Изобретение относится к ранее неизвестным соединениям, полезным в медицинской и ветеринарной практике, к их фармацевтически приемлемым солям и биообращаемым производным, к способам получения данных новых соединений, к фармацевтическим композициям, содержащим эти новые соединения, к единичным дозированным формам этих композиций и к методам лечения пациентов с использованием указанных композиций и дозированных форм.
Лейкотриены, которые образуются через 5-липоксигеназный путь в процессе обмена арахидоновой соли, вовлечены в развитие различных патофизиологических состояний, таких как сужение бронхов, плазменная эксудация, спазм коронарных артерий, лейкоцитарный хемотаксис и дегрануляция нейтрофилов (E.J. Goetzl, D. G. Payan and D.W. Godman, J. Clin. Immunol. 4 (1984) 79). Таким образом разработка соединений, которые являются антагонистами действия лейкотриена, представляет значительный интерес.
Международная патентная заявка N PCT/DK 93/00254 (Публикация N WO 94/03431) описывает ряд хинолинзамещенных N-фенилзамещенных изосеринов, обладающих лейкотриенантогонистической активностью.
Авторы с удивлением обнаружили, что новые диолсодержащие соединения, соответствующие общей формуле I, являются мощными антагонистами, особенно в присутствии человеческого сывороточного альбумина, и обладают высокой биодоступностью и пролонгированной активностью in vivo.
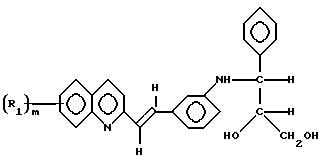
Настоящие соединения имеют общую формулу I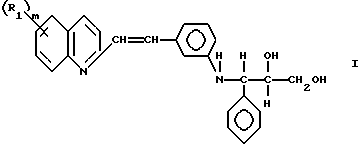
R - это водород или галоген предпочтительно фтор или хлор и m 0,1 или 2
Описанные здесь соединения имеют несколько центров асимметрии и таким образом могут образовывать большое количество стереоизомеров. Настоящее изобретение включает как все возможные стереоизомеры, так и их рацемические и стереохимические смеси.
Представленные соли соединений формулы I могут быть получены с фармацевтически приемлемыми неорганическими и органическими кислотами, такими как хлористоводородная, бромистоводородная и йодистоводородная кислоты, фосфорная кислота, серная кислота, азотная кислота, п-толуолсульфокислота, метансульфокислота, муравьиная кислота, уксусная кислота, пропионовая кислота, лимонная кислота, винная кислота и малеиновая кислота.
Ингибиторы 5-липогеназы и антагонисты лейкотриена могут представлять интерес как средства для лечения астмы, аллергии, ревматоидного артрита, спондилоартрита, падагры, атеросклероза, пролиферативных и воспалительных кожных заболеваний, таких как псориаз и атопический дерматит, хронических воспалительных кишечных заболеваний, и других воспалительных состояний, сосудистого спазма, связанных со стенокардией, легочной гипертонией, фиброзом желчного пузыря, синдромом возрастных респираторных нарушений, ишемических и реперфузионных нарушений, мигрени и др. (R.A. Lewis. K.F. Austen and R.J. Soberman, New Eng. J. Med. 323 (1990) 645.) Обнаружение специфических ингибиторов 5-липоксигеназ и антагонистов лейкотриена таким образом является новым подходом, открывающим широкие возможности в лечении разнообразных клинических расстройств.
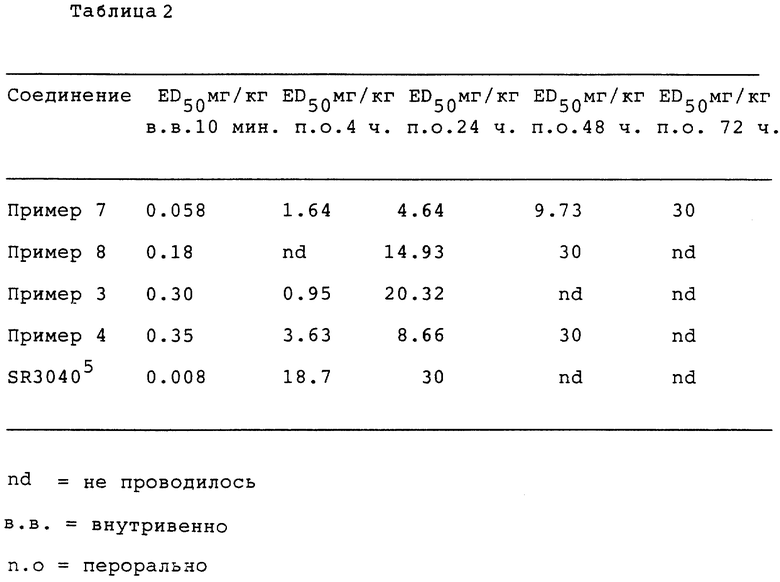
Антагонисты лейкотриена могут быть обнаружены при наблюдении сокращения, происходящего на препарате из среза подвздошной кишки морской свинки, суспендированного в физиологическом буфере, при добавлении к нему чистого лейкотриена D4(LTD)4(L. Ainfelt-Ronne, D. Kirstein and C. Kaergaard-Nielsen, European J. Pharmacol. 155 (1988) 117). Когда перед внесением LTD4 к препарату подвздошной кишки были добавлены соединения настоящего изобретения, наблюдалось значительное ингибирование специфического сокращения, вызванного LTD4. Ингибирование проявлялось при концентрации не ниже 0,1 - 1 нМ. С другой стороны, сокращение, вызванное гистамином при концентрации 10-7 М, не подавлялось этими соединениями даже при микромолярных концентрациях.
Очень важно исследовать механизм рецепторного связывания антагонистов лейкотриена при ингибировании сокращений гладкой мускулатуры. Изучение рецепторного связывания может быть проведено на легочных мембранах морской свинки при прямом конкуркурентном испытании между антагонистами лейкотриена и [3H] LTD4 в связывании LTD4 - рецепторов. (I.Ahnfelt-Ronne, D. Kirstein and C. Kaergaard-Nielsen, European J. Pharmacol. 155 (1988) 117. S. Mong. H.-L. Wu, M. O. Scott, M.A. Lewis, M.A. Clarke, B.M. Weichman, C.M. Kinzig J.G. Gleason and S.T. Crooke, J. Pharmacol. Exp. Ther. 234 (1985) 316). Величина pIC50 определяется как отрицательный логарифм молярной концентрации антагониста, ингибирующей [3H]LTD4 - связывание на 50%. Значения plC50 для соединений настоящего изобретения равны или выше, чем те же значения для контрольного соединения SR3040 (International Patent Appl. N PCT/DK/00254 (Publ. N WO 94/03431), Example 18) (См. табл. 1).
Антагонистический эффект по отношению к лейкотриену был проверен in vivo при LTD4 индуцированном бронхоспазме у находящихся под анестезией морских свинок. (I.Abnfelt-Ronne, D. Kirstein and C. Kaergaard-Nielsen, European J. Pharmacol. 155 (1988) 177). Внутривенно соединение вводилось за 10 минут, а через рот за 24, 48 и 72 часа до бронхоспазма. Значение ED50 представляет собой дозу, которая ингибирует лейкотриен-индуцированный бронхоспазм на 50%. Значение ED50 вычисляли при анализе 2-3 уменьшающихся доз. Результаты приведены в табл. 2.
Активность соединений настоящего изобретения превосходит те же значения для SR3040.
Настоящее изобретение также относится к способам получения данных соединений.
В одном из вариантов воплощения, амин формулы II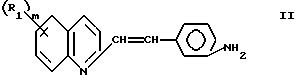
в котором R и m имеет вышеприведенное значение, реагирует с соединением формулы III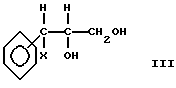
в которой X способен образовать "хорошо удаляемую группу", X таким образом может быть, например, атомом галогена, такого как хлор, бром или йод, или алкильной или арилсульфонилоксигруппой, но для получения соединений формулы I могут быть использованы и другие уходящие группы, такие как алкилсульфогруппа, хлорсульфонилоксигруппа, алкилсульфитная группа, моно- или диалкилфосфатная группа или группа нитрата.
Реакция проводится в соответствующих инертных органических растворителях, таких как диметилформамид, но могут быть использованы и другие растворители. Реакцию предпочтительно проводить при комнатной температуре, но в некоторых случаях, в зависимости от природы реагента формулы I и III, может быть удобно охладить реакционную смесь ниже комнатной температуры или нагреть выше комнатной температуры вплоть до точки кипения использованного растворителя. Полученные в результате реакции неочищенные продукты формулы I собираются путем фильтрации, или, после растворения в воде, выделяются из реакционной смеси с помощью подходящего растворителя, такого как диэтиловый эфир, этилацетат, дихлорметан или хлороформ. Продукты реакции очищаются, например, перекристаллизацией или хромотографией.
В другом варианте амин формулы II вступает в реакцию с соединением формулы IV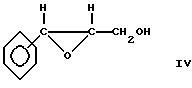
Реакция также проводится в подходящем инертном органическом растворителе, таком как метанол, этанол, диметилформамид или гексаметил фосфорный триамид, или в воде, или в их смеси. Реакция проводится при температуре, близкой к комнатной или выше нее, вплоть до точки кипения используемого растворителя. Однако в некоторых случаях, в зависимости от природы используемого соединения формулы IV, возможно охлаждение реагирующей смеси ниже комнатной температуры. Выделение и очистка продукта может быть проведена приведенными выше способами.
Настоящие соединения предназначаются для использования в фармацевтических композициях, которые могут применяться при лечении вышеприведенных заболеваний.
Количество соединения формулы I (здесь и далее называемое активным ингредиентом), необходимое для получения терапевтического эффекта будет, конечно, варьироваться в зависимости от определенного соединения, способа введения и от того, какое млекопитающее подвергается лечению. Приемлемая доза соединения формулы I для системного лечения от 0.1 до 20 мг на килограмм веса, наиболее предпочтительная доза от 0.2 до 10 мг/кг веса, при приеме один или несколько раз в день.
Для спрей-препаратов подходящая анти-астматическая доза соединения формулы (I) колеблется от 1 мкг до 5 мг на килограмм веса тела, наиболее предпочтительная доза от 1 мкг до 1 мг/кг веса тела млекопитающего, например, от 1 мкг до 0.5 мг/кг.
Хотя возможно назначение активного ингредиента самого по себе, как химического вещества, предпочтительно представлять его в виде фармацевтического препарата. Обычно активный ингредиент может составлять от 0,1% до 100% массы препарата. Удобно, если дозированная форма препарата содержит от 0.07 мг до 1 г активного ингредиента. При местном применении предпочтительное содержание активного ингредиента составляет от 1% до 2% веса препарата, но он может доходить и до 10% в/в.
Препараты, пригодные для назального или буккального применения, могут содержать от 0.1 до 20% в/в, например около 2% в/в активного ингредиента.
Под термином "дозированные формы" имеется в виду единичная, то есть разовая доза, которая может быть принята пациентом и которая удобна с точки зрения упаковки и употребления, оставаясь физически и химически стабильной единичной дозой, содержащей активный материал, как таковой, или его смесь с твердым или жидким фармацевтическим растворителем или носителем.
Препарат настоящего изобретения, предназначенный как для ветеринарных, так и для медицинских целей, содержит активный ингредиент в сочетании с фармацевтически-приемлемыми носителями и необязательно с другими терапевтическими ингредиентами. Носитель должен быть "подходящим" в том смысле, что он должен быть совместимым с другими ингредиентами препарата и не быть вредным для реципиента.
Препарат включает формы, пригодные для орального применения, офтальмического, ректального, парентерального (включая подкожный, внутримышечный и внутривенный), чрезкожного, внутрисуставного, местного, назального или буккального введения.
Препарат может быть представлен в виде дозированной формы и приготовлен любым из хорошо известных в фармации методов. Все методы включают этап объединения активного компонента с носителем, который состоит из одного или нескольких дополнительных ингредиентов. В общем случае, препараты готовятся путем однородного и тщательного соединения активного ингредиента с жидким носителем или очень мелко измельченного твердого носителя, или и тем и другим способом, и затем, если необходимо, препарату придается желаемая форма.
Препараты настоящего изобретения, предназначенные для орального применения, могут быть представлены в форме дискретных единиц, таких как капсулы, облатки, таблетки или лепешки, каждая из которых содержит определенное количество активного ингредиента; в форме порошка или гранул; в форме раствора или суспензии в водных или неводных жидкостях, или в форме эмульсии масло-в-воде или вода-в-масле. Активный ингредиент может также быть назначен в форме болюсов, лекарственной кашки или пасты.
Препарат, предназначенный для ректального применения, может быть в форме суппозитория, соединяющего активный ингредиент и носитель, или в форме клизм.
Препарат, предназначенный для парентерального применения, содержит стерильный масляный или водный раствор активного ингредиента, изотоничный по отношению к крови реципиента.
Препараты, пригодные для внутрисуставного или глазного применения, могут быть в форме стерильного водного раствора активного ингредиента, который может быть в форме микрокристаллов, например в форме водной микрокристаллической суспензии. Липосомные препараты или биодеградируемые полимерные системы также могут быть использованы, чтобы приготовить активный ингредиент как для внутрисуставного, так и для глазного применения.
Препараты, предназначенные для местного или глазного применения, включат жидкие или полужидкие препараты, такие как эмульсии масло-в-воде или вода-в-масле, мази или пасты; растворы или суспензии, такие как капли.
Препараты, предназначенные для введения в назальную или буккальную полости, включают порошок для вдыхания или спреи, такие как аэрозоли или распылители.
Другие препараты, предназначенные для назального применения, включают мелкий порошок, который принимается способом вдыхания, то есть путем быстрого вдыхания через нос из контейнера с порошком, поднесенного близко к носу.
Препараты этого изобретения, кроме вышеуказанных, могут включать один или более дополнительных ингредиентов.
Композиция может дополнительно содержать другие терапевтически активные вещества, которые обычно используются в лечении перечисленных патологических состояний, например глюкокортикоиды, антигистаминные препараты, антагонист тромбоцит-активирующего фактора (PAF), антихолинергические агенты, метилксантины, в-адренергические агенты, салицилаты, индометацин, флуфенамат, напроксен, тимегадин, соли золота, пеницилламин, сывороточные холестерин-снижающие агенты, ретиноиды, соли цинка, и салицилазосульфапиридин (Салазопирин).
Изобретение может быть дополнительно описано в следующих примерах.
Пример 1
(+)-2R, 3R-E-3-N-[3-2-(хинолин-2-ил)этенил]-фениламино-3-фенил-1,2- пропандиол
Смесь Е-3-[2-(хинолин-2-ил)этенил] анилина (0,5 г, 2 ммол) (сравни ЕР 0206751 A Merk Frosst Canada Inc) и (2R,3R-(+)-3-фенил-глицидола (Aldrich) (0,3 г 2 ммол) в этаноле (10 мл) кипятят с обратным холодильником 8 дней. После охлаждения полученный преципитат собирают фильтрацией и промывают этанолом и эфиром. Получают вышеназванное соединение (указанное в заголовке) с точкой плавления 164-166oC и [а]D 20 = +60.9o (c = 1,0, CH3OH)
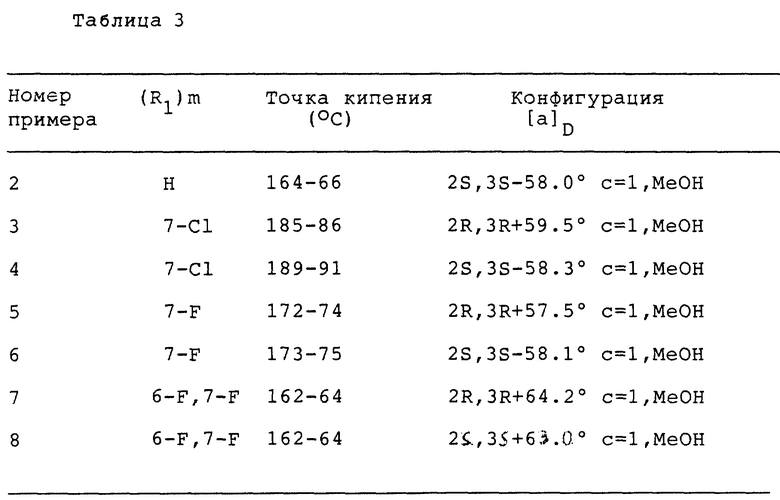
Пример 2-8
Используя процедуру, приведенную в примере 1, и используя подходящие исходные материалы, получают соединения, представленные в табл. 3.
Пример 9
Таблетка
(+)-2R, 3R-E-3-N-[3-2-(6,7-дифторохинолин-2-ил)этенил] фениламино-3-фенил-1,2-пропандиол
(активное вещество) - 100 мг
Лактоза - 75 мг
Крахмал - 12 мг
Метил целлюлоза - 2 мг
Натрий карбоксиметил целлюлоза (CMC-Na) - 10 мг
Стеарат магния - 1 мг
Активное вещество, лактозу и крахмал смешивают до гомогенного состояния в подходящем смесителе и смачивают 5%-ым водным раствором метил целлюлозы 15 сПз. Смешивание продолжают до формирования гранул. Если необходимо, влажные гранулы пропускают через подходящее сито и сушат до содержания воды менее 1% подходящим образом, например в псевдоожиженном слое или в сушильной печи. Сухой гранулят пропускают через 1 мм сито и смешивают до гомогенного состояния с натрийкарбометилцеллюлозой. Стеарат магния добавляют и смешивание продолжают короткий период времени.
Таблетки весом от 200 мг получают из гранулята с помощью подходящей таблетирующей машины.

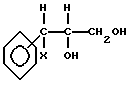
Содержащие диод производные хинолина общей формулы I,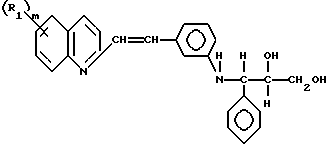
где R1 обозначает водород или галоген, предпочтительно фтор или хлор, и m-0,1 или 2. Указанные соединения получают взаимодействием амина формулы II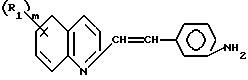
с соединением формулы III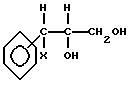
Соединения формулы I обладают антагонистической активностью и поэтому могут быть использованы в качестве фармацевтического препарата в медицине. Настоящее изобретение относится также к способу ингибирования специфического сокращения гладкой мускулатуры. 4 с. и 4 з.п. ф-лы, 3 табл.
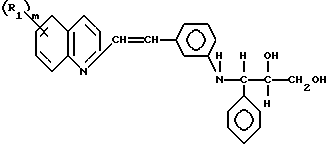
в котором R1 - это водород или галоген, предпочтительно фтор или хлор, и m = 0, 1 или 2; и его стереоизомеры.
(+)-2R, 3R-E-3-N-[3-2-(6,7-дифторохинолин-2-ил)этенил] -фениламино-3-фенил-1,2-пропандиол,
(+)-2R, 3R-E-3-N-[3-2-(хинолин-2-ил)этенил] -фениламино-3-фенил-1,2-пропандиол,
(-)-2S, 3S-E-3-N-[3-(2-хинолин-2-ил)этенил] -фениламино-3-фенил-1,2-пропандиол,
(+)-2R, 3R-E-3-N-[3-2-(7-хлорохинолин-2-ил)этенил] -фениламино-3-фенил-1,2-пропандиол,
(-)-2S, 3S-E-3-N-[3-2-(7-хлорохинолин-2-ил)этенил] -фениламино-3-фенил-1,2-пропандиол,
(+)-2R, 3R-E-3-N-[3-2-(7-фторохинолин-2-ил)этенил] -фениламино-3-фенил-1,2-пропандиол,
(-)-2S, 3S-E-3-N-[3-2-(7-фторохинолин-2-ил)этенил] -фениламино-3-фенил-1,2-пропандиол,
(-)-2S, 3S-E-3-N-[3-2-(6,7-дифторохинолин-2-ил)этенил] -фениламино-3-фенил-1,2-пропандиол,
и их энантиомерные формы.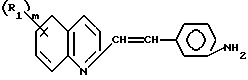
в котором R1 и m имеют вышеуказанные значения, вступает в реакцию с соединением формулы III.
 (
(
| Способ получения производных -содержащих гетероциклических соединений | 1976 |
|
SU682514A1 |
| УСТРОЙСТВО ДЛЯ ИЗМЕРЕНИЯ ЭНЕРГИИ И СОПРОТИВЛЕНИЯ СВАРОЧНОГО КОНТАКТА | 0 |
|
SU206751A1 |
| СПОСОБ ПОДГОТОВКИ ПЛАТИНОВОГО тигля | 0 |
|
SU256180A1 |
| Устройство для электростатической записи информации | 1978 |
|
SU725063A1 |
| WO 9403431 A, 23.11.1987. | |||

