Изобретение относится к неизвестным до сих пор соединениям полезным в лечении людей и животных, их фармацевтически приемлемым солям, к их биообратимым производным, способам получения указанных новых соединений, фармацевтическим композициям, содержащим новые соединения, единицам дозировки композиций и к методам лечения пациентов, использующим указанные композиции и единицы дозировок.
Лейкотриены, которые образуются через метаболизм 5-липоксигеназы арахидоновой кислоты, вовлечены во множество патофизиологических функций, таких как бронхосужение, плазменная эксудация, спазм коронарной артерии, хемотаксис лейкоцитов и нейтрофильная дегрануляция. Следовательно, соединения, которые бы тормозили 5-липоксигеназы, а поэтому и образование лейкотриенов, или противодействовали бы действию лейкотриенов, были бы очень полезны.
Международная патентная заявка (N PCT/DK 90/00201 [1]) описывает серию производных хинолил замещенного N-фенил замещенного изосерина (т.е. 3-амино-2-гидроксипропионовую кислоту) с хорошей лейкотриен-антагонистической активностью.
Теперь неожиданно было выявлено, что замена -CH2O группы -CH=CH- (транс) и сопутствующее замещение галогеном в хинолиновом кольце дает соединения даже с большей антагонистической активностью к лейкотриену, особенно в присутствии человеческого сывороточного альбумина.
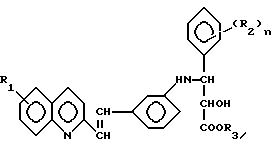
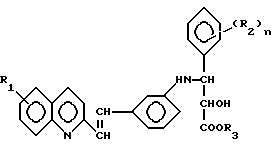
Настоящие соединения имеют общую формулу I
Y обозначает -CH=CH-;
R1 - водород или галоген, предпочтительно фтор, хлор или бром;
R2 - галоген, предпочтительно фтор, хлор или бром, CH3, OCH3, NO2 или CF3, и n = 0-3, предпочтительно 0, 1 или 2;
A представляет кислотную группу, например, карбокси, 1-H-тетразолил или группу гидроксамовой кислоты.
Среди предпочтительных соединений изобретения - предпочтительны соединения формулы I, в которых A представляет карбоксигруппу.
Соединения, описанные здесь, содержат больше центров асимметрии и могут дать начало диастереоизомерам и оптическим изомерам. Настоящее изобретение относится к таким возможным диастереоизомерам, а также к их рацемическим и разделенным оптическим активным формам.
Настоящие соли соединений формулы I можно получить с фармацевтически приемлемыми неорганическими или органическими кислотами, такими, как хлористо-водородная, бромисто-водородная и йодисто-водородная кислоты, фосфорная кислота, серная, азотная, п-толуолсульфокислота, метансульфокислота, муравьиная, уксусная, пропионовая, лимонная, винная и малеиновая кислоты.
Настоящие соли соединения формулы I можно также получить с фармацевтически приемлемыми неорганическими или органическими основаниями. В качестве примеров солей, полученных с фармацевтически приемлемыми нетоксичными основаниями, можно упомянуть соли щелочных и щелочно-земельных металлов, такие как соли лития, натрия, калия, магния, кальция, а также соли с аммиаком и подходящими нетоксичными аминами, такими как C1-C6-алкиламины, например триэтиламин, C1-C6-алканоламины, например диэтаноламин или триэтаноламин, прокаин, циклоалкиламины, например дициклогексиламин, бензиламины, например N-метилбензиламин, N-этилбензиламин, N-бензил-β-фенетиламин, N,N'-дибензилэтилендиамин или дибензиламин, и гетероциклические амины, например морфолин, N-этилпиперидин и другие.
Даже если настоящие соединения хорошо абсорбируются кишечником, в некоторых случаях лучше готовить биообратимые производные соединения изобретения, т. е. так называемые пролекарства, предпочтительно производные, физиохимические свойства которых обеспечивают улучшенную растворимость при физиологическом pH и/или абсорбцию соединения.
Такими производными являются, например, сложные эфиры N-гидроксиметил производных соединений изобретения, такие соединения готовятся реакцией вторичной аминфункции соединений изобретения с формальдегидом (R.G. Kallen and W.P. Jencks, J. Biol. Chem, 241 (1966) 5864 [2]; C.J. Martin и M.A. Marini, J. Biol. Chem. 242 (1967) 5736 [3]; M. Levy и D.E. Silberman, J. Biol. Chem. 118 (1937) 723 [4]; S. Lewin и D.A. Humphany, J. Chem. Soc. B (1966) 210 [5] ) с последующей реакцией с подходящим кислотным соединением или активированными производными таких соединений, например, с бисульфитом ( B.C. Jain, B.H. Iyer, и P.C. Guha, Science и Culture 11 (1946) 568 [6]), N,N-диметилглицином, N,N-диэтил-β-аланином, или фосфорной кислотой (S.A. Varia, S. Schuller, K.B. Sloan и V.J. Stella, J. Pharm. Sci., 73 (1985) 1068 и последующие статьи [7]), но можно также использовать другие подходящие кислоты, которые образуют биобратимые производные с нужными физико-химическими свойствами.
Другие примеры включают сложные эфиры, образованные с кислотной функцией в молекуле, такие, как, например, метил или этил, сложные эфиры ацилоксиалкила, алкоксикарбонилоксиалкила или аминоацилоксиалкила, которые легко гидролизуются ин виво или ин витро.
Среди упомянутых эфиров предпочтительны следующие: алканоилоксиметил с 3-8 атомами углерода, 1-(алканоилокси)этил с 4 - 9 атомами углерода, алкоксикарбонилоксиметил с 3 - 6 атомами углерода, 1-(алкоксикарбонилокси)этил с 4 - 7 атомами углерода, и α-аминоалканоилоксиметил с 2 - 6 атомами углерода.
Другими предпочтительными эфирами являются лактонильные эфиры, например, 3-фталидил, 4-кротонолдактонил или γ-бутиролактон-4-ил эфиры.
В объем изобретения также входят метоксиметил, цианометил, или моно- или диалкил замещенные амино-алкиловые эфиры, например 3-диметиламиноэтил, 2-диэтиламиноэтил, или 3-диметиоаминопропиловые эфиры.
В частности, предпочитаются такие эфиры, которые хорошо абсорбируются при назначении через кишечник и во время или после абсорбции гидролизуются до соединений формулы I.
Можно также использовать другие подходящие способы улучшения физико-химических свойств и растворимости соединений.
Метаболиты арахидоновой кислоты включают простагландины и лейкотриены. Обе эти группы метаболитов важны в патофизиологии воспалительных и аллергических реакций. Многие ингибиторы синтеза простагландина используются как противовоспалительные агенты (R. J. Flower, S. Moncada и J.R. Vane, в: The Pharmacological Basis of Therapeutics (1980), изд. A.G. Gilman, L.S. Goodmann и A. Gilman, (Macmillan, New York) стр. 682 [8]), но относительно небольшое число ингибиторов лейкотриена в настоящее время являются клинически приемлемым. Первой стадией в биохимическом синтезе всех лейкотриенов является окисление в перекисное соединение на 5-атоме углерода арахидоновой кислоты. Эта реакция катализируется ферментом 5-липоксигеназой, присутствующей в основном в лейкоцитах. Лейкотриен B4 - один из наиболее сильных хемоаттрактантов для полиморфоядерных лейкоцитов, и в то же время вызывает агрегацию и дегрануляцию этих воспалительных клеток. Таким образом, это сильный провоспалительный гормон. Лейкотриены C4, D4 и E4 вместе образуют агент, известный ранее как "медленно реагирующее вещество анафилаксии" (SRS-A), которое на три порядка более сильное, чем гистамин, вызывающий бронхосужение, и, кроме того, регулирует сокращение и проницаемость гладкой мускулатуры микрососудов. Поэтому он является посредником астматических, аллергических и воспалительных реакций.
Таким образом ингибирование 5-липоксигеназы ведет к сокращению образования всех этих воспалительных и аллергических посредников. Это имеет очень важные клинические последствия, поскольку специфические ингибиторы 5-липоксигеназы и антагонисты лейкотриена представляют потенциальный интерес в лечении астмы, аллергии, ревматоидного артрита, спондилоартрита, атеросклероза, подагры, пролиферативных и воспалительных кожных реакций, таких как псориаз и атопический дерматит, хронических воспалительных кишечных заболеваний и других воспалительных состояний, вазоспазма, связанного с грудной ангиной, легочной гипертензией, кистозного фиброза, синдрома респираторного нарушения у взрослых, ишемии и реперфузионных нарушений, и т.д. ( E.J. Goetzl, D. G. Payan и D.W. Godman, J. Clin. Immunol. 4 (1984) 79 [9]). Таким образом определение специфических ингибиторов 5-липоксигеназы и антагонистов лейкотриена представляет собой новый подход с очень большими осложнениями в лечении множества клинических расстройств.
Ингибиторы биосинтеза лейкотриена можно идентифицировать, используя брюшинные лейкоциты крысы, помеченные 1-14C арахинодатом и стимулированные ионофором A23187 кальция (I. Ahnfelt-Ronne, D. Kirstein и C.K. Kaergaard - Nielsen, European J. Pharmacol. 155 (1988) 117 [10]). Было отмечено, что соединения, полученные по примерам 1-5, тормозят образование лейкотриена B4 в пробе с концентрацией 10μM.
Антагонисты лейкотриена можно идентифицировать наблюдением сокращений в препаратах полосок подвздошной кишки, суспендированных в физиологическом буфере с добавлением чистого лейкотриена D4 (LTD4) [10]. Полоски подвздошной кишки связаны с изотоническим передатчиком и сокращения непрерывно записываются на многоканальное записывающее устройство. До добавления LTD4 к буферу добавляется атропин и индометацин, чтобы блокировать холинергические или эффекты сокращений, вызванные простогландином. Тестуемые соединения, исследуемые относительно антагонизма к лейкотриену, растворяются в ДМСО и добавляются в ванночку с органом за 2 минуты до добавления LTD4 с концентрацией 10-9 М (окончательная концентрация), окончательная концентрация ДМСО составляет 0,1%, концентрация, которая не влияет на реакцию подвздошной кишки на LTD4. Тестуемые соединения можно добавлять в различных концентрациях.
Когда соединения настоящего изобретения были добавлены к препарату до добавления LTD4, произошло значительное торможение специфического сокращения, вызванного LTD4. Это торможение имело место при концентрации даже таких низких, как 0,1 - 1 нМ. С другой стороны, сокращения, вызванные гистамином при 10-7 М не тормозились этими соединениями даже при микромолярных концентрациях.
Важно исследовать связующие свойства рецептора антагонистов лейкотриена относительно их тормозящей способности сокращений гладкой мускулатуры.
Исследование связи рецептора можно провести с легочными мембранами морской свинки в непосредственной сравнительной пробе между антагонистом лейкотриенами [3H] LTD4 на связывание с LTD4 рецептором [10], (S. Mong, H.-L. Wu, M. O. Scott, M.A. Lewis, M.A. Clarke, B.M. Weichman, C.M. Kinzig, J.G. Gleason и S.T. Crooke, J. Pharmacol. Exp. Ther. 234 (1985) 316 [11]). pIC50 определяется как отрицательный логарифм молярной концентрации антагониста, тормозящего [3H] LTD4 связывание на 50%. pIC50 величины для соединений по примерам 6 и 17 составили 8.7 и 8.2 и 8.6 и 7.7 соответственно в отсутствие и присутствии 0,1% альбумина человеческой сыворотки. Эти величины указывают, что сродство соединений для LTD4 рецептора очень высокое, также в присутствии альбумина.
Настоящее изобретение также относится к способу получения настоящих соединений.
В одном варианте амин формулы II
в которой R1, Y и n имеют вышеуказанные значения, реагирует с соединением формулы III
в которой R2, A и X имеют вышеуказанные значения, и X способен образовывать "хорошую уходящую группу", X означает, например, атом галогена, такой, как хлор, бром или йод, или алкил- или арилсульфонилоксигруппу, но могут использоваться и другие уходящие группы, такие, как алкилсульфатная группа, хлоросульфонилокси группа, алкилсульфитная группа, моно- или диалкилфосфатная группа или нитратная группа, чтобы образовать соединение формулы I.
Во время реакции A может быть защищена традиционными защитными группами, например, в случае карбоксильной группы, такой как сложный эфир.
Реакция проводится в подходящем инертном органическом растворителе, таком, как диметилформамид, возможно использование и других растворителей. Реакция предпочтительно проводится при температуре окружающей среды, но в некоторых случаях удобно охлаждать реакционную смесь ниже комнатной температуры до точки кипения используемого растворителя, в зависимости от характера реагентов формулы II и III. Необработанные продукты реакции формулы I собираются фильтрацией или после разбавления водой экстрагируются из реакционной смеси подходящим растворителем, таким, как диэтиловый эфир, этилацетат, дихлорметан или хлороформ. Продукты очищаются рекристаллизацией или хроматографией.
В другом варианте амин формулы II реагирует с соединением формулы IV
в которой R2, A и n имеет следующие вышеуказанные значения. Реакция проводится либо в подходящем инертном органическом растворителе, таком, как метанол, этанол, диметилформамид или гексаметил фосфорный триамид, либо в воде, или их смеси. Температура реакции около или выше комнатной до точки кипения используемого растворителя. В некоторых случаях представляется удобным охлаждать реакционную смесь ниже комнатной температуры в зависимости от характера используемого соединения формулы IV. Изоляцию и очистку продуктов можно проводить как описано выше.
Дополнительно кислотные функциональности A можно получить в соответствии со следующими общими схемами реакций из соединений формулы I, в которых A - CN или COOH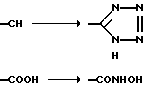
Настоящие соединения предназначены для использования в фармацевтических композициях, которые полезны в лечении упомянутых заболеваний.
Требуемое количество соединения формулы I (далее упоминается как активный ингредиент) для терапевтического эффекта будет зависеть от конкретного соединения, способа назначения и пациента (млекопитающего). Приемлемая доза соединения формулы I для систематического лечения составляет 0,1 - 20 мг на килограмм веса, наиболее предпочтительная дозировка - 0,2 - 10 мг/кг веса тела млекопитающего, назначаемая один или несколько раз в день.
В аэрозольных препаратах антиастматическая доза соединения формулы I может быть от 1 г до 5 мг соединения на кг веса тела, предпочтительный диапазон дозировки от 1 г до 0,5 мг/кг.
Хотя активный ингредиент может назначаться в виде одного только чистого соединения, лучше, если он будет входить в состав фармацевтического препарата. Обычно активный ингредиент составляет от 0,1 до 100% по весу препарата. Единица дозировки препарата содержит между 0,07 мг и 1 г активного ингредиента. Для местного назначения активный ингредиент предпочтительно включают в количестве от 1 до 2% от веса препарата, но может составлять и 10% вес/вес. Препараты для носового или щелочного назначения (так называемые самодозирующиеся порошковые препараты, описанные ниже) могут содержать 0,1 - 20% вес/вес, например 2 вес.% активного ингредиента.
Под термином "единичная доза" подразумевается единая доза, которая назначается пациенту, и которая упаковывается и представляет собой физически и химически стабильную единицу дозирования, включающую либо активный материал как таковой, либо его смесь с твердыми или жидкими фармацевтическими разбавителями или носителями.
Препараты как для ветеринарного, так и лечения людей включает активный ингредиент вместе с фармацевтически приемлемым носителем для него и возможно другими терапевтическими ингредиентами. Носитель(и) должны быть приемлемыми в смысле совместимости с другими ингредиентами препаратов и не вредными для реципиента.
Препараты включают эти компоненты в форме, приемлемой для орального, глазного, ректального, парентерального (включая подкожное, внутримышечное и внутривенное), трансдермального, внутриартикулярного, локального, носового или щечного назначения.
Препараты могут иметь форму единицы дозировки и быть приготовлены любым из способов, хорошо известных в фармацевтике. Все способы включают этап смешивания активного ингредиента с одним или несколькими дополнительными ингредиентами. В общем, препараты готовятся однородным и тщательным соединением активного ингредиента с жидким носителем или мелко измельченным твердым носителем или с тем и другим, и затем, при необходимости формованием продукта в нужный препарат.
Препараты настоящего изобретения, приемлемые для орального назначения, могут быть в форме отдельных единиц: капсул, пакетов, таблеток, пастилок, причем каждая содержит определенное количество активного ингредиента; в форме порошка или гранул; в форме раствора или суспензии в водной или не-водной жидкости; или в форме эмульсии масло-в-воде или вода-в-масле. Активный ингредиент может также назначаться в форме шариков, электуария или пасты.
Таблетка делается прессованием или формованием активного ингредиента возможно с одним или несколькими дополнительными ингредиентами. Прессованные таблетки можно готовить прессованием в машине активного ингредиента в свободно текучей форме, такой, как порошок или гранулы, возможно смешанные со связующим веществом, смазочным веществом, инертным разбавителем, поверхностно-активным или диспергирующим агентом. Формованные таблетки делаются формованием в соответствующей машине смеси порошкового активного ингредиента и подходящего носителя, увлажненного инертным жидким разбавителем.
Препараты для ректального назначения могут иметь форму свечей, включающих активный ингредиент и носитель, такой как масло какао, или быть в виде клизмы.
Препараты, приемлемые для парентерального назначения, обычно включают стерильный масляный или водный препарат активного ингредиента, который предпочтительно изотоничен к крови реципиента.
Препараты, пригодные для внутримышечного назначения, могут быть в форме стерильного водного препарата активного ингредиента, который имеет микрокристаллическую форму, например, в форме водной микрокристаллической суспензии. Липосомные препараты биораспадающихся полимерных систем могут также использоваться для введения активного ингредиента во внутриартикулярные и глазные препараты.
Препараты, пригодные для локального назначения, представляют собой жидкие или полужидкие формы, такие как линименты, лосьоны, эмульсии масло-в-воде и вода-в-масле, такие как кремы, мази или пасты; или растворы или суспензии, такие как капли. Например, для глазного назначения активный ингредиент может быть представлен в форме водных глазных капель, как, например, 0,1 - 1,0% раствора.
Препараты пригодные для назначения в нос или щечную полость включают порошковые, с реактивным выбрасыванием и распылительные препараты, такие, как аэрозол и спреи. Распыляющиеся препараты предпочтительно имеют размер частиц в диапазоне от 10 до 100μ.
Такие препараты наиболее предпочтительны в форме мелко измельченного порошка для легочного назначения из порошкового ингалятора или распылительного порошкового дозатора, где активный ингредиент в виде мелкоизмельченного порошка может включать до 99,9% вес/вес препарата. В случае распыляющегося раствора и спрея эффект может быть достигнут либо выбором клапана с нужными характеристиками распыления (т.е. способным давать струю с нужным размером частиц), либо включением активного ингредиента в виде суспендированного порошка в контролируемый размер частиц. Такие самораспыляющиеся препараты могут быть препаратами, дозирующими порошок, или препаратами, дозирующими активный ингредиент в виде капель раствора или суспензии.
Распыляющиеся препараты с дозированием порошка предпочтительно включают диспергированные частицы твердых активных ингредиентов и жидкое реактивное вещество с температурой кипения ниже 18oC при атмосферном давлении. Жидкое реактивное вещество может быть известным, пригодным для назначения в медицинских целях и может включать один или несколько C1-C6-алкилуглеводородов или галогенированных C1-C6-алкилуглеводородов или их смеси; хлорированные C1-C6-алкилуглеводороды особенно предпочтительны. Обычно реактивное вещество содержит 50 - 99,9% вес/вес препарата, а активный ингредиент составляет 0,1 - 20% вес/вес, например 2% в весовом соотношении от веса препарата.
Фармацевтически приемлемый носитель в таких распыляющих препаратах может включать другие составляющие в дополнение к реактивному веществу, в частности, ПАВ или жидкий разбавитель или и то и другое. ПАВ нужны, чтобы воспрепятствовать агломерации частиц активного ингредиента и поддерживать его в суспензии. Особенную ценность представляют жидкие неионные ПАВ и твердые анионные ПАВ и их смеси. Подходящими жидкими неионными ПАВ являются эстеры и частичные эстеры жирных кислот с алифатическими многоатомными спиртами, например, сорбитан моноолеат и сорбитан триолеат, известные под коммерческими названиями как "Span 80" (товарное наименование) и "Span 85" соответственно. Жидкое неионное ПАВ может составлять от 0,01 до 20 вес.% препарата, хотя предпочтительно его содержание менее 1 вес.%. Подходящие твердые анионные ПАВ включают соли диалкил сульфосукцината щелочного металла, аммония и амина (в которых алкиловые группы имеют 4 - 12 атомов углерода). Твердые анионные ПАВ могут составлять от 0,01 до 20 вес.% препарата, хотя лучше, если процент твердых разбавителей в таком распыляющемся препарате составит ниже 1 вес.% от композиции препарата, в котором плотность активного ингредиента существенно отличается от плотности реактивного вещества; кроме того, они помогают поддерживать активный ингредиент в суспензии. Твердый разбавитель имеет форму мелкого порошка с размером частиц того же порядка, что и частицы активного ингредиента. Подходящие твердые разбавители включают хлорид натрия, сульфат натрия и сахара. Препараты настоящего изобретения могут быть также в форме самораспыляющегося лекарства, где активный ингредиент присутствует как таковой в суспензии или в растворе. Такие препараты могут включать активный ингредиент, реактивное вещество и сорастворитель, а также антиокислительный стабилизатор. Реактивное вещество представляет собой одно или несколько из указанных выше. Сорастворители выбираются по их растворимости в реактивном веществе, их способности растворять активный ингредиент, и по наинизшей температуре кипения, совместимой с этими вышеупомянутыми свойствами. Подходящими сорастворителями являются C1-C6-алкил спирты и эфиры и их смеси. Сорастворитель может составлять 5 - 40 вес.% от веса препарата, хотя лучше, если эта цифра будет менее 20 вес.%. В такие растворы-препараты можно включить антиокислительные стабилизаторы для замедления распада активного ингредиента и это аскорбаты или бисульфаты щелочных металлов. Они присутствуют в количестве до 0,25 вес.% от веса препарата.
Такие самораспыляющиеся препараты можно приготовить любым методом, известным в области техники. Например, активный ингредиент (либо в виде частиц, как указано выше, либо в суспензии или жидкости или до 20 вес.% в растворе в приемлемом сорастворителе) смешивается с любыми другими составляющими фармацевтически приемлемого носителя. Полученная смесь охлаждается, вводится в подходящий охлажденный контейнер и в жидкой форме к ней добавляется реактивное вещество; контейнер герметизируется. В другом случае такие самораспыляющиеся препараты можно приготовить смешиванием активного ингредиента либо в частицах, либо, как указано выше в 2 - 20% вес/об. спиртовом или водном растворе с остальными составляющими фармацевтически приемлемого носителя, а не с реактивным веществом, введением полученной смеси, возможно с частью реактивного вещества, в подходящий контейнер, и закачиванием реактивного вещества под давлением в контейнер при температуре окружающей среды через клапан, который составляет часть контейнера и используется для контроля распыляемого из него препарата. Желательно контейнер продуть, чтобы удалить из него воздух на определенной стадии изготовления.
Подходящим контейнером для самораспыляющегося препарата является контейнер, снабженный ручным клапаном и выполненный из алюминия, нержавеющей стали или упорного стекла. Клапан должен, безусловно обладать нужными характеристиками распыления частиц определенного размера, как указано выше. Лучше, если клапан представляет собой тип, который выбрасывает определенное количество препарата при однократном нажатии на него, например, около 50 - 100 микролитров препарата при каждом выбросе.
Препараты настоящего изобретения могут быть также в форме водного или разбавленного спиртового раствора, также стерильного раствора активного ингредиента для использования в распылителе, где ускоренный поток воздуха используется для получения туманного облака, состоящего из мелких капель раствора. Буферный агент и ПАВ могут также включаться в такой препарат, который содержит консервант, например, бензилгидроксибензоат.
Другие препараты для назначения в нос включают мелкий порошок с размером частиц от 10 до 100 микрон, который вдыхается через носовой проход из контейнера.
Кроме указанных ингредиентов препараты изобретения могут включать один или несколько дополнительных ингредиентов, таких, как разбавители, буферы, отдушки, связующие вещества, ПАВ, сгустители, смазочные вещества, консерванты, например, метилгидроксибензоат (включая антиокислители), эмульгирующие агенты и другие.
Композиции могут далее содержать другие терапевтически активные вещества, обычно используемые в лечении указанных патологических состояний, например глюкокортикоиды, антигистамины, антагонисты активирующего фактора тромбоцитов (PAF), антихолинергические агенты, метил ксантины, β-адренергические агенты, салицилаты, индометацин, флюфенамат, напроксен, тимегадин, соли золота, пеницилламин, агенты снижения холестерола в сыворотке, ретиноиды, соли цинка и салицилазосульфапиридин (салазопирин).
В соответствии с изобретением, настоящие соединения назначаются пациенту, страдающему одним из вышеуказанных патологических состояний в ежедневной дозировке (для взрослых) от 0,2 мг до 7000 мг, предпочтительно от 1 до 3500 мг, и в ветеринарной практике соответственно в ежедневных дозировках от 0,5 до 100 мг/кг веса тела.
Далее изобретение описывается по следующим примерам.
Пример 1
E-(2R,3R,2S,3S)-3-фенил-N-3-[2-(7-хлорохинолил)-2-этенил]-фенил изозерин этиловый эстер.
Смесь E-3-[2-(7-хлорохинолил)-2-этенил] -анилина (2,8 г, 10 мМ) и (±)-транс-3-фенилоксиран-2-карбоновой кислоты этилового эстера (2,0 мл, 11 мМ) в этаноле (75 мл) нагревается с флегмацией 24 часа. После охлаждения полученный осадок собирается фильтрацией и промывается этанолом и эфиром.
Искомое соединение получается с температурой плавления 161 - 163oC.
Пример 2
E-(2R,3R,2S,3S)-3-фенил-N-3-[2-(хинолил)-2-этенил]-фенил изозерин этиловый эстер
Следуя процедура примера 1, но заменяя E-3-[2-(7-хлорохинолил)- 2-этенил] анилин E-3-[2-(хинолил)-2-этенил]анилином получают искомое соединение. Оно изолируется в виде гидрохлорида с температурой плавления 132 - 138oC.
Примеры 3 - 5
По процедуре примера 1 и используя соответствующие исходные материалы получают соединения таблицы 1 (2R, 3R, 2S, 3S) в виде E-рацематов.
Пример 6
E-(2R, 3R, 2S,3S)-N-3-[2-(7-хлорохинолил)-2-этенил]-фенил-3-фенил изозерин, натриевая соль
К раствору этилового эстера (пример 1) (1,4 г, 3 мМ) в этаноле (40 мл) добавлялся 2N NaOH (2 мл). Раствор нагревался с флегмацией 1,5 часа. После охлаждения полученный осадок собирался фильтрацией и промывался холодной водой и эфиром. Искомое соединение было получено в виде дигидрата.
Температура плавления: >250oC.
Примеры 7 - 12
Аналогично процедуре примера 6 и используя соответствующие исходные материалы были получены соединения таблицы 2 (2R, 3R, 2S, 3S) в качестве рацематов.
Пример 13
E-(2R, 3R, 2S, 3S)-3-(2,3-дифторофенил-N-3(2-(хинолил)-2-этенил-фенил изозерин
К раствору E-3-[2-(хинолил)-2-этенил] -анилина (0,5 г, 2 мМ) в этаноле (10 мл) добавлялся раствор (±)-E-3-(2,3-дифторофенил)оксиран- 2-натриевой соли карбоновой кислоты (0,56 г, 2,5 мМ) в воде (2,0 мл) и этаноле (3,0 мл). Реакционная смесь нагревалась с флегмацией 24 часа. После охлаждения до комнатной температуре осадок отфильтровывался и получалась соль натрия с примесями. Она растворялась в воде (10 мл) и pH доводилось до pH=6,0 добавлением 3N уксусной кислоты. Осадок отфильтровывался, промывался водой и получалось искомое соединение с температурой плавления 233 - 235oC.
Пример 14
E-(2R, 3R, 2S, 3S)-N-3-[2-(7-хлорохинолил)-2-этенил] -фенил- 3-(2,6-дифторфенил) изозерин
Аналогично процедуре примера 13, но с заменой E-3-[(2-хинолил)- 2-этенил] -анилина E-3-[2-(7-хлорохинолил)-2-этенил] -анилином и (±)-E-3-(2,3-дифторофенил) оксиран-2-натриевой соли карбоновой кислоты 3-(2,6-дифторофенил)оксиран-2-натриевой солью карбоновой кислоты было получено искомое соединение с температурой плавления: 215 - 217oC.
Пример 15
E-(2R,3R,2S,3S)-N-3-{2-(7-хлорохинолил)-2-этенил]-фенил- 3-(2-дифторофенил)изозерин
Аналогично процедуре примера 13, но с заменой E-3[(2-хинолил)-2-этенил] -анилина E-3-[2-(7-хлорохинолил)-2-этенил] -анилином и (±)-E-3-(2,3-дифторофенил)оксиран-2-натриевой соли карбоновой кислоты 3-(2,5-дифторофенил)оксиран-2-натриевой солью карбоновой кислоты было получено искомое соединение с температурой плавления: 196 - 198oC.
Пример 16
E-(2R, 3R, 2S,3S,)-N-3-[2-(7-хлорохинолил)-2-этенил]-фенил- 3-(4-трифторометилфенил) изозерин, соль натрия
Следуя процедуре примера 13, но заменяя E-3[(2-хинолил-2-этенил]-анилин E-3-[2-(7-хлорохинолил)-2-этенил] -анилином и (±)-E-3-(2,3-дифторофенил)оксиран-2-натриевую соль карбоновой кислоты 3-(4-трифторометилфенил)оксиран-2-натриевой солью карбоновой кислоты, было получено искомое соединение. Оно изолируется как соль натрия с температурой плавления: > 250oC.
Пример 17
E-(2R, 3R, 2S, 3S)-3-фенил-N-3-[(2-хинолил)-2-этенил] -фенил изозерин, натриевая соль
К раствору этилового эфира примера 2, и с последующей процедурой примера 6 получалось искомое соединение.
1H ЯМР (CD3)2SO: δ = 3,83 (1H, д), 4,50 (1H, д), 6,50 (1H, бд), 6,85 (2H, м), 7,02 (1H, т), 7,10 - 7,32 (4H, м), 7,43 (2H, д), 7,54 (1H, м), 7,62 (1H, д, J = 16,3 Гц), 7,74 (1H, дт), 7,82 (1H, д), 7,93 (1H, бд), 7,99 (1H, бд), 8,32 (1H, д).
Пример 18
E-(-)-(2S, 3S)-N-3-[2-(7-хлорохинолил)-2-этенил]-фенил-3-фенил изозерин, натриевая соль
Аналогично процедуре примера 14, но с заменой (±)-транс- 3-(2,6-дифторофенилхоксиран-2-натриевой соли карбоновой кислоты (-)-(2R,3S)-3-фенилоксиран-2-карбоновой кислотой, натриевой солью, было получено искомое соединение.
Температура плавления: > 250oC.
[α]D 25 = +24,0 (с = 1, MeOH).
[α]D 25 = -90,4 (c = 1, In HCl).
Пример 19
E-(+)-(2R, 3R)-N-3-[2-(7-хлорохинолил)-2-этенил]-фенил-3-фенил изозерин, натриевая соль
По процедуре примера 14, но заменяя (±)-транс-3-(2,6- дифторофенил)оксиран-2-карбоновую кислоту, натриевая соль (+)-(2S,3R)-3-фенилоксиран-2-карбоновой кислоты, натриевой солью, было получено искомое соединение.
Температура плавления: > 250oC.
[α]D 25 = -23,7 (c = 1, MeOH).
[α]D 25 = +90,1 (c = 1, In HCl).
Пример 20
E-(2R, 3R,2S,3S)-N-[2-(7-хлорохинолил)-2-этенил]-фенил-3- (4-нитрофенил) изозерин, этиловый эстер
По процедуре примера 1, но с заменой (±)-транс-3-фелилоксиран- 2-этилового эстера карбоновой кислоты (±)транс-3-(4-нитрофенил)оксиран-2-этиловым эстером карбоновой кислоты, было получено искомое соединение с температурой плавления: 175 - 177oC.
Пример 21
E-(2R, 3R, 2S,3S)-N-[2-(7-хлорохинолил)-2-этенил]-фенил-3-(4-нитрофенил) изозерин, натриевая соль
Аналогично процедуре примера 6, но заменяя E-3[(2-хинолил)-2-этенил]- анилин E-3[2-(7-хлорохинолил)-2-этенил] -анилином и E-3-(2,3-дифторофенил)оксиран-2-натриевую соль карбоновой кислоты 3-(4-нитрофенил)оксиран-2-натриевой солью карбоновой кислоты, получено искомое соединение. Оно изолируется как соль натрия с температурой плавления: > 250oC.
Пример 22
Аэрозоль
E-(-)-(2S, 3S)-N-3-[2-(7-хлорохинолил)-2-этенил]-фенил-3-фенил изозерин, соль натрия (активное вещество) - 1000 мг
Сорбитан триолеат - 700 мг
Монофторотрихлорметан - 595 г
Дифтородихлорметан - 798 г
Активное вещество измельчается в реактивной мельнице. Большая часть частиц должна быть менее 5μм в диаметре.
Лекарственный концентрат готовится растворением сорбитана триолетата в небольшом количестве монофторотрихлорметана и добавлением активного вещества. Концентрат тщательно гомогенизируется. Он переносится в герметичный резервуар, снабженный системой охлаждения. Остальные реактивные вещества добавляются при перемешивании и охлаждении до -50oC.
Подходящие аэрозольные контейнеры заполняются расчетным количеством препарата и немедленно герметизируются измерительными клапанами с соответствующими активаторами. Каждый выброс выдает 50μг активного вещества.
Пример 23
Таблетка
E-(-)-(2S, 3S)-N-3-[2-(7-хлорохинолил)-2-этенил]-фенил-3-фенил изозерин, соль натрия (активное вещество) - 100 мг
Лактоза - 75 мг
Крахмал - 12 мг
Метилцеллюлоза - 2 мг
Карбоксиметилцеллюлоза натрия (CMC-Na) - 10 мг
Стеарат магния - 1 мг
Активное вещество, лактоза и крахмал смешиваются до гомогенного состояния в подходящем смесителе и увлажняются 5% водным раствором метилцеллюлозы 15 сп. Смешивание продолжается до формирования гранул. При необходимости мокрая грануляция пропускается через подходящий экран и высушивается до содержания воды менее 1% в сушилке, например, в сушильной печи или псевдожидким слоем. Высушенная грануляция пропускается через экран в 1 мм и смешивается до гомогенного состояния с CMC-Na. Добавляется стеарат магния и смешивание продолжается еще немного. Из грануляции с помощью соответствующей таблетирующей машины получаются таблетки с весом в 200 мг.
Пример 24
Препарат для инъекций
E-(-)-(2S, 3S)-N-3-[2-(7-хлорохинолил)-2-этенил]-фенил-3-фенил изозерин, натриевая соль (активное вещество) - 1%
Хлорид натрия - q.s.
Вода для инъекций до доведения общего состава до 100%. Активное вещество растворяется в воде для инъекции. Раствор заполняется в ампулы и стерилизуется.

Производные изозерина общей формулы I, где У - -СН=СН-; R1 - водород или галоген; R2 - галоген, СН3, ОСН3, NO2 или СF3, и n =0-3, А - карбокси, или их фармацевтически приемлемые нетоксичные соли, ин виво гидролизуемые сложные эфиры, получают взаимодействием соединения II с соединением формулы III. Соединения формулы I проявляют лейкотриен антагонистическую активность. 4 с. и 4 з.п. ф-лы, 2 табл.
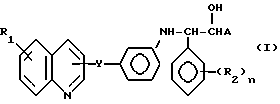


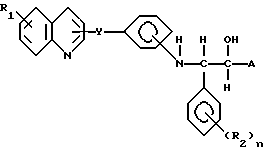
в которой У означает -СН=СН-;
R1 - водород или галоген, предпочтительно фтор, хлор или бром;
R2 - галоген, предпочтительно фтор, хлор или бром; CH3, OCH3, NO2 или CF3;
n = 0-3, предпочтительно 0,1 или 2;
А - карбокси,
или их фармацевтически приемлемые нетоксичные соли или "ин виво" гидролизуемые сложные эфиры.
в которой R1, У имеют вышеуказанные значения,
подвергают реакции с соединением формулы III
в которой R2, A и n имеют вышеуказанные значения.
| EP 228959, 1987. |
