Изобретение относится к новым иммунореагентам, в особенности к направленным радиоактивным иммунореагентам, которые находят применение, например в терапевтических и диагностических препаратах и способах, предназначенных для получения изображения. Настоящее изобретение относится также к новым комплексообразующим агентам.
До 1980 г. рядом лабораторий при различных институтах (S.E. Order et al. , "Use of Isotopic Immunoglobulin in Therapy", Cancer Research 40, 3001-7 (August 1980) была показана возможность направленной доставки радиоиммуноглобулина в участки организма, имеющие опухоли. К 1980 г. было доказано, что опухоли должны концентрировать меченые радиоактивным изотопом антитела к обусловленным опухолью антигенам и что применение меченых радиоактивным изотопом реагентов позволяет как получать диагностическое изображение опухолей, например при помощи гамма-излучения (радиоиммуносцинтиграфии) и позитронной томографии, так и проводить лечение, т.е. уменьшать размер опухоли направлением в цель радиоактивного иммунореагента.
Предыдущие работы по направленной доставке меченых радиоактивным изотопом иммунореагентов проводили с радиоактивным иодом. Однако, как отмечено Scheinberg et al., "Tumor Imaging with Radioactive Metal Chelates Corjugated to Monoclonal Antibodies", Science 215, N 19, 1511 - 13 (March 1982), применение изотопов иода ставит несколько проблем, в частности при получении изображений опухолей сканированием. Из трех обычно доступных изотопов только 123I имеет подходящую излучательную способность для получения изображения и достаточно короткий период полураспада для безопасного диагностирования.
Гамма-излучение 125I слишком слабое для получения изображения 131I часто применяют, но он нежелателен из-за его длинного периода полураспада и обладающего высокой энергией гамма-излучения и цитотоксичного бета-излучения. 131I также применяют для лечения больших опухолей, но он оказался неэффективным при лечении маленьких опухолей. Кроме того, быстрый метаболизм радиоиодированных антител допускает инкорпорирование иода в щитовидную железу и активную экскрецию иода желудком и мочевыми путями. Такая дисперсия радиоактивного иода препятствует получению изображения специфических опухолей, поскольку на опухоли оказывает влияние фоновое ионизирующее излучение.
Кроме направленной доставки радиоактивных антител в опухоли с целью получения диагностического изображения или лечения, аналогичную направленную доставку проводили для получения изображения при диагностировании инфарктов, в частности инфаркта миокарда, применяя антитела на собачий сердечный миозин (Khaw et al, "Myocardial Infarct Imaging of Antibodies to Canine Cardige Myosin Indium - 111 - Diethylenetriamine Pentaacetic Acid", Science 209, 195-7 (July 1980) и получения изображения при атеросклерозе аналогичной направленной доставкой их в атеросклеротические бляшки. Те же самые недостатки появляются при применении радиоактивного иода для получения изображения при диагностировании инфаркта, как и при применении его для получения изображения опухоли и ее лечении.
Известно, что 111In может образовать комплексы с полиаминокарбоновыми кислотами, например этилендиаминотетрауксусной кислотой (EDTA) и диэтилентриаминпентауксусной кислотой (DTPA). Однако ковалентное соединение белков (антител) с этими комплексообразующими агентами, достигаемое ацилированием активированными карбонилами, ароматическим диазониевым сочетанием или бромацетилированием, неэффективно, даже если для облегчения ковалентного соединения белков с комплексообразующими агентами были получены описанные Brechbiel et al ["Synthesis of 1-[p-Isothiocyanatobenzyl) Derivatives of DTPA and EDTA. Antibody Labeling and Tumor Imaging Studies." Inorg. Chem. 25, 2772-81 (1986)] изоцианатобензилпроизводные этих кислот.
Последние исследовательские работы были направлены на усовершенствование антител (Ab's), например моноклональных, специфических антител для специфической направленной доставки, антител, которые образуют комплекс или связываются непосредственно с радионуклидами, предпочтительных радионуклидов и комбинаций их с антителами и комплексообразующими агентами. Некоторые попытки были предприняты для усовершенствования комплексообразующих агентов.
Тем не менее, EDTA и особенно DTPA, и их производные остаются широко распространенными комплексообразующими агентами, применяемыми для ковалентного связывания антител и образования координационных комплексов с радионуклидами металлов. Однако недостатки DTPA были отмечены, например Parker et al, "Implementation of Macrocycle Conjugated Antibodies for Tumor Targeting", Pure and Appl. Chem. , 61, N 9, 1637 - 41 (1989) : "Обычно образуют комплексное соединение радионуклида металла с ациклическим хелатом образующим агентом (например EDTA или DTPA), который ковалентно соединяют с антителами. Ни один из хелатов не был полностью пригодным из-за тенденции металла к отделению от хелата in vivo, ...", u Cox et al, "Synthesis of a Kinetically stable Yttrium-90 Labelled Macrocycle- Antibody Conjugate", J. Chem. Soc., Chem. Commun. 797-8 (1989) : "Иттрий-90 является привлекательным изотопом для терапии ..., но его клиническое применение будет очень ограничено из-за токсичности его для костного мозга, являющейся результатом промотированного кислотой выделения 90Y из соединенного с антителами хелата, например диэтилентриаминпентауксусной кислоты (DTPA)".
В результате предыдущих попыток разработать улучшенные комплексообразующие агенты были получены материалы, которые имеют свои недостатки. Например Craig et al B "Towards Tumor Imaging with Indium-111 Labelled Macrocycle-Antibody Conjugates, "J. Chem. Soc. Chem. Commun. 794-6 (1989), описали макроциклические гексакоординирующие лиганды, но сообщали, что ограничивающей особенностью такого подхода является необходимость мечения макроцила 111In до образования конъюгата с антителами. Связывание индия с (4) проходит недостаточно быстро при 37oC для эффективного мечения радиоактивным изотопом . ... Поэтому были отобраны другие трехосновные триазамакроциклические лиганды по их способности связывать индий быстро в мягких условиях (20oC, pH 5 и времени менее 1 ч) и в то же время образовать кинетически стабильный комплекс in vivo ....
Однако только (6) оказался эффективным при концентрации лиганда 10 мкМ, в этих условиях содержание меченого радиоактивным изотопом продукта достигало 96% (30 мин, pH 5 20oC).
Тем не менее, тридцать минут все же неподходящее время. Желательно иметь комплексообразующие агенты, которые лучше чем EDTA и DTPA и которые координационно связывали бы предпочтительные радионуклины, например In, Y, Sc, Ga, Ge, в течение нескольких минут, т.е. в течение менее чем 5 мин, непосредственно перед введением реагента пациенту, особенно когда короткоживующий радионуклид должен обязательно генерироваться из долгоживущего радионуклида во время лечения пациента.
Следует отметить, что комплексы иттрия, предпочтительно радионуклида иттрия, применяемые в терапии, менее стабильны, чем комплексы индия [Mather et al, "Labelling Monoclonal Antibodies with Yttrium 90" Eur. J. Nucl. Med., 15, 307-312 (1989)], причем имеются в виду обычные комплексы их. Mather et al сообщает, что исследования по поиску тканей- мишеней у раковых пациентов (биораспределение вещества после его введения в организм) с применением меченых радиоактивным изотопом антител дают возможность предположить, что in vivo стабильность меченых иттрием антител не так велика, как у их 111In - меченых аналогов, и что эти данные подкрепляются другими недавними публикациями в этой области.
Когда хелатирующие агенты ковалентно связываются с белками (например Ab's), белки обычно способны акцептировать намного более чем одну молекулу хелатирующего агента, поскольку они содержат много аминогрупп и меркаптогрупп, через которые присоединяются хелатирующие агенты. Часто очень важно определить, сколько хелатирующих участков присоединено к каждой молекуле белка. Наиболее подходящий путь осуществления этого - применение спектрометрических средств. Однако ранее известные хелатирующие агенты и хелаты их имеют спектры поглощения, которые перекрываются со спектрами поглощения применяемых белков, и аналитическое определение числа хелатирующих или хелатированных участков на молекулу белка не может быть проведено однозначно спектрометрией, т.к. перекрываемые спектры поглощения маскируют друг друга. Поэтому очень желательно получить хелатирующие агенты для конъюгирования с белками, спектры поглощения которых и спектры поглощения хелатов с металлами которых не перекрываются такими спектрами белков, с которыми хелатирующие агенты химически связаны.
Другая проблема, возникающая при применении некоторых известных таких препаратов, состоит в том, что хелатирующий агент нужно активировать восстановителем до образования радионуклидного хелата. Если конъюгаты с белками образованы до получения радионуклидного хелата, то восстановитель, применяемый для активирования комплексообразующего агента, может разрушать белок. Например, предпочтительные хелатирующие агенты, обычно применяемые для комплексообразования с технецием (Tc) и рением (Re), соединяются с металлами через серосодержащие группы, которые нужно восстановить восстановителем (дитиотреитом) для активирования хелатирующего агента до образования радионуклидного хелата. Если белковый конъюгат, содержащий дисульфидные связи, образован до восстановления, то восстановитель может разрушить белок. Поэтому желательно иметь хелатирующие агенты, способные образовать конъюгаты с белками до комплексообразования с радионуклидами.
Коротко говоря, различные обычно применяемые меченые радиоактивными изотопами антитела и хелатирующие агенты, применяемые для получения иммунореактивных конъюгатов ковалентным связыванием хелатирующего агента с иммунореактивным белком, и радионуклидные комплексы их, применяемые в диагностических препаратах для получения изображений и направленных терапевтических препаратах, обладают одним или несколькими следующими недостатками : 1) токсичностью; 2) дисперсией реагента, обусловленной быстрым метаболизмом, 3) неадекватными эмиссионными характеристиками, 4) недостаточным ковалентным связыванием с белком для получения конъюгата, 5) медленным комплексообразованием с металлами, 6) нестабильными комплексами с металлами, например в отношении температуры, времени или pH, 7) неспособностью образовать конъюгаты и оставаться стабильными при хранении до комплексообразования с металлом, 8) невозможностью спектрофотопетрического анализа радионуклидного комплексного реагента и 9) невозможностью комплексообразования без стадии активирования, которая разрушает белок.
Предложены направленные (в цель) радиоактивные иммунореагенты, которые позволяют разрешить указанные выше проблемы. Направленные радиоактивные иммунореагенты изобретения содержат ион радионуклидного металла, комплексообразующий агент, который является производным пиридина, дипиридина, терпиридина, тетрапиридина, пентапиридина, гексапиридина или фенантролина, и иммунореактивную группу, ковалентно связанную через реагирующую с белком группу с компексообразующим агентом.
В соответствии с изобретением предложен направленный (в цель) радиоактивный иммунореагент, содержащий ион радионуклидного металла, комплексообразующий агент и иммунореактивную группу, ковалентно связанную с комплексообразующим агентом, который является соединением формулы A-I (см. в конце описания), R представляет собой водород, алкил, алкокси-, алкилтио-, алкиламино-, алкилформамидогруппу, арил, арилоксигруппу, гетероциклический радикал или реагирующую с белком группу; R1 представляет собой водород, алкил, алкокси-, алкилтио-, алкиламино-, алкилформамидогруппу, арил, арилоксигруппу, гетероциклический радикал или реагирующую с белком группу; R2 представляет собой гидрокси-, карбоксигруппу, гидроксиалкил, алкилтиогруппу, остаток карбонилиминодиуксусная кислота, метилениминодиуксусная кислота, метилентиоэтилениминодиуксусная кислота, карбоксиалкилтиоалкил, остаток гидразинилидендиуксусная кислота или соль такой кислоты или два R2 вместе обозначают атомы, необходимые для замыкания макроциклического ядра, содержащего по меньшей мере один координирующий гетероатом и по меньшей мере одну, предпочтительно две, алкиленовые группы, образующие часть ядра; R3 представляет собой водород, алкил, алкокси-, алкилтио-, алкиламино-, алкилформамидогруппу, арил, арилоксигруппу, гетероциклический радикал или реагирующую с белком группу; R4 представляет собой водород или реагирующую с белком группу; n = 0, 1, 2, 3 или 4; o = 0 или 1; m = 0 или 1, причем по меньшей мере один из n и m = 0 и по меньшей мере один из R, R1, R3 и R4 является реагирующей с белком группой.
Пиридины имеют формулу A-II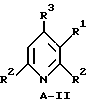
где
R1, R2 и R3 имеют указанные выше значения.
Дипиридины, терпиридины, тетрапиридины, пентапиридины и гексапиридины имеют формулу A-III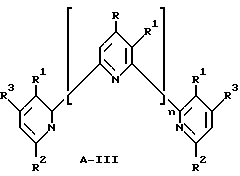
где
R, R1, R2 и R3 имеют указанные выше значения и n = 0, 1, 2, 3 или 4.
Фенантролины имеют формулы A-IV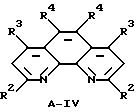
где
R2, R3 и R4 имеют указанные выше значения.
Настоящее изобретение предлагает новые терперидины, тетрапиридины, пентапиридины и гексапиридины формулы A-I. Предпочтительные терпиридины изобретения имеют формулу A-III, в которой n = 1 и R является радикалом формулы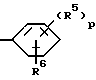
где R5 представляет собой алкоксигруппу или алкил, p = 0, 1, 2, 3 или 4 и R6 является реагирующей с белком группой.
Изобретение предлагает также новые фенантролины, предпочтительно имеющие формулу A-IV, в которой по меньшей мере один из R4 представляет собой реагирующую с белком группу.
Изобретение предлагает также терапевтические и диагностические препараты, содержащие описанный выше направленный (в цель) радиоактивный иммунореагент.
Изобретение предлагает также способ получения для целей диагностирования изображения участка организма пациента, предусматривающий a) введение пациенту эффективного количества описанного выше радиоактивного иммунореагента, способного направиться в участок-цель, и b) активирование (для получения изображения) чувствительного к радиации элемента или устройства, например пленочного или электронного сенсора, радиацией, испускаемой участком-целью.
Способ лечения участков заболевания у пациента по изобретению предусматривает введение пациенту или в образец, взятый у пациента, эффективного количества терапевтического препарата, содержащего указанный выше радиоактивный иммунореагент, способный попадать в определенный участок, и его фармацевтически пригодный носитель.
Важным признаком изобретения является то, что описанные в нем направленные в цель радиоактивные иммунореагенты, содержащие иттрий, обладающий меньшей радиационной токсичностью, чем радиоактивные, содержащие иттрий, иммунореагенты, полученные с другими хелатирующими агентами.
К важным признакам относится также то, что направленные иммунореагенты изобретения не подвергаются быстрому метаболизму и вредному диспергированию.
Другим важным признаком изобретения является то, что описанные комплексы эффективно образуют ковалентные связи с белками и другими биологическими молекулами.
Еще один важный признак изобретения состоит в том, что описанные иммунореагенты обладают хорошими излучающими характеристиками и легко анализируются спектрометрическими способами.
Кроме того, белковые конъюгаты комплексообразующих агентов можно получить и хранить до необходимости получения комплекса с металлом и комплексообразование можно проводить без стадий активирования, которые разрушают белок.
Дополнительно к этому, комплексообразующие агенты быстро образуют комплексы с металлами и полученные хелаты обладают высокой стабильностью по отношению ко времени хранения, температуре и pH.
Другие важные признаки изобретения станут очевидными при прочтении приведенного ниже описания предпочтительных вариантов с учетом предлагаемых чертежей.
Фиг. 1 изображает анализы иммунокомпетентности B 72.3 - THT - Sc+++, радиоактивного иммунореагента настоящего изобретения, B 72.3 - THT - конъюгаты и немодифицированного B 72.3.
Фиг. 2 изображает анализы иммунокомпетенции B 72.3 - THT - конъюгата, двух препаратов B 72.3 - TMT - конъюгатов и немодифицированного B 72.3.
Фиг. 3 изображает результаты излучения биораспределения B 72.3 - TMT - 111In, радиоактивного иммунореагента изобретения, и B 72.3 - DTPA - 111In.
Фиг. 4 изображает результаты изучения биораспределения B 72.3 - TMT - 90Y, радиоактивного иммунореагента изобретения, и B 72.3 - DTPA - 90Y.
Фиг. 5 изображает кривую выживаемости, т. е. число выживших мышей каждый день после первого дня иммунизации по схеме с низкой дозой B 72.3 - DTPA и B 72.3 - TMT и схеме с высокой дозой B 72.3 - TMT и B 72.3 - DTPA.
Фиг. 6 и 7 изображает спектры поглощения TMT, TMT- Y+++ и TMT - Pb++.
Приведенное ниже описание в основном относится к применению направленных (в цель) радиоактивных иммунореагентов в терапевтических и диагностических (для получения изображения) препаратах и способах. Кроме того, направленные радиоактивные иммунореагенты применяют в качестве диагностических реагентов, например реагентов для радиоиммуноэлектрофореза.
Иммунореагенты изобретения содержат ион радионуклеидного металла, комплексообразующий агент и иммунореактивную группу, ковалентно связанную с комплексообразующим агентом через реагирующую с белком группу.
Комплексообразующий агент является производным пиридина, дипиридина, терпиридина, тетрапиридина, пентапиридина, гексапиридина или фенантролина, предпочтительно структурной формулы A-I, приведенной в кратком изложении существа изобретения.
Каждый R в формуле A-I, независимо от других, представляет собой водород, алкил нормального или разветвленного строения, предпочтительно содержащий 1-20 атомов углерода, например метил, этил, пропил, изопропил, бутил, втор-бутил, трет-бутил, 2-этилгексил, децил, гекасадецил, октадецил; алкоксигруппу, алкил которой содержит 1-20 атомов углерода, как описано выше для R, алкилтиогруппу, алкил которой содержит 1-20 атомов углерода, как описано выше для R, алкиламиногруппу, алкил которой содержит 1-20 атомов углерода, как описано выше для R, алкилформамидогруппу, алкил которой содержит 1- 20 атомов углерода, как описано выше для R, замещенный или незамещенный арил, предпочтительно содержащий 6-20 атомов углерода, например фенил, нафтил, фенантрил, нитрофенил, гидроксифенил, аминофенил, гексадециламинофенил, октадециламинофенил, толил, ксилил, метоксифенил. 3-амино-4-метоксифенил, 4-метокси-3- (N-метилгидразинотиоформамидо) фенил, 3-изоцианато-4-метоксифенил, 3-изотиоцианато-4-метоксифенил, метилтиофенил, карбоксифенил, и алкиларил, например алкилфенил, алкил которого содержит 1-20 атомов углерода, как описано для R, арилоксигруппа, арил которой содержит 6 -20 атомов углерода, как описано выше для R, замещенный или незамещенный гетероциклический радикал, предпочтительно содержащий в ядре 5 или 6 атомов углерода и гетероатомов, например N, S, P или O, например, пиридил, метилпиридил, нитропиридил, метоксипиридил, оксазолил, имидазолил, пиразолил и хинолил, или реагирующую с белком группу. В особенно предпочтительных вариантах R является 4-алкокси-3-аминофенилом или 4-алкокси-3-изотиоцианатофенилом.
Каждый R1, независимо от других, имеет значение, отобранное из групп, указанных для R. Предпочтительно R1 является водородом или реагирующей с белком группой.
Каждый R2, независимо от других, представляет собой гидроксигруппу, карбоксигруппу, гидроксиалкил, алкил которого предпочтительно содержит 1-4 атома углерода, например гидроксиметил, остаток карбонилиминодиуксусная кислота [-CON(CH2COOH)2] , метилениминодиуксусная кислота [-CH2N(CH2COOH)2] ; метилентиоэтилениминодиуксусная кислота [-CH2SCH2CH2N(CH2COOH)2]; карбоксиалкилтиоалкил, алкилы которого независимо содержат 1-4 атома углерода, например CH2CH2SCH2 CH2COOH; остаток 1-гидразин-2- или дендиуксусная кислота, например 1-гидразин-2-или дендиуксусная кислота [-NHN(CH2COOH)2] и 1-метил-1-гидразин-2- или дендиуксусная кислота [-N(CH3)N(CH2COOH)2]; и 2,6-дикарбокси-4-пиперидил или соли таких кислот, включая например соли таких металлов, как Na, K, Li и т.д., и соли аммония, например незамещенного аммония, тетраэтиламмония и тетраметиламмония.
Или два R2 вместе обозначают атомы, необходимые для замыкания макроциклического кольца, содержащего a) по меньшей мере один гетероатом, координирующий ионы, и b) по меньшей мере одну, предпочтительно две, алкиленовые группы, образующие часть кольца. Образующей макроциклическое ядро группой может быть замещенный (группой с гетероатомом) алкилен, например 2,2-бис-(этоксикарбонил)-1,3-пропилен; или содержащие гетероатом группы, например оксибис (алкилен), например оксибис (этилен), оксибис (этиленоксиметилен), оксибис (этиленоксиэтилен), алкиленокси алкиленоксиалкилен, например метиленоксиэтиленоксиметилен; ариленди (оксиалкилен), например 1,4-диметил-5,6-фениленбис (оксиметилен); 2,6-пиридиленбис (метиленоксиметилен); 2-метокси-5-метил-1,3-фениленбис (метиленоксиметилен) и 1,10-фенантролин-2,9-иленбис (метиленоксиметилен); карбоксиметилиминобис триметилен (карбоксиметил) иминометилен [-CH2N(CH2COOH) (CH2)3N(CH2COOH)(CH2)3 N(CH2COOH)CH2-]; карбоксиметилтиоэтилиминобис триметилен (карбоксиметилтиоэтил) иминометилен, [-CH2N (CH2CH2SCH2COOH)(CH2)3 N(CH2CH2SCH2COOH)(CH2)3N (CH2CH2SCH2COOH)CH2-] ; карбоксиметилиминобис [Этилен (карбоксиметил) иминометилен] , [-CH2N(CH2COOH)CH2CH2N(CH2COOH) CH2CH2N(CH2COOH)CH2-]; карбоксиметилтиоэтилиминобис этилен (карбоксиметилтиоэтил) иминометилен, [-CH2N(CH2CH2SCH2COOH) CH2CH2N(CH2CH2SCH2COOH) CH2CH2N(CH2CH2SCH2COOH) CH2-]; этиленбис (карбоксиметил) иминометилен, [-CH2N (CH2COOH)CH2CH2N(CH2COOH) CH2-]; карбоксиметилиминобис (метилен) [-CH2N (CH2COOH)CH2-]; или соли указанных выше групп, содержащих карбоксигруппы, включая например соли металлов и аммония, указанные для R2. В особенно предпочтительных вариантах R2 является остатком метилениминодиуксусной кислоты или его солью.
Каждый из R3 независимо от других имеет значение, отобранное из группы, указанных для R. Предпочтительно R3 представляет собой водород.
Каждый из R4 независимо от других представляет собой водород или регулирующую с белком группу.
В приведенной выше формуле A-I n = 0, 1, 2, 3 или 4, m = 0 или 1 и o = 0 или 1, причем по меньшей мере один из n и m = 0.
По меньшей мере один из R, R1, R3 и R4 реагирующей с белком является группой. Предпочтительно не более чем один из R, R1, R3 и R4 на каждом ароматическом кольце является реагирующей с белком группой. Наиболее предпочтительно, если только один из R, R1, R3 и R4 в молекуле является реагирующей с белком группой.
Термин "реагирующая с белком группа" обозначает любую группу, которая может реагировать с любыми функциональными группами, обычно присутствующими или введенными в белки. Однако, предполагается, что реагирующая с белком группа может быть конъюгирована с небелковыми биомолекулами. Таким образом, реагирующие с белком группы, применяемые на практике настоящего изобретения, включают такие группы, которые могут реагировать с любой биологической молекулой (включая углеводы, нуклеиновые кислоты и липиды), содержащей реакционноспособную группу, или группу специфического взаимодействия рецептор-лиганд, с образованием группы, связывающей комплексообразующий агент с иммунореактивной группой.
Предпочтительными, реагирующими с белками группами являются (они не ограничиваются перечисленными группами ниже): (1) группа, которая будет реагировать непосредственно с аминогруппами или меркаптогруппами белка или биологической молекулой, содержащей иммунореактивную группу, например содержащие активный галоген группы, включая например хлорметилфенилы и хлорацетил (ClCH2 CO-), активированные, 2-замещенные удаляемой группой этилсульфонилы и этилкарбонилы, например 2-хлорэтилсульфонил и 2-хлорэтилкарбонил; винилсульфонил; винилкарбонил; эпоксигруппу; изоцианатогруппу; изотиоцианатогруппу, альдегидную группу; азиридиногруппу; сукцинимидооксикарбонил; активированные ацилы, например остатки галогенагидридов карбоновых кислот или смешанных ангидридов, и другие группы, которые применяют в обычных фотографических желатиновых отвердителях; (2) группа, которая может легко реагировать с модифицированными белками или биологическими молекулами, содержащими иммунореактивную группу, т.е. белками или биологическими молекулами, модифицированными для того, чтобы они содержали реакционноспособные группы, указанные в (1), например частичным окислением белка для введения в него альдегидной группы или карбоксигруппы, в этом случае реагирующей с белком группой может быть амино-, алкиламино-, ариламино-, гидразино-, алкилгидразино-, арилгидразино-, карбазидо-, семикарбазидо-, тиокарбазидо-, тиосемикарбазидо-, меркаптогруппа, меркаптоалкил, меркаптоарил, гидрокси-, карбоксигруппа, карбоксиалкил и карбоксиарил. Алкилы реагирующей с белками группы могут содержать 1-20 атомов углерода, как описано выше для P. Арилы реагирующей с белком группы могут содержать 6-20 атомов углерода, как описано выше для R. (3) Группа, которая может соединяться с белком или биологической молекулой, содержащей иммунореактивную группу, или модифицированным белком, как указано в (1) и (2) при помощи сшивающего средства. Некоторые применяемые сшивающие средства, такие, как например дифункциональные желатиновые отвердители, бисэпоксиды и бисизоцианаты, становятся частью, т.е. соединяющей группой, в конъюгате белок - комплексообразующий агент в процессе реакции сшивания.
Другие применяемые сшивающие средства, кроме того, облегчают сшивание, например действуя в качестве расходуемых катализаторов, но не присутствуют в конечном конъюгате. Примерами таких сшивающих средств являются карбодиимидные и карбамоилониевые сшивающие средства, описанные в U. S Patent 4421847, и дикатионные простые эфиры, описанные в U.S Patent 4877724. Описание этих патентов приводится здесь в качестве ссылок. При применении этих сшивающих средств один из реагентов должен иметь карбоксигруппу, а другой должен содержать амино- или меркаптогруппу. Сшивающее средство сначала реагирует селективно с карбоксигруппой, а затем отщепляется в процессе реакции "активированной" карбоксигруппы с амином с образованием амидной связи между белком и комплексообразующими агентами формулы A-I. Таким образом ковалентно соединяются две молекулы. Преимущество этого подхода в том, что исключается сшивание подобных молекул, например комплексообразующих агентов с комплексообразующими агентами, тогда как реакция дифункциональных сшивающих средств населективна, в результате ее образуются нежелательные сшитые молекулы. Предпочтительные, реагирующие с белком группы включают амино- и изотиоцианатогруппы.
Предпочтительные комплексообразующие агенты включают соединения формул 1-59 (см. в конце описания).
Предпочтительные классы комплексообразующих агентов включают терпиридины формулы A-III и фенантролины формулы A-IV. Конкретно предпочтительный класс комплексообразующих агентов имеет формулу A-III, в которой n=1 и R представляет собой фенил, замещенный алкилом или алкоксигруппой, или реагирующую с белком группу. Предпочтительные комплексообразующие агенты включают приведенные выше соединения 20-32. Наиболее предпочтительным комплексообразующим агентом является TMT - изотиоцианат (соединение 24).
Изобретение предлагает новые терпиридины приведенной выше формулы A-III, в которой n=1 и R представляет собой радикал формулы
где
R5 является алкоксигруппой или алкилом;
p= 0, 1, 2, 3 или 4;
R6 является группой, реагирующей с белком, R5 является алкилом, предпочтительно содержащим 1-20, более предпочтительно 1-8 атомов углерода, например метилом, этилом; или алкоксигруппой, алкил которой содержит предпочтительно 1-20, более предпочтительно 1-8 атомов углерода, например метокси-, этоксигруппой. R6 является описанной выше группой, реагирующей с белком. Предпочтительные группы, реагирующие с белком, включают амино-, алкиламино-, ариламино-, карбазидо-, семикарбазидо-, тиосемикарбазидо-, тиокарбазидо, изоцианато- и изотиоцеанато группу. Особенно предпочтительные группы, реагирующее с белком, включают амино-, изотиоцианато- и семикарбоазидогруппу. Особенно предпочтительные терпиридины включают TMT (соединение 21), TMT - изотиоцианат (соединение 24) и THT соединение 20).
Предпочтительные фенантролины настоящего изобретения имеют формулу A-IV, в которой по меньшей мере один из R4 является группой, реагирующей с белком. Предпочтительные группы, реагирующие с белком, включают группы, указанные для R6.
Полипиридиновые и фенантролиновые комплексообразующие агенты, имеющие атомы или группы, образующие комплекс металлом, например гетероатомы и иминодиацетатные остатки, можно получить известными в данной области исследования методами. Подходящие схемы реакций описаны в U.S Patent 4837169 и U.S. Patent 4859777, полное описание которых, таким образом, включено здесь в качестве ссылок.
Получение некоторые предпочтительных соединений настоящего изобретения, а именно, тетранатриевой соли 4'-(3-амино-4-метоксифенил)-6,6''-бис(N',N'-дикарбоксиметил-N- метилгидразино)-2,2':6', 2''-терпиридина (THT) и тетранатриевой соли 4'-(3-амино-4-метоксифенил)-6,6''-бис-[N,N-ди(карбоксиметил)аминометил]-2,2': 6', 2''-терпиридина (TМT) показано на реакционной схеме I (см. в конце описания).
Введение в комплексообразующие агенты изобретения описанной выше необходимой группы, реагирующей с белком, можно проводить обычными химическими реакциями. Например, аминогруппы можно ввести в фенилзамещенные полипиридины и фенантролины нитрованием с последующим восстановлением нитрогрупп в аминогруппы. Если необходимо, аминогруппы можно легко превратить в изоцианатогруппы реакцией с фосгеном с образованием хлоркарбониламиногруппы с последующим превращением ее в изоцианатогруппу нагреванием соединения для выделения HC. Карбоксигруппы можно ввести обработкой аминосодержащих полипиридинов и фенантролинов реагентами, например глутаровым ангидридом, с последующим селективным активированием функциональной карбоксигруппы. Защищенные (превращением в циклические ацетали) альдегиды можно получить реакционной последовательностью, необходимой для синтеза полипиридиновых хелатообразующих агентов, и затем удалить у них защитную группу перед конъюгированием с белком.
Класс терпиридинов формулы A-III, содержащих фенил, замещенный алкилом или алкоксигруппой, и группу, реагирующую с белком, особенно благоприятен с синтетической точки зрения. Например присутствие алкила или алкоксигруппы у дибромированного полупродукта синтеза (63) обеспечивает повышенную растворимость его в ТНГ, который является предпочтительным растворителем, применяемым для получения дигидроксизамещенного полупродукта синтеза (66).
Более конкретно, ТМТ (70) является членом подкласса пиридилсодержащих хелатирующих агентов формулы A-III, а именно терпиридином формулы (71), в которой P в положении 4' может быть группой, реагирующей с белком. Действительно, это соединение является членом еще одного подкласса (71), который можно определить как соединения семейства 4' - арилзамещенных терпиридинов (72), у которых X или Y является группой, реагирующей с белками.
Несколько синтетических путей, которые можно применять для получения соединений (72), которые содержат у X или Y группу, реагирующую с белком, в частности нескольких соединений, у которых X или Y является гидроксигруппой, алкилом, алкоксигруппой или содержащей азот или кислород группой, соединенной с группой, реагирующей с белком, описаны ниже.
Соединения (71) и (72) приведены в конце описания.
Полупродуктом синтеза TMT является тетраэфир (73). Его можно омылением превратить в тетракарбоксилат (74) при помощи едкого натра и затем деметилировать с применением H и красного фосфора для получения производного фенола (75) (см. в конце описания).
Фенол (75) можно цианоэтилировать акрилонитрилом и затем обработать спиртом, например этанолом, в присутствии безводного HCl для получения амидата (76a). По другому варианту фенол в виде фенолята можно алкилировать α-циано- ω-галогеналканом и затем обработать снова спиртовым раствором HCl для получения (76b) (см. в конце описания).
Фенол (75) можно также обработать дивинилсульфоном для получения соединения (77a), которое содержит реагирующую с белком винилсульфоновую группу. По другому варианту (75) можно алкилировать ω- -гидрокси- α- -алкилгалогенидом (или сульфонатом) или ω- -гидрокси- α- -полиалкиленоксиалкилгалогенидом (или сульфонатом) и затем обработать дивинилсульфоном для образования винилсульфона (77b). Можно также превратить галогензамещенный спирт, применяемый для получения (77b) в защищенный спирт, например тетрагидропираниловый (ТНР) эфир. Свободный спирт можно выделить обработкой кеталя водной кислотой перед реакцией его с дивинилсульфоном. Реакцию с дивинилсульфоном рекомендуется катализировать основание. В (77b) m=0 или 1 и n=0 или 1-200, но будет пригодна любая оксиалкиленовая группа (см. в конце описания).
По другой синтетической схеме (75) можно алкилировать эфиром галогенуксусной кислоты, например метилбромацетатом, и затем обработать гидразином для получения гидразида (78), который способен реагировать с альдегидной группой, например введенной окислением углеводного остатка, присоединенного к антителу.
По другому варианту гидразид (78) можно обработать водной кислотой, например HCl, и затем нитритом натрия для получения карбонилизида (79). Реакцией последнего с аминогруппами белков при pH выше семи можно получить связанные амидной группой хелаты (см. в конце описания).
Фенил (75) можно также алкилировать для образования алкиленовой или оксиалкиленовой соединяющей группы, которая имеет в концевом положении гидроксигруппу или защищенную гидроксигруппу, как в случае получения винилсульфонов (77b). Алифатический спирт можно затем обработать арилхлорформиатом, например n - нитрофенилхлорформиатом или 2,4,5-трихлорфенилхлорфомиатом для получения карбоната (80). Этот арилхлорформиат может реагировать с аминогруппами белков, например аминогруппами остатков лизина и концевыми аминогруппами пептидов (см. в конце описания).
Соединение (75) можно непосредственно реакцией с хлорангидридом циануровой кислоты превратить в цианурат (81a) или реакцией спирта, который получен из (75) алкилированием, например спиртового полупродукта синтеза (80), также с хлорангидридом циануровой кислоты можно получить цианурат (81b). Эти хлорцианураты могут реагировать с аминогруппами на белках (см. в конце описания).
По другому варианту фенол (75) можно превратить в альдегид (82a) обработкой акролеином и в альдегид (82b) обработкой галогеналкилацеталем (в качестве защитной группы альдегида) с последующим удалением защитной группы водной кислотой. Эти альдегиды пригодны для реакций восстановительного аминирования (например с применением цианоборогидрида натрия в качестве восстановителя при pH в пределах 4-7, предпочтительно при pH 6, в водной системе) с целью соединения альдегида с аминогруппами на белках (см. в конце описания).
Фенол (75) можно превратить также в амин (83а) обработкой акрилонитрилом (например в присутствии N-метилимидазола, применяемого в качестве катализатора) с последующим гидрированием нитрила в теплом растворе уксусной кислоты с применением водорода и 10% палладия на угле, применяемого в качестве катализатора. Кроме того, (75) можно превратить в амин (83b) алкилированием его ω-галогеналкилнитрилом с последующим гидрированием нитрила в теплом растворе уксусной кислоты водородом и в присутствии 10% палладия на угле, применяемого в качестве катализатора (см. в конце описания).
Указанные выше амины можно присоединить к белкам несколькими путями. Они пригодны например для реакций восстановительного аминирования (например с применением цианоборогидрида натрия в качестве восстановителя при pH 6 в водной системе) с целью соединения аминогруппы с альдегидной группой, образованной действием окислителя, например периодата натрия, на углеводный остаток, присоединенный к белку (например антителу). Альдегидные группы можно также генерировать на белках с применением таких реагентов, как 4-азофенилглиоксаль (APG, доступен от Pieree Chemical Co), который реагирует селективно с остатками аргинина на белках. Эти альдегиды могут затем реагировать с амином (83a) и (83b).
Кроме того, эти амины можно присоединять к белкам после предварительной обработки обычно доступными гетеродифункциональными реагентами (например от Pieree Chemical Company). Например амины (83b) можно обработать 2-иминотиоланом для получения (84a) или N-сукцинимидил S-ацетилтиоацетатом (SATA) и затем гидроксиламином для получения (84b). Эти соединения содержат свободные меркаптогруппы (SH), которые могут реагировать с малеинимидогруппой, которую можно ввести в белки через аминогруппы при помощи таких реагентов, как SMCC [N-(4-карбоксициклогексилметил) малеинимид N-гидроксисукцинимидат] (см. в конце описания).
Кроме того, амины формулы (83b) можно обработать SMCC [N-(4-карбоксициклогексилметил) малеинимид N-гидроксисукцинимидатом] для генерирования (84c). Эти соединения содержат малеинимидогруппу, которая может реагировать со свободными меркаптогруппами (SH), которые можно ввести в аминогруппы белков с применением таких реагентов, как 2-аминотиолан, или с N-сукцинимидил-S-ацетилтиоацетатом (SATA) с последующей обработкой гидроксиламином. Свободные меркаптогруппы можно также генерировать в белках действием содержащих меркаптогруппы восстановителей, например дитиотреита, на дисульфидные связи, которые могут присутствовать в белке или его фрагменте (см. в конце описания).
В объем изобретения включено также получение некоторых соединений из амина (85), у которого X имеет указанные выше значения. Амин (85) можно обработать водной соляной кислотой и нитритом натрия (образуют HONO, который затем реагируют с амином) для получения диазониевой соли (86). Эту соль можно соединить с остатками тирозина на белках через диазосвязи к ароматическому кольцу, содержащему гидроксигруппу. Это можно схематически изобразить как соединение (87), в котором  является остатком белка, например антитела, содержащего гидроксизамещенное ароматическое кольцо тирозина (например тирозина и в антителе, например ING-1), (см. в конце описания).
является остатком белка, например антитела, содержащего гидроксизамещенное ароматическое кольцо тирозина (например тирозина и в антителе, например ING-1), (см. в конце описания).
Кроме того, амин (85) можно обработать избытком хлорангидрида циануровой кислоты для получения дихлорцианурамида (88) и дихлорангидридом 2-метоксициануровой кислоты для получения хлорметоксицианурамида (89). Оба этих соединения могут реагировать с аминогруппами, например лизина, или концевыми аминогруппами пептидных цепей в белках. Реакция идет через замещение атома хлора в кольце триазина (см. в конце описания).
Реакцией амина (85) с эфиром N-гидроксисукцинимида и α- иодуксусной кислоты или ангидридом α- иодуксусной кислоты получают иодацетамидопроизводное (90). Это производное может реагировать с белками, содержащими меркаптогруппы, например теми, которые применяют в указанных выше реакциях малеинамидопроизводного (84c). Иодацетамид (90) будет также реагировать с аминогруппами на белках, например ε- аминогруппой лизина и концевой аминогруппой пептидной цепи (см. в конце описания).
Кроме того, амин (85) можно алкилировать нитрилозамещенными алкилгалогенидами, например Br-(CH2)n-CN для получения нитрилов (91), у которых n - целое число от 1 до 20. Амин (85) можно также алкилировать нитрилозамещенными фенилгалогенидами, например α- бромметилбензонитрилом для получения нитрилов (92).
Амин (85) можно ацилировать реакционноспособными эфирами (или аналогичными активированными карбонильными соединениями) карбоновых кислот, которые содержат нитрильную группу (например цианобензоилсоединениями и производными алифатических кислот, например производными цианоуксусной кислоты) для получения нитрилоамидов (93) и (94). Любой из этих нитрилов можно превратить в соответствующие амидаты обработкой спиртом и безводным HC и получить (95), (96), (97) и (98) соответственно. Эти амидаты будут реагировать с аминогруппами белков, например ε- аминогруппой остатка лизина и концевыми аминогруппами пептидных цепей (см. в конце описания).
Следует признать, что во всех указанных выше схемах карбоксигруппы остатков иминодиуксусных кислот могут быть протонированы или могут присутствовать в виде ионных групп (например в виде натриевых солей). Степень протонирования и депротонирования любой из этих групп является функцией pH реакционной среды, которая содержит как реагент, содержащий группу, реагирующую с белком, так и белок. Применение буферных солей, например фосфатных, боратных, цитратных и ацетатных буферов, для регулирования pH реакционной среды, является частью этой особенности изобретения.
Хотя предшествующее обсуждение синтетических путей, применяемых для получения типичных соединений, имеющих группы, реагирующие с белками, было сосредоточено на некоторых предпочтительных терпиридиновых соединениях формулы 71, подразумевается, что эти пути синтеза можно применять для более широкого круга соединений, представленных более общей формулой A-I, если это нужно для целей изобретения.
Продукты реакции любого из этих хелатирующих агентов, содержащих реагирующую с белком группу, с белками (или другими иммунореактивными группами) можно очистить обычными способами, например диафильтрованием, ЖХВР, электрофорезом и подобными способами. Белки (или другие иммунореактивные группы) можно затем модифицировать агентами, например ПЭГ (полиэтиленгликолем), т.е. реагентами, хорошо известными в настоящей области исследования и применяемыми для придания пониженной иммуногенности модифицированным белкам.
Модифицированные белки можно обработать радиоизотоппами ионов металлов, например 90Y+3 (приведен в качестве неограничивающего примера) и конъюгат радионуклид-хелат-белок можно применять для лечения опухолей, в особенности, если белок является специфичным для антигена опухоли антителом или его фрагментом.
Модифицированные белки можно также обработать радиоизотопами ионов металлов, например 111In+3 или 187Y+3 (приведены в качестве неограничивающих примеров) и полученный конъюгат радионуклид-хелат-белок можно применять для получения изображения опухолей у раковых пациентов для целей диагностирования, в особенности, если белок является специфичным для антигена опухоли антителом или его фрагментом.
Новые терпиридины и фенантролины изобретения можно получить указанными выше схемами синтезов. Дополнительные синтезы этих соединений указаны в приведенных ниже примерах.
Направленный радиоактивный иммунореагент настоящего изобретения включает радионуклидный ион. Применяют например радионуклидный ион Sc, Fe, Pb, Ga, Y, Bi, Mn, Cu, Cr, Zn, Ge, Mo, Tc, Ru, In, Sn, Sm, Sb, W, Re, Po, Ta и Te. Предпочтительные радионуклидные ионы включают 44Sc+++, 64,67Cu++, 111In+++, 212Pb++, 68Ga++, 90Y+++ и 212Bi+++. Наиболее предпочтительными из этих ионов являются ионы 90Y+++.
Комплекс иона радиоактивного металла и комплексообразующего агента легко получают простым смешиванием в водном растворе комплексообразующего агента с солью радиоактивного металла в водном растворе, предпочтительно имеющем pH 4-11. Можно применять любую растворимую соль нужного металла, например галогенид. Хелат обычно получают в водном растворе при pH 5-9, предпочтительно 6-8. Комплекс можно смешать с буферами, например ацетатным, фосфатным или боратным, для обеспечивания оптимального значения pH.
Направленный иммунореагент настоящего изобретения включает иммунореактивную группу, ковалентно связанную с комплексообразующим агентом. Направленный иммунореагент, таким образом, представляет собой конъюгат комплекса формула A-I и иммунореактивной группы. Комплекс комплексообразующего агента и радионуклидного металла можно получить до или после присоединения комплексообарзующего агента к иммунореактивной группе. Термин "иммунореактивная группа" обозначает любое органическое соединение, которое способно ковалентно связываться с комплексообразующим агентом и которое найдено в живых организмах или которое применяют для установления диагноза, лечения или генной инженерии клеточного материала или живых организмов и которое обладает способностью взаимодействовать с другим компонентом, который можно найти в биологических жидкостях или который связан с клетками, которые нужно лечить, например опухолевыми клетками.
В зависимости от предлагаемого применения иммунореактивную группу можно выбрать из различных природных или синтетических полученных материалов, включая (но не ограничиваясь приведенными примерами) ферменты, аминокислоты, пептиды, полипептиды, белки, липопротеины, гликопротеины, гормоны, лекарственные средства (например дигоксин, фенитоин, фенобарбитал, тирозин, трииодтиронин, гентамицин, карбамазепин и теофиллин), стероиды, витамин, полисахариды, вирусы, простейшие, грибы, паразиты, рикетсии, плесени и их компоненты, компоненты крови, компоненты тканей и органов, фармацевтические средства, гаптены, лецитины, токсины, нуклеиновые кислоты (включая олигонуклеотиды), антитела (включая антитела, полученные с применением генной инженерии, и их фрагменты), фрагменты антител, антигенные материалы (включая белки и углеводы), авидин и его производные, биотин и его производные и другие известные в настоящей области исследования вещества.
Предпочтительными иммунореактивными группами для практического использования изобретения являются те из них, которые имеют рецепторную молекулу, специфичную для интересующего лиганда. Таким образом, реагент, принимающий участие в реакции специфического связывания, можно применять для направления в цель - мишень. Примеры таких комплексов лиганд-рецептор (они не ограничивают применение других комплексов) включают комплексы антитело-антиген, авидин-биотин, репрессор (индуктор)-промотор оперонов и сахар-лецитин. Дополнительно, комплементарные нуклеиновые кислоты, т. е. гибридизированный продукт комплементарных цепей, также считаются специфическим связывающим материалом.
В частности предпочтительные иммунореактивные группы включают (1) любое вещество, которое при попадании к иммунокомпетентному хозяину вызывает продуцирование специфических антител, способных связываться с этим веществом, или (2) таким образом полученное антитело, которое принимает участие в реакции антиген-антитело. Таким образом, иммунореактивная группа может быть антигенным материалом, антителом или антиантителом. Применяют как моноклональные, так и поликлональные антитела. Антитела могут быть целыми молекулами или различными фрагментами их, если они содержат по меньшей мере один реакционноактивный участок для реакции с реакционноспособными группами комплексообразующего агента или со связывающими группами, как описано в патенте. Предпочтительные иммунореактивные группы включают антитела и белки, полученные способами молекулярной биологии. Такие способы хорошо известны и описаны например в U.S. Patent N 4816397 и 4816567.
В некоторых примерах осуществления изобретения иммунореактивная группа может быть ферментом, который имеет реакционноспособную группу для присоединения к комплексообразующему агенту. Типичные примеры ферментов включают (но не ограничиваются ими) аспартат-аминотрансаминазу, аланинаминотрансаминазу, лактатдегидрогеназу, креатинфосфокиназу, дигидрофолатредуктазу, γ-/ глутаминтрансферазу, щелочную и кислую фосфатазу, фосфатазу простатовой кислоты, пероксидазу хрена обыкновенного и различные эстеразы.
При желании иммунореактивную группу можно модифицировать или химически изменить для образования реакционноспособных групп, необходимых для присоединения к комплексообразующему агенту известными способами. Такие способы включают применение соединяющих остатков и химическую модификацию, например описанных в WO-A-89/02931 и WO-A-89/02932 (предложены способы модификации олигонуклеотидов) и U.S. Patent 4719182. Полное описание их, таким образом, приводится здесь в качестве ссылок.
Два наиболее предпочтительных направления применения направленных иммунореагентов настоящего изобретения - это получение изображений опухолей для целей диагностирования и радиологическое лечение опухолей. Предпочтительные иммунологические группы их включают антитела на связанные с опухолями антигены. Конкретные примеры их включают антитела B 72.3 (описаны в U.S. Patent N 4522918 и 4612282), которые распознают колоректальные опухоли, антимеланомные антитела 9.2.27, антитела D612, которые распознают колоректальные опухоли, антитела UJ13A, которые распознают мелкоклеточный рак легких, антитела NRLU-10 (Tfs -2), которые распознают мелкоклеточный рак легких и колоректальные опухоли, антитела 7E11C5, которые распознают опухоли простаты, антитела CC49, которые распознают колоректальные опухоли, антитела TNT, которые распознают некротическую ткань, антитела PR1A3, которые распознают карциному ободочной кишки [Richman, P.I. и Bodmer, W.F. 1987) Int. J. Cancer 39, pp, 317 - 328], антитела ING-1 и другие полученные с применением генной инженерии антитела, которые описаны в Международной патентной публикации WO-A-90/02569, антитела B174 (разработаны в Biomira, Inc. of Edmonton, Canada), которые распознают карционому сквамозных клеток, антитела B43, которые взаимодействуют с некоторыми лимфомами и лейкозами, и другие антитела, которые могут иметь практический интерес.
Такие антитела и другие применяемые иммунологические группы, описанные выше, являются большими, сложными молекулами, имеющими несколько сайтов для присоединения комплексообразующего агента. Следовательно, иммунореактивная группа может иметь присоединенные к ней дополнительные комплексообразующие агенты через одну из реагирующих с белками групп. Таким образом, термин иммунореактивная группа предполагает включение иммунологических групп, имеющих молекулы комплексообразующих агентов, присоединенных к ним через одну или несколько реагирующих с белком групп.
Дополнительно, антитела или их фрагменты, содержащие углеводную область, могут быть соединены с комплексообразующим агентом через углеводную область антитела, например как описано в U.S. Patent 4937183, полное описание которого, таким образом, приводится здесь в качестве ссылки. Применяемые способы присоединения антител описаны также в U. S. Patent N 4671958; 4699784; 4941900 и 4867973. Термин "реагирующая с белком группа", как определено в них, предполагает включение таких соединений.
Известны другие способы проведения ковалентного связывания иммунореактивной группы с агентами, образующими комплекс с радиоактивными металлами, они включают простое смешивание реагентов.
Радиоактивный иммунореагент настоящего изобретения может иметь любые соотношения иона радионуклидного металла и комплексообразующего агента. В предпочтительных вариантах молярное соотношение иона металла и комплексообразующего агента находится в пределах 1:100 - 1:1.
Соотношение комплексообразующего агента и иммунореактивной группы может быть в широких пределах от 0,5 : 1 до 10 : 1 или более. В некоторых примерах осуществления изобретения молярное соотношение комплексообразующего агента и иммунореактивных групп находится в пределах от 1:1 до 6:1.
На фиг. 6 и 7 приведены спектры поглощения комплексообразующих агентов и комплексов их с металлами настоящего изобретения. Часть спектров не перекрывается со спектрами белков, к которым комплексообразующие агенты присоединены химически. Аналогичные спектральные сдвиги были получены для комплексообразующих агентов настоящего изобретения и других типичных катионов, например Ga+3, Bi+3, In+3, Sc+3 и Cu+2. Таким образом, иммунореагент изобретения можно легко анализировать спектрофотометрически.
Раздел I: Комплексообразующие агенты/хелатирующие агенты
Получение 1 - Получение 4'-(3-амино-4-метоксифенил)-6,6''- [N, N-ди(карбоксиметил)аминометил] -2,2': 6', 2'' - терпиридина в виде тетранатриевой соли (ТМТ)
Часть A - Бромид пиридиния, 61.
2-Ацетил-6-бромпиридин (60) синтезировали по методу J.E. Parks, B.E. Wagner и R. E. Holm, Organometal. Chem. 56, 53 - 66 (1973). 2-Ацетил-6-бромпиридин (20,0 г, 100 ммоль) обработали бромом (6,2 мл, 0,12 моль) при кипячении в 200 мл CHCl3 в течение 45 мин. Раствор охладили до комнатной температуры и затем промыли разбавленным водным раствором NaHCO3 Na2S2O3. Органическую фазу сушили над Na2SO4, фильтровали и выпаривали для получения масла. Масло растворили в 200 мл ТГФ и добавили 30 мл пиридина. Полученный раствор кипятили в течение 30 мин. Смесь охладили и фильтровали для получения 26,1 г порошка нестандартного белого цвета (нечистого белого цвета) (73%) с т.пл. 256oC (разложение, изменяет цвет при 245oC).
Элементный состав для C12H10Br2N2O :
Рассчитано: C 40,26; H 2,82; N 7,82.
Найдено: C 40,12; H 2,85; N 7,79.
Спектры ЯМР и ИК были совместимы с предполагаемой структурой. Продукт был гомогенный при анализе ТСХ.
Часть B - Халкон, 62.
Едкий кали (18,2 г, 325 ммоль) растворили в 100 мл H2O, в раствор добавили 100 мл метанола. 2-Ацетил-6-бромпиридин (65 г, 325 ммоль) и n - анисовый альдегид (68 г, 650 ммоль) растворили вместе в 400 мл метанола и раствор вылили в раствор КОН. Осаждение продукта началось через несколько минут. Реакционную смесь оставили при комнатной температуре на ночь. Затем осадок отделили, промыли изопропанолом и сушили для получения 79 г, (76%) желтого твердого продукта с т. пл. 100 - 102oC. FDMS (т/е) 317 м. Аликвоту очищали колоночной хроматографией на силикагеле Woem, элюировали 100%-ным дихлорметаном.
Элементный состав для C15H12BrNO2:
Рассчитано: C 56,63; H 3,80; N 4,40.
Найдено: C 56,66; H 3,87; N 4,41.
ЯМР - и ИК-спектры были совместимы с предполагаемой структурой. Продукт был гомогенный при анализе ТСХ.
Часть C - Дибромтерпиридин, 63.
Бромид пиридиния, 61 (11,3 г, 31,6 ммоль) и халкон, 62 (10,0 г, 31,4 ммоль) кипятили в 100 мл уксусной кислоты с 10 г ацетата аммония 16 ч. Раствор охладили и фильтровали. Твердую часть промыли уксусной кислотой, затем этанолом для получения 13,48 г белых кристаллов (86%) с т.пл. 203 - 204oC.
FDMS (m/e) 495 (M+).
Элементный состав для C22H15Br2N3O:
Рассчитано: C 53,1; H 3,0; N 8,5.
Найдено: C 52,9; H 3,1; N 8,4.
ЯМР - и ИК-спектры были совместимы с предполагаемой структурой. Продукт был гомогенный при анализе ТСХ.
Часть D - Термиридиндиол, 66.
Дибромид 63 (7,46 г, 15,5 ммоль) в 100 мл сухого ТГФ добавили по каплям в раствор 28,1 мл 1,6 М н-бутиллития в 20 мл сухого ТГФ в атмосфере азота в течение 12 мин. Температуру во время добавления поддерживали ниже - 75oC при помощи бани ацетона с сухим льдом. Полученный темно-зеленый раствор перемешивали 10 мин затем в течение 2 мин добавляли 7,5 мл сухого диметилформамида. Через 10 мин добавили 90 мл 10%-ного раствора HCl и полученный раствор перемешивали 45 мин при постоянном охлаждении. Смесь распределили между CHCl3 и H2O (оба растворителя предварительно охладили до 4oC) в разделительной воронке. Фазы, как обычно, встряхивали и затем оставили на 15 - 90 мин при комнатной температуре. За это время цвет органической фазы постепенно менялся от зеленого до золотисто-желтого. Органическую фазу промыли насыщенным раствором NaCl, затем выпаривали для выделения окрашенного в кремовый цвет остатка. Остаток растирали с CH3CN для получения белого твердого не чисто-белого продукта (3,53 г, 60%) т. пл. 225 - 227oC.
FDMS (m/e) 395 М.
Элементный состав для C24H17N3O:
Рассчитано: C 72,90; H 4,33; N 10,63.
Найдено: C 72,44; H 4,31; N 10,56.
Неочищенный диальдегид (3,53 г, 8,93 ммоль) кипятили с 1 г NaBH4 в смеси 70 мл ТГФ и 70 мл абсолютного этанола в атмосфере азота 15 мин. После концентрирования в вакууме остаток кипятили 30 мин в разбавленном растворе NaHCO3, охладили, фильтровали, промыли H2O и затем сушили для получения диола 66 в виде белого твердого вещества (3,35 г, 94,4%) с т.пл. 187 - 189oC.
FDMS (m/e) 400 MH+, 399 М.
Элементный состав для C24H21N3O3:
Рассчитано: C 72,17; H 5,30; N 10,52.
Найдено: C 71,71; H 5,20; N 10,37.
ЯМР - и ИК-спектры были совместимы с предполагаемой структурой. Анализ ТСХ показал, что продукт гомогенный.
Часть E - Тетраэфир, 67.
Диол 66 (15,4 г, 38,5 ммоль) суспензировали в смеси 17 мл триэтиламина и 175 мл CH2Cl2 при перемешивании при 8oC. В суспензию в течение 10 мин по каплям добавили раствор (CH3SO2)2O (16,8 г, 96,5 ммоль) в 50 мл CH2Cl2. Реакционную смесь встряхивали с водой. Органический слой сушили над MgSO4, фильтровали и концентрировали почти досуха. После добавления этилацетата бисмезилат выделился в виде белых кристаллов, которые отделили и сушили (17,2 г, 80,4%). Смесь бисмезилата (0,50 г, 0,96 ммоль), диизопропилэтиламина (0,26 г, 2,0 ммоль) и диэтилиминодиацетата (0,38 г, 2,0 ммоль) перемешивали в 20 мл сухого ДМФ 16 ч. Смесь концентрировали в вакууме и остаток распределили между диэтиловым эфиром и H2O. Эфирную фазу промыли два раза водой и затем сушили над Na2SO4 и выпаривали для получения продукта в виде бледно-желтого масла (0,58 г, 82%).
Элементный состав для C40H47N5O9:
Рассчитано: C 64,76; H 6,39; N 9,44.
Найдено: C 64,35; H 6,17; N 9,39.
ЯМР - и ИК-спектры были совместимы с предполагаемой структурой. Анализ ТСХ показал, что продукт гомогенный.
Часть F - Нитротетраэфир, 68.
Тетраэфир 67 (3,27 г, 4,11 ммоль) растворили в 60 мл концентрированной H2SO4 для образования красно-оранжевого раствора. Смесь охладили до 0oC и добавили смесь (1: 10, объемные части) концентрированной HNO3 и концентрированной H2SO4 в таком количестве, чтобы 4,41 ммоль HNO3, попало в смесь. Цвет раствора после добавления стал бледно-желтым. Реакционную смесь перемешивали 15 мин при 0oC, затем вылили в измельченный лед. Добавили разбавленный раствор K2CO3 до достижения pH 8. Водный раствор экстрагировали три раза CH2Cl2. Органические слои объединили, высушили над Na2SO4 и концентрировали до образования желтого масла, которое хроматографировали на силикагеле (5% CH3 OH/CH2Cl2). Фракции, содержащие продукт, объединили и выпаривали для получения продукта, который перекристаллизовали три раза из метанола для получения нечисто-белого твердого продукта (1,84 г, 53%) с т.пл. 76 - 79oC.
FDMS (m/e) 787 MH+, 786 М.
Элементный состав для C40H46N6O11
Рассчитано: C 61,06; H 5,89; N 10,68.
Найдено: C 60,69; H 6,22; N 11,04.
ЯМР и ИК-спектры совместимы с предполагаемой структурой. Анализ ТСХ показал, что продукт гомогенный.
Часть G - Аминотетраэфир, 69.
Нитротетраэфир 68 (1,80 г, 2,29 ммоль) растворили в смеси 90 мл ТГФ и 90 мл абсолютного этанола. Добавили формиат аммония (2,89 г, 45,8 ммоль), растворенный в 16 мл H2O, затем 4,8 г 10% Pd/C (4,6 ммоль). После перемешивания при комнатной температуре в течение 2 ч реакционную смесь фильтровали через фильтр из диатомовой земли. Фильтр хорошо промыли ТГФ, абсолютным этанолом и CH2Cl2. Фильтрат концентрировали и остаток распределили между CH2Cl2 и водным раствором NaCl. Органическую фазу концентрировали, затем очищали на силикагеле с применением 10% метанол/ CHCl3 для получения продукта в виде масла соломенного цвета (0,80 г, 46%) FDMS (m/e) 757 MH+, 756 М.
Элементный состав для C40H48N6O9• 1/2H2O:
Рассчитано: C 62,73; H 6,45; N 10,97.
Найдено: C 62,98; H 6,47; N 10,67.
ЯМР - и ИК-спектры совместимы с предполагаемой структурой. Анализ ТСХ показал, что продукт гомогенный.
Часть H - Аминотетракислота, 70 (ТМТ).
Аминотетраэфир 69 (0,75 г, 0,99 ммоль) перемешивали с 4 экв. NaOH в смеси 50 мл метанола и 2 мл H2O в течение 16 ч при комнатной температуре. Смесь концентрировали для выделения продукта в виде твердого дигидрата (0,72 г, 94%).
FABMS (m/e) 640 (M+ для тетракарбоксилата).
Элементный состав для C32H28Na4O9 • 2H2O:
Рассчитано: C 50,01; H 4,20; N 10,9
Найдено: C 49,82; H 4,12; N 10,74.
ЯМР и ИК-спектры совместимы с предполагаемой структурой. Анализ ТСХ показал, что продукт гомогенный.
Получение 1a - Альтернативный способ получения 4'-амино-(4-метоксифенил)-6,6''-[N, N-ди(карбоксиметил)аминометил]- 2,2' : 6',2'' - терпиридина в виде тетранатриевой соли (ТМТ)
Стадия 1 - 2-Ацетил-6-бромпиридин, 60.
В трехгорловую колбу объемом 5 л загрузили 2,6-дибромпиридин (181 г, 0,75 моль) и 2,0 л эфира. Суспензию перемешивали в атмосфере азота при -60oC и затем добавили в течение 20 мин н-бутиллитий (300 мл, 2,5 М) из капельной воронки с уравновешенным давлением. Баню опустили и раствор оставили для нагревания до -50oC. Через 15 мин темно-желтый раствор снова охладили до - 78oC в бане эфира с сухим льдом и добавили диметилацетамид (70 г, 0,86 моль) в 70 мл эфира с такой скоростью, чтобы температура не поднималась выше - 75oC. Добавление занимало около 1 ч, после чего баню удалили и оставили смесь нагреваться до -60oC. Раствор хлористого аммония (50 г, 0,93 моль) в 150 мл H2O добавили по каплям в течение 10 мин в атмосфере азота и температуру повысили до -40oC. Смесь перемешивали 45 мин и слой разделили. Водный слой снова экстрагировали 500 мл эфира и объединенный органический слой промыли два раза H2O и один раз насыщенным раствором соли. Органический слой сушили над MgSO4 и после выпаривания его досуха получили 150 г желтого масла. Это масло после смешивания с другой порцией продукта в таком же количестве перегоняли при 0,1 мм рт.ст., собирая кипящее при 80oC бесцветное масло в количестве 257 г (86%), которое легко кристаллизируется при охлаждении. Анализ ТХС проводили с применением силикагеля и смеси гексана и эфира (90 : 10).
Стадия 2 - Иодид 1-2-(6-бром-2-пиридил)-2-оксоэтил пиридиния.
В перемешиваемый раствор 2-ацетил-6-бромпиридина (100 г, 0,5 моль) в 600 мл пиридина добавили иод (127 г, 0,5 моль). Смесь нагревали на паровой бане 45 мин и продукт кристаллизовали при охлаждении. Твердую часть отделили фильтрованием и бледно-желтую кристаллическую лепешку два раза промыли хлористым метиленом. После сушки получили 183 г (90%) продукта. Т.пл. 176-178oC. Анализ ТХС проводили на силикагеле с применением смеси ацетона и изопропанола (95:5).
Стадия 3 - 1-(2-Бромпиридил)-3-(4-метоксифенил)-2-пропенон, 62.
В перемешиваемый раствор 2-ацетил-6-бромпиридина (50 г, 0,25 моль) в 450 мл метанола добавили 4-анисовый альдегид (48 г, 0,35 моль). Смесь охладили на водяной бане до 20oC и затем быстро добавили раствор KOH (18 г, 0,28 моль) в 100 мл H2O. Через 30 с продукт закристаллизовался и температура поднялась до 35oC. Светло-желтую смесь после перемешивания в течение 30 мин отфильтровывали. Лепешку промывали два раза изопропанолом и после сушки получили 68 г (86%) продукта. Т.пл. 106,1 - 106,5oC. Анализ ТСХ проводили на силикагеле с применением гексана и этилацетата (90:10).
Стадия 4 - 6,6''-Дибром-4'-(4-метосифенил)-2,2':6',2''-терпиридин, 63.
Смесь халкона (продукт стадии 3) (64 г, 0,20 моль), иодида, пиридиния (продукт стадии 2) (81 г, 0,20 моль) и ацетата аммония (77 г, 1,0 моль) в 600 мл уксусной кислоты нагревали при 95oC 3 ч и затем выдерживали в течение ночи при 60oC. Реакционную смесь охлаждали до 15oC и кристаллическую лепешку три раза промыли водой. После сушки в течение ночи получили 76,5 г (83%) бледно-желтого кристаллического продукта. Т.пл. 205-206oC. Анализ ТХС проводили на силикагеле с применением гексана и этилацетата (90:10). Определение методом Карла Фишера показало, что продукт содержит 0,02% H2O.
Стадия 5 - 4'-(4-Метоксифенил)-2,2':6',2''-терпилидин-6,6''-дикарбоксальдегид.
В колбу объемом 3 л при перемешивании в атмосфере азота загрузили 400 мл ТГФ, охладили ее в бане эфира с сухим льдом до -78oC. Раствор н-бутиллития (55 мл, 2,5 М) перенесли при помощи азотного насоса в капельную воронку с уравновешенным давлением и добавили в колбу с ТГФ. После перемешивания в течение 15 мин температуру смеси установили - 78oC в течение 11 мин по частям добавили (при помощи трубки Gooch) твердый бисбромид (продукт стадии 4) (21 г, 42 моль). Температура в процессе добавления не превышала -75oC. Постепенно цвет суспензии изменялся от коричневого до коричневато-зеленого, но твердое соединение было еще видно через 10 мин. Баню опустили и смесь оставили для нагревания до -70oC в течение 10 мин. Темно-зеленый раствор снова охладили до -78oC и в течение 3 мин при помощи азотного насоса добавили охлажденный (-15oC) ДМФ (диметилформамид) 20 (мл) в 15 мл ТГФ. Температура поднялась до -60oC, после перемешивания в течение 10 мин в смесь (при -72oC) для прекращения реакции в течение 10 мин добавили водный раствор HCl (80 мл HCl разбавили до 240 мл H2O). В процессе добавления кислоты цвет смеси изменился до темно-желтого, что является индикатором образования продукта. Температуру подняли до -35oC и зелено-желтый продукт перемешивали 45 мин без наружного охлаждения. Затем в реакционную смесь добавили 2 л H2O и перемешивали еще 2,5 ч. Желтую суспензию фильтровали через бумажный фильтр грубой очистки и желтый осадок промывали 400 мл H2O. После просушивания в течение ночи получили неочищенный кристаллический продукт с выходом 72-79%. Очищенная проба (перекристаллизованная из этилацетата) имела т.пл. 228-229oC. Анализ ТСХ проводили на силикагеле с применением смеси толуола, метанола и изопропиламина (94:4:2).
Стадия 6 - 6,6''-Бис(гидроксиметил)-4'-(4-метоксифенил)-2,2':6,2''-терпиридин, 66.
В перемешиваемую суспензию бисальдегида со стадии 5 (18,5 г, 46,8 ммоль) в 250 мл абсолютного этанола при комнатной температуре добавили постепенно NaBH4 (2,3 и 60,8 ммоль) в течение 10 мин. Кипячением смеси в течение 15 мин получили желтый раствор, который перемешивали дополнительно в течение 30 мин и затем разбавили 150 мл H2O, в результате чего выпал осадок. После перемешивания еще 1,5 ч смесь отфильтровали.
Кристаллы рыжевато-коричневого цвета после высушивания весили 16 г (86%). Неочищенный кристаллический продукт применяли без дальнейшей очистки в следующей стадии. Т.пл. 140-141oC.
Стадия 7 - Бисмезилат терпиридина стадии 6.
Суспензию бис (гидроксиметил) терпиридина со стадии 6 (15,4 г, 38,5 ммоль) в 175 мл CH2Cl2 и триэтиламина (17 мл, 120 ммоль) перемешивали и охлаждали до 8oC. По каплям в течение 10 - 15 мин добавили раствор ангидрида метансульфокислоты (16,8 г, 96,5 ммоль) в 50 мл CH2Cl2. Смесь постепенно превращалась в раствор и почти в конце добавления наблюдали изменение цвета ее из красно-оранжевого в желто-зеленый. Анализ продукта ТХС на силикагеле с применением системы толуол-метанол-изопропиламин в соотношении 92:6:2 показал, что он не содержит исходных соединений и образует только одно пятно. Смесь вылили в 200 мл H2O и образованные слои разделили. Водный слой экстрагировали CH2Cl2 и объединенный органический слой промыли H2O. Фильтрованием через слой MgSO4 и 3/4'' - силикагеля получили почти бесцветный фильтрат, который концентрировали досуха. Неочищенную смесь разбавили этилацетатом и кристаллическую взвесь охладили и затем профильтровали. После сушки в течение ночи получили 17,2 г (82%) белых кристаллов с т.пл. 190-192oC.
Стадия 8 - Тетраэфир, 67.
В раствор бисмезилата стадии 7 (32,5 г, 58,5 ммоль) в 180 мл N-метилпирролидинона (NMP) и этилдиизопропиламинамина (17 г, 131 ммоль) добавили диэтилиминодиацетат (25 г, 131 ммоль). Смесь нагревали на паровой бане 35 мин и затем перемешивали в течение ночи при комнатной температуре. После выливания реакционной смеси в 300 мл H2O смесь экстрагировали этилацетатом (2 • 250 мл) и затем экстракт промыли H2O. Органический слой концентрировали в вакууме и сушили под высоким вакуумом для получения светло-красного сиропа (51,7 г). ЯМР - спектр (DMSO) были совместимы с целевым продуктом и NMP. Сироп фильтровали через короткую колонку с силикагелем и элюировали смесью эфира и гексана (80:20) для получения прозрачного бесцветного сиропа (41 г). Анализ его TCX с применением смеси эфира, гексана и триэтиламина (60:38:2) показал, что продукт чистый.
Стадия 9 - Нитротетраэфир, 68.
Раствор тетраэфира стадии 8 (тетраэфир 67) (3,1 г, 40,6 ммоль) в 160 мл трифторуксусной кислоты охладили до 15oC и добавили в него нитрат калия (4,4 г, 44 ммоль) в виде одной порции. Смесь перемешивали в течение нескольких минут и затем по каплям в течение 15 мин добавили концентрированную H2SO4 (22 г, 230 ммоль). Наблюдалось слабое выделение тепла. В конце добавления температура смеси поднялась до 20oC. Прозрачную желтую смесь перемешивали 30 мин. Анализ ее ТСХ показал, что она не содержит исходное соединение. Основную часть трифторуксусной кислоты отделили перегонкой при пониженном давлении и остаток декантировали в 500 мл 10% K2CO3 и 250 мл этилацетата. Слои разделили и переэкстрагировали этилацетатом. Объединенный органический слой промыли H2O, затем раствором соли и сушили над MgSO4. Фильтрат концентрировали почти досуха и затем растворили в 90 мл горячего абсолютного этанола. Смесь охладили в бане со льдом. Кристаллы отделили фильтрованием и промыли этанолом. После сушки в вакууме в течение ночи получили 29 г (90%) белого кристаллического продукта. Т.пл. 81 - 82oC. Анализ TCX проводили с применением смеси эфира, гексана и триэтиламина (70:25:5).
Стадия 10 - Аминотерпиридинтетраэфир, 69.
В раствор нитротетраэфира 68 (продукт стадии 9) (28,3 г, 36 ммоль) в 700 мл смеси (1: 1) ТГФ и абсолютного этанола в атмосфере аргона добавили 10% Pd/C (10 г, 10 ммоль). После перемешивания в течение нескольких минут по каплям в течение 3 - 5 мин добавили формиат аммония (9,4 г, 149 ммоль). В течение 10 мин наблюдалось некоторое пенообразование у вихревой воронки мешалки и слабое нагревание реакционной смеси от 22 до 28oC. Анализ ТСХ показал, что после 1 ч прореагировало около 70% реагентов. Реакционную смесь перемешивали дополнительно 1 ч и затем кипятили на паровой бане 1/2 ч. Перемешивание продолжали еще 1 ч и затем реакционную смесь охлаждали до 20oC. После охлаждения смесь осторожно фильтровали через слой солкафлока и катализатор на фильтре промыли абсолютным этанолом общим объемом 400 мл. Слабо-желтый фильтрат выпаривали досуха в вакууме для получения 25,3 г (выход 95%) продукта. В результате дальнейшего промывания катализатора 150 мл ДМФ после выпаривания получили также 1,4 г темного сиропа (выход 5%), который не отличался значительно от продукта, полученного при промывании спиртом. Объединенный продукт очищали флаш-хроматографией с применением колонки, заполненной (4,5''• 2,5'') силикагелем 60 (EM Science) в атмосфере азота с элюированием гексаном, содержащим 1% триэтиламина. Продукт растворили в минимальном количестве толуола и ввели сверху колонки. Колонку элюировали смесью эфира и гексана (30: 70) при скорости течения около 100 мл/мин. При постепенном повышении полярности до применения смеси эфира и гексана в соотношении 70: 30 и 1% триэтиламина получили около 15 г бесцветного сиропа, который при анализе ТСХ образовал одно пятно. Для анализа применяли смесь эфира, гексана, метанола и триэтиламина (70:25:4:1).
Стадия 11 - ТМГ, 70.
В раствор аминотерпиридинтетраэфира 69 стадии 10 (39,0 г, 51,4 ммоль) в 450 мл абсолютного этанола добавили раствор NaOH (8,39 г/ 100 мл трижды перегнанной воды). После перемешивания 1 мин прозрачный бледно-желтый раствор стал мутным и постепенно образовался осадок. Смесь отфильтровали через два часа и полученное светло-желтое твердое вещество промыли два раза этанолом и один раз гексаном. Исследование поляризационным микроскопом подтвердило его кристаллическую структуру. После просушивания его в вакууме в течение 15 ч получили 33,9 г, (выход 90%) продукта. ЯМР-, ИК- и УФ - спектры его совместимы с предполагаемой структурой. Анализ ТСХ с применением PAW и ацетона в соотношении 40:10 (PAW обозначает смесь пиридина, уксусной кислоты и воды в соотношении 54:16:30) показал, что продукт однородный. Анализ продукта методом Карла Фишера показал, что он содержит 6,18% H2O. Т. пл. его выше 290oC. Содержание в продукте этанола, применяемого в качестве растворителя (определяли газовой хроматографией, составило 0,07%.
Получение 2 - Получение 4'-(3-амино-4-метоксифенил)-6,6''-бис- (N',N'-дикарбоксиметил-N-метилтетразино)-2,2' : 6', 2''-терпиридина в виде тетранатриевой соли, (ТНТ), 65.
Часть A - 6,6''-Бис(N-метилгидразино)-4'-(4-метоксифенил)- 2,2' : 6', 2''-термиридин.
6,6''-Дибром-4'-(4-метоксифенил)-2,2': 6', 2''-терпиридин, 63 (1,0 г, 2 ммоль) кипятили в 20 мл метилгидразина в течение 16 ч в атмосфере азота. Раствор охладили и образованный осадок отделили фильтрованием и сушили до постоянного веса. Получили 0,68 г твердого продукта кремового цвета с т.пл. 218 - 220oC.
EDMS (m/e) 428 MH+, 427 M+.
Элементный состав для C24H25N7O • 0,25H2O:
Рассчитано: C 66,72; H 5,96; N 22,70.
Найдено: C 66,85; H 5,95; N 23,0.
ЯМР - ИК-спектры его были совместимы с предполагаемой структурой.
Часть B - 6,6''-Бис-(N',N'-ди(этоксикарбонилметил)-N- метилгидразино-4'-(4-метоксифенил)-2,2':6',2''-терпиридин.
Бис (метилгидразино) терпиридин части A (3,50 г, 82 ммоль) этилбромацетат (13,2 мл, 820 ммоль), 2,6 - лутидин (9,6 мл, 820 ммоль) и иодид натрия (0,35 г, 2 ммоль) добавили в 350 мл ацетонитрила и раствор кипятили 48 ч в атмосфере азота, когда было добавлено дополнительно 4,1 мл (370 ммоль) этилбромацетата и 4,8 мл (410 ммоль) 2,6 - лутидина. Реакционный раствор кипятили еще 48 ч и охладили. Большое количество белой соли, образованной из избытка бромацетата и лутидина, отделили фильтрованием. Фильтрат концентрировали и концентрат растворили в дихлорметане. Раствор экстрагировали два раза разбавленным водным раствором хлористого натрия. Органическую фазу концентрировали в вакууме до образования масла без запаха бромацетата и лутидина. Масло очищали на колонке (91 • 10 см) с силикагелем Woelm. Колонку элюировали сначала 100%-ным дихлорметаном, затем смесью дихлорметана и ацетона (50: 1) с постепенным увеличением концентрации ацетона до соотношения дихлорметана и ацетона 25:1. Концентрированием очищенных фракций получили 2,91 г (46%) масла, окрашенного в светло-соломенный цвет. Фракция очищенного масла при стоянии при комнатной температуре кристаллизовалась и после растирания с метанолом образовала белый твердый продукт с т.пл. 100 - 103oC.
Элементный состав для C40H49N7O9:
Рассчитано: C 62,24; H 6,40; N 12,70.
Найдено: C 62,31; H 6,32; N 12,69.
ЯМР - ИК-спектры его совместимы с предполагаемой структурой, Анализ продукта ТСХ показал, что продукт однородный.
Часть C - 6,6''-Бис(N',N'-ди(этоксикарбонилметил)-N- метилгидразино-4'-(4-метокси-3-нитрофенил)-2,2':6',2''-терпиридин.
Терпиридинтетраэфир части B (0,659 г, 0,84 ммоль) растворили в 5 мл концентрированной H2SO4 и получили красно-оранжевый раствор, который охладили до 0oC и добавили смесь концентрированной HNO3 и концентрированной H2SO4 в таком количестве, чтобы 0,84 ммоль HNO3 было в растворе. После полного добавления цвет раствора стал бледно-желтым. Реакционную смесь перемешивали 15 мин при 0oC и затем вылили в раскрошенный лед. В смесь добавили разбавленный раствор K2CO3 до достижения pH 8. Водный раствор экстрагировали три раза CH2Cl2. Органические слои объединили и сушили над Na2SO4, затем концентрировали для образования желтого масла, которое хроматографировали на силикагеле (применяли смесь хлористого метилена и ацетона в соотношении 20:1). Фракции, содержащие продукт, объединили и выпаривали для получения продукта в виде бледно-желтого стеклообразного вещества. Выход 0,15 г (22%).
Элементный состав для C40H48N8O11:
Рассчитано: C 58,82; H 5,92; N 13,72.
Найдено: C 58,76; H 5,61; N 13,84.
FDMS (m/e) 316 М.
ЯМР - и ИК-спектры его совместимы с предполагаемой структурой. Анализ продукта ТСХ показал, что он однородный.
Часть D - 4'-(3-Амино-4-метоксифенил)-6,6''-бис(N',N'-ди (этоксикарбонилметил)-N-метилгидразино-2,2' : 6', 2''-терпиридин
Нитротетраэфир части C (0,72 г, 0,88 ммоль) растворили в смеси 30 мл ТГФ и 30 мл этанола. В раствор добавили формиат аммония (1,08 г, 17,7 ммоль), растворенный в 6 мл воды, и затем 1,8 г 10% Pd/C (1,7 ммоль). После перемешивания при комнатной температуре в течение ночи реакционную смесь фильтровали через слой диатомовой земли. Слой фильтра промыли тщательно ТГФ, абсолютным этанолом и CH2Cl2. Фильтрат концентрировали и остаток растворили в хлороформе. Раствор в хлороформе промыли два раза водой. Органическую фазу концентрировали до образования масла. Добавлением в масло этанола вызвали его кристаллизацию. Смесь смешали с 20 мл метанола и фильтровали. Твердую часть сушили на воздухе, и получили 0,64 г твердого продукта кремового цвета в количестве 0,64 г (92,7%).
Элементный состав для C40H50N8O9:
Рассчитано: C 61,06; H 6,40; N 14,24.
Найдено: C 60,74; H 6,39; N 14,01.
ЯМР - и ИК-спектры его совместимы с предполагаемой структурой. Анализ продукта ТСХ показал, что он однородный.
Часть E - 4'-(3-Амино-4-метоксифенил)-6,6''-бис(N', N'- дикарбоксиметил-N-метилгидразино)-2,2': 6', 2'' - терпиридин в виде тетранатриевой соли (ТНТ), 65.
Аминотетраэфир части D (0,60 г, 0,76 ммоль) перемешивали с 4 экз. NaOH в смеси 25 мл метанола и 1 мл H2O в течение 16 ч при комнатной температуре. Смесь концентрировали для выделения твердого тетракарбоксилата с количественным выходом.
Элементный состав для C32H30N8Na4O9 • 2H2O:
Рассчитано: C 48,12; H 4,29; N 14,03.
Найдено: C 48,01; H 3,97; N 12,77.
Получение 3 - Получение 6,6''-бис(N',N'-дикарбоксиметил-N- метилгидразино)-4'-(3-изоцианато-4-метоксифенил)-2,2': 6', 2'' - терпиридина в виде натриевой соли.
Тетранатриевую соль (получение 2, часть E) (0,39 г, 0,51 ммоль) растворили в 80 мл метанола. При комнатной температуре добавили 0,58 г (0,50 ммоль) тиофосгена в 1,0 мл тетрагидрофурана и затем 0,51 г (0,50 ммоль) триэтиламина в 1,0 мл тетрагидрофурана. Реакционную смесь концентрировали и остаток после смешивания с дихлорметаном отфильтровали для выделения 0,26 г (65%) желтого твердого продукта. ИК - спектр его совместим с предполагаемой структурой. Считают, что продукт является тринатриевой солью (одна карбоксигруппа свободна).
Получение 4 - Получение 5-амино-2,9-бис[N,N-ди(карбоксиметил)-аминометил]-1,10-фенантролина в виде тетранатриевой соли.
Часть A - 2,9-Диметил-5-нитро-1,10-фенантролин.
Полугидрат хлоргидрата неокупроина (25,0 г, 99 ммоль) растворили в 100 мл концентрированной азотной кислоты. Затем добавили 200 мл концентрированной серной кислоты и реакционную смесь нагревали 2,5 ч и после этого реакционную смесь оставили на 8 дней при комнатной температуре. Затем реакционную смесь постепенно добавили в смесь 3 кг льда и 351 г (8,8 моль) LiOH•H2O при перемешивании стеклянной палочкой. В процессе нейтрализации, если необходимо, добавляли лед таким образом, чтобы в процессе нейтрализации всегда присутствовал нерасплавленный лед. Реакционную смесь при нейтрализации одновременно охлаждали баней с ацетоном и льдом. После окончания реакции pH реакционной смеси установили 12. Водную смесь экстрагировали два раза хлористым метиленом, сначала 1000 мл, затем 400 мл. Экстракты объединили и экстрагировали один раз 500 мл дистиллированной воды. Фазу хлористого метилена концентрировали и остаток растирали с 500 мл ацетонитрила. Твердую часть отделили фильтрованием, промыли ацетонитрилом и сушили в вакууме. Получили 10,3 г твердого продукта кремово-желтого цвета, который растворили в 100 мл кипящего ацетонитрила, оставили кристаллизоваться при комнатной температуре. Фильтрованием получили 8,5 г (34%) кристаллов кремово-желтого цвета с т.пл. 184 - 186oC.
FDMS (m/e) 253 М.
Элементный состав для C14H11N3O2 • 0,25H2O
Рассчитано: C 65,23; H 4,50; N 16,30.
Найдено: C 65,57; H 4,45; N 16,27.
ЯМР - и ИК-спектры его совместимы с предполагаемой структурой. Анализ продукта ТСХ показал, что он однородный.
Часть B - 2,9-Ди(бромметил)-5-нитро-1,10-фенантролин.
2,9-Диметил-5-нитро-1,10-фенантролин (1,0 г, 4 ммоль) и N-бромсукцинимид (1,42 г, 8 ммоль) смешали в стеклянном стакане для образования гомогенной смеси, которую добавили сразу (все количество) в 20 мл кипящего (181oC) о-дихлорбензола. Нагревание при температуре кипения проводили 2 мин и затем реакционную смесь оставили для охлаждения до комнатной температуры. Реакционную смесь очищали на колонке (45х5 см) с силикагелем Woelm. Элюировали 100%-ным дихлорметаном. Получили 0,47 г (29%) очищенного продукта в виде желтого масла. Rf его (ТСХ) 0,5 (применяли смесь дихлорметана и ацетона в соотношении 40: 1). Растиранием аликвоты очищенного продукта получили серовато-белое твердое вещество, которое разлагалось при 146oC.
Элементный состав для C14H9Br2N3O2 • 0,5H2O:
Рассчитано: C 40,02; H 2,39; N 10,00.
Найдено: C 40,36; H 2,67; N 9,77.
FD-MS (m/e) 409 М.
ЯМР - ИК-спектры его совместимы с предполагаемой структурой.
Часть C - 2,9-Бис-[N, N-ди(этоксикарбонилметил) аминометил]-5-нитро-1,10-фенантролин
2,9-Ди(бромметил)-5-нитро-1,10-фенантролин (2,28 г, 5,5 ммоль) и 2,08 г (11 ммоль) 1,8 - бис (диметиламино) нафталина растворили в 25 мл 1-метил-2-пирролидинона и добавили 2,08 (11 ммоль) диэтилимидиацетата. Реакционную смесь закрыли герметично притертой стеклянной пробкой и после встряхивания примерно в течение 10 мин при комнатной температуре начал образовываться белый осадок. Реакционную смесь затем поместили в холодильник (при 4oC) и выдерживали ее там 20 ч. Реакционную смесь распределяли между 400 мл диэтилового эфира и 400 мл дистиллированной воды. Эфирную фракцию экстрагировали пять раз (применяли каждый раз по 400 мл) дистиллированной воды. Затем эфирную фазу концентрировали и концентрат растворили в 400 мл хлористого метилена. Фазу хлористого метилена экстрагировали один раз дистиллированной водой, сушили смесью целита (диатомовая земля) и сульфата натрия, фильтровали и затем концентрировали до образования темного масла. Была приготовлена колонка силикагеля Woelm в смеси хлористого метилена и метанола в соотношении 10:1 (длина колонки 50 см, диаметр 5 см) и в колонку ввели неочищенное масло в хлористом метилене. В колонку ввели также 50 мл хлористого метилена и затем колонку элюировали смесью CH2Cl2 /метанол (10:1). Отбирали фракции (каждая 100 мл). Отбирали фракции (каждая 100 мл). Фракции чистого продукта (определяли ТХС с применением смеси хлористого метилена и метанола в соотношении 10:1, Rf 0,4) объединили и концентрировали для получения 1,17 г. (34%) продукта в виде масла красновато-янтарного цвета. FDMS (m/e) 628 MH+.
Элементный состав для C30H37N5O10 • 2H2O
Рассчитано: C 54,29; H 6,23; N 10,55.
Найдено: C 54,71; H 5,58; N 10,53.
ЯМР - и ИК-спектры продукта совместимы с его предполагаемой структурой.
Часть D - 5-Амино-2,9-бис-[N, N-ди(этоксикарбонилметил)аминометил] -1,10-фенантролин
2,9-Бис-[N,N-ди(этоксикарбонилметил)аминометил]-5-нитро-1,10-фенантролин 3,37 г 5,4 ммоль) растворили в растворе 100 мл тетрагидрофурана и 200 мл абсолютного этанола. В полученный раствор добавили раствор 6,8 г (108 ммоль) формиата аммония в 22 мл дистиллированной воды и затем 5,72 г палладия на угле (10%) (50% влаги для безопасности). Реакцию тормозили барботером и перемешивали 30 мин при комнатной температуре и фильтровали через слой диатомовой земли. Растворитель затем удалили в роторном испарителе. Остаток растворили в 300 мл дихлорметане и образованный раствор экстрагировали 300 мл дистиллированной воды. Органическую фазу экстрагировали 300 мл насыщенного раствора хлористого натрия, сушили смесью целита (диатомовой земли) и сульфата натрия, фильтровали и затем концентрировали в высоком вакууме до образования темно-янтарного масла в количестве 2,90 г. Масло обработали 9,0 мл пропионитрила для индуцирования кристаллизации масла и образования желтого твердого продукта, который отделили фильтрованием, промыли 13 мл пропионитрила и 15 мл гексана и затем сушили на воздухе до постоянного веса. Получили 1,04 г (32%) продукта с т.пл. 137-139oC.
FDMS (m/e) 598 MH+, 597 M.
Элементный состав для C30H39N5O9• H2O:
Рассчитано: C 58,52; H 6,71; N 11,38.
Найдено: C 58,28; H 6,55; N 11,53.
ЯМР - ИК-спектры продукта совместимы с его предполагаемой структурой.
Часть E - 5-Амино-2,9-бис-[N, N-ди(карбоксиметил)аминометил]-1,10-фенантролин в виде тетранатриевой соли
5-амино-2,9-бис-[N,N-ди(этоксикарбонилметил) аминометил-1,10-фенантролин (0,90 г, 1,5 ммоль) растворили в 200 мл метанола. В раствор добавили едкий натр (0,25 г, 6,25 ммоль), растворенный в 10,0 мл дистиллированной воды. Реакционную колбу закрыли пробкой и реакционную смесь перемешивали магнитной мешалкой 24 ч при комнатной температуре. Растворитель удалили в роторном испарителе. Твердый остаток растирали с хлористым метиленом и затем смесь фильтровали для получения 0,81 г (94%) продукта. Тетранатриевая соль гигроскопична. Значительное количество ее остается на стенках стеклянной фильтровальной воронки из-за прилипания к ним.
Элементный состав для C22H19Na4N5O8•3H2O:
Рассчитано: C 42,11; H 4,02; N 11,16
Найдено: C 42,01; H 3,71; N 11,03.
ИК-спектр и масс-спектр (ГАВ) продукта были совместимы с его предполагаемой структурой.
Получение 5 - Получение 2,9-бис-[N,N-ди(карбоксиметил)аминометил]-5-изотиоцианат-1,10-фенантролина в виде натриевой соли
Тетранатриевую соль 5-амино-2,9-бис-[N,N-ди(карбоксиметил)аминометил]-1,10-фенантролина (0,16 г, 0,28 ммоль) растворили в смеси 20 мл метанола и 4 мл дистиллированной воды. В раствор добавили тиофосген (0,28 ммоль, т.е. 1,0 мл раствора 0,32 г тиофосгена в 10,0 мл ТГФ) и затем сразу 0,28 ммоль триэтиламина (1,0 мл раствора 0,28 г триэтиламина в 10,0 мл ТГФ). Реакционную смесь перемешивали при комнатной температура и затем сразу концентрировали. Остаток (0,16 г, 94%) представлял собой желтое твердое вещество. ИК - спектр продукта совместим с его предполагаемой структурой.
Получение 6 - Получение 6,6''-[N,N-ди(карбоксиметил)аминометил]-4'-(3-изоцианато-4-метоксифенил) -2,2': 6', 2''-терпиридина в виде тетранатриевой соли
ТМТ, 70, продукт получения 1 (0,31 г 0,42 ммоль), растворили в 30 мл метанола. В раствор добавили 1 мл ТГФ, содержащего 0,42 ммоль тиофосфгена и затем 1,00 мл ТГФ, содержащего 0,42 ммоль триэтиламина. Реакционный раствор сразу концентрировали для выделения остатка, который затем растирали с триэтиламином. Твердую часть отделили фильтрованием и промыли дихлорметаном. Получили 0,30 г продукта. ИК - спектр продукта совместим с его предполагаемой структурой.
Получение 7 - Тринатриевая соль 15-амино-3,5,6,8,9,11-гексагидро-4,7,10- три(карбоксиметил)-2,17: 12,14-ди-этено-1,4,7,10,13 - пентаазабензоциклопентадецина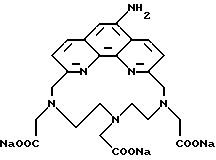
Часть A. 3,5,6,8,9,11-Гексагидро-15-нитро-4,7,10-три- (p-толуолсульфамидо)-2,17:12,14-диэтено-1,4,7,10,13- пентаазабензоциклопентадецин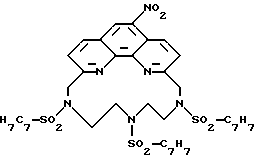
100 мл N-Метилпирролидин-2-она, содержащего 2% 2,9-бисбромметил-5-нитро-1,10-фенантролина, синтезированного в части. В получении 4, добавили через шприцовый насос в течение 2 ч в 20 мл перемешиваемого магнитной мешалкой N-метилпирролидин-2-она, который выдерживают при 20oC в атмосфере аргона. Одновременно добавляли 2%-ный раствор (в N-метилпирролидин-2-оне) 1 экв. двунатриевой соли диэтилентриамин-N, N' N''-три-p-толуолсульфамида. Реакционную смесь перемешивали после добавления при 20oC дополнительно 6 ч. В высоком вакууме отогнали 200 мл растворителя и оставшийся раствор охладили и вылили в 100 мл ледяной воды. Образованный осадок отделили фильтрованием, промыли холодной водой и растирали с ацетонитрилом для получения неочищенного три-p-толуолсульфамидопроизводного.
Часть B. 3,4,5,6,7,8,9,10,11-Нонагидро-15-нитро-2,17: 12,14-диэтено-1,4,7,10,13-пентаазабензоциклопентадецин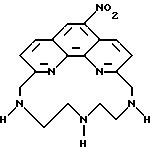
Одну часть неочищенного макролида, полученного в части A, растворяли в 10 ч концентрированной серной кислоты (96%-ная) и реакционную смесь перемешивали при нагревании до 110oC в атмосфере аргона. Через 24 ч раствор охладили при энергичном перемешивании в бане с ледяной водой и затем обработали 10%-ным раствором едкого натра до достижения pH 10. Водную фазу пять раз экстрагировали равными объемами хлороформа. Экстракты объединили и сушили над безводным сульфатом натрия. Соли затем удалили фильтрованием и фильтрат выпаривали в атмосфере аргона. Неочищенный триамин можно очистить кристаллизацией из смеси этанола и воды.
Часть C. 3,5,6,8,9,11-Гексагидро-15-нитро-4,7,10- три(этоксикарбонилметил)-2,17 : 12,14-диэтено-1,4,7,10,13-пентаазабензоциклопентадицин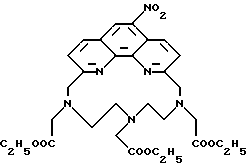
Смесь 1 ч. триаминомакроцикла, полученного в части B, с 10 ч. карбоната натрия и 3 ч. этилбромацетата в виде 3%-ного раствора в безводном ацетонитриле перемешивают 24 ч при 60oC в атмосфере аргона. Реакционную смесь затем охлаждали до комнатной температуры, профильтровали и выпаривали из него растворитель. Остаток можно далее очистить растиранием в холодном простом эфире.
Часть D. 15-Амино-3,5,6,8,9,11-гексагидро-4,7,10-три (этоксикарбонилметил)-2,17 : 12,14-диэтено-1,4,7,10,13-пентаазабензоциклопентадецин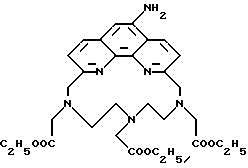
Одну часть нитротриэфира части C растворяют в 100 ч. смеси (50:50) тетрагидрофурана (ТГФ) и этанола. В раствор добавляют 2 экв. формиата аммония в шестикратном количестве (по массе) воды и затем 2 экв. 10% Pd/C. Реакционную смесь перемешивают при комнатной температуре в течение ночи в атмосфере аргона. Затем реакционную смесь фильтруют также в атмосфере аргона и отфильтрованный катализатор промывают тщательно ТГФ, этанолом и затем дихлорметаном также в атмосфере аргона. Фильтрат и промывные жидкости объединяют и концентрируют. Остаток растворяют в хлороформе и раствор в хлороформе промывают два раза водой. Органическую фазу концентрируют для выделения масла. Кристаллизацию его промотируют добавлением метанола. Продукт растирают в метаноле, фильтруют и затем сушат на воздухе.
Часть E. Продукт A
Аминотриэфир части D перемешивают при комнатной температуре с 3 экв. едкого натра в 5%-ном водном растворе метанола в течение 24 ч. Растворитель удаляют в вакууме и продукт (A) растирают с небольшим количеством холодного метанола. Продукт A выделяют фильтрованием.
Получение 8. Тринатриевая соль 15-изотиоцианато-4,7,10-три-(карбоксиметил)-2,17 : 12,14-диэтено-1,4,7,10,13-пентаазабензоциклопентадецина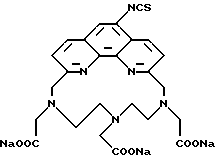
Одну часть тринатриевой соли из части E получения 7 перемешивают в 200 ч. метанола при комнатной температуре. Раствор обрабатывают при комнатной температуре 1 экв. тиофосгена, растворенного в 2 м. ч. тетрагидрофурана, затем 1 экв. триэтиламина, растворенного в 2 м. ч. тетрагидрофурана. Через 1 ч реакционную смесь концентрируют, получая остаток, который превращают в взвесь в метаноле. Продукт выделяют фильтрованием.
Получение 9. Тринатриевая соль 2-метил-3-тио -{15-[3,5,6,8,9,11-гексагидро-4,7,10-три(карбоксиметил)-2,17 : 12,14-диэтено-1,4,7,10,13-пентаазабензоциклопентадецинил]}- семикарбазид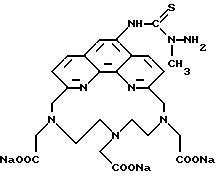
В свежеприготовленную и перемешиваемую смесь 1 м. ч. тринатриевой соли 15-изотиоцианато-4,7,10-три(карбоксиметил)- 2,17 : 12,14-диэтено-1,4,7,10,13-бензопентаазациклопентадецина (получение 8) и 200 м. ч. метанола при комнатной температуре в атмосфере аргона быстро добавляют раствор 1 экз, метилгидразина, растворенного в 10 ч. метанола. Через один час при комнатной температуре растворитель удаляют выпариванием при пониженном давлении. Остаток растирают с 10 ч. безводного, не содержащего кислорода простого эфира. Твердую часть отделяют фильтрованием.
Получение 10. Тетранатриевая соль 2-метил-3- тио-4-{5 -([6,6''-ди-бис(карбоксиметил)аминометил]-2:2', 6': 2'' - терпиридин- 4'- ил)-2-метоксифенил}-семикарбазид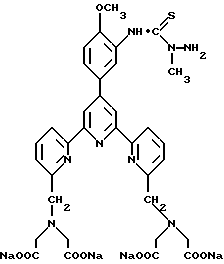
В свежеприготовленную и перемешиваемую смесь 1 м. ч. тетранатриевой соли ТМТ - изоцианата (получение 6) и 200 м. ч. метанола в атмосфере аргона при комнатной температуре быстро добавляют раствор 1 экз. метилгидразина, растворенного в 10 м. ч. метанола. Через 1 ч при комнатной температуре растворитель удаляют при пониженном давлении. Остаток растирают с 10 ч. безводного, не содержащего кислород простого эфира. Твердый продукт отделяют фильтрованием.
Получение 11. Тетранатриевая соль 2-метил-3-тио-4-{5-([6,6''- бис-(N', N'-дикарбоксиметил-N-метилгидразино)-2: 2', 6:2'' - терпиридин - 4'- ил]-2-метоксифенил)}-семикарбазид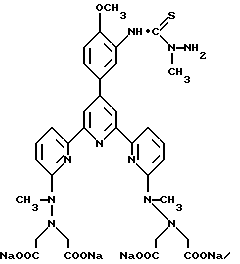
В свежеприготовленную и перемешиваемую смесь 1 м. ч. тетранатриевой соли ТНТ - изотиоцианата, полученного в разделе Получение 3, и 200 м.ч. метанола в атмосфере аргона при комнатной температуре быстро добавляют раствор 1 экз. метилгидразина, растворенного в 10 ч. метанола. После стояния 1 ч при комнатной температуре растворитель удаляют выпариванием при пониженном давлении. Остаток растирают с 10 ч. безводного, не содержащего кислорода простого эфира. Твердый продукт отделяют фильтрованием.
Получение 12. Тетранатриевая соль 2-метил-3-тио-4-[2,9-дибис (карбоксиметил)аминометил-1,10-фенантролин-5-ил]семикарбазида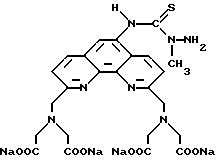
В свежеприготовленную и перемешиваемую смесь 1 м.ч. тетранатриевой соли PheMT - изоцианата (получение 5) и 200 м.ч. метанола в атмосфере аргона при комнатной температуре быстро добавили раствор 1 экв. метилгидразина, растворенного в 10 ч. метанола. После стояния 1 ч при комнатной температуре из смеси удаляют растворитель выпариванием при пониженном давлении. Остаток растирают с 10 ч. безводного, не содержащего кислород простого эфира. Твердый продукт отделяют фильтрованием.
Получение 13. Макроциклический хелатирующий агент: Двунатриевая соль 1,7-дикарбоксиметил-[4'-(3-амино-4- метоксифенил)-2,2': 6',2''-терпиридино] -1,4,7,10,13,16 -гексааза -18-крауна-6
Часть A -6,6''-Дециано-4'-(4 метоксифенил)-2,2':6',2''-терпиридин
Смесь 6,6''-дибром-4'-(4-метоксифенил)-2,2':6'',2''-терпиридина (0,0745 моль), цианида меди (I) (0,296 моль), цианида натрия (0,297 моль) и 300 мл диметилформамида выдерживали при 160oC для реакции в течение 6 ч. После охлаждения реакционную смесь переносят в 1500 мл воды и образованную смесь перемешивают 2 ч и затем фильтруют. Твердый остаток растворяют в растворе цианида натрия (200 г в 400 мл воды) и полученный раствор перемешивают всю ночь. Целевой твердый продукт отделяют фильтрованием, растирают с водой (6 х 600 мл) и сушат, получая белый порошок.
Часть B. 6,6''-Бис(аминометил)-4'-(4-метоксифенил)-2,2': 6,2''-терпиридин
Смесь 6,6''-дициано- 4'-(4-метоксифенил)-2,2': 6',2'' - терпиридина (4 ммоль) и 480 мг 10% Pd/C в 130 мл уксусной кислоты гидрируют (H2, 3,5 кг/см2) при 45oC в течение 22 ч. Реакционную смесь фильтруют и из фильтрата растворитель выпаривают для получения целевого диамина в виде ацетата, который растирают с метанолом. Растворитель удаляют и остаток сушат для получения ацетата диамина.
Часть C. Циклический дилактам 6,6''-бис(аминометил)-4'-(4-метоксифенил) -2,2': 6',2''-терпиридина и пиридин-2,6-дикарбоновой кислоты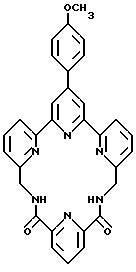
Раствор 6,6''-бис(аминометил)-4'-(4-метоксифенил)-2,2': 6',2''-терпиридина (2 ммоль) в тетрагидрофуране (41 мл) и раствор диизопропиламина (4 ммоль) в диметилформамиде (9 мл) добавляют при перемешивании в 350 мл ТГФ. В полученный раствор добавляют по каплям раствор хлорангидрида 2,6-пиридинкарбоновой кислоты в ТГФ (50 мл) и реакционную смесь перемешивают в течение ночи. Растворитель отгоняют при пониженном давлении и остаток растворяют в 250 мл CH2Cl2. Органический слой промывают последовательно водой (2х30 мл) 0,01N раствором HCl (2х25 мл), водой (1х20 мл), 5%-ным раствором NaHCO (2х25 мл) и водой (2х20 мл). После сушки (Na2SO4) растворитель отгоняют для выделения целевого дилактама, который очищают хроматографией на силикагеле.
Часть D. Двунатриевая соль 1,7-дикарбоксиметил-[4'-(3-амино-4-метоксифенил)-2,2':6',2'' -терпиридино]-1,4,7,10,13,16-гексааза-18-крауна-6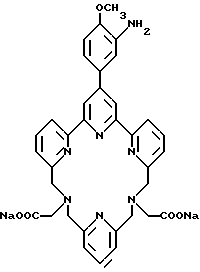
Циклический дилактам(а) части C восстанавливают дибораном в диамин, реакцией которого с этилбромацетатом получают бис(этилацетат). Последнее соединение нитруют азотной кислотой и серной кислотой, получая 4'-(4-метокси-3-нитрофенил)-2,2': 6', 2''-терпиридилпроизводное. Нитрогруппу затем восстанавливают формиатом аммония в присутствии палладия на угле и сложные эфирные группы омыляют едким натром для получения целевого макроциклического хелатирующего агента (b).
Получение 14. Макроциклический хелатирующий агент: Тринатриевая соль 1,4,7-три(карбоксиметил)-[4'-(3-амино-4-метоксифенил)-2,2': 6',2'' -терпиридино]-1,4,7,10,13,16-гексааза-18-крауна-6
Часть A. Терпиридингексаазациклический тритолуолсульфамид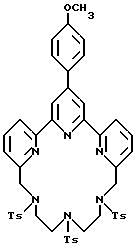
В раствор 6,6''-бис(мезилоксиметил)-4'-(4-метоксифенил)-2,2': 6',2''-терпиридина (2 ммоль) в 60 мл смеси ДМФ и ацетонитрила (1:1), который нагревают до 80oC в атмосфере азота, добавляют по частям раствор двунатриевой соли N, N',N''-тритозил(N,N-бисаминоэтил)амина в 20 мл ДМФ. Реакционную смесь оставляют реагировать при 80oC в течение 7 ч. Растворители удаляют перегонкой при пониженном давлении. Остаток растворяют в 150 мл хлористого метилена. Органический слой промывают водой, сушат. Растворитель затем отгоняют при пониженном давлении для выделения неочищенного терпиридингексаазацикличесского тритолуолсульфамина, 14(a). Его очищают колоночной хроматографией на силикагеле.
Часть B. Тринатриевая соль 1,4,7-три (карбоксиметил)-[4'-(3-амино-4-метоксифенил)-2,2':6',2''-терпиридино]- 1,4,7,10,13,16-гексааза-18-крауна-6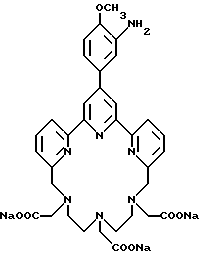
Тритолуолсульфамид, 14(a), части A гидролизуют в серной кислоте для удаления толуолсульфамидогрупп и образования макроциклического терпиридилтриаминосоединения. Этот триамин реакцией в этилбромацетатом превращают в три(этилацетат). Последний нитруют азотной кислотой и серной кислотой для получения 4'-(4-метокси-3- нитрофенил)-2,2':6',2''-терпиридилпроизводного. Затем нитрогруппу восстанавливают формиатом аммония в присутствии палладия на угле и эфирные группы омыляют едким натром и получают целевой макроциклический хелатирующий агент 14(b).
Раздел II: Иммунореагенты.
Получение конъюгатов антител с ТМТ и ТНТ
Часть A - Способ присоединения хелатирующего агента к антителам.
ТМТ или ТНТ присоединяли к антителам путем окисления углеводных остатков на антителах NaIO4 для образования на них альдегидных групп. Раствор (10 мл) 0,1M NaIO4 в очищенной воде добавили в 1 мл раствора (4 мг/мл) антител B72.3 (B72.3 являются хорошо известными антителами по отношению к связанным с колоректальными опухолями антигенам, которые впервые были описаны Национальным Институтом Здоровья (Патент США 4522918)). В PB (10 мМ фосфата натрия, 0,15 M NaCl) с pH 6,0 pH образованного раствора снова установили 6,0 и инкубировали 1 ч при комнатной температуре в темноте. После окончания инкубирования избыток периодата удалили пропусканием раствора антител через колонку Сефадекса G50, который был уравновешен PBS, pH 6,0. Отбирали фракции по 1 мл. Две фракции, имеющее наивысшее поглощение у 280 нм, объединили.
0,33 M раствор каждого хелатирующего агента (ТМГ или ТНТ) получили растворением твердого хелатирующего агента в PBS, pH 6,0. Величина pH раствора повысилась до 9,0, поэтому добавлением 6 M HCl pH снова установили около 6,8. 200 мл каждого из растворов хелатирующих средств добавили по отдельности в 2 мл часть раствора антител и pH полученного раствора установили 6,0. Эти смеси инкубировали 5 ч при комнатной температуре в темноте. Затем в каждый раствор добавили NaCNBH3 (Aldrich 29, 694-5, 5 M в I M NaOH) до конечной концентрации 10 мМ и pH установили 6,0 для восстановления основания Шиффа, образованного альдегидом. Смеси инкубировали в течение ночи при комнатной температуре. Растворы затем центрифугировали (Модель Eppendorf 5412) для удаления нерастворенного хелатообразующего агента, который выпал в осадок в течение ночи и концентрировали до объема 1 мл в микроконцентраторе Centricon 30 (Amicon). Эти растворы пропустили через колонки Сефадекса C50 для удаления избытка хелатирующего агента и NaCNBH3. Отбирали фракции по 1 мл. По две фракции каждого из растворов хелатирующего агента, имеющие наибольшее поглощение у 280 нм, объединили.
Объединенные фракции диализовали с применением 0,01 М ацетата натрия, 0,15 М (NaCl (pH 6,0) и двумя изменениями буфера. Если в процессе диализа образуется осадок, то его удаляют центрифугированием.
Обычно препараты конъюгатов, полученные этим способом, имеют среднее молярное соотношение ТМТ или ТНТ и антител около 1,7.
Часть B - Анализ соотношения хелатирующего агента и антител.
Концентрацию антител в растворах конъюгатов определяли тестом на белок Biorad с применением бычьего иммуноглобулина в качестве белкового стандарта. Концентрацию ТНТ определяли спектрофотометрически с использованием измеренного коэффициента экстинкции у 350 нм. В случае конъюгата ТМТ-В 72.3 часть (около 0,5 мг) конъюгата ввели в 1 мл 0,01 М раствора ацетата натрия, 0,15 М NaCl (буфер, pH 6,0) и добавили избыток E+3. Поглощение этого раствора у 330 нм применяли для определения количества хелатирующего средства в этих растворах.
Часть C - функциональное тестирование (анализ иммунокомпентности) in vitro конъюгата антитело-хелатирующий агент (иммунореагента)
Лунки микроитрационного планшета Linbro EIA II Plus покрыли антигеном добавлением 100 мл (на лунку) раствора 4 мг/мл бычьего субмаксиллярного муцина (Sigma М4503), т.е. антигена, реагирующего с антителами В72.3, в PBS (солевой раствор, содержащий фосфатный буфер) и инкубировали 1 ч при комнатной температуре. После промывания планшета три раза PB с pH 6,8 лунки блокировали добавлением 200 мл/лунку раствора 1% BSA (альбумин бычьей сыворотки крови, Sigma A-9306) - PBS с pH 6,8 и инкубировали 1 ч при комнатной температуре. Микропланшеты снова промыли три раза PBS с pH 6,8. Были получены пробы конъюгатов с концентрацией 1 • 10-7 М в PB с pH 6,8, содержащие 1% лошадиной сыворотки крови (Sigma S-7390). Из этих растворов получили разбавленные растворы с применение того же буфера. 100 мкл каждой пробы разбавленного раствора добавили в лунки (каждую пробу в две лунки). После инкубирования 1 ч при комнатной температуре планшеты промыли три раза PBS с pH 6,8 и затем проводили определение путем добавления 100 мкл на лунку конъюгата кроличий противомышиный фрагмент F (ab) 2 - HRP (Jackson Labs, 315 - 035-047) при разведении 1:1000 в PBS с pH 6,8, содержащего 1% лошадиной сыворотки. После инкубирования при комнатной температуре в течение 1 ч лунки промыли три раза PBS с pH 6,8. Цвет появился после добавления 100 мкл (на лунку) ABTS - HRP - субстрата (Kirkegaard and Perry Labs, Product # 506502 и 506402). Реакцию остановили добавлением равного объема 4N H2SO4. Цвет фиксировали при помощи фильтра для 414 нм в приборе Titertek Multiscan через 15 мин.
При тестировании этим способом было показано, что иммуноконъюгаты В 72.3 с ТМТ или ТНТ обладают иммунореактивностью, сравнимой с активностью природных В 72.3. Результаты представлены на фиг. 1 и 2. На фиг. 1 имеется также кривая для скандиевого комплекса конъюгата В 72.3-ТНТ, полученного так же, как описанный выше комплекс европия.
Пример 1-2. Получение и оценка радиоактивных иммунореагентов (конъюгатов, антитела-ТМТ), меченых радионуклидами (111In 90Y). Применение для сравнения конъюгата DTPA.
A. Функциональное тестирование in vivo конъюгатов хелат-антитело - эксперименты по биораспределению
1. Тестируемые материалы. В качестве тестируемых иммуноконъюгатов применяли конъюгат В 72.3-ТМТ, полученный как указано выше, или конъюгат В 72.3-DTPA, в котором DTPA соединен (через связывающий агент) с остатком окисленного углевода антителами способами, аналогичными описанным для присоединения TMT.
2. Мечение конъюгатов радиоактивными изотопами. Приготовляли растворы конъюгатов антитело-хелат, которые нужно было метить 111In или 90Y в содержащем фосфатный буфер солевом растворе с pH 6,0. В раствор конъюгата (1 мл содержит около 1 мл конъюгата антитело - хелат) добавили радионуклиды (около 1 м Ci) смешали и инкубировали 30 мин при комнатной температуре. Нехелатированный металл удалили из препарата конъюгата жидкостной гель-хроматографией высокого разрушения (колонка TSK 30000 SW). Вытекающий из колонки поток контролировали как по белку (OD280 нм), так и по радиоактивности (монитор радиоактивности). Специфическую активность очищенного меченого конъюгата рассчитывали из этих данных. Конъюгаты сразу применяли для изучения биораспределения.
3. Общий способ. Пять мышей применяли в каждой тестируемой группе для изучения биораспределения 90Y и 3 мыши в каждой группе для изучения 111In. Каждая доза для любой мыши содержала 10 мг радиоактивного конъюгата, т.е. 20-100 мкг антитела на 1 мкл и более чем 50 мк Ci. Мышам, как имеющим опухоли, так и не имеющим, дозу вводили инъекцией в день 0.
4. Инициирование роста опухоли. Каждой голой мыши (nu/nu: Swiss Background, Taconic Farms, Germantown, NY) ввели подкожной инъекцией в заднюю часть левой стороны один миллион клеток LS 174T в логарифмической фазе роста в 0,2 мл стериальной среды или физиологического раствора клеточной культуры. Мышей обследовали в отношении роста опухолей до тех пор, когда опухоли становятся измеряемыми. Затем опухоли измеряли с точностью 0,1 мм на пересечении двух перпендикулярных диаметров (длина х ширина) при помощи пальцевого циркуля до тех пор, пока произведение длины и ширины и не будет в пределах 50-100 мм2 для всех мышей.
5. Протокол инъекцирования. В каждый желаемый день после инокулирования опухолевых клеток (день 0) каждой мыши вводили инъекцией меченый конъюгат. Меченые радиоактивным изотопом антитела для каждой дозы набирали в отдельный инсулироновый шприц на 1 см3 с иглой 28 G (1,27 см) для ведения. По три аликвоты 10 мкл каждого раствора сохраняли для подсчета гамма-квантов с целью определения инъекцированной дозы.
Каждый шприц, содержащий раствор, получил номер в соответствии с порядком введения. Каждый шприц взвесили и в устройстве для калибровки определили количественное содержание радионуклида. Эта информация была зарегистрирована. Мышей анестезировали внутрибрюшинной инъекцией 0,15 мл стериального раствора, содержащего 11 мг/мл кетамина HCl и 3 мг/мл ксилазина. Инъекции вводили через ретроглазничные синусы твердой мозговой оболочки. Сразу после инъекции в каждой мыши определяли дозу радионуклида в калибраторе доз. Шприцы взвесили снова, сделали подсчеты и данные записали.
6. Способы обследования. Каждую мышь взвешивали с точностью 0,1 г в день 0 и непосредственно перед вскрытием. Непосредственно перед получением изображения и/или вскрытия в каждой мыши определяли дозу радионуклида в калибраторе доз и затем проводили стандартные операции. В каждой туше после вскрытия определяли дозу радионуклида. Непосредственно перед вскрытием в каждой мыши измеряли размер опухоли с точностью 0,1 мм по двум перпендикулярным диаметром (длина х ширина) при помощи пальцевого циркуля. У каждого животного отобрали пробу крови в тарированную пробирку с культурой и затем пробирку опять взвесили. Каждое животное умерщвляли шейным смешением. Органы (легкие, селезенку, печень, ободочную кишку, правую почку, левую почку, опухоль, костный мозг и мышцы) рассекали и обрабатывали для удаления инородной ткани и затем взвешивали с точностью до мг. Радиоактивность рассеченных органов определяли счетчиком гамма-квантов.
7. Результаты. Биораспределение, приведенное на фиг. 3, показано для 6 мышей в течение 4 дне после инъекции радиоактивных иммунореагентов. Результаты биораспределения 111In показывают, что меченый радиоактивным изотопом конъюгат ТМТ и В 72,3 осуществил направленную доставку радиоактивности в опухоль. Хотя содержание В 72.3 - ТМТ - 111In в печени и почках было незначительно выше, чем конъюгата В 72.3 - ТРА- 111In, конъюгат В 72.3 - ТМТ - 111In можно применять для доставки 111In в опухоль.
Биораспределение, приведенное на фиг. 4, получили на восьмой день после инъекции радиоактивного иммунореагента. Средняя величина биораспределения для каждой группы из пяти мышей графически представлена для изученных типов тканей. Данные биораспределения 90Y показывают, что конъюгат В 72.3-ТМТ - 90Y направленно доставлял 90Y в опухоль, так же как и конъюгат В 72.3 - DTPA 90Y. Однако значительно меньше 90Y было обнаружено в костном мозге, когда применяли конъюгат В 72.3- ТМТ - 90Y, чем при введении конъюгата В. 72.3 - DTPA - 90Y. Поскольку бедренная кость (костный мозг ее) является дозоограничивающим органом при 90Y - терапии, этот результат указывает, что ТМТ является лучшим хелатом для направленной антителами радионуклидной терапии с применением этого изотопа. Из других исследованных тканей только кровь имеет более высокое содержание 90Y, кода ТМТ применяли в качестве хелатирующего агента для получения иммуноконъюгата. Считают, что это обусловлено лучшей стабильность in vivo комплекса В 72.3 - ТМТ - 90Y по сравнению с комплексом В 72.3 - DTPA - 90Y.
B. Изучение выживаемости.
Если комплекс В 72.3 - ТМТ - 90Y уменьшает количество 90Y, которое доставляется в костный мозг, по сравнению с В 72.3 - DTPA 90Y и если костный мозг является дозолимитирующей тканью, то введение конъюгата В 72.3 - ТМТ - 90Y для мышей должно быть менее токсично, чем введение конъюгата В 72.3 - DTPA - 90Y. Общие способы проверки этой гипотезы (изучение выживаемости) были такие же, как способы, описанные для изучения биораспределения, но со следующими исключениями. В каждой изучаемой группе имелось 8 голых мышей с опухолями. Схема инъекций была следующая: 200 мкCi, 120 мкCi и 120 мкCi ввели на 0, 4 и 8 день соответственно (схема низких доз), причем вводили конъюгат B 72.3-TMT-90Y или B 72.3-DTPA-90Y. Две другие группы мышей получили 400 мкCi, 320 мкCi и 320 мкCi этих двух иммуноконъюгатов на 0, 4 на 8 день соответственно (схема высоких доз). Изучали выживаемость мышей (но не проводили биораспрелеления конъюгатов). Результаты (фиг. 5) указывают, что выживаемость мышей пролонгировалась, если 90Y вводили в составе конъюгата B 72.3 - TMT -90Y.
C. TMT - Иммуноконъюгат с антиопухолевой активностью.
TMT или его подходящее производное можно конъюгировать с молекулой антитела для образования молекулы конъюгата антитело - TMT, которая обладает способностью связываться с антигеном - мишенью, которую распознает вариабельная область антитела. Такую молекулу конъюгата можно применять для доставки радиоактивного изотопа, который хелатирован остатком TMT, для локализации и/или лечения повреждений, в которые направлен такой иммуноконъюгат. В одном из предпочтительных вариантов отбирают такие антитела, которые имеют широкий спектр реакционной способности для молекул антигена, экспрессированных опухолевыми клетками, благодаря чему образуется конъюгат антитело-TMT, который может доставить радиоактивный изотоп к опухолям для терапевтических и диагностических целей. ING-I является химерным антителом (описан в International Patent Publication NWO 90/02569, 22 March 1990) и состоит из мышиной вариабельной области и константной области иммуноглобулина человека. Молекулы антител получают культивированием мышиной миеломной клеточной линии, экспрессирующей химерные антитела, по существу как описано в указанной выше публикации. Химерные антитела ING-I применяют при концентрации 5,2 мг/мл в 50 мМ растворе буфера (ацетата натрия) с pH 5,6, в который добавлено 150 мМ NaCl. В 1,15 мл раствора антител добавляют раствор 50 мМ бората натрия с pH 9,0, в который добавлено 100 мМ хлористого натрия, до конечного общего объема 2,5 мл. Раствор вводят в хроматографическую колонку PD-10, уравновешенную буфером (ацетатом натрия), добавленным в раствор антител. Антитела элюируют из колонки 3,5 мл того же буферного раствора (бората натрия). Элюат концентрируют при помощи устройства для ультрафильтрации Centricon-30 до концентрации приблизительно 4,0 мг химерных антител ING-I на 1 мл раствора. Раствор изотиоцианатопроизводного TMT (из раздела Получение 6, далее соединение будет обозначено как TMT-NCS) получают в буферном растворе (борат натрия) при концентрации 10 мг/мл и добавляют в раствор антител до конечной концентрации 138 мкМ TMT-NCS. Раствор осторожно перемешивают и инкубируют в темноте при комнатной температуре (приблизительно 22oC) в течение ночи (приблизительно 12 ч). Конъюгат ING-I; TMT освобождают от TMT-NCS и других низкомолекулярных продуктов при помощи ЖХВР с применением колонки с суперозой 12, уравновешенной и элюированной буферным раствором (50 мМ ацетата натрия) с pH 5,6, в который добавлено 150 мМ хлористого натрия. Иммуноконъюгат TMT затем тестируют на его способность связываться с антигеном опухолевых клеток, а также связывать изотоп иттрий-90, чтобы показать, что конъюгат можно метить радиоактивным изотопом в остатке TMT и он может доставить этот изотоп в опухолевые клетки.
Пример 3. Получение конъюгата ING-I с TMT.
Антитела ING-I получали как описано выше. После очистки ING-I применяли с концентрацией 5,0 мг/мл в растворе 50 мМ ацетата натрия и 150 мМ хлористого натрия (буферный раствор) с pH 5,6.
Конъюгат ING-I и TMT-NCS получили способом, по которому сначала буферный раствор с pH 9,3, содержащий 1,0 М карбоната и 150 мМ хлористого натрия, добавляли в INC-I до достижения pH раствора антител 9,0. Пробу раствора INC-I, содержащую 5 мг белка, перенесли пипеткой в промытый кислотой конический реакционный стеклянный сосуд. Раствор TMT-NCS (Получение 6) получили растворением 100 мг этого соединения в 10 мл буферного раствора (1,0 М карбоната и 150 мМ хлористого натрия) с pH 9,3. Реакцию конъюгирования начинали добавлением 96,5 мкл раствора TMT-NCS в раствор антител до достижения 4-кратного молярного избытка TMT-C относительно ING-I. Раствор перемешивали для смешивания реагентов и затем оставили в темноте при комнатной температуре. Через 16 ч конъюгат ING-I с TMT отделили от неконъюгированного ТМТ подачей реакционной смеси в хроматографическую колонку с PD10, которую предварительно промыли и уравновесили буферным раствором (50 мМ ацетата натрия и 150 мМ хлористого натрия) с pH 5,6. Чистый конъюгат элюировали из колонки 3,5 мл того же буферного раствора.
Анализ соотношения хелатирующего агента и антитела.
Концентрацию ING-I в растворах конъюгата определяли анализом белка BioRad с применение в качестве белкового стандарта бычьего иммуноглобулина.
Для подсчета числа функциональных молекул TMT на антитело конъюгат ING-I с TMT (ING-I/TMT) был обработан для реакции раствором хлористого европия до насыщения способности TMT связывать металл; 0,5 мг. аликвоты ING-I/TMT в 2,5 мл 0,05 М Трис-HCl буферного раствора с pH 7,5 пипеткой перенесли в 5 мл кварцевую кювету. Был получен 20 мкМ раствор хлористого европия (гексагидрат хлористого европия: Aldrich) в 0,05 М Трис HCl буфере с pH 7,5. Аликвоту (50 мкл) этого раствора хлористого европия добавили в кювету, содержащую ING-I/TMT и полученный раствор медленно перемешивали магнитной мешалкой при комнатной температуре 10 мин с применением маленького магнитного стержня, помещенного в кювету. Флуоресценцию комплекса металл - ING-I/TMT определяли в спектрофотометре Перкин Эльмер LS50 с использованием длины волны возбуждения 340 нм (ширина щели 10 нм). Флуоресцентное излучение контролировали у 618 нм, используя ширину щели 10 нм и фильтр с ограниченной полосой пропускания у 430 нм. Эту процедуру повторяли и флуоресцентные показатели получали после каждого добавления. Аликвоты хлористого европия добавляли до тех пор, пока повышение интенсивности флуоресценции не было менее 5% относительно предшествующего показателя. Коррекцию разбавления применяли для интенсивности флуоресценции, измеренной для каждого молярного соотношения, чтобы компенсировать изменение в объеме тестируемого раствора. Поскольку каждый хелатирующий центр на конъюгате ING-I/TMT связывает один ион европия и ион европия должен быть в центре хелата, чтобы имела место флуоресценция, этот метод позволяет определить количественно число центров функционального хелатирования.
С применение этого метода установили, что соотношение числа молекул ТМТ и молекул антител находится в пределах 1:1 - 2:1.
Анализ иммунореактивности ING-I/TMT.
a) Методом ELISA.
Антиген, с которым антитело, ING-I, соединяется, получили из клеток LS174T или HT 29 (из ATTC) соскабливанием конфлюэнтных монослоев клеток со стенок склянок для культур клеточным скребком. Клетки из многих склянок объединили и в пробе определяли общее число собранных клеток. Все это время клетки сохраняли во льду. После центрифугирования клеток при 1500 об/мин в течение 10 мин при 4oC, их промыли один раз охлажденным во льду 50 мМ буфером фосфата натрия с pH 7,4, в который добавлено 150 мМ хлористого натрия (PBS) превратили в гранулки в тех же условиях и перенесли в охлаждаемую льдом стеклянную ступку. Клетки гомогенизировали при 4oC с использованием приводимого в движение мотором пестика и затем центрифугировали при 3000 х г в течение 5 мин. Обогащенный антигеном супернатант отделили от другого клеточного дебриса и подвергали дальнейшему центрифугированию при 100000 х г в течение 1 ч при 4oC. Гранулки (фракция антигена) из последней стадии суспензировали в 100 мкл PBS для каждого миллиона собранных клеток. После установления концентрации белка (анализ белка BioRad BCA с использованием бычьего иммуноглобулина в качестве белкового стандарта) антигены хранили при -20oC до их применения.
Каждую лунку титрационных микропланшетов Costar (96 лунок) покрыли антигеном путем добавления лизата клеток 100 мкл/лунку (10 мкг/мл), полученного описанным выше образом. Микропланшеты высушили в течение ночи в инкубаторе при 37oC. После промывания планшета пять раз, 0,05%-ным Tween-20 (Sigma), лунки блокировали добавлением 125 мкл/лунку 1%-ного раствора BSA (альбумина бычьей сыворотки, Sigma A-7906) в PBS и инкубировали 1 ч при комнатной температуре. Планшеты промыли пять раз 0,05%-ным Tween-20. Пробы (50 мкл/лунку, в двух экземплярах) конъюгатов INC-I/TMT и стандартные растворы антител ING -I получали в пределах соответствующих концентраций в PBS. Биотинилированные антитела ING-I (1,0 мг/мл в 0,1%-ном BSA) добавили в каждую лунку (50 мкл/лунку) и планшеты затем инкубировали в течение 2 ч при комнатной температуре. После промывания 5 раз 0,05%-ным Tween-20, провели сухой блоттинг планшетов и инкубировали их при комнатной температуре в течение 1 ч с разбавленной (1 : 4000 в 0,1%-ном BSA) стрептовидин-щелочной фосфатазой (Tago; # 6567). После дальнейшего пятикратного промывания цвет в каждой лунке проявили при добавлении 100 мкл на лунке субстрата фосфатазы (две таблетки фосфатазы Sigma 104, растворенные в 10 мл дистиллированной воды, и 20 мкл щелочного буфера Sigma 221). После стояния 1 ч при комнатной температуре цвет определяли с использованием фильтра 405 нм в устройстве для чтения микропланшетов Titertek Nultiscan.
При тестировании этим способом было показано, что иммуноконъюгаты ING-I c TMT обладают иммунореактивностью, сравнимой с активностью природного ING-I.
Анализ иммунореактивности ING-I/TMT
b) Проточной цитометрией
Клетки-мишени HT29 выращивали до конфлюэнции в колбе для культур тканей с применением DMEM-среды, в которую добавлено 10% сыворотки внутриутробных телят. Клетки собирали выскребыванием стенок колбы клеточным скребком. Клетки из многих отдельных колб объединили, центрифугировали для образования дебриса, пересуспензировали при 5 • 105/мл в растворе охлажденного льдом 50 мМ раствора фосфата натрия с 150 мМ хлористого натрия (pH 7,4) (PBS), в который добавлено 0,1% альбумина бычьей сыворотки (Sigma) и 0,02% азида натрия (флоубуфер). Клетки промывали тем же буфером и затем провели подсчет. Стандартную кривую антител построили разбавлением ING-I посторонними (не связывающими), совместимыми по изотипу контрольными антителами (IgGI человека) для получения ряда проб с содержанием ING-I от 10% до 100%. Стандартную кривую сделали в флоцбуфере таким образом, чтобы каждая проба содержала 1,0 мкг белка в 1 мл. Пробы стандартной кривой и неизвестные пробы затем инкубировали с 5 • 105 клетками HT29 при 4oC в течение 1 ч. После экстенсивного промывания для удаления несвязанных антител клетки пересуспензировали в 100 мкл флоубуфера и инкубировали 1 ч при 4oC с антителами коз на антитела человека, мечеными флуоресцентным изотиоцианатом (FITC). После дальнейшего промывания флуобуфером пробы анализировали проточной цитометрией на проточном цитометре Counter EPICS 753. Флуоресцентный изотиоцианат (FITC) и иодид пропидия (PI) возбуждали с применением линии испускания 488 нм аргонового лазера. Мощность на выходе устанавливали 500 мВт в режиме регулирования света. Отдельные клетки идентифицировали рассеянием света (прямым и под углом 90o). Окна анализа применяли с целью отделения отдельных клеток от агрегатов и клеточного дебриса. Флуоресценцию от FITC и пропидия разделяли с помощью многоходового дихроичного фильтра 550 нм и собирали с помощью полосного фильтра 530 нм (для FITC) и полосного фильтра 635 нм (для PI). Параметры рассеяния света отбирали в виде интегрированных импульсов, флуоресценцию представляли в виде log интегрированных импульсов. Мертвые клетки исключались из анализа помещением анализного окна на клетки, дающие отрицательный результат на поглощение PI. Среднюю флуоресценцию для пробы (средняя от 2500 клеток) подсчитали для каждой гистограммы. Калибровочные гранулы FITC анализировали в каждом эксперименте для получения стандартной кривой. Среднюю интенсивность флуоресценции для каждой пробы затем выразили в виде средних FITC - эквивалентов на клетку. Иммунореактивность подсчитывали сравнением средней интенсивности флуоресценции неизвестной пробы с величинами из стандартной кривой. Пробы ING-I/TMT имели величины иммунореактивности, сравнимые с величинами природных антител ING-I.
Определение образования агрегатов ЖХВР (с исключением по размеру)
Колонку ЖХВР с исключением по размеру (30 см x 7,5 мм, TSK - G3000, Supelco), снабженную кварцевой колонкой того же материала, уравновешивали 12-кратным количеством (от объема колонки) 20 мМ буфера (фосфата натрия (pH 6,0), в который добавлено 150 мМ хлористого натрия, при помощи системы для ЖХВР Waters 600 E со скоростью потока 1,0 мл в мин при давлении 28 - 42 кг/см2. Пробу белковых стандартов для гельфильтрации BioRad (25 мкл) вводили в колонку.
Время удерживания каждого стандарта регистрируют УФ-детектором Waters 490 у 280 нм. После извлечения последнего стандарта из колонки ее промыли еще 10 объемами 10 мМ буфера фосфата натрия (pH 6,0), в который добавлено 150 мМ хлористого натрия. Пробы (50 мкл) природных антител ING-I или ING-I/TMT ввели в колонку и регистрировали их время удерживания. Количество агрегированного материала в пробе ING-I/TMT подсчитывали из площади пиков и времени удерживания.
Природные антитела INC-I имели время удерживания 9,1 мин при определении их этим способом. ING-I/TMT имел основной пик также со временем удерживания 9,1 мин, а также небольшой пик, приписываемый агрегатом. Этот пик иногда появлялся на хроматограммах и имел время удерживания 7,3 мин. Сравнение площадей пиков показало, что содержание агрегатов было менее 5% от общего количества.
Мечение конъюгата ING-I/TMT изотопом 90Y.
Объем радиоактивного хлорида иттрия (90Y в 0,04 М соляной кислоте со специфической активностью > 500 Ci/г : Amersham - Mediphysics) нейтрализовали двумя объемам и 0,5 М раствора ацетата натрия с pH 6,0. Нейтрализованный 90Y (1,0 мCi) добавили в 1,0 мл ING-I/TMT (1 мг/мл) в 50 мМ буфере ацетата натрия, содержащем 150 мМ хлористого натрия и имеющем pH 5,6. Реакционную смесь оставили на 1 ч и затем ввели в хроматографическую колонку PD-10, которая предварительно была промыта и уравновешена буфером, содержащим 50 мМ фосфата натрия с 150 мМ хлористого натрия и имеющим pH 7,4 (PBS). Пробу элюировали из колонки 1,5 мл PBS. Фракции меченого конъюгата ING-I/TMT (0,5 мл) отобрали, проанализировали на радиоактивность и объединили. Эффективность мечения определяли отбором 1,0 мкл пробы и нанесением ее на полоску GelmanI TLC-SG. Полоску проявляли в стеклянном стакане, содержащем 0,1 М цитрата натрия (pH 6,0), в течение нескольких минут до достижения фронтом растворителя 3/4 пути до верха бумаги. Полоску поместили в сканирующее устройство для получения изображения System 200 (Bioscan), которое оптимизировали для 90Y и которое контролировали компьютером Compaq 386/20e. В этой системе свободный 90Y мигрирует у фронта растворяли, тогда как ING-I/TMT (90Y) остается в первоначальном состоянии.
С помощью этой системы было показано, что более 98% 90Y (от общего количества) ассоциировано с ING-I/TMT.
Мечение ING-I/TMT флуоресцентными металлами
Связывание ланданидов, например европия (III), с хелатирующими агентами, которые содержат ароматический остаток, расположенный близко к координационной сфере, может привести к появлению "сенсибилизированной" флуоресценции, при которой свет поглощается через ароматическую систему и энергия переносится в металл. Металл затем продуцирует излучение, характеризующееся очень большим смещением Стока и временем люминесценции до нескольких секунд. 0,5 мг аликвоты ING-I/TMT в 2,5 мл 0,05 М трис-HCl буфера (pH 7,5) перенести пипеткой в коническую реакционную колбу объемом 4 мл, содержащую небольшой перемешивающий брусок. Был получен раствор 250 мкМ хлористого европия (тексагидрата хлористого европия : Aldrich в буфере 0,05 М трис HCl с pH 7,5. Аликвоту (50 мкл) этого раствора хлористого европия добавили в реакционную колбу, содержащую ING-I/TMT и полученный раствор перемешивали очень медленно магнитной мешалкой при комнатной температуре. Для проведения реакции смесь оставили на 1 ч и затем загрузили в хроматографическую колонку PD-10, которая была предварительно промыта и уравновешена буфером, содержащим 10 мМ фосфата натрия и 150 мМ хлористого натрия и имеющим pH 6,0 (PBS). Пробу элюировали из колонки 3,5 мл PBS. Флуоресценцию 50 мкл пробы комплекса металл - ING-I/TMT определяли спектрофотометром Перкин Элмера LS50 с использованием длины волны возбуждения 340 нм (ширина щели 10 нм). Флуоресцентное испускание регистрировали у 618 нм, применяя ширину щели 10 нм и фильтр с полосой пропускания 430 нм. Каждый функциональный хелатирующий центр на конъюгате ING-I/TMT связывает один ион европия. При помощи этого метода было показано, что молекула антитела связывает 1-3 флуоресцентных иона европия.
Пример 4. Получение конъюгата PRIA3 с TMT.
PRIA3 является мышиным антителом (разработан ICRF, U.K.). Эти антитела применяли при концентрации 4,0 мг/мл в буфере 50 мМ ацетата натрия с pH 5,6, в который добавлено 150 мМ NaCl. В раствор 1,0 мг антител добавили раствор 50 мМ бората натрия с pH 9,0, в который добавлено 100 мМ хлористого натрия, до конечного общего объема 2,5 мл. Раствор ввели в хроматографическую колонку PD-10, уравновешенную буферным раствором бората натрия, добавленного в раствор антител. Антитела элюировали из колонки 3,5 мл того же буферного раствора бората натрия. Элюат концентрировали при помощи устройства для ультрафильтрации Centricon-30 до концентрации 5,0 мг PRIA3 - антител на 1 мл раствора. Раствор TMT - NCS (получение 6) получили в буфере бората натрия с концентрацией 2,0 мг/мл и добавили в раствор антител до достижения конечной концентрации 104 мкМ TMT-NCS. Раствор осторожно перемешивали и инкубировали в темноте при комнатной температуре (около 22oC) в течение ночи (14-16 ч). Конъюгат PRIA3 : TMT отделяли от свободного TMT-NCS и других низкомолекулярных продуктов при помощи колонки ЖХВР с суперозой 6, уравновешенной и затем элюированной буферным раствором 50 мМ фосфата буфера с pH 7,2, в который добавлено 150 мМ хлористого натрия. Элюат концентрировали при помощи устройства для ультрафильтрации Centricon до достижения концентрации 0,25 мг PRIA3 : TMT на 1 мл раствора. Иммуноконъюган TMT затем тестировали на способность связываться с антигеном опухолевых клеток, а также связывать изотоп иттрий-90.
Аналитические тесты на конъюгаты PRIA3/TMT.
Аналитические тесты, проведенные на конъюгатах PRIA3/TMT, были аналогичны указанным выше тестам на конъюгаты ING-I/TMT. Наиболее заметное различие состояло в том, что для определения иммунореактивности при помощи проточного цитометра применяли клетки SW1222. Эти клетки (ATCC) были выращены в MEM-среде, в которую добавили 10% сыворотки плодных телят и неэссенциальные кислоты. Тесты способом ELISA на иммунореактивность конъюгатов PRIA3/TMT не проводили.
Пример 5. Получение конъюгата NRLU-10 с TMT.
NRLU-10 представляет собой мышиные антитела (разработаны Okabe как TFS-2). Эти антитела применяли при концентрации 3,4 мг/мл в 50 мМ буферном растворе фосфата натрия с pH 7,2, в который добавлено 150 мМ NaCl. К раствору 12 мг антител добавили 1,5 мл раствора 50 мМ бората натрия с pH 9,0, в который добавили 150 мМ хлористого натрия. Две аликвоты (2,5 мл каждая) раствора ввели для разделения в хроматографические колонки PD-10, уравновешенные 50 мМ раствором бората натрия (pH 9,0), в который добавлено 150 мМ хлористого натрия. Антитела элюировали из каждой колонки 3,5 мл того же раствора бората натрия. Эти элюаты объединили. Был получен раствор TMT-NCS в буферном растворе бората натрия с концентрацией 10 мг/мл. 19 мл его добавили в препарат антител. Раствор осторожно перемешивали и инкубировали в темноте при комнатной температуре (около 22oC) в течение ночи (около 12 ч). Конъюгат NRLU-10 : TMT отделили от свободного TMT-NCS и других низкомолекулярных продуктов разделением при помощи ЖХВР с применением колонки с суперозой 12, уравновешенной и затем элюированной 50 мМ буферным раствором фосфата натрия с pH 6,0, в который добавили 150 мМ хлористого натрия.
Иммуноконъюгат TMT затем тестировали на его способность связываться с антигеном опухолевых клеток и также связывать изотоп-иттрий-90.
Аналитические тесты на конъюгаты NRLU-10/TMT.
Аналитические тесты на конъюгаты NRLU-10/TMT были аналогичны тестам, указанным выше на конъюгаты ING-I/TMT.
Пример 6. Получение иммунореактивного агента, содержащего олигонуклеотид, ковалентно соединенный с комплексообразующим средством.
Комплексообразующий агент TMT можно ковалентно связать с иммунореактивной группой другой, чем антитело, например олигонуклеотидом. Этот иммунореагент можно метить 90Y и полученный таким образом радиоактивный иммунореагент может реагировать с его комплиментом, а именно, комплиментарным олигонуклеотидом.
Часть A. Получение олигонуклеотида (CI).
20-Мерный олигонуклеотид, содержащий 3'-аминогруппу и имеющий формулу 5'-TTAGCTTCTCCGTCCATAA-C5 -амино-T-3' (cI) получают на синтезаторе ДНК (Applied Biosystem DNA synthesizer), как предложено изготовителем этого устройства, с применением аминомодификатора (Uni-link Amino Modifier, Clonetech) в качестве предшественника 3'-аминогруппы и 2-дезоксинуклеотидфосфорамидитных предшественников: 5'-диметокситритилцитидин-3'-О-фосфорамидита, 5'-диметокситритиладенозин-3'-O-фосфорамидита, 5'-диметокситритилгуанозин-3'-O-фосфорамидита и 5'-диметокситритилтимидн-3'-O-фосфорамидита (Applied Biosystems). После удаления защитных групп при помощи гидроксида аммония аминофункционализированный олигонуклеотид далее очищают элюированием из OPEC Cantridge (полимер RPI) - колонки (Clonetech) деионизированной водой. Концентрацию олигонуклеотида контролировали по поглощению у 260 нм.
Часть B. Получение олигонуклеотида, конъюгированного с TMT (CI-TMT).
В 2,5 мМ раствор олигонуклеотида части A в 0,033 мМ буфере карбоната/бикарбоната с pH 9,0 добавляют 10 мг TMT - изотиоцианата (получение 6). Реакционную смесь перемешивают с созданием завихрения и выдерживают 16 ч при 23oC. Продукт очищают хроматографией на колонке с сефадексом C-25 и элюированием деионизированной водой.
Часть C. Получение меченого радионуклидом конъюгата олигонуклеотида и TMT части B.
Раствор конъюгата олигонуклеотида с TMT части B в 0,01 мМ буферном растворе ацетата натрия при 20oC обрабатывают раствором 90YCl3 в 0,01 М буфере ацетата натрия при pH 6,0. Поглощение радиоактивной метки конъюгатом контролировали тонкослойной хроматографией.
Часть D. Получение олигонуклеотида (I), который комплементарен олигонуклеотиду CI части A.
20-Мерный олигонуклеотид, содержащий 3'-аминогруппу и имеющий формулу 5'-TCT TAT GGA CGG AGA AGC-C5-амино-T-3, получают на синтезаторе ДНК (Applied Biosystems DNA Synthesizer) как в части A с применением аминомодификатора (UniLink Amino Modifier, Clonetech) в качестве предшественника 3'-аминогруппы и 2-дезоксинуклеотидфосфорамидитных предшественников: 5'-диметокситритилцитидин-3'-O-фосформамидита, 5'-диметокситритиладенозин-3'-O-фосфорамидита, 5'-диметокситритилгуанозин-3'-O-фосфорамидита и 5'-диметокситритилтимидин-3'-O-фосфорамидита (Applied Biosystems). После удаления защитных групп при помощи гидроксида аммония аминофункционализированный олигонуклеотид далее очищали элюированием из OPEC cartridge (полимер RPI) - колонки (Clonetech) деионизированной водой. Концентрацию олигонуклеотида контролировали по поглощению у 260 нм.
Часть E. Гибридизация CI, конъюгированного с TMT и меченого 90Y, с комплементарным олигонуклеотидом, обозначенным 1.
В раствор олигонуклеотида I, полученного в части D, в содержащем фосфатный буфер соляном растворе с pH 7,4 при 4oC добавляют раствор 90Y - меченого TMT - CI, как указано выше. После 1 ч компоненты реакционной смеси анализировали электрофорезом на полиакриламидном геле. Радиоактивную метку наблюдают в положении геля, соответствующем гибридизированному олигонуклеотиду.
Изобретение было описано подробно с приведением предпочтительных примеров осуществления изобретения, однако в пределах существа и объема изобретения могут быть осуществлены его варианты и модификации.
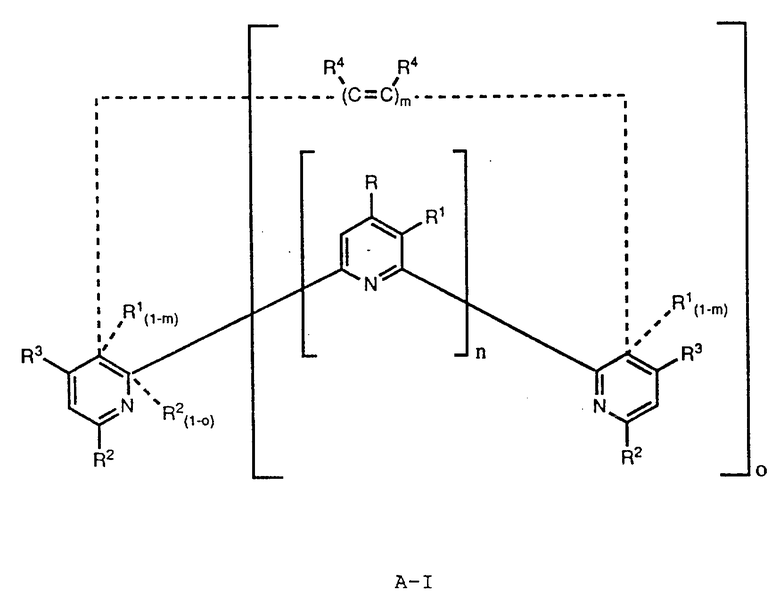
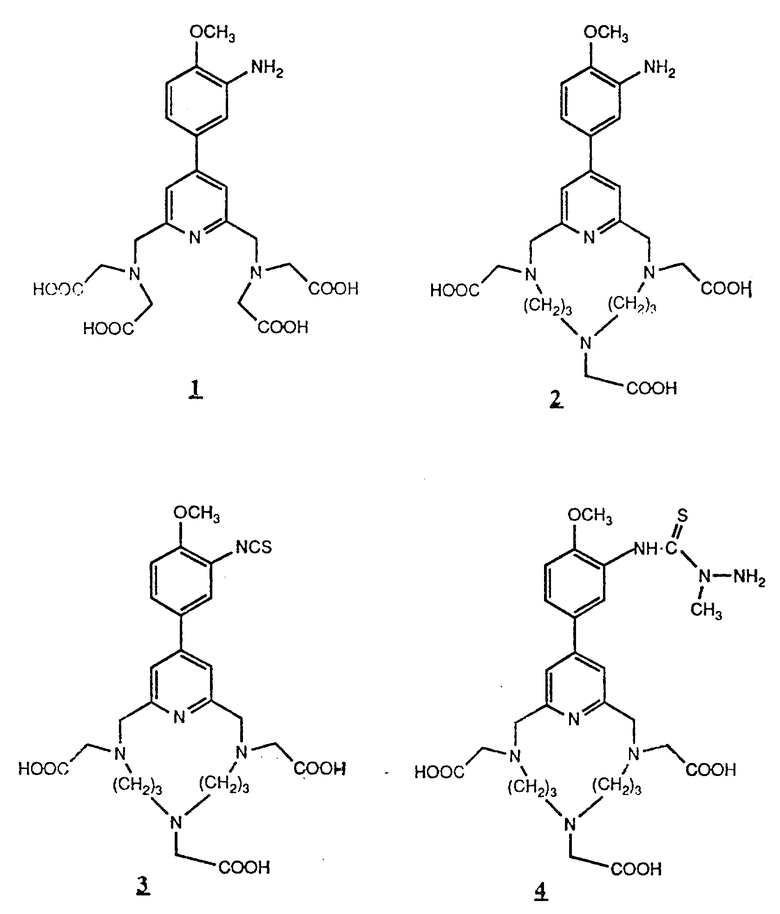
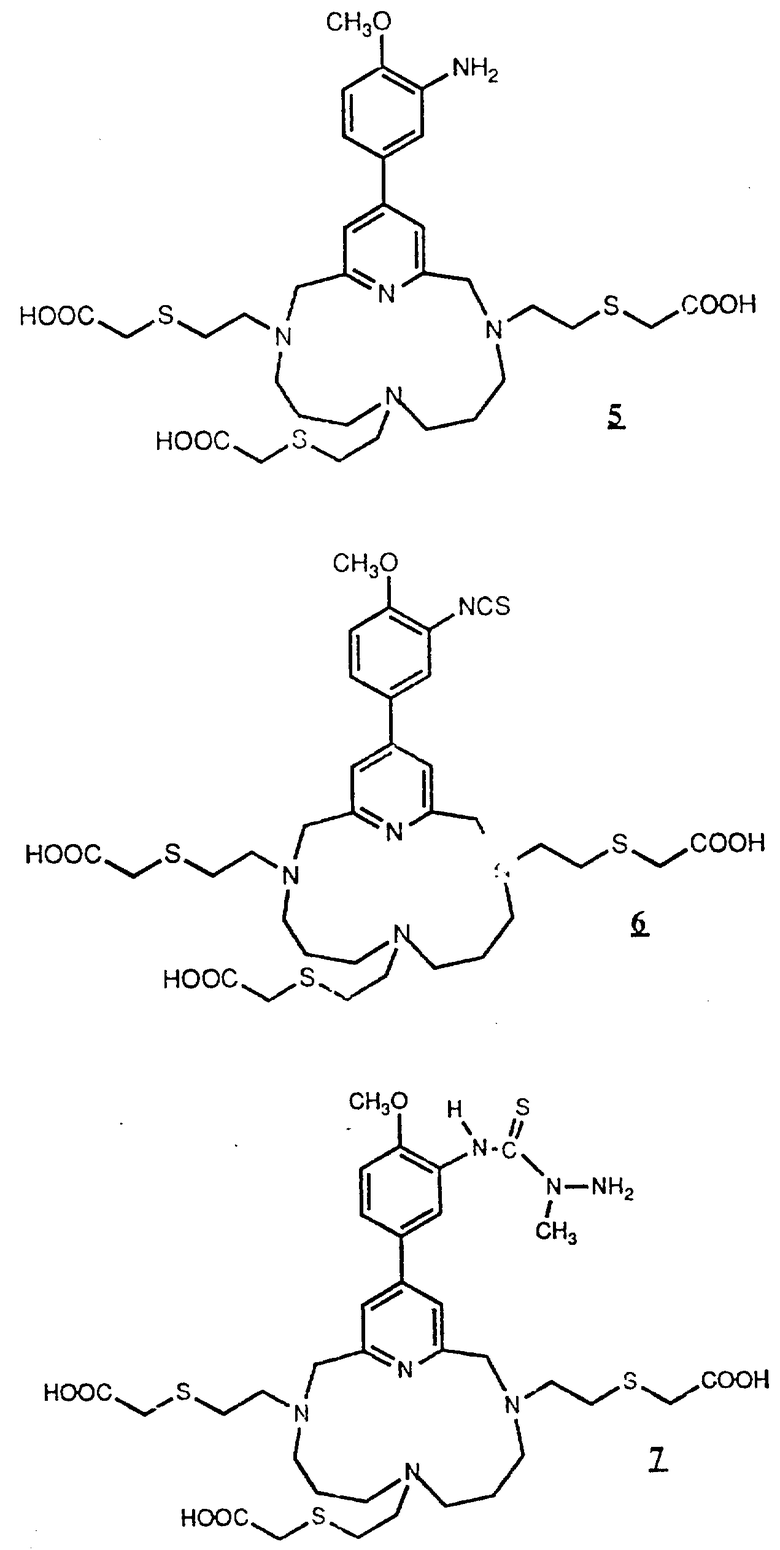
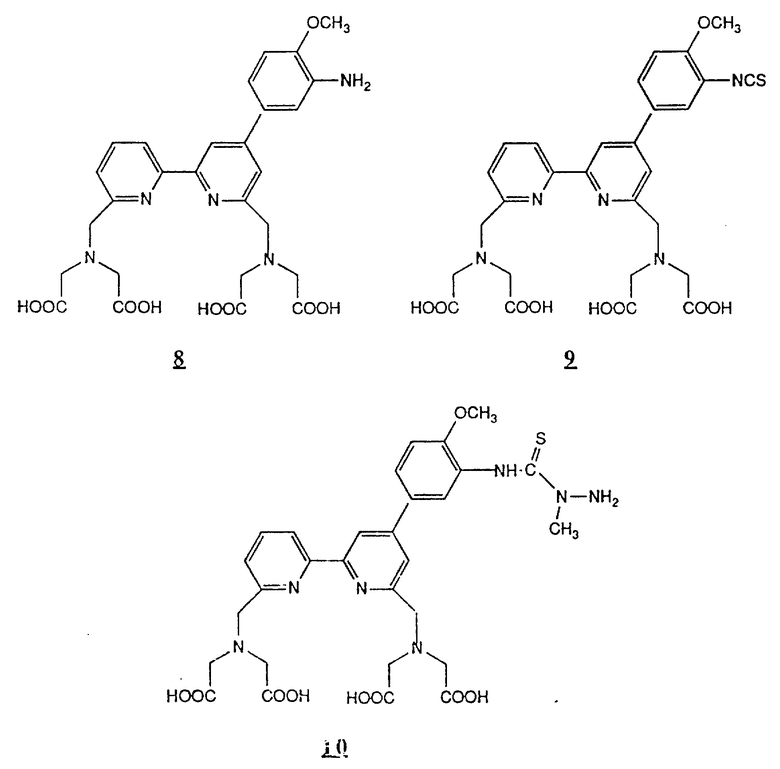
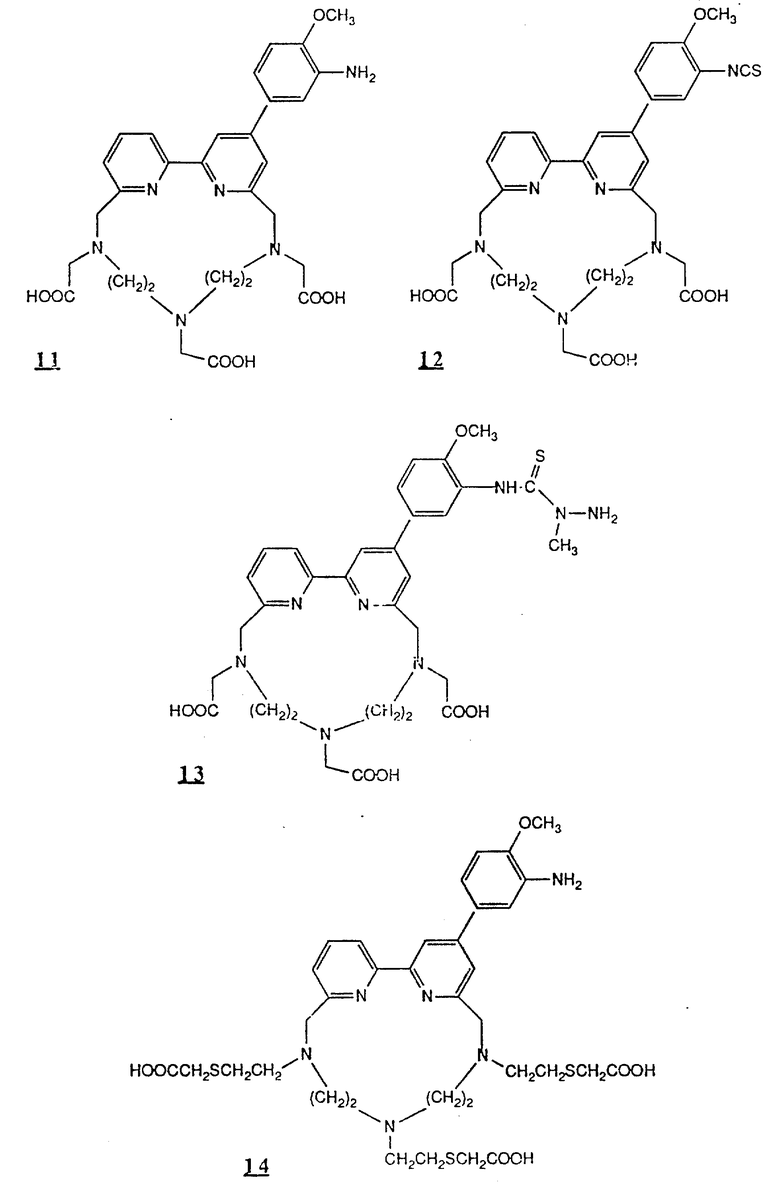

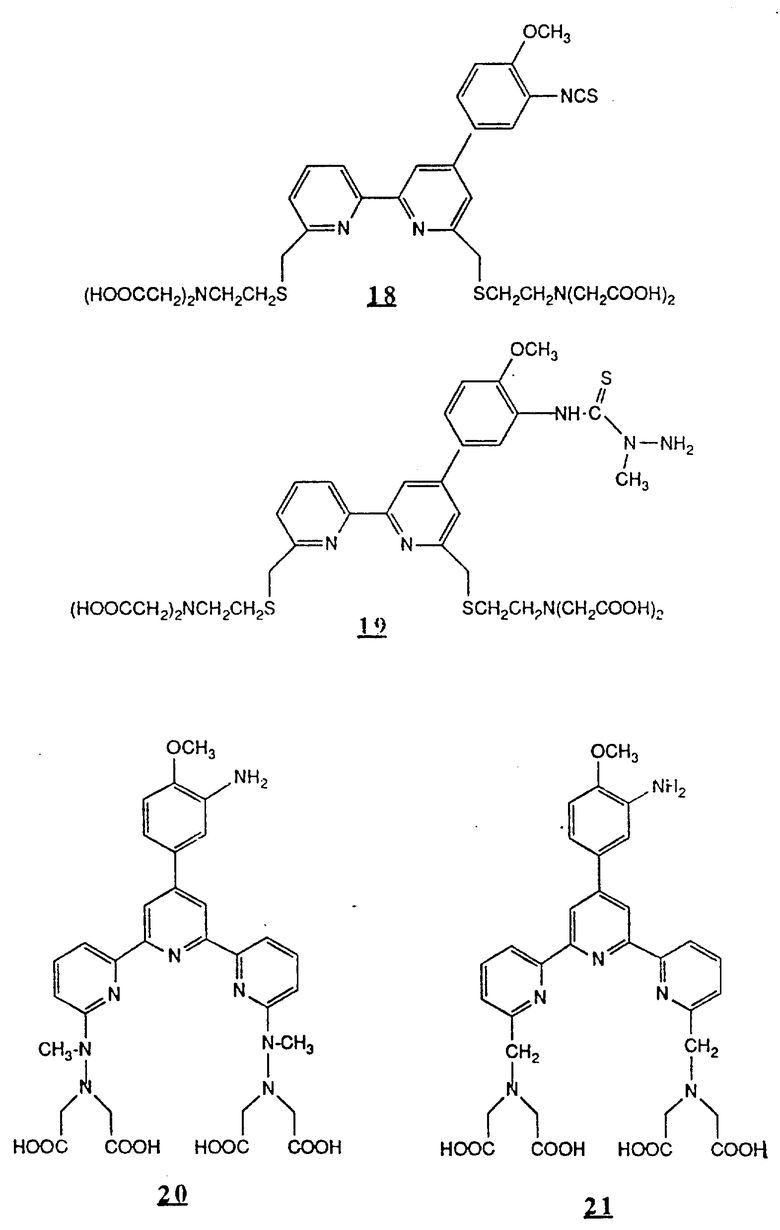
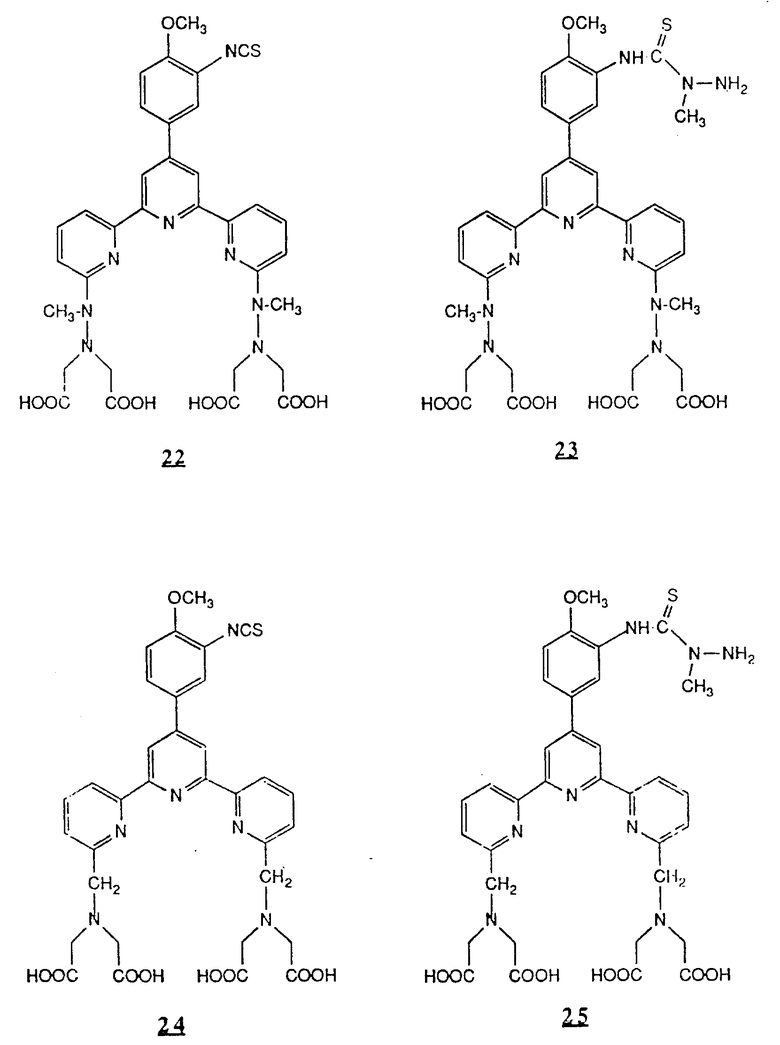
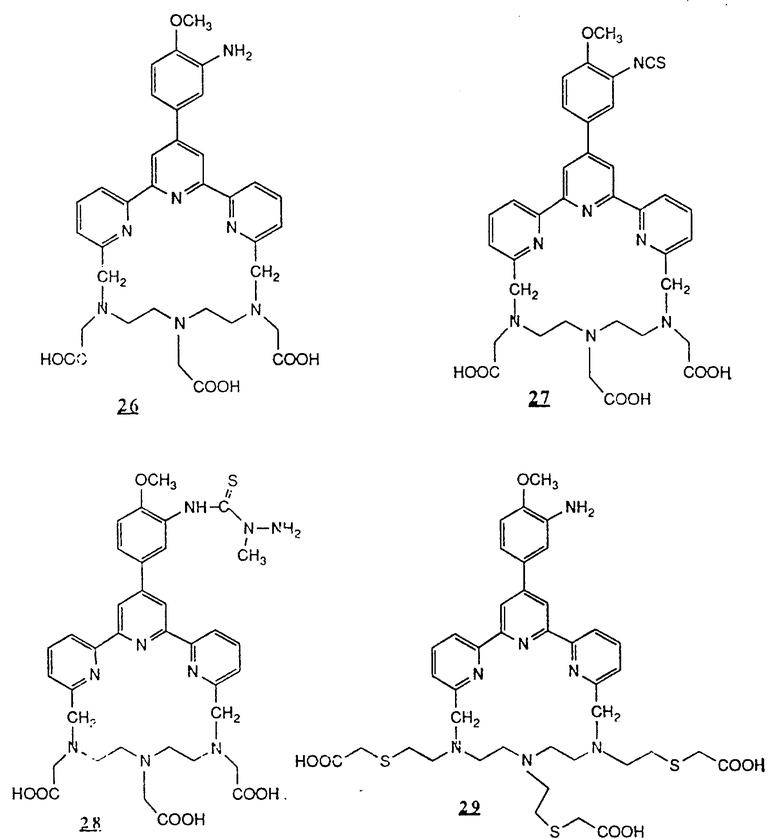
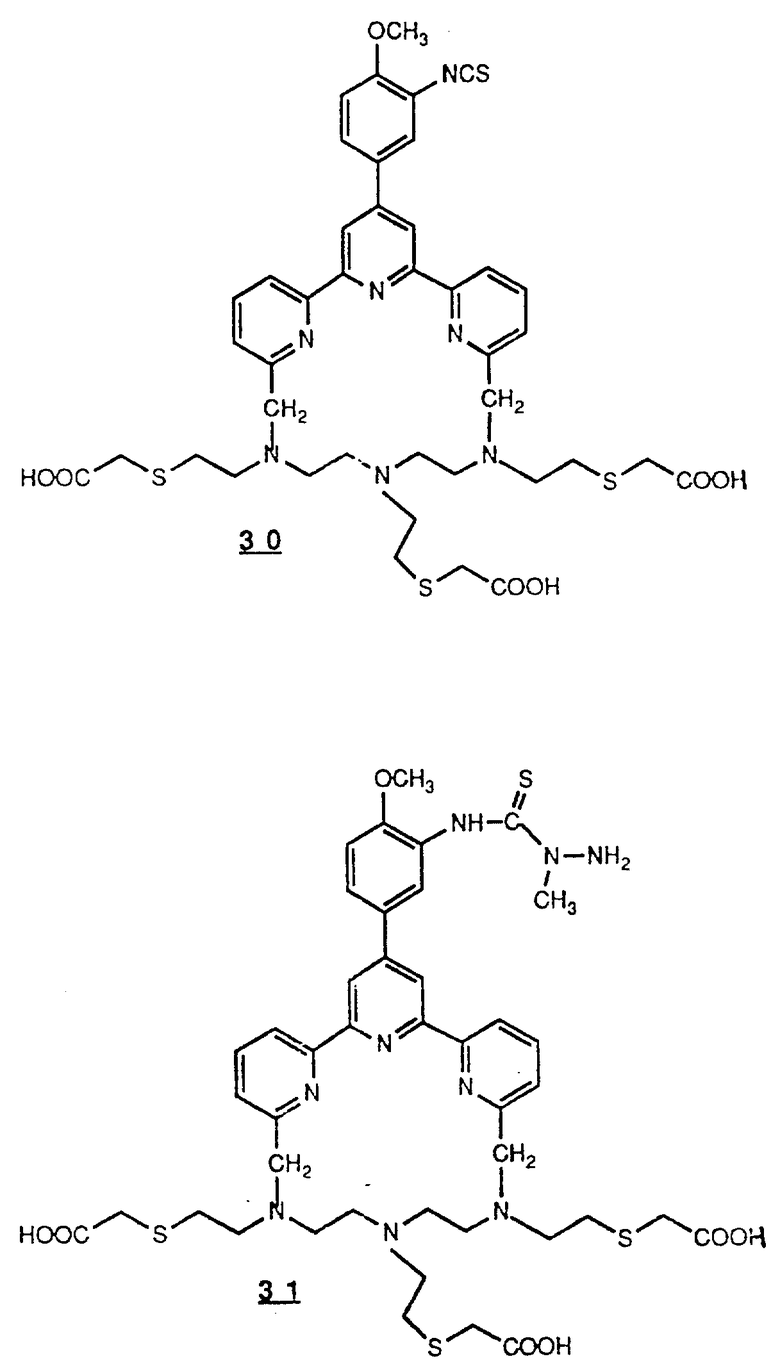
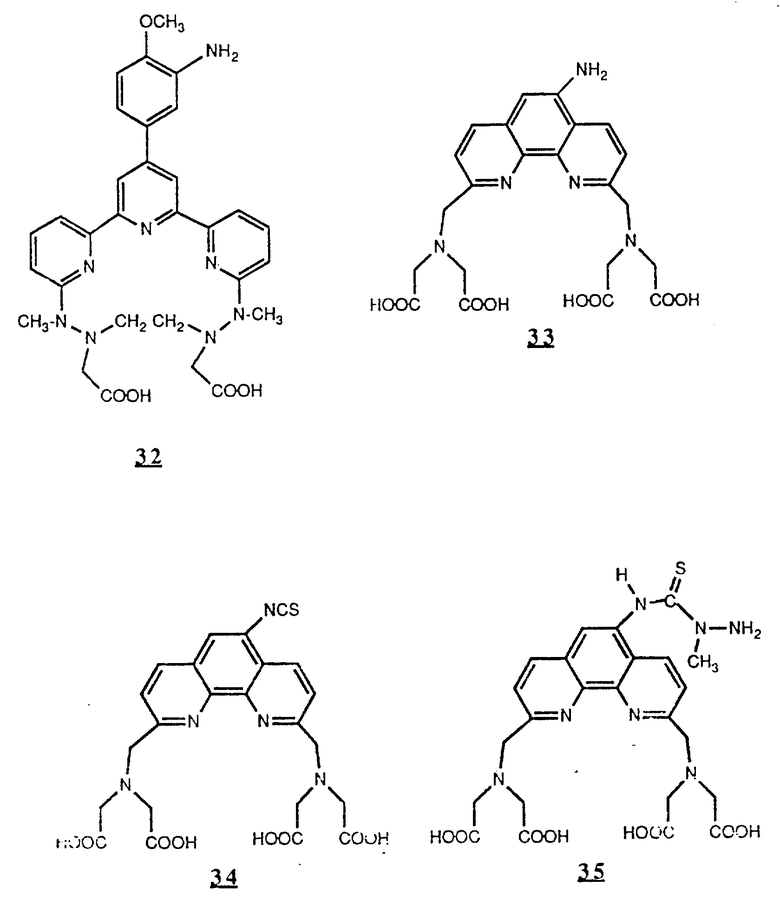
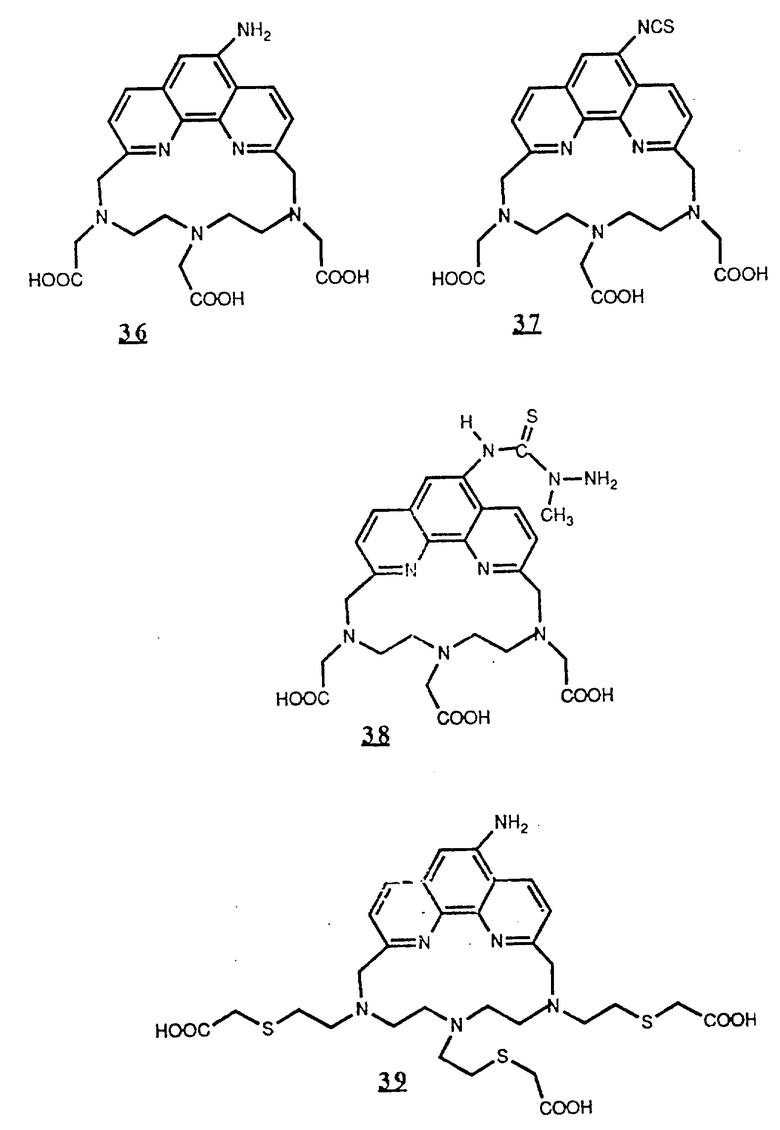
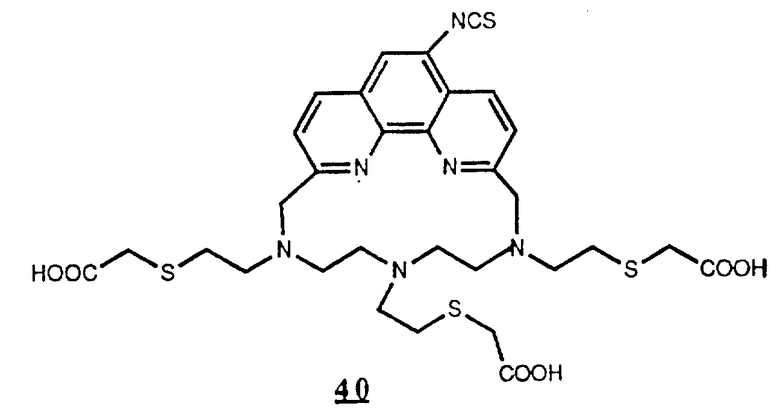
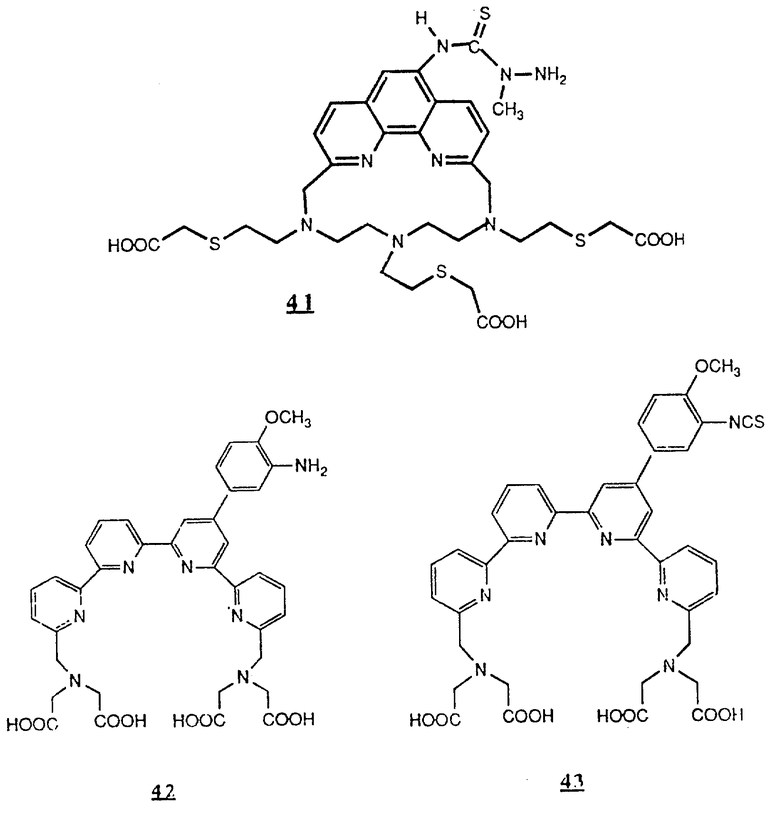
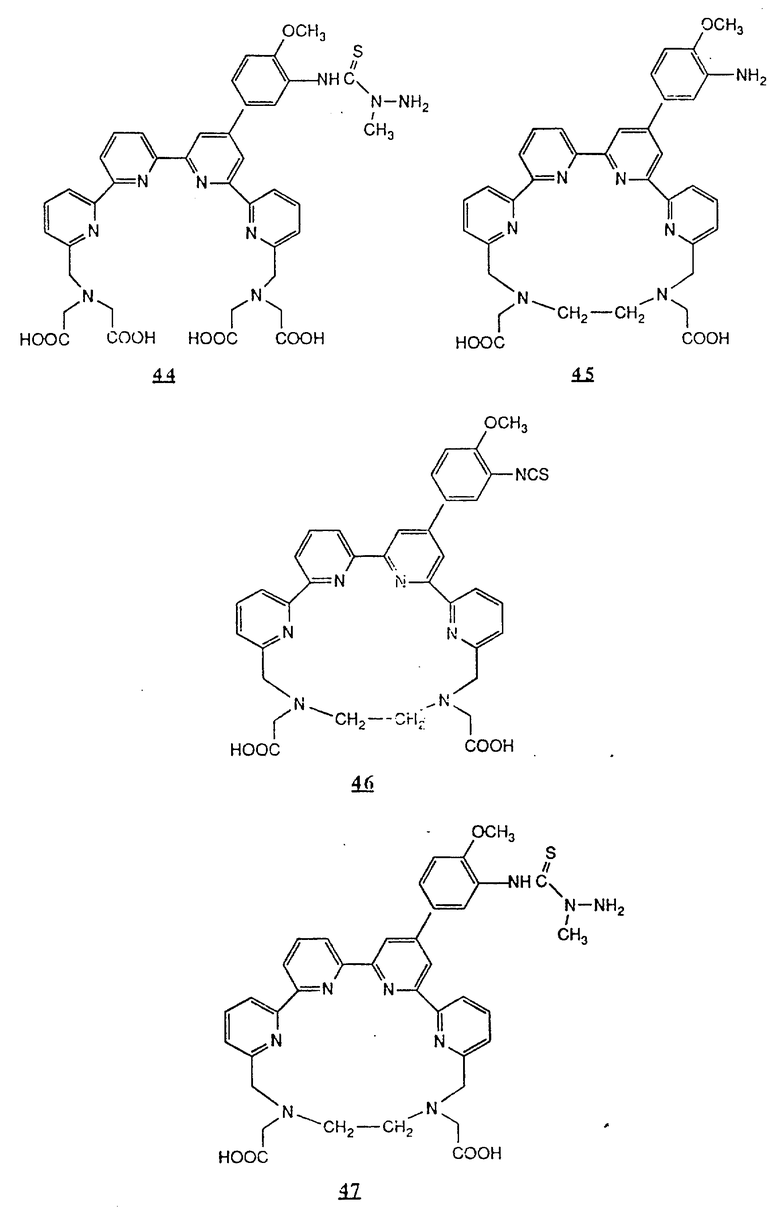
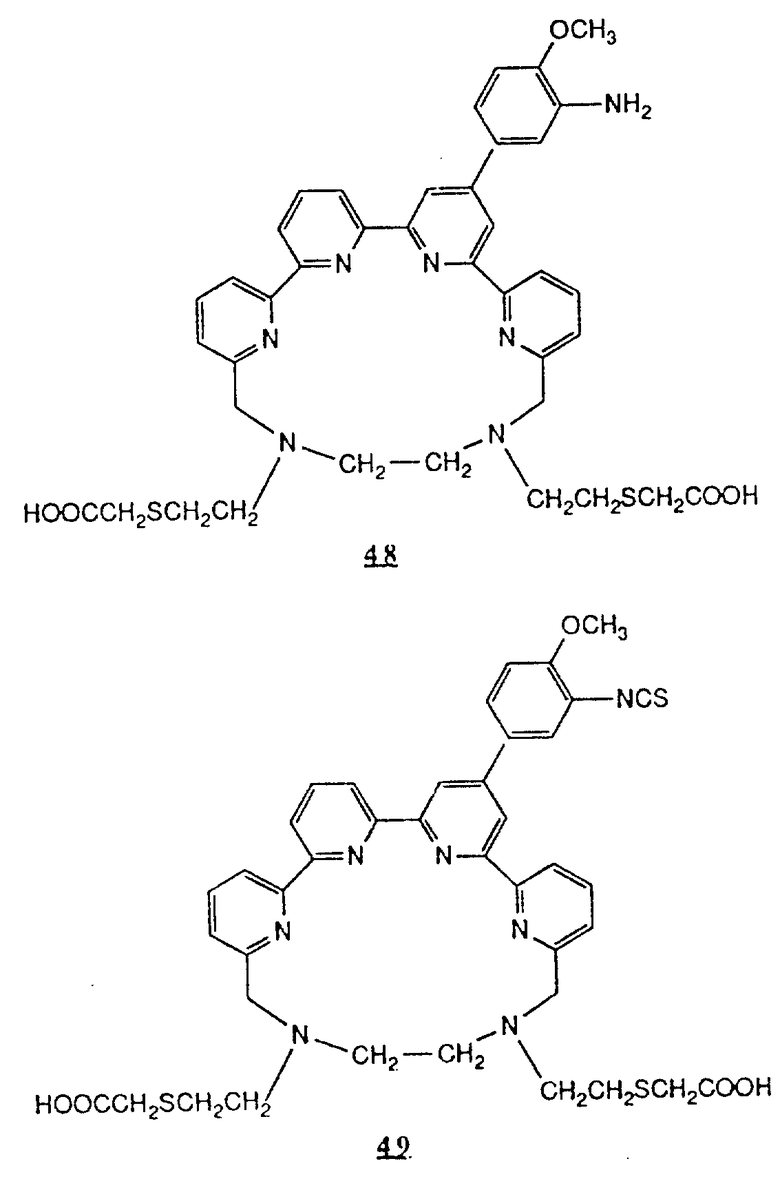
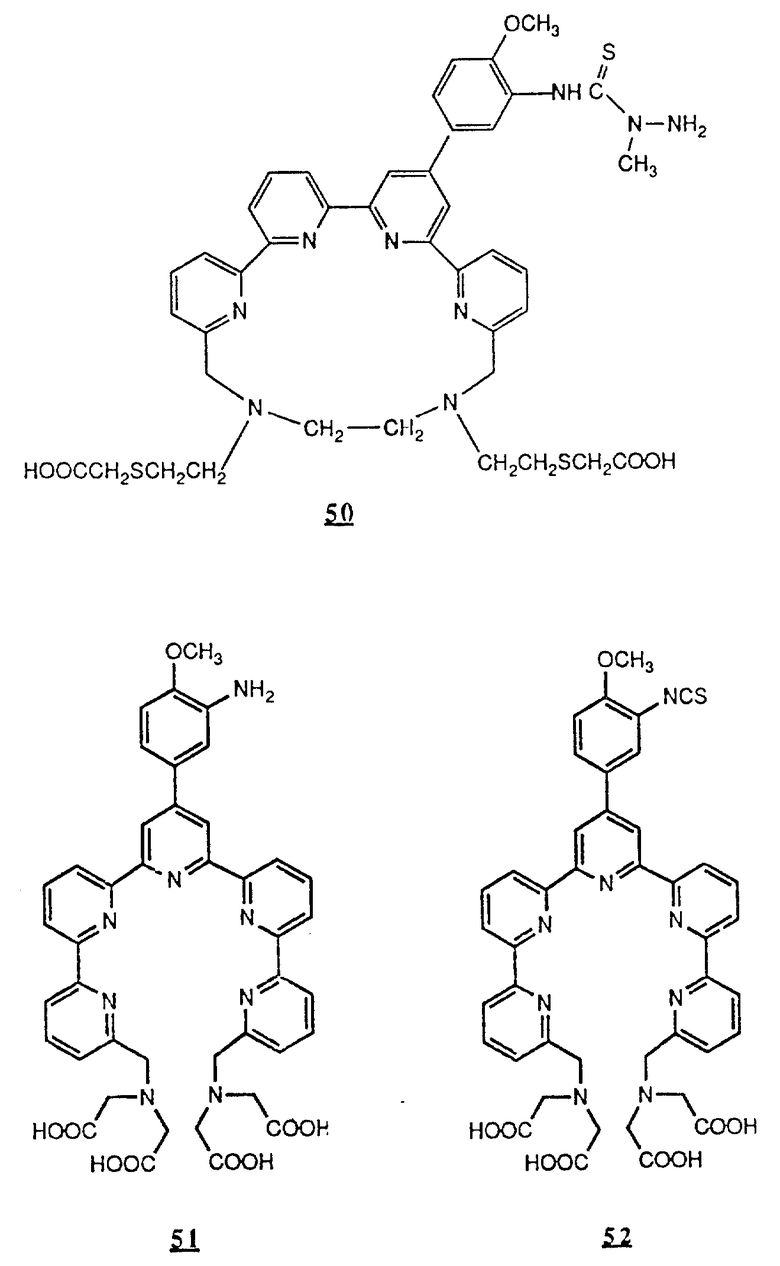
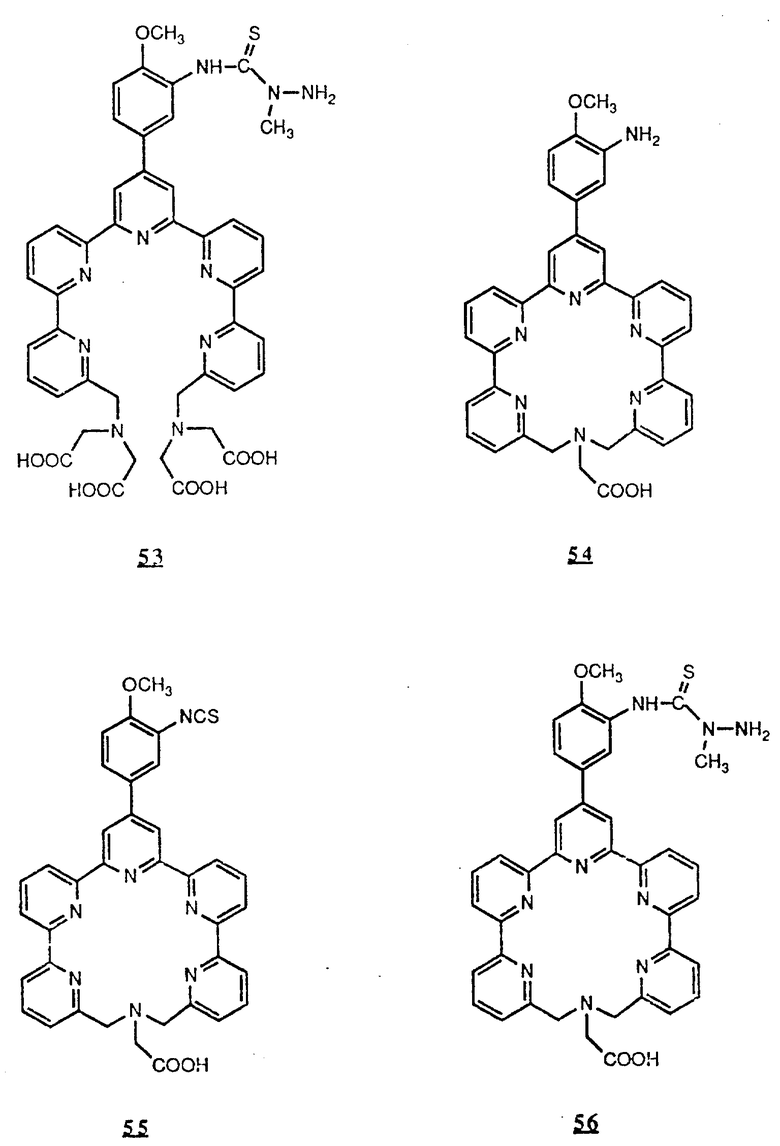
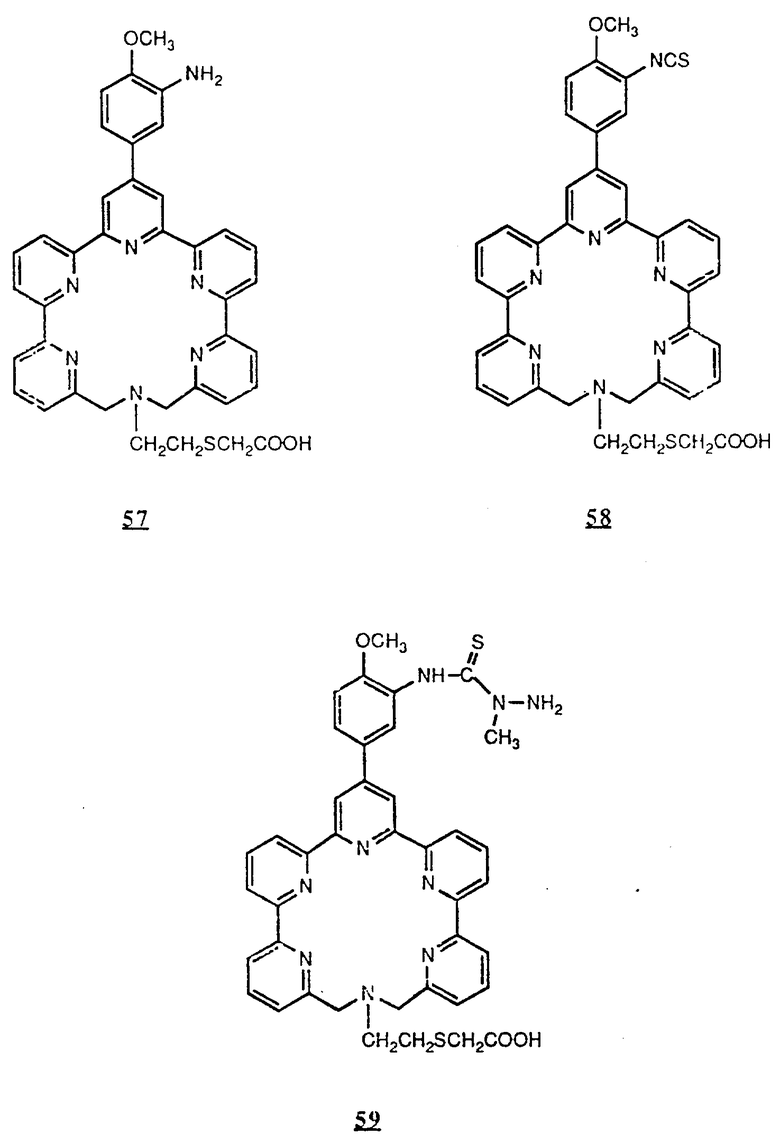
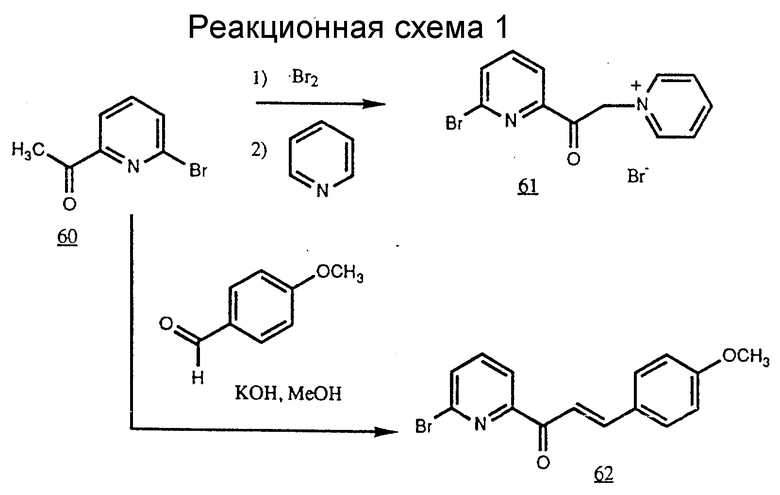
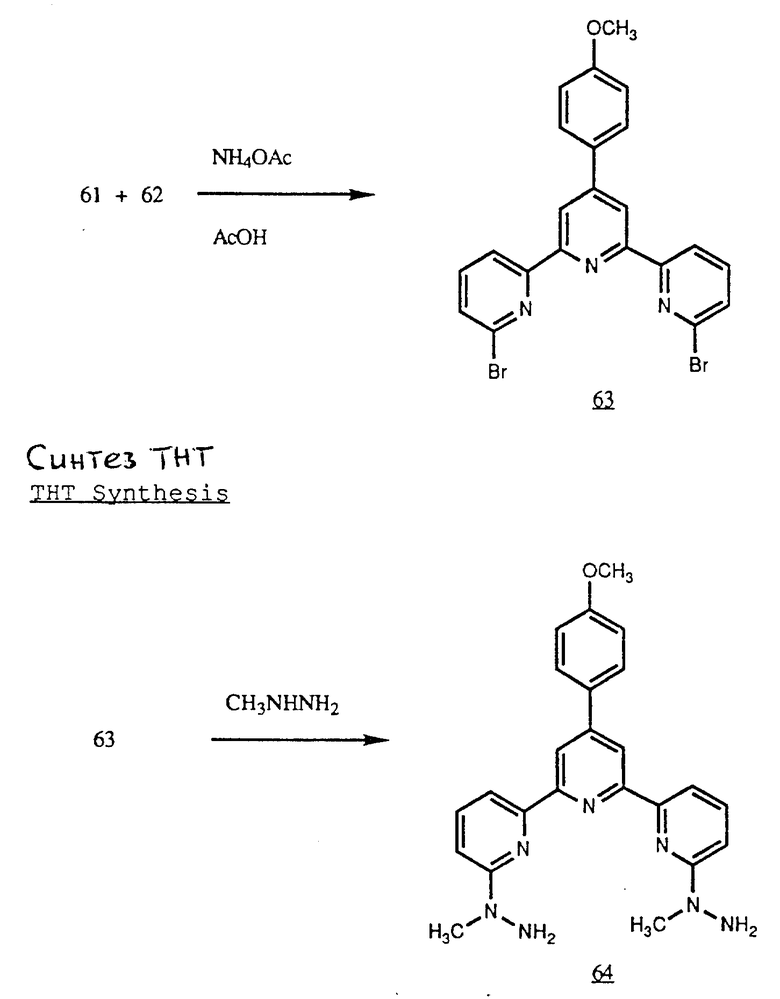
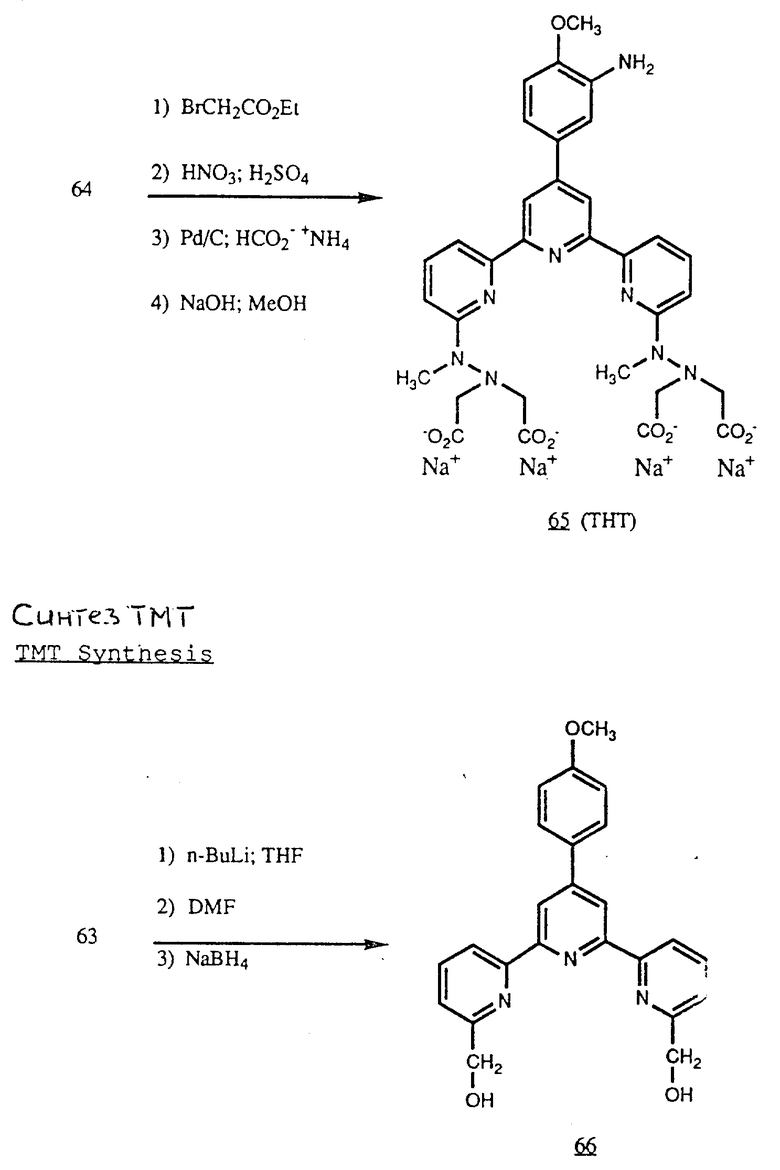
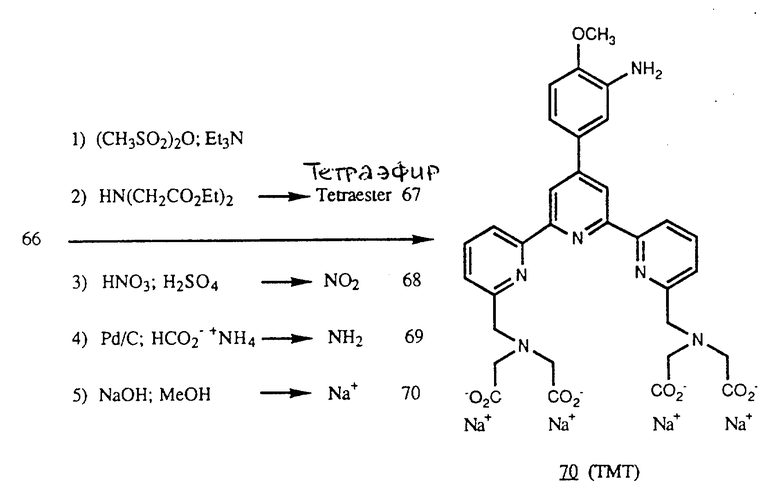
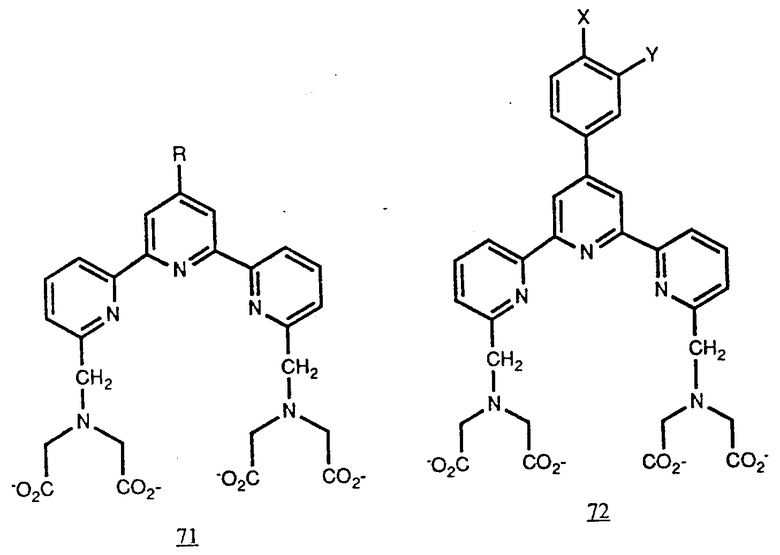
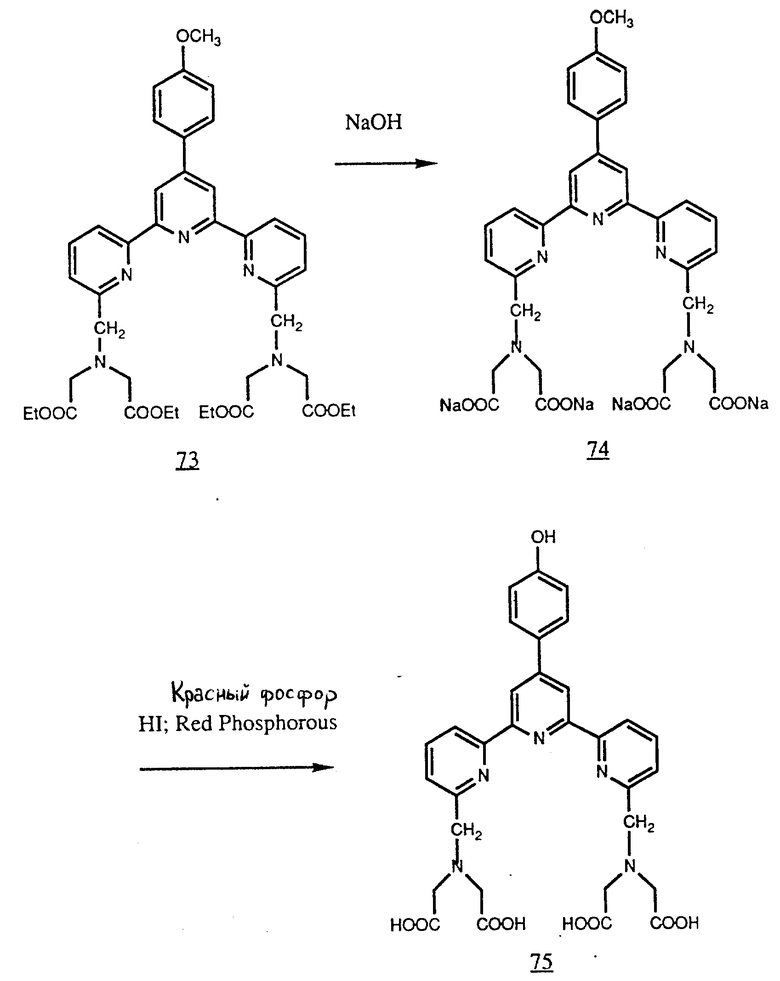
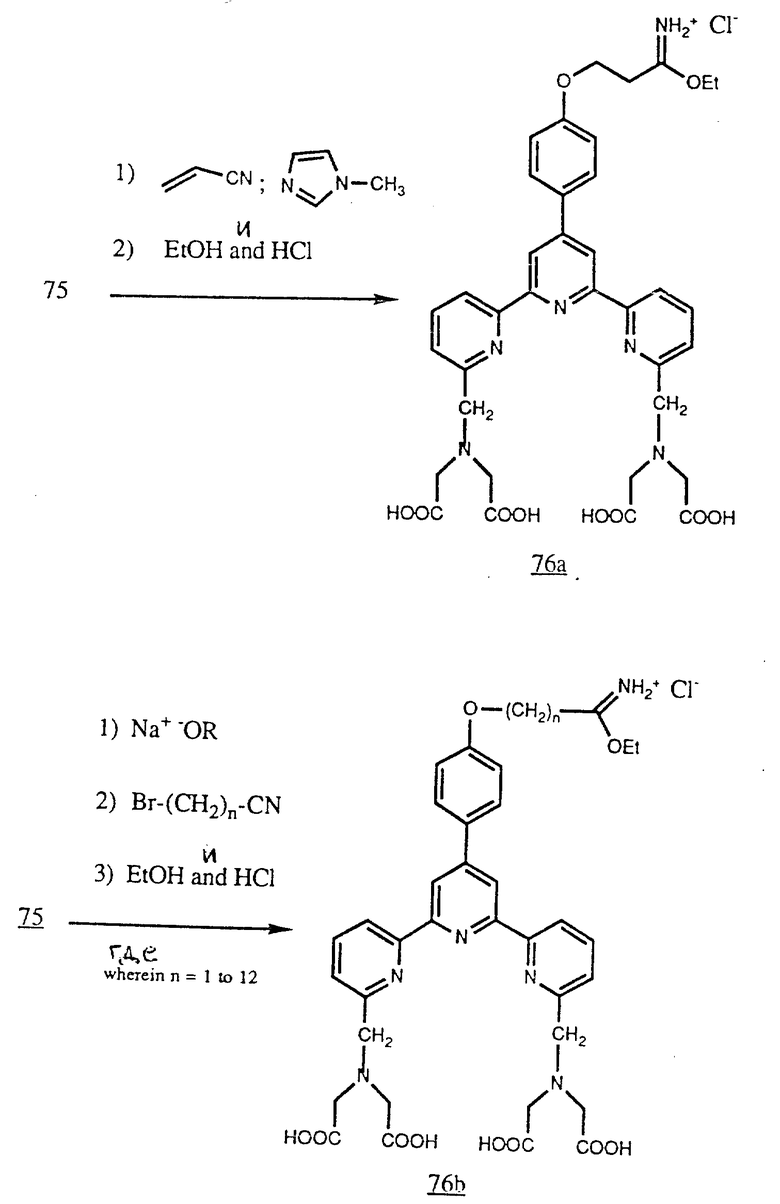
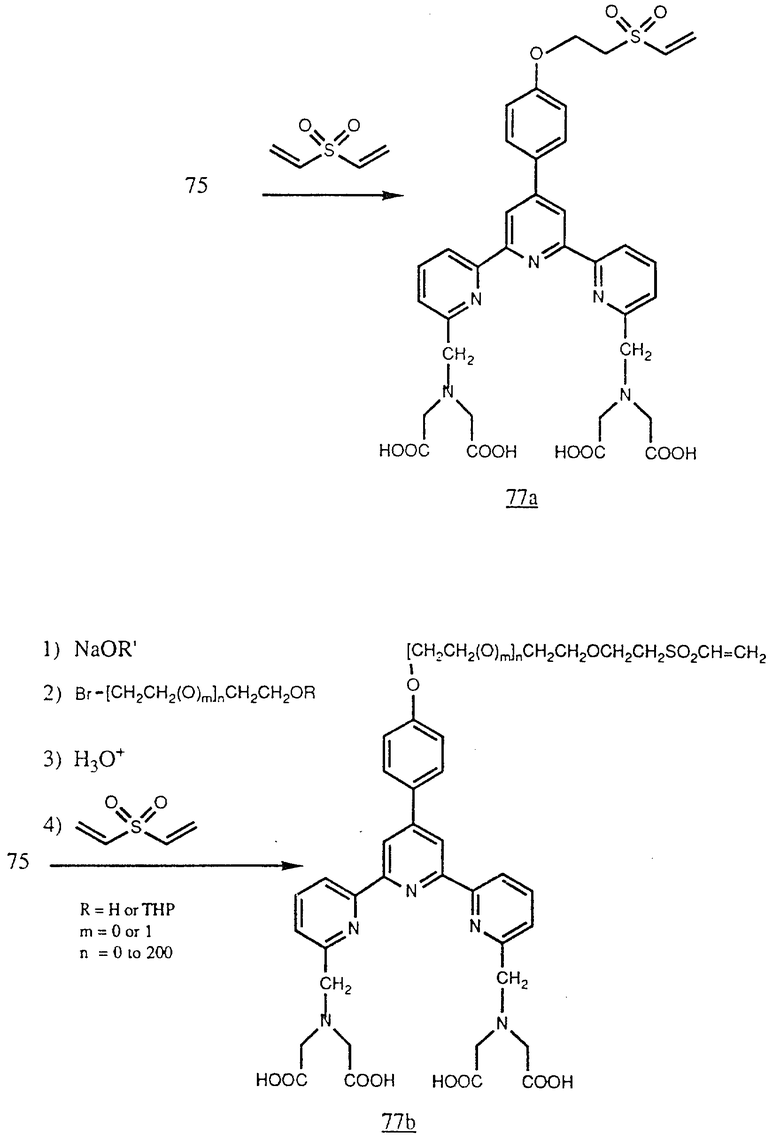
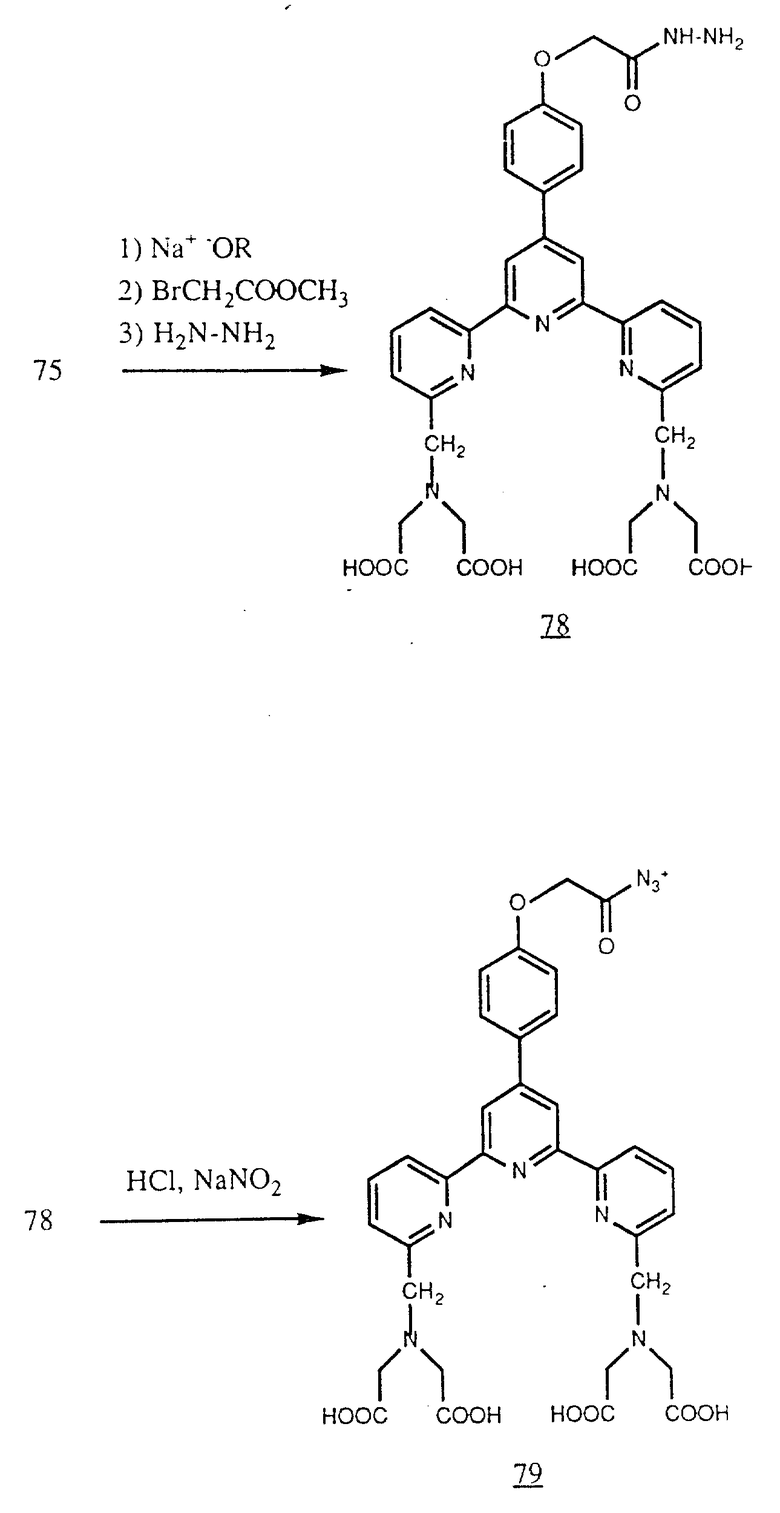
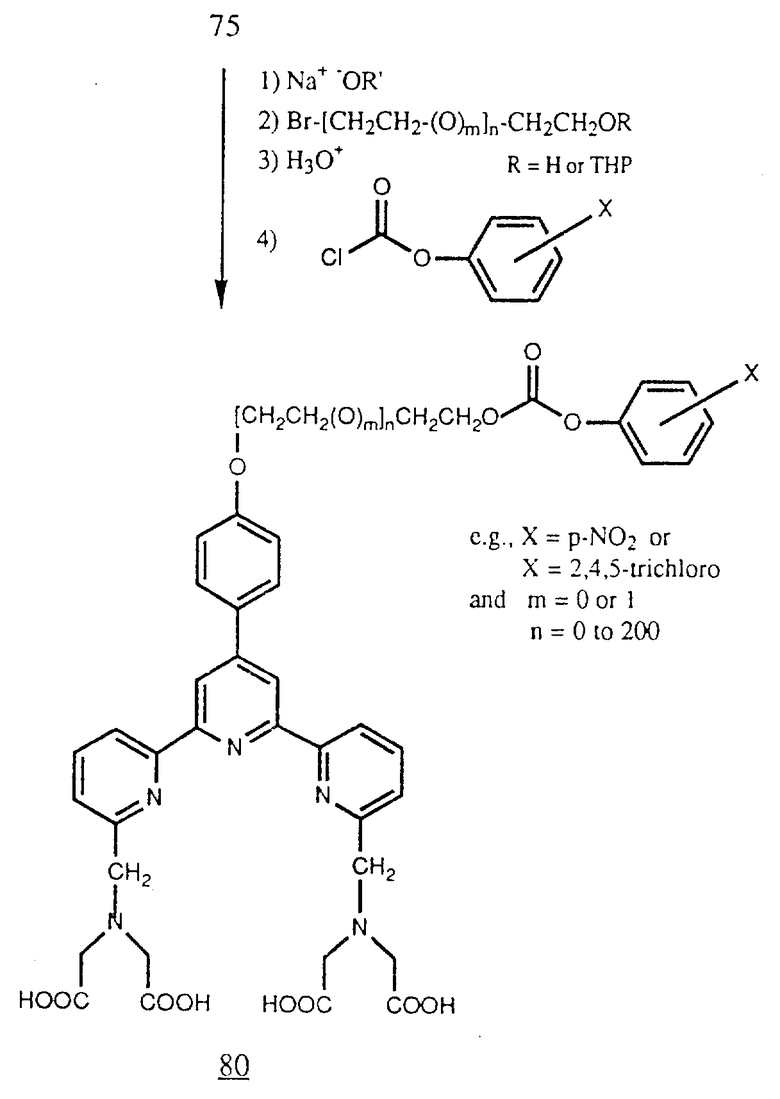
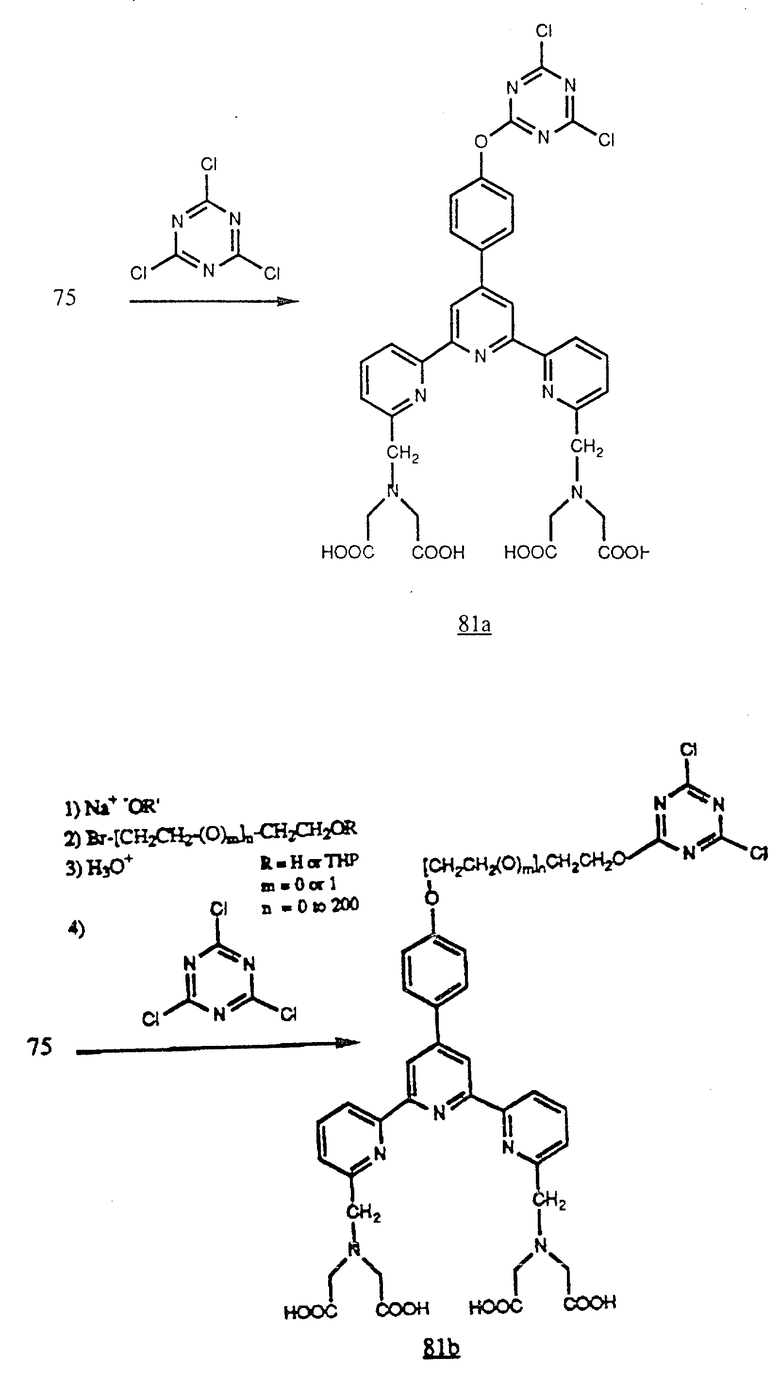
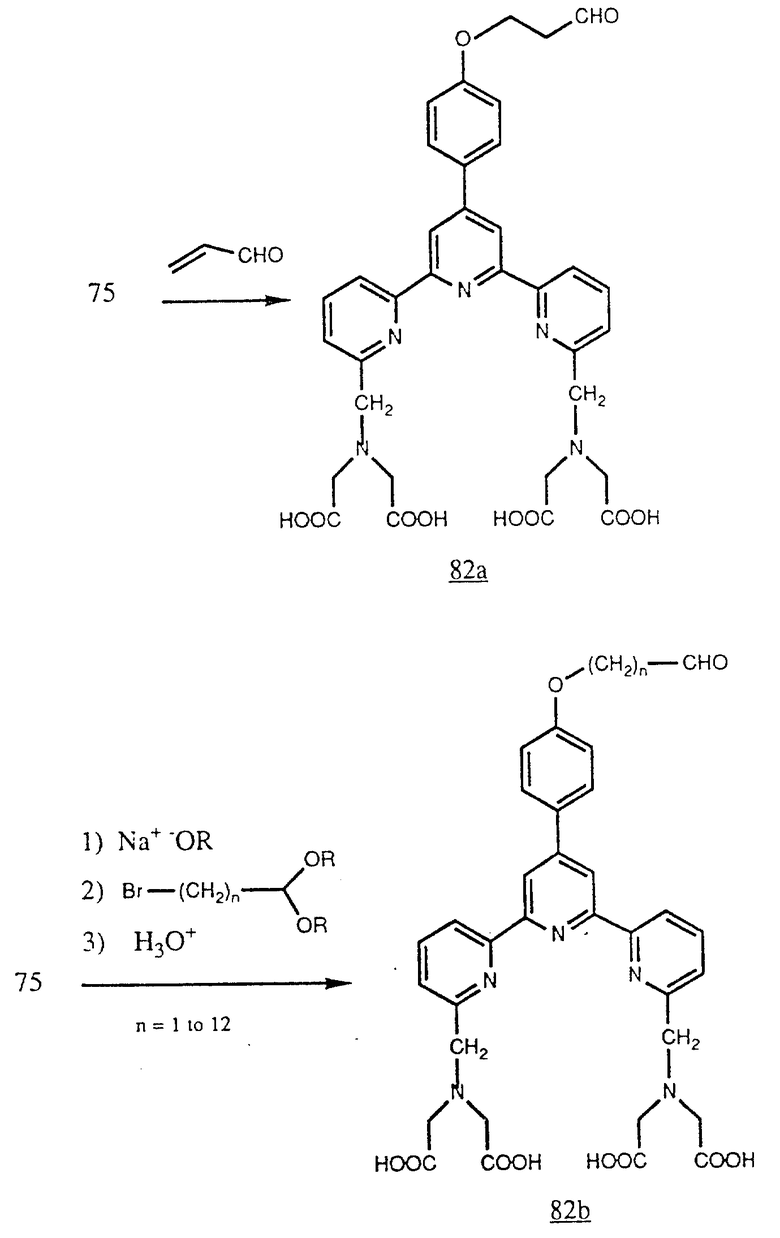
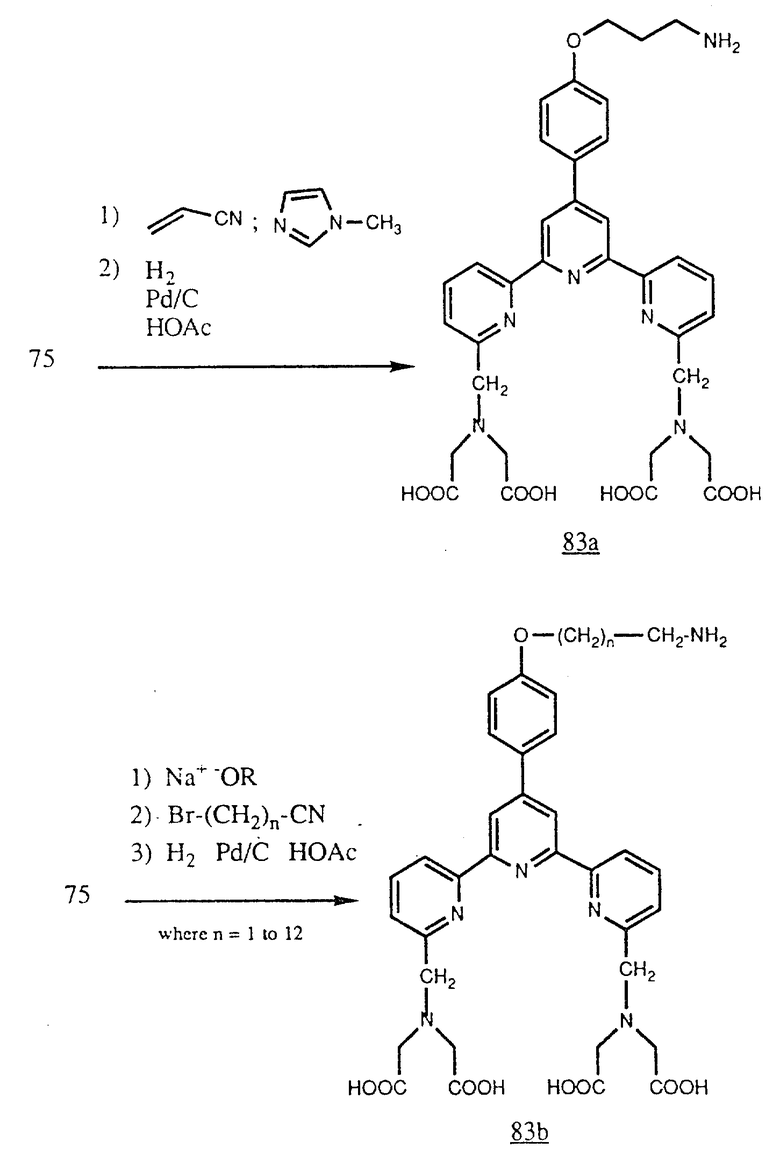
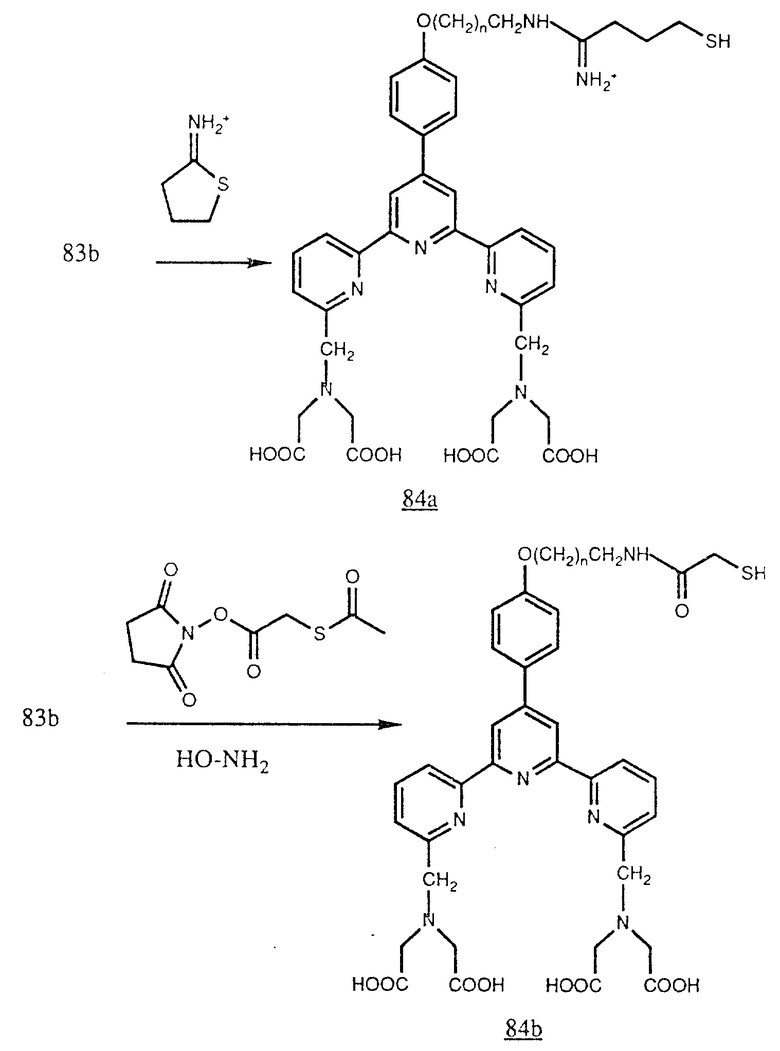

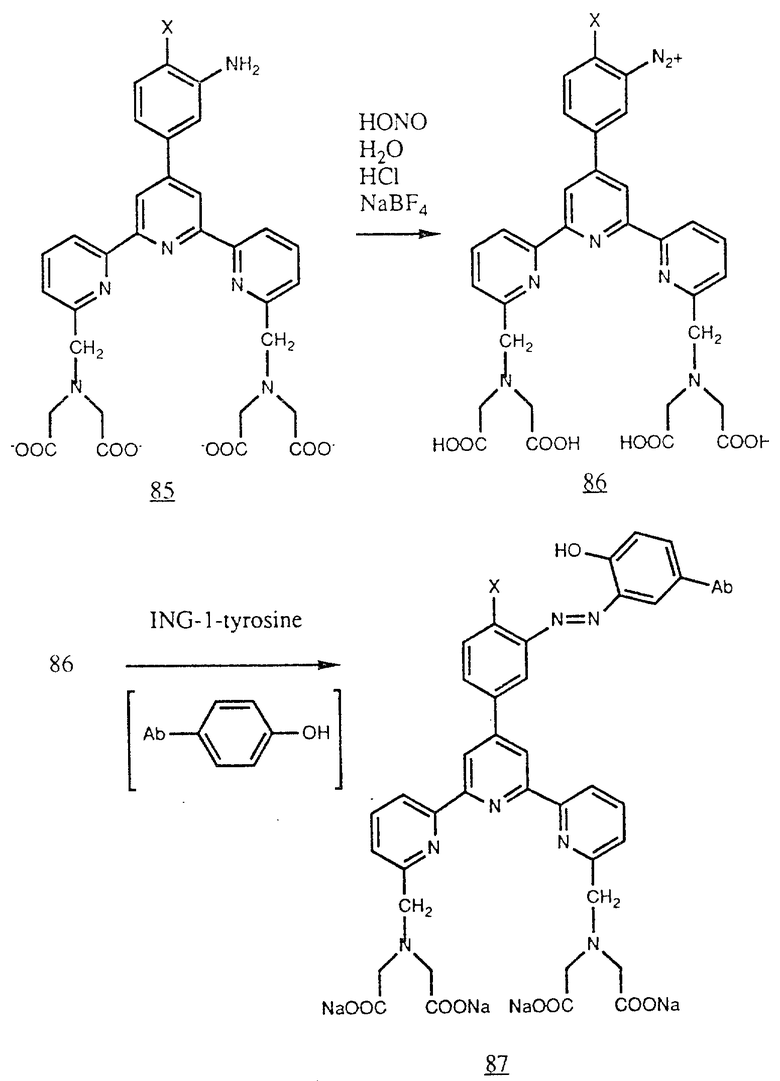
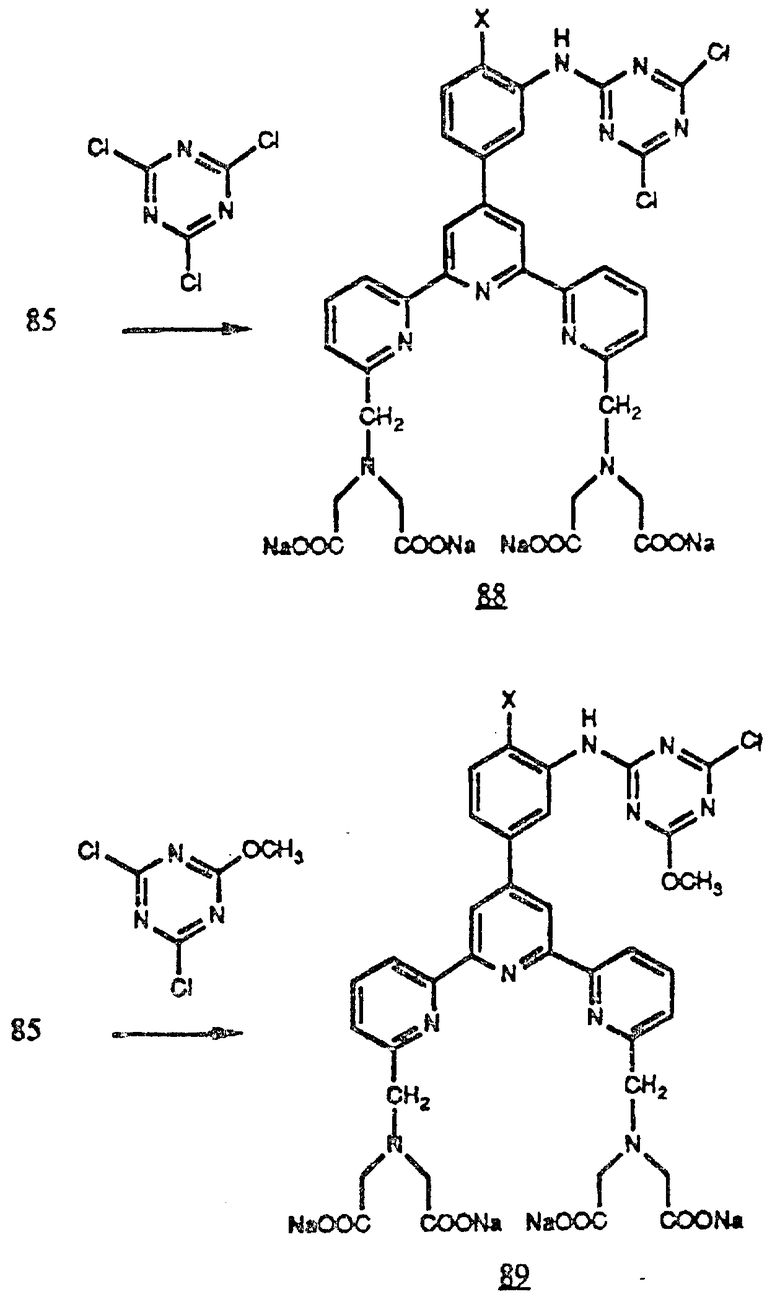
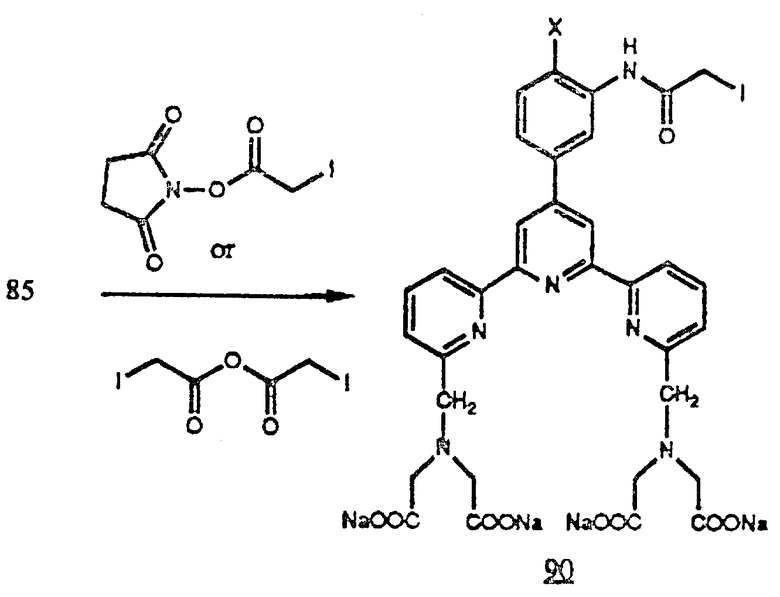
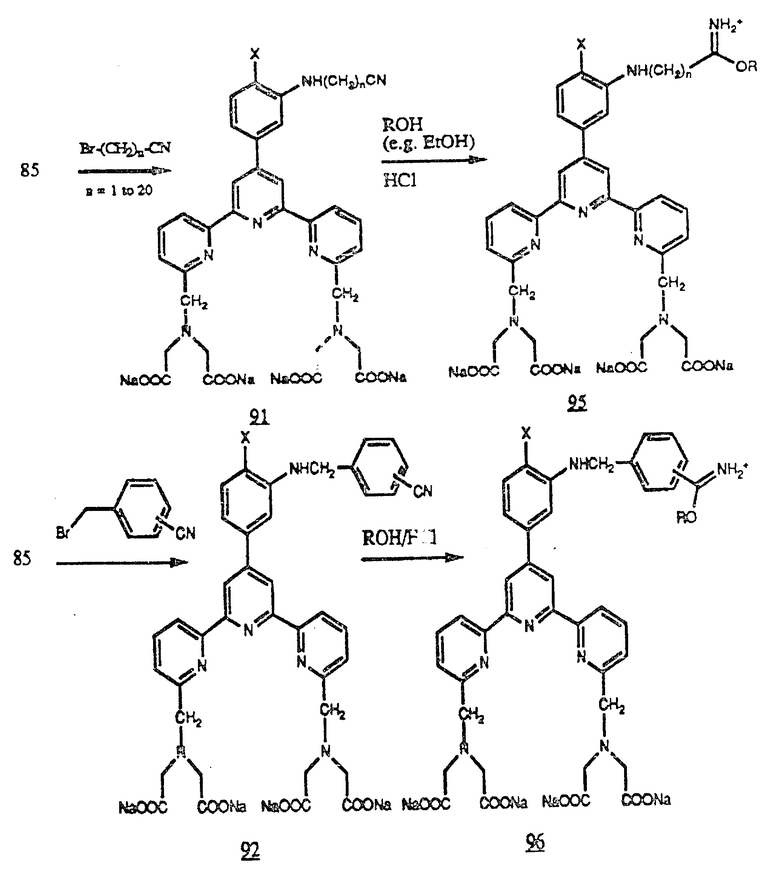
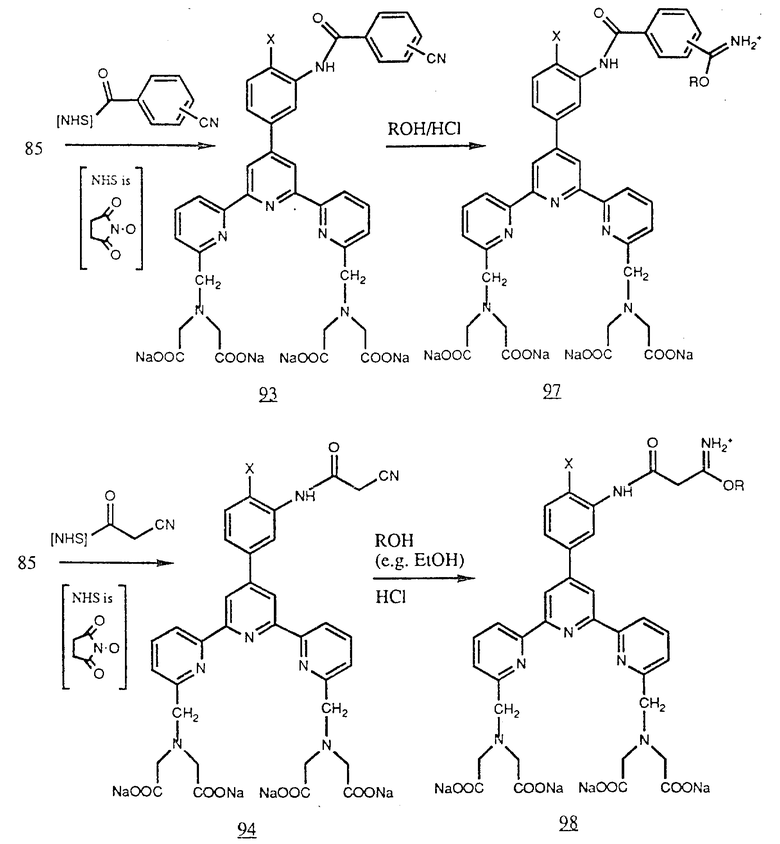
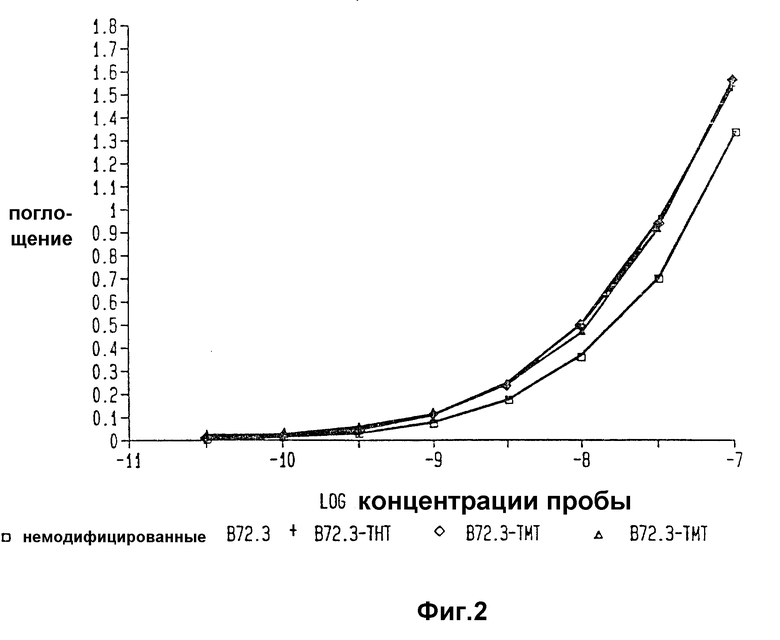
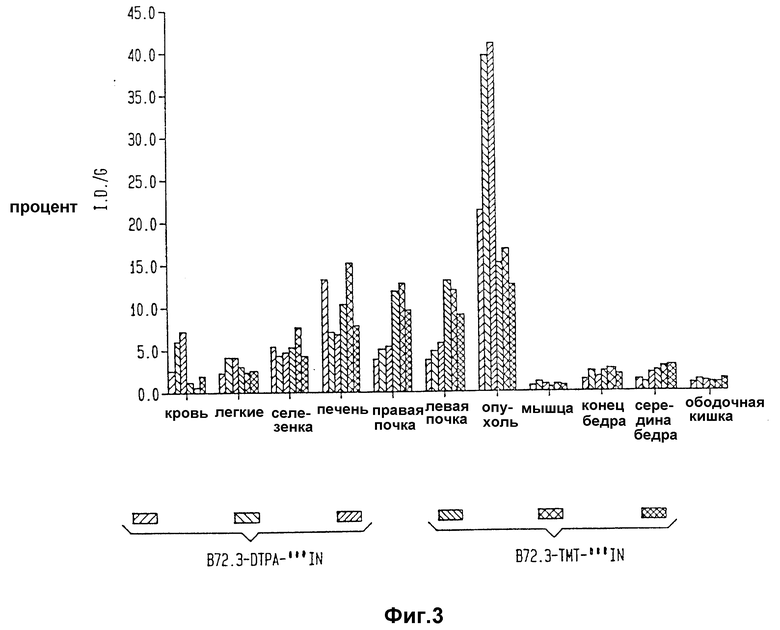
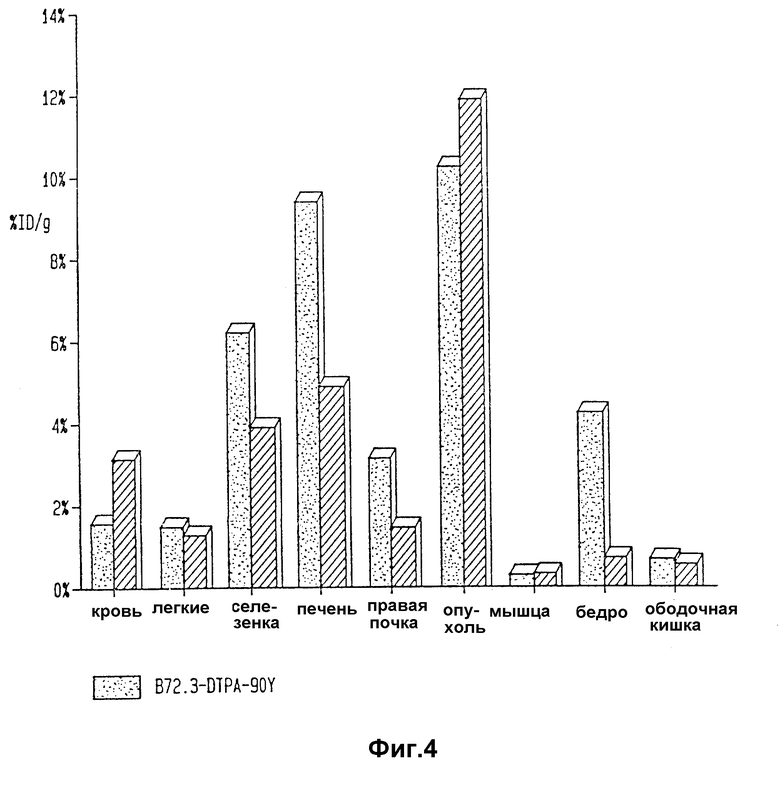
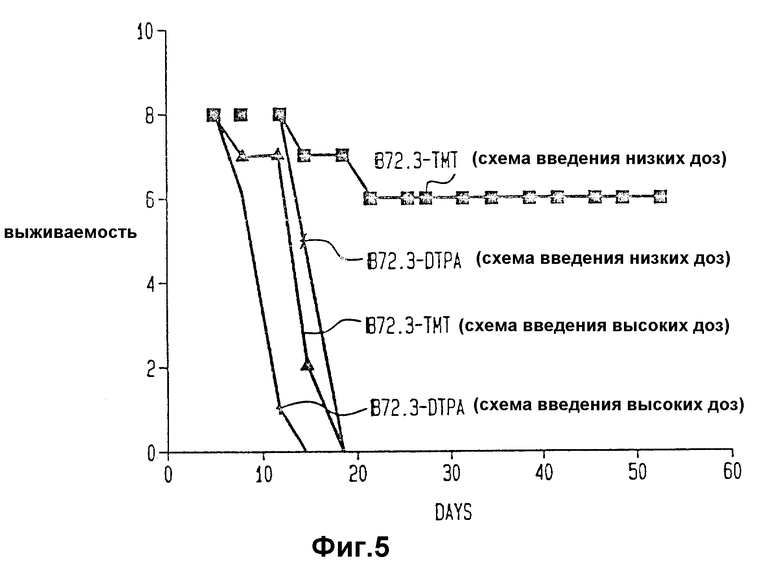
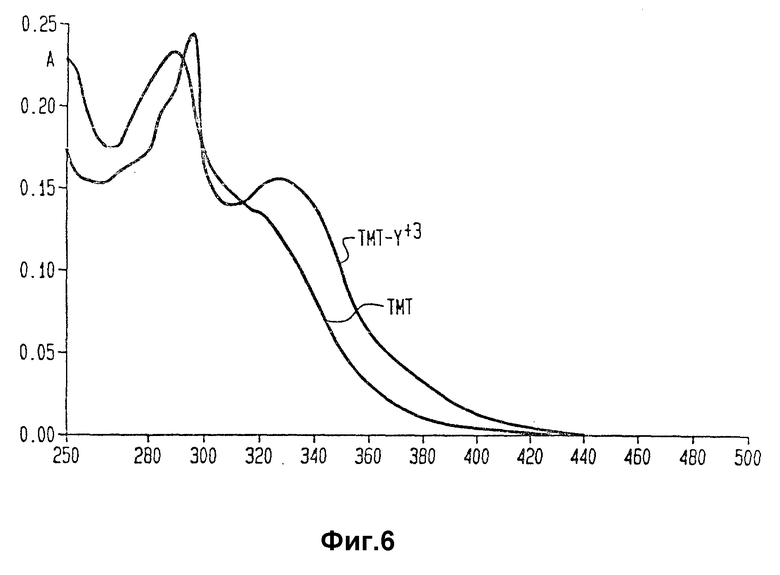
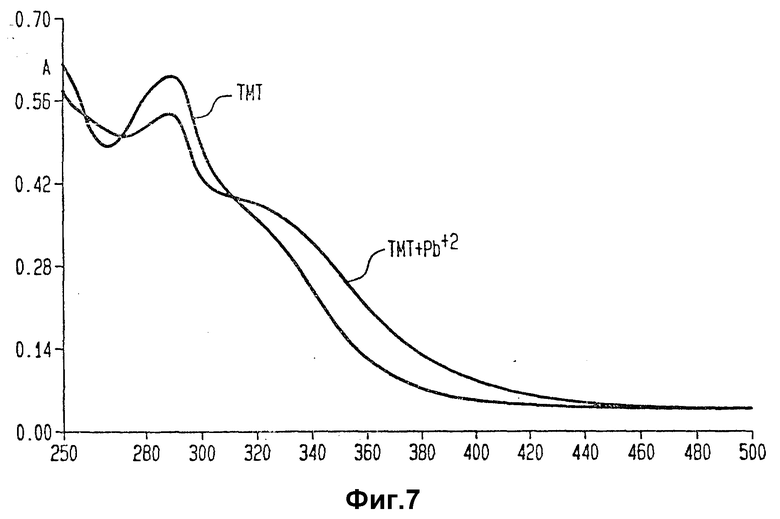
Изобретение относится к радиоактивному иммунореагенту направленного действия, который представляет собой конъюгат комплексообразующего агента и иммунореактивной группы, меченый ионом радиоактивного металла. Ион радиоактивного металла имеет атомный номер от 21 до 84. Иммунореактивная группа представляет собой антитело, олигонуклеотид или полипептид. Комплексообразующий агент является производным трипиридина. В описании изобретения приведена общая формула используемого комплексообразующего агента. Описана также композиция для получения изображения злокачественного новообразования в организме, содержащая контрастирующий радиоактивный агент приведенного выше состава в эффективном количестве и фармацевтически приемлемый разбавитель. Кроме того, изобретение относится к способу получения изображения злокачественного новообразования. Способ осуществляют введением пациенту композиции с радиоактивной меткой, включающей эффективное количество комплекса. Измеряют интенсивность излучения, создаваемого радиоактивной меткой за период времени, необходимый для аккумуляции антител на участке новообразования. Интенсивность излучения определяют с помощью специального устройства для фотосканирования. Приводится также комплексообразующий агент, входящий в состав иммунореагента направленного действия, представляющий производное трипиридина. 4 с. и 8 з.п. ф-лы, 7 ил.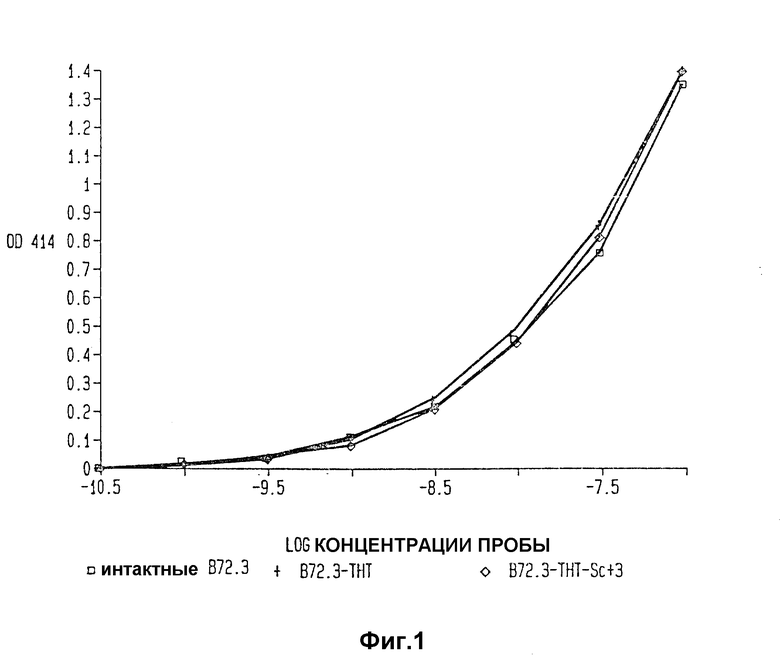
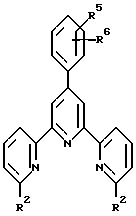
где каждый R2 независимо выбирают из метилентиоэтилениминодиуксусной кислоты, гидрозинилилидендиуксусной кислоты и солей этих кислот, или две группы R2, взятые вместе, представляют собой атомы, необходимые для замыкания макроциклической кольцевой структуры, содержащей по меньшей мере один гетероатомный координационный центр и по меньшей мере одну алкиленовую группу, образующую часть кольцевой структуры;
R5 - алкокси;
R6 - группа, реагирующая с белком,
а также его соли, комплексы и белковые конъюгаты, а иммунореактивная группа представляет собой антитело, олигонуклеотид или полипептид, при молярном соотношении комплексообразующего агента и иммунореактивной группы от 0,5 : 1 до 10 : 1.
или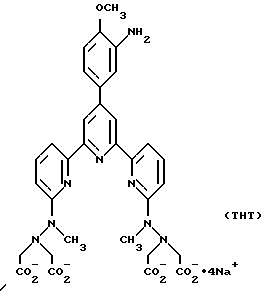
3. Иммунореагент по п. 1, где указанную группу, реагирующую с белком, выбирают из группы, состоящей из амино-, алкиламино-, ариламино-, гидразино-, алкилгидразино-, арилгидразино-, карбазидо-, семикарбазидо-, тиокарбазидо-, тиосемикарбазидо-, сульфгидрила, сульфгидрилалкила, сульфгидриларила, гидроксильной группы, карбоксильной группы, карбоксиалкила, карбоксиарила, групп, содержащих активный галоген, этилкарбонила, 2-замещенного удаляемой группой, винилсульфонила, винилкарбонила, эпокси-, изоцианато-, изотиоцианато-, альдегида, азиридина, сукцинимидоксикарбонила, активированных ацильных групп и смешанных ангидридов.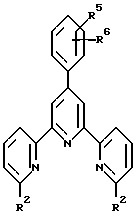
где каждый R2 независимо выбирают из группы метилентиоэтилениминодиуксусной кислоты, гидрозинилилидендиуксусной кислоты и солей этих кислот, или две группы R2, взятые вместе, представляют собой атомы, необходимые для замыкания макроциклической кольцевой структуры, содержащей по меньшей мере один гетероатомный координационный центр и по меньшей мере одну алкиленовую группу, образующую часть кольцевой структуры:
R5 - алкокси;
R6 - группа, реагирующая с белком,
а также его соли, комплексы и белковые конъюгаты.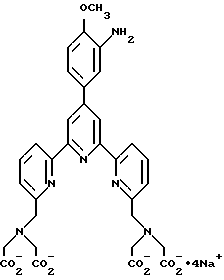 о
о
| Огнетушитель | 0 |
|
SU91A1 |
| ДИАПАЗОННЫЙ КОМПЕНСАЦИОННЫЙ ФАЗОМЕТР | 0 |
|
SU329481A1 |
| Устройство для выпрямления опрокинувшихся на бок и затонувших у берега судов | 1922 |
|
SU85A1 |
| УСТРОЙСТВО ДЛЯ ПРИВЕДЕНИЯ ВО ВРАЩЕНИЕ РАБОЧИХ | 0 |
|
SU264333A1 |
