Данное изобретение касается нового полипептида (полипептидов) или его фармацевтически приемлемой соли, проявляющих сильное сродство к липолисахаридам, в частности эндотоксинам. Полипептид можно применять в фармацевтической композиции в качестве антивирусного агента (например, агента против ВИЧ-инфекции).
Из мечехвоста были выделены два семейства антимикробных полипептидов, которые проявляют сродство к эндотоксинам (см., например, Shigenaga et al., 1990, J. Biol. Chem. 265: 21350-21354; Kawano et al., 1990, J. Biol. Chem. 265: 15365-15367; Muta et al., 1990, J. Biol. Chem. 108: 261-266; Japanese Laid-Open Patent Publication N 167230/1990; Japanese Laid-Open Patent Publication N 152987/1990; Japanese Laid-Open Patent Publication N 53799/1990; Published Searched Application 500194/1990; Miyata et al., 1989, J. Bio Chem. 106:663-668; Akaji et al., 1989, Chem. Pharm. Bull. 37:2661-2664; Tokunaga and Iwanaga, 1989, Taisha (Metabolism) 26:429-439; Shieh et al., 1989, FEBS Lett. 252: 121-124; u Nakamira et al. , 1989, J. Biol. Chem. 263: 16709-16713).
Одно семейство, семейство тахиплезинов, было выделено из Японского мечехвоста Tachypleus. Были идентифицированы три тахиплезина, I, II и III. Второе семейство, семейство полифемузинов, было выделено из Американского мечехвоста Limubus polyphemus. Были идентифицированы два полифемузина, I и II; их аминокислотные последовательности показаны на фиг. 1.
Полипептиды в обоих семействах состоят из 17 или 18 аминокислотиных остатков и имеют 4 консервативных района, общие для всех, и два дисульфидных мостика (см. фиг.1).
Было обнаружено, что как тахиплезины, так и полифемизины ингибируют рост грамотрицательных, так и грамположительных бактерий при низких концентрациях, а также грибов, таких как Candida albicans, и образуют комплексы с бактериальными диполисахаридами (Shigenaga et al.m, 1990, J.Biol. Chem. 265: 21350-21354 и Muta et al., 1990, J.Biochem. 108:261-266). Также было обнаружено, что полипептиды в семействе тахиплезинов проявляют некоторую ингибирующую активность в отношении вируса, такого, как вирус гриппа, вирус везикулярного стоматита (Murakami et al., 1991, Chem therapy, 37:327-334) или вирус иммунного дефицита человека (Morimoto et al., 1991, Chemtherapy, 37, 206-211). Очень интересно, что такой полипептид c указанными выше свойствами может быть одним из ключевых веществ, которое позволяет мечехвосту адаптироваться к изменениям в наружной среде их обитания и сохранить их виды с древних времен до настоящего времени в виде живого ископаемого.
С другой стороны, что касается выживания высокоэволюционировавших существ, людей, чрезвычайно желательно развитие таких лекарственных средств, которые обладают профилактическим или терапевтическим действием на синдром приобретенного иммунодефицита (СПИД), вызываемый инфекцией вирусом иммунодефицита человека (ВИЧ).
Авторы данной заявки и сотрудники исследовали корреляцию между молекулярной структурой и анти-ВИЧ активностью этих полипептидов с изменением или модификацией аминокислоты компонентов, обращая внимание на те полипептиды, которые имеют отношение к длительному сохранению мечехвоста. В результате нами были изобретены новые полипептиды, фундаментально отличающиеся от общей структуры таких известных полипептидов мечехвоста.
Неожиданно было обнаружено, что эти предшествующие новые полипептиды имеют превосходную биологическую активность. Их величины анти-ВИЧ активности по меньшей мере в 5 или более раз выше, чем эти величины известных полипептидов мечехвоста.
Появились следующие публикации: Nakashima et al., 1992, Antimicrob. Agents Chemother., 36:1249-1255; Masuda et al., 1992, Biochem. Biophys. Res. Commun. , 189, 845-850; Tamamura et al., 1993, Chem. Phatm. Bull., 41, 978-980; Tamamura et al., 1993, Biochem. Biophys. Acta, 1163, 209-216; Masuda et al. , 1992, J, Pharmacobio. Dyn., 15, S-90; International Laid-Open Publication WO 92/04374; Jananese Laid-Open Patent Publication N 163298/1993.
(Далее среди новых полипептидов соединение T-22 называют соединением наивысшего действия из типичных представителей этих полипептидов). После испытания структурных требований для проявления активности этого нового полипептида, состоящего из 16-18 аминокислот, представленного соединением T-22, заявители и их сотрудники пришли к концепции о минимальной существенной (основной) структуре.
Обычно при введении в тело человека экзогенного пептида с относительно высоким молекулярным весом этот пептид часто воспринимается как чуждое вещество функцией защитных сил организма человека. В результате он становится антигенным веществом. При использовании для целей медицины желательно, чтобы биологически активное вещество на основе пептида было низкомолекулярным соединением, чтобы уменьшить вероятность восприятия его в качестве чуждого вещества. Необходимо также, чтобы это вещество имело высокую биологическую активность.
Было обнаружено, что соединение T-22 является полипептидом, состоящим из 18 аминокислотных остатков. Целью нашего исследования было сохранение того же самого уровня анти-ВИЧ активности, который имеется у соединения T-22, при уменьшении числа аминокислотных остатков. В результате нам удалось уменьшить число остатков на 4 (четыре). Пока соединение имеет основную структуру, его активность не снижается, даже при модификации специфического района. Кроме того, при помощи такой модификации мы обнаружили новый полипептид, имеющий такую существенную (основную) структуру, который может обеспечить более широкий диапазон физико-химических свойств и также более широкую селекцию в терапевтических способах, т.е. увеличение/уменьшение гидрофильности и липофильности (сродство к липиду); избирательное накопление в определенном органе и (или) в определенной клетке; увеличение/уменьшение времени его нахождения внутри тела человека; и возможное развитие препаратов.
Данное изобретение касается нового полипептида (полипептидов), который произведен из уже заявленных новых полипептидов с высокой анти-ВИЧ активностью, основанных на известных полипептидах мечехвоста с высокой аффинностью по отношению к эндотоксинам, но имеет значительное отличие.
Прежние новые полипептиды состоят из 16-18 аминокислотных остатков, 4 цистеиновых или 2 цистиновых остатков и антипараллельной β пластинчатой структуры с β-витком в качестве положения поворота. Подобно прежним новым полипептидам, полипептиды данного изобретения имеют антипараллельную β пластинчатую структуру с возможным β -витком, расположенным в X при положении 7. Однако в полипептидах данного изобретения число аминокислотных остатков и число цистеиновых остатков уменьшены на четыре и два соответственно. Кроме того, биологическая активность не уменьшается, даже при модификации специфического района.
Полипептиды данного изобретения можно использовать в качестве агента против ВИЧ и в качестве компонента ДНК-трансфицирующих систем для генной терапии. Как будет детализировано в разделе 6, infra, полипептиды данного изобретения имеют анти-ВИЧ величины, которые значительно выше, чем величины прежних новых полипептидов, произведенных из известных полипептидов с высоким сродством к эндотоксинам мечехвоста.
3.1. Определения.
Пептидные последовательности, описанные здесь, представлены при помощи трехбуквенных сокращений для аминокислотных остатков и замещенных аминокислотных остатков следующим образом:
Ala (аланин); Arg (аргинин); Cys (цистеин); Ile (изолейцил); Gly (глицин); Leu (лейцин); Lys (лизин); Orn (орнитин); Phe (фенилаланин); Pro (пролин); Trp (триптофан); Tyr (тирозин); Val (валин); D Arg (D-аргинин); D Lys (D-лизин); D Orn (D-орнитин); As-Arg (N- α -ацетиларгинин); FTC-Arg (N- α -флуоресцеинтиокарбамоиларгинин); Laur-Arg (N-альфа -лауроиларгинин); Myr-Arg (N- α -миристоиларгинин); Nicot-Arg (N- α -никотиноиларгинин); Oct-Arg (N- α -октаноиларгинин); Parm-Arg (N- α -пальмитоиларгинин); Parm-Oru (N- α -пальмитоилорнитин); PCT-Arg (N- α -фенилтиокарбамоиларгинин); ∈ -N-Ac-D Lys ( ∈- N- ω -аминоацетил-D-лизин) и ∈ -N-Beet-G Lys ( ∈ -N- ω -аминобутирил-D-лизин).
Следующие термины, примененные здесь, будут иметь указанные значения: HIV = вирус иммунодефицита человека (все разновидности); MOI = множественность заражения; SI = индекс селективности (отношение CC50 к EC50).
4. Краткое описание рисунков
Фиг. 1 показывает аминокислотные последовательности Тахиплезина I, Тахиплезина II, Тахиплезина III, Полифемузина I и Полифемузина II. Консервативные аминокислоты помещены в блоки. Дисульфидные связи между Cys-3 или -4 и Cys-16 или -17 и Cys-7 или -8 и Cys-12 или -13 обозначены толстыми линиями.
Фиг. 2 представляет схему синтеза полипептида (1) данного изобретения.
Данное изобретение выполнено на основе описанных выше аспектов и касается нового полипептида, представленного формулой I (см. в конце описания), или его соли, в которых
A1 представляет собой остаток основной аминокислоты или остаток пептида, имеющий одну или по меньшей мере две основные аминокислоты, выбранные из группы, состоящей из лизина, аргинина и орнитина, причем остаток основной аминокислоты или остаток пептида, в котором N- α атом водорода амино-конца этого аминокислотного остатка может быть замещен ацильной группой или замещенной тиокарбамоильной группой, образует N- α замещенный ацилом основной аминокислотный остаток, N- α замещенный ацилом пептидный остаток, N- α замещенный замещенной тиокарбамоильной группой основной аминокислотный остаток или N- α замещенный замещенной тиокарбамоильной группой пептидный остаток;
A2 представляет собой остаток тирозина или фенилаланина;
A3 представляет собой остаток лизина или аргинина;
A4 представляет собой -OH (происходящий из карбоксильной группы) или -NH2 (происходящий из кислой амидной группы);
X представляет собой остаток пептида, выбранный из группы, состоящей из пептидов, таких как D-орнитил-пролин, пролил-D-орнитин, D-лизил-пролин, пролил-D-лизин, D-аргинил-пролин, пролил-D-аргинин, глицил-орнитин, орнитил-глицин, глицил-лизин, лизил-глицин, глицил-аргинин и аргинил-глицин, в которых атом водорода ω -аминогруппы D-лизина, L-лизина, D-орнитина или L-орнитина может быть заменен ω -аминоацильной группой, и этот пептидный остаток соединен с аминокислотными остатками в положениях 6 и 8 через пептидную связь per se;
Trp представляет собой остаток триптофана;
Cys представляет собой остаток цистеина; и остатка цистеина в положениях 3 и 11 могут быть соединены дисульфидной связью.
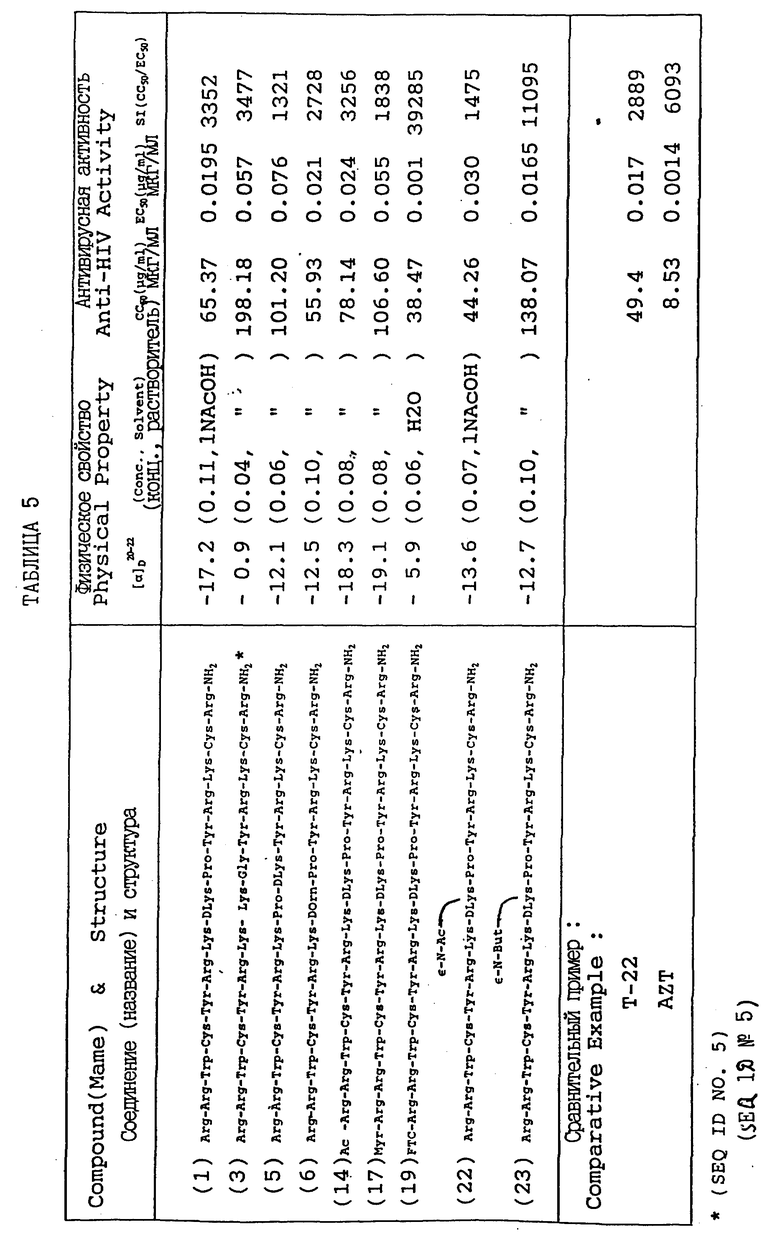
Характерные примеры полипептидов изобретения, представленных формулой (I), показаны в табл. 1 и пронумерованы (1) - (25).
Каждый символ обозначает соответствующий аминокислотный остаток при помощи международно признанного трехбуквенного выражения (см. раздел 3.0, supra).
Вышеупомянутый полипептид с высокой активностью против ВИЧ состоит из 16 - 18 аминокислотных остатков. В случае соединения наилучшего способа действия (далее сокращаемого как "n-18 полипептид" или Т-22) обращается внимание на то, что структурным фактором, существенным для проявления высокой активности, является присутствие четырех остатков цистеина, четырех или пяти остатков ароматических аминокислот, восьми остатков основных аминокислот и одного остатка глицина. Кроме того, что касается позиционной взаимосвязи n-18 полипептида, как показывает формула (A), свойства аминокислотных остатков при положениях 2-17 прочно фиксированы. И при положении 1-18 полипептида была обнаружена корреляция структуры с активностью, выражающаяся в том, что относительные величины проявляемой анти-ВИЧ активности увеличиваются с увеличением числа аминокислотных остатков.
Полипептид n-18 выражают в виде формулы (A) (см. в конце описания).
в которой A1 обозначает не более двух аминокислот, выбранных из группы, состоящей из лизина и аргинина; A2 обозначает остаток тирозина, фенилаланина или триптофана; A3 обозначает остаток аргинина или лизина; A4 обозначает по меньшей мере одну или не более двух аминокислот, выбранных из группы, состоящей из лизина и аргинина; A5 обозначает -OH (происходящий из карбоксильной группы); или -NH2 (происходящий из амидной группы); Cys обозначает остаток цистеина; Gly обозначает остаток глицина.
В характерном варианте остатки цистеина в положениях 3 и 16 и (или) остатки цистеина в положениях 7 и 12 могут быть соединены дисульфидной связью (-S-S-).
В полипептиде n-18 положение поворота с возможной структурой β -витка находится при положениях 9 и 10. И пептидная часть от положения 3 до положения 8 и часть пептида от положения 11 до положения 16 обращены друг к другу.
Подобно полипептиду n-18, полипептиды данного изобретения имеют антипараллельную β -пластинчатую структуру, обусловленную существованием внутримолекулярных водородных связей и дисульфидных связей (-S-S-) между остатками цистеина. Хотя в полипептидах данного изобретения положение поворота с возможной структурой β -витка расположено в X при положении 7, так что часть пептида от положения 3 до положения 6 и часть пептида от положения 8 до положения 11 обращены друг к другу.
В данном изобретении корреляция числа аминокислотных остатков при положении 1 формулы данного изобретения с активностью такая же, какая установлена для полипептида n-18.
Подтверждено, что замена атома водорода α -аминогруппы N-концевого остатка аминокислоты в этом положении ацильной группой или замещенной тиокарбамоильной группой важна для проявления высокой активности против ВИЧ нового полипептида, представленного указанной формулой. Путем подбора различных свойств ацила или замещенной тиокарбамоильной группы стало возможным придание новому полипептиду гидрофильности, сродства к липидам, отличающихся свойств флуоресценции и т.д. Например, характеристики флуоресценции замещенной флуоресцеином тиокарбамоильной группы в полипептидах данного изобретения можно использовать в качестве высоко чувствительного репортерного красителя для различных целей. См., например, Brand and Withold in "Metods in Euzymology", Vol. 11, p. 776 - 856, ed. by Hirr, Academic Press, New York, New York (1967); Brand and Gohlke, 1972, Annu. Rev. Biochem. 41: 843 - 868; Stryer, 1978, Annu. Rev. Biochem. 47: 819 - 846.
Кроме того, очень важно и ценно, что замещенные флуоресцеином полипептиды данного изобретения могут проявлять даже более высокую активность против ВИЧ. Таким образом, полипептиды изобретения со свойствами флуоресценции могут быть использованы в качестве важного инструмента для выяснения механизма манифестации активности против ВИЧ полипептидов изобретения. Например, судьба, метаболизм или распределение внутри тела, органа, ткани или клетки, инфицированных или неинфицированных ВИЧ, после введения может быть детектирована при помощи флуоресцентной микроскопии. На молекулярном уровне информация о тончайших конформационных изменениях этих полипептидов, взаимодействующих с рецепторной молекулой внутри клеток, может быть получена путем применения внутренних флуоресцентных зондов этих полипептидов. С применением таких флуоресцентных зондов возможны выделение и идентификация самой молекулы рецептора.
Далее, при помощи ацильной группы или замещенного тиокарбамоила стало возможным получить биологическую активность новых полипептидов у соединений, таких, как соединения с сахарной цепью, липидные соединения, нуклеиновые кислоты, другие типы пептидов или белков и т.д. Важную роль в манифестации активности нового полипептида данного изобретения играет то, что свойства аминокислотных остатков при положениях 4 - 6 и положениях 8 - 10 формулы изобретения являются одними и теми же. Что касается образования трехмерной структуры нового полипептида изобретения, важен порядок аминокислот пептида, который легко образует структурные части пептида, представленные в положениях 3 - 7 и в положениях 7 - 11 в виде одной и той же плоской структуры, расположенной напротив друг друга, а пептидный остаток, состоящий из двух аминокислот, обозначенных X, в положении 7 является точкой поворота. В случае, когда цистеины при положениях 3 и 7 соединены дисульфидной связью, трехмерная структура, образованная пептидным каркасом нового полипептида данного изобретения, является важной чертой изобретения. Следует сказать, что повернутая структура полипептидого остатка X при положении 7, состоящего из пары глицин и основная аминокислота или пролин и D-форма основной аминокислоты (в принципе, гидроксипролин может быть заменен пролином, имеющим тот же самый эффект), является необходимым фактором для образования той же самой плоскостной структуры с β -пластинчатой структурой. Соединенные дисульфидной боковой цепью положения 3 и 11 остатков цистеина и основные боковые цепи основных аминокислот в положениях 5 и 6 находятся на одной и той же стороне каркасной плоскости, тогда как ароматические боковые цепи остатков ароматических аминокислот в положениях 6 и 10 находятся на противоположной стороне плоскости каркаса. Образование этой трехмерной указанной выше структуры является важным. Поэтому новый полипептид, представленный указанной выше формулой, был изобретен с такой трехмерной структурой, приведшей к уменьшению на четыре аминокислотных остатка по сравнению с полипептидом n-18.
Кроме того, подобно полипептиду (полипептидам) n-18, полипептиды данного изобретения проявляют очень основные свойства. Благодаря их основной природе, эти полипептиды могут образовать соль с кислотой. Например, такой полипептид образует соль с неорганической кислотой (соляной кислотой, бромистоводородной кислотой, фосфорной кислотой, азотной кислотой, серной кислотой или т.п.) или с органической карбоновой кислотой (уксусной кислотой, галогенуксусной кислотой, такой, как трифторуксусная кислота, пропионовой кислотой, малеиновой кислотой, янтарной кислотой, яблочной кислотой, лимонной кислотой, винной кислотой, салициловой кислотой) и кислым сахаром (глюкуроновой кислотой, галактуроновой кислотой, глюконовой кислотой, аскорбиновой кислотой или т.п.), кислым полисахаридом (гиалуроновой кислотой, сульфатом хондроитина, альгиновой кислотой или т.п.) или органической сульфоновой кислотой (метансульфоновой кислотой, п-толуолсульфоновой кислотой или т. п. , в том числе с эфиром сульфоновой кислоты и сахара, таким, как сульфаты хондроитина).
Далее следует более детальное описание нового полипептида данного изобретения.
Получение полипептидов.
Новый полипептид изобретения может быть получен известными в этой области способами, например способами твердофазного синтеза, описанными в "Solid-Phase Peptide Synthesis", Stewart and Young, Pierce Chemical Company, Rockford, Illinois (1984). А именно, полипептид с прямой цепью данного изобретения указанной выше формулы (I) можно получить путем соединения карбоксильной группы N-защищенного аргинина при положении 12 с нерастворимой смолой, имеющей аминогруппы, присоединенные непосредственно или присоединенные через спейсер, имеющие функциональную группу, способную соединяться с карбоксильной группой (например, функциональную группу, способную превращать карбоксильную группу аргинина в п-карбоксиметилбензиловый эфир). Аминогруппа нерастворимой смолы, имеющая остаток аргинина (Arg) в положении 12, после деблокирования N-защищающей группы способна успешно соединяться, согласно твердофазному способу, с соответствующими защищенными аминокислотами положения 11 к положению 1 аминокислотной последовательности, представленной формулой (I) (см.в конце текста).
где A1, A2, A3, A4, Cys, Trp и X означают то же, что и в приведенной выше формуле (I).
В случае, когда N- α -ациламинокислотный остаток или ацилпептидный остаток выбран для A1, в которых атом водорода при N- α -положении аминоконцевого аминокислотного остатка заменен ацильными группами, N-концевая аминогруппа содержащей пептид смолы ацилируется соответствующим ангидридом карбоновой кислоты или соответствующей кислотой с применением конденсирующих агентов с ацильной группой для получения N-ацил-пептидной смолы. Путем последующего отщепления нерастворимой смолы и защитных групп на аминокислотах можно получить полипептид с прямой цепью данного изобретения, имеющий указанную выше формулу (I). В случае, когда N- α -замещенный тиокарбамоиламинокислотный остаток или N- α -замещенный тиокарбамоилпептидный остаток выбран в A1 вышеупомянутой формулы, где атом водорода в положении N- α аминоконцевого аминокислотного остатка заменен замещенными тиокарбамоильными группами, N-концевой N- α -замещенный тиокарбамоил-полипептид данного изобретения может быть получен реакцией с замещенным изотиоцианатным соединением при слабощелочных условиях.
В этом случае карбоксильный конец аминокислотного остатка в положении 12 может быть либо свободен (A4 соответствует -OH), либо превращен в амид кислоты (A4 соответствует -NH2). Далее, в полученном полипептид два цистеина в положениях 3 и 11 могут образовать дисульфидную связь (-S-S-) через меркаптогруппы.
Эта дисульфидная связь может быть образована окислением кислородом воздуха или по способу Atherton, E. et al., 1985, J. Chem. Soc. Perkin Trans. 1, 2065.
Если нет других указаний, отдельная аминокислота, применяемая в упомянутом выше твердофазном способе, представляет собой L-форму, а основная аминокислота, соединенная с пролином в положении 7, обозначенном X, может быть только в D-форме.
Любая нерастворимая смола, имеющая аминогруппы, может быть использована в синтезе нового полипептида данного изобретения, если она может соединяться через ее аминогруппу с карбоксильной группой N-защищенного аргинина или лизина при C-конце или в некоторых случаях с крабоксильной группой спейсера, соединенного с ней, и после этого может быть удалена. Примеры таких нерастворимых смол включают в себя (но не ограничены ими) аминометильные смолы (аминометилированные сополимеры стирола и бензола), бензгидриламиновые смолы и их производные. При использовании бензгидриламиновой смолы, метилбензгидриламиновой смолы, диметоксибензгидриламиновой (DMBHA) смолы или аминометилфеноксиметильной смолы амид получают непосредственно путем отщепления, но ввиду большего выхода предпочтительна аминометильная смола.
В качестве спейсера, имеющего функциональную группу, способную соединяться с карбоксильной группой или имеющую карбоксильную группу, может быть применен спейсер, способный превращать карбоксильную группу аргинина в п-карбоксиметилбензиловый эфир, однако особых ограничений в отношении спейсера нет.
Защищенная аминокислота представляет собой аминокислоту, функциональные группы которой могут быть защищены защитной группой при помощи известных способов, или различные защищенные аминокислоты могут быть приобретены в фирмах. Специалистам в данной области понятно, что способы синтеза полипептидов требуют применения защитных групп для стабилизации лабильной боковой цепи от химического изменения во время процесса синтеза.
Защитную группу для α -аминогруппы аминокислоты выбирают из группы, включающей в себя (но не ограниченной) Вoc(т-бутилоксикарбонил) или Fmoc (9-фторенилметилоксикарбонил). Защитная группа для гуанидиногруппы аргинина (Arg) выбрана из группы, включающей в себя (но не ограниченной) Tos (тозил), NO2(нитро), Mtr (4-метокси-2,3,6-триметилбензолсульфонил) или Pmc (2,2,5,7,8-пентаметилхроман-6-сульфонил). Защитная группа для меркаптогруппы цистеина (Cys) может быть выбрана из группы, включающей в себя (но не ограниченной) Bzl (бензил), MBzl (4-метоксибензил),4-MeBzl (4-метилбензил), Acm (ацетамидометил), Trt (тритил), Npys (3-нитро-2-пиридинсульфенил), t-Bu (T-бутил) или t-Bus (т-бутилтио) и Mbzl, 4-MeBzl, Trt, Acm и Npyc являются предпочтительными. Защитную группу для гидроксигруппы тирозина (Tyr) выбирают из группы, включающей в себя (но не ограниченной) Brl, C12Bzl (2,6-дихлорбензил) или t-Bu, и гидроксигруппа может быть незащищенной. Защитную группу для ∈-аминогруппы лизина (Lys) выбирают из группы, включающей в себя (но не ограниченной) Z (бензилоксикарбонил), C/Z (2-хлорбензилоксикарбонил), Boc или Npys. В случае атома водорода ∈ -аминогруппы боковой цепи D-или L-лизина, совпадающего с X в положении 7, эта аминогруппа может быть защищена Z, C/Z, Boc или Npys.
Предпочтительно выбирать из этих защитных групп такие, о которых известно, что они пригодны для условий синтеза.
Соединение защищенных аминокислот можно проводить конденсационными методами, известными в этой области, например, такими как ДСС (дициклогексилкарбодиимидный) метод, D/PCD/ (диизопропилкарбодиимидный) метод (Tartar, A. et al., 1979, J. Org. Chem. 44:5000), метод активного эфира, метод с применением смешанного D или симметричного ангидрида карбоновой кислоты, карбонилдиимидазольный метод, DCC-HOBt (1-гидроксибензотриазольный) метод (Konig W. et. al., 1970, Chem. Ber, 103: 788, 2024, 2034) или дифенилфосфорилазидный метод, но предпочтительно при помощи DCC метода, DCC-HOBt метода, D/PCD/-HOBt метода или метода с применением симметричного ангидрида карбоновой кислоты. Реакцию конденсации можно проводить в органическом растворителе, таком, как дихлорметан, диметилформамид, N-метилпирролидон (NMP) или смешанном из них растворителе. Деблокирующий реагент, такой, как трифторуксусная кислота/дихлорметан, HCl /диоксан, пиперидин/диметилформамид (DMF) или NMP применяют для деблокирования защитной группы для α -аминогруппы. За степенью прогресса реакции конденсации в каждой стадии синтеза следят при помощи способа E. Kaiser et al., 1970, Anal. Biochem 34, 595 (способ с применением нингидриновой реакции).
Согласно предшествующим способам может быть получена смола с защищенным пептидом, имеющим желаемую аминокислотную последовательность. При применении в качестве нерастворимой смолы производного аминометильной смолы защищенный полипептид может быть удален из смолы обработкой ее подходящим растворителем. Полученный защищенный пептид затем обрабатывают фтороводородом для получения амида полипептида, представленного приведенной выше формулой и свободного от всех защитных групп.
При применении в качестве нерастворимой смолы бензгидриламиновой смолы, метилбензгидриламиновой смолы, аминометилфеноксиметильной смолы или DMBHA смолы (Funakoshi, S. et al, 1988, J. Chem. Sos., Chem. Commun., 382) полипептид может быть удален из смолы и одновременно защитные группы могут быть удалены из полипептида путем обработки смолы с защищенным полипептидом фтороводородом, TPMSA (трифторметансульфоновой кислотой) (Yajima, H.et al; "The Peptids", vol. 5, p. 65 (1983), Publishem by Academic Press, edited by E. Gross), TMSTf (тиметилсилилтрифлатом) (Fujii, N, et. al., 1987, J.Chem.Soc., Chem. Commun 274) или THSBr, (триметилсилилбромидом) (Fujii, N, et al., 1987, Chem. Pharm. Ball, 35, 3880) или т.п.
В предпочтительном варианте полученный полипептид восстанавливают 2-меркаптоэтанолом, ДТТ (дитиотреитолом) или т. п. для гарантии того, что меркаптогруппы цистеинов находятся в восстановленном виде. Меркаптогруппы могут затем быть окислены для получения циклического полипептида.
Окислительную обработку можно проводить известным способом. Обычно применяют такой окислительный агент, как воздух или феррицианат (например, феррицианид калия). Альтернативно полипептид (полипептиды) данного изобретения могут быть получены при помощи технологии рекомбинантных ДНК. В соответствии с этим нуклеотидные кодирующие последовательности для полипептида (полипептидов) могут быть клонированы и экспрессированы при помощи известных способов. См. , например, Maniatis et al., Molecular Cloning, A Laboratory Manual, Cold Spring Harbor Laboratory, Cold Spring Harbor N 9, 1991.
Полипептиды изобретения могут быть выделены и очищены способами, известными в данной области для полипептидов, например при помощи экстракции, перекристаллизации, различных хроматографических способов (гель-фильтрации, ионообменной хроматографии, распределительной хроматографии, адсорбционной хроматографии, хроматографии с обращенной фазой), электрофореза, противоточного распределения и т.д. и жидкостной хроматографии высокого разрешения, которая является наиболее эффективной.
Применение полипептидов.
Полипептид (полипептиды) изобретения, представленные формулой (I), обладают способностью связываться с эндоксинами, антибактериальной активностью и активностью гемолиза чувствительных к эндотоксинам гемоцитов (форменных элементов крови). Кроме того, они обладают антивирусной активностью. В характерном варианте полипептиды изобретения имеют активность против ВИЧ. Как будет описано в деталях в разделе 6, полипептиды изобретения проявляют значительно более высокую анти-ВИЧ активность, чем известные полипептиды с высокой аффинностью в отношении эндотоксинов (например, Тахиплезины I, II или III или Полифемузины I или II).
Недавнее развитие систем доставки, способных эффективно вводить ДНК в клетку-мишень, сделало практической генную терапию человека для генетических заболеваний, рака, СПИДа и т.д. (Morgan and Anderson, Annu. Rev. Biochem., 62, 191-217 (1993)).
ДНК-трансфекционные системы предусматривают применение поликатионов, фосфата кальция, слияния липосом, ретровирусов, микроинъекции, электропорации и слияния протопластов. Однако все эти способы страдают от одной или нескольких проблем, относящихся либо к клеточной токсичности, слабой воспроизводимости, неудобству, либо к неэффективности доставки ДНК (Flegner et al., Proc. Natl. Acad. Sci, USA, 84, 7413-7417 (1987)).
Недавно было сообщено о высокоэффективном способе трансфекции ДНК с применением катионного комплекса липид-ДНК или комплекса амфипатический пептид-ДНК (Legendre and Szoka, Jr., Proc. Natl. Acad. Sci. USA, 90, 983-897 (1993)).
Пептиды, которые могут образовать трансфицирующий комплекс с ДНК, включают в себя грамицидин S, тироцидин, полимиксин В, полилизин и мелиттин, все они имеют катионную природу. Среди них наиболее эффективным катионным пептидом является грамицидин S, который известен как амфипатический циклический декапептидный антибиотик с β -пластинчатой конформацией и может делать проницаемыми и разрушать клеточные мембраны. Считают, что как положительный заряд, так и амфипатический характер грамицидина S важны для высокого уровня трансфекции. С учетом этих структурных характеристик полипептида данного изобретения могут быть альтернативным кандидатом для образования комплекса с ДНК с высокой трансфицирующей способностью, вследствие их сильной катионной и амфипатической природы с β -пластинчатой конформацией.
Действительно, тахиплезин I, одна из родительских молекул полипептидов данного изобретения, может проникать через мембраны и связываться с ДНК подобно грамицидину S (Matsuzaki et al. , Biochem. Biophys. Acta, 1070, 259-264 (1991) и Yonezawa et al., Biochemistry, 31, 2998-3004 (1992).
Кроме того, было подтверждено, что структура Т-22, одного из полипептидов этого изобретения (I), очень похожа на структуру Тахиплезина I с амфипатической антипараллельной β -пластинчатой структурой (Tamamure et al., Biochem. Biophys. Acta, 1163, 209-216 (1993)).
Таким образом, полипептиды данного изобретения могут быть использованы в качестве компонента ДНК-трансфицирующих систем для генной терапии.
Поэтому полипептиды данного изобретения могут быть использованы в фармацевтической композиции, содержащей полипептид (полипептиды) изобретения или их соль и фармацевтически приемлемый носитель, выбранный в соответствии со способом введения и формой введения этой фармацевтической композиции. Фармацевтическими носителями могут быть такие фармацевтически совместимые буферы, как раствор Хенка или Рингера, физиологический раствор, смесь, содержащая солевой раствор и глюкозу, и гепаринизированный раствор цитрата натрия - лимонной кислоты - декстрозы. Фармацевтическую композицию вводят перорально или парентерально в соответствии с объектом лечения. Композиция может быть приготовлена в виде таких препаратов, как порошок, гранулы, раствор для инъекции или для перорального введения, таблетки, суппозитории, пессарии, мазь, крем или аэрозоль, с применением подходящих носителей в соответствии со способом введения.
При прямом введении фармацевтической композиции в виде инъекции больному полипептид или его соль могут вводиться непрерывно или прерывисто в количестве 10 - 5000 мг на кг веса тела больного и на один день и при помощи капельного внутривенного вливания в виде растворов в физиологическом растворе.
6. Примеры.
Далее изобретение описано в примерах, которые не предназначены для ограничения данного изобретения.
В приведенных здесь примерах описан синтез полипептида (I). Кроме того, описаны результаты определений активности против ВИЧ для полипептидов изобретения и известных полипептидов с высокой аффинностью в отношении эндотоксинов. Полипептиды данного изобретения имеют значительно более высокую активность против ВИЧ, чем известные полипептиды с высокой аффинностью в отношении эндотоксинов.
В следующих далее примерах применяли следующие устройства и реагенты:
Прибор для жидкостной хроматографии высокого давления Shimadzu Corporation, Model LC-6A
Колонка прибора: Asahipak ODP-90 (Asahi Chemical Industry Co, Ltd).
Fmoc аминокислота и амино-смола: изготовленная Watanabe Chemical Industries, Ltd.
Конденсирующий агент: изготовленный в Peptide Institude, INC and Applied Biosystems Japan.
FAB-MS (FAB - масс-спектрометр): VC Co. (USA), Mod ZAB-SE DMF-диметилформамид
Пример 1: синтез полипептида (1)
Синтез полипептида (1)(имеющий формулу, представленную в конце описания) в разделах 6.1.1 - 6.1.7, infra.
Полипетпиды (2-13, 22, 23) и предшественники-пептиды полипетпидов (14 - 21, 24, 25) (см. табл. 1 в конце текста, supra в отношении структур) синтезированы при помощи сходных процедур.
В формуле (1). Arg, Trp, Cys, Tyr, Lys, DLys и Pro обозначают упомянутые выше аминокислотные остатки, а линия между Cys в положениях 3 и 11 обозначает дисульфидную связь.
Введение в аминометильную смолу Fmoc-DMBHA-CH2CH2COOH
6.1.1. [(3-( α -Fmoc-амино-4-метоксибензил)-4- метоксифенил)пропионовой кислоты].
270 мг (0,2 ммоль) аминометильной смолы (0,74 мэкв/г) и 268,5 мг (0,5 ммоль, 2,5 эквз.) Fmoc-DMBHA-CH2CH2COOH (мол. маса 537) помещали в колонку для твердофазного синтеза и конденсационную реакцию проводили в течение 2 часов по методу D|PCD| - HOBt в DMF согласно Guo, L. et. al., (Chem. Pharm. Bull 36, 4989, (1988)).
После завершения реакции конденсации проводили реакцию сочетания для защиты свободных аминогрупп с применением уксусного ангидрида (смола DMBHA).
6.1.2. Введение аргинина в положении 12 в смолу DMBHA.
После удаления Fmoc групп из смолы DMBHA, приготовленной в разделе 6.1.1. , supra, смесью 20% пиперидин/DMF 2,5 эквивалента (экв) Fmoc - Arg (Pmc)-OH в расчете на смолу DMBHA добавляли и проводили реакцию конденсации в DMF согласно D|PCD|-HOBt по способу.
Степень прогресса реакции конденсации прослеживали при помощи нингидринового теста Kaiser, E. et. al. [Anal. Biochem. 34, 595 (1980)].
Введение цистеина в положение 11
После удаления Fmoc групп из смолы DMBHA, приготовленной в 6.1.2, при помощи смеси 20% пиперидин/DMF 2,5 экв Fmoc-Cys(Trt)-OH в расчете на смолу DMBHA добавляли и реакцию конденсации проводили в DMF по методу D|PCD|-HOBt. Степень прогресса реакции конденсации прослеживали, как в 6.1.2, supra при помощи нингидринового теста.
Введение аминокислот в положениях 10 - 1
Так же, как описано выше, Lys(Boc), Arg(Pmc), Tyr(tBu), Pro, DLys(Boc), Lys(Boc), Arg(Pmc), Tyr(tBu), Cys(Trt), Trp, Arg(Pmc) и Arg(Pmc) последовательно вводили в смолу DMBHA для получения смолы с защищенным защитными группами пептидом (I).
Каждую реакцию конденсации аминокислот в твердофазном синтезе проводили в соответствии с рабочими условиями табл. 2.
Получение полипептида (I) путем удаления защитных групп, удаления полипептида (I) из смолы и частичной очистки
Смолу с защищенным защитными группами полипептидом (I) подвергали обработке смесью 20% пиперидин/DMF для удаления Fmoc из группы и затем подвергали реакции при 25oC в течение 2 часов в системе 1M TMSOTf-тиоанизол/TFA (трифторуксусная кислота) (10 мл трифторуксусной кислоты в присутствии м-крезола (100 экв. ) и этандитиола (300 экв.)) на 100 мг смолы. Затем смолу отфильтровывали от реакционной смеси и промывали дважды 1 мл трифторуксусной кислоты. Затем к смеси фильтрата и промывки добавляли 100 мл охлажденного на льду сухого эфира. Образовавшийся осадок центрифугировали и осадок отделяли от супернатанта декантацей. Полученный осадок промывали холодным эфиром, растворяли в 10 мл 4N AcOH и добавляли 830 мг, 80 экв. дитиотреитола. Смесь перемешивали при комнатной температуре в течение ночи.
Реакционный раствор центрифугировали, супернатант обрабатывали Сефадексом G-10 (3,7 x 5 см), подвергали гель-фильтрации с 4N уксусной кислотой (AcOH) и собирали протекающие через Сефадекс фракции в виде основной части элюата и лиофилизировали, получая в виде порошка частично очищенный нециклизованный полипептид (I).
Получение полипептида (I) окислением кислородом воздуха
Половинное количество протекающей через Сефадекс при гель-фильтрации фракции доводили до pH 7,5 концентрированным водным аммиаком и подвергали окислению кислородом воздуха путем аэрации для проведения реакции циклизации. После завершения окисления кислородом воздуха циклизованный полипептид (I) адсорбировали на 10 г смолы Diaion HP-20 и затем элюировали 60% CH3CN (в 1 N AcOH).
Элюат концентрировали при комнатной температуре при пониженном давлении для удаления CH3CN и затем лиофилизировали, получая порошок. Порошок растворяли в небольшом количестве воды и раствор наносили на колонку Asahipak ODP-90 и очищали жидкостной хроматографией высокого разрешения (HPLC-Model LC-6A, получаемая Shimadzu Corp.) с применением градиентной элюции при помощи CH3CN, получая единственный пик полипептида (I) с выходом 27% (величина рассчитана на основе смолы с защищенным защитными группами полипептидом (I).
Анализ полипептида.
Было обнаружено, что аминокислотный состав после кислотного гидролиза по методу Liu et al., [J. Biol. Chem., 251 1936 (1976)] и аминокислотный состав, полученный расщеплением лейцинаминопептидазой полипептида, очищенного, как описано в разделе 6.1.6, хорошо соответствует рассчитанным величинам аминокислотного состава, основанного на аминокислотной последовательности формулы (I).
Далее, величина мол. массы, полученная при помощи FAB-MS была 1996,3, тогда как рассчитанная величина [M + H]+ равна 1996, 1.
Удельное вращение [α]
Пример 3: синтез полипептида (14) [N- α ацетилирование аминоконцевого аминокислотного остатка полипептида (1)].
Синтез полипептида (14) формулы 9 описан в разделах 6.2.1-6.2.2.
[Формула 9](см. в конце текста).
В этой формуле (14)Ac-Arg, Arg, Trp, Cys, Tyr, Lys, D Lys и Pro обозначают упомянутые выше аминокислотные остатки и линия между Cys в положениях 3 и 11 обозначает дисульфидную связь.
Ацетилирование смолы с частично защищенным защитными группами пептидом (I).
1,301 г (0,25 ммоль) смолы с защищенным защитными группами пептидом (I), полученной в стадии (4) (Раздел 6,1.4) примера 1, брали в реакционный сосуд для проводимого вручную твердофазного синтеза. После удаления Fmoc группы N-концевую аминогруппу ацетилировали по методу Hudson [J.Org. Chem. 53, 617, (1988)] , получая 1,241 г смолы с пептидом (1), защищенным ацетилированной N-концевой α аминокислотной защитной группой (выход сухого веса 100%). Процедура суммирована в табл. 3.
Ацетилирование проводили повторением операций 1 -3 до тех пор, пока нингидриновая реакция не переставала быть положительной.
Получение пептида (14) путем удаления защитных групп и смолы из смолы с пептидом (1), защищенным ацетилированной N-концевой аминокислотной защитной группой частичной очистки и окисления
Полипептид (14) получали так же, как описано в 6.1.6 supra .Удельное вращение [α]
Результат аминокислотного анализа хорошо согласовался с рассчитанными величинами.
Пример 4: синтез полипептида (19) [N- α флуоресцеинтиокарбамоилполипептида (1)].
Синтез полипептида (19)) формулы 10 проводили путем N- α флуоресцеинтикарбамоилирования аминоконцевого аминокислотного остатка полипептида (1).
[Формула 10] (см. в конце текста).
В формуле (19) FTC-Arg, Arg, Trp, Cys, Tyr, Lys, D Lys и Pro обозначают упомянутые выше аминокислотные остатки и твердая линия между Cys в положениях 3 и 11 обозначает дисульфидную связь.
10 мг (3,9 мкмоль) соли уксусной кислоты полипептида (1), полученной в 6.1.6, растворяли в 1 мл PBS буфера (солевой раствор с фосфатным буфером, pH 7,5). К этому раствору добавляли 2,8 мг (7,2 мкмоль) изомера-1 флуоресцеинизотиоцианата (FITC) (Wako Pure Chemical Ind., Ltd), растворенного в 1 мл DMSO, при охлаждении на льду. После 6 - 7 часов перемешивания при комнатной температуре свободная аминогруппа была флуоресцеинтиокарбамоилирована.
Реакционную смесь наносили на колонку Сефадекса G-25 (fine) [уравновешенную 50 мМ PB (фосфатным буфером), pH 4,2], предварительно обессоленную, и фракционировали. Пептидную фракцию поглощали на Sep-Pak C 18 plus ENV cartridge колонке (Millipore Co), элюировали раствором 80% ацетонитрил/уксусная кислота с pH 4,2 и лиофилизировали, получая 3,93 мг соли уксусной кислоты полипептида (19) (выход 35%)
Удельное вращение [α]
Аминокислотный анализ кислотного гидролизата этого соединения по методу Liu показал на 1 меньшее число остатков аргинина, чем рассчитанная величина для политпептида (1).
При помощи тонкослойной хроматографии (н-бутанол: уксусная кислота:вода= 4: 1: 1) частичных кислотных гидролизатов этого соединения в трифторуксусной кислоте (комнатная температура, в течение 2 часов) обнаружили одно основное пятно, которое соответствует пятну FTH-Arg (флуоресцеинаргинин-тиогидантоина).
Результаты этих анализов свидетельствует о том, что α аминогруппа N-концевого остатка аргинина избирательно тиокарбамоилирована.
Пример 2 и сравнительные примеры 1 и 2: антивирусная активность против вируса иммунодефицита человека (ВИЧ)
Антивирусную активность против ВИЧ полипептида (1), синтезированного в примере 1, тестировали и оценивали согласно следующему способу.
Инфицированные ВИЧ МТ-4 клетки (2,5•104 клеток на лунку, множественность заражения (MOI):0,001) сразу же после инфицирования добавляли на 96-луночный микротитрационный планшет. После инкубирования при 37oC в течение 5 дней в инкубаторе с CO2 число выживших клеток измеряли по MТТ методу [Pauwels et al., J.Virol. Methods, 20, 309-321 (1982)]
Антивирусную активность выражали в виде концентрации, при которой смерть клеток, вызываемая инфекцией ВИЧ, ингибируется на 50% (EC50:50% эффективная концентрация). С другой стороны, для того чтобы знать цитотоксичность тест-вещества на МТ-4 клетках, не инфицированные вирусом клетки инкубировали, как описано выше, вместе с тест-соединением с различными концентрациями. Цитотоксичность выражали в виде 50% цитотоксичной концентрации (CC50), обнаруживаемой тест-веществом. Далее, приблизительное отношение CC50 к EC50 (CC50/EC50) выражали как эффективное отношение (SI).
Следующая далее формула 8 представляет пептидный антивирусный агент, применяемый для сравнения с полипептидом (1): (Arg-Arg-Trp-Cys-Tyr-Arg-Lys-Cys-Tyr-Lys-Gly-Tyr-Cys-Tyr-Arg-Lys -Cys-Arg-NH2........Т-22)
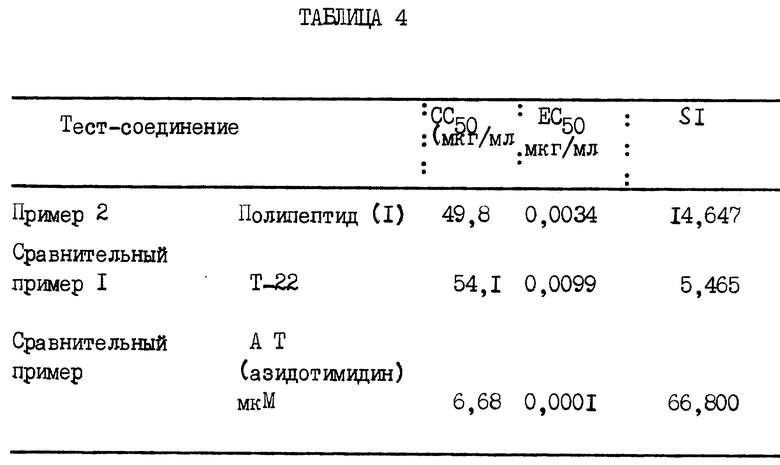
Табл. 4 показывает величины EC50, CC50 и SI полипептида (1), пептида Т-22 и анти-ВИЧ агента AZT.
Эта таблица ясно показывает, что полипептид (1) данного изобретения имеет ту же самую цитотоксичность, что и Т-22, антивирусная активность которого была обнаружена ранее, но проявляет антивирусную активность при 1/3 концентрации Т-22. Даже хотя 4 аминокислотных остатка были удалены из этого пептида и мол. масса была понижена, полипептид данного изобретения обнаруживал даже более высокую активность.
По сравнению а азидотимидином (AZT) величина EC50 полипептида (1) была несколько выше, но он проявлял чрезвычайно низкую цитотоксичность. Поэтому мы ожидаем, что он может быть использован как более безопасный анти-ВИЧ агнт.
Пример 5: Характеристики полипептидов и их анти-ВИЧ активность
Табл. 5 показывает структурные формулы и характеристики полипептидов данного изобретения, полученных при помощи процедур примеров 1,3 и 4. Табл. 5 также показывает антивирусную активность этих полипептидов, тестированную и оцениваемую по способу примера 2.
Если нет других указаний, Cys в положениях 3 и 11 приводимых в качестве примеров соединений, представленных в приведенных таблицах, соединены дисульфидной связью.
Далее, в таблице "AZT" обозначает азидотомидин (обычно известный как зидовудин), а Т-22 обозначает полипептид, представленный в формуле 8.
Эффективность изобретения
Данное изобретение обеспечивает новый полипептид, обладающий антивирусной активностью против вируса иммунодефицита человека (ВИЧ), и позволяет получить гидрофильность, аффинность в отношении липидов, более высокую активность и применимо для выяснения механизма манифестации активности против вируса иммунодефицита человека (ВИЧ).
| название | год | авторы | номер документа |
|---|---|---|---|
| СОЛИ ПЕРЕХОДНОГО МЕТАЛЛА С ПОЛИПЕПТИДОМ И СПОСОБ ПОВЫШЕНИЯ АКТИВНОСТИ ПОЛИПЕПТИДА ПРОТИВ ВИЧ | 1997 |
|
RU2179981C2 |
| ПОЛИПЕПТИДЫ ИЛИ ИХ СОЛИ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ОБЛАДАЮЩАЯ ПРОТИВОВИРУСНОЙ АКТИВНОСТЬЮ В ОТНОШЕНИИ ВИРУСА ИММУНОДЕФИЦИТА ЧЕЛОВЕКА | 1991 |
|
RU2073685C1 |
| ПОЛИПЕПТИДЫ ИЛИ ИХ СОЛИ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1992 |
|
RU2083589C1 |
| КОНЪЮГАТЫ ИНСУЛИН-ИНКРЕТИН | 2014 |
|
RU2678134C2 |
| АНАЛОГИ ГЛЮКАГОНА, ОБЛАДАЮЩИЕ ПОВЫШЕННОЙ РАСТВОРИМОСТЬЮ И СТАБИЛЬНОСТЬЮ В БУФЕРАХ С ФИЗИОЛОГИЧЕСКИМИ ЗНАЧЕНИЯМИ Ph | 2009 |
|
RU2560254C2 |
| ОСНОВАННЫЕ НА АМИДАХ ПРОЛЕКАРСТВА ПЕПТИДОВ ГЛЮКАГОНОВОГО НАДСЕМЕЙСТВА | 2009 |
|
RU2550696C2 |
| ПОЛИПЕПТИДЫ ДЛЯ ЛЕЧЕНИЯ СИНДРОМОВ СТРЕССА, ИММУННОЙ РЕАКЦИИ И ИНСУЛЬТА | 2019 |
|
RU2824567C2 |
| АНТИБИОТИЧЕСКИЕ ПЕПТИДЫ | 2008 |
|
RU2472805C2 |
| ПЕПТИДНЫЕ КОМПОЗИЦИИ | 2014 |
|
RU2725150C2 |
| МОДУЛЯЦИЯ СПЕЦИФИЧНОСТИ СТРУКТУРИРОВАННЫХ БЕЛКОВ | 2012 |
|
RU2745572C2 |
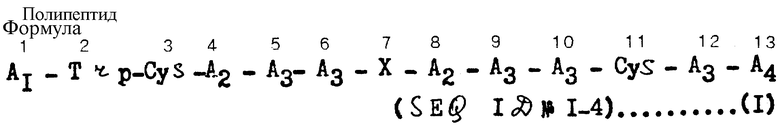


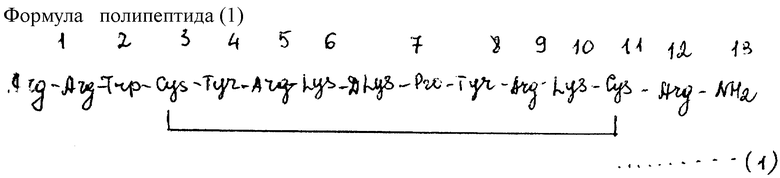

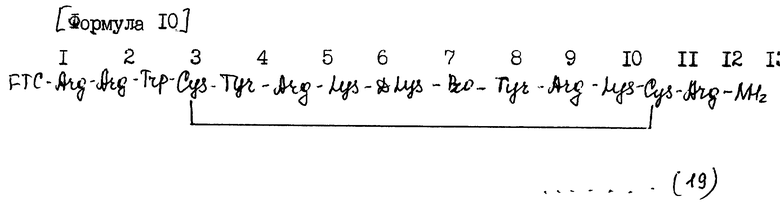




Полипептид формулы A1-Trp-Cys-A2-A3-A3-X-A2-A3-A3-Cyr-A3-А4, где A1 - пептидный остаток, имеющий две основные аминокислоты, выбранные из лизина, аргинина и орнитина, причем остаток, в котором N-α атом водорода аминоконца аминокислотного остатка может быть замещен ацильной группой или тиокарбамоильной группой, образует N-α замещенный пептидный остаток, N-α замещенный замещенной тиокарбамоильной группой пептидный остаток; А2 - остаток тирозина, А3 -остаток лизина или аргинина; А4 - NH2; Х - остаток пептида D-лизил-пролин, пролил-D-лизин, лизил-глицин, D-орнитил-пролин, в которых атом водорода: ω-аминогруппы, D-лизина, L-лизина, D-лизина, D-орнитинаил и L-орнитина может быть заменен ω-аминоацильной группой и пептидный остаток соединен с амино-кислотными остатками в положениях 6 и 8 через пептидную связь per se; Trp - остаток триптофана, Cys - остаток цистеина или его соль, можно применять в фармацевтической композиции в качестве ативирусного агента, например, против ВИЧ-инфекции. 4 c. и 1 з.п. ф-лы, 2 ил., 5 табл.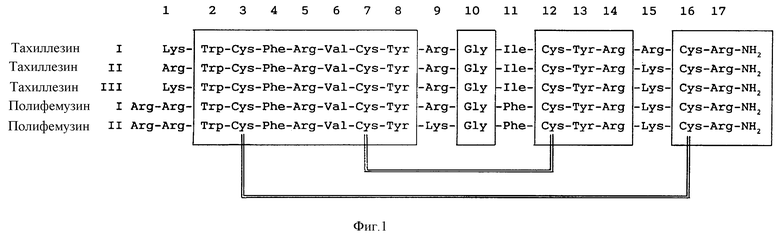
где A1 представляет собой пептидный остаток, имеющий две основные аминокислоты, выбранные из группы, состоящей из лизина, аргинина и орнитина, причем остаток, в котором N-α атом водорода амино-конца аминокислотного остатка может быть замещен ацильной группой или замещенной тиокарбамоильной группой, образует N-α замещенный ацилом пептидный остаток, N-α замещенный замещенной тиокарбамоильной группой пептидный остаток;
A2 представляет собой остаток тирозина;
A3 представляет собой остаток лизина или аргинина;
A4 представляет собой -NH2;
X представляет собой остаток пептида, выбранный из группы, состоящей из таких пептидов, как D-лизил-пролин, пролил-D-лизин, лизил-глицин, D-орнитил-пролин, в которых атом водорода ω-аминогруппы, D-лизина, L-лизина, D-орнитина или L-орнитина может быть заменен ω-аминоацильной группой, и пептидный остаток соединен с аминокислотными остатками в положениях 6 и 8 через пептидную связь per se;
Trp представляет собой остаток триптофана;
Cys представляет собой остаток цистеина или его соль.
где A1 представляет собой пептидный остаток, имеющий две основные аминокислоты, выбранные из группы, состоящей из лизина, аргинина и орнитина, причем пептидный остаток, в котором N-α атом водорода амино-конца аминокислотного остатка может быть замещен ацильной группой или замещенной тиокарбамоильной группой, образует N-α замещенный ацилом пептидный остаток, N-α замещенный замещенной тиокарбамоильной группой пептидный остаток;
A2 представляет собой остаток тирозина;
A3 представляет собой остаток лизина или аргинина;
A4 представляет собой -OH (происходящий из карбоксильной группы) или -NH2 (происходящий из кислой амидной группы);
X представляет собой пептидный остаток, выбранный из группы, состоящей из таких пептидов, как D-лизил-пролин, пролил-D-лизин, лизил-глицин, D-орнитил-пролин, в которых атом водорода ω-аминогруппы D-лизина, α-лизина, D-орнитина или L-орнитина может быть заменен ω-аминоацильной группой, и пептидный остаток соединен с аминокислотными остатками в положениях 6 и 8 при помощи пептидной связи per se;
Trp представляет собой остаток триптофана;
Cys представляет собой остаток цистеина,
где остатки цистеина в положениях 3 и 11 соединены дисульфидной связью или его соль.
| Устройство для комплектного измерения диаметров сопрягаемых деталей | 1971 |
|
SU502198A1 |
| Устройство для зажима стропы | 1973 |
|
SU513613A3 |
| Машковский М.Д | |||
| Лекарственные средства | |||
| - М.: Медицина, 1985, т | |||
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| Счетная бухгалтерская линейка | 1922 |
|
SU386A1 |

