Данная заявка является частичным продолжением находящейся в процессе одновременного рассмотрения заявки США N 08/138 820, поданной 15 октября 1993 года.
Это изобретение касается соединений, имеющих антитромботическую активность. Более конкретно, изобретение касается азациклоалкилалканоилпептидов и псевдопептидов, которые ингибируют агрегацию тромбоцитов и образование тромбов у млекопитающих и которые применимы для профилактики и лечения тромбоза, связанного с такими патологическими состояниями, как инфаркт миокарда, тромботический инсульт, поражение периферической артерии и диссеминированное внутрисосудистое свертывание.
Гемостаз, биохимия свертывания крови, представляет собой чрезвычайно сложный феномен, посредством которого нормальная цельная кровь и ткань тела самопроизвольно останавливают кровотечение из поврежденных кровеносных сосудов. Эффективный гемостаз требует комбинированной активности сосудистых, тромбоцитарных и плазматических факторов, а также регулируемого механизма для предотвращения избыточного свертывания. Дефекты, отсутствие или избыток любого из этих компонентов могут привести к геморрагическим или тромботическим последствиям.
Адгезия тромбоцитов, разрастание и агрегация на внеклеточных матриксах являются центральными событиями в образовании тромба. Эти события опосредованы семейством адгезивных гликопротеинов, т.е. фибриногена, фибронектина и фактора фон Виллебранда. Фибриноген представляет собой кофактор агрегации тромбоцитов, тогда как фибронектин поддерживает присоединение тромбоцитов и реакции разрастания, а фактор фон Виллебранда важен для присоединения тромбоцитов к субэндотелиальному матриксу и распространения на нем. Сайты связывания для фибриногена, фибронектина и фактора фон Виллебранда были обнаружены на белковом комплексе мембран тромбоцитов, известном как гликопротеин IIb/IIIa.
Адгезивные гликопротеины, подобные фибриногену, не связываются с нормальными покоящимися тромбоцитами. Однако при активации тромбоцита агонистом, таким как тромбин или аденозиндифосфат, тромбоцит изменяет свою форму, возможно, делая сайты связывания GPIIb/IIIa доступными для фибриногена. Соединения в объеме данного изобретения блокируют рецептор фибриногена, ингибируя, таким образом, агрегацию тромбоцитов, и при введении в форме фармацевтических композиций, содержащих такие соединения, применимы для предотвращения и лечения тромбообразующих заболеваний, таких как инфаркт миокарда, тромботический инсульт, поражение периферической артерии и диссеминированное внутрисосудистое свертывание.
Известно, что присутствие Arg-Gly-Asp (RGD) является необходимым в фибриногене, фибронектине и факторе фон Виллебранда для их взаимодействия с рецептором поверхности клетки (Ruoslahti Е., Pierschbacher, Cell 1986, 44, 517-18). По-видимому, две другие аминокислотные последовательности также принимают участие в функции присоединения тромбоцитов фибриногена, а именно, Gly-Pro-Arg и последовательность додекапептида His-His-Leu-Gly-Gly-Ala-Lys-Gln-Ala-Gly-Asp-Val. Показано, что небольшие синтетические пептиды, содержащие RGD или додекапептид, связываются с рецептором тромбоцитов GPIIb/IIIa и конкурентно ингибируют связывание фибриногена, фибронектина и фактора фон Виллебранда, а также ингибируют агрегацию активированных тромбоцитов (Plow, et al., Proc. Natl. Acad. Sci. USA 1985, 82, 8057-61; Ruggeri, et al. , Proc. Natl. Acad. Sci. USA 1986, 5708-12; Ginsberg, et al., J.Biol. Chem. 1985, 260, 3931-36; Gartner, et al., J.Biol.Chem. 1987, 260, 11, 891-94).
Известно, что содержащие индолил соединения, включающие в себя гуанидиноалканоил- и гуанидиноалкеноиласпартил, являются ингибиторами агрегации тромбоцитов (Tjoeng, et аl., патент США N 5037808 и N 4879313).
Патент США N 4992463 (Tjoeng et al.), выданный 12 февраля 1991 г., описывает ряд арил- и аралкилгуанидиноалкилпептидных миметических соединений, которые проявляют активность ингибирования агрегации тромбоцитов, и описывает конкретно ряд моно- и диметоксифенилпептидных миметических соединений и бифенилалкилпептидное миметическое соединение.
Патент США N 4857508 (Adams, et al), выданный 15 августа 1989 г., описывает ряд гуанидиноалкилпептидных производных, содержащих концевые аралкильные заместители, которые проявляют активность ингибирования агрегации тромбоцитов, и описывает конкретно ряд содержащих О-метилтирозин, бифенил и нафтилпроизводных, имеющих концевую амидную группу.
Haverstick, D. M. , et al., в Blood 66(4), 946-952 (1985) описывают ряд синтетических пептидов, включающих в себя Arg-Gly-Asp-Ser и Gly-Arg-Gly-Asp-Ser, способных ингибировать индуцированную тромбином агрегацию тромбоцитов.
Plow, E. F. , et ai., Proc. Natl. Acad. Sci. USA 79, 3711-3715 (1982) описывают, что тетрапептид глицил-L-пролил-L-аргинил-L- пролин ингибирует связывание фибриногена с тромбоцитами человека.
Заявка Франции N 86/17507 от 15.12.1986 г., раскрывает, что тетра-, пента- и гексапептидные производные, содержащие последовательность -Arg-Gly-Asp- применимы в качестве антитромботических средств.
Патент США N 4683291 (Zimmerman, et al.), выданный 28 июля 1987 г., сообщает, что ряд пептидов, состоящих из 6-40 аминокислот, содержащих последовательность -Arg-Gly-Asp-, является ингибиторами связывания тромбоцитов.
ЕПВ N 0319506, опубликованная 7 июня 1989 г., раскрывает ряд тетра-, пента- и гексапептидных производных, содержащих последовательность -Arg-Gly-Asp-, являющихся ингибиторами агрегации тромбоцитов.
В патенте США N 5023233 сообщается, что циклические пептидные аналоги, содержащие последовательность Gly-Asp, являются антагонистами рецептора фибриногена.
В ожидающих решения заявки США NN 07/677006, 07/534385 и 07/460777, поданных 28 марта 1991 г., 7 июня 1990 г. и 4 января 1990 г., соответственно, а также в U. S. Patent N 4952562 и International Application N PCT/US90/05448, поданной 25 сентября 1990 г., описываются пептиды и псевдопептиды, содержащие амино-, гуанидиногруппу, имидизалоил, и/или аминоалканоил и алкеноил, являются антитромботическими агентами.
Известно, что пептиды и псевдопептиды, содержащие аминогруппу и радикалы гуанидиноалкил и алкенилбензоил, фенилалканоил и фенилалкеноил, являются антитромботическими агентами (заявка США N 07/475, поданная 5 февраля 1990, N PCT/US91/02471, поданная 11 апреля 1991 г., опубликованная как International Application N WO 92/13117, 29 октября 1992).
В патенте США N 5053392, поданном 1 декабря 1989, и переуступленном тому же правопреемнику, и имеющем тех же авторов, что и данное изобретение, сообщается, что содержащие алканоил и (замещенный алканоил) азациклоалкилформилпроизводные аспарагиновой кислоты являются ингибиторами агрегации тромбоцитов.
В патенте США N 5064814, поданном 5 апреля 1990 г. теми же авторами и переуступленном тому же правопреемнику, сообщается, что производные N-замещенной азациклоалкилкарбонил (циклический аминоацил)аспарагиновой кислоты являются антитромботическими агентами. Производные азациклоалкилформилглициласпарагиновой кислоты являются антитромботическими агентами также согласно патенту США N 5051406, поданному 10 октября 1989 г. и переуступленному тому же правопреемнику, который является правопреемником данного изобретения.
ЕР заявка 0479 481, опубликованная 8 апреля 1992 г., описывает азациклоалкилалканоилглициласпартиламинокислоты в качестве антагонистов рецептора фибриногена.
ЕР заявка N 0478 362, опубликованная 1 апреля 1992 г., описывает азациклоалкилалканоилпептидил -β- аланины в качестве антагонистов рецептора фибриногена.
Данное изобретение касается азациклоалкилалканоилпептидов и псевдопептидов, которые ингибируют агрегацию тромбоцитов и тромбообразование.
Краткое изложение сущности изобретения
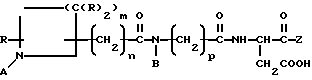
Соединения данного изобретения имеют формулу I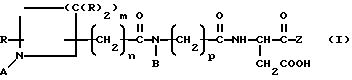
в которой А представляет собой -H, амидиногруппу или замещенную амидиногруппу;
B представляет собой алкил, циклоалкил, циклоалкилалкил, алкилциклоалкил, алкилциклоалкилалкил, арил, аралкил, алкиларил или алкиларалкил
Z представляет собой: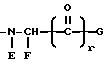
в которой E обозначает -H или, в комбинации с F, образует 4-, 5-, 6- или 7-членное азациклоалкановое кольцо,
F обозначает α- углеродную боковую цепь природной α- аминокислоты, -H, алкил, циклоалкил, циклоалкилалкил, алкилциклоалкил, алкилциклоалкилалкил, арил, замещенный арил, аралкил, замещенный аралкил, гетероциклил, замещенный гетероциклил, гетероциклилалкил, замещенный гетероциклилалкил или, в комбинации с E, образует 4-, 5-, 6- или 7-членное азациклоалкановое кольцо.
G обозначает алкил, циклоалкил, циклоалкилалкил, алкилциклоалкил, алкилциклоалкилалкил, арил, замещенный арил, аралкил, замещенный аралкил, гетероциклил, замещенный гетероциклил, гетероциклилалкил, замещенный гетероциклилалкил, OR1 или NR1R2, где R1 и R2 независимо обозначают -H, алкил, циклоалкил, циклоалкилалкил, алкилциклоалкил, алкилциклоалкилалкил, арил, аралкил, алкиларил или алкиларалкил и
r обозначает 0 или 1;
R обозначает -H, алкил, арил или аралкил;
m обозначает 1-5;
n обозначает 0-6; и
р обозначает 1-4;
или являются их фармацевтически приемлемой солью.
Кроме того, данное изобретение касается фармацевтических композиций, содержащих такие соединения, и способов профилактики или лечения тромбоза у млекопитающих, требующих такого лечения, предусматривающих введение таких соединений и фармацевтических композиций.
Данное изобретение характеризуется заметной и пролонгированной антитромботической активностью соединений формулы I, представленной выше, причем активность наблюдается после перорального введения этих соединений.
Как в предшествующем тексте, так и во всем этом изобретении следующие ниже термины будут иметь следующие значения (если нет других указаний);
"Амидино" обозначает группу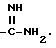
"Замещенная амидино" обозначает амидиногруппу, N-замещенную на одном или обоих атомах азота одним или более алкилом, циклоалкилом, циклоалкилалкилом, алкилциклоалкилом, алкилциклоалкилалкилом, арилом или аралкилом.
"Алкил" обозначает насыщенную алифатическую углеводородную группу, которая может быть прямой или разветвленной, имеющую приблизительно 1-20 атомов углерода в цепи. Разветвленная группа обозначает, что группа низшего алкила, такая как метил, этил или пропил, присоединена к линейной цепи алкила. Предпочтительными прямыми или разветвленными алкильными группами являются группы "низшего алкила", представляющие собой алкильные группы, имеющие от 1 до приблизительно 10 атомов углерода. Наиболее предпочтительные группы низшего алкила имеют от 1 до приблизительно 5 атомов углерода.
"Циклоалкил" обозначает насыщенную карбоциклическую группу, имеющую одно или более колец, имеющих приблизительно 3-10 атомов углерода. Предпочтительные циклоалкильные группы включают в себя циклопропил, циклобутил, циклопентил, циклогексил, циклогептил и декагидронафтил.
"Циклоалкилалкил" обозначает группу алкила, замещенную циклоалкильной группой. Предпочтительные циклоалкилалкилы включают в себя циклопентилметил, циклогексилметил, циклогексилэтил, декагидронафт-1-илметил и декагидронафт-2-илметил.
"Алкилциклоалкил" обозначает циклоалкил, замещенный алкилом. Примерами алкилциклоалкилов являются 1-, 2-, 3- или 4-метил или этилциклогексил.
"Алкилциклоалкилалкил" обозначает алкил, замещенный алкилциклоалкилом. Примеры алкилциклоалкилалкила включают в себя 1-, 2-, 3- или 4-метил- или этилциклогексилметил или 1-, 2-, 3- или 4-метил- или этилциклогексилэтил.
"Азациклоалкан" обозначает насыщенное алифатическое кольцо, содержащее атом азота. Предпочтительные азациклоалканы включают в себя пирролидин и пиперидин.
"Природная α- аминокислота" обозначает глицин, аланин, валин, лейцин, изолейцин, серин, треонин, фенилаланин, тирозин, триптофан, цистеин, метионин, пролин, гидроксипролин, аспарагиновую кислоту, аспарагин, глутамин, глутаминовую кислоту, гистидин, аргинин, орнитин и лизин.
"α- углеродная боковая цепь природной α- аминокислоты" обозначает часть молекулы, которая замещает α- углерод природной α- аминокислоты. Примеры α- углеродных боковых цепей природных α- аминокислот включают в себя изопропил, метил и карбоксиметил для валина, аланина и аспарагиновой кислоты соответственно.
"Арил" обозначает фенил или нафтил.
"Замещенный арил" обозначает фенил или нафтил, замещенные одним или более заместителями арильной группы, которые могут быть одинаковыми или различными, где "заместитель арильной группы" включает в себя алкил, алкенил, алкинил, арил, аралкил, гидроксигруппу, алкоксигруппу, арилоксигруппу, аралкоксигруппу, гидроксиалкил, ацил, формил, карбоксигруппу, алкеноил, ароил, галоген, нитрогруппу, тригалогенметил, цианогруппу, алкоксикарбонил, арилоксикарбонил, аралкоксикарбонил, ациламиногруппу, ароиламиногруппу, карбамоил, алкилкарбамоил, диалкилкарбамоил, арилкарбамоил, аралкилкарбамоил, алкилсульфонил, алкилсульфинил, арилсульфонил, арилсульфинил, аралкилсульфонил, аралкилсульфинил или - NRaRb, где Ra и Rb обозначают независимо водород, алкил, арил или аралкил.
"Аралкил" обозначает алкил, замещенный радикалом арилом. Предпочтительные аралкильные группы включают в себя бензил, нафт-1-илметил, нафт-2-илметил и фенетил.
"Замещенный аралкил" обозначает аралкил, замещенный арильной частью одного или более заместителей арильной группы.
"Гетероциклил" обозначает 4-15-членную моноциклическую или полициклическую систему, в которой один или более атомов в кольце представляют собой элемент, иной, чем углерод, например азот, кислород или серу. Предпочтительные гетероциклильные группы включают в себя пиридил, пиримидил и пирролидил.
"Замещенный гетероциклил" обозначает гетероциклил, замещенный одним или более заместителями арильной группы.
"Гетероциклоалкил" и "замещенный гетероциклилалкиk" обозначают алкил, который замещен гетероциклилом и замещенным гетероциклилом соответственно.
Предпочтительный класс соединений данного изобретения описывается формулой 1, в которой F обозначает -H, алкил, гидроксиметил, 1-гидроксиэтил, меркаптометил, 2-метилтиоэтил, карбоксиметил, 2-карбоксиэтил, 4-аминобутил, 3-гуанидинопропил, циклоалкил, циклоалкилалкил, алкилциклоалкил, алкилциклоалкилалкил, арил, замещенный арил, аралкил, замещенный аралкил, гетероциклил, замещенный гетероциклил, гетероциклилалкил, замещенный гетероциклилалкил или, в комбинации с E, образует 4-,5-, 6- или 7- членное азациклоалкановое кольцо, при условии, что гетероциклилалкил иной, чем индол-3-илметил.
Более предпочтительный класс соединений данного изобретения описан в виде предпочтительного класса соединений, в которых F обозначает -H, алкил, гидроксиметил, 1-гидроксиэтил, меркаптометил 2-метилтиоэтил, карбоксиметил, 2-карбоксиэтил, 4-аминобутил, 3-гуанидинопропил, циклоалкил, циклоалкилалкил, алкилциклоалкил, алкилциклоалкилалкил, арил, замещенный арил, аралкил, замещенный аралкил или, в комбинации с E, образует 4-, 5-, 6- или 7-членное азациклоалкановое кольцо.
Еще более предпочтительный класс соединений данного изобретения описывается более предпочтительным классом соединений, в которых F обозначает -H, алкил, гидроксиметил, 1-гидроксиэтил, меркаптометил, 2-метилтиоэтил, карбоксиметил, 2-карбоксиэтил, 4-аминобутил, 3-гуанидинопропил, циклоалкил, циклоалкилалкил, алкилциклоалкил, алкилциклоалкилалкил или, в комбинации с E, образует 4-, 5-, 6- или 7-членное кольцо азациклоалкана.
Наиболее предпочтительный класс соединений данного изобретения описывается еще более предпочтительным классом соединений, в которых В обозначает алкил, циклоалкил, циклоалкилалкил, алкилциклоалкил или алкилциклоалкилалкил.
Особый вариант данного изобретения описывается формулой II: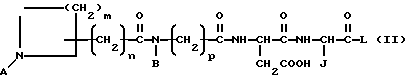
в которой A обозначает -H или амидиногруппу,
B обозначает алкил, циклоалкил, циклоалкилалкил, алкилциклоалкил, алкилциклоалкилалкил, арил, аралкил, алкиларил или алкиларалкил,
j обозначает -H, алкил, циклоалкил, циклоалкилалкил, алкилциклоалкил, алкилциклоалкилалкил, арил, замещенный арил, аралкил, замещенный аралкил,
L обозначает OR1 или NR1R2, где R1 и R2 обозначают независимо -H, алкил, циклоалкил, циклоалкилалкил, алкилциклоалкил, алкилциклоалкилалкил, арил, аралкил, алкиларил или алкиларалкил.
m обозначает 1-5,
n обозначает 2-6 и
p обозначает 1 или 2,
или является фармацевтически приемлемой солью этого соединения.
Более предпочтительный особый вариант данного изобретения описывается соединениями особого варианта, в которых А обозначает -H,
B обозначает алкил, циклоалкил, циклоалкилалкил, алкилциклоалкил, алкилциклоалкилалкил,
j обозначает -H, алкил, циклоалкил, циклоалкилалкил, алкилциклоалкил или алкилциклоалкилалкил,
m обозначает 3 и
n обозначает 3 или 4.
Наиболее предпочтительный особый вариант данного изобретения описывается соединениями более предпочтительного особого варианта, в котором
А обозначает -H,
B обозначает алкил,
j обозначает алкил, циклоалкил или циклоалкилалкил,
R1 и R2 независимо обозначают -H, алкил, циклоалкил, циклоалкилалкил, алкилциклоалкил, алкилциклоалкилалкил,
m обозначает 3,
n обозначает 3 или 4 и
p обозначает 1.
Характерные соединения данного изобретения включают в себя:
N-[N-[N-(4-(пиперидин-4-ил)бутаноил)-N-этилглицил]аспартил]-валин,
N-[N-[N-(4-(пиперидин-4-ил)бутаноил)-N-этилглицил]аспартил]-D-валин,
N-[N-[N-(3-пиперидин-4-ил)пропаноил)-N-этилглицил]аспартил]-валин,
N-[N-[N-(5-(пиперидин-4-ил)пентаноил)-N-этилглицил]аспартил]-валин,
N-[N-[N-(4-пиперидин-4-ил)бутаноил)-N-этилглицил] аспартил] -L-α- циклогексилглицин,
N-[N-[N-(4-пиперидин-4-ил)бутаноил)-N-этилглицил] аспартил] -β- циклогексилаланин,
N-[N-[N-(4-(пиперидин-4-ил)бутаноил)-N- этилглицил]аспартил]норлейцин,
N-[N-[N-(4-(пиперидин-4-ил)бутаноил)-N- этилглицил] acпapтил] -L- α- (2,2-диметил)проп-3-илглицин,
N-[N-[N-(4-(пиперидин-4-ил)бутаноил)-N- этилглицил] аспартил]-L- β- декагидронафтил-1-илаланин,
N-[N-[N-(4-(пиперидин-4-ил)бутаноил)-N- этилглицил]аспартил]-L- α- (2-циклогексилэтил)глицин,
N-[N-[N-(4-(пиперидин-4-ил)бутаноил)-N- этилглицил]аспартил]фенилаланин,
N-[N-[N-(4-пиперидин-4-ил)бутаноил)-N- этилглицил] аспартил] -L- β- нафт-1-илаланин,
N-[N-[N-(4-(пиперидин-4-ил)бутаноил)-N- этилглицил[аспартил] -L- β- нафт-2-илаланин,
N-[N-[N-(4-(пиперидин-4-ил)бутаноил)-N-этилглицил] аспартил] -L- β- циклогексилаланинамид,
Этиловый эфир N-[N-[N-(4-(пиперидин)бутаноил)-N- этилглицил]аспартил]-L- -β- циклогексилаланина,
2-циклогексил-N-[N-[N-(4-(пиперидин-4-ил)бутаноил)-N- этилглицил] аспартил]-этиламин,
N-[N-[N-(4-(пиперидин-4-ил)бутаноил)-N- этилглицил] аспартил] -L- β- цис-декагидронафт-2-илаланин,
3-aдaмaнт-1-илпpoпил-N-[N-(4-(пиперидин-4-ил)бутаноил)-N- этилглицил] аспартат,
N-[N-[N-(4-(пиперидин-4-ил)бутаноил)-N- этилглицил] аспартил] -α- аминоциклогексанкарбоновая кислота,
N-[N-[N-(4-(пиперидин-4-ил)бутаноил)-N- этилглицил] аспартил] -β- циклогексил-D-аланин,
N-[N-[N-(4-пиперидин-4-ил)бутаноил)-N- этилглицил] аспартил] -β- декагидронафт-1-илаланин,
N-[N-[N-(4-пиперидин-4-ил)бутаноил)-N- этилглицил] аспартил] -β- циклогексилаланинэтиламид,
N-[N-[N-(4-(пиперидин-4-ил)бутаноил)-N- этилглицил] аспартил] -β- циклооктилаланин,
N-[N-[N-(4-пиперидин-4-ил)бутаноил)- N-этилглицил] аспартил] -α- циклогексилметилэтаноламин,
N-[N-[N-(4-(пиперидин-4-ил)бутаноил)-N- этилглицил]аспартил] -β- циклогексилметилаланинамид,
N-[N-[N-(4-(пиперидин-4-ил)бутаноил)-N- этилглицил]аспартил] -β- адамант-1-илаланин,
N-[N-[N-(4-(пиперидин-4-ил)бутаноил)-N- этилглицил] acпapтил] -β- (1,2,3,4)-тетрагидронафт-5-илаланин,
N-[N-[N-4-(пиперидин-4-ил)бутаноил)-N-этилглицил] аспартил] -β- (4-циклогексил)циклогексилаланин,
N-[N-[N-[ (4-(пиперидин-4-ил)бутаноил)-N- этилглицил]аспартил] -β- циклогептилаланин,
N-[N-[N-(4-(пиперидин-4-ил)бутаноил)-N- этилглицил]аспартил] -β- циклооктилаланинамид,
N-[N-[N-(4-(пиперидин-4-ил)бутаноил)-N-этилглицил] аспартил] -α- циклогексилпропилглицин,
N-[N-[N-(4-(пиперидин-4-ил)бутаноил)-N-этилглицил] аспартил] -β- циклооктилметилаланин,
N-[N-[N-(4-(пиперидин-4-ил)бутаноил)-N- этилглицил]acпapтил] -β- циклопентилаланин и
этиловый эфир N-[N-[N-(4-(пиперидин-4-ил)бутаноил)-N- этилглицил]аспартил] -β- циклогексилметилаланина,
и их фармацевтически приемлемые соли.
Соединения данного изобретения содержат асимметричные центры. Эти асимметричные центры могут быть независимо в R-конфигурации или в S-конфигурации. Данное изобретение включает в себя отдельные стереоизомеры и их смеси.
Соединения данного изобретения могут применяться в форме свободного основания или кислоты или в форме их фармацевтически приемлемой соли. Все эти формы находятся в объеме данного изобретения.
Если соединение данного изобретения замещено основным радикалом, то могут быть образованы соли с кислотой, которые являются более удобной формой для использования; и на практике применение такой формы соли равнозначно применению формы свободного основания. Кислоты, которые можно применять для получения солей с кислотами, включают в себя предпочтительно те кислоты, которые дают при объединении со свободным основанием фармацевтически приемлемые соли, т. е. соли, анионы которых являются нетоксичными для организма животного в фармацевтических дозах этих солей, так что полезные антитромботические свойства, присущие свободным основаниям, не исчезают вследствие побочных эффектов, приписываемых анионам. Хотя предпочтительны фармацевтически приемлемые соли основных соединений, все соли с кислотой применимы в качестве источников формы свободного основания, даже если конкретная соль, per se, желательна только в качестве промежуточного продукта, например, при образовании соли только для целей очистки или идентификации или при использовании ее в качестве промежуточного продукта в приготовлении фармацевтически приемлемой соли при помощи ионообменной процедуры. Фармацевтически приемлемыми солями в объеме данного изобретения являются соли, полученные из следующих кислот: минеральных кислот, таких как соляная кислота, серная кислота, фосфорная кислота и сульфаминовая кислота; и органических кислот, таких как уксусная кислота, лимонная кислота, молочная кислота, винная кислота, малоновая кислота, метансульфоновая кислота, этансульфоновая кислота, бензолсульфоновая кислота, п-толуолсульфокислота, циклогексилсульфаминовая кислота, хинная кислота и т.п. Соответствующие соли с кислотами включают в себя: гидрохлорид, сульфат, фосфат, сульфамат, ацетат, цитрат, лактат, тартрат, малонат, метансульфонат, этансульфонат, бензолсульфонат, п-толуолсульфонат, циклогексилсульфамат и хиннат, соответственно.
Соли с кислотами соединений данного изобретения получают либо растворением свободного основания в водном или водно-спиртовом растворе или других подходящих растворителях, содержащих подходящую кислоту, и выделением соли выпариванием раствора, либо реакцией свободного основания и кислоты в органическом растворителе, причем в этом случае соль отделяется сразу или она может быть получена концентрированием раствора.
Если соединение изобретения замещено кислотной частью молекулы, то соли с основанием могут быть образованы и они являются абсолютно более удобной формой для применения; и на практике применение такой соли равнозначно применению формы свободной кислоты. Основания, которые можно применять для получения солей с основаниями, включают в себя предпочтительно те основания, которые при взаимодействии со свободной кислотой образуют фармацевтически приемлемые соли, т.е. соли, катионы которых являются нетоксичными для организма животного в фармацевтических дозах этих солей, так что полезные антитромботические свойства, присущие свободной кислоте, не исчезают вследствие побочных эффектов, приписываемых катионам. Фармацевтически приемлемыми солями в объеме данного изобретения являются соли, полученные из следующих оснований: гидроксида натрия, гидроксида калия, гидроксида кальция, гидроксида алюминия, гидроксида лития, гидроксида магния, гидроксида цинка, аммиака, этилендиамина, N-метилглутамина, лизина, аргинина, орнитина, прокаина, N-бензилфенетилдиамина, диэтиламина, холина, N,N'-бензилфенетиламина, пиперазина, трис(гидроксиметил)аминометана, гидроксида тетраметиламмония и т.п.
Соли металлов соединений данного изобретения можно получить контактированием гидроксида, карбоната или подобного реактивного соединения выбранного металла в водном растворителе с формой свободной кислоты данного соединения. Используемым водным растворителем может быть вода или смесь воды с органическим растворителем, предпочтительно спиртом, таким как метанол или этанол, или кетоном, таким как ацетон, алифатическим простым эфиром, таким как тетрагидрофуран, или сложным эфиром, таким как этилацетат. Такие реакции обычно проводят при комнатной температуре, но их можно проводить, при желании, с нагреванием.
Соли аминов соединений данного изобретения можно получить контактированием амина в водном растворителе со свободной кислотной формой данного соединения. Подходящими водными растворителями являются вода и смеси воды со спиртами, такими как метанол или этанол, простыми эфирами, такими как тетрагидрофуран, нитрилами, такими как ацетонитрил, или кетонами, такими как ацетон. Подобным образом можно получить также соли с аминокислотами.
Соединения этого изобретения можно получать в соответствии с описанными ниже последовательностями реакций или их можно получать способами, известными в данной области. Исходные материалы, применимые в получении соединений этого изобретения, известны или коммерчески доступны или они могут быть приготовлены известными способами или по описанным здесь характерным схемам реакций.
Соединения данного изобретения можно легко получить при помощи процедур стандартного пептидного синтеза в твердой фазе или в фазе раствора с применением исходных материалов и/или легко доступных промежуточных продуктов из поставляющих химических компаний, таких как Aldrich или Sigma (Н. Paulsen, G. Merz, V. Weichart, "Solid Phase Synthesis of O-Glycopeptide Sequences", Angew. Chem. Int. Ed. Engl. 27 (1988); Н. Mergler, R. Tanner, J. Gosteli, and P. Grogg, "Peptide Synthesis by a Combination of Solid-Phase and Soluti on Methods 1: A New Very Acid-Labile Anchor Group for the Solid-Phase Synthesis of Fully Protected Fragments. Tetrahedron letters 29, 4005 (1988); Merrifield, R. B. "Solid Phase Peptide Synthesis after 25 Years: The Design and Synthesis of Antagonists of Glucagon", Makromol. Chem. Macromol. Symp. 19, 31 (1988).
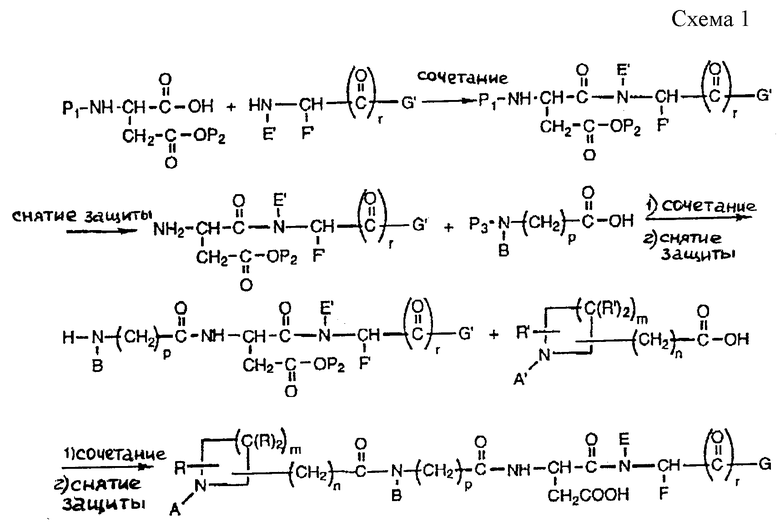
Предпочтительным способом получения соединений данного изобретения является жидкофазный способ, изображенный в схеме 1 (см. в конце описания).
в которой A, B, E, F, G, R, m, n, p и r имеют определенные выше значения;
A', E', F', G' и R' обозначают A, B, E, F, G и R соответственно, или они защищены их аналогами или замещены предшественниками заместителей; и
P1, P2, и P3 обозначают защитные группы для аминокислот.
Соединения данного изобретения получают обычно в результате первоначального присоединения подходящей аминокислоты или другой подходящей группы Z (предшественника), где Z имеет указанное выше значение, которая содержит свободный первичный и вторичный амин, со свободной частью карбоновой кислоты защищенного производного аспарагиновой кислоты.
Функциональные группы аспарагиновой кислоты или любые функциональные группы предшественника Z группы, которые не должны соединяться, защищены, если необходимо, блокирующими группами для предотвращения перекрестной реакции во время процедуры присоединения, так же как и производные аминокислот и производные азациклоалкилалкановой кислоты, применяемые в последующих стадиях синтеза. Эти блокирующие группы включают в себя N-α- трет- бутилоксикарбонил (ВОС), бензилоксикарбонил (CBZ), бензил, метил, т-бутил, 9-флуоренилметилоксикарбонил (FMOC), 2-(триметилсилил)- этил и 4-метокси-2,3,6-триметилбезолсульфонил.
Предпочтительным защищенным производным аспарагиновой кислоты является β- бензиловый эфир ВОС-аспарагиновой кислоты. Взаимодействие проводят способами, известными в этой области. Предпочтительным способом проведения соединения является объединение амина и карбоновой кислоты в подходящем апротонном органическом растворителе, например метиленхлориде или диметилформамиде (DMF), в присутствии подходящих связующих веществ. Предпочтительным связующим агентом является изопропилхлорформат в присутствии N-метилпиперидина. Другим предпочтительным связующим агентом является гидрохлорид 1-(3-диметиламинопропил)-3- этилкарбодиимида (EDC) в присутствии 1-гидроксибензотриазола (НОВТ) и триэтиламина. Еще одним предпочтительным связующим агентом является хлорангидрид бис(2-оксо-3-оксазолидинил)-фосфоновой кислоты (ВОР-С1) в присутствии триэтиламина.
Полученный защищенный продукт селективно освобождается от защитных групп известными способами с образованием N-концевого свободного амина части аспарагиновой кислоты. Предпочтительным способом для удаления ВОС группы является обработка трифторуксусной кислотой в апротонном органическом растворителе, например метиленхлориде.
Затем полученный, освобожденный от защитных групп продукт соединяют с N-защищенным N-замещенным производным глицина или β- аланина, имеющим свободную карбоксильную группу. Затем полученный продукт освобождают от N-защитных групп. Полученный свободный амин затем соединяют с подходящей защищенной азациклоалкилалкановой кислотой и этот продукт освобождают от защитных групп (депротектируют) известными способами, получая конечный продукт.
В другом предпочтительном способе соединения данного изобретения можно получать твердофазными способами, хорошо известными в этой области. В таком твердофазном способе C-концевой остаток связывают по карбоксильной части с нерастворимой смолой, например, остаток можно связать в виде сложного эфира полимера n-алкоксибензилового спирта. Способом, подобным жидкофазному способу, защищенную аминокислоту или другие остатки добавляют по одному до тех пор, пока вся последовательность не будет построена на полимере. Затем это соединение депротектируют и выделяют из смолы стандартными способами, получая конечное соединение
Во время получения соединений данного изобретения или полупродуктов их синтеза может быть желательно или необходимо предотвратить перекрестную реакцию между химически активными заместителями, иными, чем те, которые присутствуют у природных или других аминокислотах. Эти заместители могут быть защищены стандартными защитными группами, которые затем могут быть удалены или сохранены, если необходимо, известными способами для получения целевых продуктов или промежуточных продуктов (см. , например. Green, "Protective Groups in Organic Synthesis", Wiley, New York, 1981). Селективная защита или депротектирование могут быть также необходимыми или желательными, чтобы сделать возможными превращение или удаление существующих заместителей и получение в последующей реакции конечного целевого продукта.
Далее изобретение поясняется иллюстративными примерами. В следующих далее примерах, если нет других указаний, α- аминокислоты, которые могут иметь хиральные α- углероды, находятся в L-конфигурации.
Если нет других указаний, сообщаемые данные масс-спектрального анализа получали способом Lou Resolution Fast Atom Bombard - ment, проводимым на VG 70SE с "рассчитанными" величинами (М+Н)+. Спектральные данные ядерного магнитного резонанса получают на Brucker ACF 300, в D2O. Флэш-хроматографию выполняли на силикагеле. Жидкостная хроматография высокого разрешения (ЖХВР, HPLC) проведена на Dynamax 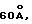 8 микрон С-18 колонке с обращенной фазой.
8 микрон С-18 колонке с обращенной фазой.
Пример 1
N-[N-[N-(4-(пиперидин-4-ил)бутаноил)-N- этилглицил] аспартил] β- циклогексилаланин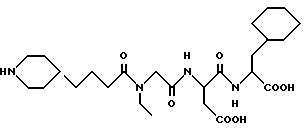
А. β- циклогексилаланин (1,12 г) растворяют в метаноле (50 мл) и пропускают газообразный хлористый водород через раствор в течение приблизительно 15 минут. Раствор выпаривают в вакууме и перегоняют в виде азеотропа с толуолом из осадка, получая метиловый эфир β- циклогексил-L-аланина в виде соли гидрохлорида.
B. β- бензиловый эфир BOC-L-аспарагиновой кислоты (1,27 г) растворяют в метиленхлориде (20 мл). Раствор охлаждают до 0oC и добавляют N-метилпиперидин (0,48 мл), а затем изопропилхлорформиат (3,94 мл). Этот раствор перемешивают при 0oC в течение приблизительно 2 минут и добавляют гидрохлорид метилового эфира β- циклогексил-L-аланина (0,88 г). Раствору дают согреться до комнатной температуры и перемешивают в течение ночи. Раствор выпаривают в вакууме и остаток распределяют между этилацетатом (200 мл) и 1 н. соляной кислотой (HCl) (50 мл). Органический слой промывают 1 н. HCl, насыщенным раствором бикарбоната натрия, солевым раствором, сушат над сульфатом магния, фильтруют и выпаривают в вакууме, получая метиловый эфир N-/BOC-L-acпapтил] (β- бeнзиловый эфир)] -β- циклогексил-L-аланина.
С. Метиловый эфир N-[ВОС-L-аспартил (β- бензиловый эфир)]- β- циклогексилаланина (2,01 г) растворяют в метиленхлориде (15 мл). Раствор охлаждают до 0oC и добавляют трифторуксусную кислоту (5 мл) в течение периода приблизительно 1 мин. Этот раствор перемешивают при 0oC в течение 2 часов, выпаривают в вакууме и остаток поглощают в этилацетат, после чего органический раствор промывают насыщенным раствором бикарбоната натрия до тех пор, пока промывки не станут щелочными. Этот органический раствор промывают солевым раствором, сушат над сульфатом магния, фильтруют и выпаривают в вакууме, получая метиловый эфир L-аспартил (β- бензиловый эфир) -β- циклогексил-L-аланина.
D. Используя процедуру примера 1В, описанную выше, с последующей процедурой удаления защитных групп согласно примеру С, получают метиловый эфир N-этилглицил-L- аспартил -(β- бензиловый эфир) -L-β- циклогексил-L-аланина из метилового эфира L- аспартил- (β- бензиловый эфир) -β- циклогексил-L-аланина и N-BOC-N- этилглицина.
Е. Смешивают 4-пиридинуксусную кислоту (10 г) и оксид платины (1,0 г) в уксусной кислоте (100 мл) и смесь качают под водородом при 50 psi (344734,85 Па) в течение приблизительно 18 часов. Смесь фильтруют и раствор выпаривают в вакууме и перегоняют в виде азеотропа с тотуолом из остатка, получая 2-(пиперидин-4-ил) уксусную кислоту.
F. 2-(пиперидин-4-ил) уксусную кислоту (11,6 г) растворяют в 1 N водном растворе гидроксида натрия (200 мл) и раствор охлаждают до 0oC. К нему по каплям добавляют раствор ди-трет- бутилдикарбоната (18,0 г) в тетрагидрофуране (THF) (100 мл) и смеси дают нагреться до комнатной температуры и перемешивают в течение приблизительно 18 часов. Далее смесь выпаривают в вакууме для удаления THF и остаток смешивают с водой и промывают этилацетатом. Этилацетат добавляют к водному слою и смесь подкисляют 1 N HCl. Органический слой отделяют и водный слой экстрагируют этилацетатом. Объединенные органические порции промывают водой, солевым раствором, сушат над сульфатом магния, фильтруют и выпаривают в вакууме, получая N-BOC-2-(пиперидин-4-ил)уксусную кислоту.
G. N-BOC-2-(пиперидин-4-ил)уксусную кислоту (15,8 г) растворяют в THF (150 мл) и добавляют каплями 1 М боран/THF (70 мл). Раствор перемешивают при комнатной температуре в течение приблизительно 20 часов и добавляют по каплям 1 N раствор гидроксида натрия (200 мл). THF выпаривают в вакууме и водный остаток экстрагируют этилацетатом. Раствор в этилацетате промывают водой, сушат над сульфатом натрия, фильтруют, выпаривают в вакууме, получая N-BOC-2-(пиперидин-4-ил)этанол.
Н. Раствор оксалилхлорида (11,8 г) в метиленхлориде (180 мл) охлаждают до -70oC и добавляют по каплям диметилсульфоксид (DMSO) (8,9 мл). Раствор перемешивают при -78oC в течение приблизительно 3 минут и добавляют раствор N-BOC-2-(пиперидин-4-ил) этанола (14,3 г) в метиленхлориде (250 мл) в течение периода приблизительно 10 минут. Раствор перемешивают в течение приблизительно 1 часа и добавляют N-метилморфолин (21,6 г) в течение приблизительно 15 минут. Раствору дают нагреться до комнатной температуры и, после приблизительно 30 минут, добавляют метил (трифенилфосфоранилиден) ацетат (68,6 г). Раствор перемешивают при комнатной температуре в течение приблизительно 18 часов, выпаривают в вакууме и остаток поглощают в этилацетате. Раствор этилацетата промывают водой, 5% HCl, 5% раствором гипохлорита натрия, водой, солевым раствором, сушат над сульфатом натрия, фильтруют и выпаривают в вакууме, получая метил-4-(N-ВОС-пиперидин-4-ил)транс-кротонат.
I. Метил-4-(N-ВОС-пиперидин-4-ил)транс-кротонат (11,5 г) растворяют в метаноле (200 мл) и добавляют палладий/уголь (3 г) и эту смесь качают под водородом при 50 psi (344737,85 Па) в течение 18 часов. Смесь фильтруют, добавляют к раствору свежий катализатор и гидрогенизацию повторяют. Смесь фильтруют и выпаривают в вакууме, получая метил-4-(N-ВОС-пиперидин-4-ил)бутират.
J. К смеси 1 N водного гидроксида натрия (100 мл) и метанола (200 мл) добавляют метил-4-(N-ВОС-пиперидин-4-ил)бутират (10,1 г) и смесь перемешивают при комнатной температуре в течение приблизительно 18 часов. Смесь выпаривают в вакууме, разбавляют водой и промывают эфиром. Водную часть подкисляют 5% HCl, экстрагируют этилацетатом и органический раствор промывают водой, солевым раствором, сушат над сульфатом натрия, фильтруют и выпаривают в вакууме, получая 4-(N-ВОС-пиперидин-4-ил)масляную кислоту.
К. Раствор 4-(N-ВОС-пиперидин-4-ил) масляной кислоты (0,91 г) в метиленхлориде (50 мл) охлаждают до 0oC и добавляют хлорангидрид бис(2-оксо-3-оксазолидинил)фосфиновой кислоты (ВОР-С1) (0,86 г) и триэтиламин (0,47 мл). Раствор перемешивают при 0oC в течение приблизительно 10 минут и добавляют метиловый эфир N-этилглицил-L-аспартил (β- бензиловый эфир) -L-β- циклогексил-L- аланина (1,52 г) в минимальном количестве метиленхлорида с последующим добавлением по каплям триэтиламина (0,47 мл) в метиленхлориде в течение приблизительно 15 минут. Смесь перемешивают при 0oC в течение приблизительно 1 часа при комнатной температуре в течение приблизительно 18 часов. Смесь выпаривают в вакууме и остаток поглощают в этилацетат. Этот органический раствор промывают 1 N HCl, насыщенным раствором бикарбоната натрия, солевым раствором, сушат над сульфатом магния, фильтруют, упаривают в вакууме и остаток очищают флаш-хроматографией с элюцией 60% этилацетатом в гексанах, получая метиловый эфир N-[N-[N-(4-(N-ВОС- пиперидин-4-ил)бутаноил)-N-этилглицил]аспартил (β- бензиловый эфир)] -β- циклогексилаланин.
L. Метиловый эфир N-[N-[N-(4-(N-ВОС-пиперидин-4-ил)бутаноил) -N-этилглицил]аспартил] (β- бензиловый эфир)] -β- циклогексилаланина, (1,79 г) растворяют в метаноле (40 мл) и добавляют 10% палладий на угле (0,25 г). Эту смесь качают под водородом при 50 psi (344737, 85 Па) в течение приблизительно 18 часов. Смесь фильтруют через целитовую подушку и фильтрат упаривают в вакууме, получая метиловый эфир N-[N-[N-(4-(N-ВОС-пиперидин-4-ил)бутаноил)-N-этилглицил]- аспартил] -β- циклогексилаланин. Эфир растворяют в метаноле (20 мл) и добавляют 1 N водный раствор гидроксида натрия (10 мл). Смесь перемешивают при комнатной температуре в течение приблизительно 4 часов, разбавляют водой (25 мл) и подкисляют 1 N HCl до pH 2. Эту смесь экстрагируют этилацетатом (3х100 мл) и раствор в этилацетате сушат над сульфатом магния, фильтруют и выпаривают в вакууме, получая N-[N-[N-(4-(N-ВОС-пиперидин-4-ил)бутаноил)- N-этилглицил]аспартил] -β- циклогексилаланин.
М. N-[N-[N-(4-(N-ВОС-пиперидин-4-ил)бутаноил)-N-этилглицил] аспартил] -β- циклогексилаланин (1,39 г) растворяют в метиленхлориде (15 мл) и раствор охлаждают до 0oC. Трифторуксусную кислоту (5 мл) добавляют и раствор перемешивают при 0oC в течение приблизительно 2,5 часов. Раствор выпаривают в вакууме и остаток разводят водой, замораживают и лиофилизируют. Остаток очищают HPLC (ЖХВР) с обращенной фазой с элюцией градиентом 40-80% метанола в воде, содержащего 1,0% трифторуксусную кислоту. Нужные фракции объединяют и лиофилизируют, получая N-[N-[N-(4-пиперидин-4-ил)-бутаноил)-N-этилглицил] аспартил] -β- циклогексилаланин в виде соли трифторуксусной кислоты. M.S., рассчитано: 525, Найдено: 525; ЯМР, δ = 4,58-4,48 (м, 1H), 4,35-4,22 (м, 1H), 3,88 (с, 2H), 3,32 (к, 2H), 3,28-3,10 (м, 2H), 2,88-2,60 (м, 4H), 2,33 (т, 2H), 1,85-1,70 (м, 2H), 1,62-1,35 (м, 10H), 1,30-1,06 (м, 5H), 1,04-0,65 (м, 8H).
Используя процедуры представленного выше примера 1, следующие соединения получают из соответствующих исходных материалов.
Пример 2
N-[N-[N-(4-(пиперидин-4-ил)бутаноил)-N- этилглицил]аспартил]валин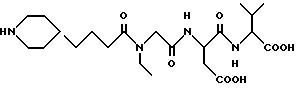
M.S. Рассчитано; 471, найдено: 471; ЯМР, δ = 4,15-4,05 (м, 2H), 3,90 (с, 2H), 3,30 (к, 2H), 3,30-3,15 (м, 2H), 2,90-2,60 (м, 4H), 2,33 (т, 2H), 2,05 (к, 1H), 1,85-1,72 (м, 2H), 1,55-1,35 (м, 3H), 1,30-1,08 (м, 4H), 1,02 (т, 3H), 0,70 (д, 6H).
Пример 3
N-[N-[N-(4-пиперидин-4-ил)-N-этилглицил]аспартил]фенилаланин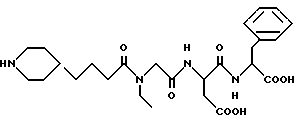
M. S. рассчитано: 519, найдено: 519; ЯМР, δ = 7,20-6,95 (м, 5H), 4,55-4,35 (м, 2H), 3,73 (с,2H), 3,30-2,40 (м, 10H), 2,25 (т, 2H), 1,75-1,60 (м, 2H), 1,45-1,25 (м, 3H), 1,20-0,97 (м, 4H), 0,92 (т, 3H).
Пример 4
N-[N-[N-4-(пиперидин-4-ил)бутаноил)-N-этилглицил] аспартил]-D-валин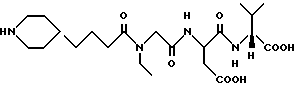
M. S. , Рассчитано: 471, найдено: 471; ЯМР, δ = 4,10-3,98 (м, 2H), 3,85 (с, 2H), 3,28 (к, 2H), 3,23-3,10 (м, 2H), 2,82-2,58 (м, 4H), 2,28 (т, 2H), 2,02 (к, 1H), 1,80-1,65 (м, 2H), 1,50-1,28 (м, 3H), 1,25-1,00 (м, 4H), 0,95 (т, 3H), 0,78-0,65 (м, 6H).
Пример 5
N-[N-[N-(4-(пиперидин-4-ил)бутаноил)-N-(этилглицил] аспартил] - L- α- 2,2-диметил)проп-3-илглицин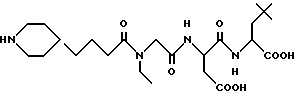
M. S. , Рассчитано: 499, найдено: 499; ЯМР δ = 4,63-4,55 (м, 1H), 4,30-4,20 (м, 1H), 3,38 (с,2H), 3,33 (к, 2H), 3,30-3,15 (м, 2H), 2,88-2,60 (м, 4H), 2,35 (т, 2H), 1,85-1,75 (м, 2H), 1,73-1,35 (м, 5H), 1,30-1,08 (м, 4H), 1,03 (т, 3H), 0,78 (с, 9H).
Пример 6
N-[N-[N-(4-пиперидин-4-ил)бутаноил)-N-этилглицил]аспартил]норлейцин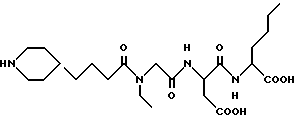
M. S., Рассчитано: 485, найдено: 485; ЯМР, δ = 4,59-4,50 (м, 1H), 4,20-4,10 (м, 1H), 3,85 (с, 2H), 3,32 (к, 2H), 3,25-3,10 (м, 2H), 2,85-2,55 (м, 4H), 2,30 (т, 2H), 1,82-1,50 (м, 4H), 1,50-1,30 (м, 3H), 1,28-1,05 (м, 8H), 0,98 (т, 3H), 0,75-0,60 (м, 3H).
Пример 7
N-[N-[N-(4-(пиперидин-4-ил)бутаноил)-N- этилглицил] acпapтил] -L -β- нафт-1-илаланин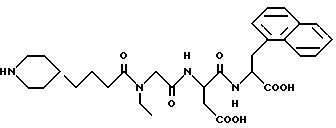
M. S., Рассчитано: 569, найдено: 569; ЯМР, δ = 8,00-7,15 (м, 7H), 4,60-4,45 (м, 2H), 3,71 (c, 2H), 3,65-3,50 (м, 2H), 3,40-2,98 (м, 4H), 2,70-2,42 (м, 4H), 2,21 (т, 2H), 1,70-1,45 (м, 2H), 1,40-0,96 (м, 7H), 0,92 (т, 3H).
Пример 8
N-[N-[N-(4-(пиперидин-4-ил)бутаноил)-N- этилглицил] аспартил] -L- -β- нафт-2-илаланин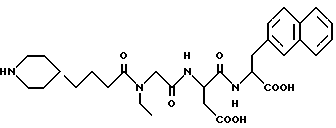
M. S., Рассчитано: 569, найдено: 569; ЯМР, δ = 7,65-7,03 (м, 7H), 4,60-4,45 (м, 2H), 3,41 (с, 2H), 3,20-2,01 (м, 3H), 3,00-2,71 (м, 3H), 2,68-2,40 (м, 4H), 1,98 (т, 2H), 1,68-1,42 (м, 3H), 1, 25-0,85 (м, 6H), 0,71 (т, 3H).
Пример 9
N-[N-[N-(3-(пиперидин-4-ил)пропаноил)-N-этилглицил]аспартил]-валин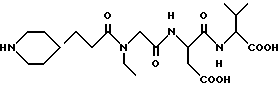
M.S., Рассчитано: 547, найдено: 457; ЯМР, δ = 4,62 (м, 2H), 3,90 (с,2H), 3,33 (м, 4H), 2,66 (м, 4H), 2,37 (т, 2H), 2,16 (м, 1H), 2,03 (м, 1H), 1,78 (м, 2H), 1,44 (м, 2H), 1,20 (м, 2H), 1,00 (м, 3H), 0,78 (д, 6H).
Пример 10
N-[N-[N-(5-(пиперидин-4-ил)пентаноил)-N-этилглицил]аспартил]-валин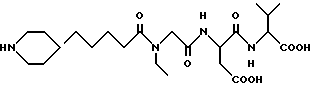
M. S. , Рассчитано: 485, найдено: 485; ЯМР, δ = 4,20-4,05 (м, 2H), 3,92 (с, 2H), 3,33 (м, 2H), 3,28-3,15 (м, 2H), 2,90-2,61 (м, 4H), 2,34 (т, 2H), 2,06 (м, 1H), 1,85-1,70 (м, 2H), 1,55-1,32 (м, 3H), 1,30- 1,12 (м, 6H), 1,06 (т, 3H), 0,81 (д, 6H).
Пример 11
Этиловый эфир N-[N-[N-(4-(пиперидин-4-ил)бутаноил)-N- этилглицил] acпapтил] -L-β- циклoгeкcилaлaнинa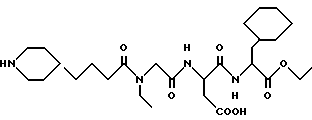
А. β- циклогексилаланин (1,5 г) растворяют в абсолютном этиловом спирте (75 мл) и раствор охлаждают до 0oC. Тионилхлорид (1,1 мл) добавляют по каплям в течение 10-15 минут, раствору дают нагреться до комнатной температуры и затем перемешивают при комнатной температуре в течение приблизительно 18 часов. Реакционную смесь выпаривают в вакууме, перегоняют в виде азеотропа с толуолом 2 раза и остаток поглощают в этилацетат. Раствор в этилацетате промывают водой, 1 N гидроксидом натрия, водой, солевым раствором, сушат над сульфатом натрия, фильтруют и выпаривают в вакууме, получая этиловый эфир β- циклогексилаланина.
В. Используя процедуру примера 1В-1М (за исключением гидролиза водным гидроксидом натрия стадии 1L), получают целевой продукт.
M. S. , Рассчитано: 553, найдено: 553; ЯМР δ = 4,4 (1H, м), 4,1 (2H, д), 3,2-3,5 (5H, м), 2,7-3,0 (5H, м), 2,4 (2H, т), 1,9 (2H, д), 1,4-1,7 (7H, м), 0,7-1,4 (18H, м).
Пример 12
N-[N-[N-(4-(пиперидин-4-ил)бутаноил)-N- этилглицил] аспартил] β- циклогексилаланинамид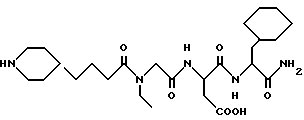
А. N-ВОС -β- циклогексилаланин (2,0 г) и триэтиламин (1,03 мл) растворяют вместе в THF (100 мл) и раствор охлаждают до -20oC. Добавляют изобутилхлорформиат (1,06 мл) и раствор перемешивают при -20oC в течение приблизительно 30 минут. Добавляют насыщенный раствор аммиака в метаноле (20 мл) и раствору дают нагреться до комнатной температуры и перемешивают его при комнатной температуре в течение приблизительно 18 часов. Раствор выпаривают в вакууме и остаток растворяют в этилацетате. Раствор в этилацетате промывают водой, 5% HCl, насыщенным раствором бикарбоната натрия, водой, солевым раствором, сушат над сульфатом натрия, фильтруют и выпаривают в вакууме, получая N-BOC -β- циклогексилаланинамид.
В. N-ВОС -β- циклогексилаланинамид (2,0 г) растворяют в этилацетате (100 мл) и газообразный хлористый водород HCl пробулькивают через раствор и перемешивают раствор при комнатной температуре в течение приблизительно 18 часов. Раствор выпаривают в вакууме и перегоняют 2 раза в виде азеотропа с толуолом, получая β- циклогексилаланинамид в виде гидрохлорида.
С. Используя процедуру примера 1В-1М (за исключением гидролиза водным гидроксидом натрия примера IL), получают целевой продукт.
M. S., Рассчитано: 524, найдено: 524; ЯМР, δ = 8,4 (1H, д), 8,1 (1H, д), 4,2 (2H, к), 4,1 (1H, с), 3,9 (4H, к), 3,4 (2H, к), 3,3 (4H, д), 2,8-3,0 (6H, м), 2,4 (2H, т), 2,2 (1H, м), 1,8 (4H, д), 1,4-1,7 (7H, м), 0,7-1,3 (10H, м).
Пример 13
N-[N-[N-(4-(пиперидин-4-ил)бутаноил)-N- этилглицил] аспартил] -L- α- циклогексилглицин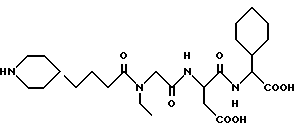
A. Раствор метилового эфира гидрохлорида α- фенилглицина (1,0 г) в THF (25 мл) охлаждают до 0oC и добавляют триэтиламин (1,38 мл). К этой смеси добавляют раствор ди-трет- бутилдикарбоната (1,08 г) в THF (25 мл) и смеси дают нагреться до комнатной температуры и перемешивают при комнатной температуре в течение приблизительно 18 часов. Раствор выпаривают и остаток растворяют в этилацетате (200 мл) и органический раствор промывают 1 N HCl, насыщенным раствором бикарбоната натрия, солевым раствором, сушат над сульфатом натрия, фильтруют и выпаривают в вакууме, получая метиловый эфир N-ВОС -α- фенилглицина.
В. Метиловый эфир N-ВОС -α- фенилглицина (1,2 г) растворяют в метаноле (50 мл), содержащем уксусную кислоту (1 мл). Добавляют 5% родий на порошке оксида алюминия (0,60 г) и смесь качают под водородом при 50 psi в течение приблизительно 18 часов. Смесь фильтруют, выпаривают в вакууме и остаток растворяют в этилацетате. Этот органический раствор промывают водой, насыщенным раствором бикарбоната натрия, водой, солевым раствором, сушат над сульфатом магния, фильтруют, выпаривают в вакууме, получая метиловый эфир N-ВОС- -α- -циклогексилглицина.
С. Используя процедуру примера 1В-1М, получают целевой продукт.
M. S. , Рассчитано: 511, найдено: 511; ЯМР, δ = 4,62-4,55 (1H, м), 4,06 (2H, м), 3,85 (2H, с), 3,30 (2H, к), 3,23-3,10 (2H, м), 2,85-2,55 (4H, м), 2,30 (2H, т), 1,83-1,60 (3H, м), 1,59-1,32 (8H, м), 1,30- 0,75 (12H, м).
Пример 14
N-[N-[N-(4-(пиперидин-4-ил)бутаноил)-N- этилглицил]аспартил]-L -β- декагидронафт-1-илаланин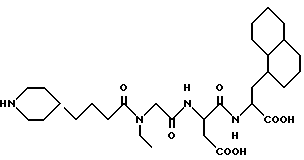
А. β- (1-нaфтил)aлaнин (2,0 г) перемешивают в насыщенном растворе HCl/метанол в течение приблизительно 2 часов при комнатной температуре. Смесь выпаривают в вакууме и дважды перегоняют в виде азеотропной смеси с толуолом из остатка. Остаток суспендируют в метиленхлориде, добавляют N-метилморфолин (1,02 мл) и смесь охлаждают до 0oC. Добавляют ди-трет-бутилдикарбонат (2,02 г) и 4- диметиламинопиридин (DMAP) (0,8 г), раствору дают нагреться до комнатной температуры и перемешивают его при комнатной температуре в течение приблизительно 2 часов. Смесь промывают 5% HCl, водой, сушат над сульфатом натрия, фильтруют и выпаривают в вакууме, получая метиловый эфир N-ВОС -β- (1-нафтил)аланина.
В. Метиловый эфир N-BOC -β- (1-нафтил)аланина (2,0 г) и 5% родия на оксиде алюминия (1,0 г) объединяют в метаноле (50 мл), содержащем уксусную кислоту (1,0 мл), и смесь качают в атмосфере водорода при 50 psi в течение приблизительно 18 часов. Смесь фильтруют, выпаривают в вакууме и остаток растворяют в этилацетате. Этот органический раствор промывают водой, 5% раствором бикарбоната натрия, водой, солевым раствором, сушат над сульфатом натрия, фильтруют и выпаривают в вакууме, получая метиловый эфир L-β- декагидронафт-1-илаланина.
С. Используя процедуру примера 1В-1М, получают целевой продукт.
M. S. , Рассчитано: 579, найдено: 579; ЯМР, δ = 4,1-4,3 (м, 1H), 3,8-4,1 (м, 2H), 2,6-2,9 (м, 4H), 2,3 (м, 1H), 2,0 (м, 1H), 1,8 (д, 3H), 0,5-1,6 (м, 33H).
Пример 15
2-циклогексил-N-[N-[N-(4-(пиперидин-4-ил)бутаноил)-N- этилглицил] аспартил]-этиламин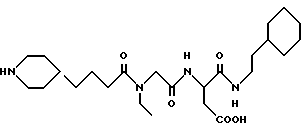
А. 2-Фенилэтиламин (2,0 г) растворяют в метиленхлориде и раствор охлаждают до 0oC. Добавляют ди-трет-бутилдикарбонат (4,0 г) и DMAP (0,4 г). Раствору дают нагреться до комнатной температуры и перемешивают его при комнатной температуре в течение приблизительно 18 часов. Раствор промывают 5% HCl, водой, фильтруют и выпаривают в вакууме, получая N-BOC-2-фенилэтиламин.
В. N-BOC-2-фенилэтиламин (3,1 г) и 5% родия на оксиде алюминия (1,1 г) объединяют в метаноле (40 мл), содержащем уксусную кислоту (1,0 мл). Смесь качают под водородом при 50 psi в течение приблизительно 20 часов. Смесь фильтруют, выпаривают в вакууме и остаток растворяют в этилацетате. Этот органический раствор промывают водой, 5% раствором бикарбоната натрия, водой, солевым раствором, сушат над сульфатом натрия, фильтруют и выпаривают в вакууме, получая N-BOC-2-циклогексилэтиламин.
С. Используя процедуру примера 1В-1М (за исключением гидролиза водным пероксидом натрия примера 1L), получают целевой продукт.
M.S., Рассчитано: 481, найдено: 481; ЯМР, δ = 3,9 (с, 2H), 3,35 (д, 4H), 3,25 (д, 4H), 2,6-2,9 (м, 8H), 2,35 (т, 2H), 2,15 (т, 1H), 1,8 (4H, д), 1,4-1,7 (м, 7H), 0,9-1,3 (м, 12H), 0,7 (т, 2H).
Пример 16
N-[N-[N-(4-(пиперидин-4-ил)бутаноил)-N- этилглицил]аспартил]-L- α- (2-циклогексилэтил)глицин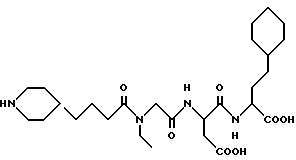
А. Используя процедуру примера 13А, метиловый эфир N-BОС- α- (2- фенилэтил) глицина получают из L-гомофенилаланина.
В. При помощи в основном процедуры примера 14В (выше) N-ВОС- -α- (2-циклогексилэтил)глицин (метиловый эфир) получают из метилового эфира N-BOC -α- (2-фенилэтил)глицина.
С. При помощи процедур Примера 1В-1М получают целевой продукт.
M. S., Рассчитано: 539, найдено: 539; ЯМР, δ = 4,60-4,55 (м, 1H), 4,20-4,08 (м, 1H), 3,85 (с, 2H), 3,29 (к, 2H), 3,25-3,12 (м, 2H), 2,84-2,55 (м, 4H), 2,29 (т, 2H), 1,83-1,65 (м, 2H), 1,63-1,32 (м, 10H), 1,28-0,81 (м, 13H), 0,79-0,56 (м, 2H).
С использованием процедур представленных выше примеров из подходящих исходных материалов получают следующие соединения.
Пример 17
N-[N-[N-(4-(пиперидин-4-ил)бутаноил)-N- этилглицил] аспартил] -L- -β- цис-декагидронафт-2-илаланин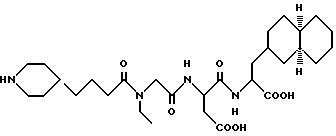
M. S. , Рассчитано: 579, найдено: 579; ЯМР, δ = 4,7(м, 1H), 4,3 (м, 1H), 4,1 (д, 2H), 3,3-3,7 (м, 5H), 2,6-2,8 (м, 5H), 2,5 (т, 2H), 2,3 (т, 1H), 1,9 (д, 2H), 1,3-1,8 (м, 14H), 0,9-1,3 (м, 14H), 0,7-0,8 (м, 3H).
Пример 18
3-адамант-1-илпропил-N-[N-(4-(пиперидин-4-ил)бутаноил)-N- этилглицил] аспартат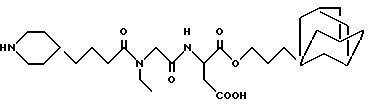
M. S. , Рассчитано: 548, найдено: 548; ЯМР (DMSO-d6), δ = 4,65-4,50 (м, 1H), 4,05-3,85 (м, 4H), 3,35-3,15 (м, 4H), 2,90-2,50 (м, 4H), 2,30 (т, 2H), 2,18 (т, 1H), 1,94 (д, 2H), 1,85-1,35 (м, 20H), 1,32-1,12 (м, 4H), 1,10-0,90 (м, 5H).
Пример 19
N-[N-[N-(4-(пиперидин-4-ил)бутаноил)-N- этилглицил] аспартил] -α- аминоциклогексанкарбоновая кислота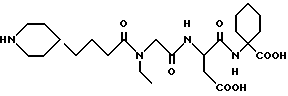
M. S. , Рассчитано: 497, найдено: 497; ЯМР, δ = 4,60-4,55 (м, 1H), 4,05 (с, 1H), 3,90 (с, 1H), 3,30 (к, 2H), 3,25-3,12 (м, 2H), 2,85-2,55 (м, 4H), 2,35 (т, 1H), 2,11 (т, 1H), 1,90-1,70 (м, 4H), 1,53-1,32 (м, 6H), 1,30-1,06 (м, 7H), 1,05-1,85 (м, 3H).
Пример 20
N-[N-[N-(4-пипepидин-4-ил)бутаноил)- этилгексил] аспартил] -β- циклогексил-D-аланин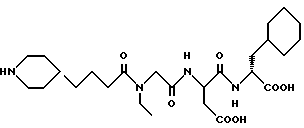
M. S., Рассчитано: 525, найдено: 525; ЯМР, δ = 4,60-4,55 (м, 1H), 4,32-4,20 (м, 1H), 4,05 (с, 1H), 3,85 (с, 1H), 3,32 (к, 2H), 2,25-3,12 (м, 24), 2,85-2,60 (м, 4H), 2,32 (т, 1H), 2,12 (т, 1H), 1,85-1,68 (м, 2H), 1,60-1,32 (м, 10H), 1,28-0,60 (м, 13H).
Пример 21
N-[N-[N-(4-(пиперидин-4-ил)бутаноил)-N-этилглицил] аспартил] -β- декагидронафт-1-илаланин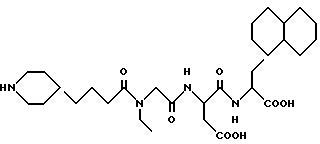
M. S. , Рассчитано: 579, найдено: 579; ЯМР, δ = 4,1-4,3 (1H, м), 3,8-4,1 (2H, м), 3,1-3,4 (4H, м), 2,6-2,9 (4H, м), 2,3 (1H, м), 2,0 (1H, м), 1,8 (3H, д), 0,5-1,6 (33H, м).
Пример 22
N-[N-[N-(4-(пиперидин-4-ил)бутаноил)-N- этилглицил] аспартил] -β- циклогексилаланинэтиламид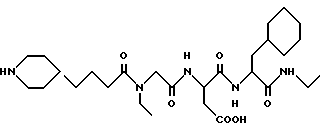
M. S., Рассчитано: 552, найдено: 552; ЯМР, δ = 4,55-4,45 (м, 1H), 4,20-4,06 (м, 1H), 4,05-3,85 (м, 2H), 3,40-3,25 (м, 2H), 3,28-3,15 (м, 2H), 3,10-2,90 (м, 2H), 2,88-2,55 (м, 4H), 2,40-2,25 (м, 1H), 2,20-2,05 (м, 1H), 1,85-1,70 (м, 2H), 1,60-1,32 (м, 9H), 1,30-0,62 (м, 17H).
Пример 23
N-[N-[N-(4-(пиперидин-4-ил)бутаноил)-N-этилглицил] аспартил] -β- циклооктилаланин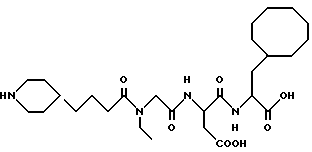
M. S. , Рассчитано: 553, найдено: 553; ЯМР, δ = 4,1-4,3 (1H, м), 3,8-4,1 (2H, м), 3,1-3,4 (4H, м), 2,6-2,9 (4H, м), 2,3 (1H, м), 2,0 (1H, м), 1,8 (2H, д), 0,5-1,6 (31H, м).
Пример 24
N-[N-[N-(4-(пиперидин-4-ил)бутаноил)-N- этилглицил] аспартил] -α- циклогексилметилэтаноламин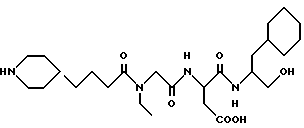
M. S., Рассчитано: 511, найдено: 511; ЯМР, δ = 4,60-4,45 (м, 1H), 4,10-3,75 (м, 3H), 3,45-3,15 (м, 6H), 2,90-2,60 (м, 4H), 2,35 (т, 1H), 2,00-2,08 (м, 1H), 1,88-1,75 (м, 2H), 1,62-1,35 (м, 8H), 1,30-1,08 (м, 7H), 1,10-0,60 (м, 8H).
Пример 25
N-[N-[N-(4-(пиперидин-4-ил)бутаноил)-N- этилглицил] аспартил] -β- циклогексилметилаланинамид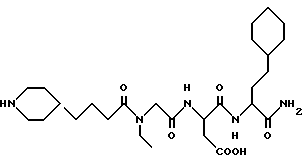
M. S., Рассчитано: 538, найдено: 538; ЯМР, δ = 4,60-4,50 (м, 1H), 4,15-4,00 (м, 1H), 4,00-3,80 (м, 2H), 3,35 (к, 2H), 3,30-3,15 (м, 2H), 2,90-2,62 (м, 4H), 2,35 (т, 1H), 2,15 (т, 1H), 1,88-1,75 (м, 2H), 1,65-1,40 (м, 9H), 1,30-0,88 (м, 14H), 0,85-0,65 (м, 2H).
Пример 26
N-[N-[N-(4-(пиперидин-4-ил)бутаноил)-N- этилглицил] аспартил] -β- адамант-1-илаланин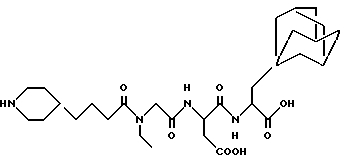
M. S. , Рассчитано: 577, найдено: 577; ЯМР, δ = 4,5-4,1 (1H, м), 4,1-4,2 (1H, м), 3,9 (2H, д), 3,2 (2H, к), 3,1-3,1 (6H, м), 2,4-2,98 (5H, м), 2,3 (1H, м), 2,0 (1H, м), 1,8 (4H, м), 1,2-1,7 (16H, м), 1,0-1,2 (6H, м), 0,8-1,0 (3H, м).
Пример 27
N-[N-[N-(4-(пиперидин-4-ил)бутаноил)-N- этилглицил] аспартил] -β- (1,2,3,4)-тетрагидронафт-5-илаланин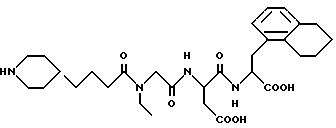
M. S., Рассчитано: 573, найдено; 573; ЯМР, δ = 6,9 (д, 4H), 4,7 (м, 1H), 4,3 (м, 1H), 4,1 (д, 2H), 3,3-3,7 (м, 6H), 2,6-3,1 (м, 12H), 2,5 (т, 2H), 2,3 (т, 1H), 1,9 (д, 2H), 1,2-1,8 (м, 16H), 1,1 (т, 2H), 1,0 (т, 2H).
Пример 28
N-[N-[N-(4-(пиперидин-4-ил)бутаноил)-N- этилглицил] аспартил] -β- (4-циклогексил)циклогексилаланин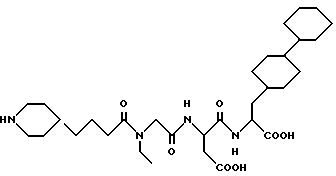
M. S. , Рассчитано: 607, найдено: 607; ЯМР, δ = 4,2-4,3 (1H, м), 3,9-4,1 (2H, м), 3,1-3,4 (5H, м), 2,6-2,9 (5H, м), 2,3 (1H, м), 2,0 (1H, м), 1,8 (3H, д), 0,9-1,6 (32H, м), 0,7-08 (3H, м).
Пример 29
N-[N-[N-(4-(пиперидин-4-ил)бутаноил)-N- этилглицил] аспартил] -β- циклогептилаланин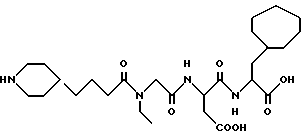
M. S., Рассчитано: 539, найдено: 539; ЯМР, δ = 4,60-4,55 (м, 1H), 4,35-4,25 (м, 1H), 4,08 (с, 1H), 3,35 (к, 2H), 3,33-3,20 (м, 2H), 2,90-2,60 (м, 4H), 2,35 (т, 1H), 2,18 (т, 1H), 1,90-1,75 (м, 2H), 1,70-1,10 (м, 22H), 1,10-0,85 (м, 3H).
Пример 30
N-[N-[N-(4-(пиперидин-4-ил)бутаноил)-N- этилглицил] аспартил] -β- циклооктилаланинамид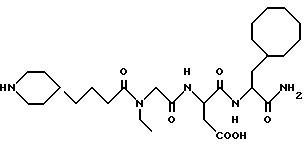
M. S., Рассчитано: 551, найдено: 551; ЯМР, δ = 4,46 (дд, 1H, H-12), 4,18 (м, 1H, H-14), 3,89 (д, 1H, H-11), 3,69 (д, 1H, H-11), 3,31 (к, 2H, H-9), 3,18 (дт, 2H, H-1а), 2,74 (дт, 2H, H-1е), 2,65 (дд, 2H, H-13), 2,25 (т, 2H, H-8), 1,85-1,10 (м, 26H, H-3 - H-7 и H-15 - H-23), 1,06 (т, 3H, H-10).
Пример 31
N-[N-[N-(4-(пиперидин-4-ил)бутаноил)-N- этилглицил] аспартил] -α- циклогексилпропилглицин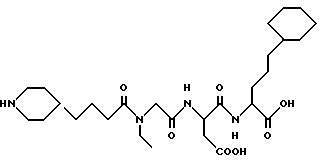
M. S., Рассчитано; 553, найдено: 553; ЯМР, δ = 4,70-4,60 (м, 1H), 4,30-4,15 (м, 1H), 4,10 (с, 1H), 3,95 (с, 1H), 3,35 (к, 2H), 3,35-3,20 (м, 2H), 2,90-2,60 (м, 4H), 2,40 (т, 1H), 2,15 (т, 1H), 1,90-1,75 (м, 2H), 1,75-1,45 (м, 10H), 1,35-1,15 (м, 6H), 1,12-0,90 (м, 9H), 0,85-0,60 (м, 2H).
Пример 32
N-[N-[N-(4-(пиперидин-4-ил)бутаноил)-N-этилглицил] аспартил] - -β- циклооктилметилаланин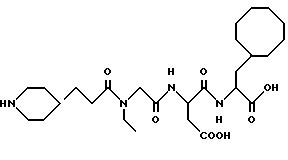
M. S. , Рассчитано: 567, найдено: 567, ЯМР, δ = 4,05-4,15 (м, 1H, 14), 3,75-4,00 (м, 2H, 11 и 18), 3,10-3,30 (м, 4H, 19 и 26 эк), 2,50-2,80 (м, 4H, 15 и 26 акс), 2,05-2,25 (м, 2H, 21) 0,75-1,75 (м, 31H, 1-10, 20 и 22-25).
Пример 33
N-[N-[N-(4-(пиперидин-4-ил)бутаноил)-N- этилглицил] acпapтил] -β- циклопентилаланин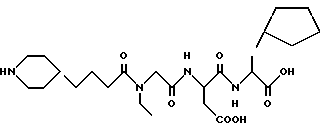
M. S., Рассчитано; 511, найдено: 511, ЯМР, δ = 4,7 (м, 1H), 4,3 (м, 1H), 4,1 (д, 2H), 3,3-3,7 (м, 5H), 2,8 (т, 2H), 2,7 (м, 3H), 2,5 (т, 2H), 2,3 (т, 1H), 1,9 (д, 2H), 1,0-1,8 (м, 16H).
Пример 34
Этиловый эфир N-[N-[N-(4-(пиперидин-4-ил)бутаноил)-N- этилглицил] аспартил] -β- циклогексилметилаланина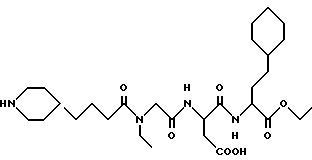
M. S., Рассчитано: 567, найдено: 567; ЯМР, δ = 4,30-4,10 (м, 1H), 4,10-3,80 (м, 3H), 3,35 (к,2H), 3,30-3,15 (м, 2H), 2,90-2,60 (м, 4H), 2,40-2,10 (т, 2H), 1,90-1,70 (м, 2H), 1,65-1,40 (м, 10H), 1,35-0,85 (м, 18H), 0,85-0,60 (м, 2H).
Соединения в объеме данного изобретения ингибируют агрегацию тромбоцитов путем ингибирования связывания фибриногена с активированными тромбоцитами и другими адгезивными гликопротеинами, участвующими в агрегации тромбоцитов и свертывании крови, и применимы в предотвращении и лечении тромбоза, связанного с некоторыми патологическими состояниями, такими как инфаркт миокарда, тромботический инсульт, поражение периферической артерии и диссеминированное внутрисосудистое свертывание у человека и других млекопитающих.
Соединения этого изобретения могут обычно вводиться перорально или парентерально в лечении или предотвращении связанных с тромбозом патологических состояний.
Соединения этого изобретения могут быть приготовлены в виде формы для введения любым стандартным путем, и изобретение включает в его объем фармацевтические композиции, содержащие по меньшей мере одно соединение изобретения, приспособленное для использования в медицине и ветеринарии. Такие композиции могут быть изготовлены общепринятыми способами с применением одного или более фармацевтически приемлемых носителей или наполнителей. Подходящие носители включают в себя разбавители или наполнители, стерильные водные среды и разнообразные нетоксичные органические растворители. Композиции могут быть приготовлены в виде таблеток, капсул, лепешек, пастилок, карамели, порошков, водных суспензий или растворов, растворов для инъекций, эликсиров, сиропов и т.п. и могут содержать один или более агентов, выбранных из группы, включающей в себя подслащивающие агенты, ароматизаторы, красители и консерванты, для обеспечения фармацевтически приемлемого препарата.
Конкретный носитель в отношении ингибирующего агрегацию тромбоцитов и тромбообразование соединения к носителю определяют на основании растворимости и химических свойств этих соединений, на основании конкретного способа введения и стандартной фармацевтической практики. Например, в производстве таблеток можно применять наполнители, такие как лактоза, цитрат натрия, карбонат кальция и кислый фосфат кальция (Ca2HPO4), и различные дезинтеграторы, такие как крахмал, альгиновая кислота и некоторые комплексные силикаты, вместе со смачивающими агентами, такими как стеарат магния, лаурилсульфат натрия и тальк. Для капсул лактоза и высокомолекулярные полиэтиленгликоли являются предпочтительными фармацевтически приемлемыми носителями. При изготовлении водных суспензий для перорального применения носитель может быть эмульгирующим или суспендирующим агентом. Разбавители, такие как этанол, пропиленгликоль, глицерин и хлороформ и их комбинации, могут быть использованы, а также и другие материалы.
Для парентерального введения можно использовать растворы или суспензии этих соединений в кунжутном или арахисовом масле или водных растворах полипропиленгликоля, а также стерильные водные растворы растворимых фармацевтически приемлемых солей, описанных здесь. Растворы солей этих соединений особенно пригодны для целей внутримышечной и подкожной инъекции. Водные растворы, в том числе растворы солей в чистой дистиллированной воде, также применимы для внутривенного введения при условии, что их pH должным образом доведен, они содержат подходящий буфер, сделаны изотоничными при помощи достаточных количеств солевого раствора или глюкозы и простерилизованы нагреванием или микрофильтрацией.
Схема приема при выполнении способа этого изобретения такова, что она обеспечивает максимальную терапевтическую ответную реакцию, пока не будет получено улучшение, и после этого минимальный эффективный уровень, при котором достигается успокаивающий эффект. В общем, пероральная доза может быть между приблизительно 0,1 мг/кг и приблизительно 100 мг/кг, предпочтительно между 0,1 мг/кг и 20 мг/кг и наиболее предпочтительно между приблизительно 1 мг/кг и 20 мг/кг, а внутривенная доза равна приблизительно 0,1 мкг/кг - 100 мкг/кг, предпочтительно между 0,1 мкг/кг и 50 мкг/кг, имея в виду, что в выборе подходящей дозы в каждом конкретном случае следует учитывать вес пациента, общее состояние здоровья, возраст и другие факторы, которые могут влиять на ответную реакцию на лекарственное средство. Лекарственное средство можно вводить перорально 1-4 раза в день, предпочтительно два раза в день.
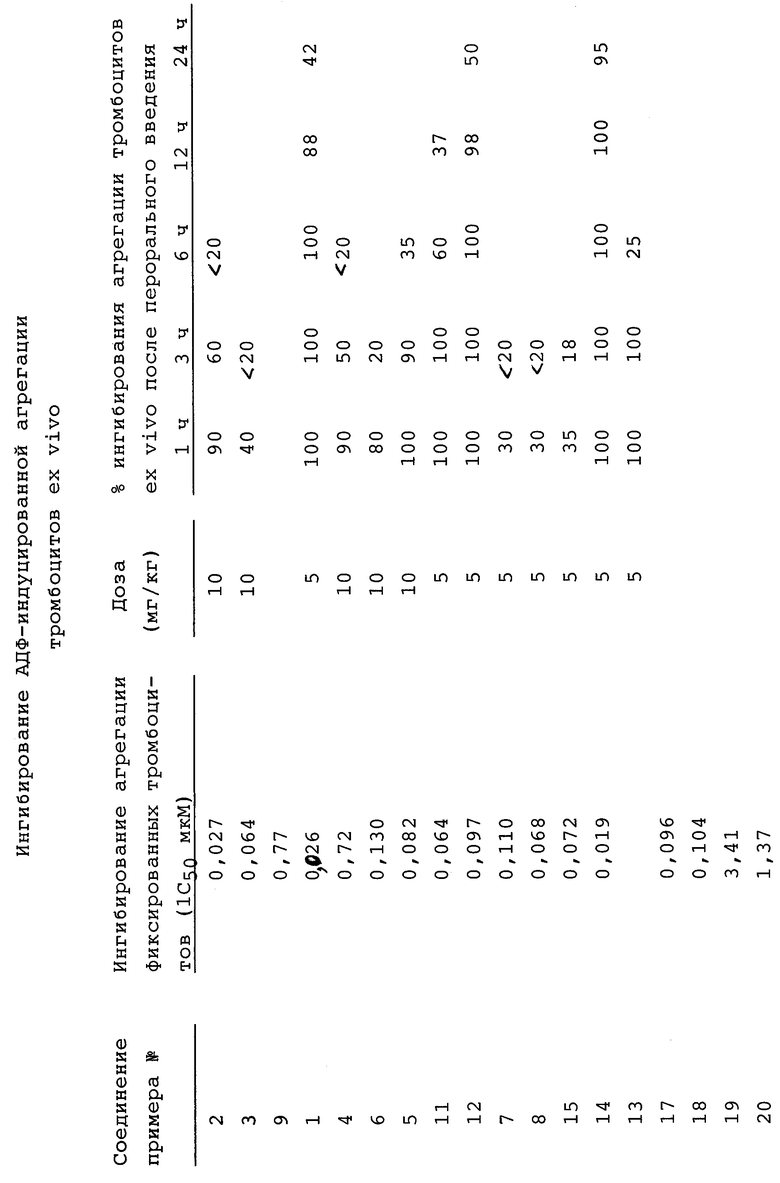
Следующие фармакологические тесты оценивают ингибиторную активность этих соединений данного изобретения в отношении медиированной фибриногеном агрегации тромбоцитов, связывания фибриногена со стимулируемыми тромбином тромбоцитами и ингибирование АДФ-индуцированной агрегации тромбоцитов ex vivo. Результаты этих тестов коррелируют с ингибиторными свойствами in vivo соединений данного изобретения.
Тест агрегации тромбоцитов основан на тесте, описанном в Blood 66(4), 946-952 (1985). Тест связывания фибриногена по существу является тестом Ruggeri, Z. M., et al., Proc. Natl. Acad. Sci. USA 83, 5708-5712 (1986) и Plow E. F. , et al., Proc. Natl. Acad. Sci. USA 82, 8057-8061 (1985). Тест ингибирования АДФ- индуцированной агрегации тромбоцитов ex vivo основан на тесте Zucker, "Platelet Aggregation Measured by the Photoelectric Method", Methods in Enzymology 169, 117-133 (1989).
Тест агрегации тромбоцитов
Получение фиксированных-активированных тромбоцитов
Тромбоциты изолировали из концентратов человеческих тромбоцитов при помощи гель-фильтрационного способа, описанного Marguerie, G.A., et al., J. Biol. Chem. 254, 5357-5363 (1979) M Ruggeri, Z.M., et al., J. Clin. Invest. 72, 1-12 (1983). Тромбоциты суспендируют при концентрации 2 • 10-8 клеток/мл в модифицированном не содержащем кальция буфере Tyrode, содержащем 137 мМ хлорид натрия, 2 мМ хлорид магния, 0,42 мМ N2HPO4, 11,9 мМ NaHCO3, 2,9 мМ KCl, 5,5 мМ глюкозу, 10 мМ HEPES, при pH 7,35 и 0,35% человеческий сывороточный альбумин (HSA). Эти промытые тромбоциты активировали добавлением человеческого α- тромбина при конечной концентрации 2 Е/мл, с последующим добавлением ингибитора тромбина I-2581 в конечной концентрации 40 мкМ. К активированным тромбоцитам добавляют параформальдегид до конечной концентрации 0,50% и инкубируют их при комнатной температуре в течение 30 минут. Фиксированные активированные тромбоциты затем собирают центрифугированием при 650 xg в течение 15 минут. Осадки тромбоцитов промывают 4 раза указанным выше буфером Tyrode с 0,35% HSA и ресуспендируют в том же буфере до концентрации 2 • 10 клеток на мл.
Анализ агрегации тромбоцитов
Фиксированные активированные тромбоциты инкубируют с выбранной дозой тестируемого соединения, испытываемого на ингибирование агрегации тромбоцитов, в течение 1 минуты и агрегацию инициируют добавлением фибриногена человека до конечной концентрации 250 мкг/мл. Для регистрации агрегации тромбоцитов использовали анализатор агрегации тромбоцитов Profiler Model РАР-4. Степень ингибирования агрегации выражали в виде процента скорости агрегации, наблюдаемой в отсутствии ингибитора. IC50, т.е. количество ингибитора, необходимое для снижения скорости агрегации на 50%, рассчитывали затем для каждого соединения (см. , например. Plow, E.F., et al., Proc. Natl. Acad. Sci. USA 82, 8057-8061 (1985).
Анализ связывания фибриногена
Тромбоциты промывают для освобождения от компонентов плазмы способом градиента плотности альбумина Walsh, P. N.et al.,Br.J. Haematol. 281-296 (1977), модифицированным Trapani-Lombardo, V., et al., J. Clin. Invest. 76, 1950-1958 (1985). В каждой экспериментальной смеси тромбоциты в модифицированном буфере Tyrode (Ruggeri,Z.M.,et al.,J.Clin.lnvest,72, 1-12 (1983) стимулируют человеческим α- тромбином при 22-25oC в течение 20 минут (3,125 • 1011 тромбоцитов на литр и тромбин при 0,1 NIH единиц/мл (NIH - Национальный институт здоровья США). Затем добавляют гирудин при 25-кратном избытке (ед./ед.) за 5 минут перед добавлением 125I-меченого фибриногена и испытуемого соединения. После этого конечное число тромбоцитов в смеси равно 1 • 1011 тромбоцитов/литр. После инкубирования в течение дополнительных 30 минут при 22-25oC связанный и свободный лиганд отделяют центрифугированием 50 мкл смеси через 300 мкл 20% сахарозы при 12000 xg в течение 4 минут. Затем осадок тромбоцитов отделяют от остальной смеси для определения связанной с тромбоцитами радиоактивности. Неспецифическое связывание измеряют в смесях, содержащих немеченный лиганд. При анализе кривых связывания с применением анализа Скетчарда неспецифическое связывание получают в виде приспособленного параметра из изотермы связывания при помощи компьютеризованной программы (Munson, P.J., Methods Enzymology, 92, 542-576 (1983)). Для определения концентрации каждого ингибиторного соединения, необходимой для ингибирования на 50% связывания фибриногена со стимулированными тромбином тромбоцитами (IC50), каждое соединение тестировали при 6 или более концентрациях с 125I-меченым фибриногеном при концентрации 0,176 мкмоль/литр (60 мкг/мл). IC50 получают путем построения кривой оставшегося связывания фибриногена в зависимости от логарифма концентрации соединения в пробе.
Ингибирование АДФ-индуцируемой агрегации тромбоцитов ex vivo
Экспериментальный протокол
Контрольные пробы крови получают за 5-10 минут перед введением тест-соединения нечистокровным собакам весом от 10 до 20 кг. Соединение вводят внутрижелудочно, через желудочный зонд, или перорально, в виде желатиновых капсул. Затем получают пробы крови (5 мл) с интервалом 30 минут в течение 3 часов и при 6, 12 и 24 часах после дозирования. Каждую пробу крови получают венепункцией головной вены и собирают непосредственно в пластиковый шприц, содержащий одну часть 3,8% цитрата натрия на девять частей крови.
Агрегация тромбоцитов собак ex vivo
Пробы крови центрифугируют при 1000 об/мин, в течение 10 минут для получения богатой тромбоцитами плазмы (PRP). После удаления PRP пробу центрифугируют еще 10 минут при 2000 об/мин, получая бедную тромбоцитами плазму (РРР). Число тромбоцитов в PRP определяют при помощи счетчика Коултера (Coulter Electronics, Hialeah,FL). Если концентрация тромбоцитов в PRP больше 300000 тромбоцитов/мкл, то PRP разбавляют РРР для доведения числа тромбоцитов до 300000 тромбоцитов на мкл. Затем аликвоты PRP (250 мкл) помещают в силиконизированные стеклянные кюветы (7,25х55 мм, Bio/Data Corp. Horsham, PA). Затем к PRP добавляют эпинефрин (конечная концентрация 1 мкМ) и проводят инкубирование в течение 1 минуты при 37oC. Затем к PRP добавляют стимулятор агрегации тромбоцитов АДФ при конечной концентрации 10 мкМ. Агрегацию тромбоцитов наблюдают спектрофотометрически с применением пропускающего свет агрегометра (Bio/Data Platelet Aggregation Profiler, Model PAP-4, Bio/Data Corp., Horsham, PA). Для испытуемого соединения скорость изменения (падения) пропускания света и максимальное пропускание света (максимум агрегации) регистрируются в двух повторностях. Данные агрегации тромбоцитов даются в виде процентного снижения (среднее ±SEM) в падении или максимальной агрегации по сравнению с данными, полученными в случае контрольных PRP, приготовленных из проб крови, взятых перед введением тест-соединения.
Соединения данного изобретения обнаруживают заметную активность в вышеописанных тестах и рассматриваются как применимые в профилактике и лечении тромбоза, связанного с некоторыми патологическими состояниями. Антитромботическая активность в тесте агрегации тромбоцитов собак ex vivo прогнозирует подобную активность в человеке (см., например, Catalfamo, J.L., and Dodds, W. Jean, "Isolation of Platelets from Laboratory Animals", Methods Enzymol. 169, Part A, 27 (1989)). Результаты тестирования соединений данного изобретения описанными выше способами представлены в таблице ниже. Также представлены в таблице результаты сравнительного теста для 4-4(пиперидил)бутаноилглициласпартилтриптофана, т.е. соединения, описанного в European Patent Application N 0479481.
Специалисту в этой области будет понятно, что данное изобретение хорошо приспособлено для выполнения целей и получения результатов и преимуществ, указанных здесь, а также и других присущих ему преимуществ. Описанные здесь соединения, композиции и способы представлены как типичные предпочтительные варианты или предназначены служить примерами, но не ограничениями объема данного изобретения. Изменения в нем и иные применения могут встретиться специалистам в этой области, причем эти изменения могут быть без отхода от сущности данного изобретения или определяться объемом прилагаемой формулы изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| КАРДИОЗАЩИТНОЕ СРЕДСТВО И ЕГО КОМПОЗИЦИЯ | 1995 |
|
RU2166319C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПЕРОКСИАЦЕТАЛЬЛАКТОНОВОГО СОЕДИНЕНИЯ (ВАРИАНТЫ) И ЛАКТОН | 1992 |
|
RU2098407C1 |
| АРИЛ-S(О)ЗАМЕЩЕННЫЕ КАРБОНОВЫЕ/ГИДРОКСАМОВЫЕ КИСЛОТЫ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1996 |
|
RU2175316C2 |
| КАРБОКСАМИДЫ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1995 |
|
RU2163232C2 |
| ПРОИЗВОДНЫЕ 3-Н-1,2,3-ТРИАЗОЛО-[4,5-D]ПИРИМИДИНА, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1996 |
|
RU2174518C2 |
| ПРОИЗВОДНЫЕ НИПЕКОТИНОВОЙ КИСЛОТЫ КАК ВЕЩЕСТВА, ПРЕПЯТСТВУЮЩИЕ ТРОМБООБРАЗОВАНИЮ | 1995 |
|
RU2135470C1 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ ТАКСАНА И ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ СИНТЕЗА | 1993 |
|
RU2118958C1 |
| АЗАЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ ДЛЯ ПРИМЕНЕНИЯ ПРИ ЛЕЧЕНИИ ОПОСРЕДОВАННЫХ СЕРОТОНИНОМ ЗАБОЛЕВАНИЙ | 2001 |
|
RU2398765C1 |
| СПИРОГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ТЕРАПЕВТИЧЕСКИХ СРЕДСТВ | 2006 |
|
RU2428424C2 |
| ДИГИДРОПИРИДОФТАЛАЗИНОНОВЫЕ ИНГИБИТОРЫ ПОЛИ(АДФ-РИБОЗА)ПОЛИМЕРАЗЫ | 2009 |
|
RU2514937C2 |

Антитромботические пептиды формулы 1, где A, R -водород, В - алкил; Z обозначает -NE-CHF-(CO)r-G; Е -водород; F - Н, алкил, гидроксиалкил, циклоалкил, циклоалкилалкил, аралкил или замещенный аралкил; G -циклоалкилалкил, OR1 или NR1R2, R1, R2 - Н или алкил, r = 0 или 1, m = 1-5, n = 0-6, р = 1-4, или их фармацевтически приемлемые соли, применимы для профилактики и лечения тромбоза. 4 с. и 9 з.п. ф-лы, 1 табл.
в которой A обозначает - H;
B обозначает алкил;
Z обозначает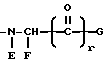
где E обозначает - H;
F обозначает - H, алкил, гидроксиалкил, циклоалкил, циклоалкилалкил, аралкил или замещенный аралкил;
G обозначает циклоалкилалкил, OR1 или NR1R2, где R1 и R2 независимо обозначают - H, алкил, и r обозначает 0 или 1,
R обозначает - H;
m = 1 - 5;
n = 0 - 6;
p = 1 - 4,
или его фармацевтически приемлемая соль.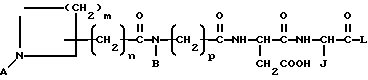
в которой A обозначает - H;
B обозначает алкил;
J обозначает - H, алкил, циклоалкил, циклоалкилалкил, аралкил, замещенный аралкил;
L обозначает OR1 или NR1R2, где R1 и R2 независимо обозначают - H, алкил,
m = 1 - 5;
n = 2 - 6;
p = 1 или 2,
или его фармацевтически приемлемая соль.
N-[N-[N-(4-пиперидин-4-ил)бутаноил)-N-этилглицил]аспартил]-D-валин,
N-[N-[N-(3-(пиперидин-4-ил)пропаноил)-N-этилглицил]аспартил]валин,
N-[N-[N-(5-пиперидин-4-ил)пентаноил)-N-этилглицил]аспартил]валин,
N-[N-[N-(4-(пиперидин-4-ил)бутаноил)-N-этилглицил]аспартил] -L-α-циклогексилглицин,
N-[N-[N-(4-(пиперидин-4-ил)бутаноил)-N-этилглицил]аспартил] -L-α-циклогексилаланин,
N-[N-[N-(пиперидин-4-ил)бутаноил)-N-этилглицил]аспартил]норлейцин,
N-[N-[N-(4-(пиперидин-4-ил)бутаноил)-N-этилглицил]аспартил]-L-α-(2,2-диметил)проп-3-илглицин,
N-[N-[N-(4-(пиперидин-4-ил)бутаноил)-N-этилглицил] аспартил]-L-β-цис-декагидронфат-2-илаланин,
N-[N-[N-(4-(пиперидин-4-ил)бутаноил)-N-этилглицил]аспартил] -α-аминоциклогексанкарбоновая кислота,
N-[N-[N-(4-(пиперидин-4-ил)бутаноил)-N-этилглицил]аспартил] -β-циклогексил-D-аланин,
N-[N-[N-(4-(пиперидин-4-ил)бутаноил)-N-этилглицил]аспартил] -β-декагидронафт-1-илаланин,
N-[N-[N-(4-(пиперидин-4-ил)бутаноил)-N-этилглицил]аспартил] -β-циклооктилаланин,
N-[N-[N-(4-(пиперидин-4-ил)бутаноил)-N-этилглицил] аспартил] -β-адамант-1-илаланин,
N-[N-[N-(4-(пиперидин-4-ил)бутаноил)-N-этилглицил] аспартил]-β-(4-циклогексил)циклогексилаланин,
N-[N-[N-(4-(пиперидин-4-ил)бутаноил)-N-этилглицил]аспартил] -β-циклогептилаланин,
N-[N-[N-(4-(пиперидин-4-ил)бутаноил)-N-этилглицил]аспартил] -α-циклогексилпропилглицин,
N-[N-[N-(4-(пиперидин-4-ил)бутаноил)-N-этилглицил]аспартил] -β-циклооктилметилаланин,
N-[N-[N-(4-(пиперидин-4-ил)бутаноил)-N-этилглицил]аспартил] -β-циклопентилаланин,
N-[N-[N-(4-(пиперидин-4-ил)бутаноил)-N-этилглицил]аспартил] -L-β-декагидронафт-1-илаланин или
N-[N-[N-(4-(пиперидин-4-ил)бутаноил)-N-этилглицил] аспартил]-L-α-(2-циклогексилэтил)глицин,
N-[N-[N-(4-(пиперидин-4-ил)бутаноил)-N-этилглицил]аспартил]-фенилаланин,
N-[N-[N-(4-(пиперидин-4-ил)бутаноил)-N-этилглицил]аспартил]-β-(1,2,3,4)-тетрагидронафт-5-илаланин,
N-[N-[N-(4-(пиперидин-4-ил)бутаноил)-N-этилглицил]аспартил]-L-β-нафт-1-илаланин или
N-[N-[N-(4-(пиперидин-4-ил)бутаноил)-N-этилглицил]аспартил]-L-β-нафт-2-илаланин,
N-[N-[N-(4-(пиперидин-4-ил)бутаноил)-N-этилглицил]аспартил] -L-β-циклогексилаланинамид,
N-[N-[N-(4-(пиперидин-4-ил)бутаноил)-N-этилглицил]аспартил] -β-циклооктилаланинамид,
N-[N-[N-(4-(пиперидин-4-ил)бутаноил)-N-этилглицил]аспартил] -β-циклогексилметилаланинамид, или
N-[N-[N-(4-(пиперидин-4-ил)бутаноил)-N-этилглицил]аспартил] -β-циклогексилаланинэтиламид,
этиловый эфир N-[N-[N-(4-(пиперидин-4-ил)бутаноил)-N-этилглициласпартил-L-β-циклогексилаланина,
этиловый эфир N-[N-[N-(4-(пиперидин-4-ил)бутаноил)-N-этилглицил]аспартил]-β-циклогексилметилаланина или
3-адамант-1-илпропил-N-[N-(4-пиперидин-4-ил)бутаноил)-N-этилглицил] аспартат,
2-циклогексил-N-[N-[N-(4-пиперидин-4-ил)бутаноил)-N-этилглицил]аспартил] этиламин или
N-[N-[N-(4-(пиперидин-4-ил)бутаноил)-N-этилглицил]аспартил] -α-циклогексилметилэтаноламин,
или их фармацевтически приемлемые соли.
| Преобразователь движения для приборов времени | 1974 |
|
SU503203A2 |
| Преобразователь переменного тока | 1974 |
|
SU530167A1 |
| US 5064814 A1, 12.11.91 | |||
| ПРОИЗВОДНЫЕ ГЛИЦИНА ИЛИ ИХ ФИЗИОЛОГИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ, ОБЛАДАЮЩИЕ СПОСОБНОСТЬЮ ТОРМОЗИТЬ СВЯЗЫВАНИЕ ФИБРИНОГЕНА У ФИБРИНОГЕННОГО РЕЦЕПТОРА ТРОМБОЦИТОВ | 1990 |
|
RU2024549C1 |

