Эта заявка является продолжением заявки, находящейся на рассмотрении на патент США, серийный номер 08/316761, поданной 3 октября 1996 г., которая является частичным продолжением заявки на патент США, серийный номер 08/229882, поданной 19 апреля 1994 г., в настоящее время отозванной, которая является частичным продолжением заявки на патент США, серийный номер 07/955783, поданной 2 октября 1992 г., в настоящее время патент США 5364562, выданный 15 ноября 1994 г., которая является частичным продолжением заявки на патент США, серийный номер 07/587884, поданной 25 сентября 1990 г., в настоящее время отозванной.
Предпосылки изобретения
1. Область изобретения
Настоящее изобретение относится к соединениям - производным аденозина и его аналогам, к фармацевтическим композициям, содержащим такие соединения, к их использованию в качестве кардиозащитных средств, которые уменьшают ишемическое поражение или размер инфаркта миокарда, вызванного ишемией миокарда, и к их использованию в качестве противолиполитических средств, которые понижают уровни липидов в плазме крови, уровни сывороточных триглицеридов и уровни холестерина в плазме крови.
Ишемия
Ишемия миокарда является результатом дисбаланса поступления кислорода в миокард и его расхода, и включает дисфункцию от физических нагрузок и вазоспазматическую дисфункцию миокарда. Ишемия, связанная с физическими нагрузками, обычно приписывается наличию критического атеросклеротического стеноза, включая большие коронарные артерии, приводящему к уменьшению субэндокадиального потока. Ангиоспастическая ишемия связана со спазмом очагового типа, возникновение которого не связано с напряжением или стрессом. Этот спазм лучше определяется как внезапное усиление тонуса сосуда. Механизмы ангиоспастической ишемии включают: (1) повышенный тонус сосуда в месте стеноза, вызванный увеличенным выходом катехоламина; (2) скоротечную внутреннюю закупорку и (3) выход вазоактивных средств, образованных тромбоцитами на месте поражений эндотелия.
Коронарная циркуляция является уникальной, поскольку она снабжает кровью орган, который создает перфузионное давление для всей циркуляционной системы. Таким образом, вмешательства, которые изменяют состояние периферической циркуляции и сужение, должны иметь значительное влияние на коронарную циркуляцию. Регуляторным компонентом мускулатуры коронарных сосудов являются мелкие коронарные артериолы, которые могут сильно изменять свой внутренний диаметр. Изменение внутреннего радиуса является результатом либо собственного сокращения гладкой мышцы сосуда (авторегуляция), либо внешнего сжатия из-за вентрикулярного сокращения. Суммарный эффект терапевтического лечения ишемической проблемы включает комплексное взаимодействие противоположных факторов, которые определяют приток и расход кислорода.
Кардиопротекция и предотвращение ишемического поражения
Разработка новых терапевтических средств, способных ограничивать размер поражения миокарда, то есть размер инфаркта миокарда, следующего за острой ишемией миокарда, является главной проблемой современной кардиологии.
Развитие тромболитической (растворяющей тромб) терапии в течение последних десяти лет демонстрирует, что раннее вмешательство во время сердечного приступа может давать в результате значительное уменьшение поражения ткани миокарда. Множество клинических опытов с тех пор показали документально, что тромболитическая терапия уменьшает риск развития расстройства сердечных сокращений, а также поддерживает способность сердца функционировать в качестве насоса. Это сохранение нормальной функции сердца, как показано, уменьшает последующую смертность после инфаркта.
Существует также интерес к разработке методов лечения, способных обеспечить дополнительную защиту миокарда, которые могли бы проводиться вместе с тромболитической терапией или сами по себе, поскольку ретроспективные эпидемиологические исследования показали, что смертность во время первых нескольких лет после инфаркта по- видимому связана с начальным размером инфаркта.
В предклинических исследованиях инфаркта, проведенных на различных животных-моделях, многие виды фармакологических средств, таких как блокаторы кальциевых каналов, аналоги простациклина и средства, способные ингибировать некоторые пути метаболизма, как было показано, способны снизить ишемическое поражение у некоторых видов животных.
Недавние исследования показали, что подвергая миокард на короткие периоды времени ишемии (прерывание потока крови к сердцу) с последующей реперфузией (восстановление притока крови) можно защитить сердце от последующего ишемического поражения, которое могло бы быть результатом более длительного периода ишемии. Это явление было названо предварительным кондиционированием миокарда, и, как предполагается, частично объяснимо высвобождением аденозина во время периода предварительного кондиционирования.
Другие исследования показали, что аденозин и агонисты аденозина уменьшают степень поражения ткани, наблюдаемое после прерывания притока крови к сердцу на различных моделях ишемического поражения у некоторых образцов (смотри, например, Toombs, С. et al., "Myocardial protective effects of adenosine. Infarct size reduction with pretreatment and continued receptor stimulation during ischemia.", Circulation 86, 986-994 (1992); Thornton, J. et al., "Intravenous pretreatment with A1-selective adenosine analogs protects the heart against infraction". Circulation 85, 659-665 (1992); и Downey, J. , "Ischemic preconditioning-nature's own cardio-protective intervention." Trends Cardiovasc. Med. 2(5), 170-176 (1992)).
Соединения по настоящему изобретению имитируют предварительное кондиционирование миокарда, тем самым уменьшая ишемическое поражение или производя уменьшение размера инфаркта миокарда, следующего за ишемией миокарда, и поэтому являются полезными в качестве кардиопротекторных средств.
Антилиполиз
Гиперлипидемия и гиперхолестериномия, как известно, являются двумя главными факторами риска для атеросклероза и коронарной болезни сердца, главной причины смерти и инвалидности в западных странах. Хотя этиология атеросклероза является многофакторной, развитие атеросклероза и состояний, включая болезнь коронарных сосудов, заболевание периферических сосудов и церебрососудистое заболевание, являющихся результатом ограничения притока крови, связаны с аномалиями уровней холестерина и липидов в плазме. Этиология гиперхолестериномии и гиперлипидимии является прежде всего генетической, хотя могут вносить свой вклад такие факторы, как диетический прием насыщенных жиров и холестерина.
Антилиполитическая активность аденозина и аналогов аденозина возрастает при активации A1 рецепторного подтипа (Lohse, M.J., Recent Advances in Receptor Chemistry, Melchiorre, С. и Gianella, Eds, Elsevier Science Publishers B. V. , Amsterdam, 1988, 107-121). Стимуляция этого подтипа рецептора понижает внутриклеточную концентрацию циклического АМФ в адипоцитах. Циклический АМФ является необходимым кофактором для фермента липопротеинлипазы, который гидролитически расщепляет триглицериды до свободных жирных кислот и глицерина в адипоцитах (Egan, J. J., et al., Proc. Natl. Acad. Sci. 1992 (89), 8357-8541). Соответственно, уменьшение внутриклеточной концентрации циклического АМФ в адипоцитах уменьшает активность липопротеинлипазы и, следовательно, гидролиз триглицеридов.
Повышенные давление крови и содержание липидов в плазме, включая триглицериды, являются двумя общепризнанными факторами риска, связанными со смертностью от сердечно-сосудистых заболеваний.
Для пациентов-диабетиков, у которых вероятность смертности от сердечно-сосудистых заболеваний является существенно более высокой, риск, связанный с этими факторами, еще более возрастает (Bierman, E.L., Arteriosclerosis и Thrombosis 1992 (12), 647-656). Кроме того, данные свидетельствуют, что избыточный липолиз является характерным для неинсулинзависимого диабета и, возможно, вносит вклад в инсулиновую невосприимчивость и гипергликемию (Swislocki, A.L., Horm. Metab. Res. 1993 (25), 90-95).
Соединения по данному изобретению используются в качестве противолиполитических средств при лечении и для уменьшения как сосудистых, так и метаболических факторов риска, и имеют особое значение и полезность.
Настоящее изобретение относится к классу агонистов аденозина и к их применению при лечении ишемии миокарда, в качестве кардиопротекторных средств, которые уменьшают ишемическое поражение или размер инфаркта миокарда вследствие ишемии миокарда, и в качестве противолиполитических средств, которые уменьшают уровни липидов в плазме, уровни триглицеридов в плазме и уровни холестерина в плазме, и к способам и промежуточным соединениям, используемым при получении таких соединений.
2. Опубликованные разработки
Аденозин имеет широкий спектр физиологического и фармакологического действия, включая заметное изменение сердечно-сосудистой и почечной функции. У животных и у человека внутривенная инъекция аденозинового нуклеотида вызывает гипотонию.
Физиологическое и фармакологическое действия аденозина опосредуются через специфические рецепторы, расположенные на клеточных поверхностях. Идентифицированы два подтипа рецепторов аденозина, обозначаемые как A1 и A2 рецепторы. A1 рецептор ингибирует образование сАМР путем подавления активности аденилатциклазы, в то время как стимуляция A2 рецепторов увеличивает активность аденилатциклазы и внутриклеточной сАМР. Каждый рецептор по-видимому опосредует специфическое действие аденозина в различных тканях: например, сосудистое действие аденозина по-видимому опосредуется через стимуляцию A2 рецепторов, которое поддерживается с помощью положительной корреляции между производством сАМР и вазорелаксацией в изолированной гладкой мышце сосудов, обработанных аденозином, тогда как стимуляция сердечных A1 рецепторов уменьшает генерацию сАМР в сердце, что вносит вклад в отрицательные дромотропные, инотропные и хронотропные сердечные эффекты. Далее, в отличие от большинства сосудорасширяющих средств, введение аденозина не вызывает рефлекса тахикардии.
Аденозин также оказывает заметное влияние на функцию почек. Интраренальная инфузия аденозина вызывает долговременное падение потока крови через почки и увеличивает невосприимчивость ренальных сосудов. При продолжении инфузии аденозина поток почечной крови возвращается к контрольным уровням, и невосприимчивость сосудов почек уменьшается. Начальные ренальные сосудосуживающие реакции на аденозин не являются вызванными прямым сосудосуживающим действием нуклеотида, но включает взаимодействие между аденозином и ренин- ангиотензинной системой.
Аденозин широко рассматривается в качестве первичного физиологического медиатора реактивной гиперемии и авторегуляции коронарного русла в ответ на ишемию миокарда. Сообщалось, что коронарный эндотелий обладает A2 рецепторами аденозина, связанными с аденилатциклазой, которая активируется параллельно с увеличением коронарного потока, и что рецепторы кардиомиоцитов являются преимущественно рецепторами аденозин A1 подтипа и связаны с брадикардией. Соответственно, аденозин дает уникальный механизм терапевтического лечения ишемии.
Сердечно-сосудистый ответ на аденозин является короткодействующими из-за быстрого извлечения и метаболизма эндогенного нуклеотида. В противоположность этому, аналоги аденозина являются более устойчивыми к метаболической деградации и, как сообщалось, вызывают длительные изменения артериального давления и частоты сердечных сокращений.
Были синтезированы несколько эффективных метаболически стабильных аналогов аденозина, которые демонстрируют различные степени селективности к двум подтипам рецепторов. Агонисты аденозина, как правило, проявляют более высокую селективность по отношению к A1 рецепторам по сравнению с A2 рецепторами. Циклопентиладенозин (СРА) и R-фенилизопропил-аденозин (R-PIA) являются стандартными агонистами аденозина, которые проявляют заметную селективность по отношению A1 рецептору (отношение A2/A1 = 780 и 106, соответственно). В противоположность этому, N-5'-этилкарбоксамидоаденозин (NECA) является сильным агонистом A2 рецептора (Ki-12 нМ), но имеет равное сродство к A1 рецептору (Ki - 6,3 нМ; соотношение A2/A1 = 1,87). До недавнего времени CV-1808 был наиболее селективным доступным агонистом A2 (A2/A1 = 0,19), даже несмотря на то, что соединение является в десять раз менее эффективным, чем NECA в его сродство к A2 рецептору. В недавних разработках не было описано никаких соединений, которые являются очень эффективными и селективными агонистами A2 (Ki= 3-8 нМ для A1; соотношение A2/A1 = 0,027 - 0,042).
Различные N6-арил- и N6-гетероарилалкилзамещенные аденозины и замещенные-(2-амино и 2-гидрокси)аденозины, как сообщалось в литературе, обладают различной фармакологической активностью, включая сердечную и циркуляторную активность. Смотри, например, патент Великобритании 1123245, патент Германии 2136624, патент Германии 2059922, патент Германии 2514284, Южно-Африканский патент 67/7630, патент США 4501735, Европейский патент 0139358 (описывающий N6-[геминал диарилзамещенные алкил] аденозины). Европейская патентная заявка 881068183 (описывающая, что N6- гетероциклзамещенные производные аденозина проявляют сердечную вазодилаторную активность), патент Германии 2131938 (описывающий арильные и гетероарилалкилгидразинильные производные аденозина), патент Германии 2151013 (описывающий N6-арил- и гетероарилзамещенные аденозины), патент Германии 2205002 (описывающий аденозины с N6-заместителями, содержащие мостиковые кольцевые структуры, связывающие N6-азот с заместителями, включающими тиенил) и Южно-Африканский патент 68/5477 (описывающий N6-индолилзамещенные-2-гидроксиаденозины).
Патент США 4954504 и Европейский патент 0267878 описывают по существу, что карбоциклические рибозные аналоги аденозина и их фармацевтически приемлемые сложные эфиры, замещенные по 2- и/или N6-положениям арилнизкоалкильными группами, включая тиенил, тетрагидрофуранил, тетрагидротиопиранил и бициклические бензоконденсированные 5- или 6-членные насыщенные гетероциклические низшийалкилпроизводные, проявляют свойства агониста аденозиновых рецепторов. Аденозиновые аналоги, имеющие заместители тиенильного типа, описаны в Европейском патенте 0277917 (описывающем N6-замещенные-2- гетероарилалкиламинозамещенные аденозины, включая 2-[(2-[тиен-2- ил]этил)амино]замещенный аденозин), патенте Германии 2139107 (описывающем N6-[бензотиенилметил]-аденозин), PCT WO 85/04882 (описывающем N6-гетероциклалкилзамещенные производные аденозина, включая N6-[2-(2-тиенил)этил]амино-9-(D-рибофуранозил)-9H- пурин, проявляют сердечно-сосудистую сосудорасширяющую активность, и что N6-хиральные заместители проявляют повышенную активность), опубликованной EP заявке 0232813 (описывающей, что N6-(1- замещенныетиенил) циклопропилметилзамещенные аденозины проявляют сердечно-сосудистую активность), патенте США 4683223 (описывающем, что N6-бензотиопиранилзамещенные аденозины проявляют противогипертонические свойства), PCT WO 88/03147 и WO 88/03148 (описывающих, что N6-[2-арил-2-(тиен-2-ил)]этилзамещенные аденозины проявляют противогипертонические свойства), патентах США 4636493 и 4600707 (описывающих, что N6-бензтиенилэтилзамещенные аденозины проявляют противогипертонические свойства).
Амиды аденозин-5'-карбоновых кислот описаны как имеющие применение в качестве противогипертонических и противостенокардических средств в патенте США 3914415, в то время как патент США 4738954 описывает, что N6-замещенные арил и арилалкиладенозин 5'-этилкарбоксамиды проявляют различные сердечные и противогипертонические свойства.
N6-алкил-2'-O-алкил аденозины описаны в публикации EP патенте 0378518 и в патентной заявке Великобритании 2226027 как имеющие противогипертоническую активность. N6-алкил-2'3'-ди-О- алкиладенозины также описаны как имеющие применение в качестве противогипертонических средств, патент США 4843066.
Аденозин-5'-(N-замещенные)карбоксамиды и их карбоксилатные эфиры и N1-оксиды описаны как коронарные сосудорасширяющие агенты. Stein, et al., J. Med. Chem. 1980, 23, 313-319 и J. Med. Chem. 19(10), 1180(1976). Аденозин-5'-карбоксамиды и их N1-оксиды также описаны в качестве слабых животных ядов в патенте США 4167565.
Противолиполитическая активность аденозина описана Dole, V.P., J. Biol. Chem.236 (12), 3125-3130(1961). Ингибирование липолиза с помощью (R)-N6 фенилизопропиладенозина описано Westermann, Е., et.al. Adipose Tissue, Regulation and metabolic Functions, Jeanrenaud, В. И Hepp, D.Eds., George Thieme, Stuttgart, 47-54 (1970). N6-моно- и дизамещенные аналоги аденозина описаны как имеющие противолиполитическую, противогиперхолестериномическую и противогипергликемическую активность в патентах США 3787391, 3817981, 3838147, 3840521, 3835035, 3851056, 3880829, 3929763, 3929764, 3988317 и 5032583.
Как полагают, обнаруженная токсичность, CNS свойства и увеличение частоты сердечных сокращений, связанные с аналогами аденозина, создают трудности, мешающие разработке коммерческих аналогов аденозина как противогипертонических/противоишемических средств. Настоящее изобретение относится к классу метаболически стабильных агонистов аденозина и их производных, описанных, например, в патентах США 4600707 и 5310731, неожиданно обладающих желаемыми фармакологическими свойствами, то есть являющихся кардиопротективными, противоишемическими и противолиполитическими средствами, имеющими уникальный терапевтический профиль.
Сущность изобретения
Соединения по настоящему изобретению описываются формулой I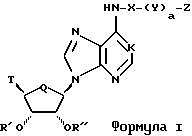
где K обозначает N, N--->O, или CH;
Q обозначает CH2 или О;
T обозначает
или R3O-CH2;
X обозначает алкилен с прямой или разветвленной цепью, циклоалкиленовую или циклоалкениленовую группу, каждый из которых необязательно замещен по меньшей мере одним CH3, CH3CH2, Cl, F, CF3 или CH3O;
Y обозначает NR4, О или S;
a = 0 или 1;
Z имеет формулу
Z1 обозначает N, CR5, (CH)m-CR5 или (CH)m-N, m равно 1 или 2;
Z2 обозначает N, NR6, О или S, n равно 0 или 1;
R1, R2 R3, R4, R5 и R6 обозначают, независимо, H, алкил, арил или гетероциклил;
Ra и Rb обозначают, независимо, H, ОН, алкил, гидроксиалкил, алкилмеркаптил, тиоалкил, алкокси, алкоксиалкил, амино, алкиламино, карбоксил, ацил, галоген, карбамоил, алкилкарбамоил, арил или гетероциклил; и
R' и R'' обозначают, независимо, водород, алкил, аралкил, карбамоил, алкилкарбамоил, диалкилкарбамоил, ацил, алкоксикарбонил, аралкоксикарбонил, арилоксикарбонил, или R' и R'' вместе могут образовывать
где Rc обозначает водород или алкил, 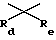 где Rd и Re обозначают, независимо, водород, алкил, или вместе с атомом углерода, которому они присоединены, могут образовывать 1,1-циклоалкильную группу;
где Rd и Re обозначают, независимо, водород, алкил, или вместе с атомом углерода, которому они присоединены, могут образовывать 1,1-циклоалкильную группу;
при условии, что, когда X обозначает алкилен с прямой цепью и Q обозначает кислород, тогда Z представляет гетероциклил, включающий, по крайней мере, два гетероатома;
или их фармацевтически приемлемыми солями.
Настоящее изобретение относится также к способам лечения сердечно-сосудистой болезни, отмеченной ишемией миокарда, используя фармацевтические композиции, содержащие противоишемически эффективное количество соединения формулы I, указанной выше, к способу уменьшения ишемического поражения или инфаркта миокарда, используя фармацевтические композиции, содержащие кардиопротекторное количество соединения формулы I. указанной выше, к способу лечения гиперлипидемии или гиперхолестеринемии, используя фармацевтические композиции, содержащие противолиполитическое количество соединения формулы I,
Подробное описание
Как используются выше и во всем описании изобретения, следующие термины, если не указано другого, должны пониматься как имеющие следующие значения:
"Ацил" обозначает алкил-C= O группу с прямой или разветвленной цепью. Предпочтительные ацильные группы являются низшими алканоилами, имеющими от 1 до 6 атомов углерода в алкильной группе.
"Алкил" обозначает насыщенную алифатическую углеводородную группу, которая может быть прямой или разветвленной и иметь от приблизительно 1 до приблизительно 20 атомов углерода в цепи. Разветвленный означает, что низшая алкильная группа, такая как метил, этил или пропил, присоединена к линейной алкильной цепи.
"Низший алкил" обозначает алкильную группу, имеющую от 1 до приблизительно 6 атомов углерода.
"Алкилен" обозначает прямую или разветвленную двухвалентную углеводородную цепь, имеющую от 1 до приблизительно 20 атомов углерода. Предпочтительные алкиленовые группы являются низшими алкиленовыми группами, имеющими от 1 до приблизительно 6 атомов углерода. Наиболее предпочтительными алкиленовыми группами являются метилен, этилен, этилэтилен, метилэтилен и диметилэтилен.
"Циклоалкилен" обозначает 1,2- или 1,3-двухвалентную карбоциклическую группу, имеющую от приблизительно 4 до приблизительно 8 атомов углерода. Предпочтительные циклоалкиленовые группы включают 4,5-цис- или транс-циклогексенилен, 1,2-циклогексанилен и 1,2-циклопентилен.
"Необязательно замещенный" обозначает, что данный заместитель или заместители могут быть как представлены, так и не представлены:
"Алкиламино" обозначает аминогруппу, замещенную одной или двумя алкильными группами, предпочтительные группы являются низшими алкиламиногруппами.
"Алкилкарбамоил" обозначает карбамоильную группу, замещенную одной или двумя алкильными группами. Предпочтительными являются низшие алкилкарбамоильные группы.
"Алкилмеркаптил" обозначает алкильную группу, замещенную меркаптильной группой. Меркаптильно - низкоалкильные группы являются предпочтительными.
"Алкокси" обозначает алкилоксигруппу, в которой "алкил" является тем, что описано выше. Низшие алкильные группы являются предпочтительными. Примерные группы включают метокси, этокси, н-пропокси, изопропокси и н-бутокси.
"Алкоксиалкил" обозначает алкильную группу, как описано выше, замещенную алкоксигруппой, как описано выше. "Аралкил" обозначает алкильную группу, замещенную арильным радикалом, где "арил" обозначает фенил или фенил, замещенный одним или более заместителями, которые могут быть алкилом, алкокси, амино, нитро, карбокси, карбоалкокси, циано, алкиламино, гало, гидрокси, гидроксиалкилом, меркаптилом, алкилмеркаптилом, карбалкилом или карбамоилом.
"Карбалкокси" обозначает карбоксильный заместитель, этерифицированный спиртом формулы CnH2n+1OH, где n равен от 1 до приблизительно 6.
"Галоген" (или "гало") обозначает хлор (хлоро), фтор (фторо), бром (бромо) или йод (йодо).
"Гетороциклил" обозначает кольцевую структуру, от приблизительно 4- до приблизительно 10-членной, в которой один или более атомов в кольце является элементом, отличающимся от углерода, например, N, О или S.
Представительные гетероциклические радикалы, содержащие N6-заместитель, соединений формулы I включают следующие:
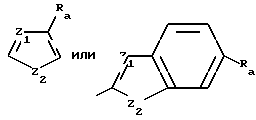
Предпочтительные гетероциклические группы включают незамещенные и замещенные тиенильную, тиазолильную и бензтиазолильную группы, где заместители могут быть одним или несколькими членами группы из алкокси, алкиламино, арила, карбалкокси, карбамоила, циано, гало, гидрокси, меркаптила, алкилмеркаптила или нитро.
"Гидроксиалкил" обозначает алкильную группу, замещенную гидроксигруппой. Группы гидроксинизшийалкил являются предпочтительными. Примерные предпочтительные группы включают гидроксиметил, 2-гидроксиэтил, 2-гидроксипропил и 3-гидроксипропил.
"Лекарство-предшественник" обозначает соединение, которое может быть или не быть биологически активным само по себе, но которое может с помощью метаболических, сольволитических или других физиологических средств быть превращенным в биологически активное химическое вещество.
"Кардиопротекция" относится к действию, от которого миокард делается менее чувствительным к ишемическому поражению и к инфаркту миокарда, следующему за ишемией миокарда.
"Уменьшение ишемического поражения" означает предотвращение или уменьшение ишемического поражения миокарда, следующего за ишемией миокарда. "Уменьшение размера инфаркта миокарда" означает уменьшение инфаркта миокарда или предотвращение инфаркта миокарда, следующего за ишемией миокарда.
Соединения формулы I включают, предпочтительно, хиральный (асимметричный) центр. Например, предпочтительные соединения, имеющие такой асимметричный центр, включают соединения, где X обозначает изопропилен и имеет либо R, либо S конфигурацию, и R конфигурация является наиболее предпочтительной. Изобретение включает индивидуальные стереоизомеры и их смеси. Индивидуальные изомеры получают и выделяют с помощью методов, хорошо известных в данной области или с помощью способов, описанных здесь.
Соединения по изобретению могут быть использованы в виде свободного основания, в виде солей кислот или в виде гидрата. Все эти формы находятся в рамках изобретения. Соли кислот являются просто более удобной формой для использования. На практике использование солевой формы изначально равнозначно использованию основной формы. Кислоты, которые могут быть использованы для получения солей кислот, включают предпочтительно те, которые образуют при соединении со свободным основанием фармацевтически приемлемые соли, то есть соли, анионы которых не являются токсичными для пациента при фармацевтических дозах солей, так что полезное кардиопротекторное, противоишемическое и противолиполитическое действие, производимое свободным основанием, не сопровождается побочными эффектами, приписываемыми анионам. Хотя фармацевтически приемлемые соли соединений по настоящему изобретению являются предпочтительными, все соли кислот используются в качестве источника формы свободного основания, даже если отдельная соль сама по себе является желательной только как промежуточный продукт, как, например, когда соль образуется только для целей очистки и идентификации или, когда она используется как промежуточный продукт при получении фармацевтически приемлемой соли путем ионного обмена. Фармацевтически приемлемыми солями в рамках изобретения являются такие, которые получают из следующих кислот: минеральные кислоты, такие как хлористоводородная кислота, серная кислота, фосфорная кислота, сульфаминовая кислота; и органические кислоты, такие как уксусная кислота, лимонная кислота, молочная кислота, винная кислота, малоновая кислота, метансульфоновая кислота, фумаровая кислота, этансульфоновая кислота, бензолсульфоновая кислота n-толуолсульфоновая кислота, циклогексилсульфаминовая кислота, хинная кислота и тому подобное. Соответствующие соли кислот включают следующие: гидрохлорид, сульфат, фосфат, сульфамат, ацетат, цитрат, лактат, тартрат, метансульфонат, фумарат, этансульфонат, бензолсульфонат, n-толуолсульфонат, циклогексилсульфонат и хиноат, соответственно.
Соли кислот соединений изобретения удобно получаются либо с помощью растворения свободного основания в водном или водно-спиртовом растворе, или в других подходящих растворителях, содержащих соответствующую кислоту, и выделения соли путем выпаривания раствора, или путем взаимодействия свободного основания и кислоты в органическом растворителе, в этом случае соль отделяется непосредственно или может быть получена с помощью концентрирования раствора.
Включенными в рамки формулы I являются классы соединений, которые могут быть охарактеризованы в общем как N6-гетероциклически замещенные аденозины; N6-гетероциклически замещенные карбоциклические аденозины (или, альтернативно, дигидрокси [N6-гетероциклическизамещенные-9-аденил] циклопентаны) и их N-оксиды; и N6-гетероциклическизамещенные-N'-1-деазааристеромицины (или, альтернативно, дигидрокси[N7-гетероциклическизамещенные [4,5-]имидазопиридил]циклопентаны). Также в рамках формулы I находятся 5'-алкилкарбоксамидные производные аденозинов, карбоциклических аденозинов и 1-деазоаристеромицинов, производные соединений упомянутых выше классов, в которых одна или обе 2- или 3-гидроксильных групп циклопентанового кольца, или в случаях классов соединений, содержащих рибозный остаток, 2'- или 3'-гидроксильные группы рибозного кольца, являются замещенными. Такие производные могут включать биологически активные химические вещества, используемые при лечении ишемии миокарда сами по себе, и в качестве кардиопротекторных и противолиполитических средств, или могут действовать в качестве лекарств-предшественников таких биологически активных соединений, которые образуются из них в физиологических условиях.
Представительные соединения изобретения включают: N6-[транс-2- (тиофен-2-ил)циклогекс-4-ен-1-ил] аденозин; N6-[транс-2- (тиофен-3-ил)циклогекс-4-ен-1-ил] аденозин; N6-[транс-2- (тиофен-2-ил)циклогекс-4-ен-1-ил] аденозин-5'-N-этилкарбоксамид; N6-[2-(2'-аминобензтиазолил)этил]аденозин; N6-[2-(2' -тиобензтиазолил)этил] аденозин; N6-[2-(6'-этоки-2'-тиобензтиазолил) этил] аденозин; N6-[2-(2'-аминобензтиазолил)-этил]аденозин-5' -N-этилкарбоксамид; N6-[2-(2'-аминотиазолил)этил] карбоциклический аденозин-5'-N-этилкарбоксамид; N6-[2-(4'-метилтиазол-5'-ил)этил] аденозин; N6-[2-(2'-тиазолил)-этил] аденозин; N6-[(R)-1-(5'-хлортиен- 2'-ил)-2-пропил] аденозин-5'-N-этилкарбоксамид; N6-[2-(2'-метил-4' -тиазолил)этил]-аденозин; N6-[(R)-1-метил-2-(2'-бенз[b] тиофенил) этил] аденозин; N6-[2-(4''-метил-5''-тиазолил)этил] карбоциклический аденозин-5'-N-этилкарбоксамид; N6-[2-(2''-тиазолил) этил] карбоциклический аденозин-5'-N-этилкарбоксамид; N6-[2- (4'-фенил-2'-тиазолил)этил] аденозин; N6-[(R)-1-(5''-хлор-2''- тиенил)про-2-ил] карбоциклический аденозин-5'-N-этилкарбоксамид; (-)-N6- [тиофен-2''-ил)этан-2-ил)карбоциклический аденозин-5'-N- этилкарбоксамид; N6-[1-(тиофен-3-ил)этан-2-ил) карбоциклический аденозин-5'-N-этилкарбоксамид; N6-[(R)-1- ((тиофен-2-ил)проп-2-ил)]карбоциклический аденозин-5'-N- этилкарбоксамид; N6-[1-(тиофен-2-ил)этан-2-ил] -N'-1- деазааристеромицин-5'-N-этилкарбоксамид; N6-[(R)-1-((тиазо-2- ил)проп-2-ил)] аденозин-5'-N-этилкарбоксамид; N6-[1-(тиофен- 2-ил)-2-метилпропил]аденозин-5'-N-этилкарбоксамид; N6-[(R)- 1-(5'-хлортиен-2-ил)-2-бутил]-карбоциклический аденозин-5'- N-этилкарбоксамид; N6-[2-(4'-метил-2'-тиазолил)этил] аденозин; N6-[4'-фенил-2'-тиазолил)-метил] аденозин; (-)-[2S-[ 2α,3α -диметилметилендиoкcи-4-β-[N6-[2-(5-хлор-2-тиенил)- (1R)-1-метилэтил] амино] -9-аденил] циклопентан] -1-β-N-этилкарбоксамид; (2S)-2α,3α-дигидрокси-4β-[N6-[2- (5-хлор-2-тиенил)-(1R)-1-метилэтил] амино] -9-аденил]циклопентан]-1β-N-этилкарбоксамид; (2S)-2α,3α-дигидрокси-4β-[N6-[2-(5- хлор-2-тиенил)-(1R)-1-метилэтил] амино]-9-аденил]циклопентaн]-1β-N-этилкapбoкcaмид-N1-oкcид;
[1S-[ 1α,2β,3β,4α (S*)]]-4-[7-[[2-(5-хлор-2-тиенил) -1-метил-этил]амино] -3H-имидазо[4,5-b] пиридин-3-ил] -N-этил- 2,3-дигидроксициклопентанкарбоксамид; [1S-[ 1α,2β,3β,4α ]]-4-[7-[[2- (3-хлор-2-тиенил)-1-этилэтил]амино]-3H-имидазо[4,5-b]пиридин-3-ил]-N-этил-2,3-дигидроксициклопентанкарбоксамид;
[1S-[ 1α,2β,3β,4α]]-4-[7-[[2-(2-тиенил)-1-изопропилэтил]-амино]- 3H-имидазо[4,5-b]пиридин-3-ил]-N-этил-2,3- дигидроксициклопентанкарбоксамид; [1S-[ 1α,2β,3β,4α (S*)]]-4- [7-[[2-(3- хлор-2-тиенил)-1-этилэтил]амино]-2H-имидазо[4,5-b]пиридин-3-ил] -N-этил-2,3-дигидроксициклопентанкарбоксамид;
[1S-[ 1α,2β,3β,4α (S*)]]-4-[7-[[2-(2-тиенил)-1-метилэтил]-амино] -3H-имидазо[4,5-b] пиридин-3-ил] -N-этил-2,3- дигидроксициклопентанкарбоксамид; [1S-[ 1α,2β,3β,4α]]-4-[7- [[2-(5-хлор-2-тиенил)-1-этилэтил]амино]-3H-имидазо [4,5-b] пиридин-3-ил] -N-этил-2,3-дигидроксициклопентанкарбоксамид; (2S)-2α,3α-бисметоксикарбонилокси-4 β -[N6-[2-(5-хлор-2-тиенил) -(1R)-1-метилэтил]амино-9-аденил]циклопентан-1β-N- этилкарбоксамид; (2S)- 2α,3α-дигидрокси-4 β-[N6-[2-(5-хлор-2-тиенил)-(1R) -1-метилэтил]амино-9-аденил] циклопентан-1β-N- этилкарбоксамидэтоксиметиленацеталь; (2S)-2α,3α-дигидрокси-4β-[N6-[2-(5-хлор-2-тиенил)-(1R)-1-метилэтил] амино-9-аденил] циклопентан-1β-N-этилкарбоксамид-2,3-карбонат; (2S)-2α,3α-бис-метилкарбамоилокси-4β-[N6-[2-(5-[хлор-2-тиенил) -(1R)-1-метилэтил] амино-9-аденил] циклопентан-1β-N-этилкарбоксамид; (2S)-2α,3α -дигидрокси-4 β-[N6-[2-(5-хлор-2-тиенил)-(1R)-1-метилэтил]амино-9-аденил]циклопентан-1β-N-этилкарбоксамид-2,3-тиокарбонат; N6-[2-(3-хлор-2-тиенил)-(1R)-1-метилэтил]-2' -О-метиладенозин; N6-[2-(5-хлор-2-тиенил)-(1R) -1-метилэтил] -2'-О-метиладенозин; и N6-[транс-5-(2-тиенил) циклогекс-1-ен-4-ил]-2'-О-метил-аденозин.
Предпочтительный класс соединений изобретения описан формулой I, где R' и R'' обозначают H.
Другим предпочтительным классом соединений изобретения являются 5'-алкилкарбоксамидные производные N6-гетероциклически замещенных карбоциклических аденозинов, другими словами, соединений формулы I, где K обозначает N, Q обозначает CH2 и T обозначает R1R2N-C=O или их фармацевтически приемлемые соли.
Еще другим предпочтительным классом соединений изобретения являются 5'-N-алкилкарбоксамидные производные N6-гетероциклически замещенных N'-1-деазааристеромицинов, то есть 4-[7- [гетероциклиламино]-3H-имидазол[4,5-b] пиридин-3-ил] алкил-2,3- дигидроксициклопенткарбоксамиды, другими словами, соединения формулы I, где K обозначает CH, Q обозначает CH2 и T обозначает R1R2N-C=O или их фармацевтически приемлемые соли.
Наиболее предпочтительный класс соединений по настоящему изобретению включает соединения формулы I, характеризуемые присутствием хирального центра α на атоме N6 пурина или 1-деазапуринового кольца, в то время как специальное осуществление этого класса включает соединения, характеризуемые хиральной этильной группой, присоединенной к атому углерода альфа на N6-азоте. Особенно предпочтительный класс соединений характеризуется N6-[1-низший алкил-2-(3-галотиен-2-ил) этил]замещающей группой.
Наиболее предпочтительные осуществления изобретения включают соединения (-)-[2S-[ 2α,3α -дигидрокси-4 β -[N6-[2-(5- хлор-2-тиенил)-(1R)-1-метилэтил] амино]-9-аденил]циклопентан]- 1 β -этилкарбоксамид; (-)-[2S-[ 2α,3α -дигидрокси-4 β [N6-[1-(R)- этил-2-(3-хлор-2-тиенил)этил]амино]-9-аденил] циклопентан] -1 β -этилкарбоксамид; [1S-[ 1α,2β,3β,4α (S*)]]-4- [7-[[2-(5-хлор-2-тиенил)-1-метилэтил] амино]-3H-имидазол[4,5-b] пиридин-3-ил]-N-этил-2,3-ди-гидроксициклопентанкарбоксамид;
[1S-[ 1α,2β,3β,4α (S*)]]-4-[7-[[2-(3-хлор-2-тиенил)-1- этилэтил]амино] -3H-имидазо[4,5-b]пиридин-3-ил]-N-этил-2, 3-дигидроксициклопентанкарбоксамид и их фармацевтически приемлемые соли.
Соединения по настоящему изобретению могут быть получены с помощью известных способов или в соответствии с последовательностями реакций, описанными ниже. Исходные материалы, используемые при получении соединений изобретения, являются известными или коммерчески доступными, или могут быть получены с помощью известных способов или конкретных схем реакций, описанных здесь.
Соединения формулы I, где К обозначает N, Q обозначает О и T обозначает R3O-CH2, могут быть получены путем взаимодействия коммерчески доступного 6-хлорпуринрибозида с различными гетероциклическими аминами, как представлено ниже.
Соединения формулы I, где K обозначает N, Q обозначает О и T обозначает R1R2N-C=O, получают подобным образом, начиная с продукта реакционной схемы A (см. в конце описания). В этой реакции 6-хлорпуринрибозид, у которого 2'- и 3'-гидроксильные группы рибозного кольца являются защищенными, обрабатывают оксидантом, например реагентом Джонса, и получаемую в результате кислоту обрабатывают либо дициклогексилкарбодиимидом (ДЦК) либо БОФ-Cl в присутствии выбранного амина с получением 5'-алкил-карбоксамидного производного.
Соответствующие исходные продукты для соединений формулы I, где K обозначает N, Q обозначает CH2 и T обозначает R1R2N-C=O могут быть получены как описано Chen et al. Tetrahedron Letters 30; 5543-46(1989). Альтернативно, для получения таких исходных продуктов может быть использована реакционная схема B. При осуществлении реакционной схемы В 4-этилкарбоксамидное производное 2,3-дигидроксициклопентиламина, получаемое, как описано Chen et al., подвергают взаимодействию с 3-амино-2,4-дихлорпиримидином. Продукт этой начальной реакции затем нагревают с альдегидиламидинацетатом, например формамидинацетатом, в диоксане и метоксиэтаноле, в течение времени, достаточного для осуществления замыкания кольца (от приблизительно 30 минут до приблизительно 4 часов), тем самым получая продукт, который может быть удобно подвергают взаимодействию с различными гетероциклическими аминами способом, описанным ниже, с получением соединения по изобретению. Порядок проведения реакции не является определяющим. Например, промежуточный продукт, получаемый по реакционной схеме B (см. в конце описания), может реагировать с гетероциклическим амином с последующим замыканием кольца, давая желаемый конечный продукт.
Различные гетероциклические амины, используемые для получения соединений по настоящему изобретению, могут быть получены с помощью одной или более реакций, представленных на реакционных схемах C-I (см. в конце описания) и в примерах получения от В до G, и от 50 до 74, здесь и далее (Het = гетероциклическая группа; Halo = галоген; R = например, H или низший алкил; Ra и Y указаны выше).
Последовательность реакций схемы I выше описана в патенте США 4321398 с соответствующей информацией, включенной здесь в качестве ссылки.
Пример B
Получение 1-(тиофен-3-ил)этиламина
3-Тиофенкарбоксальдегид (1 ммоль), нитрометан (1,5 ммоль) и бета-аланин (0,1 ммоль) подвергают взаимодействию в бутаноле в течение 6 часов, с получением 3-нитровинилентиофена, который восстанавливают литийалюминийгидридом (2,5 ммоль), с получением желаемого продукта, амина.
3-Замещенные тиенилалкиламины получают путем замены на 3-замещенные тиофены, такие как 3-хлортиофен, тиофеновых исходных продуктов в примере B, выше.
Пример C
Получение транс-2-(тиофен-2-ил)циклогекс-4-ениламина
Смесь 1,3-бутадиена (5 мл) и 2-нитровинилентиофена (7 г) в толуоле нагревают при 140oC в течение ночи в герметически закрытой пробирке. Полученный в результате нитроциклогексен гидрогенируют (~ 35 ф/дюйм2 H2) (5% Pd/C MeOH) и обрабатывают литий алюминий гидридом (2,5 г). Рацемический транс- 2-(тиофен-2-ил)циклогексиламин получают обычной обработкой.
Пример D
Общее получение 2-замещенных тиазоламинов
Бензоилхлорид и аминоэтилцианид взаимодействуют, давая N-бензоиламиноэтилцианин, который взаимодействует с гидросульфидом в аммиаке, давая тиоамид, который взаимодействует с соответствующим α -галокетоном, давая желаемый тиазол. Обработка 5 н соляной кислотой удаляет защитную бензоильную группу, давая желаемый аминовый продукт.
Пример E
Общее получение 4-замещенных тиазолиламинов
Предпочтительный синтез 2-(2'-метил-4'-тиазолил)-этиламина осуществляют путем реакции тиоацетамида с этилмоно- бромацетоацетатом, с получением тиазолового эфира, который восстанавливают, предпочтительно, боргидридом натрия, получая спирт, который преобразуют в амин. Предпочтительные приемы для получения амина включают обработку (1) диэтилазо-дикарбоксилатом, трифенилфосфином и фталимидом; и (2) гидразингидратом.
Получение 4-замещенных тиазоламинов может быть также проведено путем предшествующей реакционной схемы взаимодействием замещенного тиоамида и этилмонобромацетоацетата. Преобразование полученного в результате тиазолилового эфира в амид осуществляют водным раствором аммиака и амин получают с помощью восстановления бораном. Пример получения 2-(1,1-диметил-1'-тиофенил)этиламина описано в патенте США 4321398.
Диастереомерные смеси соединений или промежуточных продуктов, полученных по реакционным схемам A-I представленным ниже, могут быть разделены на отдельные рацемические или оптически активные энантиомеры способами, известными в данной области; например, с помощью хроматографии, фракционной дистилляции или фракционной кристаллизации d- или 1-(тартратных, дибензоил-тартратных, манделатных или камфарсульфонатных солей.
Пример F
Получение (+) и (-) транс-2-(тиофен-2-ил)циклогекс-4-ениламина
(S)-(+)-Миндальную кислоту (0,55 экв) добавляют к изопропаноловому раствору рацемического амина (3,4 г), полученного по примеру C. Осадок повторно кристаллизуют из изопропанола, получая 1,78 г соли ([α]DRT=+4,13 (с= 1,3, MeOH)). Амины выделяют путем экстракции нейтрализованных солей (насыщ. NaHCO3) с помощью CH2Cl2, высушивания (Na2SO4) и концентрирования, получая частично разделенные свободные амины.
Примерно 1 г левовращающего амина ([α]DRT=-25,8 (с=1,54, MeOH)) обрабатывают 2 г 1-(-)-дибензоилвинной кислоты в метаноле и полученную соль обрабатывают, получая 0,64 г левовращающего амина ([α]DRT=-28,8 (с=1,65, MeOH)). Анализ с помощью ЯМР высокого разрешения MPTA амида левовращающего амина показывает энантиомерный избыток >96%.
Примерно 1,6 г обогащенной правовращающим амином смеси обрабатывают 3,2 г d(+) - дибензоилвинной кислоты в метаноле. После обработки получают 0,87 г правовращающего амина ([α]DRT=+25,8 (с=1,67, MeOH)).
N6-Гетероциклическизамещенные аденозины и карбоциклические аденозины по изобретению могут быть получены путем взаимодействия 6-хлорпуринрибозида или продуктов реакционной схемы A или B с различными гетероциклическими аминами, в соответствии с последовательностью синтеза, представленным ниже на реакционной схеме J (см. в конце описания), где K, P, Q и T указаны выше.
N6-Гетероциклическизамещенные-N'алкил-деазааристеромицины по изобретению могут быть получены, как показано на реакционной схеме K (см. в конце описания).
Соединения по настоящему изобретению, которые могут действовать в качестве лекарств-предшественников, включают такие соединения, в которых гидроксильные группы рибозы или циклопентанового кольца замещены группами R' и R'', как описано выше в формуле I. Они могут быть получены известными способами и представлены с помощью примеров получения, показанных на реакционной схеме L (см. в конце описания).
Обработка дигидроксисоединений хлорформиатным эфиром в присутствии органического основания, например триэтиламина, должна давать соответствующий бис-карбонат. Алкоксиметиленацеталь может быть получен путем обработки соответствующим ортоэфиром в присутствии каталитического количества n-толуолсульфоновой кислоты. Карбонат становится доступным при обработке 1,1'- карбонилдиимидазола, а тиокарбонат - при обработке тиокарбонилдиимидизола. Алкил и диалкилкарбамоил производные могут быть получены обработкой соответствующим алкилизоцианатом или диалкилкарбамоилхлоридом, соответственно, в присутствии органического основания.
Соединения по настоящему изобретению, где K обозначает N--->O, т.е. N-оксиды, могут быть получены путем окисления соответствующего аденозина или карбоциклического аденозина известными способами, например обработкой перекисью водорода в уксусной кислоте.
2'-О-алкильные производные могут быть получены с помощью известных способов, например, взаимодействием соответствующего гетероциклиламина с 6-хлор-9-(2'-O-метил- β -D-рибофуранозил) -9H-пурином.
Функциональные группы исходных соединений и промежуточных продуктов, которые используются для получения соединений изобретения, могут быть защищены с помощью обычных защитных групп, известных в данной области. Удобные защитные группы для амина и гидроксильных функциональных групп описаны, например, в книге "Protective Groups in Organic Synthesis", T.W. Green, Wiley, New York (1984).
Гидроксильные группы могут быть защищены в виде сложных эфиров, таких как ацильные производные или в виде простых эфиров. Гидроксильные группы на соседних атомах углерода могут преимущественно быть защищены в форме кеталей или ацеталей. На практике соседние 2' и 3' гидроксильные группы исходных соединений в реакционных схемах A и B удобно защищаются с помощью, например, 2', 3'- изопропилиденовых производных. Свободные гидроксилы могут быть получены повторно с помощью, например, кислотного гидролиза или других реакций сольволиза или гидролиза, традиционно используемых в органической химии. После синтеза, соединения по изобретению обычно очищают с помощью жидкостной хроматографии среднего давления (MPLC) на хроматотроне, радиально ускоренной тонкослойной хроматографии, флэш-хроматографии или колоночной хроматографии на силикагеле или флорисиле с последующей кристаллизацией. Для соединений формулы I, где K обозначает N, Q обозначает О и T обозначает R3O-CH2, типичные системы растворителей включают хлороформ:метанол, этилацетат:гексан и метиленхлорид: метанол. Элюаты могут быть кристаллизованы из метанола, этанола, этилацетата, гексана или хлороформа.
Для соединений формулы I, где K обозначает N, Q обозначает О и T обозначает R1R2N-C=O, типичные системы растворителей включают хлороформ:метанол. Элюаты могут быть кристаллизованы из 50-100% этанола (водный раствор).
Для соединений формулы I, где Q обозначает СH2, K обозначает N или CH и T обозначает R1R2N-C=O, типичные системы растворителей включают метиленхлорид : метанол. Элюаты могут быть кристаллизованы из этилацетата с и или без метанола, этанола или гексана.
Соединения, требующие нейтрализации, могут быть нейтрализованы слабым основанием, таким как бикарбонат натрия, с последующей промывкой метиленхлоридом и насыщенным солевым раствором. Продукты, которые очищают в виде масел, иногда растирают с гексаном/этанолом перед конечной кристаллизацией.
Следующий аспект настоящего изобретения относится к усовершенствованному способу получения по существу оптически чистого 2-замещенного-2-амино-1-(гетероар-2- или 3-ил)этанового производного. 2-(Гетероарил)этиламины и их алкильные и фенильные производные получают различными путями, включая восстановление 2- β -нитровинилгетероарильных соединений, полученных из гетероарилформальдегидов (смотри, например, W. Foye и Tovivich, J. Pharm. Sci. 68(5), 591(1979), S.Conde, et al., J. Med. Chem. 21(9), 978 (1978), M. Dressler и М. Joullie, J. Het. Chem. 7,1257 (1970)); восстановление цианометил-гетероарильных соединений (смотри, например, В. Crowe и F. Nord, J. Org. Chem. 15, 81(1950), J. McFarland и Н. Howes, J. Med. Chem. 12, 1079(1969)); реакцию разложения Хофмана 2-(2-тиенил)пропиламида (смотри, например, G. Barger и A. Easson, J. Chem. Soc. 1938, 2100); и аминирование 2-(2-тиенил)этилпаратолуолсульфонатов, патент США 4128561.
Настоящий способ включает взаимодействие хирального 2-замещенного этиленоксидного производного с 2- или 3-ильного анионом гетероарильного соединения и преобразование с помощью стереоспецифических средств гидроксильной группы, образующейся в указанной реакции в аминогруппу. Способ по настоящему изобретению представлен в реакционной схеме М (см. в конце описания).
Где Sub обозначает замещающую группу указанного хирального этилен оксида, и Het обозначает гетероциклическую группу.
Преимущество способа по настоящему изобретению по сравнению со способами получения 2-замещенных-2-амино-1-(гетероар-2- или 3- ил)этановых производных, известными в данной области, состоит в том, что получение оптически чистых производных производится непосредственно, в отличие от рацемической смеси, которая должна затем быть разделена с помощью других способов, для получения оптически чистых изомеров.
Предпочтительная группа способов по настоящему изобретению является такой, в которой гетероар-2- или 3-ильная группа является замещенной или незамещенной тиен-2- или 3-илом или замещенной или незамещенной бензтиофен-2- или 3-ильной группой.
Более предпочтительной группой способов по настоящему изобретению является такая, в которой указанный анион образуется путем реакции замещенного или незамещенного тиофена или бензтиофена, имеющего водородный заместитель в 2- или 3-положении, с металлорганическим основанием в апротонном органическом растворителе.
Другой более предпочтительной группой способов по настоящему изобретению является такая, в которой указанный хиральный 2-замещенный этиленоксид является замещенным во 2-положении группой, выбираемой из группы, содержащей алкил, арил, тригалогенметил и бензилокси.
Наиболее предпочтительной группой способов по настоящему изобретению является такая, в которой указанное металлорганическое основание является алкиллитием или литийдиизопропиламидом, указанный протонный органический растворитель является тетрагидрофураном, эфиром, гексаном или смесью этих растворителей, и указанный хиральный 2-замещенный этиленоксид является 2- алкилэтиленоксидным производным.
Средства для стереоспецифического преобразования гидроксигруппы в аминогруппу являются хорошо известными в данной области (смотри, например, Mitsunobu, Synthesis 1981 (1), 1).
Следует учесть, что (R)- или (S)-2-замещенное-2-гидрокси-1- гетероарилэтановое производное может быть получено непосредственно, как описано выше, с помощью использования соответствующего (S)- или (R)-2-замещенного этиленоксидного производного в качестве исходного продукта или, если желательно или необходимо, полученный в результате (R) или (3)-2-замещенный-2-гидрокси-1-гетероарилэтан может быть преобразован в соответствующее (S) или (R)-2- замещенное-2-гидроксид-1-гетероарилэтановое производное, соответственно, с помощью средств, хорошо известных в данной области, для инверсии конфигурации по гидроксигруппе (смотри, например, Mitsunobu, Synthesis 1981 (1), 1).
Конкретное осуществление способа по настоящему изобретению является таким, в котором: (а) замещенный или незамещенный тиофен или бензтиофен, имеющий водородный заместитель во 2- или 3-положении, обрабатывают бутиллитием в смеси тетрагидрофурана и гексана при пониженной температуре, например при приблизительно -30oC, в течение времени, достаточного для образования аниона указанного тиофена или бензотиофена; (б) после этого добавляют (S) или (R)-2-алкилэтиленоксид, и смесь выдерживают при более высокой температуре, например, при около 0oC, в течение времени, достаточного для образования соответствующего (R) или (S)-2-алкил- 2-гидрокси-1-тиенильного или бензтиофенилэтанового производного; и (в) после этого преобразуют с помощью стереоспецифических средств гидроксигруппу указанного этанового производного в аминогруппу.
Способ по настоящему изобретению далее иллюстрируется и объясняется с помощью примеров от 50-74.
Примеры 1-3 описывают получение предшествующих соединений, используемых при получении соединений по настоящему изобретению, которые описаны ниже.
Пример 1
Получение 6-хлор-2', 3'-диметилметилендиоксид-N-5' -этилкарбоксамидоаденозина
Стадия 1: 2', 3'-диметилметиленовое производное 6-хлорпурин рибозида
6-Хлорпуринрибозид (31,5 г), триэтилортоформиат (73 мл) и TsOH (19,8 г) перемешивают в 600 мл ацетона в течение 2 часов при комнатной температуре. Раствор, реакционную смесь, концентрируют в вакууме, объединяют с этилацетатом и промывают насыщенным раствором NaHCO3 и насыщенным солевым раствором, сушат (Na2SO4) и концентрируют, получая 2',3'-диметилметиленовое производное 6-хлорпуринрибозида в виде белого твердого продукта.
Стадия 2: 6-хлор-2',3'-диметилметилендиоксиаденозин карбоновая кислота
Продукт со стадии 1 (10 г) подвергают окислению по Джонсу, кислоту экстрагируют из этилацетата 2,5% раствором NaOH, и водную часть промывают этилацетатом и подкисляют концентрированной HCl, и экстрагируют этилацетатом. Органический слой промывают H2O и насыщенным солевым раствором, сушат (Na2SO4), фильтруют и концентрируют в вакууме досуха, получая желаемую 5'-карбоновую кислоту.
Стадия 3: 6-хлор-2', 3'-диметилметилендиокси-N-5'-этилкарбоксамидоаденозин
Продукт со стадии 2 (5,7 г) перемешивают с БОФ-Cl (бис-(2-оксо-3- оксазолидинил)фосфонийхлорид) (4,26 г) и триэтиламином (2,33 мл) в 100 мл метиленхлорида в течение 20 минут при комнатной температуре. Этиламин (3,46 г) перемешивают до раствора, который перемешивают в течение 2 часов при комнатной температуре. Органическую часть промывают разбавленным раствором HCl, разбавленным NaOH, H2O и насыщенным солевым раствором и сушат (Na2SO4), получая конечный продукт в виде пены.
Пример 2
Получение (+)-2S-[ 2α,3α -диметилметилендиокси]-4 β -[6-хлор- 9-аденил] циклопентан-1- β -N-этилкарбоксамида
Стадия 1: 5,6-диметилендиокси-2-азабицикло[2.2.1]гептан-3-он
5,6-Дигидрокси-2-азабицикло[2.2.1] гептан-3-он (23,5 г) (Aldrich) или полученный по способу Cermak и Vince, Nucleic Acid Chemistry, Inproved и New Synthetic Procedures, Methods и Techniques, Part Three, page 26 (J.Wiley, 1986) растворяют в ацетоне (150 мл), содержащем 2,2-диметоксипропан (185 мл) и п-толуолсульфоновую кислоту (5,25 г), и смесь нагревают с обратным холодильником в течение 10 минут, охлаждают, обрабатывают NaHCO3 (9,3 г) и концентрируют в вакууме. Остаток растворяют в CH2Cl2, промывают насыщенным солевым раствором, сушат над MgSO4 и растворитель выпаривают, получая масло. Масло хроматографируют на SiO2 (4:1, этилацетат:гексан), получая 17,0 г (63%) рыжевато-коричневого прозрачного твердого продукта (т.пл. 153-154oC).
Стадия 2: (+)-4β-амино- 2α,3α диметилендиоксициклопентан -1 β -N-этилкарбоксамид
(A) 5,6-Диметилендиокси-2- азабицикло[2.2.1]гептан-3-он (5 г), полученный на стадии 1, обрабатывают этиламином (15 мл) при 140oC около 7 часов. Полученный в результате продукт очищают путем флэш-хроматографии (CH2Cl2/CH3OH/N, N-диметилэтиламин, (90/7/3)), получая (+)-4 β -амино- 2α,3α -диметилендиоксициклопентан -1 β -N-этилкарбоксамид (5,8 г).
(B) Обработка рацемического амина (13,1 г), полученного, как описано в части A, D-дибензоилвинной кислотой (21,6 г) дает 15,1 г энантиомерно чистой соли [α] DRT= +70,1 (C. 1,77, CH3OH). Соль растворяют в 10% водном NaOH, и водную фазу экстрагируют этилацетатом. Объединенные органические слои промывают насыщенным солевым раствором, сушат над MgSO4, и растворитель удаляют, получая оптически чистое соединение [α]DRT=-+31,4 [C.1,40, MeOH].
Стадия 3: 4-β-(3-амино-4-хлор-2-пиримидиниламино)-2,3- диметилендиоксициклопентан-1β -N-этилкарбоксамид
Конденсация (+)4β-амино- 2α,3α диметилендиоксициклопентан-1β-N-этилкарбоксамида (2,10 г), полученного на стадии 2, часть B, с 3-амино-2,4-дихлорпиридином (1,5 г) в н-бутаноле (70 мл), содержащем триэтиламин (3 мл), в течение около 14 часов при нагревании с обратным холодильником с последующим удалением растворителя в вакууме дает масло, которое растворяют в этилацетате и промывают водным NaHCO3. Органический экстракт сушат над Na2SO4 и концентрируют в вакууме, получая оптически чистое соединение. [ α ]DRT=+15,8 [C. 41,48, CH3OH].
Стадия 4: (+)-4 β -(3-амино-4-хлор-2-пиримидиниламино)- 2α,3α диметилендиоксициклопентан (2,10 г), формамидинацетат 1,85 г) в метоксиэтаноле (2 мл) и диоксане (80 мл) перемешивают при 70oC в течение около 3 часов. Смесь охлаждают до комнатной температуры и растворитель удаляют в вакууме. Остаток растворяют в этилацетате, который промывают водным NaHCO3 и насыщенным солевым раствором, органический экстракт сушат над Na2SO4, концентрируют в вакууме и очищают путем колоночной флэш- хроматографии (метиленхлорид/метанол 95: 5), получая чистый (+)-[ 2α,3α -диметилметилендиокси]-4 β -[6-хлор-9-аденил] циклопентан-1- β -N-этилкарбоксамид (1,45 г).
Альтернативно, оптически чистые 2α,3α -дизащищенные диокси-4 β -6-замещенные-9-аденил-циклопентан-1 β -этилкарбоксамидные производные могут быть получены по схеме реакции, приведенной в примере 3.
Пример 3
Получение 2S-[ 2α,3α -циклогесилидендиокси]-4 β -[N6- (2-тиенэтан-2-ил)-9-аденил]циклопентан-1-β-N-этилкарбоксамида
Стадия 1: 4 β -этилен-[2α,3α-[циклогесилидендиокси] циклопентанон
(-)-2α,3α-[Циклогесилидендиокси]-4-циклопентанон (2,95 г), полученный по способу Borchardt et al. , J. Org. Chem. 1987, 52, 5457, добавляют в виде раствора в ТГФ (5 мл) к смеси винилмагнийбромида (15,2 ммоль) и CuI (15,2 ммоль) в ТГФ (100 мл). Эту смесь выдерживают при -78oC в инертной атмосфере в течение около 2 часов, нагревают до 0oC и гасят насыщенным водным раствором NH4Cl. Органическую фазу промывают насыщенным солевым раствором, сушат над MgSO4 и концентрируют в вакууме, получая желтое масло, которое очищают путем флэш-хроматографии (метиленхлорид, 100%), получая 2,9 г желаемого соединения в виде масла.
Стадия 2: 4 β -этилен-1- β -гидрокси- 2α,3α [циклогесилидендиокси] циклопентан
3,95 мл 1 М раствора диизобутилалюминийгидрида в тетрагидрофуране добавляют к раствору ТГФ (75 мл) и кетона, полученного на стадии 1 (0,73 г), который охлаждают до -78oC. Смесь нагревают до -40oC в течение около 2,5 часов, обрабатывают 2 н NaOH (5 мл), нагревают до комнатной температуры и перемешивают в течение около 1,5 часов. Водную фазу экстрагируют диэтиловым эфиром и объединенные органические фазы промывают насыщенным солевым раствором, сушат над MgSO4 и концентрируют в вакууме до получения желтого масла, которое очищают путем колоночной флэш- хроматографии (метилен-хлорид/метанол, 95:5), получая 0,65 г чистого продукта в виде вязкого масла.
Стадия 3: 4 β -этилен-1 β -трифторметансульфонил- 2,3-[циклогексилидендиокси]циклопентан
Раствор 4 β -этилен-1 β -гидрокси- 2α,3α -[циклогексилидендиокси]циклопентана (0,65 г) в метиленхлориде (5 мл) и пиридине (0,24 мл) добавляют к перемешиваемому раствору трифторметилсульфонил ангидрида (0,49 мл) в метиленхлориде (25 мл) при 0oC в атмосфере аргона. Через приблизительно 20 мин насыщенный солевой раствор добавляют к реакционной смеси, органическую фазу сушат над Na2SO4, и растворитель удаляют в вакууме, получая желаемый, продукт в виде органического масла, который используют без дополнительной очистки.
Стадия 4: 1- β этилен-[ 2α,3α -циклогексилидендиокси]- 4- β -[N6-(2-тиенилэтан-2-ил)-9-аденил] циклопентан
Раствор N6-тиофенилэтилпурина (2,13 г), NaH (50% масляная дисперсия, 0,35 г) и 18-краун-6 (0,15 г) в ДМФ (60 мл) добавляют к раствору 4 β -этилен-1 β -трифторметилсульфонил- 2α,3α -[циклогексилидендиокси]циклопентана, полученному на стадии 4, в ДМФ (2 мл) при 0oC. Смесь перемешивают при 0oC в течение около 8 часов, гасят насыщенным NH4Cl, растворитель удаляют в вакууме и остаток объединяют с этилацетатом (100 мл) и насыщенным солевым раствором. Органический слой сушат над MgSO4 и концентрируют в вакууме, и сырой продукт очищают путем флэш-хроматографии (метиленхлорид/метанол (99:1)), получая 0,85 г чистого продукта.
Стадия 5: 2S[ 2α,3α -циклогексилидендиокси]-4- β -[N6- (2-тиенилэтан-2-ил)-9-аденил]циклопентан-1 β-N-этилкарбоксамид
Раствор 1β-этилен-[ 2α,3α циклогексилидендиокси]-4- β -[N6- (2-тиенилэтан-2-ил)-9-аденил] циклопентана (0,32 г) в 2 мл бензола добавляют к бензоловому раствору перманганата калия (0,29 г) и 18-краун-6 (0,016 г) при 0oC. Реакционную смесь выдерживают при комнатной температуре в течение около 6 часов, добавляют 5% водный NaOH (15 мл), и водную фазу фильтруют через Целит® и подкисляют до pH 5 1 н HCl, и экстрагируют этилацетатом. Органические экстракты сушат над MgSO4 и концентрируют в вакууме, получая 0,1 г [ 2α,3α -циклогексилидендиокси]-4-β-[N6-(2-тиенилэтан-2-ил)- 9-аденил] -циклопентан-1β-N-карбоксилата в виде желтого масла, которое растворяют в метиленхлориде (4 мл), содержащем дициклогексилкарбодимид (ДЦК) (0,044 г). Этиламин (0,4 мл) добавляют к смеси, которую перемешивают при комнатной температуре в течение около 18 часов, растворитель удаляют в вакууме и сырой продукт очищают путем флэш-хроматографии (метиленхлорид/метанол (98:2)), получая 0,077 г чистого продукта.
Пример 4
Получение N6-[транс-2-(тиофен-2-ил)циклогекс-4-ен-ил) - аденозина
Транс-2-(2'-тиофенил)циклогекс-4-ениламин (0,3 г), полученный по способу, описанному в примере C, выше, 6- хлорпуринрибозид (0,28 г) и триэтиламин (0,27 мл) в 20 мл этанола нагревают с обратным холодильником в течение ночи в атмосфере аргона. Реакционную смесь охлаждают до комнатной температуры, растворитель выпаривают и остаток очищают с помощью MPLC (хлороформ:метанол 95:5), последующая сушка в вакууме при приблизительно 80oC дает целевой продукт в виде твердого продукта, т. пл. 105-110oC; элементный анализ, C20H23N5O4S.
Пример 5
Получение N6-[транс-2-(тиофен-2-ил)циклогекс-4-ен-1-ил] аденозин-5'-N-этилкарбоксамида
Стадия 1: (+)Транс-2-(тиофен-2-ил)циклогекс-4-ениламин и 2',3'-диметилметилендиоксипроизводное 6-Cl-NECA подвергают взаимодействию в условиях, описанных в примере 4, давая 2',3'-диметилметилендиоксипроизводное целевого продукта.
Стадия 2: 2',3'-диметилметилендиоксипроизводное желаемого продукта смешивают с трифторуксусной кислотой/водой (90/10) в течение 30 мин при комнатной температуре, нейтрализуют, медленно выливая смесь в насыщенный раствор бикарбоната натрия и экстрагируют метиленхлоридом. Водный слой экстрагируют метиленхлоридом и органические слои объединяют, промывают насыщенным солевым раствором, сушат над сульфатом магния и фильтруют, отфильтрованный чистый раствор выпаривают. Остаток очищают с помощью флэш-хроматографии (метиленхлорид:метанол 9:1), сушат в вакууме, получая целевой продукт в виде белой прозрачной пены, т.пл. 112-117oC; C22H26N6O4S.
Пример 6
Получение (-)-[2S-[ 2α,3α-дигидpoкcи-4- β -[N6-[2-(5-хлор-2- тиенил)-(1R)-1-метилэтил]амино]-9-аденил]циклопентан]-1- β -N-этилкарбоксамида
Стадия 1: Оптически чистый (+)-[2S-[ 2α,3α -диметилметилендиокси] -4-β-(6-хлор-9-аденил] циклопентан-1- β -N-этилкарбоксамид, полученный как описано в примере 2, и 2'-R-(5-хлортиен-2-ил) -2-пропиламин, [α]DRT=-15,6 [C. 3,7, CH3OH], полученный как описано в примере 4, объединяют как описано в примере 4, получая 2,3-диметилметилен-диоксипроизводное целевого продукта.
Стадия 2: Диметилметилендиоксипроизводное со стадии (1) нагревают с обратным холодильником в 5 мл 50% водной муравьиной кислоты в течение около 3 часов. Охлажденную реакционную смесь выпаривают, к твердому осадку добавляют толуол и растворитель выпаривают. Остаток растворяют в этилацетате, промывают раствором бикарбоната натрия и насыщенным солевым раствором, сушат, фильтруют и выпаривают, получая после сушки в печи в течение ночи белый твердый продукт (0,240 г), т.пл. 188-4oC; C20H25N6SO3Cl, [α]DRT=-86,49 [C. 5,5, MeOH].
Примеры 7-29, 31-34
Следуя примерам 1-6 выше, получают соединения по настоящему изобретению, представленные в таблице 1. В примерах 7-21, 31 и 32 гетероциклический амин подвергают взаимодействию с коммерчески доступным 6-хлорпуринрибозидом; в примерах 22 и 23, гетероциклический амин подвергают взаимодействию с N6-хлор-5'-N-этилкарбоксамидоаденозином; и в примерах 24-31, 33 и 34, гетероциклический амин подвергают взаимодействию либо с (±), либо с (+)-[2S-[2α,3α] диметилметилендиокси-4-β-(6-хлор-9-аденил] циклопентан-1-β-N-этилкарбоксамид.
Пример 30
Получение (±)-N6-[1-(тиофенил-2-ил)этан-2-ил] -N'-1- деазааристе-ромицин-5'-N-этилкарбоксамида
Cтадия 1: 2-хлор-3-нитро-4-[2-(2-тиофенил)этил]аминопиридин
Смесь 2,4-дихлор-3-нитропиридина (1,5 г) 2-аминоэтилтиофенил (1 г) и триэтиламина (5 мл) нагревают с обратным холодильником в EtOH (60 мл). Реакционную смесь охлаждают, растворитель выпаривают, и остаток хроматографируют на силикагеле (10% гексан/CH2Cl2), получая желаемый продукт присоединения.
Стадия 2: (±)-1 β -N-этилкарбоксамид- 2α,3α -изопропилидендиокси-4 β -[2-(3-нитро-4-[2-(2-тиофенил)этил] аминопиридил)-амино]циклопентан
Смесь тиофениламинопиридина со стадии (1) (1,8 ммоль), (±)-1 β -N-этилкарбоксамид-4 β -амино- 2α,3α изопропилидендиоксициклопентана (0,3 г) и триэтиламина (0,3 мл) нагревают с обратным холодильником в нитрометане (15 мл) в течение около 5 часов. Растворитель удаляют и остаток извлекают метиленхлоридом, хроматографируют на силикагеле (2% метанол/хлороформ), получая твердый продукт, который используют как таковой на следующей стадии.
Стадия 3: (±)-1 β -N-этилкарбоксамид- 2α,3α -изопропилидендиокси-4 β -[2-(3-амино-4-[2-(2-тиофенил)этил] аминопиридил)-амино]циклопентан
Смесь нитросоединения со стадии (2) (0,39 г), Pd/C (0,01 г) в этаноле (7 мл) перемешивают в атмосфере водорода в течение около 5 часов. Катализатор отфильтровывают и после выпаривания фильтрата получают масло, которое очищают на Флорисиле (10% метанол/метиленхлорид), получая желаемый продукт в виде твердого продукта.
Стадия 4: (±)-N6-[1-(тиофенил-2-ил)этан)-2-ил] -N'-1- деазааристеромицин-5'-N-этилкарбоксамид
Смесь аминосоединения со стадии (3) (0,31 г) и формамидинацетата (0,72 г) в метоксиэтаноле (30 мл) нагревают с обратным холодильником в течение около 3 часов. Реакционную смесь охлаждают, растворитель выпаривают и к остатку добавляют воду (5 мл) и муравьиную кислоту (5 мл). Подкисленную смесь нагревают до 50oC в течение около 5 часов, после чего растворитель удаляют и остаток хроматографируют на силикагеле (10% метанол/метиленхлорид), получая масло, которое перекристаллизуют из этилацетата, получая желаемый продукт в виде кристаллического твердого вещества, т.пл. 155-156oC.
Оптически чистое соединение получают, используя + или - энантиомер циклопентанамина со стадии (2).
Пример 35
Получение (2S)- 2α,3α -дигидpoкcи-4 β [N6-[2-(5-хлор-2-тиенил)- 1(R)-1-метилэтил]амино-9-аденил]циклопентан-1 β -N- этилкарбоксамид-N1-оксида
Раствор (2S)- 2α,3α дигидрокси-4 β -[N6-[2-(5-хлор-2- тиенил)-1(R)-1-метилэтил] амино-9-аденил] циклопентан-1 β -N- этилкарбоксамида (0,25 г) и ледяной уксусной кислоты (20 мл) в 30% перекиси водорода (1 л) перемешивают в течение 4 дней при комнатной температуре и смесь концентрируют в вакууме. Остаток очищают путем флэш-хроматографии, элюируют 20% метанолом в этилацетате, затем перемешивают с горячим метанолом и фильтруют, получая желаемый продукт, т. пл. > 240oC.
Пример 36
Получение [1S-[ 1α,2β,3β,4α (S*)]]-4-[7-[[2-(5-хлор-2-тиенил)- 1-метилэтил] амино] -3H-имидазо[4,5-b]пиридин-3-ил]-N- этил-2, 3- дигидроксициклопентанкарбоксамида
Стадия 1: Получение 2-хлор-4-[2-(5-хлор-2-тиенил)-(1R)- 1-метилэтил]амино]-3-нитропиридина
Используя, по существу, процедуру примера 30, стадия 1, и очищая сырой продукт флэш-хроматографией, элюируя градиентом от 10% до 30% этилацетата в гептане, получают желаемый продукт из 2-(5-хлор-2-тиенил)-(1R)-1-метилэтиламина.
Стадия 2: Получение (-)-1β-N-этил-2α,3α-изопропилиден-диокси-4-β-[2-(5-хлор-2-тиенил)-(1R)-1-метилэтил] амино-3-нитро-2-пиридил] аминоциклопентанкарбоксамида
2-хлор-4-[2-(5-хлор-2-тиенил)-(1R)-1-метилэтил] амино-3- нитропиридин (0,68 г), (-)-1β-N-этил- 2α,3α-изопропилидендиокси -4β-аминоциклопентанкарбоксамид (0,381 г) и триэтиламин (0,85 мл) объединяют в метаноле (50 мл) и смесь нагревают с обратным холодильником в течение около 18 часов. Смесь концентрируют в вакууме и сырой продукт очищают путем флэш- хроматографии, элюируя 0,5% метанолом в метиленхлориде, получая желаемый продукт.
Стадия 3: Получение (-)-1 β -N-этил- 2α,3α -изопропилидендиокси -4β-[3-амино-4-[2-(5-хлор-2-тиенил)-(1R)-1-метилэтил] амино-2-пиридил] аминоциклопентанкарбоксамида
(-)-1 β-N-этил- 2α,3α -изопропилидендиокси-4β-[4-[2-(5- хлор-2-тиенил)-(1R)-1-метилэтил] амино-3-нитро-2-пиридил] -аминоциклопентанкарбоксамид (0,90 г) и хлорид дигидрат олова (II) (2,1 г) смешивают в этаноле (20 мл) и смесь нагревают при 70oC в течение около 30 минут. Смесь выливают на лед, слегка подщелачивают водным раствором бикарбоната натрия и водный раствор экстрагируют этилацетатом. Этилацетатный раствор сушат над сульфатом магния, фильтруют и концентрируют в вакууме, получая желаемый продукт, который используют без дополнительной обработки на следующей стадии.
Стадия 4: Получение [1S-[ 1α,2β,3β,4α (S*)]]-4- [7-[[2-(5-хлор-2-тиенил)-1-метилэтил] амино] -3H-имидазо[4, 5-b]пиридин-3-ил]-N-этил-2,3-дигидроксициклопентанкарбоксамида
Используя, по существу, процедуру примера 30, стадия 4, желаемый продукт, т. пл. 164-165oC, получают из (-)-1β-N- этил- 2α,3α -изoпpoпилидендиoкcи-4 β-[3-aминo-4-[2-(5-xлop-2- тиенил)-(1R)-1-метилэтил]амино-2-пиридил] аминоциклопентанкарбоксамида.
Используя, по существу, процедуру примера 30, получают соединения примеров из соответствующих исходных продуктов.
Пример 37
[1S-[1α,2β,3β,4α] ]-4-[7-[[2-(3-хлор-2-тиенил)-1-этилэтил]-амино] -3H-имидазо[4,5-b] пиридин-3-ил]-N-этил-2,3- дигидроксициклопентанкарбоксамид, т. пл. 79-82oC.
Пример 38
[1S-[ 1α,2β,3β,4α]]-4-[7-[[2-(2-тиенил)-1-изопропилэтил]- амино]-3Н-имидазо[4,5-b]пиридин-3-ил]-N-этил-2,3- дигидроксициклопентанкарбоксамид, т.пл. 75-85oC.
Пример 39
[1S-[1α,2β,3β,4α(S*)] ] -4-[7-[[2-(3-хлор-2- тиенил)-1-этилэтил]амино]-3Н-имидазо[4,5-b] пиридин-3-ил]-N-этил-2,3-дигидроксициклопентанкарбоксамид, т.пл. 75-78oC.
Пример 40
[1S-[ 1α,2β,3β,4α (S*)]]-4-[7-[[2-(2-тиенил)-1-метилэтил]-амино]-3Н-имидазо[4,5-b] пиридин-3-ил] -N-этил-2,3- дигидроксициклопентанкарбоксамид, т. пл. 155-60oC.
Пример 41
Получение [1S-[ 1α,2β,3β,4α ]]-4-[7-[[2-(5-хлор-2- тиенил)-1-этилэтил] амино] -3Н-имидазо[4,5-b] пиридин-3-ил]-N-этил-2,3-дигидроксициклопентанкарбоксамида.
Используя, по существу, процедуру примера 36, желаемый продукт, т.пл. 77-85oC, получают из 2-(5-хлор-2-тиенил)-(1R) -1-этилэтиламина.
Пример 42
Получение (2S)- 2α,3α-бис-метоксикарбонилокси-4β-[N6-[2-(5- хлор-2-тиенил)-(1R)-1-метилэтил]амино-9-аденил] циклопентан-1β-N-этилкарбоксамида
К раствору (2S)- 2α,3α -дигидрокси-4 β-[N6-[2-(5- хлор-2-тиенил)-(1R)-1-метилэтил] амино-9-аденил] циклопентан-1β- этилкарбоксамида (0,56 г), триэтиламина (0,5 мл) и 4-диметиламинопиридина (1 мг) в тетрагидрофуране (25 мл) добавляют метилхлорформиат (0,21 мл) и раствор перемешивают при комнатной температуре в течение 1 часа. Смесь разбавляют этилацетатом, промывают насыщенным солевым раствором и органический раствор сушат над сульфатом магния, фильтруют и концентрируют в вакууме. Сырой продукт перекристаллизуют из гексана/этилацетата, получая желаемый продукт, т.пл. 74-76oC.
Пример 43
Получение (2S)- 2α,3α -дигидрокси-4 β [N6-[2-(5-хлор-2-тиенил) (1R)-1-метилэтил] амино-9-аденил] циклопентан-1 β -N- этилкарбоксамида этоксиметиленацеталя
Раствор (2S)- 2α,3α -дигидрокси-4 β -[N6-[2-(5-хлор-2- тиенил)-(1R)-1-метилэтил] амино-9-аденил] циклопентан-1 β -N-этилкарбоксамида (0,14 г), триэтилортоформиата (3 мл) и п-толуолсульфоновую кислоту (1 мг) нагревают с обратным холодильником в течение около 1 часа и растворитель затем отделяют в вакууме. Остаток растворяют в этилацетате и раствор промывают насыщенным солевым раствором, сушат над сульфатом натрия, фильтруют, концентрируют в вакууме. Сырой продукт очищают путем флэш-хроматографии, элюируют 5% метанолом в метиленхлориде, с последующей перекристаллизацией из гексана/этилацетата, получая желаемый продукт, т.пл. 67-70oC.
Пример 44
Получение (2S)- 2α,3α -дигидрокси- 4 β -[N6-[2-(5-хлор-2- тиенил)-(1R)-1-метилэтил] амино-9-аденил] циклопентан-1 β-N-этилкарбоксамид-2,3-карбоната
Раствор (2S)- 2α,3α дигидрокси-4β[N6-[2-(5-хлор-2-тиенил -(1R)-1-метилэтил]амино-9-аденил]циклопентан-1β-N- этилкарбоксамида (0,17 г) и 1,1'-карбонилдиимидазола (0,071 г) в бензоле (5 мл) нагревают с обратным холодильником в течение 5 часов, затем перемешивают при 60oC в течение примерно 18 часов. Раствор промывают насыщенным соляным раствором, сушат над сульфатом магния, фильтруют и концентрируют в вакууме. Остаток очищают путем флэш-хроматографии, элюируют 5% метанолом в метиленхлориде с последующей кристаллизацией из гексана/этилацетата, получая желаемый продукт, т. пл. 87- 89oC.
Пример 45
Получение (2S)- 2α,3α -бис-метилкарбамоилокси-4 β -[N6-[2-(5- хлор-2-тиенил)-(1R)-1-метилэтил]амино-9-аденил] циклопентан-1 β -N-этилкарбоксамида
К раствору (2S)- 2α,3α -дигидрокси-4 β -[N6-[2-(5-хлор-2- тиенил)-(1R)-1-метилэтил] амино-9-аденил] циклопентан-1 β -N-этилкарбоксамида (0,16 г) в тетрагидрофуране (5 мл) добавляют метилизоцианат (0,05 мл) и 1,8-диазабицикло [5.4.0]ундек-7-ен (1 каплю). Раствор перемешивают при 50oC в течение около 2,5 часов, охлаждают до комнатной температуры, разбавляют этилацетатом и промывают насыщенным солевым раствором. Органический раствор промывают насыщенным солевым раствором, сушат над сульфатом магния и концентрируют в вакууме. Остаток очищают путем флэш-хроматографии, элюируют 5% метанолом в метиленхлориде, с последующей кристаллизацией из гексана/этилацетата, получая желаемый продукт, т.пл. 97-99oC.
Пример 46
Получение (2S)- 2α,3α -дигидpoкcи-4 β -[N6-[2-(5-хлор-2-тиенил)- (1R)-1-метилэтил]амино-9-аденил]циклопентан-1 β -N-этилкарбоксамид -2,3-тиокарбоната
Раствор (2S)- 2α,3α -дигидрокси-4 β -[N6-[2-(5-хлор-2- тиенил)-(1R)-1-метилэтил] амино-9-аденил] циклопентан-1 β -N- этилкарбоксамида (0,35 г) и тиокарбонилдиимидазола (0,134 г) в бензоле (10 мл) нагревают при 45oC в течение около 2 часов. Раствор промывают насыщенным солевым раствором, сушат над сульфатом магния и концентрируют в вакууме. Остаток очищают путем флэш-хроматографии, элюируют 5% метанолом в гексане, с последующей кристаллизацией из гексана, получая желаемый продукт, т.пл. 115-117oC.
Пример 47
Получение N6-[2-(3-хлор-2-тиенил)-(1R)-1-метилэтил] -2'-O-метиладенозина
Раствор 6-хлор-9-(2'-O-метил-β-D-рибофуранозил)-9Н- пурина (полученного согласно Европейской патентной публикации 0378518) (0,28 г), 2-(3-хлор-2-тиенил)-(1R)-1-метилэтиламина (0,163 г) и триэтиламин (0,5 мл) в этаноле (30 мл) нагревают с обратным холодильником в течение около 18 часов, охлаждают и концентрируют в вакууме. Остаток очищают путем флэш- хроматографии, элюируют 10% метанолом в метиленхлориде, с последующей кристаллизацией из гексана/этилацетата, получая желаемый продукт, т.пл. 75-76oC.
Пример 48
Получение N6-[2-(5-хлор-2-тиенил)-(1R)-1-метилэтил] -2'-O-метиладенозина
Используя, по существу, процедуру примера 47, желаемый продукт, т.пл. 84-85oC, получают из 2-(5-хлор-2-тиенил)-(1R)-1- метилэтиламина.
Пример 49
Получение N6-[транс-5-(2-тиенил)циклогекс-1-ен-4-ил] -2'-O-метиладенозина
Используя, по существу, процедуру примера 47, желаемый продукт, т.пл. 86-89oC, получают из транс-2-(2-тиенил) циклогекс- 4-ениламина.
Пример 50
Получение 1(R)-2-(5-хлор-2-тиенил)-1-метилэтиламина
Стадия 1: Получение 1(S)-2-(5-хлор-2-тиенил)-1- гидрокси-1-метилэтана
Раствор 2-хлортиофена (8,17 г) в тетрагидрофуране (80 мл) охлаждают до -30oC и добавляют по каплям 1,6 М н-бутиллитий в гексане (43,0 мл). Смесь перемешивают при -30oC в течение около 1 часа, добавляют (S)-пропиленоксид (4,00 г) и смесь подогревают до 0oC и перемешивают при этой температуре в течение 3 часов. Реакцию гасят насыщенным водным раствором аммоний хлорида, разбавляют эфиром и слои разделяют. Органический слой промывают насыщенным солевым раствором, сушат над сульфатом магния и концентрируют в вакууме, получая желаемый продукт.
Стадия 2: Получение 1(R)-2-(5-хлор-2-тиенил)-1-метил-1- фталимидоэтана
К раствору 1(S)-2-(5-хлор-2-тиенил)-1-гидрокси-1- метилэтана (8,8 г), трифенилфосфина (13,1 г) и фталимида (7,35 г) в тетрагидрофуране (80 мл) по каплям добавляют диэтилазодикарбоксилат (7,9 мл). Раствор перемешивают в течение около 18 часов и растворитель удаляют в вакууме. Остаток очищают путем флэш-хроматографии, элюируют 20% гексаном в метиленхлориде, получая желаемый продукт.
Стадия 3: Получение 1(R)-2-(5-хлор-2-тиенил)-1- метилэтиламина
1(R)-2-(5-хлор-2-тиенил)-1-метил-1-фталимидоэтан (13,0 г) растворяют в этаноле (75 мл) и добавляют гидразингидрат (2,5 мл), и смесь перемешивают при нагревании с обратным холодильником в течение около 1 часа. Смесь охлаждают до комнатной температуры, твердый продукт удаляют путем фильтрации и фильтрат концентрируют в вакууме. Остаток растворяют в этилацетате и этот раствор перемешивают с 5 н водным раствором соляной кислоты. Слои разделяют и у водного раствора устанавливают pH>10 10% раствором гидроксида натрия, затем экстрагируют этилацетатом. Органический раствор промывают насыщенным солевым раствором, сушат над сульфатом магния, фильтруют и концентрируют в вакууме, получая желаемый продукт [ α ]D=-22,96 (с=11,5, метанол).
Пример 51
Получение 1 (R)-2-(2-тиенил)-1-метилэтиламина
Стадия 1: Получение 1(S)-2-(2-тиенил)-1-гидрокси-1-метилэтана
Используя, по существу, процедуру примера 50, стадия 1, желаемый продукт получают из тиофена.
Стадия 2: Получение 1(R)-2-(2-тиенил)-1-метил-1-фталимидоэтана
Используя, по существу, процедуру примера 50, стадия 2, желаемый продукт получают из 1(S)-2-(2-тиенил)-1-гидрокси-1-метилэтана.
Стадия 3: Получение 1(R)-2-(2-тиенил)-1-метилэтиламина
Используя, по существу, процедуру примера 50, стадия 3, желаемый продукт [ α ] D=-15,6o (с=1, метанол) получают из 1(R)-2-(2-тиенил)-1-метил-1-фталимидоэтана.
Пример 52
Получение 1(S)-2-(5-хлор-2-тиенил)-1-метилэтиламина
Стадия 1: Получение 1(S)-2-(5-хлор-2-тиенил)-1-гидрокси-1- метилэтана
К перемешиваемому раствору 1(S)-2-(5-хлор-2-тиенил)-1- гидрокси-1-метилэтана (5,70 г) в тетрагидрофуране (100 мл) добавляют трифенилфосфин (5,34 г) и бензойную кислоту (2,49 г). Диэтилазодикарбоксилат (3,22 мл) добавляют по каплям и смесь перемешивают при комнатной температуре в течение около 18 часов. Растворитель удаляют в вакууме. Остаток очищают путем флэш- хроматографии, элюируют 30% гексаном в метиленхлориде, получая (R)-3-(5-хлор-2-тиенил)-2-пропилбензоат. Эфир (3,91 г) растворяют в диоксане (50 мл) и добавляют 20% водный раствор гидроксида натрия (15 мл). Смесь нагревают до 55oC в течение 3 часов и концентрируют в вакууме. Остаток извлекают этилацетатом (200 мл) и органический слой промывают насыщенным солевым раствором, сушат над сульфатом магния, фильтруют, концентрируют в вакууме, получая желаемый продукт.
Стадия 2: Получение 1(S)-2-(5-хлор-2-тиенил)-1-метилэтиламина
Используя, по существу, процедуру примера 50, стадия 2 и 3, желаемый продукт, [ α ]D=+21,71o (с=1,1, метанол) получают из 1(S)-2-(5-хлор-2-тиенил)-1-гидрокси-1-метилэтана.
Используя, по существу, процедуры Примеров 50, 51 и 52, следующие соединения получают из соответствующих исходных продуктов.
Пример 53
1(R)-2-(бензтиофен-2-ил)-1-метилэтиламин
Пример 54
1(S)-2-(2-тиенил)-1-метилэтиламин, [αD]= 15,5o (c=1, метанол)
Пример 55
1(R)-2-(3-бром-2-тиенил)-1-метилэтиламин
Пример 56
1(R)-2-[5-(2-пиридил)-2-тиенил]-1-метилэтиламин
Пример 57
1(R)-2-[5-(2-тиенил)-2-тиенил]-1-метилэтиламин
Пример 58
1(R)-2-(5-фенил-2-тиенил)-1-метилэтиламин
Пример 59
1(R)-2-(5-метокси-2-тиенил)-1-метилэтиламин
Пример 60
1(R)-2-(5-метил-2-тиенил)-1-метилэтиламин
Пример 61
1(R)-2-(5-бром-2-тиенил)-1-метилэтиламин
Пример 62
1(R)-2-(5-йод-2-тиенил)-1-метилэтиламин
Пример 63
1(R)-2-(5-метилтио-2-тиенил)-1-метилэтиламин
Пример 64
1(R)-2-(5-метилсульфонил-2-тиенил)-1-метилэтиламин
Пример 65
1(R)-2-(5-этил-2-тиенил)-1-метилэтиламин
Пример 66
1(R)-2-(5-н-гептил-2-тиенил)-1-метилэтиламин
Пример 67
1(R)-2-(3-метил-2-тиенил)-1-метилэтиламин
Пример 68
1(R)-2-(4-метил-2-тиенил)-1-метилэтиламин
Пример 69
1(R)-2-(3-хлор-2-тиенил)-1-метилэтиламин, [ α ]D=-6,1o (с=1, метанол).
Пример 70
1(R)-2-(4-хлор-2-тиенил)-1-метилэтиламин
Пример 71
1(R)-2-(3-хлор-5-фенил-2-тиенил)-1-метилэтиламин
Пример 72
1(R)-2-(5-бром-2-хлор-2-тиенил)-1-метилэтиламин
Пример 73
1(R)-2-(4-метил-5-хлор-2-тиенил)-1-метилэтиламин
Пример 74
1(R)-2-(2,5-дихлор-3-тиенил)-1-метилэтиламин
Соединения по настоящему изобретению увеличивают коронарный поток крови и, соответственно, используются при лечении ишемии миокарда; они также действуют в качестве кардиопротекторных средств, используемых для предотвращения или уменьшения ишемического поражения миокарда вследствие ишемии миокарда; они также действуют как противолиполитические средства, используемые для лечения гиперлипидемии и гиперхолестериномии.
Соединения в рамках по настоящему изобретению проявляют активность при стандартных анализах связывания A1/A2 рецепторами для определения активности агонистов рецепторов аденозина у млекопитающих. Примерные процедуры тестов, которые являются полезными при определении сродства к связыванию соединений по настоящему изобретению рецепторами, описаны ниже.
Определение сродства к связыванию рецептора аденозина in vitro
Сродство к связыванию A1 рецептора определяется с помощью конкурентносвязывающего анализа, основанного на лигандном замещении 3H-CHA (циклогексиладенозин) [Research Biochemicals Inc., Natick, Mass.] на рецепторе, используя препарат мембраны от целого мозга крысы в соответствии с процедурой R. F. Bruns et ali., Mol.Pharmacol., 29:331 (1986). Неспецифическое связывание оценивается в присутствии 1 ммоль теофилина.
Сродство к связыванию A2 рецептора определяется с помощью подобной методики анализа, основанной на лигандном замещении 3H-CGS 21680, специфического к A2 рецептору известного агониста аденозина, на рецепторе, используя мембраны от бороздчатого тела мозга крысы. Неспецифическое связывание оценивается в присутствии 20 мкм 2-хлораденозина.
Анализы осуществляются в стеклянных пробирках для тестов попарно при 25oC. Как только добавляют мембраны, пробирки поворачивают и инкубируют при 25oC в течение 60 минут (анализ A1) или 90 минут (анализ A2) во вращательном шейкере. Пробирки для анализа поворачивают в середине инкубации и повторно перед концом. Анализы завершают быстрой фильтрацией через 2,4 см GF/B, используя коллектор клеток Brandel Cell Harvestor. Пробирки для анализов промывают три раза холодным 50 ммоль трис-HCl (pH 7,7 или 7,4), при фильтрации, завершающейся в течение 15 секунд. Влажные фильтровальные кружочки помещают в стеклянные сцинциляционные флаконы, наполненные 10 мл Aquasoll II (New Englad Nuclear). Флаконы оставляют во вращательном шейкере и помещают в анализатор сцинцилляций в жидкости для отсчетов в течение двух минут. Значения IC50 для связывания рецепторов, то есть концентрация, при которой соединение изобретения замещается стандартной радиохимической меткой, получают используя компьютерную программу подгонки кривых (RS/1, Bolt, Beranek and Newman, Boston, MA).
Определение кардиопротекторной активности у крыс
1. Общая хирургическая подготовка
Взрослых крыс Sprague-Dawley анестезируют инактином (100 мг/кг подкожно). Трахею интубируют, и вентиляция при положительном давлении обеспечивается через маленький респиратор для животных. Катетеры помещают в бедренную вену и артерию для введения соединений по настоящему изобретению, подлежащих тестированию, и измерения кровяного давления соответственно. Надрез делают на левой стороне грудной клетки над грудными мышцами и мышцы отделяют, чтобы обеспечить доступ к четвертому межреберному пространству. Грудную полость вскрывают и обеспечивают доступ к сердцу. Отрезок пролиновой нити 4-0 помещают через вентрикулярную стенку вблизи левой большой коронарной артерии и используют для прерывания потока крови через коронарную артерию с помощью затягивания скользящего узла. Анализатор пульсирующего потока (прибор, который измеряет поток крови) помещают на поверхности сердца, чтобы подтвердить, что коронарная артерия правильно идентифицирована. Катетер также помещают в левом желудочке для мониторинга функции левого желудочка во время эксперимента.
2. Процедуры предварительного кондиционирования и тестовые процедуры
Для предварительного кондиционирования сердца коронарную артерию перекрывают (поток прерван) в течение периода в две минуты. Скользящий узел затем распускают для возобновления потока (реперфузия) в течение периода в три минуты. Эта процедура перекрывания/реперфузии повторяется дважды. Через пять минут после завершения последнего этапа предварительного кондиционирования артерию повторно перекрывают в течение 30 минут с последующей реперфузией в течение трех часов. Когда исследуется соединение по настоящему изобретению, вместо осуществления процедуры перекрывания/реперфузии инфузируют соединение в течение 30 минут. После окончания трехчасового периода реперфузии артерию повторно перекрывают, и 1 мл красителя патента синего вводят в левый вентрикулярный катетер, и сердце останавливают путем внутривенного введения хлорида калия. Эта процедура дает возможность красителю проникнуть в нормальные области сердца, в то время как часть сердца, которую сделали ишемической, не потребляет краситель (это область риска, "риск-область"). Сердце быстро удаляют для определения размера инфаркта. Область размера инфаркта определяют путем приготовления срезов сердца от верхушки до основания в виде от 4 до 5 срезов толщиной 1-2 мм. Срезы инкубируют в растворе 1%-ного трифенилтетразолия в течение 15 минут. Краситель взаимодействует с живой тканью и вызывает ее переход в кирпично-красный цвет. Инфарктная ткань не реагирует с красителем и является матово-белой на вид. Срезы ткани помещают в систему анализа видеоизображений, и размер инфаркта определяют с помощью планиметрии. Оценивают действие соединения по настоящему изобретению на размер инфаркта миокарда и используют для количественного определения степени кардиопротекторной активности.
Результаты представлены в виде процента риск-области, которая является инфарктной.
Результаты исследованного соединения по примерам, находящихся в рамках настоящего изобретения, указанными выше способами представлены в таблице II.
Соединения по настоящему изобретению проявляют активность в тестах, используемых для определения способности соединений к ингибированию липолиза. Примеры способов исследования, которые пригодны для определения противолиполитической активности соединений по настоящему изобретению, описаны ниже.
Определение противолиполитической активности в адипоцитах крыс
1. Изоляция адипоцитов из жировых подушек яичек
Жировая ткань удаляется у анестезированных крыс и промывается дважды в инкубационной среде (2,09 г бикарбоната натрия и 0,04 г ЭДТА, двунатриевой соли в 1 л буфера Кребса). Каждая крыса (300-350 г) дает приблизительно 4 мл жировой ткани. Жировую ткань (35 мл) разрезают на мелкие кусочки ножницами и промывают инкубационной средой (50 мл). Смесь наливают в корпус 50 мл шприца, к которому присоединен короткий отрезок закупоренной трубки вместо иглы. Водной фазе дают возможность высохнуть. Вторая промывка инкубационной средой проходит через шприц. Ткань добавляют к 50 мл раствора коллагеназы (коллагеназа (90 мг), бычий сывороточный альбумин (БСА) (500 мг), и 0,1 М раствор хлорида кальция (1 мл) в инкубационной среде (50 мл)) в 1 л бутылке. Смесь встряхивают при 37oC в течение приблизительно 60 минут в атмосфере 95% кислород/5% двуокись углерода для воздействия на диспергирование ткани. Диспергированные клетки выливают через два слоя марли в 100 мл пластиковую пробирку с носиком. Недиспергированные скопления клеток на ткани промывают однократно инкубационной средой (20 мл). Клетки в пробирке с носиком центрифугируют в двух пластиковых пробирках в течение 30 секунд при комнатной температуре при 300 об/мин. Водная фаза отсасывается из-под рыхлого слоя плавающих жировых клеток и удаляется. Адипоциты осторожно выливают в 250 мл пластиковую пробирку с носиком, содержащую 100 мл промывочного раствора (1 г БСА на 100 мл инкубационной среды). После осторожного перемешивания стадию центрифугирования повторяют. Следует еще одна промывка промывочным раствором. Клетки собирают, и их объем определяют мерным цилиндром. Адипоциты разводятся в два раза по объему буфером для анализа (инкубационная среда (120 мл), БСА (1-2 г), пировиноградная кислота (13 мг)).
2. Анализ липолиза in vitro
Анализ проводят в 20 мл пластиковых сцинциляционных флаконах и при общем объеме анализируемой пробы 4,2 мл. Буфер для анализа (2,5 мл), разбавленные адипоциты (1,5 мл), и раствор соединения, подлежащего исследованию (12,3 мкл) в качестве агониста аденозина (12,3 мкл; различные концентрации), инкубируют в шейкере в условиях окружающей среды в течение 15 минут, затем начинают реакцию с норпинефриновым раствором (41,1 мкл) (10 нМ) в растворе-носителе, содержащем воду (100 мл), БСА (4 мг), 0,1 М ЭДТА (20 мкл) и аденозиндеаминазу (1 мкг/мл, 41,2 мкл). Через 60 минут в шейкере реакция прекращается путем выставления флаконов на лед. Содержимое каждого флакона переносят в 12 х 75 мм стеклянную пробирку и центрифугируют при 8-10oC при 3600 об/мин в течение 20 минут. Твердый липидный слой удаляют с помощью отсасывания и водный слой анализируют на глицерин (400 мкл образца). Положительный контроль проводят в отсутствие любого агониста аденозина, заменяя водой тестируемый раствор.
Результаты испытаний соединений по настоящему изобретению представлены в таблице III ниже, и даны как % ингибирования продуцирования глицерина 1 мкМ и/или 0,1 мкМ испытуемого соединения по сравнению с положительным контролем, и как значение EC50, то есть концентрация испытуемого соединения, необходимая для осуществления 50% ингибирования производства глицерина. Для целей сравнения результаты также представлены для известных в литературе соединений N-циклопентиладенозина (CHA), N- этилкарбоксамидоаденозина (NECA), R-фенилизопропиладенозина (R- PIA), и 2-[[2-[4-[-(2-карбокс-этил)фенил]этил] амино]-N- этилкарбоксамидаденозина (CGS21680)
Связывающая и вазорелаксантная активность по отношению к A1 и A2 рецепторам аденозина для известных соединений, указанных в таблице IV, как определено способами, описанными выше, представлены в таблице IV.
Противолиполитическая активность аденозина опосредуется через активацию подтипа рецептора A1. Селективные агонисты подтипов рецепторов A2, такие как CGS21680, не проявляют противолиполитической активности. Соответственно, хотя определенные селективные агонисты A1 могут не иметь желаемой противогипертонической активности, и агонисты A2 могут не быть эффективными противолиполитическими средствами, соединения по настоящему изобретению, которые являются смешанными агонистами, являются уникально пригодными для эффективного лечения обоих факторов риска, обсуждаемых выше, то есть гиперлипедемии.
Соединения по настоящему изобретению могут быть обычным образом вводимы орально или парентерально при лечении пациентов, страдающих от ишемии миокарда или пациентов, нуждающихся в кардиопротекторной терапии или противолиполитической терапии. Как он здесь используется, термин "пациенты" включает людей и других млекопитающих.
Соединения по настоящему изобретению, предпочтительно, в форме соли, могут приготавливаться для введения любым удобным способом, и изобретение включает фармацевтические композиции, содержащие хотя бы одно соединение по изобретению, адаптированное для использования в медицине или в ветеринарии. Такие композиции могут быть приготовлены обычным способом, используя один или несколько фармацевтически приемлемых носителей или добавки. Соответствующие носители включают разбавители или наполнители, стерильные водные среды и различные нетоксичные органические растворители. Композиции могут быть приготовлены в форме таблеток, капсул, лепешек, драже, конфет, порошков, водных суспензий или растворов, растворов для инъекций, эликсиров, сиропов и тому подобное, и могут содержать одно или несколько средств, выбираемых из группы, включающей подслащивающие вещества, ароматизирующие вещества, красящие вещества и консервирующие вещества для создания фармацевтически приемлемого препарата.
Конкретный носитель и отношение агонистов аденозина и носителя определяются растворимостью и химическими свойствами соединений, конкретным способом введения и обычной фармацевтической практикой. Например, при производстве таблеток могут быть использованы наполнители, такие как лактоза, цитрат натрия, карбонат кальция и дикальций фосфат, и различные разрыхлители, такие как крахмал, альгиновая кислота и определенные комплексные силикаты, вместе со смазывающими веществами, такими как стеарат магния, натрий лаурилсульфат и тальк. Для капсульной формы лактоза и высокомолекулярные полиэтиленгликоли находятся среди предпочтительных фармацевтически приемлемых носителей. Когда приготавливают водные суспензии для орального введения, носитель может быть эмульсифицирующим или суспендирующим веществом. Наряду с другими материалами могут быть использованы разбавители, такие как этанол, пропиленгликоль, глицерин и хлороформ, и их сочетания.
Для парентерального введения могут быть использованы наряду со стерильными водными растворами растворимых фармацевтически приемлемых солей, описанных здесь, растворы или суспензии этих соединений в кунжутном или ореховом масле, или в водных растворах пропиленгликоля. Растворы солей этих соединений являются особенно пригодными для введения путем внутримышечной или подкожной инъекции. Водные растворы, включая растворы солей в чистой дистиллированной воде, являются пригодными для введения путем внутривенной инъекции при условии, что их pH является соответствующим, и что они находятся в соответствующем буфере, которому придана изотоничность достаточным количеством солевого раствора или глюкозы и который стерилизован с помощью нагрева или микрофильтрования.
Схема лечения, используемая при осуществлении способов по настоящему изобретению, является такой, которая обеспечивает терапевтический ответ до тех пор, пока не будет улучшения, и после этого обеспечивает минимальный эффективный уровень, который дает ослабление. Таким образом, обычно дозировки являются такими, которые являются терапевтически эффективными для увеличения коронарного потока крови при лечении ишемии миокарда, для получения кардиопротекторного эффекта, то есть уменьшения ишемического поражения или размера инфаркта миокарда вследствие ишемии миокарда или для получения противолиполитического эффекта. Обычно доза для перорального приема может быть между приблизительно 0,1 и приблизительно 100 (предпочтительно в диапазоне от 1 до 10) мг/кг, и внутривенная доза от приблизительно 0,01 до приблизительно 10 мг/кг (предпочтительно в диапазоне от 0,1 до 5 мг/кг), имея в виду, конечно, что при выборе соответствующей дозировки в каждом конкретном случае должны быть приняты во внимание вес пациента, общее состояние здоровья, возраст и другие факторы, которые могут оказать влияние на реакцию на лекарство.
Соединения по настоящему изобретению могут назначаться так часто, как это необходимо для достижения и поддержания желаемого терапевтического эффекта. Некоторые пациенты могут реагировать быстро на относительно большую или малую дозу и требуют малую поддерживающую дозу или вообще ее не требуют. С другой стороны, другим пациентам может требоваться поддерживающая дозировка от приблизительно 1 до приблизительно 4 раз в день, в зависимости от физиологических потребностей конкретного пациента. Обычно лекарство может приниматься перорально от приблизительно 1 до 4 раз в день. Предполагается, что многим пациентам будут необходимо не более чем от одной до двух доз каждый день.
Также ожидается, что настоящее изобретение будет использоваться в виде лекарственной формы для инъекций, которая может быть введена для срочной помощи пациенту, страдающему от ишемии миокарда, или пациенту, нуждающемуся в кардиопротекторном и противолиполитическом лечении. Такое лечение может сопровождаться затем внутривенной инфузией активного соединения, и количество соединения, введенного таким образом пациенту, должно быть эффективным для достижения и поддержания желаемого терапевтического эффекта.
Пример кардиозащитной композиции
Компонент:
(2S)-2α,3α-дигидрокси-4β-[N6-[2-(5-хлор-2-тиенил)-(1R)-1-метилэтил]амино-9-аденил]циклопентан-1β-этилкарбоксамид-N1-оксид - 0,2 мг
лактат натрия - 1,67 мг
хлорид натрия - 8,1 мг
(D,L) молочная кислота - 1,44 мг
дистиллированная вода - 1 мл
Композицию получают обычным способом смешивания всех составляющих компонентов.
Заявитель просит учесть этот пример в качестве подтверждения притязаний по п. 26 формулы изобретения.


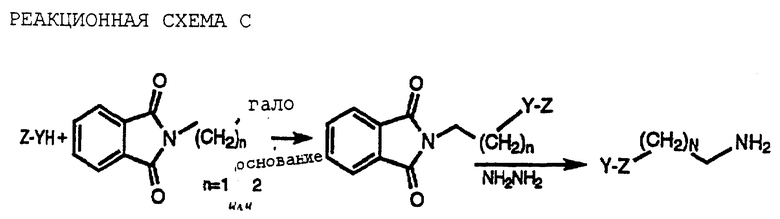


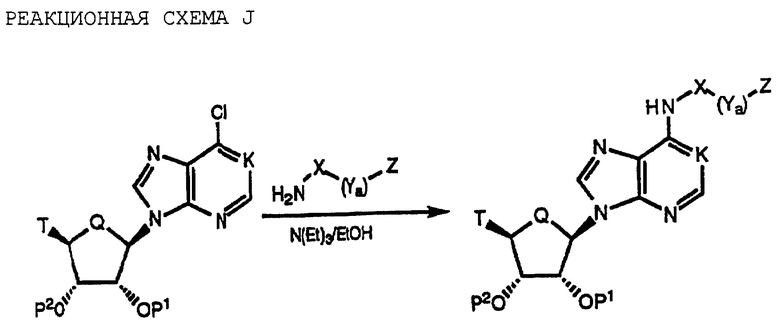
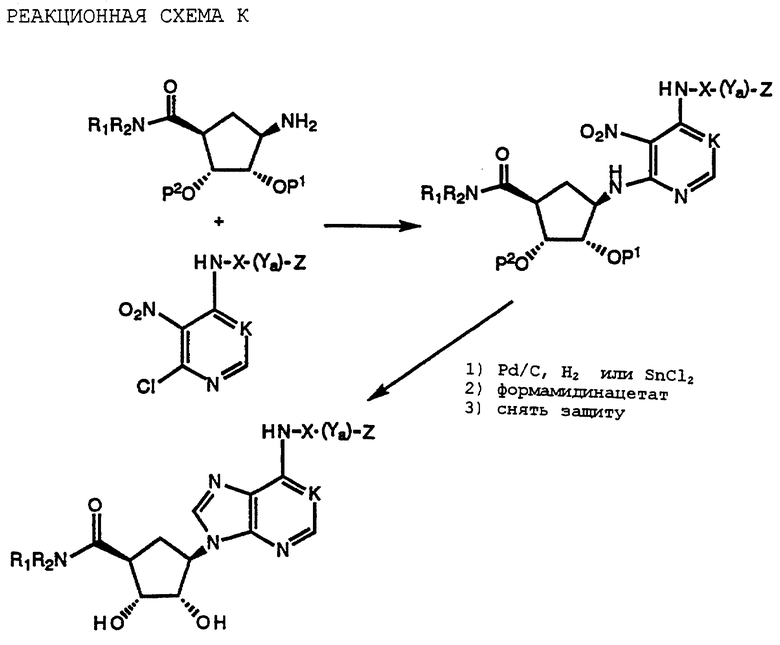
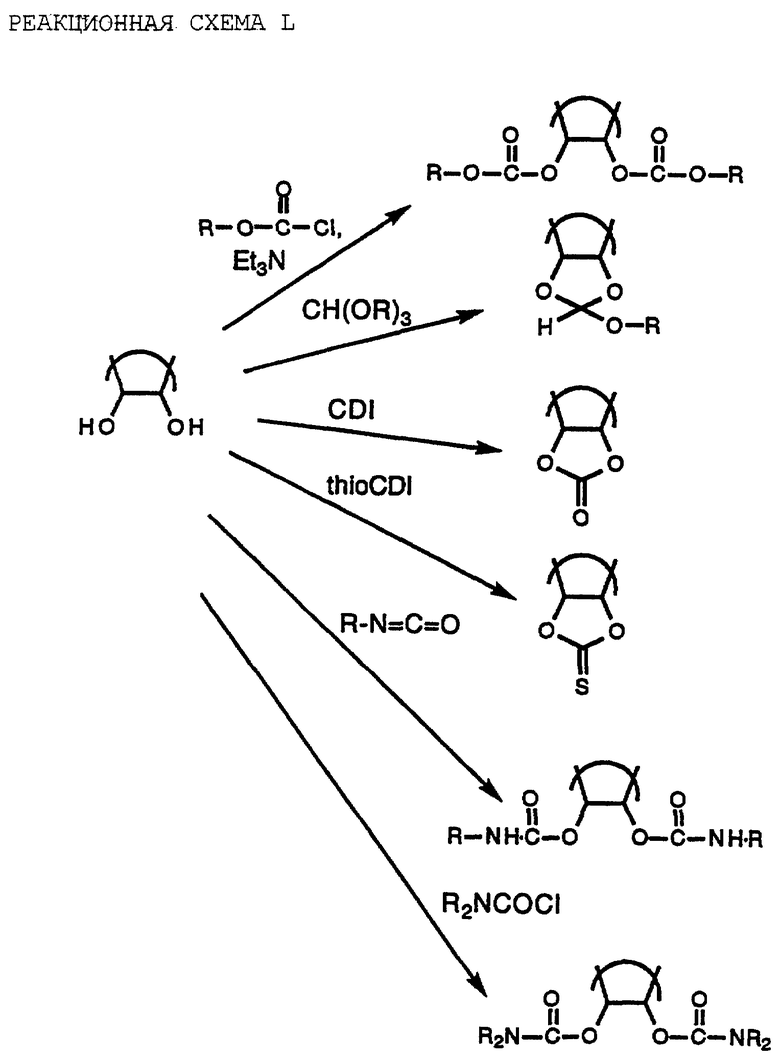

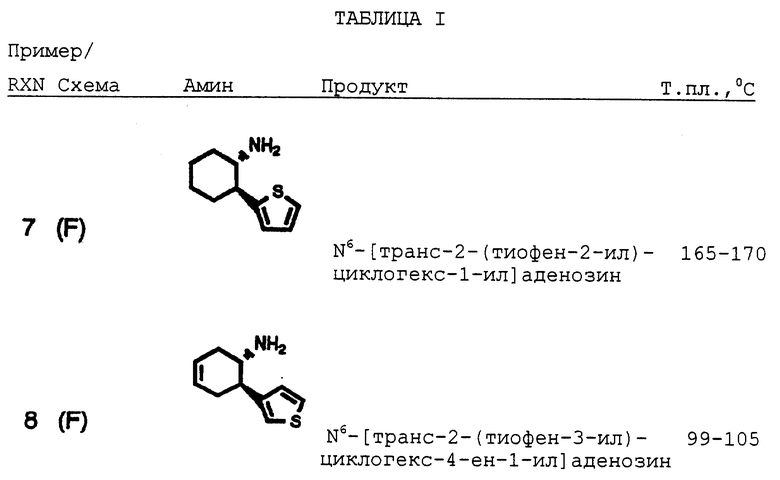

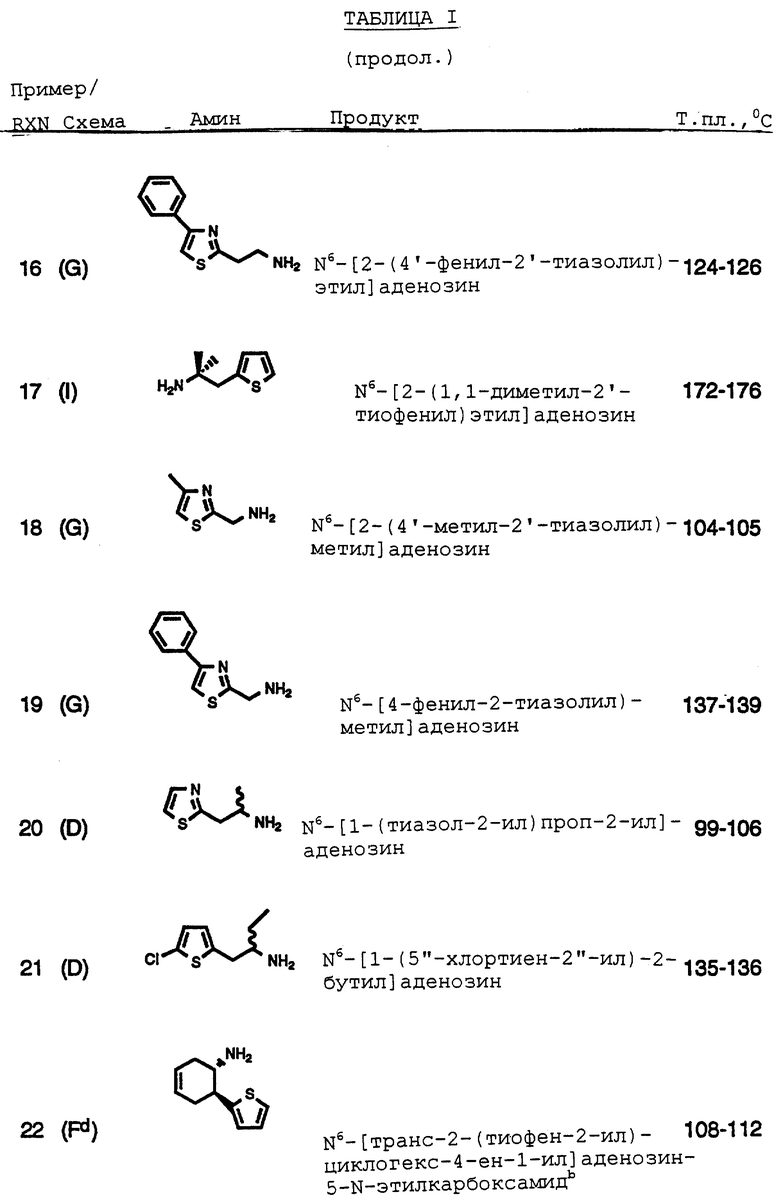
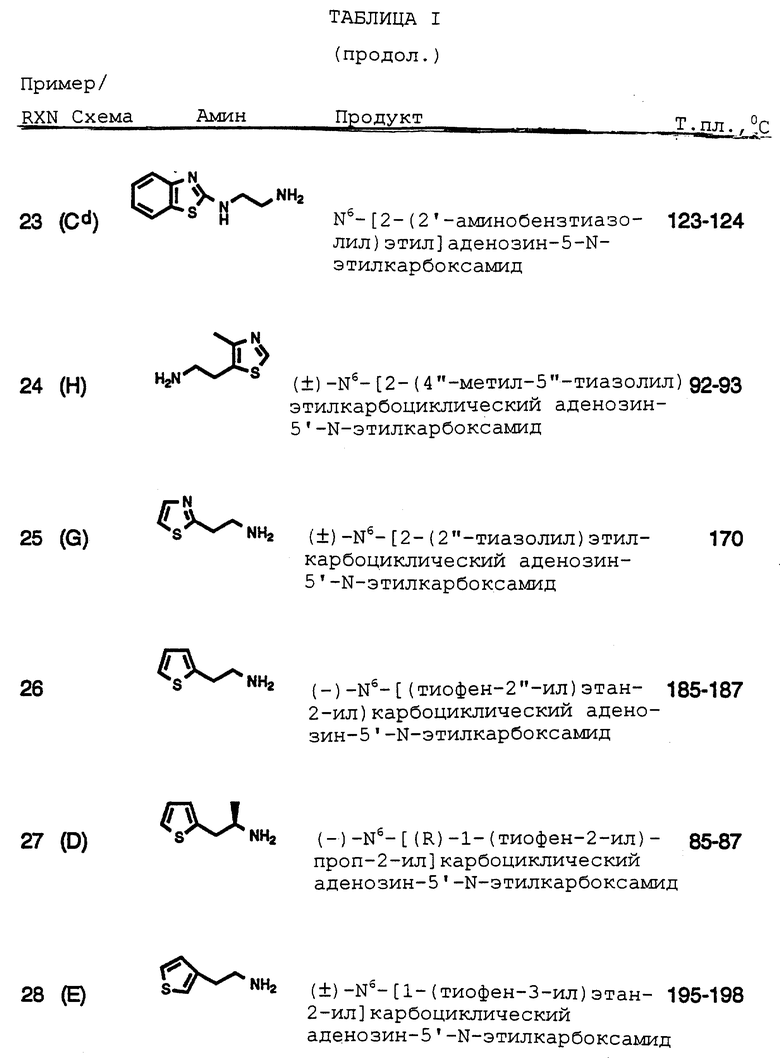
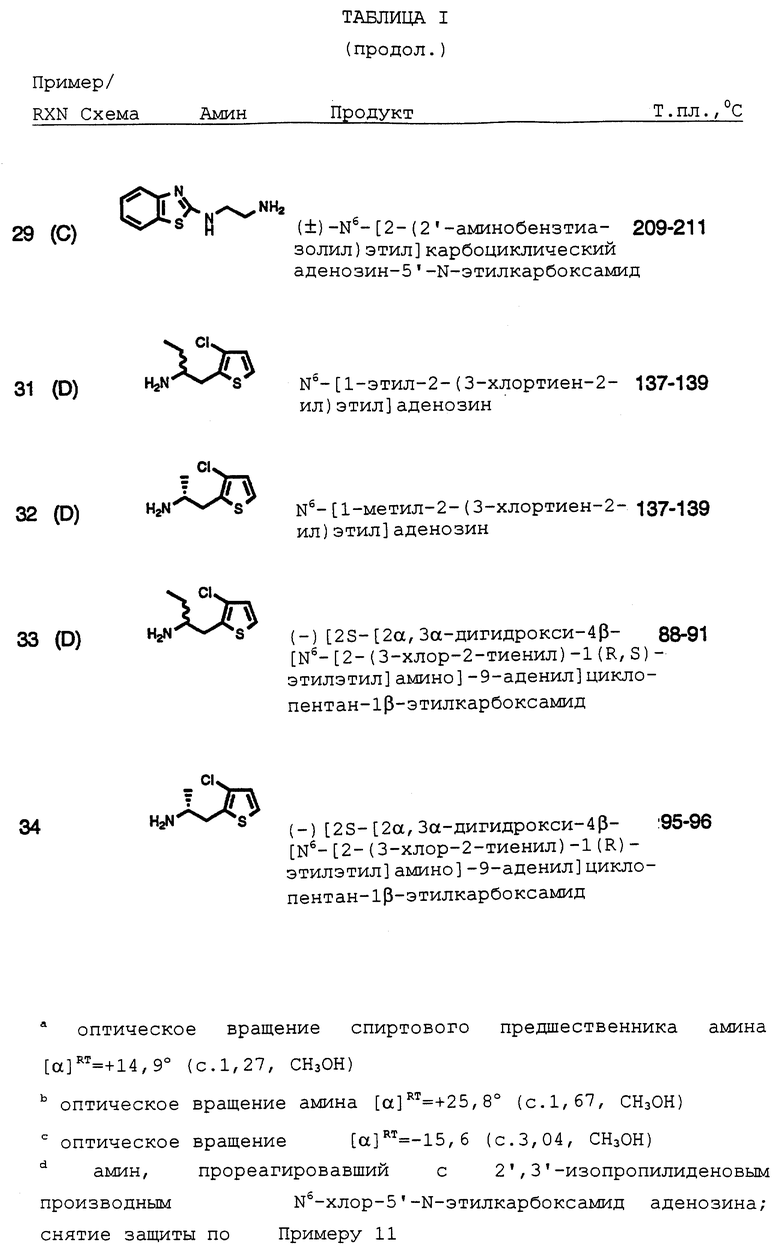



Изобретение относится к медицине, к производным и аналогам аденозина, которые обладают активностью в качестве агонистов аденозина и являются пригодными в качестве кардиопротекторных, противоишемических и противолиполитических средств, к фармацевтической композиции, содержащей такие соединения. Изобретение уменьшает ишемическое поражение или размер инфаркта миокарда. 2 с. и 24 з.п. ф-лы, 4 табл.
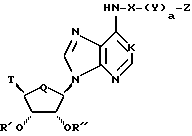
где К обозначает N, N-->O или СН;
О обозначает СН2 или О;
Т обозначает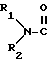
или R3O-СН2;
Х обозначает алкилен с прямой или разветвленной цепью, циклоалкиленовую или циклоалкениленовую группу, каждая из которых, необязательно, замещена низшим алкилом, таким как СН3, СН3СН2, галогеном, таким как С1, F, галогеннизшим алкилом, таким как CF3, или низшим алкокси, таким как СН3O;
Y обозначает NR4;
а равно 0 или 1;
Z имеет формулу
или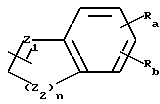
Z1 обозначает N, CR5, (CH)m-CR5 или (CH)m- N, где m равно 1 или 2;
Z2 обозначает N, NR6, О или S, где n равно 0 или 1;
R1, R2, R3, R4, R5 и R6 обозначают независимо Н, алкил, фенил, фенил, замещенный одним или несколькими заместителями, которыми могут быть алкил, алкокси, амино, нитро, карбокси, карбоалкокси, циано, алкиламино, галоген, гидрокси, гидроксиалкил, меркаптил, алкилмеркаптил, карбалкил или карбамоил; или 4-10-членное кольцо гетероциклической структуры, в котором один или несколько атомов кольца являются N, О или S;
Ra и Rb обозначают независимо Н, ОН, алкил, гидроксиалкил, алкилмеркаптил, тиоалкил, алкокси, алкоксиалкил, амино, алкиламино, карбоксил, ацил, галоген, карбамоил, алкилкарбамоил, арил или гетероциклил;
R' и R" обозначают независимо водород, алкил, аралкил, карбамоил, алкилкарбамоил, диалкилкарбамоил, ацил, алкоксикарбонил, аралкоксикарбонил, арилоксикарбонил или R' или R" вместе могут образовывать
где Rc обозначает водород или алкил,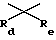
где Rd и Re обозначают независимо водород, алкил или вместе с атомом углерода, к которому они присоединены, могут образовывать 1,1-циклоалкильную группу;
при условии, что, когда Х обозначает алкилен с прямой цепью и Q обозначает кислород, тогда Z представляет гетероциклил, содержащий по крайней мере два гетероатома,
или его фармацевтически приемлемой соли в качестве кардиозащитного средства, которое уменьшает ишемию миокарда.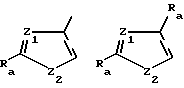
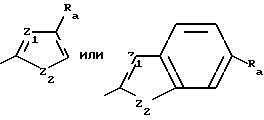
8. Применение соединения по п.5, где Z обозначает замещенную или незамещенную тиазолильную группу или замещенную или незамещенную бензитиазолильную группу.
N6 - [транс-2-(тиофен-2-ил)циклогекс-4-ен-1-ил]аденозин;
N6 - [транс-2-(тиофен-3-ил)циклогекс-4-ен-1-ил]аденозин;
N6 - [2-(2'-аминобензтиазолил)этил]аденозин;
N6 - [2-(2'-тиобензтиазолил)этил]аденозин;
N6 - [2-(6'-этокси-2'-тиобензтиазолил)этил]аденозин;
N6 - [2-(4'-метилтиазол-5'-ил)этил]аденозин;
N6 - [2-(2'-тиазолил)этил]аденозин;
N6 - [2-(4'-фенил-2' тиазолил)этил]аденозин;
N6 - [4'-фенил-2'-тиазолил)метил]аденозин;
N6 - [2-(4'-метил-2'-тиазолил)этил]аденозин;
N6 - [2-(2'-метил-4'-тиазолил)этил]аденозин;
N6 - [(R)-1-метил-2(2'-бенз[b]тиофенил) этиладенозин или
(-)-[2S-[2α,3α-дигидрокси-4β-[N6-[2-(3-хлор-2-тиенил)-(1R)-этилэтил]амино] -9-аденил] циклопентан]-1β-этилкарбоксамид или их фармацевтически приемлемые соли.
N6-[транс-2-(тиофен-2-ил)циклогекс-4-ен-1-ил] аденозин-5'-N-этилкарбоксамид,
N6-[2-(2'-аминобензтиазолил) этил]аденозин-5'-N-этилкарбоксамид;
N6-[(R) -1-(5'- хлортиен-2'-ил)-2-пропил]аденозин-5'-N-этил-карбоксамид;
N6- [ (R)-l- ((тиазо-2-ил)проп-2-ил)] аденозин-5'-N-этилкарбоксамид или N6 - [1-(тиофен-2-ил)-2-метил-2-пропил]аденозин-5'-N-этилкарбоксамид или их фармацевтически приемлемые соли.
N6- [ 2- (4"-метил-5"-тиазолил)этил]карбоциклический аденозин-5'-N-этилкарбоксамид;
N6- [2-(2'-аминобензтиазолил)этил] карбоциклический аденозин-5'-N-этилкарбоксамид;
N6-[2-(2"-тиазолил)этил]карбоциклический аденозин-5'-N-этил-карбоксамид;
(-)-[2S-[2α,3α-дигидрокси-4-β-[N6-[2-(5-хлор-2-тиенил)-1-метилэтил] амино]-9-аденил]циклопентан]-1-β-N-этилкарбоксамид;
N6-[1-тиофен-2"-ил) этан-2-ил] карбоциклический аденозин-5'-N-этилкарбоксамид;
N6-[1-(тиофен-3-ил)этан-2-ил] карбоциклический аденозин-5'-N-этилкарбоксамид;
N6- [(R )-1- ((тиофен-2-ил)проп-2-ил) ] карбоциклический аденозин-5'-N-этилкарбоксамид или
N6- [( R )-1- (5"-хлортиен-2-ил)-2-бутил]карбоциклический аденозин-5'-N-этилкарбоксамид или их фармацевтически приемлемые соли.
(-)-[2S-[2α,3α-дигидрокси-4-β-[N6- [2- (5-хлор-2тиенил) - (1R)-1-мeтилэтил]aминo]-9-aдeнил]циклoпeнтaн]-1β-N-этилкapбoкcaмид или его фармацевтически приемлемую соль.
(±)-N6-[1-(тиофен-2-ил)этан-2-ил] -N'-деазааристеромицин-5'-N'-этилкарбоксамид;
[1S-[1α,2β,3β,4α(S*)] ]-4-[7-[[2-(5-хлор-2-тиенил)-1-метил-этил]амино]-3Н-имидазо[4,5-b]пиридин-3-ил]-N-этил-2,3-дигидроксициклопентанкарбоксамид;
[1S-[1α,2β,3β,4α] ]-4-[7-[[2-(3-хлор-2-тиенил)-1-этилэтил]амино]-3Н-имидазо[4,5-b]пиридин-3-ил]-N-этил-2,3-дигидроксициклопентанкарбоксамид;
[1S-[1α,2β,3β,4α] ] -4-[7-[[2-(2-тиенил)-1-изопропилэтил]-амино]-3Н-имидазо[4,5-b]пиридин-3-ил]-N-этил-2,3-дигидроксициклопентанкарбоксамид;
[1S-[1α,2β,3β,4α(S*)] ] -4-[7-[[2-(3-хлор-2-тиенил)-1-этилэтил] амино]-3Н-имидазо[4,5-b]пиридин-3-ил]-N-этил-2,3-дигидроксициклопентанкарбоксамид;
[1S-[1α,2β,3β,4α(S*)] ] -4-[7-[[2-(2-тиенил)-1-метилэтил]-амино]-3Н-имидазо[4,5-b]пиридин-3-ил]-N-этил-2,3-дигидроксициклопентанкарбоксамид или
[1S-[1α,2β,3β,4α]]-4-[7-[[2-(5-хлор-2-тиенил)-1-этилэтил]-амино]-3Н-имидазо[4,5-b] пиридин-3-ил]-N-этил-2,3-дигидроксициклопентанкарбоксамид или их фармацевтически приемлемые соли.
N6 - [2-(3-хлор-2-тиенил)-(1R)-1-метилэтил]-2'-O-метиладенозин;
N6 - [2-(5-хлор-2-тиенил)-(1R)-1-метилэтил]-2'-O-метиладенозин или
N6 - [транс-5-(2-тиенил)циклогекс-1-ен-4-ил]-2'-O-метиладенозин или его фармацевтически приемлемую соль.
(2S)-2α,3α-бис-метоксикарбонилокси-4β-[N6-[2-(5-хлор-2-тиенил)-(1R)-1-метилэтил]амино-9-аденил]циклопентан-1β-N-этилкарбоксамид;
(2S)-2α,3α-дигидрокси-4β-[N6-[2-(5-хлор-2-тиенил)-(1R)-1-метилэтил] амино-9-аденил]циклопентан-1β-N-этилкарбоксамидэтоксиметиленацеталь;
(2S)-2α,3α-дигидроски-4β-[N6-[2-(5-хлор-2-тиенил)-(1R)-1-метилэтил] амино-9-аденил]циклопентан-1β-N-этилкарбоксамид- 2,3-карбонат;
(2S)-2α,3α-бис-метилкарбамоилокси-4β-[N6-[2-(5-хлор-2-тиенил)-(1R)-1-метилэтил]амино-9-аденил]циклопентан-1β-N-этилкарбоксамид или
(2S)-2α,3α-дигидpoкcи-4β-[N6-[2-(5-хлор-2-тиенил)-(1R)-1-метилэтил] амино-9-аденил] циклопентан-1β-N-этилкарбоксамид-2,3-тиокарбонат или их фармацевтически приемлемые соли.
Приоритет по пунктам и признакам:
19.04.94 по пп.1-3, 15-23, 26;
03.10.94 по пп.4-14, 24-25 в части снижения уровня липидов в плазме крови.
| Способ получения производных имидазо (1,2- @ ) пурина или их кислотноаддитивных солей | 1978 |
|
SU1066463A3 |
| US 4639436, 27.01.1987 | |||
| Круглый ткацкий станок | 1929 |
|
SU15148A1 |

