Изобретение относится к новым производным триазола, к способу их получения и к содержащим их лекарствам.
Более конкретно настоящее изобретение касается новых непептидных соединений, проявляющих аффинность к холецистокининовым рецепторам.
Холецистокинин (ХЦК) является пептидом, который в ответ на глотание пищи секретируется периферически и участвует в регулировании многих пищеварительных процессов (Crawley J.N. et al., Peptides, 1994, 15 (4), 731-735).
С тех пор как ХЦК был идентифицирован в мозге, он является, может быть, наиболее распространенным нейропептидом, действующим как нейромодулятор церебральных функций посредством стимулирования рецепторов типа ХЦК-В (Crawley J.N. et al., Peptides, 1994, 15 (4), 731-735). В центральной нервной системе ХЦК взаимодействует с допаминопосредованной нейронной передачей (Crawley J. N. et al., ISIS Atlas of Sci., Pharmac. 1988, 84-90). Он также играет определенную роль в механизмах, в которые вовлечены ацетилхолин, ГАМК (4-аминомасляная кислота), серотонин, опиоиды, соматостатин и вещество P, и в ионных каналах.
Его введение вызывает физиологические изменения: пальпебральный птоз, гипотермию, гипергликемию, каталепсию, и изменения в поведении, снижение способности передвижения, снижение способности к исследованию, аналгезию, изменение в способности обучаться, а также изменение в сексуальном поведении и насыщении.
ХЦК проявляет свою биологическую активность через по меньшей мере два типа рецепторов: ХЦК-А-рецепторы, локализованные преимущественно периферически, и ХЦК-В-рецепторы, присутствующие, главным образом, в коре головного мозга. ХЦК-А-рецепторы периферического типа присутствуют также в некоторых зонах центральной нервной системы, включая самую последнюю область, ядро одиночного пути и межножковое ядро (Moran T.N. et al., Brain Research, 1986, 362, 175-179; Hill D.R. et al., J. Neurosci. 1990, 10, 1070-1081); однако со специфическими различиями (Hill D.R. et al., J. Neurosci. 1990, 10, 1070-1081; Mailleux P. et al., Neurosci. lett, 1990, 117, 243-247; Barrett R.W. et al. , Mol. Pharmacol., 1989, 36, 285-290; Mercer J.G. et al., Neurosci Lett, 1992, 137, 229-231; Moran T.N. et al., Trends in Pharmacol. Sci., 1991, 12, 232-236).
На периферии, через ХЦК-А-рецепторы (Moran T.N. et al., Brain Research, 1986, 362, 175-179), ХЦК задерживает опорожнение желудка, модифицирует перистальтику кишечника, стимулирует сокращение желчного пузыря, усиливает секрецию желчи и контролирует панкреатическую секрецию (McHugh P.R. et al., Fed. Proc. , 1986, 45, 1384-1390; Pendleton R.G. et al., J. Pharmacol. Exp. Ther., 1987, 241, 110-116).
В некоторых случаях ХЦК может действовать на артериальное давление и влиять на иммунную систему.
Роль ХЦК в сигнале насыщения подтверждается тем фактом, что концентрации ХЦК в плазме крови, которые зависят от состава пищи (высокие концентрации белков или липидов), становятся после принятия пищи выше, чем концентрации, наблюдаемые до принятия пищи (Izzo R.S. et al., Regul. Pept., 1984, 9, 21-34; Pfeiffer A. et al., Eur. J. Clin. Invest., 1993, 23, 57-62; Lieverse R. J. Gut, 1994, 35, 501). У страдающих булимией имеется снижение секреции ХЦК, индуцированное принятием пищи (Geraciotti T.D. Jr. et al., N. Engl. J. Med., 1988, 319, 683-688; Devlin M.J. et al., Am. J. CIin. Nutr., 1997, 65, 114-120), и снижение концентрации ХЦК в цереброспинальной жидкости (Lydiard R.B. et al. , Am. J. Psychiatry, 1993, 150, 1099-1101). В Т-лимфоцитах, которые являются клеточным компартментом, который может отражать центральную нейронную секрецию, базальные концентрации ХЦК значительно ниже у пациентов, страдающих нервной булимией (Brambilla F. et al., Psychiatry Research, 1995, 37, 51-56).
Лечение (например L-фенилаланином или ингибиторами трипсина), которое повышает секрецию эндогенного ХЦК, вызывает снижение потребления пищи у некоторых видов, включая человека (Hill A.J. et al., Physiol. Behav. 1990, 48, 241-246; Ballinger A.B. et al., Metabolism, 1994, 735-738). Подобным образом, введение экзогенного ХЦК снижает потребление пищи у многих видов, включая человека (Crawley J.N. et al., Peptides, 1994, 15, 731-755).
Ингибирование потребления пищи ХЦК опосредуется ХЦК-А-рецептором. Девазепид - антагонист, селективный в отношении ХЦК-А-рецепторов, ингибирует анорексигенный эффект ХЦК, в то время как селективные агонисты этих рецепторов ингибируют потребление пищи (Asin К.Е. et al., Pharmacol. Biochem. Behav., 1992, 42, 699-704; Elliott R.L. et al., J. Med. Chem., 1994, 37, 1562-1568). Кроме того, крысы линии OLEFT, которые не экспрессируют ХЦК-А-рецептор, нечувствительны к анорексигенному влиянию ХЦК (Myasaka К. et al., 1994, 180, 143-146).
Основываясь на этих доказательствах ключевой роли ХЦК в периферическом сигнале насыщения, использование агонистов и антагонистов ХЦК в качестве лекарств при лечении некоторых расстройств поведения при приеме пищи, ожирения и диабета неоспоримо. Агонист ХЦК-рецепторов можно использовать также терапевтически при лечении эмоциональных и сексуальных расстройств поведения и расстройств памяти (Itoh S. et al., Drug. Develop. Res., 1990, 21, 257-276), шизофрении, психозов (Crawley J. N. et al., Isis Atlas of Sci., Pharmac., 1988, 84-90 и Crawley J.N. Trends in Pharmacol. Sci., 1991, 12, 232-265), болезни Паркинсона (Bedhar I. et al., Biogenic amine, 1996, 12 (4), 275-284), поздней дискинезии (Nishikawa T. et al., Prog. Neuropsychopharmacol. Biol. Psych. , 1988, 12, 803-812; Kampen J.V. et al., Eur. J. Pharmacol., 1996, 298, 7-15) и различных расстройств желудочно-кишечной сферы (Drugs of the Future, 1992, 17 (3), 197-206).
ХЦК-А-рецепторные агонисты ХЦК описаны в литературе. Например, некоторые продукты, обладающие такими свойствами, описаны в ЕР 383690 и WO 90/06937, WO 95/28419, WO 96/11701 или WO 96/11940.
Большая часть агонистов ХЦК-А, описанных к настоящему времени, имеют пептидную природу. Так, FPL 14294, получаемый из ХЦК-7, является мощным неселективным агонистом ХЦК-А по отношению к ХЦК-В. Он обладает высокой ингибиторной активностью в отношении потребления пищи у крыс и у собак после интраназального введения (Simmons R.D. et al., Pharmacol. Biochem. Behav., 1994, 47 (3), 701-708; Kaiser E.F. et al., Faseb, 1991, 5, A864). Точно так же, было показано, что A- 71623, тетрапептидный агонист, селективный в отношении ХЦК-А-рецепторов, является эффективным в моделях анорексии в течение периода 11 дней и приводит к значительному снижению прироста веса по сравнению с контрольными грызунами и собакоголовыми обезьянами (павианами) (Asin К. Е. et al., Pharmacol. Biochem. Behav., 1992, 42, 699-704). Точно так же, структурные аналоги А 71623, имеющие хорошую эффективность и селективность к рецепторам ХЦК-А, обладают значительной анорексигенной активностью у крыс (Elliott R.L. et al., J. Med. Chem., 1994, 37, 309-313; Elliott R.L. et al., J. Med. Chem. , 1994, 37, 1562- 1568). GW 7854 (Hirst G.C. et al. J. Med. Chem. , 1996, 38, 5236-5245), 1,5-бензодиазепин является агонистом ХЦК-А-рецептора in vitro. Эта молекула перорально активна также в отношении сокращения желчного пузыря у мышей и потребления пищи у крыс.
Теперь неожиданно обнаружено, что ряд производных триазола обладает активностью частичного или полного агониста в отношении ХЦК-А-рецепторов.
Соединения по настоящему изобретению подвергались системным исследованиям с целью охарактеризовать:
- их способность вытеснять [125I]-ХЦК с их сайтов связывания, присутствующих на панкреатических мембранах крысы (ХЦК-А-рецептор) или клетках ЗТЗ, которые экспрессируют рекомбинантный ХЦК-А-рецептор человека;
- их аффинность по отношению к ХЦК-В-рецептору, присутствующему на мембранах коры головного мозга морской свинки, причем некоторые соединения являются селективными или неселективными лигандами ХЦК-А-рецептора;
- их свойство агониста ХЦК-А-рецептора посредством их способности индуцировать in vitro мобилизацию внутриклеточного кальция в клетках ЗТЗ, которые экспрессируют ХЦК-А-рецептор человека.
Согласно настоящему изобретению производные триазола являются агонистами ХЦК-А, так как они подобно ХЦК способны стимулировать частично или полностью мобилизацию внутриклеточного кальция в клеточных линиях, которые экспрессируют рекомбинантный ХЦК-А-рецептор человека. Они, что удивительно, гораздо более сильны, чем производные тиазола, описанные в патентных заявках EP 518731 и EP 611766, чем производные тиадиазола, описанные в патентной заявке EP 620221, или чем производные бензодиазепина, описанные в патенте EP 667344.
Причиной этого является то, что эти производные тиазола, тиадиазола и бензодиазепина не способны индуцировать эту мобилизацию внутриклеточного кальция, опосредованную ХЦК-А-рецептором.
Согласно настоящему изобретению производные триазола являются более сильнодействующими, чем эти производные тиазола, тиадиазола или бензоазепина, также в силу их способности блокировать in vivo, внутрибрюшинно, опорожнение желудка у мышей.
Так, свойства агонистов ХЦК-А исследовали in vivo, оценивая их способность блокировать опорожнение желудка у мышей или вызывать, опять же in vivo, опорожнение желчного пузыря у мышей.
Некоторые производные также обладают активностью антагониста ХЦК-В-рецептора.
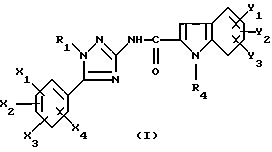
Таким образом, настоящее изобретение относится к N-триазолил-2-индолкарбоксамидам общей формулы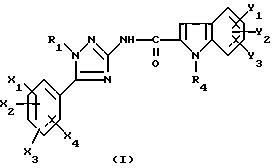
где R1 представляет собой -(C2-C6)алкил; группу -(CH2)n-G с n в пределах от 0 до 5 и G, представляющим собой неароматическую моно- или полициклическую C3-C13 углеводородную группу; фенил(C1-C3)алкил, в котором фенильная группа возможно замещена один раз галогеном или (C1-C3)алкокси; группу -(CH2)nNR2R3, в которой n представляет собой целое число от 1 до 6, а R2 и R3, которые могут быть одинаковыми или разными, представляют собой (C1-C3)алкил или составляют с атомом азота, к которому они присоединены, группу морфолино или пиперидино;
X1, X2, X3 или X4 каждый независимо представляет собой атом водорода или галогена, (C1-C6)алкил или (C1-C3)алкокси при условии, что только один из X1, X2, X3 и X4 возможно представляет собой атом водорода;
R4 представляет собой водород, группу -(CH2)nCOOR5, в которой n является таким, как определено выше, a R5 представляет собой атом водорода или (C1-C6)алкил; (C1-C6)алкил; группу -(CH2)nOR5 или группу -(CH2)nNR2R3, в которой n, R2, R3 и R5 являются такими, как определено выше; группу -(CH2)n-тетразолил, в которой n является таким, как определено выше, или R4 представляет собой одну из этих групп в форме соли щелочного металла;
Y1, Y2 и Y3 независимо представляют собой водород, галоген, (C1-C3)алкил, (C1-C3)алкокси или карбамоил;
или одной из их солей или сольватов.
Соединения формулы (I), в которых R1 представляет собой циклогексил-(C1-C3)алкил, являются предпочтительными соединениями.
Также предпочтительными являются соединения формулы (I), в которых фенил в положении 5 триазола является трехзамещенным предпочтительно метоксигруппой в положениях 2 и 6 и метилом в положении 4.
Еще более предпочтительными являются соединения формулы (I), в которых фенил в положении 5 триазола является трехзамещенным предпочтительно метоксигруппой в положениях 2 и 5 и метилом или хлором в положении 4.
В частности, соединения формулы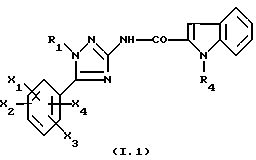
где R1, R4, X1, X2, X3 и X4 являются такими, как определено для (I),
их соль или сольват являются предпочтительными.
Среди этих соединений те из них, в которых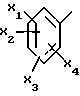
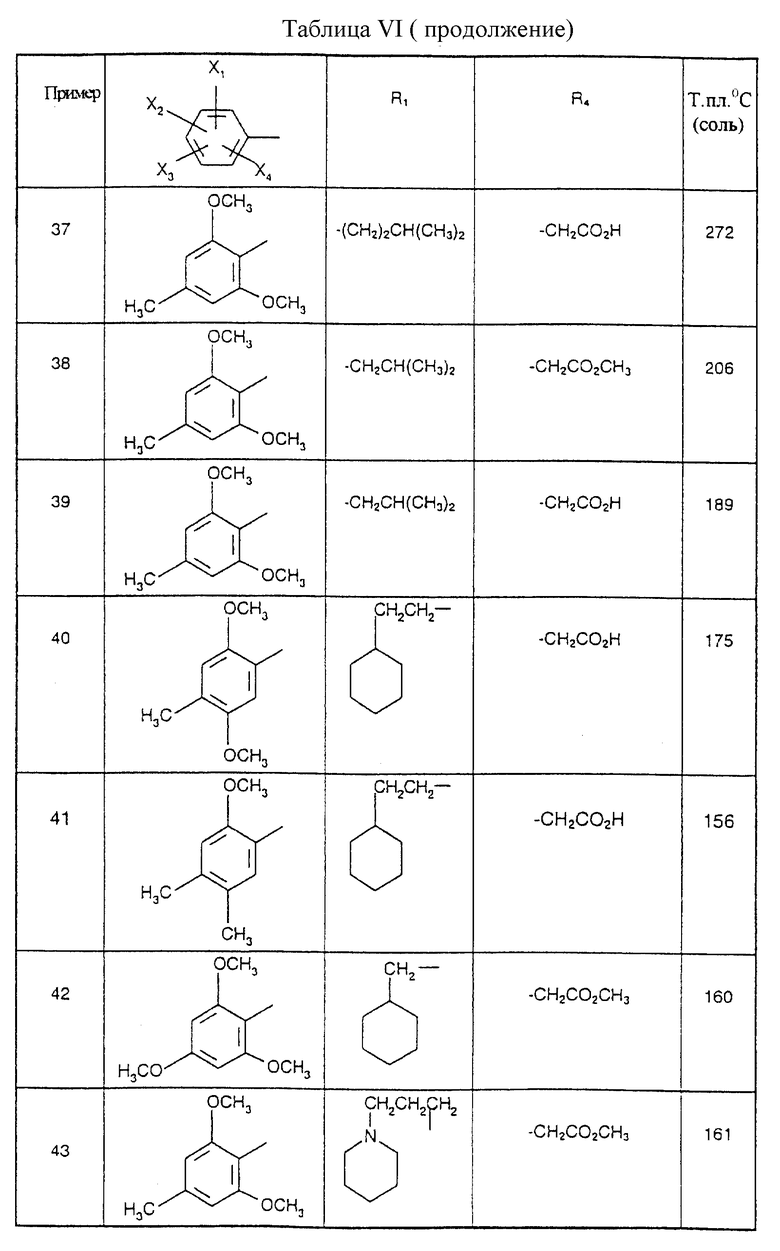
представляет собой 2,6-диметокси-4-метилфенил, являются предпочтительными.
Соединения формулы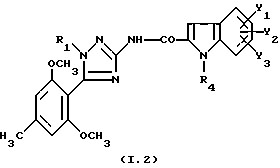
где R1 и R4 являются такими, как определено для (I),
их соль или сольват являются наиболее предпочтительными.
Соединения формулы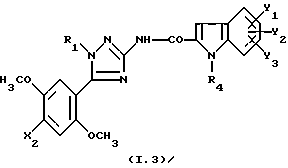
где R1, R4, Y1, Y2 и Y3 являются такими, как определено для (I), а X2 представляет собой метил или атом хлора, их соль или сольват являются наиболее предпочтительными.
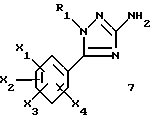
1-замещенные 3-аминотриазолы общей формулы (7) представляют собой новые ключевые промежуточные соединения, которые используют для получения соединений (I), и они также входят в задачу настоящего изобретения.
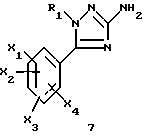
где R1, X1, X2, X3 и X4 являются такими, как определено для (I).
Задачей настоящего изобретения является также способ получения соединений формулы (I), их солей или сольватов, отличающийся тем, что аминотриазол формулы (7)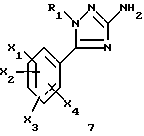
где R1, X1, X2, X3 и X4 являются такими, как определено для (I), подвергают взаимодействию с производным индолкарбоновой кислоты формулы (8)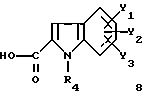
где R4, Y1, Y2 и Y3 являются такими, как определено выше для (1).
Задачей настоящего изобретения является также способ получения соединений формулы (I), их солей или сольватов, отличающийся тем, что аминотриазол формулы (7)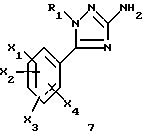
где R1, X1, X2, X3 и X4 являются такими, как определено для (I), подвергают взаимодействию с производным индолкарбоновой кислоты формулы (8')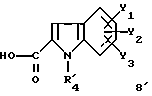
где Y1, Y2 и Y3 являются такими, как определено выше для (1), а R'4 является группой-предшественником R4, причем R4 является таким, как определено для (I), с получением соединения формулы (I')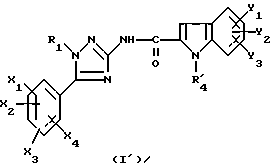
где R1, X1, X2, X3, X4, Y1, Y2 и Y3 являются такими, как определено для (I), a R'4 является группой-предшественником R4, причем полученное соединение формулы (I') преобразуют в соединение формулы (I) путем преобразования группы R'4 в R4.
В задачу настоящего изобретения также входят фармацевтические композиции, имеющие активность агониста ХЦК-А-рецептора, содержащие в качестве активного начала соединение по изобретению или одну из его фармацевтически приемлемых солей.
В соответствии с настоящим изобретением "(C1-C6)алкил" или "(C2-C6)алкил" означают прямой или разветвленный алкил, имеющий от 1 до 6 атомов углерода или от 2 до 6 атомов углерода соответственно.
Радикал алкокси обозначает радикал алкилокси, в котором алкил является таким, как определено выше.
Неароматические C3-C13 углеводородные группы включают в себя насыщенные или ненасыщенные, конденсированные или мостиковые, моно- или полициклические радикалы, которые могут быть терпеновыми. Эти радикалы являются возможно моно- или полизамещенными (C1-C3)алкилом. Моноциклические радикалы включают в себя циклоалкилы, например циклопропил, циклобутил, циклопентил, циклогексил, циклогептил, циклооктил и циклододецил. Полициклические радикалы включают в себя, например, норборнан, адамантан, гексагидроиндан, норборнен, дигидрофенален, бицикло[2.2.1] гептан, бицикло[3.3.1] нонан и трицикло[5.2.1.02,6]декан.
В соответствии с настоящим изобретением термин галоген означает атом, выбранный из атомов фтора, хлора, брома и йода, предпочтительно фтора или хлора.
Катионы щелочных металлов предпочтительно выбраны из катионов натрия или калия.
Когда соединение по изобретению имеет один или более чем один асимметрический атом углерода, оптические изомеры этого соединения образуют неотъемлемую часть данного изобретения.
Когда соединение по изобретению имеет стереоизомерию, например, аксиально-экваториального типа, данное изобретение включает в себя все стереоизомеры этого соединения. Соли соединений формулы (I) по изобретению включают в себя соли с неорганическими или органическими кислотами, которые являются подходящими для разделения или кристаллизации соединений формулы (I), такими как пикриновая кислота, оксалиновая кислота или оптически активная кислота, например винная кислота, дибензоилвинная кислота, миндальная кислота или камфорсульфоновая кислота, и с кислотами, которые образуют физиологически приемлемые соли, такие как гидрохлориды, гидробромиды, сульфаты, гидросульфаты, дигидросульфаты, малеаты, фумараты, 2-нафталинсульфонаты или пара- толуолсульфонаты.
Соли соединений формулы (I) включают в себя также соли с органическими или неорганическими основаниями, например соли щелочных металлов, таких как соли натрия или калия, или соли с амином, таким как трометамол, или, альтернативно, соли аргинина или лизина или соли любого физиологически приемлемого амина.
Функциональные группы, возможно присутствующие в молекуле соединений формулы (I) и в промежуточных соединениях, могут быть защищены либо постоянными, либо временными защитными группами, которые обеспечивают точный синтез ожидаемых соединений.
Выражение временная защитная группа для аминов, спиртов или карбоновых кислот означает такие защитные группы, которые описаны в "Protective Groups in Organic Synthesis", Greene T.W. and Wuts P.G.M., published John Wiley and Sons, 1991, и в Protecting Groups, Kocienski P.J., 1994, Georg Thieme Verlag.
Соединения (I) могут содержать группы-предшественники для других функциональных групп, которые образуются последовательно на одной или более чем одной другой стадии.
Промежуточные соединения (I') приводят к получению соединений формулы (I) преобразованием группы R'4 в R4, которое проводят способом, известным per se, согласно общепринятым способам органической химии.
Исходные материалы имеются в продаже или их получают в соответствии со способами, описанными ниже.
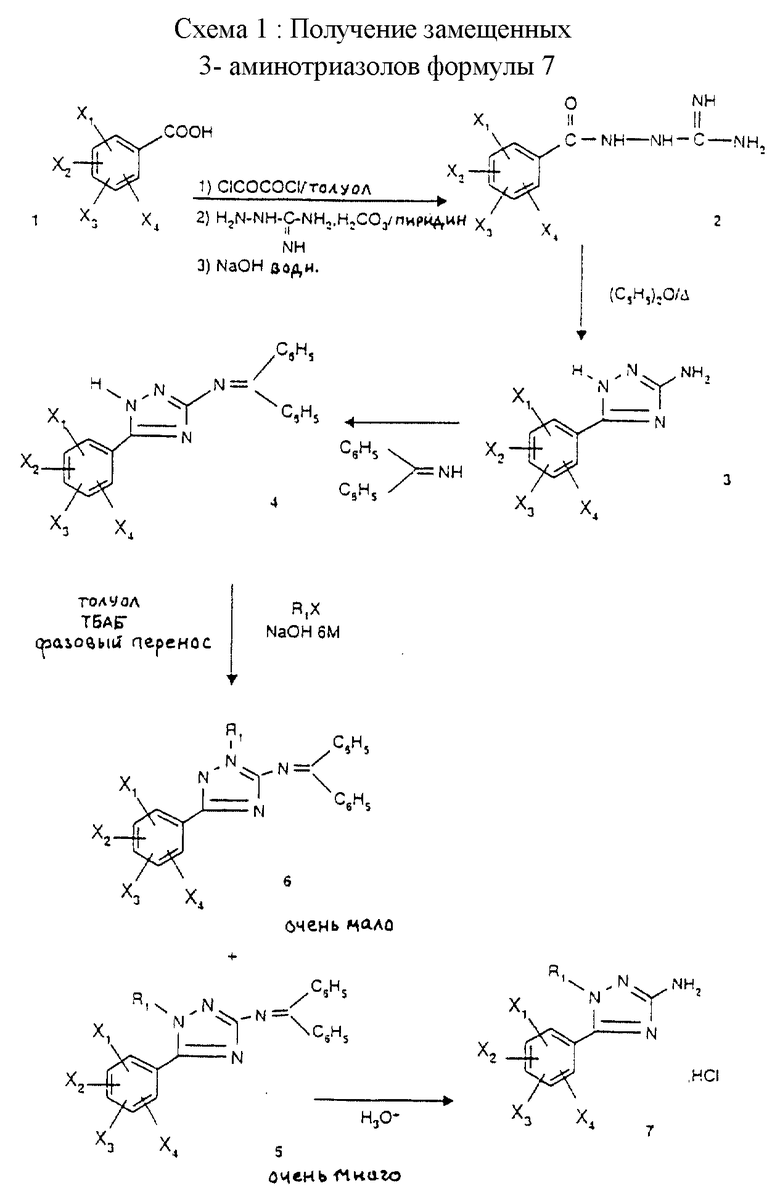
Схема 1 иллюстрирует путь синтеза соединений формулы 7, (схемы 1-12 приведены в конце описания).
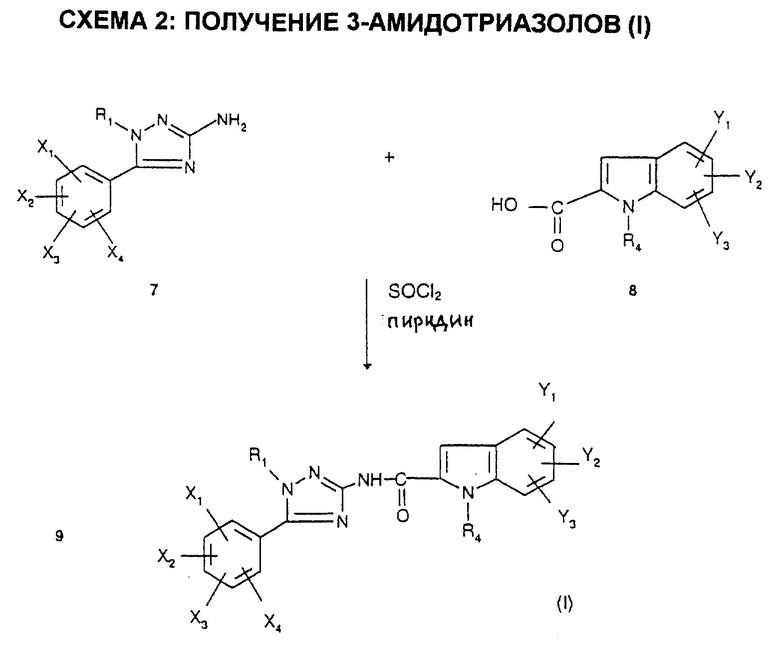
Схема 2 иллюстрирует получение соединений формулы (I) из аминотриазолов формулы 7.
Когда R4 = -(CH2)nCOOH, соединения (I) получают из соответствующих эфиров, которые в свою очередь получают по схеме 2.
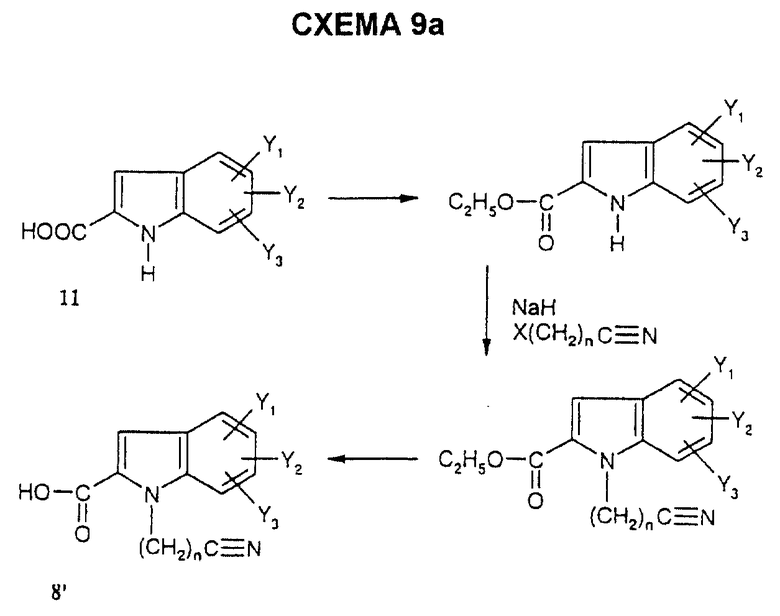
Когда R4 = -(CH2)n-тетразолил, соединения (I) получают из соответствующих нитрилов формулы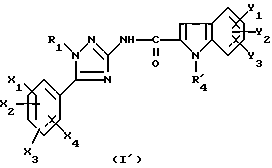
в которой R'4 = -(CH2)n-C≡N,
путем взаимодействия азидотриметилсилана в присутствии дибутилоловооксида в соответствии со способом, описанным в J.Org. Chem. 1993, 58, 4139 - 4141.
Соединения формулы (I') получают по схеме 2 из соединений 7 и 8' формулы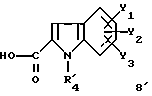
в которой R'4 = -(CH2)n-C≡N.
Замещенные бензойные кислоты имеются в продаже или их получают способами, описанными в литературе, например:
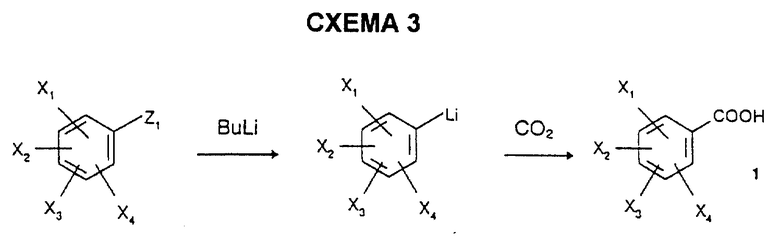
1) региоселективным литиированием замещенных бензолов с последующим карбоксилированием литиированного производного с использованием CO2 по схеме 3, где Z1 = Br или H в зависимости от природы и/или положения заместителей X1, X2, X3 и X4, в соответствии с N.S. Narasimhan et al., Indian J. Chem., 1973, 11, 1192; R.C. Cambie et al., Austr. J. Chem., 1991, 44, 1465; T. de Paulis et al., J. Med. Chem., 1986, 29, 61; или альтернативно
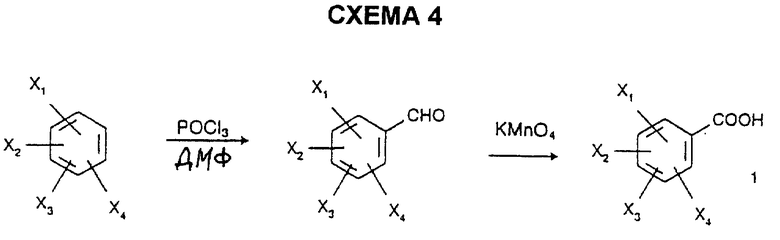
2) региоселективным формилированием замещенных бензолов с последующим окислением замещенного бензальдегида с использованием KMnO4 по схеме 4 согласно способу, описанному S.В. Matin et al. в J. Med. Chem., 1974, 17, 877; или альтернативно
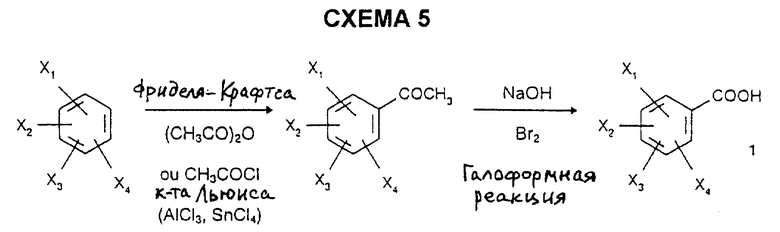
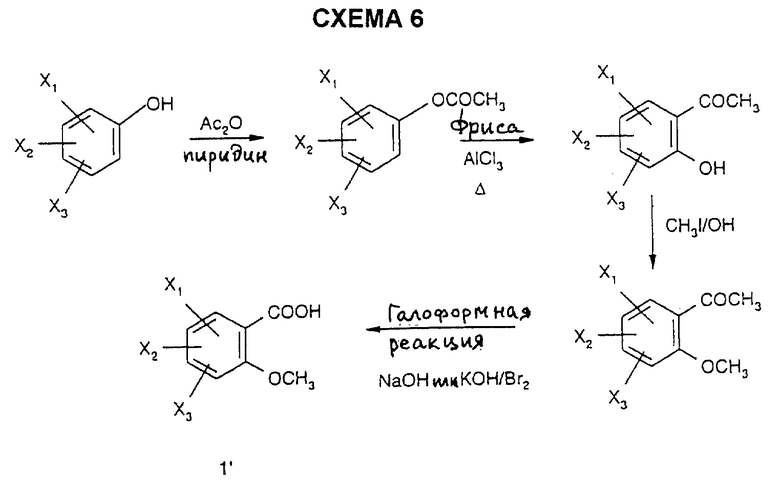
3) галоформным окислением согласно R. Levine et al., J. Am. Chem. Soc., 1959, 72, 1642, ароматических метилкетонов, полученных ацилированием Фриделя-Крафтса замещенных бензолов (C.A. Bartram et al., J. Chem. Soc., 1963, 4691) или перегруппировкой Фриса замещенных ацилоксибензолов согласно S.E. Cremer et al., J. Org. Chem., 1961, 26, 3653, согласно схемам 5 и 6.
Кислоты, замещенные метокси в положении 2, могут быть получены из замещенного фенольного производного путем взаимодействия уксусного ангидрида в пиридине с последующим проведением реакции Фриса в присутствии хлорида алюминия с получением гидроксиацетофенона, который после этого подвергают взаимодействию с метилйодидом в щелочной среде с получением, наконец, в результате галоформной реакции ожидаемой кислоты 1' в соответствии со схемой 6.
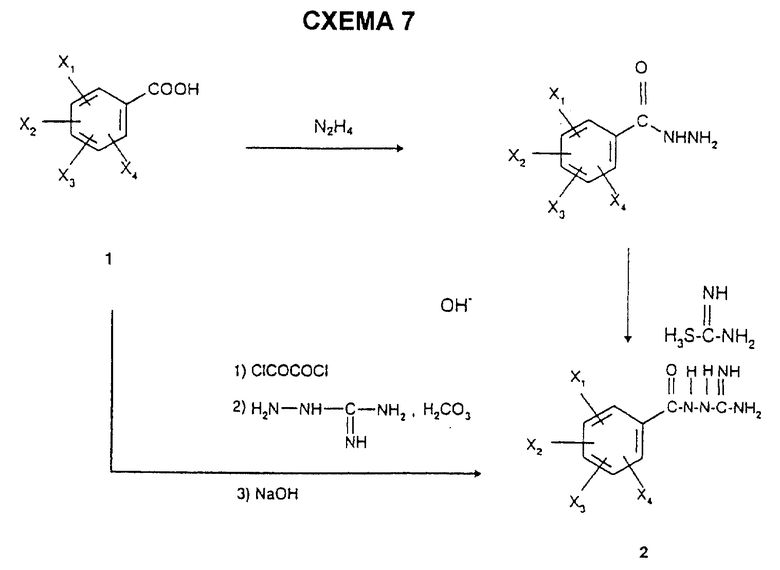
Бензамидогуанидин 2 получают ацилированием аминогуанидина гидрокарбоната с бензоилхлоридом, полученным из бензойной кислоты 1 стандартными способами (SOCl2, оксалилхлорид в инертном растворителе), с использованием известного (Hoggarth, J. Chem. Soc. , 1950, 612) способа. Его также можно получить альтернативным путем, описанным в этой же публикации, согласно схеме 7.
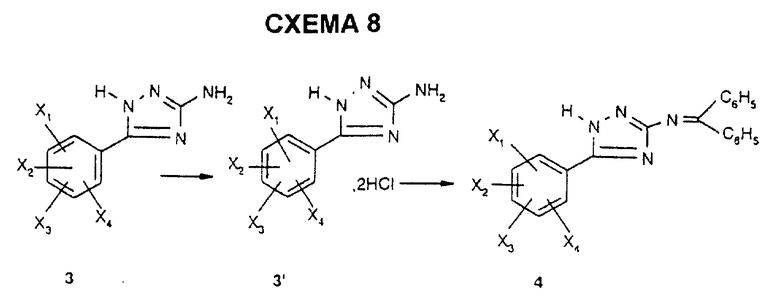
Термическая циклизация бензамидогуанидина 2 в растворителе с высокой температурой кипения, таком как дифениловый эфир, приводит к получению арил-5-амино-3-триазола 3 в соответствии со способом, описанным Е. Hoggarth в J. Chem. Soc., 1950, 612.
Защита функциональной первичной аминогруппы триазола 3 в форме дифенилимина приводит к получению N-защищенного триазола 4 в соответствии со способом, описанным M.J.O' Donnell et al. в J.Org. Chem., 1982, 47, 2663.
Соединение 4 может также быть получено альтернативным путем, который заключается в обработке триазола 3, который предварительно превращают в гидрохлорид 3', дифенилимином согласно схеме 8.
N-алкилирование дифенилиминотриазола 4 алкилгалогенидом R1X в условиях фазового переноса (сильное основание в концентрированном водном растворе в присутствии несмешивающегося органического сорастворителя и катализатора на основе четвертичного аммониевого соединения) приводит к получению преимущественно триазола 5 в сопровождении очень небольшого количества триазола 6. Используемыми сильными основаниями могут быть водные растворы NaOH или КОН в концентрациях от 6 М до 12 М. Сорастворителем может быть толуол или бензол, а четвертичное аммониевое соединение может быть выбрано из любой четвертичной аммониевой соли и, более конкретно, ТБАБ (тетрабутиламмония бромид).
а) М-Алкилирование дифенилиминотриазола 4 можно проводить в неводной среде (например диметилформамид или тетрагидрофуран) в присутствии сильного основания, такого как K2CO3 или NaH.
б) Можно выбрать альтернативный путь, такой как описан E. Akerblom в Acta Chem. Scand., 1965, 19, 1142, где используют алкилирующий агент в спирте, таком как этанол, в присутствии твердого сильного основания, такого как КОН или NaOH.
Триазол 5 очень легко отделяется от его изомера 6 хроматографией на колонке с диоксидом кремния или флэш-хроматографией в зависимости от природы группы R1. Расщепление продукта 5, полученного после отделения от меньшего изомера, проводят в водной кислотной среде, такой как 1 н. HCl, согласно способу, описанному J. Yaozhong et al. в Tetrahedron, 1988, 44, 5343, или M. J. O' Donnell et al. в J. Org. Chem., 1982, 47, 2663. Это позволяет получать амино-3- триазолы формулы 7, N-алкилированные в положении 1.
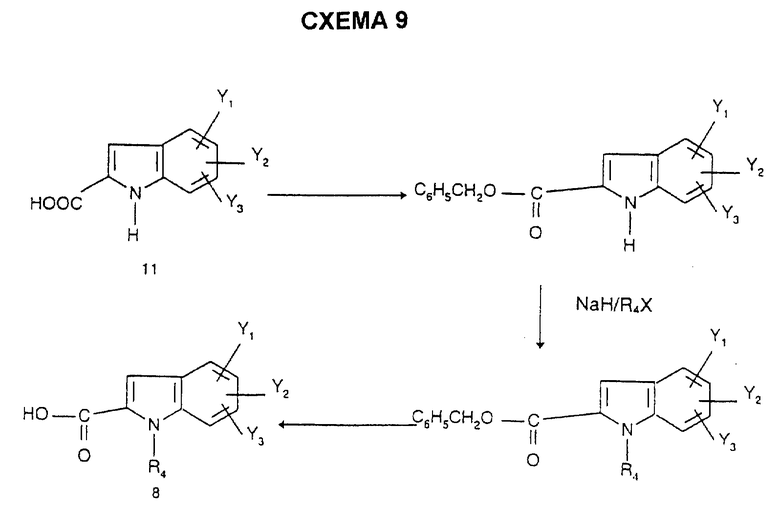
Индолкарбоновые соединения формулы 8 получают согласно способам, описанным в патенте EP 611766, по схеме 9.
Карбоксилсодержащие индолы 8', в которых R'4 = -(CH2)n-C≡N, получают аналогичными способами, представленными на схеме 9а.
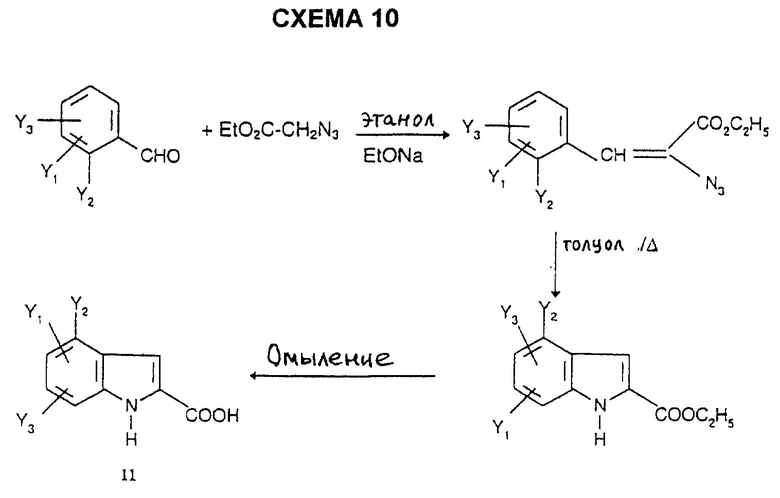
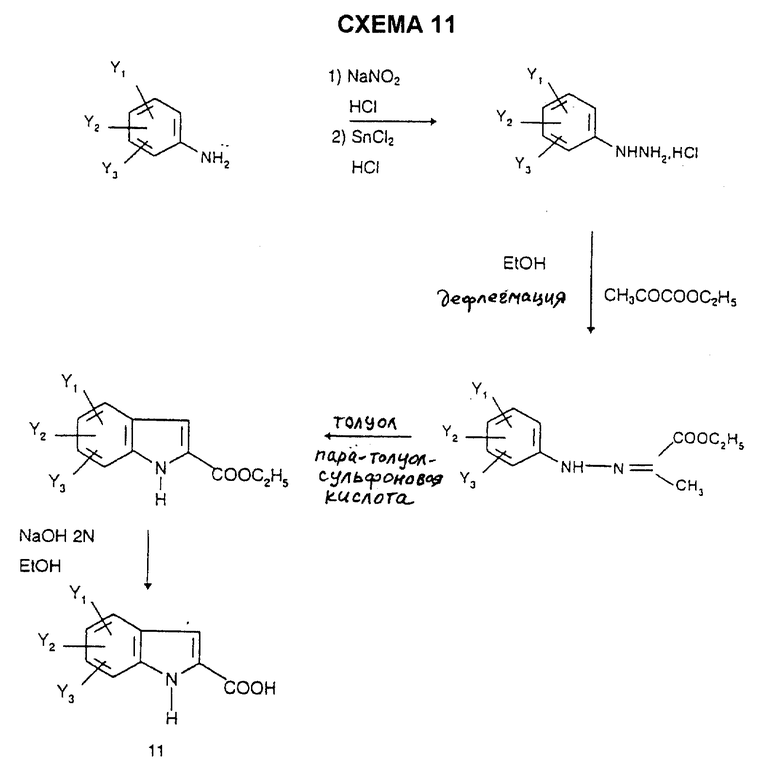
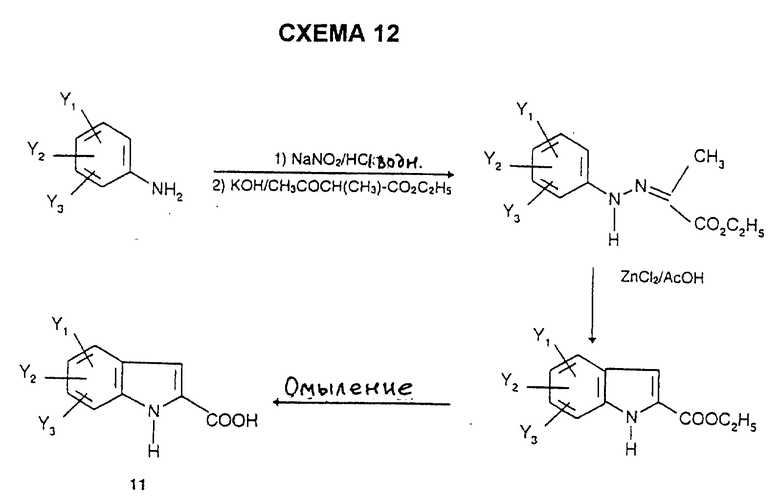
Индолы 11 имеются в продаже или их получают способами, описанными в литературе, например согласно L. Henn et al., J. Chem. Soc. Perkin Trans., 1, 1984, 2189, по схеме 10, или альтернативно, например, согласно синтезу Фишера (V. Prolog et al. Helv. Chim. Acta., 1948, 31, 1178) по схеме 11, или согласно синтезу Джеппа-Клингемана (H. Ishii et al., J. Chem. Soc. Perkin. Trans., 1, 1989, 2407) по схеме 12.
Приведенные выше соединения формулы (I) также включают в себя соединения, в которых один или более чем один водород, углерод или галоген, в частности атомы хлора или фтора, заменены их радиоактивными изотопами, например тритием или углеродом-14. Такие меченые соединения пригодны в исследовательских целях, в исследовании метаболизма или фармакокинетики, в биохимических тестах в качестве лигандов рецепторов.
Соединения формулы (I) были подвергнуты исследованиям in vitro связывания с ХЦК-А- и ХЦК-В-рецепторами с использованием метода, описанного в Europ. J. Pharmacol., 1993, 232, 13-19.
Активность агониста соединений по отношению к ХЦК-А-рецепторам оценивали in vitro на клетках ЗТЗ, экспрессирующих ХЦК-А-рецептор человека, путем измерения мобилизации внутриклеточного кальция ([Ca++]) в соответствии с методикой, производной от методики Lignon MF et al., Eur. J. Pharmacol., 1003, 245, 241-245. Концентрацию кальция [Ca++]i оценивали с использованием Fura-2 по методу длины волны двойного возбуждения. Соотношение флюоресценции, излучаемой на двух длинах волн, дает после калибровки концентрацию [Ca++]i (Grynkiewiez G. et al., J. Biol. Chem., 1985, 260, 3440-3450).
Соединения по изобретению стимулируют [Ca++]i частично или полностью, так же как ХЦК, и поэтому ведут себя как агонисты ХЦК-A-рецептора.
Исследование агонистического влияния соединений на опорожнение желудка проводили следующим образом. Самок мышей линии Swiss albino CD1 (20-25 г) ставили на голодную диету в течение 18 часов. В день эксперимента продукты (в виде суспензии в 1% растворе карбоксиметилцеллюлозы или в 0,6% растворе метилцеллюлозы) или соответствующий носитель вводили внутрибрюшинно за 30 минут до введения пищи, содержащей древесный уголь (0,3 мл суспензии 10% угольного порошка в воде на мышь, 5% гуммиарабика и 1% карбоксиметилцеллюлозы) или перорально на 1 час раньше. Мышей умерщвляли 5 минут спустя смещением шейных позвонков, и опорожнение желудка определяли по присутствию угля в кишечнике перед сфинктером привратника (Europ. J. Pharmacol., 1993, 232, 13-19). Соединения формулы (I) частично или полностью блокируют опорожнение желудка, так же как сам ХЦК, и, таким образом, ведут себя как агонисты ХЦК-рецептора. Некоторые из них имеют значения ЭД50 (эффективная доза, которая индуцирует 50% от эффекта ХЦК) менее 0,1 мг/кг внутрибрюшинно.
Исследование агонистического влияния соединений на сокращение желчного пузыря проводили следующим образом. Самок мышей линии Swiss albino CD1 (20-25 г) ставили на голодную диету в течение 24 часов. В день эксперимента продукты (в виде суспензии в 1% растворе карбоксиметилцеллюлозы или в 0,6% растворе метилцеллюлозы) или соответствующий носитель вводили перорально. Мышей умерщвляли смещением шейных позвонков через час после введения продуктов, и желчные пузыри удаляли и взвешивали. Результаты выражали в мг/кг веса тела (Europ. J. Pharmacol., 1993, 232, 13-19).
Соединения формулы (I) частично или полностью сокращают желчный пузырь, так же как сам ХЦК, и, таким образом, ведут себя как агонисты ХЦК. Некоторые из них имеют ЭД50 (эффективная доза, которая индуцирует 50% от снижения массы везикул, наблюдаемого с ХЦК) менее чем 0,1 мг/кг перорально.
Следовательно, соединения формулы (I) пригодны в качестве агонистов ХЦК-рецептора типа A для приготовления лекарств, предназначенных для борьбы с заболеваниями, лечение которых требует стимуляции полного или частичного агонизма ХЦК-А- рецепторов холецистокинина. Более конкретно, соединения формулы (I) пригодны для получения лекарств, предназначенных для лечения некоторых расстройств желудочно-кишечной сферы (предупреждение образования конкрементов в желчном пузыре, синдром раздраженного кишечника), расстройств приема пищи и ожирения и ассоциированных патологий, таких как диабет и гипертензия. Соединения (I) индуцируют состояние насыщения и, таким образом, они пригодны для лечения расстройств поведения при приеме пищи, для регулирования аппетита и для снижения потребления пищи, для лечения булимии и ожирения и для похудания. Соединения (I) также полезны при расстройствах эмоционального и сексуального поведения и расстройствах памяти, при психозе и особенно при шизофрении, болезни Паркинсона и поздней дискинезии. Их также можно применять при лечении расстройств аппетита, то есть для регулирования желания есть, в частности регулирования потребления сахара, углеводов, алкоголя или лекарственных средств и вообще возбуждающих аппетит ингредиентов.
Соединения формулы (I) имеют слабую токсичность; их токсичность совместима с их применением в качестве лекарств для лечения вышеуказанных заболеваний и расстройств.
Никаких признаков токсичности не наблюдается при использовании этих соединений в фармакологически активных дозах, и их токсичность, следовательно, совместима с их медицинским применением в качестве лекарств.
Фармацевтические композиции по данному изобретению могут содержать подходящие эксципиенты. Указанные эксципиенты выбирают в соответствии с фармацевтической композицией и желаемым способом введения.
В фармацевтических композициях по настоящему изобретению, предназначенных для перорального, подъязычного, подкожного, внутримышечного, внутривенного, наружного, внутритрахеального, интраназального, трансдермального, ректального или внутриглазного введения, активные начала формулы (I), указанные выше, или их возможные соли, могут быть введены в стандартных формах для введения, смешанные со стандартными фармацевтическими носителями, животным и людям для профилактики или лечения вышеуказанных заболеваний или расстройств. Соответствующие стандартные формы для введения включают в себя пероральные формы, такие как таблетки, желатиновые капсулы, порошки, гранулы и пероральные суспензии и растворы, подъязычные, буккальные, внутритрахеальные, интраназальные, подкожные, внутримышечные или внутривенные формы для введения и ректальные формы для введения. Соединения по изобретению могут быть использованы в кремах, мазях, лосьонах или глазных каплях для наружного введения.
Для получения требуемого профилактического или терапевтического эффекта доза активного начала может находиться в пределах между 0,01 и 50 мг на 1 кг веса тела в день. Каждая стандартная доза может содержать от 0,5 до 1000 мг, предпочтительно от 1 до 500 мг, активных ингредиентов в комбинации с фармацевтическим носителем. Эту стандартную дозу можно вводить от 1 до 5 раз в день так, чтобы ввести суточную дозу от 0,5 до 5000 мг, предпочтительно от 1 до 2500 мг.
При приготовлении твердой композиции в форме таблетки основной активный ингредиент смешивают с фармацевтическим носителем, таким как желатин, крахмал, лактоза, стеарат магния, тальк, гуммиарабик или тому подобное. Таблетки могут быть покрыты сахарозой, производным целлюлозы или другими подходящими веществами, или альтернативно они могут быть обработаны таким образом, чтобы они имели пролонгированное или замедленное действие, и таким образом, чтобы они высвобождали предопределенное количество активного начала непрерывно.
Препарат в форме желатиновых капсул получают смешиванием активного ингредиента с разбавителем и вливания полученной смеси в мягкие или твердые желатиновые капсулы.
Препарат в форме сиропа или эликсира или для введения в форме капель может содержать активный ингредиент вместе с подсластителем, предпочтительно бескалорийным подсластителем, метилпарабеном и пропилпарабеном в качестве антисептиков, а также корригентом и подходящим красителем. Диспергируемые в воде порошки или гранулы могут содержать активный ингредиент, смешанный с диспергирующими агентами или смачивающими агентами, или суспендирующими агентами, такими как поливинилпирролидон, а также с подсластителями или усилителями вкуса и аромата.
Для ректального введения используют суппозитории, которые готовят с использованием связующих, которые плавятся при ректальной температуре, например, масла какао или полиэтиленгликолей. Водные суспензии, изотонические солевые растворы или стерильные инъецируемые растворы, которые содержат фармакологически совместимые диспергирующие агенты и/или смачивающие агенты, например пропиленгликоль или бутиленгликоль, используют для парентерального введения.
Активное начало может быть также приготовлено в форме микрокапсул возможно в составе с одним или более чем одним носителем или добавкой, или альтернативно с матрицами, такими как полимер или циклодекстрин (пластырь, формы пролонгированного высвобождения).
Композиции по настоящему изобретению можно применять при лечении или предупреждении различных болезней, при которых ХЦК имеет терапевтическое значение.
Композиции по настоящему изобретению могут содержать, наряду с указанными выше продуктами формулы (I) или их фармацевтически приемлемыми солями, другие активные начала, которые могут быть использованы при лечении заболеваний или расстройств, указанных выше.
Преимущественно композиции по настоящему изобретению содержат продукт формулы (I. 1), (I. 2) или (I.3), указанной выше, или его фармацевтически приемлемую соль, сольват или гидрат.
ПОЛУЧЕНИЕ СИНТЕТИЧЕСКИХ ПРОМЕЖУТОЧНЫХ СОЕДИНЕНИЙ
A. Получение кислот 1 (варианты)
2,5-Диметокси-4-метилбензойная кислота (Соединение A1)
а) 2,5-Диметокси-4-бензальдегид
После перемешивания смеси 8,5 мл N-метилформанилида (0,068 моль) и 6,3 мл окситрихлорида фосфора (0,068 моль) при комнатной температуре в течение 40 минут вводят 17,8 г 2,5-диметокситолуола (0,117 моль). Реакционную смесь нагревают в течение 6 часов при 50oC, а затем, после возвращения к температуре 20oC, ее гидролизуют 100 мл водного 10% раствора ацетата натрия, экстрагируют дважды диэтиловым эфиром и концентрируют. Остаток переносят в водный раствор гидросульфита натрия и дважды экстрагируют диэтиловым эфиром. Водную фазу подщелачивают (pH = 12) с получением белых кристаллов; т.пл. = 83oC; выход = 67%.
б) 2,5-Диметокси-4-метилбензойная кислота
23,86 г (0,132 моль) 2,5-диметокси-4-метилбензальдегида, растворенного в 500 мл воды, нагревают до 75oC и вводят 29,3 г (0,185 моль) перманганата калия, растворенного в 500 мл воды. Реакционную смесь оставляют на 2 часа при 75oC, pH доводят до 10 с использованием 10% раствора гидроксида натрия, и нерастворимое вещество отфильтровывают, пока оно горячее, и промывают трижды 80 мл горячей воды. Фильтрат охлаждают, и образовавшийся осадок отфильтровывают и сушат под вакуумом при 40oC с получением белых кристаллов; т.пл.=120oC; выход=71%.
2,5-Диметокси-4-хлорбензойная кислота (Соединение A2)
а) 2,5-Диметокси-4-хлорфенилметилкетон
162,5 г трихлорида алюминия (1,2 моль) добавляют при комнатной температуре к 2 литрам четыреххлористого углерода с последующим, при 0oC, добавлением по каплям 82 мл ацетилхлорида (1,2 моль), а затем 200 г 1,4-диметокси-2-хлорбензола (1,2 моль). Реакционную смесь оставляют на 3,5 часа при 0oC, а затем гидролизуют с использованием 700 мл воды. Органическую фазу промывают 2 М раствором гидроксида натрия, сушат над безводным сульфатом натрия и концентрируют. Полукристаллический остаток переносят в петролейный эфир, фильтруют и сушат с получением белых кристаллов; т.пл. = 96oC; выход = 70%.
б) 2,5-диметокси-4-хлорбензойная кислота
278 г гидроксида натрия (4,96 моль) добавляют к 800 мл воды с последующим добавлением по каплям, при 5oC, 84 мл брома (1,6 моль). Реакционную смесь оставляют на 1 час при комнатной температуре. Полученный водный раствор гипобромита натрия добавляют к 107 г 2,5-диметокси-4-хлорфенилметилкетона (0,494 моль), растворенного в 1,5 литрах 1,4-диоксана. Через 1 час при 20oC реакционную смесь нагревают в течение 1 часа при температуре дефлегмации. После окончания реакции вводят 100 мл водного раствора гидросульфита натрия, и растворитель затем выпаривают. Остаток подкисляют 6 н. соляной кислотой, а затем дважды экстрагируют этилацетатом. Органическую фазу сушат над безводным сульфатом натрия и концентрируют. Остаток кристаллизуют в диизопропиловом эфире с получением белых кристаллов; т.пл.=160oC; выход = 91%.
2,6-Диметокси-4-метилфенилбензойная кислота (Соединение А3)
231,6 г (1,5 моль) 3,5-диметокситолуола растворяют в 1 литре диэтилового эфира с последующим добавлением по каплям в токе азота и при комнатной температуре 1 литра 1,6 н. раствора бутиллития (1,6 моль) в гексане. Реакционную смесь оставляют на 18 часов при комнатной температуре, а затем, после охлаждения до -30oC, добавляют 1 литр диэтилового эфира и через эту смесь в течение 1 часа барботируют диоксид углерода, поддерживая температуру -30oC. Реакционную смесь переносят в 6 литров 2 М раствора гидроксида натрия, водную фазу отделяют после отстаивания и подкисляют 6 н. раствором соляной кислоты. Осадок отфильтровывают, промывают водой и сушат под вакуумом при 40oC с получением белых кристаллов; т.пл.=187oC; выход=88%.
Б. Получение замещенных индолов и их вариантов
Получение этилового эфира 5-метил-1Н-2-индолкарбоновой кислоты (Соединение В1)
1-й способ: (Способ Джеппа-Клингемана)
7,2 г (0,104 моль) нитрита натрия, растворенного в 40 мл воды, добавляют при -5oC к смеси 10,7 г (0,1 моль) 4-метиланилина, 74 мл 12 н. соляной кислоты и 140 мл воды. Реакционную смесь перемешивают в течение 15 минут при -5oC и нейтрализуют добавлением 8,1 г ацетата натрия. 12,33 г (0,085 моль) этилового эфира α-метилацетоуксусной кислоты и 80 мл этанола вводят в трехгорлую колбу с последующим добавлением, при 0oC, 4,8 г (0,085 моль) гидроксида калия, растворенного в 20 мл воды и 100 г льда. Раствор диазония, полученный выше, добавляют по каплям при 0oC к этой реакционной смеси, и полученную смесь оставляют на 18 часов при 0oC. Водную фазу экстрагируют 4 раза 50 мл этилацетата, и органические фазы объединяют и сушат над безводным сульфатом натрия. Остаток переносят в 100 мл толуола и 16,3 г (0,085 моль) моногидрата пара-толуолсульфоновой кислоты. Смесь затем медленно нагревают до 110oC и выдерживают при этой температуре в течение 5 часов. После охлаждения и последующего добавления насыщенного раствора карбоната натрия нерастворимое вещество выделяют фильтрованием, а органическую фазу отделяют после отстаивания, сушат над безводным сульфатом натрия и концентрируют. Остаток хроматографируют на колонке с силикагелем, элюент: смесь 30/70 (об./об.) дихлорметан/циклогексан, с получением кристаллов бежевого цвета; т.пл.=94oC; выход = 25%.
Получение этилового эфира 4-метил-1Н-2-индолкарбоновой кислоты (Соединение В2)
2-й способ:
Стадия 1: Получение азида
9,3 г (0,405 моль) натрия добавляют порциями к 200 мл этанола. 16,2 г (0,135 моль) орто-толуальдегида, растворенного в 52,2 г (0,405 моль) этилазидоацетата, вводят по каплям при -20oC в этот раствор этилата в этаноле. После 2 часов при -10oC реакционную смесь заливают 400 мл воды, и образовавшийся осадок отфильтровывают. Его сушат в течение 18 часов при 40oC под вакуумом с получением белых кристаллов; т.пл. = 55oC; выход = 78%.
Стадия 2: Циклизация азида
19,5 г (0,0844 моль) азида, полученного на стадии 1, добавляют порциями к 100 мл ксилола, нагретого до 140oC. Сразу после окончания добавления реакционную смесь оставляют на 1 час при 140oC. Ксилол концентрируют, и остаток переносят в изопропиловый эфир, фильтруют и сушат в течение 18 часов под вакуумом при 40oC с получением белых кристаллов; т.пл.=141oC; выход = 62%.
Получение 5-этил-1Н-2-индолкарбоновой кислоты (по методу Фишера) - (Соединение В3)
3-й способ:
Стадия 1: 4-Этилфенилгидразина гидрохлорид
150 мл воды и 160 мл 12 н. соляной кислоты добавляют к 24,2 г (0,2 моль) 4-этиланилина. Смесь охлаждают до 0oC и по каплям вводят 14 г (0,2 моль) нитрита натрия, растворенного в 140 мл воды. После 1 часа при 0oC в реакционную смесь добавляют 112 г (0,496 моль) дигидрата хлорида олова, растворенного в 90 мл 12 н. соляной кислоты, при -10oC. После 1 часа при -10oC реакционную смесь фильтруют с получением коричневого твердого вещества; т. пл.=198oC; выход=95%.
Стадия 2: Этиловый эфир 2-[2-(4-этилфенил)-гидразоно]пропионовой кислоты
23 мл (0,2 моль) этилпирувата добавляют к 34,5 г (0,2 моль) 4-этилфенилгидразина гидрохлорида, полученного выше, в виде суспензии в 500 мл этанола, и реакционную смесь нагревают в течение 3,5 часов при температуре дефлегмации. Смесь затем охлаждают до температуры 20oC, и этанол выпаривают. Твердый остаток промывают пентаном и сушат при 40oC под вакуумом с получением бесцветной жидкости; выход = 94%.
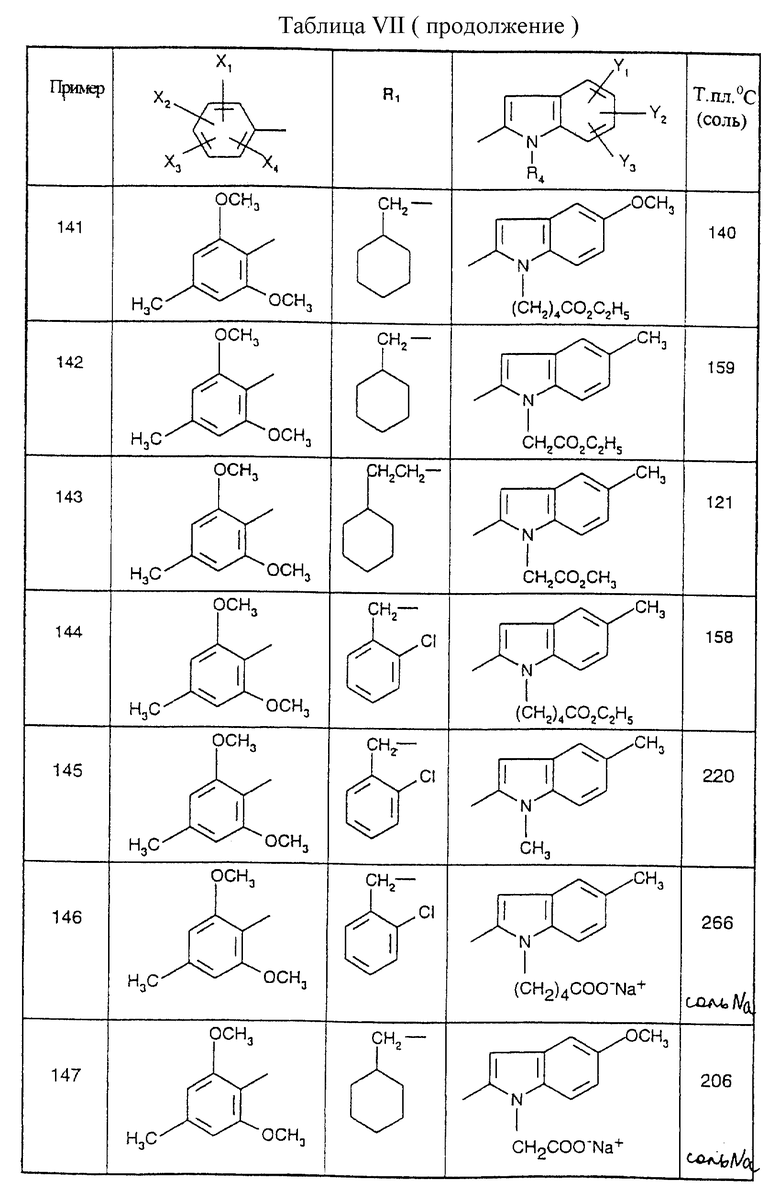
Стадия 3: Этиловый эфир 5-этил-1Н-2-индолкарбоновой кислоты
19 г (0,1 моль) моногидрата пара-толуолсульфоновой кислоты добавляют порциями в течение 7 часов при температуре дефлегмации к 44 г (0,188 моль) гидразона, полученного выше, суспендированного в 300 мл толуола. Смесь охлаждают до температуры 20oC, и нерастворимое вещество отделяют фильтрованием и промывают водой. Фильтрат промывают насыщенным водным раствором карбоната калия. Фазы разделяют после отстаивания, и органическую фазу сушат над безводным сульфатом натрия и концентрируют. Остаток очищают хроматографией на колонке с силикагелем с использованием элюента: смесь 5/5 (об./об.) дихлорметан/циклогексан, с получением бежевых кристаллов; т.пл.=94oC; выход= 51%.
Стадия 4: 5-Этил-1H-2-индолкарбоновая кислота
15,8 г (0,073 моль) этилового эфира 5-этил-2-индолкарбоновой кислоты, полученной на стадии 3, добавляют к 150 мл 1,4-диоксана с последующим добавлением 45 мл 2 М раствора гидроксида натрия (0,09 моль). Реакционную смесь оставляют на 48 часов при комнатной температуре. После выпаривания 1,4-диоксана остаток переносят в 6 н. раствор соляной кислоты, и образовавшийся осадок отфильтровывают и сушат под вакуумом при 60oC с получением 5-этил-1Н-2-индолкарбоновой кислоты в форме белых кристаллов; т.пл.=184oC; выход = 92%.
Получение N-алкил-1Н-2-индолкарбоновых кислот
5-Этил-1-(метоксикарбонилметил)-1H-2-индолкарбоновая кислота: (Соединение В4)
Стадия 1: Бензиловый эфир 5-этил-1Н-2-индолкарбоновой кислоты
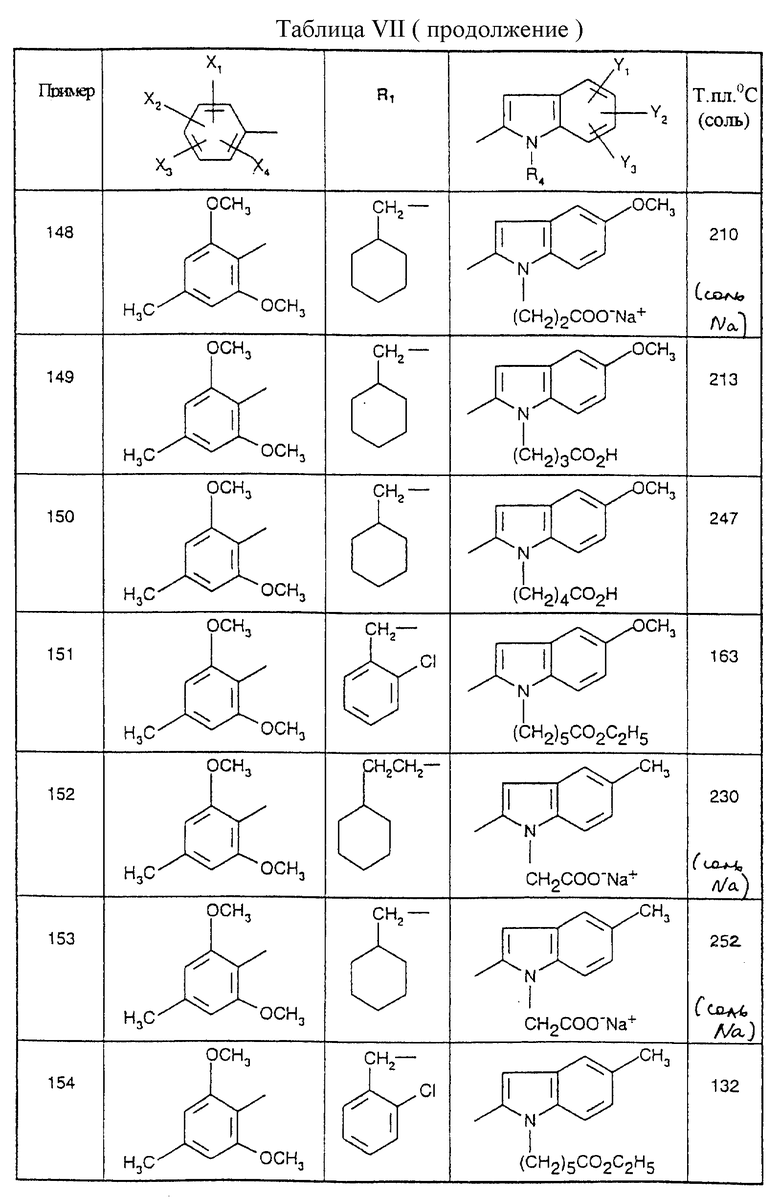
12,7 г (0,067 моль) 5-этил-1Н-2-индолкарбоновой кислоты и 10 мл 1,8- диазабицикло[5.4.0] ундец-7-ена (0,067 моль) последовательно добавляют к 70 мл диметилформамида. Реакционную смесь оставляют на 40 минут при 0oC, после чего по каплям вводят 10,6 мл бензилбромида (0,089 моль). После взаимодействия в течение 18 часов при комнатной температуре реакционную смесь заливают 300 мл воды, осадок отфильтровывают, промывают водой, а затем сушат в течение 18 часов при 50oC под вакуумом с получением желтых кристаллов; т.пл. =99oC; выход=90%.
Стадия 2: Бензиловый эфир 5-Этил-1-(метоксикарбонилметил)-1Н-2- индолкарбоновой кислоты
75 мл диметилформамида добавляют к 1,5 г (0,031 моль) гидрида натрия в виде 50% суспензии в масле с последующим добавлением порциями 7,9 г (0,0283 моль) бензилового эфира 5-этил-1Н-2-индолкарбоновой кислоты, полученного на стадии 1. После 40 минут при 0oC по каплям вводят 3,5 мл (0,0315 моль) метилбромацетата, и реакционную смесь оставляют на 2 часа при 20oC. Добавляют 300 мл этилацетата, смесь промывают 2 х 300 мл воды, фазы затем разделяют после отстаивания, и органическую фазу сушат над безводным сульфатом натрия и концентрируют, получая 9,5 г бесцветного масла; выход=95%.
Стадия 3: 5-Этил-1-(метоксикарбонилметил)-1Н-2-индолкарбоновая кислота
2,5 г 10% Pd/C добавляют к 9,5 г (0,0269 моль) бензилового эфира 5-этил-1-(метоксикарбонилметил)-1Н-2-индолкарбоновой кислоты, полученного на стадии 2, растворенного в 150 мл этанола, с последующим добавлением 40 мл циклогексена (0,395 моль). Реакционную смесь нагревают в течение 2 часов при 70oC, а затем охлаждают до 20oC. Реакционную смесь фильтруют через тальк, и фильтрат выпаривают досуха. Остаток сушат в течение 18 часов при 40oC под вакуумом с получением бежевых кристаллов; т.пл.=181oC; выход=90%.
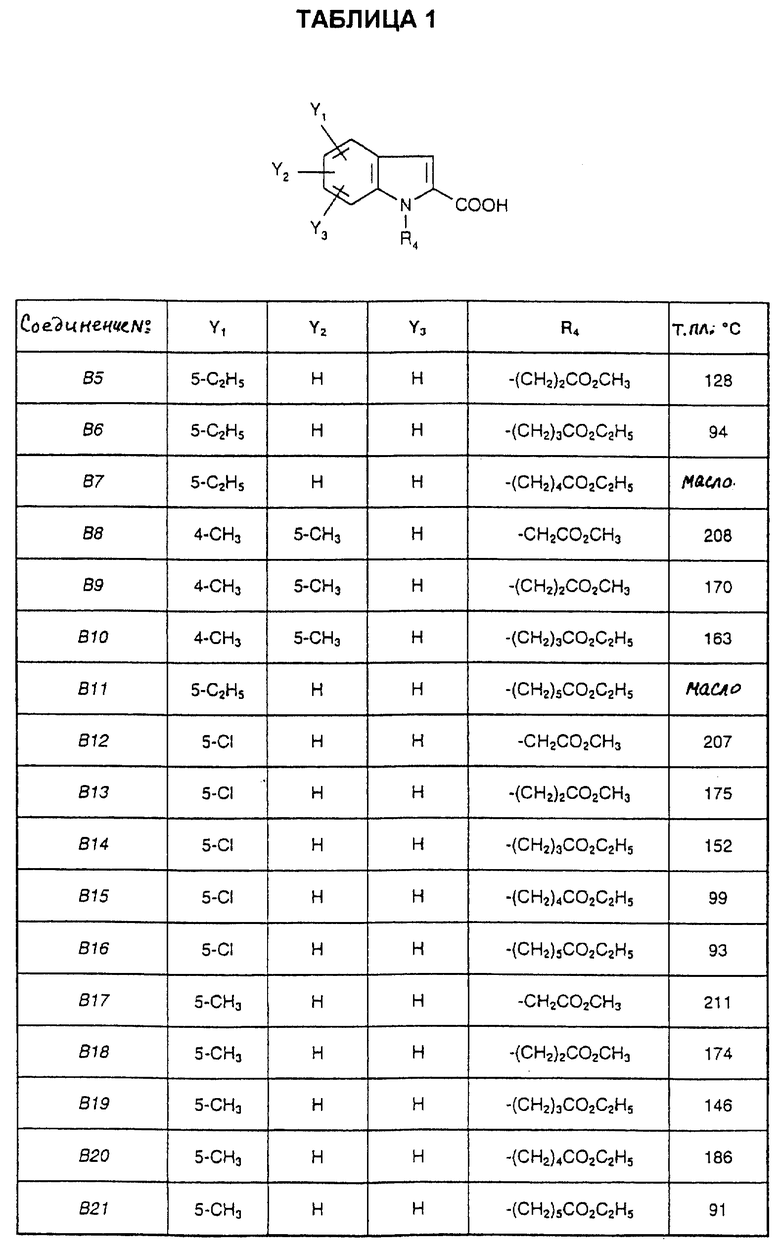
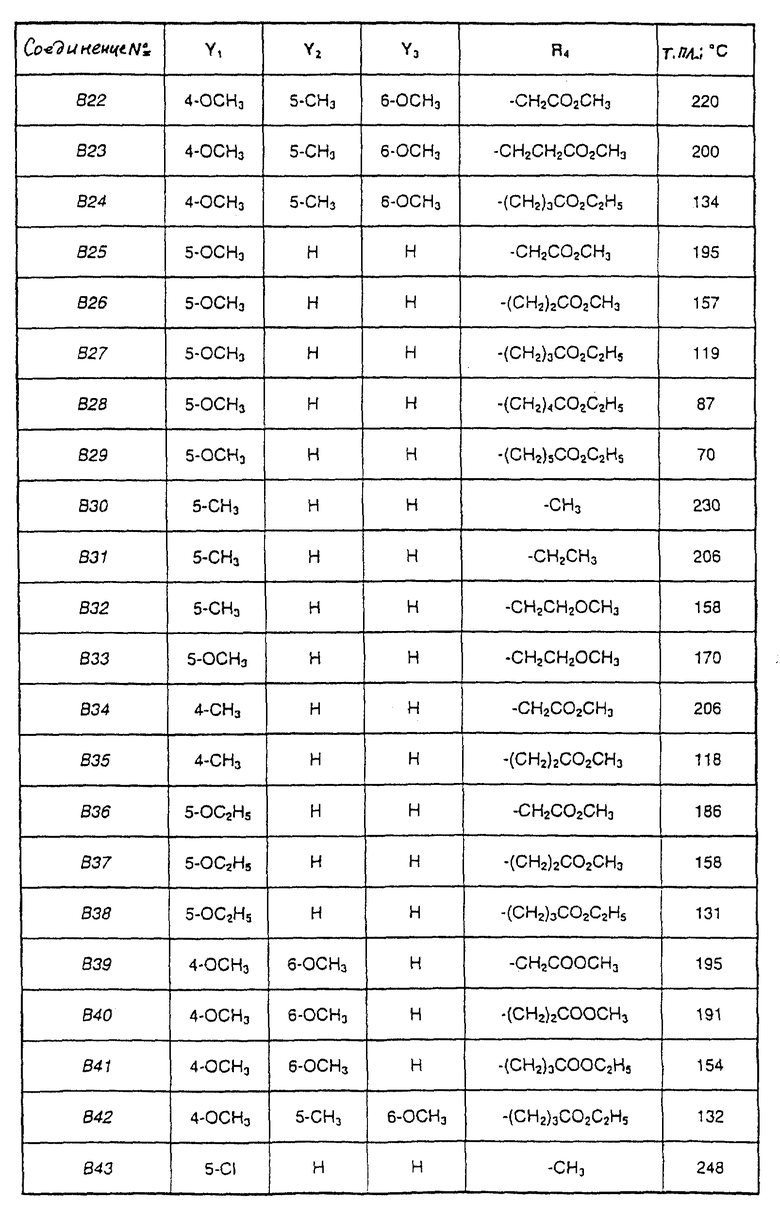
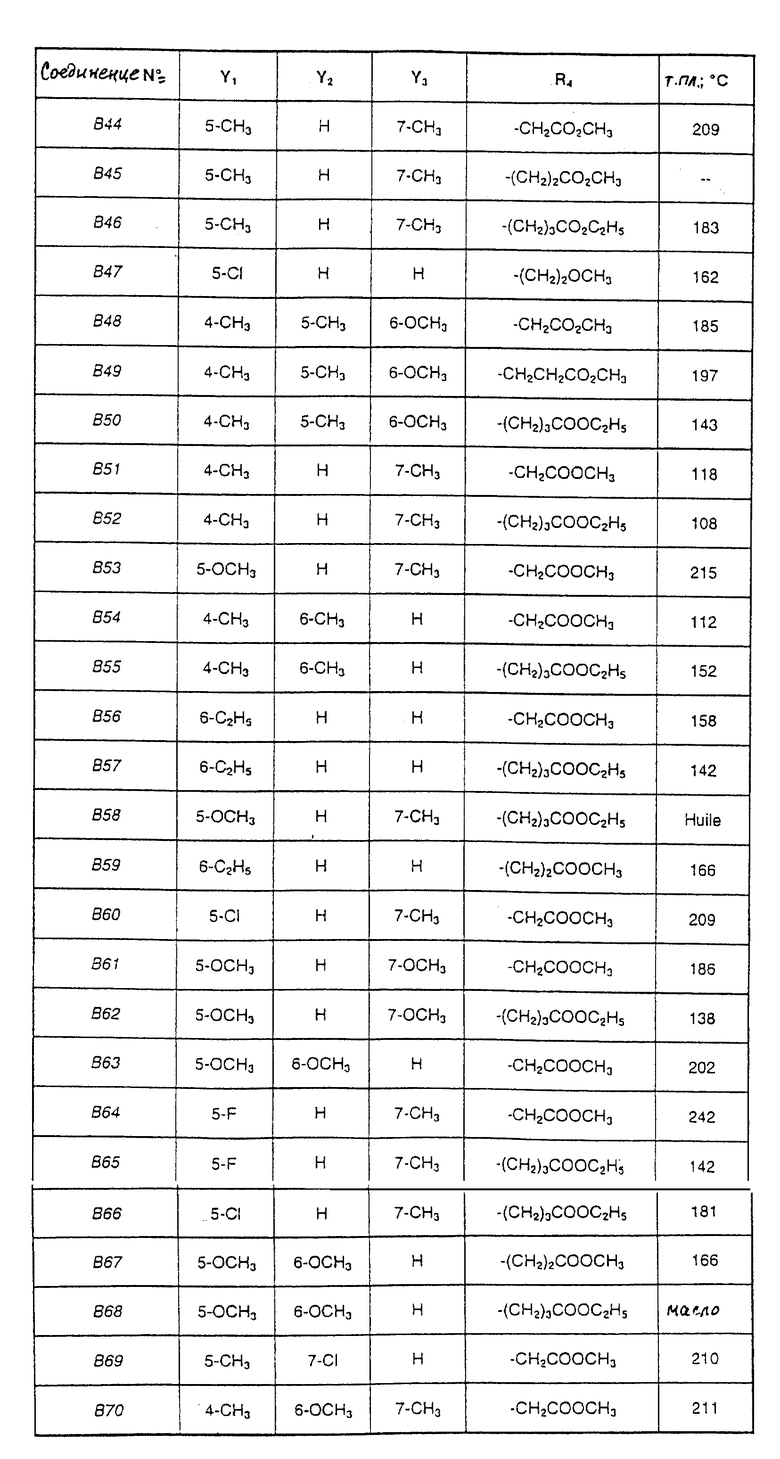
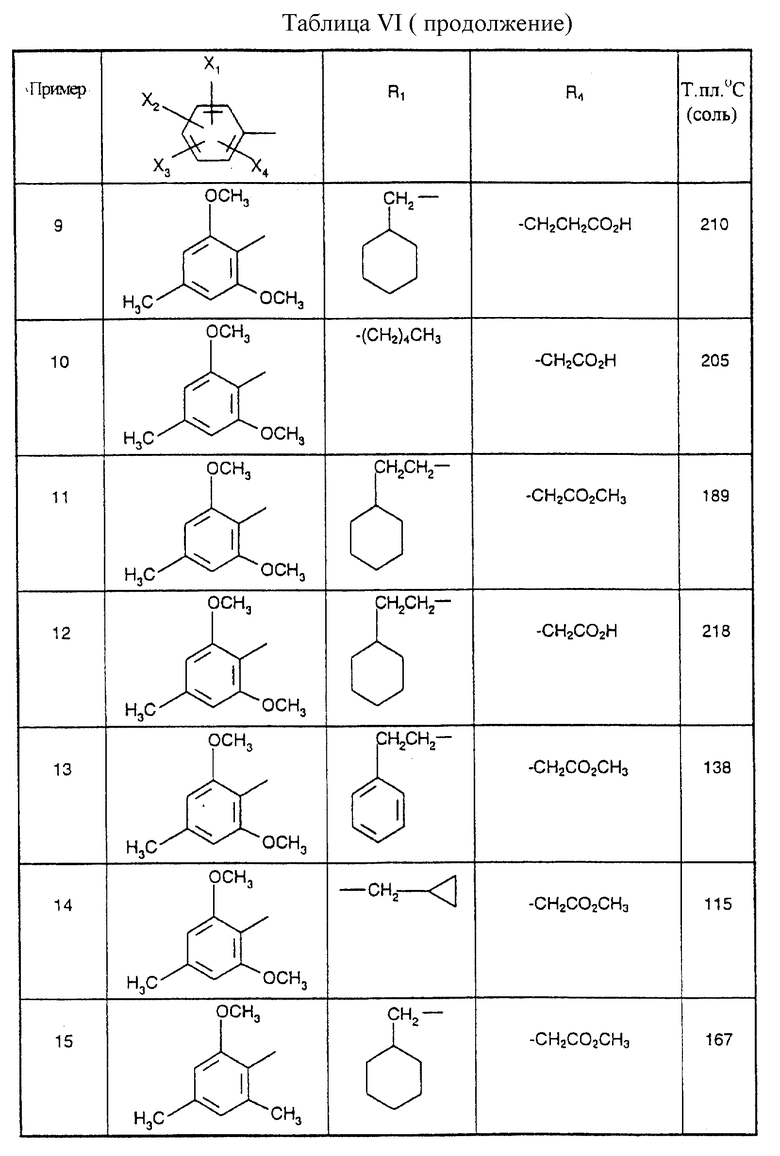
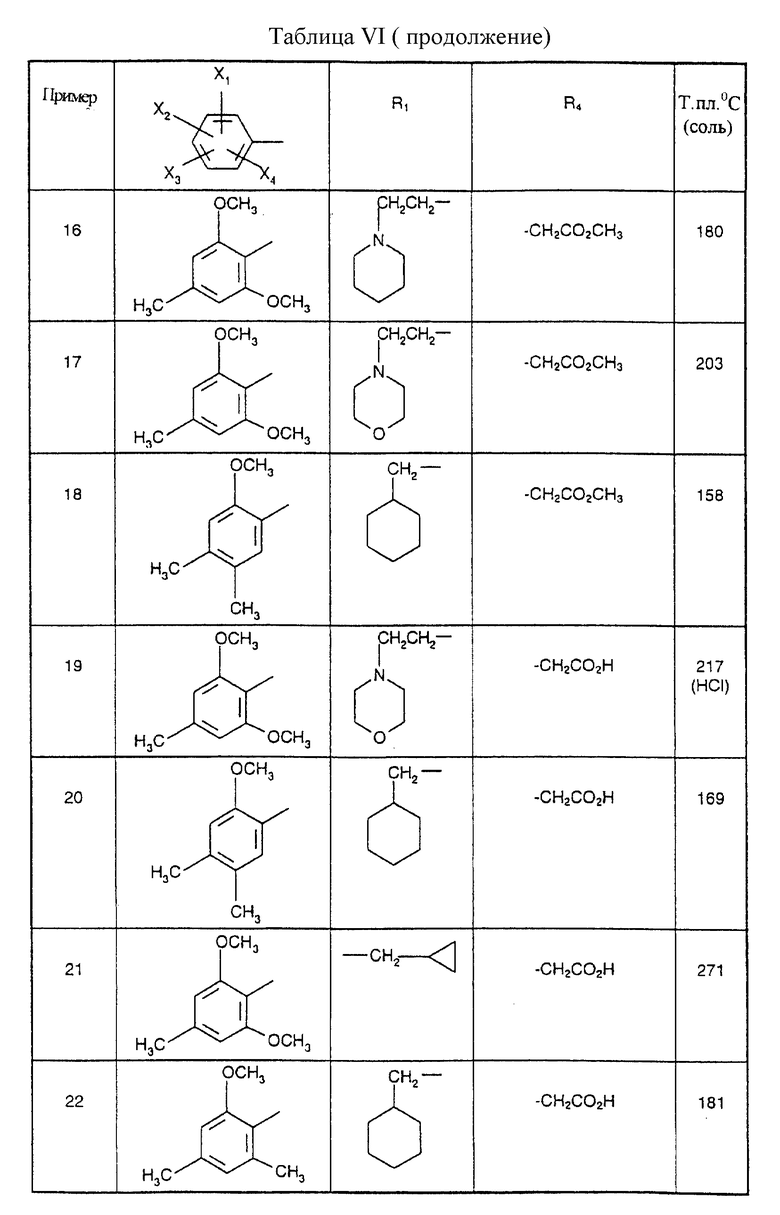
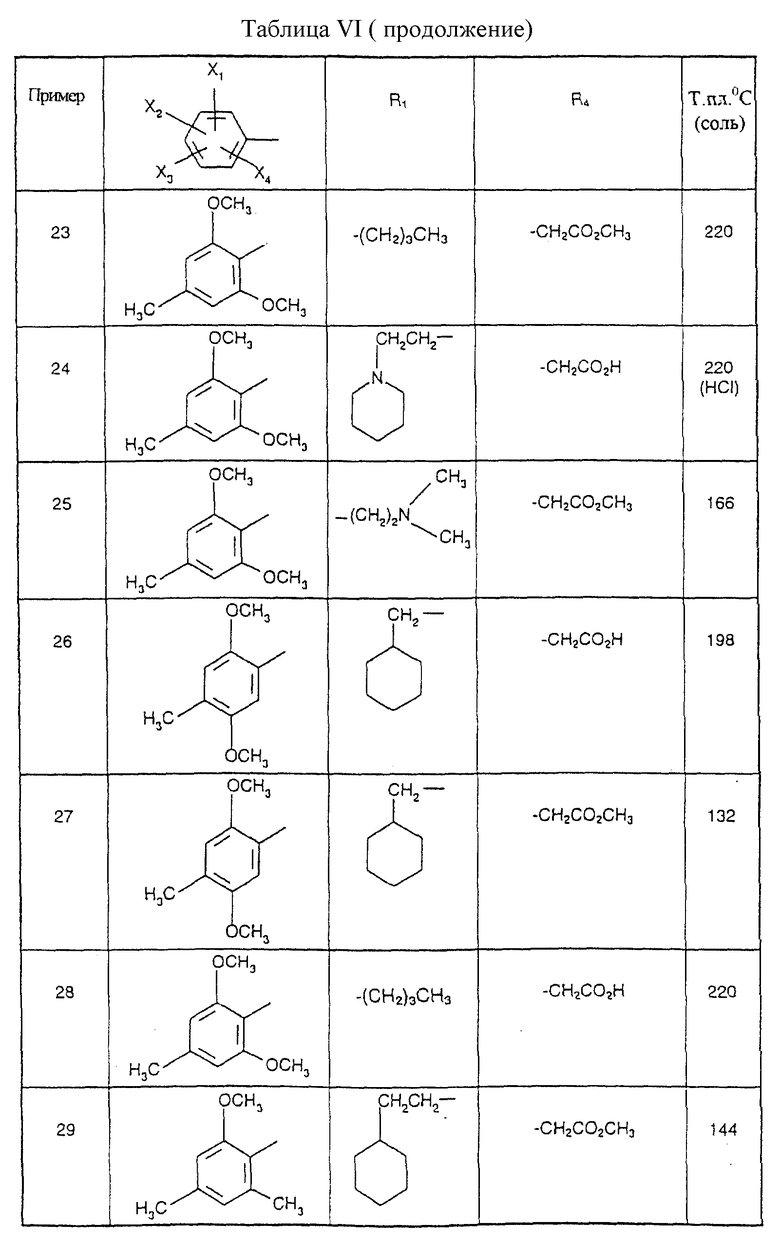
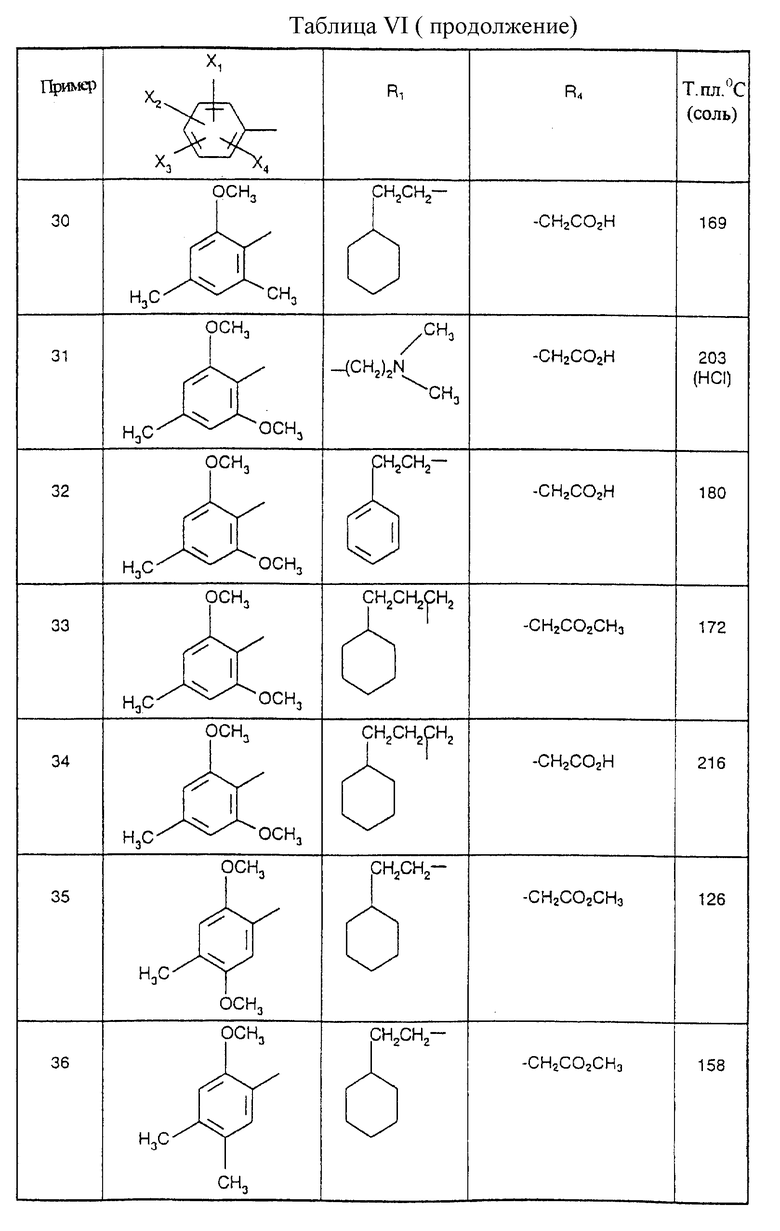
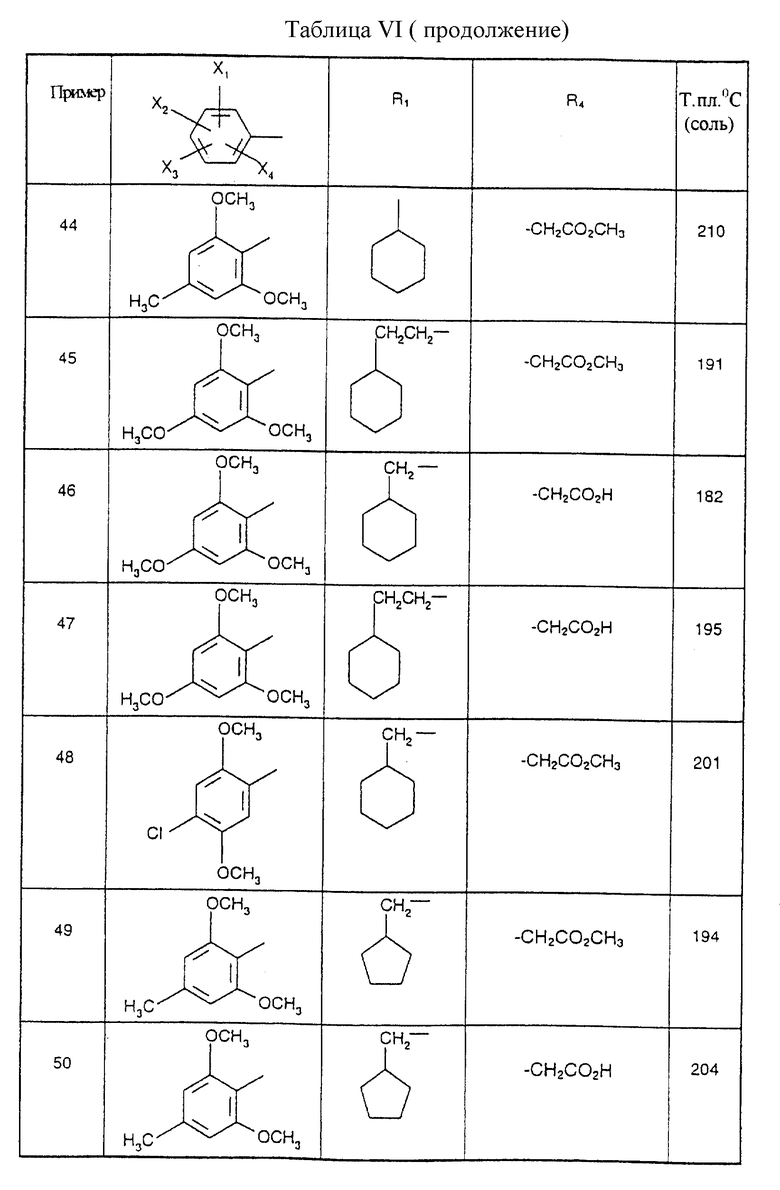
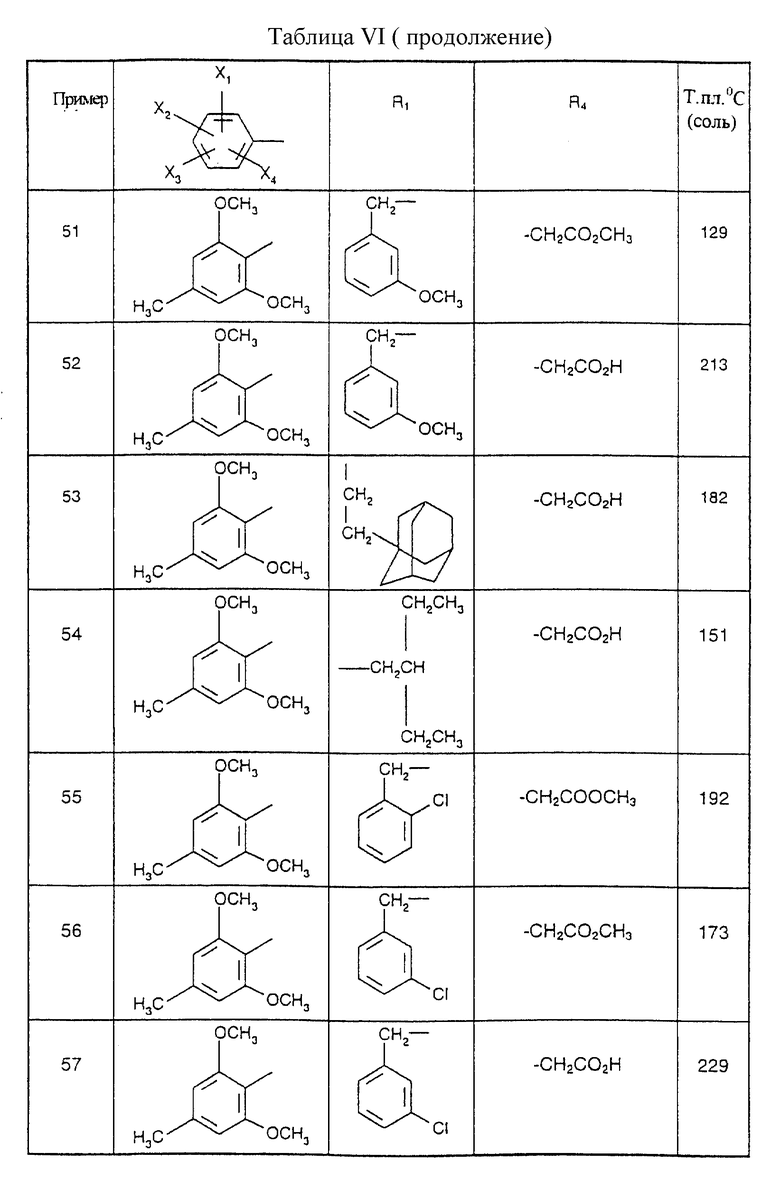
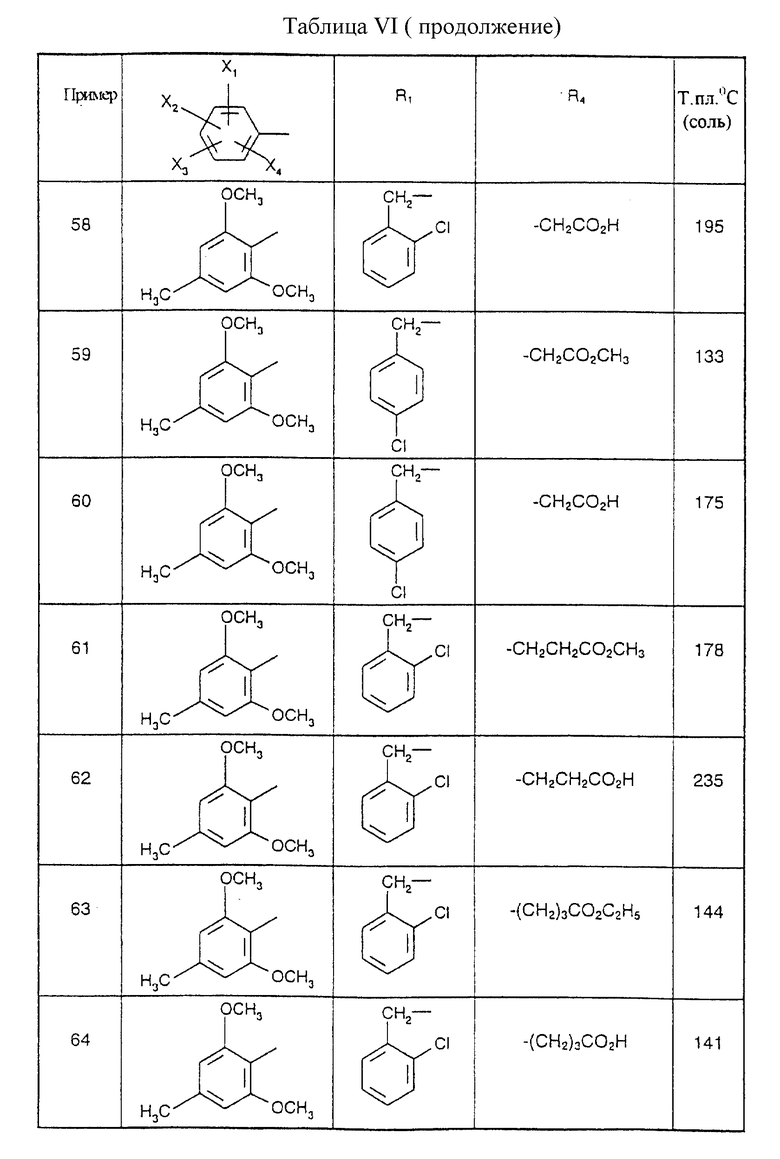
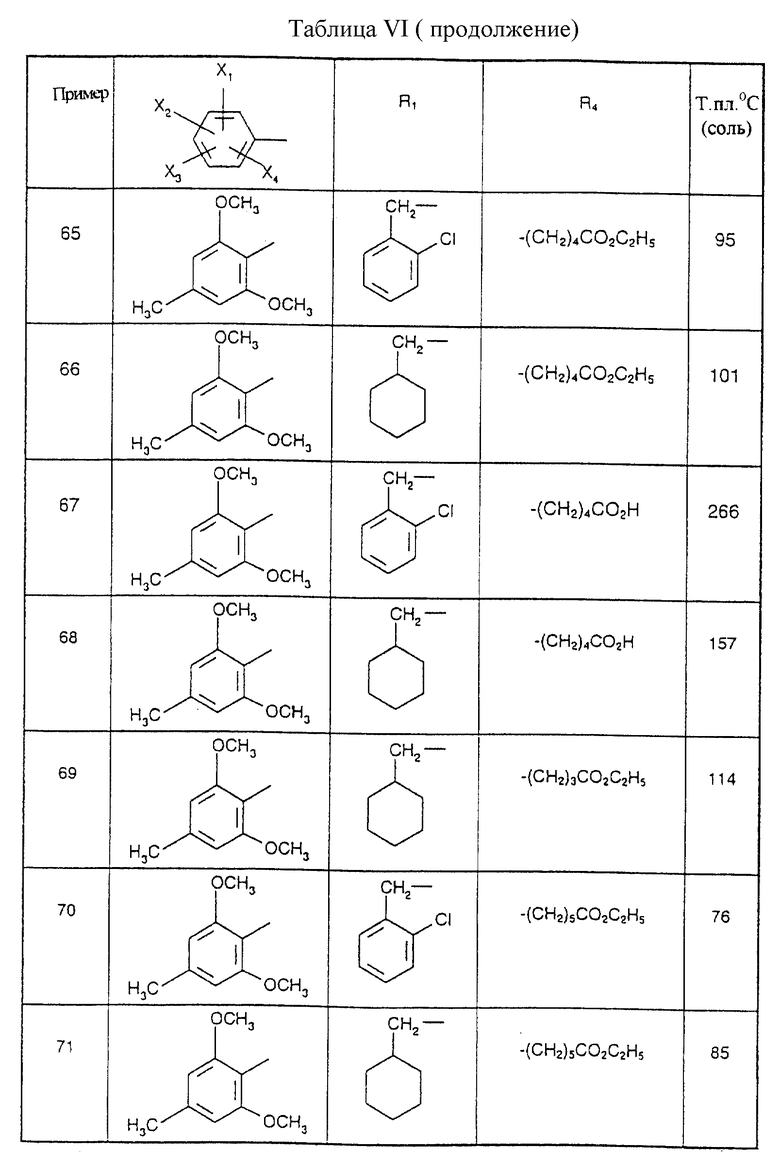
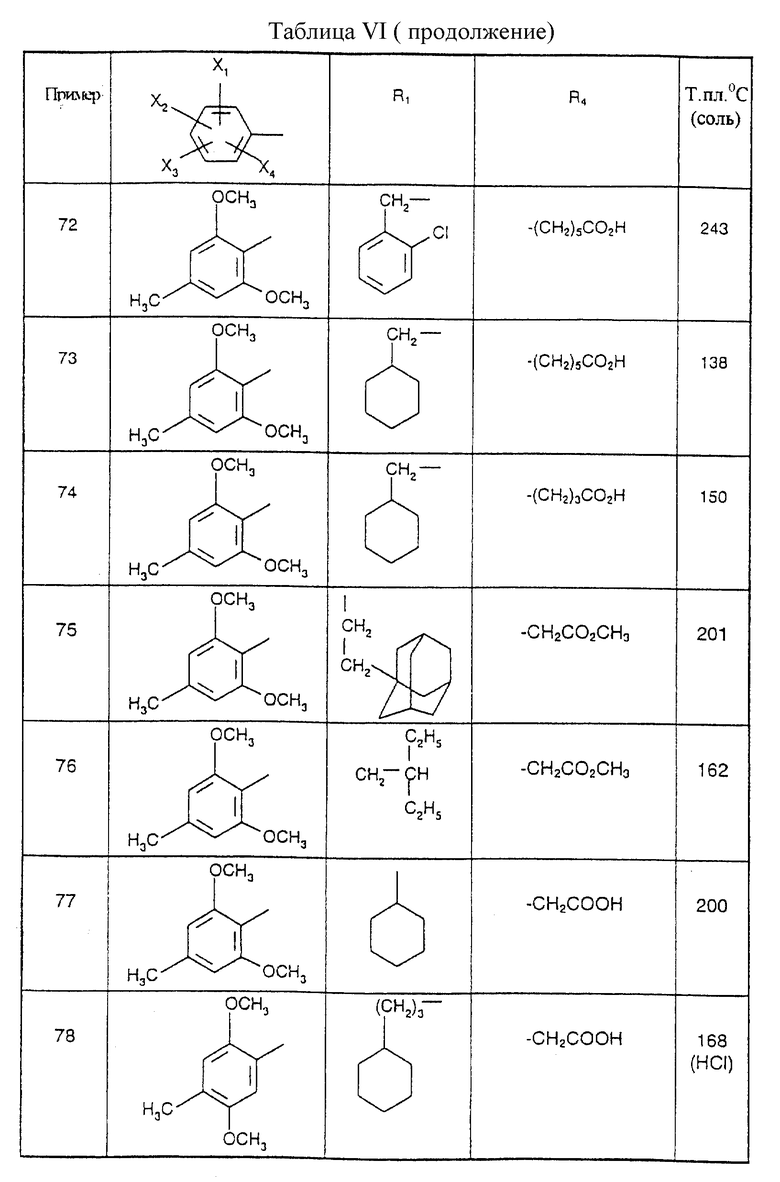
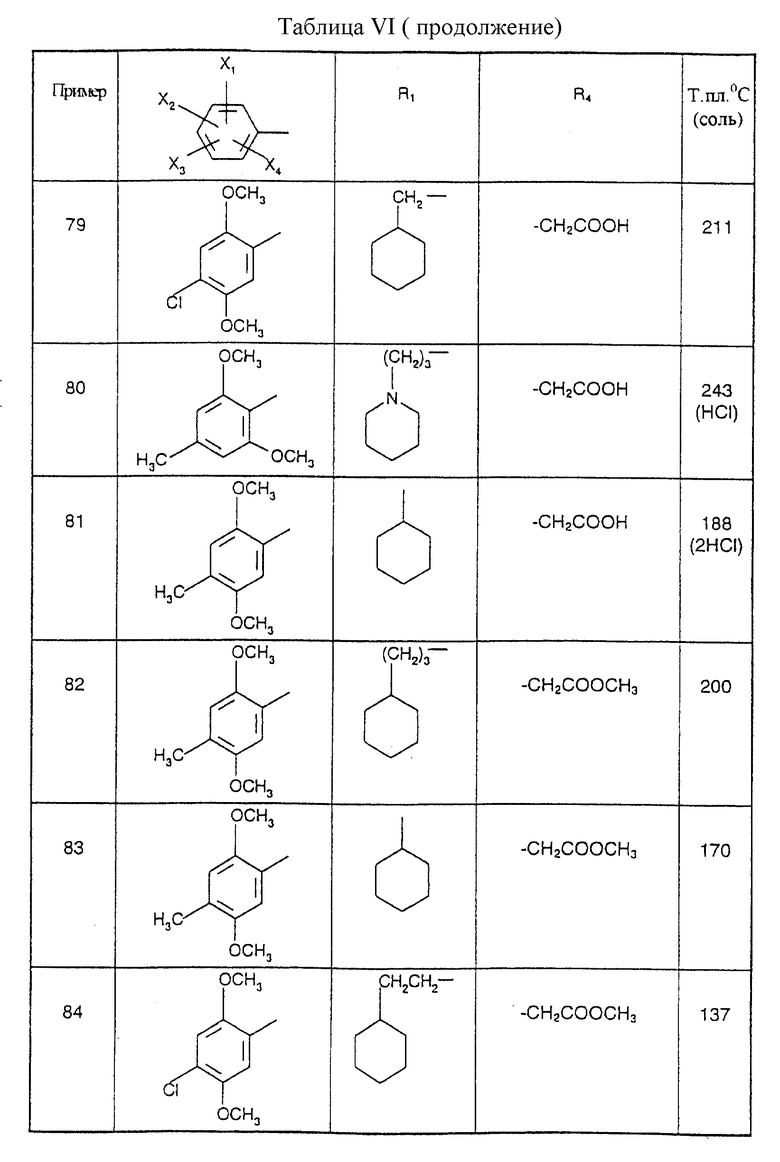
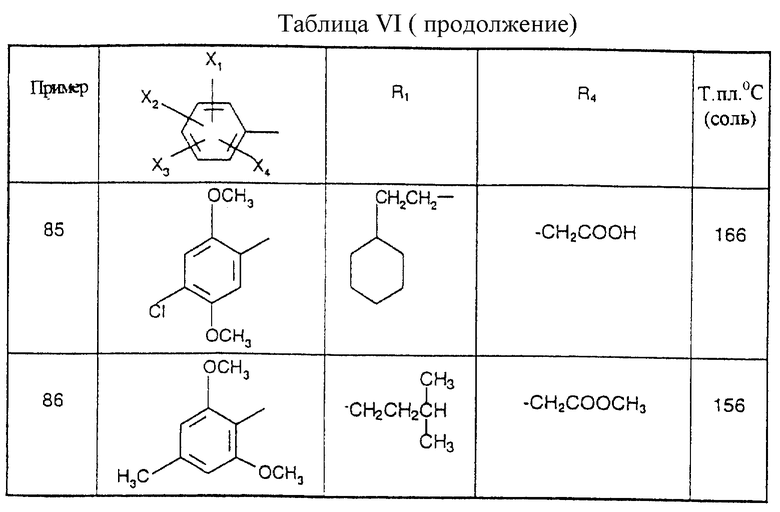
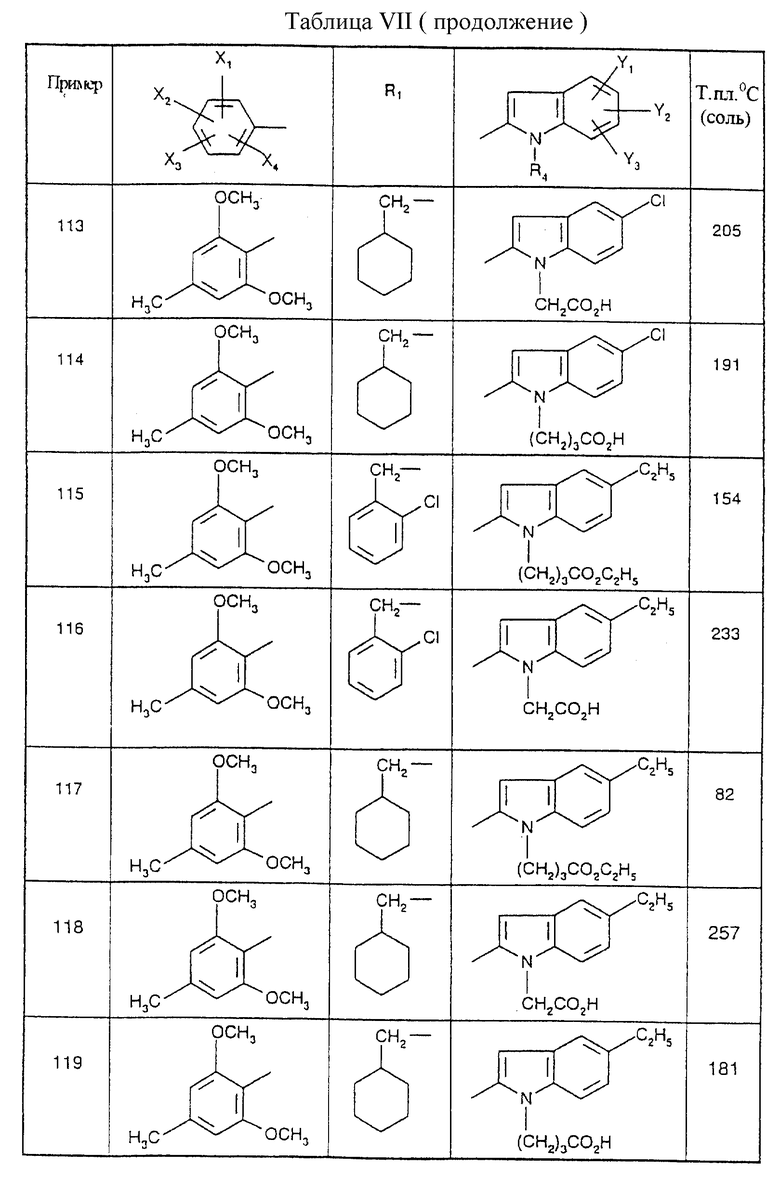
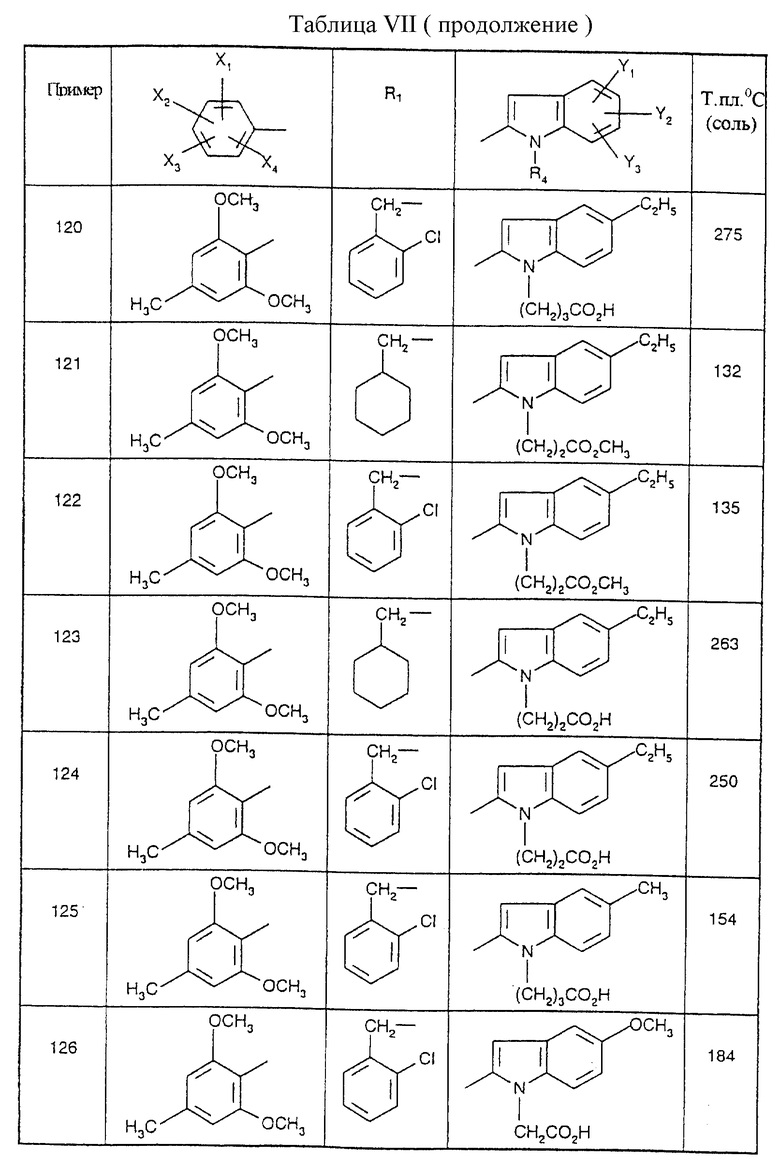
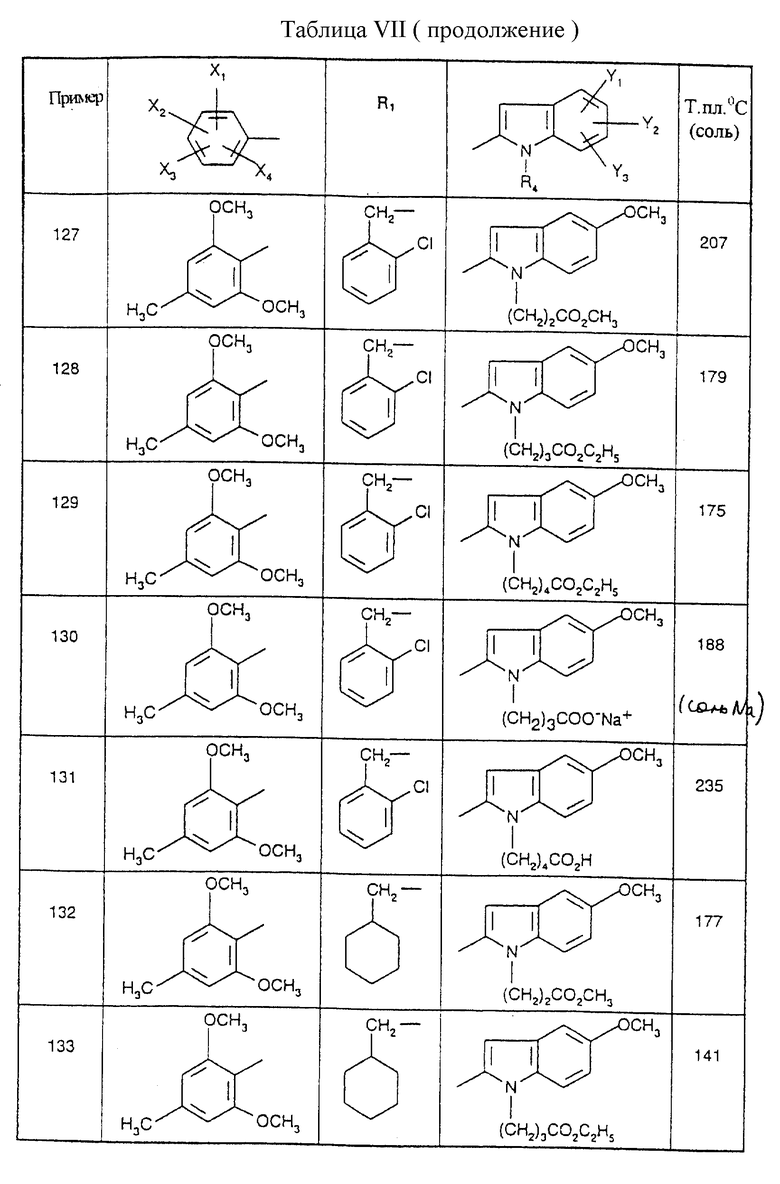
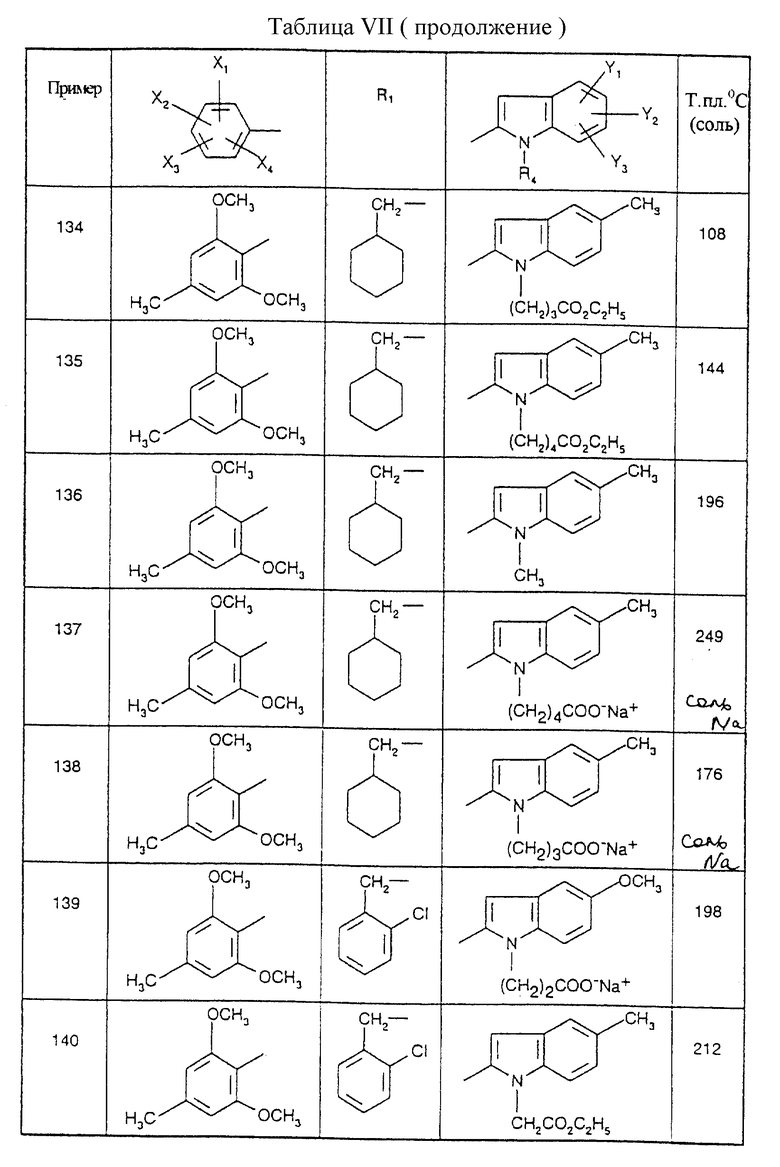
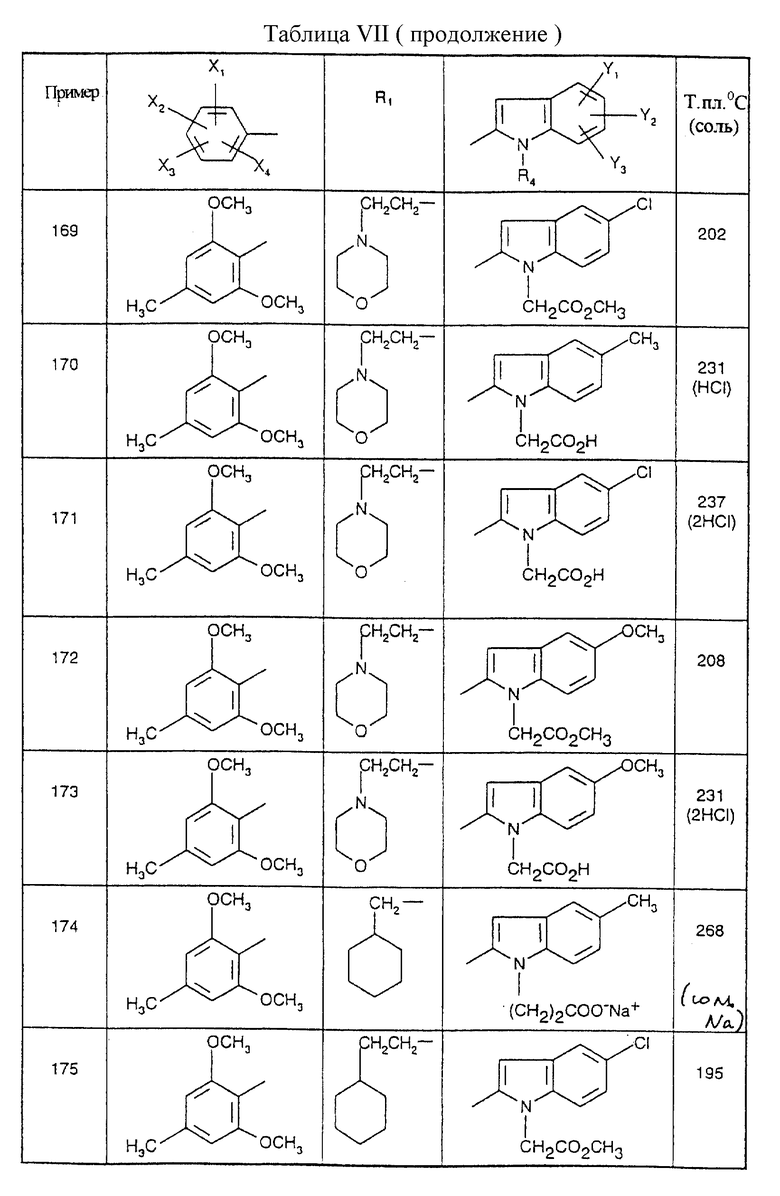
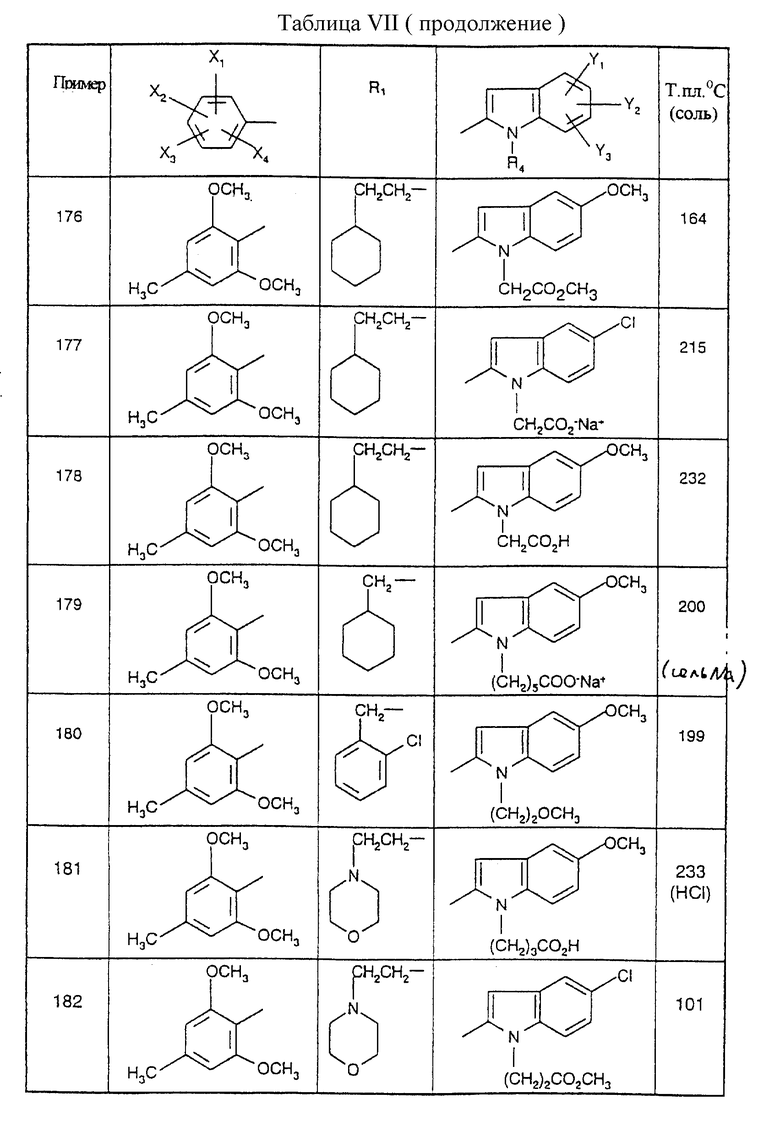
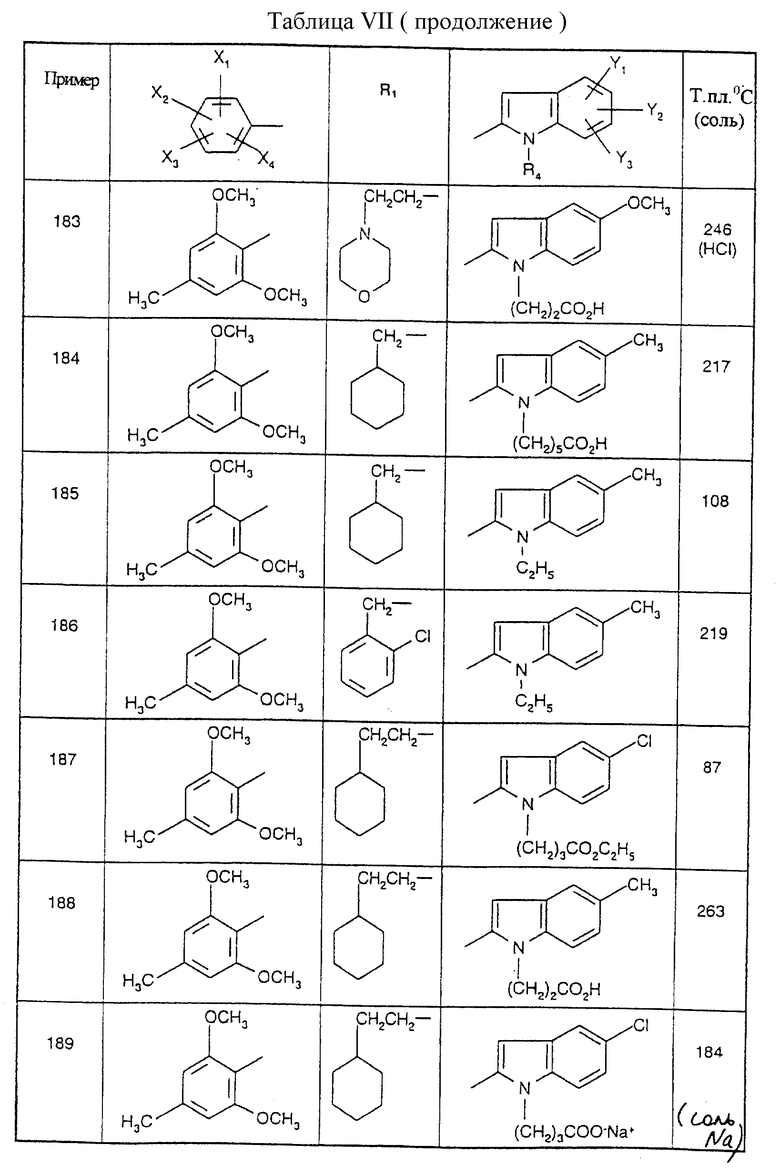
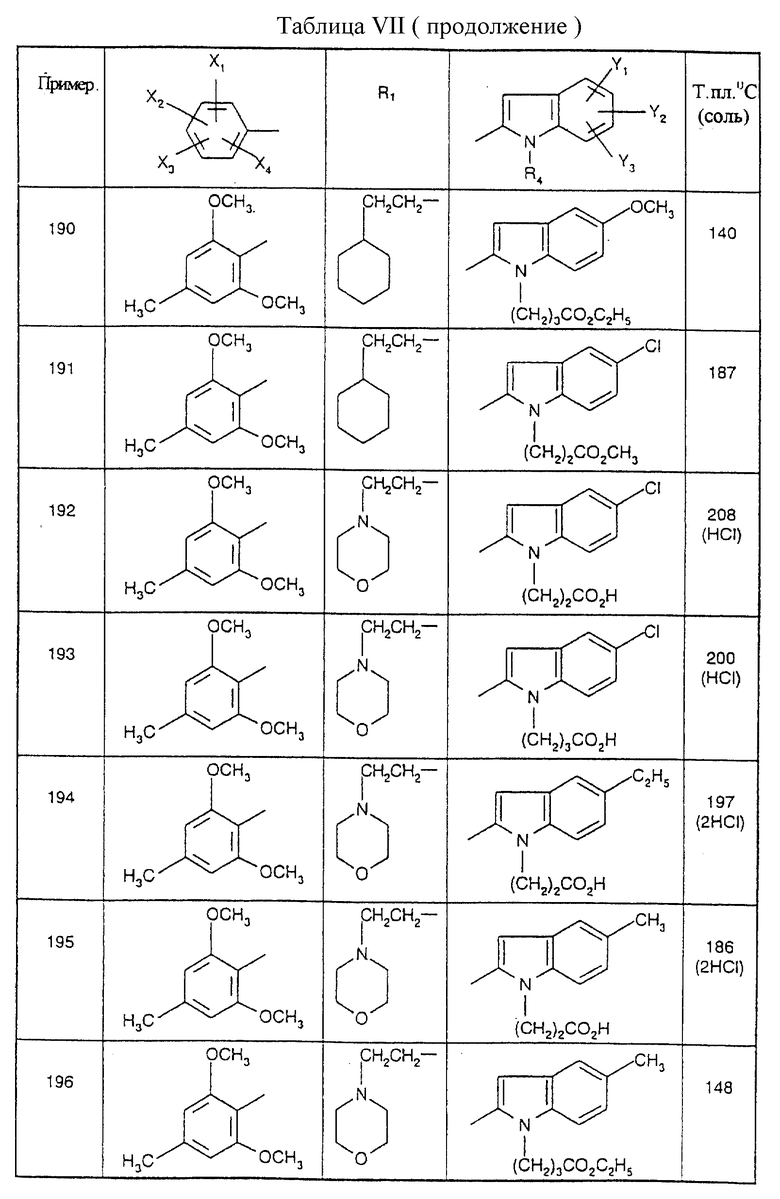
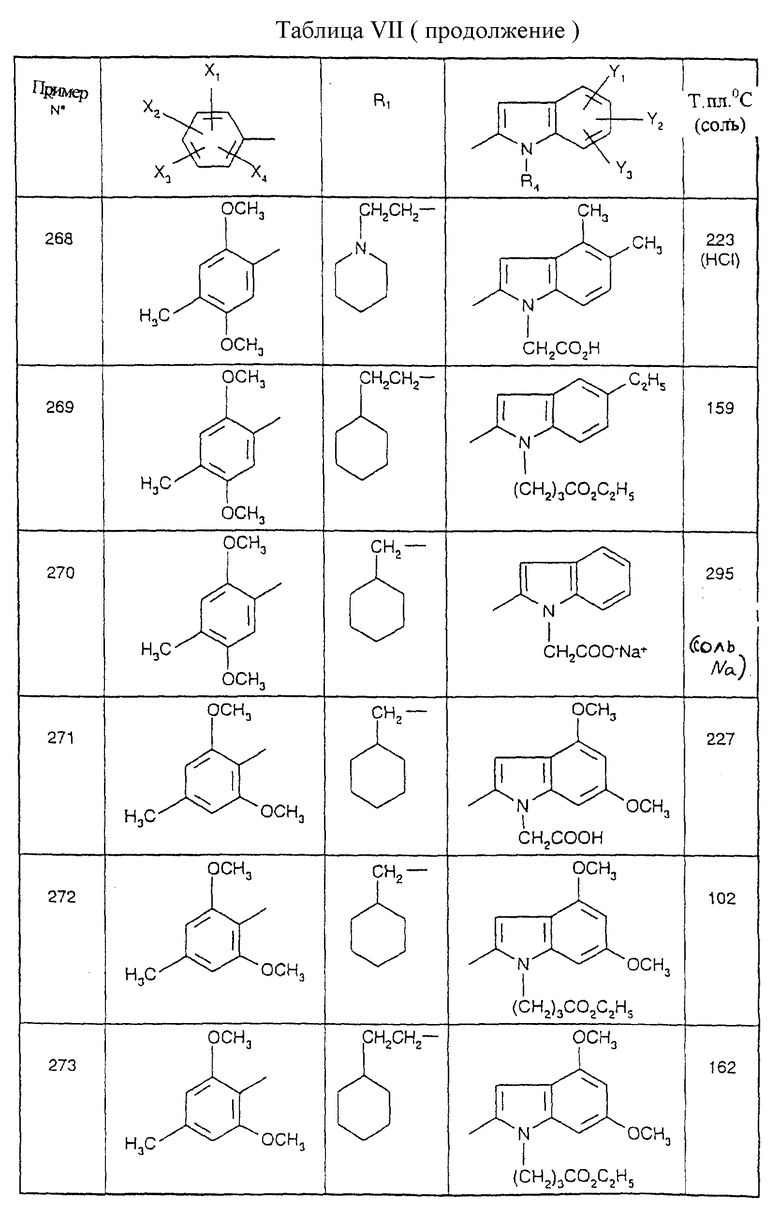
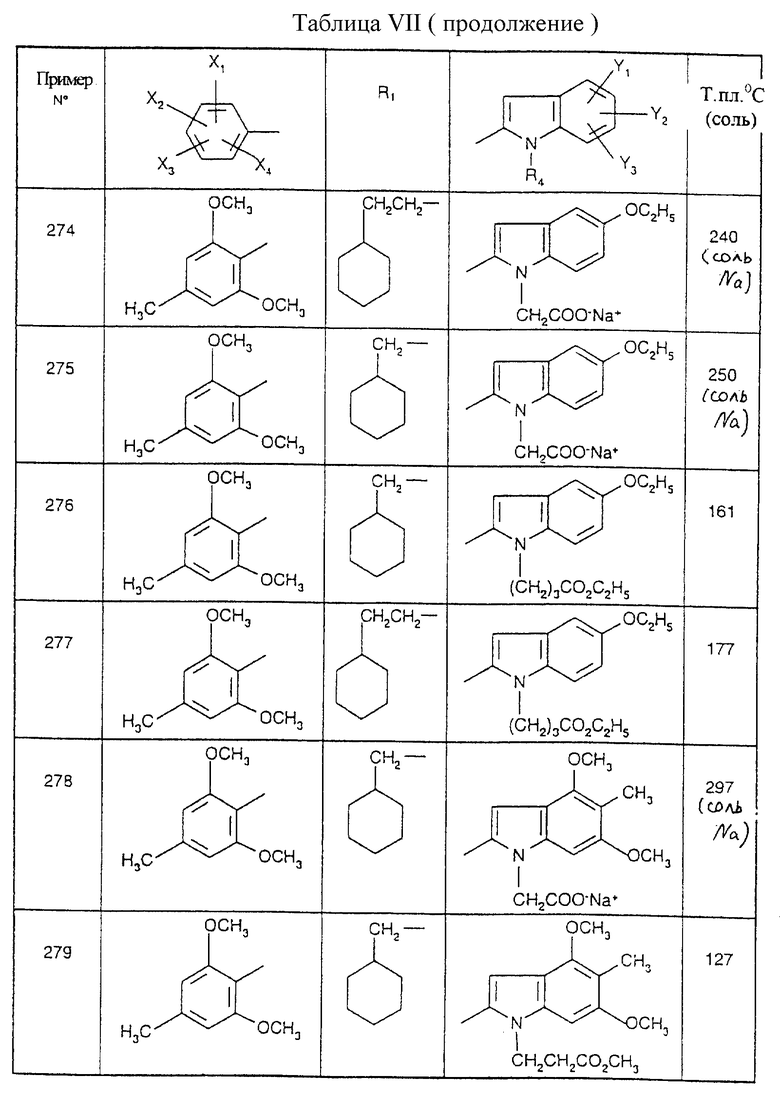
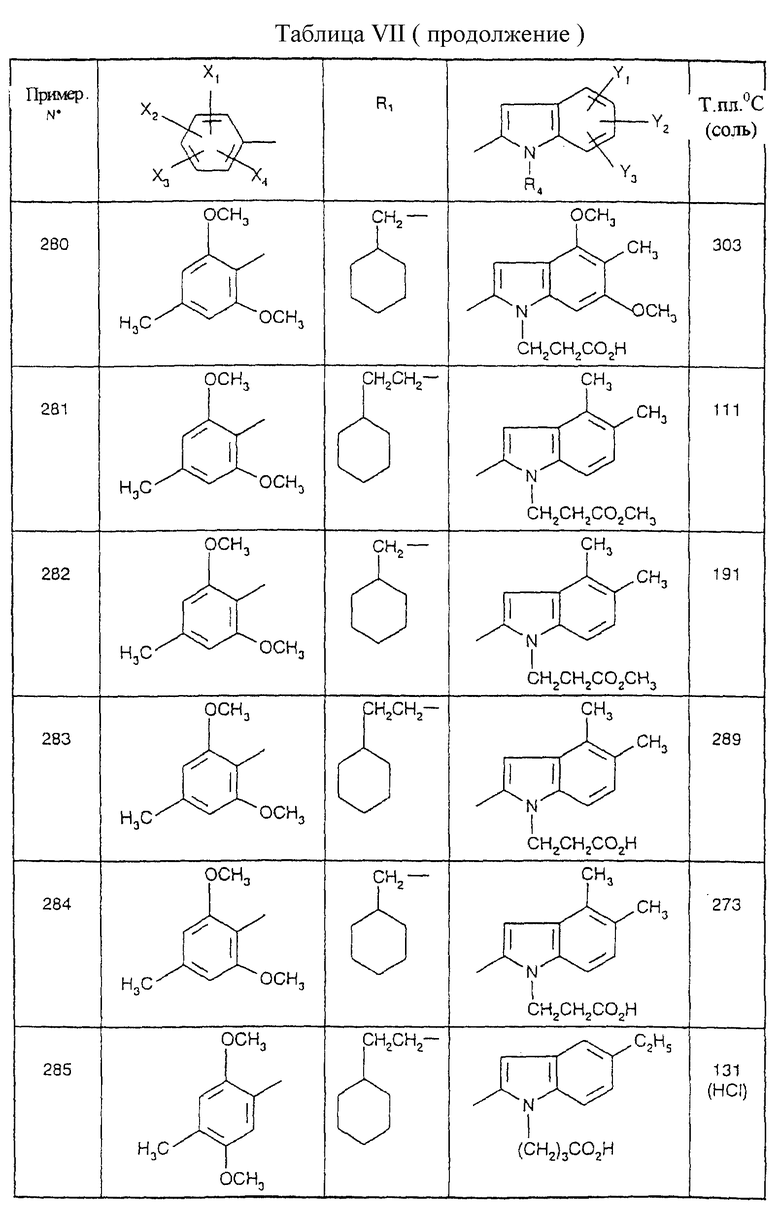
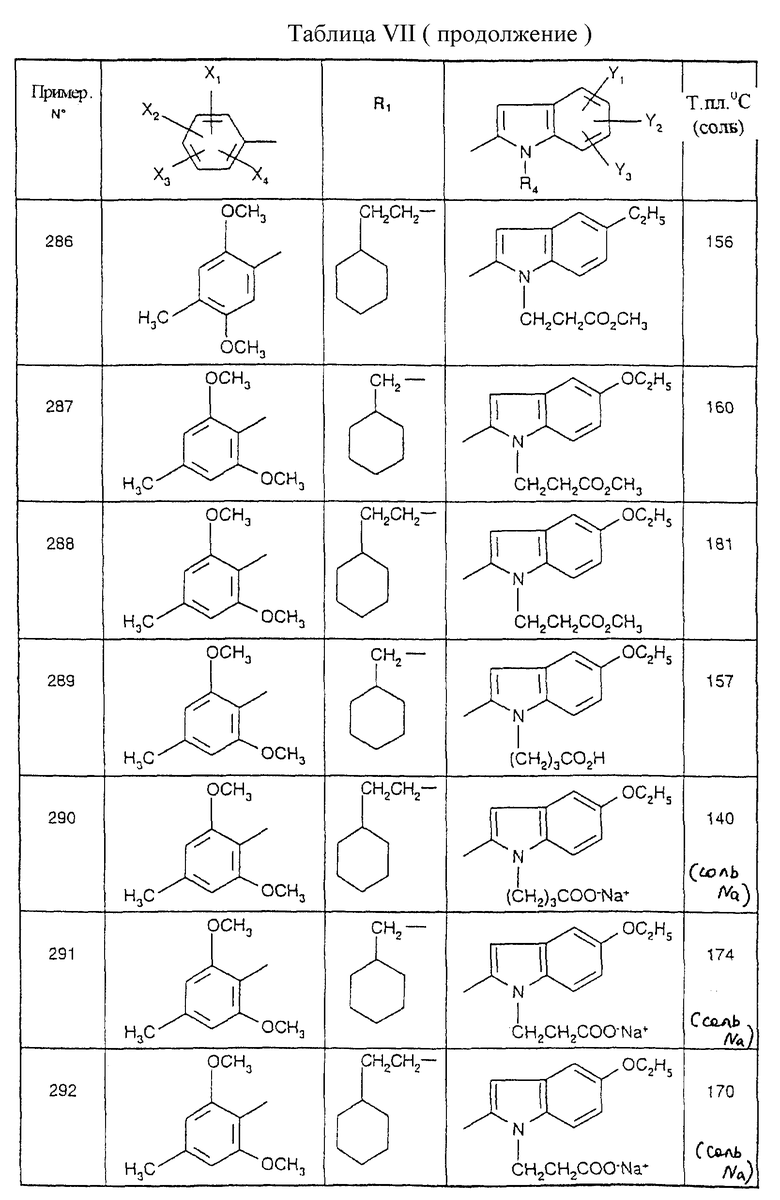
Соединения от В5 до В70, описанные в табл. 1, синтезированы в соответствии с процедурами приведенных выше получений, начиная с соответствующих синтетических промежуточных соединений.
4,5-Диметил-1-(3-цианопропил)-1Н-2-индолкарбоксильная кислота (Соединение В71)
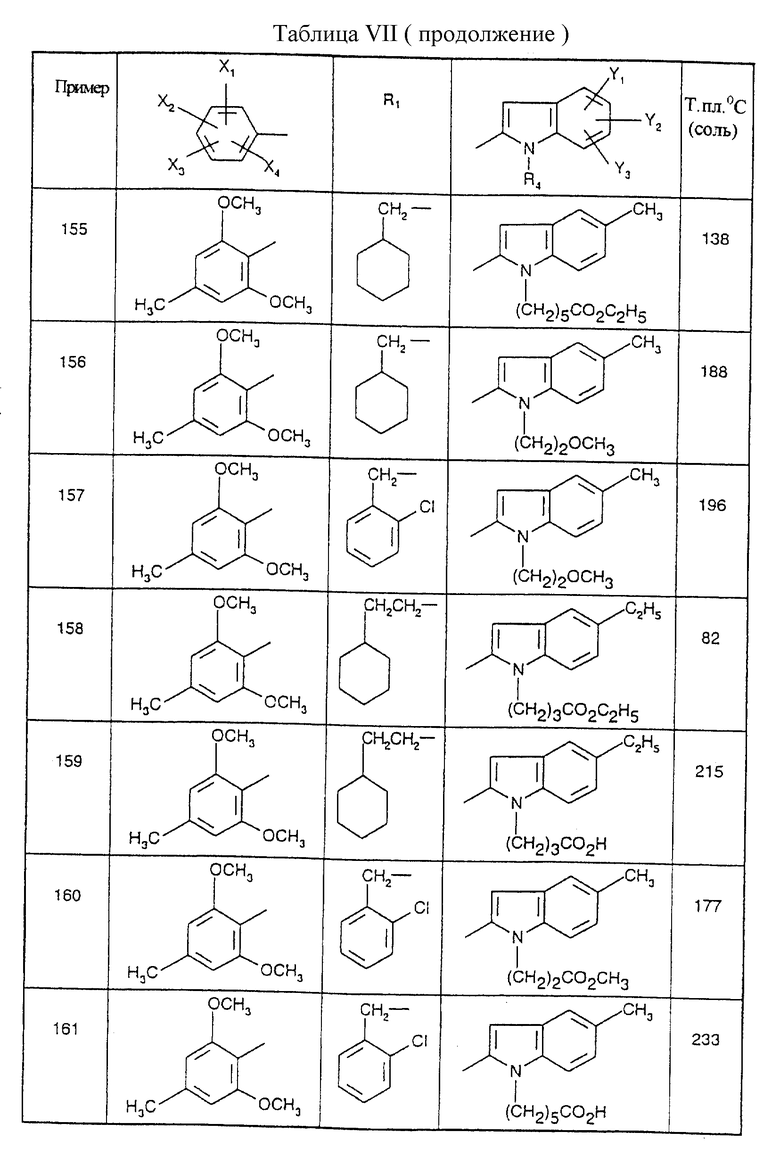
Стадия 1: Этиловый эфир 4,5-диметил-1-(3-цианопропип)-1Н-2- индолкарбоновой кислоты
75 мл диметилформамида добавляют к 1,92 г (0,040 моль) гидрида натрия в виде 50% суспензии в масле с последующим добавлением порциями 7,9 г (0,0363 моль) этилового эфира 4,5-диметил-1Н-2-индолкарбоновой кислоты. После перемешивания в течение 40 минут при 0oC вводят по каплям 4,0 мл (0,040 моль) 4-бромбутиронитрила, и реакционную смесь выдерживают в течение 2 часов при 20oC. Добавляют 300 мл этилацетата, смесь промывают дважды 300 мл воды, фазы разделяют после отстаивания, и органическую фазу сушат над безводным сульфатом натрия и концентрируют. Получают 9,8 г бесцветного масла; выход=95%.
Стадия 2: 4,5-Диметил-1-(3-цианопропил)-1Н-2-индолкарбоновая кислота
9,8 г (0,0345 моль) этилового эфира 4,5-диметил-1-(3-цианопропил)- 1Н-2-индолкарбоновой кислоты добавляют к 150 мл 1,4-диоксана с последующим добавлением 25 мл 2 М раствора гидроксида натрия (0,05 моль). Реакционную смесь выдерживают в течение 48 часов при комнатной температуре. После выпаривания 1,4-диоксана остаток переносят в 6 М раствор соляной кислоты, и полученный осадок отфильтровывают и сушат при пониженном давлении при 60oC с получением 4,5-диметил-1-(3-цианопропил)-1Н-2-индолкарбоновой кислоты в форме белых кристаллов; т.пл.=175oC; выход=92%.
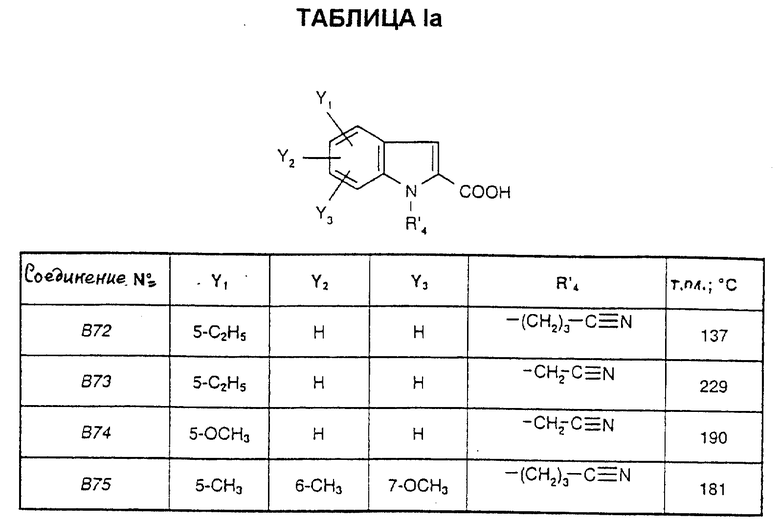
Соединения от В72 до В75, представленные в табл. Iа, получены таким же способом.
В. Получение бензамидогуанидиновых производных
Получение 2,6-диметокси-4-метилбензамидогуанидина (Соединение C1)
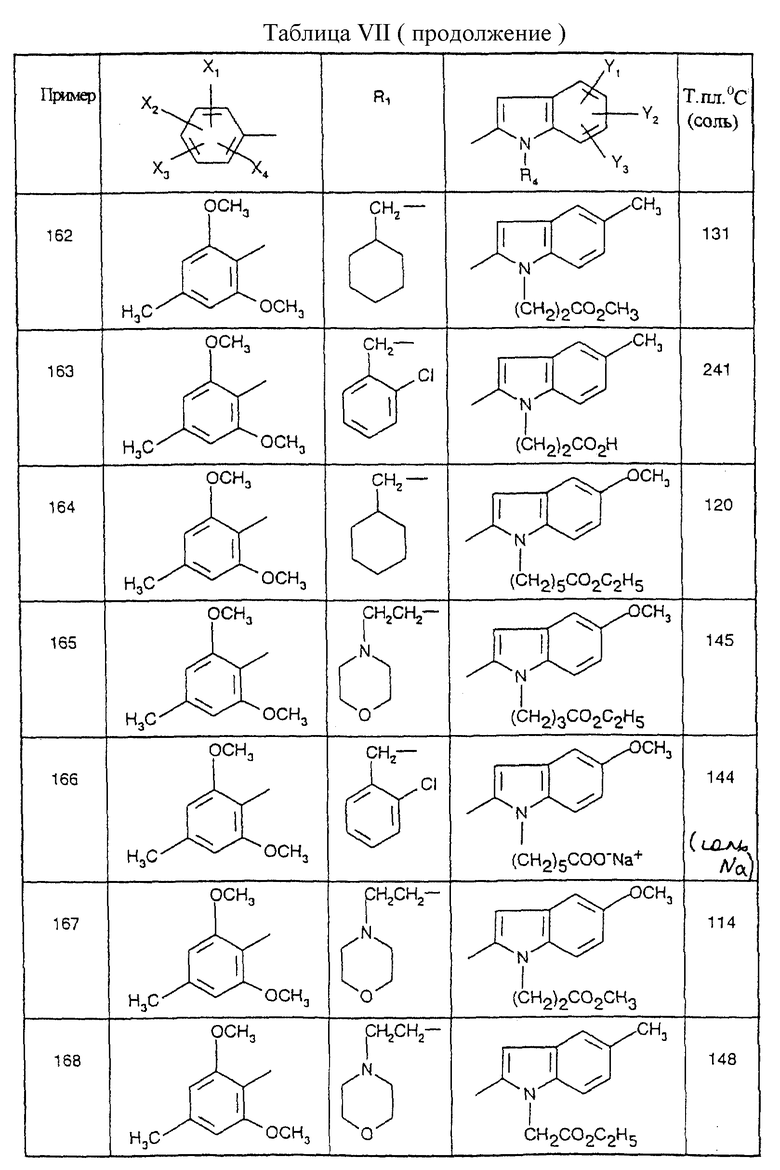
1 мл диметилформамида добавляют к 353 г (1,8 моль) 2,6-диметокси-4-метилбензойной кислоты, суспендированной в 1,5 литрах толуола, с последующим добавлением по каплям 190 мл оксалилхлорида (2,16 моль). Реакционную смесь оставляют на 2 часа при комнатной температуре, а затем выпаривают досуха. Кристаллический остаток добавляют порциями к суспензии 293,8 г аминогуанидина гидрокарбоната (2,16 моль) в 2,5 литрах пиридина при ±5oC и оставляют на 18 часов при 20oC. Реакционную смесь выпаривают досуха, и остаток затем переносят в 1 литр 2 М раствора гидроксида натрия. Осадок отфильтровывают и промывают минимальным количеством воды, а затем сушат под вакуумом при 60oC с получением кристаллического остатка; т.пл.=222oC; выход=81%.
Г. Получение производных 3-аминотриазола
3-Амино-5-(2,6-диметокси-4-метилфенил)-1,2,4-триазол (Соединение D.1)
2 литра дифенилового эфира добавляют к 230 г (0,91 моль) 2,6-диметокси-4-метилбензамидогуанидина, после чего реакционную смесь нагревают в течение 5 минут при 220oC. Смесь охлаждают до 80oC, и осадок затем отфильтровывают, промывают диизопропиловым эфиром и сушат под вакуумом при 60oC с получением кристаллов; т.пл.=286oC; выход=93%.
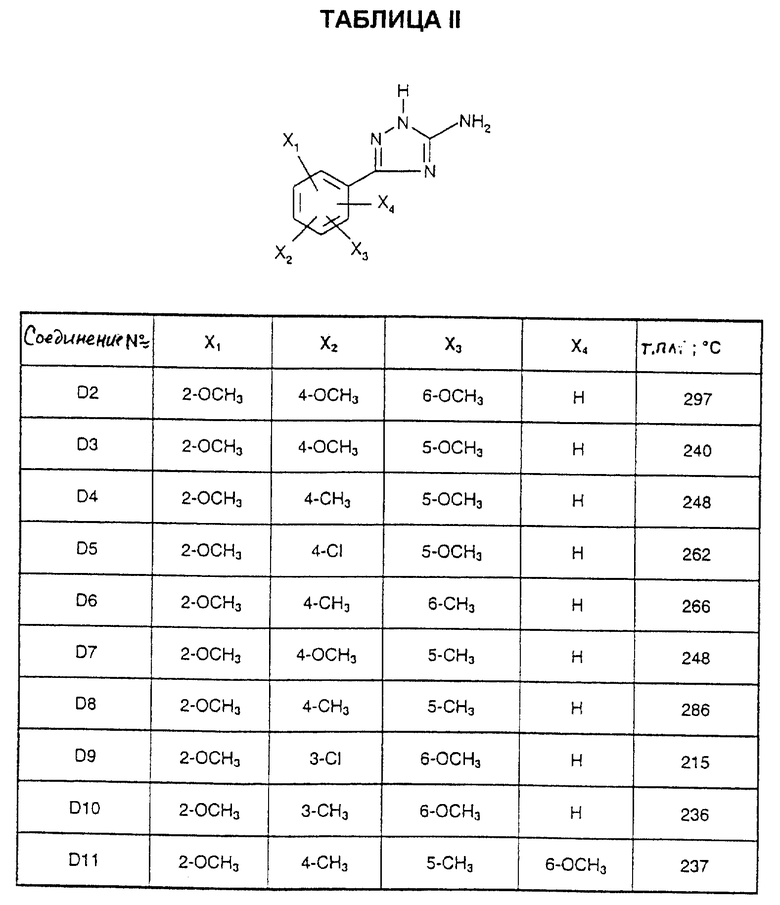
Соединения от D2 до D11, описанные в табл. II, синтезированы таким же способом согласно процедуре этого получения и с использованием соответствующих исходных материалов.
Д. Получение дифенилиминопроизводных
Получение N-[3-(2,6-диметокси-4-метилфенил)-1H-1,2,4-триазол-5-ил] -N- дифенилметиленамина (Соединение E1)
105 г (0,45 моль) 3-амино-5-(2,6-диметокси-4-метилфенил)-1,2,4-триазола, суспендированного в 200 мл ксилола и 150 г (0,9 моль) бензофенонимина нагревают при 140oC в течение 48 часов в токе аргона. Реакционную смесь охлаждают до температуры 80oC, а затем вливают в 4 литра изопропилового эфира, и образовавшийся осадок отфильтровывают, промывают диизопропиловым эфиром и сушат в течение 18 часов при 50oC; т.пл.=126oC, выход=90%.
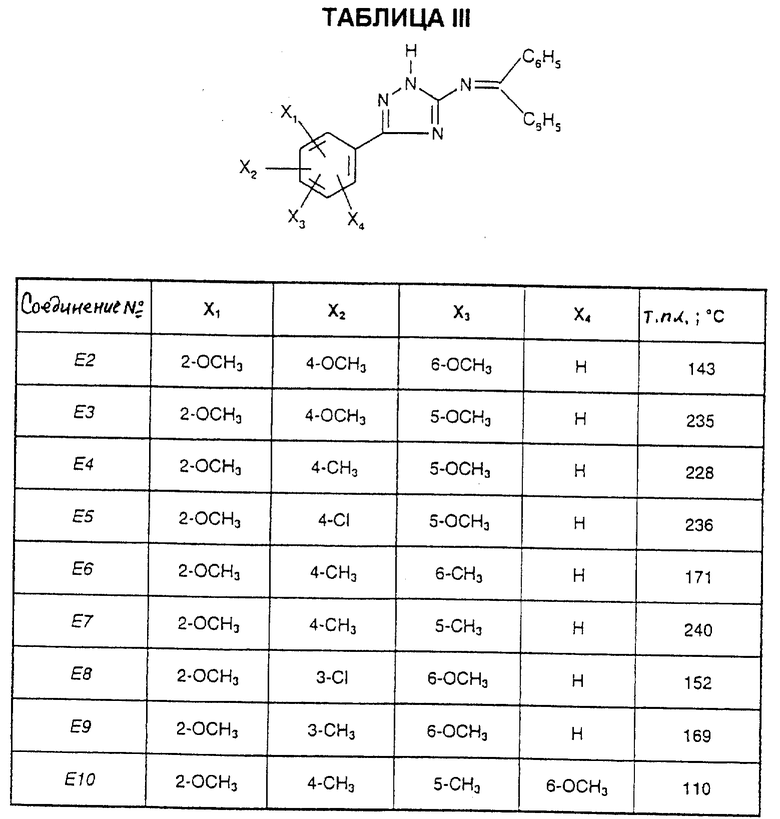
Соединения от E2 до E11, описанные в табл. III, синтезированы таким же способом согласно процедуре этого получения и с использованием соответствующих исходных материалов.
E. Получение 1-замещенных 3-аминотриазолов
Получение 1-(2-циклогексилэтил)-5-(2,6-диметокси-4-метилфенил)-1Н- 1,2.4-триазол-3-амина (Соединение F1)
а) N-Алкилирование триазола
300 мл водного 6 н. раствора гидроксида натрия, 24 г (0,06 моль) N-[3-(2,6-диметокси -4-метилфенил)-1Н-1,2,4-триазол-5-ил]-N- дифенилметиленамина и 2,7 г тетрабутиламмония бромида добавляют последовательно к 400 мл толуола. 17 г (0,09 моль) 2-бромэтилциклогексана добавляют по каплям к этой реакционной смеси, нагревают до 70oC. Взаимодействие продолжают в течение двух часов при 80oC. Органическую фазу отделяют после отстаивания и сушат над безводным сульфатом натрия и выпаривают досуха. Остаток хроматографируют на колонке с силикагелем с использованием в качестве элюента смеси 90/10 (об./об.) толуол/этилацетат. Получают 21,4 г бесцветного масла; выход=70%.
б) Гидролиз дифенилиминной функциональной группы
100 мл 1 н. раствора соляной кислоты добавляют к 10,3 г (0,02 моль) N-[1-(2-циклогексилэтил)-5-(2,6-диметокси-4-метилфенил)-1 Н- 1,2,4-триазол-3-ил] -N-дифенилметиленамина, растворенного в 200 мл метанола. Реакционную смесь оставляют на 18 часов при комнатной температуре, а затем выпаривают досуха. Маслянистый остаток отверждают в диэтиловом эфире, и полученный осадок отфильтровывают и сушат под вакуумом при 40oC; т.пл.=136oC (гидрохлорид); выход=90%.
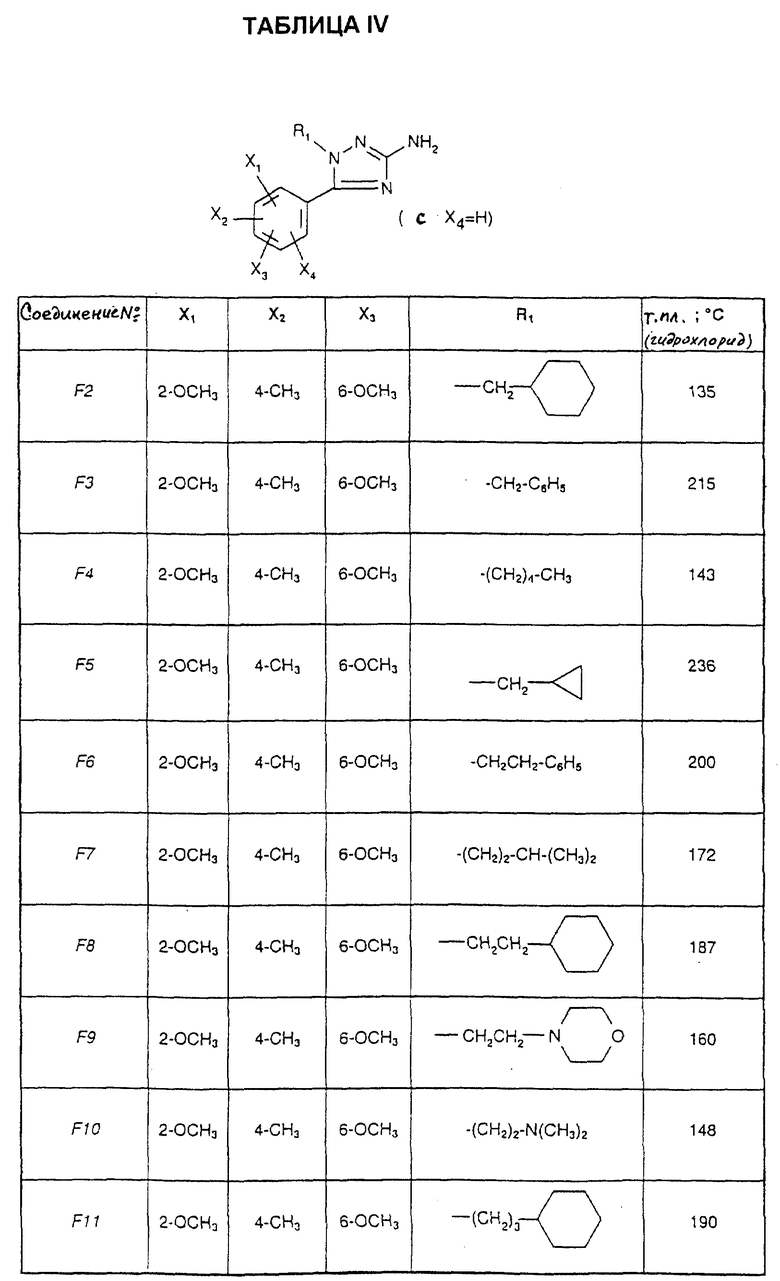
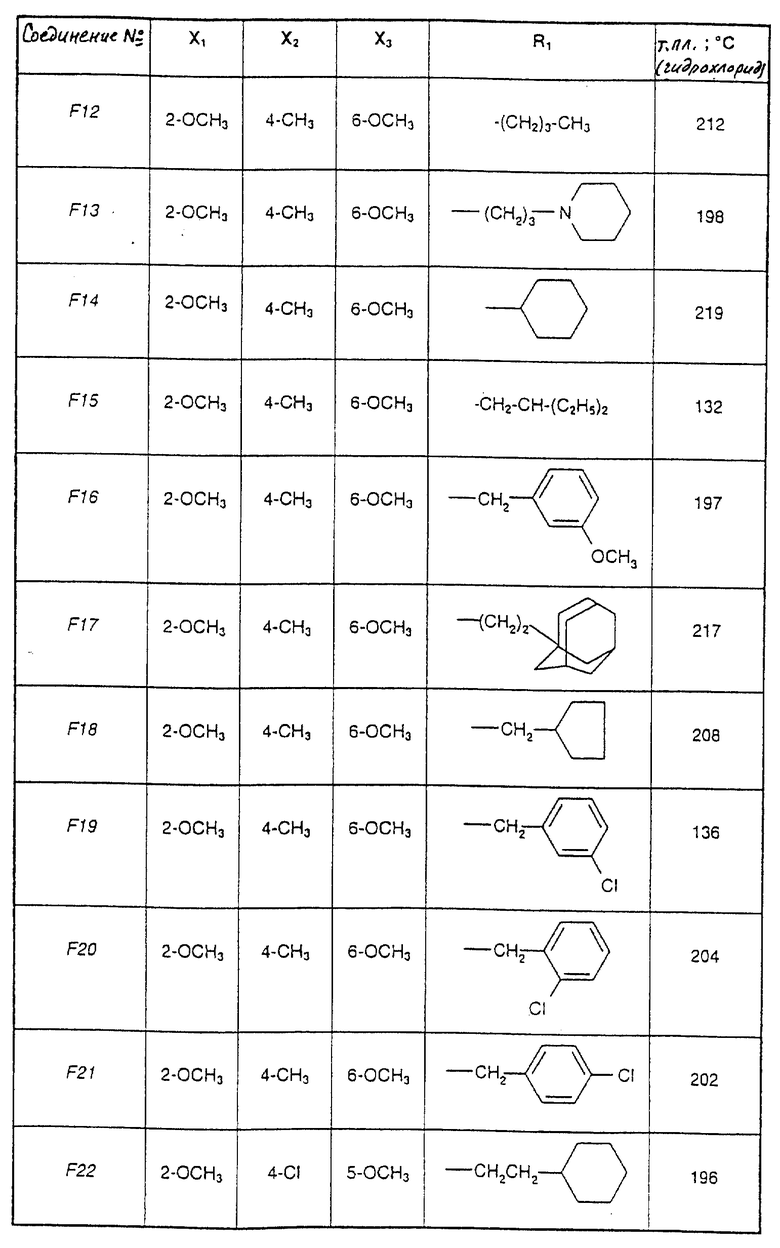
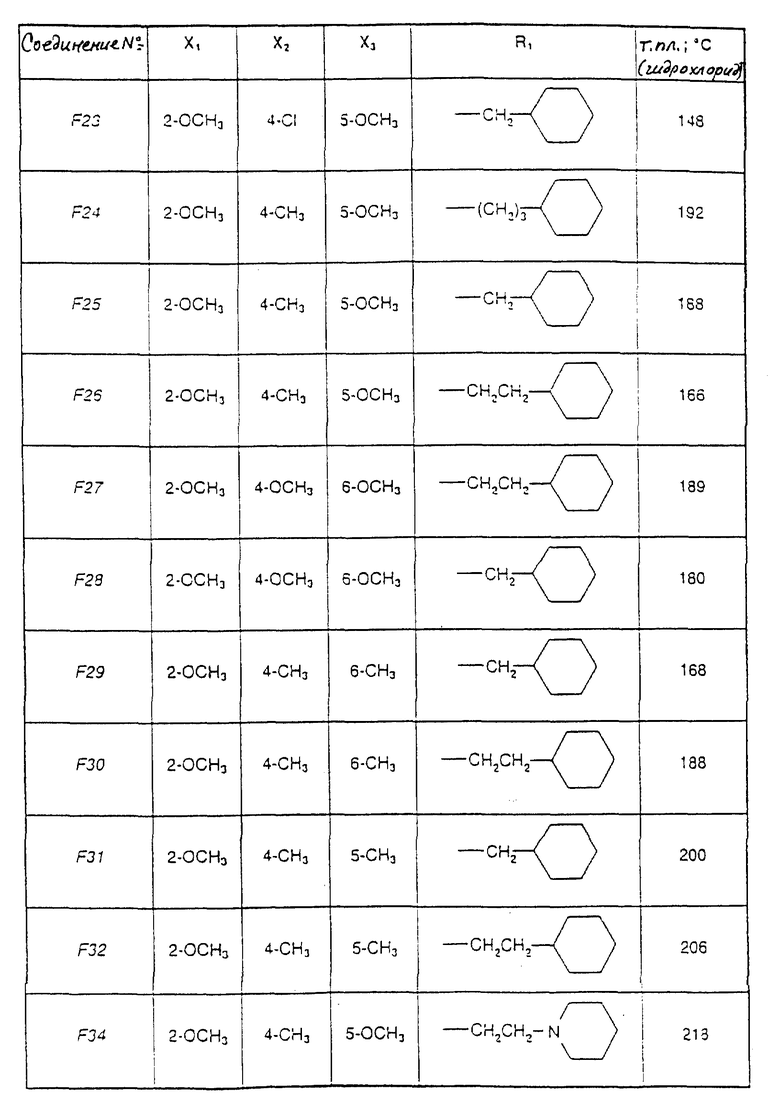
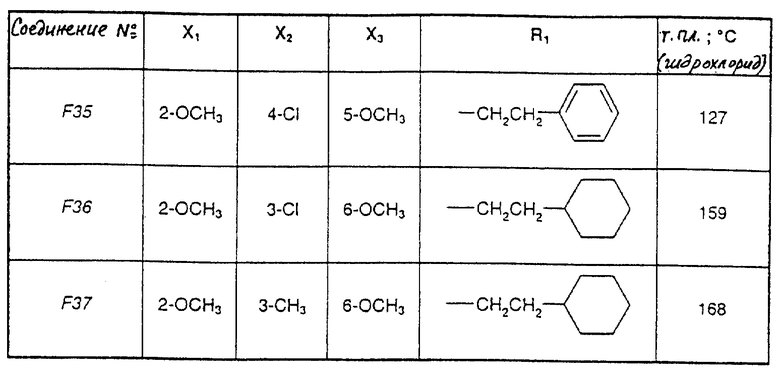
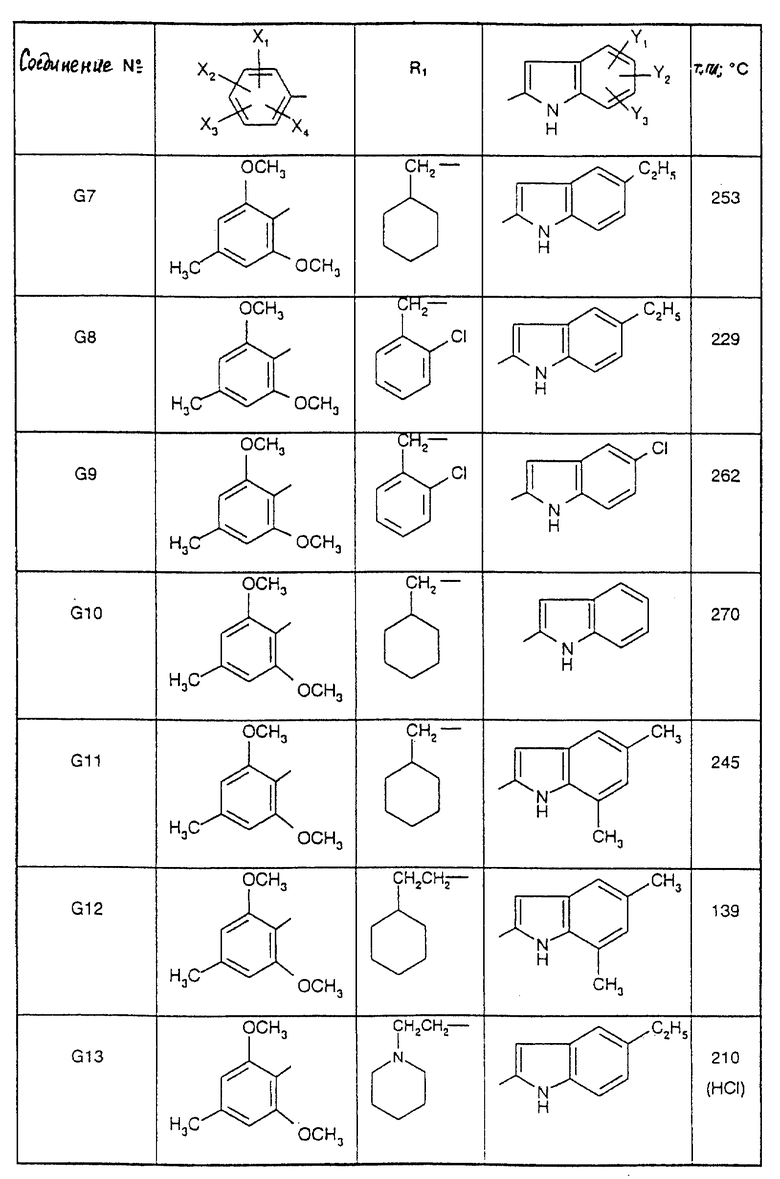
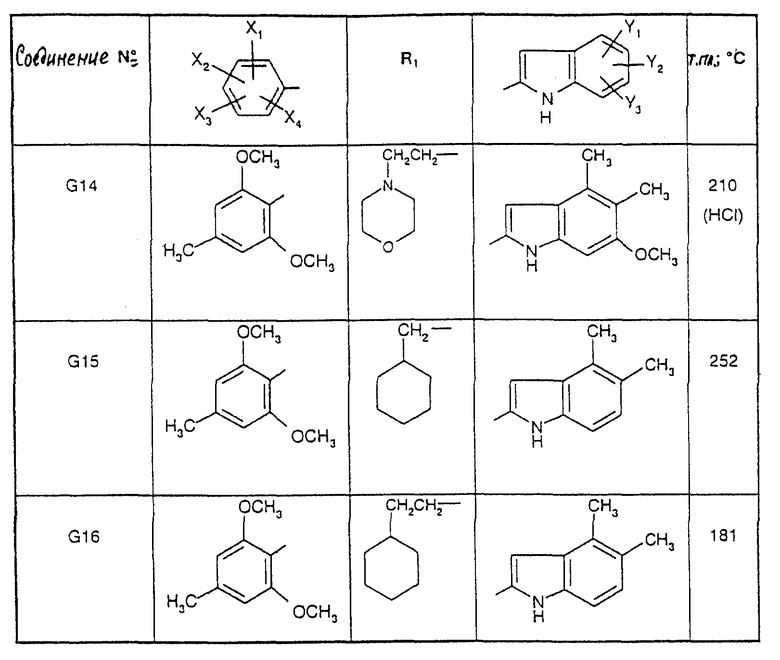
Соединения от F2 до D37, описанные в табл. IV, синтезированы таким же способом согласно процедуре этого получения и с использованием соответствующих исходных материалов.
1-(2-Циклогексилэтил)-5-(2,6-диметокси-4,5-диметилфенил)-1Н-1,2,4- триазол-3-амин (Соединение F38) получают таким же способом, начиная с Соединения Е10; т.пл.=180oC.
Ж. Получение амидотриазольных производных с не-N-замещенными индолами
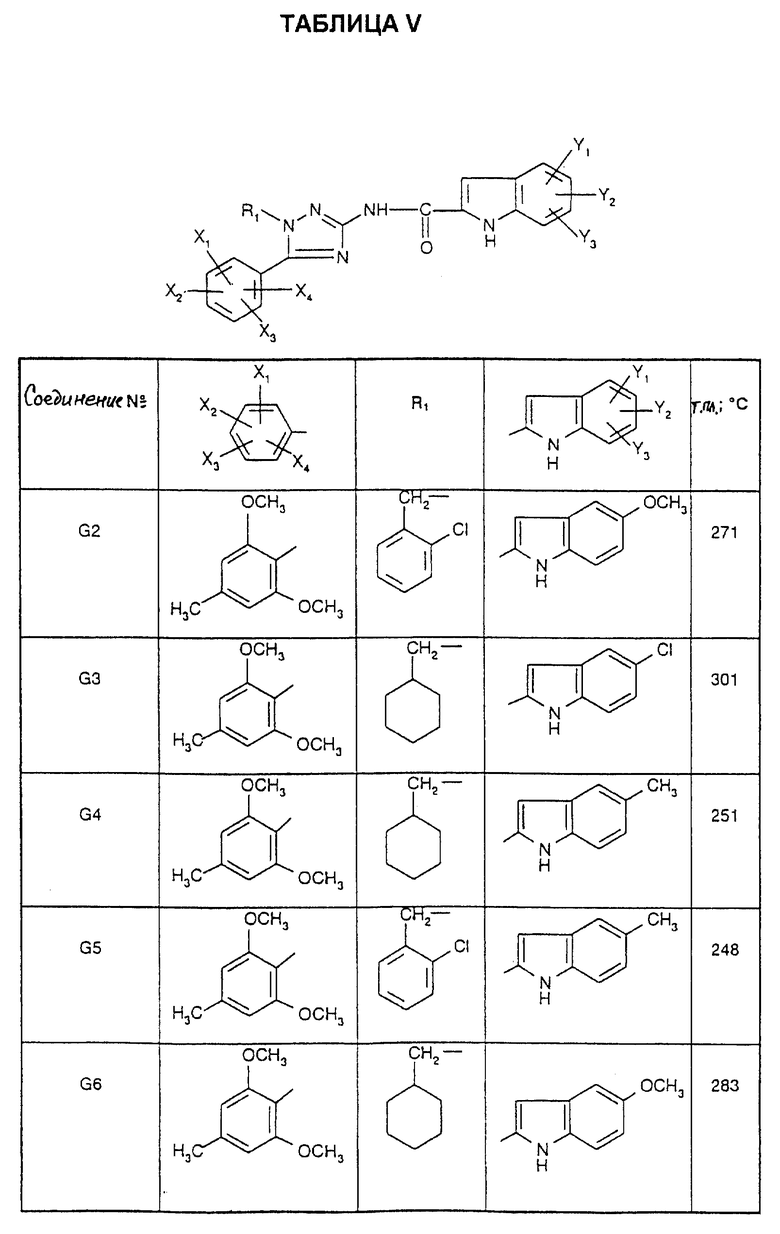
Синтез N-[1-(2-хлорбензил)-5-(2,6-диметокси-4-метилфенил)-1H-1,2,4- триазол-3-ил]-5-хлор-1Н-2-индолкарбоксамида (Соединение G1)
0,2 мл тионилхлорида (0,0028 моль) добавляют при 0oC к раствору 1 мл пиридина (0,013 моль) в 30 мл метиленхлорида. После 15 минут при 0oC вводят 500 мг (0,0025 моль) 5-хлориндолкарбоновой кислоты, и эту реакционную смесь оставляют на 30 минут при 0oC. 0,91 г (0,0028 моль) 1-[(2-хлорфенил)метил] -5-(2,6-диметокси-4- метилфенил)-1Н-1,2,4-триазол-3-амина гидрохлорида добавляют к образовавшемуся ацилхлориду, и смесь оставляют на 18 часов при 20oC.
Реакционную смесь промывают 1 М раствором гидроксида натрия. Органическую фазу сушат над безводным сульфатом натрия и выпаривают досуха. Остаток хроматографируют на силикагеле с использованием элюента: смесь 95/5 (об./об. ) дихлорметан/метанол, с получением 0,980 г кристаллов: т.пл.=262oC; выход= 73%.
Соединения от DG до D16, описанные в табл. V, синтезированы таким же способом согласно процедуре этого получения и с использованием соответствующих исходных материалов.
З. Получение аминотриазольных производных с N-замещенными индолами
Пример 1
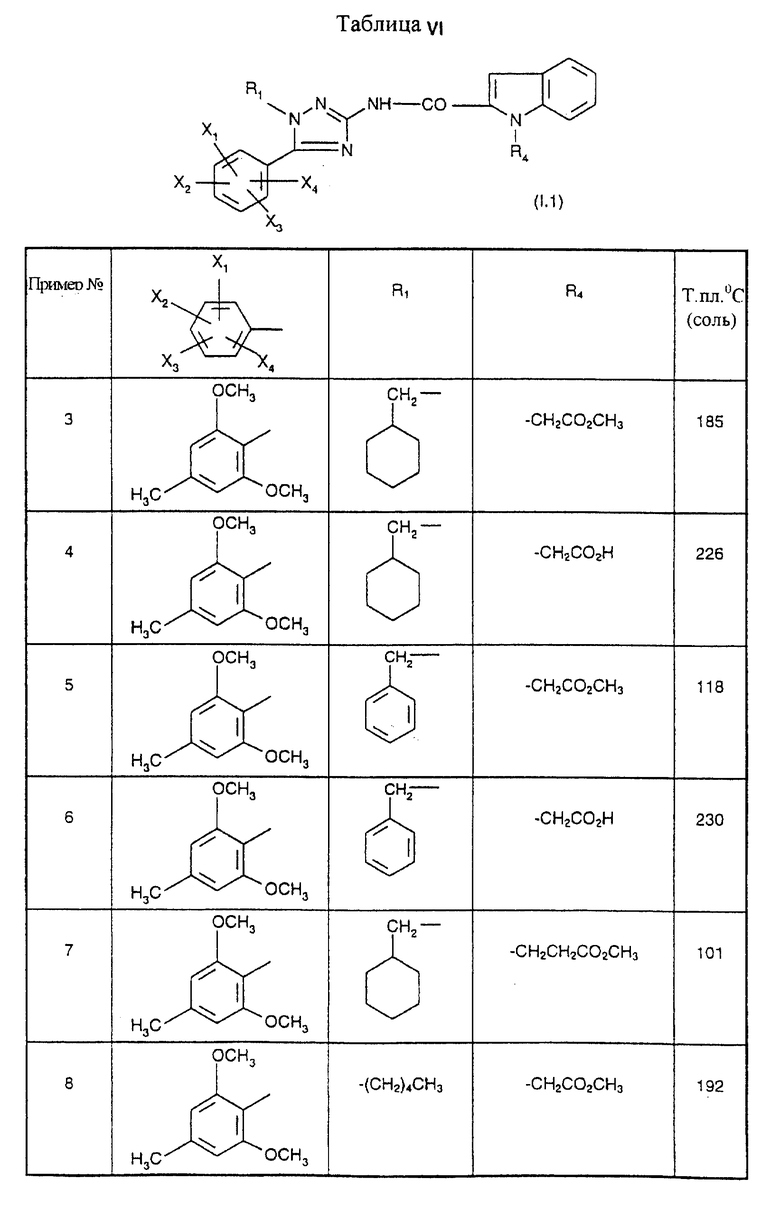
Метиловый эфир 2-[2-({ [1-(2-циклогексилэтил)-5-(2,6-диметокси-4- метилфенил)-1H-1,2,4-триазол-3-ил] амино} карбонил)-5-этил-1H-индол-1- ил]уксусной кислоты
1 мл пиридина (0,013 моль) и 0,21 мл тионилхлорида (0,00029 моль) добавляют последовательно к 15 мл дихлорметана. После 15 минут при 0oC вводят 0,627 г 5-этил-1-метоксикарбонилметил-1H-2- индолкарбоновой кислоты (0,0024 моль) с последующим добавлением 0,9 г 1-(2-циклогексилэтил)-5-(2,6-диметокси-4-метилфенил)-1H- 1,2,4-триазол-3-амина гидрохлорида. Реакционную смесь оставляют на 18 часов при комнатной температуре, после чего проводят кислотную промывку, а затем щелочную промывку. Органическую фазу сушат над безводным сульфатом натрия и концентрируют. Маслянистый остаток хроматографируют на силикагеле с использованием элюента: смесь 98,5/1,5 (об./об.) дихлорметан/метанол, с получением белого порошка; т.пл.=191oC; выход=87%.
Пример 2
2-[2-({ [1-(2-Циклогексилэтил)-5-(2,6-диметокси-4-метилфенил)-1H- 1,2,4-триазол-3-ил]амино}карбонил)-5-этил-1H-индол-1-ил]уксусная кислота
1,8 мл (0,0018 моль) 1 н. раствора гидроксида натрия добавляют к 530 мг (0,0009 моль) метилового эфира 2-[2-({[1 -(2-циклогексилэтил)-5-(2,6-диметокси -4-метилфенил)-1H-1,2,4- триазол-3-ил] амино}карбонил)-5-этил-1H-индол-1 -ил]уксусной кислоты, полученного согласно примеру 1, растворенного в 50 мл метанола. После 18 часов при комнатной температуре реакционную смесь выпаривают досуха. Остаток переносят в этилацетат и 0,5 н. раствор соляной кислоты. Органическую фазу отделяют после отстаивания, сушат над безводным сульфатом натрия и концентрируют. Остаток очищают хроматографией на колонке с силикагелем с использованием элюента: смесь 92/8 (об./об.) дихлорметан/метанол, с получением белых кристаллов; т.пл.=198oC; выход=91%.
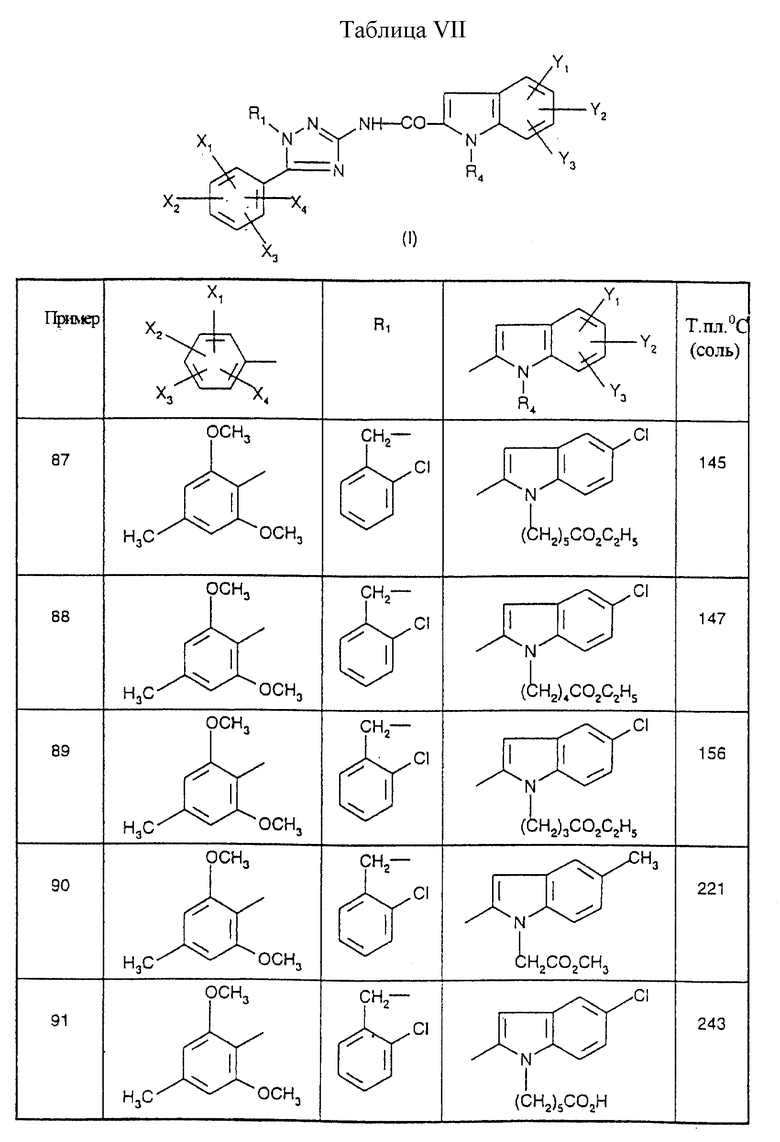
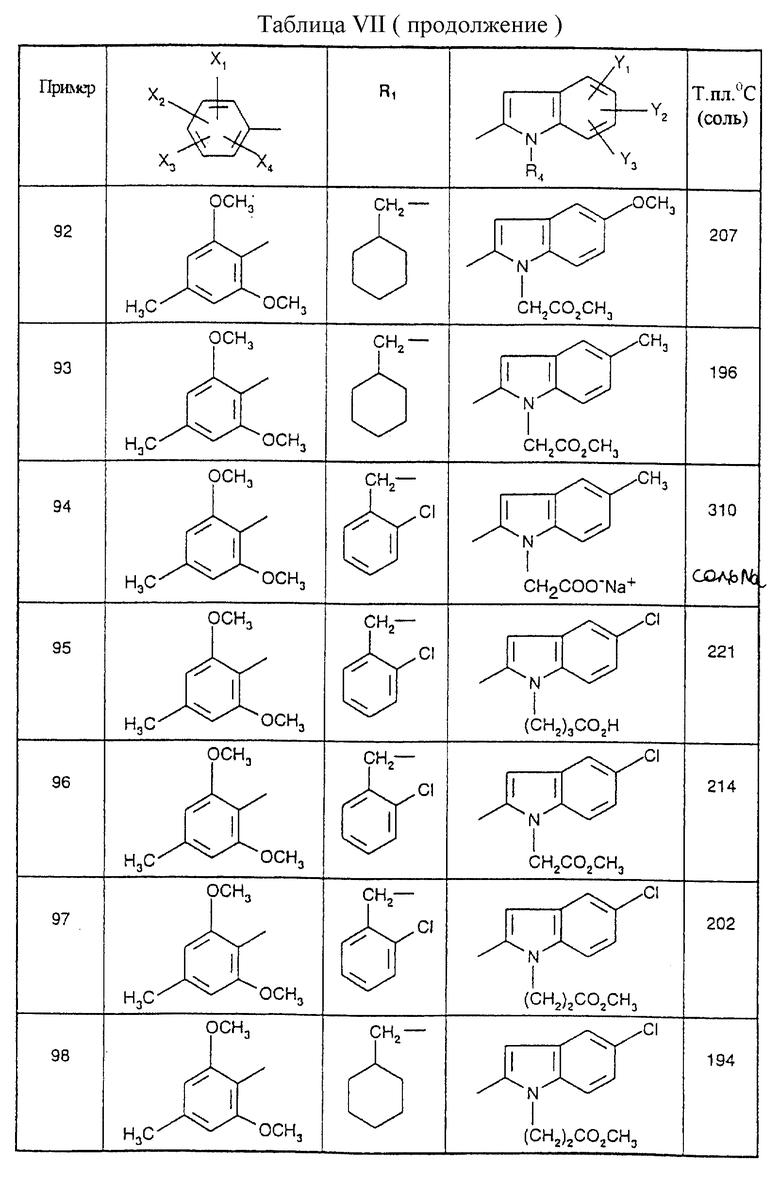
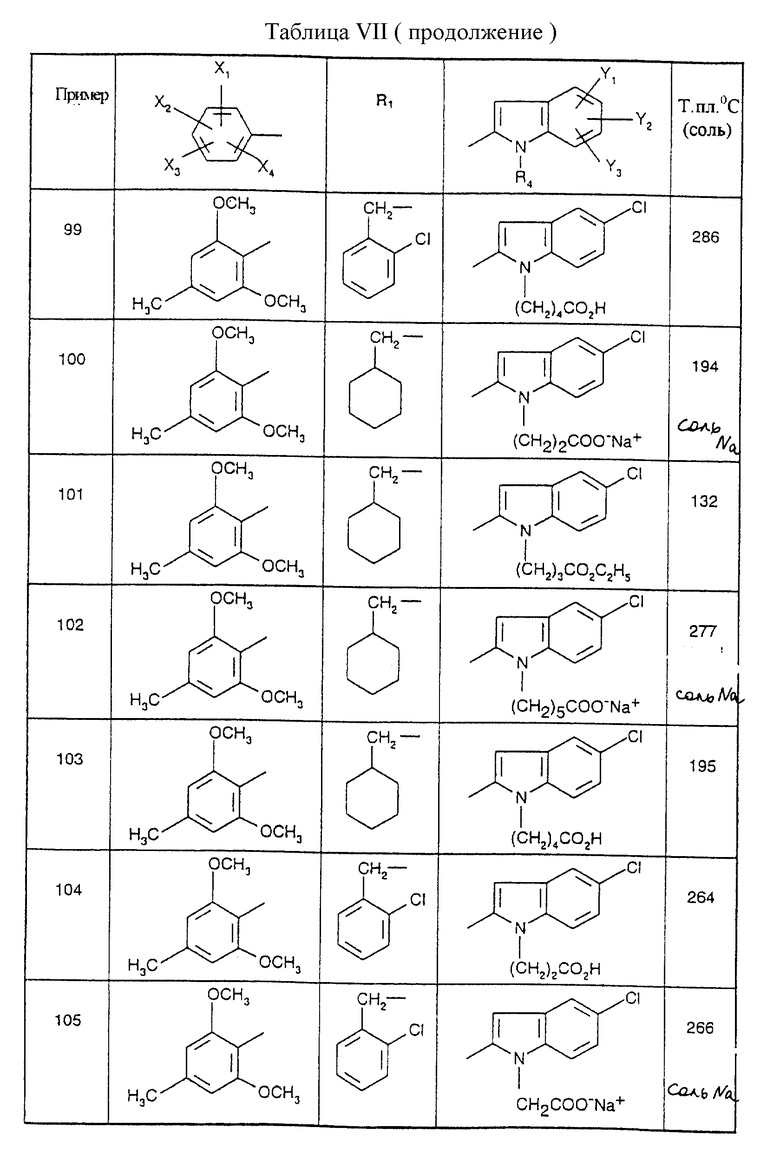
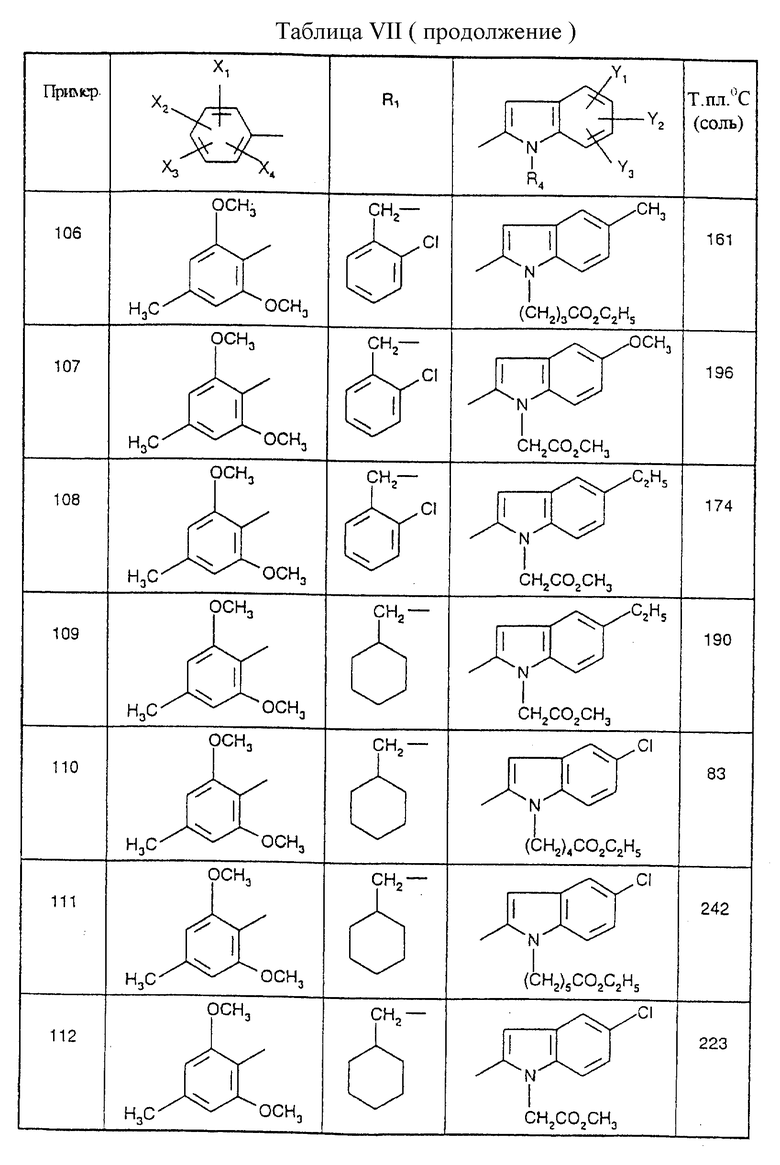
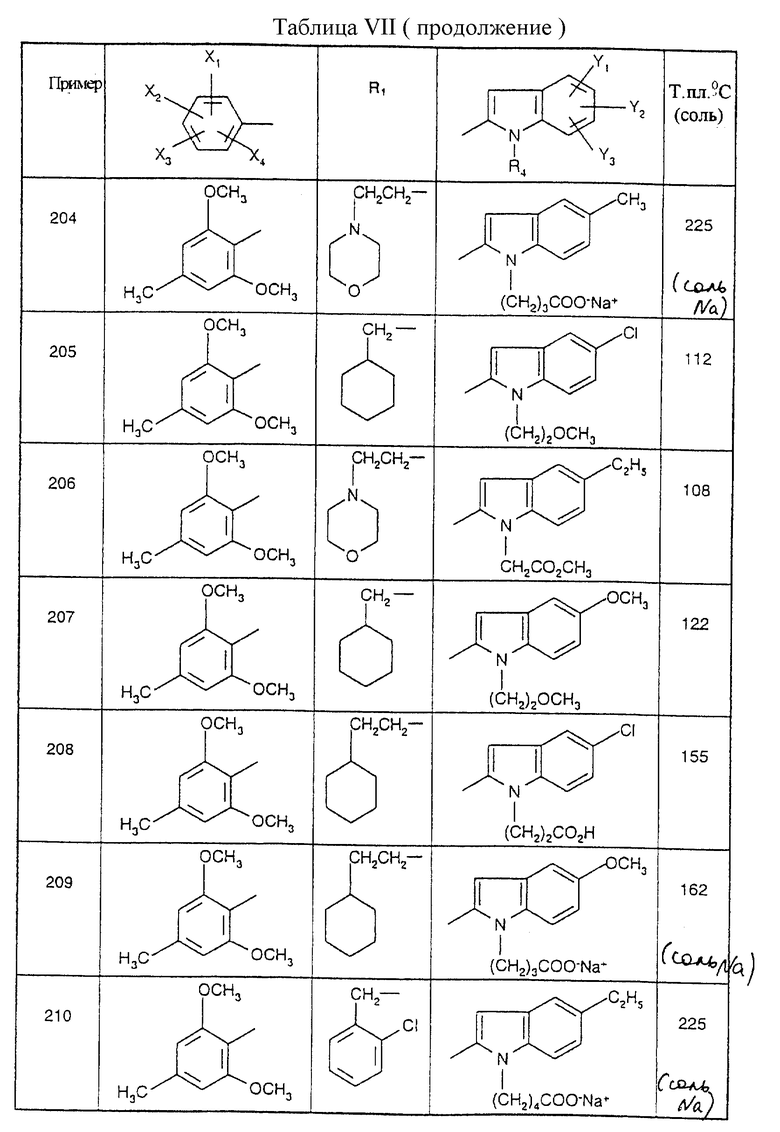
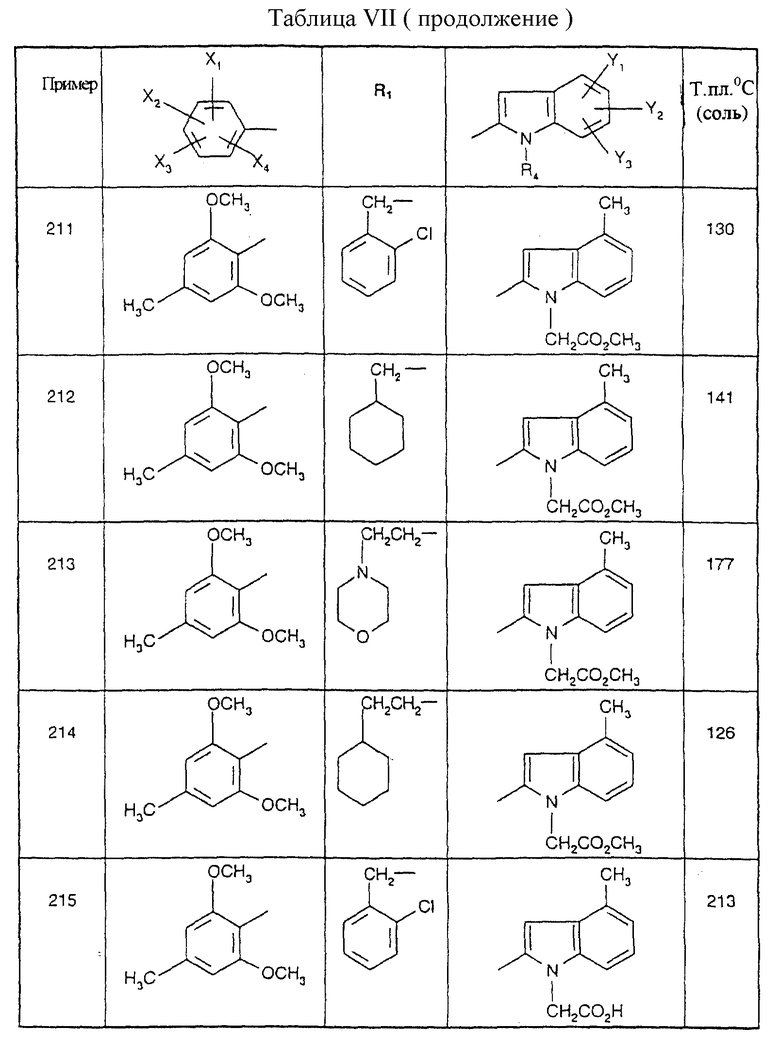
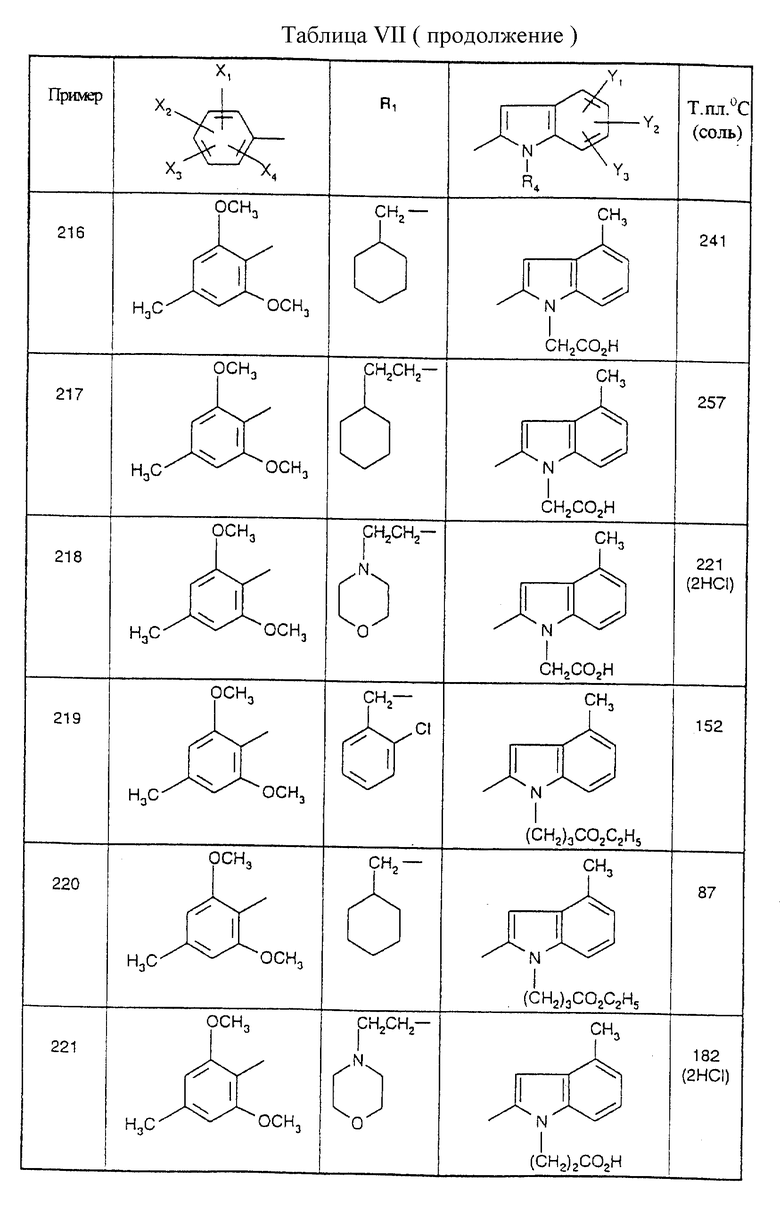
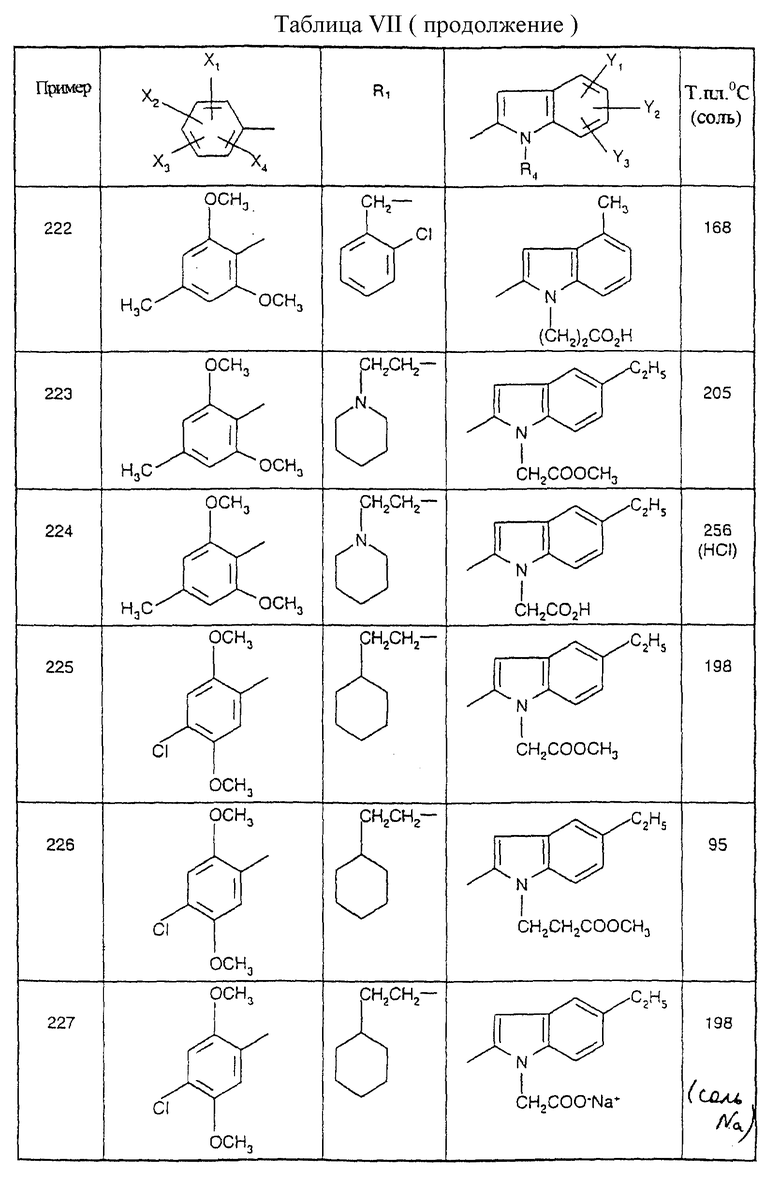
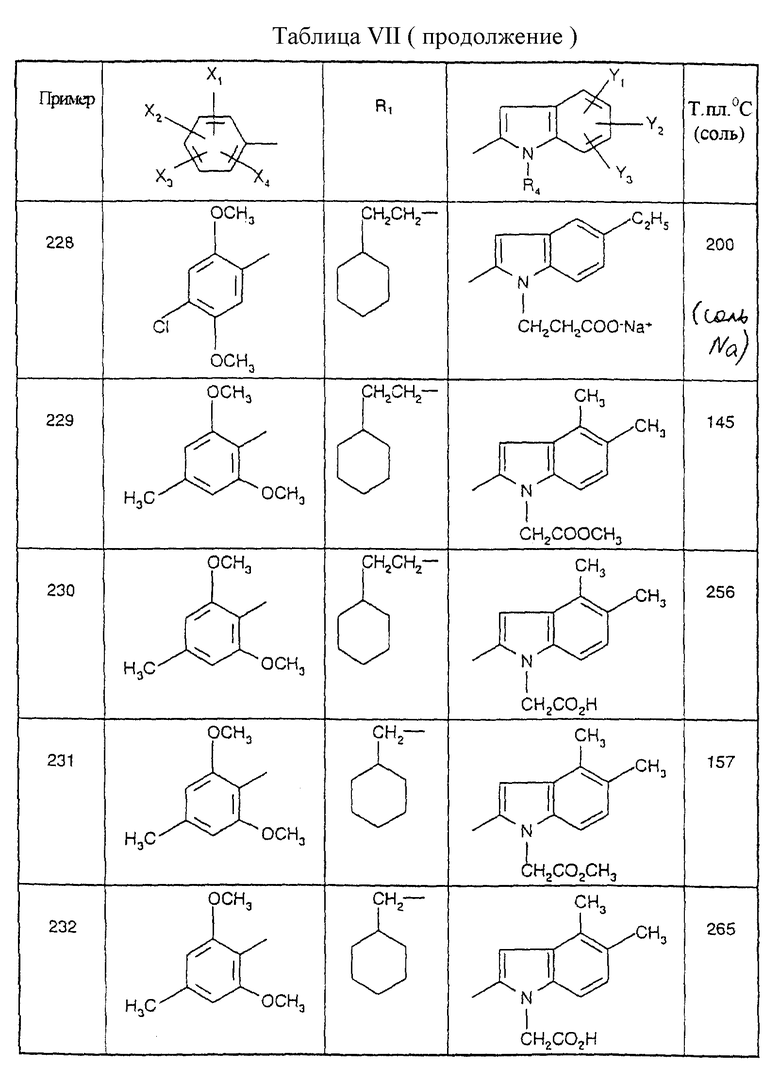
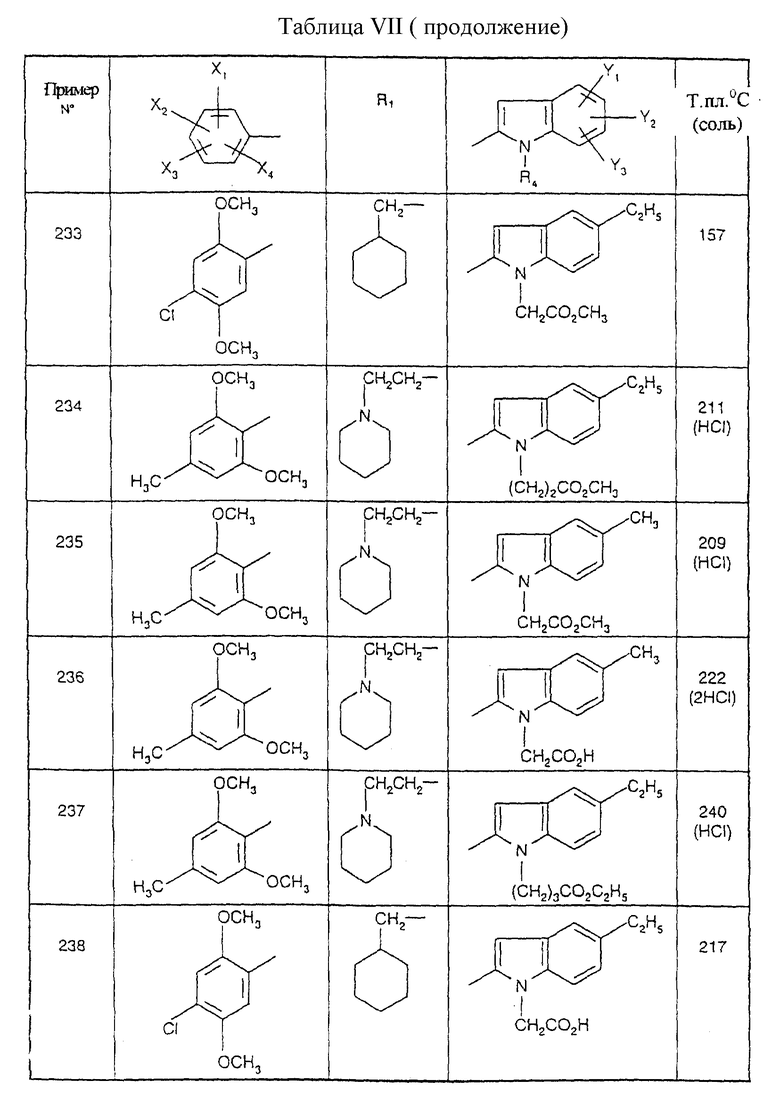
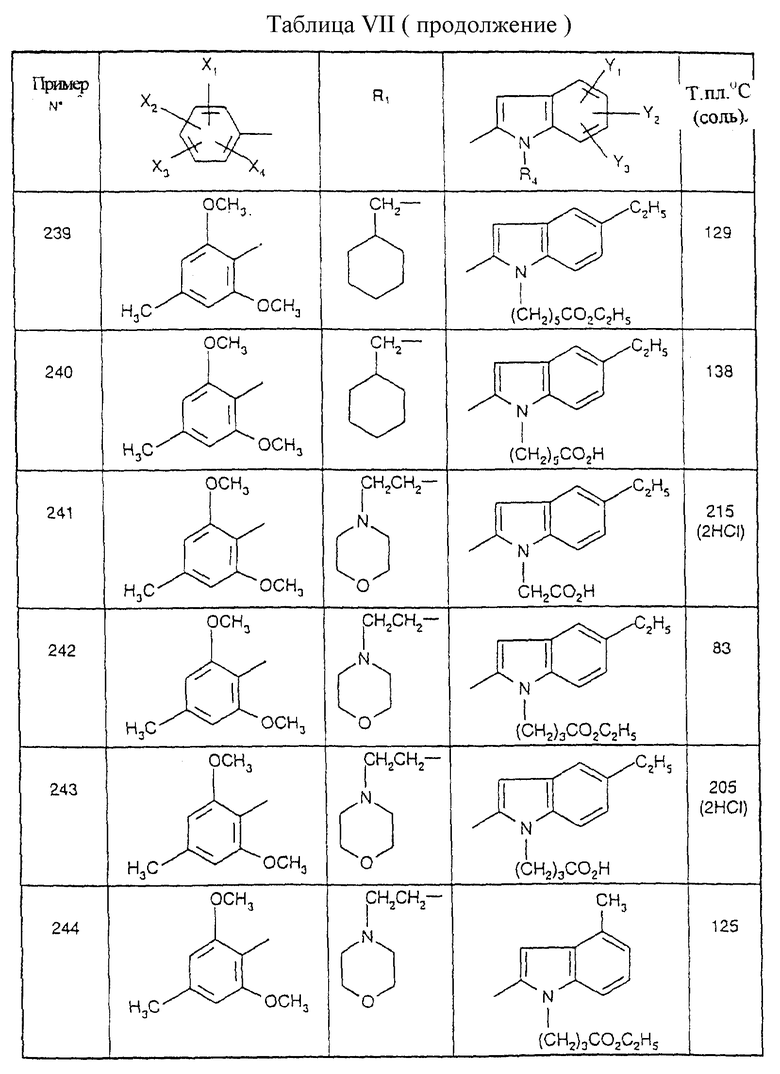
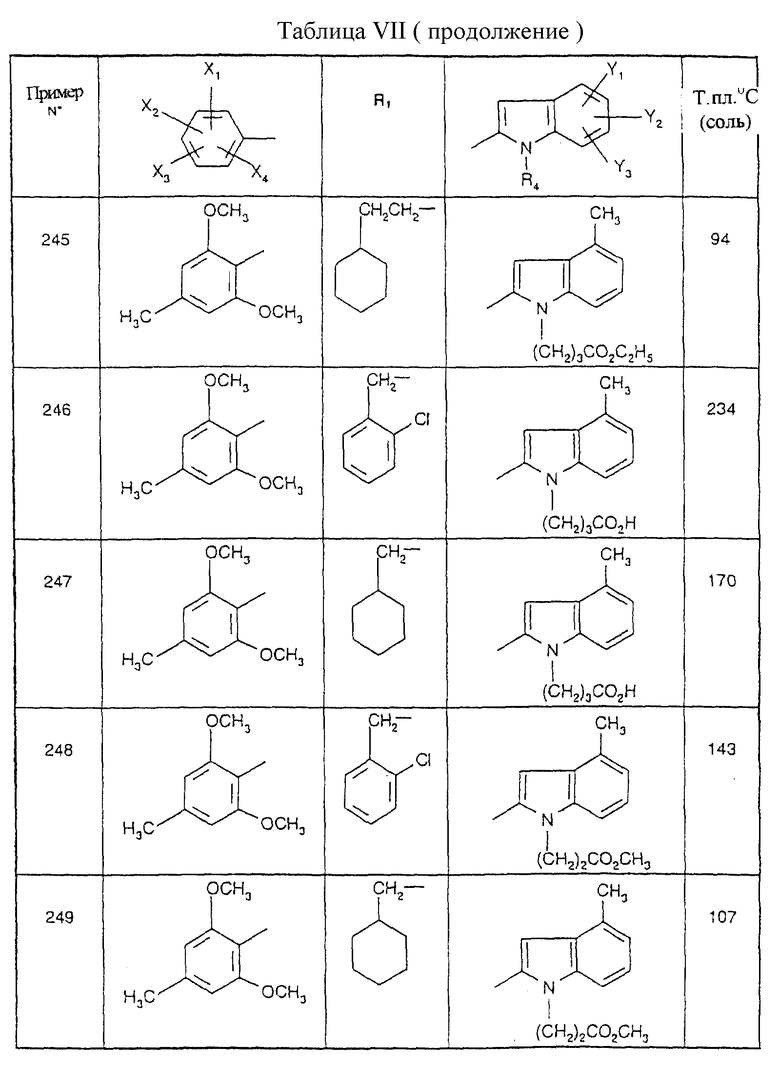
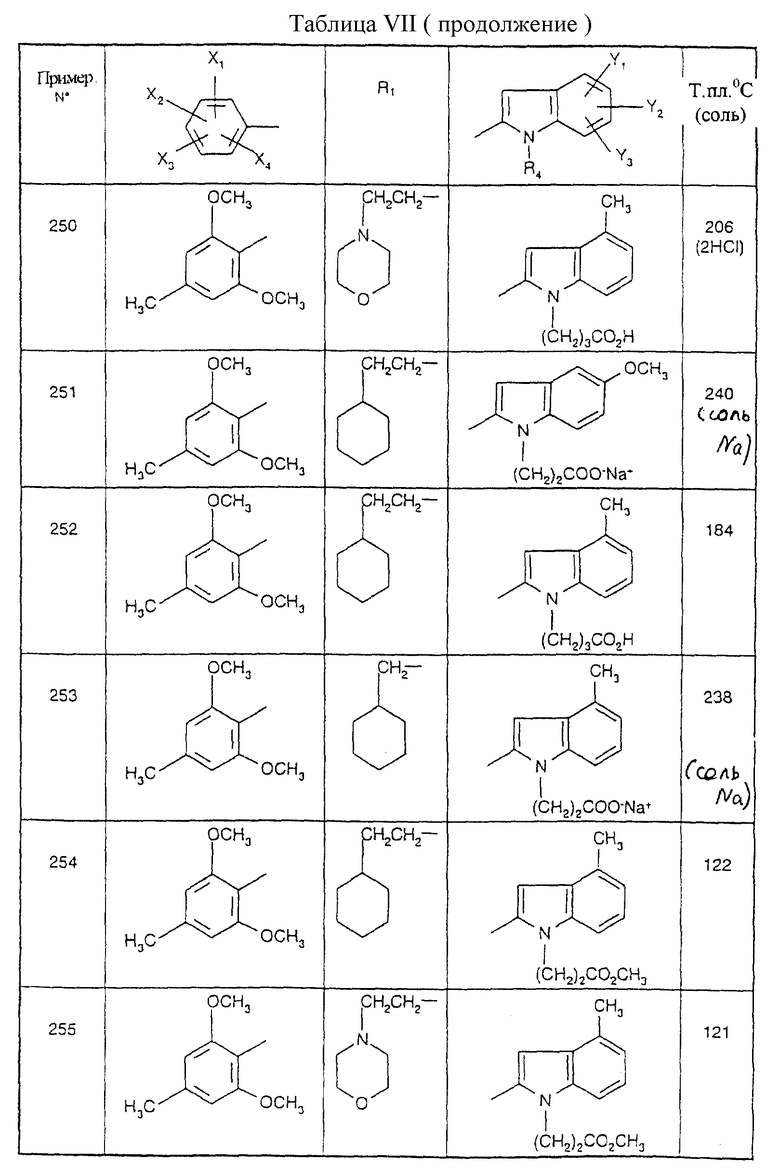
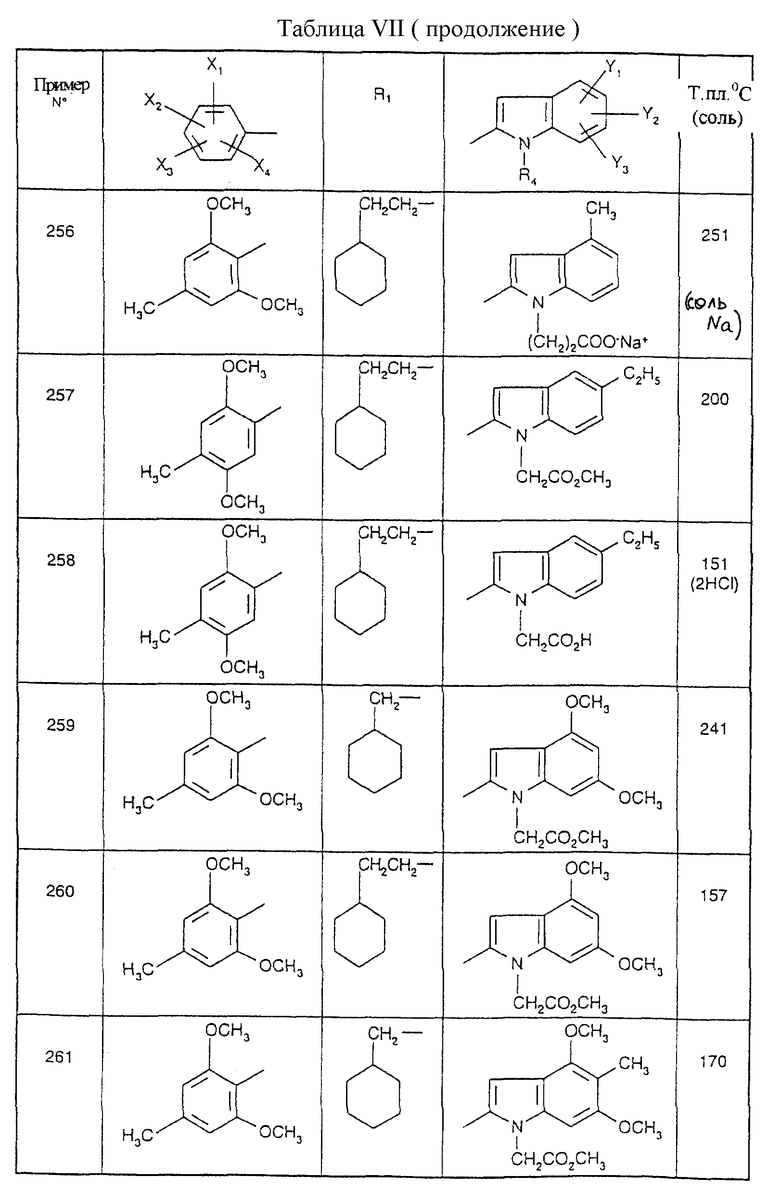
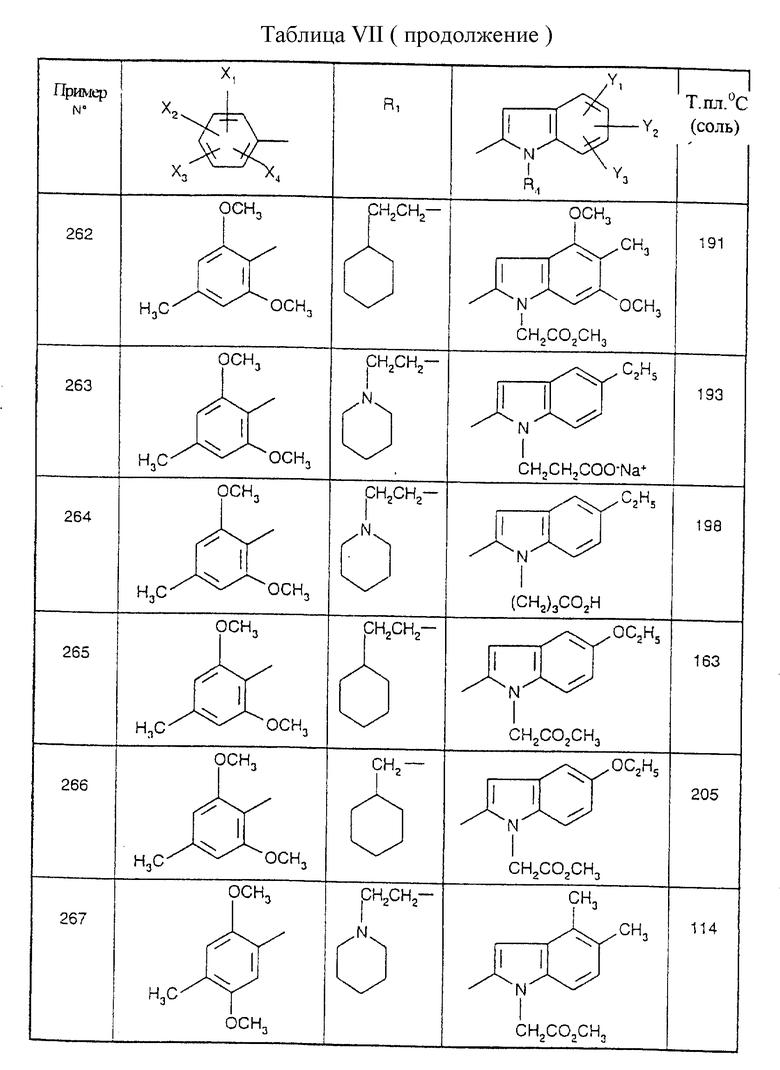
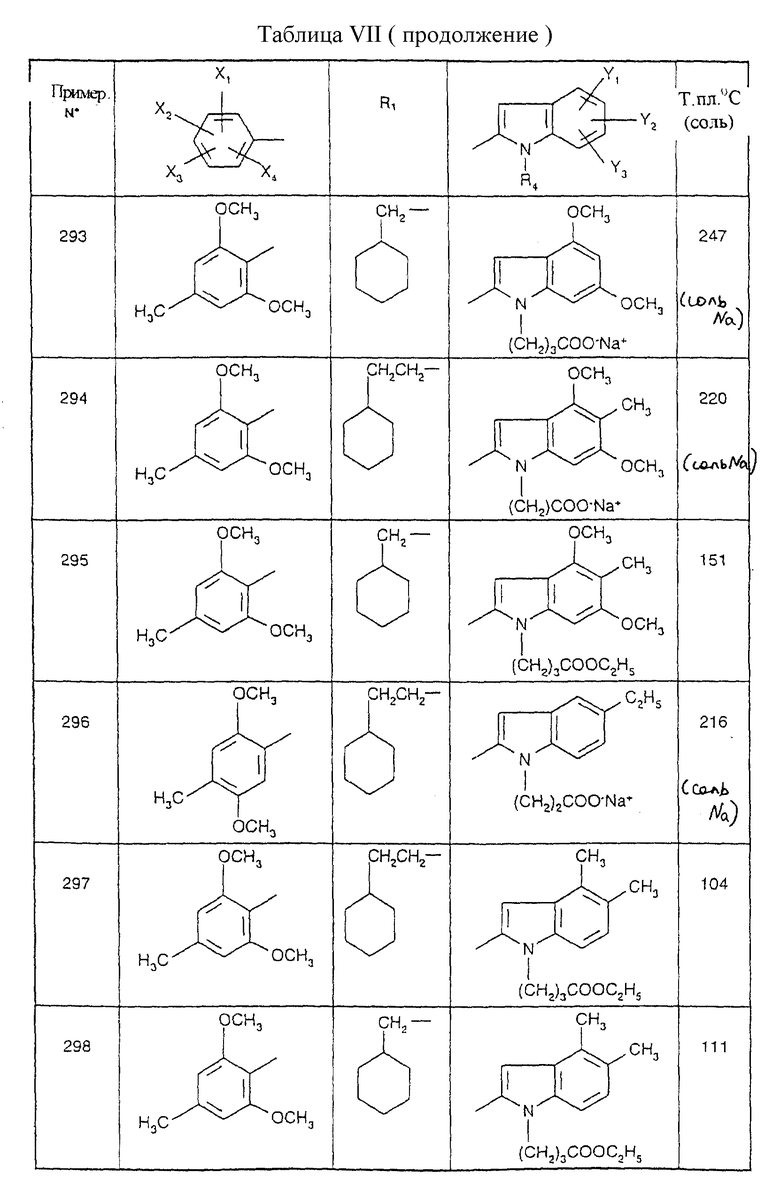
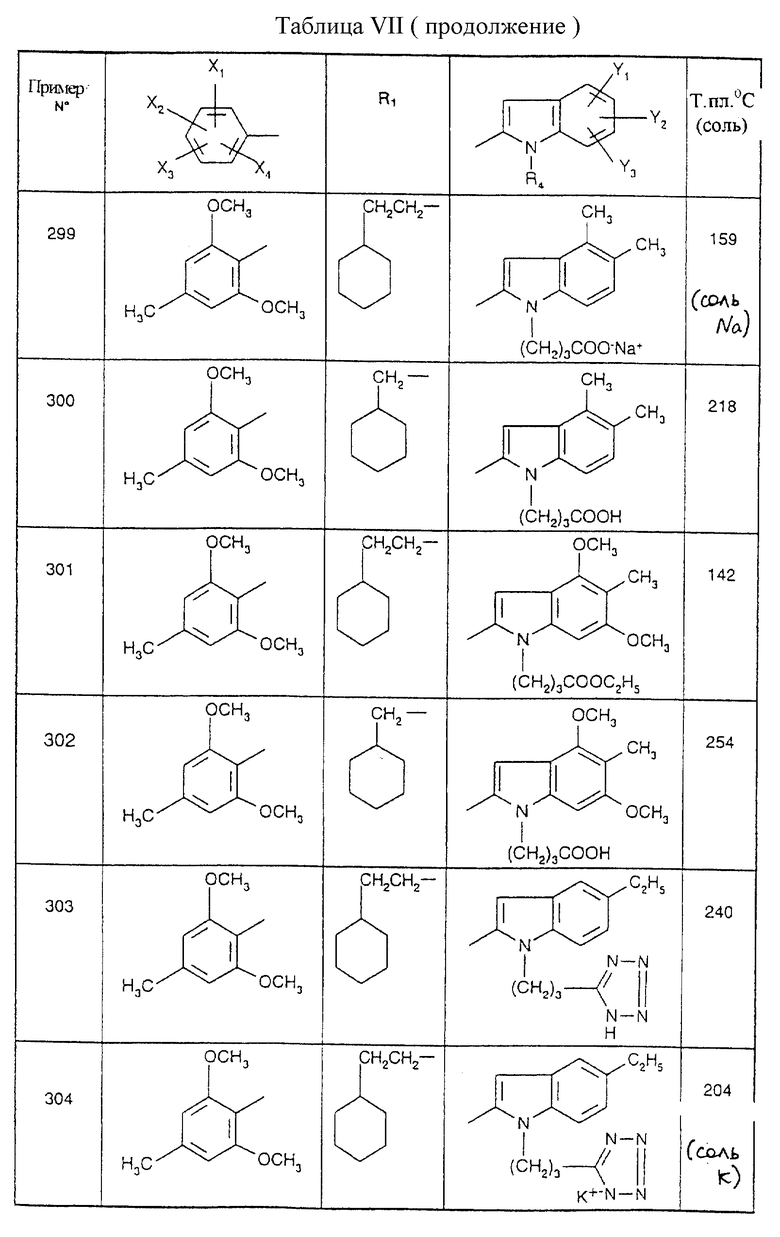
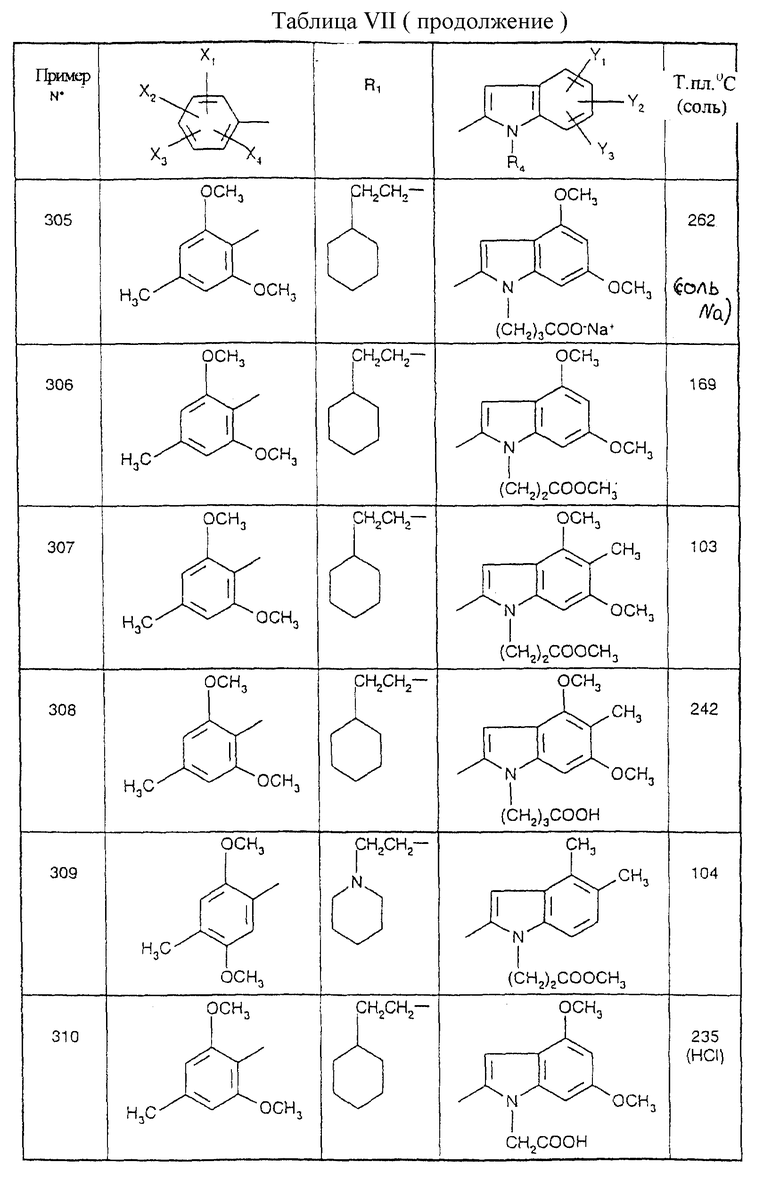
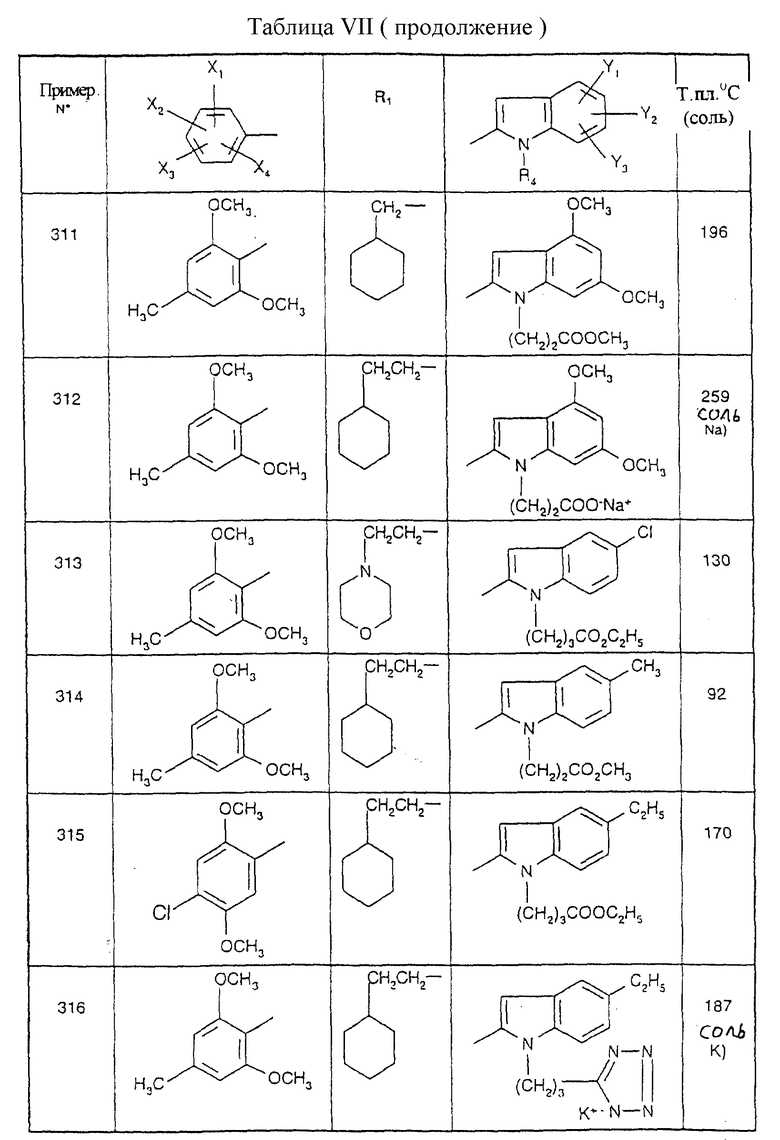
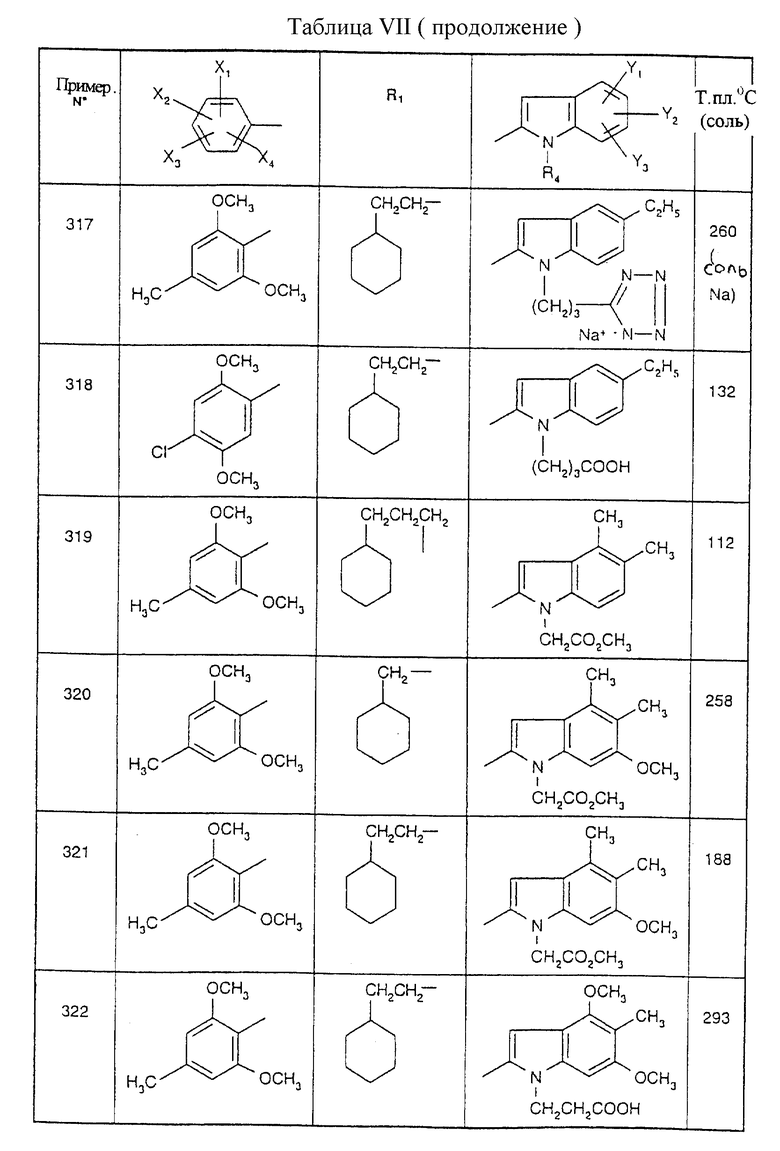
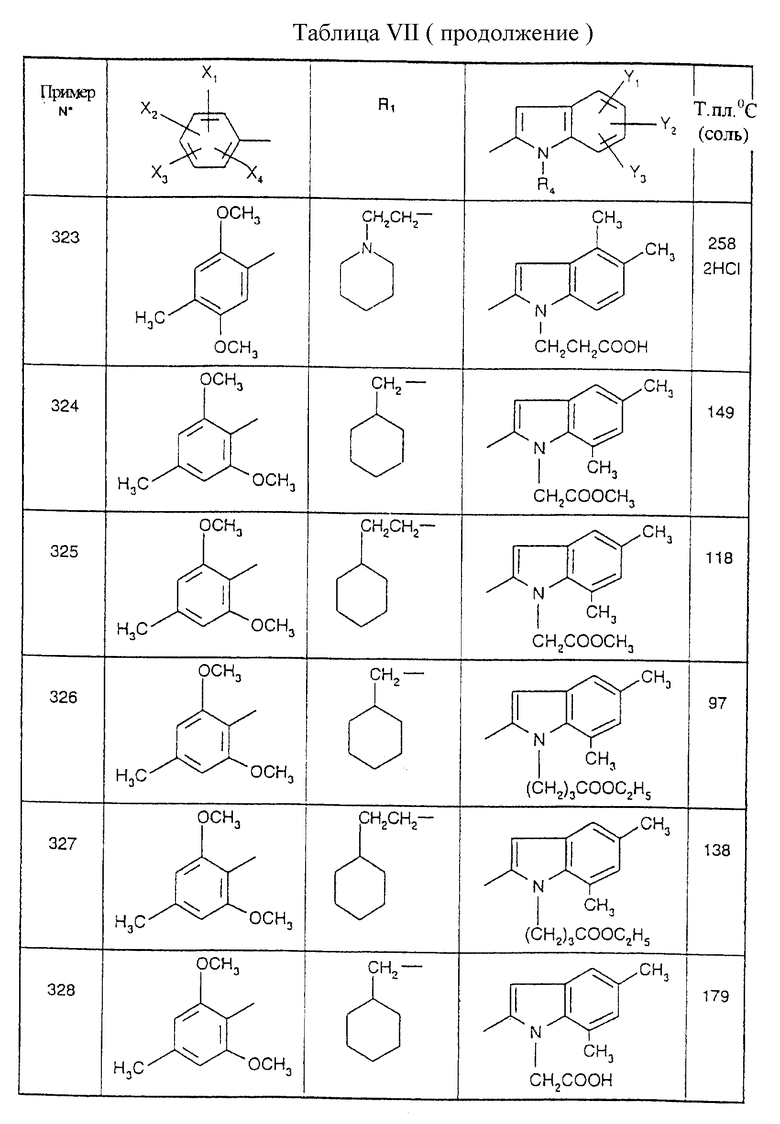
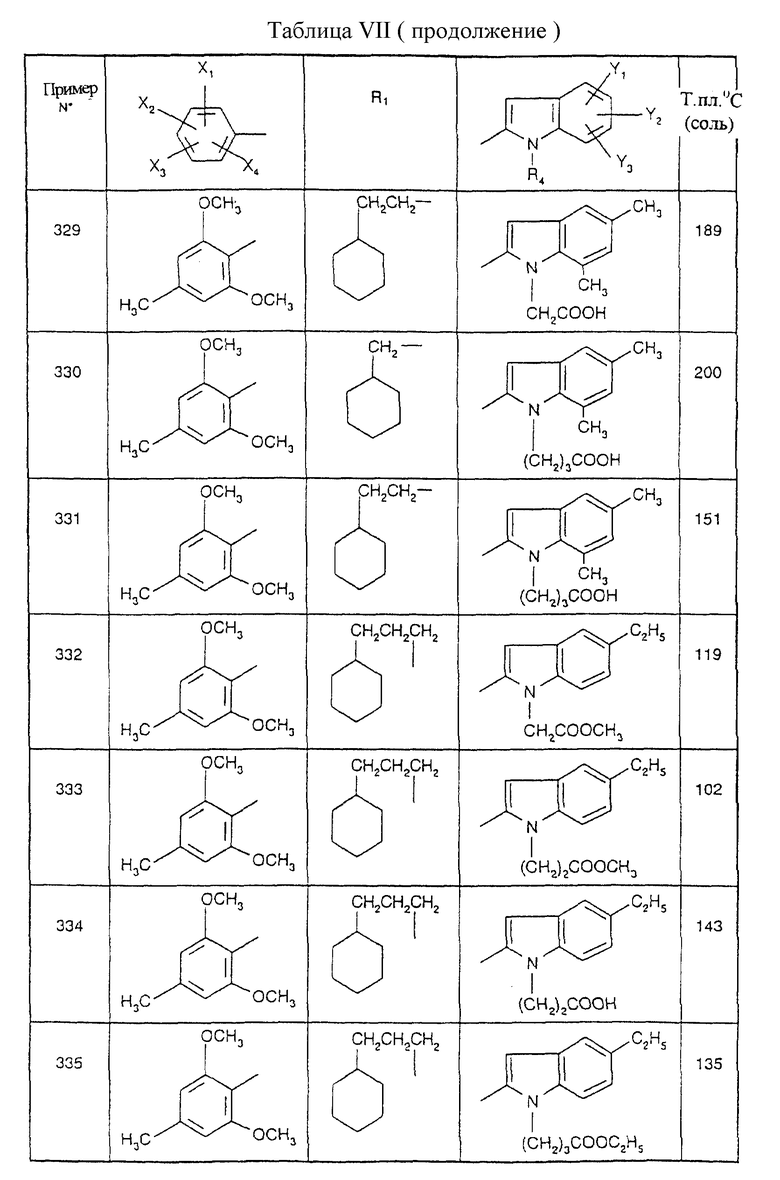
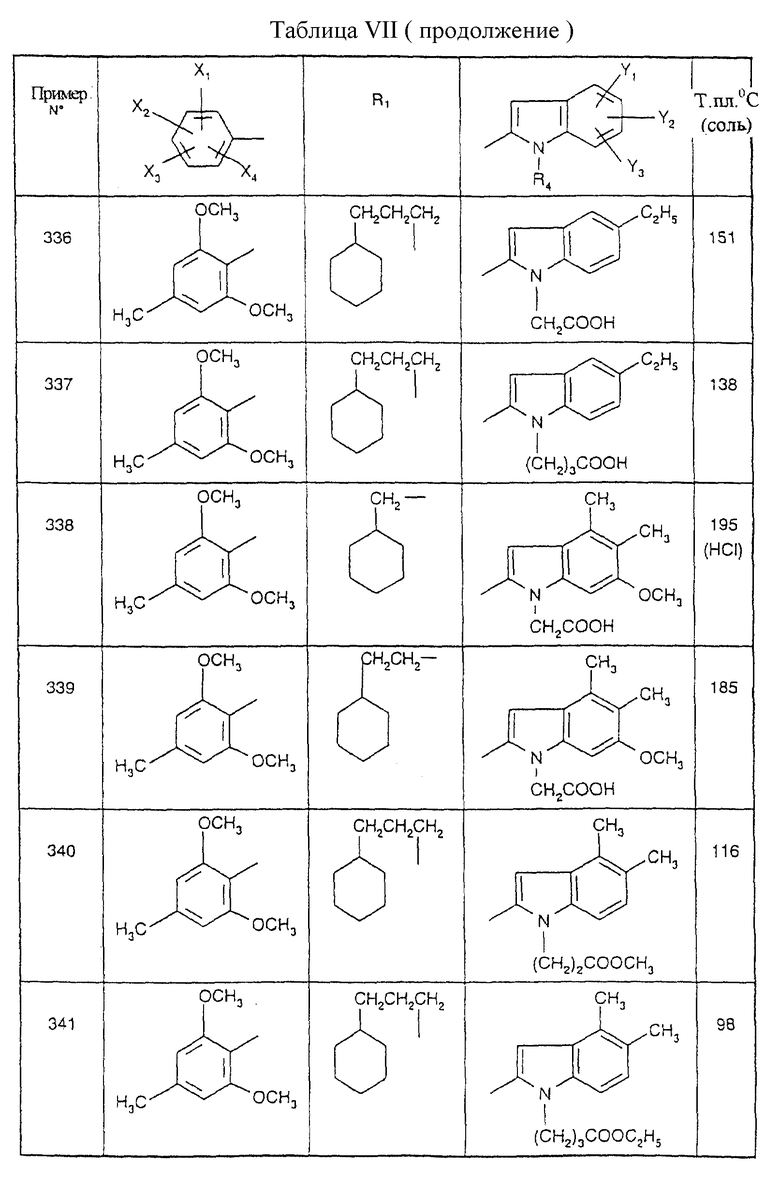
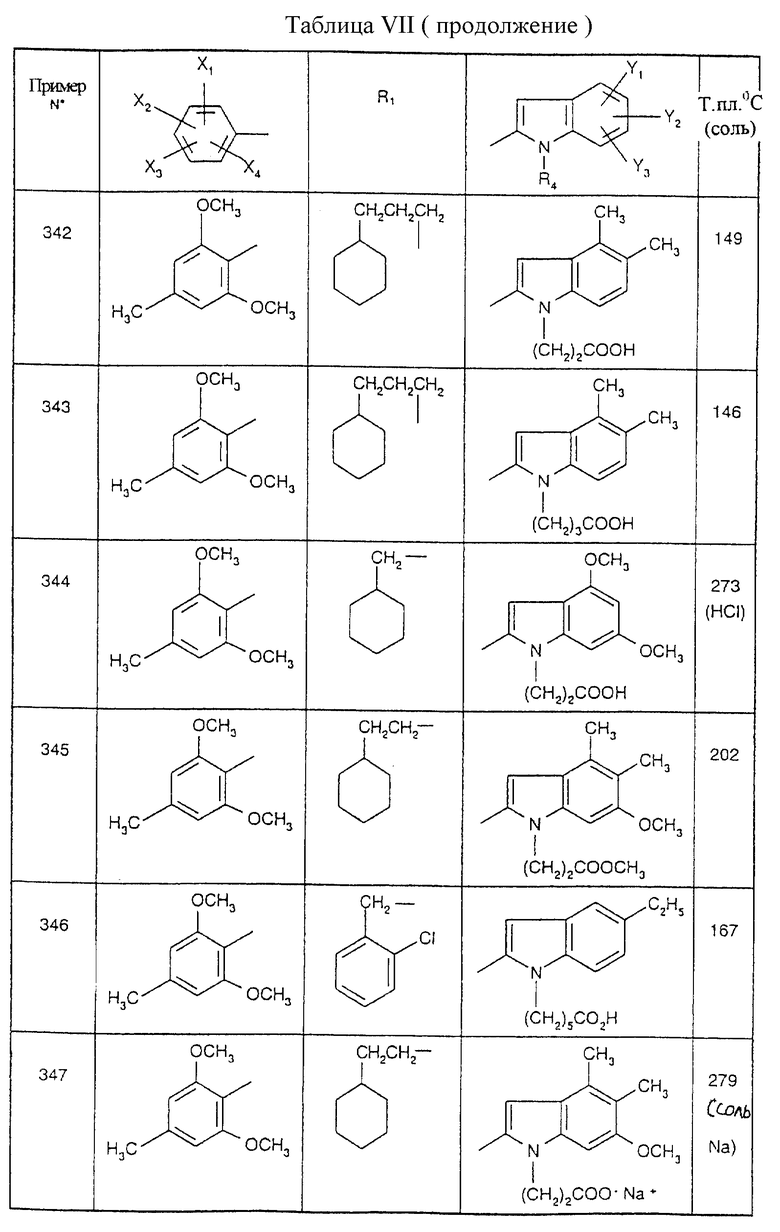
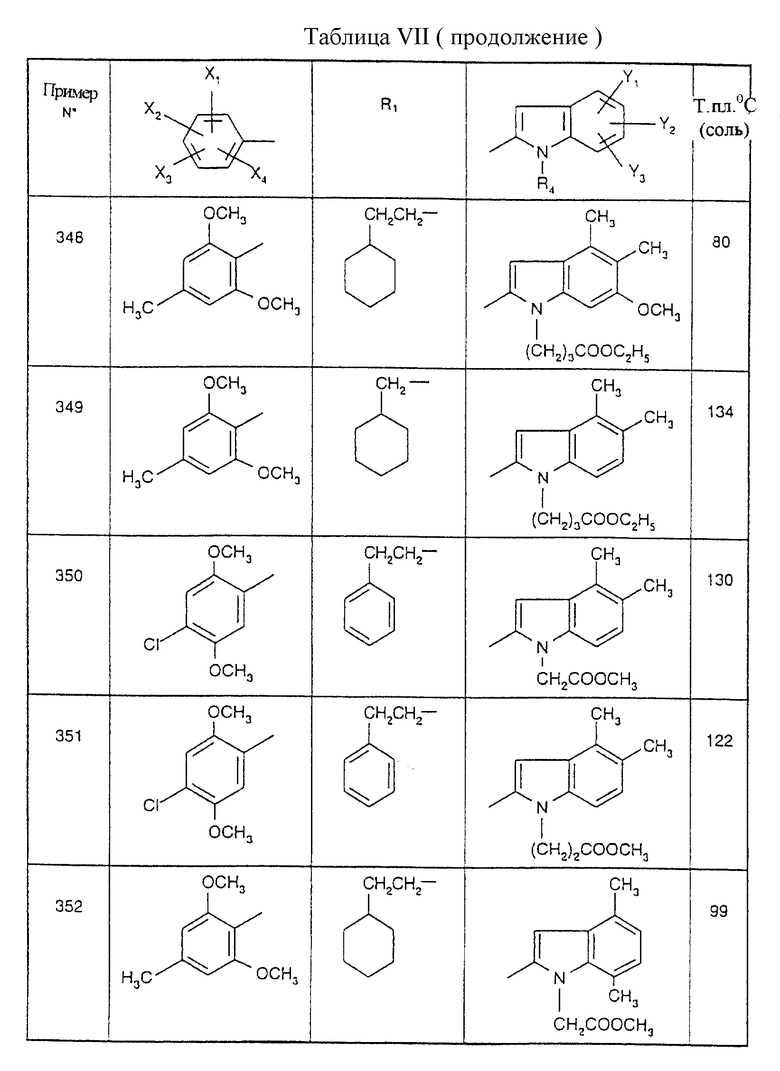
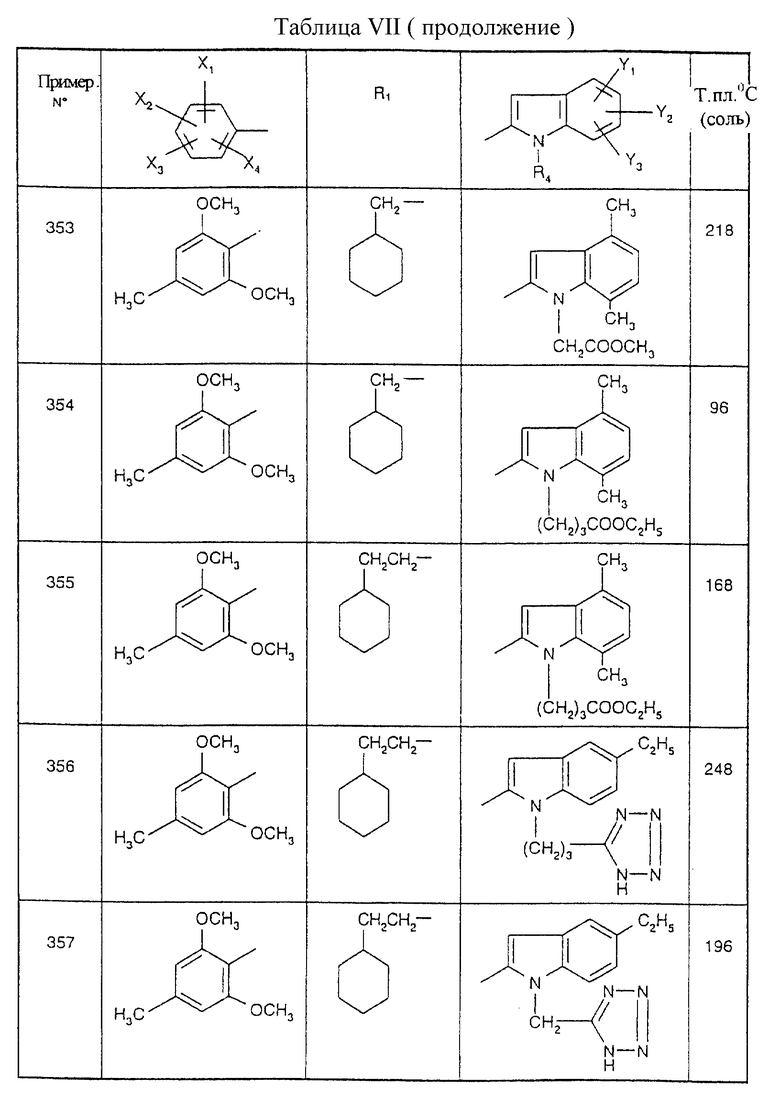
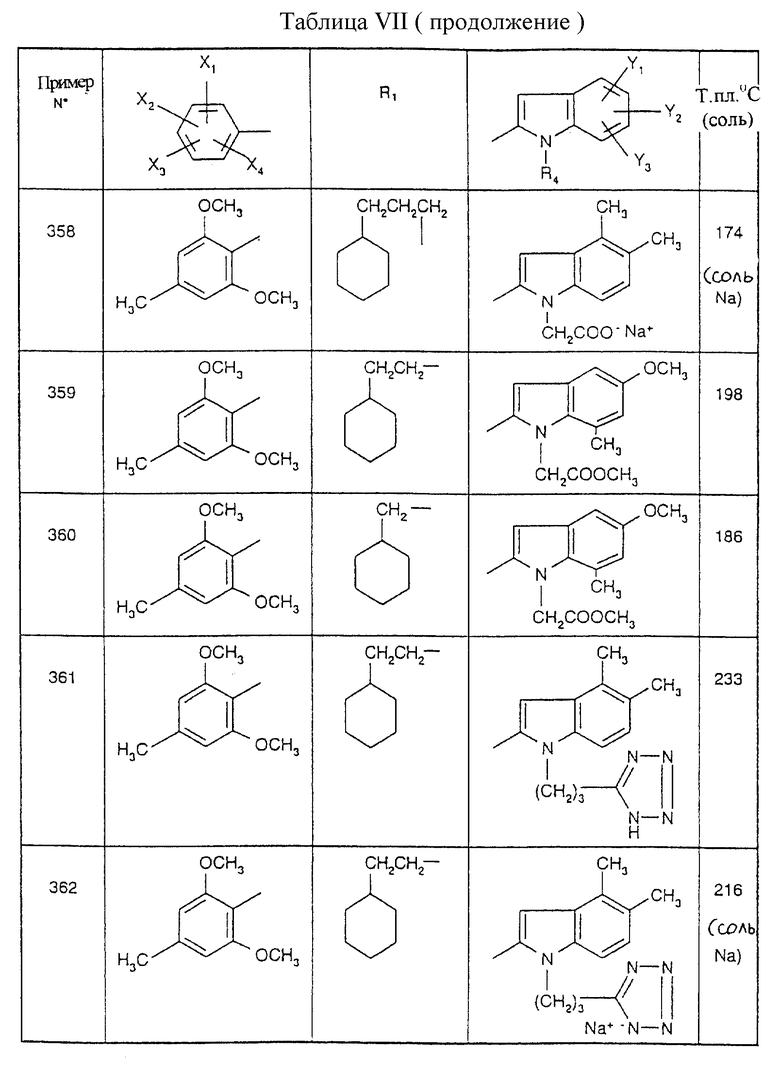
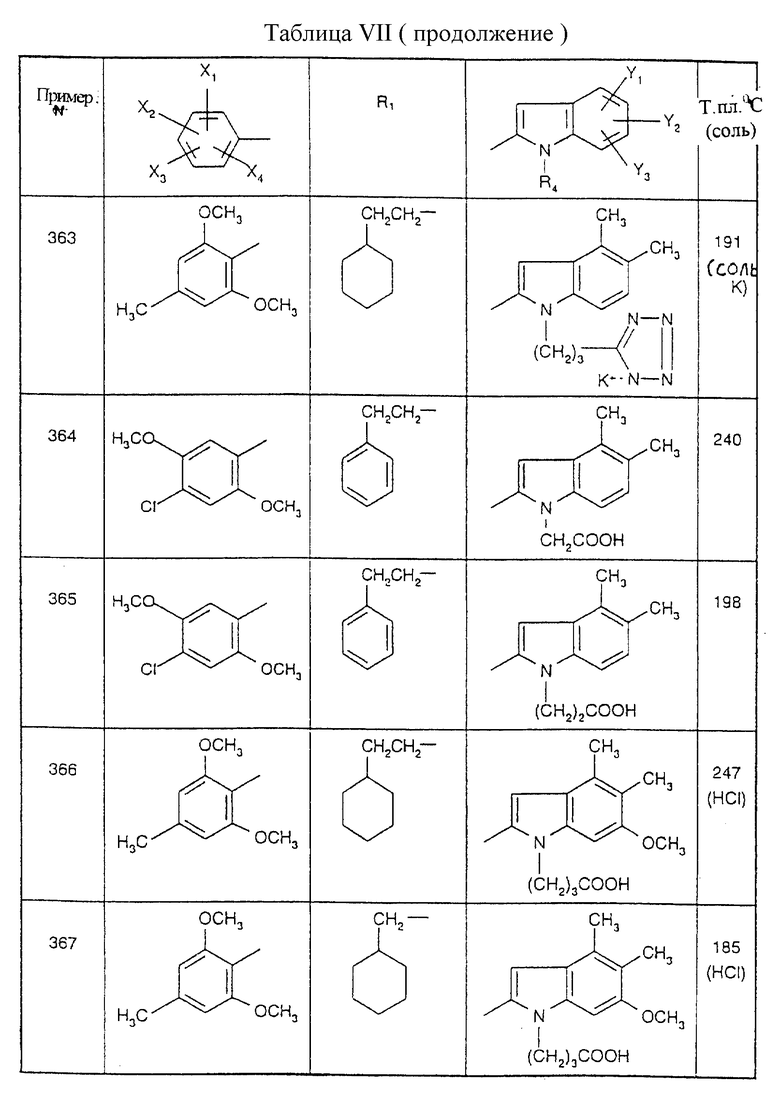
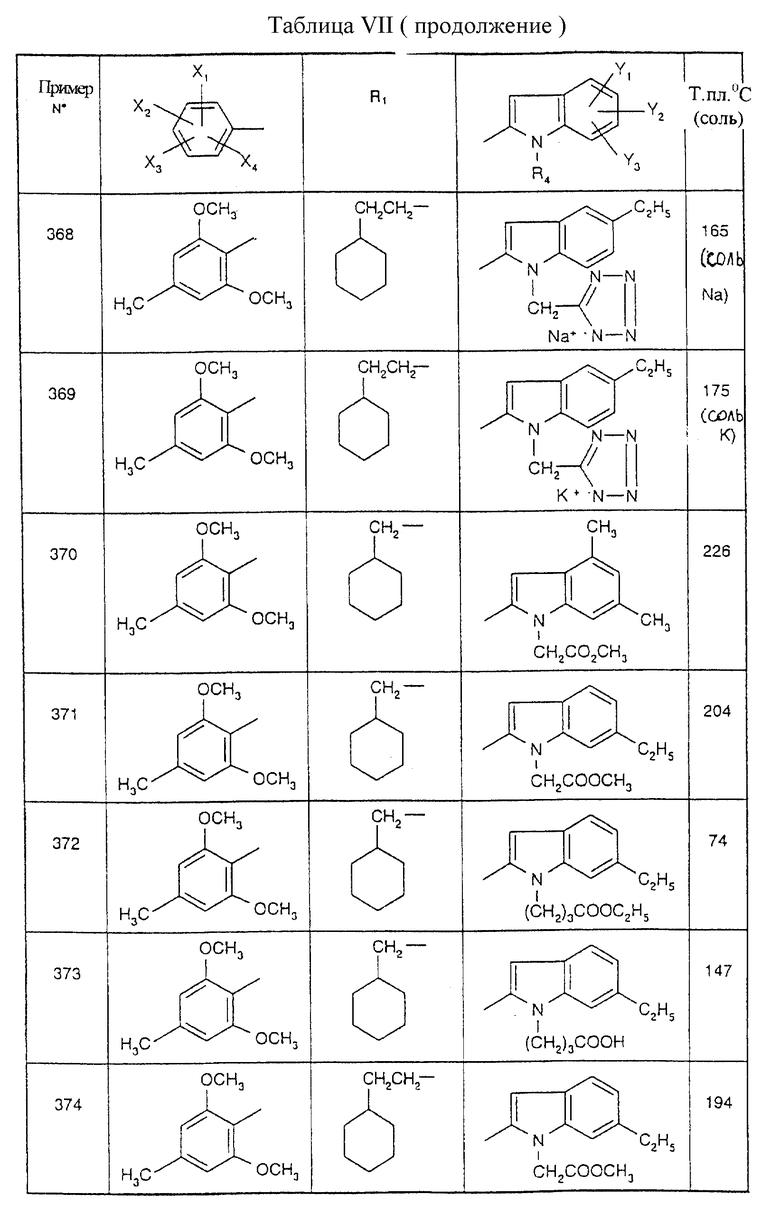
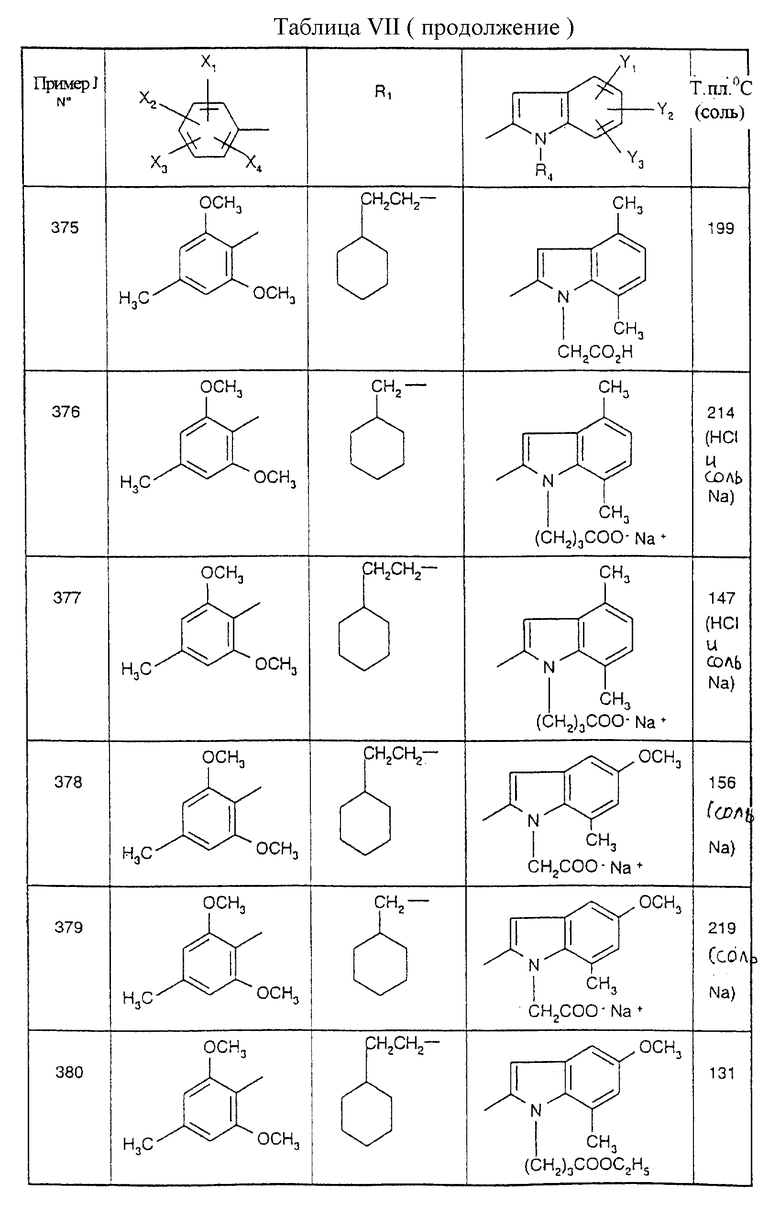
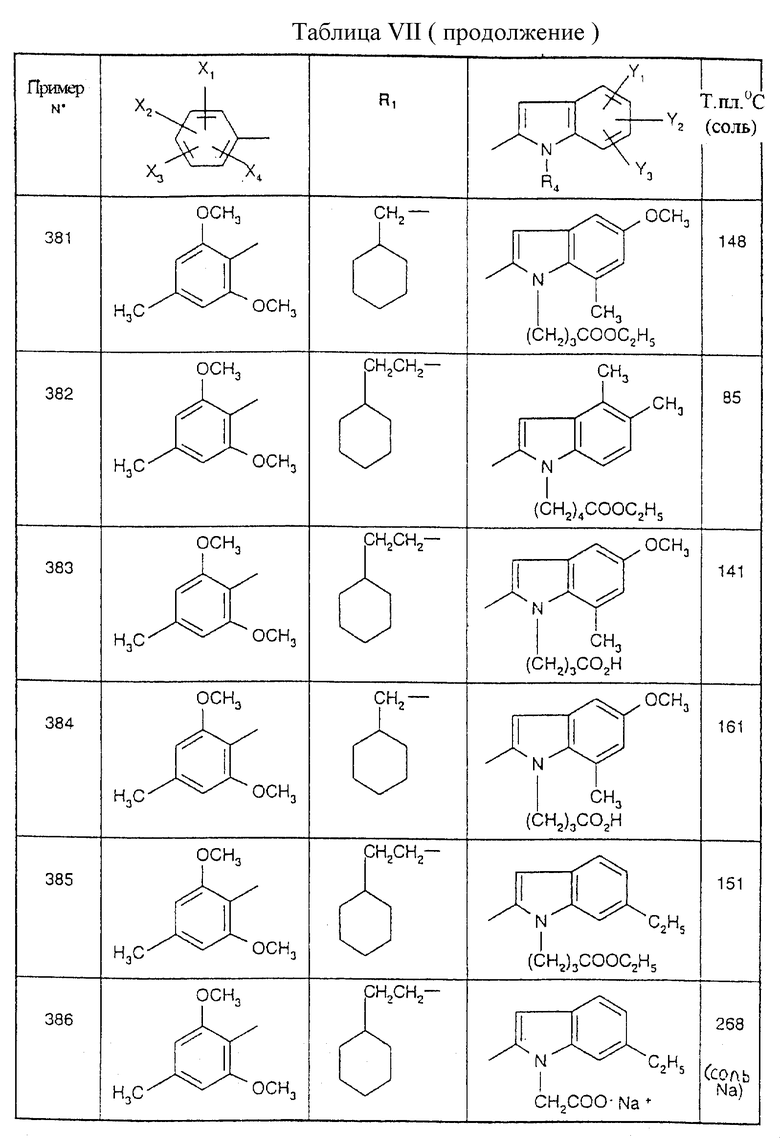
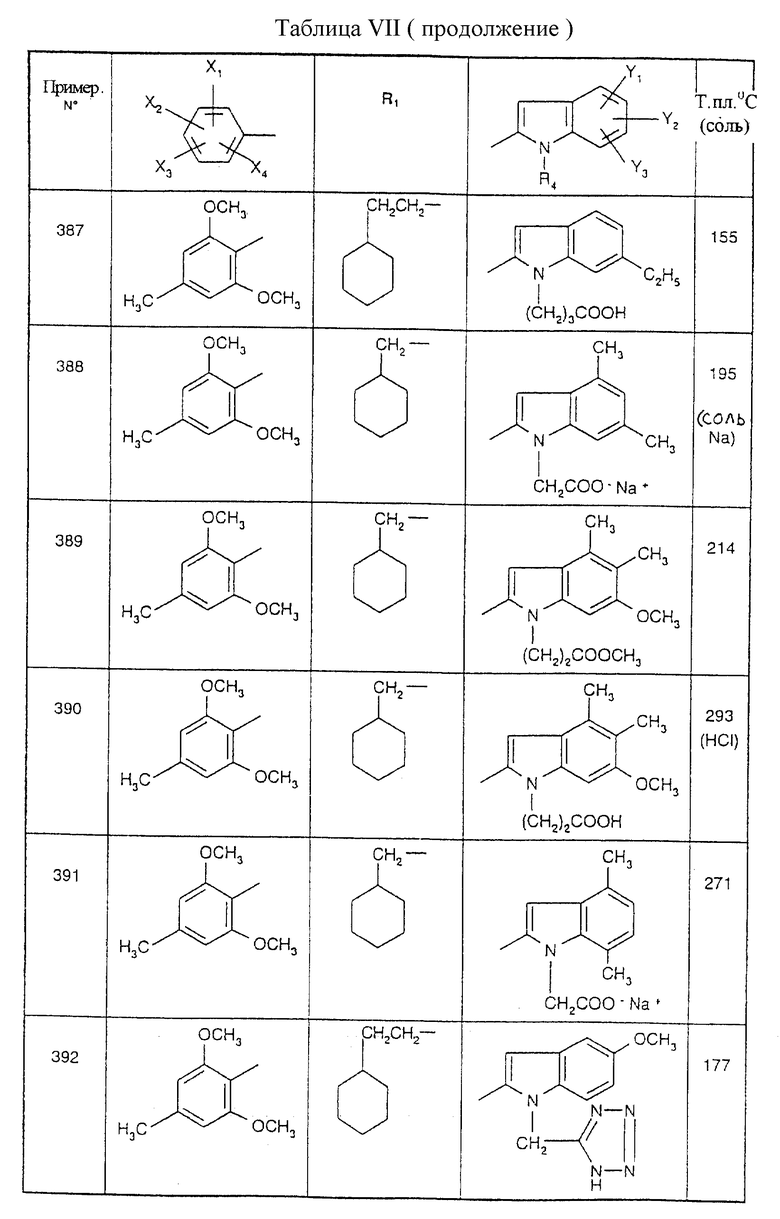
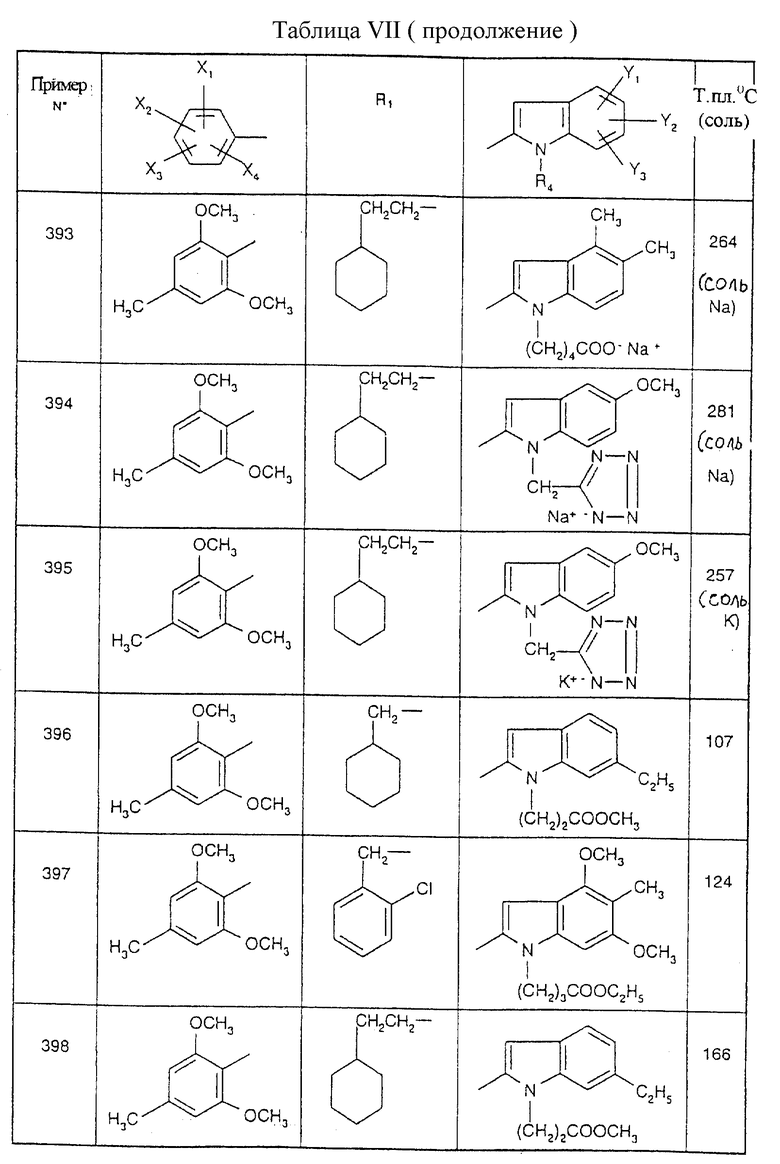
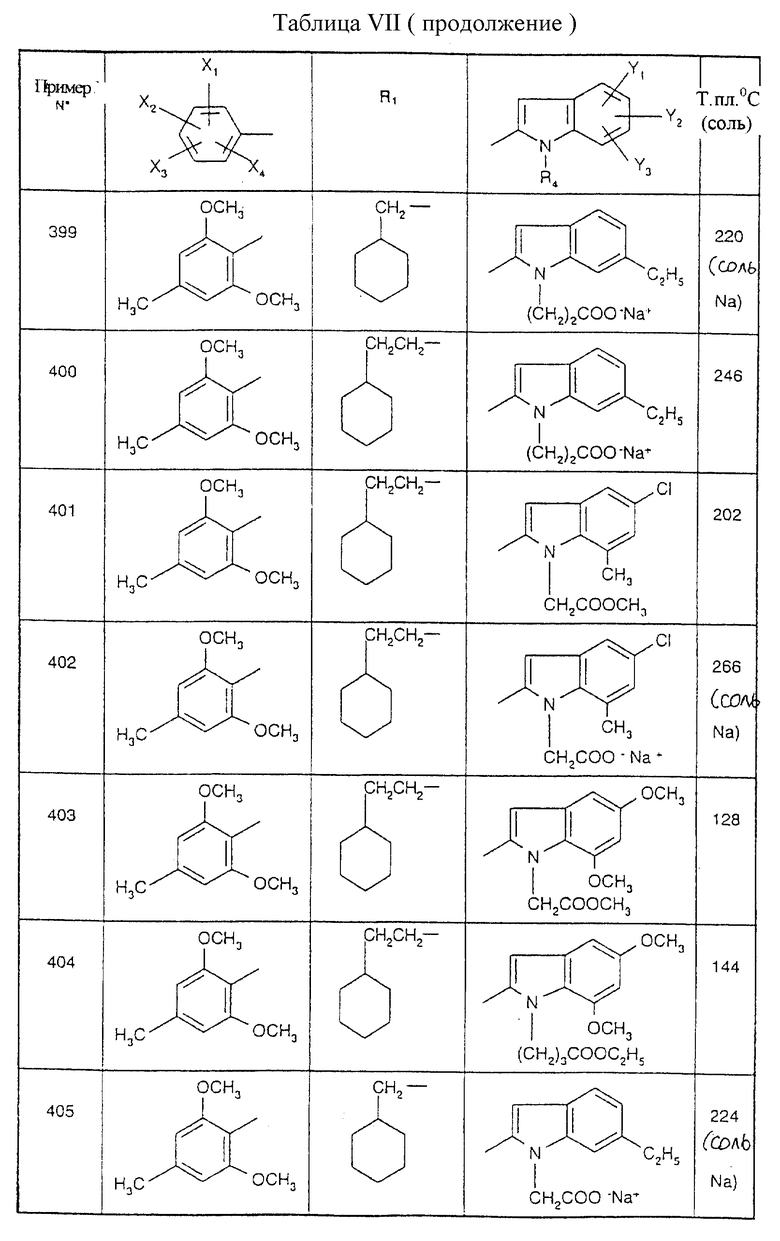
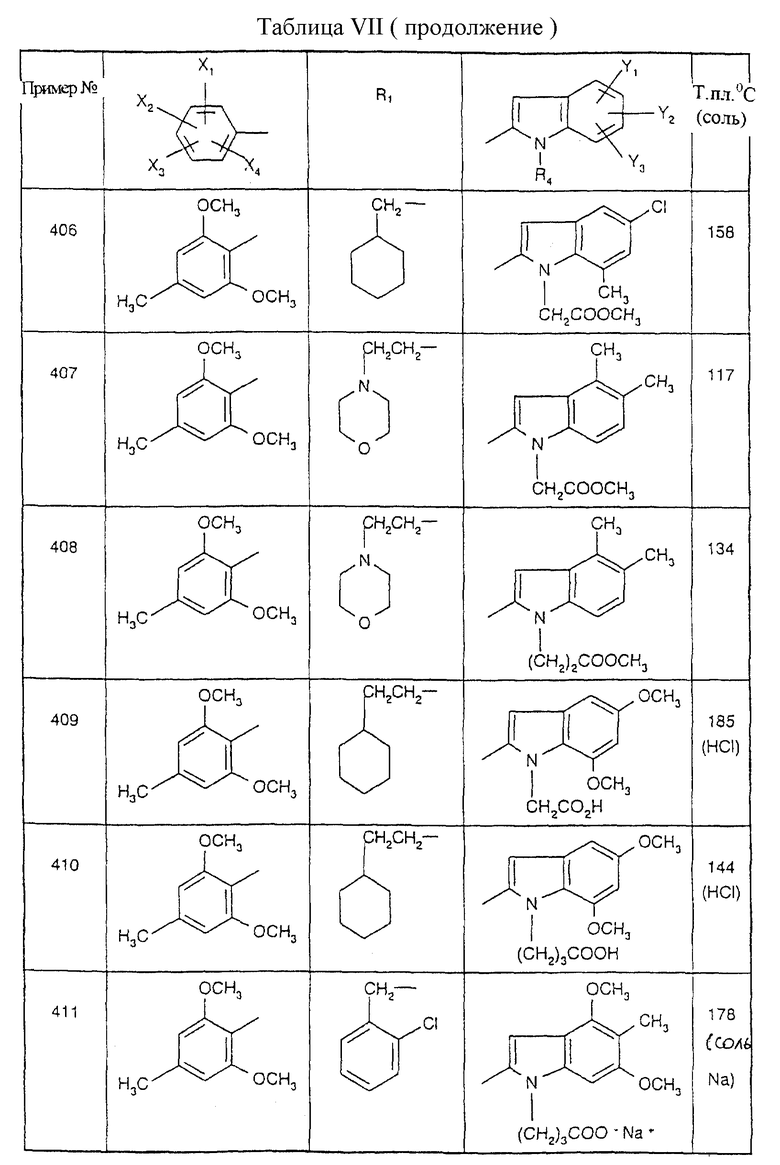
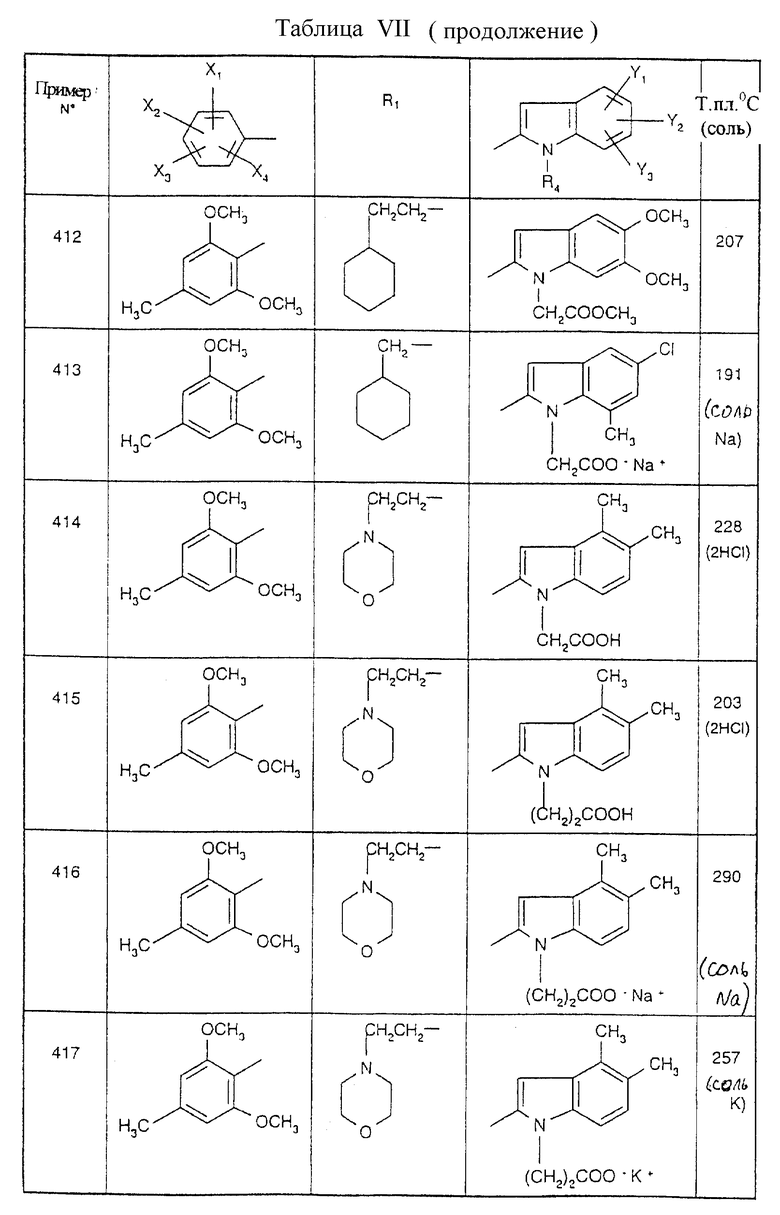
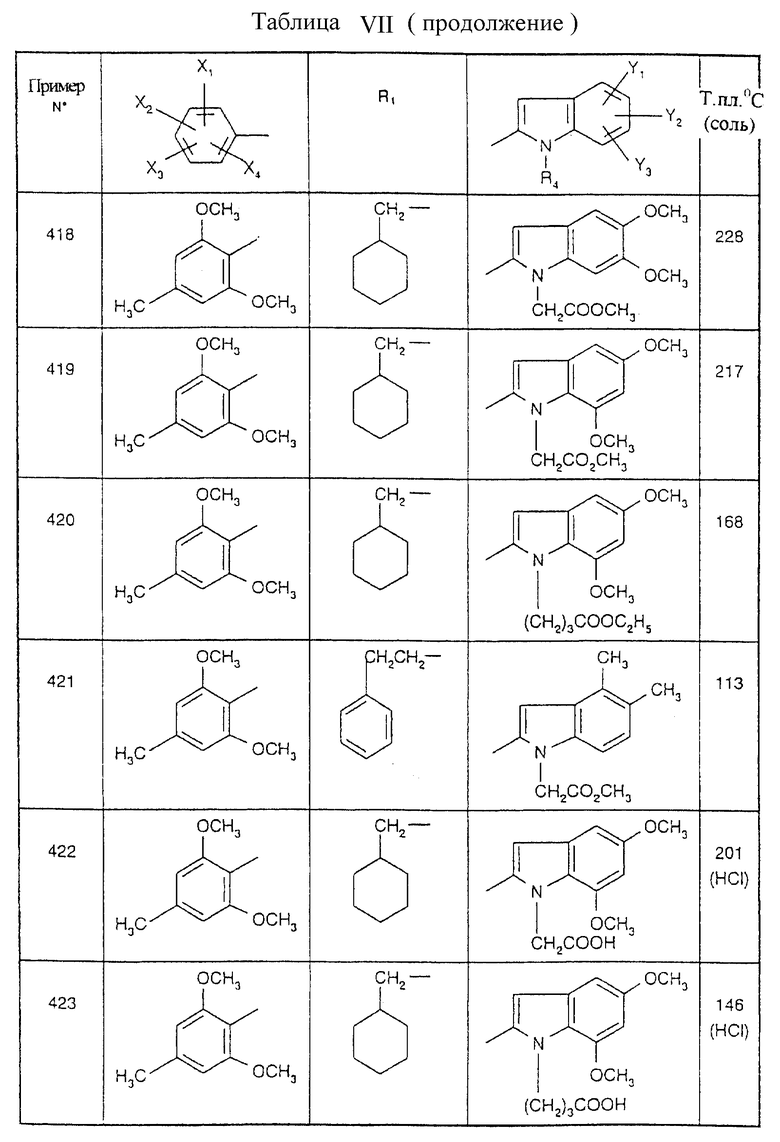
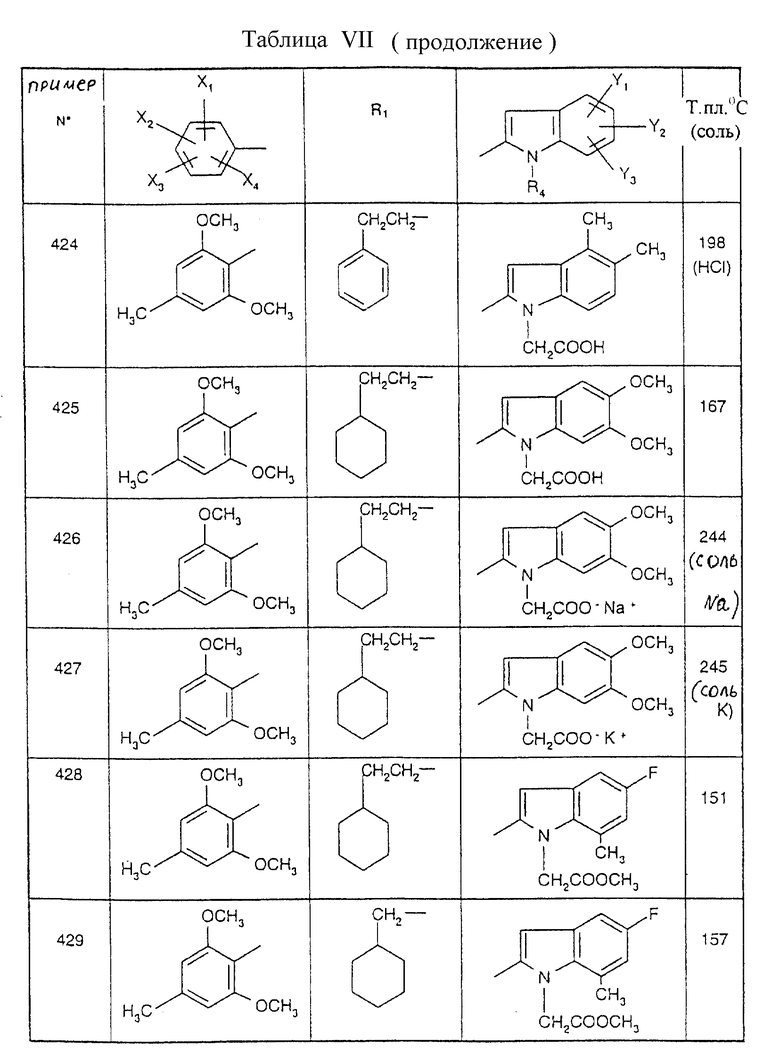
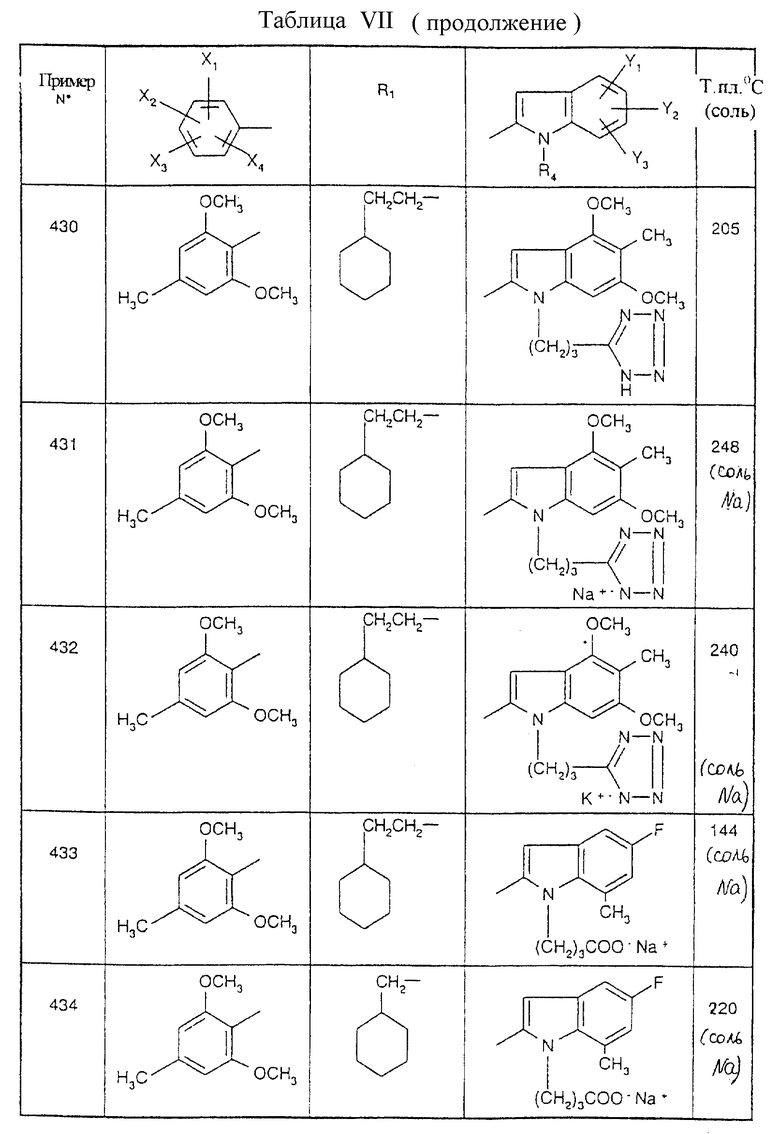
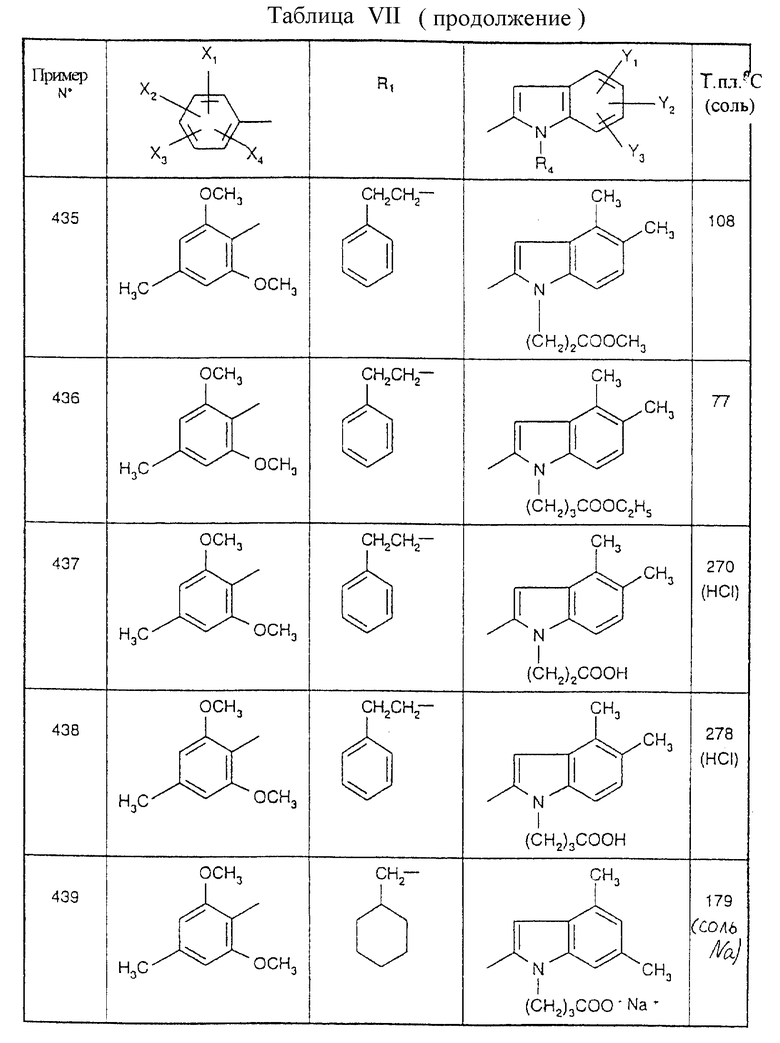
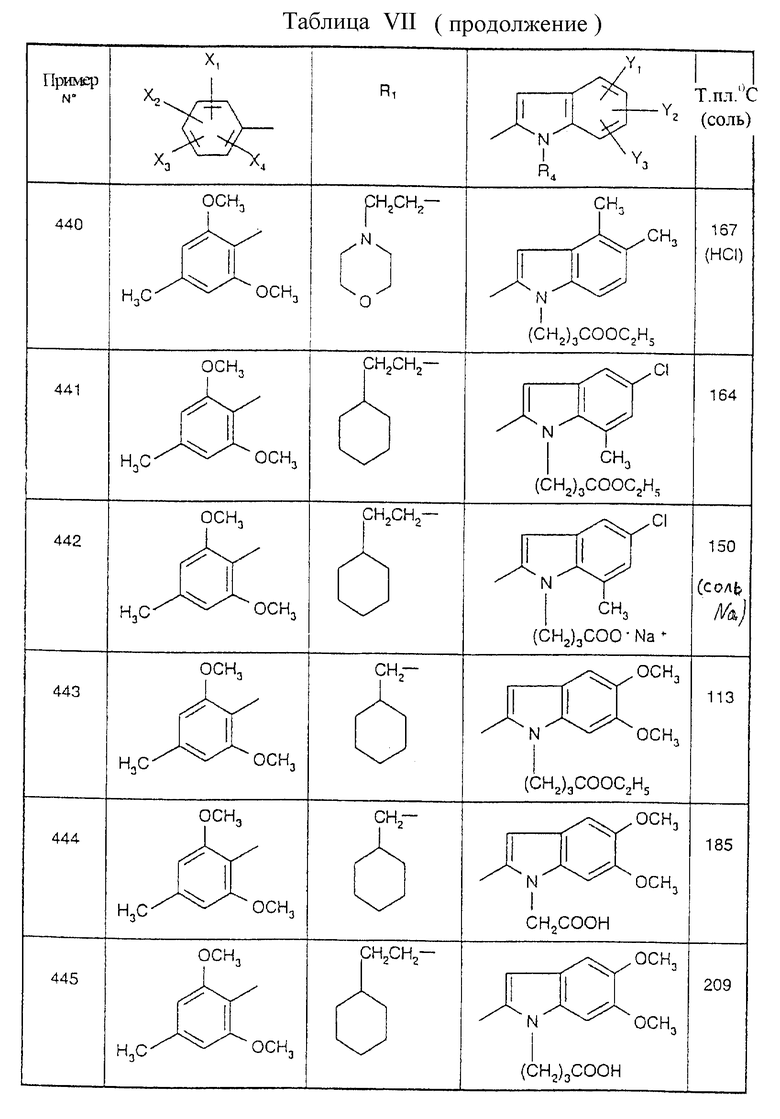
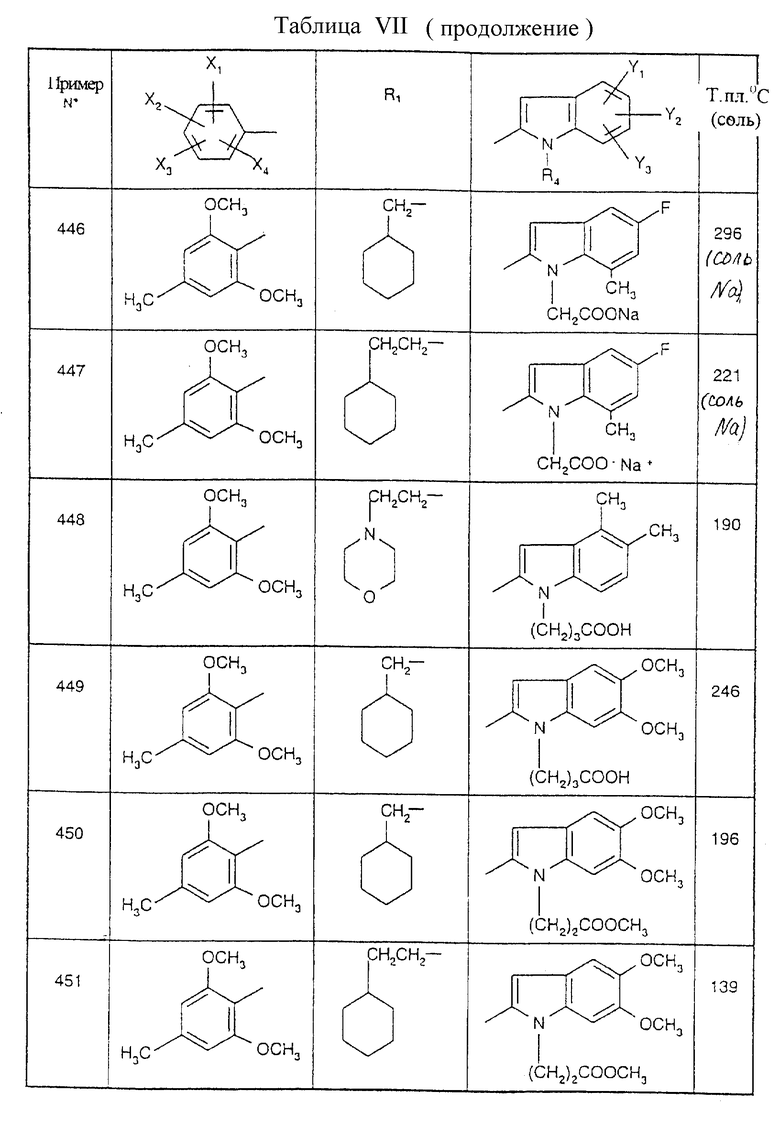
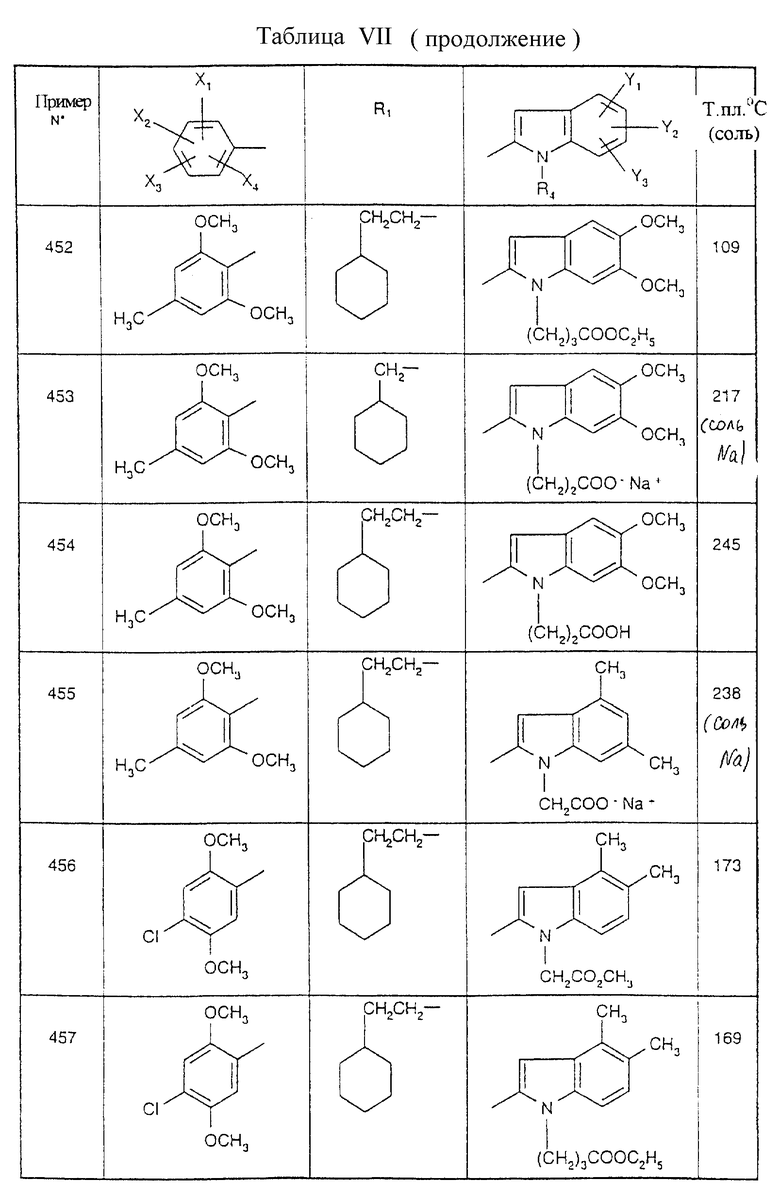
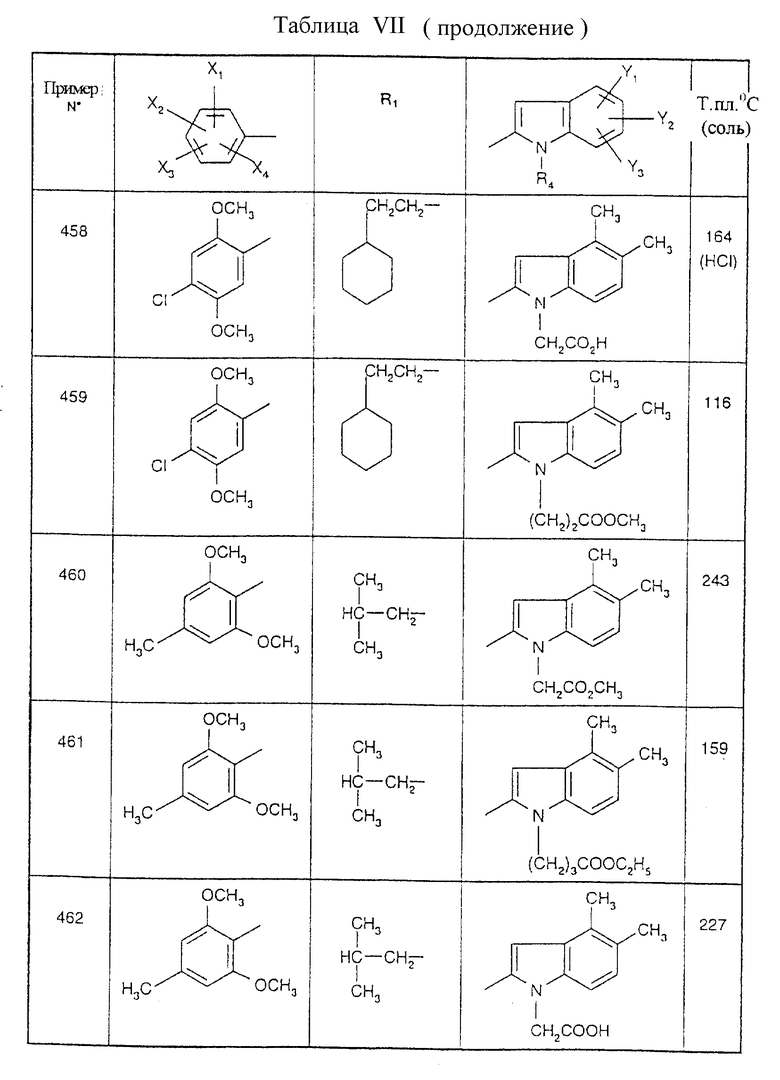
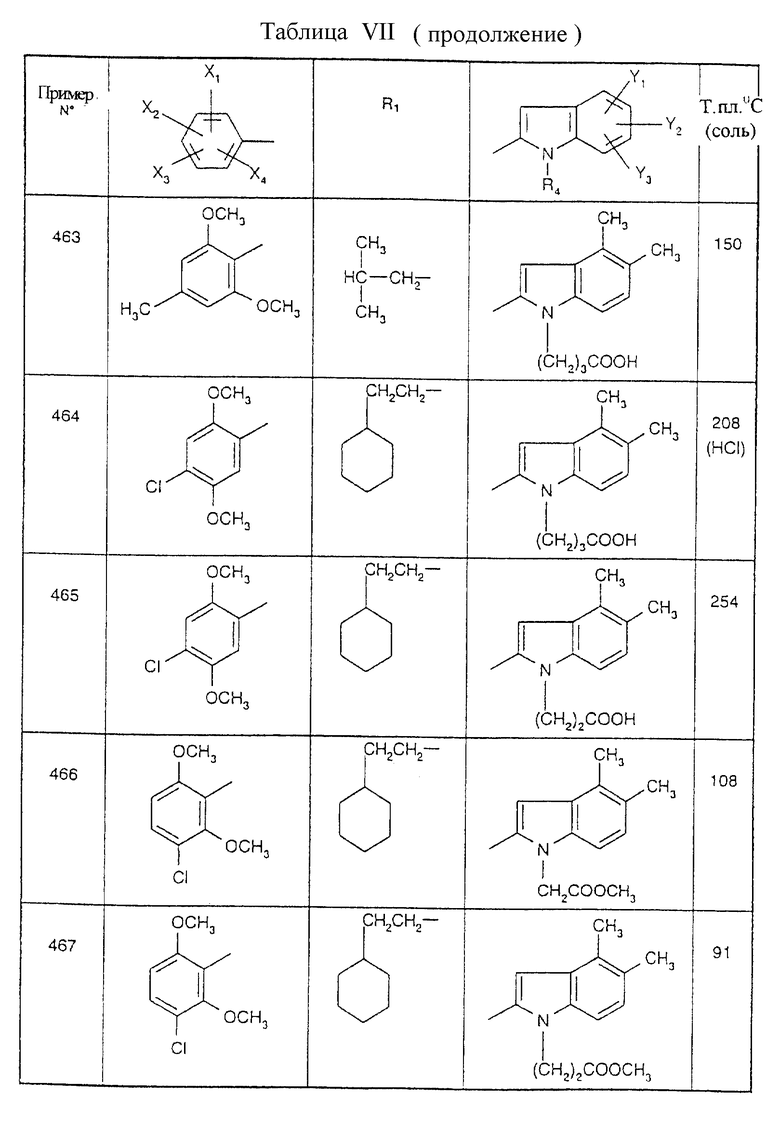
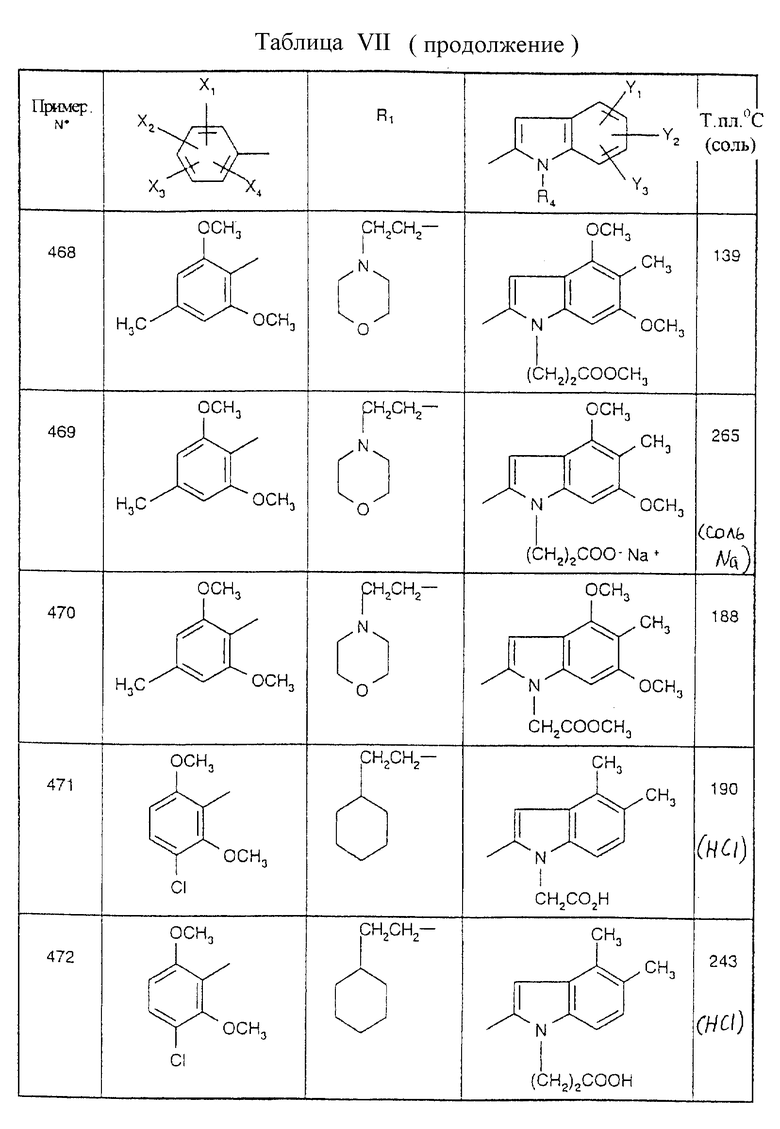
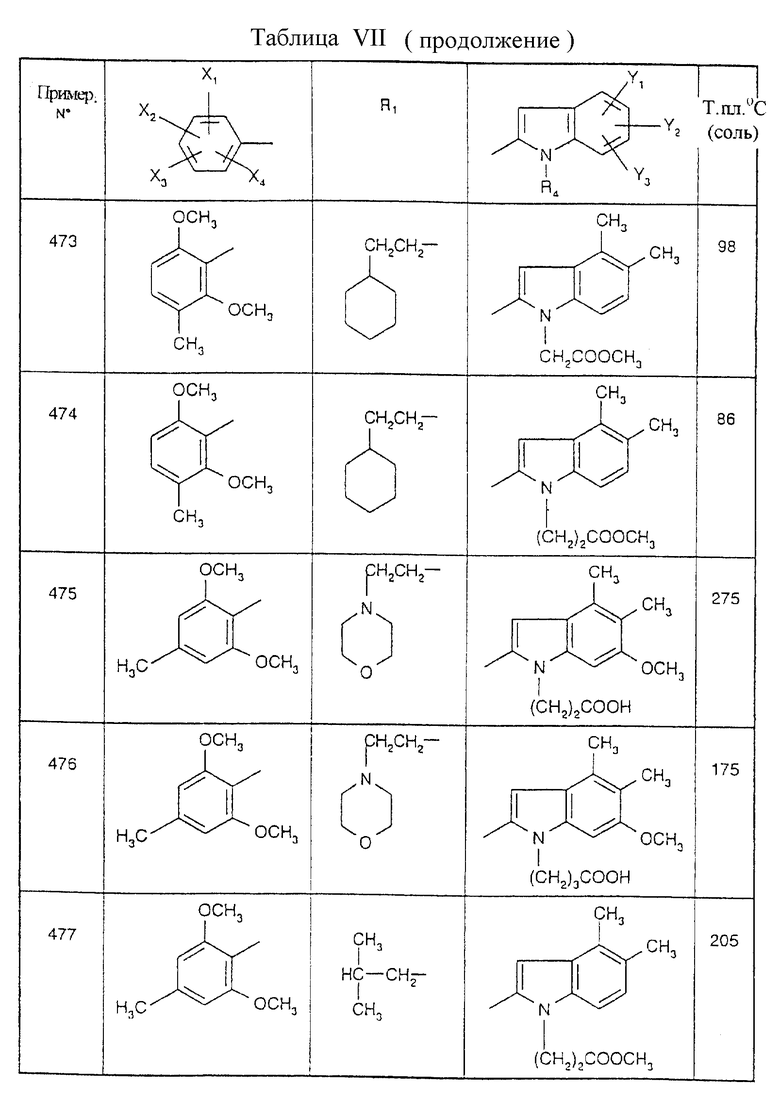
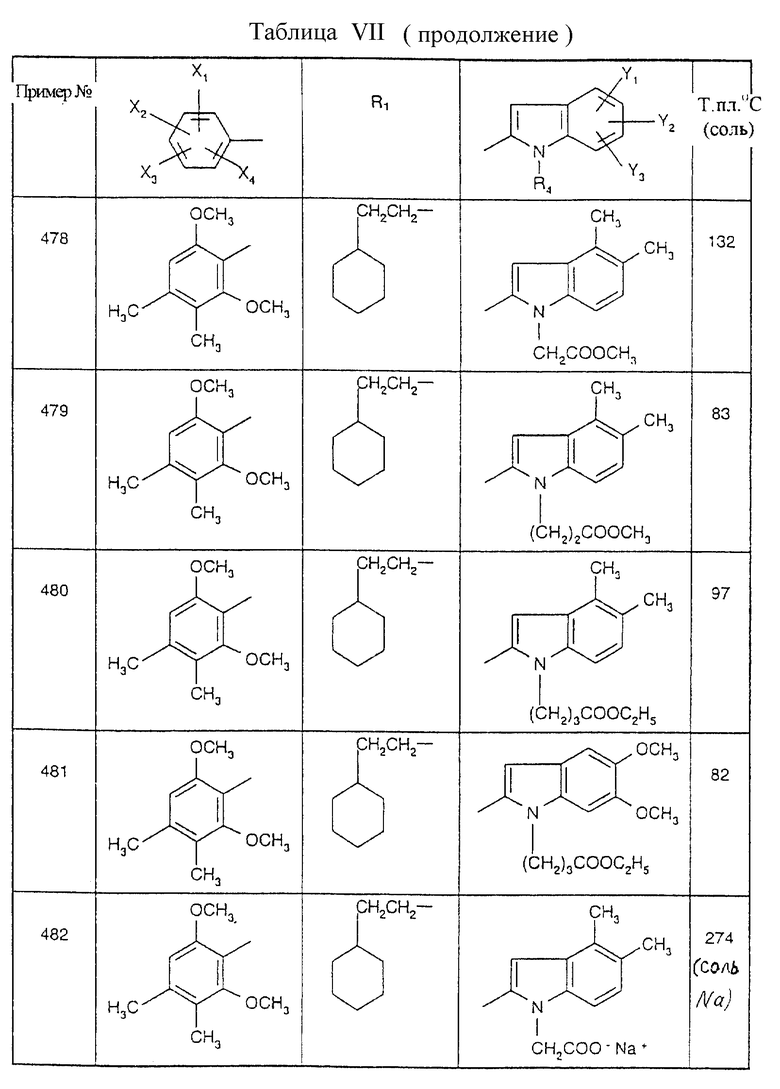
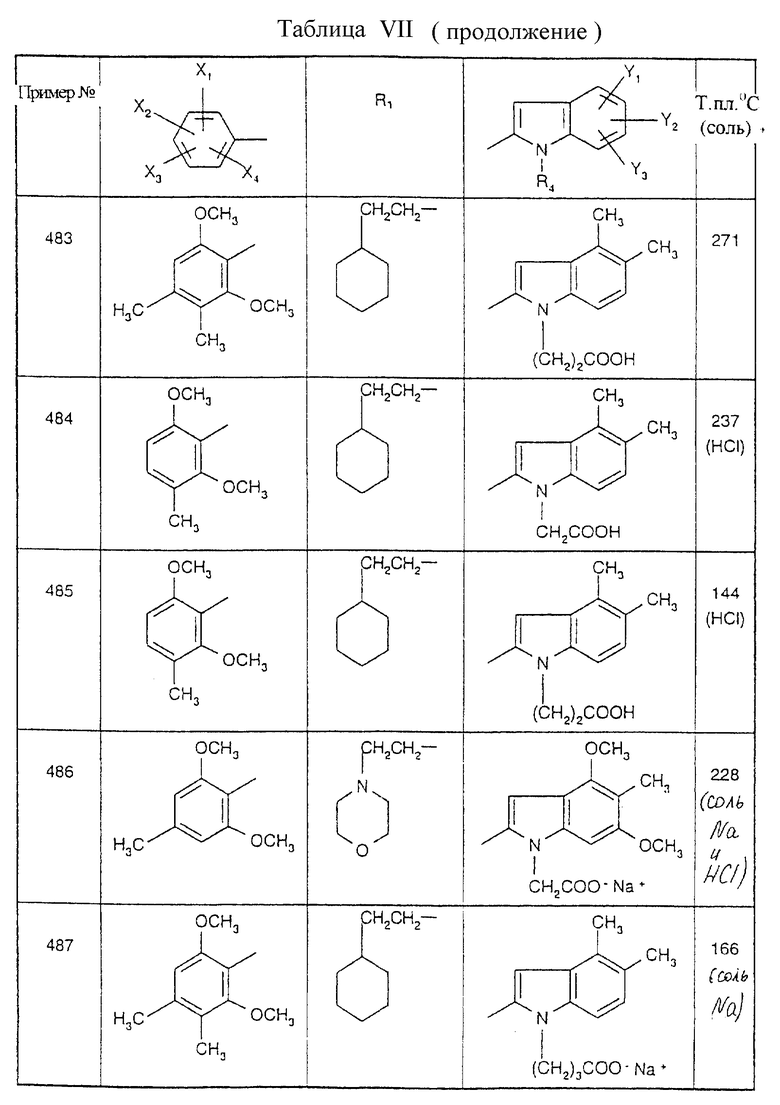
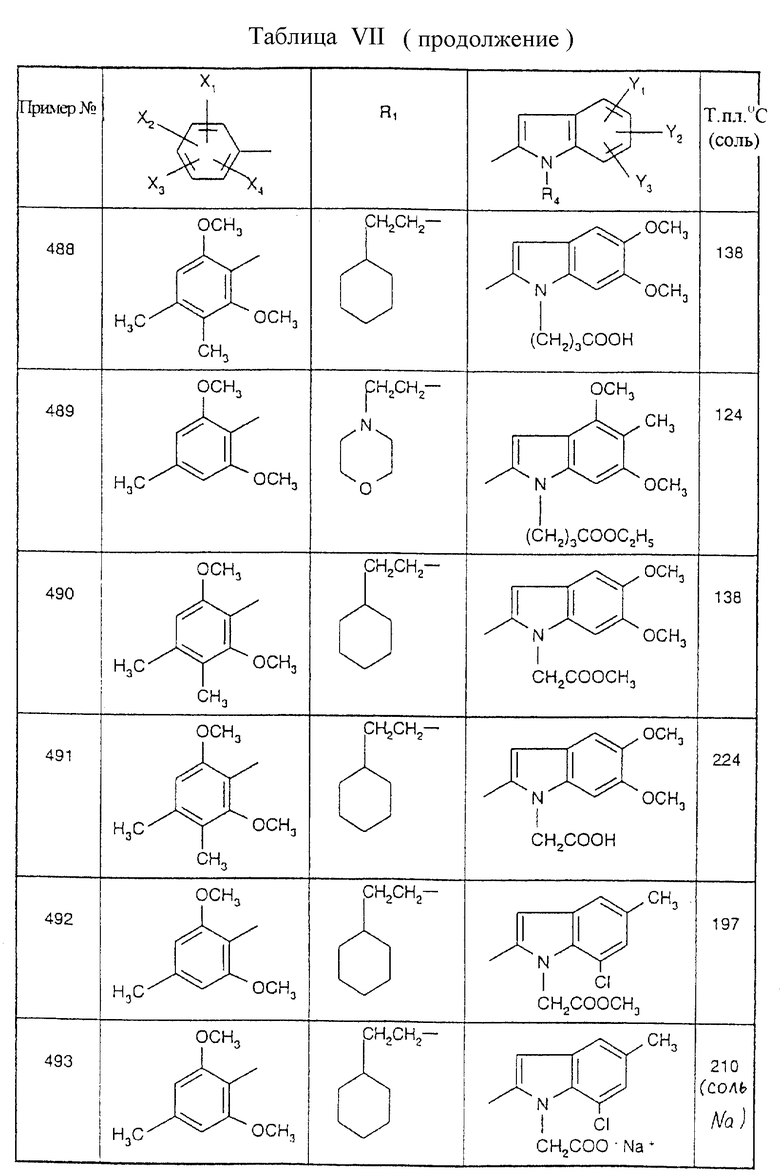
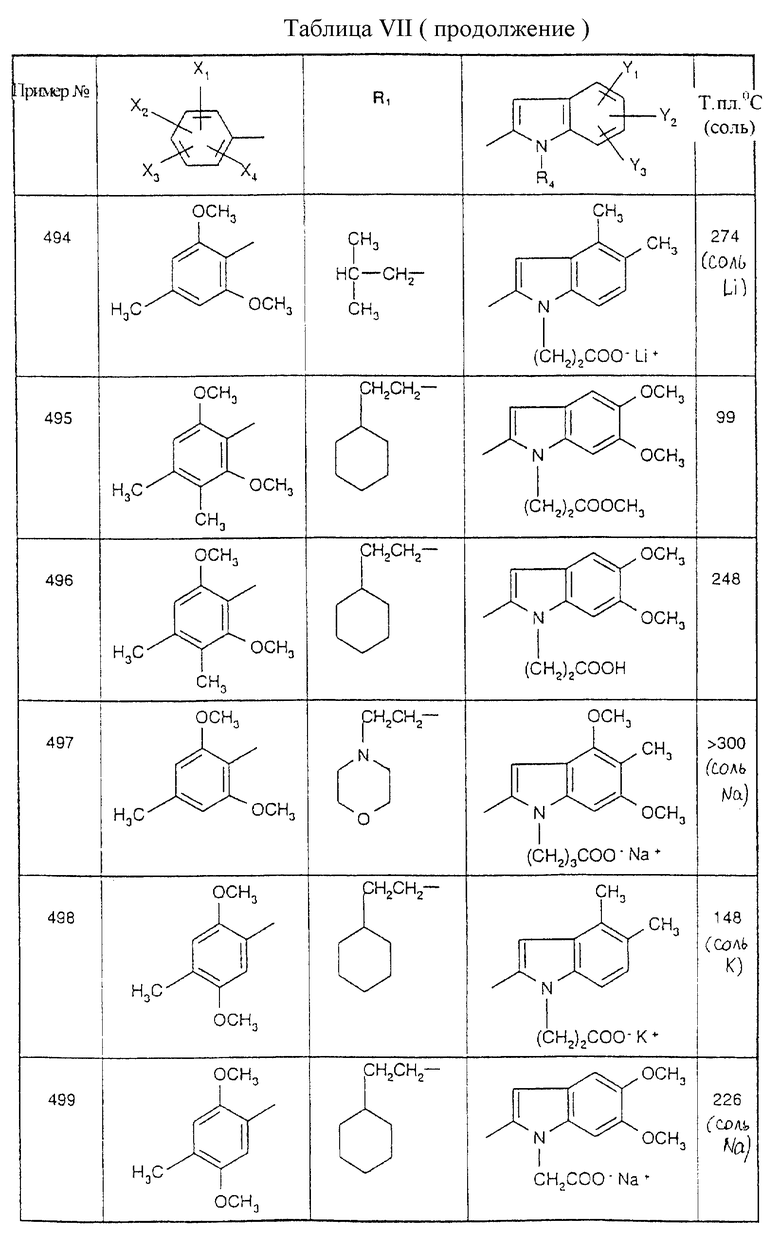
Примеры от 3 до 511, описанные в табл. VI и VII, получены таким же путем согласно процедуре примеров 1 и 2, приведенных выше, начиная с соответствующих промежуточных соединений.
Пример 512
2-{ N-[5-(4-хлор-2,5-диметоксифенил)-1-(2-циклогексилэтил)-1H-1,2,4- триазол-3-ил]карбамоил}-4,5-диметил-1-[3-(2H-1,2,3,4-тетразол-5-ил)- пропил] -1H-индол
Стадия 1: 4-[2-({[1-(-(2-Циклогексилэтил)-5-(2,5-диметокси-4-хлорфенил)- 1H-1,2,4-триазол-3-ил] амино}карбамоил)-4,5-диметил-1H-1- индолил]бутиронитрил
1 мл пиридина (0,013 моль) и 0,21 мл (0,0029 моль) тионилхлорида последовательно добавляют к 15 мл дихлорметана. После 15 минут при 0oC вводят 0,615 г 4,5-диметил-1-(3-цианопропил)-1H-2-индолкарбоновой кислоты (0,0024 моль), а затем 0,9 г 1-(2-циклогексилэтил)-5-(2,5-диметокси-4- хлорфенил)-1H-1,2,4-триазол-3-амина гидрохлорида. Реакционную смесь выдерживают в течение 18 часов при комнатной температуре, после чего проводят кислотную промывку и щелочную промывку. Органическую фазу сушат над безводным сульфатом натрия и концентрируют при пониженном давлении. Маслянистый остаток хроматографируют на колонке с силикагелем, элюируя смесью 98,5/1,5 (об./об.), с получением белого порошка; т.пл.=178oC; выход=87%.
Стадия 2: 2-{ N-[5-(4-хлор-2,5-диметоксифенил)-1-(2-циклогексилэтил)- 1H-1,2,4-триазол-3-ил] карбамоил} -4,5-диметил-1-[3-(2H-1,2,3,4-тетразол-5- ил)пропил]-1H-индол
0,5 мл азидотриметилсилана и 0,030 г дибутилоловооксида добавляют к 0,720 г (0,0012 моль) 4-{2-({[1-(2-циклогексилэтил)-5- (2,5-диметокси-4-хлорфенил)-1H-1,2,4-триазол-3-ил] амино}карбонил)-4,5- диметил-1H-1-индолил] бутиронитрила, растворенного в 15 мл тетрагидрофурана, и смесь нагревают при температуре дефлегмации в течение 18 часов. Реакционную смесь оставляют охлаждаться до комнатной температуры, тетрагидрофуран удаляют при пониженном давлении, а остаток хроматографируют на колонке с силикагелем, элюируя смесью 95/5 (об./об.) дихлорметан/метанол. Поучают белое твердое вещество; т.пл.=233oC, выход=78%.
Эту процедуру, описанную для примера 512, используют также для примеров 303, 304, 316, 317, 356, 357, 361, 362, 363, 368, 369, 392, 394, 395, 430 431 и 432.
Калиевые и натриевые соли этих соединений получают в ацетонитриле добавлением одного эквивалента основания при комнатной температуре с последующим выпариванием растворителя при пониженном давлении, а затем высушиванием. Тд
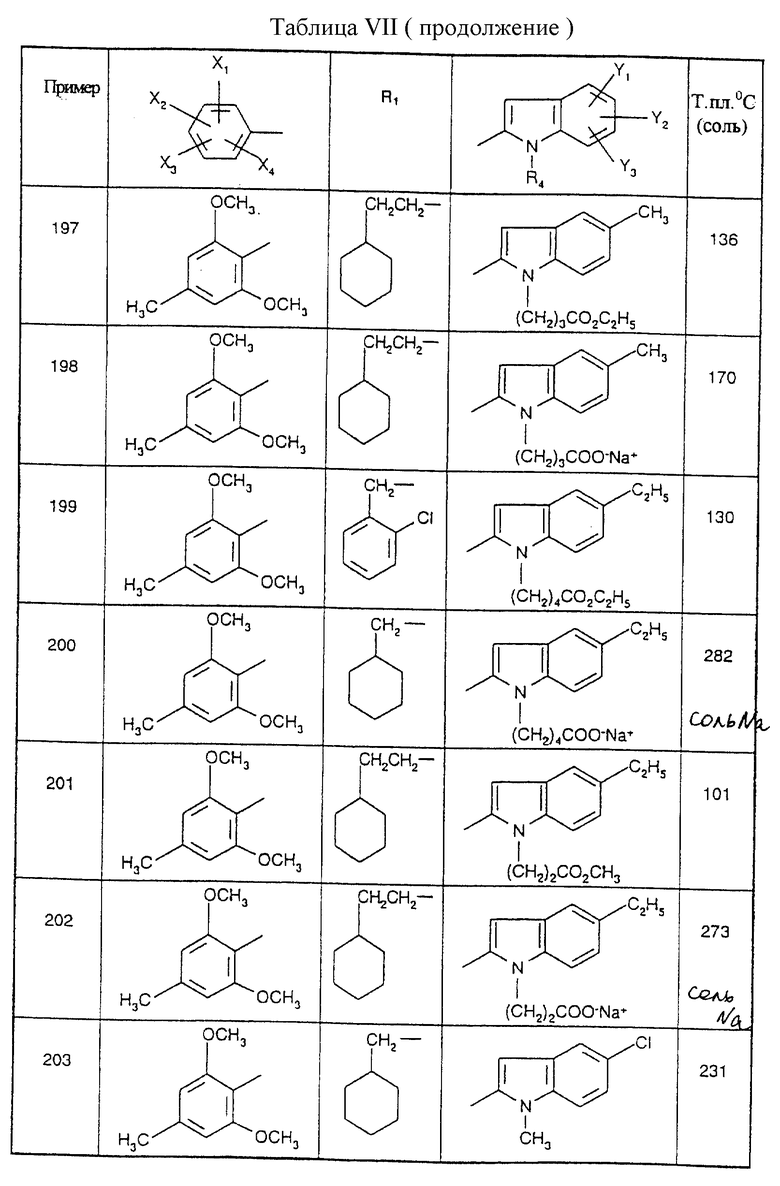
Описываются N-триазолил-2-индолкарбоксамиды общей формулы I, где R1 представляет собой -(С2-С6)алкил; группу -(CH2)n-G с n в пределах от 0 до 5 и G, представляющим собой неароматическую моно- или полициклическую С3-С13 углеводородную группу; фенил(С1-С3)алкил, в котором фенильная группа возможно замещена один раз галогеном или (С1-С3)алкокси; группу -(СН2)nNR2R3, в которой n представляет собой целое число от 1 до 6, a R2 и R3, которые могут быть одинаковыми или разными, представляют собой (С1-С3)алкил или составляют с атомом азота, к которому они присоединены, группу морфолино или пиперидино; X1, Х2, Х3 и Х4 каждый независимо представляет собой атом водорода или галогена, (С1-С6)алкил или (С1-С3)алкокси при условии, что только один из X1, Х2, Х3 и Х4 возможно представляет собой атом водорода; R4 представляет собой водород, группу -(CH2)nCOOR5, в которой n является таким, как определено выше, a R5 представляет собой атом водорода или (С1-С6)алкил; (С1-С6)алкил; группу -(CH2)nOR5 или группу -(CH2)nOR2R5, в которой n, R2, R3 и R5 являются такими, как определено выше; группу -(СН2)n-тетразолил, в которой n является таким, как определено выше, или R4 представляет собой одну из этих групп в форме соли щелочного металла; Y1, Y2 и Y3 независимо представляют собой водород, галоген, (С1-С3)алкил, (С1-С3)алкокси или карбамоил; или одна из их солей или сольватов; способы их получения, фармацевтическая композиция, их содержащая, и 1-замещенные 3-аминотриазолы формулы (7) - промежуточные соединения в синтезе соединений I. Соединения I обладают активностью частичного или полного агониста ХЦК-А-рецептора и могут быть использованы для лечения проблем приема пищи, ожирения, поздней дискинезии и расстройств желудочно-кишечной сферы. 9 с. и 4 з.п. ф-лы, 8 табл.
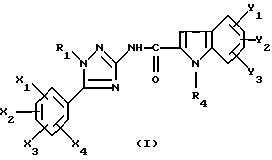
где R1 представляет собой -(С2-С6) алкил; группу -(CH2)n-G с n в пределах от 0 до 5 и G, представляющим собой неароматическую моно- или полициклическую С3-С13 углеводородную группу: фенил (С1-С3) алкил, в котором фенильная группа, возможно, замещена один раз галогеном или (С1-С3) алкокси; группу -(CH2)nNR2R3, в которой n представляет собой целое число от 1 до 6, а R2 и R3, которые могут быть одинаковыми или разными, представляют собой (С1-С3) алкил или составляют с атомом азота, к которому они присоединены, группу морфолино или пиперидино;
X1 X2, Х3 или Х4 каждый независимо представляет собой атом водорода или галогена, (С1-С6) алкил или (С1-С3) алкокси, при условии, что только один из Х1, X2, Х3 и Х4 возможно представляет собой атом водорода:
R4 представляет собой водород, группу -(CH2)nCOOR5, в которой n является таким, как определено выше, a R5 представляет собой атом водорода или (С1-С6) алкил; (С1-C6) алкил; группу -(CH2)nOR5 или группу -(СН2)nNR2R3, в которой n, R2, R3 и R5 являются такими, как определено выше; группу -(СН2)n-тетразолил, в которой n является таким, как определено выше, или R4 представляет собой одну из этих групп в форме соли щелочного металла;
Y1, Y2 и Y3 независимо представляют собой водород, галоген, (С1-С3) алкил, (С1-С3) алкокси или карбамоил,
или одна из их солей или сольватов.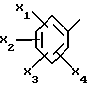
представляет собой 2,6-диметокси-4-метилфенил; его соль или сольват.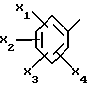
представляет собой 2,6-диметокси-4-метилфенил; его соль или сольват.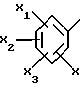
представляет собой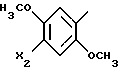
где X2 представляет метил или атом хлора;
его соль или сольват.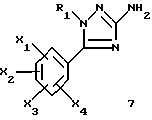
где R1, X1, Х2, Х3 и Х4 являются такими, как определено для (I) в п.1.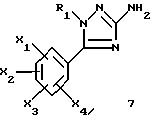
где R1, X1, X2, Х3 и X4 являются такими, как определено для (I) в п.1,
подвергают взаимодействию с производным индолкарбоновой кислоты формулы (8)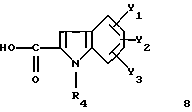
где R4, Y1, Y2 и Y3 являются такими, как определено для (I) в п.1.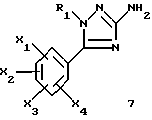
где R1, X1, Х2, Х3 и Х4 являются такими, как определено для (I) в п.1,
подвергают взаимодействию с производным индолкарбоновой кислоты формулы (8')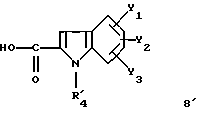
где Y1, Y2 и Y3 являются такими, как определено для (I) в п.1, a R'4 является группой-предшественником R4, причем R4 является таким, как определено для (I) в п.1,
с получением соединения формулы (I')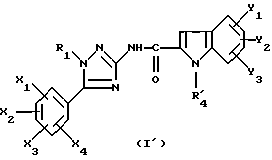
где R1, X1, X2, Х3, X4, Y1, Y2 и Y3 являются такими, как определено для (I) в п. 1, а R'4 является группой-предшественником R4, причем полученное соединение формулы (I') преобразуют в соединение формулы (I) путем преобразования группы R'4 в R4.
| Устройство для крепления конвейера скрепера | 1976 |
|
SU620221A3 |
| Устройство для обработки металлов давлением | 1976 |
|
SU611766A1 |
| RU 94016190 A1, 10.12.1995. | |||

