Изобретение касается высокомолекулярных блок-полимеров сопряженных диенов, содержащих спиртовые группы в диеновых блоках. Изобретение касается также способа получения таких полимеров из эпоксидированных диеновых блок-полимеров.
Известно, что традиционные АВА-блок-сополимеры используются в покрытиях, герметизирующих составах, адгезивах и модифицированных асфальтах, но их полезность в таких продуктах недостаточна из-за отсутствия некоторых физических свойств. Например, в патенте США N 3792005 описано, что покрытия могут быть получены с использованием блок-полимеров типа A-B-A, в которых A является моновинил-ароматическим блок-полимером, обычно полистиролом (c), и B является блоком каучукового полимера, обычно гидрированным полибутадиеном (БЕ) или гидрированным полиизопреном (ПЕ). Эти полимеры могут быть особенно полезны в эластомерных покрытиях, поскольку они могут обладать высокой эластичностью и поэтому не растрескиваются в процессе термообработки, что является важным требованием, например, для покрытий крыш или в процессе формования металлоизделий, когда покрытия разрушаются при сгибании металла. Покрытия на основе традиционных блок-сополимеров типа A-B-A не обладают достаточной адгезией и при применении в покрытиях, которые контактируют с органическими жидкостями, такими как бензин, или при высоких температурах покрытия просто растворяются или сходят с сусбтрата в виде расплава.
Было бы полезно иметь блок-полимеры такого типа, которые обладают улучшенными физическими свойствами. При введении функциональных групп в эти традиционные блок-полимеры они могут быть сшиты с образованием полиуретановых структур, имеющих лучшие физические свойства, и, таким образом, могут быть более полезными, чем традиционные блок-полимеры во многих покрытиях, герметиках, клеях и модифицированных асфальтах.
Известные отверждающиеся или сшитые покрытия на основе винил-ароматических соединений и сопряженных диенов не обладают достаточно длительной термостойкостью, водостойкостью и стабильностью к ультрафиолетовому облучению, обусловленными необходимостью использования негидрированных полимеров (трудно ввести функциональные группы без использования негидрированных полимеров). Известно, что группирование улучшает стабильность к длительным температурным, погодным и ультрафиолетовым воздействиям, но оно удаляет двойные связи, необходимые для введения функциональных групп, которые могут быть использованы при сшивке.
Предметом настоящего изобретения является получение блок-полимера, который может быть модифицирован с тем, чтобы в значительной мере не содержать остаточных олефиновых двойных связей, и может быть сшит взаимодействием с аминосмолами и изоцианатами. Далее объектом этого изобретения является композиция покрытия на основе этого сшивающегося блок-полимера.
Целью настоящего изобретения является разработка способа получения таких блок-полимеров, обеспечивающего высокую степень конверсии при относительно коротком времени реакции в хорошо идентифицируемый продукт в мягких условиях. В прошлом использовали сильные минеральные кислоты, такие как хлорная кислота, для взаимодействия эпоксидированных полимеров, как описано в патентах США NN 3555112, 3607977 и 3607982. Однако использование хлорной кислоты нежелательно, поскольку она образует взрывчатые соединения. Должны быть предусмотрены специальные меры предосторожности для работы с кислотой в ее концентрированной форме. Необходимое время реакции для достижения заметного превращения также имеет тенденцию к относительному увеличению (примеры конкретного времени реакции порядка шести часов). Эти примеры также ограничиваются полимерами, которые имеют высокую концентрацию функциональных эпоксигрупп. Неясно, смогут ли эти условия приводить к существенному количеству спиртовых функциональных групп применительно к полимерам, обладающих высокими уровнями эпоксифункциональности.
Могут быть также использованы катализаторы из числа протонных кислот и кислот Льюиса для проведения взаимодействия эпоксидированных полимеров, таких как описанные в патенте США N 5015697. Однако катализаторы из протонных кислот, таких как серная кислота, обычно требуют длительного времени реакции, тогда как многие катализаторы из кислот Льюиса приводят к побочным реакциям, таким как образование кетонов или альдегидов. Гелеобразование и обесцвечивание также являются проблемами в этих системах. Было необходимо разработать способ получения производных эпоксидированных диеновых блок-полимеров с гидроксильными функциональными группами в мягких условиях, при которых можно избежать указанных выше проблем.
Преимущества этого процесса выражаются также в существенной конверсии при времени реакции продолжительностью порядка одного часа. Далее важно то, что эти условия применимы для гидрированных диеновых блок-сополимеров, которые содержат эпоксигруппы. Настоящее изобретение предусматривает такие средства для раскрытия затрудненных эпоксиколец гидрированного диенового блок-сополимера, чтобы ввести реакционноспособные функциональные группы в основную цепь молекулы полимера.
Настоящее изобретение предусматривает способ получения эластомерных блок-полимеров сопряженных диенов, которые содержат в диеновых блоках функциональные гидроксигруппы, способные взаимодействовать с отвердителями, такими как аминосмолы и изоцианаты. Предпочтительно, чтобы конечные продукты в значительной мере не содержали остаточной ненасыщенности.
Способ включает следующие операции:
a) получение полимера-предшественника полимеризацией по меньшей мере одного сопряженного диена с получением продукта, содержащего 1,2-двузамещенные, 1,1-двузамещенные, 1,1,2-тризамещенные или тетразамещенные олефиновые двойные связи;
b) эпоксидирование полимера-предшественника с образованием продукта, в котором эпоксигруппы находятся в местах замещения, причем количество эпоксифункциональностей в полимере составляло по меньшей мере 0,1 миллиэквивалента на грамм полимера, и
c) контактирование эпоксидированного полимера со спиртом, имеющим одну незащищенную гидроксигруппу, и с соединением общей формулы MXn, где M выбран из группы, включающей водород, бор, алюминий, железо и олово, X представляет собой галоген и n - целое число, соответствующее валентности M, или с его органическим комплексом.
В одном из предпочтительных вариантов этого процесса на стадии c) эпоксидированный полимер контактирует со спиртом, имеющим одну незащищенную гидроксигруппу, в растворителе в присутствии катализатора, выбранного из группы, включающей кислоты Льюиса формулы MXn, где M выбран из группы, включающей бор, алюминий, железо и олово, и X является галогеном, а n - целое число, соответствующее валентности металла M, и органические комплексы таких кислот Льюиса.
Термин "1,2-двузамещенная олефиновая двойная связь" означает структуру, подобную (A). Аналогично термин "1,1-двузамещенная олефиновая двойная связь", "1,1,2-тризамещенная олефиновая двойная связь" и "тетразмещенная двойная связь" относятся к структурам (B), (C) и (D) соответственно:

Сокращение "мэкв/г полимера", использованное в описании, означает миллиэквивалентны на грамм полимера.
В предпочтительном варианте описанные выше сопряженные диены сополимеризованы либо беспорядочно или в блоках с сопряженными диеновыми мономерами, которые составляют только виниловые или 1,2-двузамещенные олефиновые группы. Эти группы гидрированы до или после эпоксидирования, так что конечный эпоксидированный продукт содержит по меньшей мере 0,1 и предпочтительно от 0,1 до 5 миллиэквивалентов эпоксигрупп на грамм полимера, и менее 1,0, предпочтительно менее 0,6 и наиболее предпочтительно менее 0,3 миллиэквивалента остаточного олефина на 1,0 г полимера (мэкв/г). В наиболее предпочтительном варианте изобретения полимеризацию проводят так, что от 0,3 до 3,5 миллиэквивалентов двузамещенного, тризамещенного или тетразамещенного олефина присутствует в грамме полимера-предшественника, по существу все виниловые и большинство 1,2-двузамещенных олефинов израсходованы при частичном гидрировании и эпоксидирование проводят для того, чтобы израсходовать остальные олефины с тем, чтобы в эпоксидированном полимере их оставалось не более 0,3 миллиэквивалента в 1,0 г полимера.
Предпочтительно в случае, когда соединение формулы MXn используют в качестве катализатора, этот катализатор выбирают из группы, включающей органические комплексы трифторида бора, в частности диэтилэфират трифторида бора, дибутилэфират трифторида бора, диметилэфират трифторида бора или трет-бутилметилэфират трифторида бора. Предпочтительно, чтобы спирт, имеющий одну незащищенную гидроксигруппу, был выбран из группы, включающей одноатомные спирты и производные двухатомных и трехатомных спиртов, имеющие одну незащищенную гидроксигруппу, остальные гидроксигруппы защищены как ацеталь, кеталь или карбонат. Предпочтительными примерами таких соединений являются бутанол, октанол, солкеталь и триметилолпропанкеталь (ацетонкеталь триметилолпропана).
Полимер согласно настоящему изобретению является сопряжением диеновым полимером, содержащим от 0,1 до 15 мэкв гидроксигрупп в грамме полимера, который может включать высоко реакционноспособные первичные оксигруппы в диеновых блоках.
Полимер содержит спиртовые группы формулы
или
в которых P представляет собой -OH, -Cl или -OR, где R выбран из группы, включающей алкильные радикалы, содержащие до 10 углеродных атомов, одноатомные и двухатомные спиртовые группы и карбонатные группы; Q представляет собой -OH или -Cl при условии, что по меньшей мере одни из и Q является -OH и R1 и R1 и R2 оба являются водородом или алкильным радикалом при условии, что, если X является -OR, то только один из R1 и R2 может быть водородом.
В особом случае, когда P представляет собой -OR и Q является -OH, блок-сополимер согласно настоящему изобретению содержит эфирно-спиртовые группы формул
или
в которых P, R1 и R2 имеют определенные выше значения.
Полимер может также содержать (а) гидрированный диеновый мономер(ы), который может быть диеновым мономером, являющимся источником спиртовых звеньев или другим диеновым мономером, (в) эпоксидированный диеновый мономер(ы) и/или (с) остаточный олефин. В предпочтительном варианте общее количество остаточного олефина составляет менее 1,0, предпочтительно менее 0,6 и наиболее предпочтительно менее 0,3 мэкв/г полимера.
Предпочтительно в полимеризации участвует достаточно спирта, чтобы присутствовало О от 0,25 до 9,0 мэкв гидроксильных групп на грамм полимера. В случае (III) и (IV) предпочтительно, чтобы по меньшей мере одна треть гидроксигрупп была первичной и присутствовала как часть эфирного радикала (OP). В другом варианте изобретения в полимере могут также присутствовать продукты гидрирования диенового мономера, который полимеризуется с образованием только винилового или 1,2-двузамещенного олефинового остатка. Еще в одном варианте осуществления изобретения полимер или отдельный блок полимера может содержать до 75% беспорядочно распределенных моноалкенил-ароматических углеводородных мономерных остатков. Количество таких алкилариловых мономеров в отдельном блоке полимера может достигать 99% при условии, что используют достаточно сопряженного диенового мономера для получения требуемого количества мест эпоксидирования.
Сопряженные диеновые блок-полимеры согласно настоящему изобретению должны содержать по меньшей мере 0,1 миллиэквивалента гидроксильных групп в грамме полимера.
Менее 0,1 мэкв гидроксигрупп на грамм полимера недостаточно для достижения цели изобретения, тогда как присутствие более 15 мэкв может вызвать слишком высокую растворимость полимера водой и хрупкость после отверждения. Предпочтителен предел от 0,25 до 8,0 мэкв/г.
Предпочтительным типом полимера в объеме настоящего изобретения является сопряженный диеновый блок-сополимер, содержащий функциональные гидроксигруппы, которые могут включать высоко реакционноспособные первичные гидроксигруппы в диеновых блоках, и имеющий формулу
Cq-Yp-(Bx-A)r, (V)
где B представляет собой гидрированный сопряженный диен, предпочтительно бутадиен, повторяющиеся производные элементарные звенья и x равен нулю или единице. Блок B может также содержать небольшое количество эпоксидированных спиртовых и/или ненасыщенных повторяющихся элементарных звеньев при условии, что в полимере присутствует не более одного, предпочтительно не более 0,6 и наиболее предпочтительно 0,3 мэкв ненасыщенных двойных связей на 1,0 г полимера; все блоки B, описанные ниже, могут также содержать такие элементарные звенья. А представляет собой полимерный блок, содержащий спиртовые элементарные звенья формулы (I) или (II), включая, таким образом, эфирно-спиртовые элементарные звенья формул (III) или (IV). Радикалы p могут содержать гидроксигруппы, одна или обе из них могут быть первичными. Блок A может также содержать гидрированные или эпоксидированные и/или ненасыщенные повторяющиеся элементарные звенья, так что не более 1,0, предпочтительно 0,6 и наиболее предпочтительно 0,3 мэкв ненасыщенных двойных связей остается в полимере на 1,0 г полимера (все блоки A-, описанные ниже, могут содержать такие элементарные звенья). С является таким же как A и B или полимерным блоком, производным от акрилатного или метакрилатного мономера, и Y представляет собой остаток агента сочетания, остаток инициатора или полимерный блок, производный от мономерного агента сочетания, такого как, например, дивинилбензол. Как уже отмечалось ранее, от 0,1 до 15,0 мэкв, предпочтительно от 0,25 до 9,0 мэкв оксигрупп присутствуют в одном грамме полимера, при этом предпочтительно по меньшей мере одна треть которых является высоко реакционноспособными первичными спиртовыми группами. Что касается целых чисел q, p и r, то их применение находится в пределах: r > 0, q ≤ 0, r + q равны от 1 до 100 и p равно нулю или единице.
Другой предпочтительный тип полимера в объеме изобретения имеет формулу
SZ-A-SZ', (VI)
в которой S является блоком, содержащим моноалкенил-ароматический углеводород и Z и Z' равны 0 или 1. А является полимерным блоком, содержащим спиртовые элементарные звенья формулы (I) или (II), а также эфирно-спиртовые элементарные звенья формулы (III) или (IV). Кроме того, A может также содержать эпоксидированный диеновый мономер, гидрированный диеновый мономер, который может отличаться от диенового мономера, от которого являются производными спиртовые группы, моноалкенил-ароматический углеводород и негидрированные мономерные остатки, как описано ранее. Спиртовые группы присутствуют в количестве от 0,1 до 15,0 мэкв/г полимера, предпочтительно от 0,25 до 9,0 мэкв/г, гидроксильные группы присутствуют в полимере, наиболее предпочтительно по меньшей мере одна треть которых представляет собой высокого реакционноспособные первичные группы. В предпочтительном варианте осуществления этого изобретения блок A состоит из сополимера изопрена и бутадиена и большая часть спиртовых групп являются производными от изопрена.
Изобретение включает также блок-полимеры формулы
SZ-A-SZ', (VII)
в которой S и A имеют значения, определенные в предыдущем абзаце, но блоки не гидрированы. Другой негидрированный полимер, входящий в объем настоящего изобретения, имеет формулу
CqYp-(Bx-A)r, (VIII)
в которой B представляет собой полимерный блок, производный от сопряженного диена, и A является полимерным блоком, который содержит более высокую концентрацию спиртовых групп, чем блок B. Остальные символы имеют значения, указанные для формулы (V) выше. Для всех перечисленных выше полимеров предпочтительно, чтобы по меньшей мере 1/3 гидроксигрупп была первичными группами, чтобы блок B был производным от бутадиена и блок A - производным от изопрена и чтобы количество гидроксигрупп находилось в пределах от 0,25 до 9,0 мэкв/г полимера. В другом варианте осуществления полимер содержит до 75 весовых процентов полимера винил-ароматического углеводорода.
Молекулярные массы этих полимеров обычно находятся в пределах от 2 • 103 до 5 • 106. Молекулярные массы A-блоков обычно находятся в интервале от 100 до 50000, наиболее предпочтительно от 500 до 15000. Молекулярные массы B-блоков обычно находятся в пределах от 100 до 200000, наиболее предпочтительно от 13000 до 50000. Молекулярные массы блоков С обычно находятся в пределах от 50 до 100000, наиболее предпочтительно от 500 до 50000. Молекулярные массы S-блоков обычно находятся в интервале от 100 до 50000. Чтобы получить полимеры, имеющие такие молекулярные массы, полимеры-предшественники должны быть приготовлены так, чтобы их молекулярные массы были аналогичными. Могут также быть получены полимеры с более высокими молекулярными массами, образованные сочетанием в процессе реакции со спиртом или хлористоводородный кислотой.
Молекулярные массы линейных полимеров или несвязанных линейных сегментов полимеров, таких как моно-, ди-, три-блок и так далее, или цепей звездообразных полимеров перед сочетанием обычно измеряют гель-проникающий хроматографией, при этом система для проведения гель-хроматографии соответствующим образом откалибрована. Для калибрования используют полимеры с известной молекулярной массой, которые должны иметь такие же молекулярное строение и химический состав, что и неизвестные линейные полимеры или сегменты, подлежащие измерению. Для полимеризованных по анионному типу линейных полимеров полимер является в сущности монодисперсным, и это удобно и достаточно точно описывается термином "пиковой" молекулярной массы наблюдаемого узкого распределения молекулярных масс. Измерение истинной молекулярной массы конечного связанного звездоподобного полимера не так очевидно или не так просто выполнить с использованием гель-проникающей хроматографии. Это объясняется тем, что звездоподобные молекулы не разделяются и не элюируют через упакованные колонки для гель-хроматографии таким же образом, что и линейные полимеры, применяемые для калибровки системы, и, следовательно, время поступления сигнала на УФ-детектор (или детектор) показателя преломления не является хорошим показателем молекулярной массы. Хорошим методом для звездообразных полимеров является измерение среднечисленной молекулярной массы методом светорассеяния. Образец растворяют в подходящем растворителе при концентрации менее 1,0 г образца на 100 миллилитров растворителя и фильтруют с использованием шприца и пористых мембранных фильтров с размером пор менее 0,5 микрон непосредственно в светорассеивающей ячейке. Измерения светорассеяния проводят как функцию угла рассеяния и концентрации полимера с использованием стандартных методов. Дифференциальный показатель преломления образца измеряют при той же длине волны и в том же самом растворителе, что и для измерения светорассеяния. Ниже приводятся ссылки на источники, в которых описаны вышеупомянутые методы:
1. Modern Size - Exclusion Liquid Chromatography, M.W. Yau, J.J.Kirkland, D.D.Bly, John Wiley & Sons, New York, NY, 1979.
2. Light Scattering from Polymer Solutions, M.B.Huglin, ed., Academic Press, New York, NY 1972.
3. W.Kay and A.J.Havlik, Applied Optics, 12, 541 (1973).
4. M.L.McConnell, American Laboratory, 63, May, X 1978.
Блок-сополимеры согласно изобретению предназначены для применения, требующего функциональности, чтобы придать полярность и реакционную способность молекулам без функциональных групп. Конкретно эти молекулы предназначены в качестве модификаторов каучука и основных компонентов в покрытиях и герметизирующих средствах, особенно в полиуретановых покрытиях и герметиках. Эти продукты полезны в термореактивных адгезивах, герметиках и покрытиях, особенно в уретановых, полиэфирных и отверждаемых меламином продуктах. Полимеры согласно изобретению полезны также для модификации асфальтов, где желательна полярность. Они полезны также в волокнах, пленках и печатных платах, а равно как и для модификации сложных полиэфиров, простых полиэфиров и полиамидов. Введение длинных углеводородных цепей (реакцией с нормальными спиртами) полезно при получении блок-полимеров с низкой температурой стеклования и для улучшения показателя вязкости путем снижения температуры текучести. Взаимодействие этих содержащих гидроксигруппы полимеров с молекулами, содержащими электрофильные функциональные группы, такими как изоцианаты, хлорангидриды, карбоновые кислоты и так далее, может быть полезным для введения новых функциональных групп в главную цепь полимера.
Например, акрилатные группы могут быть введены взаимодействием с метакриловой кислотой и изоцианатные группы - взаимодействием с избытком диизоцианата толуола.
Полимеры согласно настоящему изобретению особенно полезны в покрытиях. Предпочтительно эти полимеры являются сшитыми изоцианатами для создания отличных материалов для покрытий. Изоцианат используют для сшивки полимеров путем взаимодействия с функциональными оксигруппами, присутствующими в полимере. Стандартные методы химии полиуретанов используют в реакции полимера с изоцианатом.
Например, см."Coatings Based on Polyurethane Chemistry: An Overview and Recent Developments" by T.A.Potter and J.L.Williams, Journal of Coatings Technology, Vol. 59, N 749, June 1987, pps. 63-72.
Мономерные изоцианаты могут быть использованы для сшивания полимеров с функциональными группами. Мономерные изоцианаты имеют тот недостаток, что являются высокотоксичными веществами. С целью уменьшить проблемы, связанные с использованием таких материалов, часто вместо мономерных изоцианатов используют продукты присоединения изоцианатов. Оба типа изоцианатов обладают тем преимуществом, что взаимодействие протекает при комнатной температуре. Возможно также использование защищенных изоцианатов. Изоцианаты блокируют взаимодействием с летучими гидрокси-веществами с целью предохранить их от взаимодействия с гидроксигруппами блок-полимера. Изоцианат восстанавливают нагреванием. Это описано в статье, на которую имеется ссылка выше. Такие защищенные изоцианаты полезны, например, в покрытиях горячей сушки. Материал может быть нанесен на подложку, которую затем нагревают. Блокатор удаляется из покрытия под действием тепла и будет иметь место сшивки по гидроксигруппам блок-полимера.
Эти полимеры могут быть также сшиты и отверждены аминосмолами и ангидридами. Например, см. 50 Years of Amino Coating Resins, изданной и напечатанной Albert J.Kirsch и опубликованной в 1986 году American Cyanamid Company, в которой детально описана целая серия аминосмол, полезных для целей настоящего изобретения. В ней утверждается на странице 20 что полимеры с главной цепью, т.е. полимеры, которые подлежат сшиванию, "должны содержать одну или более функциональных групп: гидрокси, карбокси, амидную, перечисленные выше, чтобы быть полезным для взаимодействия с аминосмолами". Ангидриды используют для отверждения OH-групп в покрытиях горячей сушки (нагревание, например, с фталевым ангидридом).
Полимеры, содержащие олефиновые двойные связи или ароматические и олефиновые двойные связи, которые используют в качестве предшествующих полимеров в способе согласно настоящему изобретению, могут быть получены с использованием анионогенных инициаторов или катализаторов полимеризации. Такие полимеры могут быть получены полимеризацией в массе, в растворе или эмульсии. Полимеры, полученные полимеризацией в растворе, предпочтительны для последующего частичного гидрирования.
Общие методы получения блок-сополимеров рассмотрены в R.P.Quirk and J. Kim, "Recent Advances in Thermoplastic Elastome Synthesis" Rubber Chemistry and Technology, vol. 64, N 3 (1991).
Особенно полезным является метод последовательной полимеризации мономеров с анионогенным инициатором. Поскольку типы мономеров, подлежащих полимеризации с удалением защиты, относительно ограничены для анионогенного метода, при этом наиболее благоприятными являются сопряженные диолефины и стиролы, то обычно необходимо частичное негидрирование анионогенного блок-сополимера, чтобы получить предшествующий неэпоксидированный полимер. Полимеры, полученные последовательной полимеризацией подходящего диолефинового мономера и мономера, имеющего только одну углерод-углеродную двойную связь, или последовательной полимеризацией двух различных смесей (отношений) таких мономеров, используя либо монофункциональный инициатор, монофункциональный инициатор и связующее вещество или многофункциональный инициатор, могут быть не гидрированы.
Полимеры, содержащие олефиновые двойные связи или ароматические и олефиновые двойные связи, могут быть получены с использованием анионоактивного инициатора или катализатора полимеризации. Такие полимеры могут быть получены полимеризацией в масле, в растворе или в эмульсии. Полимеры, полученные в растворе, предпочтительны для последующего частичного гидрирования.
Например, предшествующие полимеры, пригодные для получения полимеров формул (V) и (VII), могут быть традиционно получены анионной полимеризацией с получением блоков A и B и необязательно боковой цепи C, состоящих из гомополимеров или сополимеров сопряженных диеновых мономеров, или сополимеров сопряженных диеновых мономеров и винил-ароматических мономеров (75% или менее винил-ароматического мономера), в которых мономеры, используемые для блоков A, таковы, что A-блоки имеют большее число 1,1-двузамещенных, тризамещенных и тетразамещенных олефиновых групп на элементарное звено массы блока, чем B-блоки. Полимер может быть частично гидрирован с подходящим катализатором и в условиях, благоприятствующих гидрированию двойных связей, которые монозамещены (виниловые) или 1,2-двузамещенные (и также составляет ароматические двойные связи нетронутыми), так что от 0,2 до 11,6 мэкв на грамм полимера 1,1-двузамещенных, тризамещенных или тетразамещенных олефиновых ненасыщенных групп остаются непреждевременными. Блоки B в среднем будут содержать меньшее количество олефиновых двойных связей. Особым случаем является полимер, в котором блок A представляет собой полиизопреновый блок, в котором все остаточные двойные связи являются 1,1-двузамещенными (3,4-изопреновые повторяющиеся элементарные звенья) или тризамещенными (повторяющиеся элементарные звенья 1,4-изопрена), и B является полибутадиеновым блоком, в котором присутствуют только монозамещенные (виниловые) или 1,2-двузамещенные остаточные двойные связи. Частичное гидрирование этого полимера осуществляется очень хорошо. Если B является полибутадиеном и этот блок подвергают взаимодействию с агентом сочетания, то обычно часто используют миниблок олигоизопрена или олигостирола, чтобы улучшить условия процесса сочетания при получении звездообразного полимера. Полимер может быть эпоксидирован, чтобы иметь от 0,2 до 10,0 мэкв эпоксигрупп/г массы блока A, в то время как блоки B будут содержать меньшее количество эпоксигрупп вследствие эпоксидирования остаточного 1,2-двузамещенного олефина.
Другой пример будет представлять последовательная полимеризация единственного сопряженного диенового мономера в двух системах условий реакции, таких как анионная полимеризация 1,3-бутадиена в циклогексане для получения 1,4-полибутадиена с последующим добавлением структурного модификатора и эфирного растворителя, и полимеризация высокомолекулярного 1,2-полибутадиена с последующим сочетанием и избирательным гидрированием 1,2-полибутадиена для получения полимера A-B-A.
В другом примере 1,1-двузамещенные, тризамещеннные и тетразамещенные олефиновые группы могут быть беспорядочно распределены среди 1,2-двузамещенных и винил-олефиновых групп, например, сополимеризацией бутадиена и изопрена в присутствии структурного модификатора и эфирного растворителя. Частичное гидрирование такого полимера позволяет введение спиртовых функциональностей в концентрациях, ограниченных количеством мономера, который полимеризуется с получением медленнее гидрирующихся олефиновых групп и который добавляют в исходное сырье для полимеризации.
В общем, если используют анионные растворы, то сопряженные диеновые полимеры и сополимеры сопряженных диенов в винил-ароматических углеводородах получают контактированием мономера или мономеров, подлежащих полимеризации, одновременно или последовательно с анионоактивным инициатором полимеризации, таким как органические соединения щелочного металла в подходящем растворителе в интервале температур от -150oC до 300oC, предпочтительно от 0oC до 100oC. Особенно эффективным инициатором анионной полимеризации являются органические соединения лития.
Сопряженные диены, которые могут полимеризоваться по анионному типу, включают сопряженные диены, содержащие от 4 до 24, предпочтительно 4 - 8 углеродных атомов, такие как 1,3-бутадиен, изопрен, 1,3-пентадиен, метилпентадиен, фенилбутадиен, 3,4-диметил-1,3-гексадиен, 4,5-диэтил-1,3-октадиен и тому подобные. Изопрены и бутадиены являются предпочтительными сопряженными диеновыми мономерами для использования в настоящем изобретении благодаря их низкой стоимости и доступности.
Винил-ароматические углеводороды, которые могут сополимеризоваться, включают винилариловые соединения, такие как стирол, различные алкилзамещенные стиролы, алкоксизамещенные стиролы, винилнафталин, алкилзамещенные винилнафталины и тому подобные. Предпочтителен стирол.
Сопряженные диены могут быть также сополимеризованы с метакрилатами, такими как трет-бутилметакрилат, как описано в патенте США N 5002676, и такие сополимеры могут быть частично гидрогенизированы и эпоксидированы, как описано в настоящей заявке. Предпочтительным положением для метакрилатов при их использовании в полимере являются С-боковые цепи, соответствующие формулам (V) и (VIII).
В общем, могут быть использованы любые растворители, известные в данной области, полезные для получения таких полимеров. Подходящие растворители, таким образом, включают углеводороды с прямой и разветвленной цепью, такие как пентан, гексан, октан и тому подобные, а также их алкилзамещенные производные; циклоалифатические углеводороды, такие как циклопентан, циклогексан, циклогептан и тому подобные, а также их алкилзамещенные производные; ароматические и алкилзамещенные ароматические углеводороды, такие как бензол, нафталин, толуол, ксилол и тому подобные; гидрогенизированные ароматические углеводороды, такие как тетралин, декалин и тому подобные; линейные и циклические простые эфиры, такие как метиловый эфир, метилэтиловый эфир, диэтиловый эфир, тетрагидрофуран и тому подобные.
Наиболее предпочтительно получать примеры согласно настоящему изобретению анионной полимеризацией сопряженных диеновых мономеров и винил-ароматических углеводородных мономеров в углеводородном растворителе в интервале температур от 0oC до 100oC, используя алкиллитиевый инициатор. Цепи "живого" полимера могут быть связаны добавлением дивинилового мономера, чтобы получить звездообразный полимер. Могут быть добавлены дополнительные мономеры, чтобы вырастить большее количество боковых цепей C или получить концевую функциональность, например, добавлением окиси этилена или двуокиси углерода для образования гидроксильных или карбоксильных групп, соответственно и реакцию в полимере и на концах "живых" цепей прекращают введением донора протонов. Полимеризация может быть также инициирована мономерами, такими как мета-дивинилбензол и мета-диизопропенилбензол, обработанными бутиллитием.
Имеется широкий выбор агентов сочетания или инициаторов, которые могут быть использованы. Любой полифункциональный агент сочетания, содержащий по меньшей мере две реакционноспособные группы, может быть применен. Примеры таких типов соединений, которые могут быть использованы, включают полиэпоксиды, полиизоцианаты, полиамины, полиальдегиды, поликетоны, полиангидриды, сложные полиэфиры, полигалогениды и тому подобные. Эти соединения могут также содержать две или более типов функциональных групп, например, сочетание эпокси- и альдегидных групп, изоцианатных и галогенидных групп и тому подобные. Многие подходящие типы этих многофункциональных соединений описаны, например, в патентах США NN 3595941, 3468972, 3135716, 3078254, 4096203 и 3594452. Если агент сочетания имеет две реакционноспособные группы, такой как дибромэтан, то полимер будет иметь линейную A-B-A-структуру. Если агент сочетания имеет три или более реакционноспособных групп, такой как тетрахлорид кремния, то полимер будет иметь разветвленное строение, такое как (AB)nY при n, равном трем или более, и Y является остатком агента сочетания. Мономерные агенты сочетания содержат несколько элементарных звеньев мономера, необходимых для каждой концевой группы цепи макромолекулы, подлежащей связыванию. Дивинилбензол является наиболее часто используемым сочетающим мономером и приводит к получению звездообразного полимера.
Полимеры необязательно могут быть частично гидрированы. Гидрирование может иметь место до или после эпоксидирования. Получение полимеров, имеющих регулируемые и заранее заданные количества остаточной олефиновой ненасыщенности, описано в патенте США N 4879349.
Вероятно, что будут достигнуты лучшие характеристики, если катализатор, описанный в патенте США N 4879349, заменить титановым катализатором, таким как описанный в патенте США N 5039755.
В предпочтительном варианте осуществления полимеры частично гидрируют перед эпоксидированием и блоки A содержат более высокие концентрации 1,1-двузамещенных, 1,1,2-тризамещенных или тетразамещенных олефиновых двойных связей, чем блоки B, и блоки A содержат от 0,2 до 11,6 мэкв таких двойных связей в грамме полимера, предпочтительно в пределах от 0,5 до 9,0 мэкв/г и наиболее предпочтительно от 0,1 до 5,4 мэкв/г. Предпочтительно, чтобы соотношение концентрации (в мэкв/г) таких двойных связей в блоках A и концентрации в блоках B было больше 3 : 1. Более предпочтительно это соотношение должно быть больше 5 : 1, так как в общем важно ограничить функциональность блоков, в частности, если желательно сохранить эластомерные свойства после сшивания. Если конечным использованием полимера являются чувствительные к давлению адгезии или эластичные покрытия, то часто полезно, чтобы блоки B содержали меньше или вообще не содержали олефиновые двойные связи. Если при полимеризации получают большие количества двойных связей в блоках A или B, то полимер должен быть эпоксидирован в большей степени, чтобы удалить большинство двойных связей. Это приводит к получению полимеров, чувствительных к воде в блоках A, излишне сшитых, особенно в блоках B, которые обладают малой эластичностью и, таким образом, непригодны для применения по назначению.
После частичного гидрирования температура стеклования полимеров, предназначенных для применения в чувствительных к давлению условиях, должна нормально быть менее 10oC, предпочтительно ниже -15oC и наиболее предпочтительно ниже -40oC. Полимеры с более высокими температурами стеклования в блоках A не являются достаточно пластичными, тогда как полимеры с более высокими температурами стеклования в блоках B не являются достаточно эластомерными. Полимеры с более высокими температурами стеклования в блоках B могут быть желательными для покрытий, в которых следует избегать липкости.
Частичное гидрирование является диеноселективным. Вообще, степень гидрирования более высока для углерод-углеродных двойных связей, в которых ни один из атомов углерода не является третичным, как найдено в моно- и 1,2-двузамещенных олефинах, чем для углерод-углеродных двойных связей, в которых один из атомов углерода является третичным углеродным атомом, как установлено в 1,1-двузамещенных, 1,1,2-тризамещенных и тетразамещенных олефинах. Степень эпоксидирования углерод-углеродных двойных связей как раз наоборот. Третичные углеродные атомы способствуют эпоксидированию перкислотами лучше, чем вторичные углеродные атомы, которые, в свою очередь, лучше, чем первичные углеродные атомы. Таким образом, описанные полимеры особенно пригодны для процессов частичного гидрирования или эпоксидирования и особенно пригодны для последовательного использования в обоих процессах на полимере. Использование одного частичного гидрирования на настоящих полимерах согласно изобретению преимущественно приводит к большему числу остаточных диеновых двойных связей на единицу массы в блоках A полимеров, тогда как использование одного эпоксидирования продуцирует большее число эпоксидированных диеновых мономеров на единицу массы в блоках A в сравнении с блоками B. Эпоксидирование также избирательно, благоприятствуя двузамещенным, тризамещенным и тетразамещенным олефинам, и полученные эпоксиды устойчивы к гидрогенизации, так что насыщенные полимеры могут быть также получены гидрированием менее замещенных олефинов после эпоксидирования.
Независимо от выбора, будет или не будет полимер подвергаться гидрированию, он должен быть эпоксидирован так или иначе с тем, чтобы по меньшей мере 0,1 миллиэкв. эпоксигрупп присутствовало в одном грамме полимера, или не будет достаточной функциональности, чтобы получить достаточное количество оксигрупп для обеспечения преимуществ настоящего изобретения. Предпочтительный интервал составляет от 0,1 до 5,0 мэкв/г и наиболее предпочтительный - от 0,25 до 3,0 мэкв/г.
Эпоксидированные сополимеры могут быть получены способами эпоксидирования, описанными или рассмотренными в Encyclopedia of Chemical Techology 19,3 rd ed., 251-266 (1980), D.N.Schultz, S.R.Turner and M.A.Golub, Rubber Chemistry and Technology, 5, 809 (1982), W-K.Huang, G-H.Hsuie and W-H. Hou, Journal of Polymer Scince, Part A:Polymer Chemistry, 26, 1867 (1988), K.A. Jprgensen, Chemical Reviews, 89, 431 (1989) and Hermann, Fischer and Marz, Angew. Chem. Int. Ed. Engl., 30 (N 12) 1638 (1991).
Например, эпоксидирование исходного полимера может быть проведено взаимодействием с органическими перкислотами, которые могут быть приготовлены заранее или образованы.
Подходящие полученные заранее надкислоты включают перуксусную и пербензойную кислоты. Образование может быть осуществлено с использованием перекиси водорода и карбоновой кислоты с низким молекулярным весом, такой как муравьиная кислота. Альтернативно перекись водорода в присутствии уксусной кислоты или перекись водорода в присутствии уксусного ангидрида и катионообменной смолы будет образовывать надкислоту. Катионообменная смола может быть необязательно замещена сильной кислотой, такой как серная кислота или пара-толуолсульфокислота. Реакцию эпоксидирования можно проводить непосредственно в полимеризационном цементе (полимерный раствор, в котором полимеризовали полимер) или альтернативно полимер может быть снова растворен в инертном растворителе, таком как толуол, бензол, гексан, циклогексан, хлористый метилен и тому подобном, и эпоксидирование проводят в этом новом растворе, или полимер может быть эпоксидирован в чистом виде. Температуры эпоксидирования составляют от 0oC до 130oC и время реакции составляет от 0,1 до 72 часов. Если применяют перекись водорода и уксусную кислоту вместе с катализатором, таким как серная кислота, то продукт может представлять собой смесь эпоксида и сложного оксиэфира. Использование перекиси и муравьиной кислоты в присутствии сильной кислоты может также привести к получению полимерных блоков, содержащих эпоксидные и оксиэфирные группы. Благодаря этим побочным реакциям, вызванным присутствием кислоты, предпочтительно проводить эпоксидирование при минимально возможных температурах и в течение минимально короткого времени, согласованных с желаемой степенью эпоксидирования. Эпоксидирование может быть также осуществлено обработкой гидроперекисями или кислородом в присутствии переходных металлов, таких как молибден, вольфрам, хром, ванадий и серебро.
Эпоксидированные и необязательно избирательно гидрированные полимеры, полезные в способе согласно настоящему изобретению для получения оксидосодержащих полимеров согласно изобретению, описаны в зарегистрированных заявках на патент США под серийным номером 629839 от 21 апреля 1991 г. и N 722172, зарегистрированной 7 октября 1991 года. Способы получения таких полимеров подробно описаны в упомянутых выше заявках на патенты.
Последняя операция способа согласно изобретению включает контактирование эпоксидированного полимера со спиртом, имеющим одну незащищенную гидроксигруппу, и с соединением обшей формулы MXn, где M выбран из группы, включающей водород, бор, алюминий, железо и олово; X является галогеном и n - целое число, соответствующее валентности M, или с его органическим комплексом.
Предпочтительно эпоксидированный полимер контактирует со спиртом, имеющим одну незащищенную гидроксигруппу, в растворителе в присутствии катализатора, выбранного из группы, включающей кислоты Льюиса формулы MXn, где M является металлом, выбранным из группы, включающей бор, алюминий, железо и олово; X является галогеном и n - целое число, соответствующее валентности металла M, органические комплексы таких кислот Льюиса.
В этом случае предпочтительными катализаторами являются органические комплексы трехфтористого бора, наиболее предпочтительным катализатором является диэтиловый эфират трифторида бора, дибутиловый эфират трифторида бора, диметиловый эфират трифторида бора и трет-бутилметиловый эфират трифторида бора. Предпочтительно, чтобы молярное отношение катализатора к эпоксигруппам находилось в пределах от 0,1:1,0 до 1,0:1,0 и наиболее предпочтительным отношением является от 0,1 до 0,2 моля катализатора на моль эпоксифункциональности. Если это отношение слишком низкое, то не наблюдается превращения. Наиболее предпочтительно при проведении этого процесса, чтобы катализатор был разбавлен до концентрации 1,0 мэкв в 0,2-2,0 миллилитрах растворителя, при этом растворитель является тем же растворителем, который используется в реакции. Предпочтительно также относительно медленно добавлять раствор катализатора, например, порядка 1,0-10,0 миллилитров в минуту в реактор, содержащий 1,0 литр 10% по весу раствора эпоксидированного полимера в циклогексане.
Предпочтительно согласно настоящему изобретению спирт, имеющий одну незащищенную гидроксигруппу, выбран из одноатомных спиртов и производных двухатомных и трехатомных спиртов, имеющих одну незащищенную гидроксигруппу, а остальные гидроксигруппы защищены как ацеталь, кеталь или карбонат. Одноатомные спирты включают нормальные и вторичные спирты, насчитывающие от 1 до 10 углеродных атомов, наиболее предпочтительны н-бутанол, н-пропанол, н-пентанол, н-гексанол, н-гептанол и н-октанол. Производные двухатомных и трехатомных спиртов, имеющие одну незащищенную гидроксигруппу и остальные гидроксигруппы, защищенные, включают ацетали, кетали и карбонаты диолов и триолов и могут иметь строение, подобное следующим формулам:


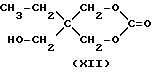
которые являются формулами для солкеталя (IX, ацетонкеталя глицерина) и триметилолпропанкеталя (X, ацетонкеталь триметилолпропана) или аналогичных карбонатов (формулы XI и XII). Получение других подходящих частично защищенных полиолов описано в Protective Groups in Organiс Synthesis, 2 nd Edition, by T.W. Green and P.G.M Wuts, Published in 1991 by J. Wiley and Sous of N.Y., N.J.
Наиболее подходящими соединениями для использования являются солкеталь, бутанол, триметилолпропанкеталь и октанол. Предпочтительно, чтобы молярное отношение эпокси к этому соединению находилось в пределах от 1:2 до 1:30. Большой избыток соединения предпочтителен для сведения к минимуму сшивания полимера.
Таким образом, предпочтительно, чтобы молярное отношение эпоксигрупп к соединению составляло от 1:15 до 1:25.
Эта реакция может быть проведена в интервале температур от 20oC до 200oC. Предпочтительный интервал температур от 25oC до 100oC. Более низкие температуры снижают скорость реакции и более высокие температуры промотируют побочные реакции, такие как сшивание и образование кетонов или альдегидов. Процесс может быть выполнен в течение времени от 15 минут до 24 часов после добавления катализатора, но почти во всех случаях требуется только 15 минут для достижения нужного результата (после полного добавления каталитического раствора).
Могут быть использованы разнообразные растворители, включая алифатические и ароматические углеводороды, галогенизированные углеводороды и ациклические простые эфиры. Эти растворители совместимы с катализаторами трифторида бора. Если используют катализаторы из других кислот Льюиса, то возможно, что потребуются другие приемлемые растворители. Неожиданно оказалось, что получение производного полимера может быть проведено добавлением спирта и катализатора из кислоты Льюиса (и необязательно добавлением внешнего растворителя для достижения желаемой концентрации твердой фазы) к среде для эпоксидирования (полимер в циклогексане, например), из которой уксусная кислота была удалена промывкой водным основанием, затем водой. Реакции, катализируемые кислотами Льюиса, часто чувствительны к влаге и растворимость воды в циклогексане значительная.
Достаточно неожиданным является то, что кетальзащищенные двух- и трехатомные спирты будут взаимодействовать с эпоксидированными полимерами с образованием растворимого полимера. Обычно считают, что защитные группы кеталя и ацеталя весьма доступны в присутствии катализатора трехфтористого бора. Следовало ожидать, что остаточные эпоксигруппы прореагируют с полученными высоко реакционноспособными первичными спиртовыми группами в присутствии трифторида бора, и это взаимодействие приводит к образованию поперечных связей. Указание на это содержится в Protective Grouрs in Organic Synthesis, 2nd Edition by P.W. Green and P.G.M. Wuts, published in 1911 by John Wiley and Sons, New York, NY.
В приведенной ссылке обсуждаются типичный ацеталь, метоксиметиленацеталь, и на страницах 411, 412, 421 и 423 показано, что этот материал высоко активен с трехфтористым бором. Основываясь на этом утверждении, можно предположить, что взаимодействия с участием солкеталя и триметилолпропанкеталя будут производить с образованием трехмерной структуры, как обсуждалось выше. В действительности защитные группы кеталя удаляются в процессе реакции или обработки; полимер-связанные кетали не выделяются. Для солкеталя это приводит к присоединению остатка, имеющего один первичный и один вторичный спирты, к главной цепи полимера через эфирную связь. В триметилолпропанкетале обе гидроксигруппы являются первичными. Неожиданно и то, что не наблюдалось увеличения побочных реакций, приводящих к сочетанию при использовании солкеталя или триметилолпропанкеталя с избытком, что наблюдается в случае одноатомных спиртов, если они взаимодействуют с тем же эпоксидировнным полимером с тем же содержанием твердого вещества и при том же молярном отношении эпокси к спирту.
В другом предпочтительном варианте осуществления настоящего изобретения последней операцией способа может быть контактирование эпоксидированного полимера со спиртовым раствором хлористоводородной кислоты.
Предпочтительно, чтобы этот спиртовой раствор хлористоводородной кислоты готовили добавлением водного раствора хлористоводородной кислоты, например 37% (по весу) водной хлористоводородной кислоты (концентрированный реактив AC), к спирту с тем, чтобы присутствовало от 0,01 до 1,0 г хлористоводородной кислоты в грамме полимера. По меньшей мере один эквивалент хлористоводородной кислоты должен присутствовать на эквивалент эпоксигрупп. Для обеспечения быстрой реакции наиболее предпочтительно иметь по меньшей мере 5 молей кислоты на моль эпоксигрупп. Далее предпочтительно, чтобы этот кислотный раствор добавляли к спирту в отношении от 1:2 до 1:10, предпочтительно от 1:5 до 1: 6 по объему, так как должно присутствовать достаточно спирта, чтобы обеспечить взаимодействие с полимером в органической фазе. Подходящие спирты содержат 1 - 4 углеродных атома, такие как метанол, этанол, н-пропанол, изопропанол, н-бутанол и вторбутанол, наиболее предпочтительным из них является метанол. Для получения спиртового раствора могут быть использованы более разбавленные растворы хлористоводородной кислоты, но слишком мало кислоты приводит к замедленным реакциям, как и слишком высокое отношение воды к спирту в реакционном растворе. Слишком высокая концентрация кислоты может приводить к побочным реакциям, ведущим к образованию трехмерной структуры. Может быть использована газообразная хлористоводородная кислота для получения спиртового реактива.
Подходящие растворители включают любой растворитель, в котором растворим полимер, однако предпочтительны углеводородные растворители (ароматические и алифатические) и наиболее предпочтителен циклогексан. Использование полярных растворителей, таких как тетрагидрофуран, может привести к проблемам эмульгирования, поскольку предпочтительно, чтобы остаточную кислоту удаляли из модифицируемого полимерного цемента промывкой водой. Также предпочтительно, чтобы раствор спиртового реактива добавляли к полимерному цементу в отношении от 1:1 до 1:10, предпочтительно от 1:5 до 1:6, так как добавление в таких отношениях обеспечивает присутствие достаточного количества спирта, чтобы облегчить взаимодействие в органической фазе.
Эта реакция может быть проведена в интервале температур от 20oC до 100oC. Предпочтителен интервал от 25 до 50oC. Более низкие температуры уменьшают скорость реакции, а более высокие температуры могут привести к побочным реакциям, таким как сшивание. Максимальная температура реакции будет также ограничена точкой кипения спирта. Предпочтительно проводить реакцию ниже температуры образования флегмы раствора. Процесс может быть проведен в течение времени от 15 минут до 24 часов после добавления хлористоводородной кислоты, но почти во всех случаях только 15 - 60 минут потребуется (после окончания добавления раствора хлористоводородной кислоты) для достижения достаточно полного гидролиза.
Использование конкретно хлористоводородной кислоты наиболее предпочтительно, так как она является наиболее практичным реактивом минеральной кислоты, способным провести эффективный гидролиз в предпочтительных условиях реакции, описанных выше. Хотя хлорная кислота может обеспечить достаточный гидролиз, но нежелательны опасности, связанные при работе с ней. Если хлористоводородную кислоту заменить серной кислотой в способе по настоящему изобретению или хлорной кислотой по способу по патенту США N 3555112, происходит конкурирующее сшивание и не получают растворимых полимеров, содержащих достаточные уровни спиртовых функциональных групп. В дополнение кислотный катализатор трудно удалить или нейтрализовать, что приводит к обесцвечиванию продукта при сушке. Реакции с применением сильных органических кислот, включая алкил- и арилсульфокислоты, создают аналогичные проблемы. Другие галогеновые кислоты, такие как бромистоводородная кислота (HBr) или иодистоводородная кислота (HI) могут привести к высокой конверсии, но известно, что даже следы этих кислот и их солей придают окрашивание продукту, особенно при выдерживании при повышенных температурах. Это изменение цвета будет вредным для многих предполагаемых применений, особенно в прозрачных покрытиях.
Следует ожидать, что значительное число спиртсодержащих повторяющихся элементарных звеньев, полученных способом согласно настоящему изобретению, являются хлоргидринами. Один из X или X в формуле (I) является атомом хлора и другой - группой ОН. Обработка сильным основанием может привести к регенерации эпоксида дегидрогалогенированием, но в большинстве случаев присутствие хлора рядом с гидроксигруппой не будет ограничивать полезности продукта.
Для обоих рассмотренных выше взаимодействий со спиртом в присутствии кислоты Льюиса и со спиртовым раствором хлористоводородной кислоты важным является концентрация эпоксидированного полимера в растворе. Она может находиться в пределах от 1,0 до 30,0 весовых процентов. Более высокие уровни твердого вещества способствуют образованию связанных (двумерных, трехмерных и так далее) полимеров вследствие межмолекулярных взаимодействий эпоксигрупп. Предпочтительны более низкие концентрации, от 10 до 20 процентов, наиболее предпочтительно 10 весовых процентов, так как это сводит к минимуму образование поперечных связей. Увеличение связывания с увеличением твердой фазы является наибольшим для высокомолекулярных линейных полимеров, но меньшим для звездообразных полимеров, имеющих относительно короткие (от 3000 до 10000 молекулярных масс) ответвления.
Отверждаемые вещества согласно настоящему изобретению полезны в клеях, герметиках, покрытиях, пленках (таких, которые удовлетворяют требованиям теплостойкости и устойчивости к растворителям) и так далее. В дополнение к полимеру с функциональными гидроксигруппами и любым отвердителям или реагентам в композиции продуктов, отвечающих требованиям конкретного применения, могут быть включены различные другие ингредиенты, такие как повышающие клейкость смолы, пластификаторы, наполнители и стабилизаторы (например, антиоксиданты и ингибиторы ультрафиолетового излучения).
Во многих применениях, особенно в клетях и герметиках, может быть необходимым добавление промотирующих адгезию или повышающих липкость смол, совместимых со средним каучуковым блоком полимера. Обычной повышающей клейкость смолой является диено-олефиновый сополимер пиперилена (1,3-пентадиена) и 2-метил-2-бутена, имеющий температуру размягчения 95oC. Эта смола поступает в продажу под товарным названием Wing tack 95 (Wing tack является товарным знаком), и ее получают катионной полимеризацией 60% 1,3-пентадиена, 10% изопрена, 5% циклопентадиена, 15% 2-метил-2-бутена и около 10% димера, как описано в патенте США N 3577398. Могут быть применены и другие смолы, повышающие клейкость, в которых содержание смолистого компонента составляет 20 - 80 весовых процентов сополимера 1,2-пентадиена и 80 - 20 весовых процентов 2-метил-2- бутена. Смолы обычно имеют температуры размягчения от 80 до 115oC (кольцо и шарик).
Другие способствующие повышению адгезии смолы могут быть также применены и включают гидрированные канифоли, сложные эфиры канифолей, олитерпены, терпенфенольные смолы и полимеризованные смешанные олефины, смолы с более низкими температурами размягчения и жидкие смолы. Примеры смол с низкой температурой размягчения и жидких смол включают Adtack, Piccolastic A5, Piccovar AP 10 и Piccolyte S 25, смолы, поставляемые Hercules (Adtak, Piccolastic, Piccovar и Piccolyte являются товарными знаками). Чтобы получить хорошую термоокислительную и цветную стабильность, желательно, чтобы повышающая клейкость смолы была насыщенной смолой, например, гидрогенизированной дициклопентадиеновой смолой, такой как серия смол Escorez 5000 (Escorez является товарным знаком), производимых фирмой Exxon, или гидрогенизированной полистироловой или полиальфаметилстироловой смолой, такой как смола Regalrez фирмы Геркулес (Regalrez является товарным знаком). Количество применяемой смолы для промотирования адгезии может варьироваться от 10 до 400 весовых частей на 100 частей каучука, предпочтительно от 20 до 350 весовых частей на 100. Выбор конкретного повышающего клейкость реагента по большей части зависит от конкретного полимера, применяемого в соответствующей клеевой композиции.
Необязательно может быть применена совместимая с ароматическим блоком смола. О совместимости судят по результатам метода, раскрытого в патенте США N 3917607. Нормально смола должна иметь температуру размягчения выше 100oC, определяемую методом E 28 Американского общества по испытаниям материалов, используя кольцевые и шариковые модели. Смеси смол, совместимых с ароматическими блоками, имеющие высокие и низкие температуры размягчения, могут быть также использованы. Полезные смолы включают кумарининденовые смолы, полистироловые смолы, винилтолуол-альфаметилстироловые сополимеры и полиинденовые смолы. Более предпочтительна кумарининденовая смола. Количество совместимой с ароматическим блоком смолы может варьироваться от 0 до 200 весовых частей на 100 частей массы блока.
Клеевая композиция может содержать пластификаторы, такие как пластификаторы для растяжения каучука или компаундные масла. Масла для компаундирования каучука хорошо известны в данной области и включают как масла с высоким содержанием насыщенных углеводородов, так и масла с высоким содержанием ароматических углеводородов. Предпочтительными пластификаторами являются высоко насыщенные масла, например масло Tufflo 6056 и 6204, поставляемое Lyondell, и переработанные масла, например масло Shellflex 371, поставляемое компанией Шелл (Tufflo и Shellflex являются товарными знаками). Масла с более высоким содержанием ароматических углеводородов включают Tufflo II и Shellflex 212. Количество масла для компаундирования каучука, применяемого в композиции согласно изобретению, может варьироваться от 0 до 500 весовых частей, предпочтительно от 0 до 100 весовых частей и наиболее предпочтительно от 0 до 60 весовых частей на 100 частей полимера.
В композицию для покрытия могут быть включены различные типы наполнителей и пигментов. Это особенно важно для внешних покрытий, в которые добавляют наполнители не только для придания привлекательного внешнего вида, но и также для улучшения характеристик покрытия, таких как погодостойкость. Могут быть использованы разнообразные наполнители. Подходящие наполнители включают карбонат кальция, глины, тальки, окись цинка, двуокись титана и тому подобные. Количество наполнителя обычно находится в пределах от 0 до 70 весовых процентов в расчете на свободную часть растворителя покрытия в зависимости от типа используемого наполнителя и предполагаемого применения покрытия. Особенно эффективным наполнителем является двуокись титана.
Если покрытия будут применять из раствора растворителя, то органическую часть покрытия следует растворить в растворителе или в смеси растворителей. Подходящими являются ароматические углеводороные растворители, такие как толуол, ксилол или Shell Cyclo Sol 53. Если желают, то можно получить обычно более низкую вязкость использованием смеси растворителей, содержащей ароматический углеводородный растворитель и полярный растворитель. Подходящие полярные растворители включают сложные эфиры, такие как изопропиловый эфир уксусной кислоты, кетоны, такие как метилизобутилкетон, и спирты, такие как изопропиловый спирт. Количество используемого полярного растворителя зависит от конкретного выбранного полярного растворителя и количества функциональных групп в функционально превращаемом гидрированном блок-сополимере. Обычно количество используемого полярного растворителя составляет от 0 до 50 весовых процентов в смеси растворителей.
Антиокислители и ингибиторы ультрафиолетового облучения могут быть добавлены в композиции для защиты составов от разложения окислением или при облучении солнечным светом в процессе приготовления и использования композиций. Сочетания стабилизаторов часто бывают более эффективными, что обусловлено различными механизмами разложения, которым подвержены различные полимеры.
Основным компонентом антиокислительной части сложного стабилизатора является антиоксидант затрудненного фенольного типа. Примерами поступающих в продажу антиоксидантов этого типа являются Etanox 330, CYANOX 2246 и SAGANOX 1010 (Etanox, Cyanox и Irganox являются товарными знаками). Разнообразные вторичные антиоксиданты и синергетики могут быть также включены в композицию. Примеры включают диалкилдитиокарбаматы цинка, такие как Butyl Zimate (Butyl Zimate является товарным знаком), сложные эфиры фосфористой кислоты, такие как WESTIN 618 (Westin является товарным знаком), и серусодержащие соединения, такие как дилаурилтиодипропионат, CYANOX LTDP (Cyanox Ltdp является товарным знаком). Антиоксиданты обычно используются в композиции в концентрациях от 0,05 до 5,0 весовых процентов.
Ингибирующая ультрафиолетовый свет часть сложного стабилизатора обычно будет состоять из сочетания соединения, абсорбирующего ультрафиолетовый свет, и стабилизатора света типа затрудненного амина. Обычные ингибиторы ультрафиолетового света абсорбирующего типа включают бензофеноновый тип, такой как CYASORB UV 531 (Cyasorb является товарным знаком), и бензотриазоловый типа, такой как TINUVIN P с TINUVIN 328 (Tinuvin является товарным знаком). Типичные стабилизаторы света из затрудненных аминов включают TINUVIN 770 и SANDUVOR 3056 (Sanduvor является товарным знаком). Эти ингибиторы ультрафиолетового света обычно включены в композицию в концентрациях от 0,05 до 10,0 весовых процентов.
Защитные пигменты и наполнители могут также повышать устойчивость к разрушению под воздействием солнечного света. Примеры включают газовую сажу, окись цинка и двуокись титана.
Композиции, содержащие полимер согласно настоящему изобретению, обычно готовят смешением компонентов при повышенной температуре, предпочтительно от 5oC до 200oC, до получения однородной смеси обычно менее трех часов. Известны различные методы смешения, и любой метод, приводящий к гомогенной смеси, удовлетворителен. Полученные композиции могут быть затем использованы для различного применения. Альтернативно ингредиенты могут быть смешаны в растворителе.
Клеевые композиции, содержащие полимеры согласно настоящему изобретению, могут быть использованы как различные виды адгезивов, например слоистые адгезивы, контактные адгезивы, монтажные клеи, чувствительные к давлению клеи, эффективные под прессом, пленочные клеи, термоплавкие клеи, устойчивые к растворителю клеи и водостойкие клеи, из которых вода удалена до отверждения. Этот клей может состоять просто из полимера с функциональными оксигруппами или, что более обычно, из составленной композиции, содержащей значительную часть полимера с функциональными оксигруппами вместе с другими известными компонентами клеевой композиции и, возможно, сшивающего средства. Наблюдалось, что введение спиртов в эпоксидные композиции, предназначенные для катионного отверждения, повышает скорость отверждения; предпочтительная композиция для катионного отверждения содержит эпоксидированный полимер с функциональными оксигруппами. Катионное отверждение может быть инициировано облучением пучком электронов или ультрафиолетовым светом в присутствии соответствующего фотоинициатора или термически активированных инициаторов.
Изобретение далее пояснено на следующих ниже примерах, однако без ограничения объема изобретения этими конкретными вариантами осуществления.
Пример 1.
В этом примере использовали два полимера-предшественника. Полимер A был линейным блок-сополимером стирола, изопрена и стирола, имеющим пиковую молекулярную массу, определенную проникающей гель-хроматографией, равную 160000, и содержание полистирола составляло 15%.
Полимер B был получен из ненасыщенного линейного блок-сополимера стирола, смеси бутадиена с изопреном и стирола, в котором 1,5 мэкв изопреновых элементарных звеньев было бессистемно распределено в среднем блоке полимера, имевшего пиковую молекулярную массу 50000 и содержание стирола 30 весовых процентов.
Полимер A не подвергали гидрированию, а полимер B был частично гидрирован так, что 1,2 мэкв олефиновых двойных связей оставалось в полимере. Частичное гидрирование проводили следующим образом: полимер в циклогексане подвергали взаимодействию с катализатором, приготовленным реакцией 2-этилгексаноата никеля с триэтилалюминием (2, 3 моля алюминия на моль никеля) и водородом при 34,5 бар (около 35 кг/см2) при температуре 70oC. Использовали 13 миллионных долей никеля (1,8 • 10-3 моля на г полимера). Катализатор и остаточный литий вымывали. Оба полимера были эпоксидированы взаимодействием с перуксусной кислотой при 45oC; кислоту добавляли в течение 60 минут, и затем раствор выдерживали при 45oC шесть часов. Добавляли 1,0 мэкв перуксусной кислоты на мэкв остаточного олефина в полимере B; один миллиэквивалент перуксусной кислоты добавляли к полимеру A на каждый мэкв эпоксигрупп в конечном продукте. Уксусная кислота и остаточная перуксусная кислота были нейтрализованы и раствор полимера тщательно промывали водой. Эпоксидированный полимер A содержал 1,30 мэкв эпоксигрупп на грамм полимера и эпоксидированный полимер B содержал 0,85 мэкв эпоксида на грамм полимера.
Чтобы превратить эпоксигруппы в гидроксигруппы, полимеры растворяли до содержания 10 весовых процентов в смеси толуола и спирта (н-бутанола или н-октанола). После продувки азотом при комнатной температуре по каплям добавляли диэтиловый эфират трифторида бора (0,15 моля на моль эпоксигрупп) в 15-25 миллилитрах толуола. Наблюдалась экзотермическая реакция 2-3oC. Раствор нагревали до 105oC требуемый период времени реакции (см. таблицу 1).
По мере охлаждения полимерный клей промывали избытком (150 объемных процентов) водного карбоната натрия и затем водой, осаждали в изопропаноле и затем сушили в вакууме. Растворимые продукты выделяли во всех случаях. Титрование остаточных эпоксидных групп осуществляли широко известным методом, добавляя раствор бромистого тетраэтиламмония в уксусной кислоте и титруя ацетат, полученный при взаимодействии эпоксигрупп со стандартизованной хлорной кислотой. О степени превращения эпоксигрупп в эфирно-спиртовые группы можно было судить по уменьшению эпоксидных групп в течение реакции. Использовали метод ядерного магнитного резонанса на 1H для контроля реакции. Анализ спектров 1H-ЯМР ненасыщенного полимера-предшественника B и эпоксидированного продукта использовали для определения распределения эпоксидированных олефиновых структур в полимере B; 45 процентов эпоксигрупп были производными от 1,4-изопреновых повторяющихся элементарных звеньев, 30% - от повторяющихся элементарных звеньев 3,4-изопрена и 25% - от повторяющихся элементарных звеньев 1,2-бутадиена; по существу вся ненасыщенность в предшественнике полимеру A была обусловлена повторяющимися элементарными звеньями 1,4-изопрена. Спектры 1H-ЯМР спиртовых производных содержали резонансы, обусловленные альфа-протонами к гидроксилу и эфирному кислороду, разрешенные от резонансов, обусловленных альфа-протонами к эпоксидному кислороду. Используя известное распределение структур повторяющихся эпоксидированных элементарных звеньев, можно оценить количество миллиэквивалентов элементарных эпоксидных звеньев (Е) и эфирно-спиртовых элементарных звеньев (ДЕ) и, таким образом, степень завершения реакции. Степень реакции (% реакции) определяли по следующей формуле: 100•ДЕЯМР/Еначальное, где Еначальное является количеством эпоксигрупп в эпоксидированном предшественнике, определенных методом ЯМР на 1H, и ДЕЯМР является содержанием ДЕ производного полимера (все измерения представлены в миллиэквивалентах на грамм).
Полимер B взаимодействовал с н-бутанолом или н-октанолом, как описано выше. Получали белое растворимое вещество, в процессе обработки на всех стадиях наблюдалось очень слабое эмульгирование. Титрование и ЯМР обнаруживали высокую степень превращения элементарных эпоксидных звеньев (Е) в эфирно-спиртовые элементарные звенья (ЕД). Исследования инфракрасного спектра методом преобразования рядов Фурье показали ожидаемые гидроксильные и эфирные полосы поглощения. Определялось небольшое количество или вообще не определялось присутствие кетоновых или альдегидных функциональностей. Гель-проникающую хроматографию использовали для контроля за изменениями молекулярной массы полимера, обусловленными распадом или связыванием (реакция между молекулами с образованием димеров, тримеров и так далее). Не было получено подтверждения образования образцов с низкой молекулярной массой (разложение), однако производные полимеры содержали около 15-20 процентов димера, 8-10% тримера, 4-5% тетрамера и 5-10% более высоко связанных продуктов.
Реакцию повторяли с использованием полимера A. Титрование показало высокую степень превращения эпоксидных групп в первые 45 минут. Продукты изменяли цвет при длительности реакции более 45 минут и в 24 часа продукт становился совсем желтым. Инфракрасный спектр поглощения, полученный методом преобразования рядов Фурье, показал уменьшение интенсивности полосы поглощения гидроксила в сравнении с интенсивностью эфирной полосы по мере увеличения времени реакции, что согласуется с дегидратацией, дающей виниловые эфирные элементарные звенья. Эти результаты ясно показывают, что более короткое время реакции благоприятно для субстратов ненасыщенного эпоксидированного полимера.
Спектр ЯМР на ядрах 13C производного A-полимера показал резонансы, наиболее согласующиеся с раскрытием колец эпоксигрупп 1,4-изопрена с образованием замещенной вторичной гидроксигруппы. Эти данные показывают, что разрыв колец эпоксида производного изопрена приводит к получению наиболее реакционноспособной (последней замещенной) оксифункциональности, как показано в формуле (I) в описании выше. Допуская, что в реакции полимера B с бутанолом также образуются вторичные спирты, результаты титрования и 1H-ЯМР анализа спиртов показывают, что около 30 относительно реакционноспособных вторичных гидроксигрупп введены в каждую молекулу полимера B.
Пример 2.
В этом примере модификации эпоксидированных полимеров A и B подвергали взаимодействию с изопропилиденглицерином: 25 г полимера растворяли в 10% по весу растворе смеси толуола и изопропилиденглицерина, реактор продували азотом и при комнатной температуре по каплям добавляли раствор диэтилового эфирата трифторида бора в 15 миллилитрах толуола. Наблюдалось повышение температуры на 3-4oC в связи с экзотермической реакцией. Раствор перемешивали при комнатной температуре или нагревали определенное время (см. таблицу 2). Затем раствор промывали избытком (150 объемных процентов) водного карбоната натрия и затем водой, осаждали в изопропаноле и затем сушили при 35-50oC в вакууме. Титрование остаточных эпоксигрупп и анализ спектра ЯМР на 1H проводили как описано в примере 1. Данные титрования сведены в таблице 2.
Временная защита реакционноспособных функциональных групп путем превращения в производное (защищенная группа), стабильное в заданных условиях, но легко переводимое в первоначальную форму в других условиях, является общей стратегией в органическом синтезе. Защита двух гидроксигрупп триола как кеталя приводит к продукту, который, как установлено, взаимодействует с эпоксидированными повторяющимися элементарными звеньями в полимере как монофункциональный спирт. Воздействие водной кислоты, например, предполагает гидролиз кеталя (снятие защиты) до диола. В случае солкеталя это приводит к одной первичной и одной вторичной спиртовой группе.
В первом эксперименте полимер B подвергали взаимодействию с солкеталем в условиях, аналогичных описанным в примере 1. После добавления кетализатора реакционную смесь нагревали до 105oC. В начальный стадии не происходило гелеобразования и титрование показывало почти полное израсходование эпоксифункциональности. Однако полимер изменял окраску при нагревании и проявляет тенденцию к сшиванию при хранении. Позднее было определено, что взаимодействие с эпоксигруппами в значительной степени завершалось после добавления катализатора при комнатной температуре. Более низкое значение для количества эфирно-спиртовых элементарных звеньев в полимере, полученном при 105oC, несмотря на сравнительное израсходование эпоксигрупп (при титровании), предполагает, что может происходить некоторое разложение смеси эфира и триола при этой температуре. Полимеры, полученные при комнатной температуре, были белыми и сохраняли стабильность. Во всех случаях инфракрасные спектры продуктов реакции, полученные преобразованием рядов Фурье, показали полосы, характерные для эфироспирта. Интенсивность полосы гидроксила предполагает присутствие более одной гидроксигруппы на повторяющуюся элементарную группу. Снова наблюдалось мало или вообще не наблюдались группы кетона или альдегида. Кетализируемый кислотой гидролиз в тетрагидрофуране не вызывает изменений в спектре. Из этих данных можно сделать вывод, что в процессе реакции или во время выделения продукта имеет место снятие защиты. Кетальные группы, как сообщалось, лабильны в присутствии кислот Льюиса. Анализы данных спектра ЯМР и титрования предполагают, что степень превращения сравнима с наблюдавшейся для реакций полимера B с одноатомными спиртами в предыдущем примере. Это свидетельствует о том, что около 90 гидроксигрупп введено в молекулу полимера, по меньшей мере 30 из них являются высоко реакционноспособными первичными группами. Гель-проникающие хроматограммы этих материалов почти идентичны хроматограммам производных одноатомного спирта; генерирование структуры триола не ведет к увеличенному связыванию.
Эксперимент с полимером A приводит к получению растворимого белого продукта. Продукт, полученный реакцией этого полимера при 105oC, нерастворим. Как и с производным полимера B, образование триола не ведет к гелеобразованию. Результаты анализов спектров ЯМР и инфракрасного спектра совпадали с ожидаемыми для эфирно-спиртового продукта. Низкий выход производных (эфироспирта) элементарных звеньев на спектре 1H-ЯМР предполагает, что побочные реакции могут израсходовать некоторую часть эпоксигрупп.
Солкетальное производное полимера A подвергали взаимодействию с уксусным ангидридом в пиридине. Спектроскопия инфракрасного спектра поглощения по методу преобразования Фурье обнаруживала значительную этерификацию, как и следовало ожидать, если присутствуют высоко реакционноспособные первичные оксигруппы. Реакция соответствующего производного бутанола приводила к меньшей этерификации. Отсюда ясно, что больше реакционноспособных оксигрупп введено реакцией с солкеталем, чем введено реакцией с бутанолом.
Пример 3.
Этот пример иллюстрирует использование способа согласно изобретению с точки зрения удовлетворения требований, предъявляемых к покрытиям. Для покрытий, в которых полимер является скорее основным связующим, чем модификатором, полимер должен иметь низкую вязкость (сохранять линейные свойства при комнатной температуре или близкой к ней) и достаточно высокую температуру стеклования, чтобы дать относительно твердое покрытие, а также обладать достаточной полярностью, чтобы придать приемлемую совместимость с относительно полярными сореактивами, такими как, например, изоцианаты и растворители.
Полимер, использованный в этом примере, имел следующее строение:
ДВБ[(ЭБ)С-ЭИ]n,
где ДВБ означает дивинилбензол, связующее вещество, ЭБ является полиэтилен-бутиленом (гидрированный полибутадиен), С является стиролом, ЭИ является эпоксидированным полиизопреном и n равно 15-20. Молекулярная масса цепи приблизительно равна 5100. Боковая цепь состоит из неупорядоченного сополимера полиэтилен-бутилена и стирола в весовом отношении 50:50, присоединенного одним концом к ядру дивинилбензола и другим концом присоединенного к полиизопреновому блоку с молекулярной массой 500. Общая молекулярная масса звездообразного полимера приблизительно равна 88000.
Этот полимер был получен обработкой звездообразного полимера с цепями из бутадиен/стирола и изопрена, частичным гидрированием полимера, как описано выше, чтобы полностью гидрировать бутадиеновые сегменты и частично гидрировать изопреновые сегменты, и затем эпоксидированием полимера, как описано выше, чтобы получить упомянутый выше полимер, который содержит 0,65 миллиэквивалента эпоксигрупп в изопреновых сегментах. Полимер подвергали взаимодействию со спиртом и диэтиловым эфиратом трифторида бора, как описано выше. Условия реакции приведены в таблице 3. Катализатор (в растворителе) по каплям добавляли к полимеру и раствору солкеталя при комнатной температуре. Через 15 минут после окончания добавления всего катализатора полимерный цемент промывали разбавленным водным основанием и затем водой. Большую часть растворителя удаляли выпариванием в вытяжном шкафу и полимер далее сушили в вакууме.
Эпоксидированный звездообразный полимер на начальной стадии подвергали взаимодействию в условиях, наиболее эффективных для получения полимера B, а именно 10% твердого вещества и смесь толуол-солкеталь в отношении 85:15 при комнатной температуре, используя в качестве катализатора диэтиловый эфират трифторида бора. Как ожидалось, результаты анализов согласовывались с существенным превращением эпоксигрупп в эфиротриоловые. Конверсия может быть также осуществлена добавлением солкеталя и катализатора к промытому цементу (эпоксидированному полимеру в растворителе, в котором он был эпоксидирован, обычно в циклогексане). В качестве катализатора использовали дибутиловый эфир трифторида бора, так как диэтиловый эфират нерастворим в циклогексане. Не было свидетельства наличия остаточной воды в цементе, мешающей каким-либо образом проведению реакции.
Титрование, спектроскопию 1H-ЯМР, спектральный анализ в ИК-области поглощения с применением преобразования рядов Фурье и гель-проникающую хроматографию проводили как описано ранее. Были получены результаты спектроскопических анализов, совпадающие с преобразованием в эфир-триол. Кроме того, резко увеличивалось связывание, как определено гель-проникающей хроматографией, в сравнении с линейными полимерами в предыдущем примере, полученными с тем же содержанием твердого вещества и отношением солкеталя к растворителю; образовывалось только около 10-20% димера и совсем немного или вообще не образовывалось более высоких сетчатых структур.
Степень реакции указывает на то, что около 1,5 производных повторяющихся элементарных звеньев DE (эфира-спирта) присутствовало в боковой цепи (22-30 на молекулу), то есть около 4,5 оксигрупп на боковую цепь (66-90 на молекулу), по меньшей мере одна треть которых являются первичными.
Пример 4.
В этом примере солкетальные производные полимера B и звездообразного полимера по примеру 3 (полимер C) отверждали ароматическими и алифатическими изоцианатами для получения полиуретановых покрытий. Покрытия, полученные из звездообразного производного, показали хорошую прозрачность и внешний вид и, хотя будучи довольно мягкими, обладали приемлемой химической стойкостью и адгезией.
Эти покрытия получали растворением полимера B (0,85 миллиэквивалента эпоксигрупп на грамм полимера) и полимера A (0,65 мэкв эпокси/г), прореагировавших с солкеталем и диэтиловым эфиратом трифторида бора, как описано ранее, до 20 весовых процентов в толуоле. Затем добавляли дифенилметандиизоцианат (MONDUR, поставляемый Mobay) или изофоронадиизоцианат (IPDI, поставляемый Huls) к растворам каждого полимера в концентрации 1 мэкв на один мэкв эпоксигрупп в исходном полимере. Добавляли 0,1 весового процента (относительно диизоцианата) дилаурата дибутилолова (BAYS/LONE 162, поставляемого Mobay) с изофорондиизоцианатом в качестве катализатора. Затем тонкие пленки (0,05 мм) наносили на стекло и стальные панели и более толстые пленки (1,3 мм) формовали литьем растворов в бумажные лодочки. Небольшое количество каждого раствора наносили также на пластинки из хлористого натрия для проведения анализа ИК-спектра методом преобразования уравнения Фурье. Пленки сушили при комнатной температуре две недели, первые три типа пленок сушили в условиях окружающей среды и образцы для спектрального анализа - в эксикаторе до измерения каких-либо физических свойств. ИК-спектры записывали спустя 1, 2, 4, 8 и 14 дней. Пленки спиртовых производных (не содержащие изоцианата) и пленки с изоцианатами с эпоксидированным полимеров B (без гидроксигрупп) отливали в качестве контрольных.
Все спиртсодержащие полимеры взаимодействовали с диизоцианатами с образованием сшитой сетчатой структуры, но образование сплошной сетки от звездообразного полимера проходило медленнее. Образцы композиций, полученные из производного полимера B, оставленные в колбах, желатинизировали в течение полусуток, тогда как образцы, полученные из звездообразного полимера, требовали по меньшей мере неделю для гелеобразования. Ни один из контрольных образцов не показал какого-либо увеличения вязкости. Через две недели, когда образцы пленок помещали в толуол, отвержденные изоцианатом спиртовые производные набухали (около 50%), сохраняя свою механическую целостность, указывающую на прочно сшитую сетку.
Пленки, полученные из производного полимера B и отлитые на стекло, показали высокий глянец, не имели отлипов и были несколько тверже, чем пленки неотвержденного контроля, но имели шероховатую структуру поверхности, "апельсиновую корку", как и у покрытий неотвержденного контроля и покрытий на основе полимера B (не содержащего гидроксигрупп), дефект, связанный со слабым увлажнением поверхности при получении покрытия. Отвержденное дифенилметандиизоцианатом покрытие было несколько мутным, указание на слабую совместимость с высшим алифатическим полимером. Соответствующая пленка, полученная из полимера B (без гидроксигрупп), была совершенно прозрачной.
Внешний вид покрытий, полученных из производного звездообразного полимера (полимер C), был гораздо лучше. Покрытия имели высокий глянец, не имели "апельсиновой корки", были более твердыми и менее липкими, чем неотвержденный контроль. Отвержденное диизоцианатом изофорона покрытие было кристально чистым, но слегка липким, тогда как отвержденное диизоцианатом дифенилметана покрытие не имело отлипов, но было слегка мутноватым в утолщенных частях. Полимер полностью совместим с алифатическим изоцианатом.
Пленки испытывали на механические свойства. Внешний вид оценивали визуально. Тесты на царапание карандашом и полукруглой стамеской выполнены стандартным методом, проводя графитом с последовательно возрастающей твердостью по покрытию до образования царапины или трещины соответственно. Тест на циклическое истирание с метилэтилкетоном проводили, протирая покрытие тканью, смоченной в метилэтилкетоне, в течение 100 циклов или до появления прорыва в подложке (один цикл включает одно протирание, выполненное вперед и назад). Результаты представлены в таблице 4.
Для звездообразного полимера канавка быстро протиралась через контрольную (неотвержденную) пленку в тексте на трение с метилэтилкетоном, тогда как оба отвержденных образца были довольно устойчивыми.
Образец, отвержденный дифенилметандиизоцианатом, выдержал свыше 100 циклов трения. Пленки производного полимера B отслаивались от подложки вследствие малой адгезии. В результате не могли быть получены достоверные данные в тексте на истирание с метилэтилкетоном.
Отверждение может также сопровождаться увеличением поглощения карбонила уретана и соответствующим снижением поглощения изоцианата, используя инфракрасный спектр поглощения. Эти результаты предполагают, что отвержденные было в значительной степени полным для покрытий, отвержденных ароматическим изоцианатом менее чем за 24 часа, тогда как требовалось около 48 часов для покрытий, отвержденных алифатическим изоцианатом.
Пример 5.
Звездообразный полимер по примеру 3 (полимер C) подвергали взаимодействию с н-бутанолом в смеси толуол-циклогексан при комнатной температуре, используя диэтиловый эфир трифторида бора, как описано в примере 3, для реакции с солкеталем.
Полимер C и полимер B подвергали также взаимодействию с кеталем ацетона триметилолпропана при комнатной температуре, как описано для реакции с солкеталем в примерах 2 и 3 соответственно. При реакции с н-бутанолом эпоксидированные повторяющиеся элементарные звенья превращаются в соответствующий эфироспирт, в котором R является н-бутилом, и только введенные гидроксигруппы присоединены к главной цепи полимера преимущественно у вторичных углеродных атомов. Реакция с ацетонкеталем триметилолпропана вводит R-группу, имеющую две первичные гидроксигруппы, приводящие к соответствующему эфиротриолу, сочетание, как определено гель-проникающей хроматографией, было сравнимо с таковым, о котором сообщалось для соответствующих производных полимеров B и C в предыдущих примерах. Условия реакции и результаты анализов сведены в таблицу 5.
Пример 6.
В этом примере производные бутанола и ацетонкеталя триметилпропана по примеру 5 отверждали ароматическими и алифатическими изоцианатами, как описано в примере 4. Все отвержденные изоцианатом пленки были сшитыми (нерастворимые). Хотя устойчивость к истиранию с метилэтилкетоном была сравнительно невысока, улучшение наблюдалось в неотвержденных контрольных пленках. Как и в примере 4, пленки полимера B отслаивались по причине слабой адгезии. Ни одна из отвержденных пленок не была липкой, хотя пленки были относительно пластичными. В большинстве случаев наблюдалась достаточно хорошая совместимость, приводящая к прозрачным пленкам.
Для покрытий, классифицированных как "слегка замутненные", помутнение было заметно только в утолщенных частях. Неожиданным явилось то, производные бутанола, не имеющие достаточного количества первичных спиртовых групп, отверждались с образованием пленок, по своим свойствам сравнимых с пленками, полученными из полимеров, содержащих первичные спиртовые группы. Так как стоимость при получении бутаноловых производных, по-видимому, значительно ниже, то такие продукты могут иметь значение для рынка. Свойства этих покрытий сведены в таблице 6.
Пример 7.
10 г эпоксидированного полимера B, полученного как и в примере 1, растворяли в 100 г циклогексана. Добавляли 24 мл раствора, содержащего метанол и концентрированную хлористоводородную кислоту в отношении 5:1 по объему (37% водная HCl, ACS реактив в концентрации 0,17 г/г полимера). Раствор перемешивали либо при комнатной температуре, либо при 50oC в течение желаемого времени (от 15 минут до 6 часов). Небольшие образцы отбирали через определенные промежутки времени для анализа спектра 1H-ЯМР и инфракрасного спектра. Реакционную смесь охлаждали и полимеризационную среду промывали водой. Полимер выделяли осаждением в изопропаноле и затем сушили в вакууме при 50oC. Получали белое растворимое вещество.
Результаты анализов сведены в таблице 7. Титрование остаточных эпоксигрупп проводили широко используемым методом добавления бромистого тетраэтиламмония в уксусной кислоте и титрованием полученного ацетата реакцией эпоксигрупп со стандартизованной хлорной кислотой. 0 степени превращения эпоксигрупп в спиртовые группы можно судить по уменьшению эпоксифункциональности в ходе реакции. Спектры 1H-ЯМР обнаруживают резонансы, идентифицированные как обусловленные протонами на атоме углерода, связанном с гидроксигруппой (H-C-OH) или хлором (H-C-Cl); протоны появляются в той же области спектра. Резонансы, обусловленные гидроксигруппами, почти полностью отсутствовали, и расчеты показали почти 96-процентную реакцию. Спектры 1H-ЯМР, полученные на 15 и 30-й минутах, показали, что реакция в значительной мере завершилась в первые 15 минут. Инфракрасные спектры, определенные с преобразованием рядов Фурье, как ожидалось, показали интенсивные резонансы при 3450 см-1 (связанный водородом OH) и более слабые полосы при 3580 см-1 (мономерный OH) и при 1070 и 1030 см-1 (полоса C-OH). Никаких новых резонансов в области C-Cl (800-600 см-1) нельзя было идентифицировать. Однако эти резонансы могут быть замаскированы в интенсивном широком поглощении при 750 см-1, присутствующем в исходном полимере. Не наблюдалось эфирных полос C-O-C. Это свидетельство предполагает, что большинство, если не все, производных повторяющихся элементарных звеньев являются диолами (продукт добавления воды) или хлоргидринами (продукт добавления хлористоводородной кислоты), а не эфироспиртами (продукт добавления метанола). Это свидетельство не исключает присутствия некоторого числа метокси-спиртовых повторяющихся элементарных звеньев. Промывка одной водой или водным карбонатом натрия, кажется, дает такую же структуру.
Рассмотрение таблицы 7 показывает, что эпоксигруппы были израсходованы очень быстро и реакция, по-видимому, в значительной степени была закончена в течение 15 минут при 50oC. Эти результаты противоречат патенту США N 3555112, согласно которому около 50% или более эпоксигрупп остаются нетронутыми после 6 часов реакции при 50oC.
Пример 8.
Связанный полимер по примеру 3 гидролизовали как описано в примере 7. Результаты анализов представлены в таблице 8. Как и в примере 7, спектроскопические данные наиболее согласуются с гидролизом эпоксигрупп до диолов и/или дополнения хлористоводородной кислоты до образования хлоргидрина. Результаты, представленные в таблице, показывают, что этот полимер более трудно гидролизуется, чем линейный полимер по примеру 7. Однако ясно, что существенное превращение происходит в предпочтительное время реакции продолжительностью один час.
Примеры для сравнения, пример 1.
В этом примере 10 г эпоксидированного полимера по примеру 7 растворяли в 100 г циклогексана. Добавляли 24 мл раствора, содержащего метанол и концентрированную серную кислоту в объемном отношении 5 : 1 (96% водная серная кислота, концентрированный реактив Американского химического общества). Раствор перемешивали при комнатной температуре шесть часов. Малые пробы для анализа спектра ЯМР и ИК-спектра отбирали после 15-, 30- и 60-минутных интервалов. Полимерный раствор промывали деионизированной водой. Полимер выделяли осаждением в изопропанол и затем сушили в вакууме при 50oC. Образцы показали увеличение степени сшивания от времени реакции. Образцы, взятые в течение первых 60 минут появления в растворе, не растворялись, а только набухали после осаждения. Реакционная смесь начала образовывать неплотный гель по истечении 360 минут. Когда полимер оставляли на ночь для просушивания, изменения окрашивания были очевидны. Промывка полимерного цемента разбавленным водным карбонатом натрия и затем водой до осаждения не оказывали воздействия на продукт. Сшивание и изменение цвета наблюдались по-прежнему.
Пример для сравнения 2.
Эксперимент, описанный в примере 1, повторяли, используя 37% (по весу) водную серную кислоту вместо концентрированного реактива. Образец отбирали через 15 минут, а остальной продукт выделяли через 60 минут. Гелеобразование по-прежнему было налицо, хотя сшивание казалось менее интенсивным. Изопропаноловые осадки, набухшие в толуоле, отливали на пластинки хлористого натрия для проведения ИК-спектроскопии. После сушки в вакууме при 40oC записывали спектры. В противоположность результатам реакции с хлористоводородной кислотой ИК-спектры показали слабые признаки реакции.
Пример для сравнения 3.
Растворимые спиртовые производные полимера по примеру 7 могут быть получены с использованием серной кислоты и алифатических и ароматических сульфокислот, если присутствуют большие количества воды, как описано в патенте США N 3607982, и использовали очень малые концентрации кислоты (от 5 до 20 молярных процентов эпоксигрупп). Реакции проводили следующим образом. 25 г полимера по примеру 7 растворяли в концентрации 10 весовых процентов в смеси изопропанол-толуол при объемном отношении 25 : 75 с последующим добавлением желаемого количества кислотного катализатора (в 20 мл изопропанола) и реакционную смесь нагревали при 50oC с перемешиванием желаемое время. Полимер выделяли осаждением в изопропанол, повторно растворяли в 10 весовых процентах толуола и промывали разбавленным водным карбонатом натрия, затем водой. Полимер снова осаждали в изопропаноле и сушили в вакууме при 50oC. Условия реакции и результаты представлены в таблице 9.
Конверсия даже через пять часов была очень низкой, если в качестве катализатора использовали серную кислоту. Более высокие концентрации метансульфокислоты (МСК) также давали существенное превращение лишь после очень длительного времени реакции. Более высокие степени превращения могут быть получены с использованием додецилбензолсульфокислоты (Cycat 600, поставляемой American Cyanamid в виде 72-процентного по весу раствора в изопропаноле, Cycat является товарным знаком). Однако этот катализатор очень трудно вымывается из полимера и остатки часто катализируют процесс сшивания при сушке полимера. Растворимый полимер может быть надежно получен только при добавлении достаточного количества нелетучего основания, такого как диизопропаноламин для нейтрализации катализатора. Полученная соль не может быть удалена и составляет значительный процент выделенного продукта. При более высоких температурах реакции сшивание, ведущее к гелеобразованию, конкурировало с гидролизом. В результате было установлено, что более высокие температуры реакций нежелательны.
Пример 9.
Гидролизованный хлористоводородный кислотой полимер по примеру 8 отверждали аминосмолой следующим образом. Полимер смешивали с гексаметоксиметилмеламином (Cynol 303, American Cyanamid) и ароматической сульфокислотой в качестве катализатора (Cycat 600, American Cyanamid) в смеси ксилол-н-бутанол в отношении 90 : 10 (Cynol является товарным знаком). Получали композиции, содержащие аминосмолу с общим количеством твердого вещества около 4%, 9%, 16% и 28 процентов. На каждый грамм аминосмолы добавляли 0,1 г катализатора. Пленки толщиной от 0,01 до 0,05 мм (0,5 - 2,0 мил) наносили на стальные и алюминиевые плиты и отверждали при
1) 150oC в течение 20 минут пли
2) 200oC в течение 10 минут.
Контрольные группы получали литьем пленок полимера и одного процента (от веса твердого вещества) ароматической сульфокислоты Cycat 600.
После термообработки все отвержденные меламином пленки были прозрачными, глянцевыми и твердыми. Пленки, отвержденные при 200oC, были слегка желтыми, тогда как пленки, спеченные при 150oC, были бесцветными. Контрольные пленки были в обоих случаях окрашены, пластичны и липки. Хотя композиции, приготовленные с одной ароматической сульфокислотой Cycat 600, и меланинсодержащие композици были полностью сшиты, ясно, что свойства пленок без меламина были значительно хуже.
Результаты испытания физических свойств сведены в таблице 10. Карандашную твердость до появления царапин и вмятин определяли с использованием стандартного метода, проводя графитом с последовательно увеличивающейся твердостью по покрытию до появления царапины или вмятины соответственно. Тест на истирание с метилэтилкетоном проводили согласно стандартному методу, проводя 200 раз по поверхности покрытия тканью, увлажненной метилэтилкетоном, или до появления прорыва к подложке. Определение адгезии методом решетчатого надреза проводили стандартным способом нанесения сетчатого образца нарезкой и контактированием его с чувствительной к давлению лентой. Оценка определяется количеством снятого покрытия. Оценки проводили по шкале от 0 до 5, при этом 5 означает, что покрытие не снимается и при 0 удаляется более 65 процентов покрытия.
Как можно видеть из таблицы 10, твердость была очень высокой, особенно для покрытий, содержащих наибольшую концентрацию аминосмол. Тест на истирание с метилэтилкетоном показывает, что степень отверждения зависит от уровня меламина. Высоко отвержденные (более плотно сшитые) покрытия получали при наиболее высокой концентрации меламина. В большинстве случаев адгезия была ниже оптимальной, но отчетливо лучше для композиций с высоким содержанием меламина. Использование смол, таких как Cymel 1125 (которая содержит группы карбоновых кислот), или реактивов совместно с реактивами, содержащими карбоновую кислоту, может далее улучшить адгезию. Покрытия обнаруживали полезные свойства в пределах рекомендованных изготовителем концентраций от 15 до 30 процентов меламина для общей обработки металла. Твердость, прозрачность и блеск показывают, что эти материалы обладают высокими потенциальными возможностями в покрытиях, отвержденных аминосмолами.


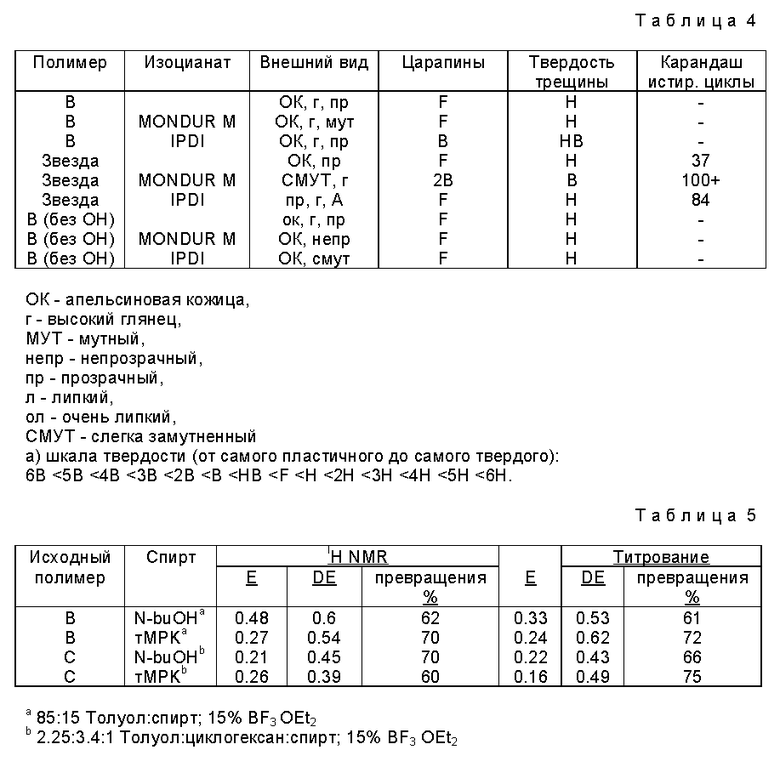



Описывается способ получения блок-сополимеров сопряженных диенов, включающий стадии: а) получение предшествующего полимера полимеризацией по меньшей мере одного сопряженного диена с получением продукта, содержащего 1,2-двузамещенные, 1,1-двузамещенные, 1,1,2-тризамещенные или четырезамещенные олефиновые двойные связи в полимере; b) эпоксидирование полимера - предшественника с образованием эпоксигрупп на местах замещения при количестве функциональных эпоксигрупп в полимере от 0,1 до 5,0 мэкв/г полимера, с) контактирование эпоксидированного полимера со спиртом, имеющим одну незамещенную гидроксигруппу, и либо с соединением общей формулы МХn, где М выбран из группы, включающей водород, бор, алюминий, железо и олово; Х является галогеном и n -целое число, соответствующее валентности М, или с его органическим комплексом. Описывается также сопряженный диеновый блок-сополимер, содержащий от 0,1 до 15 мэкв гидроксигрупп на 1 г полимера, спиртовые группы формул (I) или (II), в которых Р является -ОН, -Сl или -OR, где R выбран из группы, включающей алкильные радикалы, содержащие до 10 углеродных атомов, группы одноатомных и двухатомных спиртов и карбонатные группы; Q является -ОН или -Сl при условии, что по меньшей мере один из R и Q являлся -ОН и R1 и R2 были водородом или алкильными радикалами при условии, что если Р является -ОН, то только один из R1 и R2 может быть водородом. Техническим результатом изобретения является разработка способа получения блок-сополимеров, обеспечивающего высокую степень конверсии при относительно коротком времени реакции в хорошо идентифицируемый продукт в мягких условиях. 3 с. и 23 з.п. ф-лы, 10 табл.

или
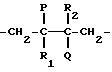 I
I
или II
II
где P = -OH, -Cl - OR,
где R выбран из группы, включающей алкильный радикал, содержащий до десяти атомов углерода и возможно имеющий в своей структуре одну или две спиртовые группы, карбонатные группы, в случае, если в структуру R входит карбонатная группа, то P относится к эфирной группе;
Q = -OH, -Cl,
при условии, что, по крайней мере, или P, или Q является -OH,
и R1 и R2 являются водородом или алкильными радикалами, при условии, что если P = -OR, то только один из R1 и R2 может быть водородом.
Cq - Yp - (Bx - A)r,
где B является полимерным блоком, включающим гидрированный сопряженный диен, возможно, содержащий мономерные звенья формул (I) или (II), и X означает 0 или 1;
где A означает полимерный блок, содержащий мономерные звенья формулы (I) или (II) при более высокой концентрации, чем в блоке B;
где C является одинаковым с A или B или является полимерным блоком, полученным из акрилатного или метакрилатного мономера,
где Y является остатком агента сочетания, остатком инициатора или полимерного блока, полученного из сочетающихся мономеров, и где r, q и p являются целыми числами, причем r равно по крайней мере 1, q больше или равно 0, r + q равно от 1 до 100 и p равно 0 или 1.
SZ - A - SZ',
где Z и Z' = 0 или 1;
S - моноалкенилароматический мономер;
A - блок, содержащий статистически распределенные звенья гидрированного или негидрированного диенового мономера и мономерные звенья структурных звеньев I и II c содержанием в них гидроксильных групп в количестве 0,1 - 15,0 мэкв/г полимера.
| US, 3607982, A, 1971 | |||
| Химическая модификация полимеров диенов, обзор | |||
| - М.: ЦНИИТЭНефтехим, 1976, с | |||
| Прибор для промывания газов | 1922 |
|
SU20A1 |
| EP, 387947, 1990 | |||
| Пожарный двухцилиндровый насос | 0 |
|
SU90A1 |
| US 3135716, A, 1964 | |||
| US 4051199, A, 1977 | |||
| US 5002676, A, 1991. | |||

