной активности и фармакокинетических свойств и на данный момент большое количество соединений используются в клинической области в качестве хемотерапевтических средств, эффективных при большинстве системных инфекционных заболеваний.
В последние годы в клинической практике наблюдается увеличение числа поколений бактерий, имеющих низкую чувствительность к синтетическим хинолоновым антибактериальным средствам. Например, как и в случае с Staphylococcus aureus (MRSA) и Streptococcus pneumococcus (PRSP), которые нечувствительны к β-лактамным антибиотикам, и Enterococcus (VRE), который нечувствителен к аминогликозидным антибактериальным средствам, увеличивается число случаев, когда грамположительные бактерии, изначально устойчивые к лекарственным препаратам, отличным от синтетических хинолоновых антибактериальных средств, также становятся низкочувствительными к синтетическим хинолоновым антибактериальным средствам. Следовательно, в клинической практике существует потребность в синтетических хинолоновых антибактериальных средствах, имеющих более высокую эффективность.
Что касается побочных эффектов синтетических хинолоновых антибактериальных средств, кроме эффекта возбуждения центральной нервной системы, который до настоящего момента представлял собой трудность, также известно о появлении судорог в результате совместного применения с нестероидными противовоспалительными средствами, фототоксичности и так далее, и, таким образом, также существует потребность в разработке более безопасных синтетических хинолоновых антибактериальных средств.
Известно, что структура заместителей в положениях 7 и 1 (или положениях, соответствующих этим положениям; это же утверждение используется в данном описании и далее) в большой степени влияет на антибактериальную активность, фармакокинетические свойства и безопасность синтетических хинолоновых антибактериальных средств.
Известно, что хинолоновые производные, имеющие в качестве заместителя в положении 7 исходного хинолонового скелета пирролидиниловую группу с аминометильной группой в положении 3, обладают сильной антибактерильной активностью в отношении грамотрицательных и грамположительных бактерий. В качестве примера можно привести производные 7-[3-(1-аминометил)пирролидин-1-ил]хинолонкарбоновой кислоты [Journal of Medicinal Chemistry, том 29, стр.445 (1986)].
Более того, известные примеры производных хинолонкарбоновой кислоты, имеющих заместитель на углеродном атоме аминометильной группы 3-(1-аминометил)пирролидин-1-ильной группы, включают производные 7-[3-(1-аминоэтил)пирролидин-1-ил]хинолонкарбоновой кислоты [Journal of Medicinal Chemistry, том 36, стр.871 (1993)]; производные 7-[3-(1-амино-1-метилэтил)пирролидин-1-ил]хинолонкарбоновой кислоты [Journal of Medicinal Chemistry, том 37, стр.733 (1994)]; и производные 7-[3-(1-аминоалкил)пирролидин-1-ил]хинолонкарбоновой кислоты [Chemical и Pharmaceutical Bulletin, том 42, стр.1442 (1994)] и так далее.
Однако несмотря на то что указанные выше хинолоновые производные, имеющие в качестве заместителя группу 3-(аминометил)пирролидин-1-ил, группу 3-(1-аминоэтил)пирролидин-1-ил или группу, имеющую подобную структуру, являются соединениями, которые обладают сильной антибактериальной активностью, было обнаружено, что в результате низкоселективной токсичности [см., например, Journal of Antimicrobial Chemotherapy, том 33, стр.685 (1994)], такие соединения действуют не только на бактерии, но и на клетки эукариотического организма и их сложно применять в качестве лекарственных средств или лекарственных препаратов в ветеринарии. Следовательно, хинолоновые соединения, имеющие такие заместители, до сих пор не использовались в существующей клинической практике.
В то же время, производные хинолонкарбоновой кислоты, имеющие в качестве заместителя группу 3-(1-аминоциклоалкил)пирролидин-1-ил и являющиеся релевантными настоящему изобретению, были подробно описаны в PCT/JP96/00208, в которой приведено описание соединений со структурой, показанной формулой А или формулой В. То есть в соединениях хинолона формулы A, заместитель (X1) в положении 6 определен как атом галогена или атом водорода. Однако в указанной выше патентной заявке конкретно описаны только хинолонкарбоновые кислоты, в которых заместителем в положении 6 является атом фтора или другой атом галогена. Следовательно, в PCT/JP96/00208 не представлено конкретного описания, касающегося хинолонкарбоновых кислот, в которых заместителем в положении 6 является водород. Кроме того, в данной публикации не представлено какого-либо конкретного описания в качестве воплощения 3-(1-аминоциклоалкил)пирролидинил-замещенных-6-водород-замещенных-хинолонкарбоновых кислот, к которым относится настоящее изобретение.

А
[В указанной выше формуле A X1 представляет атом галогена или атом водорода, и X2 представляет атом галогена. (Определения заместителей в соединении, показанном формулой A, представляют собой определения, которые даны в PCT/JP96/00208, и не относятся к определениям заместителей по настоящему изобретению, даже если используются одинаковые обозначения.)]
В указанной выше формуле A R2 представлен формулой B:
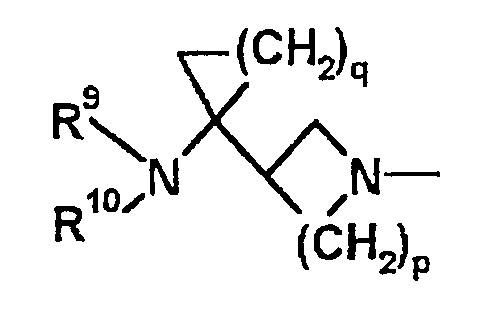
В
[В указанной выше формуле B p равно целому числу 1-3, q равно целому числу 1-3, R9 представляет атом водорода или алкильную группу, содержащую 1-6 атомов углерода, R10 представляет атом водорода, алкильную группу, содержащую 1-6 атомов углерода, алкильную группу, содержащую 1-6 атомов углерода, которая имеет гидроксильную группу, или алкильную группу, содержащую 1-6 атомов углерода, которая имеет атом галогена. (Определения заместителей в соединении, показанном формулой В, представляют собой определения, которые даны в PCT/JP96/00208, и не относятся к определениям заместителей по настоящему изобретению, даже если используются одинаковые обозначения.)]
Кроме указанного выше, примером литературного источника, в котором указано производное хинолонкарбоновой кислоты, содержащее 3-(1-аминоциклоалкил)пирролидин-1-ильную группу и являющееся релевантным настоящему изобретению, является Chemical and Pharmaceutical Bulletin, том 42, стр.1442 (1994). Однако в данном источнике нет каких-либо описаний, относящихся к 3-(1-аминоциклоалкил)пирролидинил-замещенным-6-водород-замещенным-хинолонкарбоновым кислотам, которые представляют собой соединения по настоящему изобретению.
Кроме того, например, в PCT/WO99/14214 раскрыто производное 6-водород-замещенной-хинолонкарбоновой кислоты, в котором азотсодержащий гетероциклический заместитель, например, 3-(1-аминоэтил)пирролидин-1-ильная группа введена через связь углерод-азот в 7-положение хинолонового скелета, и которое является релевантным данному изобретению. В указанной выше заявке описываются соединения, представленные формулами С и D. Однако в ней нет какого-либо описания, касающегося 3-(1-аминоциклоалкил)пирролидин-1-ильной группы, которая является релевантной настоящему изобретению, в качестве заместителя в положении 7 хинолонового скелета, показанного формулой С. Кроме того, в указанной заявке нет какого-либо описания, касающегося 3-(1-аминоциклоалкил)пирролидинил-замещенных-6-водород-замещенных-хинолонкарбоновых кислот, которые являются релевантными настоящему изобретению и содержат в качестве заместителя указанную выше группу.
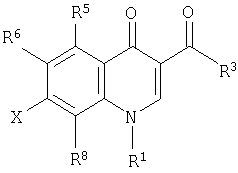
С
[В указанной выше формуле C R1 представляет циклическую алкильную группу, содержащую 3-6 атомов углерода, алкильную группу, содержащую 1 или 2 атома углерода, алкенильную группу с прямой цепью, содержащую 2-3 атома углерода, или алкильную или алкенильную группу с разветвленной цепью, содержащую 3-4 атома углерода, причем данная алкильная группа или циклическая алкильная группа может быть незамещенной, или алкильная группа или циклическая алкильная группа может быть замещена 1-3 атомами фтора или фенильной группой, которая является незамещенной или замещенной 1-3 атомами фтора или замещенной в положении 4 одной гидроксильной группой, R6 представляет атом водорода, гидроксильную группу, аминокарбонильную группу, атом брома, цианогруппу, алкильную группу, содержащую 1 или 2 атома углерода, или алкенильную группу или алкинильную группу, содержащую 2-4 атома углерода, и данная алкильная группа может быть незамещенной или данная алкильная группа может быть замещена метильной группой или этильной группой, которая является незамещенной или замещена 1-3 атомами фтора или одной гидроксильной группой или аминогруппой. (Определения заместителей в соединении, показанном в формуле С, представляют собой определения, которые даны в PCT/WO99/14214, и не относятся к определениям заместителей по настоящему изобретению, даже если используются одинаковые обозначения.)]
В указанной выше формуле X представлен формулой D:

D
[В указанной выше формуле D, R7 представляет аминогруппу, которая присоединена к углероду, рядом с которым нет атома азота пирролидинового кольца, и может быть незамещенной или замещена одной или двумя алкильными группами, содержащими 1-3 атома углерода, или аминоалкильную группу, которая присоединена к углероду на пирролидиновом кольце, и может быть незамещенной или замещена алкильной группой, содержащей 1-3 атома углерода, R9 представляет группу, выбранную из группы, содержащей атом водорода, алкильную группу, содержащую 1-4 атома углерода, алкенильную группу и алкинильную группу, содержащую 2-6 атомов углерода, и конденсированную и спироалкильную группу, содержащую 3-6 атомов углерода, алкильные части данных групп могут быть незамещенными или замещены 1-3 атомами фтора, и указанные выше заместители R7 и R9 могут быть объединены с пирролидиновым кольцом с образованием кольцевой структуры по типу конденсированной или спироструктуры, где данная конденсированная или спироциклическая часть образована из 2-5 атомов углерода и 0 или 1 атомом азота. (Определения заместителей в соединении, показанном формулой D, представляют собой определения, которые даны в PCT/WO99/14214, и не относятся к определениям заместителей по настоящему изобретению, даже если используются одинаковые обозначения.)]
Другие примеры литературных источников, в которых раскрыты производные 6-водород-замещенной-хинолонкарбоновой кислоты, которые являются релевантными настоящему изобретению, включают в себя Journal of Medicinal Chemistry, том 39, стр.4952 (1996). Однако даже в этих источниках нет какого-либо описания, касающегося 3-(1-аминоциклоалкил)пирролидинил-замещенных-6-водород-замещенных-хинолонкарбоновых кислот, которые представляют собой соединения по настоящему изобретению.
Описание изобретения
Были проведены тщательные исследования с целью получения хинолоновых соединений, которые обладают превосходной антибактериальной активностью, высокой эффективностью, а также высокой безопасностью. В результате было обнаружено, что производные 3-(1-аминоциклоалкил)пирролидинил-замещенной-6-дегалоген(водород-замещенной)хинолонкарбоновой кислоты, представленные формулой (I), показанной ниже, их соли и их гидраты обладают сильной антибактериальной активностью в отношении широкого спектра грамотрицательных и грамположительных бактерий и, в частности, обладают сильной антибактериальной активностью в отношении бактерий, устойчивых к лекарственным препаратам, таких как грамположительные кокки, включая MRSA, PRSP и VRE.
Более того, было обнаружено, что, кроме такой превосходной антибактериальной активности, соединения по данному изобретению обладают как прекрасными показателями безопасности, так и фармакокинетическими свойствами и, таким образом, могут использоваться в клинических случаях, в которых подобные эффекты не могут быть достигнуты при применении соединений, предшествующих данному изобретению, содержащих заместители той же структуры в положении 7 исходного хинолонового скелета. Настоящее изобретение было выполнено на основании данных заключений.
Сравнение производных 6-водород-замещенной-хинолонкарбоновой кислоты, представленных формулой (I), их солей и их гидратов по настоящему изобретению и хинолоновых соединений, в которых водород в положении 6 соединения по данному изобретению замещен атомом фтора, показывает, что оба типа соединений обладают превосходной антибактериальной активностью в отношении широкого спектра грамотрицательных и грамположительных бактерий, включая бактерии, устойчивые к лекарственным препаратам. Однако неожиданно было обнаружено, что производные 6-водород-замещенного-хинолона, которые представляют собой соединения данного изобретения, являются соединениями, которые по сравнению с производными 6-фтор-замещенного-хинолона, являются соединениями чрезвычайно безопасными, острая токсичность которых снижена и значительно понижена в отношении микронуклеарной индукции, и также демонстрируют хорошие фармакокинетические свойства, как, например, улучшенное выведение с мочой и так далее.
Таким образом, было обнаружено, что даже хинолоновое соединение, которое содержит 3-(1-аминоциклоалкил)пирролидин-1-ильную группу, содержащую в качестве заместителя метильной группы 3-(аминометил)пирролидин-1-ильной группы циклическую алкильную группу и которая, как было указано выше, известна своей низкой по селективности токсичностью, неожиданно будет представлять собой соединение с превосходной селективной токсичностью и соединение с прекрасными фармакокинетическими свойствами, если оно является хинолоновым соединением со структурой по настоящему изобретению.
То есть настоящее изобретение относится к соединениям, представленным следующей общей формулой (I), их солям и их гидратам:
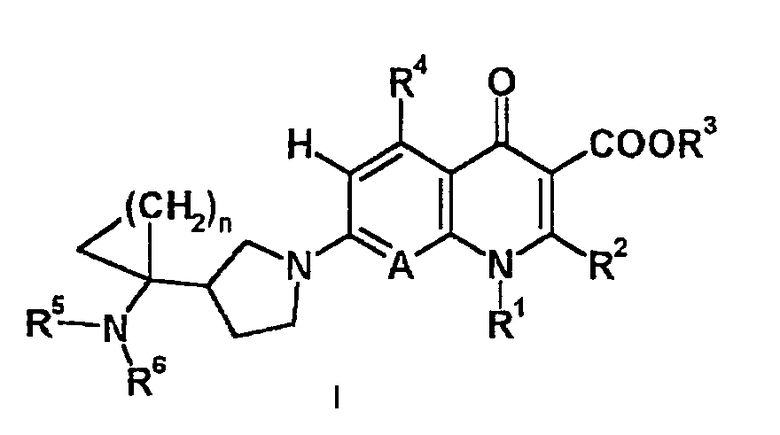
[где R1 представляет алкильную группу, содержащую 1-6 атомов углерода, алкенильную группу, содержащую 2-6 атомов углерода, галогеноалкильную группу, содержащую 1-6 атомов углерода, циклическую алкильную группу, содержащую 3-6 атомов углерода, которая может содержать заместитель, арильную группу, которая может содержать заместитель, гетероарильную группу, которая может содержать заместитель, алкоксигруппу, содержащую 1-6 атомов углерода, или алкиламиногруппу, содержащую 1-6 атомов углерода;
R2 представляет алкилтиогруппу, содержащую 1-6 атомов углерода, или атом водорода,
где R2 и указанный выше R1 могут быть объединены с образованием кольцевой структуры путем включения части исходного скелета, и образованное таким образом кольцо может содержать в качестве атома, образующего кольцо, атом серы, и кольцо может быть замещено алкильной группой, содержащей 1-6 атомов углерода, которая может иметь заместитель;
R3 представляет фенилалкильную группу, состоящую из алкиленовой группы, содержащей 1-6 атомов углерода, и фенильной группы, алкильную группу, содержащую 1-6 атомов углерода, алкоксиметильную группу, содержащую 2-7 атомов углерода, атом водорода, фенильную группу, ацетоксиметильную группу, пивалоилоксиметильную группу, этоксикарбонильную группу, холиновую группу, диметиламиноэтильную группу, 5-инданильную группу, фталидинильную группу, 5-алкил-2-оксо-1,3-диоксол-4-илметильную группу или 3-ацетокси-2-оксобутильную группу;
R4 представляет алкильную группу, содержащую 1-6 атомов углерода, алкенильную группу, содержащую 2-6 атомов углерода, алкинильную группу, содержащую 2-6 атомов углерода, алкоксигруппу, содержащую 1-6 атомов углерода, атом водорода, аминогруппу, гидроксильную группу, тиольную группу или галогенметильную группу, и
в указанном выше, аминогруппа может содержать один или более заместителей, выбранных из группы, включающей алкильную группу, содержащую 1-6 атомов углерода, ацильную группу, содержащую 2-5 атомов углерода, и формильную группу;
A представляет атом азота или часть структуры, представленную формулой (II):
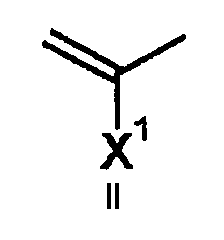
(где X1 представляет алкильную группу, содержащую 1-6 атомов углерода, алкенильную группу, содержащую 2-6 атомов углерода, алкинильную группу, содержащую 2-6 атомов углерода, алкоксигруппу, содержащую 1-6 атомов углерода, атом водорода, аминогруппу, атом галогена, цианогруппу, галогенметильную группу или галогенметоксигруппу,
в указанном выше, аминогруппа может содержать один или более заместителей, выбранных из группы, включающей алкильную группу, содержащую 1-6 атомов углерода, ацильную группу, содержащую 2-5 атомов углерода, и формильную группу,
где X1 и указанный выше R1 могут быть объединены с образованием кольцевой структуры путем включения части исходного скелета, полученное таким образом кольцо может содержать в качестве атома, образующего кольцо, атом кислорода, атом азота или атом серы, и данное кольцо может быть замещено алкильной группой, содержащей 1-6 атомов углерода, которая может содержать заместитель);
каждый R5 и R6 независимо представляет алкильную группу, содержащую 1-6 атомов углерода, атом водорода или замещенную карбоксильную группу, полученную из аминокислоты, дипептида или трипептида,
где алкильная группа может содержать один или несколько заместителей, выбранных из группы, включающей алкилтиогруппу, содержащую 1-6 атомов углерода, алкоксигруппу, содержащую 1-6 атомов углерода, гидроксильную группу и атом галогена; и
n равно целому числу 1 или 2].
Настоящее изобретение также относится к каждому нижеследующему:
к соединению указанной выше формулы (I), его солям или их гидратам, где соединение формулы (I) представляет собой стереохимически чистое соединение;
к соединению формулы (I), его солям или их гидратам, где n в формуле (I) равно 1;
к соединению формулы (I), его солям или их гидратам, где R3 в формуле (I) представляет атом водорода;
к соединению формулы (I), его солям или их гидратам, где R2 в формуле (I) представляет атом водорода;
к соединению формулы (I), его солям или их гидратам, где R4 в формуле (I) представляет атом водорода;
к соединению формулы (I), его солям или их гидратам, где A в формуле (I) представляет собой часть структуры, представленную формулой (II);
к соединению формулы (I), его солям или гидратам, где X1 в формуле (II) представляет собой метоксигруппу, метильную группу, дифторметоксигруппу, атом фтора или атом хлора;
к соединению формулы (I), его солям или их гидратам, где X1 в формуле (II) представляет собой метоксигруппу или метильную группу;
к соединению формулы (I), его солям или их гидратам, где каждый R5 и R6 в формуле (I) представляет атом водорода;
к соединению формулы (I), его солям или их гидратам, где один из R5 или R6 в формуле (I) представляет атом водорода, а другой представляет метильную группу;
к соединению формулы (I), его солям или их гидратам, где один из R5 или R6 в формуле (I) атом водорода, а другой представляет замещенную карбоксильную группу, полученную из аминокислоты, дипептида или трипептида;
к соединению формулы (I), его солям или их гидратам, где каждый R5 и R6 в формуле (I) представляет комбинацию атома водорода и метильной группы;
к соединению формулы (I), его солям или их гидратам, где R6 в формуле (I) представляет замещенную карбоксильную группу, полученную из аминокислоты, дипептида или трипептида;
к соединению формулы (I), его солям или их гидратам, где циклическая алкильная группа, включающая 3-6 атомов углерода, которая может содержать заместитель, в R1 представляет галогенциклопропильную группу;
к соединению формулы (I), его солям или их гидратам, где галогенциклопропильная группа представляет собой 1,2-цис-2-галогенциклопропильную группу;
к соединению формулы (I), его солям или их гидратам, где галогенциклопропильная группа представляет собой стереохимически чистый заместитель;
к соединению формулы (I), его солям или их гидратам, где галогенциклопропильная группа представляет собой (1R,2S)-2-галогенциклопропильную группу;
к соединению формулы (I), его солям или их гидратам, где атом галогена галогенциклопропильной группы представляет собой атом фтора;
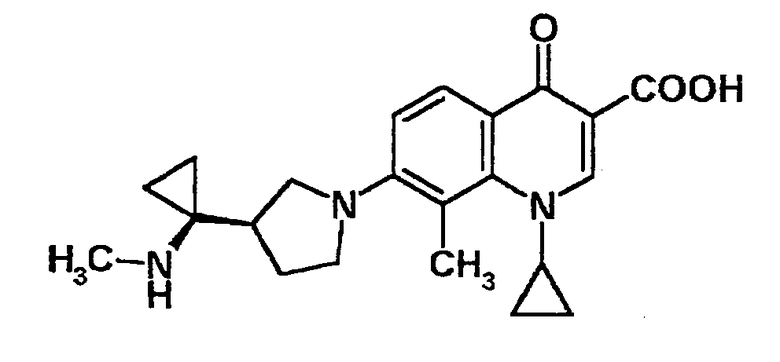
к 7-[3-(R)-(1-аминоциклопропил)пирролидин-1-ил]-1-[2-(S)-фтор-1-(R)-циклопропил]-1,4-дигидро-4-оксо-1,8-нафтиридин-3-карбоновой кислоте, ее солям или их гидратам;
к 7-[3-(R)-(1-аминоциклопропил)пирролидин-1-ил]-1-[2-(S)-фтор-1-(R)-циклопропил]-1,4-дигидро-4-оксохинолин-3-карбоновой кислоте, ее солям или их гидратам;
к 7-[3-(R)-(1-аминоциклопропил)пирролидин-1-ил]-1-[2-(S)-фтор-1-(R)-циклопропил]-8-хлор-1,4-дигидро-4-оксохинолин-3-карбоновой кислоте, ее солям или их гидратам;
к 7-[3-(R)-(1-аминоциклопропил)пирролидин-1-ил]-1-[2-(S)-фтор-1-(R)-циклопропил]-8-фтор-1,4-дигидро-4-оксохинолин-3-карбоновой кислоте, ее солям или их гидратам;
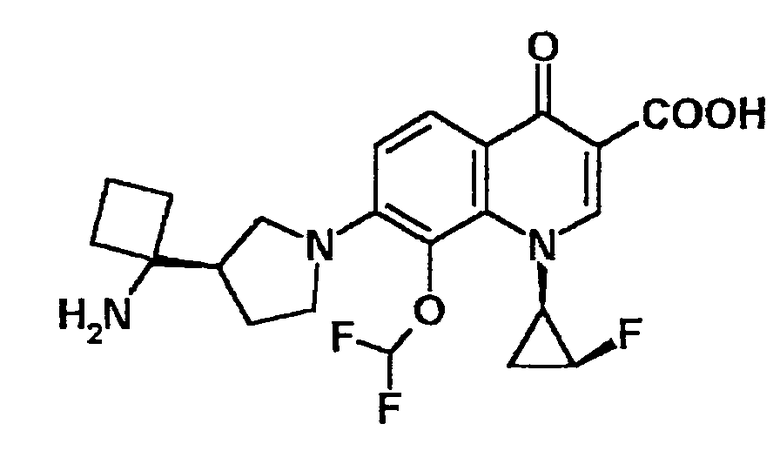
к 7-[3-(R)-(1-аминоциклопропил)пирролидин-1-ил]-1-[2-(S)-фтор-1-(R)-циклопропил]-8-дифторметокси-1,4-дигидро-4-оксохинолин-3-карбоновой кислоте, ее солям или их гидратам;
к 7-[3-(R)-(1-аминоциклопропил)пирролидин-1-ил]-1-[2-(S)-фтор-1-(R)-циклопропил]-1,4-дигидро-8-метокси-4-оксохинолин-3-карбоновой кислоте, ее солям или их гидратам;
к 7-[3-(R)-[1-(метиламино)циклопропил]пирролидин-1-ил]-1-[2-(S)-фтор-1-(R)-циклопропил]-1,4-дигидро-8-метокси-4-оксохинолин-3-карбоновой кислоте, ее солям или их гидратам;
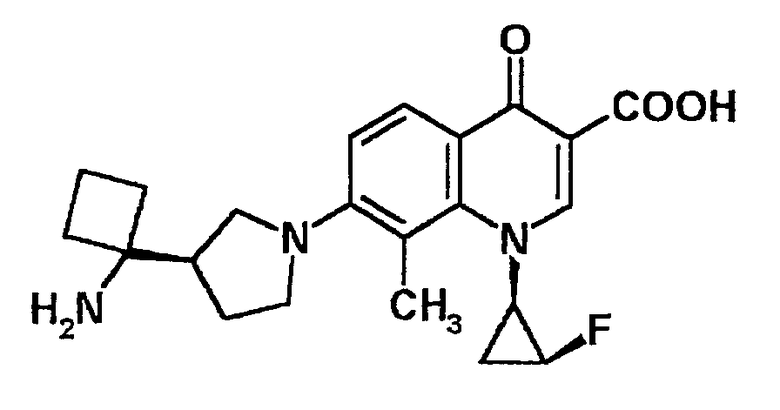
к 7-[3-(R)-(1-аминоциклопропил)пирролидин-1-ил]-1-[2-(S)-фтор-1-(R)-циклопропил]-1,4-дигидро-8-метил-4-оксохинолин-3-карбоновой кислоте, ее солям или их гидратам;
к 7-[3-(R)-[1-(метиламино)циклопропил]пирролидин-1-ил]-1-[2-(S)-фтор-1-(R)-циклопропил]-1,4-дигидро-8-метил-4-оксохинолин-3-карбоновой кислоте, ее солям или их гидратам;
к 7-[3-(R)-[1-(этиламино)циклопропил]пирролидин-1-ил]-1-[2-(S)-фтор-1-(R)-циклопропил]-1,4-дигидро-8-метил-4-оксохинолин-3-карбоновой кислоте, ее солям или их гидратам;
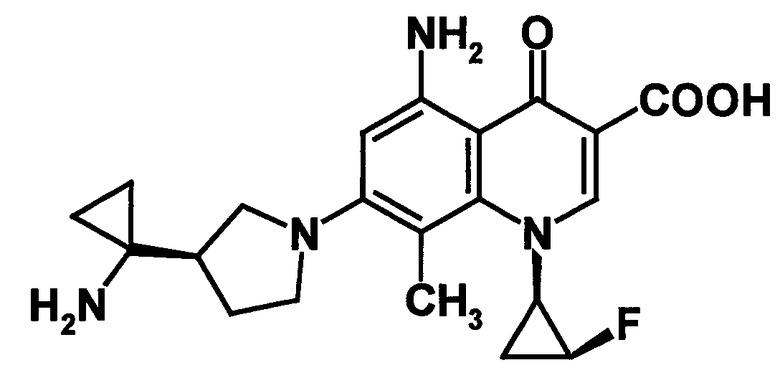
к 5-амино-7-[3-(R)-(1-аминоциклопропил)пирролидин-1-ил]-1-[2-(S)-фтор-1-(R)-циклопропил]-1,4-дигидро-8-фтор-4-оксохинолин-3-карбоновой кислоте, ее солям или их гидратам;
к 5-амино-7-[3-(R)-(1-аминоциклопропил)пирролидин-1-ил]-1-[2-(S)-фтор-1-(R)-циклопропил]-1,4-дигидро-8-метокси-4-оксохинолин-3-карбоновой кислоте, ее солям или их гидратам;
к 5-амино-7-[3-(R)-(1-аминоциклопропил)пирролидин-1-ил]-1-[2-(S)-фтор-1-(R)-циклопропил]-1,4-дигидро-8-метил-4-оксохинолин-3-карбоновой кислоте, ее солям или их гидратам;
к 5-амино-7-[3-(R)-[1-(метиламино)циклопропил]пирролидин-1-ил]-1-[2-(S)-фтор-1-(R)-циклопропил]-1,4-дигидро-8-метокси-4-оксохинолин-3-карбоновой кислоте, ее солям или их гидратам;
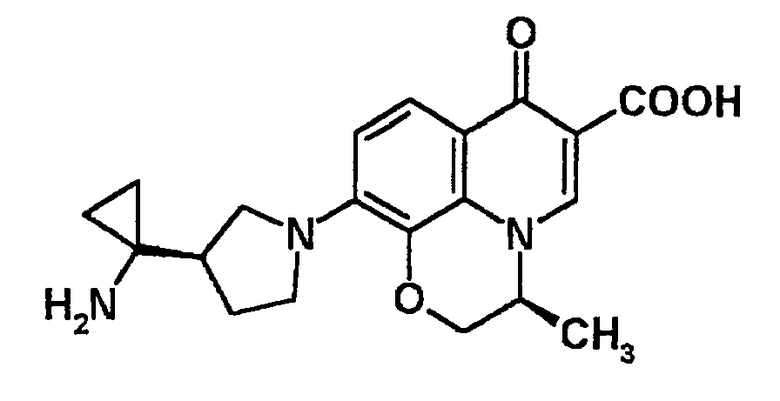
к 10-[3-(R)-(1-аминоциклопропил)пирролидин-1-ил]-2,3-дигидро-3-(S)-метил-7-оксо-7H-пиридо[1,2,3-de][1,4]бензоксазин-6-карбоновой кислоте, ее солям или их гидратам;
к 1-(циклопропил)-8-метил-7-[3-(R)-[1-(метиламино)циклопропил]пирролидин-1-ил]-1,4-дигидро-4-оксохинолин-3-карбоновой кислоте, ее солям или их гидратам;
к лекарственным средствам, которые в качестве активного компонента содержат соединение формулы (I), его соли или их гидраты;
к антибактериальному средству, которое в качестве активного компонента содержит соединение формулы (I), его соли или их гидраты;
к терапевтическому средству для лечения инфекционного заболевания, которое в качестве активного компонента содержит соединение формулы (I), его соли или их гидраты;
к способу лечения заболевания, который включает введение в качестве активного компонента соединения формулы (I), его солей или их гидратов;
к способу лечения инфекционного заболевания, который включает введение в качестве активного компонента соединения формулы (I), его солей или их гидратов;
к способу получения лекарственного средства, который включает получение фармацевтической композиции, в состав которой в качестве активного компонента входит соединение формулы (I), его соли или их гидраты;
к способу получения антибактериального средства, который включает получение композиции, в состав которой в качестве активного компонента входит соединение формулы (I), его соли или их гидраты;
к способу получения средства для лечения инфекционного заболевания, который включает получение композиции, в состав которой в качестве активного компонента входит соединение формулы (I), его соли или их гидраты;
к применению соединения формулы (I), его солей или их гидратов для получения лекарственного средства;
к применению соединения формулы (I), его солей или их гидратов для получения антибактериального средства;
к применению соединения формулы (I), его солей или их гидратов для получения средства для лечения инфекционного заболевания; и так далее.
(Способ осуществления изобретения)
Далее будут описаны различные заместители соединения данного изобретения, представленного формулой (I):
(R1, R2, R3, R4, R5, R6, n и A такие, как определено выше) теперь должны быть описаны. (Для положения 6 хинолонового исходного скелета или эквивалентного положения в структурных формулах, представленных в настоящем описании, для того чтобы подчеркнуть, что присоединен атом водорода, причем данный атом водорода, присоединенный к углероду, который обычно не указывается при обычном написании структурных формул в органической химии, указан в некоторых случаях (как «-H»). Однако структурные формулы данного описания указаны в соответствии с правилами изображения структурных формул, которые обычно используются в области органической химии, и атом водорода, присоединенный к атому углерода, не всегда будет указан и обычно будет опущен).
Заместитель R1 представляет алкильную группу, содержащую 1-6 атомов углерода, алкенильную группу, содержащую 2-6 атомов углерода, галогеналкильную группу, содержащую 1-6 атомов углерода, циклическую алкильную группу, содержащую 3-6 атомов углерода, которая может содержать заместитель, арильную группу, которая может содержать заместитель, гетероарильную группу, которая может содержать заместитель, алкоксигруппу, содержащую 1-6 атомов углерода, или алкиламиногруппу, содержащую 1-6 атомов углерода.
Где алкильная группа, содержащая 1-6 атомов углерода, может быть алкильной группой с прямой или разветвленной цепью, предпочтительно, алкильной группой, содержащей 1-4 атома углерода, более предпочтительно, этильной группой. В качестве алкенильной группы, содержащей 2-6 атомов углерода, винильная группа или 1-изопропенильная группа является предпочтительной. В качестве галогеналкильной группы, содержащей 1-6 атомов углерода, предпочтительной является 2-фторэтильная группа. В качестве циклической алкильной группы, особенно предпочтительной является циклопропильная группа. Циклическая алкильная группа может содержать заместитель, и в качестве заместителя предпочтительным является атом галогена. В качестве циклической алкильной группы предпочтительной является галогенциклопропильная группа, которая может содержать заместитель, и в качестве атома галогена в данной группе особенно предпочтительным является атом фтора. В качестве галогенциклопропильной группы предпочтительной является моногалогенциклопропильная группа и еще более предпочтительной является цис-замещенная группа.
Примеры арильной группы, которая может содержать заместитель, включают фенильную группу и так далее, которая может содержать 1-3 заместителя, выбранных из группы, состоящей из алкильной группы, содержащей 1-6 атомов углерода, алкоксигруппы, содержащей 1-6 атомов углерода, атома галогена, такого как атом фтора, атом хлора и атом брома, гидроксильной группы, аминогруппы, нитрогруппы и так далее. (В случае, когда арильная группа имеет несколько заместителей заместители могут быть одного типа или могут быть разных типов). Более конкретно, фенильная группа, 2-фторфенильная группа, 4-фторфенильная группа, 2,4-дифторфенильная группа, 2-фтор-4-гидроксифенильная группа, 3-амино-4,6-дифторфенильная группа и 4,6-дифтор-3-метиламинофенильная группа являются предпочтительными. Что касается арильной группы, к ней относится группа, полученная из ароматического углеводородного соединения. Кроме фенильной группы, арильная группа также может быть нафтильной группой или трициклической арильной группой, содержащей больше колец.
Гетероарильная группа представляет собой группу, полученную из пентациклического или гексациклического ароматического гетероциклического соединения, содержащего один или более гетероатомов, выбранных из атомов азота, атома кислорода и атома серы. Особенно предпочтительным является пентациклический или гексациклический азотсодержащий гетероциклический заместитель, содержащий 1 или 2 атома азота. Например, пиридильная группа, пиримидильная группа и так далее являются желательными. В качестве заместителей таких колец предпочтительными являются алкильная группа, атом галогена и так далее. Особенно предпочтительной является 6-амино-3,5-дифтор-2-пиридильная группа.
В качестве алкоксигруппы, содержащей 1-6 атомов углерода, предпочтительной является алкоксигруппа, полученная из указанной выше алкильной группы, и среди них предпочтительной является метоксигруппа. Что касается алкиламиногруппы, содержащей 1-6 атомов углерода, алкильная часть может быть указанной выше алкильной группой. В качестве алкиламиногруппы предпочтительной является метиламиногруппа.
В качестве заместителя R1 предпочтительной является циклическая алкильная группа или галогенциклолкильная группа. Из них предпочтительной является циклопропильная группа или 2-галогенциклопропильная группа. В качестве атома галогена во 2-й группе, предпочтительным является атом фтора.
Заместитель R2 представляет алкилтиогруппу, содержащую 1-6 атомов углерода, или атом водорода, R1 и R2 могут быть объединены с образованием кольцевой структуры, состоящей из полиметиленовой цепи, путем включения части исходного скелета (то есть так, чтобы содержать атом азота, к которому присоединен R1, и атом углерода, к которому присоединен R2). Образованное таким образом кольцо может содержать в качестве атома, образующего кольцо, атом серы, и это кольцо может также содержать алкильную группу или галогеналкильную группу, содержащую 1-6 атомов углерода, в качестве заместителя. Образованное кольцо по размеру может быть тетрациклическим-гексациклическим, и данное кольцо также может быть насыщенным или ненасыщенным. В качестве заместителя на образованном кольце предпочтительной является метильная или фторметильная группа. Примеры конденсированной кольцевой структуры, полученной данным способом, включают следующие структуры:
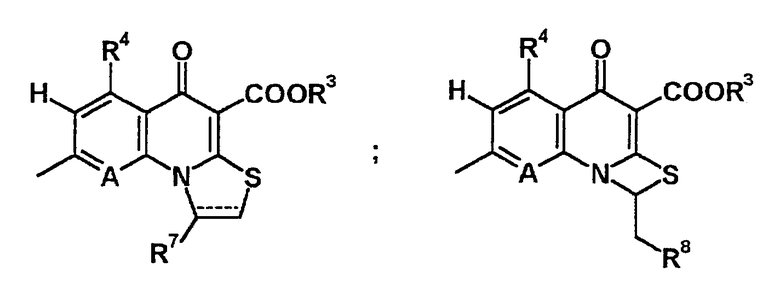
(В указанной выше формуле, R7 представляет алкильную группу, содержащую 1-6 атомов углерода, такую как метильная группа, галогеналкильная группа, содержащая 1-6 атомов углерода, такую как фторметильная группа, или атом водорода, и R8 представляет атом галогена, такой как атом фтора, или атом водорода).
В качестве заместителя R2 соединения формулы (I) предпочтительным является атом водорода.
Заместитель R3 представляет фенилалкильную группу (аралкильную группу), состоящую из алкиленовой группы, содержащей 1-6 атомов углерода, и фенильной группы, или алкильную группу, содержащую 1-6 атомов углерода, алкоксиметильную группу, содержащую 2-7 атомов углерода, атом водорода, фенильную группу, ацетоксиметильную группу, пивалоилоксиметильную группу, этоксикарбонильную группу, холиновую группу, диметиламиноэтильную группу, 5-инданильную группу, фталидинильную группу, 5-алкил-2-оксо-1,3-диоксол-4-илметильную группу или 3-ацетокси-2-оксобутильную группу.
В случае, когда соединение данного изобретения используется для антибактериальных целей, предпочтительно использовать соединение карбоновой кислоты, где R3 представляет атом водорода. Между тем, хинолоновое производное, в котором группа карбоновой кислоты образует сложный эфир, эффективно в качестве промежуточного соединения синтеза или в качестве пролекарства. Данные аспекты будут далее описаны более подробно.
R4 представляет алкильную группу, содержащую 1-6 атомов углерода, алкенильную группу, содержащую 2-6 атомов углерода, алкинильную группу, содержащую 2-6 атомов углерода, алкоксигруппу, содержащую 1-6 атомов углерода, атом водорода, аминогруппу, гидроксильную группу, тиольную группу или галогенметильную группу, и среди перечисленных выше аминогруппа может содержать один или более заместителей, выбранных из группы, содержащей алкильную группу, содержащую 1-6 атомов углерода, ацильную группу, содержащую 2-5 атомов углерода, и формильную группу. В случае, когда присутствует несколько замещающих групп, заместители могут быть все одного типа или могут быть множеством различных типов.
В качестве алкильной группы, которая может быть группой либо с прямой, либо с разветвленной цепью, содержащей 1-6 атомов углерода, предпочтительной является метильная группа, этильная группа, нормальная пропильная группа или изопропильная группа. В качестве алкенильной группы, которая может быть группой либо с прямой, либо с разветвленной цепью, содержащей 2-6 атомов углерода, предпочтительной является винильная группа. В качестве алкинильной группы, которая может быть группой либо с прямой, либо с разветвленной цепью, содержащей 2-6 атомов углерода, предпочтительной является этинильная группа. В качестве галогена галогенметильной группы особенно предпочтительным является атом фтора, и его количество может составлять 1-3. В качестве алкоксигруппы, которая может содержать 1-6 атомов углерода, предпочтительной является метоксигруппа.
Заместитель R4 предпочтительно представляет атом водорода, алкильную группу или аминогруппу, и среди них особенно предпочтительной является атом водорода, метильная группа или незамещенная аминогруппа (-NH2).
В случае, когда R4 представляет аминогруппу, гидроксильную группу или тиольную группу, она может быть защищена защитной группой, которая обычно используется в соответствующих областях.
Примеры таких защитных групп включают (замещенные) алкоксикарбонильные группы, такие как трет-бутоксикарбонильная группа, 2,2,2-трихлорэтоксикарбонильная группа и так далее; (замещенные) аралкилоксикарбонильные группы, такие как бензилоксикарбонильная группа, параметоксибензилоксикарбонильная группа, паранитробензилоксикарбонильная группа и так далее; (замещенные) ацильные группы, такие как ацетильная группа, метоксиацетильная группа, трифторацетильная группа, хлорацетильная группа, пивалоильная группа, формильная группа, бензоильная группа и так далее; (замещенные) алкильные группы или (замещенные) аралкильные группы, такие как трет-бутильная группа, бензильная группа, паранитробензильная группа, параметоксибензильная группа, трифенилметильная группа и так далее; (замещенные) простые эфиры, такие как метоксиметильная группа, трет-бутоксиметильная группа, тетрагидропиранильная группа, 2,2,2-трихлорэтоксиметильная группа и так далее; и (алкил и/или аралкил) замещенные силильные группы, такие как триметилсилильная группа, изопропилдиметилсилильная группа, трет-бутилдиметилсилильная группа, трибензилсилильная группа, трет-бутилдифенилсилильная группа и так далее. (Где "(замещенный)" означает, что группа может содержать заместитель). В качестве промежуточного продукта особенно предпочтительным является соединение, содержащее аминогруппу, гидроксильную группу или тиольную группу, защищенную таким заместителем.
A представляет атом азота или частичную структуру, выраженную формулой (II):
В случае, когда A представляет часть структуры формулы (II), X1 представляет алкильную группу, содержащую 1-6 атомов углерода, алкенильную группу, содержащую 2-6 атомов углерода, алкинильную группу, содержащую 2-6 атомов углерода, алкоксигруппу, содержащую 1-6 атомов углерода, атом водорода, аминогруппу, атом галогена, цианогруппу, галогенметильную группу или галогенметокси группу, и среди указанных выше групп аминогруппа может содержать один или более заместителей, выбранных из группы, содержащей алкильную группу, содержащую 1-6 атомов углерода, ацильную группу, содержащую 2-5 атомов углерода, и формильную группу.
В качестве атома галогена предпочтительными являются атом фтора, атом хлора и атом брома, и особенно предпочтительными являются атом фтора и атом хлора. В качестве алкильной группы, которая может быть группой с прямой или разветвленной цепью, содержащей 1-6 атомов углерода, предпочтительной является метильная группа, этильная группа, нормальная пропильная группа или изопропильная группа. В качестве алкенильной группы, которая может быть группой с прямой или разветвленной цепью, содержащей 2-6 атомов углерода, предпочтительной является винильная группа. В качестве алкинильной группы, которая может быть с прямой или разветвленной цепью, содержащей 2-6 атомов углерода, предпочтительной является этинильная группа. В качестве галогена галогенметильной группы особенно предпочтительным является атом фтора, и его количество может составлять 1-3. В качестве алкоксигруппы, которая может содержать 1-6 атомов углерода, предпочтительной является метоксигруппа. В качестве галогена галогенметоксигруппы, особенно предпочтительным является атом фтора, и его количество может составлять 1-3.
Среди таких заместителей предпочтительной является алкильная группа или алкоксигруппа. Особенно предпочтительной является метильная группа, этильная группа, метоксигруппа или дифторметоксигруппа.
Кроме того, данный X1 и указанный выше R1 могут быть объединены с образованием кольцевой структуры, включающей полиметиленовое кольцо, путем включения части исходного скелета (так, чтобы содержать атом углерода, к которому присоединен X1, и атом азота, к которому присоединен R1). Образованное таким образом кольцо в качестве атома, образующего кольцо, может содержать атом кислорода, атом азота или атом серы, и данное кольцо может также содержать в качестве заместителя алкильную группу, содержащую 1-6 атомов углерода, которая, в свою очередь, может иметь заместитель.
По размеру образованное кольцо может быть пентациклическим-гептациклическим кольцом, и атомы, образующие кольцо, не ограничиваются атомом углерода и могут включать атом кислорода, атом азота или атом серы. Кроме того, данное кольцо может быть насыщенным или ненасыщенным. Образованное таким образом кольцо может содержать в качестве заместителя алкильную группу, содержащую 1-6 атомов углерода. Данная алкильная группа может рассматриваться как группа, аналогичная описанной выше алкильной группе, и предпочтительно является метильной группой. Данная алкильная группа может быть замещена атомом галогена, алкоксигруппой и так далее.
В качестве частичной структуры, которая образует структуру кольца, построенную с помощью X1 и R1, структура следующей формулы:
-O-CH2-CH(-CH3)-
является предпочтительной (правый конец в указанной выше формуле присоединен к атому азота), и особенно предпочтительным является хинолоновый скелет следующей структуры:
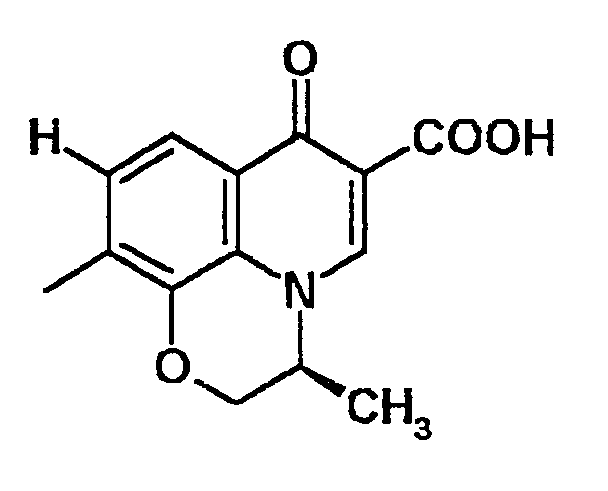
Если A представляет частичную структуры формулы (II), предпочтительными комбинациями R4 и X1 являются те, в которых R4 представляет алкильную группу, содержащую 1-6 атомов углерода, аминогруппу, атом водорода или гидроксильную группу, и X1 представляет алкильную группу, содержащую 1-6 атомов углерода, алкоксигруппу, содержащую 1-6 атомов углерода, галогенметоксигруппу или атом водорода.
Более предпочтительными комбинациями являются те, в которых R4 представляет аминогруппу, атом водорода, гидроксильную группу или метильную группу, и X1 представляет метильную группу, метоксигруппу, дифторметоксигруппу или атом водорода.
Особенно предпочтительными комбинациями являются те, в которых R4 представляет атом водорода, гидроксильную группу или метильную группу, и X1 представляет метильную группу или метоксигруппу.
Каждый из заместителей R5 и R6 независимо представляет алкильную группу, содержащую 1-6 атомов углерода, атом водорода или замещенную карбоксильную группу, полученную из аминокислоты, дипептида или трипептида.
Данная алкильная группа может содержать один или более заместителей, выбранных из группы, включающей алкилтиогруппу, содержащую 1-6 атомов углерода, алкоксигруппу, содержащую 1-6 атомов углерода, гидроксильную группу и атом галогена.
В качестве алкильной группы, которая может быть группой либо с прямой, либо с разветвленной цепью, содержащей 1-6 атомов углерода, предпочтительной является метильная группа, этильная группа, нормальная пропильная группа или изопропильная группа.
В случае, когда алкильная группа в качестве заместителя содержит гидроксильную группу, алкильная группа может быть группой с прямой или разветвленной цепью, содержащей 1-6 атомов углерода, и, более предпочтительно, гидроксильная группа является заместителем на концевом атоме углерода алкильной группы. В качестве алкильной группы, содержащей гидроксильную группу, предпочтительными являются те группы, которые содержат до 3 атомов углерода, и предпочтительными являются гидроксиметильная группа, 2-гидроксиэтильная группа, 2-гидроксипропильная группа, 3-гидроксипропильная группа и так далее.
В случае, когда алкильная группа в качестве заместителя содержит атом галогена, алкильная группа может быть группой с прямой или разветвленной цепью, содержащей 1-6 атомов углерода, и атом галогена предпочтительно является атомом фтора. Число атомов фтора может соответствовать состоянию от моно-замещенного до перфтор-замещенного. Примеры включают монофторметильную группу, дифторметильную группу, трифторметильную группу, 2,2,2-трифторэтильную группу и так далее.
В случае, когда алкильная группа в качестве заместителя содержит алкилтиогруппу, алкильная группа может быть группой с прямой или разветвленной цепью, содержащей 1-6 атомов углерода, и алкилтиогруппа может также быть группой с прямой цепью или с разветвленной цепью, содержащей 1-6 атомов углерода. В качестве алкильной группы, содержащей алкилтиогруппу, предпочтительной является алкилтиометильная группа, алкилтиоэтильная группа или алкилтиопропильная группа, и более предпочтительно, чтобы алкилтиогруппа также представляла собой группу, содержащую 1-3 атома углерода. Более предпочтительные примеры включают метилтиометильную группу, этилтиоэтильную группу и метилтиоэтильную группу.
В случае, когда алкильная группа содержит в качестве заместителя алкоксигруппу, алкильная группа может быть группой с прямой или разветвленной цепью, содержащей 1-6 атомов углерода, и алкоксигруппа может также быть группой с прямой или разветвленной цепью, содержащей 1-6 атомов углерода. В качестве алкильной группы, содержащей алкоксигруппу, предпочтительной является алкоксиметильная группа, алкоксиэтильная группа или алкоксипропильная группа, и более предпочтительно, чтобы алкоксигруппа также представляла собой группу, содержащую до 3 атомов углерода. Более предпочтительные примеры включают метоксиметильную группу, этоксиметильную группу и метоксиэтильную группу.
Предпочтительными комбинациями R5 и R6 являются те, в которых один представляет атом водорода, а другой представляет атом водорода, алкильную группу или замещенную карбоксильную группу, полученную из аминокислоты, дипептида или трипептида. Среди них предпочтительной является комбинация, в которой один из R5 или R6 представляет атом водорода, а другой представляет атом водорода или алкильную группу. В качестве алкильной группы, предпочтительной является метильная группа или этильная группа, и особенно предпочтительной является метильная группа. Таким образом, особенно предпочтительной является комбинация, в который оба R5 и R6 представляют атомы водорода, или комбинация, в которой один из R5 или R6 представляет атом водорода, а другой представляет метильную группу. Соединение такой комбинации может наибольшим образом проявлять предпочтительную физиологическую активность в качестве антибактериального средства.
В качестве пролекарства особенно эффективным является хинолоновое производное, в котором один из заместителей R5 или R6 представляет атом водорода, а другой представляет замещенную карбоксильную группу, полученную из аминокислоты, дипептида или трипептида. Конкретные примеры, относящиеся к данному производному, будут описаны ниже.
Далее приводится описание, касающееся галогенциклопропильной группы R1.
Атом галогена как заместитель предпочтительно представляет собой атом фтора или атом хлора, и особенно предпочтительным является атом фтора.
Для стереохимического окружения данной группы особенно предпочтительно, чтобы атом галогена и группа хинолонкарбоновой кислоты в циклопропановом кольце имели цис-конфигурацию. Также, несмотря на то что заместитель цис-конфигурации в данном случае может принимать форму 2-(S)-галоген-1-(R)-циклопропильной группы или 2-(R)-галоген-1-(S)-циклопропильной группы, первая из двух указанных является предпочтительной.
Было обнаружено, что несмотря на присутствие так называемых энантиоморфных изомеров, обусловленных группой цис-2-галогенциклопропила отдельно от R1, все такие изомеры обладают сильной антибактериальной активностью и высокой безопасностью.
Соединение данного изобретения обладает превосходными характеристиками в тех случаях, когда заместитель структуры, представленной следующей формулой E, находится в положении 7 хинолонового исходного скелета, в частности скелета 1-[2-(S)-фтор-1-(R)-циклопропил]-1,4-дигидро-4-оксохинолин-3-карбоновой кислоты, содержащей 2-(S)-галоген-1-(R)-циклопропильную группу.
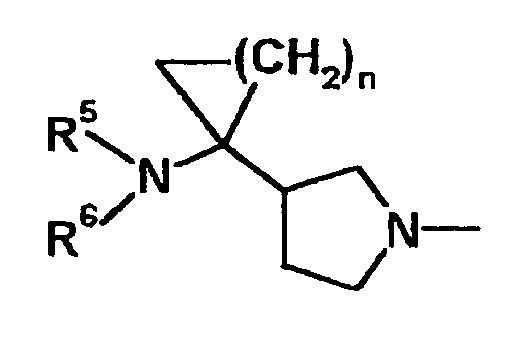
Е
Для данного заместителя, в результате присутствия асимметричного атома углерода в положении 3 пирролидинового кольца, существует два оптических изомера, которые находятся в энантиоморфном взаимоотношении связи. Чтобы быть более точными, они приведены далее:

F
Между тем, корреляция структурных активностей двух типов оптически активных соединений, которые обусловлены стерической конфигурацией заместителя в положении 7 (или его эквивалентного положения) производного 7-[3-(1-аминометил)пирролидин-1-ил]хинолонкарбоновой кислоты и корреляция структурных активностей четырех типов оптически активных веществ, которые обусловлены стерической конфигурацией заместителя в положении 7 производного 7-[3-(1-аминоэтил)пирролидин-1-ил]хинолонкарбоновой кислоты, описаны в Journal of Medicinal Chemistry, том 36, стр.1442 (1994). В данном литературном источнике указывается на то, что среди таких оптических изомеров наибольшей антибактериальной активностью обладают изомеры структур, показанных в следующей формуле.
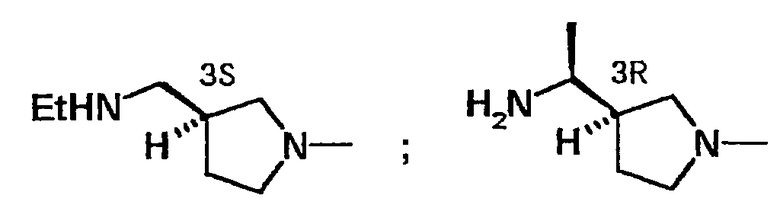
Исходя из стерических конфигураций в положении 3 данных пирролидиновых колец, было предположено, что из двух оптических изомеров, показанных в указанной выше формуле F, более предпочтительным является изомер, показанный ниже:
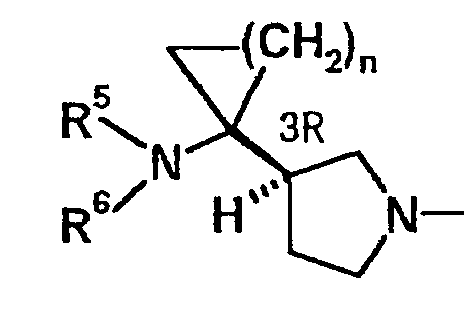
Таким образом, более предпочтительное соединение из соединений по данному изобретению имеет структуру, представленную следующей формулой:
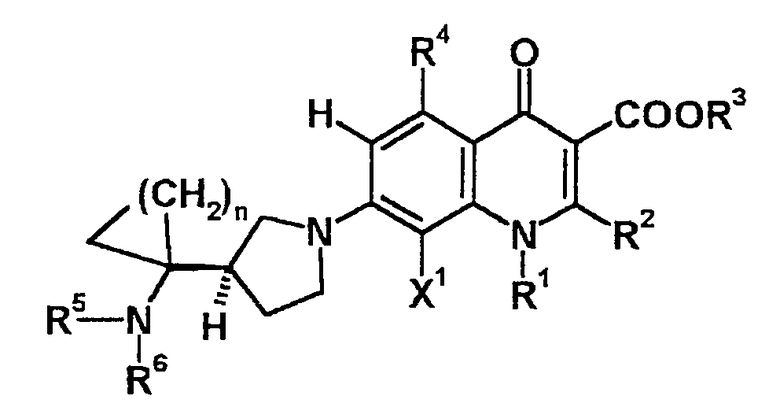
Следовательно, 3-(1-аминоциклоалкил)пирролидинил замещенные-6-водород-замещенные-хинолонкарбоновые кислоты, представленные формулой (I), их соли и их гидраты (особенно соединения, имеющие структуру, в которой положение 3 указанного выше пирролидинового кольца является R-конфигурацией, их соли и их гидраты) обладают сильной антибактериальной активностью в отношении широкого спектра грамотрицательных и грамположительных бактерий, и, в частности, характерной особенностью таких соединений данного изобретения является то, что они обладают сильной антибактериальной активностью в отношении устойчивых к антибиотикам бактерий, таких как грамположительные кокки, включая MRSA, PRSP и VRE. Кроме того, характерными особенностями соединений данного изобретения является то, что они обладают прекрасной безопасностью и хорошими фармакокинетическими свойствами, что делает возможным использование данных соединений в клинических случаях, в которых подобные эффекты не могли быть достигнуты при применении соединений, предшествующих данному изобретению, даже если они содержат заместители с той же структурой.
Такими превосходными свойствами соединений данного изобретения обладают соединения, в которых n в описанном выше заместителе равно целому числу 1 или 2, и превосходные результаты были особенно у соединений, в которых n равно целому числу 1. Следовательно, предпочтительными соединениями являются соединения, в которых циклическая часть представляет собой трициклическое кольцо.
В случае когда соединение формулы (I) данного изобретения имеет структуру, в которой присутствуют диастереомеры, при введении человеку или животным предпочтительно вводить соединение, содержащее один диастереомер. Используемый в данном описании термин «содержащее один диастереомер» обозначает не только случай, когда он полностью свободен от другого диастереомера, но и случай, когда он присутствует в химически чистом виде. Таким образом, говоря другими словами, другой диастереомер может присутствовать в таком количестве, в котором он не оказывает влияния на физические константы и физиологическую активность соединения.
Также, используемый в данном описании термин, «стереохимически чистый» обозначает, что в случае, когда соединение или тому подобное находится в различных изомерных формах, обусловленных присутствием асимметричных атомов углерода, соединение содержит только одну из них. Термин «чистый» в данном случае также может рассматриваться как описанное выше.
Тем не менее, производное хинолонкарбоновой кислоты по данному изобретению может использоваться как в его свободной форме, так и как кислотно-аддитивная соль или соль ее карбоксильной группы. Примеры кислотно-аддитивных солей включают соли неорганических кислот, такие как гидрохлориды, сульфаты, нитраты, гидробромиды, гидройодиды, фосфаты и так далее; и соли органических кислот, такие как метансульфонаты, бензолсульфонаты, толуолсульфонаты (и другие сульфонаты), ацетаты, цитраты, малеаты, фумараты, лактаты (и другие карбоксилаты) и так далее.
Примеры солей карбоксильной группы включают соли щелочных металлов, такие как соли лития, соли натрия, соли калия и так далее; соли щелочно-земельных металлов, такие как соли магния, соли кальция и так далее; соли аммония, соли триэтиламина, соли N-метилглюкамина, соли трис-(гидроксиметил)аминометана; и так далее, и они могут быть либо неорганическими солями, либо органическими солями.
Также, свободная форма, кислотно-аддитивные соли и соли карбоксильной группы производного хинолонкарбоновой кислоты могут присутствовать в виде гидратов.
Тем не менее, в случае, когда соединение по данному изобретению используется в антибактериальных целях, предпочтительно использовать соединение карбоновой кислоты, в котором заместитель R3 представляет атом водорода; в качестве промежуточного продукта синтеза или пролекарства эффективным является производное хинолона, в котором группа карбоновой кислоты представляет собой сложный эфир. Например, в качестве промежуточных продуктов синтеза эффективными являются алкиловые сложные эфиры, бензиловые сложные эфиры, алкоксиалкиловые сложные эфиры, фенилалкиловые сложные эфиры и фениловые сложные эфиры.
Также, сложный эфир, используемый в качестве пролекарства, представляет собой сложный эфир, который легко гидролизуется в живом организме и образует свободную форму карбоновой кислоты, и его примеры включают оксоалкиловые сложные эфиры, такие как ацетоксиметиловый сложный эфир, пивалоилоксиметиловый сложный эфир, этоксикарбониловый сложный эфир, холиновый сложный эфир, диметиламиноэтиловый сложный эфир, 5-инданиловый сложный эфир, фталидиниловый сложный эфир, 5-алкил-2-оксо-1,3-диоксол-4-илметиловый сложный эфир и 3-ацетокси-2-оксобутиловый сложный эфир.
Кроме того, хинолоновое производное, в котором один из заместителей R5 и R6 представляет атом водорода, а другой представляет замещенную карбоксильную группу, полученную из аминокислоты, дипептида или трипептида, эффективно в качестве пролекарства.
Аминокислота, дипептид или трипептид, используемые для получения такого пролекарства, представляют собой соединения, в которых пептидная связь, образованная карбоксильной группой, полученной из аминокислоты, дипептида или трипептида, и аминогруппой, которая присутствует у заместителя в положении 7 производного хинолонкарбоновой кислоты, может легко гидролизироваться в живом организме и образовывать свободную форму амина, и их примеры включают соединения, образованные из глицина, аланина, аспарагиновой кислоты и других аминокислот; глицин-глицина, глицин-аланина, аланин-аланина и других дипептидов; и глицин-глицин-аланина, глицин-аланин-аланина и других трипептидов.
Соединение данного изобретения, представленное формулой (I), может быть получено различными способами, и в предпочтительном случае такое соединение может быть получено, например, путем взаимодействия соединения, представленного формулой (III):

[где X2 представляет заместитель, который функционирует в качестве уходящей группы, такой как замещенная или незамещенная фенилсульфонильная группа, замещенная или незамещенная алкилсульфонильная группа, содержащая 1-3 атома углерода, атом фтора, атом хлора или атом брома;
R31 представляет R3, определенный в формуле (I), или бор-содержащую группу, представленную формулой (IV):

(где Y32 и Y33 могут быть одинаковыми или отличаться друг от друга, и каждый представляет атом фтора или алкилкарбонилоксигруппу, содержащую 2-4 атома углерода); и
R1, R2, R4, R5, R6 и A такие, как определено в формуле (I)]
с соединением следующей формулы (V) или его аддитивной солью:
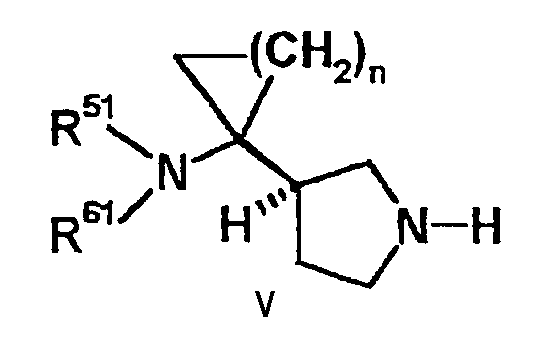
[где каждый R51 и R61 независимо представляет атом водорода, алкильную группу, содержащую 1-6 атомов углерода, или защитную группу для аминогруппы, или один из R51 или R61 представляет атом водорода, а другой представляет замещенную карбоксильную группу, полученную из аминокислоты, дипептида или трипептида, с аминогруппой, то есть незамещенной или защищенной с помощью защитной группы для аминогруппы, и
данная алкильная группа может содержать заместитель, выбранный из группы, включающей алкилтиогруппу, содержащую 1-6 атомов углерода, алкоксигруппу, содержащую 1-6 атомов углерода, гидроксильную группу, и атом галогена и
n такой, как определено в формуле (I)]
(в случае, когда используется аддитивная соль, реакцию проводят в присутствии реагентов, которые вызывают образование свободной формы аддитивной соли).
Примеры кислотно-аддитивных солей включают соли неорганических кислот, такие как гидрохлориды, сульфаты, нитраты, гидробромиды, гидройодиды, фосфаты и так далее; и соли органических кислот, такие как метансульфонаты, бензолсульфонаты, толуолсульфонаты (и другие сульфонаты), ацетаты, цитраты, малеаты, фумараты, лактаты (и другие карбоксилаты); и так далее.
Реакцию можно проводить с использованием растворителя или без него. Используемый в реакции растворитель может быть любым растворителем, который не оказывает неблагоприятного воздействия на реакцию, и его примеры включают диметилсульфоксид, пиридин, ацетонитрил, этанол, хлороформ, диметилформамид, диметилацетамид, N-метилпирролидон, тетрагидрофуран, воду, 3-метоксибутанол или их смеси.
Реакцию предпочтительно проводят в присутствии акцептора кислоты, такого как неорганическое основание или органическое основание, например соединение неорганического основания, такое как карбонат или бикарбонат щелочного металла или щелочно-земельного металла, или соединение органического основания, такое как триэтиламин, пиридин, 1,8-диазабициклоундецен, N-метилпиперидин, N,N-диизопропилэтиламин и так далее.
Температура реакции должна находиться в пределах от комнатной температуры до 200°C и, предпочтительно, в пределах от 25 до 150°C. Время реакции должно быть в пределах от 30 минут до 48 часов, и реакцию обычно проводят в течение приблизительно от 30 минут до 8 часов.
Защитная группа аминогруппы может быть любой защитной группой, которая обычно используется в соответствующей области, и примеры защитной группы включают алкоксикарбонильные группы, которые могут иметь заместитель, такие как трет-бутоксикарбонильная группа, 2,2,2-трихлорэтоксикарбонильная группа и так далее; аралкилоксикарбонильные группы, которые могут содержать заместитель, такие как бензилоксикарбонильная группа, пара-метоксибензилоксикарбонильная группа, пара-нитробензилоксикарбонильная группа и так далее; ацильные группы, которые могут содержать заместитель, такие как ацетильная группа, метоксиацетильная группа, трифторацетильная группа, хлорацетильная группа, пивалоильная группа, формильная группа, бензоильная группа и так далее; алкильные группы, которые могут содержать заместитель, и аралкильные группы, которые могут содержать заместитель, такие как трет-бутильная группа, бензильная группа, паранитробензильная группа, пара-метоксибензильная группа, трифенилметильная группа и так далее; простые эфиры, которые могут иметь заместитель, такие как метоксиметильная группа, трет-бутоксиметильная группа, тетрагидропиранильная группа, 2,2,2-трихлорэтоксиметильная группа и так далее; и замещенные силильные группы, такие как триметилсилильная группа, изопропилдиметилсилильная группа, трет-бутилдиметилсилильная группа, трибензилсилильная группа, трет-бутилдифенилсилильная группа и так далее.
В случае, когда каждый R3 и R31 представляет алкильную группу, содержащую 1-6 атомов углерода, алкоксиметильную группу, содержащую 2-7 атомов углерода, или фенилалкильную группу (аралкильную группу), состоящую из алкиленовой группы, содержащей 1-6 атомов углерода, и фенильную группу, он может быть превращен в соответствующую карбоновую кислоту обработкой в кислых или основных условиях, которые обычно используются для гидролиза сложных эфиров карбоновой кислоты.
В случае, когда R31 имеет структуру формулы (IV), он может быть превращен в соответствующую карбоновую кислоту, гидролизом в кислых или основных условиях после взаимодействия соединения (V) с соединением (III).
В случае, когда необходимо снять защиту, интересующее соединение, представленное формулой (I), может быть получено удалением защитной группы в соответствующих условиях, подходящих для защитной группы.
Соединение формулы (V) может быть получено различными способами, и хотя в качестве примера может быть приведен способ, показанный в PCT/JP96/00208, этим способом получение соединения формулы (V) не ограничивается.
Соединение формулы (V) может быть получено удалением Q из соединения, представленного следующей формулой (VI):
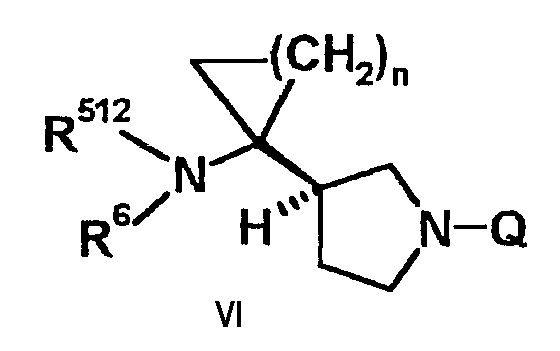
[В указанной выше формуле (VI), R512 такой же как R5, определенный в формуле (I), или представляет защитную группу аминогруппы, R6 и n такие, как определено в формуле (I), Q представляет защитную группу аминогруппы,
где защитная группа аминогруппы может быть выбрана из группы, содержащей (замещенную) алкоксикарбонильную группу, (замещенную) аралкилоксикарбонильную группу, (замещенную) ацильную группу, (замещенную) алкильную группу, (замещенную) аралкильную группу и замещенную силильную группу.]
Описанное выше соединение может присутствовать в форме его соли, гидрата или гидрата соли. Примеры кислотно-аддитивных солей включают соли неорганических кислот и соли органических кислот. Их конкретные примеры включают соли неорганических кислот, такие как гидрохлориды, сульфаты, гидробромиды, гидройодиды, фосфаты и так далее; и соли органических кислот, такие как метансульфонаты, бензолсульфонаты, толуолсульфонаты (соли сульфоновой кислоты); ацетаты, цитраты, малеаты, фумараты, лактаты (соли карбоновых кислот); и так далее.
Когда оба R512 и Q представляют защитные группы аминогруппы, они могут быть одинаковыми или отличаться друг от друга. Однако более предпочтительно при получении соединения (I), чтобы каждая защитная группа удалялась в различных реакционных условиях.
Примеры R512 и Q, которые представляют защитные группы аминогрупп, включают перечисленные выше группы. То есть их примеры включают (замещенную) алкоксикарбонильную группу, (замещенную) аралкилоксикарбонильную группу, (замещенную) ацильную группу, (замещенную) алкильную группу, (замещенную) аралкильную группу и (замещенную) силильную группу.
Их конкретные примеры включают (замещенные) алкоксикарбонильные группы, такие как трет-бутоксикарбонильная группа, 2,2,2-трихлорэтоксикарбонильная группа и так далее; (замещенные) аралкилоксикарбонильные группы, такие как бензилоксикарбонильная группа, пара-метоксибензилоксикарбонильная группа, пара-нитробензилоксикарбонильная группа и так далее; (замещенные) ацильные группы, такие как ацетильная группа, метоксиацетильная группа, трифторацетильная группа, хлорацетильная группа, пивалоильная группа, формильная группа, бензоильная группа и так далее; (замещенные) алкильные группы или (замещенные) аралкильные группы, такие как трет-бутильная группа, бензильная группа, пара-нитробензильная группа, пара-метоксибензильная группа, трифенилметильная группа и так далее; (замещенные) простые эфиры, такие как метоксиметильная группа, трет-бутоксиметильная группа, тетрагидропиранильная группа, 2,2,2-трихлорэтоксиметильная группа и так далее; и замещенные силильные группы, такие как триметилсилильная группа, изопропилдиметилсилильная группа, трет-бутилдиметилсилильная группа, трибензилсилильная группа, трет-бутилдифенилсилильная группа и так далее.
При получении соединения (I), используя указанное выше соединение, содержащее в качестве защитной группы Q, необходимо проводить реакцию путем удаления защитной группы Q. В этом случае, его взаимодействие с соединением (III) или (V) можно проводить с помощью так называемой реакции в одном сосуде, или реакцию можно проводить сразу после выделения соединения (V) путем удаления защитной группы.
Как и в случае с соединением формулы (V), соединение формулы (VI) может быть получено различными способами, и хотя в качестве примера может быть приведен способ, описанный в PCT/JP96/00208, этим способ получения соединения формулы (VI) не ограничен.
Цис-2-фторциклопропиламин, содержащий единственный изомер, который является желательным для синтеза соединения формулы (I), содержащего единственный изомер, может быть синтезирован, например, способом, описанным в JP-A-2-231475 (используемый в данном описании термин «JP-A» обозначает нерассмотренную опубликованную японскую патентную заявку). Синтез соединения формулы (I), содержащего единственный изомер, с использованием в качестве исходного соединения оптически активного производного цис-2-фторциклопропиламина, полученного способом, описанным выше, может проводиться, например, в соответствии со способом, описанным в JP-A-2-231475.
Конкретные примеры соединения данного изобретения включают перечисленные ниже соединения, такие как:
7-[3-(R)-(1-аминоциклопропил)пирролидин-1-ил]-1-[2-(S)-фтор-1-(R)-циклопропил]-1,4-дигидро-4-оксо-1,8-нафтиридин-3-карбоновая кислота [показано в следующей формуле];
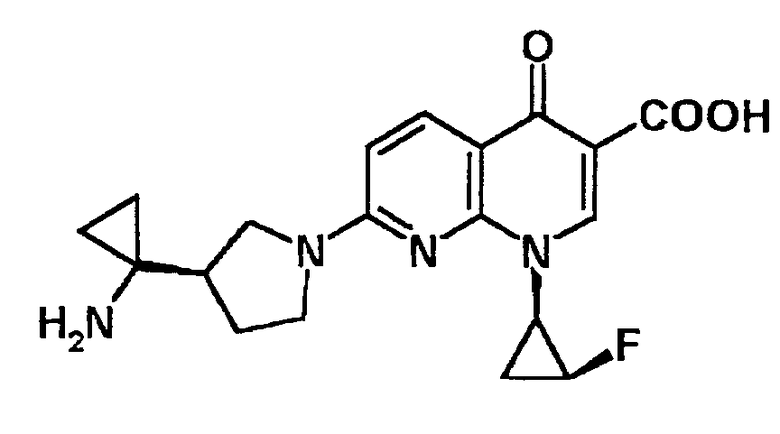
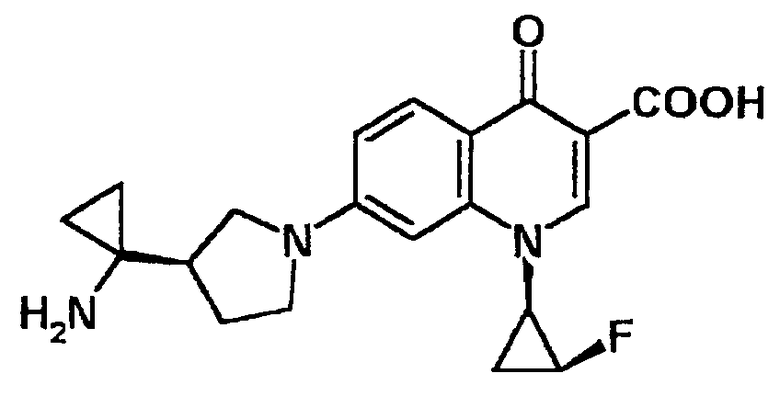
7-[3-(R)-(1-аминоциклопропил)пирролидин-1-ил]-1-[2-(S)-фтор-1-(R)-циклопропил]-1,4-дигидро-4-оксохинолин-3-карбоновая кислота [показано в следующей формуле];
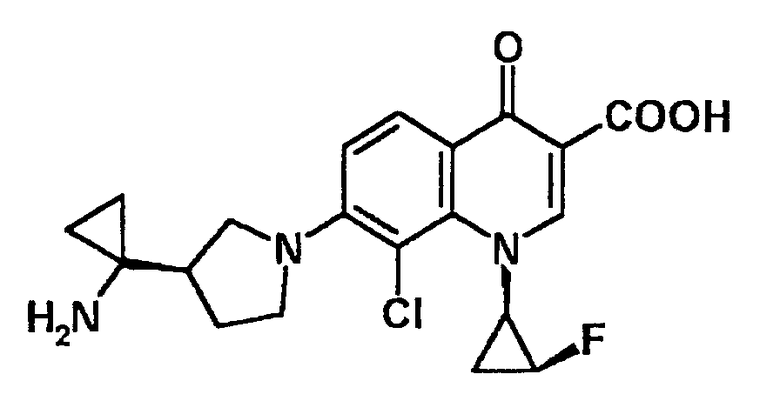
7-[3-(R)-(1-аминоциклопропил)пирролидин-1-ил]-1-[2-(S)-фтор-1-(R)-циклопропил]-8-хлор-1,4-дигидро-4-оксохинолин-3-карбоновая кислота [показано в следующей формуле];
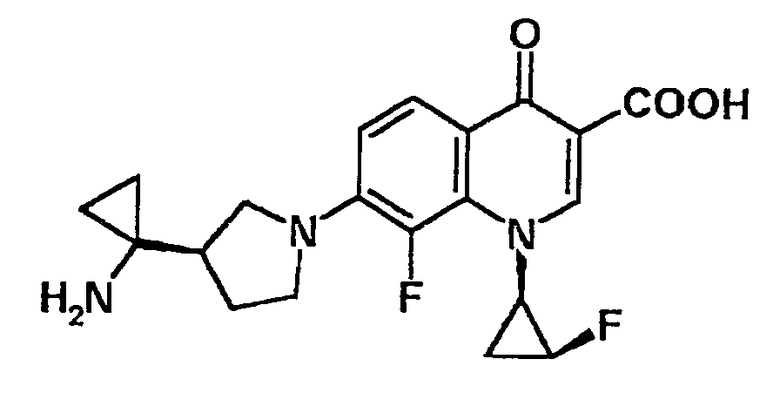
7-[3-(R)-(1-аминоциклопропил)пирролидин-1-ил]-1-[2-(S)-фтор-1-(R)-циклопропил]-8-фтор-1,4-дигидро-4-оксохинолин-3-карбоновая кислота [показано в следующей формуле];
7-[3-(R)-(1-аминоциклопропил)пирролидин-1-ил]-1-[2-(S)-фтор-1-(R)-циклопропил]-8-дифторметокси-1,4-дигидро-4-оксохинолин-3-карбоновая кислота [показано в следующей формуле];

7-[3-(R)-(1-аминоциклопропил)пирролидин-1-ил]-1-[2-(S)-фтор-1-(R)-циклопропил]-1,4-дигидро-8-метокси-4-оксохинолин-3-карбоновая кислота [показано в следующей формуле];
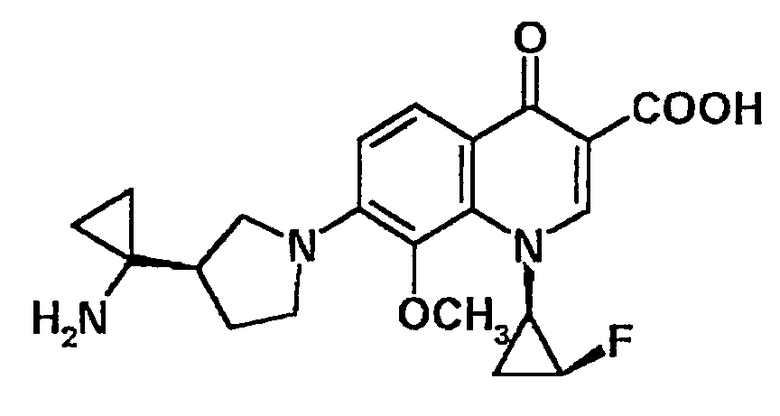
7-[3-(R)-(1-аминоциклопропил)пирролидин-1-ил]-1-[2-(S)-фтор-1-(R)-циклопропил]-1,4-дигидро-8-метил-4-оксохинолин-3-карбоновая кислота [показано в следующей формуле];
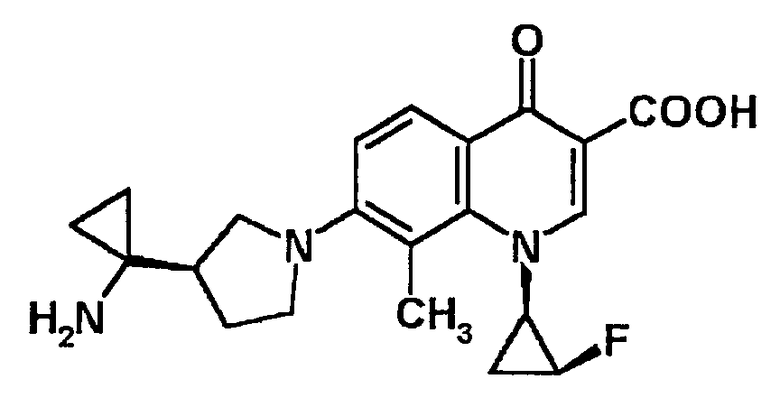
5-амино-7-[3-(R)-(1-аминоциклопропил)пирролидин-1-ил]-1-[2-(S)-фтор-1-(R)-циклопропил]-1,4-дигидро-8-фтор-4-оксохинолин-3-карбоновая кислота [показано в следующей формуле];
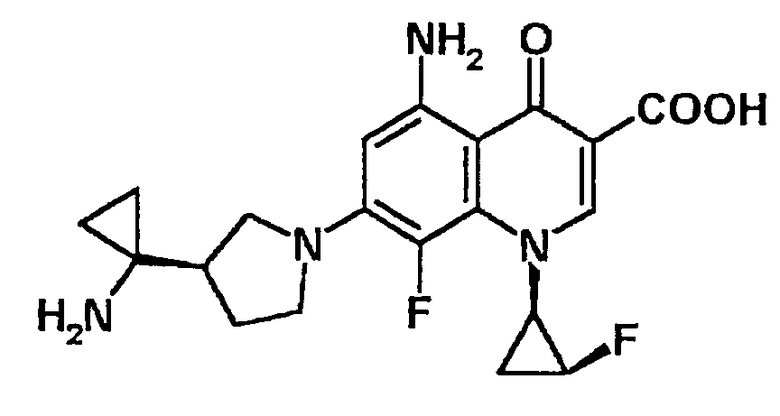
5-амино-7-[3-(R)-(1-аминоциклопропил)пирролидин-1-ил]-1-[2-(S)-фтор-1-(R)-циклопропил]-1,4-дигидро-8-метокси-4-оксохинолин-3-карбоновая кислота [показано в следующей формуле];
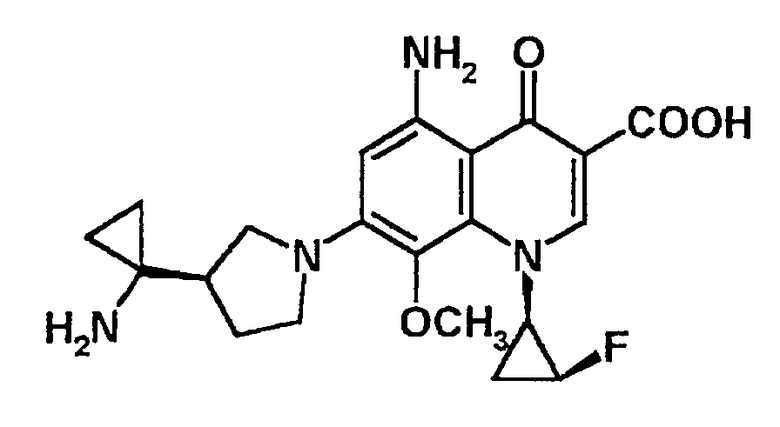
5-амино-7-[3-(R)-(1-аминоциклопропил)пирролидин-1-ил]-1-[2-(S)-фтор-1-(R)-циклопропил]-1,4-дигидро-8-метил-4-оксохинолин-3-карбоновая кислота [показано в следующей формуле];
10-[3-(R)-(1-аминоциклопропил)пирролидин-1-ил]-2,3-дигидро-3-(S)-метил-7-оксо-7H-пиридо[1,2,3-de][1,4]бензоксазин-6-карбоновая кислота [показано в следующей формуле];
7-[3-(R)-(1-аминоциклобутил)пирролидин-1-ил]-1-[2-(S)-фтор-1-(R)-циклопропил]-8-дифторметокси-1,4-дигидро-4-оксохинолин-3-карбоновая кислота [показано в следующей формуле];
7-[3-(R)-(1-аминоциклобутил)пирролидин-1-ил]-1-[2-(S)-фтор-1-(R)-циклопропил]-1,4-дигидро-8-метокси-4-оксохинолин-3-карбоновая кислота [показано в следующей формуле];

7-[3-(R)-(1-аминоциклобутил)пирролидин-1-ил]-1-[2-(S)-фтор-1-(R)-циклопропил]-1,4-дигидро-8-метил-4-оксохинолин-3-карбоновая кислота [показано в следующей формуле];
7-[3-(R)-[1-(метиламино)циклопропил]пирролидин-1-ил]-1-[2-(S)-фтор-1-(R)-циклопропил]-1,4-дигидро-8-метокси-4-оксохинолин-3-карбоновая кислота [показано в следующей формуле];
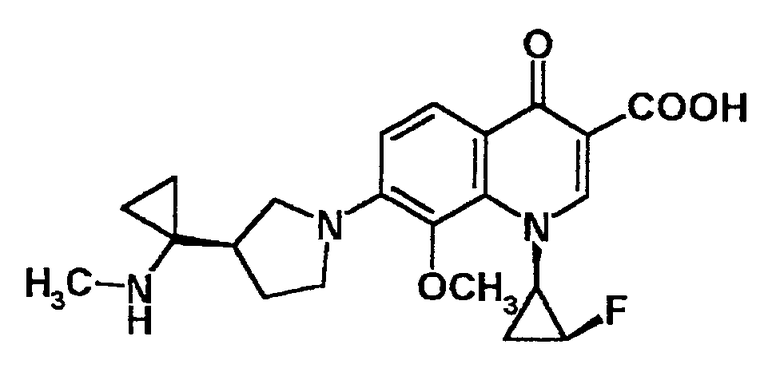
7-[3-(R)-[1-(метиламино)циклопропил]пирролидин-1-ил]-1-[2-(S)-фтор-1-(R)-циклопропил]-1,4-дигидро-8-метил-4-оксохинолин-3-карбоновая кислота [показано в следующей формуле];
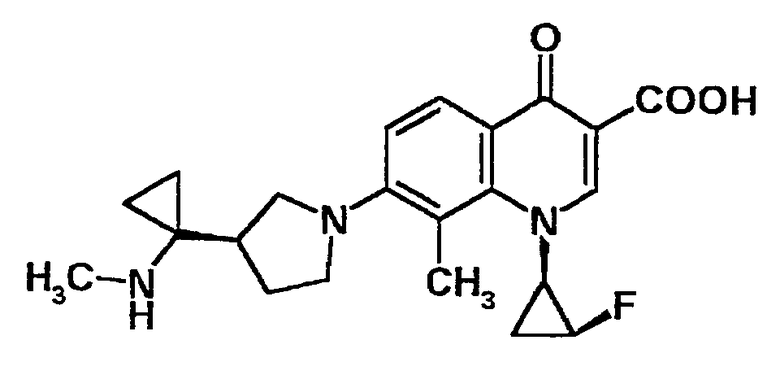
7-[3-(R)-[1-(этиламино)циклопропил]пирролидин-1-ил]-1-[2-(S)-фтор-1-(R)-циклопропил]-1,4-дигидро-8-метил-4-оксохинолин-3-карбоновая кислота [показано в следующей формуле];
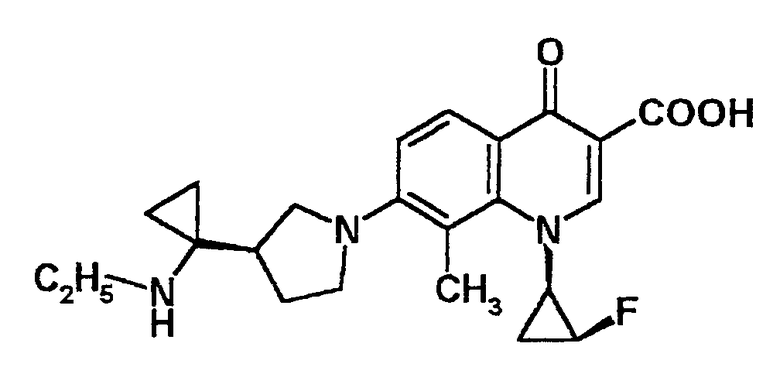
5-амино-7-[3-(R)-[1-(метиламино)циклопропил]пирролидин-1-ил]-1-[2-(S)-фтор-1-(R)-циклопропил]-1,4-дигидро-8-метил-4-оксохинолин-3-карбоновая кислота [показано в следующей формуле];

10-[3-(R)-[1-(метиламино)циклопропил]пирролидин-1-ил]-2,3-дигидро-3-(S)-метил-7-оксо-7H-пиридо[1,2,3-de][1,4]бензоксазин-6-карбоновая кислота [показано в следующей формуле];
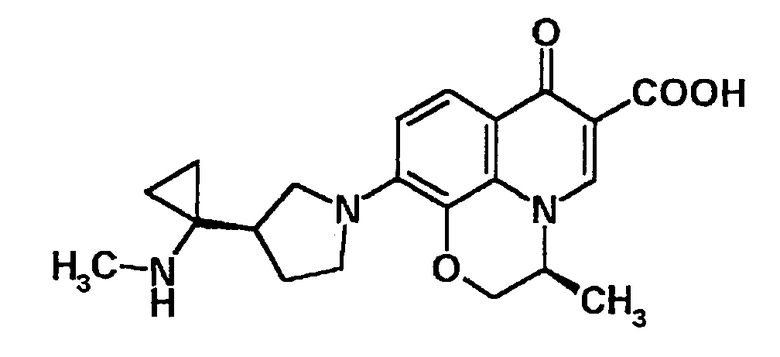
1-(циклопропил)-8-метил-7-[3-(R)-[1-(метиламино)циклопропил]пирролидин-1-ил]-1,4-дигидро-4-оксохинолин-3-карбоновая кислота [показано в следующей формуле];
Так как соединение данного изобретения обладает сильным антибактериальным действием, его можно использовать в качестве лекарственного средства для применения для человека, животных и рыб или в качестве консервантов в сельскохозяйственных химикатах и в продуктах питания.
В случае, когда соединение данного изобретения используется в качестве лекарственного средства для применения для человека, его доза для взрослого пациента находится в пределах от 50 мг до 1 г в день, предпочтительно от 100 мг до 500 мг в день.
В случае применения медицинского препарата для животного его доза изменяется в зависимости от цели введения (лечение или профилактика), вида и размера конкретного животного, нуждающегося в лечении, конкретного вида патогенного микроорганизма, вызвавшего инфекцию, и степени инфицированности, ежедневная доза обычно находится в пределах от 1 мг до 200 мг, предпочтительно от 5 мг до 100 мг на 1 кг массы тела каждого животного.
Ежедневная доза вводится за один раз или ее делят на две-четыре дозы, принимаемые в течение дня. В случае острой необходимости дневная доза может превышать указанные выше количества.
Так как соединения данного изобретения активны в отношении широкого спектра микроорганизмов, которые вызывают различные инфекционные заболевания, оно может применяться для лечения, профилактики или облегчения заболеваний, вызванных такими патогенами.
Примеры бактерий и бактероидных микроорганизмов, против которых эффективны данные соединения, включают род Staphylococcus, Streptococcus pyogenes, Hemolytic streptococcus, Enterococcus, Pneumococcus, род Peptostreptococcus, Neisseria gonorrhoeae, Escherichia coli, роды Citrobacter, род Shigella, Klebsiella pneumoniae, род Enterobacter, род Serratia, род Proteus, Pseudomonas aeruginosa, Haemophilus influenzae, род Acinetobacter, род Campylobacter, Chlamydia trachomatis и тому подобное.
Примеры заболеваний, которые вызывают указанные выше болезнетворные микроорганизмы, включают фолликулит, фурункул, карбункул, рожистое воспаление, флегмону, воспаление лимфатических узлов (лимфаденит), панариций, подкожный абсцесс, гидраденит, множественное воспаление сальных желез, инфекционную атерому, анальный абсцесс, мастит, поверхностное вторичное инфицирование ран после травм, ожогов, операционного вмешательства и так далее, фаринголарингит, острый бронхит, тонзиллит, хронический бронхит, бронхоэктазию, диффузный панбронхит, вторичную инфекцию хронического респираторного заболевания, пневмонию, пиелонефрит, цистит, простатит, эпидидимит, гонококковый уретрит, негонококковый уретрит, холецистит, холангит, бациллярную дизентерию, энтерит, воспаление придатков матки, внутриматочную инфекцию, бартолинит, блефарит, ячмень, дакриоцистит, тарзаденит, язву роговицы, средний отит, синусит, амфодонтит, перикоронит, воспаление челюсти, перитонит, эндокардит, сепсис, менингит, кожную инфекцию и так далее.
Кроме того, примеры кислотоустойчивых бактерий, по отношению к которым соединения данного изобретения эффективны, включают tubercle bacilli [Mycobacterium (далее сокращено как «M») tuberculosis, M. bovis, M. africanum], атипичные кислотоустойчивые бактерии [M. kansasii, M. marinum, M. scrofulaceum, M. avium, M. intracellulare, M. xenopi, M. fortuitum, M. chelonae] и так далее.
Инфекции, вызываемые данными кислотоустойчивыми бактериями, как правило, классифицируют в соответствии с бактериями, вызывающими инфекции, на три типа: туберкулез, атипичная инфекция, вызываемая кислотоустойчивыми бактериями, и лепра. Кроме легких, инфекция, вызываемая туберкулезной бациллой, может поражать грудную полость, трахеи/бронхи, лимфоузлы, обычно диссеминированным образом, кости и суставы, оболочки мозга/мозг, органы пищеварения (кишечник, печень), кожу, молочные железы, глаза, среднее ухо/горло, мочевой тракт, мужские гениталии, женские гениталии и так далее. Легкие наиболее часто подвергаются атипичной инфекции, вызываемой кислотостойкими бактериями (инфекции, вызываемые нетуберкулезными кислотостойкими бактериями), и другие примеры атипичной инфекции, вызываемой кислотостойкими бактериями, включают местный лимфаденит, инфекции мягких тканей, инфекции суставов, инфекции общего диссеминирующего типа и так далее.
Соединения данного изобретения также эффективны в отношении различных микроорганизмов, которые вызывают инфекции у животных. Примеры таких микроорганизмов включают Escherichia, Salmonella, Pasturella, Haemophilus, Bordetella, Staphylococcus, Mycoplasma и так далее.
Конкретные примеры заболеваний у птиц включают эшерихиоз, белую бациллярную диарею, птичью паратифоидную лихорадку, птичью холеру, инфекционный ринит, стафилококковую инфекцию, микоплазменную инфекцию и так далее, конкретные примеры заболеваний свиней включают эширихиоз, сальмонеллез, пастуреллоз, гемофильную инфекцию, атрофический ринит, экссудативный эпидермит, микоплазменную инфекцию и так далее, конкретные примеры заболеваний крупного рогатого скота включают эшерихоиз, сальмонеллез, геморрагическую септицемию, микоплазменную инфекцию, коровью контагиозную плевропневмонию, мастит и так далее, конкретные примеры заболеваний собак включают колиемию, сальмонелезную инфекцию, геморрагическую септицемию, пиометру, цистит и так далее, и конкретные примеры заболеваний кошек включают экссудативный плеврит, цистит, хронический ринит, гемофильную инфекцию, кошачью диарею, микоплазменную инфекцию и так далее.
Антибактериальный препарат, который содержит соединение данного изобретения, может быть получен путем выбора соответствующего фармацевтического препарата в соответствии со способом введения и использования, любым способом, обычно используемым для получения различных фармацевтических препаратов. Что касается дозированных форм антибактериальных препаратов, содержащих в качестве его основного агента соединение данного изобретения, то в качестве примеров форм пероральных фармацевтических препаратов можно привести таблетки, порошки, гранулы, капсулы, растворы, сиропы, эликсиры, масляные или водные суспензии и так далее.
В случае инъекций, в препарате может быть использован стабилизирующий агент, антисептический агент, солюбилизирующий агент и так далее, или раствор, который может содержать данные дополнительные агенты, может быть помещен в контейнер и затем высушен сублимационной сушкой или подобным способом до твердого препарата для повторного его растворения при использовании. Также, в одном контейнере может находиться единичная доза или в таком же контейнере могут содержаться множественные дозы.
Примеры форм препаратов для наружного применения включают растворы, суспензии, эмульсии, мази, гели, кремы, лосьоны, спреи и так далее.
Твердые препараты, наряду с активным соединением, могут содержать фармацевтически приемлемые добавки. Например, наполнители, разбавители, связующие агенты, дезинтегрирующие агенты, агенты, усиливающие растворение, увлажняющие агенты, смазывающие агенты и так далее могут быть выбраны и смешаны необходимым образом для получения препарата.
Примеры форм жидких препаратов включают растворы, суспензии, эмульсии, и в качестве добавок они могут содержать суспендирующие агенты, эмульгирующие агенты и так далее.
Примеры способов введения соединения данного изобретения животным включают способ непосредственного перорального введения или перорального введения путем подмешивания в корм, способ получения раствора и последующего осуществления перорального введения раствора непосредственно или добавлением раствора в питьевую воду или корм, способ введения путем инъекции и так далее.
Фармацевтический препарат для введения соединения данного изобретения животным может быть необязательно получен в виде порошков, мелких гранул, растворимых порошков, сиропов, растворов или инъекций с помощью способов, обычно используемых в данной области.
Примеры получения фармацевтических препаратов показаны ниже.
Пример получения 1 (капсула):
Кукурузный крахмал
CMC кальций
Гидроксиметилцеллюлоза
Стеарат магния
Всего
23,0 мг
22,5 мг
3,0 мг
1,5 мг
150,0 мг
Пример получения 2 (раствор):
Уксусная кислота или гидроксид натрия
Этил пара-гидроксибензоат
Очищенная вода
Всего
0,5-2 г
0,1 г
88,9-98,4 г
100 г
Пример получения 3 (порошок для подмешивания в корм):
Кукурузный крахмал
Легкий кремниевый ангидрид
Всего
98,5-89,5 г
0,5 г
100 г
Лучший способ осуществления изобретения
Дальнейшее описание изобретения основывается на примерах и ссылочных примерах, тем не менее данное изобретение ими не ограничивается.
Ссылочный пример 1
Этил 2-(2,4-дифтор-3-метилбензоил)-3-диметиламиноакрилат
2,4-Дифтор-3-метилбензоат (4,97 г, 28,9 ммоль) растворяли в толуоле (50 мл) и после добавления N,N-диметилформамида (0,1 мл) и тионилхлорида (3,16 мл, 43,4 ммоль) перемешивали в течение 14 часов на масляной бане при 80°C. Затем реакционный раствор охлаждали и после этого концентрировали при пониженном давлении. После добавления к остатку толуола и повторного концентрирования при пониженном давлении полученный остаток растворяли в тетрагидрофуране (10 мл). Данный раствор, охлаждая на льду, по каплям добавляли в раствор, в котором в тетрагидрофуране (20 мл) растворены этил 3-диметиламиноакрилат (4,97 г, 34,7 ммоль) и триэтиламин (5,04 мл, 36,1 ммоль). После завершения добавления реакционный раствор кипятили с обратным холодильником в течение 10 часов. После завершения реакции реакционный раствор фильтровали, удаляли соль гидрохлорида триэтиламина (промывкой диэтиловым эфиром) и фильтрат концентрировали при пониженном давлении. Полученный остаток наносили на короткую хроматографическую колонку с силикагелем, и из элюата н-гексан:этилацетата=1:1 получали 6,70 г (78%) указанного в заголовке соединения в виде желтого порошка.
1H ЯМР (400 МГц, CDCl3) δ: 0,95 (3Н, J=7,08 Гц), 2,18 (3H, т, J=1,95 Гц), 2,92-3,24 (6H, м), 3,99 (2H, кв, J=7,08 Гц), 6,86 (1H, дт, J=1,22, 8,55 Гц), 7,43 (1H, шир.с.), 7,75 (1H, c).
ИК (KBr, диск): 3055, 2985, 2933, 2875, 2814, 1942, 1693, 1630, 1593, 1477, 1431, 1379, 1277, 1255, 1221 см-1.
Точка плавления: 82-84°C.
Элементный анализ C15H17F2NO3:
Вычислено: C 60,60; H 5,76; N 4,71.
Найдено: C 60,31; H 5,73; N 4,73.
Ссылочный пример 2
Этил 7-фтор-1-[2-(S)-фтор-1-(R)-циклопропил]-1,4-дигидро-8-метил-4-оксохинолин-3-карбоксилат
Этил 2-(2,4-дифтор-3-метилбензоил)-3-диметиламиноакрилат (1,06 г, 3,57 ммоль) растворяли в тетрагидрофуране (15 мл) и, после добавления соли пара-толуолсульфоновой кислоты (1R,2S)-2-фторциклопропиламина (970 мг, 3,93 ммоль), при перемешивании добавляли по каплям раствор, в которым в тетрагидрофуране (5 мл) растворен триэтиламин (552 мкл, 3,96 ммоль), при -15°C. После перемешивания реакционного раствора в течение 2 часов при комнатной температуре добавляли карбонат калия (740 мг, 5,36 ммоль) и хлорид тетрабутиламмония (49,6 мг, 0,179 ммоль) и реакционную суспензию кипятили с обратным холодильником при перемешивании в течение 5 дней. После охлаждения реакционного раствора тетрагидрофуран упаривали при пониженном давлении. Затем к осадку добавляли дихлорметан (10 мл) и при охлаждении на льду и перемешивании постепенно по каплям добавляли 2 моль/л хлористо-водородную кислоту для доведения pH до приблизительно 3. Затем, после перемешивания в течение 15 минут при комнатной температуре экстрагировали дихлорметаном (60 мл×3). После сушки над безводным сульфатом натрия фильтровали, фильтрат концентрировали при пониженном давлении, полученные неочищенные кристаллы перемешивали и очищали во взвешенном состоянии этилацетатом. Таким образом получали 713 мг (65%) указанного в заголовке соединения в виде бесцветных кристаллов.
1H ЯМР (400 МГц, CDCl3) δ: 1,41 (3H, т, J=7,08 Гц), 1,56-1,62 (2H, т), 2,66 (3Н, д, J=2,69 Гц), 3,85-3,89 (1H, м), 4,39 (2H, кв, J=7,08 Гц), 4,78-4,79 и 4,94-4,95 (1H, дм, J=62,74 Гц), 7,13 (1H, т, J=8,91 Гц), 8,36 (1H, дд, J=6,71, 8,91 Гц), 8,56 (1H, д, J=2,93 Гц).
ИК (KBr, диск): 3438, 3097, 2983, 2939, 2902, 1907, 1720, 1630, 1593, 1566, 1460, 1429, 1387, 1367, 1311, 1250 см-1.
Точка плавления: 187-188°C.
Элементный анализ C16H15F2NO3:
Вычислено: C 62,54; H 4,92; N 4,56.
Найдено: C 62,41; H 4,87; N 4,53.
Ссылочный пример 3
7-фтор-1-[2-(S)-фтор-1-(R)-циклопропил]-1,4-дигидро-8-метил-4-оксохинолин-3-карбоновая кислота
Этил 7-фтор-1-[2-(S)-фтор-1-(R)-циклопропил]-1,4-дигидро-8-метил-4-оксохинолин-3-карбоксилат (1,40 г, 4,56 ммоль) растворяли в уксусной кислоте (4 мл), и после добавления концентрированной хлористо-водородной кислоты (4 мл) кипятили с обратным холодильником в течение 3 часов. После охлаждения реакционный раствор выливали в ледяную воду (50 мл) и выпавшие кристаллы отфильтровывали. После промывки отфильтрованных кристаллов избыточным количеством воды осуществляли промывку холодным этанолом и диэтиловым эфиром в указанном порядке, и после сушки при пониженном давлении получали 1,18 г (93%) указанного в заголовке соединения в виде белого порошка.
1H ЯМР (400 МГц, CDCl3) δ: 1,48-1,72 (2H, м), 2,75 (3H, т, J=2,56 Гц), 4,01 (1H, дд, J=2,81, 5,25 Гц), 4,83-4,84 и 4,98-5,00 (1H, дм, J=62,74 Гц), 7,31 (1H, дд, J=2,20, 8,79 Гц), 8,40-8,44 (1H, м), 8,84 (1H, д, J=2,69 Гц), 14,50 (1H, шир.c).
ИК (KBr, диск): 3097, 3014, 2956, 2642, 1957, 1728, 1618, 1566, 1508, 1469, 1435, 1389, 1321, 1254, 1200 см-1.
Точка плавления: 250-253°C.
[α]D 24,3=-50,00° (c 0,145, 0,1 моль/л NaOH).
Элементный анализ С14H11F2NO3:
Вычислено: C 60,22; H 3,97; N 5,02.
Найдено: C 59,92; H 3,98; N 4,92.
ПРИМЕР 1
7-[3-(R)-(1-аминоциклопропил)пирролидин-1-ил]-1-[2-(S)-фтор-1-(R)-циклопропил]-1,4-дигидро-8-метил-4-оксохинолин-3-карбоновая кислота
После добавления к сухому диметилсульфоксиду (2 мл) 3-(R)-[1-трет-бутоксикарбониламиноциклопропил]пирролидина (185 мг, 817 мкмоль) и триэтиламина (0,50 мл) добавляли 7-фтор-1-[2-(S)-фтор-1-(R)-циклопропил]-1,4-дигидро-8-метил-4-оксохинолин-3-карбоновую кислоту (190 мг, 681 мкмоль) и кипятили с обратным холодильником в течение 17 часов в атмосфере азота. После концентрирования реакционного раствора при пониженном давлении остаток растворяли в хлороформе (50 мл). После промывки органического слоя 10% водным раствором лимонной кислоты (25 мл) органический слой сушили над безводным сульфатом натрия. После фильтрования фильтрат концентрировали при пониженном давлении, и после добавления по каплям концентрированной хлористо-водородной кислоты (5 мл) в остаток при охлаждении на льду перемешивали при комнатной температуре в течение 30 минут. Затем в реакционный раствор добавляли 1 моль/л хлористо-водородную кислоту (5 мл), и после промывки желтого кислого водного раствора хлороформом (20 мл×3) pH доводили до 12,0 с помощью водного раствора гидроксида натрия и нерастворимый остаток удаляли фильтрованием. После доведения рН основного водного раствора до 7,4 с помощью 1 моль/л хлористо-водородной кислоты проводили экстракцию хлороформом (100 мл×4). После сушки над безводным сульфатом натрия растворитель упаривали при пониженном давлении. Остаток, полученный таким образом, очищали с помощью препаративной хроматографии (разделение в нижнем слое смесью хлороформ:метанол:вода, 7:3:1), перекристаллизовывали из этанола и сушили при пониженном давлении. Таким образом было получено 112 мг (43%) указанного в заголовке соединения в виде желтых кристаллов.
1H ЯМР (400 МГц, 0,1 моль/л NaOD) δ: 0,54 (4Н, д, J=5,61 Гц, 1,19-1,21 (1Н, м), 1,58-1,62 (1H, м), 1,66-1,69 (1H, м), 2,00-2,01 (1H, м), 2,16-2,17 (1H, м), 2,35 (3H, c), 3,16-3,23 (2H, м), 3,37-3,42 (1H, м), 3,54-3,55 (1H, м), 4,04-4,05 (1H, м), 4,94-4,95 и 5,10-5,11 (1H, дм, J=62,16 Гц), 7,01 (1H, д, J=8,78 Гц), 7,95 (1H, д, J=8,78 Гц), 8,43 (1H, c).
ИК (KBr, диск): 3375, 3062, 3006, 2925, 2864, 1728, 1610, 1508, 1475, 1431, 1394, 1348, 1315, 1257 см-1.
Точка плавления: 228-230°C.
[α]D 24,7=-235,09° (c 0,285, 0,1 моль/л NaOH).
Элементный анализ C21H24FN3O3:
Вычислено: C 65,44; H 6,28; N 10,90.
Найдено: C 65,10; H 6,32; N 10,76.
Ссылочный пример 4
Этил 7-фтор-1-[2-(S)-фтор-1-(R)-циклопропил]-1,4-дигидро-8-метокси-4-оксохинолин-3-карбоксилат
Смесь этил (2,4-дифтор-3-метокси)бензоилацетата (48,8 г, 189 ммоль), синтезированного способом, описанным в PCT/US98/19138, триэтилортоформиата (78,6 мл, 472 ммоль) и уксусного ангидрида (250 мл) перемешивали при нагревании в течение 6 часов на масляной бане с внешней температурой, равной 120°C. Затем реакционный раствор охлаждали, концентрировали при пониженном давлении и отверждали с помощью сушки. Затем полученный желтый экстракт растворяли в толуоле (800 мл), добавляли соль пара-толуолсульфоновой кислоты (1R,2S)-2-фторциклопропиламина (60,1 г, 246 ммоль) и при перемешивании при -15°C добавляли по каплям раствор, в котором в толуоле (200 мл) растворен триэтиламин (40,8 мл, 293 ммоль). После перемешивания реакционного раствора в течение 4 часов при комнатной температуре добавляли воду (500 мл) и органический слой отделяли. После промывки органического слоя насыщенным раствором соли (500 мл×2) его сушили над безводным сульфатом натрия. После фильтрования фильтрат концентрировали при пониженном давлении и сушили. Затем полученное маслянистое вещество растворяли в 1,4-диоксане (600 мл) и при охлаждении водой постепенно добавляли 60% гидрид натрия в масле (5,94 г, 242 ммоль). После перемешивания реакционной смеси в течение 30 минут при комнатной температуре концентрировали при пониженном давлении до тех пор, пока объем реакционного раствора не стал приблизительно равным 300 мл. Полученный таким образом концентрат медленно выливали в 1 моль/л хлористо-водородную кислоту, которую перемешивали и охлаждали водой, и выпавшие кристаллы отфильтровывали. После промывки кристаллов большим количеством очищенной воды, небольшим количеством этанола и избытком диэтилового эфира в указанном порядке полученные неочищенные кристаллы суспендировали в этилацетате и очищали. Таким образом получали 49,4 г (80,9%) указанного в заголовке соединения в виде бесцветных кристаллов.
1H ЯМР (400 МГц, CDCl3) δ: 1,42 (3H, т, J=7,08 Гц), 1,55-1,64 (2H, м), 3,88-3,93 (1H, м), 4,04 (3H, д, J=1,96 Гц), 4,39 (2H, кв, J=7,08 Гц), 4,78-4,79 и 4,94-4,95 (1H, дм, J=62,61 Гц), 7,22 (1H, т, J=8,79 Гц), 8,24 (1H, дд, J=5,86, 8,79 Гц), 8,60 (1H, c).
Точка плавления: 190-193°C (разложение).
Ссылочный пример 5
7-фтор-1-[2-(S)-фтор-1-(R)-циклопропил]-1,4-дигидро-8-метокси-4-оксохинолин-3-карбоновая кислота
После растворения этил 7-фтор-1-[2-(S)-фтор-1-(R)-циклопропил]-1,4-дигидро-8-метокси-4-оксохинолин-3-карбоксилата (34,0 г, 105 ммоль) в уксусной кислоте (400 мл) и последующего добавления концентрированной хлористо-водородной кислоты (400 мл) смесь кипятили с обратным холодильником в течение 3 часов. После охлаждения реакционный раствор выливали в ледяную воду (1500 мл) и выпавшие кристаллы отфильтровывали. После промывки отфильтрованных кристаллов большим количеством воды их промывали холодным этанолом и диэтиловым эфиром, в указанном порядке, и после сушки при пониженном давлении полученные неочищенные кристаллы очищали перекристаллизацией из смешанного растворителя ацетонитрил-этанол и затем сушили при пониженном давлении. Таким образом получали 27,1 г (87,4%) указанного в заголовке соединения в виде белого порошка.
1H ЯМР (400 МГц, CDCl3) δ: 1,45-1,75 (2H, м), 3,87-3,95 (1H, м), 4,03 (3H, д, J=1,95 Гц), 4,79-4,81 и 4,97-4,99 (1H, дм, J=62,68 Гц), 7,30 (1H, т, J=8,79 Гц), 8,27 (1H, дд, J=5,86, 8,79 Гц), 8,76 (1H, c).
Точка плавления: 261-263°C (разложение).
ПРИМЕР 2
7-[3-(R)-(1-аминоциклопропил)пирролидин-1-ил]-1-[2-(S)-фтор-1-(R)-циклопропил]-1,4-дигидро-8-метокси-4-оксохинолин-3-карбоновая кислота
После добавления к сухому диметилсульфоксиду (2 мл) 3-(R)-[1-трет-бутоксикарбониламиноциклопропил]пирролидина (165 мг, 731 мкмоль) и триэтиламина (0,50 мл) добавляли 7-фтор-1-[2-(S)-фтор-1-(R)-циклопропил]-1,4-дигидро-8-метокси-4-оксохинолин-3-карбоновую кислоту (180 мг, 609 мкмоль) и кипятили с обратным холодильником в течение 13 часов в атмосфере азота. После концентрирования реакционного раствора при пониженном давлении остаток растворяли в хлороформе (100 мл). После промывки органического слоя 10% водным раствором лимонной кислоты (50 мл) органический слой сушили над безводным сульфатом натрия. После фильтрования фильтрат концентрировали при пониженном давлении и после добавления по каплям в остаток концентрированной хлористо-водородной кислоты (5 мл) при охлаждении на льду перемешивали при комнатной температуре в течение 30 минут. Затем в реакционный раствор добавляли 1 моль/л хлористо-водородную кислоту (5 мл), после промывки желтого кислого водного раствора хлороформом (50 мл×4) pH доводили до 12,0 с помощью водного раствора гидроксида натрия и нерастворимые вещества удаляли фильтрованием. После доведения pH основного водного раствора до 7,4 с помощью 1 моль/л хлористо-водородной кислоты экстрагировали хлороформом (100 мл×4). После сушки над безводным сульфатом натрия растворитель упаривали при пониженном давлении. Полученный таким образом остаток очищали препаративной хроматографией (разделение в нижнем слое смесью хлороформ:метанол:вода, 7:3:1), перекристаллизовывали из изопропилового спирта и сушили при пониженном давлении. Таким образом получали 146 мг (60%) указанного в заголовке соединения в виде желтых кристаллов.
1H ЯМР (400 МГц, 0,1 моль/л NaOD) δ: 0,56 (4H, шир.c), 1,31-1,37 (1H, м), 1,50-1,56 (1H, м), 1,77-1,78 (1H, м), 2,02-2,04 (1H, м), 2,19-2,21 (1Н, м), 3,31-3,32 (1H, м), 3,49-3,51 (3H, м), 3,50 (3H, c), 4,00-4,02 (1H, м), 4,93-4,94 и 5,09-5,10 (1H, дм, J=62,87 Гц), 7,01 (1H, c), 7,90 (1H, д, J=9,03 Гц), 8,39 (1H, д, J=3,17 Гц).
ИК (KBr, диск): 3373, 3315, 3091, 3003, 2976, 2935, 2856, 1903, 1714, 1618, 1518, 1439, 1371, 1313, 1261, 1219 см-1.
Точка плавления: 189-192°C.
[α]D 24,7= -50,83° (c 0,240, 0,1 моль/л NaOH).
Элементный анализ C21H24FN3O3:
Вычислено: C 62,83; H 6,03; N 10,47.
Найдено: C 62,50; H 6,04; N 10,26.
ПРИМЕР 3
7-[3-(R)-(1-аминоциклопропил)пирролидин-1-ил]-1-циклопропил-1,4-дигидро-8-метил-4-оксохинолин-3-карбоновая кислота
После добавления в сухой диметилсульфоксид (1 мл) 3-(R)-[1-(трет-бутоксикарбониламино)циклопропил]пирролидина (132 мг, 585 мкмоль) и триэтиламина (245 мкл, 1,76 ммоль) добавляли BF2 хелат 1-циклопропил-7-фтор-1,4-дигидро-8-метил-4-оксохинолин-3-карбоновой кислоты (181 мг, 585 мкмоль) и перемешивали при комнатной температуре в течение 87 часов в атмосфере азота. После добавления в реакционный раствор холодной воды (50 мл), фильтрования выпавших твердых веществ полученные твердые вещества суспендировали в смешанном растворителе (200 мл) этанол/вода (9:1), добавляли триэтиламин (1 мл) и затем кипятили с обратным холодильником в течение 7 часов. После концентрирования реакционного раствора при пониженном давлении остаток растворяли в хлороформе (100 мл), после промывки органического слоя 10% водным раствором лимонной кислоты (50 мл) органический слой сушили над безводным сульфатом натрия. После фильтрования фильтрат концентрировали при пониженном давлении, добавления к остатку по каплям концентрированной хлористо-водородной кислоты (2 мл) при охлаждении на льду перемешивали при комнатной температуре в течение 30 минут. Затем в реакционный раствор добавляли 1 моль/л хлористо-водородной кислоты (2 мл) и после промывки желтого кислотного водного раствора хлороформом (50 мл×3) pH доводили до 12,0 с помощью водного раствора гидроксида натрия. После доведения рН основного водного раствора с помощью 1 моль/л хлористо-водородной кислоты до 7,4 экстрагировали хлороформом (100 мл×4). После сушки над безводным сульфатом натрия растворитель упаривали при пониженном давлении. Полученный таким образом остаток очищали перекристаллизацией из этанола и затем сушили при пониженном давлении. Таким образом получали 99,6 мг (46%) указанного в заголовке соединения в виде желтых кристаллов.
1H ЯМР (400 МГц, 0,1 моль/л NaOD) δ: 0,55-0,57 (4H, м), 0,74-0,76 (1H, м), 0,90-0,92 (1H, м), 1,11-1,13 (1H, м), 1,24-1,26 (1H, м), 1,75-1,77 (1H, м), 2,03-2,05 (1H, м), 2,21-2,24 (1H, м), 2,48 (3H, c), 3,29-3,38 (3H, м), 3,53-3,55 (1H, м), 4,10-4,12 (1H, м), 7,07 (1H, c), 7,96 (1H, c), 8,57 (1H, c).
Точка плавления: 230-233°C.
Удельное вращение: [α]D 24,7=-169,35° (c 0,385, 0,1 моль/л NaOH).
Элементный анализ С21Н25N3О3:
Вычислено: С 68,48%; H 6,86%; N 11,44%.
Найдено: С 68,46%; H 6,71%; N 11,38%.
Ссылочный пример 6
Этил 2-(2,6-дихлорникотиноил)ацетат
Моноэтилмалонат (6,61 г, 50,0 ммоль) растворяли в безводном тетрагидрофуране (100 мл) и после добавления этоксида магния (3,15 г, 28,0 ммоль) при охлаждении на льду перемешивали при комнатной температуре в течение 3 часов. Затем реакционный раствор концентрировали при пониженном давлении, таким образом получали магниевую соль моноэтилмалоната. Затем в безводном тетрагидрофуране (80 мл) растворяли 2,6-дихлорникотиновую кислоту (3,84 г, 20,0 ммоль) и после добавления 1,1-карбонилдиимидазола (4,87 г, 30,0 ммоль) при охлаждении на льду перемешивали при комнатной температуре в течение 1,5 часов. В данный раствор по каплям добавляли в течение 10 минут при охлаждении на льду раствор, где предварительно полученную магниевую соль моноэтилмалоната растворяли в безводном тетрагидрофуране (160 мл). После завершения добавления температуру постепенно возвращали до комнатной температуры и затем перемешивали в течение 4 часов. После добавления в реакционный раствор этилацетата (200 мл) органический слой промывали 10% водным раствором лимонной кислоты (150 мл×2), насыщенным водным раствором бикарбоната натрия (150 мл) и насыщенным раствором соли (150 мл), в указанном порядке, и затем сушили над безводным сульфатом натрия. После фильтрования остаток, полученный концентрированием фильтрата при пониженном давлении, наносили на силикагель для проведения хроматографии и из элюата н-гексан:этилацетат = 3:1 получали 4,24 г (81%) указанного в заголовке соединения в виде бледно-розового маслянистого вещества.
1H ЯМР (400 МГц, CHCl3) δ: 1,12-1,40 (3Н, м), 4,08 (1H, c), 4,15-4,35 (2H, м), 5,72 (0,5H, c), 7,37 (1H, дд, J=14,5, 8,1 Гц), 9,49 (1H, дд, J=16,4, 8,1 Гц), 12,52 (0,5H, c).
Ссылочный пример 7
Этил 2-(2,6-дихлорникотиноил)-3-[2-(S)-фтор-1-(R)-циклопропиламино]акрилат
Этил 2-(2,6-дихлорникотиноил)ацетат (7,03 г, 26,8 ммоль) растворяли в уксусном ангидриде (30 мл) и после добавления триэтилортоформиата (60 мл) перемешивали на масляной бане при 140°C в течение 2 часов. Затем, дав реакционному раствору остыть, концентрировали при пониженном давлении и после добавления толуола (50 мл) в полученный остаток концентрировали при пониженном давлении. Эти действия повторяли 3 раза, и полученный остаток сушили при пониженном давлении. Таким образом получали 8,42 г этил 2-(2,6-дихлорникотиноил)-3-этоксиакрилата в виде желтого маслянистого вещества.
Далее, неочищенный этил 2-(2,6-дихлорникотиноил)-3-этоксиакрилат (2,11 г, 6,62 ммоль) и соль пара-толуолсульфоновой кислоты 2-(S)-фтор-1-(R)-циклопропиламина (2,45 г, 9,91 ммоль) суспендировали в дихлорметане (30 мл) и постепенно добавляли по каплям триэтиламин (2,77 мл, 19,87 ммоль) при перемешивании при -15°C. После завершения добавления реакционный раствор перемешивали при комнатной температуре в течение 15 часов. После добавления в реакционный раствор этилацетата (100 мл) органический слой промывали 10% водным раствором лимонной кислоты (80 мл), насыщенным водным раствором бикарбоната натрия (80 мл) и насыщенным хлористо-водородным раствором (80 мл), в указанном порядке, и затем сушили над безводным сульфатом натрия. После фильтрования фильтрат концентрировали при пониженном давлении и таким образом получали 2,10 г (90%, 2 процесса) указанного в заголовке соединения в виде желто-коричневого маслянистого вещества (E/Z смесь). Полученное вещество использовали в следующей реакции без дополнительной очистки.
1H ЯМР (400 МГц, CHCl3) δ: 0,85-0,89 (0,7Н, м), 1,00-1,04 (2,3H, м), 1,23-1,38 (2H, м), 3,01 (1H, м), 3,94-4,05 (2H, м), 4,65-4,84 (1H, м), 7,27-7,31 (1H, м), 7,50-7,57 (1H, м), 8,29-8,38 (1H, м), 11,02 (0,8H, шир.д, J=12,5 Гц).
Ссылочный пример 8
Этил 7-хлор-1-[2-(S)-фтор-1-(R)-циклопропил]-1,4-дигидро-4-оксо-1,8-нафтиридин-3-карбоксилат
После растворения этил 2-(2,6-дихлорникотиноил)-3-[2-(S)-фтор-1-(R)-циклопропиламино]акрилата (2,07 г, 5,97 ммоль) в 1,4-диоксане (30 мл) к нему постепенно добавляли 60% гидрид натрия в масле (287 мг, 7,18 ммоль) при перемешивании при 5°C. Затем реакционную суспензию перемешивали в течение 1,5 часов при комнатной температуре и концентрировали при пониженном давлении. После растворения остатка в хлороформе (100 мл) органический слой промывали 10% водным раствором лимонной кислоты (80 мл), насыщенным водным раствором бикарбоната натрия (80 мл) и насыщенным раствором соли (80 мл), в указанной последовательности, и затем сушили над безводным сульфатом натрия. После фильтрования фильтрат концентрировали при пониженном давлении, к полученному таким образом остатку добавляли диэтиловый эфир, и выпавшие кристаллы отфильтровывали, промывали диэтиловым эфиром и затем сушили при пониженном давлении при 60°C в течение 16 часов. Таким образом получали 1,25 г (67%) указанного в заголовке соединения в виде белого порошка.
1H ЯМР (400 МГц, CHCl3) δ: 1,41 (3Н, т, J=7,1 Гц), 1,59-1,72 (2H, м), 3,58-3,63 (1H, м), 4,41 (2H, кв, J=7,1 Гц), 4,93-5,12 (1H, м), 7,39-7,41 (1H, м), 8,65-8,68 (2H, м).
MS (m/z): 310 (M+).
Ссылочный пример 9
7-хлор-1-[2-(S)-фтор-1-(R)-циклопропил]-1,4-дигидро-4-оксо-1,8-нафтиридин-3-карбоновая кислота
Смесь этил 7-хлор-1-[2-(S)-фтор-1-(R)-циклопропил]-1,4-дигидро-4-оксо-1,8-нафтиридин-3-карбоксилата (567 мг, 1,83 ммоль), уксусной кислоты (4 мл) и концентрированной хлористо-водородной кислоты (2 мл) кипятили с обратным холодильником в течение 2,5 часов. После охлаждения реакционного раствора на льду в реакционный раствор выливали ледяную воду (20 мл), выпавшие кристаллы отфильтровывали и после промывки большим количеством воды, небольшим количеством холодного этанола и избытком диэтилового эфира кристаллы сушили при пониженном давлении при 80°C в течение 18 часов. Таким образом получали 449 мг (87%) указанного в заголовке соединения в виде белых игольчатых кристаллов.
1H ЯМР (400 МГц, CHCl3) δ: 1,70-1,80 (2Н, м), 3,73-3,79 (1H, м), 4,98-5,17 (1H, м), 7,56 (1H, д, J=8,3 Гц), 8,73 (1H, д, J=8,5 Гц), 8,97 (1H, c), 14,11 (1H, шир.c).
Точка плавления: 215-220°C.
Удельное вращение: [α]D 24,5=+26,90° (c 0,422, 0,1 моль/л NaOH).
Элементный анализ С12Н8ClFN2О3:
Вычислено: С 50,99%; H 2,85%; N 9,91%.
Найдено: С 50,90%; H 2,71%; N 9,91%.
MS (m/z): 282 (M+).
ПРИМЕР 4
7-[3-(R)-(1-аминоциклопропил)пирролидин-1-ил]-1-[2-(S)-фтор-1-(R)-циклопропил]-1,4-дигидро-4-оксо-1,8-нафтиридин-3-карбоновая кислота
После добавления в сухой ацетонитрил (10 мл) 3-(R)-[1-[N-(трет-бутоксикарбонил)амино]циклопропил]пирролидина (339 мг, 1,50 ммоль) и триэтиламина (1,39 мл) добавляли 7-хлор-1-[2-(S)-фтор-1-(R)-циклопропил]-1,4-дигидро-4-оксо-1,8-нафтиридин-3-карбоновую кислоту (283 мг, 1,00 ммоль) и смесь кипятили с обратным холодильником в течение 1,5 часов в атмосфере азота. После концентрирования реакционного раствора при пониженном давлении остаток растворяли в смешанном растворителе этилацетат (100 мл) и дихлорметан (50 мл), и после промывки органического слоя 10% водным раствором лимонной кислоты (50 мл) и насыщенным раствором соли (50 мл), в указанном порядке, органический слой сушили над безводным сульфатом натрия. После фильтрования фильтрат концентрировали при пониженном давлении, и после добавления к остатку по каплям концентрированной хлористо-водородной кислоты (15 мл) при охлаждении на льду раствор перемешивали при такой же температуре в течение 30 минут. Затем в реакционный раствор добавляли 1 моль/л хлористо-водородную кислоту (10 мл), и после промывки желтого кислого водного раствора хлороформом (50 мл×4) pH доводили до 11,0 с помощью водного раствора гидроксида натрия. После доведения рН основного водного раствора с помощью 1 моль/л хлористо-водородной кислоты до 7,4 экстрагировали нижний слой (100 мл×2) смесью хлороформ:метанол:вода, 7:3:1. Затем органические слои объединяли и после сушки органического слоя над безводным сульфатом натрия растворитель упаривали при пониженном давлении. Полученный остаток очищали перекристаллизацией из этанола и затем сушили при пониженном давлении. Таким образом получали 263 мг (74%) указанного в заголовке соединения в виде белых кристаллов.
1H ЯМР (400 МГц, 0,1 моль/л NaOD) δ: 0,49-0,55 (4H, м), 1,50-1,75 (3H, м), 1,95-2,15 (2H, м), 3,00-3,80 (5H, м), 4,90-5,15 (1H, м), 6,38 (1H, дм, J=9,1 Гц), 8,01 (1H, д, J=9,1 Гц), 8,31 (1H, c).
ИК (KBr, диск) ν: 3089, 3008, 2871, 1712, 1624, 1566, 1508, 1446, 1379, 1333, 1257, 1187, 1136, 1095, 1024, 985 см-1.
Точка плавления: 216-218°C.
Удельное вращение: [α]D 24,5=+63,50° (c 0,310, 0,1 моль/л NaOH).
Элементный анализ C19H21FN4O3:
Вычислено: С 61,28%; H 5,68%; N 15,04%.
Найдено: С 61,17%; H 5,66%; N 15,04%.
MS (m/z): 373([M+H])+.
Ссылочный пример 10
Этил 2,4-дифторбензоилацетат
Моноэтилмалонат (9,25 г, 70,0 ммоль) растворяли в безводном тетрагидрофуране в атмосфере азота (150 мл), и после добавления этоксида магния (4,17 г, 36,8 ммоль) при охлаждении на льду перемешивали при комнатной температуре в течение 1 часа. Затем реакционный раствор концентрировали при пониженном давлении, получая, таким образом, магниевую соль моноэтилмалоната. Затем 2,4-дифторбензойную кислоту (7,91 г, 50,0 ммоль) растворяли в безводном тетрагидрофуране (100 мл) и после добавления 1,1-карбонилдиимидазола (8,52 г, 52,5 ммоль) при охлаждении на льду перемешивали при комнатной температуре в течение 1 часа. В раствор, при охлаждении на льду, по каплям добавляли раствор, в котором предварительно полученную магниевую соль моноэтилмалоната растворяли в безводном тетрагидрофуране (60 мл). После завершения добавления температура постепенно возвращалась до комнатной, и затем в течение 16 часов перемешивали. После добавления в реакционный раствор толуола (100 мл) органический слой промывали 10% водным раствором лимонной кислоты (200 мл), насыщенным водным раствором бикарбоната натрия (150 мл) и насыщенным хлористо-водородным раствором (150 мл), в указанном порядке, и затем сушили над безводным сульфатом натрия. После фильтрования осадок, полученный путем концентрирования при пониженном давлении фильтрата, наносили на силикагель для проведения хроматографии, и из элюата н-гексан:этилацетат = 9:1 получали 11,0 г (95%) указанного в заголовке соединения в виде бледно-желтого маслянистого вещества.
1H ЯМР (400 МГц, CHCl3) δ: 1,24-1,36 (3Н, м), 3,95 (2H×2/3, д, J=3,66 Гц), 4,20-4,30 (2H, м), 5,80 (1H×1/3, c), 6,86-7,02 (2H, м), 7,88-8,04 (1H, м), 12,72 (1H×1/3, c).
Ссылочный пример 11
Этил 7-фтор-1-[2-(S)-фтор-1-(R)-циклопропил]-1,4-дигидро-4-оксохинолин-3-карбоксилат
Смесь этил 2,4-дифторбензоилацетата (5,50 г, 24,1 ммоль), триэтилортоформиата (8,00 мл, 48,2 ммоль) и уксусного ангидрида (6,8 мл) перемешивали на масляной бане при 120°C в течение 16 часов. После того как реакционному раствору давали возможность остыть, его концентрировали при пониженном давлении, после добавления в полученный остаток толуола (30 мл) снова концентрировали при пониженном давлении и затем сушили при пониженном давлении. Таким образом получали желтое маслянистое вещество. Вещество растворяли в толуоле (100 мл) и после добавления соли пара-толуолсульфоновой кислоты 2-(S)-фтор-1-(R)-циклопропиламина (6,46 г, 26,1 ммоль) постепенно добавляли по каплям триэтиламин (4,95 мл, 35,6 ммоль), перемешивая при -15°C. После завершения добавления реакционный раствор перемешивали при комнатной температуре в течение 18 часов. После добавления в реакционный раствор воды (150 мл) экстрагировали этилацетатом (150 мл×2). Органические слои объединяли и промывали насыщенным хлористо-водородным раствором (150 мл) и затем сушили над безводным сульфатом натрия. После фильтрования фильтрат концентрировали при пониженном давлении и таким образом получали коричневое маслянистой вещество. После растворения данного вещества в диметилформамиде (35 мл) к нему добавляли карбонат калия (6,55 г, 47,4 ммоль) и затем перемешивали при комнатной температуре в течение 21 часа. Затем, охлаждая на льду и перемешивая, к нему постепенно добавляли 2 моль/л хлористо-водородную кислоту (50 мл) и затем перемешивали при комнатной температуре в течение 6 часов. Выпавшие кристаллы отфильтровывали и затем промывали большим количеством воды, небольшим количеством холодного этанола и избытком диэтилового эфира. Полученные неочищенные кристаллы затем очищали перекристаллизацией из этилацетата и затем сушили при пониженном давлении. Таким образом получали 5,92 г (84%) указанного в заголовке соединения.
1H ЯМР (400 МГц, CHCl3) δ: 1,41-1,43 (3Н, м), 1,69-1,76 (2H, м), 3,39 (1Н, шир.c), 4,37-4,43 (2H, м), 5,09 (1H, дм, J=62,46 Гц), 7,16-7,22 (1H, м), 7,41-7,44 (1H, м), 8,49-8,57 (2H, м).
Точка плавления: 227-230°C.
Ссылочный пример 12
7-фтор-1-[2-(S)-фтор-1-(R)-циклопропил]-1,4-дигидро-4-оксохинолин-3-карбоновая кислота
Смесь этил 7-фтор-1-[2-(S)-фтор-1-(R)-циклопропил]-1,4-дигидро-4-оксохинолин-3-карбоксилата (4,08 г, 13,9 ммоль), уксусной кислоты (9 мл) и концентрированной хлористо-водородной кислоты (9 мл) кипятили с обратным холодильником в течение 21 часа. После охлаждения реакционного раствора на льду в реакционный раствор выливали ледяную воду (50 мл), выпавшие кристаллы отфильтровывали и после промывки большим количеством воды, небольшим количеством холодного этанола и избытком диэтилового эфира кристаллы сушили при пониженном давлении при 80°C в течение 16 часов. Таким образом получали 3,51 г (95%) указанного в заголовке соединения в виде белого порошка.
1H ЯМР (400 МГц, CDCl3) δ: 1,78-1,84 (2H, м), 3,52-3,53 (1H, м), 5,13 (1H, дм, J=64,59 Гц), 7,31-7,36 (1H, м), 7,59 (1H, д, J=9,26 Гц), 8,54-8,53 (1H, м), 14,55 (1H, c).
Точка плавления: 302-305°C.
Удельное вращение: [α]D 24,3=+0,38° (c 0,560, 0,1 моль/л NaOH).
ПРИМЕР 5
7-[3-(R)-(1-аминоциклопропил)пирролидин-1-ил]-1-[2-(S)-фтор-1-(R)-циклопропил]-1,4-дигидро-4-оксохинолин-3-карбоновая кислота
После добавления в сухой диметилсульфоксид (1 мл) 3-(R)-[1-[N-(трет-бутоксикарбонил)амино]циклопропил]пирролидина (203 мг, 817 мкмоль) и триэтиламина (0,5 мл) добавляли 7-фтор-1-[2-(S)-фтор-1-(R)-циклопропил]-1,4-дигидро-4-оксохинолин-3-карбоновую кислоту (197 мг, 743 мкмоль), и смесь кипятили с обратным холодильником в течение 15 часов в атмосфере азота. После того как реакционному раствору давали возможность остыть, в реакционный раствор добавляли воду (30 мл) при охлаждении на льду, и выпавшие кристаллы отфильтровывали и тщательно промывали водой. После добавления к полученным кристаллам концентрированной хлористо-водородной кислоты (5 мл) при охлаждении на льду перемешивали при той же температуре в течение 30 минут. Затем в реакционный раствор добавляли 1 моль/л хлористо-водородную кислоту (10 мл), и после промывки желтого кислого водного раствора хлороформом (50 мл×2) pH доводили до 12,0 с помощью раствора гидроксида натрия. После доведения рН основного водного раствора до 7,4 с помощью 1 моль/л хлористо-водородной кислоты экстрагировали хлороформом (100 мл×3) и смесью хлороформ:метанол, 95:5 (100 мл×2). Органические слои затем объединяли и после сушки органического слоя над безводным сульфатом натрия растворитель упаривали при пониженном давлении. Полученный остаток очищали перекристаллизацией из этанола и затем сушили при пониженном давлении. Таким образом получали 203 мг (74%) указанного в заголовке соединения в виде желтых кристаллов.
1H ЯМР (400 МГц, 0,1 моль/л NaOD) δ: 0,50-0,54 (4H, м), 1,63-1,68 (3H, м), 2,00-2,12 (2H, м), 2,94-2,97 (1H, м), 3,16-3,36 (4H, м), 5,16 (1H, дм, J=62,40 Гц), 6,43 (1H, c), 6,67 (1H, д, J=9,02 Гц), 7,97 (1H, д, J=9,02 Гц), 8,32 (1H, c).
ИК (KBr, диск) ν: 3087, 3008, 2951, 2858, 1699, 1681, 1520, 1471, 1458, 1396, 1363, 1371, 1250 см-1.
Точка плавления: 251-253°C.
Удельное вращение: [α]D 24,3=+41,90° (c 0,160, 1 моль/л NaOH).
Элементный анализ С20Н22FN3О3:
Вычислено: С 64,68%; H 5,97%; N 11,31%.
Найдено: С 64,69%; H 5,96%; N 11,25%.
Ссылочный пример 13
BF2 хелат 7-бром-1-циклопропил-8-дифторметокси-1,4-дигидро-4-оксохинолин-3-карбоновой кислоты
Нагревая и перемешивая смешанный раствор этил 7-бром-1-циклопропил-8-дифторметокси-1,4-дигидро-4-оксохинолин-3-карбоксилата (2,01 г, 5,00 ммоль), уксусной кислоты (5 мл) и уксусного ангидрида (5 мл) на масляной бане при 110°C, добавляли по каплям комплекс трифторид бора-тетрагидрофуран (0,83 мл, 7,50 ммоль) в течение 5 минут. После перемешивания реакционного раствора при такой же температуре в течение 1,5 часов добавляли избыток диэтилового эфира при охлаждении на льду и выпавшие твердые вещества отфильтровывали (и промывали диэтиловым эфиром). После сушки при пониженном давлении при комнатной температуре получали 2,06 г (98%) указанного в заголовке соединения в виде бледно-серого порошка.
1Н ЯМР (400 МГц, ДМСО-d6) δ: 1,15-1,30 (4H, м), 4,43 (1H, м), 7,20 (1H, т, J=71,9 Гц), 8,25 (1H, д, J=8,8 Гц), 8,38 (1H, д, J=8,8 Гц), 9,36 (1H, c).
ПРИМЕР 6
7-[3-(R)-(1-аминоциклопропил)пирролидин-1-ил]-1-циклопропил-8-дифторметокси-1,4-дигидро-4-оксохинолин-3-карбоновая кислота
После добавления в сухой диметилсульфоксид (2 мл) 3-(R)-[1-(трет-бутоксикарбониламино)циклопропил]пирролидина (338 мг, 1,50 ммоль) и триэтиламина (209 мкл, 1,50 ммоль) добавляли хелат BF2 7-бром-1-циклопропил-8-дифторметокси-1,4-дигидро-4-оксохинолин-3-карбоновой кислоты (422 мг, 1,00 ммоль) и перемешивали при комнатной температуре в течение 39 часов в атмосфере азота. После концентрирования реакционного раствора при пониженном давлении в концентрат добавляли этанол (20 мл), триэтиламин (4 мл) и воду (4 мл) и кипятили с обратным холодильником в течение 3 часов. После концентрирования реакционного раствора при пониженном давлении остаток растворяли в хлороформе (100 мл), и после промывки органического слоя 10% водным раствором лимонной кислоты (50 мл) органический слой сушили над безводным сульфатом натрия. После фильтрования фильтрат концентрировали при пониженном давлении, и после добавления по каплям в остаток концентрированной хлористо-водородной кислоты (5 мл) при охлаждении на льду перемешивали при комнатной температуре в течение 30 минут. Затем в реакционный раствор добавляли 3 моль/л хлористо-водородную кислоту (30 мл) и после промывки желтого кислого водного раствора хлороформом (50 мл×2) pH доводили до 11,0 с помощью водного раствора гидроксида натрия. После доведения рН основного водного раствора до 7,4 с помощью 1 моль/л хлористо-водородной кислоты экстрагировали хлороформом (50 мл×2). После сушки над безводным сульфатом натрия растворитель упаривали при пониженном давлении. Полученный таким образом остаток очищали перекристаллизацией из смешанного растворителя этанол/диэтиловый эфир и затем сушили при пониженном давлении. Таким образом получали 31 мг (8%) указанного в заголовке соединения в виде желтого порошка.
1H ЯМР (400 МГц, 0,1 моль/л NaOD) δ: 0,57 (4Н, шир.с), 0,81 (1Н, м), 1,03 (1Н, м), 1,11 (1Н, м), 1,25 (1Н, м), 1,78 (1Н, м), 2,05 (1Н, м), 2,22 (1Н, м), 3,35-3,60 (4Н, м), 4,08 (1Н, м), 6,45 (1Н, дд, J=76,3, 73,8 Гц), 7,07 (1Н, д, J=9,3 Гц), 8,00 (1Н, д, J=9,3 Гц), 8,46 (1Н, с).
Точка плавления: 206-207,5°C.
Удельное вращение: [α]D 24,5=-67,70° (c 0,295, 0,1 моль/л NaOH).
Элементный анализ C21H23F2N3O4·0,25CH3CH2OH.
Вычислено: С 59,92%; H 5,73%; N 9,75%.
Найдено: С 59,85%; H 5,62%; N 9,68%.
Ссылочный пример 14
Этил 6-амино-7-фтор-1-[2-(S)-фтор-1-(R)-циклопропил]-1,4-дигидро-8-метил-5-нитро-4-оксохинолин-3-карбоксилат
Этил 6,7-дифтор-1-[2-(S)-фтор-1-(R)-циклопропил]-1,4-дигидро-8-метил-5-нитро-4-оксохинолин-3-карбоксилат (10,04 г, 27,12 ммоль) растворяли в диметилформамиде (150 мл) и добавляли по каплям 28% водный раствор аммиака (32,1 мл) при перемешивании и охлаждении на льду. Герметизируя реакционный раствор в герметичной пробирке, его перемешивали при комнатной температуре в течение 4 часов. Реакционный раствор затем растворяли в метаноле (200 мл) и концентрировали при пониженном давлении. Полученный остаток очищали перекристаллизацией из смешанного растворителя 2-пропанол/хлороформ/28% водный раствор аммиака и после сушки при пониженном давлении получали 7,07 г (71%) указанного в заголовке соединения в виде желтого порошка.
1H ЯМР (400 МГц, CDCl3) δ: 1,35-1,44 (5Н, м), 2,67 (3Н, д, J=3,41 Гц), 3,81-3,87 (1Н, м), 4,33-4,41 (3H, м), 4,75-4,78 (0,5H, м), 4,90-4,94 (0,5H, м), 8,47 (1H, д, J=3,41 Гц).
Элементный анализ C16H15F2N3O5:
Вычислено: С 52,32%; H 4,12%; N 11,44%.
Найдено: С 52,62%; H 4,16%; N 11,12%.
Ссылочный пример 15
Этил 7-фтор-1-[2-(S)-фтор-1-(R)-циклопропил]-1,4-дигидро-8-метил-5-нитро-4-оксохинолин-3-карбоксилат
Изоамилнитрит (2,56 мл, 19,1 ммоль) добавляли в диметилформамид (40 мл) и, перемешивая при 65°C, в течение 3 часов по каплям добавляли раствор, в котором этил 6-амино-7-фтор-1-[2-(S)-фтор-1-(R)-циклопропил]-1,4-дигидро-8-метил-5-нитро-4-оксохинолин-3-карбоксилат (5,00 г, 13,6 ммоль) растворен в диметилформамиде (60 мл). После завершения добавления реакционный раствор перемешивали при 65°C в течение 4 часов, оставляли охлаждаться и затем выливали в воду (500 мл). После экстракции хлороформом (200 мл×3) объединенные органические слои промывали 1 моль/л хлористо-водородной кислотой (200 мл) и насыщенным раствором соли (100 мл×2), в указанной последовательности, и затем сушили над безводным сульфатом натрия. После фильтрования фильтрат концентрировали при пониженном давлении и полученный остаток наносили на силикагель для проведения хроматографии с получением из элюата хлороформ:метанол = 30:1 2,91 г (61%) указанного в заголовке соединения в виде белого порошка.
1H ЯМР (400 МГц, CDCl3) δ: 1,37 (3Н, т, J=7,08 Гц), 1,40-1,67 (2H, м), 2,70 (3H, д, J=2,93 Гц), 3,89-3,93 (1H, м), 4,34-4,40 (2H, м), 4,79-4,83 (0,5H, м), 4,95-4,98 (0,5H, м), 8,55 (1H, д, J=2,93 Гц).
Ссылочный пример 16
Этил 5-амино-7-фтор-1-[2-(S)-фтор-1-(R)-циклопропил]-1,4-дигидро-8-метил-4-оксохинолин-3-карбоксилат
Этил 7-фтор-1-[2-(S)-фтор-1-(R)-циклопропил]-1,4-дигидро-8-метил-5-нитро-4-оксохинолин-3-карбоксилат (2,50 г, 7,10 ммоль) растворяли в ацетонитриле (20 мл) и после добавления катализатора 5% палладий на углероде (содержание воды: 50%, 1,0 г) перемешивали при комнатной температуре в течение 20 часов в атмосфере водорода при атмосферном давлении. После удаления катализатора путем фильтрования (промывка метанолом) фильтрат концентрировали при пониженном давлении, полученный остаток растворяли в этилацетате (10 мл) и кипятили с обратным холодильником в течение 30 минут. Затем добавляли н-гексан (10 мл), и после кипячения с обратным холодильником в течение 30 минут реакционный раствор оставляли при комнатной температуре. Выпавшие кристаллы затем отфильтровывали, промывали смесью н-гексан:этилацетат, 1:1 и сушили при пониженном давлении при 60°C в течение 16 часов. Таким образом получали 869 мг (38%) указанного в заголовке соединения в виде желтого порошка.
1H ЯМР (400 МГц, CDCl3) δ: 1,23-1,36 (1Н, м), 1,38 (3Н, т, J=7,08 Гц), 1,43-1,56 (1Н, м), 2,39 (3Н, д, J=2,20 Гц), 3,70-3,77 (1Н, м), 4,37 (2Н, кв, J=7,08 Гц), 4,71-4,75 (0,5Н, м), 4,87-4,90 (0,5Н, м), 6,20 (1Н, д, J=11,96 Гц), 8,37 (1Н, д, J=3,43 Гц).
Ссылочный пример 17
5-амино-7-фтор-1-[2-(S)-фтор-1-(R)-циклопропил]-1,4-дигидро-8-метил-4-оксохинолин-3-карбоновая кислота
Этил 5-амино-7-фтор-1-[2-(S)-фтор-1-(R)-циклопропил]-1,4-дигидро-8-метил-4-оксохинолин-3-карбоксилат (735 мг, 2,28 ммоль) растворяли в смешанном растворе (8 мл) уксусная кислота:вода, 1:1, и после добавления концентрированной серной кислоты (90 мкл) перемешивали в течение 4 часов на масляной бане при 120°C. После охлаждения реакционного раствора на льду в него выливали воду (20 мл), и смешанный реакционный раствор перемешивали при комнатной температуре в течение 2 часов. Выпавшие кристаллы отфильтровывали, промывали большим количеством воды, небольшим количеством холодного этанола и избытком диэтилового эфира, в указанном порядке, и затем сушили при пониженном давлении при 80°C в течение 17 часов. Таким образом получали 552 мг (82%) указанного в заголовке соединения в виде желтых кристаллов.
1Н ЯМР (400 МГц, ДМСО-d6) δ: 1,23-1,38 (1H, м), 1,56-1,66 (1H, м), 2,39 (3H, д, J=2,20 Гц), 4,14-4,22 (1H, м), 4,96-5,00 (0,5H, м), 5,12-5,16 (0,5H, м), 6,50 (1H, д, J=12,70 Гц), 8,60 (1H, д, J=3,17 Гц).
Удельное вращение: [α]D 24,3=-111,00° (c 0,510, 0,1 моль/л NaOH).
ПРИМЕР 7
5-амино-7-[3-(R)-(1-аминоциклопропил)пирролидин-1-ил]-1-[2-(S)-фтор-1-(R)-циклопропил]-1,4-дигидро-8-метил-4-оксохинолин-3-карбоновая кислота
После добавления к высушенному диметилсульфоксиду (1 мл) 3-(R)-[1-[N-(трет-бутоксикарбонил)амино]циклопропил]пирролидина (643 мг, 2,55 ммоль) и триэтиламина (0,5 мл), добавляли 5-амино-7-фтор-1-[2-(S)-фтор-1-(R)-циклопропил]-1,4-дигидро-8-метил-4-оксохинолин-3-карбоновую кислоту (250 мг, 850 мкмоль) и перемешивали при 70°C в течение 37 часов в герметичных условиях в атмосфере азота. Затем реакционному раствору давали возможность остыть, реакционный раствор концентрировали при пониженном давлении, полученный остаток растворяли в этилацетате (100 мл) и промывали 10% водным раствором лимонной кислоты (50 мл) и насыщенным раствором соли (30 мл). Органический слой затем сушили над безводным сульфатом натрия и после фильтрования фильтрат концентрировали при пониженном давлении. Полученный остаток затем наносили на короткую колонку с силикагелем для проведения хроматографии и неочищенные кристаллы получали из элюата хлороформ:метанол = 30:1. После добавления по каплям к неочищенным кристаллам концентрированной хлористо-водородной кислоты (5 мл) при охлаждении на льду раствор перемешивали при такой же температуре в течение 30 минут. Затем в реакционный раствор добавляли 1 моль/л хлористо-водородную кислоту (10 мл) и после промывки желтого кислого водного раствора хлороформом (50 мл×2) pH доводили до 12,0 с помощью водного раствора гидроксида натрия. После доведения рН основного водного раствора до 7,4 с помощью 1 моль/л хлористо-водородной кислоты экстрагировали хлороформом (100 мл×3). Органические слои затем объединяли, сушили над безводным сульфатом натрия, и затем растворитель упаривали при пониженном давлении. Полученный таким образом остаток очищали перекристаллизацией из этанола и затем сушили при пониженном давлении. Таким образом получали 32 мг (9%) указанного в заголовке соединения в виде желтых кристаллов.
1H ЯМР (400 МГц, 0,1 моль/л NaOD) δ: 0,54-0,57 (1H, м), 0,60-0,67 (1H, м), 1,23-1,55 (3H, м), 1,74-1,85 (1H, м), 1,97-2,17 (2H, м), 2,33 (3H, c), 3,18-3,27 (2H, м), 3 43-3,47 (1H, м), 3,54-3,63 (1H, м), 3,71-3,78 (1H, м), 4,77-4,79 (0,5H, м), 4,93-4,96 (0,5H, м), 6,00 (1H, c), 8,56 (1H, д, J=3,66 Гц).
ИК (KBr, диск) ν: 3402, 3344, 3276, 3097, 2918, 2864, 1724, 1616, 1548, 1506, 1477, 1441, 1408 см-1.
Точка плавления: 240-242°C (разложение).
Удельное вращение: [α]D 23,5=-225,91° (с 0,525, 0,1 моль/л NaOH).
Элементный анализ C21H25FN4О3:
Вычислено: С 62,99%; H 6,29%; N 13,99%.
Найдено: С 62,86%; H 6,38%; N 13,76%.
Ссылочный пример 18
Этил 6-амино-7-фтор-1-[2-(S)-фтор-1-(R)-циклопропил]-1,4-дигидро-8-метокси-5-нитро-4-оксохинолин-3-карбоксилат
Этил 6,7-дифтор-1-[2-(S)-фтор-1-(R)-циклопропил]-1,4-дигидро-8-метокси-5-нитро-4-оксохинолин-3-карбоксилат (13,96 г, 36,14 ммоль) растворяли в диметилформамиде (180 мл) и по каплям добавляли 28% водный раствор аммиака (60 мл) при перемешивании и охлаждении на льду. После перемешивания реакционного раствора при комнатной температуре в течение 64 часов в реакционный раствор добавляли воду (100 мл) и затем концентрировали при пониженном давлении. Полученный остаток, содержащий воду, затем экстрагировали этилацетатом (100 мл×3) и органические слои затем объединяли, промывали водой (150 мл×3) и насыщенным раствором соли (200 мл), в указанном порядке, и затем сушили над безводным сульфатом натрия. После фильтрования фильтрат концентрировали при пониженном давлении, полученный остаток наносили на силикагель для проведения хроматографии и из элюата хлороформ:метанол = 30:1, получали 8,92 г (64%) соединения, указанного в заголовке, в виде бледно-красного, маслянистого вещества.
1H ЯМР (400 МГц, CDCl3) δ: 1,37 (3H, т, J=7,0 Гц), 1,49 (1H, ддд, J=9,5, 6,0, 3,5 Гц), 1,53-1,58 (1H, м), 3,68 (1H, дт, J=8,5, 5,5 Гц), 4,11 (3H, д, J=2,5 Гц), 4,36 (2H, дкв, J=7,0, 1,5 Гц), 4,51 (2H, шир.), 4,83 (1H, дд т, J=63,3, 5,5, 3,5 Гц), 8,47 (1H, с).
ИК (KBr, диск) ν: 3379, 1724, 1608, 1525, 1471, 1323, 1259, 1063 см-1.
HRMS (FAB): C16H16F2N3O6 (M++1).
Вычислено: 384,1007.
Найдено: 384,0974.
Ссылочный пример 19
Этил 7-фтор-1-[2-(S)-фтор-1-(R)-циклопропил]-1,4-дигидро-8-метокси-5-нитро-4-оксохинолин-3-карбоксилат
Изоамилнитрил (3,81 г, 32,5 ммоль) добавляли в диметилформамид (60 мл) и при перемешивании при 70°C добавляли по каплям раствор, в котором этил 6-амино-7-фтор-1-[2-(S)-фтор-1-(R)-циклопропил]-1,4-дигидро-8-метокси-5-нитро-4-оксохинолин-3-карбоксилат (8,90 г, 23,2 ммоль) растворен в диметилформамиде (120 мл), в течение 3 часов. После завершения добавления реакционный раствор перемешивали при 70°C в течение 1 часа, оставляли охлаждаться и затем выливали в воду (500 мл). После экстрагирования этилацетатом (300 мл×3) объединенные органические слои промывали 1 моль/л хлористо-водородной кислотой (300 мл) и насыщенным раствором соли (200 мл×2), в указанном порядке, и затем сушили над безводным сульфатом натрия. После фильтрования фильтрат концентрировали при пониженном давлении, полученные неочищенные кристаллы перекристаллизовывали этанолом и затем сушили при пониженном давлении. Таким образом получали 3,81 г (45%) указанного в заголовке соединения в виде желтых кристаллов.
1H ЯМР (400 МГц, CDCl3) δ: 1,37 (3H, т, J=7,0 Гц), 1,55 (1Н, ддд, J=9,5, 6,0, 3,5 Гц), 1,59-1,69 (1Н, м), 3,93 (1Н, дт, J=8,5, 5,5 Гц), 4,11 (3Н, т, J=2,5 Гц), 4,37 (2Н, дкв, J=7,0, 1,5 Гц), 4,86 (1Н, дддд, J=63,0, 6,0, 5,5, 3,5 Гц), 7,20 (1Н, д, J=10,0 Гц), 8,55 (1Н, д, J=1,5 Гц).
ИК (KBr, диск) ν: 3062, 1722, 1639, 1602, 1544, 1425, 1328, 1259, 1057 см-1.
Точка плавления: 167-170°C (разложение).
Элементный анализ: C16H14F2N2O6:
Вычислено: С 52,18%; H 3,89%; N 7,61%.
Найдено: С 51,97%; H 3,78%; N 7,56%.
Ссылочный пример 20
Этил 5-амино-7-фтор-1-[2-(S)-фтор-1-(R)-циклопропил]-1,4-дигидро-8-метокси-4-оксохинолин-3-карбоксилат
Этил 7-фтор-1-[2-(S)-фтор-1-(R)-циклопропил]-1,4-дигидро-8-метокси-5-нитро-4-оксохинолин-3-карбоксилат (3,71 г, 10,1 ммоль) растворяли в ацетонитриле (50 мл) и затем после добавления катализатора 5% палладия на углероде (содержание воды: 50%, 1,5 г) перемешивали при комнатной температуре в течение 20 часов в атмосфере водорода при атмосферном давлении. После удаления катализатора путем фильтрования (промывка метанолом) фильтрат концентрировали при пониженном давлении, полученный остаток наносили на силикагель для поведения хроматографии и из элюата хлороформ:метанол = 30:1 получали 2,68 г (79%) указанного в заголовке соединения в виде желтого аморфного вещества.
1H ЯМР (400 МГц, CDCl3) δ: 1,38 (3H, т, J=7,0 Гц), 1,43-1,57 (2H, м), 3,75-3,82 (4H, м), 4,37 (2H, кв, J=7,0 Гц), 4,81 (1H, ддт, J=62,5, 6,5, 3,5 Гц), 6,24 (1H, д, J=13,0 Гц), 8,37 (1H, д, J=2,0 Гц).
HRMS (FAB): C16H17F2N2O4 (M++1):
Вычислено: 339,1156.
Найдено: 339,1150.
Ссылочный пример 21
5-амино-7-фтор-1-[2-(S)-фтор-1-(R)-циклопропил]-1,4-дигидро-8-метокси-4-оксохинолин-3-карбоновая кислота
Смесь этил 5-амино-7-фтор-1-[2-(S)-фтор-1-(R)-циклопропил]-1,4-дигидро-8-метокси-4-оксохинолин-3-карбоксилата (2,68 г, 7,92 ммоль), уксусной кислоты (20 мл) и концентрированной хлористо-водородной кислоты (20 мл) кипятили с обратным холодильником в течение 3 часов. После охлаждения реакционного раствора на льду в него выливали воду (200 мл) и выпавшие кристаллы отфильтровывали. После промывки большим количеством воды, небольшим количеством холодного этанола и избытком диэтилового эфира, в указанном порядке, полученные неочищенные кристаллы очищали перекристаллизацией из смешанного растворителя хлороформ/метанол и затем сушили при пониженном давлении. Таким образом получали 1,26 г (51%) указанного в заголовке соединения в виде желтых кристаллов.
1Н ЯМР (400 МГц, ДМСО-d6) δ: 1,54-1,64 (2H, м), 3,76 (3H, c), 4,02-4,07 (1H, м), 4,89-5,10 (1H, м), 6,59 (1H, д, J=14,0 Гц), 7,73 (2H, шир.), 8,57 (1H, д, J=1,5 Гц).
ИК (KBr, диск) ν: 3432, 3328, 1699, 1576, 1518, 1281, 1236 см-1.
Точка плавления: 291-298°C (разложение).
Специфическая ротация: [α]D 25,0=+40,01° (c 0,305, 0,1 моль/л NaOH).
Элементный анализ: C14Н12F2N2O4:
Вычислено: С 54,20%; H 3,90%; N 9,03%.
Найдено: С 54,10%; H 3,86%; N 9,02%.
ПРИМЕР 8
5-амино-7-[3-(R)-(1-аминоциклопропил)пирролидин-1-ил]-1-[2-(S)-фтор-1-(R)-циклопропил]-1,4-дигидро-8-метокси-4-оксохинолин-3-карбоновая кислота
После добавления к высушенному диметилсульфоксиду (1 мл) 3-(R)-[1-[N-(трет-бутоксикарбонил)амино]циклопропил]пирролидина (788 мг, 3,48 ммоль) и триэтиламина (2 мл) добавляли 5-амино-7-фтор-1-[2-(S)-фтор-1-(R)-циклопропил]-1,4-дигидро-8-метокси-4-оксохинолин-3-карбоновую кислоту (621 мг, 2,00 моль) и перемешивали при 90°C в течение 168 часов, герметизируя в герметичной пробирке в атмосфере азота. После того как реакционному раствору давали остыть и последующего концентрирования реакционного раствора при пониженном давлении полученный остаток растворяли в хлороформе (200 мл) и промывали 10% водным раствором лимонной кислоты (100 мл). Затем органический слой сушили над безводным сульфатом натрия и после фильтрования фильтрат концентрировали при пониженном давлении. После добавления по каплям к полученному остатку концентрированной хлористо-водородной кислоты (10 мл) при охлаждении на льду его перемешивали при той же температуре в течение 30 минут. Затем в реакционный раствор добавляли 1 моль/л хлористо-водородную кислоту (20 мл) и после промывки желтого кислого водного раствора хлороформом (50 мл×3) pH доводили до 12,0 с помощью водного раствора гидроксида натрия. После доведения рН основного водного раствора до 7,8 с помощью 1 моль/л хлористо-водородной кислоты экстрагировали хлороформом (100 мл×3). Затем органические слои объединяли, сушили над безводным сульфатом натрия и затем растворитель упаривали при пониженном давлении. Полученный таким образом остаток очищали перекристаллизацией из смешанного растворителя этанол и диэтиловый эфир и затем сушили при пониженном давлении. Таким образом получали 74 мг (9%) указанного в заголовке соединения в виде желтых кристаллов.
1H ЯМР (400 МГц, 0,1 моль/л NaOD) δ: 0,48-0,53 (4H, м), 1,09-1,21 (1H, м), 1,32-1,43 (1H, м), 1,64-1,75 (1H, м), 1,93-2,01 (1H, м), 2,10-2,23 (1H, м), 3,21-3,23 (1H, м), 3,21-2,27 (1H, м), 3,36-3,43 (6H, м), 3,79-3,84 (1H, м), 4,85-4,84 (1H, м), 4,85-5,04 (1H, м), 6,06 (1H, c), 8,01 (1H, д, J=3,5 Гц).
ИК (KBr, диск) ν: 3454, 3410, 1716, 1617, 1577, 1548, 1511, 1232, 1016 см-1.
Точка плавления: 172-178°C (разложение).
Элементный анализ: C21H25FN4O4·0,75 H2O:
Вычислено: С 58,66%; H 6,21%; N 13,03%.
Найдено: С 58,58%; H 6,02%; N 12,76%.
Ссылочный пример 22
Этил 3-диметиламино-2-(2,3,4-трифторбензоил)акрилат
Смешанный раствор 2,3,4-трифторбензойной кислоты (10,3 г, 58,5 ммоль), тионилхлорида (6,4 мл, 87,8 ммоль) и каталитического количества диметилформамида кипятили с обратным холодильником в течение 30 минут. Затем реакционный раствор оставляли охлаждаться, реакционный раствор концентрировали при пониженном давлении, к остатку добавляли толуол (30 мл) и вновь концентрировали при пониженном давлении. Полученный остаток затем растворяли в тетрагидрофуране (20 мл), и полученный раствор добавляли в тетрагидрофурановый (40 мл) раствор этил β-диметиламиноакрилата (9,20 г, 64,3 ммоль) и триэтиламина (10,2 мл, 73,1 ммоль) при перемешивании и охлаждении на льду. Реакционную смесь затем перемешивали при комнатной температуре в течение 1,5 часов и затем кипятили с обратным холодильником в течение 16,5 часов. После того как реакционному раствору давали остыть, выпавшие твердые вещества удаляли фильтрованием и фильтрат концентрировали при пониженном давлении. Остаток затем наносили на силикагель для проведения хроматографии и из элюата н-гексан:этилацетат = 3:1 получали 15,1 г (86%) указанного в заголовке соединения.
1H ЯМР (400 МГц, CDCl3) δ: 1,00 (3H, т, J=7,1 Гц), 2,88 (3H, шир.с), 3,33 (3H, шир.c), 4,01 (2H, кв, J=7,1 Гц), 6,95-7,01 (1H, м), 7,34 (1H, шир. c), 7,80 (1H, c).
Ссылочный пример 23
Этил 10-фтор-2,3-дигидро-3-(S)-метил-7-оксо-7H-пиридо[1,2,3-de][1,4]бензоксазин-6-карбоксилат
Этил 3-диметиламино-2-(2,3,4-трифторбензоил)акрилат (15,0 г, 49,8 ммоль) растворяли в этаноле (30 мл) и в раствор по каплям добавляли этанольный (10 мл) раствор (S)-2-амино-1-пропанола (4,50 г, 59,8 ммоль) при перемешивании и охлаждении на льду. После завершения добавления реакционный раствор перемешивали в течение 1 часа при комнатной температуре и затем концентрировали при пониженном давлении. Полученный остаток растворяли в диметилсульфоксиде (50 мл) и после добавления высушенного распылением фторида кальция (16 г) перемешивали при 120°C в течение 26 часов. После того как реакционной суспензии давали остыть, реакционную суспензию концентрировали при пониженном давлении, к остатку добавляли хлороформ (200 мл) и воду (200 мл), и после проведения разделения водный слой экстрагировали, используя хлороформ (200 мл). Затем объединенные органические слои промывали насыщенным раствором соли (100 мл) и затем сушили над безводным сульфатом натрия. После фильтрования фильтрат концентрировали при пониженном давлении, полученный остаток наносили на силикагель для проведения хроматографии и из элюата хлороформ:метанол = 50:1 получали 5,60 г (39%) указанного в заголовке соединения в виде белого порошка.
1H ЯМР (400 МГц, CDCl3) δ: 1,41 (3H, т, J=7,1 Гц), 1,61 (3H, д, J=7,1 Гц), 4,33-4,44 (5H, м), 7,18 (1H, т, J=10,0 Гц), 8,06 (1H, дд, J=10,0, 5,4 Гц), 8,39 (1H, c).
Ссылочный пример 24
10-фтор-2,3-дигидро-3-(S)-метил-7-оксо-7H-пиридо[1,2,3-de][1,4]бензоксазин-6-карбоновая кислота
Смесь этил 10-фтор-2,3-дигидро-3-(S)-метил-7-оксо-7H-пиридо[1,2,3-de][1,4]бензоксазин-6-карбоксилата (5,60 г, 19,2 ммоль), уксусной кислоты (25 мл) и концентрированной хлористо-водородной кислоты (25 мл) кипятили с обратным холодильником в течение 4 часов. После охлаждения реакционного раствора на льду добавляли воду (100 мл), выпавшие кристаллы отфильтровывали и затем промывали большим количеством воды, небольшим количеством холодного этанола и избытком диэтилового эфира, в указанном порядке. Затем полученные неочищенные кристаллы суспендировали в этаноле (40 мл) и перемешивали при комнатной температуре. Кристаллы отфильтровывали, промывали этанолом и затем сушили при пониженном давлении при 80°C в течение 17 часов. Таким образом получали 4,10 г (81%) указанного в заголовке соединения в виде белого порошка.
1Н ЯМР (ДМСО-d6) δ: 1,47 (3H, д, J=6,8 Гц), 4,44 (1H, д, J=9,6 Гц), 4,62 (1H, д, J=9,6 Гц), 4,99 (1H, подобный кв, J=6,8 Гц), 7,59 (1H, т, J=9,1 Гц), 7,95 (1H, дд, J=9,1, 5,4 Гц, 9,07 (1H, с).
Элементный анализ для C13H10FNO4:
Вычислено: С 59,32%; H 3,83%; N 7,22%.
Найдено: С 59,60%; H 3,95%; N 6,99%.
ПРИМЕР 9
10-[3-(R)-(1-аминоциклопропил)пирролидин-1-ил]-2,3-дигидро-3-(S)-метил-7-оксо-7H-пиридо[1,2,3-de][1,4]бензоксазин-6-карбоновая кислота
После добавления к высушенному диметилсульфоксиду (3 мл) 3-(R)-[1-(трет-бутоксикарбониламино)циклопропил]пирролидина (252 мг, 1,12 ммоль) и триэтиламина (0,50 мл), добавляли 10-фтор-2,3-дигидро-3-(S)-метил-7-оксо-7H-пиридо[1,2,3-de][1,4]бензоксазин-6-карбоновую кислоту (244 мг, 928 мкмоль) и перемешивали при нагревании на масляной бане при 100°C в течение 18 часов в атмосфере азота. После концентрирования реакционного раствора при пониженном давлении остаток растворяли в хлороформе (100 мл). После промывки органического слоя 10% водным раствором лимонной кислоты (50 мл) и насыщенным раствором соли (50 мл) органический слой сушили над безводным сульфатом натрия. После фильтрования фильтрат концентрировали при пониженном давлении и после добавления по каплям к остатку концентрированной хлористо-водородной кислоты (6 мл) при охлаждении на льду перемешивали при комнатной температуре в течение 30 минут. После добавления в реакционный раствор 4 мл воды и промывки кислого водного раствора хлороформом (10 мл×3) pH доводили до 12,0 водным раствором гидроксида натрия. После доведения рН основного водного раствора до 7,4 с помощью 1 моль/л хлористо-водородной кислоты экстрагировали хлороформом (100 мл×3). После сушки над безводным сульфатом натрия растворитель упаривали при пониженном давлении. Таким образом полученный остаток очищали перекристаллизацией из этанола и затем сушили при пониженном давлении. Таким образом получали 125 мг (36,5%) указанного в заголовке соединения в виде желтых кристаллов.
1H ЯМР (400 МГц, 0,1 моль/л NaOD) δ: 0,57 (4H, c), 1,54 (3H, д, J=6,80 Гц), 1,66-1,78 (1H, м), 2,01-2,11 (1H, м), 2,19-2,30 (1H, м), 3,38-3,60 (4H, м), 4,25 (1H, д, J=11,0 Гц), 4,47 (1H, д, J=11,0 Гц), 4,55-4,63 (1H, м), 7,11 (1H, д, J=9,03 Гц), 7,81 (1H, д, J=9,03 Гц), 8,32 (1H, c).
ИК (KBr, диск) ν: 1634, 1529, 1446, 1429, 1363, 1269, 1227, 798 см-1.
Точка плавления: 249-252°C (разложение).
Элементный анализ для C20H23N3O4·HCl·0,5 H2O:
Вычислено: С 57,90%; H 6,07%; N 10,13%.
Найдено: С 57,65%; H 5,87%; N 9,97%.
ПРИМЕР 10
1-[2-(S)-фтор-1-(R)-циклопропил]-8-метил-7-[3-(R)-[1-(метиламино)циклопропил]пирролидин-1-ил]-1,4-дигидро-4-оксохинолин-3-карбоновая кислота
После добавления к высушенному диметилсульфоксиду (1 мл) 3-(R)-[1-[N-(трет-бутоксикарбонил)-N-(метил)амино]циклопропил]пирролидина (118 мг, 436 мкмоль) и триэтиламина (0,50 мл), добавляли 7-фтор-1-[2-(S)-фтор-1-(R)-циклопропил]-8-метил-1,4-дигидро-4-оксохинолин-3-карбоновую кислоту (122 мг, 436 мкмоль) и кипятили с обратным холодильником на масляной бане при 100°C в течение 18 часов в атмосфере азота. После концентрирования реакционного раствора при пониженном давлении остаток растворяли в хлороформе (100 мл). После промывки органического слоя 10% водным раствором лимонной кислоты (100 мл) органический слой сушили над безводным сульфатом натрия. После фильтрования фильтрат концентрировали при пониженном давлении и после добавления по каплям в остаток концентрированной хлористо-водородной кислоты (2 мл) при охлаждении на льду перемешивали при комнатной температуре в течение 30 минут. После добавления в реакционный раствор 1 моль/л хлористо-водородной кислоты (2 мл) и промывки желтого кислого водного раствора хлороформом (50 мл×3) pH доводили до 12,0 с помощью водного раствора гидроксида натрия. После доведения как рН основного водного раствора до 7,4 с помощью 1 моль/л хлористо-водородной кислоты экстрагировали хлороформом (100 мл×3). После сушки над безводным сульфатом натрия растворитель упаривали при пониженном давлении. Полученный таким образом остаток затем очищали с помощью препаративной хроматографии (разделение в нижнем слое смесью хлороформ:метанол:вода, 7:3:1), кроме того, очищали перекристаллизацией из этанола и затем сушили при пониженном давлении. Таким образом получали 72,8 мг (42%) указанного в заголовке соединения в виде желтых кристаллов.
1H ЯМР (400 МГц, 0,1 моль/л NaOD) δ: 1,21-1,27 (1H, м), 1,50-1,64 (2H, м), 1,99-2,01 (1H, м), 2,34 (3H, c), 2,42 (3H, c), 2,87-2,89 (1H, м), 3,27-3,29 (3H, м), 3,63-3,65 1H, м), 4,06-4,07 (1H, м), 5,05 (1H, д м, J=63,72 Гц), 7,09 (1H, м), 8,00 (1H, c), 8,44 (1H, c).
ИК (KBr, диск) ν: 3348, 3086, 2939, 2844, 2789, 1711, 1614, 1518, 1435, 1354, 1315, 1257, 1221 см-1.
Точка плавления: 223-224°C.
Удельное вращение: [α]D 24,7=-119,66° (c 0,295, 0,1 моль/л NaOH).
Элементный анализ для C22H26FN3O3:
Вычислено: С 66,15%; H 6,56%; N 10,52%.
Найдено: С 65,92%; H 6,52%; N 10,40%.
ПРИМЕР 11
1-[2-(S)-фтор-1-(R)-циклопропил]-8-метокси-7-[3-(R)-[1-(метиламино)циклопропил]пирролидин-1-ил]-1,4-дигидро-4-оксохинолин-3-карбоновая кислота
После добавления к высушенному диметилсульфоксиду (1 мл) 3-(R)-[1-[N-(трет-бутоксикарбонил)-N-(метил)амино]циклопропил]пирролидина (102 мг, 379 мкмоль) и триэтиламина (0,50 мл), добавляли 7-фтор-1-[2-(S)-фтор-1-(R)-циклопропил]-1,4-дигидро-8-метокси-4-оксохинолин-3-карбоновую кислоту (112 мг, 379 мкмоль) и перемешивали при нагревании на масляной бане при 100°C в течение 15 часов в атмосфере азота. После концентрирования реакционного раствора при пониженном давлении остаток растворяли в хлороформе (100 мл). После промывки органического слоя 10% водным раствором лимонной кислоты (100 мл) органический слой сушили над безводным сульфатом натрия. После фильтрования фильтрат концентрировали при пониженном давлении и после добавления по каплям к остатку концентрированной хлористо-водородной кислоты (2 мл) при охлаждении на льду перемешивали при комнатной температуре в течение 30 минут. После добавления в реакционный раствор 1 моль/л хлористо-водородной кислоты (2 мл) и промывки желтого кислого водного раствора хлороформом (50 мл×4) pH доводили до 12,0 с помощью водного раствора гидроксида натрия. После доведения рН основного водного раствора до 7,4 с помощью 1 моль/л хлористо-водородной кислоты экстрагировали хлороформом (100 мл×3). После сушки над безводным сульфатом натрия растворитель упаривали при пониженном давлении. Полученный таким образом остаток затем очищали с помощью препаративной хроматографии (разделение в нижнем слое смесью хлороформ:метанол:вода, 7:3:1), кроме того, очищали перекристаллизацией из этанола и затем сушили при пониженном давлении. Таким образом получали 78,3 мг (50%) указанного в заголовке соединения в виде желтых кристаллов.
1H ЯМР (400 МГц, 0,1 моль/л NaOD) δ: 0,57-0,61 (4H, м), 1,33-1,40 (1H, м), 1,56-1,58 (2H, м), 1,99-2,01 (1H, м), 2,34 (3H, c), 2,87-2,89 (1H, м), 3,15-3,17 (1H, м), 3,52-3,54 (3H, м), 3,53 (3H, c), 4,00-4,02 (1H, м), 5,02 (1H, дм, J=64,45 Гц), 7,03 (1H, c), 7,92 (1H, д, J=7,03 Гц), 8,39 (1H, c).
ИК (KBr, диск) ν: 3352, 3095, 3051, 2939, 2837, 2787, 1716, 1699, 1616, 1520, 1439, 1358, 1319, 1259, 1221 см-1.
Точка плавления: 213-215°C.
Удельное вращение: [α]D 24,7=-38,46° (c 0,195, 0,1 моль/л NaOH).
Элементный анализ для C22H26FN3O4:
Вычислено: С 63,60%; H 6,31%; N 10,11%.
Найдено: С 63,36%; H 6,31%; N 9,97%.
ПРИМЕР 12
5-амино-1-[2-(S)-фтор-1-(R)-циклопропил]-8-метил-7-[3-(R)-[1-(метиламино)циклопропил]пирролидин-1-ил]-1,4-дигидро-4-оксохинолин-3-карбоновая кислота
После добавления к высушенному диметилсульфоксиду (4 мл) 3-(R)-[1-[N-(трет-бутоксикарбонил)-N-(метил)амино]циклопропил]пирролидина (690 мг, 2,25 ммоль) и триэтиламина (0,50 мл), добавляли 5-амино-7-фтор-1-[2-(S)-фтор-1-(R)-циклопропил]-8-метил-1,4-дигидро-4-оксохинолин-3-карбоновую кислоту (250 мг, 850 мкмоль) и кипятили с обратным холодильником на масляной бане при 70°C в течение 24 часов в атмосфере азота. После концентрирования реакционного раствора при пониженном давлении остаток растворяли в хлороформе (100 мл). После промывки органического слоя 10% водным раствором лимонной кислоты (100 мл) и насыщенным раствором соли (100 мл) органический слой сушили над безводным сульфатом натрия. После фильтрования фильтрат концентрировали при пониженном давлении и после добавления по каплям к остатку концентрированной хлористо-водородной кислоты (5 мл) при охлаждении на льду перемешивали при комнатной температуре в течение 30 минут. После добавления в реакционный раствор 1 моль/л хлористо-водородной кислоты (2 мл) и промывки желтого кислого водного раствора хлороформом (50 мл×3) рН доводили до 12,0 с помощью водного раствора гидроксида натрия. После доведения рН основного водного раствора до 7,4 с помощью 1 моль/л хлористо-водородной кислоты экстрагировали хлороформом (100 мл×3). После сушки над безводным сульфатом натрия растворитель упаривали при пониженном давлении. Полученный таким образом остаток затем очищали с помощью препаративной хроматографии (разделение в нижнем слое смесью хлороформ:метанол:вода, 7:3:1), кроме того, очищали перекристаллизацией из этанола и затем сушили при пониженном давлении. Таким образом получали 70,0 мг (20%) указанного в заголовке соединения в виде желтых кристаллов.
1H ЯМР (400 МГц, 0,1 моль/л NaOD) δ: 0,56-0,64 (4H, м), 1,21-1,61 (3H, м), 1,92-1,96 (1H, м), 2,22 (3H, c), 2,45 (3H, c), 2,68-2,73 (1H, м), 3,19-3,31 (3H, м), 3,59-3,66 (1H, м), 3,72-3,77 (1H, м), 4,76-4,78 (0,5H, м), 4,98-5,01 (0,5H, м), 5,97 (1H, c), 8,55 (1H, д, J=3,66 Гц).
ИК (KBr, диск) ν: 3440, 3329, 3082, 3005, 2964, 2937, 2877, 1716, 1620, 1549, 1506, 1437, 1404 см-1.
Точка плавления: 129-131°C.
Удельное вращение: [α]D 22,6=-291,90° (c 0,285, 0,1 моль/л NaOH).
Элементный анализ для C21H25FN4O3·0,25 H2O:
Вычислено: С 63,07%; H 6,62%; N 13,37%.
Найдено: С 62,89%; H 6,42%; N 13,27%.
ПРИМЕР 13
2,3-дигидро-3-(S)-метил-10-[3-(R)-[1-(метиламино)циклопропил]пирролидин-1-ил]-7-оксо-7H-пиридо[1,2,3-de][1,4]бензоксазин-6-карбоновая кислота
После добавления к высушенному диметилсульфоксиду (1 мл) 3-(R)-[1-[N-(трет-бутоксикарбонил)-N-(метил)амино]циклопропил]пирролидина (125 мг, 521 мкмоль) и триэтиламина (0,50 мл) добавляли 10-фтор-2,3-дигидро-3-(S)-метил-7-оксо-7H-пиридо[1,2,3-de][1,4]бензоксазин-6-карбоновую кислоту (132 мг, 500 мкмоль) и перемешивали при нагревании на масляной бане при 100°C в течение 20 часов в атмосфере азота. После концентрирования реакционного раствора при пониженном давлении остаток растворяли в хлороформе (100 мл). После промывки органического слоя 10% водным раствором лимонной кислоты (50 мл) и насыщенным раствором соли (50 мл) органический слой сушили над безводным сульфатом натрия. После фильтрования фильтрат концентрировали при пониженном давлении и после добавления к остатку концентрированной хлористо-водородной кислоты (3 мл) при охлаждении на льду перемешивали при комнатной температуре в течение 30 минут. После добавления в реакционный раствор воды (3 мл) и промывки желтого кислого водного раствора хлороформом (50 мл×3) рН доводили до 12,0 с помощью водного раствора гидроксида натрия. После доведения рН основного водного раствора до 7,4 с помощью 1 моль/л хлористо-водородной кислоты экстрагировали хлороформом (100 мл×3). После сушки над безводным сульфатом натрия растворитель упаривали при пониженном давлении. Полученный таким образом остаток затем очищали перекристаллизацией из смеси этанол-водный раствор аммиака и затем сушили при пониженном давлении. Таким образом получали 135 мг (70%) указанного в заголовке соединения в виде желтых кристаллов.
1H ЯМР (400 МГц, 0,1 моль/л NaOD) δ: 0,56-0,60 (4H, м), 1,45-1,50 (1H, м), 1,52 (3H, д, J=6,59 Гц), 1,99-2,01 (1H, м), 2,32 (3H, c), 2,86-2,88 (1H, м), 3,21-3,55 (4H, м), 4,22, 4,45 (каждый 1H, АВкв, J=11,36 Гц), 4,57-4,59 (1H, м), 7,04-7,08 (1H, м), 7,80 (1H, д, J=9,03 Гц), 8,32 (1H, c).
Точка плавления: 227-229°C.
Удельное вращение: [α]D 24,7=-131,00° (c 0,200, 0,1 моль/л NaOH).
Элементный анализ для C21H25N3O4:
Вычислено: С 65,78%; H 6,57%; N 10,96%.
Найдено: С 65,49%; H 6,55%; N 10,82%.
ПРИМЕР 14
7-[3-(R)-[1-(этиламино)циклопропил]пирролидин-1-ил]-1-[2-(S)-фтор-1-(R)-циклопропил]-8-метил-1,4-дигидро-4-оксохинолин-3-карбоновая кислота
После добавления к высушенному диметилсульфоксиду (10 мл) 3-(R)-[1-[N-(трет-бутоксикарбонил)-N-(этил)амино]циклопропил]пирролидина(2,16 г, 8,40 ммоль) и триэтиламина(4 мл) добавляли 7-фтор-1-[2-(S)-фтор-1-(R)-циклопропил]-8-метил-1,4-дигидро-4-оксохинолин-3-карбоновую кислоту (1,95 г, 7,00 ммоль) и кипятили с обратным холодильником на масляной бане при 100°С в течение 51 часов в атмосфере азота. После концентрирования реакционного раствора при пониженном давлении остаток растворяли в хлороформе (150 мл). После промывки органического слоя 10% водным раствором лимонной кислоты (100 мл) и насыщенным раствором соли (100 мл) органический слой сушили над безводным сульфатом натрия. После фильтрования фильтрат концентрировали при пониженном давлении и после добавления по каплям к остатку концентрированной хлористо-водородной кислоты (10 мл) при охлаждении на льду перемешивали при комнатной температуре в течение 30 минут. После добавления в реакционный раствор 1 моль/л хлористо-водородной кислоты (20 мл) и промывки желтого кислого водного раствора хлороформом (100 мл×5) рН доводили до 12,0 с помощью водного раствора гидроксида натрия. После доведения рН основного водного раствора до 7,4 с помощью 1 моль/л хлористо-водородной кислоты экстрагировали хлороформ (150 мл×4). После сушки над безводным сульфатом натрия растворитель упаривали при пониженном давлении.
Полученный таким образом остаток очищали перекристаллизацией из этанола и затем сушили при пониженном давлении. Таким образом получали 1,61 г (55%) указанного в заголовке соединения в виде желтых кристаллов.
1H ЯМР (400 МГц, 0,1 моль/л NaOD) δ: 0,57-0,63 (4H, м), 1,04 (3Н, т, J=6,95 Гц), 1,19-1,25 (1H, м), 1,47-1,64 (2H, м), 1,97-1,98 (1H, м), 2,40 (3H, c), 2,70-2,73 (2H, м), 2,86-2,87 (1H, м), 3,26-3,28 (3Н, м), 3,61-3,63 (1H, м), 4,02-4,05 (1H, м), 5,03 (1H, дм, J=64,11 Гц), 7,07 (1H, д, J=9,26 Гц), 7,98 (2H, д, J=9,26 Гц), 8,43 (1H, д, J=3,41 Гц).
ИК (KBr, диск) ν: 3294, 2964, 2848, 1699, 1612, 1508, 1473, 1431, 1396, 1389, 1350, 1308, 1261 см-1.
Точка плавления: 191-194°C.
Удельное вращение: [α]D 24,3=-236,55° (c 0,145, 0,1 моль/л NaOH).
Элементный анализ для С23Н28FN3O3:
Вычислено: С 66,81%; H 6,83%; N 10,16%.
Найдено: С 66,52%; H 6,86%; N 10,03%.
ПРИМЕР 15
1-(Циклопропил)-8-метил-7-[3-(R)-[1-(метиламино)циклопропил]пирролидин-1-ил]-1,4-дигидро-4-оксохинолин-3-карбоновая кислота
После добавления к высушенному диметилсульфоксиду (5 мл) 3-(R)-[1-[N-(трет-бутоксикарбонил)-N-(метил)амино]циклопропил]пирролидина (880 мг, 3,25 ммоль) и триэтиламина (1,0 мл) добавляли 1-циклопропил-7-фтор-8-метил-1,4-дигидро-4-оксохинолин-3-карбоновую кислоту (425 мг, 1,63 ммоль) и кипятили с обратным холодильником на масляной бане при 70°C в течение 38 часов в атмосфере азота. После концентрирования реакционного раствора при пониженном давлении остаток растворяли в этилацетате (200 мл). После промывки органического слоя 10% водным раствором лимонной кислоты (100 мл) органический слой сушили над безводным сульфатом натрия. После фильтрования фильтрат концентрировали при пониженном давлении и после добавления по каплям к остатку концентрированной хлористо-водородной кислоты (6 мл) при охлаждении на льду перемешивали при комнатной температуре в течение 30 минут. После добавления в реакционный раствор 1 моль/л хлористо-водородной кислоты (12 мл) и промывки желтого кислого водного раствора хлороформом (50 мл×3) рН доводили до 12,0 с помощью водного раствора гидроксида натрия. После доведения рН основного водного раствора до 7,4 с помощью 1 моль/л хлористо-водородной кислоты экстрагировали хлороформом (100 мл×3). После сушки над безводным сульфатом натрия растворитель упаривали при пониженном давлении. Полученный таким образом остаток затем очищали перекристаллизацией из смешанного растворителя метанол-1/2-пропанол и затем сушили при пониженном давлении. Таким образом получали 331 мг (53%) указанного в заголовке соединения в виде желтых кристаллов.
1H ЯМР (400 МГц, CDCl3) δ: 0,57-0,65 (1H, м), 0,87-0,92 (1H, м), 0,97-1,04 (1H, м), 1,11-1,18 (1H, м), 1,28-1,34 (1H, м), 1,64-1,71 (1H, м), 1,99-2,02 (1H, м), 2,45 (3H, c), 2,50 (3H, c), 2,71-2,76 (1H, м), 3,32-3,36 (3H, м), 3,64-3,70 (1H, м), 4,01-4,05 (1H, м), 6,99 (1H, д, J=9,06 Гц), 8,13 (1H, д, J=9,06 Гц), 8,85 (1H, c).
Точка плавления: 190-192°C.
Элементный анализ для С22Н27N3О3:
Вычислено: С 69,27%; H 7,13%; N 11,02%.
Найдено: С 69,00%; H 7,16%; N 10,96%.
Ссылочный пример 25
Этил 1-(2-бромацетил)циклопропанкарбоксилат
Этил 1-ацетилциклопропанкарбоксилат (200 г, 1,28 моль) растворяли в этаноле (1000 мл) и по каплям добавляли бром (72,7 мл, 1,41 моль) при перемешивании и охлаждении на льду. После завершения добавления температуру реакционного раствора повышали до 30°C и перемешивание проводили в течение 2 часов. После добавления в реакционный раствор воды (1000 мл) при охлаждении на льду концентрировали при пониженном давлении. После экстрагирования концентрата этилацетатом (750 мл×2) его промывали 10% водным раствором тиосульфата натрия (500 мл×2) и насыщенным водным раствором бикарбоната натрия (500 мл×2), в указанной последовательности, и затем сушили над безводным сульфатом натрия. После фильтрования и концентрирования фильтрата при пониженном давлении получали 291 г (97%) указанного в заголовке соединения в виде желтого, маслянистого вещества.
1H ЯМР (400 МГц, CDCl3) δ: 1,30 (3Н, т, J=7,1 Гц), 1,60-1,63 (4H, м), 4,22 (2H, кв, J=7,1 Гц), 4,49 (2H, c).
ТСХ: Rf=0,7 (н-гексан:этилацетат = 3:1).
Ссылочный пример 26
Диэтилфосфоноуксусная кислота
Этилдиэтилфосфоноацетат (100 г, 446 ммоль) растворяли в этаноле (275 мл) и после добавления по каплям 2 моль/л водного раствора гидроксида натрия (275 мл, 550 ммоль) при перемешивании и охлаждении на льду перемешивали при комнатной температуре в течение 1 часа. Реакционный раствор затем концентрировали при пониженном давлении и концентрат подкисляли с помощью концентрированной хлористо-водородной кислоты при охлаждении на льду. Экстрагировали этилацетатом (200 мл×4), хлороформом (100 мл×2) и смесью 5% метанол/хлороформ (250 мл×2). Объединенные органические слои затем сушили над безводным сульфатом натрия и после фильтрования фильтрат концентрировали при пониженном давлении. Таким образом получали 89 г (количественный выход) указанного в заголовке соединения в виде бесцветного маслянистого вещества.
1H ЯМР (400 МГц, CDCl3) δ: 1,35 (6H, т, J=6,8 Гц), 2,98 (2H, д, J=21,7 Гц), 4,19 (4H, кв, J=6,8 Гц).
ТСХ: Rf=0,1 (хлороформ:метанол = 9:1).
Ссылочный пример 27
Этил 1-[2-[N-[1-(S)-фенилэтил]амино]ацетил]циклопропанкарбоксилат
1-(S)-фенилэтиламин (12,1 г, 100 ммоль) растворяли в ацетонитриле (120 мл), добавляли по каплям ацетонитрильный (50 мл) раствор триэтиламина (15,3 мл, 110 ммоль) и добавляли по каплям этил 1-(2-бромацетил)циклопропанкарбоксилат (23,5 г, 100 ммоль) при перемешивании и охлаждении на льду. После завершения добавления реакционный раствор перемешивали при охлаждении на льду в течение 1,5 часов. Реакционный раствор затем выливали в воду (75 мл) и концентрировали при пониженном давлении. Концентрат экстрагировали диизопропиловым эфиром (75 мл×2) и затем промывали водой (75 мл). После экстрагирования органического слоя 1 моль/л хлористо-водородной кислотой (100 мл×2) кислый водный раствор промывали этилацетатом (100 мл). После добавления в кислый водный раствор 1 моль/л водного раствора гидроксида натрия (100 мл) и последующего дополнительного добавления насыщенного водного раствора бикарбоната натрия (100 мл) экстрагировали этилацетатом (100 мл). Органический слой затем промывали водой (100 мл) и насыщенным раствором соли (100 мл), в указанном порядке, и затем сушили над безводным сульфатом натрия. После фильтрования и последующего концентрирования фильтрата при пониженном давлении получали 18,6 г (68%) указанного в заголовке соединения в виде бледно-желтого маслянистого вещества.
1H ЯМР (400 МГц, CDCl3) δ: 1,17 (3H, т, J=7,1 Гц), 1,38 (3H, д, J=6,6 Гц), 1,48 (4H, c), 3,71 (1H, кв, J=6,6 Гц), 3,86 (2H, д, J=2,0 Гц), 4,10 (2H, кв, J=7,1 Гц).
ТСХ: Rf=0,6 (н-гексан:этилацетат = 1:1).
Ссылочный пример 28
Этил 1-[2-[N-(диэтилфосфоноацетил)-N-[1-(S)-фенилэтил]амино]ацетил]циклопропанкарбоксилат
Способ A:
Диэтилфосфоноуксусную кислоту (15,1 г, 76,8 ммоль) растворяли в безводном тетрагидрофуране (120 мл) и после добавления 1,1'-карбонилдиимидазола (13,7 г, 84,5 ммоль) при охлаждении на льду перемешивали при комнатной температуре в течение 1 часа. После добавления в реакционный раствор безводного тетрагидрофуранового (30 мл) раствора этил 1-[2-[N-[1-(S)-фенилэтил]амино]ацетил]циклопропанкарбоксилата (17,6 г, 64,0 ммоль) при охлаждении на льду его перемешивали при комнатной температуре в течение 1 часа. После добавления в реакционный раствор 1 моль/л хлористо-водородной кислоты (100 мл) и этилацетата (100 мл) экстрагировали и органический слой отделяли. Водный слой затем экстрагировали этилацетатом (100 мл) и объединенный органический слой промывали насыщенным водным раствором бикарбоната натрия (100 мл) и насыщенным раствором соли (100 мл), в указанном порядке, и затем сушили над безводным сульфатом натрия. После фильтрования и последующего концентрирования фильтрата при пониженном давлении, получали 28,7 г (99%) указанного в заголовке соединения в виде желтого сиропа.
Способ В
Диэтилфосфоноуксусную кислоту (32,8 г, 166 ммоль) растворяли в безводным бензоле (700 мл) и после добавления N,N'-диметилформамида (1 мл) добавляли тионилхлорид (18,2 мл, 250 ммоль) и кипятили с обратным холодильником в течение 1,5 часов. Затем реакционный раствор оставляли остывать, концентрировали при пониженном давлении и после добавления высушенного толуола (100 мл) вновь концентрировали при пониженном давлении. После повторения данной операции 3 раза концентрат растворяли в безводным тетрагидрофуране (300 мл), и после добавления по каплям безводного тетрагидрофуранового (300 мл) раствора этил 1-[2-[N-[1-(S)-фенилэтил]амино]ацетил]циклопропанкарбоксилата (45,7 г, 166 ммоль) и триэтиламина (25,1 мл, 183 ммоль) при перемешивании и охлаждении на льду его перемешивали при охлаждении на льду в течение 1,5 часов и затем перемешивали при комнатной температуре в течение 2 часов. После добавления в реакционный раствор 1 моль/л хлористо-водородной кислоты (300 мл) и этилацетата (300 мл) и проведения экстрагирования органический слой разделяли. Водный слой затем экстрагировали этилацетатом (300 мл), и объединенный органический слой промывали насыщенным водным раствором бикарбоната натрия (300 мл) и насыщенным раствором соли (300 мл), в указанном порядке, и затем сушили над безводным сульфатом натрия. После фильтрования и последующего концентрирования фильтрата при пониженном давлении получали 43,6 г (70%) указанного в заголовке соединения в виде желтого сиропа.
1H ЯМР (400 МГц, CDCl3) δ: 1,14, 1,20 (3H, т, J=7,1 Гц), 1,29-1,68 (13H, м), 2,85, 4,69 (2H, дд, J=9,5, 20,7 Гц), 3,18, 4,55 (2H, д, J=22,2 Гц), 4,06-4,22 (6H, м), 5,42, 6,05 (1H, кв, J=7,1 Гц), 7,26-7,37 (5H, м).
MS (m/z): 454 ([M+H])+.
ТСХ: Rf=0,1 (н-гексан:этилацетат = 1:1).
Ссылочный пример 29
4-(1-этоксикарбонилциклопропил)-1-[1-(S)-фенилэтил]-3-пирролин-2-он
После растворения этил 1-[2-[N-(диэтилфосфоноацетил)-N-[1-(S)-фенилэтил]амино]ацетил]циклопропанкарбоксилата (25,0 г, 55,2 ммоль) в толуоле (250 мл) постепенно добавляли трет-бутоксид калия (7,40 г, 66,2 ммоль) при перемешивании и охлаждении на льду. После перемешивания реакционного раствора в течение 15 минут при комнатной температуре добавляли 10% водный раствор лимонной кислоты (250 мл) и этилацетат (250 мл), и после проведения экстрагирования органический слой разделяли. Водный слой затем экстрагировали этилацетатом (250 мл), и после промывки объединенного органического слоя насыщенным водным раствором бикарбоната натрия (250 мл) и насыщенным раствором соли (250 мл), в указанном порядке, органический слой сушили над безводным сульфатом натрия. После фильтрования фильтрат концентрировали при пониженном давлении, концентрат наносили на силикагель для проведения хроматографии и из элюата н-гексан:этилацетат = 2:1-1:2 получали 12,1 г (73%) указанного в заголовке соединения в виде оранжевого сиропа. Данные инструментального анализа полученного соединения согласуются с данными, приведенными в PCT/JP96/00208.
1H ЯМР (400 МГц, CDCl3) δ: 1,18, (3H, т, J=7,2 Гц), 1,60-1,63 (7H, м), 3,80 (1H, дд, J=1,5, 9,0 Гц), 4,07-4,11 (2H, м), 4,13 (1H, д, J=9,0 Гц), 5,55 (1H, кв, J=7,1 Гц), 5,84 (1H, т, J=1,5 Гц), 7,24-7,36 (5H, м).
MS (m/z): 300 ([M+H])+.
ТСХ: Rf=0,5 (н-гексан:этилацетат = 1:1).
Ссылочный пример 30
4-(S)-(1-этоксикарбонилциклопропил)-1-[1-(S)-фенилэтил]пирролидин-2-он
4-(1-этоксикарбонилциклопропил)-1-[1-(S)-фенилэтил]-3-пирролин-2-он (12,1 г, 40,5 ммоль) растворяли в этилацетате (120 мл), добавляли катализатор 5% палладия на углероде (содержание воды: 50%, 2,4 г) и перемешивали при комнатной температуре в течение 17 часов в атмосфере водорода при атмосферном давлении. Реакционный раствор затем фильтровали через целит (промывали этилацетатом) и фильтрат затем концентрировали при пониженном давлении. Концентрат наносили на силикагель для проведения хроматографии и из элюата н-гексан:этилацетат = 3:2 получали 9,00 г (74%) указанного в заголовке соединения в виде бледно-желтого сиропа. Кроме того, получали 2,60 г (21%) диастереомера (4-(R)-изомера) соединения, указанного в заголовке, в виде бледно-желтого сиропа. Данные инструментального анализа полученного соединения согласуются с данными, приведенными в PCT/JP96/00208.
4-(S)-изомер:
1H ЯМР (400 МГц, CDCl3) δ: 0,63-0,65 (2H, м), 1,13 (3H, т, J=7,1 Гц), 1,12-1,19 (2H, м), 1,52 (3H, д, J=7,3 Гц), 2,17 (1H, дд, J=9,0, 16,8 Гц), 2,46 (1H, дд, J=9,3, 16,3 Гц), 2,67-2,76 (2H, м), 3,47 (1H, т, J=8,3 Гц), 3,96-4,11 (2H, м), 5,51 (1H, кв, J=7,3 Гц), 7,26-7,35 (5H, м).
ТСХ: Rf=0,45 (н-гексан:этилацетат = 1:1).
4-(R)-изомер:
1H ЯМР (400 МГц, CDCl3) δ: 0,72-0,76 (2Н, м), 1,18-1,24 (2Н, м), 1,21 (3Н, т, J=7,1 Гц), 1,52 (3H, д, J=7,1 Гц), 2,27-2,32 (1H, м), 2,44-2,52 (2H, м), 3,14 (2H, д, J=8,0 Гц), 4,10 (2H, кв, J=7,1 Гц).
ТСХ: Rf=0,5 (н-гексан:этилацетат = 1:1).
Ссылочный пример 31
1-[1-[1-(S)-фенилэтил]-2-он-4-(S)-пирролидин-4-ил]-1-циклопропанкарбоновая кислота
4-(S)-(1-этоксикарбонилциклопропил)-1-[1-(S)-фенилэтил]пирролидин-2-он (10,5 г, 34,9 ммоль) растворяли в 70 мл этанола, и после добавления 1 моль/л водного раствора гидроксида натрия (70 мл) при охлаждении на льду реакционный раствор перемешивали при комнатной температуре в течение 15,5 часов и затем при 40°C в течение 3 часов. После концентрирования реакционного раствора при пониженном давлении остаточный водный слой промывали этилацетатом (70 мл). Водный слой затем подкисляли концентрированной хлористо-водородной кислотой при охлаждении на льду и затем экстрагировали хлороформом (70 мл×3). Органический слой затем сушили над безводным сульфатом натрия, и после фильтрования фильтрат концентрировали при пониженном давлении. Таким образом получали 9,40 г (99%) указанного в заголовке соединения в виде белого твердого вещества.
1H ЯМР (400 МГц, CDCl3) δ: 0,72-0,74 (2H, м), 1,21-1,23 (2H, м), 1,52 (3H, д, J=7,3 Гц), 2,17 (1H, дд, J=8,8, 16,8 Гц), 2,48 (1H, дд, J=9,5, 16,8 Гц), 2,66-2,78 (2H, м), 3,50 (1H, т, J=9,3 Гц), 5,51 (1H, кв, J=7,3 Гц), 7,25-7,34 (5H, м).
Ссылочный пример 32
4-(R)-[1-(трет-бутоксикарбониламино)циклопропил]-1-[1-(S)-фенилэтил]пирролидин-2-он
Толуольный (15 мл) раствор триэтиламина (9,6 мл, 69 ммоль) и азид дифенилфосфорной кислоты (DPPA; 10,4 г, 37,9 ммоль) добавляли в толуольную (80 мл) суспензию 1-[1-[1-(S)-фенилэтил]-2-он-4-(S)-пирролидин-4-ил]-1-циклопропанкарбоновой кислоты (9,4 г, 34,4 ммоль), и после перемешивания при комнатной температуре в течение 1 часа в атмосфере азота кипятили с обратным холодильником в течение 1,5 часов. После охлаждения реакционного раствора до комнатной температуры добавляли трет-бутиловый спирт (95 мл) и кипятили с обратным холодильником в течение 15 часов. Затем реакционный раствор оставляли охлаждаться, реакционный раствор концентрировали при пониженном давлении и в концентрат добавляли этилацетат (95 мл) и воду (95 мл). После проведения экстрагирования органический слой разделяли и водный слой экстрагировали этилацетатом (95 мл). Объединенный органический слой затем промывали насыщенным раствором соли (95 мл) и затем сушили над безводным сульфатом натрия. После фильтрования и концентрирования фильтрата при пониженном давлении концентрат наносили на силикагель для проведения хроматографии и из элюата хлор:метанол = 50:1 получали 10,7 г (90%) указанного в заголовке соединения в виде бесцветного, аморфного вещества.
1H ЯМР (400 МГц, CDCl3) δ: 0,56-0,85 (4H, м), 1,37 (9H, c), 1,51 (3Н, д, J=7,3 Гц), 2,32-2,44 (3H, м), 2,79 (1H, дд, J=7,3, 10,0 Гц), 3,36 (1H, м), 4,66 (1H, шир.c), 5,50 (1H, кв, J=7,3 Гц), 7,26-7,34 (5H, м).
ТСХ: Rf=0,15 (н-гексан:этилацетат = 1:1).
Ссылочный пример 33
3-(R)-[1-(трет-бутоксикарбониламино)циклопропил]-1-[1-(S)-фенилэтил]пирролидин
4-(R)-[1-(трет-бутоксикарбониламино)циклопропил]-1-[1-(S)-фенилэтил]пирролидин-2-он (10,4 г, 30,2 ммоль) растворяли в безводном тетрагидрофуране (100 мл) и по каплям добавляли 1,0 M раствор комплекса бор-тетрагидрофуран/тетрагидрофуран (90,7 мл, 90,7 ммоль) в атмосфере азота при охлаждении на льду. После завершения добавления перемешивали в течение 16 часов при температуре, начиная с охлаждения на льду и заканчивая комнатной температурой. После медленного добавления в реакционный раствор водного раствора (100 мл) карбоната калия (25,0 г, 181 ммоль) при охлаждении на льду его нагревали при кипячении с обратным холодильником в течение 1,5 часов. Затем реакционный раствор оставляли охлаждаться, экстрагировали этилацетатом (100 мл×2) и затем промывали насыщенным раствором соли (100 мл). Органический слой затем сушили над безводным сульфатом натрия, и после фильтрования фильтрат концентрировали при пониженном давлении. Концентрат наносили на силикагель для проведения хроматографии, и из элюата хлороформ:метанол = 100:1-30:1 получали 8,20 г (82%) указанного в заголовке соединения в виде бесцветных кристаллов.
1H ЯМР (400 МГц, CDCl3) δ: 0,62 (2H, шир.c), 0,75-0,88 (2H, м), 1,35 (3H, д, J=6,6 Гц), 1,41 (9H, c), 1,63 (2H, м), 1,88-1,92 (1H, м), 2,14-2,17 (1H, м), 2,27-2,34 (2H, м), 2,63 (1H, шир.c), 3,15 (1H, подобный т, J=6,6 Гц), 5,10 (1H, шир.c), 7,23-7,33 (5H, м).
MS (m/z): 331 ([M+H])+.
ТСХ: Rf=0,4 (хлороформ:метанол = 9:1).
Ссылочный пример 34
3-(R)-[1-(трет-бутоксикарбониламино)циклопропил]пирролидин
3-(R)-[1-(трет-бутоксикарбониламино)циклопропил]-1-[1-(S)-фенилэтил]пирролидин (270 мг, 0,817 ммоль) растворяли в этаноле (15 мл), и после добавления катализатора 10% палладия на углероде (содержание воды: 52,0%; 270 мг) перемешивали при 40°C в течение 3 часов в атмосфере водорода при атмосферном давлении. После удаления катализатора фильтрованием (промывка этанолом) фильтрат концентрировали при пониженном давлении и в результате получали 185 мг (количественный выход) указанного в заголовке соединения в виде бесцветного сиропа.
1H ЯМР (400 МГц, CDCl3) δ: 0,69 (2H, шир.c), 0,79 (2H, шир.c), 1,42 (9H, c), 1,43-1,50 (1H, м), 1,86-1,88 (1H, м), 2,15-2,19 (1H, м), 2,68-2,72 (1H, м), 2,90-3,07 (3H, м), 4,92 (1H, шир.c).
Ссылочный пример 35
1-Бензилоксикарбонил-3-(R)-[1-(трет-бутоксикарбониламино)циклопропил]пирролидин
3-(R)-[1-(трет-бутоксикарбониламино)циклопропил]-1-[1-(S)-фенилэтил]пирролидин (1,70 г, 5,15 ммоль) растворяли в дихлорметане (34 мл) и по каплям добавляли бензилхлороформиат (1,10 мл, 7,73 ммоль) при перемешивании и охлаждении на льду. Реакционный раствор перемешивали при комнатной температуре в течение 2,5 часов и затем перемешивали при 40°C в течение 1,5 часов. Затем реакционный раствор оставляли охлаждаться, реакционный раствор концентрировали при пониженном давлении, концентрат наносили на силикагель для проведения хроматографии, и из элюата н-гексан:этилацетат = 2:1 получали 1,40 г (75%) указанного в заголовке соединения в виде бесцветного сиропа. Данные инструментального анализа полученного соединения согласуются с данными, приведенными в PCT/JP96/00208.
1H ЯМР (400 МГц, CDCl3) δ: 0,70 (2H, шир.c), 0,80 (2H, шир.c), 1,41 (9H, c), 1,63 (1H, м), 1,92 (1H, м), 2,25 (1H, м), 3,07-3,12 (1H, м), 3,29-3,31 (1H, м), 3,56 (2H, м), 4,85 (1H, шир.c), 5,12 (2H, c), 7,33-7,36 (5H, м).
ТСХ: Rf=0,4 (н-гексан:этилацетат = 1:1).
Ссылочный пример 36
1-Бензилоксикарбонил-3-(R)-[1-[N-(трет-бутоксикарбонил)-
N-(метил)амино]циклопропил]пирролидин
1-Бензилоксикарбонил-3-(R)-[1-(трет-бутоксикарбониламино)циклопропил]пирролидин (1,40 г, 3,89 ммоль), N,N'-диметилформамид (7 мл), оксид серебра (9,0 г, 39 ммоль) и метилйодид (24 мл, 389 ммоль) помещали в затемненную и герметичную пробирку, и смесь перемешивали в течение 13 часов на масляной бане при 80°C. Реакционный раствор затем фильтровали через целит (промывали этилацетатом), и фильтрат затем разбавляли этилацетатом (100 мл). Органический слой затем промывали водой (50 мл×3) и насыщенным раствором соли (50 мл), в указанном порядке, и сушили над безводным сульфатом натрия. После фильтрования фильтрат концентрировали при пониженном давлении, концентрат наносили на силикагель для проведения хроматографии и из элюата н-гексан:этилацетат = 2:1 получали 1,31 г (89%) указанного в заголовке соединения в виде бледно-желтого сиропа. Данные инструментального анализа полученного соединения согласуются с данными, приведенными в PCT/JP96/00208.
1H ЯМР (400 МГц, CDCl3) δ: 0,83 (4H, шир.c), 1,42 (9H, c), 1,55 (1H, м), 1,88 (1H, м), 2,28-2,43 (1H, м), 2,83 (3H, c), 3,02-3,04 (1H, м), 3,25-3,33 (1H, м), 3,55 (2H, м), 5,12 (2H, c), 7,32-7,35 (5H, м).
MS (m/Z): 374 ([M+H])+.
ТСХ: Rf=0,4 (н-гексан:этилацетат = 1:1).
Ссылочный пример 37
3-(R)-[1-[N-(трет-бутоксикарбонил)-N-(метил)амино]циклопропил]пирролидин
1-бензилоксикарбонил-3-(R)-[1-[N-(трет-бутоксикарбонил)-N-(метил)амино]циклопропил]пирролидин (1,31 г, 3,48 ммоль) растворяли в этаноле (13 мл), и после добавления катализатора 10% палладия на углероде (содержание воды: 50%; 0,65 г) перемешивали при комнатной температуре в течение 5 часов в атмосфере водорода при атмосферном давлении. После проведения фильтрования через целит (промывка этанолом) фильтрат концентрировали при пониженном давлении. В результате получали 0,92 г (количественный выход) указанного в заголовке соединения в виде бесцветного сиропа.
1H ЯМР (400 МГц, CDCl3) δ: 0,80 (4H, шир.c), 1,46 (9H, c), 1,81 (1H, м), 2,04 (1H, шир.c), 2,28-2,42 (1H, м), 2,54 (1H, шир.c), 2,84 (3H, c), 2,88-2,96 (3H, м).
ТСХ: Rf=0,1 (хлороформ:метанол = 9:1).
Ссылочный пример 38
3-(R)-[1-[N-(трет-бутоксикарбонил)-N-(метил)амино]циклопропил]-1-[1-(S)-фенилэтил]пирролидин-2-он
4-(R)-[1-(трет-бутоксикарбониламино)циклопропил]-1-[1-(S)-фенилэтил]пирролидин-2-он (7,87 г, 22,8 ммоль) растворяли в диметилформамиде (100 мл) при охлаждении на льду добавляли 60% гидрида натрия в масле (1,10 г, 27,4 ммоль) и перемешивали в течение 5 минут. Затем по каплям добавляли метилйодид (7,11 мл, 114 ммоль) при перемешивании при комнатной температуре. После завершения добавления реакционную суспензию перемешивали при комнатной температуре в течение 14 часов. После добавления 60% гидрида натрия в масле (296 мг, 7,40 ммоль) и метилйодида (1,00 мл, 16,1 ммоль) суспензию перемешивали при 40°C в течение 24 часов. После добавления в реакционную суспензию насыщенного водного раствора хлорида аммония (100 мл) и воды (150 мл) при перемешивании и охлаждении на льду экстрагировали этилацетатом (300 мл×2). Объединенный органический слой затем промывали водой (100 мл×2) и насыщенным раствором соли (100 мл×2), в указанном порядке, и затем сушили над безводным сульфатом натрия. После фильтрования фильтрат концентрировали при пониженном давлении, полученный остаток наносили на силикагель для проведения хроматографии и из элюата хлороформ:метанол = 50-30:1 получали 7,53 г (92%) указанного в заголовке соединения в виде бесцветного маслянистого вещества.
1H ЯМР (400 МГц, CDCl3) δ: 0,83 (4H, м), 1,32 (6H, c), 1,38 (3Н, c), 1,51 (3H, д, J=7,1 Гц), 1,61 (3H, д, J=16,6 Гц), 2,43 (1H, м), 2,68-2,81 (3H, м), 3,21 (1H, м), 5,48-5,50 (1H, м), 7,26-7,36 (5H, м).
Ссылочный пример 39
3-(R)-[1-[N-(трет-бутоксикарбонил)-N-(метил)амино]циклопропил]-1-[1-(S)-фенилэтил]пирролидин
3-(R)-[1-[N-(трет-бутоксикарбонил)-N-(метил)амино]циклопропил]-1-[1-(S)-фенилэтил]пирролидин-2-он (7,53 г, 21,0 ммоль) растворяли в безводном тетрагидрофуране (70 мл) и постепенно по каплям добавляли 1,0 М раствор комплекса бор-тетрагидрофуран/тетрагидрофуран (63,0 мл, 63,0 ммоль) при перемешивании и охлаждении на льду. После завершения добавления раствор перемешивали при комнатной температуре в течение 20 часов. После медленного добавления водного раствора (72 мл) карбоната калия (7,22 г) при охлаждении на льду смесь кипятили с обратным холодильником в течение 1,5 часов. Затем реакционный раствор оставляли охлаждаться до комнатной температуры, добавляли воду (150 мл), экстрагировали этилацетатом (200 мл×2), затем объединенный органический слой промывали насыщенным хлористо-водородным раствором (200 мл) и затем сушили над безводным сульфатом натрия. После фильтрования фильтрат концентрировали при пониженном давлении, концентрат наносили на силикагель для проведения хроматографии и из элюата хлороформ:метанол = 50:1 получали 7,19 г (99%) указанного в заголовке соединения в виде бесцветного сиропа.
1H ЯМР (400 МГц, CDCl3) δ: 0,73 (4H, м), 1,35 (9H, c), 1,36 (3Н, c), 1,61 (1H, м), 1,85 (1H, м), 1,97 (1H, м), 2,27 (1H, м), 2,50-2,58 (2H, м), 2,79 (3H, c), 2,99 (1H, м), 3,14-3,19 (1H, м), 7,27-7,30 (5H, м).
Ссылочный пример 40
3-(R)-[1-[N-(трет-бутоксикарбонил)-
N-(метил)амино]циклопропил]пирролидин
3-(R)-[1-[N-(трет-бутоксикарбонил)-N-(метил)амино]циклопропил]-1-[1-(S)-фенилэтил]пирролидин (7,19 г, 20,9 ммоль) растворяли в этаноле (78 мл) и после добавления катализатора 10% палладия на углероде (содержание воды: 50%; 3,9 г) перемешивали при 40°C в течение 4 часов в атмосфере водорода при атмосферном давлении. После проведения фильтрования через целит (промывка этанолом) фильтрат концентрировали при пониженном давлении. В результате получали 4,38 г (количественный выход) соединения, указанного в заголовке, в виде бесцветного сиропа. Данные 1H-ЯМР и значение Rf ТСХ полученного соединения согласуются с данными, показанными до этого.
1H ЯМР (400 МГц, CDCl3) δ: 0,80 (4H, шир.c), 1,46 (9H, c), 1,81 (1H, м), 2,04 (1H, шир.c), 2,28-2,42 (1H, м), 2,54 (1H, шир.c), 2,84 (3H, c), 2,88-2,96 (3H, м).
ТСХ: Rf=0,1 (хлороформ:метанол = 9:1).
Ссылочный пример 41
4-(R)-[1-[N-(трет-бутоксикарбонил)-N-(этил)амино]циклопропил]-1-[1-(S)-фенилэтил]пирролидин-2-он
4-(R)-[1-(трет-бутоксикарбониламино)циклопропил]-1-[1-(S)-фенилэтил]пирролидин-2-он (4,16 г, 12,1 ммоль) растворяли в диметилформамиде (50 мл). В смесь в атмосфере азота и при комнатной температуре добавляли 60% натрий гидрид в масле (580 мг, 14,5 ммоль), перемешивали в течение 10 минут и затем по каплям добавляли этилйодид (4,87 мл, 60,5 ммоль). После завершения добавления реакционную суспензию перемешивали при комнатной температуре в течение 15 часов. После добавления в реакционную суспензию насыщенного водного раствора хлорида аммония (150 мл) при перемешивании и охлаждении на льду экстрагировали этилацетатом (150 мл×2). Объединенный органический слой затем промывали водой (150 мл×2) и насыщенным раствором соли (150 мл), в указанном порядке, и затем сушили над безводным сульфатом натрия. После фильтрования фильтрат концентрировали при пониженном давлении полученный остаток наносили на силикагель для проведения хроматографии и из элюата н-гексан:этилацетат = 1:2 получали 4,56 г (количественный выход) указанного в заголовке соединения получали в виде бесцветного маслянистого вещества.
1H ЯМР (400 МГц, CDCl3) δ: 0,49-0,80 (4H, м), 1,02-1,04 (3H, м), 1,37 (9H, c), 1,49-1,51 (3H, м), 1,92-1,94 (1H, м), 2,04-2,06 (1H, м), 2,36-2,38 (1H, м), 2,67-2,70 (2H, м), 3,20-4,23 (2H, м), 5,48-5,50 (1H, м), 7,26-7,52 (5H, м).
Ссылочный пример 42
3-(R)-[1-[N-(трет-бутоксикарбонил)-N-(этил)амино]циклопропил]-1-[1-(S)-фенилэтил]пирролидин
4-(R)-[1-[N-(трет-бутоксикарбонил)-N-(этил)амино]циклопропил]-1-[1-(S)-фенилэтил]пирролидин-2-он (4,56 г, 12,1 ммоль) растворяли в безводном тетрагидрофуране (80 мл) и по каплям добавляли 1,0 М раствор комплекса бор-тетрагидрофурана/тетрагидрофурана (48,0 мл, 48,0 ммоль) при перемешивании и охлаждении на льду. После завершения добавления реакционный раствор перемешивали в течение 16 часов в условиях от охлаждения на льду до комнатной температуры. После концентрирования реакционного раствора при пониженном давлении добавляли смешанный раствор 9:1 (100 мл) этанол и вода и после добавления триэтиламина (5 мл) его нагревали при кипячении с обратным холодильником в течение 4 часов. После охлаждения реакционного раствора до комнатной температуры концентрировали при пониженном давлении, в остаток добавляли насыщенный водный раствор карбоната натрия (100 мл) экстрагировали хлороформом (100 мл×2), затем объединенный органический слой промывали насыщенным раствором соли (100 мл) и затем сушили над безводным сульфатом натрия. После фильтрования фильтрат концентрировали при пониженном давлении, концентрат наносили на силикагель для проведения хроматографии и из элюата хлороформ:метанол = 100:1-95:5 получали 4,26 г (99%) указанного в заголовке соединения в виде бесцветного сиропа.
1H ЯМР (400 МГц, CDCl3) δ: 0,54-0,78 (4H, м), 1,09-1,11 (3H, м), 1,33-1,43 (13H, м), 1,84-1,97 (2H, м), 2,26-2,28 (2H, м), 2,56-2,57 (2H, м), 2,86-2,96 (1H, м), 3,13-3,18 (2H, м), 7,21-7,30 (5H, м).
Ссылочный пример 43
3-(R)-[1-[N-(трет-бутоксикарбонил)-N-(этил)амино]циклопропил]пирролидин
3-(R)-[1-[N-(трет-бутоксикарбонил)-N-(этил)амино]циклопропил]-1-[1-(S)-фенилэтил]пирролидин (3,01 г, 8,40 ммоль) растворяли в этаноле (120 мл) и после добавления катализатора 10% палладия на углероде (содержание воды: 50%; 3,0 г) перемешивали при 40°C в течение 5 часов в атмосфере водорода при атмосферном давлении. После фильтрования через целит (промывали этанолом) фильтрат концентрировали при пониженном давлении. В результате получали 2,16 г (количественный выход) указанного в заголовке соединения в виде бесцветного сиропа.
1H ЯМР (400 МГц, CDCl3) δ: 0,83-0,85 (4H, м), 1,46 (9H, c), 1,74-1,82 (1Н, м), 2,16-2,18 (2H, м), 2,43-2,52 (2H, м), 2,90-2,99 (2H, м), 3,21-3,24 (2H, м).
Ссылочный пример 44
2,3,4-Трихлорбензойная кислота
Гидроксид натрия (45,42 г, 1,090 моль) растворяли в воде (220 мл) и при охлаждении на льду по каплям добавляли бром (16,85 мл, 0,327 моль) в течение 5 минут. После перемешивания реакционного раствора при 0°C в течение 15 минут по каплям добавляли раствор диоксана (220 мл) 2',3',4'-трихлорацетофенона (24,40 г, 0,109 моль) при 0°C в течение 30 минут. После перемешивания при комнатной температуре в течение 14 часов добавляли воду (350 мл) и затем промывали дихлорметаном (350 мл). Полученный водный слой постепенно подкисляли концентрированной хлористоводородной кислотой при охлаждении на льду и полученные кристаллы отфильтровывали. После промывки отфильтрованных кристаллов водой воду удаляли с помощью азеотропной перегонки с толуолом. Таким образом получали 22,33 г (91%) указанного в заголовке соединения в виде светло-желтого порошка.
1H ЯМР (400 МГц, CDCl3) δ: 7,58 (1H, д, J=8,3 Гц), 7,70 (1H, д, J=8,6 Гц).
Ссылочный пример 45
Этил (2,3,4-трихлорбензоил)ацетат
2,3,4-Трихлорбензойную кислоту (4,51 г, 20,0 ммоль) растворяли в тетрагидрофуране (80 мл), добавляли карбонилдиимидазол (3,89 г, 24,0 ммоль) при охлаждении на льду и затем перемешивали при комнатной температуре в течение 3 часов (раствор A). Тем временем, монокалиевую соль моноэтилового эфира малоновой кислоты (6,81 г, 40,0 ммоль) суспендировали в этилацетате и при охлаждении на льду добавляли триэтиламин (13,9 мл, 100,0 ммоль) и хлорид магния (5,71 г, 60,0 ммоль). После перемешивания при комнатной температуре в течение 3 часов реакционный раствор охлаждали на льду и описанный выше раствор A по каплям добавляли в данный реакционный раствор в течение 10 минут. После промывки раствора A в реакционном растворе с использованием тетрагидрофурана (10 мл) его перемешивали при комнатной температуре в течение 14 часов, и затем реакционный раствор выливали в 10% водный раствор лимонной кислоты (200 мл). Смесь затем экстрагировали этилацетатом (200 мл), промывали насыщенным хлористо-водородным раствором (200 мл) и затем сушили над безводным сульфатом натрия. Затем, после удаления агента для сушки путем фильтрования растворитель упаривали при пониженном давлении и полученный неочищенный продукт очищали хроматографией на силикагеле, таким образом получая из элюата н-гексан:этилацетат = 5:1 2,681 г (45%) указанного в заголовке соединения в виде бледно-красного масла.
1H ЯМР (400 МГц, CDCl3) δ: 1,25 (1,5H, т, J=7,2 Гц), 1,34 (1,5H, т, J=7,0 Гц), 3,99 (1H, c) 4,19 (1H, кв, J=7,2 Гц), 4,28 (1H, кв, J=7,0 Гц), 5,47 (0,5H, c), 7,37-7,49 (2H, м), 12,45 (0,5H, м).
Ссылочный пример 46
Этил 7,8-дихлор-1-[2-(S)-фтор-1-(R)-циклопропил]-1,4-дигидро-4-оксохинолин-3-карбоксилат
Смесь этил (2,3,4-трихлорбензоил)ацетата (2,681 г, 9,07 ммоль), уксусного ангидрида (10 мл) и триэтилортоформиата (20 мл) кипятили с обратным холодильником в течение 2,5 часов на масляной бане при 140°C. После упаривания растворителя при пониженном давлении проводили азеотропную перегонку, используя толуол (3 раза), с получением 3,272 г неочищенного этил 3-этокси-2-(2,3,4-трихлорбензоил)акрилата в виде бледно-красного масла.
Полученный выше неочищенный продукт этил 3-этокси-2-(2,3,4-трихлорбензоил)акрилата (3,272 г) растворяли в дихлорметане (50 мл), добавляли тозилат 2-(S)-фтор-1-(R)-циклопропиламина (2,467 г, 9,98 ммоль) и триэтиламин (1,64 мл, 11,79 ммоль), в указанном порядке, при охлаждении на льду, пропитанном солью, и перемешивали при комнатной температуре в течение 19,5 часов. В реакционный раствор добавляли этилацетат (200 мл), и после промывки 10% водным раствором лимонной кислоты (80 мл×2), насыщенным водным раствором бикарбоната натрия (80 мл) и насыщенным раствором соли (80 мл) сушили над безводным сульфатом натрия. Затем, после удаления агента для сушки путем фильтрования растворитель упаривали при пониженном давлении с получением неочищенного этил 3-[2-(S)-фтор-1-(R)-циклопропил]амино-2-(2,3,4-трихлорбензоил)акрилата (3,59 г) в виде бледно-оранжевого, смолоподобного вещества.
Указанный выше неочищенный этил 3-[2-(S)-фтор-1-(R)-циклопропил]амино-2-(2,3,4-трихлорбензоил)акрилат (3,57 г) растворяли в высушенном диоксане (45 мл) и после добавления туда гидрида натрия (60% содержание, 433 мг, 10,82 ммоль) при охлаждении на льду перемешивали в течение 14 часов при нагревании на масляной бане при 50°C. После упаривания растворителя при пониженном давлении остаток растворяли в хлороформе (150 мл), и после промывки 10% водным раствором лимонной кислоты (50 мл) и насыщенным раствором соли (50 мл) сушили над безводным сульфатом натрия. Затем, после удаления агента для сушки фильтрованием растворитель упаривали при пониженном давлении, и полученный неочищенный продукт очищали хроматографией на силикагеле, таким образом получая из элюата хлороформ:этилацетат = 1:2 1,475 г (48%) указанного в заголовке соединения в виде бледно-желтого порошка.
1H ЯМР (400 МГц, CDCl3) δ: 1,35-1,50 (1H, м), 1,41 (3H, т, J=7,1 Гц), 1,55-1,75 (1H, м), 4,08-4,13 (1H, м), 4,39 (2H, кв, J=7,1 Гц), 4,80-4,98 (1H, м), 7,53 (1H, д, J=8,5 Гц), 8,34 (1H, д, J=6,8 Гц), 8,57 (1H, д, J=2,7 Гц).
MS (m/z): 344 (M+).
Ссылочный пример 47
7,8-дихлор-1-[2-(S)-фтор-1-(R)-циклопропил]-1,4-дигидро-4-оксохинолин-3-карбоновая кислота
Смесь этил 7,8-дихлор-1-[2-(S)-фтор-1-(R)-циклопропил]-1,4-дигидро-4-оксохинолин-3-карбоксилата (1,114 г, 3,237 ммоль), уксусной кислоты (8 мл) и концентрированной хлористо-водородной кислоты (4 мл) кипятили с обратным холодильником в течение 2 часов на масляной бане при 130°C. Затем, после добавления воды (40 мл) и охлаждения на льду полученные кристаллы отфильтровывали и промывали водой (5 мл×2), 5% водным раствором этанола (5 мл×2) и диэтиловым эфиром (5 мл×2), таким образом получая 909 мг (89%) указанного в заголовке соединения в виде бледно-желтого порошка.
1H ЯМР (400 МГц, CDCl3) δ: 1,40-1,80 (2H, м), 4,23-4,28 (1H, м), 4,83-5,02 (1H, м), 7,67 (1H, д, J=8,8 Гц), 8,38 (1H, д, J=8,6 Гц), 8,86 (1H, д, J=2,7 Гц).
Точка плавления: 198-201°C.
Удельное вращение: [α]D 24,5=-24,0°.
Элементный анализ для C13H8Cl2FNO3:
Вычислено: С 49,39%; H 2,55%; N 4,43%.
Найдено: С 49,14%; H 2,40%; N 4,33%.
MS (m/z): 315 (M+), 354 [(M+K)+].
ПРИМЕР 16
7-[3-(R)-(1-аминоциклопропил)пирролидин-1-ил]-8-хлор-1-[2-(S)-фтор-1-(R)-циклопропил]-1,4-дигидро-4-оксохинолин-3-карбоновая кислота
Смесь 7,8-дихлор-1-[2-(S)-фтор-1-(R)-циклопропил]-1,4-дигидро-4-оксохинолин-3-карбоновой кислоты (253 мг, 0,80 ммоль), 3-(R)-[1-(трет-бутоксикарбониламино)циклопропил]пирролидина (272 мг, 1,20 ммоль), N-метилпиперидина (0,195 мл, 1,60 ммоль) и диметилсульфоксида (3 мл) перемешивали в течение 55 часов при нагревании на масляной бане при 80°C и в азот-замещенной атмосфере. После упаривания растворителя остаток распределяли между этилацетатом (50 мл) и 10% водным раствором лимонной кислоты (30 мл), и после отделения органического слоя органический слой промывали насыщенным хлористоводородным раствором (30 мл). Органический слой, полученный таким образом, сушили над безводным сульфатом натрия, и после удаления агента для сушки путем фильтрования растворитель упаривали при пониженном давлении. Остаток очищали хроматографией на силикагеле и из элюата хлороформ:метанол = 10:1 получали неочищенную 7-{3-(R)-[1-(трет-бутоксикарбониламино)циклопропил]пирролидин-1-ил}-8-хлор-1-[2-(S)-фтор-1-(R)-циклопропил]-1,4-дигидро-4-оксохинолин-3-карбоновую кислоту.
Указанную выше неочищенную 7-{3-(R)-[1-(трет-бутоксикарбониламино)циклопропил]пирролидин-1-ил}-8-хлор-1-[2-(S)-фтор-1-(R)-циклопропил]-1,4-дигидро-4-оксохинолин-3-карбоновую кислоту растворяли в концентрированной хлористо-водородной кислоте (5 мл) при охлаждении на льду, и после перемешивания при комнатной температуре в течение 20 минут раствор переливали в разделительную воронку и промывали хлороформом (10 мл × 10 раз или более). После промывки в водный слой добавляли насыщенный водный раствор гидроксида натрия при охлаждении на льду, таким образом доводя pH до >11, и затем рН доводили до 7,7 добавлением концентрированной хлористо-водородной кислоты и 1 моль/л хлористо-водородной кислоты. Полученный водный слой затем экстрагировали хлороформом (100 мл) и смесью хлороформ:метанол = 9:1 (100 мл×2) и объединенный органический слой сушили над безводным сульфатом натрия. После удаления агента для сушки путем фильтрования растворитель упаривали при пониженном давлении. Остаток затем очищали препаративной хроматографией (разделение в нижнем слое смесью хлороформ:метанол:вода, 7:3:1), очисткой взвеси, используя этанол-диэтиловый эфир, и затем сушили при пониженном давлении с получением 96 мг (30%) указанного в заголовке соединения в виде бледно-желтого порошка.
1H ЯМР (400 МГц, 0,1 моль/л NaOD/D2O) δ: 0,45-0,65 (4H, м), 1,15-1,30 (1H, м), 1,50-1,75 (2H, м), 2,00-2,10 (1H, м), 2,10-2,25 (1H, м), 3,25-3,40 (2H, м), 3,55-3,75 (2H, м), 4,10-4,15 (1H, м), 4,90-5,15 (1H, м), 7,05 (1H, д, J=9,0 Гц), 7,99 (1H, д, J=9,3 Гц), 8,39 (1H, д, J=3,7 Гц).
Точка плавления: 128-130°C.
Удельное вращение: [α]D 24,5=-193,0°.
Элементный анализ для C20H21ClFN3O3·1,5 H2O:
Вычислено: С 55,49%; H 5,59%; N 9,71%.
Найдено: С 55,74%; H 5,45%; N 9,57%.
MS (m/z): 406 [(M+H)+].
Ссылочный пример 48
Этил 7-фтор-1-[2-(S)-фтор-1-(R)-циклопропил]-5-гидрокси-8-метил-1,4-дигидро-4-оксохинолин-3-карбоксилат
Этил 5-амино-7-фтор-1-[2-(S)-фтор-1-(R)-циклопропил]-8-метил-1,4-дигидро-4-оксохинолин-3-карбоксилат (1,414 г, 4,39 ммоль) суспендировали в 35% водном растворе серной кислоты (15 мл), и при охлаждении на льду по каплям добавляли водный раствор нитрита натрия (394 мг, 5,70 ммоль/4,5 мл) в течение 5 минут. Затем, после перемешивания при 0°C в течение 30 минут добавляли небольшое количество мочевины и при той же температуре по каплям добавляли водный раствор тригидрата нитрата меди (II) (17,00, 70,2 ммоль/155 мл) в течение 10 минут. После перемешивания при 0°C в течение 5 минут добавляли оксид меди (I) (565 мг, 3,95 ммоль) при энергичном перемешивании реакционного раствора. Затем, после перемешивания при комнатной температуре в течение 20 минут проводили экстракцию хлороформом (200 мл×2) и после незначительного подщелачивания водного слоя с помощью бикарбоната натрия водный слой экстрагировали хлороформом (150 мл×3). Объединенный органический слой затем сушили над безводным сульфатом натрия, и после удаления агента для сушки путем фильтрования растворитель упаривали при пониженном давлении. Полученный неочищенный продукт очищали хроматографией на силикагеле, таким образом получая из элюата хлороформ:метанол = 10:1 297 мг (21%) указанного в заголовке соединения в виде желтого порошка.
1H ЯМР (400 МГц, CDCl3) δ: 1,36-1,47 (1H, м), 1,40 (3H, т, J=7,1 Гц), 1,53-1,63 (1H, м), 2,51 (3H, т, J=2,4 Гц), 3,85-3,90 (1H, м), 4,39 (2H, кв, J=7,2 Гц), 4,77-4,96 (1H, м), 6,58 (1H, д, J=11,5 Гц), 8,52 (1H, д, J=3,2 Гц).
MS (m/z): 324 [(M+H)+].
Ссылочный пример 49
7-фтор-1-[2-(S)-фтор-1-(R)-циклопропил]-5-гидрокси-8-метил-1,4-дигидро-4-оксохинолин-3-карбоновая кислота
Смесь этил 7-фтор-1-[2-(S)-фтор-1-(R)-циклопропил]-5-гидрокси-8-метил-1,4-дигидро-4-оксохинолин-3-карбоксилата (325 мг, 1,005 ммоль), уксусной кислоты (3 мл) и концентрированной хлористо-водородной кислоты (1,5 мл) кипятили с обратным холодильником в течение 2 часов на масляной бане при 120°C. После добавления воды (30 мл) и охлаждения на льду полученные кристаллы отфильтровывали и затем промывали водой, 5% водным раствором этанола и диэтиловым эфиром с получением 267 мг (90%) указанного в заголовке соединения в виде бледно-желтого порошка.
1H ЯМР (400 МГц, CDCl3) δ: 1,40-1,75 (2H, м), 2,58 (3H, т, J=2,5 Гц), 3,99-4,04 (1H, м), 4,80-5,05 (1H, м), 6,70 (1H, д, J=11,3 Гц), 8,76 (1H, д, J=3,2 Гц), 13,17 (0,7H, д, J=1,0 Гц), 13,34 (0,7H, шир.c).
Точка плавления: 209-213°C.
Удельное вращение: [α]D 24,7=-111,6°.
Элементный анализ для C14H11F2NO4:
Вычислено: С 56,95%; H 3,76%; N 4,74%.
Найдено: С 56,90%; H 3,74%; N 4,68%.
MS (m/z): 296[(M+H)+].
ПРИМЕР 17
7-[3-(R)-(1-аминоциклопропил)пирролидин-1-ил]-1-[2-(S)-фтор-1-(R)-циклопропил]-5-гидрокси-8-метил-1,4-дигидро-4-оксохинолин-3-карбоновая кислота
Смесь 7-фтор-1-[2-(S)-фтор-1-(R)-циклопропил]-5-гидрокси-8-метил-1,4-дигидро-4-оксохинолин-3-карбоновой кислоты (205 мг, 0,694 ммоль), 3-(R)-[1-(трет-бутоксикарбониламино)циклопропил]пирролидина (314 мг, 1,388 ммоль), N-метилпиперидина (0,243 мл, 1,388 ммоль) и диметилсульфоксида (1,5 мл) перемешивали в течение 66 часов при нагревании на масляной бане при 80°C в азот-замещенной атмосфере. После упаривания растворителя остаток распределяли между хлороформом (50 мл) и 10% водным раствором лимонной кислоты (30 мл) и после отделения органического слоя водный слой, кроме того, экстрагировали хлороформом (30 мл). Объединенный органический слой затем сушили над безводным сульфатом натрия, и после удаления агента для сушки путем фильтрования растворитель упаривали при пониженном давлении до получения, таким образом, неочищенной 7-{3-(R)-[1-(трет-бутоксикарбониламино)циклопропил]пирролидин-1-ил}-1-[2-(S)-фтор-1-(R)-циклопропил]-5-гидрокси-8-метил-1,4-дигидро-4-оксохинолин-3-карбоновой кислоты.
Полученную выше неочищенную 7-{3-(R)-[1-(трет-бутоксикарбониламино)циклопропил]пирролидин-1-ил}-1-[2-(S)-фтор-1-(R)-циклопропил]-5-гидрокси-8-метил-1,4-дигидро-4-оксохинолин-3-карбоновую кислоту растворяли в концентрированной хлористо-водородной кислоте (20 мл) при охлаждении на льду, и после перемешивания при комнатной температуре в течение 20 минут раствор выливали в разделительную воронку и промывали хлороформом (20 мл×5 раз). Затем в водный слой после промывки добавляли насыщенный водный раствор гидроксида натрия при охлаждении на льду, таким образом доводя pH до >11, и затем рН доводили до 7,5-7,8, добавляя концентрированную хлористо-водородную кислоту и 1 моль/л хлористо-водородную кислоту. Полученный водный слой затем экстрагировали в хлороформ (100 мл), смесью хлороформ:метанол = 9:1 (100 мл×2) и нижний слой смесью хлороформ:метанол:вода = 7:3:1 (100 мл), и объединенный органический слой сушили над безводным сульфатом натрия. После удаления агента для сушки путем фильтрования растворитель упаривали при пониженном давлении. Остаток затем очищали препаративной хроматографией (разделение в нижний слой смеси хлороформ:метанол:вода, 7:3:1), взвесь очищали, используя диэтиловый эфир, и затем сушили при пониженном давлении с получением 119 мг (43%) указанного в заголовке соединения в виде желтого порошка.
1H ЯМР (400 МГц, 0,1 моль/л NaOD) δ: 0,45-0,55 (4H, м), 1,05-1,20 (1H, м), 1,45-1,70 (2H, м), 1,90-2,00 (1H, м), 2,05-2,20 (1H, м), 2,16 (3H, c), 3,05-3,20 (2H, м), 3,25-3,35 (1H, м), 3,40-3,50 (1H, м), 3,90-3,95 (1H, м), 4,90-5,10 (1H, м), 6,16 (1H, c), 8,33 (1H, д, J=3,4 Гц).
Точка плавления: 203-206°C.
Удельное вращение: [α]D 25,1=-274,4°.
Элементный анализ для C21H24FN3O4·1,5 H2О:
Вычислено: С 58,87%; H 6,35%; N 9,81%.
Найдено: С 59,23%; H 6,20%; N 9,48%.
MS (m/z): 402[(M+H)+].
ПРИМЕР 18
7-[3-(R)-(1-аминоциклопропил)пирролидин-1-ил]-8-циано-1-[2-(S)-фтор-1-(R)-циклопропил]-1,4-дигидро-4-оксохинолин-3-карбоновая кислота
Смесь этил 8-циано-7-фтор-1-[2-(S)-фтор-1-(R)-циклопропил]-1,4-дигидро-4-оксохинолин-3-карбоксилата (250 мг, 0,785 ммоль), 3-(R)-[1-(трет-бутоксикарбониламино)циклопропил]пирролидина (267 мг, 1,18 ммоль), 1,4-диазабицикло[2.2.2]октана (132 мг, 1,18 ммоль) и диметилсульфоксида (13 мл) перемешивали в течение 2 часов при комнатной температуре в азот-замещенной атмосфере. После упаривания растворителя остаток распределяли между хлороформом (30 мл) и 10% водным раствором лимонной кислоты (30 мл) и органический слой отделяли. Полученный органический слой сушили над безводным сульфатом натрия и после удаления агента для сушки путем фильтрования растворитель упаривали при пониженном давлении. Остаток растворяли в концентрированной хлористо-водородной кислоте (4 мл) и ледяной уксусной кислоте (4 мл) при комнатной температуре и затем перемешивали в течение 12 часов при нагревании на масляной бане при 110°C. После упаривания растворителя добавляли концентрированную хлористо-водородную кислоту (2 мл) и воду (20 мл), раствор выливали в разделительную воронку и затем промывали хлороформом (50 мл). Затем в водный раствор после промывки добавляли 10 моль/л водный раствор гидроксида натрия, таким образом доводя pH до >12. Затем, после промывки хлороформом (50 мл) рН доводили до 8,3 добавлением концентрированной хлористо-водородной кислоты и 1 моль/л хлористо-водородной кислоты. Полученный водный слой затем концентрировали до 5 мл при пониженном давлении, экстрагировали смесью хлороформ:метанол = 10:1 (50 мл×3) и объединенный органический слой сушили над безводным сульфатом натрия. После удаления агента для сушки путем фильтрования растворитель упаривали при пониженном давлении. Таким образом полученное желтое твердое вещество перекристаллизовывали из этанола и затем сушили при пониженном давлении с получением 124 мг (40%) указанного в заголовке соединения в виде желтого порошка.
1Н ЯМР (400 МГц, ДМСО-d6) δ: 0,48-0,51 (4H, м), 1,66-1,90 (3Н, м), 1,99-2,03 (1H, м), 2,03-2,09 (1H, м), 3,57-3,70 (3Н, м), 3,79-3,83 (1H, м), 4,03-4,08 (1H, м), 5,18-5,35 (1H, м), 7,14 (1H, д, J=9,3 Гц), 8,16 (1H, д, J=9,3 Гц), 8,61 (1H, д, J=3,9 Гц).
Точка плавления: 138-140°C.
Удельное вращение: [α]D 24,5=+19,16°.
Элементный анализ для C21H21FN4О3·1,25 H2O:
Вычислено: С 60,21%; H 5,65%; N 13,07%.
Найдено: С 60,42%; H 5,62%; N 12,72%.
MS (m/z): 397[(M+H)+]
ПРИМЕР 19
7-[3-(R)-(1-аминоциклобутил)пирролидин-1-ил]-1-[2-(S)-фтор-1-(R)-циклопропил]-8-метокси-1,4-дигидро-4-оксохинолин-3-карбоновая кислота
Смесь 7-фтор-1-[2-(S)-фтор-1-(R)-циклопропил]-8-метокси-1,4-дигидро-4-оксохинолин-3-карбоновой кислоты (290 мг, 0,98 ммоль), 3-(R)-[1-(трет-бутоксикарбониламино)циклобутил]пирролидина (283 мг, 1,18 ммоль), триэтиламина (0,409 мл, 2,94 ммоль) и диметилсульфоксида (5 мл) перемешивали в течение 112 часов при нагревании на масляной бане при 80°C в азот-замещенной атмосфере. После упаривания растворителя остаток распределяли между хлороформом (50 мл) и 10% водным раствором лимонной кислоты (30 мл), и после отделения органического слоя его промывали насыщенным хлористо-водородным раствором (30 мл). Полученный органический слой затем сушили над безводным сульфатом натрия и после удаления агента для сушки путем фильтрования растворитель упаривали при пониженном давлении. Остаток очищали хроматографией на силикагеле и таким образом получали из элюата хлороформ:метанол = 20:1 неочищенную 7-{3-(R)-[1-(трет-бутоксикарбониламино)циклобутил]пирролидин-1-ил}-1-[2-(S)-фтор-1-(R)-циклопропил]-8-метокси-1,4-дигидро-4-оксохинолин-3-карбоновую кислоту.
Полученную выше неочищенную 7-{3-(R)-[1-(трет-бутоксикарбониламино)циклобутил]пирролидин-1-ил}-1-[2-(S)-фтор-1-(R)-циклопропил]-8-метокси-1,4-дигидро-4-оксохинолин-3-карбоновую кислоту растворяли в концентрированной хлористо-водородной кислоте (5 мл) при охлаждении на льду, и после перемешивания в течение 5 минут на ледяной бане раствор выливали в разделительную воронку и промывали хлороформом (10 мл×3 раза). Затем в водный слой после промывки добавляли 10 ммоль/л водный раствор гидроксида натрия при охлаждении на льду, таким образом доводя pH до >12, и затем рН доводили до 7,4, добавляя концентрированную хлористо-водородную кислоту и 1 моль/л хлористо-водородную кислоту. Полученный водный слой затем экстрагировали хлороформом (100 мл×3), и объединенный органический слой сушили над безводным сульфатом натрия. После удаления агента для сушки путем фильтрования растворитель упаривали при пониженном давлении. Остаток затем перекристаллизовывали в смеси этилацетат-гексан и сушили при пониженном давлении с получением 99 мг (24%) указанного в заголовке соединения в виде желтого порошка.
1Н ЯМР (400 МГц, d6-ДМСО) δ: 1,30-1,45 (1H, м), 1,45-1,60 (1H, м), 1,60-1,70 (1H, м), 1,70-2,50 (8H, м), 3,30-3,40 (1H, м), 3,40-3,50 (1H, м), 3,50 (3H, c), 3,56-3,60 (2H, м), 4,03-4,09 (1H, м), 5,00-5,22 (1H, м), 7,09 (1H, д, J=9,1 Гц), 7,92 (1H, д, J=9,1 Гц), 8,57 (1H, д, J=3,4 Гц).
Точка плавления: 174°C.
Элементный анализ для C22H26FN3O4·0,25 H2O:
Вычислено: С 62,92%; H 6,36%; N 10,01%.
Найдено: С 63,20%; H 6,22%; N 10,10%.
MS (m/z): 416[(M+H)+].
Ссылочный пример 50
3-Циано-2,4-дифторбензойная кислота
После растворения диизопропиламина (56,0 мл, 395 ммоль) в безводном тетрагидрофуране (400 мл) раствор перемешивали при -15°C в атмосфере азота. После добавления к нему гексанового раствора н-бутиллития (1,52 M, 260 мл, 395 ммоль) раствор перемешивали при охлаждении на льду в течение 1 часа. После охлаждения раствора до -78°C по каплям добавляли раствор, в котором 2,6-дифторбензонитрил (25,0 г, 180 ммоль) растворен в безводном тетрагидрофуране (100 мл), в течение 1 часа. После завершения добавления реакционный раствор перемешивали при -78°C в течение 1 часа и затем высушенный диоксид углерода барботировали в реакционный раствор в течение 30 минут. Реакционный раствор перемешивали при -78°C в течение 1 часа, затем постепенно повышали температуру и затем перемешивали при комнатной температуре в течение 12 часов. Затем в реакционный раствор добавляли 100 мл 1 моль/л хлористо-водородной кислоты при охлаждении на льду и экстрагировали диэтиловым эфиром (500 мл×2). Объединенный органический слой промывали насыщенным хлористо-водородным раствором (500 мл) и затем сушили над безводным сульфатом натрия. После фильтрования фильтрат концентрировали при пониженном давлении, таким образом получая 29,7 г (90%) желтого, аморфного соединения, указанного в заголовке. Данный продукт использовали в последующей реакции без дополнительного очищения.
1H ЯМР (400 МГц, CDCl3) δ: 7,10 (1Н, м), 8,32 (1Н, м).
Ссылочный пример 51
Этил 8-циано-7-фтор-1-[2-(S)-фтор-1-(R)-циклопропил]-l,4-дигидро-4-оксохинолин 3-карбоксилат
3-Циано-2,4-дифторбензойную кислоту (29,6 г, 162 ммоль) растворяли в высушенном толуол (250 мл), и после добавления каталитического количества N,N-диметилформамида по каплям добавляли тионилхлорид (17,7 мл, 243 ммоль) при перемешивании при комнатной температуре. После завершения добавления реакционный раствор перемешивали в течение 1 часа на масляной бане при 80°C. Затем, после того как реакционному раствору давали остыть, реакционный раствор концентрировали при пониженном давлении, к остатку добавляли толуол (100 мл) и снова проводили вакуумное концентрирование. Эту операцию проводили 3 раза. Полученный таким образом концентрат растворяли в безводном тетрагидрофуране (200 мл) и раствор по каплям добавляли при перемешивании и охлаждении на льду в раствор, в котором триэтиламин (30 мл) и этил 3-диметиламиноакрилат (24,3 г, 170 ммоль) растворены в безводном тетрагидрофуране (100 мл). После завершения добавления реакционный раствор кипятили с обратным холодильником в течение 12 часов. Затем, после фильтрования реакционного раствора через целит (промывка диэтиловым эфиром) фильтрат концентрировали при пониженном давлении и полученный остаток очищали с помощью хроматографии на короткой колонке с силикагелем. Затем из элюата хлороформ:метанол = 100:1-100:3 с помощью вакуумного концентрирования получали коричневое, маслянистое вещество.
Данное вещество затем растворяли в безводном тетрагидрофуране (300 мл), добавляли 2-(S)-фтор-1-(R)-циклопропиламинпаратолуолсульфонат (28,2 г, 114 ммоль) и при перемешивании при -15°C, постепенно по каплям добавляли раствор, в котором триэтиламин (23 мл, 165 ммоль) растворен в безводном тетрагидрофуране (50 мл). После завершения добавления реакционный раствор перемешивали при охлаждении на льду в течение 2 часов и затем перемешивали при комнатной температуре в течение 12 часов. Затем в реакционный раствор добавляли воду (300 мл) и проводили вакуумное концентрирование для упаривания тетрагидрофурана. Кроме того, добавляли воду (300 мл) и затем экстрагировали этилацетатом (400 мл×3). После промывки объединенного органического слоя насыщенным хлористо-водородным раствором (500 мл) его сушили над безводным сульфатом натрия. После фильтрования фильтрат концентрировали при пониженном давлении. Таким образом получали желто-коричневое маслянистое вещество.
Данное вещество растворяли в высушенном 1,4-диоксане (400 мл) и при перемешивании и охлаждении на льду постепенно добавляли 60% гидрид натрия в масле (4,35 г). Реакционную суспензию затем перемешивали при комнатной температуре в течение 1 часа. Затем, после концентрирования реакционного раствора до приблизительно 1/3 от первоначального объема при пониженном давлении постепенно вливали 0,5 моль/л хлористо-водородную кислоту (50 мл) при охлаждении на льду. Выпавшие твердые вещества отфильтровывали, промывали водой и затем промывали небольшим количеством холодного этанола и диэтилового эфира, в указанном порядке. Полученные неочищенные кристаллы очищали перекристаллизацией из изопропанола и сушили при пониженном давлении с получением 10,6 г (49%) указанного в заголовке соединения в виде желтовато-белых кристаллов.
Точка плавления: 172-177°C (разложение).
1H ЯМР (400 МГц, CDCl3) δ: 1,23-1,30 (1H, м), 1,41 (3H, т, J=7,1 Гц), 1,61-1,99 (1H, м), 4,00 (1H, м), 4,40 (2H, кв, J=7,1 Гц), 5,10 (1H, дм, J=63,5 Гц), 7,31 (1H, м), 8,52 (1H, д, J=2,6 Гц), 8,77 (1H, м).
[Экспериментальный пример 1]
Антибактериальную активность соединений данного изобретения определяли в соответствии со стандартными способами, разработанными Japan Society of Chemotherapy, и полученные результаты показаны в виде значений MIC (микрограмм/мл) в приведенной ниже таблице. Для сравнения со значениями MIC соединения данного изобретения в таблице также показаны значения MIC левофлоксацина (LVFX), ципрофлоксацина (CPFX) и 7-[3-(R)-(1-аминоциклопропил)пирролидин-1-ил]-6-фтор-1-[2-(S)-фтор-1-(R)-циклопропил]-8-метил-1,4-дигидро-4-оксохинолин-3-карбоновой кислоты (ссылочный препарат 1), который описан в PCT/JP96/00208.
[Экспериментальный пример 2]
Для соединения, описанного в примере 1 данного изобретения, микронуклеарный анализ в костном мозге мышей проводили следующим образом.
Использовались группы мышей, каждая из которых состояла из пяти самцов мышей Slc:ddY, возрастом шесть недель. Соединение данного изобретения, описанное в примере 1, и 7-[3-(R)-(1-аминоциклопропил)пирролидин-1-ил]-6-фтор-1-[2-(S)-фтор-1-(R)-циклопропил]-8-метил-1,4-дигидро-4-оксохинолин-3-карбоновую кислоту (ссылочный препарат 1), описанную в PCT/JP96/00208, растворяли в 0,1 моль/л NaOH/солевой раствор. В качестве контроля использовали растворитель 0,1 моль/л NaOH/солевой раствор, и в качестве положительного ссылочного препарата использовали раствор препарата, полученный путем растворения и разведения в солевом растворе циклофосфамида (CP). Все растворы препаратов дезинфицировали фильтрованием через фильтры Mylex GS 0,22 мкм. Каждый раствор препарата вводили единичной внутривенной дозой 10 мл/кг со скоростью введения 0,2 мл/мин. Через 24 часа после введения из бедренной кости брали миеломные клетки, готовили мазки и окрашивали их акриловым оранжевым. У каждой мыши было обнаружено, используя флуоресцентный микроскоп, 1000 полихроматических эритроцитов и была подсчитана частота появления микронуклеарных полихроматических эритроцитов и соотношение ортохроматических и полихроматических эритроцитов на 1000 эритроцитов.
В результате, между контролем и любой группой, которой вводили 25, 50 и 100 мг/кг соединения, описанного в примере 1, не наблюдалось значительной разницы в степени индукции микроядер, и результат оценки, таким образом, был отрицательный. То есть было обнаружено, что соединение, описанное в примере 1, незначительно индуцирует появление микроядер и является безопасным.
Наоборот, для сравнительного соединения 7-[3-(R)-(1-аминоциклопропил)пирролидин-1-ил]-6-фтор-1-[2-(S)-фтор-1-(R)-циклопропил]-8-метил-1,4-дигидро-4-оксохинолин-3-карбоновой кислоты (ссылочный препарат 1), описанного в PCT/JP96/00208, микронуклеарная индукция по сравнению с контролем была четко выражена у групп, которым вводили 50 и 100 мг/кг.
Данные результаты показывают, что соединение, описанное в примере 1 данного изобретения, где атом фтора в положении 6 сравнительного соединения 7-[3-(R)-(1-аминоциклопропил)пирролидин-1-ил]-6-фтор-1-[2-(S)-фтор-1-(R)-циклопропил]-8-метил-1,4-дигидро-4-оксохинолин-3-карбоновой кислоты, описанного в PCT/JP96/00208, заменен атомом водорода, обладает сильной антибактериальной активностью в отношении широкого спектра, как грамотрицательных бактерий, так и грамположительных бактерий, включая бактерии, устойчивые к антибиотикам, и, кроме того, является высокобезопасным.
[Экспериментальный пример 3]
Для соединения данного изобретения, описанного в примере 1, с помощью описанных ниже способов определяли концентрацию в крови и концентрацию в органах после перорального введения. Те же способы измерения также проводили для ссылочного препарата 1.
Способ 1: Исследование на животных
Вводимый раствор получали растворением исследуемого соединения до концентрации 2 мг/мл (в свободной форме) в дистиллированной воде и, используя 2,5 мл-овый одноразовый шприц или металлический пероральный зонд, раствор перорально вводили при дозе 20 мг/кг крысам (Crj: CD IGS; самцы; в возрасте 7 недель; Charles River Japan, Inc.) на голодный желудок.
Крыс из групп, исследуемых на поглощение (4 крысы на группу; всего 6 групп), умерщвляли обескровливанием под эфирным наркозом через 0,25, 0,5, 1, 2, 4 или 6 часов после введения, и кровь, печень, почки и легкие брали для образцов. После коагуляции кровь центрифугировали и брали образец сыворотки (3000 об/мин × 15 минут, 4°C). После добавления 3-5 мл 0,1 М фосфатного буферного раствора (рН 7,0) ткани гомогенизировали, и после центрифугирования собирали супернатанты (3000 об/мин × 15 минут, 4°C).
Крыс из групп, исследуемых на экскрецию (4 крысы на группу), после введения препарата помещали в метаболическую клетку и брали пробы при охлаждении на льду из образцов мочи, которые собирали в течение 0-4 часов и 4-24 часов, и в момент взятия проб внутреннюю часть клетки промывали приблизительно 15 мл 0,1 моль/л раствора фосфатного буфера (pH 7,0) для того, чтобы смыть мочу с внутренней части клетки. Также, для того чтобы оценить глюкуронид и другие конъюгированные соединения, брали часть пробы, гидролизовали эквивалентным количеством 1 моль/л водного раствора гидроксида натрия, после этого нейтрализовали 0,5 моль/л хлористо-водородной кислотой, и проводили измерения концентрации образцов, полученных этим способом.
Способ 2: Измерения концентрации препарата
Определение концентраций препарата в жидких образцах проводили с помощью биоаналитического способа агарных лунок, используя штамм B. subtilis ATCC6051 в качестве исследуемого организма. Исследуемую среду получали затравкой суспензии, содержащей 5×107 CFU/мл спор исследуемой бактерии в пропорции 1%, в пищевом агаре (Eiken Kagaku), который стерилизовали при 121°C в течение 15 минут и затем охлаждали до приблизительно 50°C. После того как в стерильную чашку Петри выливали 10 мл каждой данной среды и среда горизонтально застывала, в ней проделывали четыре отверстия диаметром 8 мм и получали систему лунок для исследования. Для получения системы лунок для исследования использовали систему Bioassay System TDA-1 (Dainippon Seiki). Для измерения готовили исследуемые образцы (разведенные сывороткой или раствором фосфатного буфера, если необходимо), серийные разведения растворов препарата для калибровки (двухкратные серийные разведения получали таким образом, чтобы диаметр ингибиторного кольца был приблизительно 10-30 мм) и раствор ссылочного препарата (раствор препарата данной концентрации для корректировки ошибки лунок; обычно, использовали концентрацию, образующую ингибиторное кольцо диаметром приблизительно 20 мм), и 50 мкл исследуемого образца (или раствора препарата для калибровки) помещали в две из четырех лунок из каждой чашки и 50 мкл ссылочного раствора помещали в две другие лунки. После добавления образцов среду чашек оставляли стоять в течение 1 часа при 4°C для осуществления предварительной дисперсии, затем проводили культивирование при 37°C в течение приблизительно 18 часов и измеряли диаметры ингибиторных колец, используя CA-400 (Dainippon Seiki). Концентрации исследуемых образцов измеряли, используя калибровочную кривую, полученную с помощью регрессии второго порядка из логарифмических значений концентрации препарата калибровочной кривой серийных разведений и диаметров ингибиторных колец.
Для определения тканевой концентрации (мкг/мл) концентрацию (мкг/мл) гомогенизированного супернатанта определяли из массы ткани (г) и количества добавленного фосфатного буфера (мл), используя следующее уравнение:
[Тканевая концентрация]=[концентрация гомогената]×([масса ткани]+[количество буферного раствора])/[масса ткани]
Экскрецию с мочой крысы (%) определяли из количества (мкг) введенного препарата, количества (мл) мочи (или промывочного раствора) и концентрации (мкг/мл) в моче (промывочном растворе):
[Экскреция с мочой крысы]=100×([количество мочи]× [концентрация в моче])/[количество введенного препарата]
Способ 3: Расчет фармакокинетических параметров
Для каждого препарата подсчитывали фармакокинетические параметры у крыс, на основе средней концентрации, с помощью некомпарментного анализа и используя программу по анализу фармакодинамики, PSAG-CP (Asmedica).
Сывороточная концентрация и концентрация в органах, в случае печени, почек и легких, соединения примера 1 и ссылочного препарата 1, которые определяли указанными выше способами, показаны в таблице 2.
(1 HCl,
0,25 IPA, 0,25 H2O
(1 MsOH,
1 H2O)
Легкие
10,0 (3,2)
5,3 (3,2)
после
добавления конъюгированных
соединений
8,7
2,3
Как видно из таблицы 2, было обнаружено, что соединение данного изобретения распределяется при более высоких концентрациях, как в сыворотке, так и в тканях, по сравнению с ссылочным препаратом 1. Таким образом, понятно, что соединение данного изобретения обладает превосходным пероральным поглощением. Также видно, что соединение данного изобретения превосходно проникает в ткани.
Ниже представлены структуры соединений, активность которых сравнивали.
примере 1
Ссылочный препарат 1
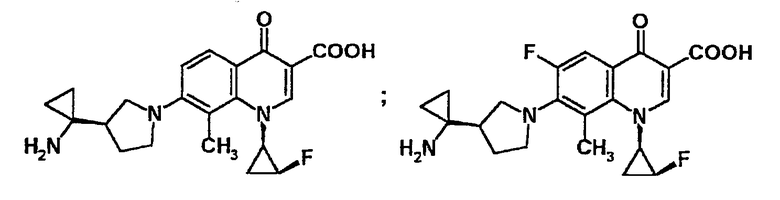
Промышленная применимость
Соединения данного изобретения обладают превосходной антибактериальной активностью в отношении широкого ряда как грамотрицательных, так и грамположительных бактерий и, в частности, обладают сильной антибактериальной активностью даже в отношении грамположительных бактерий, устойчивых к антибиотикам, таких как метициллинустойчивые Staphylococcus aureus (MRSA), пеннициллинустойчивые Streptococcus pneumoniae (PRSP), ванкомицинустойчивые Enterococcus (VRE) и так далее, и хинолонустойчивых бактерий, и также имеет прекрасные характеристики безопасности, например, является отрицательным в микроядерных тестах, и имеет превосходные фармакокинетические свойства, например прекрасно выводится с мочой у крыс и превосходно абсорбируется при пероральном введении и проникает в ткани и так далее. Соединения данного изобретения являются, таким образом, полезными в качестве антибактериальных соединений для использования в химиотерапии против микробных инфекций.
| название | год | авторы | номер документа |
|---|---|---|---|
| ЦИКЛОАЛКИЛЗАМЕЩЕННЫЕ ПРОИЗВОДНЫЕ АМИНОМЕТИЛПИРРОЛИДИНА И АНТИБАКТЕРИАЛЬНЫЙ АГЕНТ НА ИХ ОСНОВЕ | 1999 |
|
RU2248970C2 |
| СРЕДСТВО ПРОТИВ КИСЛОТОУСТОЙЧИВЫХ БАКТЕРИЙ, СОДЕРЖАЩЕЕ ГЕТЕРОЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ БЕНЗОКСАЗИНА В КАЧЕСТВЕ АКТИВНОГО КОМПОНЕНТА | 2001 |
|
RU2297420C2 |
| ПРОИЗВОДНЫЕ ТРИ- ИЛИ ТЕТРА-ЗАМЕЩЕННОГО-3-АМИНОПИРРОЛИДИНА | 2006 |
|
RU2420524C2 |
| СРЕДСТВО ПРОТИВ КИСЛОТОУСТОЙЧИВЫХ БАКТЕРИЙ, СОДЕРЖАЩЕЕ ПИРИДОНКАРБОНОВЫЕ КИСЛОТЫ В КАЧЕСТВЕ АКТИВНОГО КОМПОНЕНТА | 2001 |
|
RU2299205C2 |
| СПИРОСОЕДИНЕНИЕ ИЛИ ЕГО СОЛИ, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ЕГО ОСНОВЕ, ОБЛАДАЮЩАЯ ПРОТИВОМИКРОБНОЙ АКТИВНОСТЬЮ | 1989 |
|
RU2094432C1 |
| КОНДЕНСИРОВАННОЕ ЗАМЕЩЕННОЕ ПРОИЗВОДНОЕ АМИНОПИРРОЛИДИНА | 2007 |
|
RU2443698C2 |
| БИЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ 1,4-ДИГИДРО-4-ОКСОХИНОЛИН-3-КАРБОНОВОЙ КИСЛОТЫ ИЛИ ИХ СОЛИ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ОБЛАДАЮЩАЯ АНТИМИКРОБНОЙ АКТИВНОСТЬЮ | 1993 |
|
RU2125046C1 |
| ПРОИЗВОДНЫЕ АМИНОМЕТИЛПИРРОЛИДИНА, ИМЕЮЩИЕ АРОМАТИЧЕСКИЕ ЗАМЕСТИТЕЛИ | 2000 |
|
RU2255938C2 |
| ПРОИЗВОДНЫЕ ПИРИДОНКАРБОНОВОЙ КИСЛОТЫ И КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 1993 |
|
RU2100351C1 |
| ПРОИЗВОДНЫЕ ХИНОЛИН- ИЛИ НАФТИРИДИНКАРБОНОВОЙ КИСЛОТЫ И СПОСОБЫ ИХ ПОЛУЧЕНИЯ | 1995 |
|
RU2120940C1 |
Данное изобретение относится к синтетическому хинолоновому антибактериальному средству, которое эффективно в качестве лекарственных средств, ветеринарных препаратов, лекарств, применяемых в рыболовстве, или в качестве антибактериальных консервантов. Описывается соединение, представленное следующей общей формулой (I), в виде его отдельных изомеров или их смеси, его соли и их гидраты: 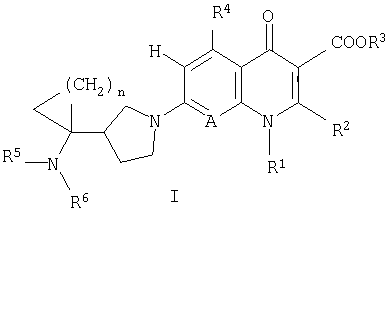 [где R1 представляет циклическую алкильную группу, содержащую 3-6 атомов углерода, которая может содержать заместитель, выбранный из галогена; R2 представляет атом водорода; R3 представляет атом водорода; R4 представляет атом водорода, аминогруппу, гидроксильную группу; А представляет атом азота или часть структуры, такой как указано в формуле изобретения, каждый R5 и R6 независимо представляет алкильную группу, содержащую 1-6 атомов углерода, или атом водорода; и n равно целому числу 1 или 2]. Также описываются антибактериальное средство и терапевтическое средство для лечения инфекционного заболевания на основе соединений формулы (I), способ получения антибактериального средства, способ получения лекарственного средства для лечения инфекционного заболевания и применение соединения формулы (I), для получения антибактериального средства и применение соединения формулы (I), для получения лекарственного средства для лечения инфекционного заболевания. Технический результат - получены новые соединения, обладающие полезными биологическими свойствами. 7 н. и 28 з.п. ф-лы, 2 табл.
[где R1 представляет циклическую алкильную группу, содержащую 3-6 атомов углерода, которая может содержать заместитель, выбранный из галогена; R2 представляет атом водорода; R3 представляет атом водорода; R4 представляет атом водорода, аминогруппу, гидроксильную группу; А представляет атом азота или часть структуры, такой как указано в формуле изобретения, каждый R5 и R6 независимо представляет алкильную группу, содержащую 1-6 атомов углерода, или атом водорода; и n равно целому числу 1 или 2]. Также описываются антибактериальное средство и терапевтическое средство для лечения инфекционного заболевания на основе соединений формулы (I), способ получения антибактериального средства, способ получения лекарственного средства для лечения инфекционного заболевания и применение соединения формулы (I), для получения антибактериального средства и применение соединения формулы (I), для получения лекарственного средства для лечения инфекционного заболевания. Технический результат - получены новые соединения, обладающие полезными биологическими свойствами. 7 н. и 28 з.п. ф-лы, 2 табл.
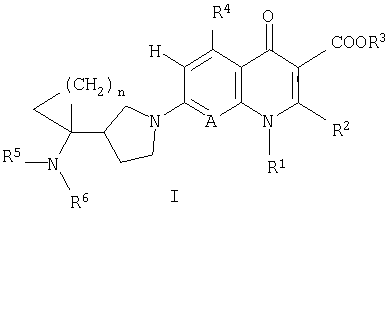
где R1 представляет циклическую алкильную группу, содержащую 3-6 атомов углерода, которая может содержать заместитель, выбранный из галогена;
R2 представляет атом водорода;
R3 представляет атом водорода;
R4 представляет атом водорода, аминогруппу, гидроксильную группу;
А представляет атом азота или часть структуры, представленную формулой (II)
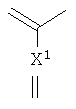
где X1 представляет алкильную группу, содержащую 1-6 атомов углерода, алкоксигруппу, содержащую 1-6 атомов углерода, атом водорода, аминогруппу, атом галогена, цианогруппу или галогенметоксигруппу,
где X1 и указанный выше R1 могут быть объединены с образованием кольцевой структуры путем включения части исходного скелета, где полученное таким образом кольцо может содержать в качестве атома, образующего кольцо, атом кислорода и данное кольцо может быть замещено алкильной группой, содержащей 1-6 атомов углерода);
каждый R5 и R6 независимо представляет алкильную группу, содержащую 1-6 атомов углерода или атом водорода; и
n равно целому числу 1 или 2.
| Прибор, замыкающий сигнальную цепь при повышении температуры | 1918 |
|
SU99A1 |
| ЕР 0230295 А2, 19.01.1987 | |||
| ПРОИЗВОДНЫЕ 5-АМИНО-8-МЕТИЛ-7-ПИРРОЛИДИНИЛХИНОЛИН-3-КАРБОНОВОЙ КИСЛОТЫ, СПОСОБЫ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СПОСОБ ЛЕЧЕНИЯ, ПРОМЕЖУТОЧНЫЙ ПРОДУКТ | 1994 |
|
RU2130932C1 |
| ПРОИЗВОДНЫЕ АЗЕТИДИНА ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ, ОБЛАДАЮЩИЕ АНТИМИКРОБНОЙ АКТИВНОСТЬЮ | 1991 |
|
RU2044735C1 |

