Изобретение касается получения по существу чистых энантиомеров фенилпропионовых кислот, выбранных из ибупрофена, флурбипрофена и их фармацевтически приемлемых солей, в частности их α-метилбензиламиновых, лизиновых и натриевых солей.
Ибупрофен, химическое название которого 2-(4-изобутилфенил) пропионовая кислота, и флурбипрофен, химическое название которого 2-(2-фтор-4-бифенилил)пропионовая кислота, являются хорошо известными лекарственными средствами противовоспалительного, жаропонижающего и анальгетического действия. К известным применениям ибупрофена и флурбипрофена относятся устранение боли и воспаления при скелетно-мышечных нарушениях, таких как ревматизм, и устранение боли при разнообразных иных недомоганиях, например при головной боли, невралгии и дисменореи.
И ибупрофен и флурбипрофен, оба содержат единственный хиральный центр на асимметрично замещенном углеродном атоме, и, следовательно, оба они существуют в двух энантиомерных формах. Известно, что S(+)- ибупрофен является активным средством и что в организме человека R(-)-ибупрофен может неполностью превращаться в S(+)-ибупрофен. Известно также, что S(+)-флурбипрофен является активным средством. У людей R(-)-флурбипрофен не превращается в (S)-энантиомер, хотя и существует мнение, что анальгезирующим действием обладает только R(-)-флурбипрофен (международная патентная заявка WO 92/04018 (Paz)). Ибупрофен и флурбипрофен поступали ранее в продажу в виде рацемической смеси. Однако в некоторых случаях может оказаться целесообразным вводить по существу один лишь энантиомер. Следовательно, желательно располагать улучшенными способами получения продукта, обогащенного требуемым энантиомером фенилпропионовой кислоты, выбранной из ибупрофена и флурбипрофена.
В европейской патентной заявке 0362476 (Paz) описано разделение энантиомерных форм арилпропионовых кислот селективной кристаллизацией диастереоизомерной соли в полярном растворителе. Утверждается, что использование полярных растворителей оказывается более благоприятным, чем неполярных, что расходится с использованием специфической смеси растворителей в способе, отвечающем настоящему изобретению.
Патент США N 5015764 (Manimaran) касается получения алифатических карбоновых кислот, включая ибупрофен и флурбипрофен, обработкой раствора их солей хиральным органическим основанием для селективного осаждения менее растворимого диаcтереоизомера. Отсутствует указание на использование специфической смеси растворителей, использованной в способе, отвечающем настоящему изобретению.
В европейской патентной заявке 0437369 описано получение (S)-ибупрофен-(S)-лизиновых солей контактированием рацемического ибупрофена с эквимолярным количеством (S)-лизина в водной смеси органических растворителей, отделением любого суспендированного твердого вещества от смеси, охлаждением прозрачной смеси до достижения пересыщенного состояния в отношении как (R)-ибупрофен-(S)-лизиновой, так и (S)-ибупрофен-(S)-лизиновой солей, контактированием пересыщенного раствора со шламом (S)-ибупрофен-(S)-лизиновой соли и отделением образованной кристаллической (S)-ибупрофен-(S)-лизиновой соли.
В международной патентной заявке WO 92/20334 (Boots) описано получение натриевой соли (S)-ибупрофена.
Настоящее изобретение дает способ получения продукта, который обогащен желательным энантиомером фенилпропионовой кислоты, выбранной из ибупрофена и флурбипрофена, который включает в себя следующие этапы (стадии):
а) этап разделения, на котором готовят α-метилбензиламиновую соль фенилпропионовой кислоты, которая обогащена желательным энантиомером посредством контактирования в смеси толуола и метанола, играющей роль растворителя, по существу рацемической смеси фенилпропионовой кислоты с энантиомером α- метилбензиламина, причем соответствующее молярное отношение содержаний по существу рацемической фенилпропионовой кислоты к α-метилбензиламину находится в области примерно от 1:0,25 и примерно до 1:1,
b) этап перекристаллизации, на котором результирующую обогащенную соль подвергают перекристаллизации из смеси метанола и толуола для получения α-метилбензиламиновой соли фенилпропионовой кислоты, которая дополнительно обогащена желательным энантиомером,
с) необязательный этап выделения, на котором фенилпропионовую кислоту, которая дополнительно обогащена желательным энантиомером, выделяют из перекристаллизованной соли,
d) необязательный этап получения соли, на котором твердую соль фенилпропионовой кислоты, дополнительно обогащенную желательным энантиомером, выделяют, причем твердая соль может оказаться, что не является обязательным, еще более энантиомерно обогащенной желательным энантиомером.
В предпочтительных вариантах осуществления способа, отвечающего настоящему изобретению, желательным энантиомером фенилпропионовой кислоты является (S)-энантиомер и
a) на первом этапе разделения по существу рацимическую фенилпропионовую кислоту и (S)-α-метилбензиламин используют при соответствующем молярном отношении, находящемся в области примерно от 1:0,35 и примерно до 1:0,8, например, в области примерно от 1:0,4 и примерно до 1:0,6, и приготовление происходит в смеси метанола с толуолом, в которой толуол содержится в количестве, составляющем, по крайней мере, примерно 50%, более предпочтительно в количестве, составляющем примерно от 60% и примерно до 90%, и наиболее предпочтительно в количестве, составляющем примерно от 70% и примерно до 80% от суммарного объема смеси, температура смеси находится в области примерно от 30oC и примерно до 70oC, предпочтительно в области примерно от 40oC и примерно до 60oC с целью образования пересыщенного раствора, из которой (S)-α-метилбензиламиновую соль фенилпропионовой кислоты, обогащенную (S)-энантиомером фенилпропионовой кислоты, кристаллизуют, например, охлаждением раствора до температуры, находящейся в области примерно от -10oC и примерно до 30oC, предпочтительно в области примерно от 0oC и примерно до 5oC,
b) на этапе перекристаллизации предпочтительным растворителем является смесь метанола с толуолом, в которой толуол содержится, по крайней мере, в количестве, составляющем примерно 25%, более предпочтительно в количестве, составляющем примерно от 50% и примерно до 80%, и наиболее предпочтительно в количестве, составляющем примерно от 60% и примерно до 70% от суммарного объема смеси, из которой (S)-α-метилбензиламиновую соль фенилпропионовой кислоты, дополнительно обогащенной (S)-энантиомером фенилпропионовой кислоты, кристаллизуют, например, охлаждением раствора до температуры в области примерно от -10oC и примерно до 30oC, предпочтительно в области примерно от 0oC и примерно до 5oC,
c) на этапе выделения (S)-α-метилбензиламиновую соль фенилпропионовой кислоты, дополнительно обогащенную (S)-энантиомером и полученную на этапе перекристаллизации, подкисляют (например, хлористоводородной кислотой) в несмешиваемом с водой растворителе для получения раствора выделенной фенилпропионовой кислоты, обогащенной (S)-энантиомером, в несмешиваемом с водой растворителе и водного раствора (S)-α-метилбензиламиновой соли (например, соли хлористоводородной кислоты), из которого может быть выделен (S)-α-метилбензиламин, в результате чего он может быть вновь использован на последующем этапе расщепления (а), например, подщелачиванием раствора экстракцией высвобожденного основания в толуол,
d) на необязательном этапе получения соли раствор дополнительно обогащенной (S)-энантиомером фенилпропионовой кислоты в несмешиваемом с водой растворителе, полученный на этапе выделения (c), может быть дополнительно обработан одним или несколькими следующими способами:
I) кристаллизацией и отделением твердой фенилпропионовой кислоты, обогащенной (S)-энантиомером, от раствора,
II) удалением растворителей отгонкой для получения расплава, который может быть использован на стадии (V), описанной ниже,
III) если фенилпропионовая кислота представляет собой ибупрофен, то тогда контактированием с водным раствором содержащего натрий основания (например, гидроксидом натрия) для получения водного раствора натриевой соли ибупрофена, которую отделяют от несмешиваемого с водой растворителя, причем водный раствор разбавляют ацетоном для кристаллизации дополнительно обогащенной (S)-энантиомером натриевой соли ибупрофена, и
IV) если фенилпропионовая кислота представляет собой ибупрофен, то тогда контактированием с (S)-лизином и водой, в которой соответствующее молярное отношение содержаний ибупрофена к (S)-лизину находится в области от 1:0,5 до 1: 1, для получения водного раствора (S)-лизиновой соли, которую отделяют от несмешиваемого растворителя, причем тогда уже к водному раствору добавляют этанол для кристаллизации (S)-лизиновой соли ибупрофена, дополнительно обогащенной (S)-энантиомером.
Продукт описанных выше стадий от d(I) до d(III) может быть использован на одной или нескольких последующих стадиях:
V) твердое вещество с описанной выше стадии d(I) или расплав со стадии d(II), который отделяют и подвергают взаимодействию в водном этанольном растворе с (S)-лизином, в котором молярное отношение содержаний фенилпропионовой кислоты к (S)-лизину находится в области от 1:0,5 до 1:1, используют для получения после кристаллизации и отделения (S)-лизиновой соли фенилпропионовой кислоты, дополнительно обогащенной (S)-энантиомером,
VI) подкисления дополнительно обогащенной (S)-энантиомером натриевой соли ибупрофена с описанной выше стадии d(III) в присутствии несмешиваемого с водой растворителя, такого как гептан для получения раствора дополнительно обогащенного (S)-энантиомером ибупрофена в несмешиваемом с водой растворителе, который отделяют. Твердый обогащенный (S)-энантиомером ибупрофен затем кристаллизуют и выделяют, и
VII) подкисления водного раствора дополнительно обогащенной (S)-энантиомером натриевой соли ибупрофена с описанного выше этапа d(III) при повышенной температуре (например, при 60oC) с получением расплава, который отделяют от водного слоя и обрабатывают так, как это описано выше на этапе (V).
В более предпочтительных вариантах осуществления способа, отвечающего настоящему изобретению, фенилпропионовая кислота представляет собой ибупрофен и желательный энантиомер - (S)-энантиомер. В этом более предпочтительном варианте осуществления
a) на этапе разделения получают (S)-α-метилбензиламиновую соль обогащенного по (S)-энантиомеру ибупрофена с энантиомерной чистотой в области примерно от 80% и примерно до 95% по весу и первый маточный раствор, содержащий обогащенный (R)-энантиомером ибупрофен, который используют на этапе рацемизации (e) для получения по существу рацемического ибупрофена, который вводят как часть исходного материала, используемого на следующем этапе разделения (a),
b) этап перекристаллизации включает две стадии:
I) первую стадию перекристаллизации, включающую в себя перекристаллизацию (S)-α-метилбензиламиновой соли обогащенного (S)-энантиомером ибупрофена, полученной на этапе разделения (a), для получения (S)-метилбензиламиновой соли обогащенного (S)-энантиомером ибупрофена предпочтительно с энантиомерной чистотой в области примерно от 90 и примерно до 99,9%, более предпочтительно в области примерно от 94 и примерно до 99% по весу и второй маточной жидкости, содержащей (S)-α-метилбензиламиновую соль обогащенного (S)-энантиомером ибупрофена с энантиомерной чистотой примерно от 40 и примерно до 70%, более предпочтительно в области примерно от 40 и примерно до 60% по весу, причем вторую маточную жидкость вводят в виде части растворителя, используемого на следующей стадии разделения (a),
II) вторую стадию перекристаллизации, включающую в себя перекристаллизацию (S)-α-метилбензиламиновой соли обогащенного (S)-энантиомером ибупрофена, полученной на первой стадии перекристаллизации (b)(I), для получения по существу энантиомерно чистого (S)-ибупрофен-(S)-метилбензиламина предпочтительно с энантиомерной чистотой примерно 99% и третьей маточной жидкости, содержащей (S)-α-метилбензиламиновую соль обогащенного (S)-энантиомером ибупрофена с энантиомерной чистотой в области примерно от 85 и примерно до 95% по весу, предпочтительно в области примерно от 88 и примерно до 95% по весу, причем третью маточную жидкость вводят как часть растворителя, используемого на следующей первой стадии перекристаллизации (b)(I).
В наиболее предпочтительном варианте осуществления изобретения первую маточную жидкость с этапа разделения (a) подвергают азеотропной перегонке для удаления по существу всего метанола при температурах, при которых по существу исключается образование побочных продуктов, причем дистиллят вновь используют как часть растворителя на последующем этапе разделения (a). Остаток, оставшийся после упомянутой выше перегонки, может быть подкислен, например, хлористоводородной кислотой для образования водного раствора (S)-α-метилбензиламиновой соли, например, солянокислого (S)-α-метилбензиламина и органической фазы, содержащей ибупрофен, обогащенный (R)-энантиомером. Водный раствор затем отделяют и подкисляют для получения свободного (S)-α-метилбензиламина, который экстрагируют толуолом и вновь используют в качестве агента разделения на начальной стадии следующего проведения этапа разделения (a) наряду с (S)-α-метилбензиламином, извлеченным на этапе (этапах) выделения. Органическая фаза, содержащая ибупрофен, обогащенный (R)-энантиомером, может быть рацемизирована на этапе рацемизации (e) любым известным способом для получения по существу рацемического ибупрофена, который может быть тогда введен как часть растворителя, используемого на начальной стадии последующего этапа разделения (a).
Целесообразно, чтобы на каждом этапе осуществления способа, отвечающего настоящему изобретению, жидкости, не содержащие фенилпропионовую кислоту, обогащенную желательно энантиомером, подвергались рециркуляции с использованием их на предшествующих этапах осуществления способа. Сочетание этапа разделения (a) и этапа перекристаллизации (b) с этапом рацемизации (e) и этапом извлечения агента разделения -(S)-α-метилбензиламина, дает преимущества за счет исключения необходимости обработки многочисленных потоков жидкостей, получаемых на каждом этапе, и понижения производственных затрат за счет экономии времени, энергии и исходных материалов.
Этап перекристаллизации в способе, описанном выше, может включать, хотя и не обязательно, третью и/или последующую стадию перекристаллизации.
Этот предпочтительный объединенный способ иллюстрируется посредством ссылки на чертеж, на котором приведена схематическая технологическая схема предпочтительного способа осуществления изобретения для получения (S)-ибупрофена, на которой буквы относятся к этапам, или стадиям, помеченным обозначениями (a), (b)(I), (b)(II), (c), (d) и (e) в описанных выше способах, и числа от 1 до 3 указывают на первую-третью маточные жидкости соответственно, число 4 указывает на рециркулированный (S)-α-метилбензиламин и число 5 - на рециркулированный рацемизированный ибупрофен. На чертеже пунктирными линиями показаны рециркулированные материалы и сплошными линиями - материал, возрастающий при обогащении (S)-ибупрофена в направлении, показанном стрелкой.
На этапе разделения (a) получают продукт в виде (S)-α-метилбензиламиновой соли обогащенного (S)-энантиомером ибупрофена, который используют в качестве исходного материала на первой стадии перекристаллизации (b)(I). Первую маточную жидкость (I) с этапа разделения (a) подают на этап рацемизации (e), с которого рацемический ибупрофен (5) возвращают обратно в виде части исходного материала для последующего проведения этапа разделения (a), и извлеченный (S)-α-метилбензиламин (4) подвергают рециркуляции для использования в виде части разделяющего агента для последующего проведения этапа разделения (a). Вторую маточную жидкость (2) с первого этапа перекристаллизации (b)(II) подвергают рециркуляции для использования на последующем этапе расщепления (a). Продукт первого этапа перекристаллизации (b)(I) обрабатывают на втором этапе перекристаллизации (b)(II) для получения (S)-ибупрофен-(S)-α-метилбензиламина повышенной энантиомерной чистоты и третьей маточной жидкости, которая претерпела рециркуляцию с образованием части растворителя, используемого на последующем первом этапе перекристаллизации (b)(I). Продукт со второго этапа перекристаллизации (b)(II) используют затем на этапе выделения (c) для получения (S)-ибупрофена высокой энантиомерной чистоты. (S)-α-Метилбензиламин (4), который также выделяют на этапе выделения (c), подвергают рециркуляции для использования в качестве части разделяющего агента для последующего этапа разделения (a). Высвобожденный (S)-ибупрофен может быть затем использован на необязательном этапе получения соли (d) для образования солей (например, соли натрия или (S)-лизина), содержащих (S)-ибупрофен еще более высокой энантиомерной чистоты.
Продукт описанных выше способов, когда фенилпропионовая кислота представляет собой ибупрофен и желательный энантиомер представляет собой (S)-энантиомер, может быть использован для приготовления (S)-лизиновой соли обогащенного (S)-энантиомером ибупрофена посредством контактирования выделенного ибупрофена, обогащенного (S)-энантиомером, с (S)-лизином, предпочтительно со стехиометрическим количеством или с меньшим количеством (S)-лизина для образования (S)-лизиновой соли ибупрофена, дополнительно обогащенного (S)-энантиомером, причем более предпочтительно, чтобы молярное отношение содержаний ибупрофена к (S)-лизину находилось в области примерно от 1:0,5 и примерно до 1:1, предпочтительно в области примерно от 1:0,5 и примерно до 1:0,95. Выделенный ибупрофен, обогащенный (S)-энантиомером, может быть также приведен в контакт с гидроксидом натрия для получения дополнительно обогащенного (S)-энантиомером ибупрофена натрия (см. например, международную патентную заявку WO 92/20334).
В другом предпочтительном варианте осуществления способа, отвечающего настоящему изобретению, получают (S)-ибупрофен и его соли высоко энантиомерной чистоты.
Неожиданно было также обнаружено, что флурбипрофен, обогащенный (S)-энантиомером, может быть выкристаллизован из толуола с эффективным удалением другого энантиомера и получением (S)-флурбипрофена высокой энантиомерной чистоты.
Совершенно очевидно, что при необходимости получения R(-)-ибупрофена, R(-)-флурбипрофена или их фармацевтически приемлемых солей описанные выше способы могут быть легко приспособлены для получения R(-)-ибупрофена, R(-)-флурбипрофена или их фармацевтически приемлемых солей использованием (R)-α-метилбензиламина вместо (S)-α-метилбензиламина в качестве агента разделения на этапе разделения (a) способов, отвечающих настоящему изобретению, с соответствующей модификацией последующих этапов.
Изобретение далее будет проиллюстрировано посредством следующих примеров.
Пример 1.
Разделение ибупрофена посредством приготовления ибупрофен-(S)-α-метилбензиламина, обогащенного (S)-энантиомером (этап разделения (а)).
Рециклированный рацемический ибупрофен (530 кг) растворяли в толуоле (1335 л), метанол (900 л) добавляли к раствору и смесь нагревали с перемешиванием до 66oC. Рециркулированный (S)-α-метилбензиламин (247 кг), находящийся в толуоле (200 л), добавляли на протяжении 3 ч, поддерживая при этом температуру в области 65-70oC. Смесь, наконец, охлаждали до температуры, находящейся в области от 0 до 5oC при перемешивании, и перемешивали при этой температуре в течение одного часа. Требуемый продукт собирали фильтрованием, промывали толуолом (600 л). Продукт содержал (S)-α-метилбензиламиновую соль обогащенного (S)-энантиомером ибупрофена с энантиомерной частотой 89,3% по весу. Маточные жидкости подвергали переработке по способу, аналогичному описанному в примере 6.
Пример 2.
Этап перекристаллизации (S) -α- метилбензиламиновой соли ибупрофена, обогащенного (S)-энантиомером (этап перекристаллизации (b)).
Пример 2(a) (Первый этап перекристаллизации (b)(I)).
(S)-α-Метилбензиламиновую соль обогащенного (S)-энантиомером ибупрофена (635 кг) с энантиомерной чистотой 85,5% по весу, полученную по способу, аналогичному описанному выше (пример 1), толуол (598 л) и рециклизованную вторую маточную жидкость (2350 л), полученную в приведенном ниже примере B2 и содержащую (S)-α-метилбензиламиновую соль обогащенного (S)-энантиомером ибупрофена (214 кг) и метанол (800 л), перемешивали, грели и растворяли при 67oC, а затем, наконец, охлаждали до температуры, находящейся в области от 0 до 5oC. Результирующее твердое вещество собирали фильтрованием. Продукт представлял собой (S)-α-метилбензиламиновую соль обогащенного (S)-энантиомером ибупрофена с энантиомерной чистотой 94,1% по весу.
Пример 2(b) (Второй этап перекристаллизации (b)(II)).
В способе, аналогичном использованному на первом этапе перекристаллизации, описанном выше в примере 2(a), (S)-α-метилбензиламиновую соль обогащенного (S)-энантиомером ибупрофена с энантиомерной чистотой 91,4% по весу (629 кг, полученных по способу, аналогичному описанному выше в примере 2(a)) перекристаллизовывали из смеси толуола (115 л) и промывали толуолом, в результате чего достигалась энантиомерная чистота величиной 98,5% по весу.
Примеры 2(a) и 2(b) свидетельствуют о том, что существенный рост энантиомерной чистоты у (S)-α-метилбензиламиновой соли обогащенного (S)-энантиомером ибупрофена может быть достигнут на стадии перекристаллизации в способе, отвечающем настоящему изобретению.
Пример 3(a).
Выделение ибупрофена, обогащенного (S)-энантиомером, в толуольном растворе (этап выделения (c)).
(S)-α-Метилбензиламиновую соль обогащенного (S)-энантиомером ибупрофена (485 кг соли, приготовленной так, как это описано выше в примере 2 (b)), толуол (814 л), воду (300 л) и концентрированную хлористоводородную кислоту с плотностью 1,18 (170 кг) перемешивали в течение 30 мин. Нижний водный слой, содержащий солянокислый (S)-α-метилбензиламин, отделяли, соединяли с водными жидкостями так, как это описано ниже в примере 6, делая это перед рециркуляцией, проводимой так, как это описано выше в примере 1. Верхний слой, содержащий толуольный раствор ибупрофена, обогащенного (S)-энантиомером, промывали водой (100 л), в результате чего получали 920 кг раствора, содержащего 300 кг ибупрофена, обогащенного (S)-энантиомером, с энантиомерной чистотой 98,5% по весу.
Пример 3(b).
Очистка ибупрофена, обогащенного (S)-энантиомером
Раствор ибупрофена, обогащенного (S)-энантиомером, с энантиомерной чистотой 98,2% (180 кг) в толуоле (1221 кг) промывали водой. Воду (220 л) и водный раствор гидроксида натрия (47 л с плотностью 1,5) добавляли, и смесь нагревали до 60oC и оставляли для осаждения на 4 ч. Нижний водный слой отделяли, и толуольный слой промывали водой. Водный промывочный раствор соединяли с водным слоем. Остаточный толуол удаляли перегонкой и добавляли гептан (250 л) и концентрированную хлористоводородную кислоту (78 кг с плотностью 1,18). Гептановый слой отделяли, промывали водой и охлаждали до -10oC. Ибупрофен, обогащенный (S)-энантиомером, с энантиомерной чистотой выше 99% собирали фильтрованием и сушили в вакууме. (Выход составлял 166 кг).
Пример 4(a).
Получение (S)-лизиновой соли обогащенного (S)-энантиомером ибупрофена (этап получения соли (d)).
Примеры с 4.1 до 4.12 проводили так, как это описано ниже со ссылкой на табл. 1. Ибупрофен, обогащенный (S)-энантиомером (100 г материала, содержащего "a"% (S)-энантиомера), растворяли в этаноле (900 мл) при температуре окружающей среды. Готовили раствор моногидрата (S)-лизина ("b" г) в смеси воды ("c" мл) и этанола ("d" мл). Раствор ибупрофена и раствор (S)-лизина добавляли в эквимолярном соотношении на протяжении одного часа, одновременно добавляли к суспензии (S)-ибупрофен-(S)-лизиновой соли (9,5 г) в воде (11 мл) и этаноле (125 мл), которую перемешивали при 20oC в течение 10 мин. Смесь затем охлаждали до 0oC за один час и затем охлаждали до -10oC. Смесь перемешивали при -10oC в течение двух часов. Результирующее твердое вещество собирали фильтрованием, промывали этилацетатом и сушили в вакууме при 35oC, в результате чего получили (S)-лизиновую соль обогащенного (S)-энантиомером ибупрофена с энантиомерной чистотой "e"% по весу.
Примеры с 4.1 по 4.12 свидетельствуют о том, что на этапе приготовления по способу, отвечающему настоящему изобретению, может быть достигнуто существенное повышение энантиомерной чистоты у (S)-лизиновой соли ибупрофена, обогащенного (S)-энантиомером.
Пример 4(b).
Получение (S)-лизиновой соли ибупрофена, обогащенного (S)-энантиомером (этап получения соли (d)).
Раствор обогащенного (S)-энантиомером ибупрофена (30 г) в толуоле (20 г) грели при температуре в области от 60 до 70oC с (S)-лизином (40 г 50%-ного по весу водного раствора) и водой (20 мл). Нижний водный слой отделяли, и остаточный растворитель удаляли отгонкой. Добавляли этанол (460 мл), и смесь грели до 50 - 55oC и затем охлаждали до температуры в области от 0oC до -10oC в течение 30 мин. Кристаллическую (S)-лизиновую соль обогащенного (S)-энантиомером ибупрофена собирали, промывали этилацетатом (50 мл) и сушили в вакууме.
Пример 5.
Приготовление натриевой соли ибупрофена, обогащенного (S)-энантиомером (этап получения соли (d)).
Раствор обогащенного (S)-энантиомером ибупрофена с энантиомерной чистотой 95,5% (211 кг) в толуоле (797 кг) грели до 60oC с водой (300 л) и водным раствором гидроксида натрия (52 л, плотность 1,5), и смесь оставляли на 4 ч для расслоения. Водный слой отделяли, и толуольный слой промывали водой. Водные промывки и водный слой соединяли, и остаточный толуол удаляли отгонкой. Добавляли ацетон (1684 кг), и смесь охлаждали до 20oC. Выпавший дигидрат натриевой соли обогащенного (S)-энантиомером ибупрофена (с энантиомерной чистотой 99,9%) собирали фильтрованием и сушили в вакууме. (Выход составлял 143,5 кг).
Пример 6.
Обработка первой маточной жидкости с этапа разделения.
Смесь первой маточной жидкости с этапа разделения концентрировали дистилляцией для удаления метанола и толуола с целью извлечения и последующего повторного использования. Добавляли воду (300 л) и концентрированную хлористоводородную кислоту (170 кг с плотностью 1,18), и смесь перемешивали. Водный слой, содержащий солянокислый (S)-α-метилбензиламин, отделяли и соединяли с водным раствором солянокислого (S)-α-метилбензиламина из примера 3(a). Объединенные растворы подщелачивали водным раствором гидроксида натрия (340 л с плотностью 1,5). Добавляли толуол (500 л), и результирующий раствор (S)-α-метилбензиламина, используемый на последующем этапе разделения, обрабатывали по способу, аналогичному описанному в примере 1.
Метанол (300 л) и концентрированную серную кислоту грели в сосуде с обратным холодильником в течение 2 ч. Верхний органический слой отделяли и грели в сосуде с обратным холодильником с метанолом (75 л) и концентрированной серной кислотой (15 л) в течение 2 ч. Верхний слой отделяли и грели с твердым гидроксидом натрия (175 кг). Метанол удаляли отгонкой, и остаток подкисляли смесью концентрированной хлористоводородной кислоты (353 кг) и воды (1750 л). Верхний толуольный слой, содержащий рацемический ибупрофен, промывали водой и использовали на этапе разделения при последующем приготовлении, поступая так, как это описано в примере 1.
Пример 7.
Разделение флурбипрофена приготовления (S)-α-метилбензиламиновой соли обогащенного (S)-энантиомером флурбипрофена.
Рацемический флурбипрофен (61,0 г) растворяли в смеси метанола (40 мл) и толуола (160 мл). Смесь нагревали до 60oC, и на протяжении 10 мин добавляли (S)-α-метилбензиламин в количестве 16,9 мл. К реакционной смеси добавляли затравочный кристалл (S)-флурбипрофен-(S)-α-метилбензиламина, которую затем охлаждали до температуры в области от 0 до 5oC и выдерживали при этой температуре в течение одного часа. Осадок собирали фильтрованием, в результате чего получали (S)-α-метилбензиламиновую соль обогащенного (S)-энантиомером флурбипрофена с энантиомерной чистотой 92,2%. После перекристаллизации осадка из смеси метанола (48 мл) и толуола (192 мл) получали (S) -α- метилбензиламиновую соль дополнительно обогащенного (S)-энантиомером флурбипрофена с энантиомерной чистотой 98,5%.
Маточную жидкость, полученную при перекристаллизации, подкисляли концентрированной HCl (10 мл) с водой (25 мл) и перемешивали при 25oC в течение 15 мин. Нижний водный слой, содержащий (S)-α-метилбензиламин, собирали и повторно использовали. Верхний органический слой содержал смесь энантиомеров флурбипрофеновой кислоты, находившихся в весовом соотношении 41,5:50,5% (S(+)-энантиомер к R(-)-энантиомеру соответственно). Этот флурбипрофен превращали в его метиловый сложный эфир, который был рацемизирован, превращали обратно в рацемический флурбипрофен гидроксидом натрия и вновь подавали на описанную выше стадию разделения.
Пример 8
Приготовление (R)-флурбипрофен-(R)-α-метилбензиламина
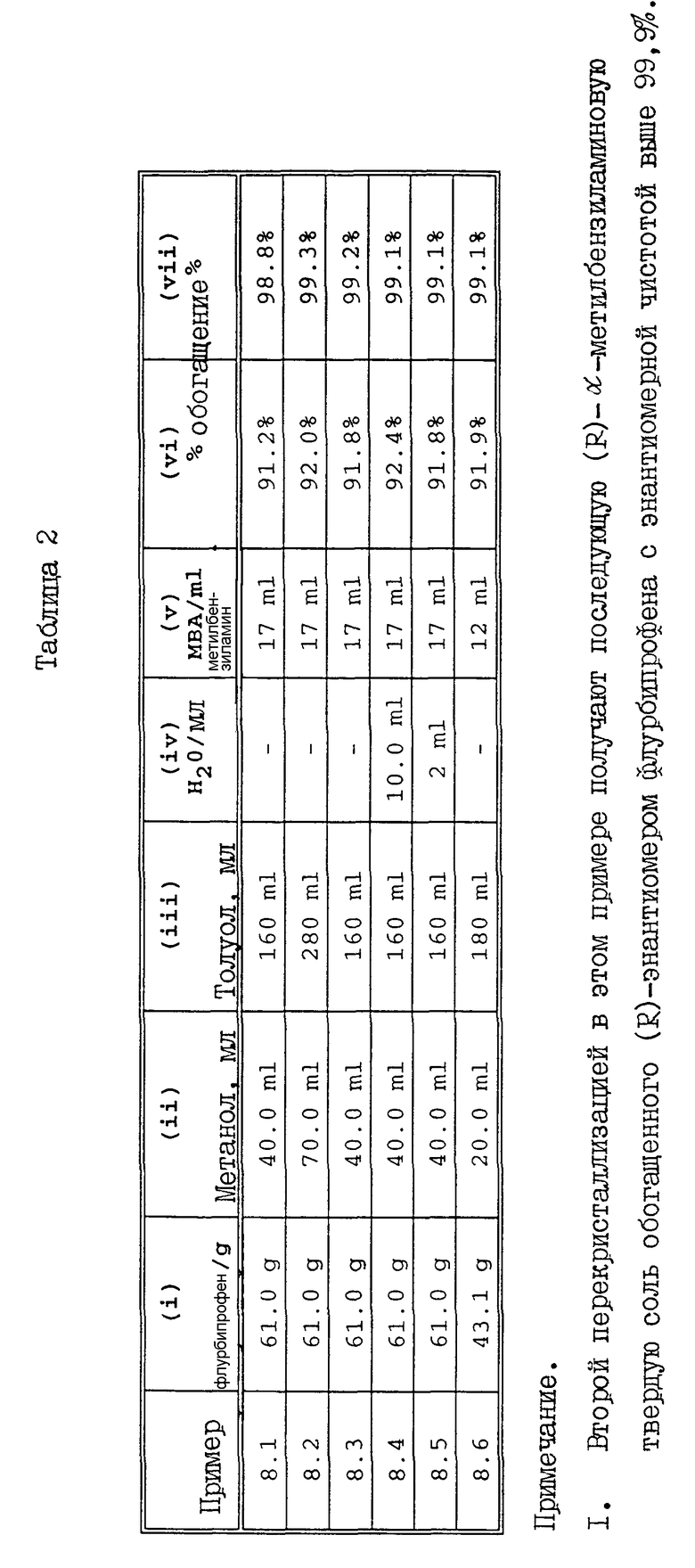
Приведенные ниже примеры с 8.1 до 8.6 проводили согласно данным табл. 2. Рацемический флурбипрофен в количестве (I) г растворяли в смеси метанола (II) мл с толуолом (III) мл и, что не является обязательным, водой (IV) мл. Смесь нагревали до 55oC для образования раствора, и на протяжении 10 мин вводили (V) мл (R)-α-метилбензиламина. Затравочный кристалл (R)-флурбипрофен-(R)-α-метилбензиламина добавляли к смеси, которую охлаждали до 25oC. Осадок собирали фильтрованием, в результате чего получали (R)-α-метилбензиламиновую соль обогащенного (R)-энантиомером флурбипрофена с энантиомерной чистотой (VI)%. После перекристаллизации осадка из смеси метанола, толуола и воды, находящихся в том же соотношении, что и использование на стадии разделения, получали твердое вещество, представляющее собой дополнительно обогащенный (R)-энантиомером (R)-флурбипрофен-(R)-α-метилбензиламин с энантиомерной чистотой (VII)%.
Пример 9(a).
Выделение флурбипрофена, обогащенного (R)-энантиомером.
(R)-α-Метилбензиламиновую соль обогащенного R-энантиомером флурбипрофена (58,7 г, получали тем же способом, что и описанный в примере 8) с энантиомерной чистотой 99,1%, грели при 80oC в течение 15 мин со смесью н-гептана (160 мл), воды (200 мл) и концентрированной хлористоводородной кислоты (17 мл с плотностью 1,18). Органический слой отделяли и охлаждали до температуры в области от 0 до 5oC. Выкристаллизованный флурбипрофен, обогащенный (R)-энантиомером, собирали, промывали н-гептаном и сушили в вакууме.
Пример 9(b).
Выделение флурбипрофена, обогащенного (S)-энантиомером.
Способом, аналогичным описанному в примере 9 (a), выделяли флурбипрофен, обогащенный (S)-энантиомером, из (S)-α-метилбензиламиновой соли обогащенного (S)-энантиомером флурбипрофена, приготовленной по способу, аналогичному описанному в примере 7.
Пример 10(a).
Энантиомерная очистка флурбипрофена, обогащенного (S)-энантиомером, проведением перекристаллизации.
Флурбипрофен, обогащенный (S)-энантиомером (47,2 г), с энантиомерной чистотой 98,9%, добавляли к толуолу (132 мл) и нагревали до 50oC. Добавляли кристаллы (S)-флурбипрофена с температурой 42oC, и раствор охлаждали до -5oC. Твердый флурбипрофен, обогащенный (S)-энантиомером, собирали фильтрованием и находили, что энантиомерная чистота составляет 99,9%.
Пример 10(b).
Энантиомерная очистка флурбипрофена, обогащенного (S)-энантиомером, проведением перекристаллизации.
Флурбипрофен, обогащенный (S)-энантиомером (13,5 г) с энантиомерной чистотой 98,4%, добавляли к толуолу (26 мл) и нагревали до 50oC. Раствор затем охлаждали до -10oC. Твердый флурбипрофен, обогащенный (S)-энантиомером, собирали фильтрованием и находили, что энантиомерная чистота составляет 99,8%. Выход составлял 13,0 г.

Описывается способ получения фенилпропионовой кислоты, выбранной из 2-(4-изобутилфенил)пропионовой или 2-(2-фтор-4-бифенилил)пропионовой кислот или их солей, таких как метилбензиламиновая, или лизиновая, или натриевая соли, которые обогащены требуемым энантиомером, включающий стадию разделения, которую проводят обработкой соответствующей рацемической кислоты энантиомером α-метилбензиламина в растворителе, при нагревании, с образованием соответствующей соли фенилпропионовой кислоты, которая обогащена требуемым энантиомером и которую отделяют с последующим превращением в фенилпропионовую кислоту или в одну из вышеуказанных солей, обогащенную требуемым энантиомером, и стадию перекристаллизации, отличающийся тем, что на стадии разделения (а) используют смесь таких растворителей, как толуол и метанол, и ее проводят при молярном отношении по существу рацемической фенилпропионовой кислоты к α-метилбензиламину, равном 1:0,25 - 1:1, а затем проводят стадию перекристаллизации (в), на которой результирующую обогащенную соль перекристаллизовывают из смеси метанола с толуолом для получения α-метилбензиламиновой соли фенилпропионовой кислоты, которая дополнительно обогащена требуемым энантиомером, после чего проводят необязательную стадию высвобождения (с), на которой фенилпропионовую кислоту, которая дополнительно обогащена требуемым энантиомером, выделяют из перекристаллизованной соли, (d) необязательную стадию получения соли, на которой твердую соль фенилпропионовой кислоты, дополнительно обогащенной требуемым энантиомером, выделяют, причем твердая соль необязательно еще более обогащена требуемым энантиомером. Технический результат - упрощение процесса. 23 з.п. ф-лы, 2 табл. , 1 ил.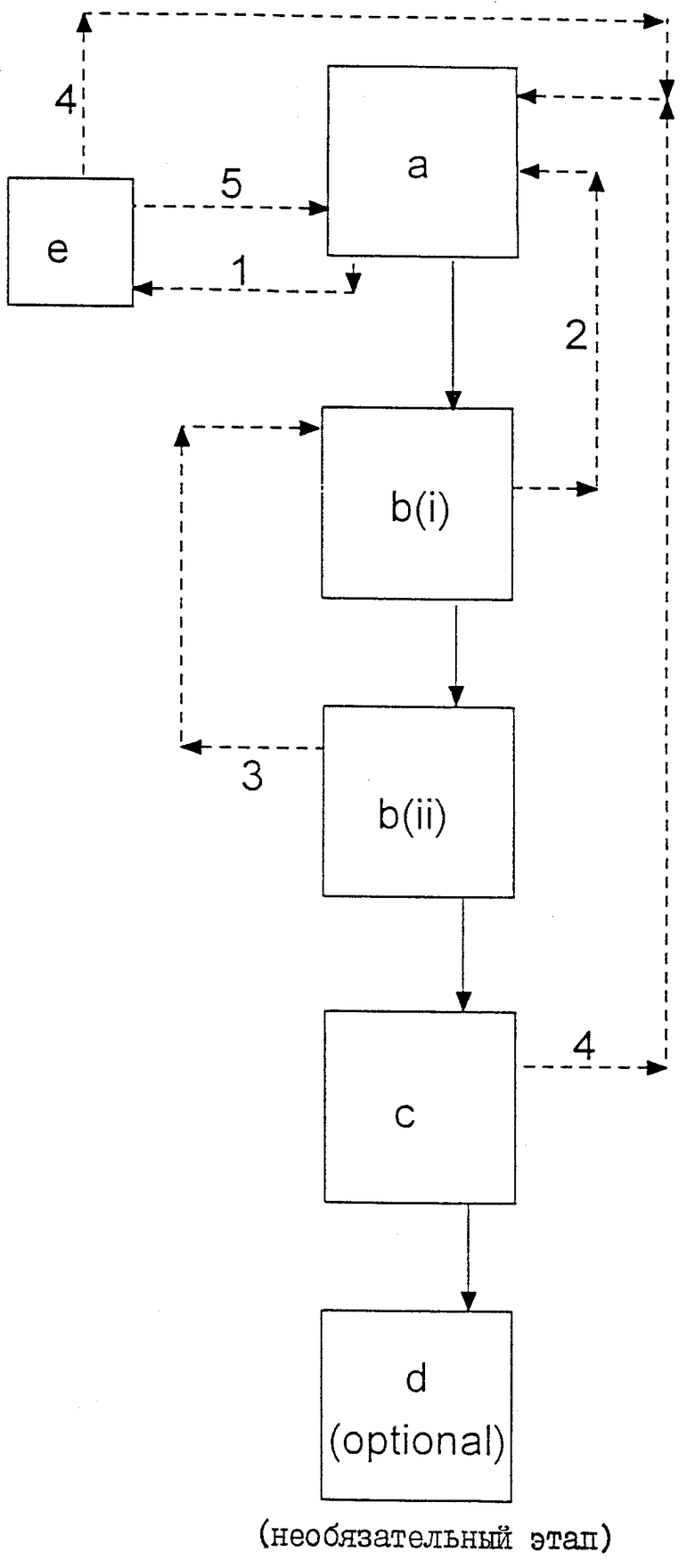
| EP, 0362476, A2, 1989 | |||
| SU, 1029836, A, 1979 | |||
| US, 5260482, A, 1993 | |||
| US, 5248813, A, 1993. |

