Настоящее изобретение относится к фармацевтической композиции, содержащей pH-чувствительное лекарственное вещество, которая обладает повышенной стабильностью при хранении.
Некоторые производные HMG-CoA редуктазы, то есть ингибиторы биосинтеза холестерина, используемые при лечении гиперлипопротеинемии и атеросклероза, представленные формулой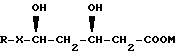
где R - органический радикал,
X - группу -CH=CH-, предпочтительно (Е)-СН=СН-, и
М - физиологически приемлемый катион щелочного металла или аммония, предпочтительно натрия или калия, и наиболее предпочтительно натрия,
обладают в значительной степени способностью к деградации при значении pH около 8. В качестве примера такого соединения можно привести соединение, обозначенное как USAN, то есть натрия флувастатин (называемого далее в описании как "флувастатин"), имеющего следующее химическое название:
R*, S*-(E)-(±)-7-[3-(4-фторфенил)-1-(1-метилэтил)-1Н-индол-2-ил] -2,5-дигидрокси-6-гептеновой кислоты натриевая соль (см. заявку на Европатент А-114027).
Например, кинетика деградации флувастатина, как установлено в водных растворах при различных значениях pH может проиллюстрирована табл. А.
Вышеуказанная неустойчивость флувастатина и родственных соединений HMG-CoA редуктазы, как полагаем, обусловлена чрезмерной подвижностью β,δ-гидроксилов в цепи гептеновой кислоты и наличием двойной связи, в результате чего при значениях pH от нейтрального до кислотного указанные соединения претерпевают реакции элиминирования, или изомеризации, или окисления с образованием сопряженных насыщенных ароматических соединений, а также трео-изомера, соответствующих лактонов и других продуктов из разложения.
Для получения пригодных для продажи лекарственных форм, содержащих указанное соединение, необходимо обеспечить соответствующую его защиту от pH-зависимой дестабилизации.
Кроме того, чувствительность к воздействию тепла и света, а также гигроскопичность предлагаемых соединений предъявляют определенные требования к технологии изготовления и хранению лекарственных форм.
Авторы смогли получить такие композиции, обладающие устойчивостью при длительном хранении, то есть в результате чего по крайней мере примерно 95% от исходного количества лекарственного вещества обладает активностью спустя два года при температуре 25 и 30oС и в течение еще больших промежутков времени.
Предлагаемые композиции при пероральном введении могут обеспечить быстрое и практически полное всасывание кишечником лекарственного препарата.
Дополнительным преимуществом является то, что предлагаемые стабилизированные композиции можно легко приготовить методами с использованием водной основы или других растворителей, например методом влажного гранулирования.
В другом аспекте настоящего изобретения предложена фармацевтическая композиция, содержащая HMG-CoA соединение формулы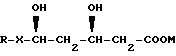
где R - органический радикал,
Х - -СН=СН- и
М - физиологически приемлемый катион и щелочную среду, способную обеспечивать pH водного раствора или дисперсии предлагаемой композиции по меньшей мере 8.
Предлагаемые композиции содержат лекарственное вещество и "щелочную среду", причем указанная щелочная среда способна стабилизировать композицию путем обеспечения pH водного раствора или дисперсии указанной композиции или по меньшей мере до значения 8. В предпочтительном варианте для обеспечения оптимальной стабильности лекарственного препарата соединение формулы I и щелочная среда находятся в непосредственном взаимодействии друг с другом в предлагаемой композиции.
Полученная композиция, как установлено, обеспечивает длительный срок хранения соединений формулы I даже в присутствии влаги, или в случае, когда такие композиции дополнительно содержат другие потенциально реакционноспособные наполнители, например как лактоза. Стабильность лекарственного вещества в предлагаемой композиции может составлять по меньшей мере 95%, и, как правило, в интервале от 98 до 99% через 18 месяцев при температуре 25oС и даже еще более длительное время.
Термин "щелочная среда" или "основание", используемые в данном описании, обозначают одно или несколько фармацевтически пригодных веществ, способных обеспечивать pH водного раствора или дисперсии предлагаемой композиции по меньшей мере до 8, предпочтительно по меньшей мере до 9 и вплоть до примерно 10. В частности, щелочная среда создает "микро- pH" (по меньшей мере 8) вокруг частичек композиции при адсорбировании на них воды, или при прибавлении воды в небольших количествах к указанной композиции. Щелочная среда должна при прочих условиях быть инертной относительно соединений формулы I. Значение pH можно определить путем подбора унифицированной дозы заявляемой композиции, содержащей, например, 20 мг флувастатина или эквивалентного количества другого соединения в рамках объема соединений формулы I с последующим диспергированием или растворением указанной композиции в 10 - 100 мл воды.
Фармацевтически приемлемое щелочное вещество (вещества), обеспечивающие щелочную среду, могут находиться в диапазоне от водорастворимых и умеренно растворимых вплоть до практически водонерастворимых веществ.
К примерам водорастворимых щелочных веществ, способных обеспечивать соответствующую основность композиции, относятся некоторые фармацевтически приемлемые неорганические карбонатные соли, например карбонат натрия или калия, бикарбонат натрия, или бикарбонат калия; фосфаты, выбранные из, например, безводного вторичного кислого фосфата натрия, калия или кальция или тринатрий фосфата; а также гидроокиси щелочных металлов, например как гидроокись натрия, калия или лития; и смеси вышеназванных веществ.
В качестве примера стабилизированная композиция предлагаемого изобретения может содержать: от 0,5 до 60 мас.%, обычно от 0,5 до 40 мас.% лекарственного вещества (например, флувастатина); и от 0,1 до 35 мас.%, предпочтительно 1 - 15 мас.% растворимого карбоната, например, выбранного из бикарбоната натрия, карбоната натрия и их смесей.
Примеры водорастворимых или умеренно растворимых щелочных веществ, которые также пригодны в качестве стабилизирующей щелочной среды в предлагаемых композициях содержат соединения, обычно используемые в антацидных составах (например окись, гидроокись или карбонат магния; бикарбонат магния; гидроокись или карбонат алюминия или кальция; смешанные соединения алюминия и магния, например магнийалюминийгидроксид); а также фармацевтически приемлемые соли фосфорной кислоты, например средний фосфат кальция и их смеси.
Из вышеуказанных щелочных веществ, как установлено, особенно эффективны для создания щелочной среды "фармацевтически приемлемые соли карбоновой кислоты", под которыми имеется в виду фармацевтически пригодные неорганические карбонаты и бикарбонаты, например карбонат натрия, бикарбонат натрия, карбонат кальция, и их смеси.
Композиции, также обладающие особенно высокой стабильностью при хранении, включают в свой состав в качестве щелочной среды как водорастворимый, так и водонератсовримый или умеренно растворимый щелочной наполнитель.
Так, например, значительное повышение стабильности и другие преимущества достигнуты при использовании щелочной среды, содержащей водорастворимую карбонатную соль и водонерастворимую карбонатную соль, особенно смесь бикарбоната натрия (или карбоната) и карбоната кальция.
Бикарбонат натрия преимущественно действует для нейтрализации кислотных групп в предлагаемой композиции в присутствии влаги, которая может адсорбироваться на частицах композиции во время ее хранения. Карбонат кальция оказывает буферное действие в композиции при хранении, не оказывая ощутимого влияния на высвобождение лекарственного вещества при приеме внутрь. Установлено также, что карбонатные соли обеспечивают в достаточной степени стабилизацию лекарственного вещества, в результате чего для получения устойчивых композиций настоящего изобретения можно использовать традиционные препаративные методы с использованием водной основы, например как метод порошкования водой или мокрого гранулирования.
Карбонат кальция модно использовать в виде преципитата или основы, но предпочтительно в осажденном виде.
Щелочная среда содержится в предлагаемых композициях в количестве, достаточном для установления pH, например по крайней мере 8, и предпочтительно по крайней мере 9, и вплоть до 10 водного раствора или дисперсии указанной композиции. Обычно предлагаемые композиции содержат примерно от 0,1 до 60 мас.% (как правило, от 0,5 до 40 мас.%) лекарственного вещества и примерно от 0,1 до 60 мас.%, предпочтительно от 20 до 35 мас.% щелочной среды. Используемое количество конкретного стабилизирующего наполнителя зависит в некоторой степени от технологии изготовления. В композициях, предназначенных для получения таблеток, карбонат кальция не должен превышать количества, которое можно для удобства подвергать уплотнению, и его обычно используют в комбинации с более легкоуплотняемым щелочным веществом, например бикарбонатом натрия. С другой стороны, лекарственные формы в виде капсулы могут содержать повышенные количества слабоуплотняемых наполнителей при условии, что вся композиция сохраняет в достаточной степени текучесть и обрабатываемость.
Композиция, выполненная в унифицированной дозе в виде твердого вещества, может иметь отношение водорастворимого карбоната к нерастворимому карбонату в интервале, например, от 1 : 40 до 2 : 1.
В качестве примера, таблетка предлагаемого изобретения может иметь весовое соотношение карбоната кальция к бикарбонату натрия примерно в интервале от 2 : 1 до 1 : 2. Предлагаемая композиция в виде капсулы может содержать указанные наполнители при весовом соотношении, например, от 25 : 1 до 35 : 1.
Кроме лекарственного вещества и щелочной среды, в предлагаемых композициях также обычно используют любой наполнитель, облегчающий обрабатываемость композиций. Потенциально пригодные материалы в качестве наполнителя общеизвестны в данной области (см. например, Remingtou's Pharmaceutical Sciences 18, издание, 1990, Mack Publishing Co., Easton, рA 1635-1636), включая лактозу и другие углеводы, предварительно оклейстеризованный крахмал, например крахмал 1500R (фирмы Colorcon Corp.), кукурузный крахмал, дикальция фосфат, целлюлозу, микрокристаллическую целлюлозу, сахара, хлористый натрий и их смеси, предпочтительными из которых являются лактоза, микрокристаллическая целлюлоза, предварительно оклейстеризованный крахмал и их смеси.
Благодаря своей высокой дезинтегрирующей и уплотняющей способности микрокристаллическая целлюлоза (торговое название АвицелR фирма FMC Corp.) и смеси, содержащие микрокристаллическую целлюлозу и один или несколько дополнительных наполнителей, например предварительно оклейстеризованный крахмал - особенно предпочтительны.
Общее содержание наполнителя в предлагаемых композициях составляет в интервале от 1 до 65 мас.% в расчете на всю композицию.
К другим компонентам, которые можно включать в состав предлагаемых композиций для облегчения обрабатываемости и/или обеспечения более эффективного действия лекарственной формы препарата, относятся и общеизвестные связующие для получения таблеток (например, желатина, сахаров, природных и синтетических смол (например, как карбоксиметилцеллюлоза, метилцеллюлоза, поливинилпирролидон, гидроксипропилметилцеллюлоза, микрокристаллическая целлюлоза и смеси вышеуказанных веществ); дезинтеграторов (например, карбоксиметилцеллюлозы сетчатой структуры, кроскармелозы, кросповидона, крахмал - гликолят натрия), смазывающих веществ (например, стеарата магния, гидрированного растительного масла, воска из карнауба и т.д.); добавки для повышения текучести (например, двуокись кремния, вещества, препятствующие склеиванию, или вещества, повышающие скольжение (например, тальк), а также подслащивающие вещества, красители (например, окись железа, алюминиевые красители), отдушки, противоокислители и тому подобное. Подбор конкретного ингредиента или ингредиентов и используемые количества их может легко определить специалист при обращении к стандартным методикам и практическому опыту получения лекарственных форм в виде таблеток или капсул или других лекарственных форм. Как правило, эффективное количество связующего при изготовлении таблеток составляет от 1 до 10 мас.%, предпочтительно от 1 до 5 мас.%; количество веществ, препятствующих связыванию, или диспергаторов составляет примерно от 1 до 10 мас.%; дезинтеграторов - примерно от 1 до 5 мас.% и смазывающих веществ примерно от 0,1 до 2 мас.% в расчете от всей композиции.
Из предлагаемых композиций можно приготавливать лекарственные препараты известными методами с обеспечением унифицированных доз заявляемого соединения для перорального приема, например 5 мг, 10 мг, 20 мг, 40 мг и т.д. в виде капсул, таблеток, пилюль и тому подобное.
Таблетки, капсулы и пилюли при желании можно покрывать пленкой растворимого в кишечнике вещества для защиты лекарственного вещества от его преждевременного разложения под действием желудочного сока до всасывания кишечником. Такие вещества хорошо известны и к ним относятся оксипропилметилцеллюлозы фталат, ацетилцеллюлозы фталат, поливинилацетата фталат, метилцеллюлозы фталат, сополимеризованная метакриловая кислота или сложные метиловые эфиры метакриловой кислоты (например, EudragitR, Rohm Pharma). Указанное покрытие предпочтительно наносят на капсулу, пилюлю или таблетку с увеличением их массы примерно на 5 - 12, предпочтительно на 8 - 10 мас.%.
Фармацевтические композиции предлагаемого изобретения, изготовленные в виде таблетки, при желании покрывают энтеросолюбильным покрытием для защиты ее от влаги и обеспечивания при действии света, а также от горького вкуса лекарственного вещества. Энтеросолюбильное покрытие может либо содержать вещества, обеспечивающие ее непрозрачность и красители, либо традиционное покрытие на основе непрозрачной пленки можно наносить на ядро таблетки при желании после того, как уже нанесено энтеросолюбильное вещество.
К примерам пригодных пленкообразующих материалов при технологии приготовления лекарственных препаратов с пленочным покрытием предлагаемого изобретения относятся, например, полиэтиленгликоль, поливинилпирролидон, поливиниловый спирт, гидрофильные полимеры, например как гидроксипропилцеллюлоза; гидроксиметилцеллюлоза и гидроксипропилметилцеллюлоза или тому подобное, из которых предпочтительно использование гидроксипропилметилцеллюлозы (например Opadry YellowT, Colorcon Corp.)
Гидрофобные пленкообразователи, которые можно наносить при использовании носителя на основе органического растворителя включают, например, этилцеллюлозу, ацетилцеллюлозу, сополимеры поливинилового спирта и малеинового ангидрида и т.д.
Пленочное покрытие можно обычно наносить с обеспечением увеличения массы пилюли или лекарственной основы таблетки примерно от 1 до 10 мас.% и предпочтительно примерно на 2 - 6 мас.%.
К другим общеизвестным компонентам лекарственного средства с использованием энтеросолюбильного или пленочного покрытия относятся смягчители (пластификаторы), например полиэтиленгликоль (полиэтиленгликоль 6000), триэтилцитрат, диэтилфталат, пропиленгликоль, глицерин, бутилфталат, которые используют в традиционных количествах, а также и вышеуказанные вещества, обеспечивающие непрозрачность лекарственного препарата, например как двуокись титана и красители, например окись железа, алюминиевые красители и т.д.
Энтеросолюбильные или пленочные покрытия можно наносить общеизвестными методами в соответствующем чане для нанесения покрытий или аппарате с псевдоожиженным слоем при использовании воды и/или традиционных органических растворителей (например, метанола, этанола, изопропанола), кетонов (ацетона, этилметилкетона), хлорзамещенных углеводородов (метиленхлорида, дихлорэтана) и т.д.
В соответствии с настоящим изобретением предлагаемая композиция содержит (в мас.% в расчете от всего состава композиции); от 0,1 до 60 мас.% (обычно от 0,5 до 40 мас.%) активного соединения (например флувастатина), от 0,1 до 60 мас. % щелочной среды (например, солей карбоновых кислот) и от 1 до 65 мас.% наполнителя (например, микрокристаллической целлюлозы).
В качестве одного примера состав предлагаемой фармацевтической композиции содержит (в мас.% в расчете на всю композицию):
от 0,5 до 60 мас.% соединения в качестве ингибитора гидроксиметилглутарил-СоА редуктазы (например, флувастатина), от 10 до 55 мас.% щелочной среды (например, карбонатных солей) и от 10 до 65 мас.% наполнителя (например, микрокристаллической целлюлозы).
В качестве другого примера состав предлагаемого изобретения содержит (в мас.% в расчете на всю фармацевтическую композицию):
от 0,5 до 60 мас. % соединения в качестве лекарственного вещества (например, флувастатина), от 5 до 40 мас.% карбоната кальция, от 0,5 до 20 мас. % бикарбоната натрия и от 10 до 65 мас.% наполнителя (например, микрокристаллической целлюлозы).
В качестве примера в состав для капсулы предлагаемого изобретения входит (в мас.% в расчете на всю фармацевтическую композицию):
от 0,5 до 60 мас.% (обычно от 0,5 до 40 мас.%) лекарственного вещества (например, флувастатина), от 25 до 40 мас.% карбоната кальция, от 0,5 до 10 мас.% бикарбоната кальция и от 20 до 35 мас.% микрокристаллической целлюлозы и возможно дополнительный наполнитель (например, предварительно оклейстеризованный крахмал) в количестве от 15 до 30 мас.%.
В качестве примера в состав таблетки в соответствии с настоящим изобретением (в мас.% от всей композиции) входит: от 0,5 до 60 мас.% лекарственного вещества (например, флувастатина), от 5 до 20 мас.% карбоната кальция, от 5 до 20 мас.% бикарбоната натрия и от 50 до 65 мас.% микрокристаллической целлюлозы.
Заявляемые стабилизированные композиции можно получить различными методами и технологиями, которые общеизвестны в данной области.
В технологии приготовления предлагаемых композиций необходимо, чтобы лекарственное вещество и щелочная среда находились бы в непосредственном контакте друг с другом. Требуемую степень их непосредственного взаимодействия друг с другом можно обеспечить методом сухого смешивания указанных компонентов для получения практически гомогенной смеси (предпочтительно до введения наполнителя или других добавок с последующим их уплотнением).
Однако для получения очень устойчивых составов предпочтительно использовать технологию их приготовления, основанную на использовании водной среды или другого растворителя, в результате которой лекарственное вещество и щелочное вещество перемешиваются в присутствии незначительных количеств, например, воды с получением частичек, содержащих лекарственное и щелочное вещество в полученной смеси в тесном сцеплении друг с другом. Из-за гигроскопичности и влагочувствительности соединений в качестве ингибитора HMG-CoA редуктазы, например как флувастатин нельзя ожидать, что лекарственное вещество будет в достаточной степени стабилизировано указанной щелочной средой, чтобы противостоять разложению при использовании такой технологии.
В другом варианте осуществления указанного способа, лекарственное и щелочное вещество порошкуют в присутствии воды, и полученные частицы после этого сушат. Наполнитель и остальные добавки, которые не использовали для включения во "внешнюю фазу" (непрерывную фазу) указанных частиц, затем смешивали с высушенными частицами с получением в результате композиции, пригодной для микрокапсулирования, таблетирования или тому подобное.
В другом варианте осуществления способа, основанного на растворителе с возможностью последующей сушки в псевдоожиженном слое, лекарственное вещество и щелочной носитель гранулируют в мокром состоянии известными методами, то есть смешивают во влажном состоянии вместе с некоторым количеством наполнителя. Полученные таким образом гранулы после просушки затем смешивают с любым оставшимся наполнителем и другими добавками, например связующим, замасливателем, после чего из них можно получать таблетки, капсулы или другие лекарственные формы.
Для обеспечения длительного срока годности предлагаемых композиций важно, чтобы частицы, полученные порошкованием или мокрым гранулированием, либо другим методом на водной основе, были практически полностью высушены, то есть до состояния, чтобы потери в массе от сушки (L.O.D.) не превышали 3%, и предпочтительно не более 2%.
Процесс сушки обычно осуществляют в лотковой сушилке, или сушкой в псевдоожиженном слое, причем последняя предпочтительнее. Сушку обычно проводят при температуре на входе около 50oС и относительной влажности менее 50%.
При получении предлагаемых фармацевтических композиций лекарственное вещество и остальные компоненты лекарственного препарата (кроме замасливателя) предпочтительно пропускают через сито размером 30 - 40 меш до измельчения в порошок или мокрого гранулирования, при этом обычно вначале просеивают лекарственное вещество, которое затем смешивают с просеянными наполнителями. Кроме этого высушенные частицы или гранулы также пропускают через сито 18 - 20 меш для обеспечения надлежащего смешивания их с адъювантами.
Композиции, предназначенные для таблетирования обычно просеивают через сита с меньшим количеством отверстий, например 24 меш до смешивания их со смазывающими веществами, а затем подвергают прессованию; причем для этой операции просеивания обычно необходимо проведение дополнительной сушки, в результате которой мокрые частицы или гранулы, полученные методом порошкования или гранулирования, сушат до потери их массы порядка 6 - 8%, после чего их пропускают через сито размером 12 - 14 меш, а затем опять сушат до потери их массы на 2 - 3%.
В альтернативном варианте относительно вышеуказанных методов порошкования и мокрого гранулирования лекарственное вещество и щелочной стабилизатор можно подвергнуть совместной лиофилизации, то есть сушке сублимацией из водного раствора, преимущественно в виде одной операции, осуществляемой in situ в процессе получения лекарственного препарата.
Как проиллюстрировано в патенте США N 4739073, включенном в данное описание в качестве противопоставляемого источника флувастатин-натрий, а также натриевые соли или другие фармацевтически приемлемые соли других предлагаемых соединений в качестве ингибитора HMG-CoA редуктазы обычно получают методом гидролиза соответствующего сложноэфирного производного, например, с едким натром в этанольном растворе. Этанол или другую органическую фракцию затем упаривают и к остатку, содержащему лекарственное вещество, прибавляют воду для получения водного раствора из которого (как правило, после экстрагирования органическим растворителем) извлекают лиофилизацией предлагаемый ингибитор HMG-CoA редуктазы. Установлено, что водорастворимый щелочной стабилизатор, например, как карбонат или бикарбонат натрия, или другое щелочное вещество можно прибавлять in situ к вышеуказанной водной фазе, содержащей флуваcтатин или другое соединение в качестве ингибитора HMG-CoA редуктазы, а после сублимационной сушки указанной водной фазы можно получить частицы, содержащие лекарственное соединение, которое совместно лиофилизированно с введенным щелочным агентом.
В результате этого можно обеспечить очень хорошее соприкосновение лекарственного вещества и стабилизатора до такой степени, что устойчивые композиции настоящего изобретения можно получить, например, лекарственного начала и карбоната натрия при весовом отношении в интервале примерно от 10 : 1 до 100 : 1. Так, например, совместно лиофилизированная композиция предлагаемого изобретения, содержащая до 0,1 мас.% карбоната натрия, как установлено, эффективна для получения высокоустойчивого лекарственного препарата.
Лиофилизацию осуществляют традиционными методами и оборудованием, первоначально снижая температуру раствора от комнатной до температуры ниже точки замерзания, обычно до температуры в интервале примерно -45oС в условиях высокого вакуума, например при давлении в интервале примерно 3 мм ртутного столба или ниже, после чего повышая температуру его до комнатной или выше комнатной температуры, приводя тем самым к упариванию водного растворителя. Извлеченные частицы практически не содержат растворителя и оптимально представляют собой гомогенную смесь из лекарственного вещества и стабилизатора.
Полученные частицы затем можно смешать с другими адъювантами, например, наполнителем, связующим, замасливателем и т.д.
Предлагаемые композиции, полученные любым из вышеуказанных методов, можно формовать в любую лекарственную форму при использовании общеизвестных в данной области технологий и методов, например таблетированием, включением в желатиновую капсулу, гранулированием, прессованием и т.д.
Как указывалось ранее, состав в качестве энтеросолюбильного и/или пленочного покрытия можно наносить на лекарственную форму в целях обеспечения конкретного положительного эффекта.
Энтеросолюбильное или пленочное покрытие таблетки на основе микрокристаллической целлюлозы при использовании водного пленкообразующего состава желательно осуществлять при подповерхностной температуре от 30 до 50oС, температуре на входе 50 - 80oС и относительной влажности менее 50%.
Для обеспечения оптимальной устойчивости лекарственного препарата важно, чтобы лекарственная форма с энтеросолюбильным и/или пленочным покрытием была высушена до влагосодержания не более 4% и предпочтительно не более 3%.
Полученные таблетированные или капсульные лекарственные формы должны быть защищены во время хранения от окисления, индуцированного действием тепла или света, а также и проникновения влаги.
Капсулы и таблетки, полученные из предлагаемых фармацевтических композиций, как установлено, обладают превосходной устойчивостью при хранении.
Эти лекарственные формы пригодны для целевого использования. Покрытые пленкой таблетки или капсулы имеют время высвобождения примерно от 10 до 30 минут. Таблетки или капсулы с энтеросолюбильной оболочкой имеют время высвобождения обычно от примерно 30 мин до примерно 6 часов.
Кроме фармацевтических композиций, содержащих флувастатин-натрий, настоящее изобретение охватывает также составы, в которые входят другие соединения формулы I в качестве ингибиторов HMG-CoA редуктазы. Эти соединения раскрыты, например, в нижеследующих патентах, опубликованных заявках на патент и публикациях, которые включены все в данное описание в качестве ссылки:
патенте США N 4739073 и заявке на Европатент N 114027 (где R означает индолил и его производные); заявке на Европатент 367895 (R означает пиримидинил и его производные);
патентах США NN 5001255 (R означает инденил и его производные); 4613610 (R означает пиразонил и его производные); 4851427 (R означает пирролил и его производные); 4755606 и 4808607 (R означает имидазолил и его производные); 4751235 (R означает индолизинил и его производные); 4939159 (R означает азаиндолил и его производные); 4822799 (R означает пиразолопиридинил и его производные); 4804679 (R означает нафтил и его производные); 4876280 (R означает циклогексил и его производные); 4829081 (R означает тиенил и его производные); 4927851 (R означает фурил и его производные); 4588715 (R означает фенилсилил и его производные) и публикации F.G.Kathawala, Medicinal Research Reviews, том 11(2), стр.121-146, 1991 г.
Другие соединения формулы I раскрыты, например, в заявке на Европатент N 304063 (R означает хинолинил и его производные); заявке на Европатент N 330057 и патентах США NN 5023708 и 4868185 (R означает пиримидинил и его производные); заявке на Европатент N 324347 (R означает пиридазинил и его производные); заявке на Европатент N 300278 (R означает пирролил и его производные) и патенте США N 5013749 (R означает имидазолил и его производные).
К соединениям, пригодным для использования в качестве активных компонентов в предлагаемых композициях относятся соединения, где R выбран из групп пирролила, имидазолила, индолизинила, пирролпиридина, пиразолпиридина, хинолила, фенилсилилфенила, нафтила, циклогексила, фенилтиенила, фенилфурила и пиридазинила и их производных. Среди указанных соединений формулы I предпочтительны соединения, где R выбран из группы индолила, пиримидинила и инденила и их производных, а Х означает (Е)-СН=СН-.
К конкретным примерам соединений, раскрываемых в вышеуказанных публикациях, которые являются производными HMG-CoA редуктазы, пригодными для использования в качестве активного компонента в предлагаемых фармацевтических композициях, относятся нижеследующие натриевые соли, или другие фармацевтически приемлемые соли:
3R, 5S-(E)-7-[4-(4-фторфенил)-6-(1-метилэтил)-2-диметиламинопиримидин-5-ил]-3,5-дигидрокси-6-гептеновой кислоты натриевая соль;
эритро-(±)-(E)-7-[3-(4-фторфенил)-спиро[циклопентан-1,1'-1Н-инден]-2-ил] -3,5-дигидрокси-6-гептеновой кислоты натриевая соль;
3R, 5S-(E)-7-[3-(4-фторфенил)-1-(1-метилэтил)-индолизин-2-ил] -3,5-дигидрокси-6-гептеновой кислоты натриевая соль;
3R, 5S-(E)-7-[3-(4-фторфенил)-1-(1-метилэтил)-1Н-пирроло] -2,3-b)-пиридин-2-ил]-3,5-дигидрокси-6-гептеновой кислоты натриевая соль;
3R, 5S-(E)-7-//4-фторфенил)-2-(1-метилэтил)-хинолин-3-ил)-3,5-дигидрокси-6-гептеновой кислоты натриевая соль;
3R, 5S-(E)-7-/1-(4-фторфенил)-3-(1-метилэтил)-4-оксо-1,4-дигидрохинолин-2-ил)-3,5-дигидрокси-6-гептеновой кислоты натриевая соль;
3R, 5S-(E)-7-[4-(4-фторфенил)-6-(1-метилэтил)-3-метил-1Н-пиразоло(3,4-b)пиридин-5-ил]-3,5-3,5-дигидрокси-6-гептеновой кислоты натриевая соль;
3R, 5S-(E)-7-/3-(1-метилэтил)-5,6-дифенилпиридазин-4-ил/-3,5-дигидрокси-6-гептеновой кислоты натриевая соль;
3R, 5S-(E)-7-/(4-фторфенил)-6-(1-метилэтил)-2-фенилпиримидин-5-ил)-3,5-дигидрокси-6-гептеновой кислоты натриевая соль;
3R, 5S-(E)-7-/4-(4-фторфенил)-2-(1-метилэтил)-1-оксо-1,2-дигидрохинолин-3-ил/-3,5-дигидрокси-6-гептеновой кислоты натриевая соль;
эритро-(±)-(E)-7-/4-(4-фторфенил)-2-(1-метилэтил)-хинолин-3-ил/-3,5-дигидрокси-6-гептеновой кислоты натриевая соль;
эритро-(±)-(E)-7-/1-(4-фторфенил)-3-(1-метилэтил)-пирроло-(2,1-а)изохинолин-2-ил)-3,5-дигидрокси-6-гептеновой кислоты натриевая соль;
эритро-(±)-(E)-7-/4-циклопропил-6-(4-фторфенил)-2-(4-метоксифенил)пиримидин-5-ил)-3,5-дигидрокси-6-гептеновой кислоты натриевая соль;
3R, 5S-(E)-7-/4-(4-фторфенил)-2,6-диметилпиримидин-5-ил/-3,5-дигидрокси-6-гептеновой кислоты натриевая соль;
3R, 5S-(E)-7-/4-(4-фторфенил)-6-метил-2-фенил/-пиримидин-5-ил/3,5-дигидрокси-6-гептеновой кислоты натриевая соль;
3R, 5S-(E)-7-/3,5-диметилфенил)-6-метил-2-фенилпиримидин-5-ил/3,5-дигидрокси-6-гептеновой кислоты натриевая соль;
эритро-(±)-(E)-7-/3,4-бис(4-фторфенил)-6-(1-метилэтил)-пиридазин-5-ил/-3,5-дигидрокси-6-гептеновой кислоты натриевая соль;
эритро-(±)-(E)-7-/1-(4-фторфенил)-3-(1-метилэтил)-5-фенил-1Н-пиррол-2-ил/-3,5-дигидрокси-6-гептеновой кислоты натриевая соль;
эритро-(±)-(E)-9,9-бис(4-фторфенил)-3,5-дигидрокси-8-(1-метил-1Н-тетразол-5-ил)-6,8-нонадиеновой кислоты натриевая соль;
эритро-(±)-(E)-3,5-дигидрокси-9,9-дифенил-6,8-нонадиеновой кислоты натриевая соль;
эритро-(±)-(E)-7-/4-(4-фторфенил)-1,2-бис(1-метилэтил)-3-фенил-пиррол-2ил/-3,5-дигидрокси-6-гептеновой кислоты натриевая соль;
3R, 5S-(E)-7-/4,5-бис(4-фторфенил)-2-(1-метилэтил)-1Н-имидазол-1-ил)3,5-дигидрокси-6-гептеновой кислоты натриевая соль;
3R, 5S-(E)-7-/4-(4-фторфенил-2,6-бис(1-метилэтил)-5-метоксиметилпиридин-3-ил/3,5-дигидрокси-6-гептеновой кислоты натриевая соль;
эритро-(±)-(E)-/4-(4-фторфенил)-2-(1-метилэтил)-6-фенилпиридин-3-ил)-3,5-дигидрокси-6-гептеновой кислоты натриевая соль;
эритро-(±)-(E)-/2-(4-фторфенил)-4,4,6,6-тетраметилциклогексан-1-ил/-3,5-дигидрокси-6-гептеновой кислоты натриевая соль;
эритро-(±)-(E)-7-/4-(4-фторфенил)-2-циклопропилхинолин-3-ил)-3,5-дигидрокси-6-гептеновой кислоты натриевая соль;
эритро-(±)-(E)-7-/4-(4-фторфенил)-2-(1-метилэтил)-хинолин-3-ил/-3,5-дигидрокси-6-гептеновой кислоты натриевая соль.
Соединения формулы I являются ингибиторами НМG-СоА редуктазы, то есть ингибиторами биосинтеза холестерина и, следовательно? они пригодны для лечения пиперлипопротеиномии и атеросклероза, как раскрыто в вышеуказанных патентах, опубликованных заявках и публикациях, включенных в качестве ссылки в данное описание.
Приводимые ниже примеры предназначены для иллюстрации различных вариантов осуществления настоящего изобретения и ни в коем случае не ограничивают его объем.
Пример 1.
20 мг капсулу, размер N 3 для перорального приема, содержащую флувастатин получают на основе рецептуры, указанный в табл.1.
(а) Флувастин, 2 мг бикарбоната натрия, 62,84 карбоната кальция, 23,35 мг микрокристаллической целлюлозы и 20,95 мг оклейстеренного крахмала перемешивают в течение 5 мин, и полученную смесь просеивают через сито 40 меш и перемешивают еще 3 мин.
(b) К полученной смеси прибавляют воду при непрерывном перемешивании в течение 4 мин с последующим формованием микрогранул методом мокрого гранулирования.
(с) Мокрые микрогранулы сушат в сушилке с псевдоожиженным слоем при температуре на входе 50oС до потери общего веса от сушки 1,59%.
(d) Просушенные гранулы пропускают через сито размером 20 меш, после чего смешивают с адъювантами на основе микрокристаллической целлюлозы и прожелатинизированным крахмалом в течение 10 минут. Тальк и стеарат магния (каждый предварительно просеянный через сито 60 меш) прибавляют к полученной смеси с перемешиванием в течение примерно 5 мин.
Полученная композиция имеет показатель потери веса от сушки 2,65%.
Дисперсия указанной композиции в 10 - 100 мл воды имеет pH 10.
(е) Непрозрачную капсулу голубого цвета заполняют предлагаемой композицией и полируют ручным способом солью.
Полученная капсула отвечает спецификации 75% ее растворения в организме за 30 минут при использовании теста на смешиваемость фирмы USp.
Лекарственный препарат, как установлено, обладает 99% устойчивостью к разложению спустя 18 месяцев при его хранении при 30oС в защищенных от света и от проникновения влаги условиях.
Пример 2.
По аналогичной методике, описанной в примере 1, 40 мг гранулы двойного размера N 3 получают при использовании ингредиентов, приведенных в табл. 1 в двухкратном количестве.
Пример 3.
По аналогичной методике, описанной в примере 1, получают 10 мг капсулы, размер N 3, содержащие флувастин, за исключением того, что используют дополнительно 10 мг микрокристаллической целлюлозы.
Пример 4.
20 мг таблетку флувастатина для перорального приема получают на основе рецептуры, указанной в табл.2.
(а) Флувастин, карбонат кальция, бикарбонат кальция, микрокристаллическую целлюлозу, поливинилпирролидон и кроскармелозунатрия каждый просеивают через сито 40 меш, а затем соединяют и перемешивают в течение 3 мин, после чего полученную смесь просеивают через сито 40 меш и перемешивают еще раз в течение 2 мин.
(b) К полученной смеси прибавляют воду при одновременном перемешивании в течение примерно 5 мин с получением мокрого гранулята.
(с) Полученный гранулят сушат в сушилке с псевдоожиженным слоем при входной температуре 50oС до просушивания гранул с потерей их веса на 6 - 8%. Гранулы затем пропускают через сито 14 меш и опять сушат до потери веса при сушке не более 2,5%. Просушенные гранулы после этого пропускают через сито 24 меш и перемешивают в течение 3 мин.
(d) Стеарат магния, просеянный через сито размером 60 меш, перемешивают вместе с полученной смесью в течение 5 минут.
Полученная в результате композиция имеет показатель потерь массы при сушке не более 2%.
Дисперсия полученной композиции в 10 - 100 мл воды имеет pH 10.
(е) Из полученной композиции светло-желтого цвета формуют таблетки при использовании пресса 8 мм с получением таблетки массой 200 мг.
(f) Пленочное покрытие на основе оксипропилметилцеллюлозы, Opadry Yellow, YS-1-6347-0, фирмы Colorcon Corp. (10% водную суспензию) наносят на основу таблетки в псевдоожиженном слое при температуре на входе в интервале от 70 до 75oС с увеличением массы таблетки на 5 - 6%.
Полученная в результате таблетка удовлетворяет спецификации 75% степени растворения за 30 мин при использовании теста на смешиваемость фирмы U Sp.
Полученный лекарственный препарат, как установлено, обладает 99% устойчивостью при хранении спустя 18 месяцев при температуре 30oС в условиях без доступа света и влаги.
Пример 5.
По аналогичной методике, описанной в примере 4, получают 40 мг таблетки флувастатина, за исключением того, что компоненты основы таблетки берут в двухкратном количестве, которые указаны в примере 4.
Пример 6.
По аналогичной методике, описанной в примере 4, получают 10 мг таблетки флувастатина, за исключением того, что компоненты основы таблетки, указанные в примере 4, берут в половинном количестве.
Пример 7.
Основу таблетки или капсулы флувастатина, полученных по любой из методик вышеуказанных примеров, покрывают в псевдоожиженном слое при температуре этого слоя в интервале от 30 до 50oС, температуре на входе в аппарат от 50 до 80oС и относительной влажности менее 50% при использовании рецептуры для энтеросолюбильного покрытия на основе EudragitR (фирмы Pohm Pharma или в альтернативном варианте оксипропилметилцеллюлозы фталата с увеличением их массы на примерно 5 - 12%.
Пример 8.
По любой из методик вышеуказанных примеров получают композицию настоящего изобретения на основе 3R, 5S-(E)-7-/4-(4-фторфенил)-6-(1-метилэтил)-2-диметиламинопиримидин-5-ил/-3,5-дигидрокси-гептеновой кислоты в виде натриевой соли в качестве активного компонента.
Пример 9.
По любой из методик, описанных в вышеуказанных примерах получают композицию предлагаемого изобретения, содержащую в качестве активного компонента эритро-(±)-(E)-7-[3-(4-фторфенил)спиро[циклопентан-1,1'-1Н-инден] -2'-ил]-3,5-дигидрокси-6-гептеновую кислоту в виде натриевой соли.





Изобретение относится к химико-фармацевтической промышленности и касается фармацевтической композиции для лечения гиперлипопротеинимии и атеросклероза. Изобретение заключается в том, что композиция содержит соединение в качестве ингибитора HMG-CoA редуктаза, а именно, например, флувастатин натрия, стабилизированный щелочным стабилизатором от рН-связанной деградации, который способен доводить рН водного раствора или дисперсии указанного состава до по меньшей мере 8. Изобретение обеспечивает быстрое и практически полное всасывание кишечником лекарственного препарата. 10 з.п. ф-лы, 2 табл.
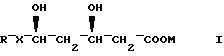
где R - органический радикал;
X - CH = CH;
M - физиологически приемлемый катион,
и щелочную среду, способную доводить pH водного раствора или дисперсии указанной композиции до величины по крайней мере 8.
| US 4671953 A, 09.06.87 | |||
| US 4739073 A, 19.04.88. |
