Предпосылки к созданию изобретения.
Настоящая заявка является продолжением заявки N 08/364896, поданной 27 декабря 1994 г. , которая в свою очередь является продолжением заявки N 08/213772, поданной 16 марта 1994 г.
Агрегация тромбоцитов представляет собой начало гемостатической реакции, направленной на прекращение кровотечения, вызванного повреждением сосуда. Однако патологическое продление этого нормального гемостатического процесса может привести к образованию тромба.
Обычно завершающей стадией агрегации тромбоцитов является связывание фибриногена с активированным незащищенным (открытым) ГПIIb/IIa тромбоцитов. Агенты, которые препятствуют связыванию фибриногена с гликопротеином тромбоцитов IIa/IIIa (ГПIIb/IIIa), следовательно, ингибируют агрегацию тромбоцитов. Таким образом, эти агенты применимы для лечения нарушений свертывания крови, опосредованных тромбоцитами, таких как тромбоз артерий и вен, острый инфаркт миокарда, нестабильная стенокардия, реокклюзия после тромболитической терапии и ангиопластики, воспаление и ряд вазоокклюзивных нарушений. Рецепторы фибриногена (ГПIIb/IIIa) активируются соответствующим стимулом, таким как АДФ, коллаген и тромбин, обнажая связывающие домены для двух различных пептидных участков фибриногена: α- цепь Арг-Гли-Асп (RGD) и γ- цепь Гис-Гис- Лей-Гли-Гли-Ала-Лиз-Глн-Ала-Гли-Асп-Вал (HHLGG AKQ AGDY, γ 400-411). Поскольку показано, что эти пептидные фрагменты вне фибриногена являются антагонистами (ингибиторами) связывания фибриногена с ГПIIb/IIIa, миметики этих фрагментов также являются такими антагонистами. На самом деле, до появления настоящего изобретения были открыты мощные антагонисты, являющиеся RGD- миметиками или миметиками на основе RGD, которые ингибируют связывание фибриногена с ГПIIb/IIIa и агрегацию тромбоцитов.
Некоторые их этих агентов показали также активность in vivo в качестве противосвертывающих агентов и в некоторых случаях применялись в комбинации с фибринолитической терапией (например, t-PA или стрептокиназой).
Описание изобретения.
Настоящее изобретение относится к соединениям, представленным общей формулой (I):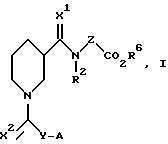
где X1, X2, Y, Z, R2 и A определены далее в тексте.
Такие соединения, основанные на структуре фибриногена 400-411, являются ингибиторами агрегации тромбоцитов, полезными для лечения нарушений свертывания крови, опосредованных тромбоцитами, таких как тромбоз артерий и вен, острый инфаркт миокарда, реокклюзия после тромболитической терапии и ангиопластики, воспаление и нестабильная стенокардия, а также ряд вазоокклюзивных нарушений. Эти соединения полезны также в качестве антитромботических агентов при использовании в сочетании с фибринолитической терапией (например, t-PA или стрептокиназой). Фармацевтические композиции, содержащие такие соединения, также являются частью настоящего изобретения.
Подробное описание изобретения.
Настоящее изобретение относится к соединениям следующей формулы (I):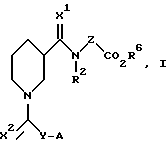
где X1 и X2 одинаковы или различаются и представляют H2 или О. Предпочтительно, каждый из X1 и X2 представляет О;
Y представляет (CH2)m, CH(NHCOR3)(CH2)m или CH(NH2)(CH2)m;
A представляет NHR1 С(NH)NH2 или циклоалкильное кольцо, содержащее азот и выбранное из группы: пиперидин-2-ил, пиперидин-3- ил, пиперидин-4-ил, пирролидин-2-ил и пирролидин-3-ил. Более предпочтительно, кольцо выбирают из группы: пиперидин-2-ил, пиперидин-3-ил и пиперидин-4-ил;
Z представляет (CH2)n или CH(CO2R4)(CH2)n. Предпочтительно Z представляет (CH2)2;
R1 представляет H, алкил или CH(NH)NH2. Более предпочтительно R1 представляет H или алкил. Наиболее предпочтительно R1 представляет водород;
R2 представляет H или алкил. Предпочтительно R2 представляет водород;
R3представляет алкокси или алкил. Предпочтительно R3 представляет т-бутокси или метил. Наиболее предпочтительно R3 представляет т-бутокси;
R4 - представляет алкил или арилалкил, такой как бензил. Предпочтительно R4 представляет метил;
R6 представляет H, алкил или арилалкил, такой как бензил. Когда R6 не является водородом, он находится в форме пролекарства;
m представляет целое число 0, 1, 2 или 3;
n представляет целое число 0, 1 или 2.
Если не указано иное, алкил или алкокси, как самостоятельно, так и в составе замещающей группы, включают прямые и разветвленные цепи, состоящие из 1-8 атомов углерода. Например, алкильные радикалы включают метил, этил, пропил, изопропил, н-бутил, втор-бутил, т-бутил, н-пентил, 3-(2-метил) бутил, 2-пентил, 2-метилбутил, неопентил, н-гексил, 2-гексил и 2-метилпентил. Алкокси радикалы представляют кислородные эфиры, образованные ранее описанными прямыми или разветвленными цепями алкильных групп. Циклоалкильные группы содержат в кольце 5-8 атомов углерода, предпочтительно 6-7 атомов углерода.
Термин "арил", применяемый здесь самостоятельно или в комбинации с другими терминами, обозначает ароматические углеводородные группы, такие как фенил или нафтил.
Термин "арилалкил" означает алкильную группу, замещенную арильной группой.
Соединения настоящего изобретения также могут быть представлены в форме фармацевтически приемлемой соли. Фармацевтически приемлемая соль обычно принимает форму, в которой азот в 1-пиперидине протонирован неорганической или органической кислотой. Однако в случае, если X2 представляет H2, азот кольца также может образовывать соль. Примеры органических или неорганических кислот включают хлористоводородную, бромистоводородную, иодистоводородную, хлорную, серную, азотную, фосфорную, уксусную, пропионовую, гликолевую, молочную, янтарную, малеиновую, фумаровую, яблочную, винную, лимонную, бензойную, миндальную, метансульфоновую, гидроксиэтансульфоновую, бензолсульфоновую, щавелевую, памоевую, 2-нафталин-сульфоновую, п-толуолсульфоновую, циклогексансульфаминовую, салициловую, сахарную или трифторуксусную кислоты.
Особенно предпочтительные соединения настоящего изобретения включают соединения, представленные формулой: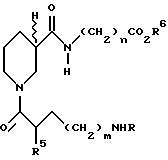
где R= H; m=3; n=2; R5=L-NHБок; R6 представляет бензил (Бн) (соединение 1);
R=H; m=3; n=2; R5=L-NHБок; R6 представляет H (соединение 2);
R=H; m=3; n=2; R5=D-NHБок; R6 представляет H (соединение 3);
R=H; m=3; n=2; R5=L-NH2; R6 представляет H (соединение 4);
R=H; m=3; n=2; R5=H; R6 представляет H (соединение 5);
R=H; m=3; n=1; R5=L-NHАц; R6 представляет H (соединение 6);
R=H; m=3; n=2; R5=L-NHАц; R6 представляет H (соединение 7);
R=C(NH)NH2 ; m=2; n=2; R5=L-NHБок R6 представляет H (соединение 8);
R=H; m=3; n=3; R5=L-NHБок; R6 представляет H (соединение 9);
R=H; m=3; n=2; R5=D-NH2; R6 представляет H (соединение 10);
R=H; m=3; n=3; R5=D-NHБок; R6 представляет H (соединение 11);
R=H; m=3; n=1; R5=D-NHБок; R6 представляет H (соединение 12);
R=H; m=3; n=2; R5=D-NHАц; R6 представляет H (соединение 13);
3-S-изомер соединения 3 R6 представляет H (соединение 14);
R-изопропил, m=3; n=2; x=L-NHБок; R6 представляет H (соединение 15);
3-R-изомер соединения 3; R6 представляет H (соединение 16);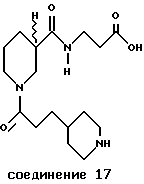
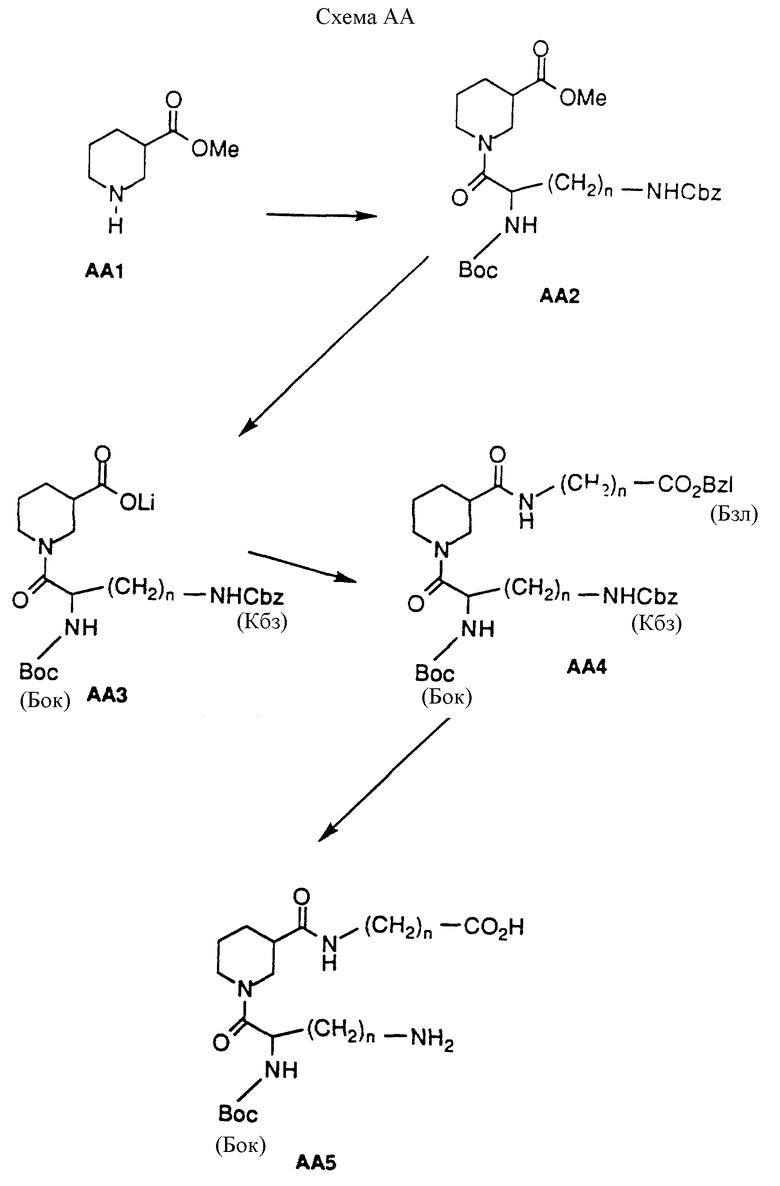
Соединения настоящего изобретения можно приготовить из коммерческих исходных материалов, согласно следующим схемам реакций AA, AB, АС и AD.
Соединения настоящего изобретения, в которых X1 и X2 каждый представляет кислород, можно приготовить согласно следующей схеме AA. В этой схеме на нипекотиновую кислоту (рацемическую смесь или отдельный энантиомер) можно действовать низшим алкиловым спиртом и каталитическим количеством кислоты, от комнатной температуры до нагревания с вертикальным холодильником, для получения эфирного производного AA1 в форме кислой соли. Типичные спирты включают этанол, метанол, изопропанол и бутанол и могут объединяться с кислотными катализаторами, такими как п-толуолсульфоновая кислота, HCl или серная кислота. Предпочтительными реагентами являются метанол и HCl. Производное AA1 может быть ацилировано по азоту кольца различными ацилирующими агентами для получения производного AA2. Типичные условия реакции включают действие на AA1 ацилирующего агента и эквивалентного количества органического основания в инертном растворителе при комнатной температуре в течение от 15 минут до 2 часов. Предпочтительными ацилирующими агентами являются аминозащищенные аминокислоты или аминозащищенные аминоалкилкарбоновые кислоты, активированные сопрягающими реагентами, такими как ДЦК (1,3-дициклогексилкарбодиимид) и БОФ-С1 (бис(2-оксо-3-оксазолидинил)фосфин хлорид). Однако можно использовать и аминозамещенные производные кислот, такие как ангидриды, N-оксисукцинимиды и кислые хлориды. Удобные защитные группы включают низшие алкилкарбаматы, разветвленные алкилкарбаматы, бензилкарбаматы, ацетамиды и замещенные ацетамиды. Выбор ацилирующего агента и его аминозащищающей группы (групп) является фактором, определяющим заместители Y и R1 - в соединениях формулы I, где X1 и X2 представляют О. В схеме AA защищенная аминокислота является диаминокислотой формулы NH(Бок)CHCO2H(CH2)n-N(Кбз), что делает возможным избирательную депротекцию двух аминогрупп на последнем этапе схемы. Этот выбор служит только целям иллюстрации и не является ограничительным.
На производное AA2 можно действовать основанием и подходящей смесью растворителей для получения солевого производного AA3. Подходящие неорганические основания включают NaOH, KOH, Mg(OH)2, LiOH, Na2CO3 и NaHCO3, которые можно комбинировать с ТГФ и водой при комнатной температуре в течение 1-6 часов для получения желаемого продукта. Органические основания, которые можно использовать, включают триэтиламин, трибутиламин, диизопропилэтиламин и тетраметилгуанидин. Эти основания можно использовать с органическими растворителями при комнатной температуре и до нагревания с вертикальным холодильником в течение 1-6 часов для получения соли AA3.
Предпочтительными условиями реакции (которые проиллюстрированы) являются действие на AA2 LiOH, водой и ТГФ при комнатной температуре в течение 1 часа. Можно использовать другие подходящие неорганические основания, такие как NaOH, KON, Mg(OH)2, Na2CO3 и NaHCO2. Если используется такое другое основание, то Li в AA3 будет, разумеется, заменен на другой соответствующий металл. На производное AA3 можно подействовать карбоксизащищенным карбоксиалкиламином или карбоксизащищенной аминокислотой при стандартных для аминокислот условиях сопряжения, для получения двузамещенного нипекотинового производного AA4. Подходящие условия сопряжения включают применение пептидных сопрягающих агентов, таких как ДЦК, БОФ-С1 и ЭДК (этилдиметиламино- пропилкарбодиимид•HCl). Подходящие карбоксизащищающие группы включают бензилкарбаматы, замещенные бензилкарбаматы, алкилкарбаматы и разветвленные алкилкарбаматы, а выбор защищающей группы очевиден для специалиста в области химического синтеза. Проиллюстрированный пример использует NH2(CH2)n CO2-(Бзл) в качестве защищенной аминокислоты. И снова выбор аминокислоты и ее карбоксизащищающей группы определяет заместители R2 и Z в соединениях формулы I, где X1 и X2 представляют О. Производное AA4 может быть избирательно лишено защиты в соответствии с требованиями амино- или карбоксизащищающей группы. В проиллюстрированном примере защищающие группы на 3-карбокси-группе и одной из аминогрупп одновременно удаляются путем каталитической гидрогенизации с помощью Pd/C в атмосфере H2 для получения производного AA5.
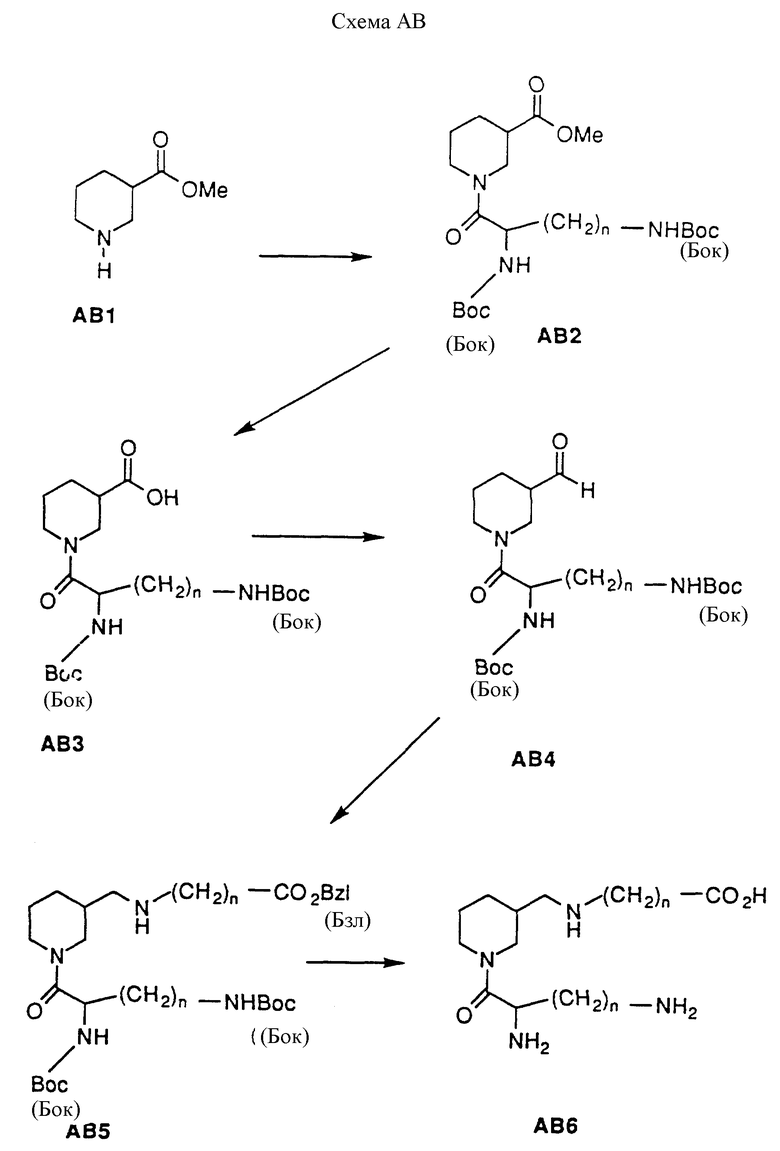
Схема AB иллюстрирует приготовление соединений формулы 1, в которых X1 представляет O, а X1 представляет H2. На нипекотиновую кислоту (рацемическую смесь или отдельный энантиомер) можно действовать алкиловым спиртом и каталитическим количеством кислоты при температуре от комнатной до нагревания с вертикальным холодильником для получения эфирного производного AB1 в форме кислой соли. Типичные спирты включают этанол, метанол, изопропанол и бутанол. Кислотные катализаторы включают п-толуолсульфоновую кислоту, HCl и серную кислоту; предпочтительные реагенты - метанол и HCl. Производное AB1 можно ацилировать по азоту кольца различными ацилирующими агентами для получения производного AB2. Типичные условия реакции включают действие на AB1 ацилирующим агентом и эквивалентным количеством органического основания в инертном растворителе при комнатной температуре в течение от 15 минут до 2 часов. Предпочтительными ацилирующими агентами являются амино защищенные аминокислоты или аминозащищенные аминоалкилкарбоновые кислоты, активированные сопрягающими агентами, такими как ДЦК (1,3- дициклогексилкарбодиимид) и БОФ-С1 (бис(2-оксо-3- оксазолидинил)фосфин хлорид). Также можно применять аминозащищенные производные кислот, такие как ангидриды, N-оксисукцинимиды и кислые хлориды. Подходящие защищающие группы включают низшие алкилкарбаматы, разветвленные алкилкарбаматы, бензилкарбаматы, ацетамиды и замещенные ацетамиды. Выбор аминокислоты и ее аминозащищающей группы (групп) является фактором, определяющим заместители Y и R1 в соединениях формулы I. В схеме AB защищенная аминокислота является диаминокислотой формулы: NH(Бок)CHCO2H(CH2)N-N(Бок); этот выбор служит только целям иллюстрации настоящего изобретения и не ограничивает его. Производной AB2 можно гидролизовать основанием и подходящей смесью растворителей для получения производного AB3. Подходящие неорганические основания включают NaOH, KON, Mg(OH)2, LiOH, Na2CO3 и NaHCO3, которые можно комбинировать со смесями ТГФ и воды при комнатной температуре в течение 1-6 часов для получения желаемого продукта. Органические основания, которые могут быть использованы, включают триэтиламин, трибутиламин, диизопропилэтиламин и тетраметилгуанидин. Эти основания можно использовать с органическими растворителями при комнатной температуре и до нагревания с вертикальным холодильником в течение 1-6 часов для получения AB3. 3-карбоксигруппу производного AB3 можно восстановить для получения альдегидного производного AB4, используя ряд условий реакции. Эти условия включают использование литий-т-диизопропиламида с ГМПТ/ТГФ в качестве растворителя, от -78o до 0oC, N,N-диметилхлорметилениминийхлорида и гидрида литий-т- бутоксиалюминия с пиридином в качестве растворителя при -78oC и стандартных условиях восстановления Розенмунда. Предпочтительные условия реакции - применение N, N'-карбонилдиимидазола, затем гидрида диизобутилалюминия при -10oC, для получения альдегидного производного AB4. На AB4 можно подействовать карбоксизащищенным карбоксиалкиламином или карбоксизащищенной аминокислотой, а затем восстановителем для получения двузамещенного нипекотинового производного AB5. Подходящие карбоксизащищающие группы включают бензилкарбаматы, замещенные бензилкарбаматы, низшие алкилкарбаматы и разветвленные алкилкарбаматы; выбор защищающей группы является очевидным для специалиста в области химического синтеза. Восстановители включают цианоборогидрид натрия, цианоборогидрид лития, натрий-9-циан-9-гидрид-боробицикло [3,3,1] нонан, тетрабутиламмония цианоборогидрид и Pd/С с кислотным растворителем; выбор восстановителя определяется используемыми защитными группами. Проиллюстрированный пример использует NH2(CH2)nCO2(Бзл) в качестве защищенной аминокислоты и цианоборогидрид натрия в качестве восстановителя. Этот выбор аминокислоты и ее карбоксизащищающей группы определяет заместители R2 и Z в соединении и служит целям иллюстрации, но не ограничения. Производное AB5 может быть избирательно лишено защиты в соответствии с требованиями амино- или карбоксизащищающей группы. В проиллюстрированном примере защищающие группы на 3-карбоксильной группе и обеих аминогруппах одновременно удаляются путем каталитической гидрогенизации в присутствии Pd/C в атмосфере H2 для получения производного AB6.
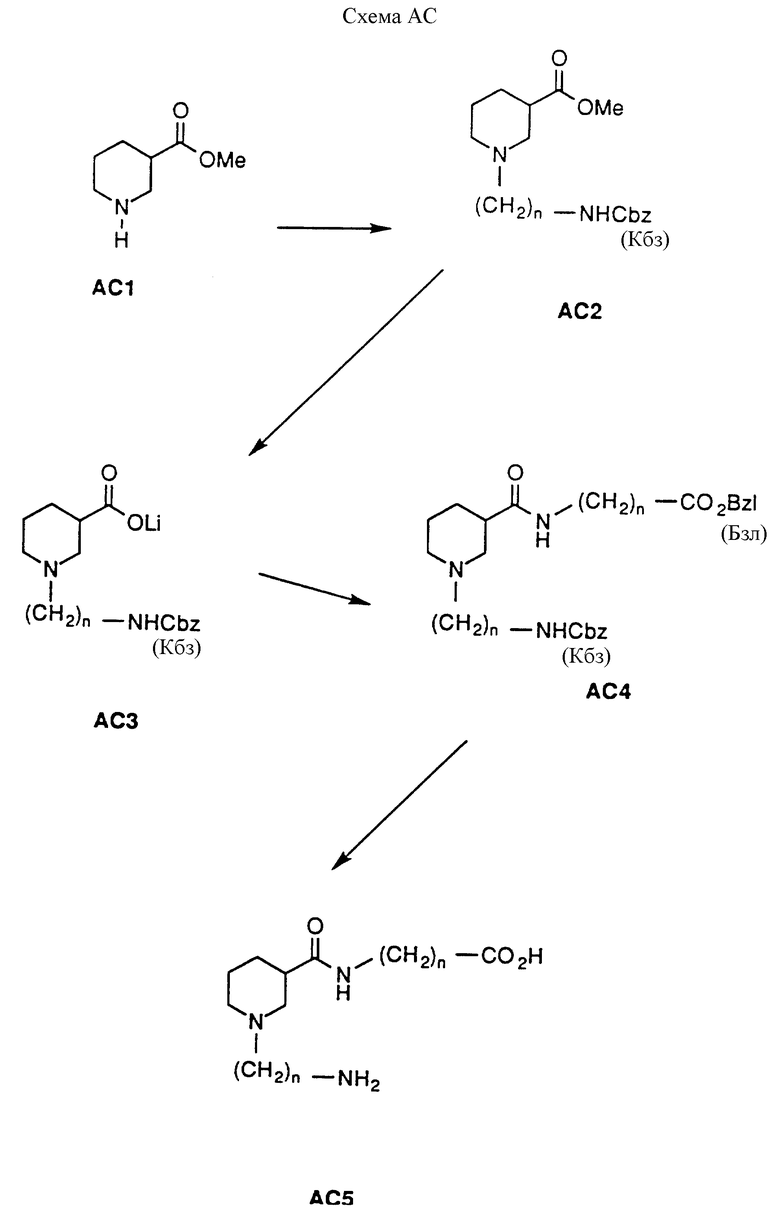
Соединения настоящего изобретения, в которых X1 представляет кислород, а X2 представляет H2, можно приготовить согласно следующей схеме AC. В этой схеме на нипекотиновую кислоту (рацемическую смесь или отдельные энантиомеры) можно подействовать низшим алкиловым спиртом и каталитическим количеством кислоты, от комнатной температуры до нагревания с вертикальным холодильником, для получения сложноэфирного производного AC1 в форме кислой соли. Типичные спирты включают этанол, метанол, изопропанол и бутанол. Кислотные катализаторы включают п-толуолсульфоновую кислоту, HCl и серную кислоту; реагентами выбора являются метанол и HCl. Производное AC1 можно алкилировать по азоту кольца алкилирующим агентом для получения производного AC2. Алкилирующие реагенты включают синтоны галоалкиламинов, такие как бромалкилфталимиды и бромалкилнитрилы, или защищенные аминоальдегиды, посредством процесса восстановительного аминирования (условия смотри в схеме AD). Типичные условия реакции включают действие на AC1 основанием, таким как гидрид натрия, или катализатором переноса между фазами, таким как тетрабутиламмония фторид, и алкилирующим агентом в инертном растворителе, при комнатной температуре в течение от 15 минут до 2 часов, а затем рутинную защиту аминогруппы 3-заместителя любой из упомянутых подходящих защищающих групп. Выбор алкилирующего агента и его аминозащищающей группы является фактором, определяющим заместители Y и R1. В схеме AC 1 положение замещается (CH2)NH(Кбз); этот выбор лишь иллюстрирует настоящее изобретение, но не ограничивает его. На производное AC2 можно подействовать основанием и подходящей смесью растворителей для получения производного AC3 в виде соли. Как и схема AA, схема AC показывает использование предпочтительного LiOH. Однако можно использовать и другие подходящие неорганические основания, включающие NaOH, KOH, Mg(OH)2, Na2CO3 и NaHCO3, которые можно объединять со смесями ТГФ и воды при комнатной температуре в течение 1-6 часов для получения желаемого продукта. Органические основания, которые можно использовать, включают триэтиламин, трибутиламин, диизопропилэтиламин и тетраметилгуанидин. Эти основания можно использовать с органическими растворителями при комнатной температуре и до нагревания с вертикальным (обратным) холодильником, в течение 1-6 часов, для получения соли AC3. Предпочтительные условия реакции (проиллюстрированные) предусматривают действие на AC2 LiOH, водой и ТГФ при комнатной температуре в течение 1 часа. На производное AC3 можно подействовать карбоксизащищенным карбоксиалкиламином или карбоксизащищенной аминокислотой при стандартных условиях сочетания для аминокислот для получения двузамещенного нипекотинового производного AC4. Приемлемые условия сопряжения включают применение пептидных сопрягающих агентов, таких как ДЦК, БОФ-С1 и ЭДК (этилдиметиламинопропил•HCl). Подходящие карбоксизащищающие группы включают бензилкарбаматы, замещенные бензилкарбаматы, алкилкарбаматы и разветвленные алкилкарбаматы; выбор защищающей группы является очевидным для специалиста в области химического синтеза. Проиллюстрированный пример использует NH2(CH2)n CO2(Бзл) в качестве защищенной аминокислоты. И вновь выбор аминокислоты и ее карбоксизащищающей группы определяет заместители R2 и Z в соединениях формулы I. Производное АС4 может быть избирательно лишено защиты в соответствии с требованиями амино- или карбоксизащищающей группы. В проиллюстрированном примере защищающие группы на 3-карбоксильной группе и 1-аминогруппе удаляются одновременно посредством каталитической гидрогенизации с применением Pd/C в атмосфере H2 для получения производного АС5.
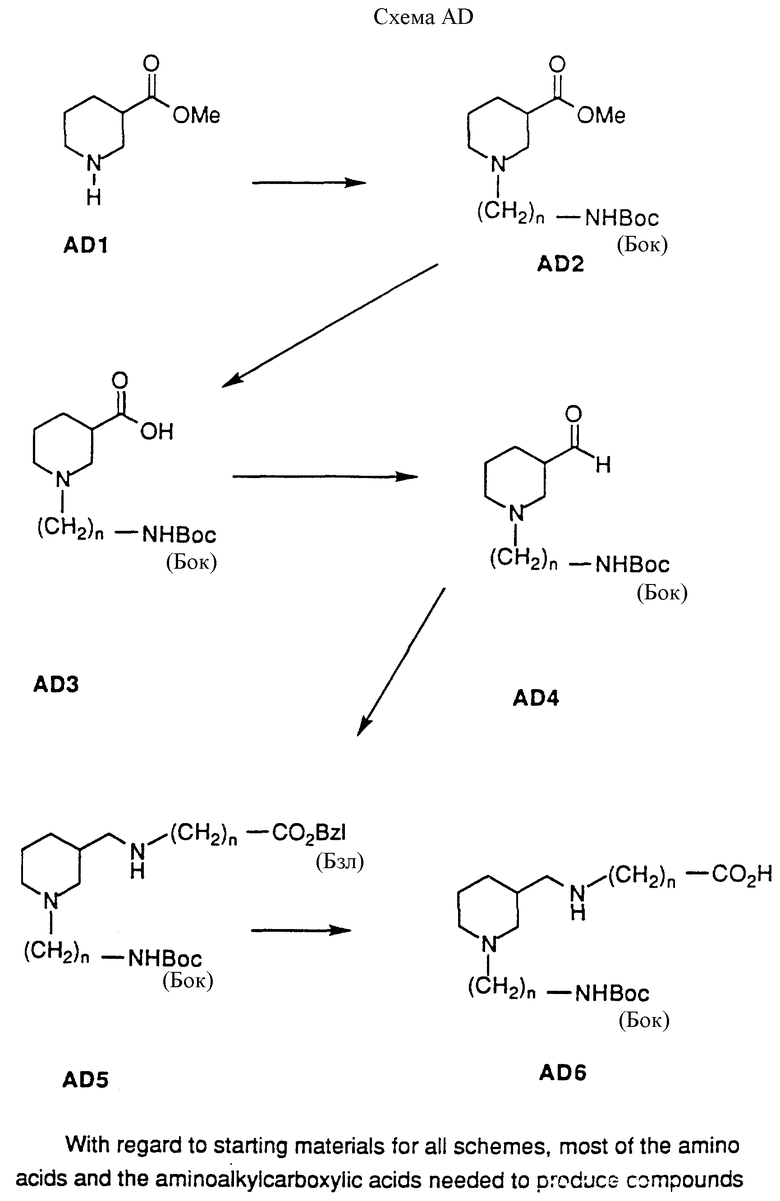
Соединения настоящего изобретения, в которых X1 и X2 каждый представляет H2, можно приготовить согласно следующей схеме AD. В этой схеме на нипекотиновую кислоту (рацемическую смесь или отдельные энантиомеры) можно подействовать низшим алкиловым спиртом и каталитическим количеством кислоты от комнатной температуры до нагревания с вертикальным холодильником для получения сложноэфирного производного AD1 в форме кислой соли. Типичные спирты включают этанол, метанол, изопропанол и бутанол. Кислотные катализаторы включают п-толуолсульфоновую кислоту, HCl и серную кислоту. Предпочтительными реагентами являются метанол и HCl. Производное AD1 можно алкилировать по азоту кольца алкилирующим агентом для получения производного AD2. Алкилирующие реагенты включают синтоны галоалкиламинов, такие как бромалкилфталимиды и бромалкилнитрилы или защищенные аминоальдегиды, посредством процесса восстановительного аминирования (условия смотри в схеме AD). Типичные условия реакции включают действие на AD1 основанием, таким как гидрид натрия, или катализатором переноса между фазами, таким как тетрабутиламмония фторид, и алкилирующим агентом в инертном растворителе, при комнатной температуре в течение от 15 минут до 2 часов, а затем рутинную защиту аминогруппы из вышеупомянутых подходящих защищающих групп. Выбор алкилирующего агента и его амино-защищающей группы является фактором, определяющим заместители Y и R1. В схеме AD1 положение замещено (CH2)NH(Кбз); этот выбор служит только целям иллюстрации настоящего изобретения. Производное AD2 можно гидролизовать основанием и подходящей смесью растворителей для получения производного AD3. Подходящие неорганические основания включают NaOH, KOH, Mg(OH)2, LiOH, Na2CO3 и NaHCO3, которые можно комбинировать со смесями ТГФ и воды при комнатной температуре в течение 1-6 часов для получения желаемого продукта. Органические основания, которые можно использовать, включают триэтиламин, трибутиламин, диизопропилэтиламин и тетраметилгуанидин. Эти основания можно использовать с органическими растворителями при комнатной температуре и до нагревания с вертикальным холодильником в течение 1-6 часов для получения AD3. 3-карбоксильную группу производного AD3 можно восстановить для получения альдегидного производного AD4 посредством использования ряда условий реакции. Эти условия включают применение литий-диизопропиламида с ГМПТ/ТГФ в качестве растворителя, при температуре от -78oC до 0oC, N,N-диметилхлорметилениминий хлорида и гидрида литий-т-бутоксиалюминия с пиридином в качестве растворителя при температуре -78oC и стандартных условиях восстановления Розенмунда. Предпочтительными условиями реакции являются применение N,N'-карбонилдиимидазола, а затем применение гидрида диизобутилалюминия при температуре -10oC для получения альдегидного производного AD4.
На производное AD4 можно подействовать карбоксизащищенным карбоксиалкиламином или карбоксизащищенной аминокислотой, а затем восстановителем для получения двузамещенного нипекотинового производного AD5. Подходящие карбоксизащищающие группы включают бензилкарбаматы, замещенные бензилкарбаматы, низшие алкилкарбаматы и разветвленные алкилкарбаматы; выбор защищающей группы является очевидным для специалиста в области химического синтеза. Восстановители включают цианоборогидрид натрия, цианоборогидрид лития, натрий-9-циан-9-гидрид- боробицикло[3,3,1]нонан, тетрабутиламмония цианоборогидрид и Pd/C в кислотном растворителе; выбор восстановителя определяется используемыми защищающими группами. Проиллюстрированный пример использует NH2(CH2)nCO2(Бзл) в качестве защищенной аминокислоты и цианоборогидрид натрия в качестве восстановителя. Этот выбор аминокислоты и ее карбоксизащищающей группы определяет заместители R2 и Z в соединении и служит целям иллюстрации, но не ограничения. Производное AD5 может быть избирательно лишено защиты в соответствии с требованиями амино- или карбоксизащищающей группы. В проиллюстрированном примере защищающие группы на 3-карбоксильной группе и аминогруппе удаляются одновременно путем каталитической гидрогенизации с применением Pd/C в атмосфере H2 для получения производного AD6.
Исходные материалы для всех схем, большинство аминокислот и аминоалкилкарбоновых кислот, необходимых для получения соединений, в которых А представляет NHR1, являются коммерчески доступными и требуют лишь введения защитных групп для получения желаемых соединений формулы I. Однако для получения соединений настоящего изобретения, в которых А представляет циклоалкильное кольцо, содержащее атом азота, заместитель в положении 1 (пиперидин) должен быть модифицирован после добавления для получения желаемых соединений формулы I. Для получения соединений, в которых заместитель в 1 положении представляет C(O)(CH2)2-4-ил-пиперидин, производные AA1 или AB1 ацилируют 3-(4-пиридил)акриловой кислотой для получения ацилированных производных AA2 и AB2, используя вышеупомянутые процессы ацилирования. Эти производные подвергаются преобразованиям, как описано в схемах, для получения AA4 и AB5. Производные AA5 и AB6 можно получить действием на AA4 и AB5 подходящим восстановителем, который в этом случае удаляет защищающую группу на карбоксильной группе в 3 положении и восстанавливает этилензамещенный пиридин для получения желаемого соединения. Предпочтительным восстановителем, удаляющим защиту, является PtO2. 2- и 3-ил-пиперидины можно получить путем модификации производного акриловой кислоты с помощью стандартной методики.
Для получения соединений, в которых заместитель в 1 положении представляет C(O)(CH2)2-3-ил-пиррол, производные AA1 или AB1 ацилируют 3-(1-бензилпирролидин-3-ил) акриловой кислотой для получения ацилированных производных AA2 и AB2 с помощью вышеупомянутых процессов ацилирования. Это замещенное производное пиррола и акриловой кислоты можно получить посредством гидролиза соответствующего нитрильного производного водным раствором кислоты, 3-(1-бензилпирролидин-3-ил)акрилонитрил был синтезирован согласно способам, описанным в патенте США N 4002643, включенном в настоящий документ в качестве ссылки. На эти производные воздействовали, как описано выше (для шестичленного случая), для получения соединений настоящего изобретения, в которых А представляет пятичленное кольцо, содержащее атом азота.
Для получения диастереометрически обогащенных конечных соединений, содержащих Бок-D-Лиз и одну из R- или S-нипекотильных групп (смотри соединения 14 и 16), в начале синтеза применяли соответствующие энантиометрически обогащенные сложные метиловые эфиры нипекотиновой кислоты. Энантиометрически обогащенные сложные метиловые эфиры нипекотиновой кислоты были выделены путем хирального разделения рацемического материала, как опубликовано (A.M. Akkerman, Rec. Traw. Chim. Pays-Bas 1951, 70 899).
Для приготовления фармацевтических композиций настоящего изобретения одно или более соединений формулы I или их соли настоящего изобретения в качестве активного ингредиента смешивали до однородности с фармацевтическим носителем с помощью стандартной фармацевтической техники; этот носитель может представлять различные формы в зависимости от желаемого способа введения препарата, например, перорального или парентерального (внутримышечного и т. п.). При приготовлении композиций для перорального приема можно использовать любой из стандартных фармацевтических носителей. Так, для жидких препаратов, предназначенных для перорального приема, например, суспензий, эликсиров или растворов, подходящие носители и добавочные вещества включают воду, гликоли, масла, спирты, отдушки, консерванты, красители и т.п.; для твердых препаратов для перорального приема, таких, например, как порошки, капсулы, пастилки, желатиновые капсулы и таблетки, подходящие носители и добавочные вещества включают крахмалы, сахара, разбавители, гранулирующие агенты, лубриканты, связующие агенты, разрыхлители и т.п. Таблетки и капсулы, вследствие простоты их введения, представляют наиболее удобную лекарственную форму для перорального введения, для которой, разумеется, используют твердые фармацевтические носители. Если это желательно, таблетки можно покрывать сахарной или энтеросолюбильной оболочкой с помощью стандартных методик. Для парентеральных форм носители обычно включают стерильную воду, хотя могут быть включены и другие ингредиенты, например для повышения растворимости или для консервации. Можно приготовить также суспензии для инъекций, для чего применяют жидкие носители, суспендирующие агенты и т.п. Фармацевтические композиции настоящего изобретения в лекарственной форме на один прием, например в таблетке, капсуле, порошке, инъекции, чайной ложке и т.п., будут содержать количество активного ингредиента, необходимое для доставки эффективной дозы, как описано выше. Фармацевтические композиции настоящего изобретения в лекарственной форме на один прием, например в таблетке, капсуле, порошке, инъекции, суппозитории, чайной ложке и т.п., будут содержать приблизительно от 0,03 мг до 100 мг/кг (предпочтительно 0,1-30 мг/кг) и могут назначаться в дозе приблизительно от 0,1 до 300 мг/кг в день (предпочтительно 1-50 мг/кг в день). Дозы, однако, можно изменять в зависимости от того, сколько требуется конкретному пациенту, тяжести состояния, требующего лечения, и применяемого соединения.
Введение может быть ежедневным или периодическим.
Фармакология.
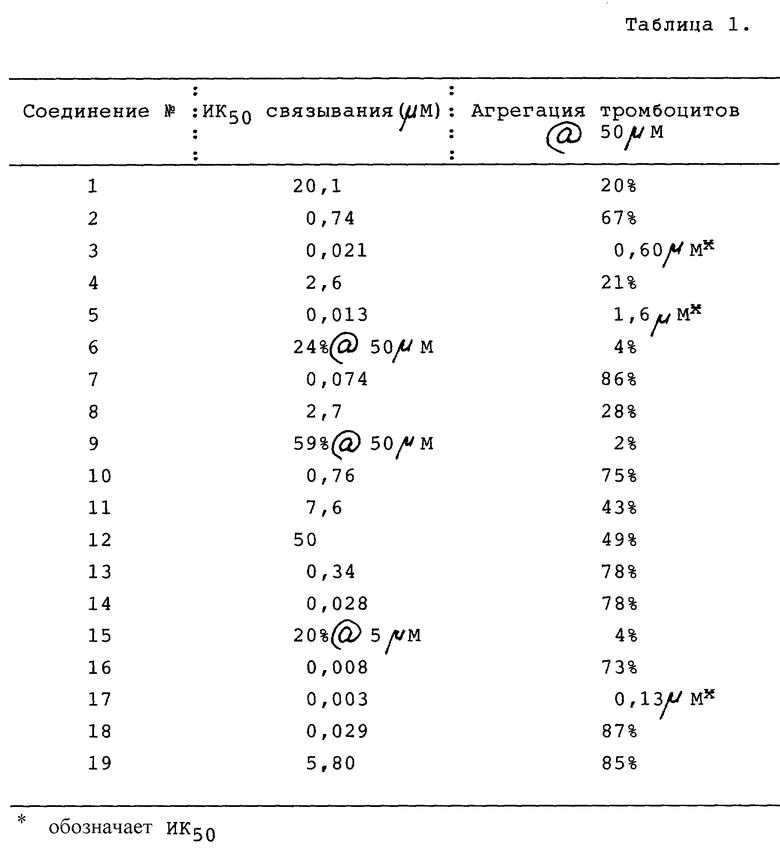
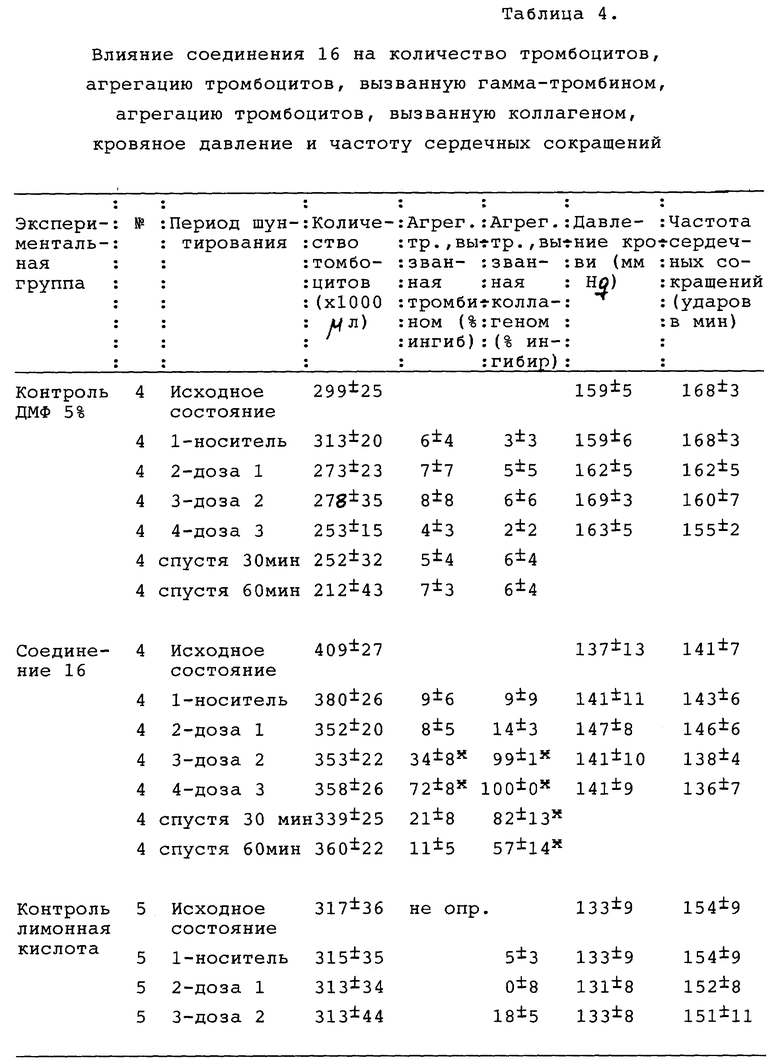
Соединения настоящего изобретения препятствуют связыванию фибриногена с гликопротеином IIb/IIIa (ГП IIb/IIIa) тромбоцитов и таким образом ингибируют агрегацию тромбоцитов. Такие соединения, следовательно, полезны для лечения нарушений свертывания крови, опосредованных тромбоцитами, таких как тромбоз артерий и вен, острый инфаркт миокарда, реокклюзия после тромболитической терапии и ангиопластики и ряда вазоокклюзивных нарушений. Поскольку конечной стадией нормальной агрегации тромбоцитов является связывание фибриногена с активированным обнажившимся ГП IIb/IIIa, ингибирование такого связывания представляет вполне состоятельный подход к предотвращению тромбообразования. Рецепторы активируются различными стимулами, такими как АДФ, коллаген и тромбин, обнажая связывающие домены для соединения с двумя различными пептидными участками фибриногена: α- цепь Арг-Гли-Асп (RGD) и γ- цепь 400-411. Как показали результаты фармакологических исследований, описанные далее в настоящем документе, соединения настоящего изобретения продемонстрировали способность блокировать связывание фибриногена с изолированным ГПIIb/IIIa (ИК50 3-5800 нМ), ингибировать агрегацию тромбоцитов in vitro в присутствии различных агентов, стимулирующих тромбоциты, и помимо этого ингибировали агрегацию тромбоцитов ex vivo на экспериментальных животных.
Исследование связывания in vitro гликопротеина IIb/IIIa, очищенного твердофазным способом.
96-луночный микротитрационный планшет Immulon-2 (Dynatech-Immulon) покрывали ГПIIb/IIIa, очищенным по сродству к RGD, в количестве 50 мкл на лунку (эффективный диапазон 0,5-10 мкг/мл) в 10 мМ ГЭПЭС, 150 мМ NaCl, 1 мМ при pH 7,4. Планшет закрывали и инкубировали в течение ночи при 4oC. Раствор ГПIIb/IIIa удаляли, добавляли 150 мкл 5% АБС и инкубировали при комнатной температуре в течение 1-3 часов. Планшет продолжительно отмывали буфером, модифицированным по способу Tyrodes. Биотинилированный фибриноген (25 мкл на лунку) в двойной конечной концентрации добавляли в лунки, содержавшие испытуемые соединения (25 мкл на лунку) в двойной конечной концентрации. Планшет закрывали и инкубировали при комнатной температуре в течение 2-4 часов. За двадцать минут до окончания инкубации добавляли по одной капле реактива А (набор с пероксидазой хрена Vecta Stain ABC Horse Radish, Vector Laboratories, Inc.) и реактива B в смеси с 5 мл буфера, модифицированного по способу Tyrodes, и оставляли стоять. Раствор лиганда удаляли и планшет отмывали (5х200 мкл на лунку) модифицированным по способу Tyrodes буфером, добавляли реагент ПX-биотин-авидин Vecta Stain HRP (50 мкл на лунку, как было приготовлено выше) и инкубировали при комнатной температуре в течение 15 минут. Раствор Vecta Stain удаляли и отмывали (5х200 л на лунку) модифицированным по способу Tyrodesf буфером. Добавляли проявляющий буфер (10 мл 50 мМ цитрат-фосфатного буфера @ pH 5.3, 6 мг о-фенилендиамина, 6 мкл 30% H2O2; 50 мкл на лунку) и инкубировали при комнатной температуре в течение 3-5 минут, а затем добавляли 2 NH2SO4 (50 мкл на лунку). Учитывали поглощение на 490 нМ.
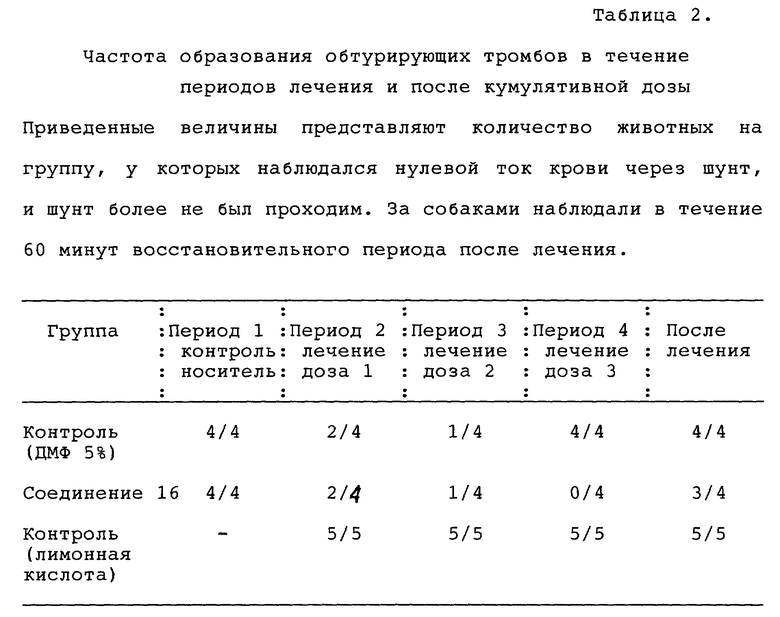
Результаты представлены в таблице 1.
Исследование in vitro тромбин-индуцированной агрегации тромбоцитов после гель-фильтрации.
Процент агрегации тромбоцитов подсчитывали как увеличение прохождения света через тромбоцитарный концентрат, обработанный испытуемым соединением, по сравнению с тромбоцитарным концентратом, обработанным как контроль. Кровь получали от нормальных доноров, не принимавших лекарственных препаратов, в пробирки, содержавшие 0,13 М цитрата натрия. Плазму, обогащенную тромбоцитами (ПОТ), собирали путем центрифугирования цельной крови на 200 х g в течение 10 минут при 25oC. ПОТ (5 мл) фильтровали через гель сефарозы 2В (объем гранул 50 мл) и количество тромбоцитов доводили до 2•107 тромбоцитов на образец. В силиконизированную кювету добавляли следующие составляющие: концентрированный тромбоцитарный фильтрат и буфер Tyrode's (0,14 М NaCl, 0,0027 М KCl, 0,012 М NaHCO3, 0,76 мМ Na2HPO4, 0,0055 М глюкозы, 2 мг/мл АБС и 5,0 мМ ГЭПЭС @ pH 7.4) в количестве, равном 350 мкл, 50 мкл 20 мМ кальция и 50 мкл испытуемого соединения. Агрегацию определяли агрегометром BIODATA в течение 3 минут после добавления агониста (тромбина в количестве 50 мкл при концентрации 1 единица на мл). Результаты представлены в таблице 1.
Исследование на собаках ex vivo
Взрослым нечистопородным собакам (8-13 кг) производили обезболивание пентобарбиталом натрия (внутривенно 35 мг/кг) и переводили их на искусственную вентиляцию легких. Артериальное давление и частоту сердечных сокращений измеряли с помощью катетера Милара, введенного в бедренную артерию. Другой датчик Милара помещали в левый желудочек (ЛЖ) через сонную артерию для измерения конечного диастолического давления в ЛЖ и индексов сократимости миокарда. Электрокардиограмму во втором отведении записывали с электродов на конечностях. В бедренную артерию и вену ввели катетеры для отбора проб крови и инфузии лекарственных препаратов соответственно. Реакции организма контролировались продолжительное время с помощью системы сбора данных Modular Instruments.
Образцы артериальной крови (5-9 мл) отбирали в пробирки, содержавшие 3,8% цитрата натрия, для приготовления плазмы, обогащенной тромбоцитами (ПОТ) и для определения влияний на параметры свертывания: протромбинового времени (ПВ) и времени активированного неполного тромбопластина (ВАНТ). Отдельные образцы крови (1,5 мл) помещали в ЭДТУ для определения гематокрита и количества клеток крови (тромбоцитов, красных кровяных клеток и белых кровяных клеток). Время кровотечения измеряли с помощью скарифицирующего устройства и ватмановской фильтровальной бумаги на щечной поверхности.
Агрегацию ПОТ выполняли на агрегометре BioData. Агрегацию цельной крови выполняли на агрегометре импеданса Chronolog. ПВ и ВАНТ определяли с помощью BioData или анализаторе AC 3000 + коагуляция. Клетки подсчитывали с помощью Sysmex K-1000.
Соединение 17 растворяли в малом объеме диметилформамида (ДМФ) и разводили физиологическим раствором до конечной концентрации 10% ДМФ. Соединение 17 вводили внутривенно с помощью автоматического шприца Harvard. Каждому животному вводили дозы 0.3, 1.3 и 10 мг/кг по кумулятивной схеме. Каждую дозу вводили с интервалом в 15 минут при постоянной скорости введения 0,33 мл/мин. Данные получали после каждой дозы и через 30 и 60 минут после окончания введения лекарства.
Соединение 17 вызывало выраженное ингибирование агрегации тромбоцитов в ex vivo. Так, в цельной крови соединение 17 ингибировало вызванную коллагеном агрегацию в дозах 0,3-10 мг/кг с выраженным ингибированием вызванного коллагеном высвобождения АТФ тромбоцитами в дозе 10 мг/кг. В ПОТ соединение 17 также ингибировало вызванную коллагеном агрегацию тромбоцитов с выраженной активностью в дозе 0,3 мг/кг. Агрегация ПОТ, индуцированная гамма-тромбином, угнеталась в дозах 3,0 мг/кг и выше. Как в случае ПОТ, так и в случае цельной крови, функция тромбоцитов начинала восстанавливаться в течение 30-60 минут, что свидетельствовало об относительно короткой продолжительности действия лекарства. Соединение 17 не имело измеримого гемодинамического действия в дозах до 10 мг/кг внутривенно. Это лекарственное средство вызывало увеличение стандартного времени кровотечения в дозах 3 и 10 мг/кг с быстрым восстановлением после лечения. Во время воздействия лекарственного средства не наблюдалось эффектов на свертывание крови (ПВ или ВАНТ), и количество тромбоцитов, лейкоцитов и эритроцитов не изменялось при любых дозах соединения 17.
Результаты (см. табл. 1 в конце описания) свидетельствуют о том, что соединение 17 является высокоэффективным ингибитором агрегации тромбоцитов ex vivo, выступая антагонистом как коллагенового, так и тромбинового пути, после внутривенного введения доз, варьирующих от 0,3 до 10 мг/кг. Антиагрегантный эффект был относительно непродолжительным и сопровождался увеличением времени кровотечения при более высоких дозах. Иных гемодинамических или гематологических эффектов не было.
Исследование на собаках in vivo
Соединение 16 испытывалось на собаках in vivo для определения терапевтической активности. Хирургическая подготовка животных.
Взрослым нечистопородным собакам обоего пола весом 9-13 кг производили анестезию пентобарбиталом натрия (35 мг/кг, внутривенно) и переводили на искусственную вентиляцию легких атмосферным воздухом с помощью эндотрахеальной трубки (12 дыхательных движений в минуту, 25 мг/кг). Для определения артериального давления в левую сонную артерию вводили наполненный физиологическим раствором полиэтиленовый катетер (РЕ-200) и соединяли его с преобразователем давления Statham (P231D, Oxnard, CA). Определяли артериальное давление диастолическое крови. За частотой сердечных сокращений наблюдали с помощью кардиотахометра (Biotach, Gould Electronics, Cleveland, OH); данные получали в 11 отведении электрокардиограммы через электроды на конечностях. В яремную вену также вводили катетер (РК-200) для введения лекарства. Левую бедренную артерию и левую бодренную вену катетеризировали обработанным силиконом (Sigmacote, Sigma Chemical Co., St. Louis, MO), наполненным физиологическим раствором катетером (РЕ-200) и соединяли с 5-сантиметровым отрезком обработанной силиконом трубки (РЕ-240) для образования экстракорпорального артериовенозного шунта (A-B). Проходимость шунта контролировали с помощью проточной системы Допплера (модель VF-1, Crystal Biotech Inc., Hopkinton, MA) проксимальнее от места шунтирования. Все параметры продолжительно наблюдались с помощью полиграфа (Gould TA-4000, Oxnard, CA) при скорости движения бумаги 10 мм в минуту.
Протокол.
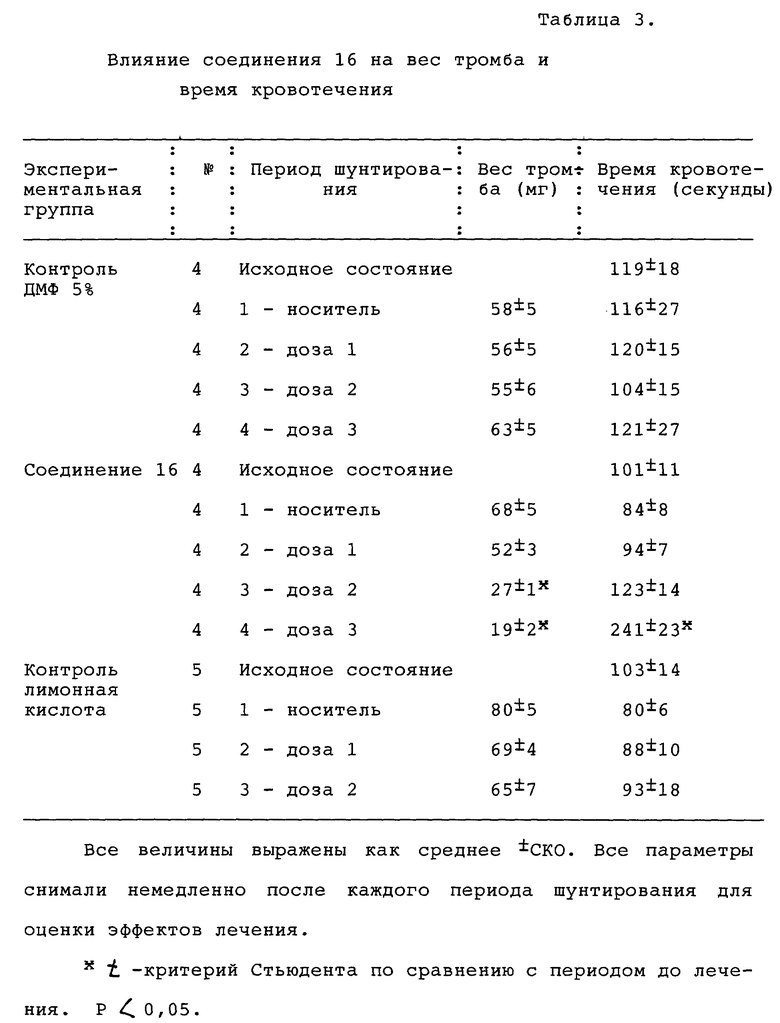
По окончании 15-минутного послеоперационного периода стабилизации был сформирован обтурирующий тромб путем введения в шунт тромбогенной поверхности (завязанная петлей шелковая нить длиной 5 см, Eticon Inc., Somerville, NJ). Было предпринято 4 последовательных периода шунтирования, первый их которых состоял из инфузии носителя, а затем возрастающих концентраций соединения 16, SC-47643, физиологического раствора с ДМФ или физиологического раствора с лимонной кислотой, введенных в виде болюса, за которыми следовала 5-минутная инфузия до введения тромбогенной поверхности и затем дополнительная 15-минутная инфузия. В конце каждого периода шунтирования шелковую нить осторожно удаляли и взвешивали. Для оценки устойчивости проходимости шунта использовали пятое шунтирование немедленно после введения общей кумулятивной лечебной дозы, что выражалось временем до полной обтурации. Вес тромба вычисляли вычитанием веса шелка до помещения в шунт из общего веса шелка после удаления его из шунта. Артериальную кровь брали до первого шунтирования и после каждого периода шунтирования для определения агрегации тромбоцитов, вызванной коллагеном, в цельной крови, дегрануляции тромбоцитов, вызванной тромбином (высвобождение АТФ тромбоцитов), протромбинового времени и количества тромбоцитов. Стандартное время свертывания крови определялось в первые 10 минут каждого периода шунтирования.
Гематологические исследования.
Количество тромбоцитов, лейкоцитов и эритроцитов и гематокрит определяли в цельной крови, собранной в 2 мг/мл динатрий-ЭДТА с помощью SysmexTM K 1000 (Baxter, Laboratories, McGraw Park, IL).
Агрегацию тромбоцитов и высвобождение АТФ в цельной крови измеряли с помощью люмиагрегации, а высвобождение АТФ измеряли с помощью люмиагрегометра (Chrono-log, Havertown, PA) путем определения изменения импеданса (агрегация тромбоцитов) и прохождения света (высвобождение АТФ) через перемешанную (1000 об/мин) суспензию цельной крови с поддерживаемой температурой 37oC. Образцы крови отбирали в 0,01 М цитрат натрия и разводили пополам физиологическим раствором с 0,5 мМ Ca (25 мкм 0,02 М CaCl2 и 20 мкм люциферола (Chrono-log, Havertown, PA)). Конечный объем составлял 1 мл. Агрегацию индуцировали коллагеном (2 мкг/мл) в отдельном образце, дегрануляцию тромбоцитов наблюдали с помощью тромбина (0,5 ед./мл) (Chrono-log, Havertown, PA), а изменения импеданса и люминесценции фиксировали в течение 6 минут. Протромбиновое время (ПВ) определяли с помощью анализатора коагуляции микрообразцов (Ciba Corning 512, Corning NY). Стандартное время кровотечения определяли, нанося порез на десну (Surgicutt, ITC Corp, Edison, NJ) и фиксируя время образования сгустка.
Лекарства.
Соединение 16; 1+0.03, 3+0.1 и 5+0,3 мг/кг, вв (болюс) + мг/кг/час, вв (инфузия) растворяли в физиологическом растворе + ДМФ (5%) и серийно разводили для достижения соответствующих концентраций, выраженных как родительское соединение.
Статистический анализ.
Результаты представлены в табл. 2 в конце описания. Все величины выражены как среднее и стандартная ошибка среднего. Статистическую достоверность изменений оценивали с помощью основного дисперсионного анализа и t-критерия Стьюдента. Различия между величинами считали достоверными при P < 0,05.
Примеры.
Защищенные аминокислоты приобретали у фирмы Bachem Bioscience Inc. Применение защищенных сложных эфиров амино- N-гидроксисукцинимида устраняет применение БОФ-С1 (см. синтез соединения 14). Энантиометрически обогащенные сложные метиловые эфиры нипекотиновой кислоты выделяли с помощью хирального разделения рацемического материала, как опубликовано (A.M. Akkerman, Rec. Traw. Chim. Pays-Bas. 1951, 70, 899). Все остальные химические вещества приобретали у фирмы Aldrich Chemical Company, Inc. Спектры интенсивного поля 1H ЯМР фиксировали на спектрометре Bruker AC-360 на 360 МГц, а сопрягающие константы даны в герцах. Температуры плавления определяли прибором для определения температур плавления Mel-Temp// и не корректировали. Микроанализы выполняли в Robertson Microlit Laboratories, Inc., Madison, New Jersy или R. W. Jonson Phamaceutical Research Institute Analitical Department. Конечные соединения очищали посредством рекристаллизации/преципитации из обычных органических растворителей и/или хроматографии на колонке с силикагелем-60 Merck. Чистоту оценивали посредством комбинации системы ЖВХР Beckman/waters и колонки Phenomenex-Ultracarb 5 ODS (30) (100 х 4,6 мм) с применением ацетонитрильной подвижной фазы (обычно 10% MeCN/90% воды). В примерах и во всей настоящей заявке следующие аббревиатуры имеют значения, приведенные ниже:
Ац = ацетил
Бн или Бзл = бензил
Бок = т-бутоксикарбонил
БОФ-С1 = бис(2-оксо-3-оксазолидинил)фосфин хлорид
Кбз = бензилоксикарбонил
ДиБАЛ = диизобутилалюминия гидрид
ЭДК = этилдиметиламинопропилкарбодиимид
ЭДТА = этилендиаминтетраацетат
ЭДТУ = этилендиаминтетрауксусная кислота
ГОБТ = гидроксибенхотриазол
и-Пр = изопропил
NMM = N-метилморфолин
Нип = нипекотил (если не указано иное, рацемат в 3 положении)
ПТСК = п-толуолсульфокислота
ТФУ = трифторуксусная кислота
Пример 1. Nα- Бок-D-Лиз-S-(+)-Нип -β- Ала-OH (соединение 14)
К смеси Nα- Бок-D-Лиз(Кбз)-OH (2,9 г, 7,74 ммоль) и CH2Cl2 (80 мл) при 5oC добавляли БОФ-С1 (1,96 г, 7,7 ммоль) и NMM (0,83 мл, 7,7 ммоль). Эту смесь перемешивали в течение 30 минут, действовали на нее гидрохлоридом метилового эфира S-(+)- нипекотиновой кислоты (1,39 г, 7,7 ммоль) и NMM (0,83 мл), перемешивали в течение 2 часов при 5oC и разводили насыщенным NH4Cl (50 мл). Органический слой отделяли от водного слоя, высушивали MgSO4 и выпаривали до получения стекловидного твердого вещества. Это твердое вещество очищали посредством проточной хроматографии (4% этанол/CH2Cl2) для получения Nα- Бок-D-Лиз(Кбз)-S-(+)-Нип-OMe в виде белой пены:
1H ЯМР(CDCl3) δ: 7.30 (m, 5H), 5.50 (m, 1H), 5.09 (s, 2H), 4.61 (m, 1H), 3.92 (m, 1H), 3.66 (s, 3H), 3.20 (m, 4H), 2.79 (m, 1H), 2.51 (m, 1H), 2.51(m1H), 2.12 (m, 1H), 1.50-1.80 (m, 10H), 1.39 (s, 9H).
МС м/e 506 (МН+).
К раствору Nα- Бок-D-Лиз(Кбз)-S-(+)-Нип-OMe (3,52 г, 7,0 ммоль) в ТГФ (25 мл) при комнатной температуре добавляли гидроксид лития (0,19 г, в 15 мл воды) по каплям в течение 3 минут. Этот раствор перемешивали 6 часов и выпаривали до получения белой пены. Эту пену смешивали с CH2Cl2 (80 мл) при комнатной температуре и действовали на нее последовательно H -β- Ала-ОБн•ПТСК (2,34 г, 7,0 ммоль), ГОБТ (5 мг), ЭДК•HCl (1,98 г, 10,4 ммоль) и NММ (0,76 мл, 7,0 ммоль). Эту смесь перемешивали 13 часов, разводили насыщенным NH4Cl (50 мл) и разделяли слои. Органический слой высушивали MgSO4 и выпаривали до получения белой пены. Эту пену очищали посредством проточной хроматографии (3-4% этанол/CH2Cl2) для получения Nα- Бок-D-Лиз(Кбз)-S-(+)Нип -β- Ала-ОБн в виде белой пены:
1H ЯМР (CDCl3) δ: 7.35 (m, 10), 6.29 (m, 1H), 5.45 (m, 1H), 5.12 (s, 2H), 5.05 (s, 2H), 5.00 (m, 1H), 4.55 (m, 1H), 4.32 (m, 1H), 3.48 (m, 2H), 3.19 (m, 4H), 2.53 (m, 3H), 2.21 (m, 1H), 1.84 (m, 1H), 1.48-1.72 (m, 9H), 1.42 (s, 9H).
МС м/e 653 (MH+).
К раствору Nα- Бок-D-Лиз(Кбз)-S-(+)-Нип -β- Ала-Обн (0,80 г, 1,22 ммоль) в ТГФ (15 мл) в колбе Парра под атмосферой азота добавляли уксусную кислоту (5 мл), воду (10 мл) и Pd/C (10%, 0,09 г). Эту смесь гидрогенизировали при 50 psi и комнатной температуре в течение 21 часа, фильтровали через целитовый фильтр и выпаривали приблизительно до 5 мл. На этот раствор действовали диэтиловым эфиром (60 мл) до получения белого осадка, который отфильтровывали и лиофилизировали для получения 14 в виде бесцветных чешуек с температурой плавления 52o-60oC.
1H ЯМР (ДМСО-d6) δ: 7.85 (m, 1H), 6.83 (d, J=7.1 H), 4.34 (d, J=12.1 H), 4.22 (m, 1H), 3.60 (m, 2H), 3.41 (m, 2H), 2.98 (t, J=11.1 H), 2.88 (m,1H), 2.69 (m, 2H), 2.35 (m, 2H), 2.12 (m, 1H), 2.03 (m, 1H), 1.70 (m, 2H), 1.4-1.6 (m, 8H), 1.35 и 1.38 (pr.s 8.5:1.9 H), 1.16 (m, 2H),
ИК (KBr): 3450-2860, 1709, 1641 см-1,
МС м/e 429 (MH+),
[α]
Анализ для C20 H36N4O6 • C2H4O2 (488,6)
Вычислено: С 54.08, H 8.25, N 11.47
Обнаружено: С 54.64, H 8.26, N 10.79.
Пример 2. Nα- Бок-L-Лиз (Кбз)-Нип -β- Ала-Обн (соединение 1)
Соединение 1, приготовленное, начиная Nα- Бок-L-Лиз(Кбз)-ОН и рацемического метилового эфира нипекотиновой кислоты, как в примере 1, было выделено в виде стекловидного вещества:
1H ЯМР (CDCl3) δ: 7.29 (m, 10H), 6.51 (m,1H), 5.39 (m, 1H), 5.11 (s, 2H), 5.06 (s, 2H), 4.94 (m, 1H), 4.54 (m, 2H), 4.18 (m, 1H), 4.02 (d, J=10.1 H), 3.61 (m, 1H), 3.48 (m, 2H), 3.17 (m, 4H), 2.54 (m, 3H), 2.20 (m, 1H), 1.83 (m, 1H), 1.67 (m, 2H), 1.51 (m, 4H), 1.39 (s, 9H),
МС м/e 653 (MH+),
Анализ для C35H48N4O8 • 1,5 H2O: (679.8)
Вычислено: С 61.84, H 7.56, N 8.24
Обнаружено: С 62.00, H 7.25, N 8.23
Пример 3. Nα- Бок-L-Лиз-Нип -β- -Ала-ОН (соединение 2)
Соединение 2, приготовленное гидрогенолизом соединения 1, как в примере 1, было выделено в виде белой пены:
1H ЯМР (ДМСО-d6) δ: 8.00 (m, 1H), 7.86 (m, 1H), 4.29 (m, 2H), 3.82 (m, 1H), 3.11 (m, 3H), 2.70 (m, 2H), 2.53 (m, 1H), 2.31 (m, 2H), 2.17 (m, 2H), 1.4-1.9 (m,10 H), 1.34 и 1.36 (ps.s, 1:1.9 H), 1.23 (m, 2H),
МС м/e 429 (MH+),
[α]
Анализ для C20H36N4O6 • 1,5H2O: (518,6):
Вычислено: С 53.27, H 8.16, N 10.80
Обнаружено: С 53.61, H 8.18, N 10.47
Пример 4. Nα- Бок-D-Лиз-Нип -β- Ала-OH (соединение 3)
Nα- Бок-D-Лиз(Кбз)-Нип -β- Ала-ОБн, приготовленный, начиная с рацемического метилового эфира нипекотиновой кислоты и Nα- Бок-D-Лиз(Кбз)-ОН, как в примере 1, был выделен в виде белой пены:
1H ЯМР (CDCl3) δ: 7.32 (m, 10H), 6.59 (m, 1H), 5.45 (m, 1H), 5.12 (s, 2H), 5.07 (s, 2H), 4.94 (m, 1H), 4.56 (m, 1H), 4.12 (m, 1H), 3.51 (m, 2H), 3.17 (m, 3H), 2.57 (m, 2H), 2.21 (m, 1H), 1.89 (m, 1H), 1.45-1.79 (m, 11H), 1.41 (S, 9H),
МС м/e 653 (MH+).
Соединение 3, приготовленное посредством гидрогенолиза Nα- Бок-D-Лиз(Кбз)-Нип -β- Ала-Обн, как в примере 1, было выделено в виде бесцветных чешуек с температурой плавления 48 - 54oC.
1H ЯМР (ДМСО-d6) δ: 7.96 (m, 1H), 6.82 (m, 1H), 4.30 (m, 2H), 3.81 (m, 1H), 3.12 (m, 4H), 2.69 (m, 2H), 2.56 (m, 1H), 2.33 (m, 2H), 2.14 (m, 2H), 1.80 (m, 2H), 1,4-1,7 (m, 9H), 1.32 и 1.34 (pr.s. 1:1.9 H), 1.22 (m,2H),
ИК (KBr): 3580-2770, 1711, 1642 см-1,
МС м/e 429 (MH+),
[α]
Анализ для C20H36N4O6• 2C2H4O2•5H2O (557,6)
Вычислено: С 51.69, H 8.13, N 10.05
Обнаружено: С 51.46, H 8.11, N 10.10
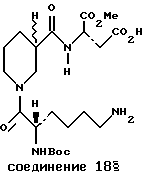
Пример 5. Nα- Бок-D-Лиз-Нип-L-Асп-OMe (соединение 18)
Nα- Бок-D-Лиз(Кбз)-Нип-L-Асп(ОБн)-OMe, приготовленный из H-L-Асп(ОБн)-OMe и Nα- Бок-D-Лиз(Кбз)-Нип-ОН, как в примере 1, был выделен в виде стекловидного вещества:
1H ЯМР (CDCl3) δ: 7.36 (m, 10H), 6.84 (m, 1H), 5.40 (m, 1H), 5.14 (s, 2H), 5.09 (s, 2H), 4.88 (m, 2H), 4.54 (m, 1H), 4.30 (m, 1H), 3.68 (m, 3H), 3.19 (m, 3H), 3.03 (m, 1H), 2.89 (m, 1H), 2.29 (m, 1H), 1.43-2.06 (m, 12H), 1.40 (s, 9H);
МС м/e 711 (MH+);
Соединение 18, приготовленное посредством гидрогенолиза Nα- Бок-D-Лиз(Кбз)-Нип-L-Асп(ОБн)-OMe, как в примере 1, было выделено в виде белой пены:
1H ЯМР (ДМСО-d6) δ: 8.33 (m, 1H), 6.77 (d, J=7.1 H), 4.32 (m, 3H), 3.82 (m, 1H), 3.59 (s, 3H), 2.96 (m, 2H), 2.73 (m, 3H), 2.46 (m, 2H), 2.34 (m, 1H), 1.79 (m, 3H), 1.4-1.7 (m, 8H), 1.34 и 1.37 (pr.s, 1:1.9 H), 1.27 (m, 2H);
МС м/e 487 (MH+); [α]
Анализ для C22H38N4O8 • C2H4O2•H2O (564,6):
Вычислено: С 51.05, H 7.85, N 9.92
Обнаружено: С 50.89, H 7.88, N 9.74
Пример 6. Н-L-Лиз-Нип -β- Ала-ОН (соединение 4)
К раствору соединения 2 (0,30 г, 0,70 ммоль) в MeOH (10 мл) и воде (10 мл) при комнатной температуре добавляли HCl (0,50 мл, конц.). Этот раствор перемешивали 1 час и выпаривали до получения приблизительно 2 мл маслянистой жидкости. К этой маслянистой жидкости добавляли MeCN (20 мл), фильтровали, отмывали диэтиловым эфиром (3х20 мл) и высушивали до получения соединения 4 в виде белого порошка с температурой плавления 65o-75oC.
1H ЯМР (ДМСО-d6) δ: 8.23 (m, 3H), 8.06 (m, 3H), 4.33 (m, 2H), 3.73 (m, 4H), 3.25 (m, 2H), 3.01 (m, 1H), 2.72 (m, 2H), 2.44 (m, 1H), 2.34 (m, 2H), 1.5-1.8 (m, 6H), 1.35 (m, 4H);
МС м/e 329 (MH+);
Анализ для C15H28N4O4 • 2HCl • 2H2O (437,4):
Вычислено: С 41.19, H 7.84, N 12.81
Обнаружено: С 40.97, H 7.75, N 12.44
Пример 7. N-(Nε- аминокапроил)-Нип -β- Ала-ОН (соединение 5)
N-(Nε- аминокапроил)-Нип -β- Ала-ОБн, приготовленный начиная с рацемического метилового эфира нипекотиновой кислоты и эфира N-оксисукцинимида и Nε- Бок-аминокапроевой кислоты, как в примере 1, был выделен в виде маслянистого твердого вещества:
1H ЯМР (CDCl3) δ: 1.34 (m, 5H), 6.51 (m, 1H), 5,12 (s, 2H), 4.60 (m, 1H), 4.39 (m, 1H), 3.90 (m, 1H), 3.71 (t, 1H), 3.52 (m, 3H), 3.19 (m, 4H), 2.59 (m, 2H), 2.30 (m, 2H), 1.85 (m, 3H), 1.63 (m, 2H), 1.51 (m, 2H), 1.42 (s, 9H), 1.34 (m, 2H);
МС м/e 504 (MH+).
Соединение 5, приготовленное посредством гидрогенолиза, а затем кислотного гидролиза N-(Nε- Бок-аминокапроил)- Нип -β- Ала-ОБн, как в примере 1, было выделено в виде стекловидного вещества:
1H ЯМР (ДМСО-d6) δ: 8.18 (t, J=5.1 H), 8.04 (br.s, 3H), 4.28 (m, 2Н), 3.78 (m, 2H), 3.20 (m, 3H), 2.99 (t, J=12.1 H), 2.71 (d, J=6.2 H), 2.39 (m, 2H), 2.31 (m, 2H), 2.16 (m, 1H), 1.79 (m, 1H), 1.61 (m, 4H), 1.42 (t, J=6.2 H), 1.28 (m, 2H), 1.19 (m, 1H);
МС м/e 314 (MH+).
Анализ для C15H27N3O4 • 2HCl (386,3):
Вычислено: С 46.04, H 7.57, N 10.88
Обнаружено: С 45.91, H 7.63, N 11.17
Пример 8. N-[3-(4-пиперидинпропионил)]-Нип- -β- Ала-ОН (соединение 17)
N-[3-(4-пиридинакрилоил)] -Нип -β- Ала-ОБн, приготовленный начиная с 3-(4-пиридин)акриловой кислоты и рацемического метилового эфира нипекотиновой кислоты, как в примере 1, был выделен в виде стекловидного вещества:
1H ЯМР (CDCl3) δ: 8,61 (d, J=4 Гц, 2H), 7.52 (d, J=15 Гц, 1H), 7.35 (m, 7H), 7.03 (d, J=15 Гц, 1H), 6.58 (m, 1H), 5.12 (m, 2H), 4.40 (m, 1H), 3.89 (m, 1H), 3.51 (m, 2H), 3.38 (m, 2H), 2.60 (t, J=6 Гц, 2H), 2.31 (m, 1H), 1.97 (m, 2H), 1.74 (m, 1H), 1.56 (m, 1H);
МС м/e 422 (MH+).
К раствору N-[3-(4-пиридинакрилоил)]-Нип -β- Ала-ОБн (0,56 г, 1,33 ммоль) в этаноле (20 мл) и воде (10 мл) под атмосферой азота добавляли HCl (0,66 мл, 4,0 М в диоксане) и оксид платины IV (0,060 г). Эту смесь гидрогенизировали при 50 psi/RT при комнатной температуре в течение 22 часов, фильтровали через целитовый фильтр и выпаривали приблизительно до 5 мл. На этот раствор действовали MeCN (30 мл), фильтровали, отмывали диэтиловым эфиром (3х20 мл) и высушивали до получения 17 в виде бледно-желтой пены:
1H ЯМР (ДМСО-d6) δ: 9.02 (br.s, 2H), 8.03 (m, 1H), 7.46 (m, 1H), 4.28 (t, J= 7.1 H), 4.11 (m, 1H), 3.79 (m, 1H), 3.44 (t, J=7.1 H), 3.19 (m, 3H), 3.06 (t, J=12.1 H), 2.75 (d, J=11.1 H), 2.53 (m, 1H), 2.32 (m, 4H), 2.12 (m, 1H), 1.77 (m, 2H), 1.4-1.7 (m, 7H), 1.27 (m, 2H), 1.18 (t, J=6, 1H);
MC м/e 340 (MH+);
Анализ для C17H29N3O4 • 2HCl (412,4):
Вычислено: С 49.52, H 7.58, N 10.19
Обнаружено: С 49.15, H 7.02, N 10.48
Точная протонированная масса, вычисленная для C17H29N3O4: 340,2236 amu, обнаружено: 340,2245 amu.
Пример 9. Nα- Ац-L-Лиз-Нип-Гли-ОН (соединение 6)
Nα- Ац-L-Лиз(Бок)-Нип-Гли-ОБн, приготовленный начиная с Nα- Ац-L-Лиз(Бок)-ОН и рацемического метилового эфира нипекотиновой кислоты (смотри 14), был выделен в виде стекловидного вещества:
1H ЯМР (CDCl3) δ: 7.35 (m, 5H), 6.97 (m, 1H), 6.38 (m, 1H), 5.14 (m, 2H), 4.70 (m, 1H), 4.46 (m, 1H), 4.06 (dd, J=5, 16 Гц, 2H), 3.71 (m, 1H), 3.10 (m, 2H), 1.99 (s, 3H), 1.91 (m, 2H), 1.64 (m, 1H), 1.41-1.60 (m, 1H), 1.39 (s, 9H);
МС м/e 547 (MH+).
Соединение 6, приготовленное посредством гидрогенолиза Nα- АЦ-L-Лиз(Бок)-Нип-Гли-ОБн, как в примере 2, а затем ТФУ-опосредованного удаления Бок (способ смотри у M.Bodanszky The Practice. of Peptide Synthesis Springer-Verlag: New York, 1984), было выделено в виде желтовато- коричневого порошка с температурой плавления 40o-55oC.
1H ЯМР (ДМСО-d6) δ: 8.24 (t, J=6, 1H), 8.03 (d, J=8, 1H), 7.75 (br.s, 3H), 4.24 (m, 1H), 3.72 (t ,J=6, 2H), 3.61 (m, 2H), 2.72 (m, 2H), 1.83 (s, 3H), 1.78 (m, 2H), 1.63 (m, 2H), 1.4-1.6 (m, 8H), 1.28 (m, 4H);
МС м/e 357 (MH+);
Анализ для C16H28N4O5 • 3C2HF3O2 (698,5):
Вычислено: С 37.83, H 4.47, N 8.02
Обнаружено: С 37.91, H 4.89, N 8.47
Пример 10. Nα- Ац-L-Лиз-Нип -β- Ала-ОН 1(Соединение 7)
Nα- Ац-L-Лиз(Бок)-Нип -β- Ала-ОБн, приготовленный, начиная с Nα- Ац-L-Лиз(Бок)-ОН и рацемического метилового эфира нипекотиновой кислоты, как в примере 1, был выделен в виде белой пены:
1H ЯМР (CDCl3) δ: 7.34 (m, 5H), 6.53 (m, 2H), 5.12 (s, 2H), 4.58 (m, 1H), 4.10 (m, 1H), 3.72 (m, 1H), 3.54 (m, 2H), 3.11 (m, 3H), 2.59 (m, 2H), 2.24 (m, 1H), 2.01 (s, 3H), 1.88 (m, 1H), 1.73 (m, 2H), 1.52 (m, 8H), 1.40 (s, 9H), 1.31 (m, 1H);
МС м/e 561 (MH+).
Соединение 7, приготовленное посредством гидрогенолиза Nα- Ац-L-Лиз(Бок)-Нип -β- Ала-ОБн, как в примере 1, а затем кислотного гидролиза, как в примере 6, было выделено в виде белой пены с температурой плавления 53-67oC.
1H ЯМР (ДМСО-d6) δ: 8.13 (m, 1H), 8.00 (m, 1H), 7.91 (d, J=15, 3H), 4.64 (m, 1H), 4.36 (m, 1H), 3.87 (m, 1H), 3.66 (m, 2H), 3.23 (m, 3H), 2.99 (m, 1H), 2.68 (m, 2H), 2.59 (m, 1H), 2.38 (m, 2H), 2.11 (m, 1H), 1.80 (s, 3H), 1.63 (m, 1H), 1.4-1.6 (m, 5H), 1.24 (m, 3H);
МС м/e 371 (MH+);
Анализ для C17H30N4O5 •2HCl•2H2O (479,4)
Вычислено: С 42.59, H 7.57, N 11.69
Обнаружено: С 43.83, H 7.79, N 10.91
Пример 11. Nα- Бок-L-Арг-Нип -β- Ала-ОН (соединение 8)
Nα- Бок-L-Арг(Кбз)-Нип -β- Ала-ОБн, приготовленный начиная с Nα- Бок-L-Арг(Кбз2)-OSu и рацемического метилового эфира нипекотиновой кислоты, как в примере 1, был выделен в виде стекловидного вещества:
1H ЯМР (CDCl3) δ: 7.33 (m, 10H), 6.69 (m, 1H), 5.70 (m, 1H), 5.13 (s, 2H), 5.03 (s, 2H), 4.59 (m, 1H), 4.29 (m, 1H), 3.52 (m, 2H), 3.28 (m, 1H), 3.09 (m, 3H), 2.60 (m, 3H), 2.18 (m, 1H), 1.49-1.90 (m, 11H), 1.42 (s, 9H);
МС м/e 681 (MH+).
Соединение 8, приготовленное посредством гидрогенолиза Nα- Бок-L-Арг(Кбз)-Нип -β- Ала-ОБн, как в примере 1, было выделено в виде белой пены с температурой плавления 47-55oC.
1H ЯМР (ДМСО-d6) δ: 9.53 (m, 1H), 7.85 (m, 2H), 6.96 (m, 1H), 4.32 (m, 2H), 3.84 (m, 1H), 3.38 (m, 2H), 3.03 (m, 4H), 2.20 (m, 3H), 1.74 (m, 2H), 1.4-1.7 (m, 8H), 1.35 (s, 9H), 1.24 (m, 2H);
МС м/e 457 (MH+);
Анализ для C20H36N6O6 • 1,5 C2H4O2 (546,6):
Вычислено: С 50.54, H 7.74, N 15.37
Обнаружено: С 50.24, H 7.96, 15.26
Пример 12. Nα- Бoк-L-Лиз-Hип -γ- aминoмасляная кислота (соединение 9)
Бензиловый эфир Nα- Бoк-L-Лиз(Kбз)-Hип -γ- аминoмacлянoй кислоты, приготовленный начиная с Nα- Бок-L-Лиз(Кбз)-ОН и рацемического метилового эфира нипекотиновой кислоты (смотри 1-1 и 1-2), был выделен в виде стекловидного вещества: 1H ЯМР (CDCl3) δ: 7.33 (m, 10H), 6.48 (m, 1H), 6.16 (m, 1H), 5.40 (m, 1H), 5.11 (s, 2H), 5.08 (s, 2H), 4.89 (m, 1H), 4.58 (m, 1H), 4.07 (m, 1H), 3.22 (m, 5H), 2.52 (m, 1H), 2.40 (m, 2H), 1.50-2.30 (m, 12H), 1.42 (s, 9H), 1.33 (m, 1H);
МС м/e 667 (MH+).
Соединение 9, приготовленное посредством гидрогенолиза бензилового эфира Nα- Бок-L-Лиз(Кбз)-Нип -γ- аминомасляной кислоты, как в примере 1, было выделено в виде белой пены с температурой плавления 65-71oC.
1H ЯМР (ДМСО-d6) δ: 8.25 (m, 1H), 6.87 (m, 1H), 4.31 (m, 3H), 3.74 (m, 2H), 3.15 (m, 2H), 2.98 (m, 3H), 2.69 (m, 2H), 2.10 (m, 3H), 1.76 (m, 3H), 1.4-1.7 (m, 9H), 1.31 (s, 9H), 1.21 (m, 2H);
МС м/e 443 (MH+);
Анализ для C21H38N4O6 • 2 C2H4O2 (562,7):
Вычислено: С 53.37, H 8.24, N 9.96
Обнаружено: С 53.94, H 8.17, N 9.70
Пример 13. H-D-Лиз-Нир -β- Ала-ОН (соединение 10)
Соединение 10, приготовленное посредством кислотного гидролиза 3, как в примере 6, было выделено в виде кремового порошка с температурой плавления 108-128oC.
1H ЯМР (ДМСО-d6) δ: 8.28 (m, 3H), 8.05 (m, 3H), 4.31 (m, 2H), 3.84 (m, 2H), 3.25 (m, 2H), 3.09 (m, 2H), 2.72 (m, 3H), 2.37 (m, 3H), 1.80 (m, 1H), 1.5-1.7 (m, 6H), 1.33 (m, 4H);
МС м/e 329 (MH+);
Анализ для C15H28N4O4 • 2HCl • C2H4O2 (461,4):
Вычислено: С 44.26, H 7.43, N 12.14
Обнаружено: С 43,98, H 7.27, N 12.29
Пример 14. Nα- Бок-D-Лиз-Нип -γ- аминомасляная кислота (соединение 11)
Бензиловый эфир Nα- Бок-D-Лиз(Кбз)-Нип -γ- аминомасляной кислоты, приготовленный начиная с Nα- Бок-D-Лиз(Кбз)-ОН и рацемического метилового эфира нипекотиновой кислоты, как в примере 1, был выделен в виде стекловидного вещества:
1H ЯМР (CDCl3) δ: 7.31 (m, 10H), 6.51 (m, 1H), 5.48 (m, 1H), 5.10 (s, 1H), 5.06 (s, 2H), 4.90 (m, 1H), 4.55 (m, 1H), 4.10 (m, 1H), 3.59 (m, 1H), 3.23 (m, 5H), 2.39 (m, 2H), 2.23 (m, 2H), 1.84 (m, 2H), 1.45-1.80 (m, 10H), 1.38 (s, 9H), 1.32 (m, 1H);
МС м/e 667 (MH+).
Соединение 11, приготовленное посредством гидрогенолиза бензилового эфира Nα- Бок-D-Лиз(Кбз)-Нип- -γ- аминомасляной кислоты, как в примере 1, было выделено в виде желтовато-коричневого порошка с температурой плавления 50-57oC.
1H ЯМР (ДМСО-d6) δ: 7.97 (m, 1H), 6.91 (m, 1H), 4.32 (m, 1H), 4.22 (m, 1H), 3.82 (m, 1H), 3.02 (m, 3H), 2.71 (m, 2H), 2.52 (m, 1H), 2.29 (m, 1H), 2.17 (m, 2H), 1.84 (m, 5H), 1.4-1.7 (m, 9H), 1.33 (s, 9H), 1.19 (m, 2H);
МС м/e 443 (MH+);
Анализ для C21H38N4O6 • C2H4O2 • 0,5 H2O (571,7):
Вычислено: С 52.53, H 8.29, N 9.80
Обнаружено: С 52.91, H 8.21, N 9.39
Пример 15. Nα- Бoк-D-Лиз-Hип-Гли-OH (соединение 12)
Nα- Бок-D-Лиз(Кбз)-Нип-Гли-ОБн, приготовленный, начиная с Nα- Бок-D-Лиз(Кбз)-ОН и рацемического метилового эфира нипекотиновой кислоты, как в примере 1, был выделен в виде стекловидного вещества:
1H ЯМР (CDCl3) δ: 7.39 (m, 10H), 6.87 (m, 1H), 5.42 (m, 1H), 5.19 (s, 2H), 5.13 (s, 2H), 4.93 (m, 1H), 4.60 (m, 1H), 4.20 (m, 1H), 4.09 (m, 1H), 3.40-4.00 (m, 3H), 3.21 (m, 2H), 2.61 (m, 1H), 2.43 (m, 1H), 1.45-2.20 (m, 10H), 1.39 (s, 9H);
МС м/e 639 (MH+).
Соединение 12, приготовленное посредством гидрогенолиза Nα- Бок-D-Лиз(Кбз)-Нип-Гли-ОБн, как в примере 1, было выделено в виде белых чешуек с температурой плавления 66-80oC:
1H ЯМР (ДМСО-d6) δ: 7.82 (m, 1H), 6.81 (d, J=4, 1H), 4.34 (m, 2H), 4.09 (m, 1H), 3.77 (m, 1H), 3.48 (m, 1H), 3.16 (m, 2H), 2.70 (m, 3H), 2,44 (m, 2H), 2.28 (m, 1H), 1.78 (m, 2H), 1.4-1.7 (m, 8H), 1.32 и 1.35 (pr.s, 1:1, 9H), 1.23 (m, 2H);
МС м/e 415 (MH+);
Анализ для C19H34N4O6 • 2 C2H4O2 (534,6):
Вычислено: С 51.67, H 7.92, N 10.48
Обнаружено: С 52.06, H 8.33, N 10.19
Пример 16. Nα- Ац-D-Лиз-Нип -β- Ала-ОН (соединение 13)
Nα- Ац-D-Лиз(Кбз)-Нип -β- Ала-ОБн, приготовленный начиная с Nα- Ац-D-Лиз(Кбз)-ОН и рацемического метилового эфира нипекотиновой кислоты, как в примере 1, был выделен в виде стекловидного вещества:
1H ЯМР (CDCl3) δ: 7.32 (m, 10H), 6.54 (m, 1H), 6.36 (m, 1H), 5.10 (s, 2H), 5.02 (s, 2H), 4.89 (m, 2H), 4.48 (m, 1H), 4.04 (m, 1H), 3.69 (m, 1H), 3.52 (m, 2H), 3.17 (m, 3H), 2.57 (m, 2H), 2.20 (m, 1H), 1.98 (s, 3H), 1.25-1.90 (m, 10H);
МС м/e 595 (MH+).
Соединение 13, приготовленное посредством гидрогенолиза  Ац-D-Лиз(Кбз)-Нип -β- АлА-ОБн, как в примере 1, было выделено в виде стекловидного вещества с температурой плавления 46-59oC.
Ац-D-Лиз(Кбз)-Нип -β- АлА-ОБн, как в примере 1, было выделено в виде стекловидного вещества с температурой плавления 46-59oC.
1H ЯМР (ДМСО-d6) δ: 8.11 (m, 3H), 4.70 (m, 1H), 4.33 (m, 1H), 3.74 (m, 1H), 3.38 (m, 1H), 3.19 (m, 4H), 3.00 (m, 1H), 2.68 (m, 2H), 2.21 (m, 4H), 1.82 (s, 3H), 1.76 (m, 2H), 1.4-1.7 (m, 7H), 1.24 (m, 2H);
МС м/e 371 (MH+);
Анализ для C17H30N4O5 • 2,5 C2H4O2 (520,6):
Вычислено: С 50.76, H 7.74, N 10,76
Обнаружено: С 51.12, H 8.04, N 10.75
Пример 17. Nα- Бок-L-Лиз(и-Пр)-Нип -β- Ала-ОН (соединение 15)
Nα- Бок-L-Лиз(и-Пр)(Кбз)-Нип -β- Ала-ОБн, приготовленный начиная с Nα- Бок-L-Лиз(и-Пр)(Кбз)-ОН и рацемического метилового эфира нипекотиновой кислоты, как в примере 1, был выделен в виде стекловидного вещества.
1H ЯМР (CDCl3) δ: 7.33 (m, 10H), 6.58 (m, 1H), 5.10 (s, 2H), 5.08 (s, 2H), 4.55 (m, 1H), 4.21 (m, 1H), 3.73 (m, 1H), 3.50 (m, 2H), 3.17 (m, 3H), 2.55 (m, 2H), 2.18 (m, 1H), 1.50-2.00 (m, 13H), 1.40 (s, 9H), 1.13 (d, J=8 Гц, 6H);
МС м/e 695 (MH+).
Соединение 15, приготовленное посредством гидрогенолиза Nα- Бoк-L-Лиз(и-Пp)(Kбз)-Hип -β- Алa-OБн, как в примере 1, было выделено в виде белых чешуек с температурой плавления 90-123oC:
H ЯМР (ДМСО-d6) δ: 7.93 (m, 1H), 6.81 (d, J=7, 1H), 4.36 (m, 1H), 4.24 (m, 1H), 3.60 (m, 1H), 3.37 (m, 1H), 3.10 (m, 1H), 2.91 (m, 3H), 2.62 (m, 3H), 2.39 (m, 2H), 2.14 (m, 1H), 2.05 (m, 1H), 1.4-1.8 (m, 9H), 1.34 и 1.37 (pr.s, 1:1, 9H), 1.26 (m, 3H), 1.13 (d, J=5, 6H);
ИК (KBr) 3500-2830, 1704, 1638 см-1;
МС м/e 471 (MH+);
Анализ для C23H42N4O6 • 2 C2H4O2 (590,7)
Вычислено: С 54.90, H 8.53, N 9.48
Обнаружено: С 54.67, H 8.65, N 9.79
Пример 18. Nα- Бок-D-Лиз-R-(-)-Нип -β- Ала-ОН (соединение 16)
Соединение 16, приготовленное начиная с Nα- Бок-D Лиз(Кбз)-ОН и метилового эфира R-(-)-нипекотиновой кислоты, как в примере 1, было выделено в виде бесцветных чешуек с температурой плавления 42-51oC.
1H ЯМР (ДМСО-d6) δ: 7.95 (m, 1H), 6.82 (d, J=7, 1H), 4.33 (m, 1H), 4.19 (m, 1H), 3.79 (m, 1H), 3.25 (m, 1H), 3.04 (t, J=10, 2H), 2.69 (m, 2H), 2.34 (m, 1H), 2.21 (m, 1H), 2.14 (m, 2H), 1.78 (m, 2H), 1.71 (m, 2H), 1.4-1.6 (m, 9H), 1.34 и 1.38 (pr.s, 1:8, 9H), 1.20 (m, 2H);
МС м/e (MH+) 429;
Анализ для C20H36N4O4 • 2,5 C2H4O2 (578,7)
Вычислено; С 51.89, H 8.01, N 9.68
Обнаружено: С 52.05, H 7.98, N 9.58
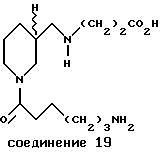
Пример 19. N-(Nε- аминокапроил)-3-пиперидин- метиламинопропионовая кислота (соединение 19)
К раствору N-(Nε- Бок-аминокапроил)нипекотиновой кислоты (3,1 г, 9,0 ммоль) и ТГФ (50 мл) добавляли 1,1-карбонилдиимидазол (1,45 г, 9,0 ммоль). Этот раствор перемешивали в течение 1 часа, охлаждали до -10oC, добавляли по каплям ДиБАЛ (36,0 мл, 1,0 М в толуоле) в течение 20 минут и перемешивали еще 2 часа. На этот раствор действовали водным раствором лимонной кислоты (5,0 г в 40 мл воды), разводили CHCl3 (200 мл) и полученные слои разделяли. Водный слой экстрагировали CHCl3 (100 мл), а объединенные органические слои высушивали, выпаривали и очищали посредством проточной хроматографии (4% этанол/CH2Cl2) до получения N-(Nε- Бок-аминокапроил)-пиперидин-3-карбоксальдегида в виде стекловидного вещества.
1H ЯМР (CDCl3) δ: 9.65 (d, J=7 Гц, 1H), 4.58 (m, 1H), 4.10 (m, 1H), 3.65 (m, 1H), 3.45 (m, 1H), 3.22 (m, 1H), 3.14 (m, 2H), 2.46 (m, 2H), 2.33 (t, J= 7 Гц, 1H), 2.09 (m, 1H), 1.5-1.8 (m, 7H), 1.39 (s, 9H), 1.33 (m, 2H);
МС м/e 327 (MH+).
K раствору N-(Nε- Бок-аминокапроил)пиперидин-3- карбоксальдегида (0,69 г, 2,12 ммоль) в метаноле (10 мл) при комнатной температуре добавляли H-β- Ала-ОБн•ПТСК (0,74 г, 2,12 ммоль) и NaC NBH3 (0,13 г, 2,12 ммоль). Эту смесь перемешивали в течение 2,5 часов и выпаривали до получения белого твердого вещества. Это твердое вещество разделяли между насыщенным раствором NaHCO3(10 мл) и CH2Cl2 (50 мл) и слои разделяли. Водный слой экстрагировали CH2Cl2 (2х50 мл) и объединенные органические слои высушивали, выпаривали и очищали посредством проточной хроматографии (0,5% NH4OH/ 4-10% этанол/CH2Cl2) до получения бензилового эфира N-(Nε- Бок-аминокапроил)-3-пиперидинметиламинопропионовой кислоты в виде стекловидного вещества.
1H ЯМР (CDCl3) δ: 7.33 (m, 5H), 5.13 (s, 2H), 4.61 (m, 1H), 4.28 (m, 1H), 3.70 (m, 1H), 3.11 (m, 3H), 2.85 (m, 3H), 2.5 (m, 4H), 2.31 (t, J=4 Гц, 2H), 1.5-1.9 (m, 8H), 1.42 (s, 9H), 1.29 (m, 3H), 0.89 (m, 1H);
МС м/e 490 (MH+).
К раствору бензилового эфира N-(Nε- Бок-аминокапроил)-3- пиперидинметиламинопропионовой кислоты (0,28 г, 0,57 ммоль) и ТГФ (10 мл) при комнатной температуре добавляли водную HCl (3,4 мл, 1,0 H). Эту смесь перемешивали в течение 22 часов, выпаривали до стекловидного твердого вещества, растирали с диэтиловым эфиром (3х25 мл) и высушивали до получения белого порошка. Этот порошок растворяли в ТГФ (5 мл) и воде (10 мл), переносили в колбу Парра под атмосферу азота и действовали Pd/C (0,04 г, 10%). Эту смесь гидрогенизировали при 50 psi и комнатной температуре в течение 20 часов, фильтровали через целитовый фильтр и выпаривали приблизительно до 5 мл. На этот раствор действовали MeCN (25 мл), фильтровали, отмывали диэтиловым эфиром (2х25 мл) и высушивали до получения 19 в виде бесцветного стекловидного вещества (ЖВХР чистота > 95%) с температурой плавления 65oC-7 oC.
1H ЯМР (ДМСО-d6) δ: 9.31 (m, 2H), 8.12 (br.s, 3H), 4.18 (m, 2H), 3.70 (m, 1H), 3.04 (m, 2H), 2,67 (m, 5H), 2.51 (m, 1H), 2.35 (m, 3H), 1.87 (m, 2H), 1.58 (m, 4H), 1.42 (m, 2H), 1.30 (m, 4H);
МС м/e 300 (MH+).
Точная протонированная масса, вычисленная для C15H29N3O3 • 2HCl (372,3): 300,2287 amu. Обнаружено: 300,2306 amu.

Соединения, основанные на структуре фибриногена формулы I, где Х1 и Х2 - Н2 или О; Y - (СН2)m, СН(NНСОR3)-(СН2)m, CH(NH2(CH2)m; А - NHR1, C(:NH)NH2 или пиперидин или пирролидин; Z - (СН2)n , CH(CO2R4) (СН2)n; R1 - Н, алкил или CH(NH )NH2; R2 - Н, алкил; R3 - алкокси или алкил, R4 - алкил или арилалкил, R6 - Н, алкил или арилалкил, m - целое число 0-3, n - целое число 0-2, являются ингибиторами агрегации тромбоцитов, полезными для лечения нарушения свертывания крови. 7 з.п. ф-лы, 4 табл.
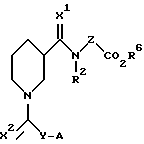
в которой x1 и x2 одинаковы или различаются и представляют H2 или O;
Y выбирают из (CH2)m, CH(NHCOR3)(CH2)m или CH(NH2)(CH2)m;
A выбирают из NHR1, C(:NH)NH2 или циклоалкильного кольца, содержащего атом азота, которое выбирают из пиперидин-2-ила, пиперидин-3-ила, пиперидин-4ила, пирролидин-2-ила и пирролидин-3-ила;
Z выбирают из (CH2)n или CH(CO2R4) (CH2)n;
R1 выбирают из H, алкила или CH(NH)NH2;
R2 выбирают из H или алкила;
R3 выбирают из алкокси или алкила;
R4 представляет алкил или арилалкил;
R6 представляет H, алкил или арилалкил;
m представляет целое число 0, 1, 2 или 3;
n представляет целое число 0, 1 или 2,
или его энантиомеры или его фармацевтически приемлемые соли.
Nα-Бок-L-Лиз(Кбз)-Нип-β-Ала-ОБн (соединение 1);
Nα-Бок-L-Лиз-Нип-β-Ала-ОН (соединение 2);
Nα-Бок-D-Лиз-Нип-β-Ала-ОН (соединение 3);
H-L-Лиз-Нип-β-Ала-ОН (соединение 4);
N-(Nε-аминокапроил)-Нип-β-Ала-ОН (соединения 5);
Nα-Ац-L-Лиз-Нип-Гли-ОН (соединение 6);
Nα-Ац-L-Лиз-Нип--β-Ала-ОН (соединение 7);
Nα-Бок-L-Арг-Нип-β-Ала-ОН (соединение 8);
Nα-Бок-L-Лиз-Нип-γ-аминомасляная кислота (соединение 9);
H-D-Лиз-Нип-β-Ала-ОН (соединение 10);
Nα-Бок-D-Лиз-Нип-γ-аминомасляная кислота (соединение 11);
Nα-Бок-D-Лиз-Нип-Гли-ОН (соединение 12);
Nα-Ац-D-Лиз-Нип-β-Ала-ОН (соединение 13);
Nα-Бок-D-Лиз-S-(+)-Нип-β-Ала-ОН (соединение 14);
Nα-Бок-L-Лиз(и-Пр)-Нип-β-Ала-ОН (соединение 15);
Nα-Бок-D-Лиз-R-(-)Нип-β-Ала-ОН (соединение 16);
N-[3-(4-пиперидинпропионил)]-Нип-β-Ала-ОН (соединение 17);
Nα-Бок-D-Лиз-Нип-L-Асп-ОМе (соединение 18) или
N-(Nε-аминокапроил)-3-пиперидинметиламинопропионовая кислота (соединение 19).
Приоритет по пунктам:
16.03.94 - по п.1;
27.12.94 - по пп.2 - 8 (уточнение признаков).
| Автоматический огнетушитель | 0 |
|
SU92A1 |
| Генератор равномерно распределенных псевдослучайных чисел | 1973 |
|
SU468231A1 |
| US 5219867 A, 15.06.93 | |||
| Машковский М.Д | |||
| Лекарственные средства, ч | |||
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| - М.: Медицина, 1987, с | |||
| Горный компас | 0 |
|
SU81A1 |

