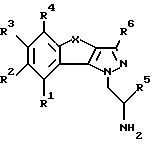
Настоящее изобретение относится к трициклическим производным пиразола, в частности оно относится к трициклическим производным 1-амино-этилпиразола общей формулы I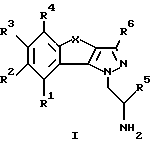
где R1-R4 обозначают соответственно водород, гидроксильную группу, галоген, низший алкил, низший алкокси или фенил;
R5 обозначает водород или низший алкил;
R6 обозначает водород, низший алкил или низший алкокси;
X обозначает -(CR7R8)n- или -CH=CH-;
R7 и R8 обозначают водород или низший алкил;
n = 1 или 2,
а также к фармацевтически применимым солям основных соединений общей формулы I.
Названные соединения и соли являются новыми и отличаются ценными фармакологическими свойствами.
Предметом настоящего изобретения являются соединения общей формулы I и их фармацевтически применимые соли как таковые и в качестве фармацевтических биологически активных веществ, получение соединений общей формулы I и их солей, далее, лекарственные средства, содержащие эти соединения и соли, и получение этих лекарственных средств, а также применение соединений общей формулы I и их фармацевтически приемлемых солей для лечения, соответственно предупреждения заболеваний и для оздоровления организма, прежде всего для лечения или предупреждения расстройств центральной нервной системы, таких как депрессии, биполярные расстройства, состояния страха, нарушения сна, сексуальные расстройства, психозы, шизофрения, мигрени и другие связанные с головными болями или обусловленные болями другого типа состояния, расстройства, обусловленные личностными факторами, расстройства компульсивно-навязчивого характера, социальная фобия или приступы паники, психические органические расстройства, психические расстройства в детском возрасте, агрессивность, возрастные нарушения памяти и поведения, непреодолимое патологическое влечение и пагубные привычки, ожирение, булимия и т.п., повреждений нервной системы, вызванных травмами, инсультом, нейродегенеративными заболеваниями и т.п., сердечно-сосудистых заболеваний, таких как артериальная гипертония, тромбоз, инсульт и т.д., и желудочно-кишечных расстройств, таких как дисфункция моторики желудочно-кишечного тракта, соответственно для изготовления соответствующих лекарственных средств.
Предметом изобретения являются далее соединения общей формулы II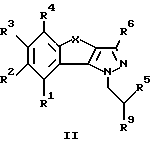
где R1-R6 имеют указанные выше значения, a R9 обозначает азидную группу, гидроксильную группу или защищенную аминогруппу.
Соединения формулы II представляют собой важные промежуточные продукты для получения фармацевтически ценных соединений общей формулы I.
Если в формуле I ни один из символов R1-R6 не имеет асимметрического центра, то соединения по изобретению могут быть представлены в виде энантиомеров, в других случаях возможно наличие различных диастереомеров. Изобретение включает в себя все возможные стереоизомеры, равно как и их смеси.
Используемое в описании заявки определение "низший" обозначает радикалы с числом атомов углерода максимум 7, предпочтительно до 4, понятие "алкил" обозначает линейные, разветвленные либо циклические насыщенные углеводородные радикалы, такие как метил, этил, пропил, изопропил или трет.-бутил, и "алкокси" обозначает связанную через атом кислорода алкильную группу, "галоген" может обозначать Cl, Br, F либо I.
Понятие "фармацевтически применимые соли" включает в себя соли, образуемые неорганическими и органическими кислотами, такими как, например, соляная кислота, бромистоводородная кислота, азотная кислота, серная кислота, фосфорная кислота, лимонная кислота, муравьиная кислота, фумаровая кислота, малеиновая кислота, уксусная кислота, янтарная кислота, винная кислота, метансульфокислота, p-толуолсульфокислота и т.п.
R5 может обозначать преимущественно низший алкил, предпочтительно метил.
Особенно предпочтительны в этом случае соединения, в которых R2 обозначает метил или метокси, X обозначает -CH2- либо -C(CH3)2-, а R1, R3, R4 и R6 являются водородом.
Некоторыми из особо предпочтительных в рамках настоящего изобретения представителями класса веществ, определяемых общей формулой I, являются следующие:
фумарат (1: 1) (RS)-2-(7-метокси-1,4-дигидроиндено [2,1-c] пиразол-1-ил)-1-метилэтиламина,
фумарат (1: 1) (S)-2-(7-метокси-1,4-дигидроиндено [2,1-c] пиразол-1-ил)-1-метилэтиламина,
фумарат (1: 1) (S)-2-(4,4,7-триметил-1,4-дигидроиндено [2,1-c] пиразол)-1-ил) -1-метилэтиламина,
фумарат (1:1) (S)-2-(7-метокси-4,4-диметил-1,4-дигидроиндено [2,1-с] пиразол-1-ил)-1-метилэтиламина,
фумарат (1: 1) (RS)-2-(7-метокси-4,4-диметил-1,4-дигидроиндено [2,1-с] пиразол-1-ил)-1-метилэтиламина,
фумарат (1: 1) (RS)-2-(7-этокси-1,4-дигидроиндено [2,1-c] пиразол-1-ил)-1-метилэтиламина;
фумарат (1: 1) (R)-2-(7-метокси-1,4-дигидроиндено [2,1-c] пиразол-1-ил)-1-метилэтиламина,
фумарат (1: 0,5) (RS)-2-(8-метокси-1-H-бенз[g]индазол-1-ил)-1- метилэтиламина.
Соединения общей формулы I и их фармацевтически применимые соли могут быть получены согласно изобретению следующим путем:
а) соединение общей формулы IIa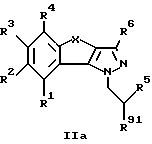
где R1-R6 имеют указанные выше значения, a R91 обозначает переводимый в аминогруппу радикал, переводят в соответствующее аминосоединение и
б) при необходимости полученное соединение формулы I переводят в фармацевтически применимую соль.
Соединения общей формулы IIa, в которой R91 обозначает переводимый в аминогруппу радикал, предпочтительно азидную группу, ацетиламиногруппу или какую-либо другую защищенную аминогруппу, могут быть получены, как это описано ниже, с помощью известных методов.
Если радикал R91 представляет собой азидную группу, то получение соединений формулы I осуществляют путем восстановления. Это восстановление может проводиться по известной методике с помощью комплексных гидридов, таких как, например, алюмогидрид лития, либо путем каталитического гидрирования с использованием металлических катализаторов, таких как, например, платина или палладий.
Если в качестве восстановителя используют алюмогидрид лития, то в качестве растворителей пригодны прежде всего безводный простой эфир или тетрагидрофуран.
Каталитическое гидрирование с помощью металлических катализаторов, например, платины или палладия, целесообразно осуществлять при комнатной температуре. В качестве растворителей для этой цели наиболее пригодны вода, спирты, уксусный эфир, диоксан или же смеси этих растворителей. Гидрирование проводят в атмосфере водорода, предпочтительно в автоклаве или в аппаратуре для встряхивания.
Если R91 представляет собой ацетиламиногруппу или какую-либо другую защищенную аминогруппу, такую как, например, трифторацетиламиногруппа, то, перевод в соответствующее аминосоединение осуществляют путем гидролиза.
Гидролиз до соответствующих аминосоединений общей формулы I проводят с помощью известных методов. Для этой цели пригодны гидроксиды металлов, например, гидроксид натрия или гидроксид калия, которые в присутствии воды и смешиваемого с водой органического растворителя, такого как, например, спирт, этиленгликоль и т.п., гидролизуют до соединений формулы I.
Перевод соединений формулы I в их аддитивные соли кислот осуществляют на последней стадии, т. е. после проведения гидрирования или гидролиза соединений формулы I.
В силу их стабилизирующих свойств соли фумаровой кислоты наиболее пригодны для использования в фармацевтике. Но и все другие упомянутые в описании кислоты также образуют применимые в фармакологии соли. Эти соли получают по известным каждому специалисту методам при комнатной температуре, в качестве растворителей при этом используют преимущественно спирто-эфирные смеси.
Получение промежуточных продуктов формулы II, необходимых для синтеза соединений общей формулы I, представлено на схемах 1 и 2.
При этом все заместители R1-R5 имеют значения, указанные в формуле I, R61 обозначает водород или низший алкил и Me обозначает метил. X1 имеет значение, также указанное в формуле I для X, за исключением соединений с X, обозначающим -CH=CH-, получение которых представлено на схеме 3.
Схема 1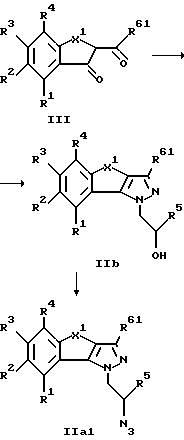
Схема 2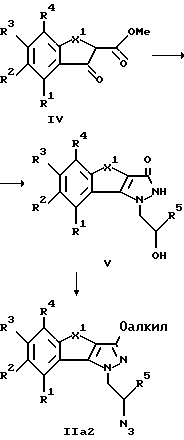
На реакционной схеме 1 представлено получение соединений формулы IIa1, где R61 обозначает водород или низший алкил, а все другие заместители имеют значение, указанное выше, за исключением X=-CH=CH-.
При этом целесообразно работать следующим образом.
Известное либо получаемое по известным способам соединение формулы III с помощью 1-гидразино-2-пропанола и p-толуолсульфокислоты в безводном толуоле трансформируют в водоотделителе в соответствующее пиразоловое соединение общей формулы IIb1. Затем гидроксильная группа с помощью известных методов может переводиться в отщепляемую группу, например, взаимодействием с хлоридом сульфокислоты, предпочтительно хлоридом метансульфокислоты, трансформируясь в сульфонат. Обработкой азидом, предпочтительно азидом натрия, в полярном растворителе, например, в ДМФА, соединения формулы IIb1 можно трансформировать в соответствующие азидные соединения формулы IIa1, которые, как описано выше, путем восстановления азидной группы могут быть переведены в соединения формулы I по изобретению.
На реакционной схеме 2 представлено получение соединений общей формулы IIa2, в которой заместители R1-R5 и X имеют указанное выше значение, за исключением X=-CH=CH-.
При этом соединение формулы IV, известное из существующих публикаций или которое может быть получено по известным способам, с помощью 1-гидразино-2-пропанола, как описано выше, трансформируют в соединение формулы V. Затем это соединение алкилируют в безводном растворителе. В качестве алкилирующих средств могут использоваться предпочтительно диалкилсульфаты или диазометан. После этого OH-группа соединения V с помощью описанных выше методов может переводиться в отщепляемую группу и затем заменяться на азидную группу.
На приведенной ниже реакционной схеме 3 представлено получение соединений формулы Ib, в которой заместители R1-R6 имеют указанное выше значение.
СХЕМА 3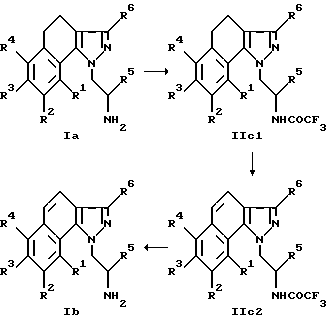
Целесообразно при этом работать следующим образом:
Соединение формулы Ia трансформируют в растворе, состоящем из триэтиламина и этилового эфира трифторуксусной кислоты в безводном растворителе, предпочтительно метаноле. После удаления растворителя остаток растворяют в диоксане, смешивают с ДДБ (2,3-дихлор-5,6-дициан-1,4-бензохинон) и нагревают с обратным холодильником. После этого дегидрирования защитную группу, как описано выше, можно отщеплять от аминогруппы. Для осуществления этой обменной реакции наиболее пригодна аминозащитная группа -COCF3, но возможно также использование других защитных групп.
Как уже упоминалось выше, соединения общей формулы I и их фармацевтически применимые соли обладают ценными фармакологическими свойствами. Они способны связываться с серотонин-рецепторами и в силу этого пригодны для лечения или предупреждения заболеваний или расстройств упомянутого выше типа, а также для изготовления соответствующих лекарственных средств.
Способность предлагаемых согласно изобретению соединений формулы I связываться с серотонин-рецепторами определялась стандартными методами in vitro. Препараты проходили экспериментальную проверку в описанных ниже опытах.
а) Для выявления сродства того или иного соединения к 5НТ1A-рецептору проводились опыты по вытеснению с помощью [3Н]-5НТ (1 нмоль) в качестве радиолиганда на рекомбинированных 5НТ1A-рецепторах человека, экспримированных в 3Т3-клетках мышей. При этом использовали мембраны, полученные из клеток 2 • 105. Каждое из испытывавшихся соединений исследовали в различных концентрациях.
б) Для связывания с 5НТ2C-рецептором согласно тесту на связывание [3Н] -5НТ по методу S.J.Peroutka et al., Brain Research 584. 191-196 (1992).
в) Для связывания с 5НТ2A-рецептором согласно тесту на связывание [3Н] -DOB по методу T.Branchek et al., Molecular Pharmacology 38, 604-609 (1990).
Ниже указаны значения pki (pki=-log10Ki) испытуемых субстанций. Значение Ki определяется следующей формулой: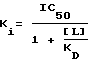
где значения IC50 представляют собой такие концентрации испытуемых соединений в нмолях, благодаря которым вытесняются 50% связанного с рецептором лиганда. [L] обозначает концентрацию лиганда, а значение КD представляет собой константу диссоциации лиганда.
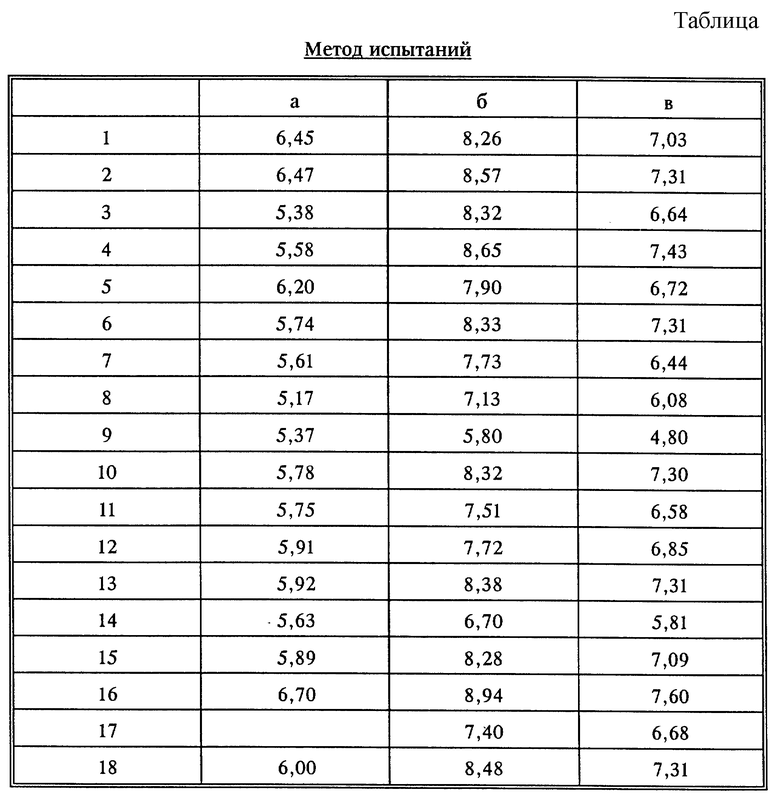
Выявленная таким путем активность некоторых соединений по изобретению представлена в таблице.
1 = фумарат (1:1) (RS)-2-(7-метокси-1,4-дигидроиндено [2,1-с] пиразол-1-ил)-1-метилэтиламина,
2 = фумарат (1:1) (S)-2-(7-метокси-1,4-дигидроиндено[2,1-с]пиразол-1-ил)- 1-метилэтиламина,
3 = фумарат (1:1) (S)-2-(4,4,7-триметил-1,4- дигидроиндено[2,1-с]пиразол-1-ил)-1-метилэтиламина,
4 = фумарат (1:1) (S)-2-(7-метокси-4,4-диметил-1,4-дигидроиндено[2,1-с] пиразол-1-ил) -1-метилэтиламина,
5 = фумарат (1:1) (RS)-2-(4,4,7-триметил-1,4-дигидроиндено[2,1-с] пиразол-1-ил)-1-метилэтиламина,
6 = фумарат (1:1) (RS)-2-(7-метокси-4,4-диметил-1,4-дигидроиндено[2,1-с] пиразол-1-ил)-1-метилэтиламина,
7 = фумарат (1:1) (R)-2-(7-метокси-1,4-дигидроиндено[2,1-с]пиразол-1-ил) -1-метилэтиламина,
8 = фумарат (1:1) (RS)-2-(7-метокси-4-метил-1,4-дигидроиндено[2,1-с]пиразол-1-ил)- 1-метилэтиламина,
9 = фумарат (1:0,5) (RS)-2-(3,7-диметокси-1,4-дигидроиндено [2,1-с]пиразол-1-ил)-метилэтиламина,
10 = фумарат (1:1) (RS)-2-(7-метил-1,4-дигидроиндено[2,1-c] пиразол-1-ил)-1-метилэтиламина,
11 = фумарат (1:1) (RS)-2-(7-фтор-1,4-дигидроиндено[2,1-c] пиразол-1-ил)-1-метилэтиламина,
12 = фумарат (1:1) (RS)-2-(7-фтор-4,4-диметил-1,4-дигидроиндено [2,1-с] пиразол-1-ил)-1-метилэтиламина,
13 = фумарат (1:1) (RS)-2-(7-этокси-1,4-дигидроиндено[2,1-c] пиразол-1-ил)-1-метилэтиламина,
14 = фумарат (1:1) (RS)-2-(6-метокси-4-метил-1,4-дигидроиндено[2,1-с]пиразол-1-ил)-1- метилэтиламина,
15 = фумарат (1:1) (RS)-2-(8-метoкcи-4,5-дигидpo-1H-бенз[g]индaзoл-1-ил)-1- метилэтиламина,
16 = фумарат (1:0,5) (RS)-2-(8-метокси-1H-бенз[g]индазол-1-ил)-1-метилэтиламина,
17 = фумарат (R)-2-(7-этокси-1,4-дигидроиндено[2,1-c]пиразол-1-ил)-1- метилэтиламина,
18 = фумарат (S)-2-(7-этокси-1,4-дигидроиндено[2,1-с]пиразол- 1-ил)-1-метилэтиламина.
Эрекция полового члена (у крыс)
В ходе экспериментов было установлено, что эрекция полового члена зависит от стимулирования 5НТ2C-рецептора (ср. Berendsen & Broekkamp, Eur. Journ. Pharmacol. 135, 179-184 (1987)).
В течение 45 мин после введения испытуемой субстанции подопытным животным определяли число эрекций полового члена. ED50 обозначает дозу, вызывающую 50% таких эрекций.
Пример N - ED50 (мг/кг), s.c.)
1 - 0,32 s.c./3,2 p.o.
2 - 0,32 s.c./1,4 p.o.
13 - 0,5 s.c./2,7 p.o.
18 - 0,7 s.c./2,3 p.o.
Соединения формулы I и фармацевтически применимые аддитивные соли кислот соединений формулы I могут находить применение в качестве лекарственных средств, например, в виде фармацевтических препаратов. Фармацевтические препараты могут вводиться орально, например, в виде таблеток, лаковых таблеток, драже, мягкожелатиновых и твердожелатиновых капсул, растворов, эмульсий или суспензий. Введение, однако, может проводиться также ректально, например, в виде суппозиториев, парентерально, например, в виде растворов для инъекций, или же через нос.
Для изготовления фармацевтических препаратов в соединения формулы I и фармацевтически применимые аддитивные соли кислот соединений формулы I могут добавляться инертные в фармацевтическом отношении, неорганические или органические наполнители. В качестве таковых для таблеток, лаковых таблеток, драже и твердожелатиновых капсул используют, например, лактозу, кукурузный крахмал или его производные, тальк, стеариновую кислоту или ее соли и т.п. В качестве наполнителей для мягкожелатиновых капсул пригодны, например, растительные масла, воск, жиры, полутвердые и жидкие полиолы и т.п. Однако в зависимости от тех или иных свойств активного вещества для мягкожелатиновых капсул вообще может не потребоваться никаких наполнителей. Для изготовления растворов и сиропов в качестве наполнителей пригодны, например, вода, полиолы, глицерин, растительное масло и т.п. В качестве наполнителей для суппозиториев пригодны, например, природные или отвержденные масла, воск, жиры, полужидкие либо жидкие полиолы и т.п.
Фармацевтические препараты могут содержать, кроме того, также консерванты, вещества, способствующие растворимости, стабилизаторы, увлажняющие средства, эмульгаторы, вещества, улучшающие вкус, красители, ароматические вещества, соли для изменения осмотического давления, буферы, оболочки или антиоксиданты. Они могут содержать также другие терапевтически ценные вещества.
Лекарственные средства, содержащие соединение формулы I или его фармацевтически применимую аддитивную соль кислоты и терапевтически инертный наполнитель, также являются предметом настоящего изобретения, равно как и способ их изготовления, отличающийся тем, что одно или несколько соединений формулы I и/или фармацевтически применимые аддитивные соли кислот и ценные в терапевтическом отношении вещества вместе с одним или несколькими терапевтически инертными наполнителями преобразуют в галеновую форму.
Согласно изобретению соединения общей формулы I, а также их фармацевтически применимые аддитивные соли кислот могут применяться для лечения или предупреждения расстройств центральной нервной системы, таких как депрессии, биполярные расстройства, состояния страха, нарушения сна, сексуальные расстройства, психозы, шизофрения, мигрени и другие связанные с головными болями или обусловленные болями другого типа состояния, расстройства, обусловленные личностными факторами, расстройства компульсивно-навязчивого характера, социальная фобия или приступы паники, психические органические расстройства, психические расстройства в детском возрасте, агрессивность, возрастные нарушения памяти и поведения, непреодолимое патологическое влечение и пагубные привычки, ожирение, булимия и т.п., повреждений нервной системы, вызванных травмами, инсультом, нейродегенеративными заболеваниями и т.п., сердечно-сосудистых заболеваний, таких как артериальная гипертония, тромбоз, инсульт и т.д., и желудочно-кишечных расстройств, таких как дисфункция моторики желудочно-кишечного тракта. Дозировку можно варьировать в широких пределах, с учетом, естественно, в каждом отдельном случае индивидуальных особенностей. При оральном введении дозу назначают в пределах от 0,01 мг (на одну дозу) до 500 мг (суточная доза) соединения общей формулы I или соответствующего количества его фармацевтически применимой аддитивной соли кислоты, причем верхняя граница, если это окажется целесообразным, также может превышаться.
Ниже настоящее изобретение поясняется более подробно на примерах, которые никоим образом не ограничивают объем изобретения. Все температуры указаны в градусах Цельсия.
Пример 1
а) Раствор из 0,95 г (5 ммолей) 2-гидроксиметилен-6-метокси-1-инданона, 0,55 г (6 ммолей) (RS)-1-гидразино-2-пропанола и 60 мг p-толуолсульфокислоты в 60 мл безводного толуола нагревали в течение 2 ч в водоотделителе. После концентрирования в вакууме реакционную смесь очищали с помощью колоночной хроматографии на силикагеле (уксусный эфир/гексан 4/1). В результате получали 0,9 г (74%) (RS)-1-(7-метокси-1,4-дигидроиндено [2,1-c]пиразол-1-ил)-пропан-2-ола в виде желтого масла, которое непосредственно использовали в последующей реакции.
б) В охлажденный до 0oС раствор из 0,9 г (3,7 ммоля) (RS)-1-(7-метокси-1,4-дигидроиндено [2,1-c] пиразол-1-ил)-пропан-2-ола и 2 мл (14,8 ммолей) триэтиламина в 40 мл дихлорметана по каплям добавляли при перемешивании 0,6 мл (7,4 ммолей) метансульфонилхлорида и продолжали перемешивать еще в течение 1,5 ч при вышеуказанной температуре. Затем реакционную смесь разбавляли 100 мл дихлорметана, дважды промывали соответственно порциями по 50 мл насыщенного раствора гидрокарбоната натрия и соединенные водные фазы один раз экстрагировали 50 мл дихлорметана. Соединенные органические фазы промывали 50 мл насыщенного раствора хлорида натрия, сушили над сульфатом магния и упаривали в вакууме.
Полученное масло желтого цвета растворяли в 40 мл безводного диметилформамида, смешивали с 0,48 мг (7,4 ммолей) азида натрия и реакционную смесь в течение 15 ч нагревали при перемешивании до температуры 70oС. После охлаждения раствор сливали на 80 мл полунасыщенного раствора хлорида натрия и дважды экстрагировали соответственно порциями по 80 мл простого диэтилового эфира. Соединенные органические фазы промывали по одному разу соответственно 80 мл воды и 80 мл насыщенного раствора хлорида натрия, сушили над сульфатом магния и раствор концентрировали в вакууме. Полученное желтое масло очищали посредством колоночной хроматографии на силикагеле (уксусный эфир/толуол 1/1). В результате получали 0,87 г (87%) (RS)-1-(2-азидопропил)-7-метокси- 1,4-дигидроиндено[2,1-c]пиразола в виде бледно-желтого масла.
в) 0,85 г (3,2 ммоля) (RS)-1-(2-азидопропил)-7-метокси-1,4- дигидроиндено[2,1-c]пиразола, растворенного в 50 мл безводного этанола, гидрировали в присутствии 85 мг оксида платины в течение 2 ч. Затем отфильтровывали от катализатора, промывали этанолом и растворитель отгоняли в вакууме. Полученное бесцветное масло растворяли в 70 мл безводного простого диэтилового эфира, фильтровали и при перемешивании смешивали с раствором из 371 мг (3,2 ммоля) фумаровой кислоты в 10 мл метанола. Затем перемешивали в течение 15 ч при комнатной температуре, после чего отфильтровывали белые кристаллы. Таким путем получали 0,9 г (78%) фумарата (1:1) (RS)-2-(7-метокси-1,4-дигидроиндено[2,1-c] пиразол-1-ил)-1-метилэтиламина с Тпл 182oС.
Пример 2
а) Раствор из 1,5 г (7,9 ммолей) 2-гидроксиметилен-6-метокси-1-инданона, 0,78 г (8,6 ммолей) (R)-1-гидразино-2-пропанола и 100 мг p-толуолсульфокислоты в 100 мл безводного толуола нагревали в течение 1,5 ч в водоотделителе. После концентрирования в вакууме реакционную смесь очищали посредством колоночной хроматографии на силикагеле (уксусный эфир/гексан 4/1). В результате получали 1,3 г (68%) (R)-1-(7-метокси-1,4-дигидроиндено[2,1-с] пиразол-1-ил)-пропан-2-ола в виде твердого вещества желтого цвета, которое непосредственно использовали в последующей реакции.
б) В охлажденный до 0oС раствор из 1,3 г (5,3 ммолей) (R)-1-(7-метокси-1,4-дигидроиндено[2,1-с] пиразол-1-ил)-пропан-2-ола и 3,05 мл (21,4 ммолей) триэтиламина в 50 мл дихлорметана при перемешивании добавляли по каплям 0,85 мл (10,7 ммолей) метансульфонилхлорида и продолжали перемешивать при вышеуказанной температуре еще в течение 1,5 ч. Затем реакционную смесь разбавляли 150 мл дихлорметана, дважды промывали соответственно порциями по 70 мл насыщенного раствора гидрокарбоната натрия и соединенные водные фазы один раз экстрагировали 70 мл дихлорметана. Соединенные органические фазы промывали 70 мл насыщенного раствора хлорида натрия, сушили над сульфатом магния и упаривали в вакууме. Полученное желтое масло растворяли в 40 мл безводного диметилформамида, смешивали с 0,83 г (12,5 молей) азида натрия и реакционную смесь при перемешивании нагревали в течение 15 ч до температуры 70oС. После охлаждения раствор сливали на 100 мл полунасыщенного раствора хлорида натрия и дважды экстрагировали соответственно порциями по 100 мл простого диэтилового эфира. Соединенные органические фазы промывали по одному разу соответственно 100 мл воды и 100 мл насыщенного раствора хлорида натрия, сушили над сульфатом магния и концентрировали раствор в вакууме. Полученное коричневое масло очищали посредством колоночной хроматографии на силикагеле (уксусный эфир/толуол 1/1). В результате получали 1,3 г (90%) (S)-1-(2-азидопропил)- 7-метокси-1,4-дигидроиндено[2,1-c] пиразола в виде бледно-желтого масла.
в) 1,3 г (4,8 ммоля) (S)-1-(2-азидопропил)-7-метокси-1,4- дигидроиндено [2,1-c] пиразола, растворенного в 50 мл безводного этанола, гидрировали в присутствии 130 мг оксида платины в течение 2 ч. Затем отфильтровывали от катализатора, промывали этанолом и растворитель отгоняли в вакууме. Полученное бесцветное масло растворяли в 80 мл безводного простого диэтилового эфира, фильтровали и при перемешивании смешивали с раствором из 560 мг (4,8 ммоля) фумаровой кислоты в 10 мл метанола. Перемешивание продолжали в течение 4 ч при комнатной температуре, после чего отфильтровывали белые кристаллы. Таким путем получали 1,4 г (81%) фумарата (1:1) (S)-2-(7-метокси-1,4-дигидроиндено [2,1-с] пиразол-1-ил)-1-метилэтиламина с Тпл180oС.
Пример 3
а) Раствор из 0,7 г (3,5 ммоля) 2-гидроксиметилен-3,3,6-триметил-1-инданона, 0,37 г (4,1 ммоля) (R)-1-гидразино-2-пропанола и 50 мг p-толуолсульфокислоты в 50 мл безводного толуола нагревали в течение 2 ч в водоотделителе. После концентрирования в вакууме реакционную смесь очищали посредством колоночной хроматографии на силикагеле (уксусный эфир). В результате получали 0,8 г (89%) (R)-1-(4,4,7-триметил-1,4- дигидроиндено[2,1-c]пиразол-1-ил)-пропан-2-ола в виде желтого масла, которое непосредственно использовали в последующей реакции.
б) В охлажденный до 0oС раствор из 0,8 г (3,1 ммоля) (R)-1-(4,4,7-триметил-1,4-дигидроиндено[2,1-с] пиразол-1-ил)-пропан-2-ола и 1,75 мл (12,5 ммолей) триэтиламина в 50 мл дихлорметана при перемешивании добавляли по каплям 0,5 мл (6,24 ммолей) метансульфонилхлорида и продолжали перемешивать еще в течение 1,5 ч при вышеуказанной температуре. Затем реакционную смесь разбавляли 150 мл дихлорметана, дважды промывали соответственно порциями по 70 мл насыщенного раствора гидрокарбоната натрия и соединенные водные фазы один раз экстрагировали с помощью 70 мл дихлорметана. Соединенные органические фазы промывали 70 мл насыщенного раствора хлорида натрия, сушили над сульфатом магния и упаривали в вакууме. Полученное желтое масло растворяли в 40 мл безводного диметилформамида, смешивали с 0,41 г (6,3 ммолей) азида натрия и реакционную смесь при перемешивании нагревали в течение 15 ч до температуры 70oС. После охлаждения раствор сливали на 100 мл полунасыщенного раствора хлорида натрия и дважды экстрагировали соответственно порциями по 100 мл простого диэтилового эфира. Соединенные органические фазы промывали по одному разу соответственно 100 мл воды и 100 мл насыщенного раствора хлорида натрия, сушили над сульфатом магния и концентрировали раствор в вакууме. Полученное масло коричневого цвета очищали посредством колоночной хроматографии на силикагеле (уксусный эфир/толуол 1/1). В результате получали 0,7 г (80%) (S)-1-(2-азидопропил)- 4,4,7-триметил-1,4-дигидроиндено[2,1-c] пиразола в виде бледно-желтого масла.
в) 0,7 г (2,5 ммоля) (S)-1-(2-азидопропил)-4,4,7-триметил-1,4-дигидроиндено[2,1-c] пиразола, растворенного в 50 мл безводного этанола, гидрировали в присутствии 70 мг оксида платины в течение 2 ч. Затем отфильтровывали от катализатора, промывали этанолом и растворитель отгоняли в вакууме. Полученное бесцветное масло растворяли в 70 мл безводного простого диэтилового эфира, фильтровали и при перемешивании смешивали с раствором из 290 мг (2,5 ммоля) фумаровой кислоты в 5 мл метанола. Затем продолжали перемешивать еще в течение 4 ч при комнатной температуре, после чего отфильтровывали белые кристаллы. Таким путем получали 0,5 г (54%) фумарата (1:1) (S)-2- (4,4,7-триметил-1,4-дигидроиндено[2,1-c]пиразол-1-ил)-1-метилэтиламина с Тпл 158oС.
Пример 4
а) Раствор из 1,5 г (6,8 ммолей) 2-гидроксиметилен-6-метокси-3,3-диметил-1-инданона, 0,74 г (8,2 ммолей) (R)-1-гидразино-2- пропанола и 100 мг p-толуолсульфокислоты в 100 мл безводного толуола нагревали в течение 1,5 ч в водоотделителе. После концентрирования в вакууме реакционную смесь очищали посредством колоночной хроматографии на силикагеле (уксусный эфир/гексан 4/1). В результате получали 1,41 г (76%) (R)-1-(7-метокси-4,4-диметил-1,4-дигидроиндено[2,1-c] пиразол-1-ил)-пропан-2-ола в виде желтого масла, которое непосредственно использовали в последующей реакции.
б) В охлажденный до 0oС раствор из 1,41 г (5,2 ммолей) (R)-1-(7-метокси-4,4-диметил-1,4-дигидроиндено[2,1-с] пиразол-1-ил)- пропан-2-ола и 2,9 мл (20,4 ммолей) триэтиламина в 50 мл дихлорметана при перемешивании добавляли по каплям 0,8 мл (10,2 ммолей) метансульфонилхлорида и продолжали перемешивание еще в течение 1,5 ч при вышеуказанной температуре. Затем реакционную смесь разбавляли 150 мл дихлорметана, дважды промывали порциями по 70 мл насыщенного раствора гидрокарбоната натрия и соединенные водные фазы один раз экстрагировали с помощью 70 мл дихлорметана. Соединенные органические фазы промывали 70 мл насыщенного раствора хлорида натрия, сушили над сульфатом магния и упаривали в вакууме. Полученное желтое масло растворяли в 40 мл безводного диметилформамида, смешивали с 0,76 г (11,4 ммолей) азида натрия и реакционную смесь при перемешивании нагревали в течение 15 ч до температуры 70oС. После охлаждения раствор сливали на 100 мл полунасыщенного раствора хлорида натрия и дважды экстрагировали соответственно порциями по 100 мл простого диэтилового эфира. Соединенные органические фазы промывали по одному разу соответственно 100 мл воды и 100 мл насыщенного раствора хлорида натрия, сушили над сульфатом магния и концентрировали раствор в вакууме. Полученное масло коричневого цвета очищали посредством колоночной хроматографии на силикагеле (уксусный эфир/толуол 1/1). В результате получали 1,38 г (89%) (S)-1-(2-азидопропил)-7-метокси- 4,4-диметил-1,4-дигидроиндено[2,1-с]пиразола в виде желтого масла.
в) 1,38 г (4,6 ммоля) (S)-1-(2-азидопропил)-7-метокси-4,4-диметил-1,4-дигидроиндено [2,1-c] пиразола, растворенного в 50 мл безводного этанола, гидрировали в присутствии 140 мг оксида платины в течение 1,5 ч. Затем отфильтровывали от катализатора, промывали этанолом и растворитель отгоняли в вакууме. Полученное бесцветное масло растворяли в 80 мл безводного простого диэтилового эфира, фильтровали и при перемешивании смешивали с раствором из 534 мг (4,6 ммоля) фумаровой кислоты в 10 мл метанола. Перемешивание продолжали в течение 18 ч при комнатной температуре, после чего отфильтровывали белые кристаллы. Таким путем получали 1,23 г (69%) фумарата (1:1) (S)-2-(7-метокси-4,4-диметил-1,4-дигидроиндено[2,1-с] пиразол-1-ил)- 1-метилэтиламина с Тпл 160-162oС.
Пример 5
а) Раствор из 1,5 г (7,4 ммолей) 2-гидроксиметилен-3,3,6-триметил-1-инданона, 0,55 г (6,1 ммолей) (RS)-1-гидразино-2-пропанола и 100 мг p-толуолсульфокислоты в 100 мл безводного толуола нагревали в течение 2 ч в водоотделителе. После концентрирования в вакууме реакционную смесь очищали посредством колоночной хроматографии на силикагеле (уксусный эфир/гексан 4/1). В результате получали 1,6 г (84%) (RS)-1- (4,4,7-триметил-1,4-дигидроиндено[2,1-с] пиразол-1-ил)-пропан-2-ола в виде желтого масла, которое непосредственно использовали в последующей реакции.
б) В охлажденный до 0oС раствор из 1,6 г (6,2 ммолей) (RS)-1-(4,4,7-триметил-1,4-дигидроиндено[2,1-с] пиразол- 1-ил)-пропан-2-ола и 3,5 мл (25 ммолей) триэтиламина в 60 мл дихлорметана при перемешивании добавляли по каплям 1 мл (12,5 ммолей) метансульфонилхлорида и продолжали перемешивание еще в течение 1,5 ч при вышеуказанной температуре. Затем реакционную смесь разбавляли 150 мл дихлорметана, дважды промывали соответственно порциями по 70 мл насыщенного раствора гидрокарбоната натрия и соединенные водные фазы один раз экстрагировали с помощью 70 мл дихлорметана. Соединенные органические фазы промывали 70 мл насыщенного раствора хлорида натрия, сушили над сульфатом магния и упаривали в вакууме. Полученное желтое масло растворяли в 60 мл безводного диметилформамида, смешивали с 0,81 г (12,5 ммолей) азида натрия и затем реакционную смесь при перемешивании нагревали в течение 15 ч до температуры 70oС. После охлаждения раствор сливали на 100 мл полунасыщенного раствора хлорида натрия и дважды экстрагировали соответственно порциями по 100 мл простого диэтилового эфира. Соединенные органические фазы промывали по одному разу соответственно 100 мл воды и 100 мл насыщенного раствора хлорида натрия, сушили над сульфатом магния и концентрировали раствор в вакууме. Полученное масло желтого цвета очищали посредством колоночной хроматографии на силикагеле (уксусный эфир/толуол 1/1). В результате получали 1,1 г (63%) (RS)-1-(2-азидопропил)- 4,4,7-триметил-1,4-дигидроиндено[2,1-c]пиразола в виде бледно-желтого масла.
в) 1,1 г (3,9 ммоля) (RS)-1-(2-азидопропил)- 4,4,7-триметил-1,4-дигидроиндено[2,1-c] пиразола, растворенного в 60 мл безводного этанола, гидрировали в присутствии 110 мг оксида платины в течение 3 ч. Затем отфильтровывали от катализатора, промывали этанолом и растворитель отгоняли в вакууме. Полученное бесцветное масло растворяли в 150 мл безводного простого диэтилового эфира, фильтровали и при перемешивании смешивали с раствором из 453 мг (3,9 ммоля) фумаровой кислоты в 10 мл метанола. Перемешивание продолжали в течение 4 ч при комнатной температуре, после чего отфильтровывали белые кристаллы. Таким путем получали 1 г (69%) фумарата (1:1) (RS)-2-(4,4,7-триметил- 1,4-дигидроиндено[2,1-c]пиразол-1-ил)-1-метилэтиламина с Тпл 167oС.
Пример 6
а) Раствор из 1,5 г (6,8 ммолей) 2-гидроксиметилен-6-метокси-3,3-диметил-1-инданона, 0,74 г (8,2 ммолей) (RS)-1-гидразино-2-пропанола и 100 мг p-толуолсульфокислоты в 100 мл безводного толуола нагревали в течение 2 ч в водоотделителе. После концентрирования в вакууме реакционную смесь очищали посредством колоночной хроматографии на силикагеле (уксусный эфир/гексан 4/1). В результате получали 1,4 г (75%) (RS)-1-(7-метокси-4,4- диметил-1,4-дигидроиндено[2,1-с]пиразол-1-ил)-пропан-2-ола в виде желтого масла, которое непосредственно использовали в последующей реакции.
б) В охлажденный до 0oС раствор из 1,4 г (5,1 ммолей) (RS)-1-(7-метокси-4,4-диметил-1,4-дигидроиндено[2,1-с] пиразол-1- ил)-пропан-2-ола и 2,9 мл (20,4 ммолей) триэтиламина в 50 мл дихлорметана по каплям добавляли при перемешивании 0,8 мл (10,2 ммолей) метансульфонилхлорида и продолжали перемешивание еще в течение 1,5 ч при вышеуказанной температуре. Затем реакционную смесь разбавляли 150 мл дихлорметана, дважды промывали соответственно порциями по 70 мл насыщенного раствора гидрокарбоната натрия и соединенные водные фазы один раз экстрагировали с помощью 70 мл дихлорметана. Соединенные органические фазы промывали 70 мл насыщенного раствора хлорида натрия, сушили над сульфатом магния и упаривали в вакууме. Полученное желтое масло растворяли в 40 мл безводного диметилформамида, смешивали с 0,66 г (10,2 ммолей) азида натрия и реакционную смесь при перемешивании нагревали в течение 15 ч до температуры 70oС. После охлаждения раствор сливали на 100 мл полунасыщенного раствора хлорида натрия и дважды экстрагировали соответственно порциями по 100 мл простого диэтилового эфира. Соединенные органические фазы промывали по одному разу соответственно 100 мл воды и 100 мл насыщенного раствора хлорида натрия, сушили над сульфатом магния и концентрировали раствор в вакууме. Полученное коричневое масло очищали посредством колоночной хроматографии на силикагеле (уксусный эфир/толуол 1/1). В результате получали 1,03 г (68%) (RS)-1-(2-азидопропил) -7-метокси-4,4-диметил-1,4-дигидроиндено [2,1-с] пиразола в виде желтого масла.
в) 1,03 г (3,5 ммоля) (RS)-1-(2-азидопропил)-7-метокси-4,4- диметил-1,4-дигидроиндено[2,1-с] пиразола, растворенного в 50 мл безводного этанола, гидрировали в присутствии 100 мг оксида платины в течение 1,5 ч. Затем отфильтровывали от катализатора, промывали этанолом и растворитель отгоняли в вакууме. Полученное масло желтого цвета растворяли в 80 мл безводного простого диэтилового эфира, фильтровали и при перемешивании смешивали с раствором из 440 мг (3,5 ммоля) фумаровой кислоты в 15 мл метанола. Перемешивание продолжали еще в течение 15 ч при комнатной температуре, после чего отфильтровывали белые кристаллы. Таким путем получали 0,72 г (53%) фумарата (1: 1) (RS)-2-(7- метокси-4,4-диметил-1,4-дигидроиндено[2,1-с] пиразол-1-ил)-1- метилэтиламина с Тпл 178-180oС.
Пример 7
а) Раствор из 0,51 г (2,7 ммоля) 2-гидроксиметилен-6-метокси-1-инданона, 0,29 г (3,2 ммоля) (S)-1-гидразино-2-пропанола и 50 мг p-толуолсульфокислоты в 50 мл безводного толуола нагревали в течение 2 ч в водоотделителе. После концентрирования в вакууме реакционную смесь очищали посредством колоночной хроматографии на силикагеле (уксусный эфир). В результате получали 0,6 г (92%) (S)-1-(7-метокси-1,4-дигидроиндено[2,1-c] пиразол-1-ил)-пропан-2-ола в виде твердого вещества желтого цвета, которое непосредственно использовали в последующей реакции.
б) В охлажденный до 0oС раствор из 0,6 г (2,46 ммоля) (S)-1-(7-метокси-1,4-дигидроиндено[2,1-с]пиразол-1-ил)-пропан-2-ола и 1,4 мл (9,84 ммолей) триэтиламина в 60 мл дихлорметана по каплям добавляли при перемешивании 0,4 мл (4,92 ммоля) метансульфонилхлорида и продолжали перемешивать еще в течение 1,5 ч при вышеуказанной температуре. Затем реакционную смесь разбавляли 100 мл дихлорметана, дважды промывали соответственно порциями по 50 мл насыщенного раствора гидрокарбоната натрия и соединенные водные фазы один раз экстрагировали с помощью 50 мл дихлорметана. Соединенные органические фазы промывали 50 мл насыщенного раствора хлорида натрия, сушили над сульфатом магния и упаривали в вакууме. Полученное коричневое масло растворяли в 40 мл безводного диметилформамида, смешивали с 0,32 г (4,92 ммоля) азида натрия и реакционную смесь при перемешивании нагревали в течение 15 ч до температуры 80oС. После охлаждения раствор сливали на 80 мл полунасыщенного раствора хлорида натрия и дважды экстрагировали соответственно порциями по 80 мл простого диэтилового эфира. Соединенные органические фазы промывали по одному разу соответственно 80 мл воды и 80 мл насыщенного раствора хлорида натрия, сушили над сульфатом магния и концентрировали раствор в вакууме. Полученное коричневое масло очищали посредством колоночной хроматографии на силикагеле (уксусный эфир/толуол 1/1). В результате получали 0,5 г (75%) (R)-1-(2-азидопропил)-7-метокси-1,4-дигидроиндено[2,1-c] пиразола в виде желтого масла.
в) 0,5 г (1,85 ммоля) (R)-1-(2-азидопропил)-7-метокси-1,4-дигидроиндено[2,1-c] пиразола, растворенного в 50 мл безводного этанола, гидрировали в присутствии 50 мг оксида платины в течение 2 ч. Затем отфильтровывали от катализатора, промывали этанолом и растворитель отгоняли в вакууме. Полученное бесцветное масло растворяли в 70 мл безводного простого диэтилового эфира, фильтровали и при перемешивании смешивали с раствором из 215 мг (1,85 ммоля) фумаровой кислоты в 5 мл метанола. Перемешивание продолжали еще в течение 15 ч при комнатной температуре, после чего отфильтровывали белые кристаллы. Таким путем получали 0,55 г (83%) фумарата (1:1) (R)-2-(7-метокси-1,4-дигидроиндено[2,1-с]пиразол-1-ил)-1- метилэтиламина с Тпл 180oС.
Пример 8
а) Раствор из 0,9 г (4,41 моля) 2-ацетил-6-метокси-1-инданона, 0,51 г (5,73 ммолей) (RS)-1-гидразино-2-пропанола и 70 мг p-толуолсульфокислоты в 70 мл безводного толуола нагревали в течение 2 ч в водоотделителе. После концентрирования в вакууме реакционную смесь очищали посредством колоночной хроматографии на силикагеле (уксусный эфир). В результате получали 1,1 г (96%) (RS)-1-(7-метокси-3-метил-1,4- дигидроиндено[2,1-с] пиразол-1-ил)-пропан-2-ола в виде твердого вещества желтого цвета, которое непосредственно использовали в последующей реакции.
б) В охлажденный до 0oС раствор из 1,1 г (4,26 ммоля) (RS)-1-(7-метокси-3-метил-1,4-дигидроиндено[2,1-c] пиразол-1-ил)-пропан-2-ола и 2,4 мл (17 ммолей) триэтиламина в 60 мл дихлорметана при перемешивании добавляли по каплям 0,7 мл (8,52 ммолей) метансульфонилхлорида и продолжали перемешивать еще в течение 1,5 ч при вышеуказанной температуре. Затем реакционную смесь разбавляли 130 мл дихлорметана, дважды промывали соответственно порциями по 60 мл насыщенного раствора гидрокарбоната натрия и соединенные водные фазы один раз экстрагировали с помощью 50 мл дихлорметана. Соединенные органические фазы промывали 70 мл насыщенного раствора хлорида натрия, сушили над сульфатом магния и концентрировали в вакууме. Полученное масло коричневого цвета растворяли в 60 мл безводного диметилформамида, смешивали с 0,55 г (8,46 ммолей) азида натрия и реакционную смесь при перемешивании нагревали в течение 15 ч до температуры 80oС. После охлаждения раствор сливали на 80 мл полунасыщенного раствора хлорида натрия и дважды экстрагировали соответственно порциями по 80 мл простого диэтилового эфира. Соединенные органические фазы промывали по одному разу соответственно 80 мл воды и 80 мл насыщенного раствора хлорида натрия, сушили над сульфатом магния и концентрировали раствор в вакууме. Полученное коричневое масло очищали посредством колоночной хроматографии на силикагеле (уксусный эфир/толуол 1/1). В результате получали 1 г (83%) (RS)-1-(2-азидопропил)-7- метокси-3-метил-1,4-дигидроиндено[2,1-c] пиразола в виде бледно-коричневого масла.
в) 1,1 г (3,88 ммоля) (RS)-1-(2- азидопропил)-7-метокси-3-метил-1,4-дигидроиндено[2,1-c]пиразола, растворенного в 60 мл безводного этанола, гидрировали в присутствии 110 мг оксида платины в течение 2 ч. Затем отфильтровывали от катализатора, промывали этанолом и растворитель отгоняли в вакууме. Полученное бесцветное масло растворяли в 120 мл безводного простого диэтилового эфира, фильтровали и при перемешивании смешивали с раствором из 450 мг (3,88 ммоля) фумаровой кислоты в 5 мл метанола. Перемешивание продолжали еще в течение 15 ч при комнатной температуре, после чего отфильтровывали белые кристаллы. Таким путем получали 1,2 г (83%) фумарата (1:1) (RS)-2-(7-метокси-3-метил-1,4-дигидроиндено[2,1-с]пиразол-1-ил)-1- метилэтиламина с Тпл 184oС.
Пример 9
а) Раствор из 4,54 г (20,6 ммолей) 2-метоксикарбонил-6-метокси-1-инданона, 2,3 г (25,5 ммолей) (RS)-1-гидразино-2-пропанола и 150 мг p-толуолсульфокислоты в 150 мл безводного толуола нагревали в течение 4 ч в водоотделителе. После концентрирования в вакууме реакционную смесь растворяли в этаноле и выпавшее в осадок твердое вещество отфильтровывали. Фильтрат концентрировали и очищали посредством колоночной хроматографии на силикагеле (дихлорметан/метанол 9/1). В результате получали 2,44 г (46%) (RS)-1-(7-метокси-1,4-дигидроиндено[2,1-c] пиразол-3- он-1-ил)-пропан-2-ола в виде масла коричневого цвета, которое непосредственно использовали в последующей реакции.
б) В раствор из 2,44 г (9,37 ммолей) (RS)-1-(7-метокси-1,4-дигидроиндено[2,1-c] пиразол-3-он-1-ил)-пропан-2-ола в 80 мл безводного простого диэтилового эфира и 50 мл безводного метанола добавляли при перемешивании раствор из 0,79 г (18,8 ммолей) диазометана в 56 мл безводного простого диэтилового эфира. Перемешивание продолжали еще в течение 15 ч при комнатной температуре, после чего концентрировали в вакууме. В результате получали 2,08 г (81%) (RS)-1-(3,7-диметокси-1,4-дигидроиндено[2,1-с]пиразол-1-ил)- пропан-2-ола в виде твердого вещества коричневого цвета, которое непосредственно использовали в последующей реакции.
в) В охлажденный до 0oС раствор из 2,08 г (7,6 ммолей) (RS)-1-(3,7-диметокси-1,4-дигидроиндено[2,1-с] пиразол-1-ил)-пропан-2-ола и 4,3 мл (30,4 ммолей) триэтиламина в 60 мл дихлорметана при перемешивании добавляли по каплям 1,21 мл (15,2 ммолей) метансульфонилхлорида и продолжали перемешивать еще в течение 1,5 ч при вышеуказанной температуре. Затем реакционную смесь разбавляли 150 мл дихлорметана, дважды промывали соответственно порциями по 100 мл насыщенного раствора гидрокарбоната натрия и соединенные водные фазы один раз экстрагировали с помощью 70 мл дихлорметана. Соединенные органические фазы промывали 100 мл насыщенного раствора хлорида натрия, сушили над сульфатом магния и упаривали в вакууме. Полученное масло коричневого цвета растворяли в 50 мл безводного диметилформамида, смешивали с 1 г (15,4 ммолей) азида натрия и реакционную смесь при перемешивании нагревали в течение 20 ч до температуры 70oС. После охлаждения раствор сливали на 80 мл полунасыщенного раствора хлорида натрия и дважды экстрагировали соответственно порциями по 80 мл простого диэтилового эфира. Соединенные органические фазы промывали по одному разу соответственно 80 мл воды и 100 мл насыщенного раствора хлорида натрия, сушили над сульфатом магния и концентрировали раствор в вакууме. Полученное коричневое масло очищали посредством колоночной хроматографии на силикагеле (уксусный эфир/толуол 1/1). В результате получали 1,11 г (49%) (RS)-1-(2-азидопропил)- 3,7-диметокси-1,4-дигидроиндено[2,1-c] пиразола в виде масла коричневого цвета.
г) 1,11 г (3,71 ммоля) (RS)-1-(2-азидопропил)- 3,7-диметокси-1,4-дигидроиндено[2,1-c] пиразола, растворенного в 60 мл безводного этанола, гидрировали в присутствии 110 мг оксида платины в течение 1,5 ч. Затем отфильтровывали от катализатора, промывали этанолом и растворитель отгоняли в вакууме. Полученное бесцветное масло растворяли в 50 мл безводного простого диэтилового эфира, фильтровали и при перемешивании смешивали с раствором из 430 г (3,71 ммоля) фумаровой кислоты в 5 мл метанола. Перемешивание продолжали еще в течение 15 ч при комнатной температуре, после чего отфильтровывали кристаллы бежевого цвета. Таким путем получали 0,56 г (46%) фумарата (1: 0,5) (RS)-2-(3,7- диметокси-1,4-дигидроиндено[2,1-с]пиразол-1-ил)-1-метилэтиламина с Тпл 209oС.
Пример 10
а) Раствор из 1,4 г (8,04 ммолей) 2-гидроксиметилен-6-метил-1-инданона, 0,87 г (9,65 ммолей) (RS)-1-гидразино-2-пропанола и 100 мг p-толуолсульфокислоты в 100 мл безводного толуола нагревали в течение 2 ч в водоотделителе. После концентрирования в вакууме реакционную смесь очищали посредством колоночной хроматографии на силикагеле (уксусный эфир/гексан 4/1). В результате получали 1,7 г (93%) (RS)-1-(7-метил-1,4- дигидроиндено[2,1-с]пиразол-1-ил)-пропан-2-ола в виде желтого масла, которое непосредственно использовали в последующей реакции.
б) В охлажденный до 0oС раствор из 1,7 г (7,45 ммолей) (RS)-1-(7-метил-1,4-дигидроиндено[2,1-с] пиразол-1-ил)-пропан-2-ола и 4,12 мл (29,7 ммолей) триэтиламина в 60 мл дихлорметана при перемешивании по каплям добавляли 1,15 мл (14,8 ммолей) метансульфонилхлорида и продолжали перемешивать еще в течение 1,5 ч при вышеуказанной температуре. Затем реакционную смесь разбавляли 100 мл дихлорметана, дважды промывали соответственно порциями по 50 мл насыщенного раствора гидрокарбоната натрия и соединенные водные фазы один раз экстрагировали с помощью 50 мл дихлорметана. Соединенные органические фазы промывали 50 мл насыщенного раствора хлорида натрия, сушили над сульфатом магния и упаривали в вакууме. Полученное желтое масло растворяли в 40 мл безводного диметилформамида, смешивали с 0,96 г (14,8 ммолей) азида натрия и реакционную смесь при перемешивании нагревали в течение 15 ч до температуры 70oС. После охлаждения раствор сливали на 100 мл полунасыщенного раствора хлорида натрия и дважды экстрагировали соответственно порциями по 100 мл простого диэтилового эфира. Соединенные органические фазы промывали по одному разу соответственно 80 мл воды и 80 мл насыщенного раствора хлорида натрия, сушили над сульфатом магния и концентрировали раствор в вакууме. Полученное масло коричневого цвета очищали посредством колоночной хроматографии на силикагеле (уксусный эфир/толуол 1/1). В результате получали 1,38 г (73%) (RS)-1-(2-азидопропил)- 7-метил-1,4-дигидроиндено[2,1-с] пиразола в виде бледно-желтого твердого вещества с Тпл 70-72oС.
в) 1,38 г (5,45 ммолей) (RS)-1-(2- азидопропил)-7-метил-1,4-дигидроиндено[2,1-c]пиразола, растворенного в 60 мл безводного этанола, гидрировали в присутствии 140 мг оксида платины в течение 2 ч. Затем отфильтровывали от катализатора, промывали этанолом и растворитель отгоняли в вакууме. Полученное бесцветное масло растворяли в 80 мл безводного простого диэтилового эфира, фильтровали и при перемешивании смешивали с раствором из 633 мг (5,45 ммолей) фумаровой кислоты в 10 мл метанола. Перемешивание продолжали еще в течение 15 ч при комнатной температуре, после чего отфильтровывали белые кристаллы. Таким путем получали 1,62 г (87%) фумарата (1:1) (RS)-2-(7-метил-1,4-дигидроиндено[2,1-c]пиразол-1-ил)-1- метилэтиламина с Тпл 205oС.
Пример 11
а) Раствор из 1,42 г (8,0 ммолей) 6-фтор-2-гидроксиметилен-1-инданона, 0,87 г (9,65 ммолей) (RS)-1-гидразино-2-пропанола и 100 мг p-толуолсульфокислоты в 100 мл безводного толуола нагревали в течение 1,5 ч в водоотделителе. После концентрирования в вакууме реакционную смесь очищали посредством колоночной хроматографии на силикагеле (уксусный эфир). В результате получали 1,5 г (81%) (RS)-1-(7-фтор-1,4- дигидроиндено[2,1-с]пиразол-1-ил)-пропан-2-ола в виде твердого вещества желтого цвета, которое непосредственно использовали в последующей реакции.
б) В охлажденный до 0oС раствор из 1,5 г (6,46 ммолей) (RS)-1-(7-фтор-1,4-дигидроиндено[2,1-с] пиразол-1-ил)- пропан-2-ола и 3,6 мл (25,8 ммолей) триэтиламина в 60 мл дихлорметана при перемешивании добавляли по каплям 1 мл (12,9 ммолей) метансульфонилхлорида и продолжали перемешивать еще в течение 1,5 ч при вышеуказанной температуре. Затем реакционную смесь разбавляли 100 мл дихлорметана, дважды промывали соответственно порциями по 50 мл насыщенного раствора гидрокарбоната натрия и соединенные водные фазы один раз экстрагировали с помощью 50 мл дихлорметана. Соединенные органические фазы промывали 50 мл насыщенного раствора хлорида натрия, сушили над сульфатом магния и упаривали в вакууме. Полученное твердое вещество желтого цвета растворяли в 50 мл безводного диметилформамида, смешивали с 0,84 г (12,9 ммолей) азида натрия и реакционную смесь при перемешивании нагревали в течение 5 ч до температуры 90oС. После охлаждения раствор сливали на 70 мл полунасыщенного раствора хлорида натрия и дважды экстрагировали соответственно порциями по 100 мл уксусного эфира. Соединенные органические фазы промывали по одному разу 80 мл воды и 80 мл насыщенного раствора хлорида натрия соответственно, сушили над сульфатом магния и концентрировали раствор в вакууме. Полученное коричневое масло очищали посредством колоночной хроматографии на силикагеле (уксусный эфир). В результате получали 1,59 г (96%) (RS)-1-(2-азидопропил)-7-фтор-1,4-дигидроиндено[2,1-c] пиразола в виде бледно-желтого масла.
в) 1,59 г (6,18 ммолей) (RS)-1-(2-азидопропил)-7-фтор-1,4- дигидроиндено[2,1-c] пиразола, растворенного в 50 мл безводного этанола, гидрировали в присутствии 160 мг оксида платины в течение 14 ч. Затем отфильтровывали от катализатора, промывали этанолом и растворитель отгоняли в вакууме. Полученное бесцветное масло растворяли в 100 мл безводного простого диэтилового эфира, фильтровали и при перемешивании смешивали с раствором из 717 мг (6,18 ммолей) фумаровой кислоты в 10 мл метанола. Перемешивание продолжали еще в течение 5 ч при комнатной температуре, после чего отфильтровывали белые кристаллы. Таким путем получали 1,68 г (78%) фумарата (1:1) (RS)-2-(7-фтор-1,4-дигидроиндено[2,1-c] пиразол-1-ил)-1-метилэтиламина с Тпл 168-170oC.
Пример 12
а) Раствор из 1,4 г (6,8 ммолей) 6-фтор-2-гидроксиметилен-3,3- диметил-1-инданона, 0,74 г (8,2 ммолей) (RS)-1-гидразино-2-пропанола и 100 мг p-толуолсульфокислоты в 100 мл безводного толуола нагревали в течение 2 ч в водоотделителе. После концентрирования в вакууме реакционную смесь очищали посредством колоночной хроматографии на силикагеле (уксусный эфир/гексан 4/1). В результате получали 1,7 г (96%) (RS)-1-(7-фтор-4,4- диметил-1,4-дигидроиндено[2,1-c]пиразол-1-ил)-пропан-2-ола в виде желтого масла, которое непосредственно использовали в последующей реакции.
б) В охлажденный до 0oC раствор из 1,7 г (6,5 молей) (RS)-1-(7-фтор-4,4-диметил-1,4-дигидроиндено[2,1-c]пиразол-1-ил)- пропан-2-ола и 3,6 мл (26 ммолей) триэтиламина в 60 мл дихлорметана при перемешивании добавляли по каплям 1 мл (13 ммолей) метансульфонилхлорида и продолжали перемешивать еще в течение 1,5 ч при вышеуказанной температуре. Затем реакционную смесь разбавляли 150 мл дихлорметана, дважды промывали соответственно порциями по 70 мл насыщенного раствора гидрокарбоната натрия и соединенные водные фазы один раз экстрагировали с помощью 70 мл дихлорметана. Соединенные органические фазы промывали 70 мл насыщенного раствора хлорида натрия, сушили над сульфатом магния и упаривали в вакууме. Полученное желтое масло растворяли в 40 мл безводного диметилформамида, смешивали с 0,85 г (13 ммолей) азида натрия и реакционную смесь при перемешивании нагревали в течение 15 ч до температуры 70oC. После охлаждения раствор сливали на 100 мл полунасыщенного раствора хлорида, натрия и дважды экстрагировали порциями по 100 мл простого диэтилового эфира соответственно. Соединенные органические фазы промывали по одному разу 100 мл воды и 100 мл насыщенного хлорида натрия соответственно, сушили над сульфатом магния и концентрировали раствор в вакууме. Полученное коричневое масло очищали посредством колоночной хроматографии на силикагеле (уксусный эфир/толуол 1/1). В результате получали 1,66 г (90%) (RS)-1-(2-азидопропил)-7-фтор-4,4-диметил-1,4-дигидроиндено[2,1-с] пиразола в виде желтого масла.
в) 1,66 г (5,82 ммолей) (RS)-1-(2- азидопропил)-7-фтор-4,4-диметил-1,4-дигидроиндено[2,1-c] пиразола, растворенного в 80 мл безводного этанола, гидрировали в присутствии 160 мг оксида платины в течение 1,5 ч. Затем отфильтровывали от катализатора, промывали этанолом и растворитель отгоняли в вакууме. Полученное желтое масло растворяли в 80 мл безводного простого диэтилового эфира, фильтровали и при перемешивании смешивали с раствором из 676 мг (5,82 ммолей) фумаровой кислоты в 10 мл метанола. Перемешивание продолжали еще в течение 15 ч при комнатной температуре, после чего отфильтровывали белые кристаллы. Таким путем получали 1,81 г (83%) фумарата (1:1) (RS)-2-(7-фтор-4,4-диметил-1,4-дигидроиндено [2,1-с] пиразол-1-ил)-1-метилэтиламина с Тпл 144-146oC.
Пример 13
а) Раствор из 1,63 г (8,0 ммолей) 2-гидроксиметилен-6-этокси-1-инданона, 0,87 г (9,65 ммолей) (RS)-1-гидразино-2-пропанола и 100 мг p-толуолсульфокислоты в 100 мл безводного толуола нагревали в течение 1 ч в водоотделителе. После концентрирования в вакууме реакционную смесь очищали посредством колоночной хроматографии на силикагеле (уксусный эфир). В результате получали 2 г (97%) (RS)-1-(7-этокси-1,4-дигидроиндено[2,1-c]пиразол-1-ил)- пропан-2-ола в виде твердого вещества желтого цвета, которое непосредственно использовали в последующей реакции.
б) В охлажденный до 0oC раствор из 2 г (7,7 ммолей) (RS)-1-(7-этокси-1,4-дигидроиндено[2,1-с] пиразол-1-ил)-пропан-2-ола и 4,3 мл (31 ммоль) триэтиламина в 50 мл дихлорметана по каплям добавляли при перемешивании 1,2 мл (15,5 ммолей) метансульфонилхлорида и продолжали перемешивать еще в течение 50 мин при вышеуказанной температуре. Затем реакционную смесь разбавляли 130 мл дихлорметана, дважды промывали соответственно порциями по 70 мл насыщенного раствора гидрокарбоната натрия и соединенные водные фазы один раз экстрагировали с помощью 70 мл дихлорметана. Соединенные органические фазы промывали 70 мл насыщенного раствора хлорида натрия, сушили над сульфатом магния и упаривали в вакууме. Полученное желтое масло растворяли в 50 мл безводного диметилформамида, смешивали с 1 г (15,5 ммолей) азида натрия и реакционную смесь при перемешивании нагревали в течение 15 ч до температуры 75oC. После охлаждения раствор сливали на 80 мл полунасыщенного раствора хлорида натрия и дважды экстрагировали соответственно порциями по 100 мл простого диэтилового эфира. Соединенные органические фазы промывали по одному разу 80 мл воды и 80 мл насыщенного раствора хлорида натрия соответственно, сушили над сульфатом магния и концентрировали раствор в вакууме. Полученное коричневое масло очищали посредством колоночной хроматографии на силикагеле (уксусный эфир). В результате получали 2,06 г (94%) (RS)-1-(2-азидопропил)-7-этокси-1,4-дигидроиндено [2,1-c] пиразола в виде бледно-желтого масла.
в) 2,05 г (7,2 ммолей) (RS)-1-(2-азидопропил)-7-этокси-1,4-дигидроиндено[2,1-c]пиразола, растворенного в 50 мл безводного этанола, гидрировали в присутствии 200 мг оксида платины в течение 1,5 ч. Затем отфильтровывали от катализатора, промывали этанолом и растворитель отгоняли в вакууме. Полученное бесцветное масло растворяли в 100 мл безводного простого диэтилового эфира, фильтровали и при перемешивании смешивали с раствором из 836 мг (7,2 ммолей) фумаровой кислоты в 10 мл метанола. Перемешивание продолжали еще в течение 15 ч при комнатной температуре, после чего отфильтровывали белые кристаллы. Таким путем получали 2,35 г (87%) фумарата (1:1) (RS)-2-(7-этокси-1,4-дигидроиндено[2,1-с]пиразол-1-ил)-1-метилэтиламина с Тпл 191oC.
Пример 14
а) Раствор из 1,4 г (7,36 ммолей) 2-гидроксиметилен-5-метокси-1-инданона, 0,8 г (8,83 ммолей) (RS)-1-гидразино-2-пропанола и 100 мг p-толуолсульфокислоты в 100 мл безводного толуола нагревали в течение 1,5 ч в водоотделителе. После концентрирования в вакууме реакционную смесь очищали посредством колоночной хроматографии на силикагеле (уксусный эфир/гексан 4/1). В результате получали 1,74 г (97%) (RS)-1-(6- метокси-1,4-дигидроиндено[2,1-c] пиразол-1-ил)-пропан-2-ола в виде желтого масла, которое непосредственно использовали в последующей реакции.
б) В охлажденный до 0oC раствор из 1,74 г (7,12 ммолей) (RS)-1-(6-метокси-1,4-дигидроиндено[2,1-с] пиразол-1- ил)-пропан-2-ола и 3,85 мл (28,5 ммолей) триэтиламина в 60 мл дихлорметана по каплям добавляли при перемешивании 1,15 мл (14,2 ммолей) метансульфонилхлорида и продолжали перемешивать еще в течение 50 мин при вышеуказанной температуре. Затем реакционную смесь разбавляли 100 мл дихлорметана и дважды промывали соответственно порциями по 70 мл насыщенного раствора гидрокарбоната натрия и соединенные водные фазы один раз экстрагировали с помощью 70 мл дихлорметана. Соединенные органические фазы промывали 70 мл насыщенного раствора хлорида натрия, сушили над сульфатом магния и упаривали в вакууме. Полученное желтое масло растворяли в 40 мл безводного диметилформамида, смешивали с 0,92 г (14,2 ммолей) азида натрия и реакционную смесь при перемешивании нагревали в течение 5 ч до температуры 90oC. После охлаждения раствор сливали на 80 мл полунасыщенного раствора хлорида натрия и дважды экстрагировали соответственно порциями по 100 мл простого диэтилового эфира. Соединенные органические фазы промывали по одному разу 80 мл воды и 80 мл насыщенного раствора хлорида натрия соответственно, сушили над сульфатом магния и концентрировали раствор в вакууме. Полученное масло коричневого цвета очищали посредством колоночной хроматографии на силикагеле (уксусный эфир/толуол 1/1). В результате получали 1,58 г (82%) (RS)-1-(2-азидопропил)-6-метокси- 1,4-дигидроиндено[2,1-c]пиразола в виде бледно-желтого масла.
в) 1,58 г (5,86 ммолей) (RS)-1-(2-азидопропил)-6-метокси- 1,4-дигидроиндено[2,1-c]пиразола, растворенного в 50 мл безводного этанола, гидрировали в присутствии 160 мг оксида платины в течение 2 ч. Затем отфильтровывали от катализатора, промывали этанолом и растворитель отгоняли в вакууме. Полученное бесцветное масло растворяли в 80 мл безводного простого диэтилового эфира, фильтровали и при перемешивании смешивали с раствором из 680 мг (5,86 ммолей) фумаровой кислоты в 10 мл метанола. Перемешивание продолжали еще в течение 15 ч при комнатной температуре, после чего отфильтровывали белые кристаллы. Таким путем получали 1,81 г (86%) фумарата (1:1) (RS)-2-(6-метокси- 1,4-дигидроиндено[2,1-с] пиразол-1-ил)-1-метилэтиламина с Тпл 192-194oC.
Пример 15
а) Раствор из 1,63 г (7,98 ммолей) 2-гидроксиметилен-7-метокси-1-тетралона, 0,87 г (9,65 ммолей) (RS)-1-гидразино-2-пропанола и 100 мг p-толуолсульфокислоты в 100 мл безводного толуола нагревали в течение 1,5 ч в водоотделителе. После концентрирования в вакууме реакционную смесь очищали посредством колоночной хроматографии на силикагеле (уксусный эфир/гексан 1/1). В результате получали 1,52 г (74%) (RS)-1-(4,5-дигидро-8- метокси-1H-бенз[g] индазол-1-ил)-пропан-2-ола в виде желтого масла, которое непосредственно использовали в последующей реакции.
б) В охлажденный до 0oC раствор из 1,52 г (5,88 ммолей) (RS)-1-(4,5- дигидpo-8-метoкcи-1H-бенз[g] индaзoл-1-ил)-пpoпaн-2-oлa и 3,27 мл (23,5 ммоля) триэтиламина в 60 мл дихлорметана при перемешивании добавляли по каплям 0,89 мл (11,8 ммолей) метансульфонилхлорида и продолжали перемешивать еще в течение 1,5 ч при вышеуказанной температуре. Затем реакционную смесь разбавляли 100 мл дихлорметана, дважды промывали соответственно порциями по 50 мл насыщенного раствора гидрокарбоната натрия и соединенные водные фазы один раз экстрагировали с помощью 50 мл дихлорметана. Соединенные органические фазы промывали 50 мл насыщенного раствора хлорида натрия, сушили над сульфатом магния и упаривали в вакууме. Полученное масло коричневого цвета растворяли в 50 мл безводного диметилформамида, смешивали с 0,76 г (11,8 ммолей) азида натрия и реакционную смесь при перемешивании нагревали в течение 15 ч до температуры 85oC. После охлаждения раствор сливали на 80 мл полунасыщенного раствора хлорида натрия и дважды экстрагировали соответственно порциями по 80 мл простого диэтилового эфира. Соединенные органические фазы промывали по одному разу 80 мл воды и 80 мл насыщенного раствора хлорида натрия соответственно, сушили над сульфатом магния и концентрировали раствор в вакууме. Полученное коричневое масло очищали посредством колоночной хроматографии на силикагеле (уксусный эфир/толуол 1/1). В результате получали 1 г (60%) (RS)-1-(2-азидопропил)-4,5-дигидро- 8-метoкcи-1H-бенз[g] индaзoлa в виде бледно-желтого масла.
в) 1 г (3,5 ммоля) (RS)-1-(2-азидопропил)-4,5-дигидро-8-метокси-1H-бенз[g] индазола, растворенного в 50 мл безводного этанола, гидрировали в присутствии 100 мг оксида платины в течение 2 ч. Затем отфильтровывали от катализатора, промывали этанолом и растворитель отгоняли в вакууме. Полученное бесцветное масло растворяли в 70 мл безводного простого диэтилового эфира, фильтровали и при перемешивании смешивали с раствором из 406 мг (3,5 ммоля) фумаровой кислоты в 10 мл метанола. Перемешивание продолжали в течение 15 ч при комнатной температуре, после чего отфильтровывали белые кристаллы. Таким путем получали 0,98 г (75%) фумарата (1:1) (RS)-2-(4,5-дигидpo-8-метoкcи-1H-бенз[g]индaзoл-1- ил)-1-метилэтилaминa с Тпл 174-176oC.
Пример 16
а) Раствор из 0,86 г (3,34 ммоля) (RS)-2-(4,5-дигидро-8-метокси-1Н-бенз[g]индазол-1-ил)-1-метилэтиламина, 0,56 мл (4 ммоля) триэтиламина и 0,56 мл (4 ммоля) этилового эфира трифторуксусной кислоты в 60 мл безводного метанола перемешивали в течение 50 ч при комнатной температуре. После отгонки растворителя в вакууме остаток растворяли в 70 мл безводного диоксана, добавляли 0,8 г (3,5 ммоля) ДДБ (2,3-дихлор-5,6-дициан-1,4-бензохинон) и кипятили в течение 3 ч с обратным холодильником. Затем реакционную смесь концентрировали в вакууме и остаток очищали посредством колоночной хроматографии на силикагеле (дихлорметан/ацетон 4/1). В результате получали 0,97 г (82%) (RS)-N-[2-(8-метoкcи-1H-бенз[g] индaзoл-1-ил)-1-метилэтил]-трифторацетамида в виде твердого вещества светло-коричневого цвета, которое без дальнейшей перекристаллизации использовали в последующей реакции.
б) Смесь из 0,97 г (2,76 ммоля) (RS)-N-[2-(8-метокси-1H-бенз[g]индазол-1-ил)-1-метилэтил]- трифторацетамида, 1 г (17,5 ммолей) гидроксида калия в 3 мл воды и 50 мл метанола кипятили в течение 5 ч с обратным холодильником. Затем реакционную смесь сливали на 100 мл 1Н едкого натра, трижды экстрагировали соответственно порциями по 100 мл простого диэтилового эфира и соединенные органические фазы сушили над сульфатом магния. После концентрирования в вакууме остаток очищали посредством колоночной хроматографии на силикагеле (дихлорметан/метанол 9/1). В результате получали 0,62 г (2,43 ммоля) желтого масла, которое растворяли в 50 мл простого диэтилового эфира и при перемешивании смешивали с раствором из 280 мг (2,43 ммоля) фумаровой кислоты в 5 мл безводного метанола. Перемешивание продолжали еще в течение 17 ч при комнатной температуре, после чего отфильтровывали белые кристаллы. Таким путем получали 640 мг (74%) фумарата (1:0,5) (RS)-2-(8-метокси-1H-бенз[g]индазол-1-ил)-1-метилэтиламина с Тпл 196-198oC.
Пример 17
а) Раствор из 1,6 г (7,83 ммолей) 2-гидроксиметилен-6-этокси-1 инданона, 0,85 г (9,40 ммолей) (S)-1-гидразино-2-пропанола и 70 мг p-толуолсульфокислоты в 80 мл безводного толуола нагревали в течение 2 ч в водоотделителе. После концентрирования в вакууме реакционную смесь очищали посредством колоночной хроматографии на силикагеле (уксусный эфир). В результате получали 1,73 г (86%) (S)-1-(7-этокси-1,4-дигидроиндено[2,1-c] пиразол-1-ил)-пропан-2-ола в виде твердого вещества желтого цвета, которое непосредственно использовали в последующей реакции.
б) В охлажденный до 0oC раствор из 1,73 г (6,7 ммолей) (S)-1-(7-этокси-1,4-дигидроиндено[2,1-с] пиразол-1-ил)-пропан-2-ола и 3,72 мл (26,8 ммолей) триэтиламина в 50 мл дихлорметана по каплям добавляли при перемешивании 1,01 мл (13,4 ммолей) метансульфонилхлорида и продолжали перемешивать еще в течение 90 мин при вышеуказанной температуре. Затем реакционную смесь разбавляли 130 мл дихлорметана, дважды промывали соответственно порциями по 70 мл насыщенного раствора гидрокарбоната натрия и соединенные водные фазы один раз экстрагировали с помощью 70 мл дихлорметана. Соединенные органические фазы промывали 70 мл насыщенного раствора хлорида натрия, сушили над сульфатом магния и концентрировали в вакууме. Полученное твердое вещество желтоватого цвета растворяли в 50 мл безводного диметилформамида, смешивали с 0,86 г (13,4 ммолей) азида натрия и реакционную смесь при перемешивании нагревали в течение 16 ч до температуры 90oC. После охлаждения раствор сливали на 80 мл полунасыщенного раствора хлорида натрия и дважды экстрагировали соответственно порциями по 100 мл простого диэтилового эфира. Соединенные органические фазы промывали по одному разу 80 мл воды и 80 мл насыщенного раствора хлорида натрия соответственно, сушили над сульфатом магния и концентрировали раствор в вакууме. Полученное желтое масло очищали посредством колоночной хроматографии на силикагеле (уксусный эфир). В результате получали 1,76 г (93%) (R)-1-(2-азидопропил)-7-этокси- 1,4-дигидроиндено[2,1-с] пиразола в виде твердого вещества бледно-желтого цвета.
в) 1,76 г (6,21 ммолей) (R)-1-(2- азидопропил)-7-этокси-1,4-дигидроиндено[2,1-c] пиразола, растворенного в 100 мл безводного этанола, гидрировали в присутствии 180 мг оксида платины в течение 17 ч. Затем отфильтровывали от катализатора, промывали этанолом и растворитель отгоняли в вакууме. Полученное бесцветное масло растворяли в 100 мл безводного простого диэтилового эфира, фильтровали и при перемешивании смешивали с раствором из 721 мг (6,21 ммолей) фумаровой кислоты в 10 мл метанола. Перемешивание продолжали еще в течение 15 ч при комнатной температуре, после чего отфильтровывали белые кристаллы. Таким путем получали 2,0 г (86%) фумарата (1:1) (R)-2-(7-этокси-1,4-дигидроиндено[2,1-с]пиразол-1-ил)-1- метилэтиламина с Тпл 161oC.
Пример 18
а) Раствор из 0,5 г (2,45 ммоля) 2-гидроксиметилен-6-этокси-1-инданона, 0,27 г (2,94 ммоля) (R)-1-гидразино-2-пропанола и 50 мг p-толуолсульфокислоты в 50 мл безводного толуола нагревали в течение 1 ч в водоотделителе. После концентрирования в вакууме реакционную смесь очищали посредством колоночной хроматографии на силикагеле (уксусный эфир). В результате получали 0,49 г (77%) (R)-1-(7-этокси-1,4- дигидроиндено[2,1-с]пиразол-1-ил)-пропан-2-ола в виде твердого вещества желтого цвета, которое непосредственно использовали в последующей реакции.
б) В охлажденный до 0oC раствор из 0,49 г (1,9 ммоля) (R)-1-(7-этокси-1,4-дигидроиндено[2,1-с] пиразол-1-ил)- пропан-2-ола и 1,06 мл (7,6 ммолей) триэтиламина в 30 мл дихлорметана при перемешивании добавляли по каплям 0,29 мл (3,79 ммоля) метансульфонилхлорида и продолжали перемешивать еще в течение 50 мин при вышеуказанной температуре. Затем реакционную смесь разбавляли 100 мл дихлорметана, дважды промывали соответственно порциями по 50 мл насыщенного раствора гидрокарбоната натрия и соединенные водные фазы один раз экстрагировали с помощью 50 мл дихлорметана. Соединенные органические фазы промывали 70 мл насыщенного раствора хлорида натрия, сушили над сульфатом магния и упаривали в вакууме. Полученное твердое вещество желтого цвета растворяли в 25 мл безводного диметилформамида, смешивали с 0,25 г (3,8 ммоля) азида натрия и реакционную смесь при перемешивании нагревали в течение 22 ч до температуры 70oC. После охлаждения раствор сливали на 70 мл полунасыщенного раствора хлорида натрия и дважды экстрагировали соответственно порциями по 70 мл простого диэтилового эфира. Соединенные органические фазы промывали по одному разу 50 мл воды и 50 мл насыщенного раствора хлорида натрия соответственно, сушили над сульфатом магния и концентрировали раствор в вакууме. Полученное коричневое масло очищали посредством колоночной хроматографии на силикагеле (уксусный эфир). В результате получали 0,53 г (99%) (S)-1-(2-азидопропил)-7-этокси- 1,4-дигидроиндено[2,1-c] пиразола в виде твердого вещества бледно-желтого цвета.
в) 0,53 г (1,87 ммоля) (S)-1-(2-азидопропил)- 7-этокси-1,4-дигидроиндено[2,1-c]пиразола, растворенного в 25 мл безводного этанола, гидрировали в присутствии 55 мг оксида платины в течение 1,5 ч. Затем отфильтровывали от катализатора, промывали этанолом и растворитель отгоняли в вакууме. Полученное бесцветное масло растворяли в 50 мл безводного простого диэтилового эфира, фильтровали и при перемешивании смешивали с раствором из 217 мг (1,87 ммоля) фумаровой кислоты в 10 мл метанола. Перемешивание продолжали еще в течение 15 ч при комнатной температуре, после чего отфильтровывали белые кристаллы. Таким путем получали 0,54 г (77%) фумарата (1:1) (S)-2-(7- этокси-1,4-дигидроиндено[2,1-с]пиразол-1-ил)-1-метилэтиламина с Тпл 157oC.
Пример А
По обычной методике изготавливают таблетки следующего состава: - мг/таблетку
Активное вещество - 100
Лактоза порошкообразная - 95
Кукурузный крахмал белый - 35
Поливинилпирролидон - 8
Na-карбоксиметилкрахмал - 10
Стеарат магния - 2
Вес таблетки - 250
Пример Б
По обычной методике изготавливают таблетки следующего состава: - мг/таблетку
Активное вещество - 200
Лактоза порошкообразная - 100
Кукурузный крахмал белый - 64
Поливинилпирролидон - 12
Na-карбоксиметилкрахмал - 20
Стеарат магния - 4
Вес таблетки - 400
Пример В
Ниже представлено изготовление капсул следующего состава: - мг/капсулу
Активное вещество - 50
Лактоза кристаллическая - 60
Микрокристаллическая целлюлоза - 34
Тальк - 5
Стеарат магния - 1
Вес содержимого капсулы - 150
Активное вещество с соответствующим размером частиц, кристаллическую лактозу и микрокристаллическую целлюлозу тщательно перемешивают друг с другом до получения однородной массы, пропускают через сито, после чего добавляют тальк и стеарат магния. Из полученной смеси изготавливают капсулы соответствующего размера с твердожелатиновой оболочкой.
Изобретение относится к трициклическим производным пиразола общей формулы I, где R1-R4 обозначают водород, галоген, низший алкил или низший алкокси; R5 обозначает низший алкил; R6 обозначает водород, низший алкил или низший алкокси; Х обозначает -(CR7R8)n или -СН=СН-; R7 и R8 обозначают водород или низший алкил и n = 1 или 2, или к фармацевтически применимым солям основных соединений общей формулы (I). Соединения обладают способностью связываться с серотониновыми рецепторами и могут применяться для лечения или предупреждения заболеваний центральной нервной системы, в частности для лечения сексуальных расстройств и других заболеваний. Предлагается также лекарственное средство на основе соединений формулы (I), связывающее серотониновые рецепторы, предназначенное, в частности, для лечения сексуальных расстройств. 2 с. и 10 з.п. ф-лы, 3 ил., 1 табл.
 (I)
(I)
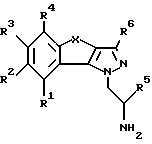
где R1 - R4 обозначают водород, галоген, низший алкил или низший алкокси;
R5 обозначает низший алкил;
R6 обозначает водород, низший алкил или низший алкокси;
Х обозначает -(CR7R8)n- или -СН=СН-;
R7 и R8 обозначают водород или низший алкил;
n обозначает 1 или 2,
или фармацевтически применимые соли основных соединений общей формулы I.
| МАШИНА И СПОСОБ ДЛЯ ПОДБИВКИ ШПАЛ РЕЛЬСОВОГО ПУТИ | 2003 |
|
RU2249644C1 |
| EP 0683155 A, 22.11.1995 | |||
| US 3957816 A, 18.05.1976 | |||
| Способ получения 1,3-дифенил-5-арил4,2 -диметилен- -пиразолинов | 1973 |
|
SU472937A1 |
| Способ получения конденсированных производных пиразола или их фармацевтически приемлемых солей | 1988 |
|
SU1676453A3 |

