Настоящее изобретение относится к 5-ациламино-1,2,4- тиадиазолам, замещенным в положении 3 ароматической группой и связанным за счет карбонильной функции с ароматическим азотсодержащим гетероциклом; эти соединения обладают сродством к биологическим рецепторам холецистокинина.
Холецистокинин (CCK) представляет собой полипептидный гормон, в котором ин виво находят несколько фрагментов 4-39 аминокислот. Они обладают многочисленными физиологическими активностями, особенно они оказывают воздействие на желчные пути, желудочно- кишечный тракт или на центральную и периферическую нервную систему, как описано J.E.Morley в Life Sciences, 30, 479-493(1982).
Показано 2 типа рецепторов, А и Б, не исключено существование других типов или подтипов. Агонисты и антагонисты воздействия холецистокинина на эти рецепторы известны; можно назвать производные 3-амино-бензодиазепинонов, J.Med.Chem. 32, 13-16 (1989), или производные 2-ациламинотиазолов, европейская патентная заявка А-432 040 и европейская патентная заявка А-518 731, которые, в зависимости от природы заместителей, являются более или менее селективными рецепторами типа А или Б.
Различные антагонисты и агонисты CCK в настоящее время изучаются в клинике для людей, особенно в качестве регуляторов аппетита, с целью лечения желудочно-кишечных расстройств, для борьбы с болью, для уменьшения беспокойства, при шизофрении или для ликвидации симптомов абстиненции у зависимых от лекарств людей.
Соединения изобретения, которые, в зависимости от их структуры, обладают агонистической или антагонистической активностью, оказываемой на холецистокинин в отношении его рецепторов типа А или Б, отвечают формуле (I)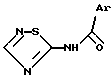
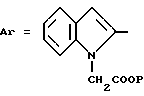
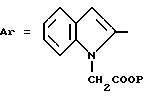
в которой Ar обозначает ароматический азотсодержащий гетероцикл, выбираемый среди хинолинила, изохинолинила, бензимидазолила и индолила, причем этот последний в известных случаях замещен на азоте группой W, где W выбирается из группы, включающей:
(I) -СО-(С1-С4)-алкил;
(II) -(CH2)nCOR, где R обозначается OR1 или NR1R2, причем R1 и R2, одинаковые или нет, выбираются среди водорода и (C1-C4)- алкила, и n = 1, 2;
(III) гидрокси (C1-C4)-алкил;
(IV) алкоксиалкил с 2-6 C-атомами;
(V) тетрагидропиранил;
(VI) цепь -(CH2)3-, последний атом углерода который фиксирован на фенильном ядре индола с образованием 6-членного гетероцикла;
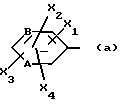
Z обозначает группу(а)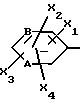
где А и В, независимо друг от друга, обозначают С, CH или N;
X1-X4, одинаковые или разные, обозначают атом водорода, (C1-C4)-алкильную группу, (C1-C3)-алкоксильную группу, галоген, в особенности хлор или бром, или трифторметил,
или нафтильную группу, в известных случаях замещенную (C1-C3)-алкильной группой, (C1-C3)-алкоксильной группой или атомом галогена.
Изобретение относится также к фармацевтически приемлемым солям соединений формулы (I).
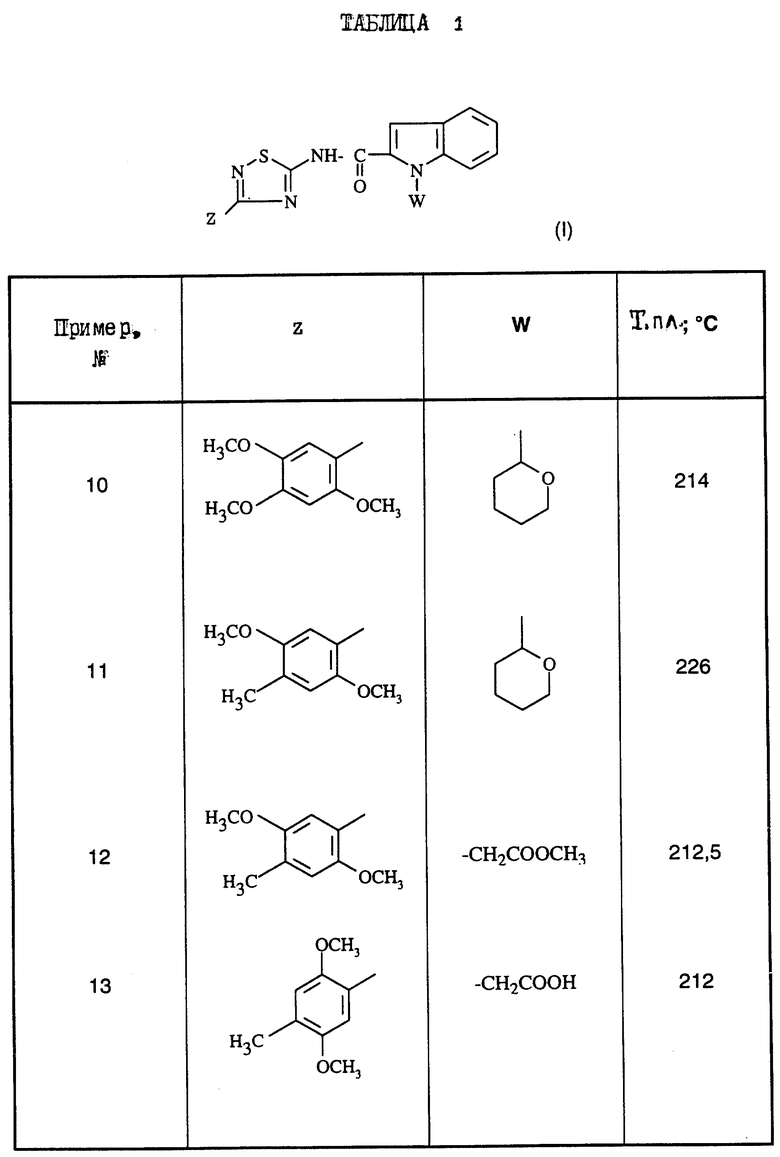
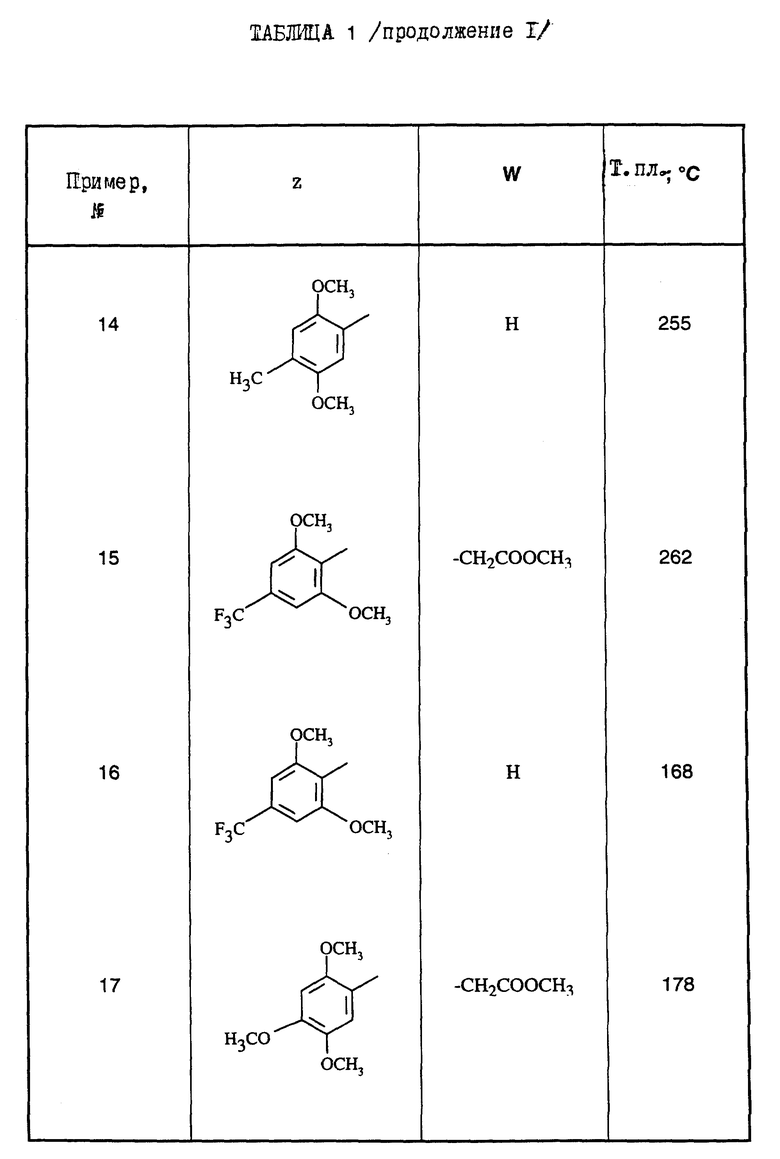
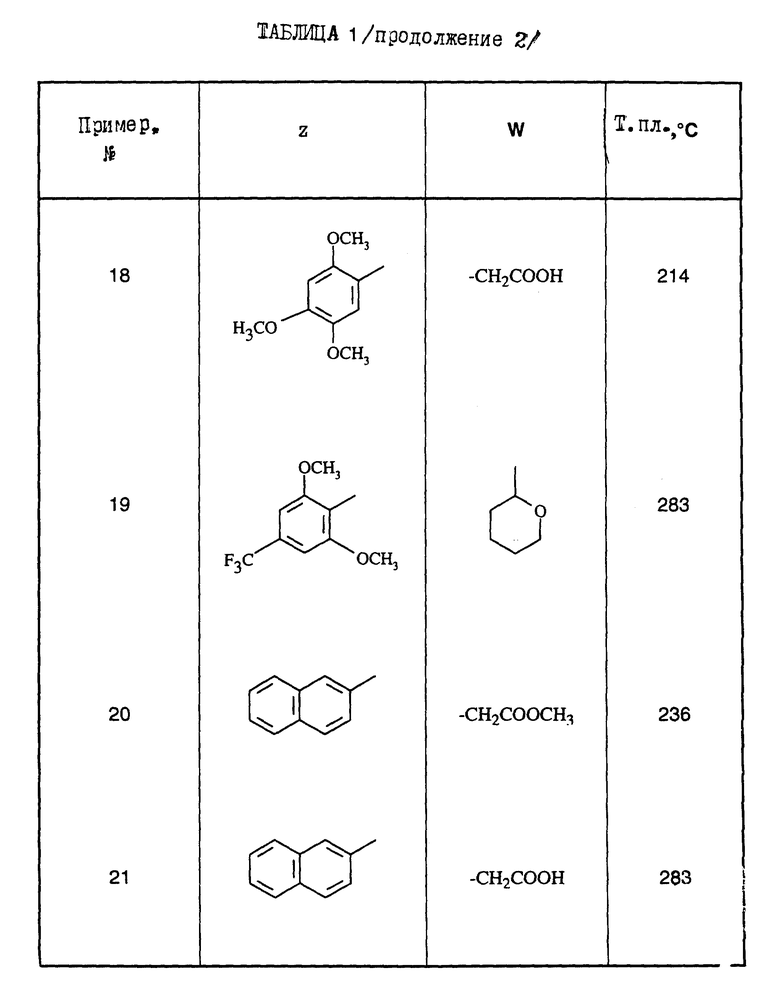
Из этих соединений предпочтительны такие, в которых по крайней мере 3 из заместителей X1-X4 обозначают атом водорода или (C1-C2)- алкильную группу или (C1-C2)-алкоксильную группу, и в особенности таковые, выбираемые среди метильных или метоксильных групп в положениях 2, 4, 6 ароматического ядра; кроме того, предпочтительно, чтобы Ar обозначал замещенный или замещенный индолил или преимущественно замещенную или нет 2-индолильную группу.
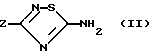
Соединения формулы (I) могут быть получены конденсацией аминотиадиазола формулы (II)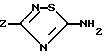
с кислотой или реакционноспособным производным кислоты формулы Ar'COOH, в которой Ar' обозначает Ar или производное Ar, в котором чувствительные к обычным условиям ацилирования функции защищены. Из пригодных производных кислот можно назвать хлорангидриды и ангидриды кислоты, в известных случаях смешанные, или активированные сложные эфиры, используемые обычно в синтезе пептидов.
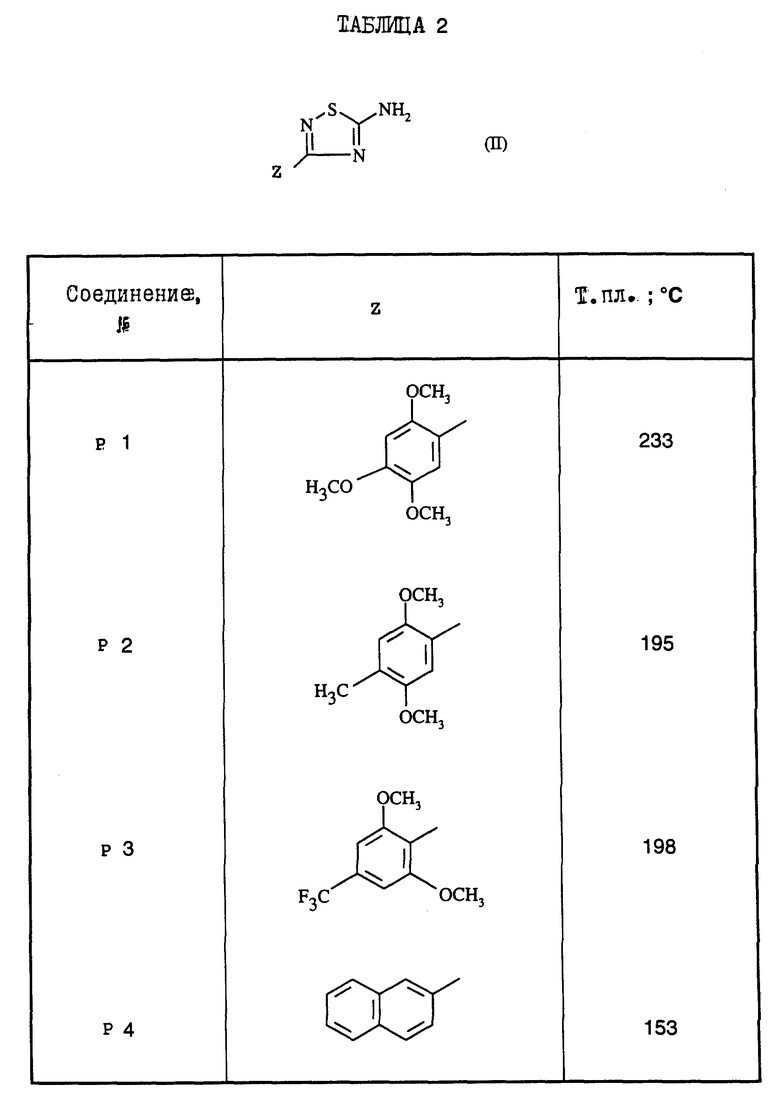
Некоторые соединения формулы (II) известны; можно сослаться, например, на европейский патент А-455 356, или на патенты ФРГ 842 346 или 955 684, где описывается их получение путем воздействия тиоцианата щелочного металла на соответствующий N-галогеноамидин. Другие соединения формулы (II) являются новыми и получаются известными способами. Их можно получать, например, путем воздействия жидкого аммиака на 5-хлортиадиазол, обычно замещенный в положении 3 и получаемый путем воздействия трихлорметансульфенилхлорида на надлежащим образом замещенный амидин. Амидины получают при применении известных способов из соответствующих нитрилов формулы (III): Z-C=N (III), или из оксима, который можно восстанавливать до амидина водородом в присутствии катализатора, или из сложного иминоэфира, который обрабатывают с помощью NH4Cl.
Условия всех этих реакций хорошо известны специалисту.
Когда нитрилов нет в продаже, их можно получать путем воздействия реактива Лавессона на соответствующий первичный амид, получаемый из карбоновой кислоты.
Для получения кислот Ar-СООН и их производных и в случае метода конденсации с амином можно сослаться на европейский патент А-432 040 и патент ФРГ 3 907 390.
Соединения изобретения вытесняют иодированный холецистокинин или его иодированные биологические фрагменты из их рецепторов типа А или Б.
Сродство к рецепторам типа А изучалось ин витро на гомогенате поджелудочной железы крысы по отношению к иодированному CCK 8 S согласно методу, описанному особенно в Endocrinology, 109, 1746 (1981); в этих условиях описанные в нижеследующих примерах продукты имеют Cl50 в пределах 10-8 М - 10-10 М.
Сродство к рецепторам типа Б изучалось методом, описанным в J.Neurochem. , 37, 443 (1981), на гомогенате коры головного мозга морской свинки, причем соединения примеров имеют Cl50 порядка 10-7 М.
Наконец, для определения, являются ли соединения агонистами или антагонистами рецепторов типа А, специалист знает, что он может изучить воздействие соединений на секрецию амилазы панкреатическими ацинусами (acini) крыс, согласно способу, описанному особенно в J.Biol. Chem. 254 (12), 5321-5327 (1979); в этих условиях агонист стимулирует секрецию амилазы, тогда как антагонист снижает секрецию, вызываемую фрагментом CCK 8S. Из соединений формулы (I) те, для которых находящийся в положении 2 X1 обозначает OCH3, а X2 и X3, независимо друг от друга, обозначают метил или метоксигруппу, являются предпочтительными агонистическими соединениями.
В соответствии со своей способностью вытеснять холецистокинин из его рецепторов А или Б, соединения изобретения предпочтительно могут быть использованы для лечения или профилактики заболеваний, в которые вовлечены холецистокинин или его фрагменты.
Когда эти соединения являются антагонистами CCK, их можно использовать против язв, рака поджелудочной железы и рака селезенки, панкреатитов, гиперинсулинемии, синдрома раздражительной ободочной кишки или еще в случае психозов, беспокойства, болезни Паркинсона, для уменьшения замедленных дискинезий, в качестве регуляторов аппетита или при лечении болей или для борьбы против синдрома абстиненции у зависимых от лекарств людей.
Когда эти соединения являются агонистами CCK к рецепторам типа А, их используют в качестве анорексигенов или еще при лечении психозов, для улучшения памяти или в качестве антишоковых средств.
Фармацевтические композиции, которые содержат в качестве действующего начала по крайней мере одно соединение формулы (I) или одну из его фармацевтически приемлемых солей, составляют другой предмет изобретения.
В фармацевтических композициях настоящего изобретения для орального, подъязычного, подкожного, внутримышечного, внутривенного, топического, через нос, чрескожного или ректального введения, действующие начала вышеуказанной формулы (I) или их возможные соли можно выпускать в единичных дозах, в виде смеси с классическими фармацевтическими основами, животным и людям для профилактики или лечения вышеуказанных расстройств или заболеваний. Соответствующие единичные формы введения включают пероральные формы, такие, как таблетки, желатиновые капсулы с лекарством, порошки, гранулы и оральные растворы или суспензии; подъязычные, ротовые, интратрахеальные, внутриносовые формы введения; подкожные, внутримышечные или внутривенные формы введения и ректальные формы введения. Для топического применения можно использовать соединения согласно изобретению в кремах, помадах или лосьонах.
Когда готовят твердую композицию в форме таблеток, основной активный ингредиент смешивают с фармацевтическим эксципиентом, таким, как желатина, крахмал, лактоза, стеарат магния, тальк, гуммиарабик или аналогичные вещества.
Таблетки можно покрывать оболочкой из сахарозы, производного целлюлозы, или других соответствующих материалов, или можно их обрабатывать таким образом, чтобы они обладали пролонгированной или замедленной активностью и чтобы они высвобождали непрерывно заданное количество действующего начала.
Препарат в виде желатиновых капсул с лекарством получают путем смешения активного ингредиента с разбавителем и внесения полученной смеси в мягкие или жесткие желатиновые капсулы.
Препарат в форме сиропа или эликсира или для введения в виде капель может содержать активный ингредиент вместе с подслащивающим, предпочтительно некалорийным, средством, метилпарабеном или пропилпарабеном в качестве антисептиков, также, как с агентом, придающим вкус и соответствующий цвет.
Порошки или гранулы, диспергируемые в воде, могут содержать активный ингредиент в виде смеси с диспергаторами или смачивателями, или с суспендирующими агентами, например, с поливинилпирролидоном, точно также, как с подслащивающими веществами или улучшающими вкус веществами.
Для ректального введения прибегают к свечам, которые готовят со связующими, плавящимися при ректальной температуре, например, как масло какао или полиэтиленгликоли.
Для парентерального введения используют водные суспензии, солевые изотонические растворы или стерильные и инъекцируемые растворы, которые содержат фармакологически приемлемые диспергаторы и/или смачиватели, например, как пропиленгликоль или бутиленгликоль.
Действующее начало также может выпускаться в виде микрокапсул, в известных случаях с одной или несколькими основами или добавками.
Композиции настоящего изобретения, наряду с продуктами вышеприведенной формулы (I) или одной из их фармацевтически приемлемых солей, могут содержать другие действующие начала, которые могут быть пригодными для лечения вышеуказанных расстройств или заболеваний.
Вводимые дозы зависят от природы и значительности заболевания, соединения и пути введения. Они обычно составляют 20-100 мг в день для взрослого перорально и 3-10 мг в виде инъекции.
Ниже описываются примеры получения образцов соединений изобретения.
Пример 1. 2-[3-(2-Хлорфенил)-1,2,4-тиадиазолил-5-амино- карбонил]индолил-1-уксусная кислота и ее сложный метиловый эфир.
Формула (I):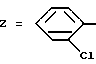
где P=H или CH3-.
a) 2-Хлорбензамидин.
К 200 мл насыщенного газообразным хлорводородом метанола добавляют 56 г 2-хлорбензонитрила при температуре около 0oC. Смесь выдерживают при +5oC в течение ночи (в холодильнике). Затем реакционную смесь выпаривают без нагревания, остаток обрабатывают с помощью 200 мл безводного метанола. Раствор охлаждают до температуры около 0oC и вводят туда газообразный аммиак вплоть до достижения щелочного pH-значения. Реакционную среду нагревают в течение 3-х часов. После выпаривания досуха, 2-хлорбензамидин очищают путем колоночной хроматографии на диоксиде кремния (элюирующее средство: дихлорметан/метанол = 8/2).
Т. пл. 236oC (бромгидрат). Выход 70%.
б) 5-Хлор-3-(2-хлорфенил)-1,2,4-тиадиазол.
К охлажденной до -10oC смеси из 37 г 2-хлорбензамидин-бромгидрата в 160 мл дихлорметана прикапывают 36 г трихлорметансульфенил-хлорида, растворенных в 160 мл дихлорметана. После перемешивания в течение 30 минут в среду, поддерживая температуру -10oC, прикапывают 65 мл водного 50%-ного раствора NaOH. Раствор оставляют стоять для повышения температуры до комнатной и перемешивают в течение 2-х часов. В реакционную среду добавляют воду и декантируют. Органическую фазу промывают водой, сушат над сульфатом натрия и выпаривают досуха.
в) 5-Амино-3-(2-хлорфенил)-1,2,4-тиадиазол.
В автоклаве полученные в предыдущей стадии кристаллы суспендируют в 100 мл метанола и при охлаждении добавляют большой избыток жидкого аммиака. Оставляют температуру повышаться до комнатной и перемешивают в течение 24-х часов, перед тем, как концентрировать досуха. Целевой продукт очищают путем колоночной хроматографии на диоксиде кремния (элюирующее средство: дихлорметан/гексан = 80/20).
Т. пл. 136oC. Общий выход стадий б) и в) составляет 61%.
г) (Метил)-2-[3-(2-хлорфенил)-1,2,4-тиадиазолил-5- аминокарбонил]-индолил-1-ацетат.
3,6 мл Пиридина растворяют в 40 мл дихлорметана и добавляют 0,90 мл тионилхлорида при температуре около -5oC. Оставляют на 30 минут при -5oC и порциями добавляют 3 г 1-(метоксикарбонилметил)- индолил-2-карбоновой кислоты. Перемешивают в течение 30 минут при той же самой температуре, затем порциями добавляют 2,4 г 5-амино-3- (2-хлорфенил)-1,2,4-тиадиазола. Оставляют температуру реакционной смеси повышаться до комнатной и перемешивают в течение 18 часов. Реакционную среду промывают водой, раствор декантируют, органическую фазу сушат над сульфатом натрия и выпаривают досуха. Продукт очищают путем колоночной хроматографии на диоксиде кремния [элюирующее средство: дихлорметан/метанол = 95/5], затем перекристаллизуют из диизопропилового эфира.
Т. пл. 197oC. Выход 69%.
д) 2-[3-(2-Хлорфенил)-1,2,4-тиадиазолил-5-аминокарбонил] - индолил-1-уксусная кислота.
1 г (Метил)-2-[3-(2-хлорфенил)-1,2,4-тиадиазолил-5- аминокарбонил]-индолил-1-ацетата суспендируют в 20 мл метанола и при комнатной температуре добавляют 7 мл 1 н. водного раствора NaOH. Смесь перемешивают в течение 3-х часов при комнатной температуре; реакционную среду концентрируют; добавляют туда воду и доводят до pH = 3 путем добавления KHSO4. Выделяют осадок.
Т. пл. 255oC. Выход 92%.
Пример 2. 2-[3-(2,4,6-Триметоксифенил)-1,2,4-тиадиазолил-5- аминокарбонил]индолил-1-уксусная кислота и ее сложный метиловый эфир.
Формула (I):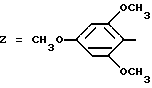
где P=H или CH3.
а) 2,4,6-Триметоксибензамидоксим.
К суспензии 13,8 г гидроксиламинхлоргидрата в 100 мл этанола при температуре около 15oС добавляют раствор этилата натрия, полученный путем растворения 4,3 г натрия в 100 мл этанола. В среду вводят 12 г 2,4,6-триметоксибензонитрила, затем, до выпаривания досуха, кипятят с обратным холодильником в течение 41 часа. Кристаллы промывают водой и дихлорметаном.
Т. пл. 205oC. Выход 71%.
б) 2,4,6-Триметоксибензамидин.
В автоклаве, 3,4 г 2,4,6-триметоксибензамидоксима растворяют в 120 мл смеси метанол/дихлорметан/уксусная кислота [2:2:1 по объему] и гидрируют под давлением 2•106 Па в присутствии 1 г никеля Ренея. После гидрирования в течение 2-х часов, катализатор отделяют и концентрируют досуха.
в) 5-Хлор-3-(2,4,6-триметоксифенил)-1,2,4-тиадиазол.
Полученную в предыдущей стадии смолу растворяют в 20 мл дихлорметана, охлажденного до -10oC, и прикапывают 2,8 мл трихлорметансульфенилхлорида, растворенных в 20 мл дихлорметана.
Реакционную среду перемешивают в течение 30 минут при -10oC, прикапывают 5 мл водного 30%-ного раствора NaOH и перемешивают затем 2 часа при комнатной температуре, после чего в реакционную среду добавляют воду; после декантации органическую фазу промывают водой, сушат над сульфатом натрия и выпаривают досуха.
г) 5-Амино-3-(2,4,6-триметоксифенил)-1,2,4-тиадиазол.
В автоклаве полученные в предыдущей стадии кристаллы суспендируют в 100 мл метанола и при охлаждении добавляют большой избыток жидкого аммиака. Смесь оставляют для повышения температуры до комнатной и перемешивают в течение 18 часов, после чего выпаривают досуха. К остатку добавляют 100 мл водного 2н. раствора соляной кислоты; образовавшийся осадок отфильтровывают, промывают водой, затем ацетоном.
Т. пл. 215oC (хлоргидрат). Выход всех трех стадий 41%.
д) (Метил)-2-[3-(2,4,6-триметоксифенил)-1,2,4- триадиазолил-5-аминокарбонил]индолил-1-ацетат.
1,5 мл Пиридина растворяют в 20 мл дихлорметана и при температуре около -5oC добавляют 0,34 мл тионилхлорида. Оставляют на 30 минут при -5oC и порциями добавляют 1 г 1-(метоксикарбонилметил)-индолил-2-карбоновой кислоты. Смесь перемешивают в течение 30 минут при той же температуре, затем порциями добавляют 1,24 г 5-амино-3-(2,4,6-триметоксифенил)- 1,2,4-тиадиазол-хлоргидрата; оставляют стоять для повышения температуры до комнатной и смесь перемешивают в течение 18 часов. Реакционную среду промывают водой. После декантации, органическую фазу сушат над сульфатом натрия и выпаривают досуха. Целевой продукт очищают путем колоночной хроматографии на диоксиде кремния (элюирующее средство: дихлорметан/метанол = 95:5) и кристаллизуют из диизопропилового эфира.
Т. пл. 205oC. Выход 78%.
е) 2-[3-(2,4,6-Триметоксифенил)-1,2,4-тиадиазолил-5- аминокарбонил] -индолил-1-уксусная кислота.
0,7 г (Метил)-2-[3-(2,4,6-триметоксифенил)-1,2,4-тиадиазолил- 5-аминокарбонил] индолил-1-ацетата суспендируют в 15 мл метанола, и при комнатной температуре добавляют 4,4 мл водного 1н. раствора NaOH. Раствор перемешивают при комнатной температуре в течение 3-х часов. Реакционную среду выпаривают. Добавляют воду и доводят pH-значение до 3 путем добавления KHS04. Образовавшийся осадок отделяют.
Т. пл. 260oC. Выход 65%.
Пример 3. 2-[3-(2,6-Диметокси-4-метилфенил)-1,2,4-тиадиазолил-5- аминокарбонил]-индолил-1-уксусная кислота и ее сложный метиловый эфир.
Формула (I):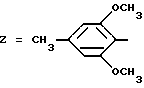
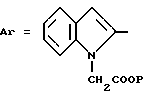
где P=H или CH3.
а) 2,6-Диметокси-4-метил-бензойная кислота.
К 200 мл тетрагидрофурана добавляют 104 мл 1,6 М раствора н-бутиллития в гексане, затем при температуре 0-5oC прикапывают 25 мл 3,5-диметокси-толуола. Перемешивают при 5oC в течение 1,5 часов, затем в течение 30 минут в реакционную среду вводят избыток газообразного CO2. Смесь вводят в 200 мл водного 0,5н. раствора HCl и водную фазу экстрагируют этилацетатом. Остаток концентрируют и хроматографируют на колонке с диоксидом кремния (элюирующее средство: дихлорметан/метанол = 93/7).
Т. пл. 178oC. Выход 72%.
б) 2,6-Диметокси-4-метил-бензамид.
7 г вышеполученной кислоты растворяют в 100 мл дихлорметана и к раствору прикапывают 6,7 мл оксалилхлорида, поддерживая температуру 10oC. Смесь оставляют на 4 часа при комнатной температуре, после чего выпаривают досуха; на остаток выливают 100 мл жидкого аммиака и смесь оставляют при 20oC в течение 18 часов в автоклаве. После выпаривания досуха, на остаток выливают смесь воды с этилацетатом и целевой продукт экстрагируют в органической фазе. Продукт очищают путем колоночной хроматографии на диоксиде кремния (элюирующее средство: дихлорметан/метанол = 9:1).
Т. пл. 201oC. Выход 73%.
в) 2,6-Диметокси-4-метилбензонитрил.
3 г Амида суспендируют в 60 мл толуола и добавляют 3,7 г реактива Лавессона, после чего смесь нагревают при 90oC в течение 1,5 часов. Реакционную среду выпаривают досуха и остаток обрабатывают этилацетатом; органический раствор промывают водным раствором NaOH, сушат и концентрируют. Остаток очищают путем колоночной хроматографии на диоксиде кремния (элюирующее средство: дихлорметан/толуол = 1:1).
Т. пл. 122oC. Выход 92%.
г) 2,6-Диметокси-4-метилбензамидоксим.
К раствору 2,6 г вышеполученного бензонитрила в 30 мл этанола добавляют 2,2 г гидроксиламин-хлоргидрата, затем 1,34 г NaOH в виде таблеток. Кипятят с обратным холодильником в течение 48 часов и реакционную среду выпаривают досуха. Обрабатывают с помощью 100 мл водного 1н. раствора HCl и промывают этилацетатом; путем добавления водного 1н. раствора NaOH pH-значение раствора доводят до 5 и затем амидоксим-хлоргидрат экстрагируют этилацетатом.
Т. пл. 140oC. Выход 75%.
д) 5-Амино-3-(2,6-диметокси-4-метилфенил)-1,2,4-тиадиазол.
В автоклав вводят 3,2 г 2,6-диметокси-4-метил-бензамидоксима в виде раствора в 50 мл метанола и добавляют 400 мг никеля Ренея; гидрируют при 20oC под давлением 1,4•106 Па водорода в течение 12 часов. Катализатор отфильтровывают на слое талька и метанольный фильтрат выпаривают досуха. Остающееся масло желтого цвета растворяют в 50 мл дихлорметана и при 20oC добавляют 2,84 г трихлорметансульфенил-хлорида, затем, при -10oC, прикапывают 20 мл раствора 3 г NaOH в 20 мл воды. По окончании добавления оставляют температуру реакционной смеси повышаться до 20oC и смесь перемешивают в течение 3-х часов. После этого из реакционной смеси экстрагируют с помощью дихлорметана целевой продукт. Это масло растворяют в смеси 40 мл метанола и 10 мл дихлорметана и, после охлаждения, вводят в автоклав, содержащий 200 мл жидкого аммиака; после выдерживания 12 часов при комнатной температуре остаток концентрируют и обрабатывают его дихлорметаном; органическую фазу промывают водой, концентрируют ее и целевой продукт очищают путем колоночной хроматографии на диоксиде кремния (элюирующее средство: дихлорметан/метанол = 96/4).
Т. пл. 117oC. Выход общий 58%.
е) (Метил)-2-[3-(2,6-диметокси-4-метилфенил)-1,2,4- тиадиазолил-5-аминокарбонил]индолил-1-ацетат.
Вводят 1 мл пиридина в 10 мл дихлорметана при 0oC, затем 0,25 мл тионилхлорида; спустя 30 минут в реакционную среду порциями добавляют 0,2 г хлорангидрида 1-/метокси-карбонилметил/индолил-2-карбоновой кислоты, затем прикапывают 0,8 г 5-амино-3-/2,6-диметокси-4-метилфенил/-1,2,4-тиадиазола в виде раствора в 10 мл дихлорметана. Реакционную смесь оставляют на 5 часов при 20oC, после чего вводят в нее один объем воды. Отделяют от смеси органическую фазу, сушат ее и растворитель выпаривают. Полученное желтое масло очищают путем колоночной хроматографии на диоксиде кремния (элюирующее средство: дихлорметан/диэтиловый эфир = 95/5). Продукт перекристаллизуют из изопропанола.
Т. пл. 122oC. Выход 65%.
ж) 2-[3-(2,6-Диметокси-4-метилфенил)-1,2,4-тиадиазолил-5- аминокарбонил] -индолил-1-уксусная кислота.
0,35 г вышеполученного сложного метилового эфира вводят в 5 мл метанола, затем вводят 1,5 мл водного 1н. раствора NaOH. После перемешивания в течение 6 часов метанол выпаривают, на остаток выливают 50 мл воды, и среду подкисляют с помощью водного 5%-ного [вес/объем] раствора KHSO4. Водную фазу экстрагируют дихлорметаном и органическую фазу концентрируют. Продукт получают в виде желтых кристаллов.
Т. пл. 235oC. Выход 82%.
Пример 4. N-[3-(2,6-Диметокси-4-метилфенил)-1,2,4-тиадиазол-5-ил] -1-(2-тетра-гидропиранил)-индолил-2-карбоксамид.
Формула (I):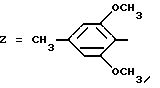
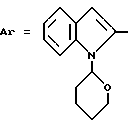
1,8 мл Пиридина вводят в 20 мл дихлорметана. Доводят смесь до 0oC и добавляют туда 0,36 мл тионилхлорида. Спустя 30 минут порциями вводят 1,2 г хлорангидрида 1-(2-тетрагидропиранил)- индолил-2-карбоновой кислоты, затем прикапывают 1,2 г 5-амино-3- (2,6-диметокси-4-метилфенил)-1,2,4-тиадиазола в виде раствора в 10 мл дихлорметана. Спустя 3 часа выдерживания при -20oC, в реакционную среду вводят один объем воды и водную фазу экстрагируют дихлорметаном. Органические фазы затем сушат и концентрируют. Остаток очищают путем колоночной хроматографии на диоксиде кремния (элюирующее средство: дихлорметан/метанол = 99/1).
Т. пл. 142oC. Выход 80%.
Пример 5. N-[3-(2-Хлорфенил)-1,2,4-тиадиазол-5-ил]-1-(2- тетрагидропиранил)-индолил-2-карбоксамид.
Формула (I):
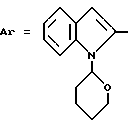
Получают, как в примере 4, из 5-амино-3-(2-хлорфенил)-1,2,6,4- тиадиазола, который плавится при 136oC.
Т. пл. 182oC. Выход 90%.
Пример 6. N-[3-(2,6-Диметокси-4-метилфенил)-1,2,4-тиадиазол- 5-ил]-идолил-2-карбоксамид.
Формула (I):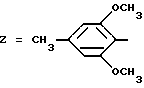
0,4 г соединения примера 4 растворяют в 25 мл метанола и добавляют 0,8 мл водного 6н. раствора HCl. Кипятят с обратным холодильником в течение 6 часов, затем растворитель выпаривают, целевой продукт экстрагируют из водной среды дихлорметаном.
Т. пл. 247oC. Выход 94%.
Пример 7. N-[3-(2-Хлорфенил)-1,2,4-тиадиазол-5-ил] - индолил-2-карбоксамид.
Формула (I):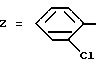
Получают, как в примере 6, из соединения примера 5.
Т. пл. 291oC. Выход 86%.
Пример 8. N-[3-(2-Хлорфенил)-1,2,4-тиадиазол-5-ил] хинолил-2-карбоксамид.
Формула (I):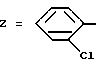
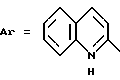
Целевое соединение получают при использовании способа примера 1 и исходя из соответствующей карбоновой кислоты.
Т. пл. 214oC. Выход 85%.
Пример 9. N-[3-(2-Хлорфенил)-1,2,4-тиадиазол-5-ил] [5,6-дигидро-(/4Н/- пиррол-/3,2,1-ij/хинолил)]-2-карбоксамид.
Формула (I):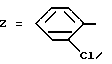
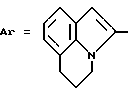
Используя способ примера 1 и исходя из соответствующей карбоновой кислоты получают целевое соединение.
Т. пл. 195oC. Выход 79%.
Поступая согласно предыдущим примерам 1-9, получают соединения формулы (I) примеров 10-21, перечисленные в нижеприведенной табл.1. Промежуточные 5-амино-1,2,4-тиадиазолы Р1-Р4, приводящие к соединениям примеров 10-21, перечислены в табл.2.
Фармацевтические испытания.
Для иллюстрации биологической активности новых соединений был использован метод испытаний in vitro, описанный в журнале European Journal of Pharmacology, 232 (1993).
Испытания новых соединений были проведены на предмет их влияния на связь холецистокинина с его рецепторами.
Первое испытание относится к рецепторам CCK типа А, в котором определялась связь холецистокинина с рецептором CCK типа А на мембранном препарате поджелудочной железы крысы, с использованием 40 мкг протеина/трубку мембран поджелудочной железы и 40 рМ радиомеченого CCK. После инкубации при 25oC в течение 40 мин инкубационную смесь нанесли на инкубационный буфер объемом 1 мл, охлажденный льдом, с добавлением 0,5% BSA, поместили в микроцентрифугу и центрифугировали при 10 000 g в течение 5 минут. Супернатант снимали и определяли радиоактивность нижней части на гамма-сцинтилляторном счетчике.
Для определения воздействия исследуемого соединения на рецептор CCK измеряли связь холецистокинина с его рецептором в отсутствии и в присутствии исследуемого соединения, взятого в концентрации 0,5 нМ. Результаты анализировались с помощью повторяющейся нелинейной регрессии. Каждая точка представляла собой среднее значение тройных замеров, и линии определялись путем регрессивного анализа.
Второе испытание относилось к определению связи CCK с рецепторами типа В на мембранном препарате коры головного мозга морской свинки с использованием 300 мкг протеина/трубку мембран коры головного мозга и 25 рМ радиомеченого лиганда. После 90 мин инкубации при 25oC реакцию останавливали и определяли связь с CCK, как описано выше для рецепторов CCK типа А.
Результаты сведены в табл. 3, в которой показатель IC50 означает концентрацию, которая ингибирует 50% связи CCK с его рецепторами.
Для иллюстрации композиций предложены несколько возможных формулировок:
Пример рецептуры для таблетки, мг:
Активное соединение формулы I - 5
Лактоза - 60
Поливинилпирролидон - 5
Микрокристаллическая целлюлоза - 25
Натриевая соль карбоксиметилкрахмала - 4
Стеарат магния - 1
Пример рецептуры для желатиновой капсулы размер n1, мг:
Активное соединение формулы I - 50
Стеарат магния - 10
Лактоза - 140
Пример препарата для инъекций, мг:
Активное соединение формулы I - 10
Инозитол - 100
Бензиловый спирт - 20

Описываются новые производные 5-ациламино-1,2,4-тиадиазолов формулы (I), в которой Ar означает ароматический азотсодержащий гетероцикл, выбираемый среди хинолинила, изохинолинила, индолила, причем последний может быть замещен на азоте группой W, где W выбирается из группы, включающей: a)-(CH2)nCOR, где n = 1 или 2 и R обозначает OR1, где R1 выбирается из водорода и (C1-C4)-алкила; б) тетрагидропиранил и в) цепь (CH2)3-, последний атом углерода которой фиксирован на фенильном ядре индола с образованием 6-членного гетероцикла; Z обозначает группу (а), где А и В означают -С, Х1-Х4, одинаковые или разные, означают атом водорода, атом хлора или брома, (С1-С3)-алкильную группу, (С1-С3)-алкоксильную группу или трифторметильную группу; нафтильную группу, а также их фармацевтически приемлемые соли. Соединения обладают сродством к биологическим рецепторам холецистокинина. Описывается также способ получения производных 5-ациламино-1,2,4-тиадиазолов формулы I по любому из пп. 1-6, заключающийся в том, что производное 5-амино-тиадиазола формулы (II), в которой Z имеет указанное в п. 1 значение, вводят во взаимодействие с реакционноспособным производным кислоты формулы Ar'СООН, в которой Ar' имеет значения, указанные в п. 1 для Ar, или обозначает производное Ar, в котором реакционноспособные в условиях ацилирования центры защищены, затем, если необходимо, удаляют защитные группы с указанных реакционноспособных центров ароматического ядра Ar. 2 c. и 6 з.п. ф-лы, 3 табл.
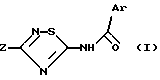
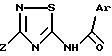
в которой Ar - ароматический азотсодержащий гетероцикл, выбираемый среди хинолинила, изохинолинила, индолила, причем последний может быть замещен на азоте группой W, где W выбирается из группы, включающей: а) -(CH2)nCOR, где n = 1 или 2, R обозначает OR1, где R1 выбирается из водорода и (C1 - C4) - алкила; б) тетрагидропиранил и в) цепь -(CH2)3-, последний атом углерода которой фиксирован на фенильном ядре индола с образованием 6-членного гетероцикла;
Z обозначает группу (а)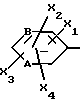
где A и B означают C;
X1 - X4, одинаковые или разные, означают атом водорода, атом хлора или брома, (C1 - C3)-алкильную группу, (C1 - C3)-алкоксильную группу или трифторметильную группу;
нафтильную группу,
а также их фармацевтически приемлемые соли.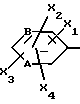
где A и B означают C;
X1 - X4, одинаковые или разные, - водород, (C1 - C3)-алкил, (C1 - C3)-алкоксил, Cl, Br или трифторметил,
а также их фармацевтически приемлемые соли.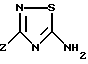
в которой Z имеет указанное в п.1 значение,
вводят во взаимодействие с реакционноспособным производным кислоты формулы Ar'COOH, в которой Ar' имеет значения, указанные в п.1 для Ar, или обозначает производное Ar, в котором реакционноспособные в условиях ацилирования центры защищены, затем, если необходимо, удаляют защитные группы с указанных реакционноспособных центров ароматического ядра Ar.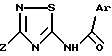
где радикалы Ar и Z имеют значения, указанные в п.1,
в эффективном количестве.
| EP, 0432040 A1, 1991 | |||
| SU, 1122224 A1, 1984 | |||
| EP, 0518731 A1, 1992. |
