ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
Настоящее изобретение относится к композициям и к твердым пероральным лекарственным формам, содержащим усилитель. В частности, изобретение относится к композициям и к твердым лекарственным формам для перорального применения, содержащим фармацевтически активный ингредиент в комбинации с усилителем, которое усиливает биодоступность и/или всасывание активного ингредиента.
ПРЕДПОСЫЛКИ СОЗДАНИЯ ИЗОБРЕТЕНИЯ
Эпителиальные клетки, выстилающие поверхность просвета желудочно-кишечного тракта (GIT), могут представлять собой главный барьер для доставки лекарственных средств путем перорального введения. Однако существует четыре признанных пути транспорта, которые могут быть использованы для облегчения доставки и транспорта лекарственных средств: трансцеллюлярный, парацеллюлярный, опосредованный носителем и трансцитотический пути транспорта. Способность лекарственного средства, такого как традиционное лекарственное средство, пептид, белок, макромолекула или система на основе нано- или микрочастицы, «взаимодействовать» с одним или более из этих путей транспорта, может приводить к увеличенной доставке этого лекарственного средства из желудочно-кишечного тракта в основное кровообращение.
Некоторые лекарственные средства используют транспортные системы для питательных веществ, которые находятся в апикальных клеточных мембранах (опосредованный носителем путь). Макромолекулы также могут быть транспортируемы через клетки в эндоцитозных везикулах (трансцитотический путь). Однако многие лекарственные средства транспортируются через кишечный эпителий посредством пассивной диффузии либо через клетки (трансцеллюлярный путь), либо между клетками (парацеллюлярный путь). Большая часть перорально введенных лекарственных средств всасываются путем пассивного транспорта. Лекарственные средства, которые являются липофильными, проникают в эпителий посредством трансцеллюлярного пути, тогда как лекарственные средства, которые являются гидрофильными, ограничиваются парацеллюлярным путем.
Парацеллюлярные пути занимают менее чем 0,1% общей площади кишечного эпителия. Кроме того, непроницаемые перегородки, которые образуют непрерывный пояс (зону) вокруг апикальной части клеток, ограничивают проникновение между клетками посредством создания уплотняющего слоя между соседними клетками. Таким образом, всасывание при пероральном применении гидрофильных лекарственных средств, таких как пептиды, может быть сильно ограничено. Другие барьеры для всасывания лекарственных средств могут включать гидролазы в щеточной каемке просвета или в эпителиальных клетках кишечника, наличие водного граничного слоя на поверхности эпителиальной мембраны, который может обеспечивать дополнительный диффузионный барьер, слизистый слой, связанный с водным граничным слоем, и кислотный микроклимат, который создает градиент протонов через апикальную мембрану.
Всасывание и в конечном счете биодоступность лекарственного средства также может быть снижена посредством других процессов, таких как регулируемый P-гликопротеином транспорт лекарственного средства обратно в просвет кишки и метаболизм цитохрома Р450. Присутствие пищи и/или напитков также может оказывать влияние на всасывание и биодоступность.
Бисфосфонаты представляют собой семейство лекарственных средств, используемых для предотвращения и лечения переломов костей, остеопороза, болезни Педжета, метастатического рака костей, и других заболеваний костей с высокой резорбцией кости. Бисфосфонаты связываются с гидроксиапатитом кости и замедляют клетки, приводящие к разрушению кости, известные как остеокласты. Этот эффект дает возможность строительным клеткам кости, известным как остеобласты, работать более эффективно.
Некоторые из ограничений в отношении традиционно используемых бисфосфонатов включают раздражение верхнего желудочно-кишечного тракта (GIT), такое как язва пищевода, и низкая биодоступность. В результате традиционно используемые бисфосфонаты требуют особого режима дозирования для того, чтобы больной мог получать при всасывании некоторую часть лекарственного средства надлежащим образом и избегать побочных эффектов. Так как пища, напитки, лекарственные препараты и кальций мешают всасыванию, то традиционно используемые бисфосфонаты должны вводиться на пустой желудок и в зависимости от конкретного бисфосфоната пациенту следует выжидать от 30 минут до двух часов прежде чем потреблять какую-либо пищу, напитки (кроме воды), лекарственные препараты или кальцийсодержащие добавки. Поскольку язва пищевода дает известный побочный эффект, традиционно используемые бисфосфонаты принимают с целым стаканом воды и избегают принимать горизонтальное положение в пределах 30-60 минут после введения лекарственной формы.
Особые характеристики алендроната оказались полезными в том, чтобы показать на его примере членов класса бисфосфонатов и спорные вопросы, связанные с ними. Алендронат представляет собой белый, кристаллический, без запаха, негигроскопичный бисфосфонат, приготовленный химическим синтезом. Алендронат натрий тригидрат имеет молекулярную массу 325,1. Алендронат одобрен в США для предупреждения и лечения остеопороза у мужчин и у женщин в период постменопаузы и для лечения болезни Педжета (деформирующий остоз кости) и вызванного глюкокортикоидом остеопороза у представителей обоих полов. Подобно другим бисфосфонатам алендронат связывается с гидроксиапатитом кости и специфично ингибирует активность остеокластов. Алендронат ослабляет процесс обновления костной ткани (костный метаболизм) у человека и на моделях заболевания на животном и сокращает повторяемость активизирования, уменьшая резорбцию кости как в трубчатой, так и в трабекулярной кости, и в конечном счете увеличивая плотность кости и прочность.
Биодоступность алендроната при пероральном введении алендроната является очень низкой и зависит от дозы (5-80 мг), в среднем составляя 0,76% у женщин и 0,59% у мужчин. Пресистемный метаболизм не происходит. После перорального введения традиционно используемых форм алендроната 40% поглощенной дозы выделяются в моче в пределах 8 часов и дополнительные 5% выделяются за последующие 64 часа. От шестидесяти до семидесяти процентов всасывания происходит в пределах 1 часа от момента введения дозы. Биодоступность значительно снижается при одновременном потреблении пищи (85%-90%) и даже потребление кофе или апельсинового сока будет ухудшать всасывание на 60% или более. Одновременное введение лекарственных препаратов также будет снижать всасывание, поскольку любые кальцийсодержащие соединения и многовалентные катионы будут связываться с бисфосфонатом. Повышение рН среды желудка выше 6 ставят в соответствие с двоекратным увеличением всасывания алендроната. Алендронат не усваивается и выделяется неизмененным с почечным клиренсом, сравнимым со скоростью клубочковой фильтрации.
Бисфосфонатные композиции и лекарственные формы для перорального применения с улучшенной системной биодоступностью, которые не подвергаются ограничениям в отношении дозирования как традиционно используемые бисфосфонаты, могли бы принести значительную пользу для больных. В результате необходимы новые стратегии доставки лекарственных средств через слои клеток желудочно-кишечного тракта, особенно для бисфосфонатов.
Были идентифицированы многочисленные усилители всасывания. Например, глицериды со средней длиной цепи продемонстрировали способность усиливать всасывание гидрофильных лекарственных средств через слизистую оболочку кишечника (Pharm. Res. (1994), 11,1148-54). Однако важность длины цепи и/или композиции является неясной и, следовательно, их механизм действия остается в значительной степени неизвестным. Было сделано сообщение о том, что капрат натрия усиливает всасывание лекарственного средства в кишечнике и в толстой кишке посредством парацеллюлярного пути (Pharm. Res. (1993), 10, 857-864; Pharm. Res. (1988), 5, 341-346). Патент U.S. No. 4656161 (BASF AG) раскрывает способ увеличения энтеральной (зондовой) всасываемости гепарина и гепариноидов путем добавления неионогенных поверхностно-активных веществ, таких как неионогенные поверхностно-активные вещества, которые могут быть приготовлены посредством реакционного взаимодействия этиленоксида с жирной кислотой, со спиртом жирного ряда, со сложным эфиром алкилфенола или сорбита, или глицерина с жирной кислотой.
Патент U.S. No.5229130 (Cygnus Therapeutics Systems) раскрывает композицию, которая увеличивает проницаемость кожи в отношении к трансдермально введенному фармакологически активному агенту, составленную с одним или более растительными маслами в качестве усилителей проницания внутрь через кожу. Также известно, что проникновение через кожу улучшается посредством ряда карбоксилатов натрия [Int. J. of Pharmaceutics (1994), 108, 141-148]. Кроме того, известно использование эфирных масел для усиления биодоступности (патент U.S. No.566386, AvMax Inc. and others). Патент дает идею о том, что эфирные масла действуют с уменьшением либо метаболизма цитохрома Р450 либо регулируемого Р-гликопротеином транспорта лекарственного средства из кровотока обратно в кишку.
Однако часто усиление всасывания лекарственного средства коррелирует с разрушением стенки кишечника. Следовательно, ограничения в отношении широко распространенного применения усилителей всасывания через желудочно-кишечный тракт зачастую определяются их потенциальными токсичностями и побочными эффектами. Дополнительно и особенно в отношении пептидных, белковых или макромолекулярных лекарственных средств «взаимодействие» усилителя всасывания через желудочно-кишечный тракт с одним из путей транспорта должно быть преходящим или обратимым, таким как преходящее взаимодействие с непроницаемыми перегородками или преходящее взаимодействие, размыкающее непроницаемые перегородки для того, чтобы усилить транспорт посредством парацеллюлярного пути.
Как упомянуто выше, известны многочисленные потенциальные усилители. Однако это не привело к соответствующему числу продуктов, включающих усилители. Один такой продукт, в настоящее время апробированный/разрешенный для применения в Швеции и Японии, представляет собой Doktacillin™, суппозиторий [Lindmark et al. Pharmaceutical Research (1997), 14, 930-935]. Суппозиторий содержит ампициллин и жирную кислоту со средней длиной цепи, капрат натрия (С10).
Предоставление твердой лекарственной формы для перорального применения, которая могла бы облегчить введение лекарственного средства вместе с усилителем, является желательным. Преимущества твердых лекарственных форм для перорального применения по сравнению с другими лекарственными формами включают удобство производства, возможность рецептировать различные составы с контролируемым восвобождением и с пролонгированным высвобождением и удобство введения. Введение лекарственных средств в виде раствора не облегчает контроль профиля концентрации лекарственного средства в кровотоке. С другой стороны, твердые лекарственные формы для перорального применения являются многофункциональными/многоцелевыми и могут быть модифицированы, например, с максимальным увеличением степени и длительности высвобождения лекарственного средства и с высвобождением лекарственного средства в соответствии с терапевтически желательным профилем высвобождения. Также могут иметь место преимущества, связанные с твердыми лекарственными формами для перорального применения, относящиеся к удобству введения, увеличивающему контактность больного, и к себестоимости.
КРАТКОЕ ИЗЛОЖЕНИЕ СУЩНОСТИ ИЗОБРЕТЕНИЯ
В соответствии с одной особенностью настоящего изобретения, композиции и изготовленные из них лекарственные формы настоящего изобретения содержат лекарственное средство и усилитель для облегчения всасывания бисфосфоната на выстилке клеток желудочно-кишечного тракта, где усилитель представляет собой жирную кислоту со средней длиной цепи или производное жирной кислоты со средней длиной цепи, имеющее длину углеродной цепи от 6 до 20 атомов углерода; с оговорками, что (i) в том случае, когда усилитель представляет собой сложный эфир жирной кислоты со средней длиной цепи, указанная длина цепи от 6 до 20 атомов углерода относится к длине цепи карбоксилатного фрагмента, и (ii) в том случае, когда усилитель представляет собой простой эфир жирной кислоты со средней длиной цепи, по меньшей мере, одна алкоксигруппа имеет длину углеродной цепи от 6 до 20 атомов углерода, и где усилитель и композиция представляют собой твердые вещества при комнатной температуре.
В соответствии с еще одной особенностью настоящего изобретения, композиции и изготовленные из них лекарственные формы содержат лекарственное средство и усилитель для облегчения всасывания бисфосфоната на выстилке клеток желудочно-кишечного тракта, где единственный усилитель, присутствующий в композиции, представляет собой жирную кислоту со средней длиной цепи или производное жирной кислоты со средней длиной цепи, имеющее длину углеродной цепи от 6 до 20 атомов углерода.
В вариантах осуществления, в которых лекарственное средство содержит бисфосфонат, лекарственное средство может быть выбрано из группы, которая включает формы свободной кислоты и биологически приемлемые соли алендроната, клодроната, этидроната, инкадроната, ибандроната, минодроната, неридроната, олпадроната, памидроната, ризедроната, тилудроната, золедроната и их производных. Бисфосфонатная лекарственная форма может представлять собой твердую лекарственную форму для перорального применения, покрытую энтеросолюбильной оболочкой, с мгновенным высвобождением, которая обеспечивает улучшенную биодоступность при пероральном введении и минимизирует риск локального раздражения верхней части желудочно-кишечного тракта.
Лекарственные формы могут представлять собой таблетку, лекарственную форму, состоящую из множества частиц, или капсулу. Лекарственная форма, состоящая из множества частиц, может быть в форме таблетки или содержаться в капсуле. Таблетка может представлять собой одно- или многослойную таблетку, спрессованную из множества частиц, в одном, во всех или ни в одном из слоев. Предпочтительно, лекарственная форма представляет собой лекарственную форму с контролируемым высвобождением, и более предпочтительно, лекарственную форму с отсроченным высвобождением. Лекарственная форма может быть покрыта полимером, предпочтительно полимером, контролирующим скорость, или с отсроченным высвобождением. Полимер также может быть спрессован с усилителем и лекарственным средством с образованием матричной лекарственной формы, такой как матричная лекарственная форма с контролируемым высвобождением. Затем на матричную лекарственную форму может быть нанесено полимерное покрытие.
Другие варианты осуществления этого изобретения включают способы изготовления лекарственных форм, и способы лечения или предупреждения состояний, подлежащих лечению, посредством введения лекарственных форм больному.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
Фиг.1 показывает влияние натриевых солей, имеющих С8, С10, С12, С14, С18 и С18:2, с 3Н-TRH (тиротропин-высвобождающий гормон, тиролиберин) на трансэпителиальное электрическое сопротивление (TEER) (Ωсм2) в монослоях кишечных клеток Caco-2 за время от 0 вплоть до 2 часов с 30-минутными интервалами, что описано в Примере 1.
Фиг.2 показывает влияние натриевых солей, имеющих С8, С10, С12, С14, С18 и С18:2, на постоянную (эффективной) проницаемость, Рарр, для транспорта 3Н-TRH (тиротропин-высвобождающий гормон, тиролиберин) в монослоях кишечных клеток Caco-2, что описано в Примере 1.
Фиг.3 показывает профили концентрация - время для сывороточного TRH (тиротропин-высвобождающий гормон, тиролиберин) после болюсного введения внутрь двенадцатиперстной кишки 500 мкг TRH с усилителем NaC8 или NaC10 (35 мг), присутствующим в соответствии с экспериментальной моделью на крысе с замкнутой системой регулирования, описанной в Примере 1.
Фиг.4 показывает профили концентрация - время для сывороточного TRH после болюсного введения внутрь двенадцатиперстной кишки 1000 мкг TRH с усилителем NaC8 или NaC10 (35 мг), присутствующим в соответствии с экспериментальной моделью на крысе с замкнутой системой регулирования, описанной в Примере 1.
ФИГ.5 показывает реакцию Активированного Частичного Тромбопластинового/Протромбинового Времени (APTT/APPT response) за период времени 4 часа после введения USP-гепарина (гепарин, отвечающий требованиям фармакопеи США) (1000 МЕ) с различными уровнями капрата натрия (С10) (10 и 35 мг) в соответствии с экспериментальной моделью на крысе c замкнутой системой регулирования, описанной в Примере 2.
ФИГ.6 показывает реакцию анти-фактора Xa за период времени 5 часов после введения USP-гепарина (1000 МЕ) в присутствии различных уровней (10 мг и 35 мг) каприлата натрия (С8) в соответствии с экспериментальной моделью на крысе с замкнутой системой регулирования, описанной в Примере 2.
ФИГ.7 показывает реакцию анти-фактора Xa за период времени 5 часов после введения USP-гепарина (1000 МЕ) в присутствии различных уровней (10 мг и 35 мг) капрата натрия в соответствии с экспериментальной моделью на крысе с замкнутой системой регулирования Примера 2.
Фиг.8 показывает среднюю реакцию анти-фактора Xa у собак за период времени вплоть до 8 часов после введения: а) подкожно раствора USP-гепарина (5000 МЕ); b) таблетированного состава для перорального применения, непокрытого оболочкой, с мгновенным высвобождением, содержащего USP-гепарин (90000 МЕ) и NaС10; с) таблетированного состава для перорального применения, непокрытого оболочкой, с мгновенным высвобождением, содержащего USP-гепарин (90000 международных единиц) и NaС8; и d) таблетированного состава для перорального применения, непокрытого оболочкой, с замедленным высвобождением, содержащего USP-гепарин (90000 МЕ) и капрат натрия, приготовленного в соответствии с изобретением так, как описано в Примере 2.
Фиг.9 показывает реакцию анти-фактора Xa за период времени три часа после введения в двенадцатиперстную кишку крыс забуференных фосфатом физиологических растворов парнапарин-натрия (гепарин с низкой молекулярной массой (LMWH)) (1000 МЕ), в присутствии 35 мг различных усилителей, таких как каприлат натрия (С8), нонаноат натрия (С9), капрат натрия (С10), ундеканоат натрия (С11), лаурат натрия (С12), и различных бинарных смесей (50:50) усилителей, крысам (n=8) в экспериментальной модели с незамкнутой системой регулирования. Эталонный продукт содержал вводящийся подкожно парнапарин-натрий (250 МЕ). Контрольный раствор содержал вводящийся в двенадцатиперстную кишку раствор, содержащий 1000 МЕ парнапарин-натрия без какого-либо усилителя.
Фиг.10 показывает средние плазменные уровни лейпролида за период времени восемь часов после введения в двенадцатиперстную кишку растворов лейпролида (20 мг), содержащих различные уровни капрата натрия (0,0 г (контрольный), 0,55 г, 1,1 г), собакам.
Фиг.11 показывает среднюю реакцию анти-фактора Xa у собак за период времени 8 часов после перорального введения парнапарин-натрия (90000 МЕ) в присутствии 550 мг капрата натрия как в форме раствора (10 мл), так и в таблетированной лекарственной форме с мгновенным высвобождением.
Фиг.12 показывает среднюю реакцию анти-фактора Xa у людей за период времени 24 часа после перорального введения парнапарин-натрия (90000 МЕ) в присутствии капрата натрия как в форме раствора (240 мл), так и в таблетированной лекарственной форме с мгновенным высвобождением.
Фиг.13 показывает среднюю реакцию анти-фактора Xa у людей за период времени 24 часа после введения в тощую кишку 15 мл растворов, содержащих различные дозы парнапарин-натрия (20000 МЕ, 45000 МЕ, 90000 МЕ) в присутствии различных доз капрата натрия (0,55 г, 1,1 г, 1,65 г).
Фиг.14 показывает среднюю реакцию анти-фактора Xa у собак за период времени 8 часов после перорального введения 45000 МЕ парнапарин-натрия, в форме: (а) капсул с мгновенным высвобождением, содержащих 0,55 г капрата натрия, (b) покрытых Эудрагитом L и быстро распадающихся таблеток, содержащих 0,55 г капрата натрия, и (с) покрытых Эудрагитом L и быстро распадающихся таблеток, не содержащих усилитель.
Фиг.15 показывает среднюю реакцию анти-фактора Xa у собак за период времени 8 часов после одновременного введения 45000 МЕ гепарина с низкой молекулярной массой (LMWH) и 0,55 г капрата натрия перорально в тощую кишку и в толстую кишку в сравнении с подкожным введением.
Фиг.16 показывает ненормированное по дозе количество алендроната, выделяемое в моче за период времени 36 часов после перорального введения алендроната (17,5 мг) с различными количествами капрата натрия (0,5 г и 0,25 г) в состояниях на голодный желудок и после приема пищи, в сравнении со средними плазменными уровнями Fosamax® (35 мг) после введения в состоянии на голодный желудок.
Фиг.17 показывает среднее кумулятивное количество золедроновой кислоты, выделяемой в моче за период 48 часов после перорального введения золедроновой кислоты в 10-мг и 20-мг таблетках в сравнении с количеством, выделяемым после внутривенной инъекции (раствора) золедроновой кислоты (1 мг), приготовленного из жидкого концентрата Zometa®.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Как использовано в этом описании изобретения и в прилагаемой формуле изобретения, формы единственного числа включают объекты во множественном числе, если в содержании не предписано ясно иное. Таким образом, например, ссылка на усилитель включает смесь двух или более усилителей, ссылка на «лекарственное средство» включает смесь двух или более лекарственных средств и тому подобное.
Как использовано в этом документе, термин «лекарственное средство» включает любое лекарственное средство, включая традиционные лекарственные средства и их аналоги, приемлемые для введения через полость рта животному, в том числе человеку. Термин «лекарственное средство» также эксплицитно включает те объекты, которые являются слабо всасываемыми через полость рта, включающие гидрофильные или макромолекулярные лекарственные средства, такие как пептиды, белки, олигосахариды, полисахариды или гормоны, включающие инсулин, кальцитонин, регуляторный белок гена кальцитонина, атриальный натрийуретический белок, колониестимулирующий фактор, бетасерон, эритропоэтин (EPO), интерфероны, соматропин, соматотропин, соматостатин, инсулиноподобный фактор роста (соматомедины), рилизинг-фактор лютеннизирующего гормона (LHRH), активатор тканевого плазминогена (TPA), тиротропин-высвобождающий гормон (TRH), фактор, высвобождающий гормон роста (GHRH), антидиуретический гормон (ADH) или вазопрессин и их аналоги, такие как, например, десмопрессин, паратиреоидный гормон (PTH), окситоцин, эстрадиол, гормоны роста, лейкопролид-ацетат, гозерелин-ацетат, наферелин, бусерелин, фактор VIII, интерлейкины, такие как интерлейкин-2, и их аналоги, но не ограниченные этим, и антикоагулянты (вещества, задерживающие свертывание крови), такие как гепарин, гепариноиды, гепарин с низкой молекулярной массой, гирудин и их аналоги, бисфосфонаты, включающие алендронат, клодронат, этидронат, инкадронат, ибандронат, минодронат, неридронат, олпадронат, памидронат, ризедронат, тилудронат и золедронат, пентасахариды, включающие антикоагулянтные пентасахариды, антигены, адъюванты и тому подобное. В тех вариантах осуществления, в которых лекарственное средство представляет собой бисфосфонат, лекарственное средство выбирают из группы, состоящей из алендроната, клодроната, этидроната, инкадроната, ибандроната, минодроната, неридроната, олпадроната, памидроната, ризедроната, тилудроната и золедроната. Как использовано в этом документе, термины «лекарственное средство» и «бисфосфонат» включают все их формы, включающие оптически чистые энантиомеры или смеси, рацемические или иные, энантиомеры, а также производные формы, такие как, например, соли, кислоты, сложные эфиры и тому подобное. Лекарственное средство может быть обеспечено в любом подходящем фазовом состоянии, включающем твердое вещество, жидкое вещество, раствор, суспензию и тому подобное. При обеспечении в твердой порошковой форме частицы могут иметь любой подходящий размер или любую подходящую морфологию и могут принимать одну или более кристаллических, полукристаллических и/или аморфных форм.
Лекарственное средство может быть включено в системы доставки лекарственных средств на основе нано- или микрочастиц, в которых лекарственное средство представляет собой нано- или микрочастицу, или вкраплено в пределах нано- или микрочастицы, инкапсулировано посредством нано- или микрочастицы, прикреплено к нано- или микрочастице, или иначе связано с нано- или микрочастицей.
Как использовано в этом документе, «терапевтически эффективное количество лекарственного средства» относится к количеству лекарственного средства, которое позволяет достигнуть терапевтически полезного эффекта в лечении существующего медицинского состояния и/или в предупреждении или в затягивании возникновения проявления медицинского состояния у животного, предпочтительно млекопитающего, наиболее предпочтительно человека.
Как использовано в этом документе, термин «усилитель» относится к соединению (или к смеси соединений), которое способно усилить транспорт лекарственного средства, особенно гидрофильного и/или макромолекулярного лекарственного средства через желудочно-кишечный тракт у животного, такого как человек, где усилитель представляет собой жирную кислоту со средней длиной цепи или производное жирной кислоты со средней длиной цепи, имеющее длину углеродной цепи от 6 до 20 атомов углерода; с оговорками, что (i) в том случае, когда усилитель представляет собой сложный эфир жирной кислоты со средней длиной цепи, то указанная длина цепи от 6 до 20 атомов углерода относится к длине цепи карбоксилатного фрагмента, и (ii) в том случае, когда усилитель представляет собой простой эфир жирной кислоты со средней длиной цепи, по меньшей мере, одна алкоксигруппа имеет длину углеродной цепи от 6 до 20 атомов углерода. Предпочтительно, усилитель представляет собой натриевую соль жирной кислоты со средней длиной цепи. Наиболее предпочтительно, усилитель представляет собой капрат натрия. В одном варианте осуществления, усилитель представляет собой твердое вещество при комнатной температуре.
Как использовано в этом документе, термин «производное жирной кислоты со средней длиной цепи» включает соли жирных кислот, сложные эфиры жирных кислот, простые эфиры жирных кислот, галогенангидриды жирных кислот, амиды жирных кислот, ангидриды жирных кислот, карбоксилатные сложные эфиры жирных кислот, нитрилы жирных кислот, а также глицериды, такие как моно-, ди- или три-глицериды жирных кислот. Углеродная цепь может быть охарактеризована как углеродная цепь с различными степенями насыщения. Другими словами, углеродная цепь может быть, например, полностью насыщенной или частично ненасыщенной (то есть содержащей одну или более углерод-углеродных кратных связей). Подразумевается, что термин «производное жирной кислоты со средней длиной цепи» также охватывает жирные кислоты со средней длиной цепи, где конец углеродной цепи, противоположный кислотной группе (или группе - производной кислотной группы), является также функционализированным одним из указанных выше фрагментов (то есть сложноэфирный, эфирный, галогенангидридный, амидный, ангидридный, карбоксилатный сложноэфирный, нитрильный или глицеридный фрагмент). Такие дифункциональные производные жирных кислот, таким образом, включают, например, дикислоты и ди-сложные эфиры (где функциональные фрагменты являются одного типа) и также дифункциональные соединения, содержащие различные функциональные фрагменты, такие как аминокислоты и производные аминокислот, например, жирную кислоту со средней длиной цепи или сложный эфир или соль жирной кислоты со средней длиной цепи, содержащие амидный фрагмент с противоположного конца углеродной цепи жирной кислоты относительно кислотного или сложноэфирного или солевого фрагмента.
Как использовано в этом документе, «терапевтически эффективное количество усилителя» относится к количеству усилителя, которое усиливает доставку лекарственного средства через кишечник в основное кровообращение и позволяет поглощать терапевтически эффективное количество лекарственного средства посредством перорального введения. Было показано, что эффективность усилителя в усилении доставки через желудочно-кишечный тракт слабо проницаемых лекарственных средств зависит от места введения (см. Примеры 6, 7 и 12), где место оптимальной доставки зависит от лекарственного средства и усилителя.
Усилитель настоящего изобретения взаимодействует преходящим и обратимым образом с выстилкой клеток желудочно-кишечного тракта, увеличивая проницаемость и облегчая всасывание иным способом слабо проницаемых молекул. Предпочтительные усилители включают (i) жирные кислоты со средней длиной цепи и их соли, (ii) сложные эфиры жирных кислот средней длины цепи с глицерином и пропиленгликолем, и (iii) соли желчных кислот. В одном варианте осуществления усилитель представляет собой соль жирной кислоты со средней длиной цепи, сложный эфир, простой эфир или другое производное жирной кислоты со средней длиной цепи, которое предпочтительно представляет собой твердое вещество при комнатной температуре и которое имеет длину углеродной цепи от 8 до 14 атомов углерода; с оговорками, что (i) в том случае, когда усилитель представляет собой сложный эфир жирной кислоты со средней длиной цепи, то указанная длина цепи от 8 до 14 атомов углерода относится к длине цепи карбоксилатного фрагмента, и (ii) в том случае, когда усилитель представляет собой простой эфир жирной кислоты со средней длиной цепи, то, по меньшей мере, одна алкоксигрупа имеет длину углеродной цепи от 8 до 14 атомов углерода. В другом варианте осуществления усилитель представляет собой натриевую соль жирной кислоты со средней длиной цепи, жирной кислоты со средней длиной цепи, имеющей длину углеродной цепи от 8 до 14 атомов углерода; где натриевая соль является твердой при комнатной температуре. В дополнительном варианте осуществления усилитель представляет собой каприлат натрия, капрат натрия или лаурат натрия. Лекарственное средство и усилитель могут присутствовать в соотношении от 1:100000 до 10:1 (лекарственное средство: усилитель), предпочтительно от 1:1000 до 10:1.
Как использовано в этом документе, термин «полимерный материал, контролирующий скорость» включает гидрофильные полимеры, гидрофобные полимеры и смеси гидрофильных и/или гидрофобных полимеров, которые способны регулировать или замедлять высвобождение лекарственного соединения из твердой лекарственной формы для перорального применения настоящего изобретения. Подходящие полимерные материалы, контролирующие скорость, включают полимерные материалы, контролирующие скорость, выбранные из группы, состоящей из гидроксиалкилцеллюлозы, такой как гидроксипропилцеллюлоза и гидроксипропилметилцеллюлоза; поли(этилен)оксида; алкилцеллюлозы, такой как этилцеллюлоза и метилцеллюлоза; карбоксиметилцеллюлозы, гидрофильных производных целлюлозы; полиэтиленгликоля; поливинилпирролидона; ацетата целлюлозы; бутиратацетата целлюлозы; фталатацетата целлюлозы; тримеллитатацетата целлюлозы; поливинилацетатфталата; фталата гидроксипропилметилцеллюлозы; сукцинатацетата гидроксипропилметилцеллюлозы; поливинилацетальдиэтиламиноацетата; поли(алкилметакралат)а и поли(винилацетат)а. Другие подходящие гидрофобные полимеры включают полимеры и/или сополимеры, полученные из акриловой или метакриловой кислоты и их соответствующих сложных эфиров, зеина, восков, шеллака и гидрогенизированных растительных масел.
Особенно полезными в практическом использовании настоящего изобретения являются полиакриловая кислота, полиакрилатные полимеры, полиметакриловая кислота и полиметакрилатные полимеры, такие как вышеупомянутые полимеры, продаваемые под торговым наименованием Eudragit® (Rohm GmbH, Darmstadt, Germany), особенно материалы для покрытий (оболочек) Eudragit® L, Eudragit® S, Eudragit® RL, Eudragit® RS и их смеси. Некоторые из этих полимеров могут быть использованы в качестве полимеров с отсроченным высвобождением для регулирования места высвобождения лекарственного средства. Они включают полиметакрилатные полимеры, такие как полиметакрилатные полимеры, продаваемые под торговым наименованием Eudragit® (Rohm GmbH, Darmstadt, Germany), особенно материалы для покрытиий (оболочек) Eudragit® L, Eudragit® S, Eudragit® RL, Eudragit® RS и их смеси.
Твердая пероральная лекарственная форма в соответствии с настоящим изобретением может представлять собой таблетку, лекарственную форму, состоящую из множества частиц, или капсулу. Предпочтительная твердая лекарственная форма для перорального применения представляет собой лекарственную форму с отсроченным высвобождением, которая минимизирует высвобождение лекарственного средства и усилителя в желудке, и, следовательно, разбавление локальной концентрации усилителя в нем, и высвобождает лекарственное средство и усилитель в кишечном тракте. Особенно предпочтительной твердой лекарственной формой для перорального применения является лекарственная форма с отсроченным высвобождением быстрого проявления. Такая лекарственная форма минимизирует высвобождение лекарственного средства и усилителя в желудке, и, следовательно, разбавление локальной концентрации усилителя в нем, но высвобождает лекарственное средство и усилитель быстро, как только достигнуто соответствующее место в кишечном тракте, максимально увеличивая доставку слабо проницаемого лекарственного средства путем максимального увеличения локальной концентрации лекарственного средства и усилителя в месте всасывания.
Как использовано в этом документе, термин «таблетка» включает таблетки с немедленным высвобождением (IR), таблетки с замедленным высвобождением (SR), матричные таблетки, многослойные таблетки, многослойные матричные таблетки, таблетки с пролонгированным высвобождением, таблетки с отсроченным высвобождением и таблетки с импульсным высвобождением, любые или все из которых могут быть необязательно покрыты одним или более материалами для покрытий, включая полимерные материалы для покрытий, такие как покрытия-оболочки для кишечного всасывания, покрытия, контролирующие скорость, полупроницаемые покрытия и тому подобное, но не ограничивается этим. Термин «таблетка» также включает системы с осмотической доставкой, в которых лекарственное соединение является связанным с осмотическим агентом (и необязательно другие эксципиенты) и покрыто полупроницаемой мембраной, где полупроницаемая мембрана определяет отверстие, через которое лекарственное соединение может быть высвобождено. Таблетированные твердые лекарственные формы для перорального применения, особенно полезные в практическом использовании этого изобретения, включают таблетированные твердые лекарственные формы для перорального применения, выбранные из группы, состоящей из таблеток с немедленным высвобождением (IR), таблеток с замедленным высвобождением (SR), таблеток с немедленным высвобождением (IR), покрытых оболочкой, матричных таблеток, матричных таблеток, покрытых оболочкой, многослойных таблеток, многослойных таблеток, покрытых оболочкой, многослойных матричных таблеток и многослойных матричных таблеток, покрытых оболочкой. Предпочтительная таблетированная лекарственная форма представляет собой таблетированную лекарственную форму с энтеросолюбильным покрытием. Особенно предпочтительной таблетированной лекарственной формой является таблетированная лекарственная форма, покрытая оболочкой для кишечного всасывания, быстрого проявления.
Как использовано в этом документе, термин «капсула» включает капсулы с мгновенным высвобождением, капсулы с замедленным высвобождением, капсулы, покрытые оболочкой, с мгновенным высвобождением, капсулы, покрытые оболочкой, с замедленным высвобождением, капсулы с отсроченным высвобождением и капсулы, покрытые оболочкой, с отсроченным высвобождением. В одном варианте осуществления инкапсулированная лекарственная форма представляет собой инкапсулированную лекарственную форму, покрытую оболочкой для кишечного всасывания. В другом варианте осуществления инкапсулированная лекарственная форма представляет собой инкапсулированную лекарственную форму, покрытую оболочкой для кишечного всасывания, быстрого проявления.
Термин «лекарственная форма, состоящая из множества частиц», используемый в этом документе, означает множество дискретных частиц, шариков, мини-таблеток и их смесей или комбинаций. Если лекарственная форма для перорального применения представляет собой капсулу, состоящую из множества частиц, то твердые или мягкие желатиновые капсулы могут быть подходящим образом использованы для вмещения множества частиц. Альтернативно пакет-саше может быть подходящим образом использован для вмещения множества частиц. Лекарственная форма, состоящая из множества частиц, может быть покрыта слоем, содержащим контролирующий скорость полимерный материал. Лекарственная форма для перорального применения, состоящая из множества частиц, может содержать смесь двух или более совокупностей частиц, шариков или мини-таблеток, имеющих различные характеристики высвобождения in vitro и/или in vivo. Например, лекарственная форма для перорального применения, состоящая из множества частиц, может включать смесь компонента с мгновенным высвобождением и компонента с отсроченным высвобождением, содержащуюся в подходящей капсуле. В одном варианте осуществления лекарственная форма, состоящая из множества частиц, заключает в себе капсулу, вмещающую мини-таблетки с отсроченным высвобождением быстрого проявления. В еще одном варианте осуществления лекарственная форма, состоящая из множества частиц, заключает в себе капсулу с отсроченным высвобождением, вмещающую мини-таблетки с мгновенным высвобождением. В дополнительном варианте осуществления лекарственная форма, состоящая из множества частиц, заключает в себе капсулу, вмещающую гранулы с отсроченным высвобождением. В еще одном варианте осуществления лекарственная форма, состоящая из множества частиц, заключает в себе капсулу с отсроченным высвобождением, вмещающую гранулы с мгновенным высвобождением.
В другом варианте осуществления лекарственная форма, состоящая из множества частиц, вместе с одним или более веществами вспомогательных эксципиентов могут быть спрессованы в таблетированную форму, такую как однослойная или многослойная таблетка. Типично многослойная таблетка может включать два слоя, содержащих одинаковые или различные уровни одного и того же активного ингредиента, имеющего одинаковые или различные характеристики высвобождения. Альтернативно многослойная таблетка может содержать различный активный ингредиент в каждом слое. Такая таблетка, однослойная или многослойная, может быть необязательно покрыта полимером с контролируемым высвобождением для того, чтобы обеспечить свойства дополнительного контролируемого высвобождения.
Теперь будет описан ряд вариантов осуществления этого изобретения. В каждом случае лекарственное средство может присутствовать в любом количестве, которое является достаточным для достижения терапевтического эффекта. Фактическое количество используемого лекарственного соединения будет зависеть, среди прочих факторов, от эффективности действия лекарственного средства, от особенностей больного и от терапевтической цели, для достижения которой используется лекарственное средство, что будет оценено специалистами в данной области. Количество лекарственного соединения подходящим образом может находиться в диапазоне от приблизительно 0,5 мкг до приблизительно 1000 мг. Усилитель присутствует подходящим образом в любом количестве, достаточном для того, чтобы сделать возможным поглощение терапевтически эффективных количеств лекарственного средства посредством перорального введения. В одном варианте осуществления лекарственное средство и усилитель присутствуют в соотношении от 1:100000 до 10:1 (лекарственное средство: усилитель). В еще одном варианте осуществления соотношение лекарственного средства к усилителю составляет от 1:1000 до 10:1. Используемое фактическое соотношение лекарственного средства к усилителю будет зависеть, среди прочих факторов, от эффективности действия конкретного лекарственного средства и от усиливающей активности конкретного усилителя.
В одном варианте осуществления обеспечивают фармацевтическую композицию и твердую лекарственную форму для перорального применения, изготовленную из нее, содержащую бисфосфонат, и в качестве усилителя для облегчения всасывания бисфосфоната на выстилке клеток желудочно-кишечного тракта жирную кислоту со средней длиной цепи или производное жирной кислоты со средней длиной цепи, имеющее длину углеродной цепи от 6 до 20 атомов углерода, где усилитель и композиция являются твердыми веществами при комнатной температуре.
В другом варианте осуществления обеспечивают фармацевтическую композицию и лекарственную форму для перорального применения, изготовленную из нее, содержащую бисфосфонат, и в качестве усилителя для облегчения всасывания бисфосфоната на выстилке клеток желудочно-кишечного тракта жирную кислоту со средней длиной цепи или производное жирной кислоты со средней длиной цепи, имеющее длину углеродной цепи от 6 до 20 атомов углерода, где усилитель, присутствующий в композиции, является единственным.
В дополнительном варианте осуществления обеспечивают многослойную таблетку, содержащую композицию настоящего изобретения. Обычно такая многослойная таблетка может включать первый слой, содержащий лекарственное средство и усилитель в форме с мгновенным высвобождением, и второй слой, содержащий лекарственное средство и усилитель в форме с модифицированным высвобождением. Как использовано в этом документе, термин «модифицированное высвобождение» включает замедленное, отсроченное или иным способом контролируемое высвобождение лекарственного средства при введении больному. В альтернативном варианте осуществления многослойная таблетка может включать первый слой, содержащий лекарственное средство, и второй слой, содержащий усилитель. Каждый слой может независимо включать дополнительные эксципиенты, выбираемые для модифицирования высвобождения лекарственного средства или усилителя. Таким образом, лекарственное средство и усилитель могут быть высвобождены из соответственных первого и второго слоев со скоростями, которые являются одинаковыми или различными. Альтернативно каждый слой многослойной таблетки может содержать как лекарственное средство, так и усилитель в одинаковых или различных количествах.
В еще одном варианте осуществления обеспечивают лекарственную форму, состоящую из множества частиц, содержащую композицию настоящего изобретения. Лекарственная форма, состоящая из множества частиц, может содержать частицы, шарики, мини-таблетки или их комбинации, и лекарственное средство и усилитель могут содержаться в одинаковых или в различных совокупностях частиц, шариков или мини-таблеток, составляющих эту лекарственную форму, состоящую из множества частиц. В вариантах осуществления лекарственной формы, состоящей из множества частиц, для вмещения множества частиц могут быть подходящим образом использованы пакеты-саше и капсулы, такие как твердые или мягкие желатиновые капсулы. Лекарственная форма, состоящая из множества частиц, может содержать смесь двух или более совокупностей частиц, шариков или мини-таблеток, имеющих различные характеристики высвобождения in vitro и/или in vivo. Например, лекарственная форма, состоящая из множества частиц, может включать смесь компонента с немедленным высвобождением и компонента с отсроченным высвобождением, содержащуюся в подходящей капсуле.
В случае любого из вышеуказанных вариантов осуществления покрытие с контролируемым высвобождением может быть нанесено на конечную лекарственную форму (капсула, таблетка, многослойная таблетка и так далее). Покрытие с контролируемым высвобождением обычно может включать контролирующий скорость полимерный материал, который определен выше. Характеристики растворения такого материала для покрытий могут зависеть от рН или могут не зависеть от рН.
Различные варианты осуществления твердых лекарственных форм для перорального применения этого изобретения могут дополнительно содержать вещества вспомогательных эксципиентов, такие как, например, разбавители, лубриканты, разрыхлители, пластификаторы, антиадгезивы, вещества, вызывающие помутнение, пигменты, ароматизирующие вещества и тому подобное. Точный выбор эксципиентов и их относительные количества будут зависеть в некоторой степени от конечной лекарственной формы, что будет оценено специалистами в данной области.
Подходящие разбавители включают, например, фармацевтически приемлемые инертные наполнители, такие как микрокристаллическая целлюлоза, лактоза, гидроортофосфат кальция, сахариды, и/или смеси любых наполнителей из вышеприведенных. Примеры разбавителей включают микрокристаллическую целлюлозу, такую как микрокристаллическую целлюлозу, продаваемую под товарным знаком Avicel (FMC Corp., Philadelphia, Pa.), например, Avicel™ pH101, Avicel™ pH102 и Avicel™ pH112; лактозу, такую как моногидрат лактозы, безводная лактоза и Pharmatose DCL21; гидроортофосфат кальция, такой как Emcompress® (JRS Pharma, Patterson, NY); маннит; крахмал; сорбит; сахарозу; и глюкозу.
Подходящие лубриканты, включая агенты, которые оказывают влияние на сыпучесть порошка, который должен быть спрессован, представляют собой, например, коллоидный диоксид кремния, такой как Aerosil™ 200; тальк; стеариновую кислоту, стеарат магния и стеарат кальция.
Подходящие разрыхлители включают, например, слегка сшитый поливинилпирролидон, кукурузный крахмал, картофельную муку, маисовый крахмал и модифицированные крахмалы, натрий-кроскармелозу (натрий-карбоксиметилцеллюлоза, поперечно сшитая), кросповидон, натрий-крахмалгликолят и их комбинации и смеси.
ПРИМЕР 1. Таблетки, содержащие тиротропин-высвобождающий гормон (TRH)
(а) Монослои кишечных клеток Caco-2
Клеточная культура: Клетки Caco-2 выращивают в среде для выращивания клеток Dulbecco's Modified Eagles Medium (DMEM): 4,5 г/л глюкозы, дополненные посредством 1% (объем/объем) заменяемых аминокислот; 10% фетальной телячьей сыворотки и 1% пенициллина/стрептомицина. Клетки выращивают при 37°С и в среде с 5% СО2 и с 95% влажностью. Клетки растут и увеличиваются в объеме в стандартных колбах для тканевой культуры и после однократного пассажа достигают 100% слияния. Затем клетки Caco-2 высевают на поликарбонатные фильтрующие элементы-вставки (Costar; с диаметром 12 мм, с размером пор 0,4 мкм) с плотностью 5 х 105 клеток/см2 и инкубируют на шестилуночных культуральных планшетах при смене среды каждый второй день. Во всех исследованиях используют непрерывные монослои на 20 день - 30 день высевания на фильтрах и с 30-40 пассажами.
Исследования трансэпителиального транспорта: Влияние натриевых солей различных жирных кислот со средней длиной цепи (MCFA) на транспорт 3Н-TRH (апикальный - базолатеральный поток) изучают следующим образом: 15,0 мкКи/мл (0,2 мкМ) 3Н-TRH добавляют апикально в нулевой момент времени для экспериментов по переносу TRH. Эксперименты по изучению транспортных свойств осуществляют в сбалансированном солевом растворе Хэнка (HBSS), содержащем 25 мМ буферного раствора N-[2-гидроксиэтил]-пиперазин-N'-[2-этансульфокислота] (HEPES), рН 7,4 при 37°С. Вследствие вариаций растворимостей разные концентрации натриевых солей различных жирных кислот со средней длиной цепи (MCFA) и разнообразные апикальные буферные растворы используют так, как показано в Таблице 1. Во всех случаях базолатеральная камера содержит стандартный раствор HBSS+HEPES.
Концентрации и буферные растворы, используемые для натриевых солей различных жирных кислот со средней длиной цепи (MCFA)
NaC8:0
NaC10:0
NaC12:0
NaC14:0
NaC18:0
NaC18:2
0,32
0,40
3,77
1,44
0,16
0,16
HBSS+HEPES
HBSS без Ca2+
PBS**
PBS**
HBSS+HEPES
HBSS+HEPES
*В номенклатуре СХ:Y для соли жирной кислоты со средней длиной цепи (MCFA), X обозначает длину углеродной цепи, и Y обозначает положение ненасыщенной связи, если таковая имеется.
**PBS - фосфатный буферный раствор.
После удаления среды клеточной культуры монослои помещают в лунки, содержащие предварительно нагретый раствор HBSS (37°С); 1 мл апикально и 2 мл базолатерально. Монослои инкубируют при 37°С в течение 30 минут. Затем в нулевой момент времени апикальный раствор HBSS заменяют релевантным апикальным раствором для испытаний, содержащим соединения с радиоактивной меткой с соединением усилителя и соединения с радиоактивной меткой без соединения усилителя. Трансэпителиальное электрическое сопротивление (TEER) монослоя измеряют от нулевого момента времени вплоть до 120 минут с 30-минутными интервалами с использованием прибора: Millicell ERS chopstix apparatus (Millipore (U.K.) Ltd., Hertfordshire, UK) с электронным вольтомметром (Evom) для отслеживания целостности монослоя. Планшеты помещают на орбитальный шейкер в инкубаторе (37°С). После осуществления транспорта через слои проводят базолатеральный отбор проб (1 мл) с 30-минутными интервалами вплоть до 120 минут. Через каждый 30-минутный интервал каждый элемент-вставку перемещают в новую лунку, содержащую 2 мл свежеприготовленного предварительно нагретого раствора HBSS. Радиоактивность апикального исходного раствора определяют посредством отбора проб объемом 10 мкл в момент времени t=0 и t=120 минут. Сцинтилляционную жидкость (10 мл) добавляют в каждую пробу и для каждой пробы определяют разрушения в минуту в сцинтилляционном счетчике Wallac System 1409. Средние значения для концентраций 3H-TRH вычисляют для апикального и базолатерального растворов в каждый момент времени. Постоянные эффективной проницаемости вычисляют с использованием способа, описанного Артурсоном (Artursson P., J. Pharm. Sci. 79:476-482 (1990)).
ФИГ.1 показывает влияние натриевых солей С8, С10, С12, С14, С18 и С18:2 с 3Н-TRH на трансэпителиальное электрическое сопротивление (TEER) (Ω см2) в монослоях кишечных клеток Caco-2 в течение 2 часов. Данные для С8, С10, С14 и С18 указывают минимальное снижение трансэпителиального электрического сопротивления по сравнению с контрольным образцом. Хотя данные для С12 указывают на некоторое разрушение клеток (снижение TEER), это снижение, вероятно, является результатом более высокой концентрации усилителя, используемого в этом образце.
ФИГ.2 показывает влияние натриевых солей С8, С10, С12, С14, С18 и С18:2 на постоянную эффективной проницаемости (Рарр) для 3Н-TRH через монослои кишечных клеток Caco-2. В сравнении с контрольным образцом натриевые соли С8, С10, С12 и С14 показывают значительные повышения постоянной эффективной проницаемости, Рарр, при используемых концентрациях. Отмечают, что высокое значение Рарр, наблюдаемое для соли С12, может служить признаком разрушения клеток при такой высокой концентрации усилителя.
Митохондриальное Токсикологическое Исследование: Определяют Активность митохондриальной дегидрогеназы (MDH) как индикатор жизнеспособности клеток с использованием способа, основанного на изменении цвета тетразолиевой соли в присутствии митохондриальной дегидрогеназы (MDH). Клетки собирают, подсчитывают и высевают на 96-луночные планшеты с приблизительной плотностью 106 клеток/мл (100 мкл суспензии клеток на лунку). Затем клетки инкубируют при 37°С в течение 24 часов в увлажненной атмосфере, с 5% СО2. Ряд лунок обрабатывают каждым раствором натриевой соли MCFA с концентрациями, показанными в Таблице 1, и планшет инкубируют в течение 2 часов. После инкубирования в каждую лунку добавляют 10 мкл реагента для введения радиоактивной MTT-метки (3-(4,5-диметилтиазол-2-ил)-2,5-дифенилтетразолия бромид) в течение 4 часов. Повышающий растворимость - буферный раствор (100 мкл; см. Таблицу 1) добавляют в каждую лунку, и планшет инкубируют в течение дополнительных 24 часов. Поглощение в области 570 нм для каждой пробы измеряют с использованием спектрофотометра (Dynatech MR7000).
(b) Введение in vivo (Экспериментальная модель на крысе с замкнутой системой регулирования)
Исследования in vivo с использованием экспериментальной модели на крысе с замкнутой системой регулирования проводят с некоторыми изменениями способов Doluisio et al. (Doluisio J.T., et al.: Journal of Pharmaceutical Science (1969), 58, 1196-1200) и Brayden et al. (Brayden D.: Drug Delivery Pharmaceutical News (1997) 4(1)). Крысам-самцам Wistar (диапазон масс 250 г - 350 г) делают анестезию посредством кетамингидрохлорида/ацепромазина. Делают срединный разрез на животе и отделяют сегмент двенадцатиперстной кишки (7-9 см ткани), удаленный от центра сфинктера привратника желудка приблизительно на 5 см, заботясь о том, чтобы избежать повреждения окружающих кровеносных сосудов. Растворы проб (растворы PBS, содержащие С8 или С10 (35 мг) и TRH (500 мкг и 1000 мкг)) и контрольный раствор (раствор PBS, содержащий только TRH (500 мкг и 1000 мкг)), нагретые до 37°С, вводят непосредственно в просвет сегмента двенадцатиперстной кишки с использованием иглы калибром 26 G. Все объемы доз, вводимых в двенадцатиперстную кишку (для проб и для контрольного раствора), составляют 1 мл/кг. Проксимальный конец сегмента перевязывают и петлю опрыскивают изотоническим солевым раствором (37°С) для обеспечения влаги, и затем помещают обратно в брюшную полость, избегая растяжения. Разрез закрывают посредством хирургических зажимов. Группе животных вводят TRH в растворе PBS (100 мкг в 0,2 мл) путем подкожного впрыскивания в качестве эталонной модели.
ФИГ.3 показывает профили концентрация - время для сывороточного тиротропин-высвобождающего гормона (TRH) после болюсного введения в двенадцатиперстную кишку 500 мкг TRH с присутствующим усилителем NaC8 или NaC10 (35 мг), в соответствии с экспериментальной моделью на крысе с замкнутой системой регулирования. ФИГ.4 показывает профили концентрация - время для сывороточного тиротропин-высвобождающего гормона (TRH) после болюсного введения в двенадцатиперстную кишку 1000 мкг TRH с присутствующим усилителем NaC8 или NaC10 (35 мг), в соответствии с экспериментальной моделью на крысе с замкнутой системой регулирования. Из ФИГ.3 и 4 можно увидеть, что присутствие усилителя в каждом случае значительно повышает уровни TRH в сыворотке по сравнению с контрольным раствором TRH, что указывает на увеличенное всасывание лекарственного средства в присутствии усилителя.
(с) Таблетирование
После установления усиливающего эффекта NaC8 и NaC10 на TRH в растворе могут быть приготовлены таблетки с немедленным высвобождением (IR) и с замедленным высвобождением (SR) TRH и тому подобное. Составы таблеток с немедленным высвобождением (IR) и с замедленным высвобождением (SR) подробно описаны в Таблицах 2 и 3 ниже:
Детали составов таблеток с немедленным высвобождением (IR) TRH (все количества в масс.%)
Mg
1,27
1,23
2,42
2,42
69,73
-
-
-
-
67,64
66,45
66,45
0,5
0,5
0,5
0,5
0,5
0,5
0,5
0,5
20
20
-
20
8
8
8
8
-
-
20
-
-
2,13
2,13
2,13
Детали составов таблеток с замедленным высвобождением (SR) TRH (все количества в масс.%)
1,05
2,68
57,95
73,94
0,5
0,5
0,5
0,5
20
20
20
-
-
2,37
ПРИМЕР 2. Таблетки, содержащие гепарин
(а) Сегмент Крысы с замкнутой системой регулирования
Методику, выполняемую в Примере 1(а) выше, повторяют с использованием USP-гепарина вместо TRH и с введением дозы в подвздошную кишку вместо введения в двенадцатиперстную кишку. Делают срединный разрез в брюшной стенке и определяют местоположение для дистального конца подвздошной кишки (приблизительно в 10 см, проксимальное к месту анатомического соединения подвздошной кишки и слепой кишки). Отделяют 7-9 см ткани и дистальный конец перевязывают, заботясь о том, чтобы избежать повреждения окружающих кровеносных сосудов. Всасывание гепарина, показателем которого является реакция активированного протромбинового времени (APTT), измеряют путем размещения капли цельной крови (свежевзятой из хвостовой артерии) в цилиндрическом контейнере для испытаний монитора свертывания (коагуляции) Biotrack 512. Измерения активированного протромбинового времени (APTT) делают в различные моменты времени. ФИГ.5 показывает реакцию APTT для USP-гепарина (1000 МЕ) с различными уровнями (10 и 35 мг) капрата натрия (С10). Из использования реакции APTT в качестве показателя всасывания гепарина в кровоток ясно, что имеет место значительное увеличение всасывания в присутствии капрата натрия в сравнении с контрольным раствором гепарина, не содержащим усилитель.
Пробы цитратной крови центрифугируют при скорости 3000 оборотов в минуту (rpm) в течение 15 минут с получением плазмы для анализа анти-фактора Ха. ФИГ.6 показывает реакцию анти-фактора Ха для USP-гепарина (1000 МЕ) в присутствии каприлата натрия (С8, 10 мг и 35 мг). ФИГ.7 показывает реакцию анти-фактора Ха для USP-гепарина (1000 МЕ) в присутствии капрата натрия (С10, 10 мг и 35 мг). Контрольный раствор в каждом случае представляет собой раствор с аналогичной концентрацией гепарина, не содержащий усилитель. Значительное увеличение анти-фактор-Ха-активности, наблюдаемое для NaC8 (при дозе 35 мг) и NaC10 (как при дозе 10 мг, так и при дозе 35 мг), служит признаком увеличения всасывания гепарина относительно контрольного раствора гепарина.
(b) Таблетирование
(i) Таблетки с мгновенным высвобождением (IR)
Таблетки с мгновенным высвобождением (IR), содержащие USP-гепарин-натрий (197,25 МЕ/мг, поставляемый компанией Scientific Protein Labs., Waunkee, Wis.) и усилитель (каприлат натрия, NaC8; капрат натрия, NaC10, поставляемые компанией Napp Technologies, New Jersey), приготавливают в соответствии с рецептами составов, подробно описанными в Таблице 4, путем прямого прессования смеси с использованием однотаблеточного пресса Manesty (E). Смесь приготавливают следующим образом: гепарин, усилитель и эксципиенты для приготовления таблеток (исключая те случаи, где применяют коллоидный диоксид кремния и стеарат магния) отвешивают в контейнер. Коллоидный диоксид кремния, когда присутствует, просеивают через сито с размером отверстий 425 мкм в контейнер, после чего смесь перемешивают в течение четырех минут прежде чем вводить стеарат магния и перемешивать в течение дополнительной одной минуты.
Данные составов для таблеток с мгновенным высвобождением (IR), содержащих гепарин и усилитель (все количества в масс.%)
№
2
3
4
5
6
7
62,2
57,49
75,66
-
-
-
-
-
-
62,0
49,43
31,29
16,8
21,91
15,34
37,5
30,07
25,94
0,5
0,1
0,5
0,5
0,5
0,5
0,5
0,5
0,5
-
-
0,5
20,0
20,0
-
-
20,0
40,0
-
-
8,0
-
-
-
-
-
-
-
-
1,77
(а) Используемое вещество, способствующее распадаемости, представляет собой натрий-крахмалгликолят;
(b) PVP = поливинилпирролидон
Эффективность таблеток, приготовленных выше, испытывают с использованием количественного анализа гепарина, основанного на определении гепарина посредством красителя азура. Пробу, которая должна быть подвергнута анализу, добавляют к раствору красителя Azure A, и содержание гепарина вычисляют по поглощению раствора пробы в области длины волны 626 нм. Данные по таблеткам и значения эффективности действия для выбранных партий, подробно описанных в Таблице 4, приведены в Таблице 5.
Профили растворения для таблеток с мгновенным высвобождением (IR), в соответствии с этим Примером, в фосфатном буферном растворе при рН 7,4 определяют посредством гепариновых образцов для анализа, отбираемых в различные моменты времени.
Гепарин/каприлат натрия: Таблетки из партий 1 и 2 дают выход с быстрым высвобождением 100% лекарственного соединения через 15 минут. Таблетки из партии 4 дают выход с быстрым высвобождением 100% лекарственного соединения через 30 минут.
Гепарин/капрат натрия: Таблетки из серий 5 и 6 дают быстрое высвобождение 100% лекарственного соединения через 15 минут.
Данные по таблеткам и значения эффективности действия для таблеток с мгновенным высвобождением гепарина
№
2
3
4
5
6
7
NaC8
NaC8
NaC8
NaC10
NaC10
NaC10
414±14
650±4
377±2
408±21
490±6
584±12
82±9
71±12
58±10
79±7
124±10
69±22
-
552
-
-
-
485
175,79
166,4
168,04
394,47
323,33
143,0
105
119
110
105
108
102
(ii) Таблетки с замедленным высвобождением (SR)
Используя методику, аналогичную методике, используемой в разделе (i) выше, приготавливают таблетки с замедленным высвобождением (SR) в соответствии с рецептами составов, показанными в Таблице 6. Эффективность действия таблеток с контролируемым высвобождением определяют с использованием методики, аналогичной методике раздела (i), описанной выше. Подробные данные по таблеткам и эффективность действия для выбранных партий показаны в Таблице 7. Профили растворения для таблеток с замедленным высвобождением (SR) в соответствии с этим Примером определяют посредством гепариновых образцов для анализа при рН 7,4, отбираемых в различные моменты времени.
Гепарин/каприлат натрия: Данные по растворению для партий 8, 9 и 11 показаны в Таблице 8. Из этих данных видно, что таблетки с замедленным высвобождением (SR) гепарина/каприлата натрия, содержащие 15% гипромеллозы (Метоцел K-100LV) c 5% натрий-крахмалгликолята и без 5% натрий-крахмалгликолята (партии 8 и 9), дают замедленное высвобождение со 100% высвобождением, происходящим через 3-4 часа. Партия 11, содержащая 10% маннита, дает более быстрое высвобождение.
Гепарин/капрат натрия: Данные по растворению для партий 13 и 14 показаны в Таблице 8. Из этих данных видно, что таблетки с замедленным высвобождением (SR) гепарина/капрата натрия, содержащие 20% Метоцел K-100LV (партия 13), дают замедленное высвобождение лекарственного соединения в течение 6-часового периода времени. В том случае когда вместо Метоцел K-100LV используют Метоцел K-15М (партия 14), то высвобождение лекарственного соединения является незавершенным через 8 часов.
Данные рецептов составов для таблеток с замедленным высвобождением (SR), содержащих гепарин и усилитель (все количества в масс.%)
№
9
10
11
12
13
14
15
65,68
65,68
65,68
53,77
-
-
-
-
-
-
-
56,2
56,2
41,63
13,32
13,32
13,32
20,48
23,3
23,3
34,52
0,5
0,5
0,5
-
0,5
0,5
0,5
0,5
0,5
0,5
1,0
-
-
1,0
15
12
10,0
14,85
20,0
20,0*
20,0
5,0
8,0
-
-
-
-
-
-
-
10,0
-
-
-
-
-
-
-
9,9
-
-
-
-
-
-
-
-
-
2,35
Данные по таблеткам и значения эффективности действия для таблеток с замедленным высвобождением (SR), содержащих гепарин
№
9
10
11
12
13
14
15
NaC8
NaC8
NaC8
NaC8
NaC10
NaC10
NaC10
436±11
384±4
400±8
683±9
491±14
456±13
470±29
40±10
42±12
72±16
84±17
69±7
47±4
-
-
-
-
3318
-
-
2982
140,08
-
129,79
147,10
-
-
148,20
Данные по растворению для выбранных серий для таблеток с замедленным высвобождением
(NaC8)
(NaC8)
(NaC8)
(NaC10)
(NaC10)
15
30
60
120
240
360
480
22,9
37,3
57,8
92,2
109,5
-
-
21,2
30,8
54,5
90,8
105,8
-
-
45,3
72,3
101,9
109,4
96,4
-
-
18,8
45,0
44,8
65,2
83,1
90,3
102,7
5,7
11,6
11,2
20,0
33,9
66,0
82,8
(iii) Таблетки, покрытые энтеросолюбильной оболочкой
Таблетки из партий 7 и 15 покрывают оболочкой для кишечного всасывания посредством раствора для нанесения покрытия, который подробно описан в Таблице 9. Таблетки покрывают посредством 5%-ного (масса/масса) раствора для нанесения покрытия с использованием ванны для нанесения покрытия, вентилируемой сбоку (Freund Hi-Coater). Тест на распадаемость проводят в тестере распадаемости VanKel VK100E4635. Среда для теста на распадаемость представляет собой в течение одного часа первично смоделированную жидкость желудка с рН 1,2 и затем фосфатный буферный раствор с рН 7. Регистрируемое время распада являлось временем от введения в фосфатный буфер с рН 7для завершения дезинтеграции. Регистрируемое время распада для таблеток, покрытых оболочкой для кишечного всасывания, из партии 7 составляет 34 минут 24 секунд, тогда как для таблеток, покрытых оболочкой для кишечного всасывания, из партии 15 время распада составляет 93 минут 40 секунд.
Раствор для нанесения покрытия в виде оболочки для кишечного всасывания
Диэтилфталат
Изопропиловый спирт
Тальк
Вода
1,26
43,33
2,46
3,06
(с) Исследование на собаках
Таблетки из партий 3, 7 и 15 в вышеприведенных Таблицах 5 и 6 дают дозированно перорально группам из пяти собак в перекрестном исследовании с однократной дозой. Каждой группе дают дозированно (1) перорально вводимые таблетки, непокрытые оболочкой, с мгновенным высвобождением (IR), содержащие 90000 МЕ гепарина и 550 мг усилителя NaC10 (партия 7); (2) перорально вводимые таблетки, непокрытые оболочкой, с мгновенным высвобождением (IR), содержащие 90000 МЕ гепарина и 550 мг усилителя NaC8 (партия 3); (3) перорально вводимые таблетки, непокрытые оболочкой, с замедленным высвобождением (SR), содержащие 90000 МЕ гепарина и 550 мг усилителя NaC10 (партия 15); и (4) подкожно вводимый раствор гепарина (5000 МЕ, контрольный опыт). Пробы крови для проведения анализа на анти-фактор Ха отбирают из яремной вены в различные моменты времени. Клиническая оценка всех животных до лечения и после лечения показывает отсутствие неблагоприятных результатов у объектов исследования. ФИГ.8 показывает среднюю реакцию анти-фактора Ха для каждого лечения наряду с лечением при использовании эталона в виде раствора гепарина для подкожного введения. Данные на ФИГ.8 показывают увеличение плазменной анти-фактор-Ха-активности для всех составов в соответствии с изобретением. Этот результат указывает на успешную доставку биоактивного гепарина при использовании как усилителя NaC8, так и усилителя NaС10. При использовании составов с мгновенным высвобождением (IR) и с эквивалентной дозой гепарина более сильную реакцию анти-фактора Ха наблюдают с усилителем NaC10 вопреки более низкой вводимой дозе NaC10 относительно NaC8 (доза NaC10 составляет половину дозы NaC8). Реакция анти-фактора Ха может поддерживаться в течение более длительных временных профилей посредством использования таблеток с замедленным высвобождением (SR) относительно составов с мгновенным высвобождением (IR).
ПРИМЕР 3. Влияние усилителей на Системную Биодоступность Гепарина с низкой молекулярной массой (LMWH) после Введения в двенадцатиперстную кишку у Крыс
Крысам-самцам Wistar (250 г - 350 г) делают анастезию смесью кетамин-гидрохлорида (80 мг/кг) и ацепромазин-малеата (3 мг/кг), вводимой посредством внутримышечной инъекции. Животным также подают газ галотан, если требуется. Делают срединный разрез в брюшной стенке и отделяют двенадцатиперстную кишку.
Растворы для испытаний, содержащие натрий-парнапарин (гепарин с низкой молекулярной массой = LMWH) (Opocrin SBA, Modena, Italy) с усилителем и без усилителя, перерастворенный в забуференном фосфатом солевом растворе (рН 7,4), вводят (1 мл/кг) через канюлю, вставленную в кишечник приблизительно в 10-12 см от привратника. Во время этой процедуры кишечник поддерживают влажным посредством солевого раствора. После введения лекарственного средства сегмент кишечника осторожно помещают обратно в брюшную полость, и разрез закрывают с использованием хирургических зажимов. Парентеральный эталонный раствор (0,2 мл) вводят подкожно в кожную складку в загривке.
Пробы крови берут из хвостовой артерии через различные промежутки времени и определяют плазменную анти-фактор-Ха-активность. ФИГ.9 показывает среднюю реакцию анти-фактора Ха в течение периода времени 3 часа после введения в двенадцатиперстную кишку крыс забуференных фосфатом солевых растворов натрий-парнапарина (LMWH) (1000 МЕ), в присутствии 35 мг различных усилителей [каприлат натрия (С8), нонаноат натрия (С9), капрат натрия (С10), ундеканоат натрия (С11), лаурат натрия (С12)] и различных бинарных смесей усилителей 50:50, крысам (n=8) в экспериментальной модели с незамкнутой системой регулирования. Эталонный продукт заключает в себе подкожное введение 250 МЕ натрий-парнапарина. Модель с контрольным раствором заключает в себе введение раствора, содержащего 1000 МЕ натрий-парнапарина, без какого-либо усилителя в двенадцатиперстную кишку.
ФИГ.9 показывает, что системная доставка LMWH в отсутствии усилителя после введения крысам в двенадцатиперстную кишку является относительно плохой; однако, совместное введение натриевых солей жирных кислот со средней длиной цепи значительно улучшает системную доставку LMWH из кишечника крыс.
ПРИМЕР 4. Влияние усилителей на Системную Биодоступность Лейпролида после Введения в двенадцатиперстную кишку у Собак
Собак Beagles (10-15 кг) подвергают седации посредством медетомидина (80 мкг/кг), и вставляют эндоскоп через рот, пищевод и желудок в двенадцатиперстную кишку. Растворы для испытаний (10 мл), содержащие лейпролид-ацетат (Mallinckrodt Inc, St. Louis, Mo.) c усилителем или без усилителя, перерастворенный в деионизированной воде, вводят в двенадцатиперстную кишку через эндоскоп. После удаления эндоскопа, седацию снимают с использованием атипамезола (400 мкг/кг). Парентеральные эталонные растворы, содержащие 1 мг Лейпролида, перерастворенного в 0,5 мл стерильной воды, вводят соответственно внутривенно и подкожно.
Пробы крови отбирают из яремной вены через различные промежутки времени, и определяют уровни лейпролида в плазме. Получающиеся в результате средние уровни лейпролида в плазме показаны на ФИГ.10. Результаты показывают, что хотя системная доставка лейпролида при введении в двенадцатиперстную кишку без усилителя является незначительной, совместное введение с усилителем приводит к значительному улучшению системной доставки лейпролида, зависимому от дозы усилителя; для верхней дозы усилителя наблюдают средний % относительной биодоступности 8%.
ПРИМЕР 5. Влияние Усилителей на Системную Биодоступность Гепарина с низкой молекулярной массой (LMWH) после Перорального введения у Собак
(а) Изготовление Гранулята
200 г смеси, содержащей натрий-парнапарин (47,1%), капрат натрия (26,2%), маннит (16,7%) и Explotab™ (Roquette Freres, Lestrem, France) (10,0%), гранулируют в смесителе Kenwood Chef с использованием воды в качестве растворителя для гранулирования. Получающиеся в результате гранулы сушат на лотке в сушильном шкафу при 67-68°С и измельчают посредством сит с размером ячеек сита 1,25 мм, 0,8 мм и 0,5 мм соответственно в вибрационном грануляторе. Фактическую эффективность, получающуюся в результате гранулята, определяют как 101,1% от заявленной эффективности.
(b) Изготовление таблеток с мгновенным высвобождением, содержащих 30000 МЕ LMWH/183 мг капрата натрия
Гранулят, описанный выше, смешивают в мешке с 0,5% стеарата магния в течение 5 минут. Получающуюся в результате смесь таблетируют с использованием инструментальной оснастки с 13-миллиметровой круглой вогнутой поверхностью на таблетировочном прессе Riva Piccalo до целевого содержания в таблетке 30000 МЕ натрий-парнапарина и 183 мг капрата натрия. Таблетки имеют среднюю твердость таблеток 108 Н и среднюю массу таблеток 675 мг. Фактическое содержание LMWH в таблетках определяют как 95,6% от заявленного содержания.
На этих таблетках проводят тест на распадаемость. По одной таблетке помещают в каждую из шести пробирок испытательной корзины в тестере распадаемости табетированных препаратов. Прибор для испытания (тестер) распадаемости работает в режиме 29-30 циклов в минуту с использованием деионизированной воды при 37°С. Распад таблеток является завершенным через 550 сек.
(с) Изготовление Раствора, содержащего 90000 МЕ LMWH/0,55 г капрата натрия
90000 МЕ натрий-парнапарина и 0,55 г капрата натрия, отдельно отвешивают в стеклянные бутылочки, и получающуюся в результате порошковую смесь перерастворяют посредством 10 мл воды.
(d) Оценка изучения биоэквивалентности лекарственных препаратов на собаках
90000 МЕ натрий-парнапарина и 550 мг капрата натрия вводят как в лекарственной форме в виде раствора (эквивалентная 10-ти мл вышеупомянутой композиции в виде раствора), так и в виде быстрораспадающейся таблетированной лекарственной формы (эквивалентная 3-м таблеткам из вышеупомянутой таблетированной композиции), в нерандомизированном перекрестном исследовании с однократной дозой группы из шести собак-самок породы бигль (9,5-14,4 кг) с семидневным промыванием между лечениями. В качестве эталона используют раствор для подкожной инъекции, содержащий 5000 МЕ натрий-парнапарина.
Пробы крови берут из яремной вены через различные промежутки времени и определяют анти-фактор-Ха-активность. Данные корректируют для исходного уровня анти-фактор-Ха-активности. Получающиеся в результате средние плазменные уровни анти-фактора Ха суммированы на ФИГ.11. Как таблетированная лекарственная форма, так и лекарственная форма в виде раствора показывают хорошие реакции при сравнении с эталонным этапом с подкожной инъекцией. Средняя доставка, определяемая уровнями анти-фактора Ха в плазме, натрий-парнапарина из твердой лекарственной формы, является значительно лучше, чем средняя доставка из соответствующей лекарственной формы в виде раствора.
ПРИМЕР 6. Влияние Усилителей на Системную биодоступность LMWH после Перорального введения у Людей
(а) Изготовление Гранулята
Натрий-парнапарин (61,05%), капрат натрия (33,95%) и поливинилпирролидон (Kollidon 30, BASF AG, Ludwigshafen, Germany) (5,0%) смешивают в течение 5 минут в смесителе Gral 10 прежде чем вводить воду, которую затем постепенно добавляют, при помешивании, с использованием шлангового насоса, до тех пор, пока весь материал не станет очевидно гранулированным.
Получающиеся в результате гранулы сушат на лотке в сушильном шкафу, например, при 50°С в течение 24 часов. Осушенные гранулы измельчают посредством сита с номером 30 с использованием мельницы Fitzmill M5A.
(b) Изготовление таблеток с мгновенным высвобождением, содержащих 45000 МЕ LMWH/275 мг капрата натрия
Гранулят (78,3%), содержащий натрий-парнапарин/капрат натрия/поливинилпирролидон, смешивают в течение 5 минут с маннитом (16,6%), эксплотабом (5,0%) и стеаратом магния (1,0%) в 10-литровом V-образном конусном смесителе. Эффективность получающейся в результате смеси (480,41 мг/г) составляет 100,5% от заявленной эффективности. Смесь таблетируют с использованием инструментальной оснастки с 13-миллиметровой круглой нормально вогнутой поверхностью на стационарном прессе Piccоlа 10 в автоматическом режиме до целевого содержания 45000 МЕ LMWH и 275 мг капрата натрия. Получающиеся в результате таблетки с мгновенным высвобождением имеют среднюю массу таблеток 1027 мг, среднюю твердость таблеток 108 Н и эффективность 97% от заявленной эффективности. Таблетки показывают время распада вплоть до 850 секунд и 100% растворение в буферном растворе с рН 1,2 за 30 минут.
(с) Изготовление Раствора, содержащего 90000 МЕ LMWH/550 мг капрата натрия
Две таблетки с мгновенным высвобождением (быстро распадающиеся таблетки), каждая содержащая 45000 МЕ LMWH и 275 мг капрата натрия, перерастворяют в 30 мл воды.
(d) Оценка исследования биоэквивалентности лекарственных препаратов на людях
90000 МЕ LMWH и 550 мг капрата натрия перорально вводят 12-ти здоровым добровольно участвующим в исследованиях людям как лекарственной формы в виде раствора (эквивалентная 30-ти мл вышеупомянутой лекарственной формы в виде раствора), так и в виде твердой лекарственной формы (эквивалентная 2-м таблеткам из вышеупомянутой композиции), в открытом (все участники исследования информированы о названии и о дозировке лекарственного препарата) трехэтапном исследовании с тремя видами лечения с семидневным промыванием между введением каждой дозы; Лечение А (Таблетки с Мгновенным Высвобождением) и Лечение В (Раствор для перорального введения) проводят в режиме перекрестного исследования рандомизированным способом, тогда как Лечение С (подкожное введение 6400 МЕ Fluxum™ (Hoechst Marion Roussel), коммерчески доступного продукта LMWH для инъекций) назначают тем же субъектам в виде одного блока.
Пробы крови берут через различные промежутки времени и определяют анти-фактор-Ха-активность. Получающиеся в результате средние уровни анти-фактора Ха показаны на ФИГ.12. Лечения А и В показывают неожиданно низкие эффекты при сравнении с эталонным лечением с подкожным введением. Однако следует отметить, что средняя доставка LMWH, которая измерена посредством уровней анти-фактора Ха в плазме, является значительно более высокой из твердой лекарственной формы, чем средняя доставка LMWH из соответствующей лекарственной формы в виде раствора, для которой наблюдают средний % биодоступности только 0,9%.
ПРИМЕР 7. Влияние Усилителей на Системную доступность LMWH после Введения в тощую кишку у Людей
(а) Изготовление Раствора
Следующие комбинации LMWH/капрата натрия приготавливают с 15 мл деионизированной воды:
(i) 20000 МЕ LMWH, 0,55 г капрата натрия;
(ii) 20000 МЕ LMWH, 1,1 г капрата натрия;
(iii) 45000 МЕ LMWH, 0,55 г капрата натрия;
(iv) 45000 МЕ LMWH, 1,1 г капрата натрия;
(v) 45000 МЕ LMWH, 1,65 г капрата натрия.
(b) Оценка исследования биоэквивалентности лекарственных препаратов на людях
15 мл каждого из вышеуказанных растворов вводят в тощую кишку посредством назоеюнальной интубации, в открытом перекрестном исследовании с шестиэтапным лечением, вплоть до 11-ти здоровых добровольно участвующих в исследовании людей. Подкожное введение 3200 МЕ Fluxum™ включают в исследование в качестве эталонного этапа с подкожным введением. Пробы крови берут через различные промежутки времени и определяют анти-фактор-Ха-активность. Получающиеся в результате средние уровни анти-фактра Ха показаны на ФИГ.13.
Следует отметить, что средний процент относительной биодоступности для каждого вида лечения в данном исследовании является значительно более высоким, чем средний % биодоступности, наблюдаемый для лекарственной формы в виде раствора в Примере 6; для вариантов лечения в данном исследовании наблюдают средние % биодоступности, колеблющиеся в диапазоне от 5% до 9%, что позволяет предположить, что для минимизирования высвобождения лекарственного средства и усилителя в желудке и для максимального повышения высвобождения лекарственного средства и усилителя в тонкой кишке должна быть разработана предпочтительная лекарственная форма для перорального введения LMWH, содержащая капрат натрия.
ПРИМЕР 8. Изготовление таблетированной лекарственной формы с отсроченным высвобождением, содержащей LMWH и усилитель
(а) Изготовление гранулята, содержащего LMWH/капрат натрия
500 г состава из партии с соотношением натрий-парнапарин:капрат натрия (0,92:1) гранулируют в смесителе Gral 10 c использованием 50%-ного водного раствора Kollidon 30 в качестве растворителя для гранулирования. Получающийся в результате грунулят сушат в течение 60 минут в аэросушилке с псевдоожиженным слоем Niro при температуре конечного продукта 25°С. Осушенный гранулят измельчают через сито с номером 30 в мельнице Fitzmill M5A. Эффективность получающегося в результате осушенного гранулята определяют как 114,8% от заявленной эффективности.
(b) Изготовление Таблеток с мгновенным высвобождением, содержащих 22500 МЕ LMWH/275 мг Капрата натрия
Вышеупомянутый гранулят (77,5%) добавляют к манниту (16%), Polyplasdone XLTM (ISP, Wayne, N.J.) (5,0%) и к аэросилу (1,0%) (Degussa, Rheinfelden, Germany) в 10-литровый V-образный конусный смеситель и смешивают в течение 10 минут. К получающейся в результате смеси добавляют стеарат магния (0,5%), и смешивание продолжают в течение дополнительных 3 минут. Получающуюся в результате смесь таблетируют на таблетировочном прессе Piccоlа с использованием инструментальной оснастки с 13-миллиметровой круглой нормально вогнутой поверхностью до средней массы таблетки 772 мг и средней твердости таблетки 140 Н.
Фактическую эффективность получающихся в результате таблеток определяют как 24017 МЕ LMWH на таблетку.
(с) Изготовление таблеток с отсроченным высвобождением, содержащих 22500 МЕ LMWH/275 мг Капрата натрия
Вышеупомянутые таблетки покрывают оболочкой посредством раствора для нанесения покрытия, содержащего Eudragit L 12,5 (50%), изопропиловый спирт (44,45%), дибутилсебацинат (3%), тальк (1,3%), воду (1,25%), в машине для нанесения покрытий Hi-coater до конечного % привеса 5,66%.
Получающиеся в результате таблетки, покрытые энтеросолюбильной оболочкой, остаются интактными после 1-часового проведения теста на распадаемость в растворе с рН 1,2; полный распад наблюдают в среде с рН 6,2 по истечении 32-33 минут.
ПРИМЕР 9
Изготовление Инкапсулированной лекарственной формы с мгновенным высвобождением, содержащей LMWH и усилитель
(а) Изготовление Капсулы с мгновенным высвобождением, содержащей 22500 МЕ LMWH/275 мг Капрата натрия
Гранулят из предыдущего примера, часть а, вручную насыпают в твердые желатиновые капсулы размером 00 до целевой массы насыпки, эквивалентной содержанию гранулята в таблетках предыдущего примера.
ПРИМЕР 10
Изготовление Таблетированной лекарственной формы с отсроченным высвобождением, содержащей LMWH без усилителя
(а) Изготовление Гранулята из LMWH
500 г состава с натрий-парнапарином из партии: Avicel™ pH 101 (0,92:1) (FMC, Little Island, Co. Cork, Ireland) гранулируют в смесителе Gral 10 с использованием 50% водного раствора Kollidon 30 в качестве растворителя для гранулирования. Получающийся в результате гранулят сушат в течение 60 минут в аэросушилке с псевдоожиженным слоем Niro при температуре выходящего воздуха 38°С. Высушенный гранулят измельчают посредством сита с номером 30 в мельнице Fitzmill M5A. Эффективность получающегося в результате высушенного гранулята определяют как 106,5% от заявленной эффективности.
(b) Изготовление Таблеток с мгновенным высвобождением, содержащих 22500 МЕ LMWH
Вышеупомянутый гранулят (77,5%) добавляют к манниту (21%) и аэросилу (1%) в 25-литровый V-образный конусный смеситель и смешивают в течение 10 минут. К получающейся в результате смеси добавляют стеарат магния (0,5%), и смешивание продолжают в течение дополнительной 1 минуты.
Получающуюся в результате смесь таблетируют на таблетировочном прессе Piccоlа с использованием инструментальной оснастки с 13-миллиметровой круглой нормально вогнутой поверхностью до средней массы таблетки 671 мг и средней твердости таблетки 144 Н.
Фактическую эффективность получающихся в результате таблеток определяют как 21651 МЕ LMWH на таблетку.
(с) Изготовление таблеток с отсроченным высвобождением, содержащих 22500 МЕ LMWH
Вышеупомянутые таблетки покрывают оболочкой посредством раствора для нанесения покрытия, содержащего Eudragit L 12,5 (50%), изопропиловый спирт (44,45%), дибутилсебацинат (3%), тальк (1,3%), воду (1,25%), в машине для нанесения покрытий Hi-coater до конечного % привеса 4,26%.
Получающиеся в результате таблетки, покрытые оболочкой для кишечного всасывания, остаются интактными после 1-часового проведения теста на распадаемость в растворе с рН 1,2; полный распад наблюдают в среде с рН 6,2 по истечении 22 минут.
ПРИМЕР 11. Влияние лекарственной формы с контролируемым высвобождением, содержащей усилитель, на системную доступность LMWH после перорального введения собакам
(а) Оценка исследования на собаках
45000 МЕ LMWH вводят 8 собакам породы бигль (10,5-13,6 кг), в открытом нерандомизированном исследовании с перекрестным блок-дизайном, в виде (а) инкапсулированной лекарственной формы с мгновенным высвобождением, содержащей 550 мг капрата натрия (эквивалентная 2 капсулам, изготовленным в соответствии с Примером 9), (b) таблетированной лекарственной формы с отсроченным высвобождением, содержащей 550 мг капрата натрия (эквивалентная двум таблеткам, изготовленным в соответствии с Примером 8) и (с) таблетированной лекарственной формы с отсроченным высвобождением, не содержащей никакой усилитель (эквивалентная 2 таблеткам, изготовленным в соответствии с Примером 10). Подкожное введение 3200 МЕ Fluxum™ SC включают в исследовании в качестве эталонного этапа с подкожным введением.
Пробы крови берут из яремной вены через различные промежутки времени, и определяют анти-фактор-Ха-активность. Средние уровни анти-фактора Ха показаны на ФИГ.14.
Следует отметить, что в отсутствие капрата натрия системная доставка LMWH является минимальной из твердой лекарственной формы с отсроченным высвобождением без усилителя. В противоположность этому хорошую реакцию анти-фактора Ха наблюдают после введения твердой лекарственной формы LMWH с отсроченным высвобождением, содержащей капрат натрия. Средняя реакция анти-фактора Ха для доставки LMWH из лекарственной формы с отсроченным высвобождением, содержащей капрат натрия, является значительно более высокой, чем средняя реакция анти-фактора Ха для доставки LMWH из лекарственной формы с мгновенным высвобождением, содержащей аналогичный уровень лекарственного средства и усилителя.
ПРИМЕР 12. Влияние Места Введения на Системную доступность LMWH у собак после совместного введения с усилителем
Четырем собакам породы бигль (10-15 кг) хирургическим путем устанавливают катетеры в тощую кишку и толстую (ободочную) кишку соответственно. Растворы для испытаний (10 мл), содержащие LMWH с капратом натрия, перерастворенный в деионизированной воде, вводят собакам либо перорально либо через катетеры, вставленные в кишечник. Подкожное введение 3200 МЕ Fluxum™ SC включают в исследовании в качестве эталонного этапа с подкожным введением.
Пробы крови берут из плечевой вены через различные промежутки времени, и определяют анти-фактор-Ха-активность. Получающиеся в результате средние уровни анти-фактора Ха показаны на ФИГ.15. Результаты показывают, что кишечное всасывание LMWH в присутствии усилителя является значительно более высоким, чем всасывание из желудка.
ПРИМЕР 13. Таблетки, содержащие Лейпролид
Следуя типу подхода, аналогичному типу подхода, используемому в Примерах 1 и 2, могут быть приготовлены лейпролид-содержащие таблетки с мгновенным высвобождением в соответствии с рецептами составов, подробно описанными в Таблице 10.
Составы с мгновенным высвобождением, содержащие Лейпролид
(все количества в масс.%)
кремния
магния
ПРИМЕР 14. Введение Алендроната в тощую кишку
Исследование проводят как открытое, рандомизированное, 6-этапное исследование, с 7 видами лечения, с введением в тощую кишку (IJ) или с пероральным (PO) введением, и с, по меньшей мере, 48-часовым периодом промывания между каждым введением дозы. Девятнадцать (19) здоровых субъектов мужского пола принимают к участию в этом исследовании, и 15 субъектов, которым вводят дозы, по меньшей мере, однократно, включают в фармакокинетический анализ. Фармакокинетический анализ является основанным на выделении с мочой алендроната. Таблица 11 показывает виды лечения, кумулятивное количество, и % введенной дозы, выделившийся в моче (рассчитан исходя из кумулятивного количества) в этом исследовании.
Средние фармакокинетические (PK) параметры (Среднее ±стандартное квадратичное отклонение(SD) - CV%)
(CV%)
181,3
181,3
(CV%)
83,9
83,9
(CV%)
74,9
74,9
(CV%)
48,6
48,6
(CV%)
34,7
34,7
Как показано с помощью этих данных, всасывание алендроната в желудочно-кишечном тракте значительно усиливается при введении в виде болюсного раствора в тощую кишку с капратом натрия, в сравнении с коммерчески доступной в настоящий момент эталонной таблеткой Fosamax®, непокрытой оболочкой, c мгновенным высвобождением.
ПРИМЕР 15. Введение в тощую кишку и пероральное введение Алендроната
В открытом, частично рандомизированном, 3-этапном исследовании, с тремя видами лечения, с по меньшей мере, 48-часовым промыванием между каждым введением дозы, двенадцати (12) субъектам мужского пола дают дозы лекарственных препаратов, по меньшей мере, однократно в ходе исследования и включают в фармакокинетический анализ. В этом исследовании назначают следующие виды лечения, предоставленные в Таблице 12.
Средние фармакокинетические (PK) параметры (Среднее ±стандартное квадратичное отклонение(SD) - CV%)
17,5 мг Алендроната +
0,5 г С10 (Вливание в тощую кишку (IJ) в течение 25 мин)
n 12
17,5 мг Алендроната +
1,1 г С10 (Вливание в тощую кишку (IJ) в течение 25 мин)
n 12
35 мг Fosamax®
(Пероральное введение (PO))
n 12
Как показано с помощью этих данных, системное всасывание алендроната значительно усиливается после совместного введения, в виде водного вливания в тощую кишку (в течение 25 минут), с капратом натрия. Это обнаружение указывает на то, лекарственная форма, содержащая алендронат и капрат натрия (С10), покрытая оболочкой для кишечного всасывания, с мгновенным высвобождением, должна быть преимущественной в сравнении с коммерчески доступной в настоящий момент лекарственной формой с улучшенным всасыванием алендроната в полости рта.
ПРИМЕР 16. Пероральное Введение Алендроната
Исследование проводят для того, чтобы сравнить относительную биодоступность алендроната, введенного в виде твердых лекарственных форм для перорального применения, содержащих усилитель всасывания, с дозой лекарственного средства (алендроната) для перорального применения, введенной в виде коммерчески доступной эталонной лекарственной формы Fosamax®. Это исследование проводят как открытое, частично рандомизированное, 5-этапное исследование, с 5 видами лечения, с однократной дозой, с, по меньшей мере, 48-часовым промыванием между каждым введением дозы. Шестнадцать (16) здоровых добровольцев (13 субъектов мужского пола и 3 субъекта женского пола в возрасте от 20 до 34 лет и имеющие вес от 64,1 до 81,5 кг) принимают к участию и они проходят полностью все 5 видов лечения, которые изложены в Таблице 13 ниже.
(PO)
введенные в виде 1 таблетки с 250 мл водопроводной воды - на голодный желудок
(PO)
введенные в виде 2 таблеток с 250 мл водопроводной воды - на голодный желудок
таблетки, содержащие алендронат/С10, покрытые оболочкой из HPMC P-55/Опадри
(PO)
введенные в виде 2 таблеток с 250 мл водопроводной воды - после приема пищи (с высокой жирностью)
(8,75 мг Алендроната и 0,25 г С10 на таблетку)
таблетки, содержащие алендронат/С10, покрытые оболочкой из HPMC P-55/Опадри
(PO)
введенные в виде 2 таблеток с 250 мл водопроводной воды - на голодный желудок
(8,75 мг Алендроната и 0,125 г С10 на таблетку)
таблетки, содержащие алендронат/С10, покрытые оболочкой из HPMC P-55/Опадри
(PO)
введенные в виде 1 таблетки с 250 мл водопроводной воды - на голодный желудок
(17,5 мг Алендроната и 0,25 г С10 на таблетку)
таблетки, содержащие алендронат/С10, покрытые оболочкой из HPMC P-55/Опадри
Образцы мочи людей собирают на протяжении 36-часового этапа сбора образцов и анализируют методом высокоэффективной жидкостной хроматографии с флуоресцентным детектированием (диапазон анализа: 2 - 2000 нг/мл). Средний % введенной дозы, выделенный с мочой (рассчитан исходя из кумулятивного количества), для видов лечений с испытанием лекарственных препаратов, является следующим (см. Таблицу 14).
выделенный с мочой (CV%)
Анализ с проверкой по парному критерию Стьюдента (t-test) проводят, сравнивая % Дозы, выделенной для опытных образцов прототипов в зависимости от % Дозы, выделенной для Fosamax® (см. Таблицу 15).
Уровень значимости менее чем 0,1%
Уровень значимости 5%
Уровень значимости 1%
Уровень значимости 1%
Статистически значимое увеличение процента (%) введенной дозы алендроната, выделенного с мочой, наблюдают для опытных образцов прототипов, введенных на голодный желудок (дозированные по 1 или по 2 таблетки), в сравнении с процентом (%) введенной дозы алендроната, выделенного с мочой, наблюдаемым для эталонного продукта, Fosamax®.
Статистически значимое уменьшение процента (%) введенной дозы алендроната, выделенного с мочой, наблюдают для опытного образца прототипа, введенного после приема пищи (Лечение С - уровень значимости на 5%), в сравнении с процентом (%) введенной дозы алендроната, выделенного с мочой, наблюдаемым для Fosamax®. Кумулятивное количество введенной дозы, извлеченное из мочи, для введений в испытаниях лекарственных препаратов, является в 4,6 - 6,4 раз больше, чем кумулятивное количество введенной дозы, извлеченное из мочи, наблюдаемое для Fosamax®.
Увеличение количества С10, совместно введенного с алендронатом, от 0,25 г до 0,5 г не изменяет процент (%) введенной дозы, извлеченный в моче (1,6 ± 1,7% и 1,5 ± 0,6%, соответственно). Введение 17,5 мг алендроната с 0,25 г С10 в виде 2 таблеток (Лечение D) приводит к более высокому % введенной дозы, извлеченному в моче алендроната (1,6 ± 1,7%), чем при введении в виде 1 таблетки в соответствии с Лечением Е (1,2 ± 0,9%). В том случае, когда 17,5 мг алендроната и 0,5 г С10 вводят в виде 2 таблеток в состоянии после приема пищи (Лечение С), то в моче определяют 0,2 ± 0,2% алендроната.
Следует отметить, что в опубликованной литературе утверждают, что биодоступность Fosamax® является незначительной в том случае, когда алендронат вводят через 2 часа или вплоть до 2 часов после стандартного завтрака.
ПРИМЕР 17. Исследование Биодоступности лекарственных форм Золедроновой кислоты для перорального применения
Выполняют перекрестное исследование, с однократной дозой, для того чтобы сравнить биодоступность золедроновой кислоты в лекарственной форме для перорального применения настоящего изобретения с продаваемой в настоящий момент формой золедроновой кислоты, то есть обеспеченной в виде жидкого концентрата для внутривенного вливания под названием Zometa® от компании Novartis. Рассматриваемая лекарственная форма для перорального применения представляет собой таблетку, покрытую оболочкой для кишечного всасывания, содержащую капрат натрия и либо 10 мг, либо 20 мг золедроновой кислоты, сформованную в соответствии со способами Примеров 6, 8 и 13. Жидкий концентрат вводят в виде раствора для внутривенного вливания, содержащего 1 мг золедроновой кислоты.
Все имеющиеся в распоряжении данные, полученные от 12 субъектов, которые полностью прошли исследование, используют в фармакокинетическом анализе. (Нет данных по эталонному продукту для Субъекта 1). Все фармакокинетические вычисления проводят с использованием программного обеспечения Института системного статистического анализа (SAS) (Компьютерная версия 6.12). Уровень золедроновой кислоты для каждого сбора мочи для каждого субъекта в каждом этапе в аналитической лаборатории сообщают в показателях как концентрации (нг/мл), так и общего выделенного количества (нг). Для любого образца присвоенное ему значение концентрации меньшее, чем аналитический предел количественного анализа, относят к нулевому количеству, выделенному для использования в фармакокинетическом анализе.
До проведения фармакокинетического анализа присвоенное количество выделенной золедроновой кислоты в нанограммах (г × 10-9) переводят в миллиграммы (г × 10-3) путем умножения каждого значения на 10-6. Это делают для упрощения статистических выходных данных и для выражения общего выделенного количества в единицах (мг), аналогичных единицам введенных доз. Выделенные количества в течение времени 0-12, 12-24, 24-36 и 36-48 с часовыми интервалами для каждого субъекта в каждом этапе суммируют с последовательным приращением с получением кумулятивных выделенных количеств в течение времени 0-12, 0-24, 0-36 и 0-48 с часовыми интервалами.
Статистический анализ выполняют с использованием подпрограммы на основе Общих Линейных Моделей (GLM) статистической программы SAS (Компьютерная версия 6.12). Кумулятивные количества выделенной золедроновой кислоты и преобразованные логарифмически кумулятивные количества выделенной золедроновой кислоты оценивают посредством анализа дисперсии. Проверку правильности гипотезы для эффектов видов лечения в этом анализе проводят при α=0,05.
Попарные сравнения, которые представляют интерес, проводят между 10-мг таблеткой и раствором для инъекций, 20-мг таблеткой и раствором для инъекций, и между 10-миллиграммовой и 20-миллиграммовой таблетками. Статистическую модель использовали в анализах для определения эффективности лечения субъектов. F-коэффициенты для проверки эквивалентности эффектов видов лечения высчитывают с использованием вектора среднеквадратичной ошибки для эффекта в качестве числителя и вектора среднеквадратичной ошибки из анализа дисперсии в качестве знаменателя.
В дополнение к проверкам правильности гипотезы вычисляют доверительные интервалы (90%) для попарных сравнений видов лечения посредством подхода с проверкой по критерию Стьюдента (t-test) (2, 1-сторонний) при α = 0,10 в случае полной проверки, при α = 0,05 в случае проверки с каждой стороны:
Нижний предел Интервала = (ХТ - ХR) - Se·tα/2
Верхний предел Интервала = (ХТ - ХR) + Se·tα/2,
где ХТ представляет собой среднее значение, полученное по методу наименьших квадратов, для лечения с испытанием лекарственного средства, и ХR представляет собой среднее значение, полученное по методу наименьших квадратов, для эталонного лечения. В сравнении двух таблеток настоящего изобретения ХТ представляет собой среднее значение, полученное по методу наименьших квадратов, для 20-миллиграммовой таблетки настоящего изобретения, и ХR представляет собой среднее значение, полученное по методу наименьших квадратов, для 10-миллиграммовой таблетки настоящего изобретения.
Se представляет собой стандартную (среднеквадратическую) ошибку оценки разности между средними значениями, полученную по алгоритму оценки программного обеспечения SAS.
tα/2 представляет собой критическое значение из t-распределения со степенью свободы критического значения в виде вектора ошибок в статистическом анализе при уровне α = 0,10.
Для логарифмически преобразованных данных вычисляют интервал из результатов для преобразованных значений и затем возводят в степень для перевода в непреобразованный масштаб:
Предел интервала = e(ln-преобразованный предел интервала)
Вычисляют доверительный интервал для «истинных» средних разностей видов лечения, выраженный в виде процента эталонного среднего значения (непреобразованные результаты), и для соотношения истинных средних геометрических значений (логарифмически преобразованные результаты). Аналогично возведенный в степень критерий и эталонные средние значения, полученные методом наименьших квадратов, из логарифмически преобразованных результатов обеспечивают оценку геометрических средних значений для этих видов лечения.
Статистический анализ на результатах осуществляют для того, чтобы сравнить 10-миллиграммовые таблетки, 20-миллиграммовые таблетки, и 1-миллиграммовую инъекцию Zometa®, в том случае, когда каждую лекарственную форму вводят после ночного воздержания от пищи. Таблетки 16-18 ниже суммируют результаты попарных сравнений выделения с мочой золедроновой кислоты в этих видах лечения. Фигура 17 показывает среднее кумулятивное выделение для трех видов лечения. Никакой статистически значимой разности не обнаруживают в отношении среднего кумулятивного мочевыделения. 10-миллиграммовая и 20-миллиграммовая таблетки имеют среднее 48-часовое выделение с мочой, приблизительно равное 0,5 мг. Лечение посредством 1-мг инъекции Zometa® имеет аналогичное среднее количество, выделяемое в течение этого периода времени. Для всех трех лекарственных форм большая часть выделения золедроновой кислоты (85% - 87%) происходит в пределах первых 12-ти часов после введения.
Обобщение статистических сравнений выделения с мочой золедроновой кислоты после введения однократной дозы в виде 10-миллиграммовой таблетки и 1-миллиграммовой инъекции 12-ти женщинам в постменопаузе на голодный желудок представлено ниже в Таблице 16.
(часы)
(мг)1
2. Соотношение, вычисленное как среднее значение по методу наименьших квадратов для 10-миллиграммовой таблетки, поделенное на среднее значение по методу наименьших квадратов для инъекции Zometa®. Ни одно из сравнений не выявляют как статистически значимое посредством модели дисперсионного анализа ANOVA (α = 0,05).
3. Оценка внутрисубъектного коэффициента вариации, CV% = 100*SQRT(eMSE-1), где MSE представляет собой вектор среднеквадратичных ошибок из данных дисперсионного анализа ANOVA.
4. Доверительный интервал для соотношения.
Сравнение 20-миллиграммовых таблеток и 1-миллиграммовой инъекции Zometa®.
Обобщение статистических сравнений выделения с мочой золедроновой кислоты после введения однократной дозы в виде 20-миллиграммовой таблетки и 1-миллиграммовой инъекции 12-ти женщинам в постменопаузе на голодный желудок представлено ниже в Таблице 17.
(часы)
(мг)1
2. Соотношение, вычисленное как среднее значение по методу наименьших квадратов для 20-миллиграммовой таблетки, поделенное на среднее значение по методу наименьших квадратов для инъекции Zometa®. Ни одно из сравнений не выявляют как статистически значимое посредством модели дисперсионного анализа ANOVA (α = 0,05).
3. Оценка внутрисубъектного коэффициента вариации, CV% = 100·SQRT(eMSE-1), где MSE представляет собой вектор среднеквадратичных ошибок из данных дисперсионного анализа ANOVA.
4. Доверительный интервал для соотношения.
Сравнение 20-миллиграммовой и 10-миллиграммовой таблеток
Обобщение статистических сравнений выделения с мочой золедроновой кислоты после введения однократных доз в виде 10-миллиграммовой и 20-миллиграммовой таблеток 12-ти женщинам в постменопаузе на голодный желудок представлено ниже в Таблице 18.
(часы)
(мг)1
2. Соотношение, вычисленное как среднее значение по методу наименьших квадратов для 20-миллиграммовой таблетки, поделенное на среднее значение по методу наименьших квадратов для 10-миллиграммовой таблетки. Ни одно из сранений не выявляют как статистически значимое посредством модели дисперсионного анализа ANOVA (α = 0,05).
3. Оценка внутрисубъектного коэффициента вариации, CV% = 100·SQRT(eMSE-1), где MSE представляет собой вектор среднеквадратичных ошибок из данных дисперсионного анализа ANOVA.
4. Доверительный интервал для соотношения.
ПРИМЕР 18. Исследование Биодоступности Лекарственных форм Алендроната для перорального применения
Это исследование представляет собой открытое, 4-этапное, рандомизированное перекрестное исследование, с 4 видами лечения, с, по меньшей мере, 7-дневным промыванием между каждым введением дозы. Целью этого исследования является изучение фармакокинетики и биодоступности лекарственных форм натрий-алендроната настоящего изобретения после введения однократных доз женщинам в постменопаузе в состояниях после приема пищи и на голодный желудок для определения соответствующей дозы для применения в лечении остеопороза, и для определения той степени, в какой такие лекарственные формы преодолевают (не требуют) утренние ритуалы введения доз, сопутствующие приему таблеток Fosamax®, продаваемых компанией Merck & Co., Inc.
Всего принимают к участию в исследовании 17 субъектов и им вводят дозы, по меньшей мере, в одном случае, и 16 субъектов полностью проходят исследование и получают, по меньшей мере, три вида лечения. Виды лечения, назначаемые в этом исследовании, представляют собой следующее:
Лечение А 35 мг Таблетка Fosamax®, принимаемая в соответствии с рекламным вкладышем в упаковке (после ночного воздержания от пищи, субъект сохраняет вертикальное положение в течение 4 часов после введения дозы).
Лечение В 6 мг Таблетка, принимаемая в соответствии с режимом дозирования Fosamax® (после ночного воздержания от пищи, субъект сохраняет вертикальное положение в течение 4 часов после введения дозы).
Лечение С 6-мг Таблетка, принимаемая в 10.30 после полудня после приема пищи в 6 после полудня (воздержание от пищи от 6.30 после полудня до завтрака; субъект сохраняет горизонтальное положение (лежит) в течение, по меньшей мере, 2 часов после введения дозы).
Лечение D 6 мг таблетка, принимаемая до полудня с завтраком высокой жирности в соответствии со стандартами Управления по контролю за продуктами и лекарствами США (FDA) (субъект сохраняет вертикальное положение в течение 4 часов после введения дозы).
Алендронат измеряют в образцах мочи посредством проверенной на точность измерений высокоэффективной жидкостной хроматографии с флуоресцентным способом детектирования. Предел количественной оценки анализа мочи по алендронату составляет 2 нг/мл (диапазон анализа 2 - 500 нг/мл). Образцы мочи собирают до введения дозы и в течение 0-12, 12-24, 24-36 и 36-48 часов после введения дозы.
Основываясь на анализе дефинитивных данных, введение 6-мг таблетки, принимаемой после полудня после 4-часового воздержания от пищи (Лечение С) или принимаемой до полудня, после 10-часового воздержания от пищи (Лечение В), приводит к 15,4- и 11,8-кратному увеличению соответственно биодоступности алендроната по сравнению с эталонной таблеткой, 35-мг таблеткой Fosamax® (Лечение А). Введение 6-мг таблетки, принимаемой до полудня, после приема пищи (Лечение D), приводит к 2,8-кратному увеличению биодоступности алендроната по сравнению с эталонной таблеткой, 35-мг таблеткой Fosamax® (Лечение А). Самая высокая относительная биодоступность алендроната по сравнению с введением 6-мг таблетки, принимаемой до полудня, после 10-часового воздержания от пищи (Лечение В), составляет 127±104% для Лечения С (6-мг таблетка, принимаемая после полудня, после 4-часового воздержания от пищи), 20±35% для Лечения D (6-мг таблетка, принимаемая до полудня, после приема пищи), и затем 10±5% для Лечения А (таблетка Fosamax®, принимаемая до полудня, после 10-часового воздержания от пищи).
Основываясь на предыдущих данных, таблетка, содержащая усилитель, содержащая 5,65 мг алендроната, является эквивалентной 35-мг таблетке Fosamax®, которую для целей этого исследования округляют до 6 мг. Задачей этого исследования является сравнить таблетку алендроната, содержащую усилитель с таблеткой Fosamax® в перекрестном исследовании с четырьмя факторами, с однократной дозой, биодоступности у вплоть до 16 женщин в постменопаузе. Между каждым этапом лечения существует, по меньшей мере, 7-дневное промывание.
Используемый способ представляет собой высокоэффективную жидкостную хроматографию с флуоресцентным детектированием в соответствии со способом испытаний. Способ основан на совместном осаждении алендроната с фосфатами кальция. Первичную аминогруппу молекулы тогда превращают в производную группу посредством 2,3-нафталиндикарбоксиальдегида и (NDA)-N-ацетил-D-пеницилламина (NAP) с образованием флуоресцентного производного. Затем осуществляют градиентную высокоэффективную жидкостную хроматографию на производной молекуле, и детектирование происходит при Возбуждении волн на длине волны: 420 нм, при испускании: 490 нм. Предел количественной оценки в анализе алендроната в моче составляет 2 нг/мл (диапазон анализа 2 - 500 нг/мл).
Фармакокинетические параметры вычисляют с использованием программного обеспечения WinNonlin™, Версия 4.0.1 (Pharsight Corporation, USA). Следующие параметры получают из данных по концентрации алендроната в моче с использованием некомпартментных способов:
Кумулятивное количество, выделяемое в каждый момент времени (Аet) и общее выделяемое количество (АеТ).
Скорость выделения в каждый момент времени (Аеt/t), общая скорость выделения (АеT/T).
Наблюдаемую максимальную скорость выделения (max rate) и последнюю измеряемую скорость (rate last).
Относительную биодоступность (F%) для видов лечения с испытанием лекарственного средства по сравнению с эталонным лечением вычисляют на базе отдельно взятого субъекта (скорректированное по дозе количество алендроната, выделенное в испытании каждым отдельно взятым субъектом, поделенное на скорректированное по дозе количество алендроната, выделенное в эталонном этапе тем же самым субъектом),
= скорректированное по дозе кумулятивное выделенное количество (испытательный этап) / скорректированное по дозе кумулятивное выделенное количество (эталонный этап) × 100%.
Относительную биодоступность вычисляют с использованием Лечения А (35-мг таблетка Fosamax®, принимаемая до полудня, после 10-часового воздержания от пищи) или Лечения В (6-мг таблетка, принимаемая до полудня, после 10-часового воздержания от пищи) в качестве эталонного лечения. Поскольку Субъект 08 не получил введения эталона (Лечение А), то среднее кумулятивное выделенное количество, полученное для группы субъектов в Лечении А, используют в качестве эталонного значения для вычисления относительной биодоступности для этого отдельно взятого субъекта. Среднее значение этих вычисленных величин представлено в качестве средней относительной биодоступности.
До формального анализа фармакокинетические данные подвергают обзорному анализу. Это включает в себя проверку на пропущенные данные и на выбросы. Субъект под номером 12 не проходил полного исследования, так как она добровольно сняла согласие и была заменена Субъектом под номером 17; поэтому Субъект под номером 12 не включен в фармакокинетический анализ. Субъект 8 не принимал лекарства во время Лечебного Этапа Два (Лечение А), но был включен в фармакокинетический анализ, и среднее кумулятивное выделенное количество, полученное для группы субъектов в Лечении А, используют в качестве эталонного значения для вычисления относительной биодоступности для этого отдельно взятого субъекта.
Всего 17 субъектов женского пола принимают к участию в этом исследовании и им вводят дозы, по меньшей мере, один раз в ходе исследования. Пятнадцать субъектов полностью проходят исследование и получают все 4 вида лечения. Один субъект (Субъект 12) снимает согласие на исследование после получения дозы в Лечебном Этапе Один (Лечение А), и один субъект (Субъект 8) не принимает дозу во время Лечебного Этапа Два (Лечение А) вследствие семейного непредвиденного случая, но он получает разрешение от Спонсора вернуться и закончить другие этапы исследования. У одного добровольца, Субъекта 6, отсутствует всасывание сколь-нибудь поддающегосяся оценке количества алендроната в том случае, когда субъект (она) принимает таблетку Fosamax®. Ее рассматривают в качестве нереспондента (лицо, не участвующее в исследовании по какой-либо причине), так как отсутствие у нее способности всасывать алендронат из таблетки Fosamax® не могло позволить ей проходить лечение в качестве больного. Следует отметить, что в том случае, когда она получает улучшенную таблетку, дозированную аналогичным образом, у нее происходит всасывание обыкновенного количества алендроната. Следовательно, улучшенная таблетка настоящего изобретения может подходить для лечения таких нереспондентов.
Количественные показатели распределения вычисляют исходя из полного набора данных и набора данных с опускаемыми данными для Субъекта S06 (нереспондент в отношении таблетки Fosamax®). Следующие результаты основаны на дефинитивном наборе данных, то есть на наборе данных, где данные для Субъекта S06 (нереспондент в отношении эталонного лечения) опускают из описательной статистики.
Ранговый порядок биодоступности алендроната из таблетированных составов алендроната, содержащих усилитель в сравнении с эталонной таблеткой, Fosamax® (Лечение А), является следующим: 1536±1554% для Лечения С (доза, принимаемая после полудня, после 4-часового воздержания от пищи), 1180±536% для Лечения В (доза, принимаемая до полудня, после 10-часового воздержания от пищи), и затем 283±559% для Лечения D (доза, принимаемая до полудня, после приема пищи).
Самая высокая относительная биодоступность алендроната в сравнении с таблетированными составами алендроната, содержащих усилитель, дозированными до полудня, после 10-часового воздержания от пищи (Лечение В), составляет 127±104% для Лечения С (доза, принимаемая после полудня, после 4-часового воздержания от пищи), 20±35% для Лечения D (доза, принимаемая до полудня, после приема пищи), и затем 10±5% для Лечения А (Fosamax®, дозированная до полудня, после 10-часового воздержания от пищи).
Самое высокое общее кумулятивное количество алендроната, выделенное с мочой после введения, составляет 220±163 мкг для Лечения С (доза, введенная после полудня, после 4-часового воздержания от пищи), 203±87 мкг для Лечения В (доза, введенная до полудня, после 10-часового воздержания от пищи), и затем 33±54 мкг для Лечения D (доза, введенная до полудня, после приема пищи), в сравнении с 113±55 мкг для эталонной таблетки, Fosamax®.
Самая высокая скорость полного выделения алендроната, определяемая после введения, составляет 5,2±3,9 мкг/час для Лечения С (доза, введенная после полудня, после 4-часового воздержания от пищи), 4,8±2,1 мкг/час для Лечения В (доза, введенная до полудня, после 10-часового воздержания от пищи), и затем 0,8±1,3 мкг/час для Лечения D (доза, введенная до полудня, после приема пищи), в сравнении 2,7±1,3 мкг/час для эталонной таблетки, Fosamax®.
Общий процент алендроната, извлекаемого после каждого ранжированного введения, является самым высоким после введения лекарственного средства в соответствии с Лечением С (доза, введенная после полудня, после 4-часового воздержания от пищи) и составляет 3,7±2,7%, 3,4±1,5% для Лечения В (доза, введенная до полудня, после 10-часового воздержания от пищи), и затем 0,6±0,9% для Лечения D (доза, введенная до полудня, после приема пищи), в сравнении с 0,3±0,2% для эталонной таблетки, Fosamax®.
Эти результаты демонстрируют не только превосходную биодоступность таблетированных составов алендроната настоящего изобретения в сравнении с существующими лекарственными формами алендроната, но также более высокую гибкость в условиях/состояниях, при которых введение алендроната может происходить без потери биодоступности. Режимы дозирования для традиционно используемых составов бисфосфата требуют: (1) утреннего введения; (2) введения в состоянии на голодный желудок/воздержания от пищи; и (3) избежания всех видов пищи, напитков и других лекарств в течение времени вплоть до 2 часов после введения. В противоположность тому таблетированные составы алендроната, содержащие усилитель настоящего изобретения, делают возможным введение не только в соответствии с режимом дозирования традиционно используемых составов бисфосфата, но также в другие периоды времени дня, кроме утра, после периодов времени меньших, чем ночное воздержание от пищи, и безотносительно какой-либо последующей отсрочки приема пищи и/или напитков. Таблетированные составы алендроната, содержащие усилитель настоящего изобретения, также обеспечивают уровни биодоступности, эквивалентные существенно более высоким дозам алендроната в существующих лекарственных формах. Такое улучшение биодоступности, проявленное лекарственными формами настоящего изобретения, позволяет применять более низкие дозы бисфосфонатов для достижения эквивалентной биодоступности или применять эквивалентные дозы в достижении более высокой биодоступности.
Композиции и лекарственные формы настоящего изобретения также включают использование других усилителей, кроме жирных кислот со средней длиной цепи и производных жирных кислот со средней длиной цепи, описанных выше. Для увеличения биодоступности и для допущения дозирования в другие периоды времени, кроме утра, при пробуждении или в пределах двух часов после приема пищи, напитков (других, кроме воды), кальцийсодержащих добавок и/или лекарств, могут быть использованы усилители всасывания, такие как другие жирные кислоты, кроме жирных кислот со средней длиной цепи; ионогенные, неионогенные и липофильные поверхностно-активные вещества; спирты жирного ряда; соли желчных кислот и желчные кислоты; мицеллы; вещества, способствующие хелатированию, и тому подобное.
Неионогенные поверхностно-активные вещества, рассматриваемые в пределах объема изобретения, включают алкилглюкозиды; алкилмальтозиды; алкилтиоглюкозиды; лаурил-макроголглицериды; полиоксиалкиленовые простые эфиры; полиоксиалкилен-алкиловые простые эфиры; полиоксиалкиленовые алкилфенолы; полиоксиалкиленовые сложные эфиры жирной кислоты и алкилфенола; полиэтиленгликолевые сложные эфиры жирной кислоты и глицерина; сложные эфиры жирной кислоты и полиглицерина; полиоксиалкиленовые сложные эфиры сорбита и жирной кислоты; сложные эфиры сорбита и жирной кислоты; гидрофильные продукты транс-этерификации полиола посредством, по меньшей мере, одного члена группы, состоящей из глицеридов, растительных масел, гидрогенизированных растительных масел, жирных кислот, и стеринов; полиоксиэтиленовые стерины, производные, и их аналоги; полиоксиэтилированные витамины и их производные; блоксополимеры полиоксиэтилен-полиоксипропилен, полиэтиленгликоль-10 лаурат (ПЭГ-10 лаурат), полиэтиленгликоль-12 лаурат, полиэтиленгликоль-20 лаурат, полиэтиленгликоль-32 лаурат, полиэтиленгликоль-32 дилаурат, полиэтиленгликоль-12 олеат, полиэтиленгликоль-15 олеат, полиэтиленгликоль-20 олеат, полиэтиленгликоль-20 диолеат, полиэтиленгликоль-32 олеат, полиэтиленгликоль-200 олеат, полиэтиленгликоль-400 олеат, полиэтиленгликоль-15 стеарат, полиэтиленгликоль-32 дистеарат, полиэтиленгликоль-40 стеарат, полиэтиленгликоль-100 стеарат, полиэтиленгликоль-20 дилаурат, полиэтиленгликоль-25 глицерилтриолеат, полиэтиленгликоль-32 диолеат, полиэтиленгликоль-20 глицериллаурат, полиэтиленгликоль-30 глицериллаурат, полиэтиленгликоль-20 глицерилстеарат, полиэтиленгликоль-20 глицерилолеат, полиэтиленгликоль-30 глицерилолеат, полиэтиленгликоль-30 глицериллаурат, полиэтиленгликоль-40 глицериллаурат, косточковое пальмовое масло, модифицированное ПЭГ-40, гидрогенизированное касторовое масло, модифицированное ПЭГ-50, касторовое масло, модифицированное ПЭГ-40, касторовое масло, модифицированное ПЭГ-35, касторовое масло, модифицированное ПЭГ-60, гидрогенизированное касторовое масло, модифицированное ПЭГ-40, гидрогенизированное касторовое масло, модифицированное ПЭГ-60, кукурузное масло, модифицированное ПЭГ-60, полиэтиленгликоль-6 капрат/каприлатглицериды, полиэтиленгликоль-8 капрат/каприлатглицериды, полиглицерил-10 лаурат, ПЭГ-30 холестерин, ПЭГ-25 фитостерин, соевый стерин, модифицированный ПЭГ-30, ПЭГ-20 триолеат, ПЭГ-40 сорбитолеат, ПЭГ-80 сорбитлаурат, полисорбаты, включающие полисорбат 20, полисорбат 40, полисорбат 60, полисорбат 65, полисорбат 80, полисорбат 85, ПОЭ-9 (полиоксиэтиленовый) эфир лаурилового спирта, ПОЭ-23 эфир лаурилового спирта, ПОЭ-10 эфир олеилового спирта, ПОЭ-20 эфир олеилового спирта, ПОЭ-20 эфир стеарилового спирта, токоферил-ПЭГ-100 сукцинат, ПЭГ-24 холестерин, полиглицерил-10 олеат, моностеарат сахарозы, монолаурат сахарозы, монопальмитат сахарозы, нонилфенольные серии с ПЭГ 10-100, октилфенольные серии с ПЭГ 15-100, и полоксамеры.
Ионогенные поверхностно-активные вещества, рассматриваемые в пределах объема изобретения, включают алкиламмониевые соли; соли фусидовой кислоты; производные аминокислот на основе жирных кислот, олигопептиды, и полипептиды; глицеридные производные аминокислот, олигопептиды, и полипептиды; лецитины и гидрогенизированные лецитины; лизолецитины и гидрогенизированные лизолецитины; фосфолипиды и их производные; лизофосфолипиды и их производные; соли сложных эфиров жирных кислот и карнитина; соли алкилсульфатов; соли жирных кислот; докузат натрия; ациллактилаты; моно- и ди-ацетилированные сложные эфиры винной кислоты с моно- и ди-глицеридами; сукцинилированные моно- и ди-глицериды; сложные эфиры лимонной кислоты и моно- и ди- глицеридов; лаурилсульфат натрия; и четвертичные аммониевые соединения.
Липофильные поверхностно-активные вещества, рассматриваемые в пределах объема изобретения, включают жирные спирты; сложные эфиры глицерина и жирной кислоты; ацетилированные сложные эфиры глицерина и жирной кислоты; сложные эфиры низшего спирта и жирных кислот; сложные эфиры пропиленгликоля и жирной кислоты; сложные эфиры сорбита и жирной кислоты; полиэтиленгликолевые сложные эфиры сорбита и жирной кислоты; стерины и производные стеринов; полиоксиэтилированные стерины и производные полиоксиэтилированных стеринов; алкиловые эфиры полиэтиленгликоля; сложные эфиры сахара; простые эфиры сахара; молочнокислые производные моно- и ди-глицеридов; гидрофобные продукты транс-этерификации полиола посредством, по меньшей мере, одного члена группы, состоящей из глицеридов, растительных масел, гидрогенизированных растительных масел, жирных кислот и стеринов; маслорастворимые витамины/производные витаминов; и их смеси. В пределах этой группы предпочтительные липофильные поверхностно-активные вещества включают сложные эфиры глицерина и жирной кислоты, сложные эфиры пропиленгликоля и жирной кислоты, и их смеси, или представляют собой гидрофобные продукты транс-этерификации полиола посредством, по меньшей мере, одного члена группы, состоящей из растительных масел, гидрогенизированных растительных масел, и триглицеридов.
Соли желчных кислот и кислоты, рассматриваемые в пределах объема настоящего изобретения, включают соли дигидроксижелчных кислот, такие как дезоксихолат натрия, соли тригидроксижелчных кислот, такие как холат натрия, холевую кислоту, дезоксихолевую кислоту, литохолевую кислоту, хенодезоксихолевую кислоту (также именуемая как «хенодиол» или «хеновая кислота»), урсодезоксихолевую кислоту, таурохолевую кислоту, тауродезоксихолевую кислоту, тауролитохолевую кислоту, таурохенодезоксихолевую кислоту, тауроурсодезоксихолевую кислоту, гликохолевую кислоту, гликодезоксихолевую кислоту, гликолитохолевую кислоту, гликохенодезоксихолевую кислоту, и гликоурсодезоксихолевую кислоту.
Солюбилизаторы, рассматриваемые в пределах объема настоящего изобретения, включают спирты и полиолы, такие как этанол, изопропанол, бутанол, бензиловый спирт, этиленгликоль, пропиленгликоль, бутандиолы и их изомеры, глицерин, пентаэритрит, сорбит, маннит, транскутол, диметилизосорбид, полиэтиленгликоль, полипропиленгликоль, поливиниловый спирт, гидроксипропилметилцеллюлозу и другие производные целлюлозы, моно-, ди- и три-глицериды жирных кислот со средней длиной цепи и их производные; глицериды циклодекстрины и производные циклодекстрина; простые эфиры полиэтиленгликолей, имеющих среднемассовую молекулярную массу от приблизительно 200 до приблизительно 6000, такие как эфир полиэтиленгликоля или метокси-полиэтиленгликоля и тетрагидрофурфурилого спирта; амиды и другие азотсодержащие соединения, такие как 2-пирролидон, 2-пиперидон, ε-капролактам, N-алкилпирролидон, N-гидроксиалкилпирролидон, N-алкилпиперидон, N-алкилкапролактам, диметилацетамид и поливинилпирролидон; сложные эфиры, такие как этилпропионат, трибутилцитрат, ацетилтриэтилцитрат, ацетилтрибутилцитрат, триэтилцитрат, этилолеат, этилкаприлат, этилбутират, триацетин, пропиленгликольмоноацетат, пропиленгликольдиацетат, эпсилон-капролактон и его изомеры, дельта-валеролактон и его изомеры, бета-бутиролактон и его изомеры; и другие солюбилизаторы, известные в данной области, такие как диметилацетамид, диметилизосорбид, N-метилпирролидоны, монооктаноин, моноэтиловый эфир диэтиленгликоля, и воду.
Дополнительные другие подходящие поверхностно-активные вещества будут очевидны для специалистов в данной области и/или описаны в пертинентных текстах и в литературе.
Настоящее изобретение не следует ограничивать в объеме конкретными вариантами осуществления, описанными в этом документе. Фактически различные модификации этого изобретения в дополнение к модификациям, описанным в этом документе, станут очевидны специалистам в данной области из предшествующего описания и сопроводительных фигур. Подразумевается, что такие модификации попадают в объем прилагаемой Формулы изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| КОМПОЗИЦИИ ПЕПТИДОВ И СПОСОБЫ ИХ ПОЛУЧЕНИЯ | 2009 |
|
RU2517135C2 |
| ТРАНСБУККАЛЬНАЯ СИСТЕМА ДОСТАВКИ | 2006 |
|
RU2406480C2 |
| КОМПОЗИЦИИ ДЛЯ ВВЕДЕНИЯ ЛЕКАРСТВЕННЫХ СРЕДСТВ | 2009 |
|
RU2554814C2 |
| ЛЕКАРСТВЕННЫЕ ФОРМЫ НА ОСНОВЕ БИСФОСФОНАТОВ | 2005 |
|
RU2359678C2 |
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ С ДЛИТЕЛЬНЫМ ПОСТЕПЕННЫМ ВЫСВОБОЖДЕНИЕМ, СОДЕРЖАЩАЯ ВОДНУЮ СУСПЕНЗИЮ БИСФОСФОНАТА | 2007 |
|
RU2453316C2 |
| ПОКРЫТАЯ ПЛЕНКОЙ ТАБЛЕТКА УЛУЧШЕННОЙ БЕЗОПАСНОСТИ ДЛЯ ВЕРХНИХ ОТДЕЛОВ ЖЕЛУДОЧНО-КИШЕЧНОГО ТРАКТА | 1998 |
|
RU2193880C2 |
| СТАБИЛЬНЫЕ СОСТАВЫ ШИПУЧЕГО БИСФОСФОНАТА СО СВОЙСТВАМИ БЫСТРОЙ СОЛЮБИЛИЗАЦИИ | 2011 |
|
RU2608724C2 |
| ЛЕКАРСТВЕННЫЕ ФОРМЫ РИЗЕДРОНАТА | 2005 |
|
RU2381791C2 |
| ПЕРОРАЛЬНЫЕ КОМПОЗИЦИИ, СОДЕРЖАЩИЕ СЛОЖНЫЙ ЭФИР 17-ГИДРОКСИПРОГЕСТЕРОНА, И СООТВЕТСТВУЮЩИЕ СПОСОБЫ | 2012 |
|
RU2640912C2 |
| ТВЕРДАЯ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ ДЛЯ ПЕРОРАЛЬНОГО ПРИМЕНЕНИЯ, СОДЕРЖАЩАЯ ПРОИЗВОДНОЕ БИСФОСФОНОВОЙ КИСЛОТЫ | 2001 |
|
RU2288705C2 |
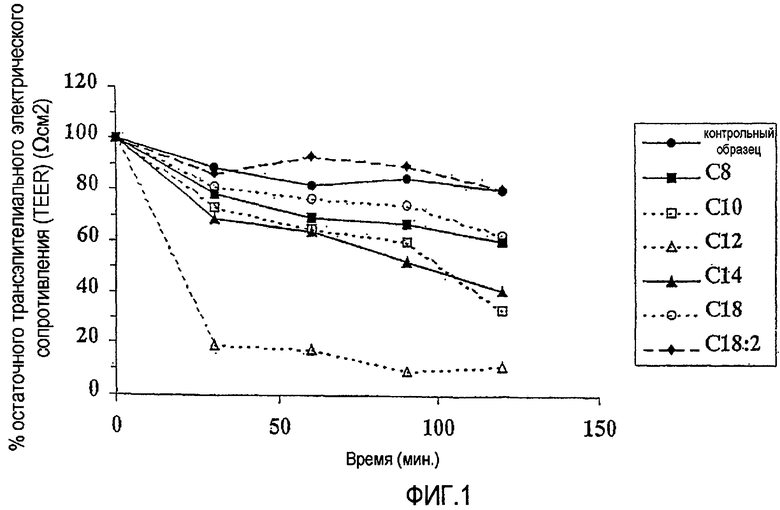
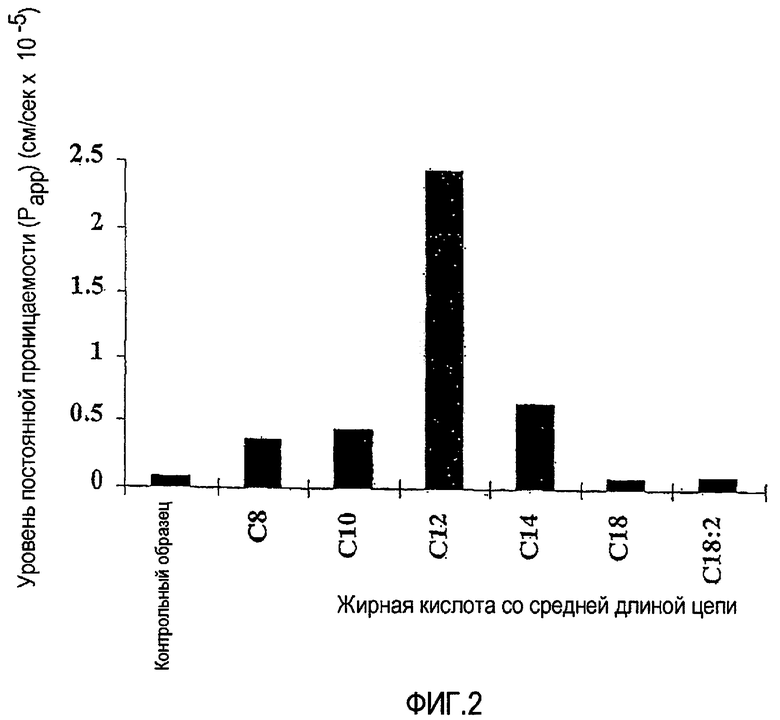
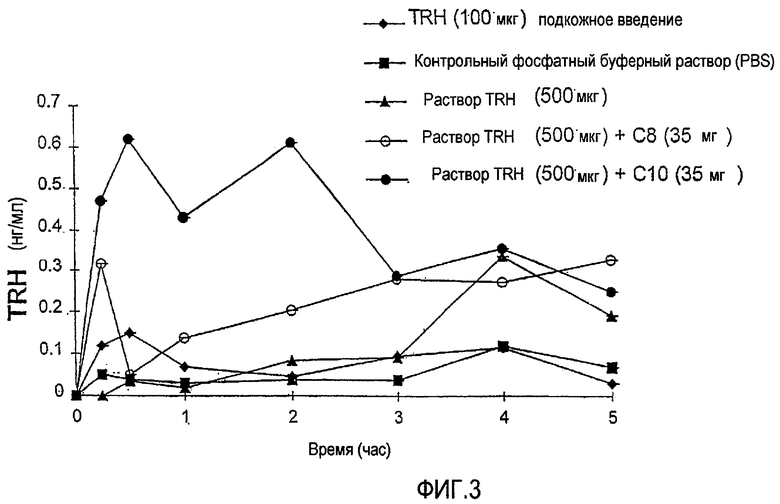
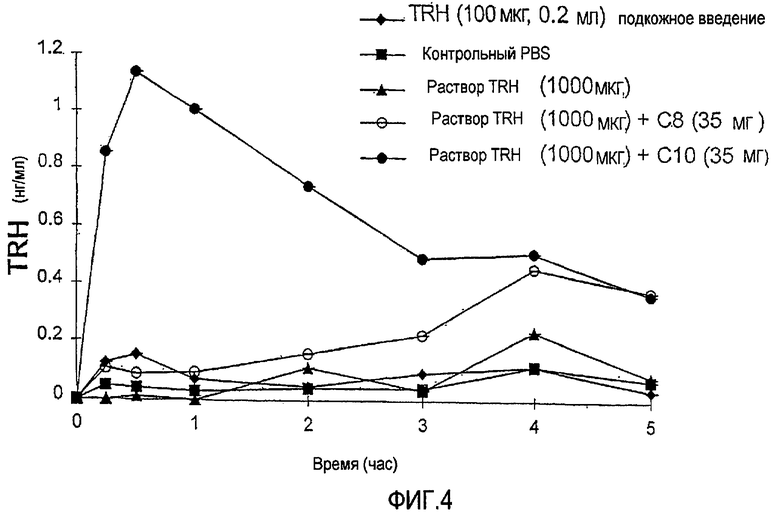
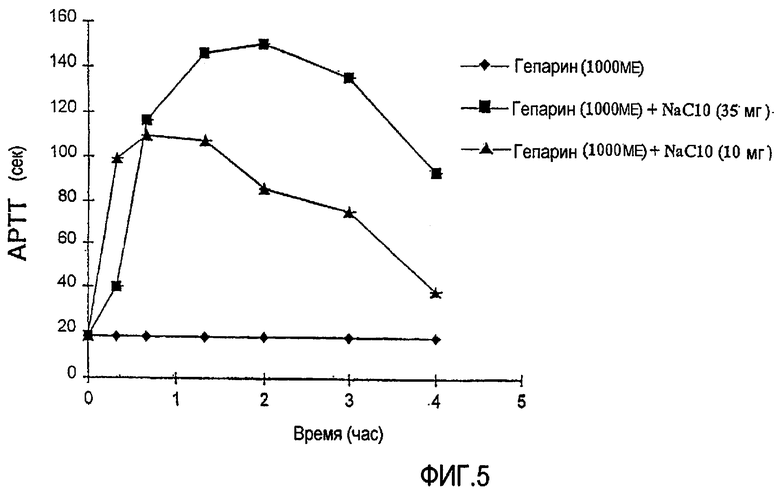
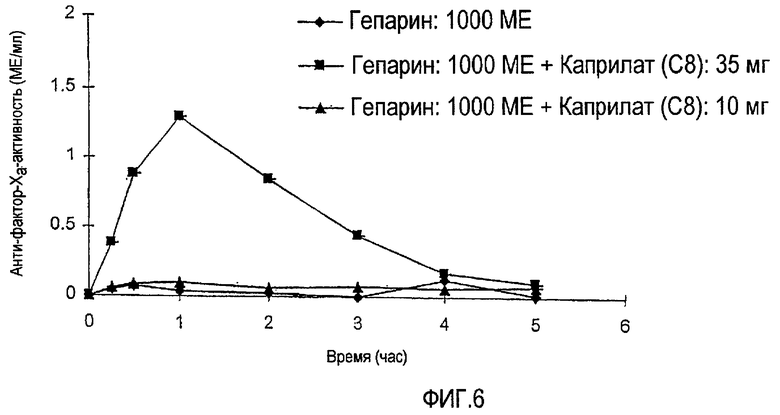
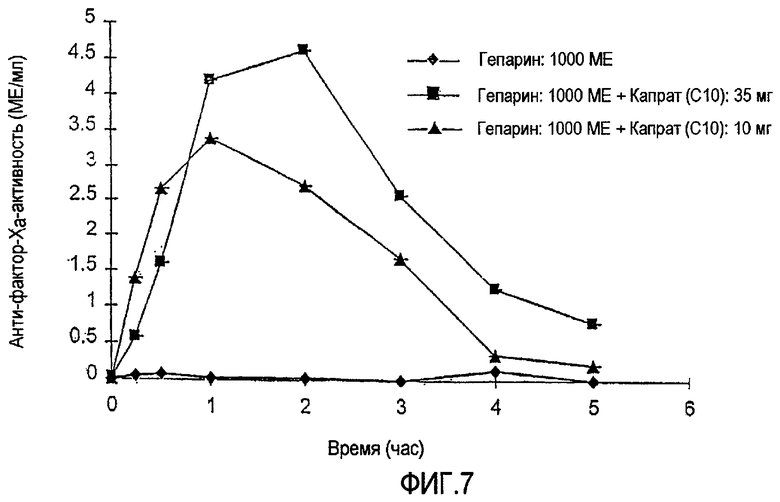
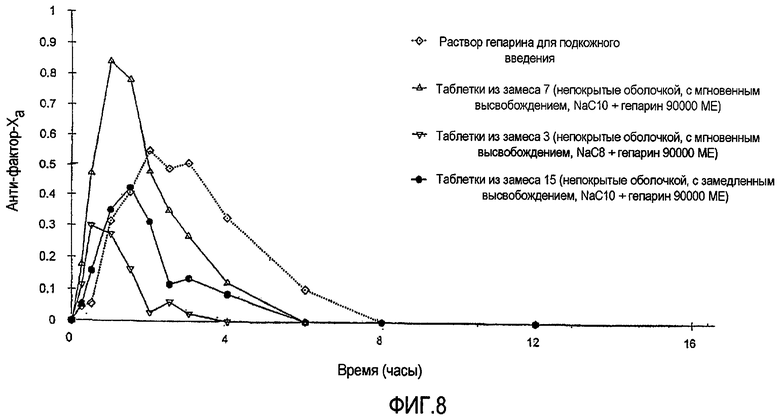

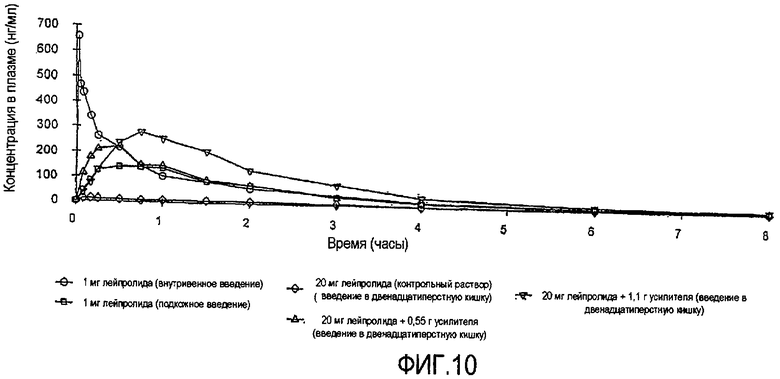
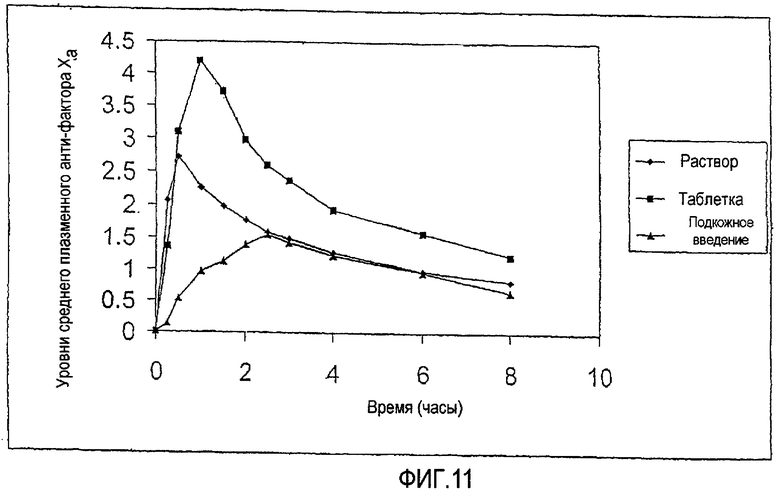
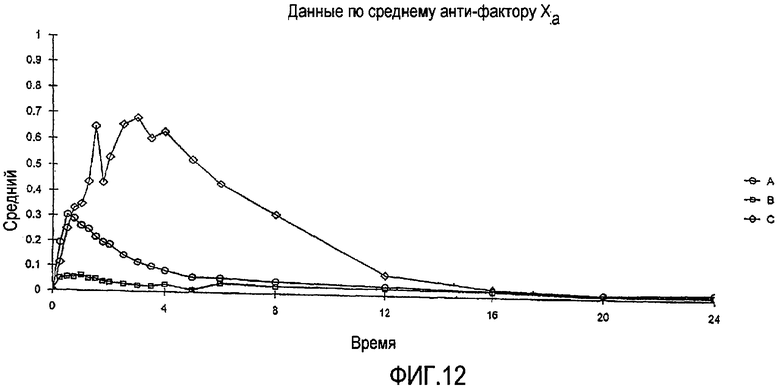
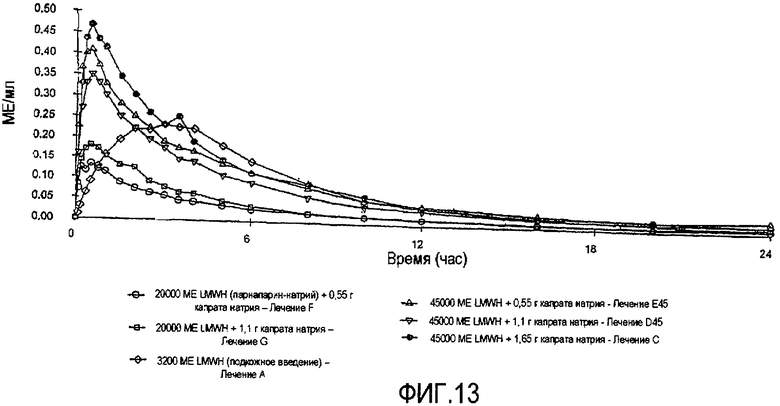
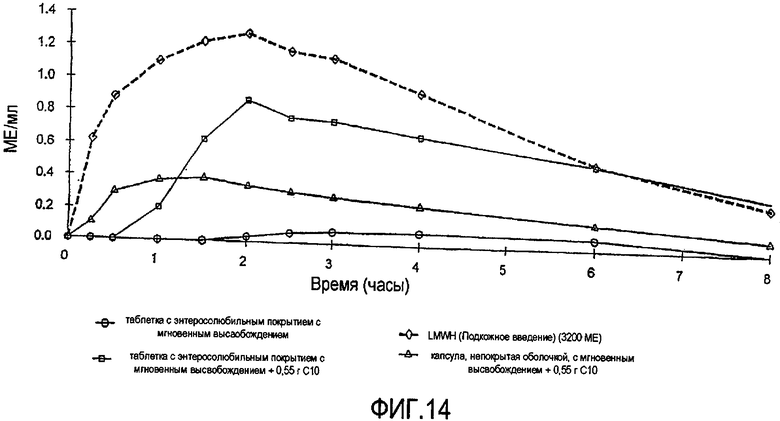
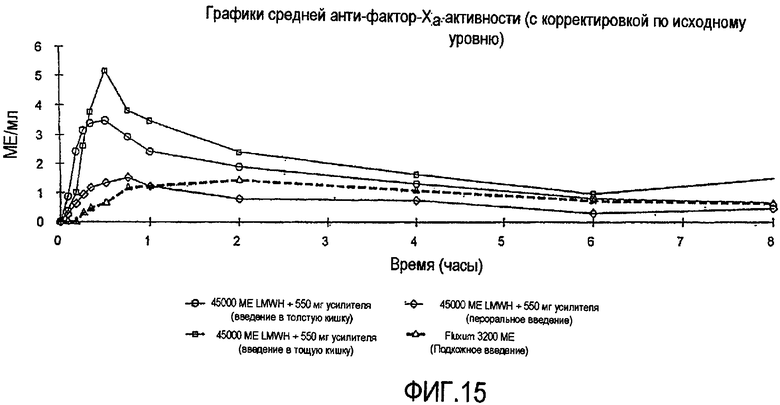

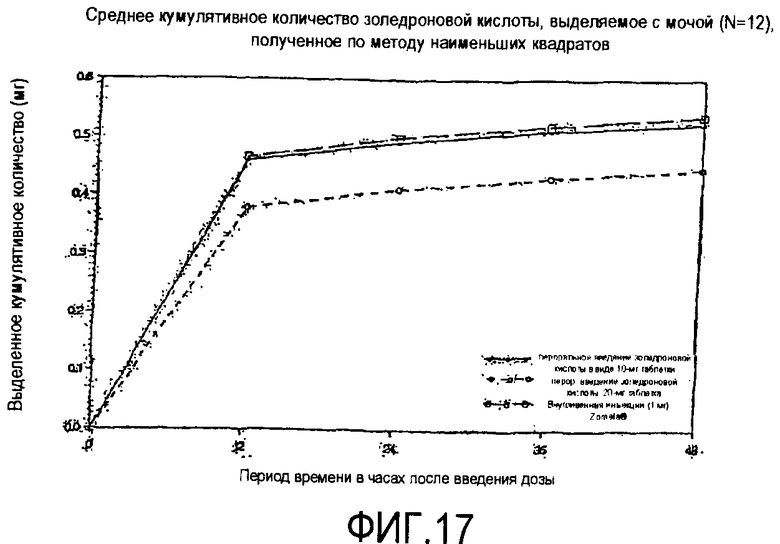
Заявленное изобретение относится к химико-фармацевтической промышленности и касается фармацевтической композиции и лекарственных форм для перорального применения, содержащих бисфосфонат в комбинации с усилителем абсорбции бисфосфоната для улучшения доставки через кишечник бисфосфоната в основное кровообращение. Предпочтительно усилитель представляет собой жирную кислоту со средней длиной цепи или производное жирной кислоты со средней длиной цепи, имеющее длину углеродной цепи от 6 до 20 атомов углерода, и усилитель является единственным усилителем абсорбции, присутствующим в композиции, а твердая пероральная лекарственная форма представляет собой лекарственную форму с контролируемым высвобождением, такую как лекарственная форма с отсроченным высвобождением. 13 н. и 6 з.п. ф-лы, 18 табл., 17 ил.
1. Применение фармацевтической композиции для лечения или предупреждения медицинского состояния, где фармацевтическую композицию вводят перорально пациенту в терапевтически эффективном количестве, где стадия введения происходит в пределах 30 мин до потребления больным пищи, лекарственных препаратов, или напитков, отличных от воды, где фармацевтическая композиция содержит бисфосфонат и усилитель, который усиливает абсорбцию бисфосфоната, где усилителем является жирная кислота со средней длиной цепи или производное жирной кислоты со средней длиной цепи, имеющее длину углеродной цепи от 6 до 20 атомов углерода, которое является твердым при комнатной температуре и является единственным усилителем абсорбции, присутствующим в композиции.
2. Применение фармацевтической композиции по п.1, где медицинское состояние представляет собой остеопороз, метастатический рак костей, ревматоидный артрит и перелом кости.
3. Применение фармацевтической композиции по п.1, где стадия введения происходит в другое время дня, чем утро.
4. Применение фармацевтической композиции по п.3, где стадия введения происходит вечером.
5. Применение фармацевтической композиции по п.1, где стадия введения происходит в пределах 30 мин до того, как пациент ложится.
6. Фармацевтическая композиция, содержащая бисфосфонат и усилитель, который усиливает абсорбцию бисфосфоната, где усилителем является жирная кислота со средней длиной цепи или производное жирной кислоты со средней длиной цепи, имеющее длину углеродной цепи от 6 до 20 атомов углерода, и является твердым при комнатной температуре, и является единственным усилителем абсорбции, присутствующим в композиции, где усилитель улучшает доставку через кишечник бисфосфоната в основное кровообращение, и где композиция доставляет перорально терапевтически эффективное количество бисфосфоната при введении пациенту в пределах 30 мин до потребления больным пищи, лекарственных препаратов или напитков, отличных от воды.
7. Пероральная лекарственная форма, содержащая композицию по п.6.
8. Фармацевтическая композиция, содержащая бисфосфонат и усилитель, который усиливает абсорбцию бисфосфоната, где усилителем является жирная кислота со средней длиной цепи или производное жирной кислоты со средней длиной цепи, имеющее длину углеродной цепи от 6 до 20 атомов углерода, и является твердым при комнатной температуре, и является единственным усилителем абсорбции, присутствующим в композиции, где усилитель улучшает доставку через кишечник бисфосфоната в основное кровообращение, и где композиция доставляет перорально терапевтически эффективное количество бисфосфоната при введении пациенту в другое время дня, чем утро.
9. Пероральная лекарственная форма, содержащая композицию по п.8.
10. Фармацевтическая композиция, содержащая бисфосфонат и усилитель, который усиливает абсорбцию бисфосфоната, где усилителем является жирная кислота со средней длиной цепи или производное жирной кислоты со средней длиной цепи, имеющее длину углеродной цепи от 6 до 20 атомов углерода, и является твердым при комнатной температуре, и является единственным усилителем абсорбции, присутствующим в композиции, где усилитель улучшает доставку через кишечник бисфосфоната в основное кровообращение, и где композиция доставляет перорально терапевтически эффективное количество бисфосфоната, когда ее вводят пациенту в пределах 30 мин до того, как пациент ложится.
11. Пероральная лекарственная форма, содержащая композицию по п.10.
12. Фармацевтическая композиция, содержащая бисфосфонат и усилитель, который усиливает абсорбцию бисфосфоната, где усилителем является жирная кислота со средней длиной цепи или производное жирной кислоты со средней длиной цепи, имеющее длину углеродной цепи от 6 до 20 атомов углерода, и является твердым при комнатной температуре, и является единственным усилителем абсорбции, присутствующим в композиции, где усилитель улучшает доставку через кишечник бисфосфоната в основное кровообращение, и где композиция доставляет перорально терапевтически эффективное количество бисфосфоната, когда ее вводят пациенту в пределах 6 ч после приема пациентом пищи, лекарственных препаратов или напитков, отличных от воды.
13. Пероральная лекарственная форма, содержащая композицию по п.12.
14. Пероральная фармацевтическая композиция, которая является эффективной в доставке терапевтически эффективных количеств лекарственного средства и усилителя в кишечник, где указанная композиция содержит алендронат или золедроновую кислоту и усилитель, который усиливает абсорбцию алендроната и золендроновой кислоты, где усилитель является единственным усилителем абсорбции, присутствующим в композиции, и где композиция доставляет перорально терапевтически эффективное количество алендроната или золедроновой кислоты в том случае, когда ее вводят пациенту непосредственно перед сном.
15. Пероральная фармацевтическая композиция, которая является эффективной в доставке терапевтически эффективных количеств лекарственного средства и усилителя в кишечник, где указанная композиция содержит алендронат и усилитель, который усиливает абсорбцию алендроната, где усилитель является единственным усилителем абсорбции, присутствующим в композиции, и где композиция является эффективной в предупреждении остеопороза или рака костей при еженедельной дозе приблизительно от 1 до приблизительно 10 мг, при ежедневной дозе приблизительно от 0,1 до приблизительно 2 мг или при ежемесячной дозе приблизительно от 3 до приблизительно 45 мг.
16. Пероральная фармацевтическая композиция, которая является эффективной в доставке терапевтически эффективных количеств лекарственного средства и усилителя в кишечник, где указанная композиция содержит алендронат или золедроновую кислоту и усилитель, который усиливает абсорбцию алендроната и золендроновой кислоты, где усилитель является единственным усилителем абсорбции, присутствующим в композиции, где композиция доставляет терапевтически эффективное количество алендроната или золедроновой кислоты в том случае, когда ее вводят больному в пределах 6 ч после приема пищи, лекарственных препаратов или напитков, отличных от воды.
17. Пероральная фармацевтическая композиция, которая является эффективной в доставке терапевтически эффективных количеств лекарственного средства и усилителя в кишечник, где указанная композиция содержит золедроновую кислоту и усилитель, который усиливает абсорбцию золендроновой кислоты, и усилитель является единственным усилителем абсорбции, присутствующим в композиции, и где композиция является эффективной при лечении или предупреждении остеопороза или рака костей, и где лекарственная форма содержит от 1 до приблизительно 25 мг золедроновой кислоты.
18. Композиция по п.14, где композиция является эффективной в лечении рака костей.
19. Композиция по п.14, где композиция является эффективной в лечении или предупреждении остеопороза.
| US 6200602 B1, 13.03.2001 | |||
| Лесняк O.M | |||
| Медикаментозные методы лечения остеопороза// Гинекология | |||
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
| Способ обработки целлюлозных материалов, с целью тонкого измельчения или переведения в коллоидальный раствор | 1923 |
|
SU2005A1 |

