Изобретение относится к новому способу получения абсолютно чистого окситетрациклина, не содержащего ацетилдекарбоксамидоокситетрациклина, а также к новому промежуточному продукту для осуществления этого способа - ацетату гидрохлорида окситетрациклина.
Известно, что в результате ферментативного получения окситетрациклина формулы (I), указанной ниже, в качестве побочного продукта получается также 2-ацетил-2-декарбоксамидоокситетрациклин в количестве от 3 до 12,5% (далее ацетилдекарбоксамидоокситетрациклин) формулы II указанной ниже.

[Antibiotiki 11, 598 (1966)].
Используя обычные способы выделения окситетрациклина из бульона брожения и дальнейшую его очистку, содержание в нем ацетилдекарбоксамидоокситетрациклина снижается лишь незначительно, и он остается как загрязнение в получаемом конечном продукте - окситетрациклине. Наличие ацетилдекарбоксамидоокситетрациклина как побочного продукта в окситетрациклине впервые было доказано путем хроматографического разделения этих двух соединений методом тонкослойной хроматографии [Archiv der Pharmazie 300, 840 (1967)]. Было проведено испытание 15 образцов окситетрациклина различных изготовителей антибиотиков и было подтверждено методом качественного анализа наличие в них ацетилдекарбоксамидоокситетрациклина. Содержание ацетилдекарбоксамидоокситетрациклина в образцах окситетрациклина было определено методом жидкостной хроматографии высокого давления (ЖХВД) [Journal of Chromatography 405, 229 (1987)].
Ацетилдекарбоксамидоокситетрациклин был получен ранее ферментативным путем. Продукт выделяли и очищали методом противоточного распределения потока и идентифицировали его химическим методом [Journal of American Chemical Society, 82, 5934 (1960)], и этот продукт был запатентован как антибиотик [патент США 3022347 (1962)]. Исследование биологической активности ацетилдекарбоксамидоокситетрациклина на 33 микроорганизмах показало наличие такого же антибактериального спектра, как и у окситетрациклина, в то время как сила его действия не достигала даже 10% от силы действия окситетрациклина.
Благодаря чрезвычайно большому сходству молекул окситетрациклина с молекулами ацетилдекарбоксамидоокситетрациклина до настоящего времени отсутствует способ выделения окситетрациклина, который полностью не содержит примеси ацетилдекарбоксамидоокситетрациклина.
Известно, что чистый окситетрациклин может быть получен из чистого дигидрата окситетрациклина, полученного ферментацией или одной из его гидрогалидных солей. Один из этих процессов описан в патентах США N 2915555 (1959) и в GB N 834579 (1960).
Способ предусматривает растворение неочищенного дигидрата окситетрациклина или гидрохлорида в C1-C6 алифатическом спирте в присутствии растворимого в спирте галогенида цинка, щелочного или щелочноземельного металла (за исключением кальция). При использовании неочищенного дигидрата окситетрациклина в качестве исходного соединения также применяются органический амин в незначительном молярном избытке, рассчитанном на окситетрациклин, и небольшое количество соответствующего гидрохлорида; сохранившиеся нерастворимые примеси отделяются от конечного продукта фильтрацией, а чистый продукт в фильтрате осаждается добавлением концентрированного раствора соответствующего гидрохлорида.
Этот метод пригоден для выделения примесей, обычно присутствующих в неочищенном окситетрациклине, полученном путем ферментации. Однако ни в этом способе, ни в других способах, известных до даты приоритета данной заявки, при получении окситетрациклина ацетилдекарбоксамидоокситетрациклин (ADOTC) не упоминался как примесь, хотя он присутствовал в образце полученном в результате ферментации.
Известно также, что окситетрациклин образует соли в виде молекулярных комплексов с различными органическими кислотами и основаниями (японский патент N 17044/63; Chem.Abstr., 60. 2875а). Кристаллическая структура некоторых из этих солей определяется методом дифракции рентгеновских лучей (Bull. Chem. Soc. Japan, 36, 1163-8(1963)). Были описаны соли в виде молекулярных комплексов гидрохлорида окситетрациклина с щавелевой кислотой, пиразином и монохлоруксусной кислотой.
Согласно настоящему изобретению предлагается способ очистки окситетрациклина от присутствующего в нем в качестве примеси ацетилдекарбоксамидоокситетрациклина, заключающийся в том, что гидрохлорид окситетрациклина содержащий ацетилдекарбоксамидоокситетрациклин в качестве примеси, суспендируют в ледяной уксусной кислоте, смесь перемешивают в течение пяти часов, образующийся осадок ацетата гидрохлорида окситетрациклина фильтруют, промывают ледяной уксусной кислотой и ацетоном, после чего высушивают при пониженном давлении и температуре до 40oC до достижения постоянного веса и переводят в основание окситетрациклина.
Суспендировать можно в ледяной уксусной кислоте гидрохлорид окситетрациклина, полученный в результате добавления эквимолярного количества по отношению к содержащемуся окситетрациклину хлористого водорода в виде концентрированной соляной кислоты.
Обычно эквимолярное по отношению к содержащемуся окситетрациклину количество хлористого водорода в виде концентрированной соляной кислоты добавляют непосредственно к раствору дигидрата окситетрациклина в ледяной уксусной кислоте.
Впервые неожиданно было обнаружено, что гидрохлорид окситетрациклина, растворенный в ледяной уксусной кислоте, образует соль в виде молекулярного комплекса, а именно ацетата гидрохлорида окситетрациклина. Значительное различие растворимости соли в виде молекулярных комплексов окситетрациклина и ацетилдекарбоксамидоокситетрациклина в ледяной уксусной кислоте обеспечивает эффективное отделение одного продукта от другого. В частности, если гидрохлорид окситетрациклина, содержащий гидрохлорид ацетилдекарбоксамидоокситетрациклина в качестве примеси (примерно 3%) суспендируется в ледяной уксусной кислоте, то эта соль сначала полностью растворяется, после чего окситетрациклин осаждается в виде соли в виде молекулярного комплекса. Соль в виде молекулярного комплекса ацетилдекарбоксамидоокситетрациклина при данных условиях является высокорастворимой и остается практически в растворе. После осаждения и фильтрации ацетата гидрохлорида окситетрациклина, окситетрациклин получают из него с чрезвычайно высоким выходом, в котором содержание ацетилдекарбоксамидоокситетрациклина согласно данным ЖХВД снижается до 0,3-0,5%. Фильтрат содержит практически весь ацетилдекарбоксамидоокситетрациклин и примерно 1-2% окситетрациклина от исходного количества, который может быть регенерирован путем выпаривания фильтрата. Окситетрациклин может быть подвергнут такой очистке также в виде дигидрата основания (далее - дигидрат окситетрациклина). В данном случае также добавляется эквимолярное количество хлористого водорода в виде концентрированной соляной кислоты в раствор дигидрата окситетрациклина в ледяной уксусной кислоте, что обеспечивает возможность образования соли в виде молекулярного комплекса ацетата гидрохлорида окситетрациклина. После начала осаждения этого комплекса может добавляться толуол в количестве до 80 об.% от присутствующей ледяной уксусной кислоты, с целью снижения растворимости продукта. Если в качестве исходного соединения используют сырой дигидрат окситетрациклина, то желательно, чтобы он был предварительно превращен - с использованием хорошо известных способов - в гидрохлорид окситетрациклина, который может далее без сушки превращаться в ацетат гидрохлорида окситетрациклина. Соотношение ледяной уксусной кислоты к окситетрациклину обычно находится в пределах 1,5-5 мл/ммоль окситетрациклина. Наилучший результат достигается при соотношении 2,5 мл/ммоль окситетрациклина.
Способ согласно настоящему изобретению осуществляется следующим образом - в ледяной уксусной кислоте суспендируется при перемешивании гидрохлорид окситетрациклина или, как другой возможный вариант, дигидрат окситетрациклина при добавлении эквимолярного количества хлористого водорода в виде концентрированной соляной кислоты. Перемешивание продолжается в течение еще пяти часов. В течение первого часа перемешивания окситетрациклин полностью растворяется, после чего осаждается соль в виде молекулярного комплекса. По прошествии пяти часов перемешивания осадок фильтруется, промывается ледяной уксусной кислотой и ацетоном, после чего он высушивается при пониженном давлении при температуре до 40oC до постоянного веса. Выход выделенного ацетата гидрохлорида окситетрациклина различного веса. Выход выделенного ацетата гидрохлорида окситетрациклина различается в зависимости от чистоты исходного продукта. Чем чище исходный продукт, тем выше выход. Ацетат гидрохлорида окситетрациклина является высококристаллической и стойкой солью желтого цвета, которая без значительных потерь превращается в гидрохлорид окситетрациклина или дигидрат окситетрациклина. Это превращение в гидрохлорид окситетрациклина осуществляется путем растворения ацетата гидрохлорида окситетрациклина в минимальном количестве воды и добавления смеси метанола с концентрированной соляной кислотой. Повторяя полностью весь этот процесс, из такого очищенного гидрохлорида окситетрациклина получается совершенно чистый ацетат гидрохлорида окситетрациклина, не содержащий ацетилдекарбоксамидоокситетрациклина. Превращение ацетата гидрохлорида окситетрациклина в дигидрат окситетрациклина осуществляется путем растворения ацетата гидрохлорида окситетрациклина в таком количестве воды, что концентрация окситетрациклина в растворе составляет примерно 50000 межд.ед./мл, и при доведении величины pH раствора до 5, в результате чего происходит осаждение дигидрата окситетрациклина, который в конечном итоге извлекается путем фильтрации.
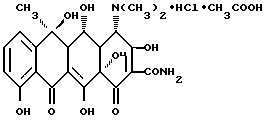
Другим объектом настоящего изобретения является ацетат гидрохлорида окситетрациклина формулы III
Ацетат гидрохлорида окситетрациклина, который выделяется из раствора окситетрациклина в ледяной уксусной кислоте, является солью в виде молекулярного комплекса, который ранее не был описан. Его молекулярный состав определяется путем титрования гидратом окиси натрия и нитратом серебра и по определению содержания уксусной кислоты методом газовой хроматографии. Этот солевой комплекс, как обнаружено, состоит из окситетрациклина.
Молекула данной композиции содержит теоретически 6,55% хлористого водорода и 10,78% уксусной кислоты, в то время как биологическая активность, рассчитанная на сухую массу, составляет 826,7 межд.ед./мг. Данные значения, получаемые путем анализа приготовленных образцов ацетата гидрохлорида окситетрациклина, отличаются от теоретически рассчитанных значений в пределах нескольких процентов. Измеренное таким образом на различных образцах содержание хлористого водорода составляло от 6,2 до 6,7%, содержание уксусной кислоты составляло от 10,2 до 11,5%, и биологическая активность составляла от 800 до 836 межд. ед./мг. Как и все тетрациклиновые соединения, ацетат гидрохлорида окситетрациклина не имеет четко выраженной точки плавления. После нагревания вещество изменяет свой цвет при температуре, превышающей 170oC, в то время как оно полностью разлагается при температуре, превышающей 200oC.
Присутствие уксусной кислоты в молекуле обнаруживается по спектральным характеристикам нового соединения. Так, например, ультрафиолетовый спектр соответствует максимальному распределению, идентичному спектру гидрохлорида окситетрациклина, с той разницей, что светопоглощение снижается за счет увеличения молекулярной массы. По этой же самой причине величина удельного оптического вращения также ниже. В спектре 1H-ЯМР и в спектре 13C-ЯМР ацетат гидрохлорида окситетрациклина показывает все сигналы окситетрациклина в их обычных сайтах в дополнение к сигналам уксусной кислоты. При масс-спектрографическом анализе ацетата гидрохлорида окситетрациклина наряду с фрагментами, характерными для окситетрациклина, обнаружены масс-фрагменты 43 и 60, характерные для уксусной кислоты.
Ввиду чрезвычайно низкой биологической активности ацетилдекарбоксамидоокситетрациклина его присутствие в окситетрациклине является неактивным балластом. При удалении этого балласта его ненужное отложение в костях человеческого организма устраняется, в то время как окситетрациклин повышает свою микробиологическую активность. Это означает, что при введении лекарства тот же эффект достигается при меньшей дозе препарата.
Данное изобретение иллюстрируется нижеследующими примерами, которые не ограничивают его объем.
Пример 1. Получение ацетата гидрохлорида окситетрациклина из гидрохлорида окситетрациклина.
Гидрохлорид окситетрациклина (100 г; 890 межд.ед./мг; согласно данным ЖХВД содержание окситетрациклина 91,9%, ацетилдекарбоксамидоокситетрациклина 2,6% в перерасчете на гидрохлорид) суспендировали в 500 мл ледяной уксусной кислоты, и суспензию перемешивали в течение 5 ч при комнатной температуре. Спустя примерно 1 ч получали прозрачный раствор, после чего начинал выпадать белый осадок. После пятичасового перемешивания осадок фильтровали, промывали в 50 мл охлажденной ледяной уксусной кислоты и 50 мл ацетона, и высушивали при температуре до 40oC и пониженном давлении до достижения постоянного веса.
Выход продукта 110 г (98%).
Анализ методом ЖХВД показал, что продукт содержит окситетрациклин и ацетилдекарбоксамидоокситетрациклин в соотношении 99,6:0,4.
Данные биоанализа: 829 межд.ед./мг.
Содержание уксусной кислоты: 10,23%.
Вода (определяемая методом К.Фишера): 1,0%.
HCl (титрование с AgNO3): 6,6%.
Температура плавления 203-208oC (разложение).
[α]
УФ λмакс: 218 нм (A1% 248,41), 268 нм (A1% 336,63) и 360 нм (A1% 251,87) (0,01 N HCl/EtOH) (Et - этил).
Пример 2. Получение ацетата гидрохлорида окситетрациклина из сырого дигидрата окситетрациклина с промежуточным продуктом гидрохлоридом окситетрациклина. Сырой дигидрат окситетрациклина (согласно ЖХВД содержание дигидрата окситетрациклина 95,6% и дигидрата ацетилдекарбоксамидоокситетрациклина 3,0%) обычным способом превращали в гидрохлорид окситетрациклина: сырой дигидрат (100 г) при перемешивании и нагревании на водяной бане до 30oC растворяли в смеси 750 мл метанола и 12 мл концентрированной соляной кислоты. После обработки 0,5 г динатриевой соли этилендиаминотетрауксусной кислоты и 1,5 г активного угля суспензию фильтровали с добавлением 1,5 г земли для фильтрования и остаток на фильтре промывали 100 мл метанола. Гидрохлорид окситетрациклина осаждался в фильтре при добавлении при перемешивании и охлаждении 55 мл концентрированной соляной кислоты. После фильтрации и сушки в печи с воздушной циркуляцией при 50oС получалось 97,06 г гидрохлорида окситетрациклина. Весь продукт растворяли при перемешивании в 500 мл ледяной уксусной кислоты. Через 10 мин в полученный прозрачный раствор добавляли для затравки 0,1 г ацетата гидрохлорида окситетрациклина и перемешивали в течение 5 ч при комнатной температуре. После осуществления такой же процедуры, как и в примере 1, получалось 98,5 г ацетата гидрохлорида окситетрациклина. Содержание в этом продукте ацетата гидрохлорида окситетрациклина, определяемое ЖХВД, составляло 99,07%, содержание ацетата гидрохлорида ацетилдекарбоксамидоокситетрациклина составляло 0,56%.
Выход регенерированного окситетрациклина составил 90,98%.
Пример 3. Получение ацетата гидрохлорида окситетрациклина непосредственно из сырого дигидрата окситетрациклина.
Сырой дигидрат окситетрациклина (200 г; 913 межд.ед./мг; вода 6,9%, согласно данным ЖХВД содержание дигидрата окситетрациклина 98,5% и дигидрата ацетилдекарбоксамидоокситетрациклина 2,1%) суспендировали в 600 мл ледяной уксусной кислоты, и в эту суспензию вводили по каплям при перемешивании 32 мл концентрированной соляной кислоты. После перемешивания получали раствор, который затравливали 0,1 г ацетата гидрохлорида окситетрациклина. После перемешивания в течение 1,5 ч добавляли 400 мл толуола, и перемешивание продолжали. После перемешивания в течение в общей сложности 5 ч полученный осадок фильтровали и образующийся на фильтре осадок перемешивали с 400 мл ацетона, еще раз промывали и фильтровали с добавлением 400 мл ацетона. Продукт высушивали при комнатной температуре и при пониженном давлении. Получали 188,06 г ацетата гидрохлорида окситетрациклина, содержащего (определено методом ЖХВД) 98,5% ацетата гидрохлорида окситетрациклина и 0,45% ацетата гидрохлорида декарбоксамидоокситетрациклина.
Выход регенерированного окситетрациклина составил 83,8%.
Пример 4. Получение дигидрата окситетрациклина из ацетата гидрохлорида окситетрациклина.
Ацетат гидрохлорида окситетрациклина (9,85 г), полученный как описано в примере 2, растворяли в 155 мл дистиллированной воды. Концентрированный аммиак добавляли по каплям в кислотный раствор (pH = 2,3) с одновременным перемешиванием, и дигидрат окситетрациклина выпадал в осадок. После того как величина pH смеси достигала 5, перемешивание продолжалось в течение 1 ч, осадок фильтровали, промывали 50 мл воды и высушивали при пониженном давлении при температуре 40-50oC. Получали 8,6 г дигидрата окситетрациклина биологической активности 921 межд.ед./мг (вода: 7,1%; 991 межд.ед./мг, в расчете на сухую массу). Продукт содержал (согласно ЖХВД) 98,9% дигидрата окситетрациклина и 0,4% дигидрата ацетилдекарбоксамидоокситетрациклина.
Выход регенерированного окситетрациклина составил 87,16%.
Пример 5. Получение гидрохлорида окситетрациклина из ацетата гидрохлорида окситетрациклина.
Ацетат гидрохлорида окситетрациклина (95,7 г), полученный как описано в примере 2, растворяли при перемешивании в 100 мл дистиллированной воды. В раствор добавляли по каплям при перемешивании смесь 600 мл метанола и 110 мл концентрированной соляной кислоты. Добавление осуществляли при комнатной температуре до тех пор, пока не начиналась кристаллизация гидрохлорида окситетрациклина (примерно 1/3 смеси), а оставшаяся смесь вводилась по каплям при охлаждении до примерно +2oC. После добавления смеси осажденные кристаллы выдерживались при охлаждении без перемешивания в течение одного часа, фильтровались, промывались 100 мл охлажденного метанола и высушивались при пониженном давлении при 40oC. Получали 80,64 г гидрохлорида окситетрациклина с биологической активностью 918 межд.ед./мг (вода: 0,6%; 924 межд.ед./мг в расчете на сухую массу). Продукт содержал (по данным ЖХВД) 93,9% гидрохлорида оксотетрациклина и 0,5% гидрохлорида ацетилдекарбоксамидоокситетрациклина.
Выход регенерированного окситетрациклина составил 89,6%.
Гидрохлорид окситетрациклина обрабатывали так же, как описано в примере 3. Продукт примера 3 (188,06 г) растворяли в 185 мл воды. Минимальное количество нерастворенных примесей фильтровали через слой земли для фильтрования и осадок на фильтре промывали смесью 10 мл метанола и 1 мл воды. После добавления по каплям смеси 1050 мл метанола и 192 мл концентрированной соляной кислоты в условиях, описанных для обработки продукта в примере 2, из фильтрата осаждался гидрохлорид окситетрациклина. После фильтрации и сушки получали 134,3 г гидрохлорида окситетрациклина с биологической активностью 933 межд.ед./мг с содержанием (по данным ЖХВД) 97,2% гидрохлорида окситетрациклина и 0,4% гидрохлорида ацетилдекарбоксамидоокситетрациклина.
Выход регенерированного окситетрациклина составил 79,07%.
Пример 6. Получение чистого гидрохлорида окситетрациклина.
Гидрохлорид окситетрациклина (50 г), полученный как описано в примере 5, согласно способу, описанному в примере 1, превращали в ацетат гидрохлорида окситетрациклина. Полученный ацетат гидрохлорида окситетрациклина (49 г) затем превращали в гидрохлорид окситетрациклина согласно способу, описанному в примере 5. Получали 39,36 г гидрохлорида окситетрациклина с биологической активностью 924 межд. ед./мг (вода 0,6%), и с содержанием (по данным ЖХВД) 97,7% гидрохлорида окситетрациклина и 0% гидрохлорида ацетилдекарбоксамидоокситетрациклина.
Описывается способ очистки окситетрациклина от присутствующего в нем в качестве примеси ацетилдекарбоксамидоокситетрациклина, отличающийся тем, что гидрохлорид окситетрациклина, содержащий ацетилдекарбоксамидоокситетрациклин в качестве примеси, суспендируют в ледяной уксусной кислоте, смесь перемешивают в течение 5 ч, образующийся осадок ацетата гидрохлорида тетрациклина фильтруют, промывают ледяной уксусной кислотой и ацетоном, после чего высушивают при пониженном давлении и температуре до 40oC до достижения постоянного веса и переводят в основание окситетрациклина. Технический результат - повышение степени очистки целевого продукта. 2 с. и 2 з.п. ф-лы.
| Анализатор спектра вибраций | 1978 |
|
SU834579A1 |
| US 2915555, 1959 | |||
| Способ повышения видимости слабых изображений | 1957 |
|
SU113855A1 |
