Настоящее изобретение относится к новому антибиотическому пептиду и пептидным производным, в частности для применения в медицине.
Дополнительно, изобретение относится к композициям и способам уничтожения микроорганизмов, таких как бактерии или грибы, и способам лечения инфекций, вызванных микроорганизмами. Изобретение дополнительно относится к способу скринингового анализа лекарственных средств.
Несмотря на значительные достижения в лечении антибиотиками увеличивается число случаев тяжелых бактериальных и грибковых инфекций. Каждый год в Соединенных Штатах имеет место более чем 40 миллионов случаев госпитализации, и приблизительно 2 миллиона пациентов заражаются внутрибольничными инфекциями, 50-60% которых вызывают бактерии, устойчивые к действию антибиотиков [1]. Ежегодное количество смертей, связанных с внутрибольничными инфекциями, оценивают как около 60000-70000 в США и вплоть до 10000 в Германии [2]. Тогда как устойчивые грамотрицательные бактерии представляли собой большую проблему в 1970-х, прошедшее десятилетие показало рост числа инцидентов с грамположительными штаммами, устойчивыми к действию многих лекарственных средств [3]. В настоящее время в быстрое возникновение устойчивых штаммов вовлечены как грамположительные, так и грамотрицательные патогенные микроорганизмы [4]. Устойчивость впервые проявилась у видов, которым было достаточно единичных мутаций, чтобы вызвать клинически значимые уровни, таких как Staphylococcus aureus и Pseudomonas aeruginosa, за ними последовали бактерии, которым необходимы множественные мутации, такие как E. coli и Neisseria gonorrhoeae. Это происходило, в основном, в результате широкого применения фторхинолонов [5]. Важные причины устойчивости грамотрицательных штаммов включают расширенный спектр лактамаз в Escherichia coli и Klebsiella pneumoniae [6]. Почти половина клинических штаммов Haemophilus ducreyi, которые обуславливают мягкий шанкр, несут гены для придания устойчивости к амоксициллину, ампициллину и ряду других β-лактамов [7]. Кроме того, у Salmonella enteric, серовара Typhimurium, устойчивость к тетрациклинам увеличилась от нуля в 1948 г. до уровня в 98% в 1998 г. [8].
Это влечет за собой необходимость продолжения поиска новых антибиотиков. Индуцибельные антибактериальные пептиды составляют область исследования, где сходятся вместе современные биохимия, иммунология и разработка лекарственных средств. Пептидные антибиотики, варьирующие по размеру от 13 до более чем сотни аминокислотных остатков, уже получены из растений, животных и микроорганизмов [9]. От одного животного получают приблизительно 6-10 пептидных антибиотиков, где каждый пептид часто демонстрирует разнообразный спектр активности [10]. Точно установлено, что подавляющее большинство бактерицидных пептидов, включая хорошо изученные дефензины, цекропины и магаинины, функционируют посредством «литического/ионофорного» механизма. Общим свойством всех «литических» пептидов является воздействие на проницаемость бактериальных цитоплазматических мембран [11, 12, 13]. Основой для такого действия является катионная, амфипатическая структура, которая способствует образованию каналов для гидрофильных ионов (протонов) в липидном бислое; утечка протонов приводит к исчезновению трансмембранного потенциала, необходимого для многих жизненно важных процессов, и, таким образом, служит причиной гибели клетки. Поскольку нарушение в структуре мембран этими пептидами не зависит от распознавания хиральными молекулами, функционально допустимыми являются аминокислотные замены, которые не изменяют общую амфипатическую структуру или общий суммарный заряд [14, 15]. Пептиды, действующие непосредственно на бактериальную мембрану, в повышенных концентрациях часто также оказывают токсическое действие на клеточные мембраны млекопитающих, что ограничивает их потенциал в качестве перспективных лекарственных средств. Когда в последовательности α-спиралей антибактериальных пептидов вводят пролины, способность пептидов к изменению проницаемости цитоплазматической мембраны E. coli значительно снижается, в зависимости от количества включенных остатков пролина. В связи с этим любопытным является то, что некоторые из наиболее активных природных антибактериальных пептидов, отобранные по меньшей мере против грамотрицательных патогенов, принадлежат к семейству пептидов, обогащенных пролином [16].
Подобные побочные эффекты можно преодолеть посредством других антибактериальных пептидов, мишенью которых являются специфические бактериальные белки или другие внутри- или внеклеточные соединения, без перекрестной активности к аналогам млекопитающих. Этому, видимо, соответствуют антибактериальные пептиды, обогащенные пролином, включающие апидецин, первоначально выделенный из насекомых. При огромной вариации в размере и биохимических свойствах не удивительно, что целью исследований антибактериальных пептидов являются взаимосвязи структура - активность и конформация - активность. Полная доработка природного репертуара антибактериального пептида до его биологических возможностей является важной не только для общей биохимии, но и представляет постоянный интерес для фармацевтической промышленности. Несмотря на проблемы, связанные с in vitro тестированием антибиотиков, основанных на пептидах, некоторое количество природных антибактериальных пептидов уже дошло до стадии клинических испытаний [17]. Тогда как некоторые из них показали эффективность на начальной стадии клинических испытаний как средства для местного действия, другие показывают активность как средства для системной терапии. Например, недавно завершил фазу III клинических испытаний катионный белок rBPI 21 для парентерального применения при менингококкемии [17].
Апидецин, который исходно выделили из медоносной пчелы, относится к семейству антибактериальных пептидов, обогащенных пролином, и демонстрирует последовательность, сходную с последовательностью пиррхокорицина, дрозоцина и формецина (таблица 1) [18]. Апидецины представляют собой пептиды длиной в 18-20 аминокислотных остатков, содержащие только немодифицированные L-аминокислоты с высококонсервативными PRPPHPRI/L C-концами и с относительно высоким содержанием пролина (33%). Их можно легко синтезировать на твердой фазе, используя стандартную стратегию Fmoc/tBu. Пептид в наномолярных дозах ингибирует жизнеспособность многих грамотрицательных бактерий, при этом грамположительные бактерии являются не восприимчивыми к его действию. Летальное действие является почти мгновенным и, как было показано, не зависит от общепринятого «классического» механизма [19]. Кроме того, у мутантов, устойчивых к действию апидецина, чувствительность к «порообразующим» пептидам не уменьшена, и D-энантиомер лишен антибактериальной активности. На сегодняшний день существует модель, связывающая антагонистическое действие апидецина на бактерии со стереоселективным распознаванием хиральных мишеней [19]. Антибактериальные пептиды, обогащенные пролином, включая в качестве члена этого семейства апидецин, не просто убивают бактерии посредством нарушения проницаемости их мембран, но связываются стереоспецифическим образом с белком-мишенью, который не идентифицирован для апидецина, что, в конечном счете, приводит к гибели клетки. Кроме того, в отличие от AMP, таких как меллитин или грамдицидин S, с определенными вторичными структурами, пептиды, обогащенные пролином, по-видимому, нетоксичны для эукариотических клеток in vitro и не являются гемолитическими. В сыворотке млекопитающих апидецин полностью разрушается в течение одного часа из-за расщеплений на N- и C-концах, которые могут быть результатом расщепления амино- и карбоксипептидазами, эндопротеазного расщепления или их всех. Ранее упомянутые метаболиты теряют антибактериальную активность при значениях MIC обычно выше 125 мкг/мл.
Все еще сохраняется потребность в новых антибактериальных и противогрибковых соединениях, новых антибактериальных и фунгицидных фармацевтических композициях, способах их применения и новых соединениях, которые можно использовать в скрининговом анализе лекарственных средств для выявления новых фармацевтических антибиотиков.
Одной целью изобретения является получение новых антибиотических пептидов, предпочтительно с повышенной стабильностью. Другой целью изобретения является обеспечение способа для скринингового анализа лекарственных средств.
Первая цель изобретения достигается пептидом или пептидным производным по изобретению с по меньшей мере 16 аминокислотными остатками и общей формулой

где
X1 представляет собой нейтральный остаток или фрагмент с суммарным положительным зарядом или с положительно заряженной в физиологических условиях боковой цепью, которая все еще несет положительный заряд после модификации с Sub1. Предпочтительные остатки X1 предпочтительно выбраны из группы нейтральных остатков (таких как цис-4-гидроксипролин, транс-4-гидроксипролин, цис-3-гидроксипролин, транс-3-гидроксипролин, цитруллин, N-метилсерин, N-метилглицин, дигидроксифенилаланин, N-этиласпарагин, N-этилглицин, гомосерин, пеницилламин, тетрагидропиранилглицин, алло-треонин, 3,5-динитротирозин) или положительно заряженных остатков, включая аргинин, δ-гидроксилизин, гомоаргинин, β-гомоаргинин, D-аргинин, метиларгинин (предпочтительно альфа-N-метиларгинин), нитроаргинин (предпочтительно N(G)-нитроаргинин), нитрозоаргинин (предпочтительно N(G)-нитрозоаргинин), аргинал (-COOH в аргинине замещена на -CHO), гуанидинопропионовая кислота, 2,4-диаминомасляная кислота, β-гомоаргинин, ε-N-метиллизин, алло-гидроксилизин, 2,3-диаминопропионовую кислоту, 2,2'-диаминопимелиновую кислоту, лизин, орнитин, симметричный диметиларгинин, асимметричный диметиларгинин, 2,6-диаминогексановую кислоту, гистидин, 1-метилгистидин, 3-метилгистидин, п-аминобензойную кислоту и 3-аминотирозин;
X2 представляет собой остаток с полярной боковой цепью (такой как аспарагин, серин, цитруллин или глутамин) или фрагмент с суммарным положительным зарядом или с положительно заряженной в физиологических условиях боковой цепью (такой как лизин или аргинин, орнитин или гомоаргинин). Предпочтительные остатки X2 выбраны из групп, включающих аспарагин, глутамин, серин, треонин, цитруллин, цис-4-гидроксипролин, транс-4-гидроксипролин, цис-3-гидроксипролин, транс-3-гидроксипролин, цитруллин, N-метилсерин, N-метилглицин, дигидроксифенилаланин, N-этиласпарагин, N-этилглицин, гомосерин, пеницилламин, тетрагидропиранилглицин, алло-треонин, 3,5-динитротирозин, а также аргинин, лизин, δ-гидроксилизин, гомоаргинин, 2,4-диаминомасляную кислоту, гомоаргинин, β-гомоаргинин, D-аргинин, метиларгинин (предпочтительно альфа-N-метиларгинин), нитроаргинин (предпочтительно N(G)-нитроаргинин), нитрозоаргинин (предпочтительно N(G)-нитрозоаргинин), аргинал, гуанидинопропионовую кислоту, ε-N-метиллизин, алло-гидроксилизин, 2,3-диаминопропионовую кислоту, 2,2'-диаминопимелиновую кислоту, орнитин, симметричный диметиларгинин, асимметричный диметиларгинин, 2,6-диаминогексановую кислоту, гистидин, 1-метилгистидин, 3-метилгистидин, п-аминобензойную кислоту и 3-аминотирозин;
X3 представляет собой фрагмент с суммарным положительным зарядом или с положительно заряженной в физиологических условиях боковой цепью. Предпочтительные остатки X3 выбраны из группы, включающей аргинин, лизин, δ-гидроксилизин, гомоаргинин, 2,4-диаминомасляную кислоту, гомоаргинин, β-гомоаргинин, D-аргинин, метиларгинин (предпочтительно альфа-N-метиларгинин), нитроаргинин (предпочтительно N(G)-нитроаргинин), нитрозоаргинин (предпочтительно N(G)-нитрозоаргинин), аргинал, гуанидинопропионовую кислоту, ε-N-метиллизин, алло-гидроксилизин, 2,3-диаминопропионовую кислоту, 2,2'-диаминопимелиновую кислоту, орнитин, симметричный диметиларгинин, асимметричный диметиларгинин, 2,6-диаминогексановую кислоту, гистидин, 1-метилгистидин, п-аминобензойную кислоту, 3-метилгистидин и 3-аминотирозин;
X4 представляет собой нейтральный остаток с полярной боковой цепью, такой как аспарагин или цитруллин, или фрагмент с суммарным положительным зарядом или с положительно заряженной в физиологических условиях боковой цепью. Предпочтительные остатки X4 выбраны из групп, включающих аспарагин, гомоглутамин, цис-4-гидроксипролин, транс-4-гидроксипролин, цис-3-гидроксипролин, транс-3-гидроксипролин, цитруллин, N-метилсерин, N-метилглицин, дигидроксифенилаланин, N-этиласпарагин, N-этилглицин, гомосерин, пеницилламин, тетрагидропиранилглицин, алло-треонин и 3,5-динитротирозин, а также аргинин, лизин, δ-гидроксилизин, гомоаргинин, 2,4-диаминомасляную кислоту, β-гомоаргинин, D-аргинин, метиларгинин (предпочтительно альфа-N-метиларгинин), нитроаргинин (предпочтительно N(G)-нитроаргинин), нитрозоаргинин (предпочтительно N(G)-нитрозоаргинин), аргинал, гуанидинопропионовую кислоту, ε-N-метиллизин, алло-гидроксилизин, 2,3-диаминопропионовую кислоту, 2,2'-диаминопимелиновую кислоту, орнитин, симметричный диметиларгинин, асимметричный диметиларгинин, 2,6-диаминогексановую кислоту, гистидин, п-аминобензойную кислоту, 1-метилгистидин, 3-метилгистидин и 3-аминотирозин;
X5 представляет собой пролин или производное пролина, такое как цис-4-гидроксипролин, транс-4-гидроксипролин, цис-3-гидроксипролин, транс-3-гидроксипролин, β-циклогексилаланин, 3,4-цис-метанопролин, 3,4-дегидропролин, гомопролин или псевдопролин;
Нейтральный остаток определяется как аминокислотный остаток, имеющий незаряженную в физиологических условиях боковую цепь. Физиологические условия обозначают pH 7,4, 37°C и осмотическое давление, равное 300 мосмоль/кг. Полярную боковую цепь определяют как боковую цепь, несущую по меньшей мере одну полярную группу (например, гидроксил, амино, амид или сульфгидрильную группу), которые обеспечивают образование водородной связи с другой полярной группой. Производное пролина представляет собой структуру, полученную из пролина, содержащую замещенное или незамещенное кольцо пирролидина. Фрагмент, имеющий суммарный положительный заряд или положительно заряженную в физиологических условиях боковую цепь, является основным аминокислотным остатком. Гидрофобный фрагмент представляет собой нейтральный остаток без полярных групп в боковой цепи алифатической или ароматической аминокислоты, предпочтительно являющийся более гидрофобным, чем аланин.
Sub1 представляет собой свободную N-концевую аминогруппу аминокислоты X1 или модификацию N-концевой аминогруппы (замещение в последовательности N-концевой аминогруппы аминокислоты X1 посредством Sub1) с общей формулой NR1R2. Sub1=NR1R2, где R1 и R2 являются независимыми друг от друга, выбранными предпочтительно из водорода или групп, состоящих из:
(i) неразветвленной цепи, разветвленной, циклической или гетероциклической алкильной группы, такой как метил, этил, н-пропил, изопропил, н-бутил, изобутил, циклогексил;
(ii) неразветвленной цепи, разветвленной, циклической или гетероциклической алканоильной группы, такой как ацетил или метаноил (формил), пропионил, н-бутирил, изобутирил, пентаноил, гексаноил или циклогексаноил;
(iii) репортерной группы, такой как флуоресцентный краситель (например, флуоросцеин, Alexa488) или биотин;
(iv) линкера, вместе с COR3 (смотрите ниже) связывающего соответствующие N- и C-концы для получения циклического пептида, на основе, например, гуанидина, олигомеров этиленгликоля, 2,4-диаминобутановой кислоты, 2,3-диаминопропионовой кислоты, 2,2'-диаминопимелиновой кислоты, десмозина или изодесмозина.
Sub2 представляет собой свободную C-концевую карбоксильную группу С-концевой аминокислоты или модификацию C-концевой карбоксильной группы, предпочтительно с общей формулой COR3 (R3 замещенная гидроксильная группа последней аминокислоты) Х6-COR3 или X7-COR3 или Х6Х7-COR3.
COR3 предпочтительно выбран из группы, состоящей из:
(i) карбоксила (R3 представляет собой свободный гидроксил), сложного эфира (R3 представляет собой алкокси), амида (R3 представляет собой амин) или имида;
(ii) линкера, вместе с Sub1 связывающего N- и C-концы с получением циклического пептида;
(iii) COR3 с R3 представляет собой дополнительный аминокислотный остаток, выбранный из группы, состоящей из Pro, Ile, Leu, Arg, Val, или R3, является пептидом предпочтительно из 2-3 аминокислот, содержащим, по меньшей мере, одну аминокислоту, выбранную из группы, состоящей из Pro, Ile, Leu, Arg, Val, замещенную членом группы, состоящей из карбоксила (R3 является свободным гидроксилом), сложного эфира (R3 является спиртом, таким как метанол, этанол, пропанол, изопропанол или бутанол), амидом (R3 является амином) или имидом (R3 является алкиламином или диалкиламином, таким как метиламин, этиламин, диметиламин или циклогексиламин).
(iv) COR3 с R3 представляет собой дополнительную разветвленную аминокислоту для образования димерной или олигомерной структуры, такую как лизин, гидроксилизин, орнитин, 2,4-диаминобутановая кислота, 2,3-диаминопропионовая кислота, 2,2'-диаминопимелиновая кислота, десмозин, изодесмозин или комбинации этих разветвленных аминокислот.
Таким образом, на С-конце производные пептидов можно, в частности, создавать в виде сложного эфира (R3 = алкокси), амида (R3 = амид), имида или пептида, удлиненного посредством дополнительных аминокислот, выбранных из группы, состоящей из Pro, Ile, Arg, Val, модифицированных вновь на C-конце в виде сложного эфира, амида или имида. Дополнительно производные пептидов можно создавать посредством модификаций на N-конце или C-конце пептида. Эти изменения могут, например, представлять собой добавление алкила, или алканоильной группы (каждый с прямой цепью, или являющиеся разветвленными, или циклическими, или гетероциклическими), или гуанидиногруппы, или добавление макромолекулы, или репортерной группы как посредством устойчивого соединения, так и соединения, которое можно расщепить в определенных условиях (такого как дисульфидные мостики или кислотонеустойчивые линкеры).
Все природные аминокислоты, неприродные аминокислоты или производные аминокислот (такие как иминокислоты), образующие пептиды или производные пептидов по изобретению, могут быть как в L-, так и в D-конфигурации. Однако, если не указано иначе, аминокислотные строительные блоки в последовательности находятся предпочтительно в L-конфигурации.
X6 и X7 являются необязательными дополнительными остатками. Таким образом, в случае если X6 и X7 отсутствуют, последний пролин (Р) в последовательности, указанной выше, имеет свободную C-концевую карбоксильную группу или соединен с Sub2.
Таким образом, в случае если присутствует по меньшей мере один из X6 и X7, пептид имеет, например, одну из следующих общих формул:
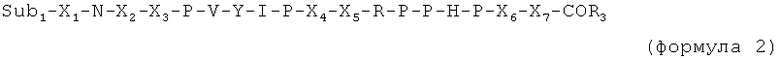
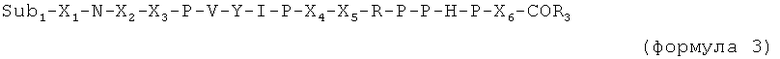
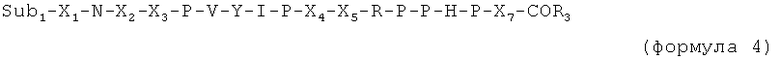
X6 выбран из пролина, или производного пролина, или фрагмента, имеющего суммарный положительный заряд или положительно заряженную в физиологических условиях боковую цепь. Предпочтительные остатки X6 выбраны из групп, включающих пролин, цис-4-гидроксипролин, транс-4-гидроксипролин, цис-3-гидроксипролин, транс-3-гидроксипролин, β-циклогексилаланин, 3,4-цис-метанопролин, 3,4-дегидропролин, гомопролин, псевдопролин, а также аргинин, предпочтительно D-аргинин, δ-гидроксилизин, гомоаргинин, 2,4-диаминомасляную кислоту, β-гомоаргинин, ε-N-метиллизин, алло-гидроксилизин, 2,3-диаминопропионовую кислоту, 2,2'-диаминопимелиновую кислоту, лизин, аргинин, орнитин, метиларгинин (предпочтительно альфа-N-метиларгинин), нитроаргинин (предпочтительно N(G)-нитроаргинин), нитрозоаргинин (предпочтительно N(G)-нитрозоаргинин), аргинал, гуанидинопропионовую кислоту, симметричный диметиларгинин, асимметричный диметиларгинин, 2,6-диаминогексановую кислоту, гистидин, 1-метилгистидин, 3-метилгистидин или 3-аминотирозин.
X7 выбран из пролина или производных пролина, полярного фрагмента (такого как серин) или гидрофобного фрагмента. Предпочтительные остатки X7 выбраны из групп, включающих пролин, цис-4-гидроксипролин, транс-4-гидроксипролин, цис-3-гидроксипролин, транс-3-гидроксипролин, β-циклогексилаланин, 3,4-цис-метанопролин, 3,4-дегидропролин, гомопролин или псевдопролин, серин, треонин, δ-гидроксилизин, цитруллин, гомосерин, или алло-треонин, а также фенилаланин, N-метиллейцин, лейцин, изолейцин, валин, метионин, трет-бутилглицин, циклогексилаланин, аланин, β-аланин, 1-аминоциклогексилкарбоновую кислоту, N-метилизолейцин, норлейцин, норвалин, N-метилвалин, или короткую пептидную последовательность, предпочтительно длиной от одного до 3 остатков, выбранных предпочтительно из пролина, изолейцина или любых фрагментов, упомянутых ранее, или разветвленного линкера, содержащего несколько пептидных единиц, таких как лизин, гидроксилизин, орнитин, 2,4-диаминобутановая кислота, 2,3-диаминопропионовая кислота, 2,2'-диаминопимелиновая кислота, десмозин, изодесмозин.
C-концевая аминокислота представляет собой, например, последний пролин (P) в формуле 1, X6 (в формуле 3) или X7 (в формуле 2 и 4).
Природные последовательности на основе немодифицированного апидецина 1a и 1b с SEQ ID № 1 (GNNRPVYIPQPRPPHPRI-OH) и SEQ ID № 2 (GNNRPVYIPQPRPPHPRL-OH), где OH замещает свободную карбоксильную группу на C-конце (Sub1 = NH2 и Sub2 = RI-OH или RL-OH, R3 = -OH), исключены из объема изобретения.
Пептиды или производные пептидов по изобретению обладают по меньшей мере одним из следующих преимуществ по сравнению с природными пептидами апидецина:
(i) увеличенным временем полужизни в сыворотке млекопитающих вследствие повышенной протеазной устойчивости, и
(ii) увеличенной антибактериальной активностью против одного или нескольких бактериальных штаммов, особенно патогенных для человека, или грибковых или других микробных инфекций, и
(iii) пептиды не являются токсичными для клеток человека, включая эритроциты.
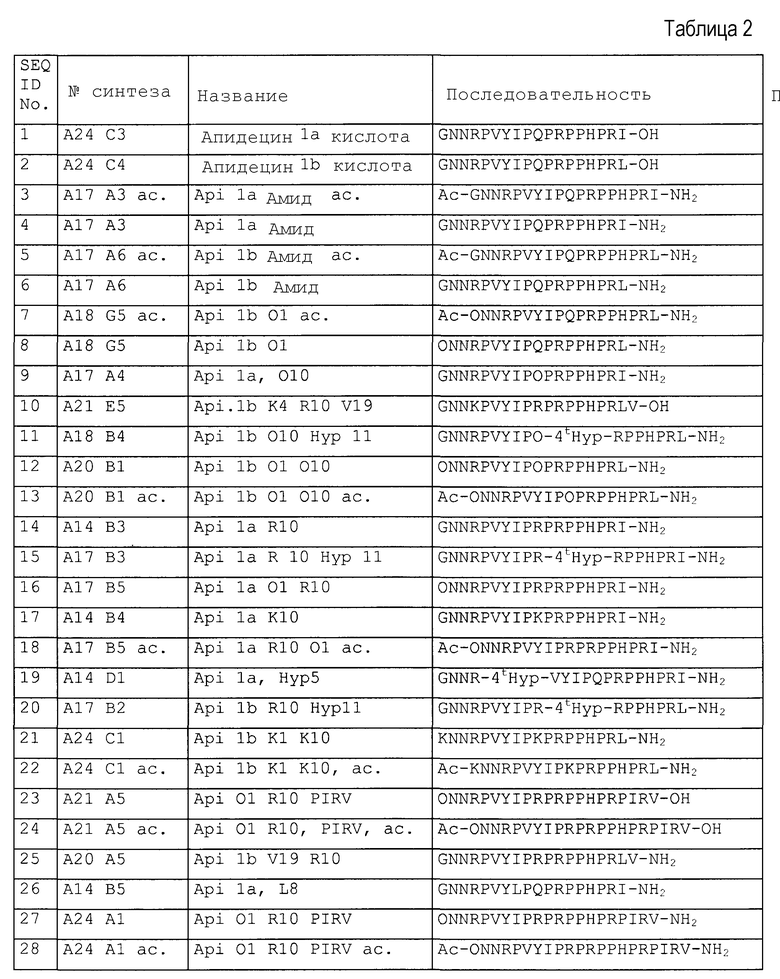
Примеры пептидов и производных пептидов по изобретению имеют последовательности согласно SEQ ID № 3-121, а также 122-159 (смотрите также таблицу 2 в примере 1). Менее предпочтительными являются пептиды и производные пептидов с SEQ ID № 92, 141 и 142 (смотрите также таблицу 2 в примере 1).
Пептиды и/или мультимерные пептидные конструкции по данному изобретению, которые модифицируют для повышения антибактериальной или противогрибковой активности, и для расширения спектра активности по отношению к другим бактериям или грибам, и для повышения устойчивости к протеазам и пептидазам, характеризуются высокой антибактериальной и/или противогрибковой эффективностью и хорошей метаболической стабильностью в сыворотке млекопитающих.
Соответствующие модификации в положениях 3 (X2), 4 (X3) и 10 (X4) повышают антибактериальную активность последовательности апидецина дикого типа по отношению к различным бактериям, как обсуждается ниже и показано в примерах.
Первые положения (Sub1, X1, X2 и X3), как предполагают, ответственны за надлежащий транспорт через мембрану внутрь клетки, тогда как положение 10 (X4) может также вносить вклад в ингибирование внутриклеточной мишени. Кроме того, остаток X2 может дополнительно придавать устойчивость N-концу последовательности пептида против деградации и, таким образом, увеличивать время полужизни в сыворотке.
Предпочтительные примеры по изобретению представляют собой последовательности с положительно заряженными остатками X4 (положение 10), такие как последовательности, выбранные из последовательностей согласно SEQ ID № 9-18, 20-25, 27-31, 34-40, 45-49, 55 и 56, 64-69, 81-83, 86-91, 97-100, 102-104, 112, 113, 117, 119, 122-128, 131, 134, 137, 138, 145-147 и 150-159. Менее предпочтительными являются последовательности с глутамином (Q) в X4 (положение 10).
Предпочтительные примеры по изобретению представляют собой последовательности с положительно заряженными остатками вместо X3 (положение 4), такие как последовательности, выбранные из последовательностей согласно SEQ ID № 10, 68, 95, 97, 100, 122-131, 134, 137 и 155.
Кроме того, свободную N-концевую аминогруппу и свободную C-концевую карбоксильную группу, которые, по-видимому, крайне важны для антибиотической активности пептидов, пептидных производных и их мультимеров, предпочтительно модифицируют, поскольку эти концы подвергаются петидазной и протеазной деградации в сыворотке, в большинстве жидкостей организма, тканях, органах или клетках. Увеличение протеазной устойчивости увеличивает время полужизни пептида в сыворотке. Дополнительно, модификация концов также позволяет пептиду образовывать связи с другими фрагментами, такими как другие аминокислотные последовательности (таким образом, возможно образование мультимерных пептидов или белков), или с другими биомолекулами, которые могут функционировать как носитель или метка. В конкретном варианте осуществления молекула носителя также функционирует в виде нацеливающей молекулы, которая способна локализовать бактериальную инфекцию и может связаться с бактерией для того, чтобы доставить антибиотическое соединение в место нахождения клетки (бактериальной) для воздействия или даже перенести его через бактериальную мембрану в бактериальную клетку. Подобные нацеливающие фрагменты могут представлять собой молекулы, которые, как известно, связываются с молекулами липополисахарида (LPS), которые образуют клеточную стенку грамотрицательных бактерий. Известными соединениями для подобного использования являются, например, якорные пептиды, такие как мотив AcmA Lactobacillus или антитела против липополисахаридов. Последние предпочтительны, так как они также обладают присущим им антибиотическим действием и, таким образом, могут быть использованы для усиления действия пептида по изобретению.
В виде N-концевой аминокислоты, которая представляет собой Sub1-X1, очень важно иметь фрагмент, который несет положительный заряд в физиологических условиях, т.е. в организме (человека). Физиологические условия, таким образом, обозначают pH, равное приблизительно 6-8, и температуру, равную приблизительно 30-40°C. Этот положительный заряд или в Sub1, или в X1 положении пептида или пептидного производного является, по всей вероятности, необходимым для осуществления антибактериальной функции.
Одним примером достижения стабильности N-конца против протеолитического расщепления является ацилирование (Sub1=ацил-NH-), такое как ацетилирование (Sub1=ацетил-NH-) α-аминогруппы положительно заряженной аминокислоты, такой как орнитин или лизин (Sub1-X1=ацил-Orn или ацил-Lys). Это ацилирование (предпочтительно ацетилирование) позволяет сохранить положительный заряд боковой цепи аминокислоты.
Другим примером достижения стабильности N-конца против протеолитического расщепления является гуанидинирование (предпочтительно Sub1=N(СН3)2-(C-N+(CH3)2)-NH-), которое вводит наряду с этим положительно заряженную группу в положение 1.
Более предпочтительными примерами по изобретению являются последовательности с положительно заряженными остатками, такими как X1 и X4 (положение 1 и 10), такие как последовательности, выбранные из последовательностей согласно SEQ ID №12 и 13, 16, 18, 21-24, 27-30, 35-40, 64, 65, 66, 81-83, 86-88, 112, 113, 117, 119, 124-128, 131, 134 и 137.
Дополнительными предпочтительными примерами по изобретению являются последовательности с пролином, производным пролина или положительно заряженным остатком вместо X6 (положение 17), такие как последовательности, выбранные из последовательностей согласно SEQ ID №10, 23, 24, 27, 28, 70, 109-113, 121, 127, 128, 131-134, 136, 138, 139, 145, 146 и 148-155.
Другими предпочтительными примерами по изобретению являются последовательности с пролином, производным пролина, полярным фрагментом или гидрофобным фрагментом вместо X7 (положение 18), такие как последовательности, выбранные из последовательностей согласно SEQ ID №10, 24, 27-37, 42, 44, 46, 47, 68-77, 83, 93, 94, 96, 107-110, 114, 115 и 126, 144.
Более предпочтительными примерами являются пептиды с положительно заряженной аминокислотой в положении 1 (X1) или 10 (X4) (такой как орнитин, аргинин или лизин) и модифицированным С-концом, в особенности пептиды согласно SEQ ID №7-8, 11-13, 20, 21, 22, 25, 38-40, 45, 65-67, 131, 134 и 137.
Наиболее предпочтительные пептиды содержат орнитин в положении 1 (остаток X1), аргинин в положении 10 (остаток X4), пролин или гидроксипролин в положении 11 (остаток X5), аргинин или его производное (такое как орнитин, гомоаргинин) в положении 17 (остаток X6 в Sub2) и ацетилирование или гуанидирование N-конца, такое как с SEQ ID №40, 88, 131, 134 и 137.
Из примеров можно видеть, что уже небольшая модификация С-конца на амид (Sub2=-NH2) значительно повышает ингибиторный эффект, направленный против Е. coli и S. typhi. Предпочтительные последовательности с амидом на С-конце представляют собой SEQ ID № 4, 6, 7, 8, 14, 20, 38, 39 и 134.
Предпочтительными также являются модификации, такие как метил, пропил, амид и пролин в положениях 17 (Х6), и/или 18 (X7), и/или на С-конце (Sub2), выполненные для снижения C-концевого разрушения. Результаты экспериментов позволили выяснить, что аминокислоты в положениях 17 и 18, по-видимому, также являются очень важными для антибиотического действия, так как замена одной аминокислоты в этих положениях на аланин приводит к полной потере эффективности относительно антибиотической активности апидецина дикого типа.
Наиболее предпочтительными примерами по изобретению являются пептиды, которые соответствуют всем преимуществам:
(i) увеличенное время полужизни в сыворотке млекопитающих вследствие повышенной протеазной устойчивости, и
(ii) повышенная противомикробная активность по отношению к одному или нескольким бактериальным штаммам, в частности патогенов человека, или грибам, или другим микробным инфекциям, и
(iii) пептиды не являются токсичными для клеток человека, включая эритроциты.
Действие противомикробных пептидов является чрезвычайно комплексным, так, они должны пройти сквозь мембрану бактериальной клетки и попасть в цитоплазму, чтобы специфически ингибировать внутриклеточную бактериальную мишень, не будучи при этом токсичными для клеток млекопитающих и клеток крови. Другим важным моментом для рассмотрения является стабильность пептидов или пептидных производных к деградации посредством петидаз или протеаз. Таким образом, идеальный пептид имеет высокую антибактериальную активность (низкие значение MIC), не токсичен для клеток, не обладает гемолитическим действием, и время его полужизни в крови составляет несколько часов. По сравнению с природной последовательностью апидецина самые подходящие производные пептидов, описанные в настоящем изобретении, имеют противомикробную активность, увеличенную более чем в десять раз. Этого частично достигали посредством амидирования C-конца и замещением Gln в положении 10 (X4) на основной остаток, наиболее предпочтительны аргинин или орнитин. Так как время полужизни этих пептидов в сыворотке относительно короткое, предпочтительным является модифицированный N-конец (посредством формилирования, ацетилирования или гуанидирования) для снижения деградации аминопептидазами или аминопротеазами. На N-конце предпочтительным является положительный заряд для того, чтобы обеспечить достаточную антибактериальную активность. Глицин в положении 1 (X1) природной последовательности апидецина предпочтительно замещают на основной остаток, такой как аргинин, лизин или орнитин, наиболее предпочтительно на орнитин, так как его C-концевую пептидную связь не расщепляют трипсин и родственные ферменты. По той же причине Arg-17 (X6) природной последовательности апидецина предпочтительно заменяют, чтобы уменьшить расщепление пептидной связи между Arg-17 (X6) и Leu/Ile-18 (X7), эндопротеазами. Существуют примеры замен Arg-17 (Х6) на орнитин или N-метилирования Leu-18 (X7), которые в обоих случаях повышали стабильность в сыворотке более чем в 24 раза, поскольку время полужизни изменилось с 15 мин до более чем 360 мин. Хотя ни один из пептидов не проявляет токсичного действия на клеточные линии COS-7 или эритроциты, замена Pro-11 (X5) на транс-4-Нур создает потенциал для дополнительного снижения возможных побочных эффектов, так как замены на полярные остатки уменьшают тенденцию пептидов или пептидных производных связываться с мембранами клеток млекопитающего, не снижая при этом антибактериальной активности.
Предпочтительно, пептиды или производные пептидов по изобретению не вызывают устойчивость или вызывают меньшую устойчивость, чем пептиды дикого типа.
Предпочтительно, ряд пептидов или пептидных производных по изобретению показывает расширенный спектр противомикробной активности, демонстрируя активность против бактерий, таких как Baccilus subtilis и Mycobacterium vaccae, которую не наблюдают у природных пептидов апидецина дикого типа. Общим свойством этих последовательностей с активностью против B. subtilis является положительно заряженный N-конец. Общим свойством этих последовательностей с активностью против M. vaccae является положительно заряженный N-конец, заряженный остаток (предпочтительно аргинин или орнитин) на X4 (положение 10) и заряженный остаток (предпочтительно орнитин) на X1 (положение 1). Примеры этих предпочтительных последовательностей выбраны из SEQ ID № 65, 83, 131 и 155.
В настоящем документе термин «пептид» обозначает последовательность аминокислот, соединенных посредством пептидной связи, где аминокислоты предпочтительно представляют собой одну из двадцати природных аминокислот, образующих пептиды, и где аминокислоты могут быть в L-конфигурации, или в D- конфигурации, или, для изолейцина и треонина, в D-алло-конфигурации (с инверсией только в одном из хиральных центров).
Термин производное пептида (или пептидомиметики) по изобретению включает не только пептиды, которые модифицируют, например на N- или С-конце посредством указанных выше групп Sub1 и Sub2. Он дополнительно включает пептиды, которые изменяют посредством замен и/или модификаций одного или нескольких аминокислотных остатков на химические группы, отличные от природных аминокислотных остатков, образующих протеины, такие как непротеиногенные α-аминокислоты, β-аминокислоты или пептиды с измененной главной цепью. Измененная главная цепь обозначает, что по меньшей мере одну пептидную связь заменили, например, на нерасщепляемую связь, такую как восстановленная амидная связь, алкилированная амидная связь или тиоамидная связь.
Включены также фрагменты, которые могут образовывать ковалентную связь как с COOH-группой предыдущего аминокислотного остатка, так и с NH2-группой последующего аминокислотного остатка, и которые, таким образом, не являются обязательно необходимыми для поддерживания структуры пептидного остова, такие как дипептидные изостеры на основе сахарной аминокислоты, азапептиды, 6-гомополимеры, y-пептиды, Y-лактамные аналоги, олиго(фениленэтилен)ы, винилогические сульфонопептиды, поли-N-замещенные глицины или олигокарбаматы. Эти модификации являются преимущественными в положениях, подверженных ферментативной деградации, в частности в трех N-концевых остатках (позиции 1-3 - X1-N-X2) и двух C-концевых остатках (после положения 16 - X6-X7). Таким образом, предпочтительно, чтобы по меньшей мере одна из связей между X1-N (например, Gly-Asn), N-X2 (например, Asn-Asn), X2-X3 (например, Asn-Arg), X6-X7 (например Arg-Leu или Arg-Ile) являлась нерасщепляемой связью.
Эту нерасщепляемую связь определяют как связь, которая не подвергается расщеплению протеазами и предпочтительно выбрана из группы, состоящей из восстановленной амидной связи, алкилированной амидной связи или тиоамидной связи. Восстановленная амидная связь представляет собой пептидную связь, в которой карбонильную группу (C=O) восстанавливают или до гидроксильной группы (HCOH), или до метиленовой группы (CH2). Алкилированная амидная связь представляет собой пептидную связь, в которой или азот (N-альфа), или углерод (C-альфа) замещают алкилом, предпочтительно с атомами C в количестве от 1 до 3, предпочтительным примером является N-метилирование.
Пептид или производное пептида по изобретению может являться линейным, то есть где первая и последняя аминокислоты последовательности имеют свободную NH2- и COOH-группу или модифицированы на Sub1 и Sub2 соответственно, или они могут являться циклическими, то есть когда первая и последняя аминокислоты связаны посредством пептидной связи или линкера.
Дополнительной целью изобретения являются способы получения указанных выше новых антибиотических соединений.
Пептиды или производные пептидов по изобретению можно получать синтетически или, в соответствующих случаях, рекомбинированием посредством стандартных способов. Конкретные варианты осуществления изобретения в отношении антибиотических пептидов, полученных на основе апидецина или пептидных производных, подробно раскрыты ниже, в экспериментальной части. Предпочтительно, пептиды или пептидные производные по изобретению получают стандартно посредством известных способов химического синтеза, таких как, например, описанные Merrifield [23].
Альтернативно, пептиды по изобретению можно получить способами с рекомбинантной ДНК, посредством клонирования и экспрессии внутри микроорганизма-хозяина или клетки, несущей фрагмент ДНК с последовательностью нуклеиновой кислоты, кодирующей один из пептидов, описанный выше. Нуклеиновую кислоту, кодирующую последовательность, можно получить синтетически [24] или можно получить из имеющейся последовательности нуклеиновой кислоты (например, последовательности, кодирующей апидецин дикого типа) посредством сайт-направленного мутагенеза. Полученные таким образом кодирующие последовательности можно амплифицировать на РНК (или ДНК) в полимеразной цепной реакции (ПЦР), посредством известных способов, используя соответствующие сконструированные праймеры. После очистки, например, электрофорезом в агарозном геле, продукт ПЦР встраивают в вектор и в клетку-хозяина, в конечном счете, трансформированную соответствующей рекомбинантной плазмидой. Различные клетки-хозяева хорошо известны в рекомбинантной технологии, это такие как E. coli, Bacillus, Lactobacillus, Streptomyces, клетки млекопитающих (например, клетки яичников китайского хомяка (CHO) или клетки COS-1), дрожжи (например, Saccharomyces, Schizophyllum), клетки насекомых или экспрессирующие системы на основе вирусов (например, бакуловирусные системы). Выбор других подходящих клеток-хозяев и способов трансформации, культур, амплификации, скрининга, получения и очистки продукта может выполнить специалист в данной области посредством обращения к известным способам [25]. Полученные посредством традиционных рекомбинантных способов пептиды по данному изобретению можно выделять или из клетки-хозяина посредством традиционных способов лизиса клеток, или из клеточной среды посредством традиционных способов, таких как жидкостная хроматография, предпочтительно аффинная хроматография. Противомикробные пептиды могут быть экспрессированы в виде одного пептида или олигомера нескольких пептидных последовательностей, соединенных N- или C-концами, или даже с N- или C-концевой меткой для обеспечения более легкой очистки рекомбинантного пептида или белковых конструкций. Традиционные способы молекулярной биологии и сайт-направленного мутагенеза можно использовать далее для модифицирования последовательности и предоставления желаемой неприродной пептидной последовательности. Все эти рекомбинантные способы известны специалистам в данной области и были описаны для многих противомикробных пептидов, включая апидецин [26], перинерин [27] и дефензин [28].
Также возможно включение неприродных аминокислот в пептиды посредством способов генной инженерии. Это было подробно описано Noren et al. и Ellman et al. [29, 30].
Далее, пептид можно выделить из культуры клеток-хозяев или системы трансляции in vitro. Это можно осуществить посредством стандартных способов очистки и выделения белков, которые установлены данной областью. Такие способы могут, например, включать иммуноадсорбцию или иммунохроматографию. Также возможно во время синтеза получение пептидов с меткой (такой как гистидиновая метка), которая позволяет быстрое связывание и очистку, после чего метку удаляют ферментативным способом для получения активного пептида.
Если сам пептид нельзя кодировать или экспрессировать, но существует очень похожий пептид, который можно кодировать или экспрессировать, можно применять способ получения пептида, на который этот пептид является похожим, с последующей одной или несколькими стадиями, на которых указанный пептид модифицируют химическими или ферментативными способами для получения конечного пептида или пептидомиметика. Более подробные обзоры способов, которые можно применять при получении пептидов, описаны в литературе [31, 32, 33, 34, 35].
Пептиды и производные пептидов по изобретению можно использовать отдельно, или в комбинации, или в форме мультимеров, или в форме разветвленных мультимеров. Подходящие комбинации пептидов по изобретению содержат конкатемеры пептидов по изобретению, последовательно связанные друг с другом с помощью спейсеров, например, в форме пептидного димера, пептидного тримера и так далее, где отдельные пептиды последовательно выстроены в линию. Эти мультимеры могут состоять из пептидов и пептидных производных с тождественными последовательностями или из нескольких последовательностей по формуле 1.
Один пептид или производные пептидов можно соединить с биологически совместимым белком, таким как сывороточный альбумин человека, гуманизированное антитело, липосома, мицелла, синтетический полимер, наночастица и фаг. Альтернативно, мультимеры отдельно связанных пептидов или пептидных производных по изобретению получают в форме дендримеров или кластеров, где три или более пептидов присоединены к одному общему центру.
В одном из вариантов осуществления мультиплетные пептиды или производные пептидов согласно формуле 1, приведенной выше, группируют в мультимерные конструкции или композиции. Например, необязательные аминокислоты (например, -Gly-Ser-) или другую аминокислоту, или химические соединения спейсеры включают в N- или C-концы пептидов с целью соединения вместе двух или более пептидов или присоединения к носителю. Эти композиции могут принимать форму одного или нескольких пептидов, описанных выше, выраженных в виде синтетического пептида, соединенного с белком-носителем. Альтернативно, композиция содержит мультиплетные пептиды, каждый из которых выражен в виде мультиплетного антигенного пептида, необязательно соединенного с белком-носителем. Альтернативно, выбранные пептиды соединяют последовательно и экспрессируют в виде белка или пептида, продуцируемого рекомбинантным способом. Как один из вариантов осуществления изобретения, мультиплетные пептиды соединяют последовательно, посредством спейсерной аминокислоты между ними или без нее, для составления большего рекомбинантного белка. Альтернативно, рекомбинантный белок может являться слитым в рамке считывания с белком-носителем.
В другом варианте осуществления мультимерная конструкция содержит, по меньшей мере, два из обозначенных выше пептидов (которые могут являться тождественными или различными пептидами формулы 1), где один пептид присоединен к любой аминокислоте другого пептида(ов). В композиции любое количество дополнительных пептидов может быть присоединено к любой аминокислоте других пептидов. В другом варианте осуществления мультимерная композиция содержит по меньшей мере два пептида, второй или дополнительные пептиды присоединены к разветвленной конструкции других пептидов в композиции. Альтернативно, каждый дополнительный пептид ковалентно связан с Sub1 или Sub2 другого пептида в композиции.
В другом варианте осуществления мультимерная конструкция или композиция содержит по меньшей мере два пептида, по меньшей мере один или несколько пептидов присоединены к носителю. В другом варианте осуществления один или несколько указанных пептидов представляют собой синтетический пептид, слитый с белком-носителем. Опять же, альтернативно, мультиплетные пептиды, описанные выше, с фланкирующей последовательностью или без нее, могут быть последовательно соединены в полипептид. Пептиды или этот полипептид, каждый, присоединяют к одному и тому же носителю, или различные пептиды можно присоединять отдельно в виде пептидов к одному или различным иммунологически неактивным белкам-носителям.
Подходящие носители могут повышать стабильность или улучшать доставку, увеличивать продукцию или изменять спектр активности пептида. Примеры носителей представляют собой альбумин человека, полиэтиленгликоль, другие биополимеры или другие природные или неприродные полимеры. В одном из вариантов осуществления фрагмент представляет собой желательный белок или другую молекулу, которая может повышать стабильность пептида. Специалист в данной области может легко выбрать подходящий для конъюгирования фрагмент.
Предпочтительные примеры мультимерных композиций по данному изобретению имеют структуру (Ас-OrnAsnAsnArgProValTyrIleProArgProArgProProHisProArgLeu)2-Dab и (OrnAsnAsnArgProValTyrIleProArgProArgProProHisProArgLeu)2-Dab с SEQ ID №83 и 119.
Дополнительные предпочтительные димеры выбирают из:
В еще одном варианте осуществления пептиды находятся в форме мультиплетных антигенных пептидов («MAP»), которые можно сконструировать, например, согласно «МАР-системе», как описано Tam et al. [36]. В этой системе используют как основную матрицу лизиновые остатки, на которых синтезируют мультиплетные копии одного пептида по изобретению, как описано [смотри, например, 37]. Каждый MAP содержит мультиплетные копии одного или нескольких пептидов по данному изобретению. В одном из вариантов осуществления MAP содержит по меньшей мере три, а предпочтительно, четыре или более пептидов. Специалист в данной области может легко получить любое количество мультимерных конструкций из пептидов формулы, приведенной выше, прибегнув только к стандартным профессиональным способам и знаниям в свете данного описания. Все подобные мультимерные композиции и конструкции подлежат включению в настоящее изобретение.
Кроме того, другие комбинации в форме мультимеров создают посредством шариков, на поверхности которых экспонируют пептиды или пептидомиметики по изобретению. Шарик может затем функционировать как носитель для пептида или пептидомиметика и может функционировать подобно метке для обнаружения. Мультимеры можно, например, получить посредством биотинилирования N-конца цепей пептида или пептидомиметика и последующим комплексообразованием со стрептавидином. Так как стрептавидин способен связать четыре молекулы биотина или соединений с высоким сродством, то таким способом можно создать очень стабильные тетрамерные пептидные комплексы. Мультимеры могут состоять из одинаковых или различных пептидов или пептидомиметиков по изобретению. Предпочтительно, однако, чтобы мультимеры по изобретению состояли из двух или более пептидов или пептидомиметиков, в которых каждый компонент вносит свой вклад в общую биоцидную активность (направленное воздействие, противомикробная активность, захват).
Другой целью изобретения является применение пептидов или пептидных производных по изобретению в медицине или фармакологии, например для терапии антибиотиками или в противомикробной композиции (в частности, в бактерицидной).
Дополнительной целью изобретения являются фармацевтические композиции, содержащие один или несколько пептидов или пептидных производных по изобретению или мультимерных конструкций, в присутствии или в отсутствие других фармацевтически активных соединений.
Также частью изобретения является применение пептида по изобретению в качестве фармацевтического средства и/или для получения лекарственного средства, которое можно использовать в качестве антибиотика.
Пептиды можно использовать в фармацевтических композициях отдельно. Альтернативно, с целью увеличения фармакокинетических параметров или биодоступности без образования иммунного ответа, один или несколько пептидов объединяют или соединяют с другими фрагментами, как описано выше. Любое количество отдельных пептидов или мультимерных конструкций можно соединить вместе для формирования отдельной композиции.
Фармацевтическая композиция по изобретению содержит терапевтически эффективное количество одного или нескольких пептидов или пептидных производных по настоящему изобретению. После получения фармацевтические композиции по изобретению можно вводить непосредственно субъекту по способу лечения микробной инфекции (в частности, бактериальной), включающему введение нуждающемуся в этом субъекту терапевтически эффективного количества композиции по изобретению.
Композиции по изобретению создают для лечения инфекций в соответствии с выбранными бактериями или грибами, которые инфицируют млекопитающего, например человека, где по меньшей мере один или, альтернативно, несколько пептидов или мультимерных конструкций по настоящему изобретению можно формулировать в противомикробные композиции (в частности, антибактериальные или противогрибковые) с фармацевтически приемлемым носителем и другими дополнительными компонентами. Для применения в таких композициях выбранный пептид можно получать предпочтительно синтетически, а также рекомбинантным способом, как раскрыто выше.
Прямая доставка композиций, как правило, будет осуществляться посредством местного применения, или другими способами введения, или перорально, парентерально, подкожно, подъязычно, внутрь пораженных тканей, интраперитонеально, внутривенно или внутримышечно, внутрилегочно, или доставляться в интерстициальное пространство тканей.
Фармацевтическая композиция может также включать подходящий фармацевтически приемлемый носитель или разбавитель и может быть в форме капсулы, таблетки, пастилки, драже, пилюли, капель, суппозитория, порошка, спрея, вакцины, мази, пасты, крема, средства для ингаляции, пластыря, аэрозоля и тому подобного. В качестве фармацевтически приемлемого носителя можно использовать любой растворитель, разбавитель или другие жидкие носители, дисперсионные или суспендирующие средства, поверхностно-активное средство, изотоническое средство, загуститель или эмульгатор, консервант, средство для капсулирования, твердое связующее средство или лубрикант, которые являются наиболее подходящими для конкретной лекарственной формы и которые являются совместимыми с пептидом, пептидомиметиками (пептидным производным), конъюгатом пептида или конъюгатом пептидомиметиков.
Фармацевтическая композиция, таким образом, содержит предпочтительно фармацевтически приемлемый носитель. Термин «фармацевтически приемлемый носитель» также включает носитель для введения терапевтического средства, такого как антитела или полипептид, гены и другие терапевтические средства. Термин относится к любому фармацевтическому носителю, который, как таковой, не вызывает продукцию антител, вредных для индивидуума, получающего композицию, и который можно вводить без побочной токсичности. Подходящие фармацевтически приемлемые носители могут быть большими, медленно метаболизирующимися макромолекулами, такими как белки, полисахариды, полимолочные кислоты, полигликолевые кислоты, полимерные аминокислоты, сополимеры аминокислот и неактивные вирусные частицы. Такие носители хорошо известны специалистам в данной области.
Соли пептидов или функциональные эквиваленты получают посредством известных способов, которые, как правило, включают смешивание пептида, или пептидомиметика, или конъюгата пептида, или конъюгата пептидомиметиков либо с фармацевтически приемлемой кислотой для получения кислотно-аддитивной соли, либо с фармацевтически приемлемым основанием для получения основно-аддитивной соли. Что является фармацевтически приемлемым, кислота или основание, может легко решить специалист в данной области после принятия решения о конкретном предполагаемом использовании соединения. Например, не все кислоты и основания, которые являются допустимыми для применений ex vivo, можно использовать для терапевтических композиций. В зависимости от предполагаемого применения, фармацевтически приемлемые кислоты включают органические и неорганические кислоты, такие как муравьиная кислота, уксусная кислота, пропионовая кислота, молочная кислота, гликолевая кислота, щавелевая кислота, пировиноградная кислота, янтарная кислота, малеиновая кислота, малоновая кислота, коричная кислота, серная кислота, соляная кислота, бромистоводородная кислота, азотная кислота, перхлорная кислота, фосфорная кислота и тиоциановая кислота, которые образуют аммонийные соли со свободными аминогруппами пептидов и функциональных эквивалентов. Фармацевтически приемлемые основания, которые образуют карбоксилатные соли со свободной карбоксильной группой пептидов и функциональных эквивалентов, включают этиламин, метиламин, диметиламин, триэтиламин, изопропиламин, диизопропиламин и другие моно-, ди- и триалкиламины, а также ариламины. Кроме того, [они] также включают фармацевтически приемлемые сольваты.
В данном документе можно использовать фармацевтически приемлемые соли, например соли неорганической кислоты, такие как гидрохлориды, гидробромиды, фосфаты, сульфаты, и тому подобное; и соли органических кислот, такие как ацетаты, пропионаты, малонаты, бензоаты, и тому подобное. Всестороннее обсуждение фармацевтически приемлемых эксципиентов приведено в Remington's Pharmaceutical Sciences (Mack Pub. Co., N.J., 1991).
Фармацевтически приемлемые носители в терапевтических композициях могут содержать жидкости, такие как вода, физиологический раствор, глицерин и этанол. Дополнительно, в таких носителях могут быть представлены вспомогательные средства, такие как увлажнители или эмульгаторы, средства, поддерживающие pH и тому подобное. Как правило, терапевтические композиции получают в виде композиций для инъекций, или в виде жидких растворов, или суспензий; могут быть также получены твердые формы, пригодные для растворения или суспендирования в жидком носителе перед инъекцией. В определение термина "фармацевтически приемлемый носитель" включают липосомы.
Для терапевтического воздействия пептид, производное пептида, конъюгат пептида или конъюгат пептидного производного можно получать, как описано выше, и их может применять субъект, нуждающийся в этом. Пептид, производное пептида, конъюгат пептида или конъюгат пептидного производного можно вводить субъекту посредством любого подходящего способа, предпочтительно в форме фармацевтической композиции, приспособленной для такого способа введения, и в дозировке, которая является эффективной для намеченного лечения.
Фармацевтические композиции по настоящему изобретению могут содержать другие активные средства, такие как традиционные антибиотики (такие как, например, ванкомицин, стрептомицин, тетрациклин, пенициллин) или другие противомикробные соединения, такие как противогрибковые, например итраконазол или миконазол. Также могут быть добавлены соединения (например, салициловая кислота), облегчающие другие симптомы инфекции, такие как лихорадка или кожная сыпь.
Также в биологической борьбе, вслед за терапевтическим применением при лечении инфекций, возможно использование пептидов или пептидных производных по изобретению в дезинфицирующих или очищающих средствах (например, бактерицидная композиция), которые можно использовать для дезинфекции или очистки поверхностей и/или оборудования. Другой областью применения является упаковывание, где пептиды можно связать с материалом для упаковки или включить в материал для упаковки, или включить в качестве консерванта для другого материала, который легко подвергается разложению под действием микроорганизмов. Пептиды или производные пептидов по изобретению являются, в частности, пригодными для упаковывания пищевого продукта, так как они не оказывают токсического действия при контакте или приеме внутрь.
Другая часть изобретения относится к способу лечения микробной инфекции у млекопитающих (в частности, бактериальной или грибковой), где способ включает введение млекопитающему с указанной инфекцией эффективного против микробной инфекции количества фармацевтической композиции, описываемой в настоящем документе.
В настоящем документе термин «терапевтически эффективное количество» относится к количеству лекарственного средства, которое представляет собой пептид, пептидомиметик, конъюгат пептида или конъюгат пептидомиметика по настоящему изобретению, которое используют для снижения или предотвращения роста и образования колоний бактерий, или для демонстрации наглядного терапевтического или профилактического эффекта. Эффект можно определять посредством, например, биоптата с микроорганизмами и исследования бактериальной активности или посредством любого другого подходящего способа для оценки развития или тяжести бактериальной инфекции. Точное эффективное количество для субъекта будет определяться на основании размеров субъекта и состояния его здоровья, природы и степени тяжести состояния и лекарственных средств или комбинации лекарственных средств, выбранных для введения. В частности, композиции по настоящему изобретению можно использовать для снижения или предотвращения бактериальной инфекции и/или для сопутствующего воздействия на биологические или физические симптомы болезни, такие как уменьшение лихорадки. Способы, которые позволяют врачу назначить начальные дозировки, известны в данной области. Необходимые для введения дозы должны быть безопасными и действенными.
Количество белка, пептида или последовательности нуклеиновой кислоты по изобретению, присутствующие в каждой дозе, эффективной против бактерий, выбирают исходя из оценки патогена, вызывающего инфекцию, тяжести инфекции, возраста пациента, веса, пола, общего физического состояния и тому подобного. Количество активного компонента, требуемого для обеспечения эффективного антибактериального или противогрибкового действия, которое не несет значительных отрицательных побочных эффектов, меняется в зависимости от используемой фармацевтической композиции и наличия других необязательных компонентов, например антибиотиков, противогрибковых средств и тому подобного. Для целей настоящего изобретения эффективные дозы, которые вводят индивидууму, могут составлять приблизительно от 0,01 мкг/кг до 50 мг/кг, предпочтительно от 0,5 мкг/кг до приблизительно 10 мг/кг пептида, пептидомиметика, конъюгата пептида или конъюгата пептидомиметика.
Необязательно начальные дозы пептидов, пептидомиметиков, мультимеров или пептидных конъюгатов, конъюгатов пептидомиметиков по данному изобретению сопровождают повторным введением. Частота введения доз зависит от факторов, определенных выше, и варьирует предпочтительно от 1 до 6 доз в сутки на протяжении от приблизительно 3 суток до максимума, равного приблизительно 1 неделе.
Кроме того, в другом альтернативном варианте осуществления пептид или пептидомиметик, или конъюгат пептида или конъюгат пептидомиметика, или композиции по изобретению вводят из матрицы с контролируемым или замедленным высвобождением, которая введена в организм субъекта.
В одном из вариантов осуществления соединение по изобретению вводят в трансмукозальной лекарственной форме. Этот путь введения является неинвазивным и безвредным для пациента; в то же время он, возможно, приводит к улучшенной биодоступности соединения по сравнению с пероральным введением, особенно если соединение не устойчиво в жидкостях пищеварительной системы или если соединение является слишком большим для того, чтобы эффективно всасываться из пищеварительного тракта. Трансмукозальное введение возможно, например, посредством назальных, буккальных, сублингвальных, гингивальных или вагинальных лекарственных форм. Эти лекарственные формы можно получать известными способами; их можно формулировать для предоставления в виде капель в нос или спреев, вставок, пленок, пластырей, гелей, мазей или таблеток. Предпочтительно, эксципиенты, используемые для трансмукозальной лекарственной формы, включают одно или несколько веществ, обеспечивающих мукоадгезивное свойство, таким образом, увеличивая время контакта лекарственной формы с местом всасывания и в связи с этим теоретически увеличивая степень всасывания.
В дополнительном варианте осуществления соединения вводят через легкие, используя дозирующий ингалятор, распылитель, распыляемый аэрозоль или ингалятор сухого порошка. Подходящие составы можно получить посредством известных способов и технических приемов. В некоторых случаях можно также выполнить трансдермальное, ректальное или внутриглазное введение.
Для более эффективной доставки соединения по изобретению можно выгодно использовать усовершенствованные способы доставки лекарственного средства или способы нацеливания. Например, если выбирают не парентеральный способ введения, то подходящая лекарственная форма может содержать средство, улучшающее биодоступность, которое может представлять собой любое вещество или смесь веществ, которые повышают доступность соединения. Этого достигают, например, защитой соединения от деградации, такой защитой, как посредством ферментативного ингибитора или антиоксиданта. Более предпочтительно, усиливающее средство увеличивает биодоступность соединения посредством повышения проницаемости абсорбционного барьера, который, как правило, представляет собой слизистую оболочку. Усилители проницаемости могут действовать посредством разнообразных механизмов; некоторые увеличивают текучесть мембран слизистых оболочек, тогда как другие открывают или расширяют щелевидные соединения между клетками слизистой оболочки. Еще одни также снижают вязкость слизи, покрывающей слой клеток слизистой оболочки. В числе предпочтительных усилителей биодоступности представлены амфифильные вещества, такие как производные холевой кислоты, фосфолипиды, этанол, жирные кислоты, олеиновая кислота, производные жирной кислоты, ЭДТА, карбомеры, поликарбофил и хитозан.
Показания, при которых можно использовать пептиды, производные пептидов, конъюгаты или мультимеры по изобретению, представляют собой бактериальные инфекции, вызванные и грамположительными, и грамотрицательными бактериями, такими как Escherichia coli, Enterobacter cloacae, Erwinia amylovora, Klebsiella pneumoniae, Morganella morganii, Salmonella typhimurium, Salmonella typhi, Shigella dysenteriae, Yersinia enterocolitica, Acinetobacter calcoaceticus, Agrobacterium tumefaciens, Francisella tularensis, Legionella pneumophila, Pseudomonas syringae, Rhizobium meliloti, Haemophilus influenzae.
Другой целью изобретения является применение пептида или пептидного производного, или мультимера по изобретению в биотехнологическом, или фармацевтическом, или скрининговом исследовании, в частности, для идентификации соединения, которое имеет потенциальный бактерицидный или противогрибковый эффект.
В связи с этим изобретение относится к способу идентификации соединения, которое имеет возможный бактерицидный или противогрибковый эффект, включающему:
(i) проведение конкурентного анализа:
(a) с микроорганизмом, чувствительным к действию пептида или пептидного производного, или мультимера по изобретению,
(b) с пептидом или пептидным производным, или мультимером по изобретению,
(c) по меньшей мере с одним тестируемым соединением;
посредством воздействия (b) и (c) на (a); и
(ii) выбор тестируемого соединения, которое конкурентно замещает связанный с микроорганизмом пептид или производное пептида, или мультимер.
Этот способ скрининга идентифицирует тестируемые соединения, которые конкурируют с пептидами, пептидными производными или мультимерными композициями по данному изобретению за связывание неизвестного рецептора на патогене. Таким образом, низкомолекулярные соединения, специфически связывающиеся с тем же участком, который является мишенью и для пептида, можно эффективно идентифицировать в высокопроизводительном скрининге. Таким образом, тестируемые соединения имеют, по всей вероятности, тот же механизм действия, что и оригинальная пептидная последовательность и, таким образом, будут также активны против микроорганизмов, устойчивых ко многим препаратам, которых уничтожают посредством апидецина или одного из его аналогов, описанных в настоящем изобретении.
Этот способ скрининга осуществляют посредством известных способов, используя, однако, по меньшей мере один пептид или производное пептида, или мультимер по изобретению. В одном из вариантов осуществления пептид или производное пептида, или мультимер метят флуоресцентной, радиоактивной или другой меткой и связывание меченого пептида или пептидного производного, или мультимера с микроорганизмом определяют и сравнивают в присутствии или в отсутствие средства (средств), подлежащего(их) тестированию.
Впоследствии идентифицируют и скринируют тестируемые соединения, которые конкурируют с пептидами или мультимерными конструкциями по данному изобретению за связывание с рецептором, предпочтительно для антибактериального или противогрибкового применения.
В одном из вариантов осуществления в конкурентном анализе используют способ бимолекулярного флуоресцентного дополнения (BIFC). Этот способ позволяет непосредственное наблюдение внутриклеточных белковых взаимодействий, примером которых является взаимодействие SH3 домена тирозинкиназы c-Abl как с природными, так и сконструированными мишенями в E. coli [38]. Анализ является достаточно чувствительным, что позволяет выявление взаимодействий между белками, которые слабо экспрессируются в бактериях. Он основывается на взаимодействии двух фрагментов функционального желтого флуоресцентного белка (YFP), после того как SH3 домен связывается со своим партнером. Как только эти два белка свяжутся друг с другом, два фрагмента YFP образуют комплекс, очень похожий на строение природного белка. Это можно контролировать посредством полученной флуоресценции комплекса YFP, так как отдельные фрагменты не обладают какой-либо флуоресцентной активностью. Похожую конструкцию можно создать для скрининга соединений, конкурирующих с пептидами и пептидными производными, описанными в этом изобретении. Специалист в данной области может легко приспособить высокопроизводительный скрининг под 386-луночные планшеты для микротитрования.
В другом варианте осуществления пептиды используют в подходящем способе конкурентного анализа с тестируемыми соединениями, чтобы оценить способность тестируемого соединения к конкурентному вытеснению пептида из соединения с его на данный момент неизвестным рецептором на патогене. Если необходимо, и в зависимости от выбранного анализа, микроорганизм (например, бактерия, вирус или грибы), про который известно, что с ним связывается выбранный(ые) пептид(ы), например штаммы E. coli или K. pneumoniae, можно прямо или опосредованно иммобилизовать на подходящей поверхности, например, в формате ELISA. Такие поверхности для иммобилизации хорошо известны. Например, можно использовать инертные гранулы. Дополнительно, лиганд можно связывать в 96-луночном планшете. Впоследствии выбранные количества тестируемых соединений и пептидов по настоящему изобретению подвергают действию иммобилизованного микроорганизма и выбирают те тестируемые соединения, которые могут конкурировать с пептидами за связывание с иммобилизованным микроорганизмом. Определяют те тестируемые соединения, которые конкурируют с пептидами за связывание с рецептором бактерии или гриба, которые в дальнейшем можно отбирать для антибактериального или противогрибкового действия в способах, описанных в примерах ниже.
В еще одном дополнительном аспекте изобретение относится к выделенной молекуле нуклеиновой кислоты, содержащей нуклеотидную последовательность, кодирующую пептид или мультимер по изобретению. Нуклеиновая кислота кодирует антибактериальный или противогрибковый пептид, или мультимерную композицию по изобретению в функциональном соединении с регуляторной последовательностью, управляющей его экспрессией в клетке-хозяине.
В еще одном аспекте изобретение относится к клетке-хозяину, трансфицированной или трансформированной вышеописанной молекулой нуклеиновой кислоты.
Изобретение проиллюстрировано следующими примерами, но не ограничено ими.
Пример 1: Твердофазный синтез пептидов
Все пептиды и производные пептидов синтезировали посредством стандартного твердофазного синтеза пептидов, используя стратегию Fmoc/tBu [39]. Производные аминокислоты получали от MultiSynTech GmbH (Witten, Germany). Пептиды и пептидные производные со свободным C-концом (COOH-группа) синтезировали на смоле Ванга на основе полистирола (допустимая нагрузка 1,33 ммоль/г) от Merck Biosciences (Schwalbach, Germany). Пептиды и производные пептидов с C-концевым амидом (-CONH2-группа) синтезировали на смоле с 4-метилбензгидриламином (MBHA) на основе полистирола (допустимая нагрузка 0,64 ммоль/г) от Merck Biosciences. Пептиды и производные пептидов синтезировали на приборе для синтеза Syro2000 multiple peptide (MultiSynTech GmbH), используя 4 эквивалента аминокислотного производного, активированного 2-(1-H-бензотриазол-1-ил)тетраметилурониумгексафторфосфатом (HBTU; MultiSynTech GmbH) и N,N'-диизопропилэтиламином (DIPEA; (Fluka, Buchs, Switzerland) в диметилформамиде (DMF; Biosolve V.B., Valkenswaard, The Netherlands). Защитные группы боковой цепи представляли собой трифенилметил (тритил) для Asn, His и Gln, простой трет-бутиловый эфир для Tyr, Ser и Thr, сложный трет-бутиловый эфир для Asp и Glu, N-омега-2,2,4,6,8-пентаметилдигидробензофуран-5-сульфонил для Arg и βHar и трет-бутилоксикарбонил для Lys и Orn. Временную защитную группу Fmoc отщепляли 40% пиперидином в DMF (об./об.) в течение 3 мин и процедуру повторяли снова в свежеприготовленном 40% пиперидине в DMF (об./об.) в течение 10 мин.
Проводили ацетилирование N-конца пептидов или пептидных производных десятью эквивалентами уксусной кислоты, активированной HBTU и DIPEA в DMF, как описано выше для аминокислотных производных Fmoc. Гуанидирование N-концов пептидов или пептидных производных выполняли десятью эквивалентами HBTU и DIPEA в DMF, как описано Gausepohl et al. [40].
После завершения синтеза пептида или пептидных производных смолы тщательно промывали DMF и DCM и высушивали под вакуумом. Пептиды, связанные со смолой, отщепляли от твердого носителя и одновременно снимали защиту с боковых цепей смесью 5% воды, 4% м-крезола, 5% тиоанизола и 2% (по объему) этандитиола в трифторуксусной кислоте (TFA) при комнатной температуре в течение 4 ч. Пептиды или производные пептидов осаждали холодным простым диэтиловым эфиром и центрифугировали при 3000 g. Осадок дважды промывали холодным простым эфиром, высушивали и растворяли в 0,1% водном TFA (УФ-спектроскопической чистоты, Fluka). Образцы хранили при -20°C.
Пептиды и производные пептидов, метилированные по С-концу, то есть содержащие метиловый (CO-OMe) или пропиловый (CO-OPr) сложный эфир, синтезировали на смоле AM с 4-гидроксиметилбензойной кислотой (HMBA-AM смола, допустимая нагрузка 1,1 ммоль/г, Novabiochem, Merck-Biosciences, Darmstadt, Germany). Первую аминокислоту вручную связывали со смолой в виде симметричного ангидрида, используя 10 экв. аминокислотного производного Fmoc, 5 экв. диизопропилкарбодиимида (DIC) и 0,1 экв. N,N-диметил-4-аминопиридина в DCM. Допустимую нагрузку определяли в анализе с пиперидином и фульвеном посредством расщепления группы Fmoc 50% пиперидином в DMF в течение 1 ч исходя из оптической плотности, регистрируемой при 301 нм [41]. Типичные допустимые емкости составляли приблизительно 0,8 ммоль/г. Автоматический синтез проводили, как описано выше. После завершения пептидного синтеза смолу HMBA-AM с пептидом тщательно промывали DMF и DCM и высушивали под вакуумом. Защитные группы пептидов, связанных со смолой, отщепляли в смеси 5% воды, 4% м-крезола, 5% тиоанизола и 2% (по объему) этандитиола в трифторуксусной кислоте (TFA) при комнатной температуре в течение 4 ч. Смолу промывали TFA и DMF. Смола набухала в DMF, и пептид или производное пептида отщепляли от смолы DIPEA/MeOH/DMF (1:5:5 по объему; 50 мл раствора на грамм смолы) для получения C-концевого сложного метилового эфира. Для получения C-концевого сложного пропилового эфира пептид или производное пептида синтезировали на AM-смоле с 4-сулфамилбутирилом (допустимая нагрузка 1,1 ммоль/г, Novabiochem, Merck-Biosciences, Darmstadt, Germany) и отщепляли 50 экв. пропиламина в DMF после активации триметилсилилдиазометаном в THF. Растворители удаляли в вакууме, пептиды (или производные пептидов) осаждали холодным простым диэтиловым эфиром и центрифугировали при 3000 g. Осадок дважды промывали холодным простым эфиром, высушивали и растворяли в 0,1% водном TFA (УФ-спектроскопической чистоты, Fluka). Образцы хранили при -20°C. Для получения димерных пептидных производных Fmoc2-Dab соединяли со смолой. После отщепления защитной группы Fmoc получали два свободных N-конца, и производные пептидов синтезировали, как описано выше. Пептидные производные PEG3000 синтезировали посредством активации PEG3000-ОН с HBTU и DIPEA в DMF и присоединения их к N-концу пептида или пептидных производных, как описано выше для Fmoc аминокислотных производных.
Неочищенные пептиды и производные пептидов очищали на ВЭЖХ Äkta System (Amersham Bioscience GmbH, Freiburg, Germany), используя колонку Jupiter C18 (20 мм × 250 мм, Phenomenex Inc., Torrance, USA). Элюирование осуществляли в линейном градиенте ацетонитрила, которое, как правило, начиналось с 5% водного ацетонитрила с возрастанием концентрации ацетонитрила на 1% в минуту в присутствии 0,1% TFA в качестве ион-парного реагента. Скорость потока составляла 10 мл/мин, и пептиды определяли посредством измерения поглощения при 220 нм. Чистоту пептидов определяли посредством аналитической ОФ-ВЭЖХ, используя колонку Jupiter C18 (4,6 мм × 150 мм, Phenomenex Inc., Torrance, USA) и ассоциированную с матрицей лазерную десорбционную ионизацию с время-пролетной масс-спектрометрией (MALDI-TOF-MS; протеомный анализатор 4700, Applied Biosystems GmbH, Darmstadt, Germany).
Синтезировали следующие пептиды, представленные в таблице 2.
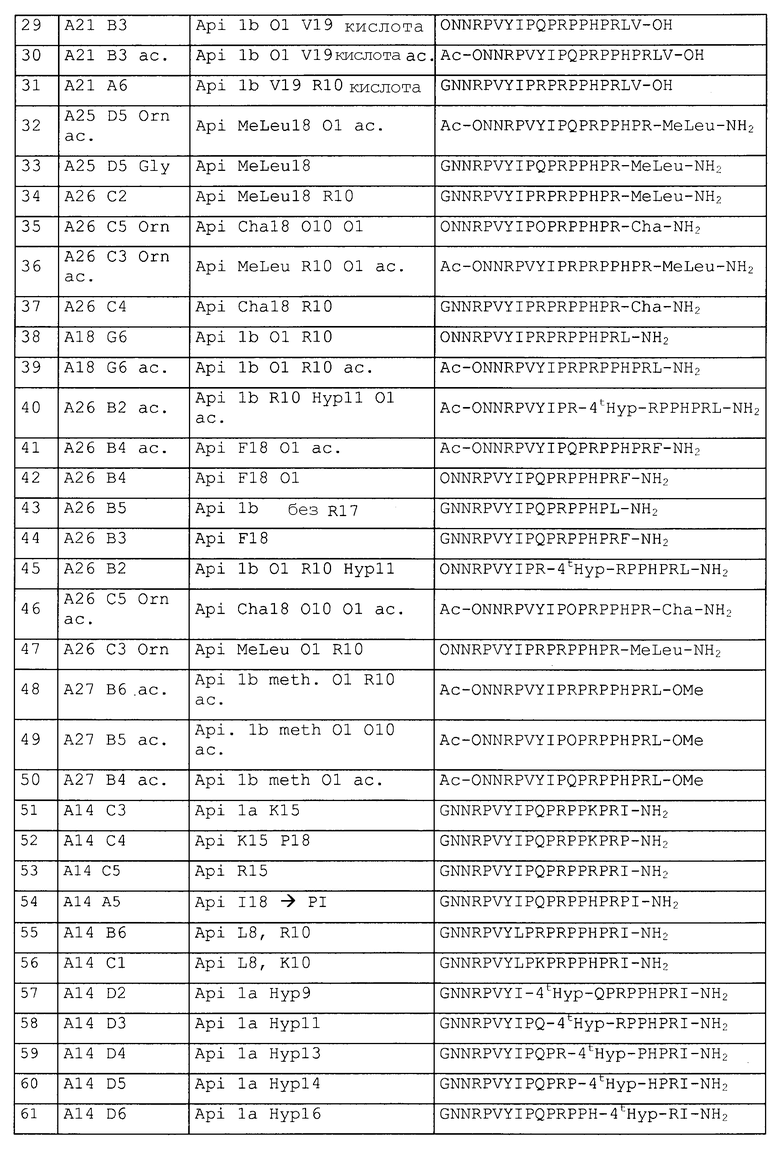


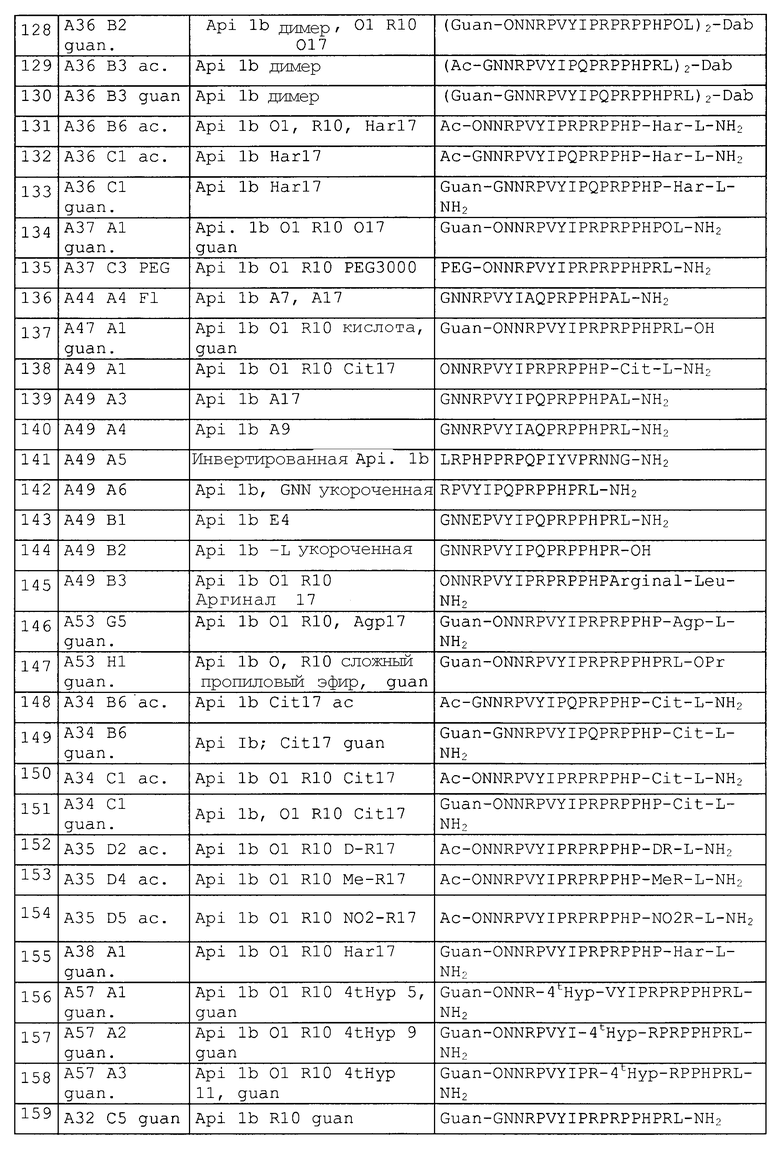

DR: D-аргинин, MeR = метиларгинин, предпочтительно альфа-N-метиларгинин (5-(диаминометилиденамино)-2-метиламинопентановая кислота), NO2R = нитроаргинин, предпочтительно N(G)-нитроаргинин(2-амино-5-[(аминонитрамидометилиден)амино]пентановая кислота), Cit: цитруллин; Ac = ацетил, For = формил, Guan = гуанидогруппа являются примерами для модифицированных N-концов (модифицированная альфа-аминогруппа N-концевой аминокислоты, Sub1 = ацетил-NH, формил-NH или гуанидогруппа).
Agp: альфа-амино-бета-гуанидинопропионовая кислота, аргинал: -COOH в аргинине замещают на -CHO, Cha: циклогексилаланин, Chex: 1-аминоциклогексилугольная кислота, Cit: цитруллин, OMe представляет собой сложный метиловый эфир на C-конце (Sub2 = OR3 = OMe), OPr представляет собой сложный пропиловый эфир на C-конце (Sub2 = OR3 = O-Pr); MeLeu = N-Метиллейцин - лейцин с метилированием пептидной связи; Ac = ацетил, For = формил, Guan = гуанидогруппа являются примерами модифицированных N-концов (модифицированная альфа-аминогруппа N-концевой аминокислоты, Sub1 = ацетил-NH, формил-NH или гуанидогруппа); βAla и βHar яаляются примерами бета-аминокислот.
Пример 2: Стабильность в сыворотке
Исследования стабильности в сыворотке проводили, повторяя два раза, как описано Hoffmann et al. [42]. В кратком изложении, 28 мкл водного пептидного раствора или раствора пептидомиметиков (0,5 мг/мл) добавляли к 0,2 мл свежевзятой смешанной 25% сыворотки мышей в воде (Sigma-Aldrich GmbH, Taufkirchen, Germany). Смеси инкубировали при 37°C с аккуратным перемешиванием. После времени инкубации, равного 0, 0,5, 1, 2, 4 и 6 ч, белки осаждали 40 мкл 15% водной TCA при 0°C в течение 20 мин перед центрифугированием в течение 5 мин при 4°C (13500 × g). Супернатанты всех образцов (240 мкл) нейтрализовали водной NaOH с концентрацией 1 моль/л и немедленно помещали на хранение при -20°C. Супернатанты анализировали посредством ОФ-ВЭЖХ, используя линейный градиент ацетонитрила, как описано выше, с 0,1% TFA в качестве ион-парного реагента. Далее, собранные фракции основных пиков анализировали посредством тандема масс-спектрометрии (MALDI-TOF/TOF-MS, 4700 Proteomics Analyzer, Applied Bio systems GmbH, Weiterstadt, Germany) со способом отражения положительно заряженных ионов, используя α-цианогидроксикоричную кислоту (50% CH3CN в 0,1% водной TFA) в качестве матрицы для идентификации продуктов деградации пептидов и пептидомиметиков, то есть метаболитов, в различные моменты времени. Контрольные образцы сыворотки состояли из 200 мкл смешанной 25% сыворотки мышей в воде, которую также осаждали 40 мкл 15% водной TCA, как описано ранее. Контрольные образцы пептидов состояли из 28 мкл полученного пептидного раствора, разведенного в 0,2 мл воды и 40 мкл 15% водной трихлоруксусной кислоты (TCA).
Стабильность в сыворотке некоторых пептидов представлена в таблице 3.
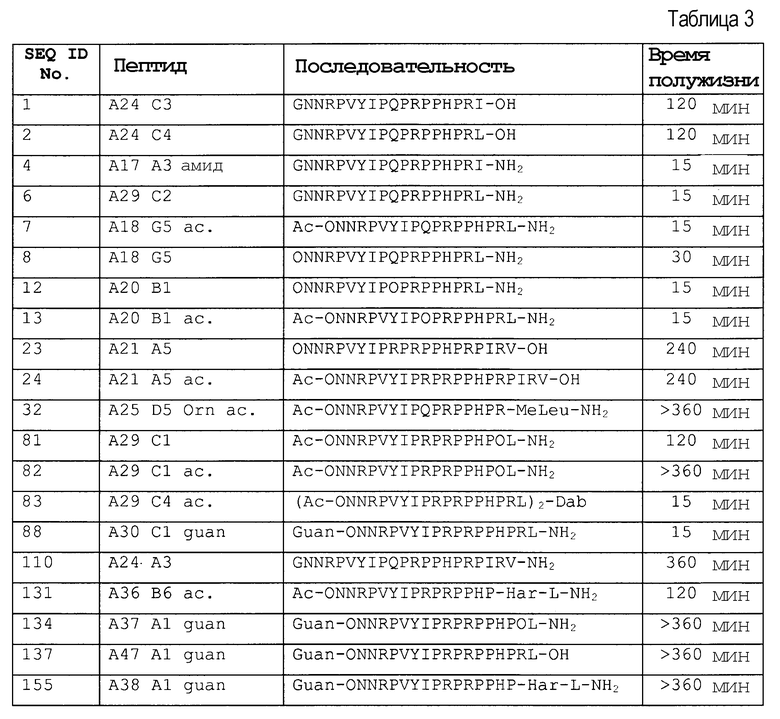
Природные последовательности апидецина дикого типа (wt) SEQ ID № 1 и 2 разрушались с обоих концов, то есть N-концевые и C-концевые остатки или пептиды отщепились.
Таким образом, пептидная последовательность на обоих концах подлежит стабилизированию предпочтительно против экзопептидаз или экзопротеаз, а также эндопротеаз. Связью, особенно чувствительной к эндопротеазам, является связь между аргинином в положении 17 и изолейцином или лейцином в положении 18. За деградацией наблюдали посредством MALDI-MS, определяя молекулярные массы пептидных метаболитов и тандемный масс-спектр соответствующих пептидов. В результате N-концевой деградации образуется последовательность, укороченная на длину от одного до трех остатков.
N-концевое ацетилирование, например (SEQ ID No. 7, 13, 24, и 32) значительно уменьшало этот путь деградации, не воздействуя значительно на деградацию C-конца. Например, пептид Ac-ONNRPVYIPQPRPPHPRL-NH2 (SEQ ID № 7) и Ac-ONNRPVYIPRPRPPHPRL-NH2 (SEQ ID № 13) не разрушался с N-конца, а разрушался только с C-конца, при этом происходило расщепление или C-концевого лейцинамида, или двух остатков с C-конца (фиг. 1).
N-концевое гуанидирование даже превосходит ацетилирование в снижении N-концевой деградации (SEQ ID № 134, 137 и 155).
На фиг.1 показано количество пептида Ac-ONNRPVYIPQPRPPHPRL-NH2 (SEQ ID № 7), находящегося в 25% сыворотке в воде после 30, 60, 120, 240 и 360 мин, а также двух выявленных метаболитов Ac-ONNRPVYIPQPRPPHPR-OH (SEQ ID № 164 - расщепление C-концевого лейцинамида) и Ac-ONNRPVYIPQPRPPHP-OH (SEQ ID № 165 - отщепление двух C-концевых остатков), выраженное через площади пиков, полученных ОФ-ВЭЖХ, с использованием УФ-детектирования.
Аналогично, C-концевую деградацию снижали, например, посредством амидирования C-конца, которое также не влияло на N-концевую деградацию. Таким образом, комбинация модификаций на N- и C-концах значительно снижала деградацию экзопептидазами или экзопротеазами (SEQ ID № 7, 13 и 32). Расщепление C-конца для Arg-17, то есть Arg-Leu или Arg-Ile, эффективно уменьшали посредством метилирования N-конца этой пептидной связи. Предпочтительно, пептид стабилизируют против всех трех возможных путей деградации, как проиллюстрировано для Ac-ONNRPVYIPQPRPPHPRMeLeu-NH2 (SEQ ID № 32), который показал время полужизни, равное более чем 6 часам (25% сыворотки в воде, 37°C). Подобную устойчивость получали для последовательности Ac- ONNRPVYIPRPRPPHPOL-NH2 (SEQ ID № 82) посредством замены Arg-17 на орнитин, поскольку орнитин не имеет место расщепления для трипсина.
Пример 3: Антибактериальные исследования
1. Анализ зон ингибирования (анализ на агаровых планшетах)
Очищенные пептиды на основе апидецина и пептидные производные разводили водой до конечной концентрации, равной 500 мкг/мл. Аликвоты в количестве 10 мкл и контроли (10 мкл в воде или в растворе антибиотика) наносили на планшет с агаром, засеянный суспензией бактериальной культуры в среднелогарифмической фазе роста. Планшеты инкубировали в темноте при 37°C. Диаметр зон ингибирования измеряли после 20 часов роста. В основном бактерии росли в 1% триптическом соевом бульоне (TSB, Fluka, Neu-Ulm, Germany) и 1,2% агаре (Fluka). Все тесты проводили в аэробных условиях.
Аланиновое сканирование природной последовательности апидецина позволило идентифицировать несколько остатков, ответственных за противомикробную активность в отношении трех различных штаммов E. coli (фиг. 2) и K. pneumoniae (фиг. 3).
На фиг. 2 показана антибактериальная активность аналогов апидецина 1a (аланиновое сканирование) по отношению к различным штаммам E. Coli, с использованием анализа на агаровых планшетах.
На фиг. 3 показана антибактериальная активность аналогов апидецина 1a (аланиновое сканирование) по отношению к Klebsiella pneumoniae штамм ATCC 10031, с использованием анализа на агаровых планшетах.
На графиках, представленных на фиг. 2 и фиг. 3, природная пептидная последовательность апидецина 1a GNNRPVYIPQPRPPHPRI (SEQ ID № 1) отложена по оси X. Каждый остаток означает соответствующий пептид, в котором этот остаток заменен на аланин, например G в положении 1, замененный в: ANNRPVYIPQPRPPHPRI (SEQ ID № 166), N в положении 2, замененный в GANRPVYIPQPRPPHPRI (SEQ ID № 167), и так далее.
Столбик означает диаметр зоны ингибирования. Таким образом, самый высокий столбик соответствует самой высокой антибактериальной активности пептида, модифицированного аланином.
Посредством аланинового сканирования определяли все положения, ответственные за антибактериальную активность в апидецине 1. Антибактериальная активность сильно снижалась, если проводили замены на аланин в положениях от Pro11 до Ile18, Arg4, Tyr7 и Pro9. В дальнейших экспериментах в конкретных положениях аминокислот проводили замену на другие похожие аминокислоты для увеличения активности и протеазной устойчивости. В большинстве случаев наблюдали сильное снижение антибактериальной активности.
Во всех протестированных штаммах обоих видов бактерий обнаружили очень похожую реакцию на различные одноточечные мутации Pro в положении 11 и Arg в положении 17 природной последовательности, являющихся обязательными для сохранения по меньшей мере частичной активности.
2. Анализ на основе ингибирования роста.
Минимальные ингибирующие концентрации (MIC) пептидов, полученных на основе апидецина и пептидных производных, определяли с помощью анализа ингибирования роста, используя последовательное разведение пептидов в стерилизованных круглодонных планшетах с 96 лунками с конечным объемом 100 мкл на лунку (полистирол, U-образное дно, Greiner Bio-One GmbH). Бактерии, например, E. coli BL21 AI, растили в питательном бульоне (NB, Carl Roth GmbH + Co. KG, Karlsruhe, Germany) при 37°C в течение ночи. В каждую лунку 96-луночного планшета добавляли 50 мкл ночной культуры, доведенной 1% TSB до концентрации 5×106 КОЕ/мл. Лиофилизованные пептиды и пептидомиметики растворяли в 1% TSB (триптический соевый бульон) или воде до получения конечной концентрации, равной 250 мкг/мл. Пятьдесят мкл раствора пептида или раствора пептидомиметиков добавляли в первую лунку ряда на планшете и перемешивали. Пятьдесят мкл из первой лунки, содержащие раствор пептида, переносили во вторую лунку, перемешивали и 50 мкл снова переносили в следующую лунку и так далее. Таким образом, получали серии разведений в два раза, начиная с концентрации 250 мкг/мл в первой лунке до концентрации 120 нг/мл в 12-й лунке ряда, что позволило получить конечную концентрацию пептида или пептидомиметика, равную от 125 мкг/мл (лунка 1) до 60 нг/мл (лунка 12). Планшеты инкубировали при 37°C в течение 20 часов. Поглощение измеряли при 595 нм, используя спектрофотометр для микропланшета TECAN (Tecan, Germany). Все измерения для пептидов и пептидомиметиков повторяли 3 раза. В качестве отрицательного контроля использовали стерилизованную воду. Значение MIC определяет наименьшую концентрацию, при которой не наблюдался рост бактерий после инкубации при 37°C в течение по меньшей мере 20 часов.
Значение MIC исследуемых пептидов и пептидных производных по отношению к бактериям, имеющим устойчивость ко многим лекарственным средствам, суммированы в таблице 4.
Исследовали следующие штаммы бактерий, устойчивые ко многим лекарственным средствам:
- Eschericha coli 45849 и D31,
- Klebsiella pneumoniae 123132 и K6,
- Salmonella typhi S5 и Salmonella enterica subsp. enterica серовар Typhimurium (ATCC 700408),
- Rhizobium radiobacter (ATCC 15955, описанная как Agrobacterium tumefaciens).
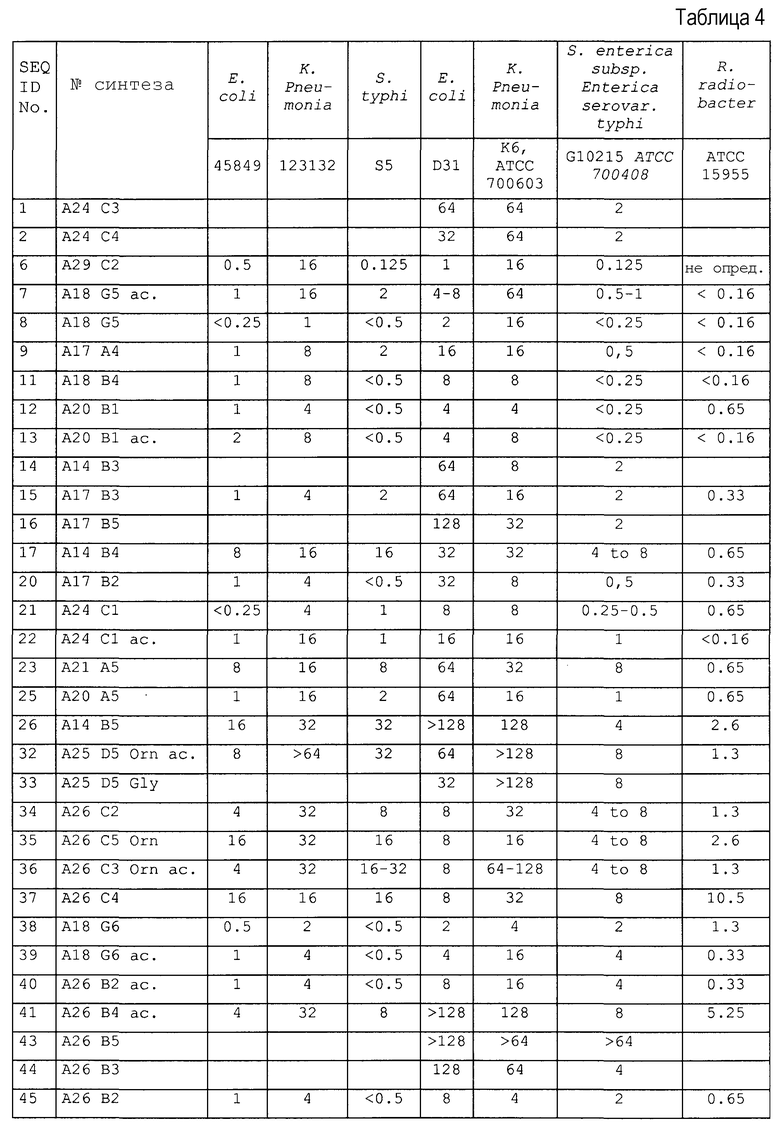
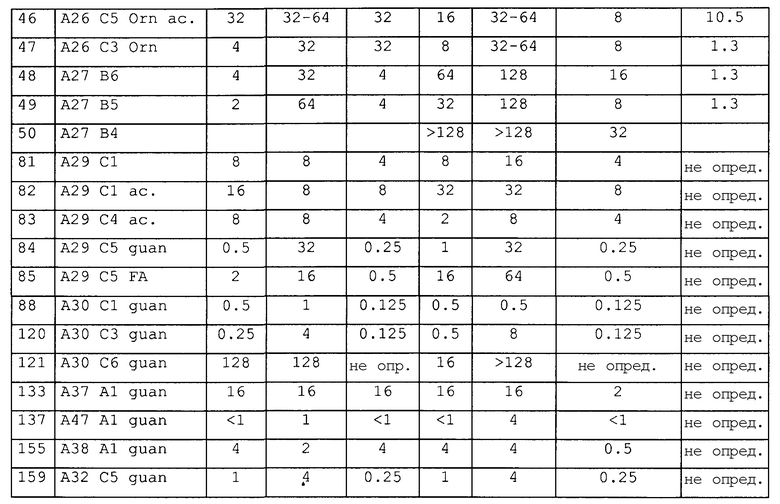
SEQ ID № 1, 2 представляют собой последовательности дикого типа как примеры для сравнения, SEQ ID № 4, 19, 26, 32, 33, 41, 43, 44, 50, 121 и 133 представляют собой менее предпочтительные примеры по изобретению (с Q на месте X4 - в положении 10). Другие представленные SEQ ID № являются предпочтительными пептидами или пептидными производными по изобретению. Наиболее предпочтительными примерами аявляются SEQ ID № 6, 7-8, 11-13, 20-22, 25, 39, 40, 45, 65, 67, 84, 85, 88, 137, 155 и 159.
Амидирование C-конца приводит не только к улучшенной противомикробной активности, то есть снижает значения MIC, но также уменьшает деградацию с С-конца под действием экзопротеаз. Аналогично, модификации, предпочтительно ацетилирование или гуанидирование аминогруппы N-конца, уменьшают N-концевую деградацию, как описано выше. Однако N-концевое ацетилирование снижает значения MIC для некоторых исследуемых бактериальных штаммов (например, SEQ ID № 21 по сравнению с SEQ ID № 22 и SEQ ID № 38 по сравнению с SEQ ID № 39), что показывает, что эти N-концевые остатки должны нести положительный заряд. Таким образом, глицин в положении X1 замещают остатками, несущими положительно заряженную боковую цепь, такими как лизин (например, SEQ ID № 21), аргинин и орнитин (например, SEQ ID № 12). Другой возможностью для введения положительного заряда в N-конец является N-концевое гуанидирование, которое, в то же время, защищает N-конец от деградации (например, SEQ ID № 133 и 137). Такие модифицированные на N-конце пептиды, имеющие остатки с положительно заряженной боковой цепью в положении X1, показали лучшие значения MIC, чем ацетилированные последовательности апидецина дикого типа. Другим важным для увеличения противомикробной активности остатком является остаток в положении 10 (остаток X4), где глутамин замещают на остаток, несущий положительно заряженную боковую цепь, такой как орнитин (например, SEQ ID № 12 и 13), лизин (например, SEQ ID № 21 и 22) или аргинин (например, SEQ ID № 38 и 39). Такая замена в положении 10 (остаток X4) на остаток, несущий положительно заряженную боковую цепь, не снижает устойчивость по отношению к протеазам, поскольку следующее положение содержит пролин, который эффективно снижает протеолитическое расщепление эндопротеазами пептидной связи с N-конца. Замена пролина в положении 11 (остаток X5) на гидроксипролин как не повлияла на значения MIC в пределах интервала погрешности анализа, так и не уменьшила устойчивость к протеазам его пептидной связи с N- конца. Исходя из того что эта замена на гидроксипролин в положении 11 (остаток X5) не повлияла как на значения MIC, так и на стабильность в сыворотке, повышенная полярность дополнительно снижает токсичное действие на клетки и гемолитическую активность, поскольку более полярные пептиды в меньшей степени связываются с клеточными мембранами. Таким образом, наиболее предпочтительный пептид содержит орнитин в положении 1 (остаток X1), аргинин в положении 10 (остаток X4) и гидроксипролин в положении 11 (остаток X5, ацетилированный N-конец и C-концевой амид, такие как SEQ ID № 40). Кроме того, пептидную связь между положениями 17 и 18, то есть Arg и Leu/Ile исходной последовательности, модифицируют для повышения ее устойчивости к протеазам или пептидазам таким образом, как метилирование N-конца (например, SEQ ID № 33, 34). Альтернативно, аргинин в положении 17 (остаток X6) замещают на основной остаток, не подверженный протеолизу, такой как орнитин (например, SEQ ID № 38, 131-133, 146-155).
Значения MIC исследуемых пептидов и пептидных производных по отношению к бактериям некоторых видов суммированы в таблице 5.
Исследуемые штаммы бактерий:
- Eschericha coli 1103, 10233, BL 21 AI, DC 2,
- Klebsiella pneumoniae 681,
- Micrococcus luteus ATCC 10240,
- Mycobacterium vaccae,
- Bactillus subtillis 347.



SEQ ID № 1, 2 представляют собой последовательности дикого типа, приведенные в качестве примеров для сравнения. SEQ ID № 32, 33 и 70-80, а также SEQ ID № 132, 133, 135, 136, 139, 140, 143, 144 и 148 являются менее предпочтительными примерами по изобретению (с Q на месте X4 - положение 10). Иневертированная последовательность апидецина (SEQ ID № 141) и укороченные пептиды апидецина (SEQ ID № 92 и 142) также являются менее предпочтительными. Предпочтительными пептидами или пептидными производными по изобретению являются остальные SEQ ID №. Наиболее предпочтительными примерами являются SEQ ID № 7, 8, 9, 11-17, 20-23, 25, 38 40, 45, 65-67, 84, 85, 88, 134 и 137.
Как правило, измеренная в способе микроразведений антибактериальная активность тестируемых пептидных последовательностей в значительной степени совпадает с данными, представленными в таблице 4, и обсуждается ниже. В кратком изложении, желательным является амидирование C-конца, поскольку оно значительно снижает значения MIC у тестируемых бактерий. Ацетилирование N-конца снижает степень N-концевой деградации пептидазами или протеазами, как описано выше. Наилучшие значения MIC получали для пептидов и пептидомиметиков, несущих положительно заряженную группу на N-конце пептида, таким образом, выгодной является замена глицина в положении 1 на лизин (например, SEQ ID № 21 и 22), аргинин или орнитин (например, SEQ ID № 7 и 8), которые все несут положительно заряженную боковую цепь. Орнитин является предпочтительным, поскольку не вносит в последовательность место расщепления для трипсина. Данные пептиды, ацетилированные по N-концу, показывали лучшие значения MIC, чем ацетилированные последовательности дикого типа. Другой заменой, важной для увеличения противомикробной активности, является замена глутамина в положении 10 (остаток X4) на остатки, несущие положительно заряженную боковую цепь, такие как орнитин (например, SEQ ID № 12 и 13), лизин (например, SEQ ID № 21 и 22) или аргинин (например, SEQ ID № 38 и 39). Эта замена не снижает устойчивости по отношению к протеазам, поскольку следующее положение содержит пролин, наличие которого эффективно снижает протеолитическое расщепление N-концевой пептидной связи. Гуанидирование (например, SEQ ID № 88, 134 и 137) в качестве альтернативы N-концевому ацетилированию было также очень эффективным и дополнительно расширяло спектр активности по отношению к B. Subtilis, которая является устойчивой к действию последовательности апедицина дикого типа, а также к большинству аналогов. Замена пролина в положении 11 (остаток X5) на гидроксипролин не снижала ни значения MIC, ни устойчивости к протеазам, но, поскольку это более полярная аминокислота, она дополнительно снижает клеточную токсичность и гемолитическую активность. Таким образом, предпочтительный пептид содержит орнитин в положении 1 (остаток X1), аргинин или орнитин в положении 10 (остаток X4) и гидроксипролин в положении 11 (остаток X5), ацетилированный или гуанидированный N-конец и амид на C-конце, такой как в SEQ ID № 40, 103.
Интересно, что некоторые модификации приводили к возникновению активности по отношению к B. subtilis, которую не наблюдают у природных пептидов апидецина дикого типа. Общим свойством этих последовательностей, имеющих активность по отношению к B. Subtilis, является положительно заряженный N-конец, то есть свободная аминогруппа или гуанидированный N-конец, аргинин или орнитин в положении 10 (остаток X4) и, предпочтительно, орнитин в положении 1 (остаток X1) последовательности апидецина 1b (лейцин в положении 18 - например, SEQ ID № 12, 38, 45, 66, 131-134 и 155). Аналогичные пептиды, гомологичные апидецину 1a (изолейцин в положении 18), не обладали активностью по отношению к B. subtilis. Такие же модификации последовательности апидецина 1a (заряженный N-конец, аргинин или орнитин в положении 10 и орнитин в положении 1) расширяли спектр активности по отношению к M. vaccae (SEQ ID № 65).
Пример 4: Исследования токсичности для клеток млекопитающих
1. Исследование гемолитической активности
Для определения того, являлись ли пептиды и производные пептидов по изобретению токсичными для клеток млекопитающих, исследовали гемолитическую активность нескольких пептидов и пептидомиметиков из примера 1, приведенного выше, положительного контроля и отрицательного контроля. Исследование гемолитической активности проводили на эритроцитах человека [43]. Концентрат эритроцитов человека был предоставлен Leipzig University Hospital (Leipzig, Germany) в буфере аденин-глюкоза-маннит хлорид натрия. Эритроциты центрифугировали при 1000 g и промывали 3 раза 10 объемами холодного фосфатно-солевого буфера (PBS, pH 7,4). Эритроциты разводили до конечной концентрации 1% в PBS. Сто микролитров этой суспензии эритроцитов человека в PBS добавляли в каждую лунку V-образного 96-луночного полипропиленового планшета для микротитрования (Greiner Bio-One GmbH). В каждую лунку добавляли 100 мкл пептида, растворенного в PBS, для получения серии разведений, с концентрациями начиная с 600 мкг/мл и до 4,8 мкг/мл, полученных семью последовательными разведениями. Планшет для микротитрования инкубировали при 37°C в течение 1 часа и затем центрифугировали в течение 10 мин при 1000 g. Сто микролитров супернатанта добавляли в лунки 96-луночного плоскодонного планшета для микротитрования из полистирола (Greiner Bio-One GmbH) и регистрировали поглощение при 405 нм на микропланшетном спектрофотометре (Tecan) для оценки высвобождения гема. В качестве отрицательного и положительного контролей использовали PBS и 0,1% Тритон X-100. Процент гемолиза подсчитывали посредством следующего уравнения [44]:
(Епептид-EPBS)/(ЕТритон-EPBS)×100% с E=экстинкция при 405 нм
Все исследования гемолитической активности проводили с повторениями. На фиг. 4 и в таблице 6 показано среднее значение, полученное из трех независимых экспериментов.
На фиг. 4 показан результат исследования гемолитической активности для пептидов A30 C1 guan. (SEQ ID № 88), A18 G6 ac. (SEQ ID № 39) и A17 A6 (SEQ ID № 6), концентрация пептидов 600 мкг/мл, PBS и Тритон X-100® (п-трет-октилфенокси)полиэтоксиэтанол, 0,1%) приведены как контроли.
Результаты для дополнительных пептидов и пептидных производных суммированы в таблице 6.

Ни один из исследуемых пептидов не показал какой-либо гемолитической активности вплоть до концентрации пептидов, равной 600 мкг/мл, то есть даже в концентрации, от 100 до 1200 раз более высокой, чем полученные значения MIC. Степень гемолиза для всех пептидов представляла собой только приблизительно 1% по сравнению с тритоном, что представляет собой фоновое значение для этого исследования. Неионное поверхностно-активное вещество Тритон X-100® использовали в качестве положительного контроля, поскольку оно, в экспериментальной модели, в течение одного часа полностью разрушало эритроциты. Это клеточное исследование показывает, что пептиды и производные пептидов могут присутствовать в крови в высоких концентрациях, не оказывая какого-либо побочного действия на эритроциты. В заключение, все исследуемые пептиды и производные пептидов соответствуют требованию о том, что идеальные противомикробные соединения не должны иметь какой-либо гемолитической активности при концентрации, в 100 раз более высокой, чем значения MIC.
2. Исследование токсичного действия на COS-клетки
COS-7 клетки выращивали в среде Игла, модифицированной Дульбекко (Cellgro, Meidatech Inc., Herndon, VA, USA), содержащей 10% эмбриональную телячью сыворотку, при 37°C в атмосфере 10% CO2. Клетки (5×103 клеток в лунке) высевали на 24-луночную чашку Петри и инкубировали в течение 24 ч перед добавлением пептида. Пептиды растворяли в 0,5 мл воды и инокулировали в 1 мл среды до конечных концентраций, равных 60, 200 и 600 мкг/мл, с дублированием. Чашки дополнительно инкубировали в течение 24 ч при 37°C в атмосфере 10% CO2. Среду аспирировали и исследование завершали добавлением 100 мкл трипсина в этилендиаминтетраацетате (ЭДТА) (0,25% трипсин/0,1% ЭДТА в сбалансированном солевом растворе Хенкса; Cellgro). Опытные и контрольные образцы собирали, промывали в PBS и фиксировали холодным 70% водным этанолом в течение 2 часов. Затем клетки ресуспендировали в PBS, содержащем 10 мкг/мл пропидиумиодида (PI) и 250 мкг/мл рибонуклеазы, и инкубировали в течение 30 мин при 37°C. Степень некроза/апоптоза клеток оценивали посредством способа проточной цитометрии, выполненного на Guava® EasyCyte™ Mini System (Guava Technologies, Hayward, CA, USA).
Этот способ изучает действие исследуемых пептидов и пептидных производных при различных концентрациях, с дублированием, на клеточный цикл клеточной культуры COS-7, то есть процент клеток в фазе Gl, S, G2/M, а также некротических и апоптотических клеток. Данные оцениваются относительно отрицательного контроля (в него не добавляли пептиды и пептидные производные) и раствора 10% DMSO, который полностью разрушает клетки.
Результаты суммированы в таблице 7. Так же, как показано в предыдущей таблице, ни один из пептидов и пептидных производных не обладал каким-либо заметным влиянием на распределение по фазам клеточного цикла, поскольку все значения находились внутри границы погрешностей двух отрицательных контролей, даже при самой высокой концентрации, равной 600 мкг/мл. Наиболее важные параметры: некроз и апоптоз не были повышены по сравнению с отрицательным контролем. Эти данные в сочетании с данными по гемолитической активности (таблица 6) доказывают, что исследованные пептиды и производные пептидов не оказывают цитотоксичного действия на клеточном уровне даже при концентрациях в пределах вплоть до 600 мкг/мл, которая выше значений MIC более чем в 100 раз.
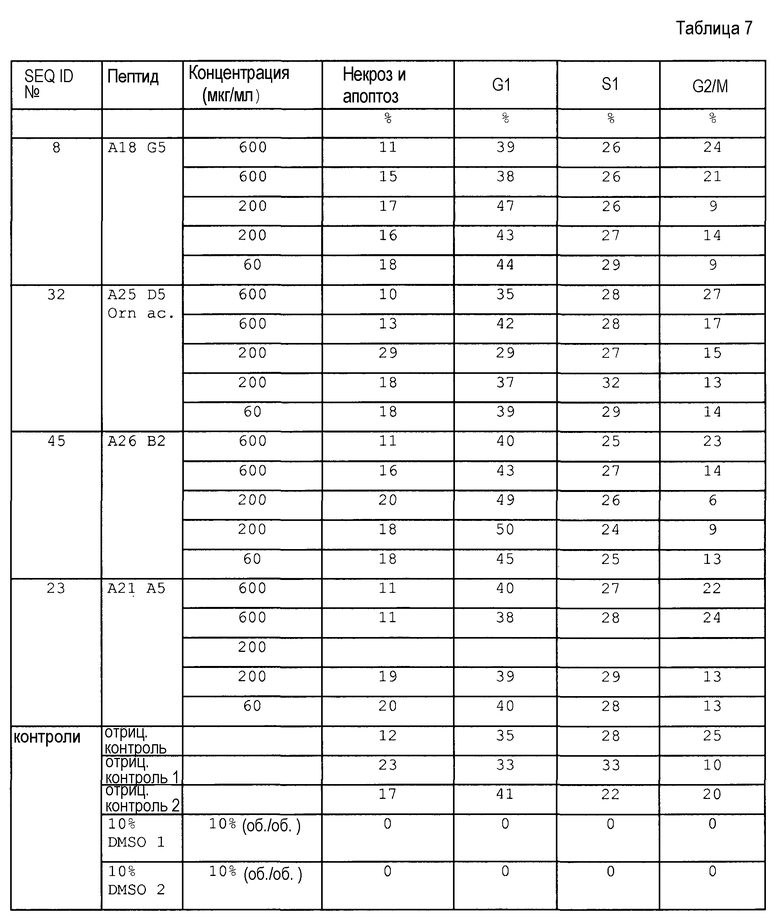
Пример 5: Возникновение устойчивости
Возникновение устойчивых штаммов определяли согласно способу макроразведений в условиях NCCLS (National Committee for Clinical Laboratory Standards - сейчас Clinical и Laboratory Standards Institute Wayne, PA, USA, NCCLS-Guideline, M7-A5, Vol. 20, Nr. 2; 2000).
Во-первых, для пептидов, подлежащих исследованию, определяли минимальные ингибирующие концентрации (MIC), как описано в примере 3.2. Для определения возникновения устойчивости при серийных разведениях пептида выбирали концентрации в диапазоне от 3 раз до 4 раз выше и ниже MIC. Тест проводили в стерильных 96-луночных круглодонных планшетах для микротитрования (полистирол, U-образное дно, Greiner Bio-One GmbH, Germany) и с конечным объемом, равным 200 мкл на лунку. Лиофилизированный пептид или производные пептидов растворяли в воде и разводили, в случае MIC, в 1% TSB (триптический соевый бульон) до получения объема, равного 100 мкл. В качестве примера, для значения MIC 2 мкг/мл выбирали серию разведений от 32 мкг/мл до 0,25 мкг/мл. Разведения внутри серий являлись двукратными.
Бактерии, E. coli BL21 AI, выращивали в течение ночи при 37°C в питательном бульоне (NB, Carl Roth GmbH + Co. KG, Karlsruhe, Germany) и разводили до количества 5×106 колониеобразующих единиц (КОЕ)/мл в 1% TSB. Для начала исследования 100 мкл разведенных бактерий добавляли в лунку планшета для микротитрования с внесенным пептидом. После инкубирования планшетов в течение 24 ч при 37°C определяли поглощение при 595 нм, используя микропланшетный ридер TECAN (Tecan, Germany).
Значения MIC подсчитывали, как описано в примере 3.2. Тест повторяли, используя новую серию разведений с концентрациями пептида, адаптированными под вновь определенные значения MIC, и используя бактериальную культуру, которая росла несмотря на наличие ингибиторного пептида. Эту бактериальную культуру (1-й пассаж) разводили до 5×106 КОЕ/мл в 1% TSB. После инкубации с новыми сериями разведений пептида в течение 24 ч при 37°C определяли поглощение при 595 нм и снова подсчитывали значения MIC. Тест повторяли, используя новую серию разведений с концентрациями пептида, адаптированными под вновь определенные значения MIC, и используя бактериальную культуру, которая росла несмотря на наличие ингибиторного пептида (2-й пассаж). Такой же способ применяли приблизительно для 8 дополнительных пассажей.
В качестве контроля выбирали интактную культуру E.coli BL21 AI и пересеивание без ингибирующих пептидов. Все измерения для пептидов и пептидных производных повторяли три раза. Число пассажей выбирали в зависимости от скорости возникновения устойчивости, и обычно, за 10 суток без перерыва, было 10 пассажей.
На фиг. 5 показано возникновение устойчивости по сравнению с апидецином дикого типа 1b (SEQ. ID № 2) и двумя оптимизированными последовательностями по изобретению (SEQ. ID № 88 и 137):
Для последовательности апидецина дикого типа первые устойчивые штаммы получали уже после 2-го пассажа. Устойчивость увеличилась до значения MIC, равного 128 мкг/мл в 10-м пассаже.
Однако для двух последовательностей по изобретению (SEQ ID № 88 и 137) совсем не наблюдали возникновения устойчивости. Таким образом, возникновение устойчивости не зависит от C-конца.
Значения MIC интактного контрольного штамма E.coli BL21 AI, который пересеивали параллельно, не изменялись во время исследования.
Эти результаты иллюстрируют, что соединения по изобретению можно применять в течение длительного периода времени как новые антибиотики, к которым не возникает устойчивости у E. coli.
Пример 6: Определение распределения in vivo
Распределение in vivo измеряли у мышей, используя флуоресцентно меченные производные пептидов.
Флуоресцентный краситель Dy675, поглощающий в ближней инфракрасной области (Dyomics GmbH, Jena, Germany), активировали DIC и соединяли с N-концом пептидных производных после завершения твердофазного пептидного синтеза (смотрите, пример 1). Затем пептиды расщепляли TFA и очищали посредством ОФ-ВЭЖХ (смотрите, пример 1).
Сорок микрограммов меченых пептидных производных вводили самкам мышей линии Balb/c, подвергнутым анестезированию изофлураном, в выбритый участок подкожно (sc) или интраперитонеально (i.p). Животных, подвергаемых непрерывному действию изофлурана, помещали в отсек флуоресцентного микроскопа. Флуоресцентные снимки получали каждую минуту в первые 10 минут после введения пептидов и затем каждые 5 мин до 65 мин с помощью микроскопа IVIS с длиной волны излучения 695 нм.
На фиг. 6 показаны изображения и биораспределение флуоресцентно меченного пептида Dy675-ONNRPVYIPRPRPPHPRL-NH2 (SEQ ID № 160) in vivo, через 60 и 65 минут после интраперитонеального введения. Представлены снимки после 60 мин (фиг. 6A, область живота, низкая интенсивность), 65 мин (фиг. 6B, область живота, высокая интенсивность) и после 65 мин (фиг. 6C, спина, высокая чувствительность). Цветные зоны показывают интенсивность флуоресценции. Флуоресцентно меченное производное пептида вводили i.p., и его распределение изучали посредством получения в определенные интервалы времени изображений in vivo (фиг. 6). На фиг. 6C, на снимке, полученном со спины, ясно видно, что пептиды скапливались в мозге, печени и обеих почках. На фиг. 6B мозг также окрашен, тогда как почки и печень не видны из-за высокой пептидной концентрации в месте введения лекарственного средства.
Через 60 мин. большинство инокулированных пептидов оставались в месте введения. Небольшое количество пептидов распределялось по всему телу.
Фармацевтический потенциал новых пептидных производных заключается в том, что в течение 60 мин. они проникают в различные органы животных, включая мозг, печень и почки, что ясно показывает, что пептиды распределяются кровью ко всем органам тела.
Сокращения
Для обозначения аминокислот на всем протяжении документа используют следующие сокращения:
Другие сокращения включают следующие:
Ac: ацетил,
Api: апидецин,
AMP: противомикробные пептиды,
БСА: бычий сывороточный альбумин,
KOE: колониеобразующая единица,
DCM: дихлорметан,
DMF: диметилформамид,
ЭДТА: этилендиаминтетраацетат,
ESI: ионизация электрораспылением,
DIPEA: N,N'-диизопропилэтиламин,
For: формил (метаноил),
Fmoc: 9-фторенилметоксикарбонил,
Guan: гуанидиногруппа,
HBTU: 2-(1-H-бензотриазол-1-ил)тетраметилурониум гексафторфосфат,
ВЭЖХ: высокоэффективная жидкостная хроматография,
HMBA: 4-гидроксиметилбензойная кислота,
MALDI: ассоциированная с матрицей лазерная десорбционная ионизация,
Me: метил,
MeCN: ацетонитрил,
MeOH: метанол,
MIC: минимальная ингибиторная концентрация,
MS: масс-спектрометрия,
NB: питательный бульон,
OMe: сложный метиловый эфир
PBS: фосфатно-солевой буфер,
PI: пропидиумиодид,
ОФ: обратнофазовая,
RT: комнатная температура,
tBu: трет-бутил,
TCA: трихлоруксусная кислота,
TFA: трифторуксусная кислота,
TOF: время-пролетная,
TSB: триптический соевый бульон,
УФ: ультрафиолетовое излучение.
Список литературы
1. Thomasz, A. (1994) Multiple-antibiotic-resistant bacteria. New Engl. J. Med. 330: 1247-1251.
2. Wenzel, R.P. (1988) The mortality of hospital-acquired bloodstream infections: need for a vital statistic? Int. J. Epidemiol. 17: 225-227.
3. Moellering, R.C, Jr. (1998) Problems with antimicrobial resistance in Gram-positive cocci. Clin. Infect. Dis. 26: 1177-1178.
4. Hand, W.L. (2000) Current challenges in antibiotic resistance. Adolesc. Med. 11: 427-438.
5. Hooper, B.C. (2001) Emerging mechanisms of fluoroquinolone resistance. Emerg. Infect. Dis. 7:337-341.
6. Jones, R.N. (2001) Resistance patterns among nosocomial pathogens: trends over the past few years. Chest 119: 397S-404S.
7. Prachayasittikul, V., Laming, R., and Bulow, L. (2000) Episome profiles and mobilizable beta lactamase plasmid in Haemophilus ducreyi. Southeast Asian J. Trop. Med. Public Health 31: 80-84.
8. Teuber, M. (1999) Spread of antibiotic resistance with food-borne pathogens. Cell. Mol. Life Sci. 30: 755-763.
9. Boman, H.G. (1995) Peptide antibiotics and their role in innate immunity. Annu. Rev. Immunol. 13: 61-92.
10. Barra, D., Simmaco, M., and Boman, H.G. (1998) Gene encoded peptide antibiotics and innate immunity. Do 'animacules' have defense budgets? FEBS Lett. 430: 130-134.
11. Ludtke, S., He, K., and Huang, H. (1995) Membrane thinning caused by magainin 2. Biochemistry, 34: 16764-16769.
12. Wimley, W.C., Selsted, M.E., and White, S.H. (1994) Interactions between human defensins and lipid bilayers: evidence for formation of multimeric pores. Protein Sci. 3: 1361-1373.
13. Shai, Y. (1995) Molecular recognition between membrane-spanning polypeptides. Trends Biochem. Sci. 20: 460-464.
14. Wade, D., Boman, A., Wahlin, B., Drain, C. M., Andreu, D., Boman, H. G., and Merrifield, R. B. (1990) Proc. Natl. Acad. Sci. USA 87: 4761-4765.
15. Steiner, H., Andreu, D., and Merrifield, R. B. (1988) Biochim. Biophys. Acta 939: 260-266.
16. Otvos, L., Jr., Bokonyi, K., Varga, I., Otvos, B.I., Hoffmann, R., Ertl, H.C.J., Wade, J.D., McManus, A.M., Craik, D.J., and Bulet, P. (2000) Insect peptides with improved protease-resistance protect mice against bacterial infection. Protein Sci. 9: 742-749.
17. Boman, H.G. (1995) Peptide antibiotics and their role in innate immunity. Annu. Rev. Immunol. 13: 61-92.
18. Casteels, P., Ampe, C., Jacobs, F., Vaeck, M., and Tempst, P. (1989) Apidaecins: antibacterial peptides from honeybees. EMBO J. 8: 2387-2391.
19. Casteels, P., and Tempst, P. (1994) Apidaecin-type peptide antibiotics function through a non-poreforming mechanism involving stereospecificity. Biochem. Biophys. Res. Commun. 199: 339-45.
20. Bulet, P., Dimarcq, J.-L., Hetru, C., Lagueux, M., Charlet, M., Hegy, G., van Dorsselaer, A., and Hoffmann, J.A. (1993) A novel inducible antibacterial peptide from Drosophila carries an O-glycosylated substitution. J. Biol Chem. 268: 14893-14897.
21. Mackintosh, J.A., Veal, D.A., Beattie, A.J., and Gooley, A.A. (1998) Isolation from an ant Myrmecia gulosa of two inducible O-glycosylated proline-rich antibacterial peptides. J. Biol. Chem. 273: 6139-6143.
22. Cociancich, S., Dupont, A., Hegy, G., Lanot, R., Holder, F., Hetru, C., Hoffmann, J.A., and Bulet P. (1994) Novel inducible antibacterial peptides from a hemipteran insect, the sap sucking-bug Pyrrhocoris apterus. Biochem. J. 300: 567-575.
23. Merrifield, R.B. (1963) Solid Phase Peptide Synthesis. I. The Synthesis of a Tetrapeptide J. Am. Chem. Soc. 85: 2149-2154.
24. Stemmer, W.P., Crameri, A., Ha, K.D., Brennan, T.M., Heyneker, H.L. (1995) Single-step assembly of a gene and entire plasmid from large numbers of oligodeoxyribonucleotides. Gene 164: 49-53.
25. Gething, M.J. and Sambrook, J. (1981) Cell-surface expression of influenza haemagglutinin from a cloned DNA copy of the RNA gene. Nature. 293: 620-625.
26. Maeno, M., Taguchi, S., Momose, H. (1993) Production of antibacterial peptide 'apidaecin' using the secretory expression system of Streptomyces. Biosci. Biotechnol. Biochem. 57: 1206-1207.
27. Zhou, Q.F., Luo, X.G., Ye, L., Xi, T. (2007) High-level production of a novel antimicrobial peptide perinerin in Escherichia coli by fusion expression. Curr. Microbiol. 54: 366-370.
28. Si, L.G., Liu, X.C., Lu, Y.Y., Wang, G.Y., Li, W.M. (2007) Soluble expression of active human beta-defensin-3 in Escherichia coli and its effects on the growth of host cells. Chin. Med. J. (Engl). 120: 708-713.
29. Noren, C.J., Anthony-Cahill, S.J., Griffith, M.C. and Schultz, P.O. (1989) A general method for site-specific incorporation of unnatural amino acids into proteins. Science 244: 182-188.
30. Ellman, J., Mendel, D., Anthony-Cahill, S., Noren, C.J., Schultz, P.O. (1991) Biosynthetic method for introducing unnatural amino acids site-specifically into proteins. Meth. Enzymol. 202: 301-336.
31. W. F. Anderson (1998) Human gene therapy. Nature 392 Supp., 25-30.
32. Pharmaceutical Biotechnology (Ed. D. J. A. Crommelin and R. D. Sindelar), Harwood Academic Publishers, 1997, pp. 8-20, 53-70, 123-152, 167-180.
33. Protein Synthesis: Methods and Protocols, Ed. R. Martin, Humana Press, 1998, pp. 1-442.
34. Amino Acid and Peptide Synthesis, Oxford University Press, 1997, pp. 1 -89.
35. Solid-Phase Peptide Synthesis (Ed. G. B. Fields), Academic Press, 1997, p. 1-780.
36. Tam, J.P. (1998) Synthetic Peptide Vaccine Design: Synthesis and Properties of a High-Density Multiple Antigenic Peptide System, Proc. Natl. Acad. Sci. USA 85: 5409-5413.
37. Posnett, D.N., McGrath, H. and Tam, J.P. (1988) A novel method for producing anti-peptide antibodies. Production of site-specific antibodies to the T cell antigen receptor beta-chain. J. Biol. Chem. 263: 1719-1725.
38. Morell, M., Espargaro, A., Aviles, F.X. and Ventura, S. (2007) Detection of transient protein-protein interactions by bimolecular fluorescence complementation: The Abl-SH3 case. Proteomics 7: 1023-1036.
39. G. B. Fields and Noble, R. (1990) Solid phase peptide synthesis utilizing 9-fluorenylmethoxycarbonyl amino acids. Int. J. Pept. Protein Res. 35: 161-214.
40. Gausepohl, H., Pieles, H., Frank, R.W. (1992) in "Peptides: chemistry, structure and biology" (ed. J. A. Smith and J.E. Rivier), p.523, ESCOM, Leiden.
41. Fmoc Solid Phase Peptide Synthesis (Ed. W.C. Chan and P.O. White) in The Practical Approaches Series (Ed. B.D. Hames), University Press, Oxford, p. 63.
42. Hoffmann, R., Vasko M. and Otvos, L., Jr. 1997. Serum stability of phosphopeptides. Anal. Chim.Acta352: 319-325.
43. Ryge, T.S. and Hansen, P.R. (2006) Potent antibacterial lysine-peptoid hybrids identified from a positional scanning combinatorial library. Bioorg. Med. Chem. 14: 4444-4451.
44. Park, Y., Lee, D.G., Jang, S.H., Woo, E.R., Jeong, H. G., Choi, C.H., and Hahm, V.S. (2003). A Leu-Lys-rich antimicrobial peptide: activity and mechanism. Biochim. Biophys. Acta 1645: 172-182.
| название | год | авторы | номер документа |
|---|---|---|---|
| АНТИМИКРОБНЫЕ ПЕПТИДЫ | 2013 |
|
RU2660351C2 |
| ПЕПТИДЫ, ВЫДЕЛЕННЫЕ ИЗ ЧЕЛОВЕЧЕСКОГО ЛАКТОФЕРРИНА, И ИХ ПРИМЕНЕНИЕ | 2012 |
|
RU2593757C2 |
| АНТИБАКТЕРИАЛЬНЫЕ ПЕПТИДЫ | 2007 |
|
RU2468033C2 |
| ПЕПТИДНЫЕ АНТАГОНИСТЫ ПЕПТИДНЫХ ГОРМОНОВ ИЗ СЕМЕЙСТВА КАЛЬЦИТОНИНА (КАЛЬЦИТОНИН ГЕН-РОДСТВЕННЫХ ПЕПТИДОВ (CGRP)) И ИХ ПРИМЕНЕНИЕ | 2013 |
|
RU2624016C2 |
| НОВЫЕ СИНТЕТИЧЕСКИЕ ПЕПТИДЫ И ИХ ПРИМЕНЕНИЕ | 2012 |
|
RU2596393C2 |
| ВАКЦИННАЯ КОМПОЗИЦИЯ | 2001 |
|
RU2328305C2 |
| ПРОТИВОВОЗРАСТНЫЕ И РАНОЗАЖИВЛЯЮЩИЕ СОСТАВЫ | 2002 |
|
RU2318873C2 |
| ИНГИБИТОРЫ АПОПТОЗА И ИХ ПРИМЕНЕНИЕ | 2011 |
|
RU2582247C2 |
| АГОНИСТЫ АПОЛИПОПРОТЕИНА А-1 И ИХ ПРИМЕНЕНИЕ ДЛЯ ЛЕЧЕНИЯ ЗАБОЛЕВАНИЙ, СВЯЗАННЫХ С ДИСЛИПИДЕМИЕЙ | 1998 |
|
RU2219941C2 |
| КОНЪЮГАТЫ ИНСУЛИН-ИНКРЕТИН | 2014 |
|
RU2678134C2 |


































































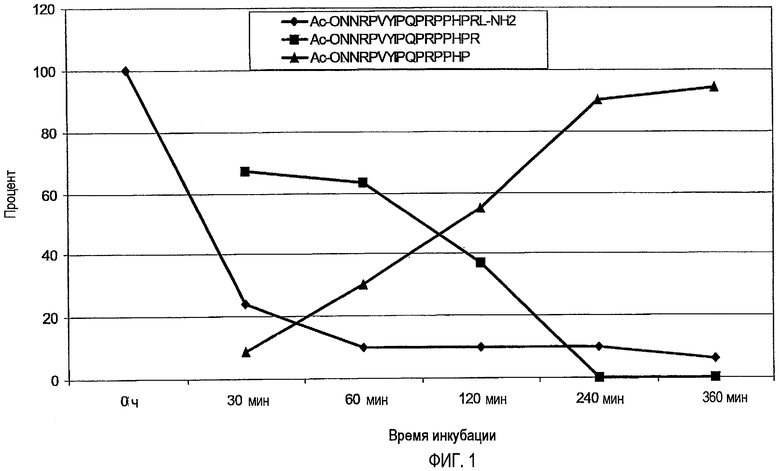

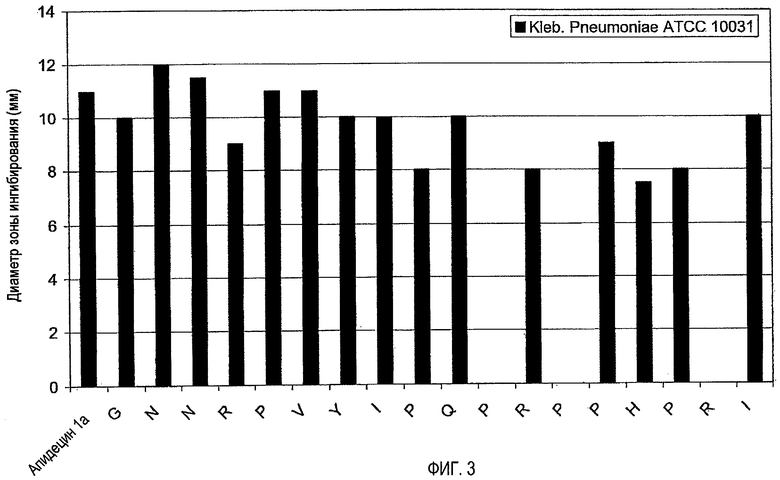
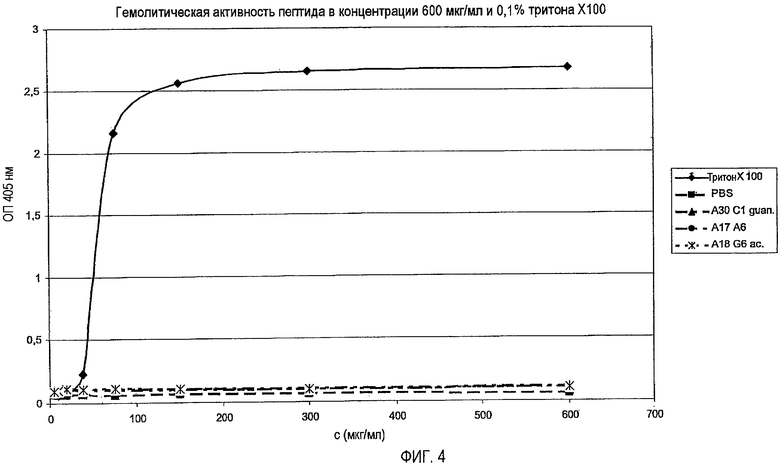
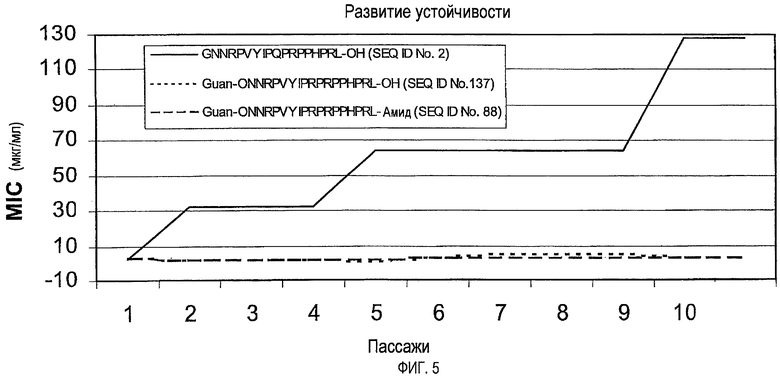
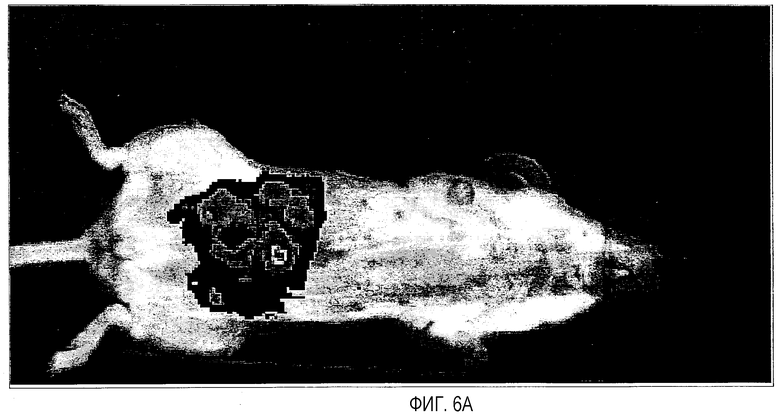
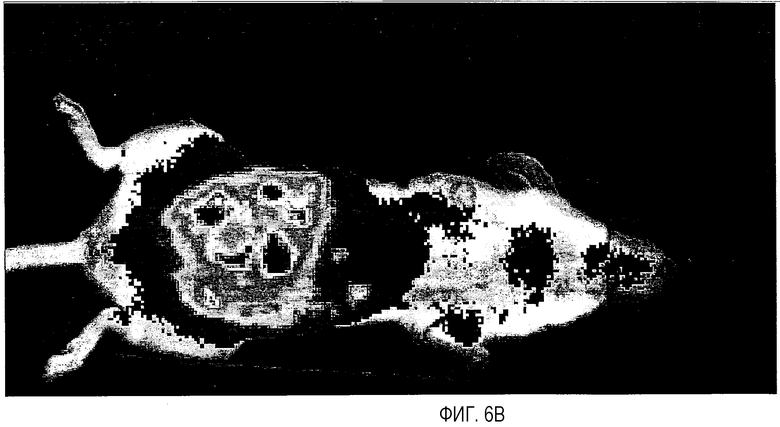
Изобретение относится к новому антибиотическому пептиду и производным пептидов, в частности, для применения в медицине. Дополнительно, изобретение относится к композициям и к способу скринингового анализа лекарственных средств. Пептиды и производные пептида имеют общую формулу Sub1-X1NX2X3PVYIPX4X5RPPHP-Sub2, где Sub1, X1, X2, X3, X5, Sub2 указаны в формуле изобретения. Пептиды или производные пептидов по изобретению обладают по меньшей мере одним из следующих преимуществ по сравнению с пептидами апидецина, встречающимися в природе: (i) увеличенное время полужизни в сыворотке млекопитающих вследствие более высокой протеазной устойчивости, (ii) повышенная противомикробная активность по отношению к одному или нескольким бактериальным штаммам, в частности к патогенам человека, или грибам, или другим микробным инфекциям, (iii) демонстрируют расширенный спектр противомикробной активности, (iv) вызывают меньшее развитие устойчивости у микроорганизмов и (v) не являются токсичными для клеток человека, включая эритроциты. 5 н. и 16 з.п. ф-лы, 7 табл, 8 ил., 6 пр.
1. Пептид или производное пептида с по меньшей мере 16 аминокислотными остатками и общей формулой
Sub1-X1NX2X3PVYIPX4X5RPPHP-Sub2,
где X1 представляет собой положительно заряженный остаток;
X2 представляет собой остаток с полярной боковой цепью или фрагмент с суммарным положительным зарядом или положительно заряженной боковой цепью в физиологических условиях;
X3 представляет собой фрагмент с суммарным положительным зарядом или положительно заряженной боковой цепью в физиологических условиях;
X4 представляет собой нейтральный остаток с полярной боковой цепью, предпочтительно не глутамин, или фрагмент с суммарным положительным зарядом или положительно заряженной боковой цепью в физиологических условиях;
X5 представляет собой пролин или производное пролина;
Sub1 является модификацией N-концевой аминогруппы, где модификация представляет собой гуанидирование;
Sub2 является свободной C-концевой карбоксильной группой C-концевой аминокислоты или модификацией C-концевой карбоксильной группы;
за исключением природных последовательностей SEQ ID NO: 1 (GNNRPVYIPQPRPPHPRI-OH) и SEQ ID NO: 2 (GNNRPVYIPQPRPPHPRL-OH).
2. Пептид или производное пептида по п.1, включающие по меньшей мере один дополнительный остаток X6 и/или X7 в Sub2, где X6 выбран из пролина или производного пролина или фрагмента, имеющего в физиологических условиях суммарный положительный заряд или положительно заряженную боковую цепь, а X7 выбран из пролина или производных пролина, полярного фрагмента или гидрофобного фрагмента.
3. Пептид или производное пептида по п.1 или 2, где остаток X1 выбран из группы, состоящей из аргинина, лизина, δ-гидроксилизина, гомоаргинина, D-аргинина, метиларгинина, нитроаргинина, N(G)-нитроаргинина, нитрозоаргинина, N(G)-нитрозоаргинина, аргинала, гуанидинопропионовой кислоты, 2,4-диаминомасляной кислоты, β-гомоаргинина, ε-N-метиллизина, алло-гидроксилизина, 2,3-диаминопропионовой кислоты, 2,2'-диаминопимелиновой кислоты, орнитина, симметричного диметиларгинина, асимметричного диметиларгинина, 2,6-диаминогексановой кислоты, гистидина, 1-метилгистидина, 3-метилгистидина и 3-аминотирозина.
4. Пептид или производное пептида по п.1 или 2, в котором остаток X2 выбран из группы, состоящей из аспарагина, глутамина, серина, треонина, цис-4-гидроксипролина, транс-4-гидроксипролина, цис-3-гидроксипролина, транс-3-гидроксипролина, цитруллина, N-метилсерина, N-метилглицина, дигидроксифенилаланина, N-этиласпарагина, N-этилглицина, гомосерина, пеницилламина, тетрагидропиранилглицина, алло-треонина, 3,5-динитротирозина, аргинина, лизина, δ-гидроксилизина, гомоаргинина, 2,4-диаминомасляной кислоты, β-гомоаргинина, D-аргинина, метиларгинина, альфа-N-метиларгинина, нитроаргинина, N(G)-нитроаргинина, нитрозоаргинина, N(G)-нитрозоаргинина, аргинала, гуанидинопропионовой кислоты, ε-N-метиллизина, алло-гидроксилизина, 2,3-диаминопропионовой кислоты, 2,2'-диаминопимелиновой кислоты, орнитина, симметричного диметиларгинина, асимметричного диметиларгинина, 2,6-диаминогексановой кислоты, гистидина, 1-метилгистидина, 3-метилгистидина, п-аминобензойной кислоты и 3-аминотирозина.
5. Пептид или производное пептида по п.1 или 2, в котором остаток X3 выбран из группы, состоящей из аргинина, лизина, δ-гидроксилизина, гомоаргинина, 2,4-диаминомасляной кислоты, β-гомоаргинина, ε-N-метиллизина, алло-гидроксилизина, 2,3-диаминопропионовой кислоты, 2,2'-диаминопимелиновой кислоты, лизина, аргинина, орнитина, метиларгинина, симметричного диметиларгинина, асимметричного диметиларгинина, нитроаргинина, нитрозоаргинина, аргинала, гуанидинопропионовой кислоты, 2,6-диаминогексановой кислоты, гистидина, 1-метилгистидина, 3-метилгистидина и 3-аминотирозина.
6. Пептид или производное пептида по п.1 или 2, в котором остаток X4 выбран из группы, состоящей из аспарагина, N-метилсерина, N-метилглицина, дигидроксифенилаланина, N-этиласпарагина, N-этилглицина, гомосерина, пеницилламина, тетрагидропиранилглицина, алло-треонина, 3,5-динитротирозина и цитруллина, а также аргинина, лизина, δ-гидроксилизина, гомоаргинина, 2,4-диаминомасляной кислоты, β-гомоаргинина, ε-N-метиллизина, алло-гидроксилизина, 2,3-диаминопропионовой кислоты, 2,2'-диаминопимелиновой кислоты, орнитина, метиларгинина, симметричного диметиларгинина, асимметричного диметиларгинина, нитроаргинина, нитрозоаргинина, аргинала, гуанидинопропионовой кислоты, 2,6-диаминогексановой кислоты, гистидина, 1-метилгистидина, 3-метилгистидина и 3-аминотирозина.
7. Пептид или производное пептида по п.1 или 2, в котором остаток X5 представляет собой пролин или производное пролина, выбранное из цис-4-гидроксипролина, транс-4-гидроксипролина, цис-3-гидроксипролина, транс-3-гидроксипролина, β-циклогексилаланина, 3,4-цис-метанопролина, 3,4-дегидропролина, гомопролина и псевдопролина.
8. Пептид или производное пептида по п.1 или 2, в котором остаток X6 выбран из группы, состоящей из пролина, цис-4-гидроксипролина, транс-4-гидроксипролина, цис-3-гидроксипролина, транс-3-гидроксипролина, β-циклогексилаланина, 3,4-цис-метанопролина, 3,4-дегидропролина, гомопролина, псевдопролинов, аргинина, предпочтительно D-аргинина, δ-гидроксилизина, гомоаргинина, 2,4-диаминомасляной кислоты, β-гомоаргинина, ε-N-метиллизина, алло-гидроксилизина, 2,3-диаминопропионовой кислоты, 2,2'-диаминопимелиновой кислоты, лизина, орнитина, метиларгинина, симметричного диметиларгинина, асимметричного диметиларгинина, нитроаргинина, нитрозоаргинина, аргинала, гуанидинопропионовой кислоты, 2,6-диаминогексановой кислоты, гистидина, 1-метилгистидина, 3-метилгистидина и 3-аминотирозина.
9. Пептид или производное пептида по п.1 или 2, в котором остаток X7 выбран из группы, состоящей из пролина, цис-4-гидроксипролина, транс-4-гидроксипролина, цис-3-гидроксипролина, транс-3-гидроксипролина, β-циклогексилаланина, 3,4-цис-метанопролина, 3,4-дегидропролина, гомопролина, псевдопролинов, серина, треонина, цитруллина, N-метилсерина, N-метилглицина, дигидроксифенилаланина, N-этиласпарагина, N-этилглицина, гомосерина, пеницилламина, тетрагидропиранилглицина, аллотреонина, 3,5-динитротирозина, δ-гидроксилизина, фенилаланина, лейцина, изолейцина, валина, метионина, аланина, 1-аминоциклогексилкарбоновой кислоты, N-метиллейцина, N-метилизолейцина, трет-бутилглицина, β-аланина, норлейцина, норвалина, N-метилвалина, 6-аминокапроновой кислоты, 2-аминогептановой кислоты, 2-аминоизомасляной кислоты, 3-аминоизомасляной кислоты, аминовалериановой кислоты, 2-аминопимелиновой кислоты, пипеколиновой кислоты, триптофана, иодтирозина, 3,5-дииодтирозина, 3,5-дибромтирозина, β-циклогексилаланина, п-аминобензойной кислоты, ε-аминокапроновой кислоты, 3,4-цис-метанопролина, фенилглицина, 3,4-дегидропролина, 4-амино-5-циклогексил-3-гидроксипентановой кислоты, O-фосфотирозина, O-сульфотирозина, аминоэтилпирролкарбоновой кислоты, 4-аминопиперидин-4-карбоновой кислоты, α-аминоадипиновой кислоты, гомопролина, гомофенилаланина, п-фторфенилаланина, 3,4-дихлорофенилаланина, п-бромфенилаланина, п-иодфенилаланина, п-нитрофенилаланина, короткой пептидной последовательности и разветвленного линкера, содержащего несколько пептидных единиц.
10. Пептид или производное пептида по п.1 или 2, выбранные из группы, состоящей из последовательностей SEQ ID NO: 88, 120, 125, 128, 134, 137, 146, 147, 151, 155, 156, 157 и 158.
11. Пептид или производное пептида по п.1 или 2, в котором по меньшей мере одна из пептидных связей в пептидном остове замещена на нерасщепляемую связь.
12. Пептид или производное пептида по п.11, в котором связь между X6-X7 является нерасщепляемой связью.
13. Пептид или производное пептида по п.1 или 2, в котором нерасщепляемая связь выбрана из группы, состоящей из восстановленной амидной связи, алкилированной амидной связи или тиоамидной связи.
14. Пептид или производное пептида по п.1 или 2, являющиеся слитыми с белком или соединенные с полимером.
15. Пептид или производное пептида по п.1 или 2, где указанный пептид присоединен к носителю.
16. Мультимер, в котором по меньшей мере два пептида или пептидных производных соединены вместе, где по меньшей мере один из пептидов или пептидных производных является пептидом или пептидным производным по любому из пп.1-15.
17. Фармацевтическая композиция для лечения микробной, бактериальной или грибковой инфекции у млекопитающего, содержащая в терапевтически эффективном количестве по меньшей мере один из пептидов или пептидных производных или мультимер по любому из пп.1-16.
18. Пептид или производное пептида по п.1 или 2, или мультимер по п.16, или фармацевтическая композиция по п.17 для применения для лечения микробной, бактериальной или грибковой инфекции у млекопитающего.
19. Применение пептида или производного пептида или мультимера по любому из пп.1-16 в способе скрининга.
20. Способ идентификации соединения, которое обладает потенциальным противомикробным, бактерицидным или противогрибковым действием, включающий:
(i) проведение конкурентного анализа:
(a) с микроорганизмом, чувствительным к пептиду или пептидному производному или мультимеру по любому из пп.1-16;
(b) с пептидом или пептидным производным или мультимером по одному из пп.1-16; и
(c) по меньшей мере с одним исследуемым соединением;
посредством воздействия (b) и (c) на (a); и
(ii) выбор исследуемого соединения, которое конкурентно замещает связанный с микроорганизмом пептид или производное пептида или мультимер.
21. Способ по п.20, где микроорганизм является видом, который принадлежит к одному из родов, выбранным из Escherichia coli, Enterobacter cloacae, Erwinia amylovora, Klebsiella pneumoniae, Morganella morganii, Salmonella typhimurium, Salmonella typhi, Shigella dysenteriae, Yersinia enterocolitica, Acinetobacter calcoaceticus, Agrobacterium tumefaciens, Francisella tularensis, Legionella pneumophila, Pseudomonas syringae, Rhizobium meliloti и Haemophilus influenzae.
| Устройство для указания и автоматического регулирования уровня жидкости | 1938 |
|
SU78956A1 |
| US 5300629 A, 05.04.1994 | |||
| WO 9523513 A1, 08.09.1995 | |||
| Способ обработки глины для употребления в необожженом виде в качестве строительного материала | 1925 |
|
SU2084A1 |
| АППАРАТ ДЛЯ РАЗРЕЗАНИЯ ПЕРЕДВИГАЮЩЕЙСЯ НА ПОДВИЖНОМ СТОЛЕ ТОРФЯНОЙ ПОЛОСЫ ПРИ ПОМОЩИ НОЖЕЙ, УКРЕПЛЕННЫХ НА ПОВЕРХНОСТИ БАРАБАНА | 1922 |
|
SU602A1 |
| KNAPPE D ET AL: "Chemical modifications of short antimicrobial peptides from insects and vertebrates to fight multi-drug resistant | |||

