Настоящее изобретение относится к новым 3(2H)-пиридазиноновым производным и их фармацевтически приемлемым солям, обладающим бронхорасширяющей активностью, противоаллергической активностью и/или антитромботической активностью.
1) Бронхорасширяющие средства
При лечении хронических обратимых обструктивных респираторных заболеваний, таких как бронхиальная астма, бронхит и респираторный дистресс-синдром у взрослых, во время приступов очень важно обеспечить восстановление дыхания. Для этих целей используют бронхорасширяющие средства. Большинство бронхорасширяющих средств, используемых в настоящее время в клинических целях, может быть, в общих чертах, классифицировано как β-стимуляторы, например, сальбутамил; и ксантиновые лекарственные средства, представленные теофиллином. β-Стимуляторы имеют тот недостаток, что при длительном применении, в случае трудноизлечимых заболеваний, эффективность их действия снижается, а при лечении бронхиальной астмы, требующем продолжительного введения, часто наблюдается даже ухудшение симптомов (The New England Journal of Medicine, vol. 321, p. 1517-1527, 1989).
С другой стороны, теофиллиновые лекарственные средства имеют ограниченное применение из-за узкого диапазона их безвредности.
2) Противоаллергические лекарственные средства
Очевидно, что различные химические in vivo-медиаторы принимают участие в аллергических реакциях немедленного типа, таких как бронхиальная астма, аллергический ринит, крапивница и сенная лихорадка. Одним из наиболее важных медиаторов является гистамин, а поэтому уже с давнего времени антигистаминные агенты используются в качестве противоаллергических средств. Однако многие противоаллергические средства антигистаминного типа обладают неблагоприятным побочным действием на центральную нервную систему, таким как сонливость. Для лечения астмы желательно как с терапевтической, так и с экономической точки зрения использовать такое лекарственное средство, которое обладало бы не только противоаллергической, но также и значительной бронхорасширяющей активностью, однако, лекарственное средство, обладающее такими функциями, пока еще не разработано.
3) Антитромботические агенты
Известно, что тромбоциты играют важную роль в процессе тромбообразования, связанного с определенным патологическим состоянием и опосредованного активацией тромбоцитов путем их стимуляции, адгезии к стенкам сосудов и агрегации. Тромбообразование вызывает различные тромботические заболевания, главными из которых являются, например, церебральный тромбоз, тромбоз легких, инфаркт миокарда, стенокардия и окклюзия периферической артерии; и для лечения всех этих заболеваний необходимо разработать нужные лекарственные средства. При разработке профилактического или терапевтического средства особое внимание было уделено антитромботическому агенту, обладающему активностью, направленной на ингибирование агрегации тромбоцитов. Так, например, было широко исследовано действие аспирина, после чего были клинически разработаны тиклопедин и цилостазол. Однако было бы крайне желательно получить более сильнодействующее средство по сравнению с указанными лекарственными средствами.
Помимо вышеупомянутых тромботических заболеваний с тромбоцитами связаны и другие различные заболевания. Примерами таких заболеваний являются нефрит, метастазы рака и т.п., и в целях поиска профилактических или терапевтических средств для лечения этих заболеваний, достигаемого, главным образом, с помощью антитромботического агента, обладающего активностью, регулирующей функцию тромбоцитов, в последнее время были проведены различные исследования ("Journal of Royal College of Physicians", vol. 7, N 1, p. 5-18, 1972; "Japan Clinics (Nihon Rinsho)", vol. 4, N 6, p. 130-136, 1988; Anticancer Research, vol. 6, p. 543-548, 1986).
Ниже будет описана взаимосвязь производных 5-ω-аминоалкиленокси- или ω-аминокарбонилалкиленокси-замещенная бензиламино-3(2H)-пиридазинона формулы (I) и их фармацевтически приемлемых солей настоящего изобретения с соединениями, раскрываемыми в опубликованных материалах.
Тип соединений, в которых замещенная бензиламиногруппа связана с 3(2H)-пиридазиноновым кольцом в положении 5 и которые являются относительно схожими с соединениями настоящего изобретения, раскрывается в нижеследующих работах.
a) В публикации японского патента N 41455/1994, в EP-186817B или в патенте США 5098900 (обозначаемых далее ссылочным материалом (a)) раскрываются соединения, представляющие собой 3(2H)-пиридазиноновые производные, где в положении 2 находится низшая алкильная группа, в положении 4 находится атом хлора или атом брома, в положении 5 находится бензиламиногруппа, имеющая бензольное кольцо, замещенное заместителем, таким как ω-аминоалкильная группа, ω- карбамоилалкиленоксигруппа, ω-N- моно (низшая) алкиламинокарбонилалкиленоксигруппа и аминокарбонильная группа; а также раскрываются фармацевтическое использование этих соединений в качестве анти-MPC-A-агентов (MPC-A-медленно реагирующая субстанция анафилаксии) и их фармакологическая активность.
(b) В публикации нерассмотренной японской заявки 030769/1987, EP201765B или в патенте США 4 892 947 (обозначаемых далее ссылочным материалом (b)) раскрываются соединения, представляющие собой 3(2H)-пиридазиноновые производные, где в положении 2 находится атом водорода, в положении 4 находится атом хлора или атом брома, в положении 5 находится бензиламиногруппа, имеющая бензольное кольцо, замещенное таким заместителем, как алкилоксигруппа, ω-фенилалкиленоксигруппа и диалкиламиногруппа, а в положении 6 находится атом углерода; а также фармацевтическое использование указанных соединений в качестве анти-MPC-A-агентов и их фармацевтическая активность.
c) В публикации нерассмотренной японской заявки N 301870/1988, в EP275997B или в патенте США 4978665 (обозначаемых далее ссылочным материалом (c)) раскрываются соединения, представляющие собой 3(2H)-пиридазиноновые производные, где в положении 2 находится атом водорода или низшая алкильная группа, в положении 4 находится атом хлора или атом брома, в положении 5 находится бензиламиногруппа, имеющая бензольное кольцо, замещенное таким заместителем, как алкилоксигруппа, ω-фенилалкиленоксигруппа и диалкиламиногруппа, а в положении 6 находится атом галогена, нитрогруппа, аминогруппа или алкоксигруппа; а также фармацевтическое использование указанных соединений в качестве анти-MPC-A-агентов и их фармацевтическая активность.
(d) В 091/16314, в EP482208A или в патенте США 5202323 (обозначаемых далее ссылочным материалом (d)) раскрываются соединения, представляющие собой 3(2H)-пиридазиноновые производные, где в положении 2 находится атом водорода или низшая алкильная группа, в положении 4 находится атом хлора или атом брома, в положении 5 находится бензиламиногруппа, имеющая бензольное кольцо, замещенное таким заместителем, как алкилоксигруппа, ω-фенилалкиленоксигруппа, где бензольное кольцо может быть замещено алкильной группой или атомом галогена, ω-алкилоксикарбонилалкиленоксигруппа и ω- аминокарбонилалкиленоксигруппа, а в положении 6 находится алкиленоксигруппа, имеющая какую-либо функциональную группу в ω-положении; а также фармацевтическое использование этих соединений в качестве антитромботических агентов, кардиотонических агентов, сосудорасширяющих факторов и анти-MPC-A-агентов агентов и их фармацевтическая активность.
В результате обширных исследований авторы настоящего изобретения пришли к выводу, что 3(2H)-пиридазиноновые производные, рассматриваемые в данной заявке, и их фармацевтически приемлемые соли, которые отличаются от всех соединений, раскрываемых в вышеуказанных ссылочных материалах (a)-(d), являются ценными сосудорасширяющими, антиаллергическими и/или антитромботическими средствами; причем эти соединения обладают особенно высокой активностью при пероральном введении, что позволяет использовать их в качестве активных ингредиентов в профилактических или терапевтических препаратах для лечения вышеупомянутых респираторных заболеваний, аллергических реакций немедленного типа и/или тромботических заболеваний. Этот вывод был положен в основу настоящего изобретения.
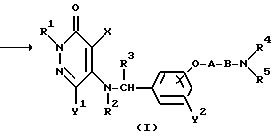
Таким образом, настоящее изобретение относится к 3(2H)-пиридазиноновым соединениям формулы (I) и их фармацевтически приемлемым солям; к способу получения этих соединений; и к фармацевтической композиции, содержащей указанные соединения в качестве активного ингредиента: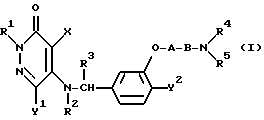
в которой R1 является атомом водорода или C1-4- алкилом; каждый из радикалов R2 и R3 является атомом водорода; X является атомом хлора или брома; Y1 является атомом водорода, атомом галогена, нитрогруппой или C1-4- алкокси; Y2 является C1-4- алкокси; A является C1-5 алкиленовой цепью, которая может быть замещена гидроксильной группой; B является карбонильной группой или метиленовой цепью, которая может быть замещена C1-4- алкилом, и (a) R4 является атомом водорода, R5 является группой Z-Ar, в которой Z является C1-5 алкиленовой цепью, а Ar является пиридилом, или (b) R4 и R5 образуют вместе с близлежащим атомом азота 4-замещенное пиперазиновое кольцо формулы: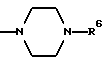
в которой R6 является C1-4- алкилом, причем алкил может быть замещен фенилом, который может быть замещен Y3, где Y3 является атомом галогена, амино, N-формиламино или C1-4- алкилкарбониламино, пиридилом, хинолилом или группой формулы: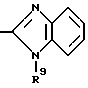
в которой R9 является C1-4- алкилом или бензилом, который может быть замещен атомом галогена, или (c) R4 и R5 образуют вместе с соседним атомом азота 4-замещенное пиперидиновое кольцо формулы: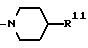
в которой R11 является C1-4- алкилом, который может быть замещен фенилом, и их фармацевтически приемлемые кислотно-аддитивные соли.
Ниже будут описаны более подробно радикалы R1, R2, R3, R4, R5, A, B, X, Y1 и Y2 в соединении формулы (I) согласно настоящему изобретению. Конкретные примеры радикала R1 включают водород, метил, этил, н-пропил, изо-пропил, н-бутил, изо-бутил, втор-бутил и трет-бутил. Предпочтительным значением для R1 является атом водорода. Каждый из радикалов R2 и R3 является атомом водорода.
X может быть атомом хлора или атомом брома.
Y1 может быть, например, атомом водорода, атомом хлора, атомом брома, атомом иода, группой нитро, метокси, этокси, н-пропокси, изо-пропокси, н-бутокси, изо-бутокси, втор-бутокси или трет-бутокси.
Y2 может быть, например, группой метокси, этокси, н-пропокси, изо-пропокси, н-бутокси, изо-бутокси, втор-бутокси и трет-бутокси.
A является алкиленовой цепью с общим числом атомов углерода от 1 до 5. Эта цепь может быть замещена гидроксильной группой и может являться, например, разновидностью связи, такой как метилен, этилен, пропилен, бутилен или пентилен. Наиболее предпочтительна линейная алкиленовая группа, содержащая от 1 до 4 атомов углерода.
B может быть карбонильной группой или связывающей метиленовой цепью.
R4 и R5 могут иметь следующие значения:
(a) R4 является атомом водорода, a R5 является группой формулы Z-Ar, в которой Z является C1-5 алкиленовой цепью, а Ar является группой пиридила,
(b) R4 и R5 образуют вместе с соседним атомом азота, с которым они связаны, 4-замещенное пиперазиновое кольцо формулы: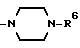
в которой R6 является C1-5- алкилом, который может быть замещен фенилом, который, в свою очередь, может быть замещен Y3, причем Y3 является атомом галогена, группой амино, N-формиламино или C1-4- алкилкарбониламино; пиридилом, хинолилом или группой формулы: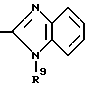
в которой R9 является C1-4- алкилом или бензилом, который может быть замещен атомом галогена.
Конкретно, радикал R6 может быть бензилом, содержащим атом галогена, замещенным в любой произвольной позиции орто-, мета- или пара-позиции бензольного кольца, пиридилметилом, хинолилметилом и бензимидазолилметилом, содержащим группу бензила, которая может быть замещена атомом галогена в бензольном кольце или же замещена C1-4- алкилом в N-позиции.
(c) Радикалы R4 и R5 образуют вместе с соседним атомом азота 4-замещенное пиперидиновое кольцо формулы: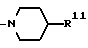
в которой R11 является C1-4- алкилом, в котором алкильная группа может быть замещена фенильной группой.
Конкретные примеры, иллюстрирующие R11, включают бензил.
Предпочтительные примеры, иллюстрирующие радикалы R4 и R5, включают вышеуказанные 4-замещенный пиперазин-1-ил и 4-замещенный пиперидин-1-ил.
Нижеследующие соединения могут быть указаны как предпочтительные из числа соединений формулы (I) согласно настоящему изобретению.
Вышеуказанное соединение, в котором R4 и R5 образуют вместе с соседним атомом азота, с которым они связаны, 4-замещенное пиперазиновое кольцо формулы: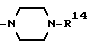
в которой R14 является группой формулы: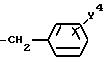
в которой Y4 является атомом водорода, атомом галогена, аминогруппой, N-формиламино или C1-4- алкилкарбониламино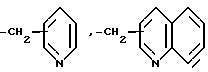
или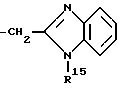
в которых R15 является бензилом, который может быть замещен атомом галогена.
Соединения формулы (I) могут существовать в виде оптических изомеров и стереоизомеров, исходя из наличия 1-5 асимметричных атомов углерода.
Соединения формулы (I) настоящего изобретения могут быть превращены в фармацевтически приемлемые нетоксичные соли с использованием соответствующих кислот, если это необходимо. Соединения формулы (I) могут быть использованы в целях настоящего изобретения либо в свободной форме, либо в форме фармацевтически приемлемых солей. Такими солями могут быть, например, соли минеральных кислот (такие как гидрохлорид, гидробромид, сульфат, бисульфат, нитрат, фосфат, бифосфат или первичный кислый фосфат), соли органических кислот (такие как формиат, ацетат, пропионат, сукцинат, малонат, оксалат, малеат, фумарат, малат, цитрат, тартрат, лактат, глутамат, аспартат, пикрат или карбонат) и соли сульфоновой кислоты (такие как метансульфонат, бензолсульфонат или толуолсульфонат). Эти соли могут быть получены соответствующими стандартными методами.
В табл. I приводятся типичные примеры 3(2H)-пиридазинонового производного формулы (I) настоящего изобретения и его фармацевтически приемлемой соли. Однако при этом следует отметить, что настоящее изобретение не ограничивается указанными конкретными примерами.
В табл. I n означает "нормальный", i означает изо, t означает "третичный", Me означает метильную группу, Et означает этильную группу, Pr означает пропильную группу, Bu означает бутильную группу, a Ph означает фенильную группу. В табл.I Q - Q42 представляют собой группы, имеющие формулы, представленные в конце описания.
Ниже описаны методы получения соединений настоящего изобретения.
3(2H) - пиридазиноновые производные формулы (I) настоящего изобретения и их фармацевтически приемлемые соли могут быть получены методами, проиллюстрированными нижеследующими реакционными схемами (1) - (7).
Реакционная схема (1)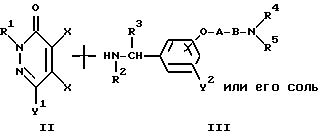
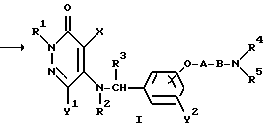
где R1, R2, R3, R4, R5, X, Y1, Y2 A и B являются такими, как они была определены выше.
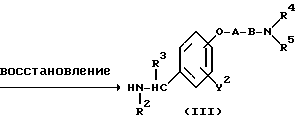
Метод получения в соответствии с реакционной схемой 1) предусматривает реакцию 4,5-дигалогено-3(2H)-пиридазинонового соединения формулы (II) и ω-аминоалкиленокси- или ω-аминокарбонилалкиленокси-замещенного бензиламинового производного формулы (III) или его соли необязательно в присутствии дегидрогалогенирующего агента в инертном растворителе с получением соединения формулы (I) настоящего изобретения.
В вышеуказанной реакционной схеме (I) изомер положения соединения формулы (I), т.е. соединение формулы (IV), имеющее оксибензиламиногруппу, замещенную в 4-положении: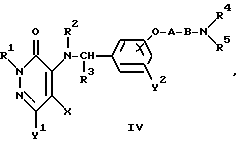
где R1, R2, R3, R4, R5, X, Y1, Y2 A и B являются такими, как они были определены выше, образуется как побочный продукт. Соотношение продуцируемых соединений формул (I) и (IV) зависит, главным образом, от полярности используемого растворителя.
Так, например, если используемый растворитель имеет высокую степень полярности, то содержание соединения формулы (I) настоящего изобретения будет более высоким. Поэтому в качестве растворителя, подходящего для эффективного продуцирования соединения формулы (I) настоящего изобретения, который в то же время будет способствовать подавлению побочной реакции, приводящей к образованию соединения формулы (IV), может быть использован эфирный растворитель (такой, как тетрагидрофуран или 1,4-диоксан), амидный растворитель (такой, как формамид, N,N-диметилформамид, N,N-диметилацетамид, или N-метилпирролидон), ацетонитрил, диметилсульфоксид, спиртовой растворитель (такой, как метанол, этанол или пропанол), органический аминовый растворитель (такой, как пиридин, триэтиламин, N,N-диметиламиноэтанол или триэтаноламин), или вода, либо смеси указанных растворителей. Для выделения и очистки соединения формулы (I) настоящего изобретения из вышеуказанной смеси соединения формулы (I) и соединения формулы (IV) могут быть использованы известные по существу методы органического синтеза, например, такие, как фракционированная перекристаллизация, или различных типов хроматография на силикагеле.
В процессе реакции между соединением формулы (II) и соединением формулы (III) образуется хлороводород или бромоводород. Для увеличения выхода к реакционной системе может быть добавлен дегидрогалогенирующий агент, который захватывает указанный галогеноводород.
Для этих целей может быть использован любой дегидрогалогенирующий агент, если только он не оказывает неблагоприятного действия на реакцию и обладает способностью захватывать галогеноводород. В качестве такого дегидрогалогенирующего агента может быть использовано неорганическое основание, такое, как карбонат калия, карбонат натрия, бикарбонат калия, или бикарбонат натрия; либо органическое основание, такое, как N,N-диметиланилин; N,N-диэтиланилин, триметиламин, триэтиламин, N,N-диметиламиноэтанол, N-метилморфолин, пиридин, или 2,6-диметил-4-N,N-диметиламинопиридин.
Альтернативно, в качестве дегидрогалогенирующего агента может быть использовано избыточное количество исходного бензиламинового производного формулы (III). В результате этого во многих случаях может быть увеличен выход нужного соединения.
Реакционная температура может составлять, в основном, от 10oC до точки кипения растворителя, используемого в реакции.
Молярное отношение исходных соединений может быть, но необязательно, установлено заранее. Однако, в основном, бензиламиновое производное формулы (III) или его соль могут быть использованы в количестве от 1 до 10 М, а предпочтительно от 1,2 до 5 М на 1 М 4,5-дигалогено-3(2H)-пиридазинового производного формулы (II).
4,5-дигалогено-3(2H)-пиридазиноновое производное формулы (II) может быть получено с использованием стандартной органической реакции или с помощью нижеописанного традиционного метода получения соединения такого типа. Так, например, соединение, в котором заместитель Y1 в 6-положении является атомом водорода, может быть получено методом, описанным в работах (a) и (b); а соединение, в котором заместитель Y1 является атомом галогена, нитрогруппой, аминогруппой, или алкоксигруппой, может быть получено методом, описанным в работах (c).
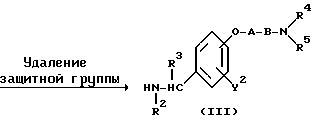
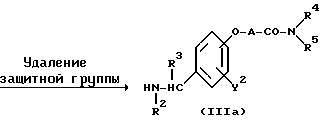
ω- Аминоалкиленокси- или ω- аминокарбонилалкиленокси-замещенное бензиламиновое производное формулы (III) или его соль могут быть получены, например, в соответствии с реакционными схемами (A) - (E) с использованием методов, описанных в работах (a).
(a). Схема (A)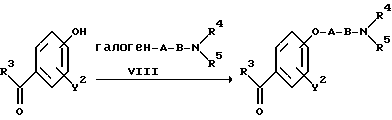
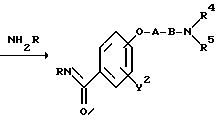
где "hal" представляет собой уходящую группу, такую, как атом хлора, атом брома, атом йода, метансульфонилоксигруппа, или п-толуолсульфонилоксигруппа, R представляет собой атом галогена, гидроксильную группу, C1-4-алкильную группу, или C1-4-алкоксигруппу, а R2, R3, R4, R5, Y2, A и B являются такими, как они были определены выше.
Схема (B)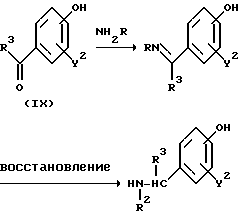
где T представляет собой аминозащитную группу, такую, как бензилоксикарбонильная группа, трет-бутоксикарбонильная группа, формильная группа, ацетильная группа, бензоильная группа, метоксикарбонильная группа или этоксикарбонильная группа; a R2, R3, R4, R5, Y2, A, B, R и "hal" являются такими, как они были определены выше.
Схема (C)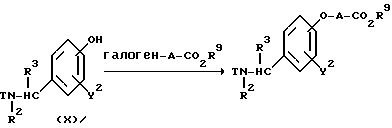
где R9 представляет собой атом водорода или низшую алькильную группу, а R2, R3, R4, R5, Y2, A, T и "hal" являются такими, как они были определены выше.
Схема (D)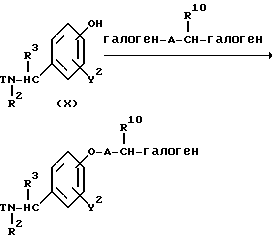
где R10 представляет собой атом водорода или C1-4-алкильную группу; "hal" представляет собой уходящую группу, определенную выше для реакционной схемы (A) в том же самом объеме, за исключением того, что, в данном случае присутствует заместитель, имеющий аналогичную или более низкую способность к отщеплению, чем "hal" в указанной ранее комбинации; a R2, R3, R4, R5, Y2, A, T и "hal" являются такими, как они были определены выше.
Схема (E)
Scheme (E)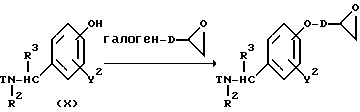
где D представляет собой C1-4-алкиленовую группу, а R2, R3, R4, R5, Y2 и "hal" являются таким, как они были определены выше.
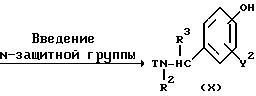
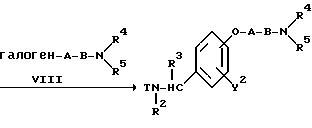
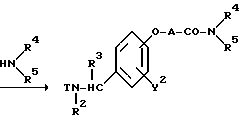
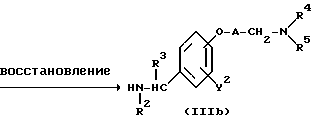
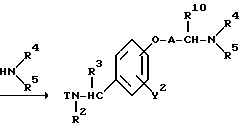
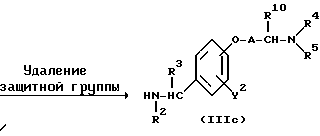
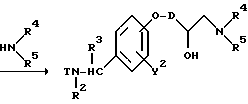
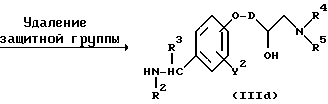
Реакционная схема (A) иллюстрирует метод, в котором в качестве исходного соединения используется гидроксикарбонильное производное (IX), и в котором, сначала, соединение формулы (VIII) реагирует с фенольным центром с введением соответствующей боковой алкоксицепи, а затем карбонильную группу превращают в аминогруппу с помощью реакции восстановления. Реакционная схема (B) иллюстрирует метод, в котором реакции были осуществлены в другом порядке по сравнению с реакционной схемой (A). Реакционная схема (C) иллюстрирует метод, в котором в качестве исходного соединения использовали N-защищенное гидроксибензиламиновое производное формулы (X), являющееся промежуточным соединением в методе, проиллюстрированном реакционной схемой (B), и в котором боковую цепь фенольной части этого соединения постадийно удлиняют, и из ω-аминокарбонилалкиленоксибензиламинового производного формулы (IlIa) получают его восстановленный продукт формулы (Illb), имеющий восстановленный участок амидной связи соединения формулы (IlIa). Реакционная схема (D) иллюстрирует метод получения омега-аминоалкиленоксибензиламинового производного формулы (Illc), т.е., бензиламинового производного формулы (III), содержащего разветвленную метиленовую цепь, где B замещен низшей алкильной группой. Реакционная схема (E) иллюстрирует метод получения соединения формулы (IIId), т.е., бензиламинового производного формулы (III), в котором A представляет собой метиленовую цепь, имеющую гидроксильную группу.
Используя легко доступные и изготавливаемые промышленностью исходные продукты, или исходные продукты, полученные из готовых продуктов, можно из вышеуказанных методов (A) - (E) легко выбрать любой подходящий метод для использования.
Для реакции гидроксикарбонильного производного (IX) с соединением (VIII) в схеме (A) используют условия, которые обычно широко используются для алкилирования фенолов. Эта реакция обычно протекает относительно быстро при использовании неорганического основания, такого, как карбонат натрия, карбонат калия, гидроксид натрия, гидроксид калия, бикарбонат натрия, или бикарбонат калия в кетоновом растворителе (таком, как ацетон, метилэтилкетон, или диэтилкетон), амидном растворителе (таком, как формамид, N,N-диметилформамид, N,N-диметилацетамид или N-метилпирролидон), спиртовом растворителе (таком, как метанол, этанол, или н-пропанол), или в воде, либо в смеси указанных растворителей, при нагревании до температуры от 40 до 150oC.
Последующая реакция для превращения карбонильной группы (формильной группы или кетоновой группы) в аминометильную группу может быть осуществлена посредством реакции конденсации, которой подвергают амин формулы RNH2, с получением имино-соединения, которое, в свою очередь, подвергают реакции восстановления. В этом методе имино-соединение может быть образовано в реакционной системе и подвергнуто последующей реакции восстановления без его выделения. В некоторых случаях этот метод может оказаться более предпочтительным с точки зрения увеличения выхода и снижения материальных затрат.
В данном случае получение первичного амина как одного из бензиламиновых производных формулы (III), где R2 является атомом водорода, может быть осуществлено с использованием в качестве RNH2 такого амина, как аммиак, гидроксиламин, или O-алкилгидроксиламин, с последующим восстановлением полученного таким образом имина.
Для такого восстановления широко применяется реакция гидрирования с использованием скелетного никелевого катализатора Ренея, катализатора "палладий-на-угле", или т.п. В случае, если при получении иминового соединения использовали О-алкилгидроксиламин, то данная реакция может быть осуществлена с использованием гидрида металла, такого, как трифтороацетоксиборогидрид натрия [NaBH3(OCOCF3)] или бис-метоксиэтоксиалюмогидрид натрия [NaAlH2(OCH2CH2OCH3)2] (Chemical and Pharmaceutical Bulletin, vol. 26, p. 2897-2898, 1978).
Последний из указанных методов восстановления может иногда оказаться более предпочтительным для получения соединения, которое относится к бензиламиновым производным формулы (III), и которое содержит в Y2 и R4 или R5 атом галогена или бензильную группу, являющуюся относительно нестабильной в условиях восстановления путем гидрирования. Для получения вторичного амина, представляющего собой бензиламиновое производное формулы (III), в котором R2 представляет собой C1-4-алкильную группу, в качестве RNH2 может быть использован соответствующий первичный алкиламин формулы R2NH2, а затем в реакции восстановления иминового производного, полученного посредством указанной реакции конденсации, в качестве восстановителя может быть добавлен не только восстановитель, описанный в вышеуказанном методе получения первичного амина, но также и гораздо более мягкий металлосодержащий восстановитель гидрирования, такой, как борогидрид натрия или цианоборогидрид натрия (NaCNBH3), которые являются наиболее подходящими и широко используемыми восстановителями.
В реакционной схеме (B) бензиламин формулы (III) получают с помощью реакций, осуществляемых в другой последовательности по сравнению с реакциями в реакционной схеме (A).
В соответствии с этим превращение карбонильной группы в аминометильную группу и реакция алкилирования фенольной части могут быть проведены в соответствующих реакционных условиях, описанных для Схемы (A). В этом методе требуется стадия введения защитной группы для атома бензиламиноазота. Для этой цели в качестве защитной группы формулы T могут быть использованы защитные группы широкого ряда, которые обычно используются для аминогрупп в пептидном синтезе, например, такие, как бензилоксикарбонильная группа, т-бутоксикарбонильная группа, формильная группа, ацетильная группа, бензоильная группа, метоксикарбонильная группа и этоксикарбонильная группа. При выборе конкретной группы из указанных различных защитных групп нет каких-либо жестких ограничений. Однако в некоторых случаях правильный выбор защитной группы или условий для ее удаления зависит от типов заместителей Y2, B, R4 и R5. Например, для получения бензиламинового соединения (III), содержащего в Y2 или R4 и R5 атом галогена или бензильную группу, в некоторых случаях необходимо правильно выбрать заместителей и реакционные условия так, чтобы реакция удаления защитной группы могла быть проведена с высокой степенью эффективности и селективности даже при использовании вместо каталитического гидрирования другого метода. Для получения бензиламина формулы (III), где B представляет собой карбонильную цепь, во многих случаях, предпочтительно использовать бензилоксикарбонильную группу или т-бутоксикарбонильную группу, поскольку удаление такой защитной группы может быть более легким в негидролизующих условиях. При этом могут быть использованы стандартные реакционные условия, которые обычно используются для введения и удаления различных защитных групп указанного типа.
Реакционная схема (C) иллюстрирует метод, в котором, используя в качестве исходного соединения гидроксибензиламин формулы (X), защищенный защитной группой T, постадийно наращивают эфирную боковую цепь, в результате чего получают соединение формулы (IlIa), где B представляет собой карбонильную цепь, и соединение формулы (IlIb), где B представляет собой линейную метиленовую цепь, полученную путем восстановления карбонильной части; причем указанные соединения относятся к ряду бензиламинов формулы (III). Для образования амидной связи у эфирной части боковой цепи в случае, если R9 является атомом водорода, могут быть использованы методы конденсации путем дегидратации, которые обычно широко используются для пептидного синтеза. Если используемый амин является относительно богатым по своей нуклеофильной природе, то может быть использован сложный эфир, где R9 представляет собой низшую алкильную группу и в этом случае, обычно, используют условия нагревания в инертном растворителе. Для получения бензиламина формулы (IIIb) в качестве восстановителя может быть использован гидрид металла, такой, как алюмогидрид лития. Реакция алкилирования фенольной части и реакция удаления защитной группы в других стадиях могут быть осуществлены в соответствии с методами, описанными для соответствующих реакций в схемах (A) и (B).
Реакционная схема (D) иллюстрирует метод получения аминоалкиленоксибензиламинового производного формулы (Illc), где α- углерод аминогруппы на конце фенольной боковой цепи представляет собой метиленовую цепь, замещенную линейным или низшим алкилом. Стадия введения радикала аминогруппы может быть осуществлена в стандартных реакционных условиях, которые обычно используются в реакциях замещения алкиламина с алкилгалогенидом.
Реакционная схема (E) предназначена для введения гидроксильной группы в фенольную боковую цепь соединения формулы (IIIb) и иллюстрирует метод, в котором эпоксигруппу вводят в фенольную боковую цепь посредством реакции с различными эпоксиалкилгалогенидными соединениями, а соединение формулы (Illb) получают посредством реакции с различными аминами.
Реакционная схема (2)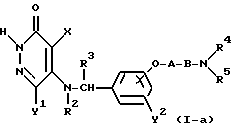
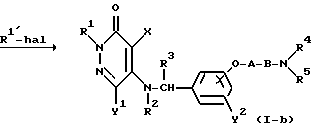
где R1' представляет собой C1-4-алкильную группу, "hal" представляет собой атом хлора, атом брома, или атом йода, а R2, R3, R4, R5, X, Y1, Y2, A и B определены выше.
Реакционная схема (2) иллюстрирует способ получения замещенного во 2-положении пиридазинонового продукта формулы (I-b) как соединения настоящего изобретения посредством реакции соединения формулы (I-a), которое представляет собой соединение настоящего изобретения формулы (I), где во 2-положении пиридазинона находится атом водорода, с галогеновым производным формулы R1'-hal.
Для этой реакции используют неорганическое основание, такое, как карбонат калия, карбонат натрия, карбонат лития, бикарбонат калия, бикарбонат натрия, или гидроксид лития; органическое основание, такое, как триэтиламин или три-н-пропиламин; либо гидрид металла или металлоорганическое соединение, такое, как гидрид натрия или н-бутиллитий.
В случае, если используется неорганическое или органическое основание, то в этой реакции в качестве растворителя может быть использован кетоновый растворитель (такой, как ацетон, метилэтилкетон или диэтилкетон), амидный растворитель (такой, как формамид, N,N-диметилформамид, или N,N-диметилацетамид), спиртовой растворитель (такой, как метанол, или этанол), вода, или смесь указанных растворителей. В случае, если используется гидрид металла, то предпочтительно использовать эфирный растворитель.
В случае, если используется неорганическое или органическое основание, то температура реакции может составлять от 0oC до точки кипения растворителя. В случае, если в данной реакции используется гидрид металла или металлоорганическое соединение, то температура реакции может составлять в пределах от -78oC до 60oC.
Молярное соотношение исходных соединений может быть, но необязательно, заранее установлено. Однако обычно реактивное производное формулы R1' - hal может быть использовано в соотношении, составляющем от 1 до 5 М на 1 М соединения формулы (I-a).
Для выделения и очистки нужного продукта могут быть использованы стандартные методы органического синтеза, такие, как перекристаллизация, громатография различных типов на силикагеле и дистилляция.
Реакционная схема (3)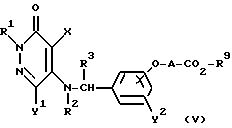
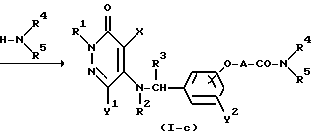
где R1, R2, R3, R4, R5, R9, X, Y1, Y2 и A определены выше.
Реакционная схема (3) иллюстрирует метод, в котором 5- ω-/ карбоксиалкиленокси)бензиламиновое производное или 5-ω-алкоксикарбонилалкиленокси)бензиламиновое производное формулы (V) подвергают вместе с аминовым соединением формулы (VI) реакции конденсации путем дегидратации или деалкоголизации, в результате чего получают амидное производное формулы (I-c).
В случае, если R9 является атомом водорода, то реакция конденсации может быть осуществлена в соответствии с традиционными методами конденсации, широко используемыми в пептидном синтезе. Так, например, данная реакция конденсации может быть проведена методом с использованием хлорангидрида, методом с использованием смешанного ангидрида кислоты, а также методом с использованием конденсирующих агентов, таких, как ди-циклогексилкарбодиимид, карбонилдиимидазол, и N-гидроксисукцинимид; причем подходящий метод конденсации может быть выбран в зависимости от реакционной способности амина формулы (VI). В качестве реакционных условий могут быть использованы обычные для таких реакций условия.
В случае, если в качестве амина формулы (VI) используется амин, являющийся богатым по своей нуклеофильной природе, то реакция конденсации будет протекать даже со сложным эфиром, где R9 является алкильной группой. В этом случае в качестве растворителя может быть использован любой растворитель без каких-либо конкретных ограничений, если только он является инертным по отношению к данной реакции. Во многих случаях эта реакция может быть проведена в отсутствие растворителя. Температура реакции может варьироваться в пределах от комнатной температуры до 200oC, но, в основном, она составляет от 50oC до 150oC.
Реакционная схема (4)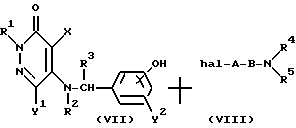
где R1, R2, R3, R4, R5, X, Y1, Y2, A, B и "hal" определены выше.
Реакционная схема (4) иллюстрирует способ получения соединения настоящего изобретения формулы (I) посредством реакции соединения формулы (VII) с галогеновым производным формулы (VIII).
Для этой реакции может быть использовано в основном неорганическое основание, такое, как карбонат калия, карбонат натрия, карбонат лития, бикарбонат натрия, бикарбонат калия, или гидроксид лития; либо органическое основание, такое, как триэтиламин или три-н-пропиламин.
В качестве растворителя для этой реакции может быть использован кетоновый растворитель (такой, как ацетон, метилэтилкетон, или диэтилкетон), амидный растворитель (такой, как формамид, N,N-диметилформамид, или N,N-диметилацетамид) спиртовой растворитель (такой, как метанол или этанол), вода или смесь указанных растворителей.
Реакционная температура может, в основном, составлять от 0oC до точки кипения растворителя.
Реакционная схема (5)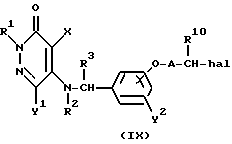
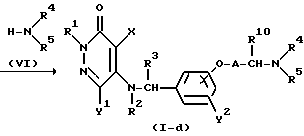
где R1, R2, R3, R4, R5, R10, X, Y1, Y2, A и "hal" являются такими, как они были определены выше, а R7 представляет собой атом водорода или C1-4-алкильную группу.
Реакционная схема (5) иллюстрирует метод получения аминового производного формулы (I-d) в качестве соединения настоящего изобретения посредством реакции соединения формулы (IX), полученного методом, описанным выше для реакционной схемы (4), с аминовым соединением формулы (VI).
Эта реакция может быть осуществлена методом, описанным для реакционной схемы (4).
Реакционная схема (6)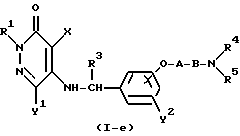
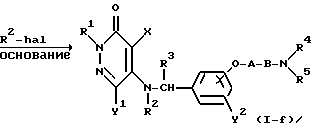
где R2' представляет собой C1-4-алкильную группу, а R1, R2, R3, R4, R5, X, Y1, Y2, A, B и "hal" являются такими, как они были определены выше.
Реакционная формула (6) иллюстрирует метод получения соединения настоящего изобретения, в котором R2 является C1-4 алкильной группой, посредством реакции соединения формулы (I-e) (которое представляет собой соединение настоящего изобретения формулы (I), где R2 является атомом водорода) с алкилгалогенидом формулы R2' - hal в присутствии основания.
Для получения хороших результатов в качестве органического растворителя может быть использован амидный растворитель, такой, как диметилформамид; эфирный растворитель, такой, как тетрагидрофуран или диэтиловый эфир; или апротонный органический растворитель, такой, как н-гексан, бензол, или толуол; а в качестве основания может быть использован гидрид металла, такой, как гидрид натрия; н-бутиллитий, диизопропиламид лития, или амид натрия.
При осуществлении реакции в присутствии основания реакционная температура может составлять в пределах от -78oC до 10oC, а при осуществлении реакции с использованием алкилгидрида реакционная температура может составлять от -15oC до 70oC.
Реакционная схема (7)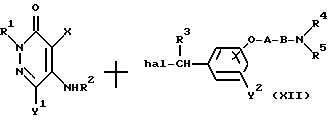
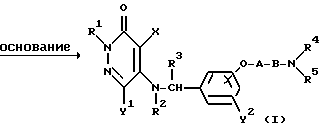
где R1, R2, R3, R4, R5, X, Y1,Y2, A, B и "hal" являются такими, как они были определены выше.
Реакционная схема (7) иллюстрирует метод получения соединения формулы (I) настоящего изобретения посредством реакции 3(2Н)-пиридазинона формулы (XI), содержащего группу - NHR2 в 5-положении, с бензилгалогенидным производным формулы (XII) в присутствии основания.
Реакционные условия для данной реакции могут быть такими же, как и для реакционной схемы (6).
3(2Н)-пиридазиноны настоящего изобретения формулы (I) или их фармацевтически приемлемые соли могут быть введены непероральным способом в виде препаратов для инъекций (подкожных, внутривенных, внутримышечных или внутрибрюшинных), мазей, суппозиториев, или аэрозолей; либо пероральным способом в виде таблеток, капсул, гранул, драже, сиропов, жидких растворов, эмульсий, или суспензий.
Вышеуказанная фармакологическая композиция содержит соединение настоящего изобретения в количестве, составляющем от около 0,1 до 99,5 мас. %, а предпочтительно от около 0,5 до 95 мас.% исходя из полной массы композиции.
С соединением настоящего изобретения или с композицией, содержащей соединение настоящего изобретения, могут быть смешаны другие фармакологически активные соединения.
Соединение настоящего изобретения может быть изготовлено в виде различных препаратов, подходящих для введения, в соответствии со стандартной методикой, обычно используемой для изготовления фармацевтических препаратов.
Так, например, таблетки, капсулы, гранулы или драже для перорального введения могут быть получены с использованием наполнителя, такого, как сахар, лактоза, глюкоза, крахмал, или маннит; связующего вещества, такого, как сироп, аравийская камедь, желатин, сорбит, трагакантовая камедь, метилцеллюлоза, или поливинилпирролидон; дезинтегрирующего агента, такого, как крахмал, карбоксиметилцеллюлоза, или ее кальциевая соль, измельченная в порошок кристаллическая целлюлоза, или полиэтиленгликоль; агента для уменьшения трения, такой, как тальк, стеарат магния или кальция, или кремнезем; или замасливателя, такого, как лаурат натрия, или глицерин.
Инъекции, растворы, эмульсии, суспензии, сиропы, или аэрозоли могут быть получены с использованием растворителя для активного ингредиента, например, такого, как вода, этиловый спирт, изопропиловый спирт, пропиленгликоль, 1,3-бутиленгликоль, или полиэтиленгликоль; поверхностно-активного вещества, такого, как сложный сорбитанэфир жирных кислот, сложный полиоксиэтиленсорбитанэфир жирных кислот, сложный полиоксиэтиленэфир жирных кислот, простой полиоксиэтиленэфир гидрогенизованного касторового масла, или лецитина; суспендирующего агента, такого, как натриевая соль карбоксиметилцеллюлозы, производное целлюлозы, такое, как метилцеллюлоза, или натуральный каучук, такой, как трагакантовая камедь или аварийская камедь; или консерванта, такого, как сложный эфир параоксибензойной кислоты, бензалконийхлорид, или соль сорбиновой кислоты.
Аналогичным образом могут быть изготовлены суппозитории, например, с использованием полиэтиленгликоля, ланолина,или кокосового масла.
Наиболее предпочтительные варианты осуществления изобретения. Примеры (сравнительные примеры, примеры получений соединений, примеры получения композиций и примеры испытаний).
Ниже приводится более подробное описание настоящего изобретения со ссылками на примеры (включая сравнительные примеры, примеры получения соединений, примеры получения композиций и примеры испытаний). Однако при этом следует отметить, что настоящее изобретение не ограничивается этими конкретными примерами. В сравнительных примерах, примерах получений или в табл. II, аббревиатуры "ЯМР" и "МС" означают спектроскопия с использованием метода ядерного магнитного резонанса" и "масс-спектроскопию" соответственно. Если это не оговорено особо, то ЯМР измеряли в дейтерохлороформе.
В МС-данных, в табл. II, приводятся только главные пики или пики характерных фрагментов.
Сравнительный пример 1
N-Бензилоксикарбонил-3-гидрокси-4-метоксибензиламин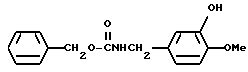
Смесь, содержащую 150 г изованилина, 93,2 г гидроксида натрия, 99 г сульфата гидроксиламина, 600 мл этанола, и 1500 мл воды, нагревали с обратным холодильником при перемешивании в течение 30 минут, а затем охлаждали до температуры 40oC. После добавления 93,2 г гидроксида натрия, к смеси в течение 30 минут добавляли 180 г катализатора Ренея. Полученную смесь перемешивали в течение одного часа. Нерастворившиеся вещества отфильтровывали и промывали 100 мл этанола и 200 мл воды. Фильтрат и промывочные растворы объединяли, а затем добавляли 53,6 г гидроксида натрия. Затем, к смеси по капле добавляли 186 г бензилоксикарбонилхлорида, охлаждая при этом льдом. Эту смесь перемешивали в течение 4 часов. К полученному реакционному раствору добавляли соляную кислоту до тех пор, пока pH не становился равным 1-2, , а затем экстрагировали этилацетатом. Органический слой промывали водой и насыщенным водным раствором хлорида натрия, после чего осушали безводным сульфатом натрия. Затем растворитель отгоняли. Полученный остаток кристаллизовали из диэтилового эфира, и получали 95,11 г N-бензилоксикарбонил-3-гидрокси-4-метоксибензиламина в виде белых кристаллов.
ЯМР: δ 7,34 (с, 5Н), 6,79 (с, 3Н), 5,78 (с, 1Н), 5,12 (шир. с, 2Н), 4,25 (д, 2Н), 3,84 ( с, 3Н).
МС (m/z): 287 (М+), 196, 152, 137, 91(100%).
Сравнительный пример 2
т-Бутилоксикарбонил-3-гидрокси-4-метоксибензиламин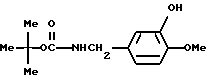
Смесь, содержащую 150 г изованилина, 91 г гидроксида натрия, 89 г сульфата гидроксиламина, 500 мл этанола и 1300 мл воды, нагревали с обратным холодильником в течение 1 часа при перемешивании, а затем охлаждали до температуры 40oC. После добавления 91 г гидроксида натрия к смеси постепенно добавляли 150 г катализатора Ренея при температуре 30-50oC. Полученную смесь перемешивали в течение 1 часа. Нерастворившиеся вещества отфильтровывали и промывали 150 мл этанола и 150 мл воды. Фильтрат и промывочные растворы объединяли и нейтрализовали при охлаждении с использованием концентрированной соляной кислоты до тех пор, пока pH не становился равным 8. После добавления 1 л ацетонитрила к смеси при комнатной температуре в течение одного часа по капле добавляли 215 г ди-т-бутилдикарбоната. Эту смесь перемешивали в течение ночи. Органический слой промывали насыщенным водным раствором хлорида натрия, а затем осушали безводным сульфатом натрия. Затем растворитель отгоняли. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (элюент: этилацетат/бензол, 1:5), и получали 126 г т-бутилоксикарбонил-3-гидрокси-4-метоксибензиламина в виде маслянистого вещества.
ЯМР: δ 6,54-6,85 (м, 3Н), 6,14-6,47 (шир. с, 1Н), 4,92-5,34 (м, 1Н), 4,09 (д, 2Н), 3,25 (с, 3Н), 1,44 (с, 9Н).
МС (m/z) : 153 (М+ - 100), 137 (100%).
Сравнительный пример 3
N-Бензилоксикарбонил-3-этоксикарбонилметилокси-4-метоксибензиламин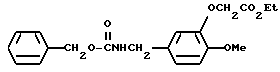
Смесь, содержащую 20 г N-бензилоксикарбонил-3-гидрокси-4-метоксибензиламина, 17,43 г этилбромоацетата, 17,43 г карбоната калия и 200 мл 2-бутанона, нагревали с обратным холодильником, перемешивая при этом, в течение ночи. Затем смесь охлаждали до комнатной температуры. После этого неорганические вещества отфильтровывали, а фильтрат подвергали дистилляции при пониженном давлении. Полученный остаток экстрагировали хлороформом, а затем, органический слой промывали водой и насыщенным водным раствором хлорида натрия, после чего осушали безводным сульфатом натрия. После отгонки растворителя полученный остаток кристаллизовали из диэтилового эфира/ н-гексана, в результате чего получали 17,83 г N-бензилоксикарбонил-3-этоксикарбонилметилокси-4-метоксибензиламина в виде белых кристаллов.
ЯМР: δ 7,33 (с, 5Н), 6,85 (с, 3Н), 5,12 (с, 2Н), 4,63 (с, 2Н), 4,26 (д, 2Н), 4,25 (кв., 2Н), 3,84 (с, 3H), 1,26 (т, 3Н).
МС (m/z): 373 (М+), 282, 239 (100%), 210, 164, 136, 91.
Аналогичным способом были получены соединения, такие
как:
N-Бензилоксикарбонил-3-этоксикарбонилпропокси-4-метоксибензиламин;
ЯМР: δ 7,25-7,55 (м, 5H), 6,72-7,06 (м, 3H), 5,14 (с,2Н), 3,71-4,52 (м, 10Н), 1,90-2,80 (м,4Н), 1,24 (т, 3Н); и
N-Бензилоксикарбонил-3-этоксикарбонилпентилокси-4-метоксибензиламин.
Сравнительный пример 4
N-Бензилоксикарбонил-3-карбоксиметилокси-4-метоксибензиламин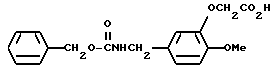
Смесь, содержащую 23,56 г N-бензилоксикарбонил-3-этоксикарбонилметилокси-4-метоксибензиламина, 7,29 г гидроксида натрия, 300 мл метанола, и 30 мл воды, перемешивали в течение одного часа при температуре 60oC. Реакционный раствор нейтрализовали путем добавления соляной кислоты, а растворитель отгоняли при пониженном давлении. К полученному остатку добавляли разбавленную соляную кислоту, и эту смесь экстрагировали хлороформом. Экстрактный слой промывали водой и насыщенным водным раствором хлорида натрия, а затем осушали безводным сульфатом натрия. После этого растворитель отгоняли. Полученный остаток кристаллизовали из диэтилового эфира/н-гексана, и получали 21,55 г N-бензилоксикарбонил-3-карбоксиметилокси-4-метоксибензиламина в виде белых кристаллов.
ЯМР: δ 7,34 (с, 5Н), 6,84 (с, 3Н), 5,13 (с, 3Н), 4,62 (с, 2Н), 4,25 (д, 2Н), 3,83 (с, 3Н).
МС (m/z): 345 (М+), 254, 210 (100%), 91.
Аналогичным способом были получены следующие соединения:
N-Бензилоксикарбонил-3-карбоксипропилокси-4-метоксибензиламин; и
N-Бензилоксикарбонил-3-карбоксипентилокси-4-метоксибензиламин.
Сравнительный пример 5
N-Бензилоксикарбонил-3-(2,З-эпоксипропилокси)-4-метоксибензиламин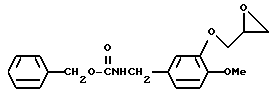
Смесь, содержащую 2 г N-бензилоксикарбонил-3-гидрокси-4-метоксибензиламина, 20 мл диметилформамида, 1,4 г карбоната калия, и 1,4 г эпибромогидрина, перемешивали в течение ночи при температуре 60oC. После отгонки растворителя при пониженном давлении реакционную смесь экстрагировали этилацетатом. Полученный органический слой промывали водным раствором карбоната калия, а затем насыщенным водным раствором хлорида натрия, после чего осушали безводным сульфатом натрия. После отгонки растворителя получали 2,6 г N-бензилоксикарбонил-3-(2,3-эпоксипропилокси)-4-метоксибензиламина в виде маслянистого вещества.
ЯМР: δ 7,32 (с, 5Н), 6,81 (с, 3H), 5,0-5,5 (м, 3Н), 3,9-4,6 (м, 7Н), 3,8 (с, 3Н).
МС (m/z): 343 (М+), 252, 208, 19 (100%).
Сравнительный пример 6
N-Бензилоксикарбонил-3-(4-метилпиперазин-1-ил)карбонилметокси- 4-метоксибензиламин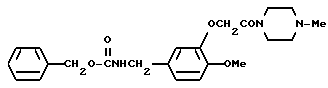
Смесь, содержащую 5 г N-бензилоксикарбонил-3-карбоксиметилокси-4-метоксибензиламина, 1,67 г триэтиламина, и 40 мл тетрагидрофурана, охлаждали льдом, а затем, по капле добавляли 1,79 г этилхлороформата, растворенного в 10 мл тетрагидрофурана. Полученную смесь перемешивали в течение 2 часов. Затем к реакционному раствору добавляли 1,65 г метилпиперазина, растворенного в 10 мл тетрагидрофурана, и эту смесь перемешивали в течение 4,5 часа при комнатной температуре. После отфильтровывания осадка фильтрат подвергали дистилляции при пониженном давлении. К полученному остатку добавляли воду, и эту смесь экстрагировали хлороформом. Раствор экстракта промывали водой и насыщенным водным раствором хлорида натрия, а затем осушали безводным сульфатом натрия. После этого, растворитель отгоняли. Полученный остаток кристаллизовали из этилацетата/диэтилового эфира/н-гексана, и получали 3,53 г идентифицированного выше соединения в виде белых кристаллов.
ЯМР: δ 7,25 (с, 5Н), 6,78 (с, 3Н), 5,03 (с, 3Н), 4,62(с, 2Н), 4,23 (д, 2Н), 3,78 (с, 3Н), 3,40-3,72 (м, 4Н), 2,11-2,60 (м, 7Н).
МС (m/z): 427 (М+), 292, 235, 141, 91 (100%).
Аналогичным способом были получены следующие соединения:
N-Бензилоксикарбонил-3-[4-(3-пиридилметил)-пиперазин-1-ил] карбонилметокси-4-метоксибензиламин
МС (m/z): 504 (М+), 92 (100%).
N-Бензилоксикарбонил-3-(4-бензилпиперазин-1-ил)-карбонилметокси- 4-метоксибензиламин;
ЯМР: δ 7,15-7,43 (м, 10Н), 6,7-6,92 (м, 3Н), 4,85-5,24 (м, 3Н), 4,62 (с, 2Н), 4,22 (д, 2Н), 3,4-3,96 (м, 9Н), 2,25- 2,7 (м, 4Н).
N-Бензилоксикарбонил-3-[4-(4-фторбензил)-пиперазин-1-ил] карбонилметокси-4-метоксибензиламин;
ЯМР: δ 6,60-7,50 (м, 12Н), 5,0-5,5 (м, 3Н), 4,62 (с, 2Н), 4,22 (д, 2Н), 3,22-3,95 (м, 9Н), 2,2-2,7 (м, 4Н).
N-Бензилоксикарбонил-3-4-(3-пиридилметил)-пиперазин-1-ил- карбонилпропокси-4-метоксибензиламин;
МС (m/z): 532 (М+), 92(100%).
N-Бензилоксикарбонил-3-(4-бензилпиперазин-1-ил)-карбонилпропокси- 4-метоксибензиламин;
ЯМР: δ 7,0-7,40 (м, 10Н), 6,60-6,90 (м, 3Н), 5,50-5,51 (м, 3Н), 3,22-4,37 (м, 13Н), 2,0-2,68 (м, 8Н); и
N-Бензилоксикарбонил-3-(4-бензилпиперазин-1-ил)- карбонилпентилокси-4-метоксибензиламин;
ЯМР: δ/ 7,0-7,35 (м, 10Н), 6,60-6,80 (м, 3Н), 5,0-5,50 (м, 3Н), 3,20-4,32 (м, 13Н), 1,1-2,48 (м, 12Н).
Сравнительный пример 7
N-Бензилоксикарбонил-3-[{4-(4-фторобензил)-пиперазин-1-ил] -β- гидроксипропилокси}-4-метоксибензиламин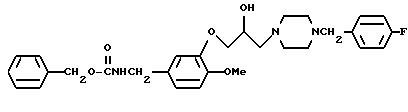
Смесь, содержащую 2,4 г N-бензилоксикарбонил-3-(2,3-эпоксипропилокси)-4-метоксибензиламина, 30 мл этанола, и 1,4 г 4-фторобензил-пиперазина, нагревали с обратным холодильником в течение ночи при перемешивании. Полученную смесь охлаждали до комнатной температуры, а затем реакционный раствор концентрировали при пониженном давлении и экстрагировали хлороформом. Органический слой промывали водным раствором карбоната калия и осушали безводным сульфатом натрия. Затем растворитель отгоняли при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (элюент: этилацетат/метанол=19:1) и получали 2,6 г идентифицированного выше соединения.
ЯМР: δ 6,75-7,42 (м, 12H), 5,0-5,5 (м, 3Н), 4,26 (д, 2Н), 3,82-4,10 (м, 2Н), 3,77 (с, 3Н), 3,20-3,60 (м, 3Н), 2,20-2,85 (м, 10Н).
МС (m/z): 537 (М+), 207 (100%), 109.
Аналогичным способом были получены следующие соединения:
N-Бензилоксикарбонил-3-[{ 4-(2-хинолилметил)-пиперазин-1-ил} -β- гидроксипропилокси]-4-метоксибензиламин;
ЯМР: δ 7,03-8,12 (м, 11Н), 6,60-6,87 (м, 3Н), 5,30-5,70 (м, 1Н), 5,05 (с, 2Н), 3,22-4,37 (м, 11H), 2,22-2,80 (м, 10Н).
N-Бензилоксикарбонил-3-[{ 4-(4-аминобензил)-пиперазин-1-ил} -β- гидроксипропилокси]-4-метоксибензиламин:
ЯМР: δ 6,45-7,41 (м, 12H), 5,40-6,78 (м, 1Н), 5,04 (с, 2Н), 3,50-4,38 (м, 11Н), 3,30 (c, 2H), 2,10-2,80 (м, 8Н).
Сравнительный пример 8
3-(4-Метилпиперазин-1-ил)-карбонилметокси-4-метоксибензиламин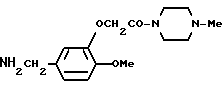
Смесь, содержащую 3,26 г N-бензилоксикарбонил-3-(4-метилпиперазин-1-ил)-карбонилметокси-4 -метоксибензиламина, 0,5 г 5% палладия-на-угле, и 70 мл этанола, перемешивали в течение 6 часов при 60oC в атмосфере водорода, а затем в течение ночи при комнатной температуре. Палладий-на-угле отфильтровывали, а затем фильтрат отгоняли при пониженном давлении, и получали 2,45 г идентифицированного выше соединения в виде слегка коричневатого маслообразного вещества.
ЯМР: δ 6,88 (с, 3Н), 4,74 (с, 2Н), 3,50-4,10 (м, 9Н), 2,29-2,58 (м, 7Н), 1,65 (с,2Н).
МС (m/z): 293 (М+), 152, 299, 70 (100%).
Аналогичным способом были получены следующие соединения:
3-[4-(3-Пиpидилметил)-пипеpaзин-1-ил] карбонилметокси- -4-метоксибензиламин;
МС (m/z): 370 (М+), 92 (100%).
3-(4-Бензилпиперазин-1-ил)-карбонилметокси-4-метоксибензиламин;
МС (m/z): 369 (М+), 91 (100%).
3-[4-(4-фторобензил)-пиперазин-1-ил]карбонилметокси-4-метоксибензиламин;
МС (m/z): 387 (М+), 109 (100%).
3-[4-(3-Пиридилметил)-пиперазин-1-ил] карбонилпропокси-4- метоксибензиламин;
МС (m/z): 398 (М+), 92 (100%).
3-(4-Метилпиперазин-1-ил)-карбонилпропокси-4-метоксибензиламин;
МС (m/z): 321 (М+), 99 (100%).
3-(4-Бензилпиперазин-1-ил)-карбонилпропокси-4-метоксибензиламин;
МС (m/z): 397 (М+), 91 (100%).
3-[4-(4-фторобензил)-пиперазин-1-ил)-1-оксо-2-метилэтилокси] -4-метоксибензиламин;
МС (m/z): 401 (М+), 109 (100%); и
3-(4-Бензилпиперазин-1-ил)-карбонилпентилокси-4-метоксибензиламин;
МС (m/z): 425 (М+), 91 (100%).
Пример получения 1
4-Хлоро-5-[3-(4-метилпиперазин-1-ил)-карбонилметокси-4-метоксибензиламино]-3(2Н)-пиридазинон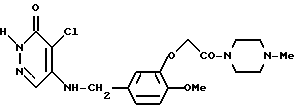
Смесь, содержащую 1,16 г 3-(4-метилпиперазин-1-ил)- карбонилметокси-4-метоксибензиламина, 0,5 г 4,5-дихлоро-3(2Н)-пиридазинона, 0,46 г триэтиламина, 10 мл этанола, и 10 мл воды, нагревали с обратным холодильником в течение ночи, перемешивая при этом. Растворитель отгоняли при пониженном давлении, и к остатку добавляли водный раствор карбоната калия. Полученную смесь экстрагировали хлороформом. Экстрактный раствор промывали водой и насыщенным водным раствором хлорида натрия, а затем осушали безводным сульфатом натрия. После этого растворитель отгоняли. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле, а затем кристаллизовали из хлороформадиэтилового эфира, в результате чего получали 0,61 г идентифицированного выше соединения в виде белых кристаллов.
ЯМР: δ 12,66 (шир. с., 1Н), 7,44 (с, 1Н), 6,78 (с, 3Н), 5,43 (т, 1Н), 4,68 (с, 2Н), 4,39 (д, 2Н), 3,77 (с, 3Н), 3,30-3,75 (м, 4Н), 2,0-2,60 (м, 7Н).
MC (m/z ): 421 (М+), 386, 140, 99, 70 (100%).
Сравнительный пример 9
4-Хлоро-5-(З-карбоксиметилокси-4-метоксибензиламино)-3(2Н)- пиридазинон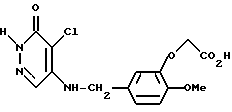
Смесь, содержащую 0,3 г {4-хлоро-5-[3-(4-метилпиперазина-1-ил)- карбонилметокси-4-метоксибензиламино}-3(2Н)-пиридазинона, 2,0 г гидроксида калия, 10 мл этанола и 2 мл воды, нагревали с обратным холодильником в течение ночи, перемешивая при этом. Реакционный раствор нейтрализовали путем добавления водного раствора соляной кислоты. Затем растворитель отгоняли при пониженном давлении. К полученному остатку добавляли воду, и смесь экстрагировали хлороформом. Раствор экстракта промывали водой и насыщенным водным раствором хлорида натрия, а затем осушали безводным сульфатом натрия. После отгонки растворителя получали 212 мг идентифицированного выше соединения в виде белого твердого вещества.
МС (m/z): 281 (M+-CHCO2H), 246, 209, 159, 145 (100%), 116.
Пример получения 2
4-Хлоро-5-[3-(З-пиридилметиламинокарбонилметокси)-4-метоксибензиламино -3(2Н)-пиридазинон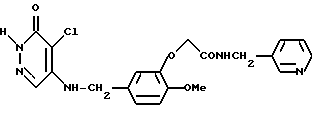
Смесь, состоящую из 200 мг 4-хлоро-5-(3-карбоксиметокси-4-метоксибензиламино)-3(2Н)-пиридазинона, 65 мг триэтиламина и 10 мл N,N-диметилформамида, охлаждали льдом, а затем добавляли 88 мг изобутилхлороформата. Полученную смесь перемешивали при той же температуре в течение одного часа, а затем добавляли 140 мг 3-пиколиламина. Эту смесь перемешивали в течение ночи при комнатной температуре. После отгонки растворителя при пониженном давлении к полученному остатку добавляли воду. Затем смесь экстрагировали хлороформом. Раствор экстракта промывали водой и насыщенным водным раствором хлорида натрия, а затем осушали безводным сульфатом натрия. После этого растворитель отгоняли. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (элюент:хлороформ/-метанол=9/1), и получали 129 мг идентифицированного выше соединения в виде белого твердого вещества.
ЯМР: δ 8,35-8,58 (м, 2Н), 7,81-8,33 (м, 1Н), 7,72 (с, 1Н), 7,45-7,60 (м, 2Н), 6,88 (c, 3H), 6,40-6,80 (м, 1Н), 4,31-4,62 (м, 6Н), 3,75 (с, 3Н).
МС (m/z): 429 (+), 394, 298, 137, 121, 107, 92 (100%).
Сравнительный пример 10
N-Бензилоксикарбонил-3-(3-хлоропропокси)-4-метоксибензиламин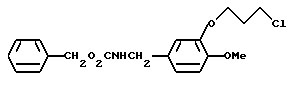
Смесь, содержащую 20 г N-бензилоксикарбонил-3-гидрокси-4-метоксибензиламин, 14,43 г карбоната калия, 16,44 г бромохлоропропана, и 200 мл 2-бутанона нагревали с обратным холодильником при перемешивании в течение 16 часов. Полученную смесь охлаждали до комнатой температуры. Затем неорганические вещества отфильтровывали, а фильтрат подвергали дистилляции при пониженном давлении. Полученный остаток экстрагировали хлороформом, а органический слой промывали водой и насыщенным водным раствором хлорида натрия, после чего осушали безводным сульфатом натрия. Затем, растворитель отгоняли. Полученный остаток кристаллизовали из диэтилового эфира/н-гексана, и получали 23,19 г идентифицированного выше соединения в виде белых кристаллов.
ЯМР: δ/ 7,21 (с, 5Н), 6,71 (с, 3Н), 5,04 (с, 3Н), 4,20 (д, 2Н), 4,02 (т, 2Н), 3,75 (с, 3Н), 3,67 (т, 2Н), 1,94-2,47 (м, 2Н).
МС (m/z): 363 (М+), 316, 273 (100%), 228, 152, 137, 125, 91.
Аналогичным способом были получены следующие соединения:
N-Бензилоксикарбонил-3-(2-хлороэтокси)-4-метоксибензиламин; и
N-Бензилоксикарбонил-3-(2-диэтиламиноэтокси)-4-метоксибензиламин.
Сравнительный пример 11
N-Бензилоксикарбонил-3-[3-(4-фopмилпипеpaзин-1-ил)пpoпокси] -4-метоксибензиламин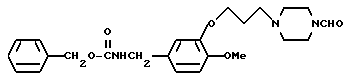
Смесь, содержащую 23,1 г N-бензилоксикарбонил-3-(3-хлоропропокси)-4-метоксибензиламина, 8,7 г N-формилпиперазина, 13,16 г карбоната калия, 0,95 г иодида натрия, и 300 мл N,N-диметилформамида, перемешивали в течение 16 часов при температуре 80oC. Полученную смесь охлаждали до комнатной температуры. Затем неорганические вещества отфильтровывали, а фильтрат отгоняли при пониженном давлении. Полученный остаток экстрагировали хлороформом, а органический слой промывали водой и насыщенным водным раствором хлорида натрия, после чего осушали безводным сульфатом натрия. После отгонки растворителя получали 30,67 г идентифицированного выше соединения в виде слегка коричневатого маслообразного вещества.
ЯМР: δ 7,97 (с, 1Н), 7,32 (с,5Н), 6,81 (с, 3Н), 5,36 (шир. т., 1Н), 5,11 (с, 2Н), 4,26 (д, 2Н), 4,02 (т, 2Н), 3,81 (с, 3Н), 3,12- 3,66 (м,4Н), 1,78-2,78 (м, 8Н).
МС (m/z): 441 (М+), 383, 306, 155 (100%), 128, 91.
Аналогичным способом были получены следующие соединения:
N-Бензилоксикарбонил-3-(3-диэтиламинопропокси)-4- метоксибензиламин;
N-Бензилоксикарбонил-3-[2-(4-бензилпиперазин-1-ил)-этокси] -4-метоксибензиламин;
N-Бензилоксикарбонил-3-[2-(4-(4-хлоробензил)-пиперазин-1-ил] -этокси]-4-метоксибензиламин;
N-Бензилоксикарбонил-3-{ 2-[4-(4-фторобензил)пиперазин-1-ил] -этокси}-4-метоксибензиламин;
N-Бензилоксикарбонил-3-[3-(4-бензилпиперазин-1-ил)-пропокси] -4-метоксибензиламин; и
N-Бензилоксикарбонил-3-[3-(4-метилпиперазин-1-ил)-пропокси] -4-метоксибензиламин.
Сравнительный пример 12
3-[3-(4-Формилпиперазин-1-ил)-пропокси]-4-метoкcибензиламин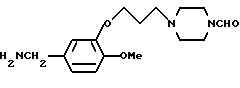
Смесь, состоящую из 30,4 г N-бензилоксикарбонил-3-[3-(4- формилпиперазин-1-ил)-пропокси-4-метоксибензиламина, 3,1 г 5% палладия-на-угле и 300 мл этанола, перемешивали в течение 9 часов при температуре 60oC в атмосфере водорода. После отфильтровывания палладия-на-угле, фильтрат отгоняли при пониженном давлении, и получали 17,99 г идентифицированного выше соединения в виде слегка коричневатого маслообразного вещества.
ЯМР: δ 8,03 (с, 1Н), 6,86 (с, 3Н), 4,11 (т, 2Н), 3,84 (с, 3Н), 3,25-3,71 (м, 4Н), 2,30-2,82 (м, 4Н), 1,82-2,30 (м, 4Н).
MC (m/z): 307 (М+), 292, 246, 171, 155, 125, 99 (100%).
Аналогичным способом были получены следующие соединения:
3-(2-Диэтиламиноэтокси)-4-метоксибензиламин;
3-(3-Диэтиламинопропокси)-4-метоксибензиламин;
3-[2-(4-Бензилпиперазин)-1-ил]-этокси-4-метоксибензилпиперазин;
3-[2-{4-(4-Хлоробензил)-пиперазин-1-ил}-этокси]-4- метоксибензиламин;
3-[2-{4-(4-фторобензил)-пиперазин-1-ил}-этокси]-4- метоксибензиламин;
3-[3-(4-Бензилпиперазин-1-ил)-пропокси]-4-метоксибензиламин; и
3-[3-(4-Метилпиперазин-1-ил)-пропокси]-4-метоксибензиламин.
Пример получения 3
4-Хлоро-5-[3-{ 3-(4-формилпиперазин-1-ил)-пропокси} -4- метоксибензиламино]-3(2Н)-пиридазинон (Соединение N 50)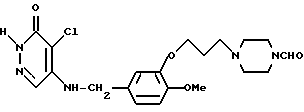
Смесь, содержащую 11,58 г 3-[3-(4-формилпиперазин1-ил)-пропокси] -4-метоксибензиламина, 5,0 г 4,5-дихлоро-3(2Н)-пиридазинона, 4,6 г триэтиламина, 50 мл н-пропанола, и 50 мл воды, нагревали с обратным холодильником при перемешивании в течение 14 часов. После отгонки растворителя при пониженном давлении к полученному остатку добавляли водный раствор карбоната калия, и эту смесь экстрагировали хлороформом. Органический слой промывали водой и насыщенным водным раствором хлорида натрия, а затем осушали безводным сульфатом натрия. После этого растворитель отгоняли, а полученный остаток очищали с помощью колоночной хроматографии на силикагеле, в результате чего получали 6,21 г идентифицированного выше соединения в виде желтовато-белого твердого вещества.
ЯМР: δ 12,49 (шир. с., 1Н), 8,06 (с, 1Н), 7,65 (с, 1Н), 6,88 (с, 3Н), 5,37 (т, 1Н), 4,51 (д, 2Н), 4,08 (т, 2Н), 3,87 (с, 3Н), 3,19-3,74 (м, 4Н), 2,30-2,84 (м, 6Н), 1,76-2,30 (м, 2Н).
Пример получение 4
4-Хлоро-5-[3-{ 3-(4-этилпиперазин-1-ил)-пропокси} -4-метоксибензиламино -3(2Н)-пиридазинон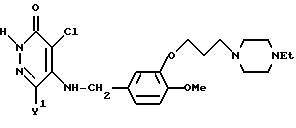
Смесь, содержащую 1,0 г 4-хлоро-5-[3-{3-(4-формилпиперазин-1-ил)- пропокси} -4-метоксибензиламино]-3(2Н)-пиридазинона, 0,62 г гидроксида калия, 7 мл этанола, и 7 мл воды, нагревали с обратным холодильником в течение 3,5 часа при перемешивании, а затем к этой смеси добавляли 0,32 г карбоната калия и 570 мг этилбромида. Полученную смесь перемешивали в течение 4 часов при температуре 60oC. После отгонки растворителя при пониженном давлении к полученному остатку добавляли воду. Эту смесь экстрагировали хлороформом. Раствор экстракта промывали водой и насыщенным водным раствором хлорида натрия, а затем осушали безводным сульфатом натрия. Затем растворитель отгоняли. Остаток очищали с помощью колоночной хроматографии на силикагеле и получали 0,50 г идентифицированного выше соединения в виде слегка коричневатого твердого вещества.
ЯМР: δ 7,65 (с,1Н), 6,89 (с, 3Н), 5,41 (коллапсированная, 1Н), 4,50 (д, 5Н), 4,08 (т, 2Н), 3,87 (с, 3Н), 1,73-3,10 (м, 14Н), 1,08 (т, 3Н).
МС (m/z): 435(М+), 365, 343, 206, 127 (100%), 99.
Аналогичным способом было получено соединение, а именно 4-хлоро-5-[3-{ 3-(4-(4-фторобензил)-пиперазин-1-ил)-пропокси} -4-метоксибензиламино] -3(2Н)-пиридазинон.
МС (m/z): 515 (М+), 109(100%).
Сравнительный пример 5
2-Этил-4-хлоро-5-[3-{ 2-(4-(4-фторобензил)-пиперазин-1-ил этокси)-4-метoкcибензилaминo}-3(2Н)-пиридазинон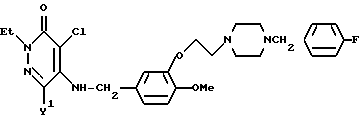
Смесь, содержащую 500 мг 4-хлоро-5-[3-{2-(4-(4-фторобензил)- пиперазин-1-ил)-этокси}- 4-метоксибензиламино]-3(2Н)-пиридазинона, 130 мг этилбромида, 190 мг карбоната калия, и 10 мл 2-бутанона, нагревали с обратным холодильником при перемешивании в течение 5 часов. Неорганические вещества отфильтровывали, а затем растворитель отгоняли при пониженном давлении. К остатку добавляли воду, и эту смесь экстрагировали хлороформом. Раствор экстракта промывали водой и насыщенным водным раствором хлорида натрия, после чего осушали безводным сульфатом натрия. Затем, растворитель отгоняли. Полученный остаток очищали с помощью хроматографии на колонке с силикагелем (элюент: хлороформ/-этанол=19/1), и получали 429 мг идентифицированного выше соединения в виде бесцветного прозрачного вязкого вещества.
ЯМР: δ 7,47 (с,1Н), 7,00-7,31 (м,4Н), 6,88 (с, 3Н), 5,20 (т,1Н), 4,46 (д, 2Н), 4,14 (т, 2Н), 4,12 (кв., 2Н), 3,85 (с, 3Н), 3,47 (с, 2Н), 2,73 (т, 2Н), 2,21-3,05 (м, 10Н), 1,32 (т, 3Н).
МС (m/z): 574 (М+), 493, 273, 221, 192 (100%), 164, 111, 84.
Сравнительный пример 13
1-Хлороацетил-4-(2-хинолилметил)-пиперазин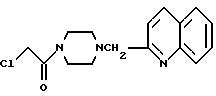
Раствор, содержащий 600 мг 14-хинолилметилпиперазина и 20 мл безводного тетрагидрофурана, охлаждали до температуры -60oC, а затем в течение 10 минут по капле добавляли перемешанный раствор из 330 мг ацетилхлорида и 5 мл безводного тетрагидрофурана. Полученную смесь перемешивали в течение 1 часа при температуре -60oC, а затем добавляли воду. Эту смесь перемешивали в течение 20 минут при комнатной температуре. Реакционный раствор подвергали дистилляции при пониженном давлении, а затем экстрагировали хлороформом. Органический слой промывали водным раствором карбоната калия и осушали безводным сульфатом натрия. После отгонки растворителя при пониженном давлении получали 750 мг идентифицированного выше соединения в виде маслообразного вещества.
ЯМР: δ 7,32-8,20 (м, 6Н), 4,01 (с, 2Н), 3,20-3,90 (м, 6Н), 2,30-2,74 (м, 4Н).
МС (m/z): 143 (М+ -160).
Аналогичным способом были получены следующие соединения:
1-Хлороацетил-4-(4-хлоробензил)-пиперазин;
МС (m/z): 286 (М+), 125 (100%).
1-Хлороацетил-4-[1-(4-фторобензил)-2-метилбензоимидазолил)-пиперазин;
ЯМР: δ 6,66-7,40 (м, 8Н), 5,44 (с, 2Н), 3,95 (с, 2Н), 3,74 (с, 2Н), 3,04-3,60 (м, 4Н), 2,24-2,66 (м, 4Н).
1-Хлороацетил-4-бензилпиперазин;
MC (m/z): 252(М+), 91 (100%).
1-Хлороацетил-4-бензилпиперидин;
МС (m/z): 251 (М+), 91 (100%), и
1-Хлороацетил-4-(т-бутилоксикарбониламинобензил)-пиперазин ;
MC (m/z): 368 (M+), 150 (100%).
Сравнительный пример 14
N-т-Бутилоксикарбонил-3-[4-(2-хинолилметилпиперазин)-1-ил] карбонилметокси-4-метоксибензиламин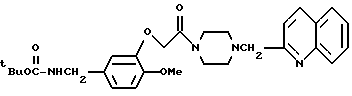
Смесь, содержащую 660 мг т-бутилоксикарбонил-3-гидрокси- 4-метоксибензиламина, 10 мл диметилформамида, 510 мг карбоната калия, и 750 мг 1-хлороацетил-4-(2-хинолилметил)-пиперазина, нагревали в течение ночи при температуре 80oC, перемешивая при этом. Нерастворившиеся вещества отфильтровывали, а затем реакционный раствор подвергали дистилляции при пониженном давлении и экстрагировали хлороформом. Раствор экстракта промывали водным раствором карбоната калия, а затем очищали с помощью колоночной хроматографии на силикагеле (элюент: этилацетат/метанол, 19:1), в результате чего получали 1,2 г идентифицированного выше соединения в виде маслообразного вещества.
ЯМР: δ 7,32-8,03 (м, 6Н), 6,63-6,93 (м, 3Н), 5,15-5,50 (м, 1Н), 4,64 (с, 2Н), 4,16 (д, 2Н), 3,38-3,93 (м, 9Н), 2,30-2,73 (м, 4Н), 1,43 (с, 9Н).
МС (m/z ): 520 (М+), 144 (100%).
Аналогичным способом были получены следующие соединения:
N-т-Бутилоксикарбонил-3-[4-(4-хлоробензил)-пиперазин-1-ил]- карбонилметокси-4-метоксибензиламин;
МС (m/z): 503 (М+), 125 (100%).
N-т-Бутилоксикарбонил-3-[4-{1-(4-фторобензил)-2- метилбензоимидазол}-пиперазин-1-ил]-карбонилметокси-4- метоксибензиламин;
ЯМР: δ/ 6,10-7,35 (м, 11Н), 5,45 (с, 2Н), 4,80-5,17 (м, 1H), 4,10 (c, 2H), 4,15 (д, 2Н), 3,76 (с, 3H), 3,70 ((c, 12H), 3,26- 3,65 (м, 4Н), 3,27-2,65 (м, 4Н)
N-т-Бутилоксикарбонил-3-(4-бензилпиперидин-1-ил)- карбонилметокси-4-метоксибензиламин;
МС (m/z): 468 (М+), 91 (100).
N-т-Бутилоксикарбонил-3-(4-т-бутилоксикарбониламинобензилпиперазин-1-ил)-карбонилметокси-4-метоксибензиламин.
МС (m/z): 585 (М+), 150 (100%).
Сравнительный пример 15
3-[4-(2-Хинолилметил)-пиперазин-1-ил]-карбонилметокси-4- метоксибензиламин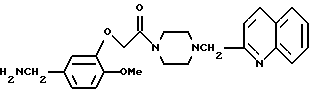
Смесь, содержащую 1,3 г т-бутилоксикарбонил-3-[4-(2-хинолилметил) -пиперазин-1-ил]-карбонилметокси-4-метоксибензиламина, 14 мл хлороформа и 2,8 г трифтороуксусной кислоты, перемешивали в течение одного дня при комнатной температуре. К этому реакционному раствору добавляли 50 мл хлороформа и 50 мл 0,5 н. соляной кислоты, и полученную смесь подвергали обратному экстрагированию. Водный слой доводили до pH = 12 путем добавления водного раствора гидроксида натрия, а затем экстрагировали хлороформом. Органический слой промывали водным раствором карбоната калия и осушали безводным сульфатом натрия. После отгонки растворителя при пониженном давлении получали 850 мг идентифицированного выше соединения в виде маслянистого вещества.
ЯМР: δ 7,39-8,20 (м, 6Н), 6,72-7,0 (м, 3Н), 4,7 (с, 2Н), 3,40-4,00 (м, 11Н), 2,32-2,70 (м, 4Н), 2,05 (шир.с., 2Н).
МС (m/z): 420 (М+), 143 (100%).
Аналогичным способом были получены следующие соединения:
3-[4-(4-Хлоробензил)-пиперазин-1-ил] -карбонилметокси- 4-метоксибензиламин;
MC (m/z): 403 (М+), 125 (100%).
3-[3-{4-(4-фторобензил)-пиперазин-1-ил}-2,2-диметилпропокси)- 4-метоксибензиламин;
МС (m/z): 429 (М+), 109 (100%).
3-(4-Бензилпиперазин-1-ил)-карбонилметокси-4-метоксибензиламин;
МС (m/z): 368 (М+), 91 (100%).
3-[4-{ 1-(4-Фторобензил)-2-бензимидазолилметил} -пиперазин-1-ил] -карбонилметокси-4-метоксибензиламин.
MC (m/z): 517 (М+), 109 (100%).
Пример получения 6
4-Хлоро-5-[3-{ 4-(2-хинолилметил)-пиперазин-1-ил} - карбонилметокси-4-метоксибензиламино]-6-этокси-3(2Н)-пиридазинон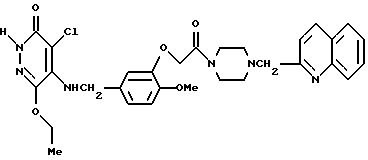
Смесь, содержащую 2,4 г 3-[4-(2-хинолилметил)-пиперазин-1-ил] -карбонилметокси-4-метоксибензиламина, 1 г 4,5-дихлоро-6-этокси-3(2Н)-пиридазинона, 580 мг триэтиламина, 10 мл пропанола и 10 мл воды, нагревали с обратным холодильником при перемешивании в течение ночи. После отгонки растворителя при пониженном давлении остаток экстрагировали хлороформом. Органический слой промывали водным раствором карбоната калия, а затем осушали безводным сульфатом натрия. После этого растворитель отгоняли. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (элюент, этилацетат/метанол (6: 1) хлороформ/метанол (12:1), а затем кристаллизовали из диэтилового эфира, в результате чего получали 1,5 г идентифицированного выше соединения в виде белых кристаллов.
ЯМР: δ 7,40-8, 28(м, 6Н), 6,72-7,05 (м, 3Н), 4,62-5,40 (м, 5Н), 3,48-4,50 (м, 11Н), 2,32-2,70 (м, 4Н), 1,31 (т, 3Н).
МС (m/z): 592 (М+), 143 (100%).
Сравнительный пример 16
1-Формил-4-(4-аминобензил)-пиперазин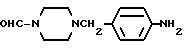
Смесь, содержащую 9 г 1-формил-4-(4-нитробензил)-пиперазина, 180 мл метанола и 14,6 г гексагидрата хлорида никеля, охлаждали в ледяной бане, и медленно добавляли 4,6 г борогидрида натрия. Полученную смесь перемешивали в течение 30 минут при температуре 0oC, а затем в течение 30 минут при комнатной температуре. Реакционный раствор отгоняли при пониженном давлении, и образовавшийся остаток растворяли путем добавления 200 мл 10% соляной кислоты, а затем доводили до pH=10 путем добавления 28% водного раствора аммиака. Полученную смесь экстрагировали этилацетатом. Раствор экстракта промывали насыщенным водным раствором хлорида натрия, после чего осушали безводным сульфатом натрия. Растворитель отгоняли при пониженном давлении. Остаток кристаллизовали из диэтилового эфира, и получали 8,0 г идентифицированного выше соединения в виде белых кристаллов.
ЯМР: δ 7,82 (с, 1Н), 6,97 (д, 2Н), 6,47 (д, 2Н), 3,01-3,91 (м, 8Н), 2,11-2,48 (м,4Н).
MC (m/z): 263 (М+), 218 (100%).
Сравнительный пример 17
1-Формил-4-(4-т-бутилоксикарбониламинобензил)-пиперазин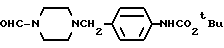
Смесь, содержащую 4 г 1-формил-4-аминобензилпиперазина, 50 мл толуола и 4,8 г ди-т-бутилдикарбоната, нагревали с обратным холодильником в течение 5 часов. Реакционный раствор концентрировали при пониженном давлении, а остаток очищали с помощью колоночной хроматографии на силикагеле (элюент: этилацетат/метанол, 9: 1), а затем кристаллизовали из диэтилового эфира, в результате чего получали 5,1 г идентифицированного выше соединения в виде белых кристаллов.
ЯМР: δ 7,87(с, 1Н), 6,97-7,42 (м, 5Н), 3,15-3,65 (м, 6Н), 2,15-2,57 (м, 4Н), 1,45 (с, 9Н).
МС (m/z): 319 (М+), 106(100%).
Сравнительный пример 18
1-(4-т-Бутилоксикарбониламинобензил)-пиперазин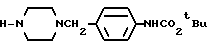
4 г 1-Формил-4-(т-бутилоксикарбониламинобензил)-пиперазина растворяли в 50 мл метанола, и к этому раствору добавляли раствор, состоящий из 1,5 г гидроксида натрия, растворенного в 10 мл воды. Полученную смесь нагревали в течение 5 часов при температуре 60oC. Реакционный раствор концентрировали при пониженном давлении, а затем экстрагировали хлороформом. Органический слой промывали водным раствором карбоната калия и осушали безводным сульфатом натрия. После этого растворитель отгоняли при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле (элюент: хлороформ/метанол= 5: 1), а затем кристаллизовали из диэтилового эфира, в результате чего получали 3,2 г идентифицированного выше соединения в виде белых кристаллов.
ЯМР: δ 7,0-7,7 (м, 5Н), 3,38 (с, 2Н), 2,60-3,12 (м, 4Н), 1,90- 2,60 (м, 5Н), 1,50 (с, 9Н).
MC (m/z): 291 (М+), 206, 106 (100%).
Пример получения 7
4-Хлоро-5-[3-(4-(4-аминобензил)-пиперазин-1-ил) -карбонилметокси-4-метоксибензиламино]-6-изопропокси-3(2Н)-пиридазинон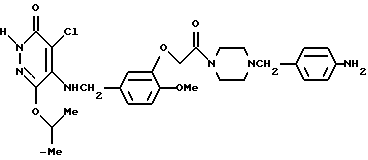
Смесь, содержащую 1,6 г 3-[4-(4-аминобензил) пиперазин-1-ил] -карбонилметокси-4-метоксибензиламина, 770 мг 4,5-дихлоро-6-изопропокси-3(2Н)-пиридазинона, 460 мг триэтиламина и 20 мл метанола, нагревали с обратным холодильником при перемешивании в течение 2 дней. После отгонки растворителя при пониженном давлении остаток экстрагировали хлороформом. Органический слой промывали водным раствором карбоната калия, а затем осушали безводным сульфатом натрия. После этого растворитель отгоняли. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (элюент: этилацетат/метанол=9:1 --->хлороформ/метанол=15:1), а затем кристаллизовали из диэтилового эфира, в результате чего получали 1,6 г идентифицированного выше соединения в виде белых кристаллов.
ЯМР: δ 6,55-7,15 (м, 7Н), 4,45-5,33 (м, 6Н), 3,13-3,88 (м, 11Н), 2,13-2,58 (м, 4Н), 1,28 (д, 6Н).
МС (m/z): 465 (М+ -106), 430, 106 (100%).
Пример получения 8
4-Xлор-5-[3{ 4-(4-формилбензил)-пиперазин-1-ил]карбонилметокси- 4-метоксибензиламино]-6-изопропокси-3(2Н)-пиридазинон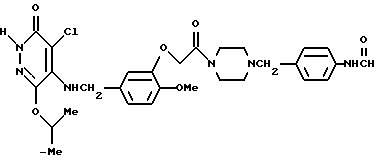
400 мг 4-Хлоро-5-[3-(4-аминобензил)пиперазин-1-ил]- карбонилметокси-4-метоксибензиламино-6-изопропокси-3(2Н)-пиридазинона растворяли в 3 мл фенилформата. Затем раствор перемешивали при комнатной температуре в течение ночи. Реакционный раствор подвергали дистилляции при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (элюент: хлороформ/метанол, 9:1), а затем кристаллизовали из диэтилового эфира, в результате чего получали 830 мг идентифицированного выше соединения в виде белых кристаллов.
ЯМР: δ 11,75 (шир. с., 1Н), 8,2-8,85 (м, 2Н), 6,75-7,62 (м, 7Н), 4,58-5,30 (м, 6Н), 3,77 (с, 3Н), 3,20-3,75 (м, 6Н), 2,05-2,60 (м, 4Н), 1,27 (д, 6Н).
МС (m/z): 464 (М+ -134), 137 (100%).
Пример получения 9
4-Хлоро-5-[3-{ 4-(4-N-ацетиламинобензил)-пиперазин-1-ил} - карбонилметокси-4-метоксибензиламино]-6-изопропокси-3(2Н)-пиридазинон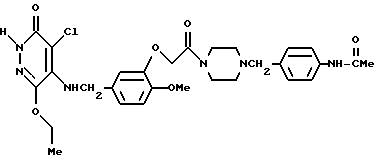
400 мг 4-хлоро-5-[3-(4-аминобензил)-пиперазин-1-ил]- карбонилметокси-4-метоксибензиламино-6-изопропокси-3(2Н)-пиридазинона растворяли в 400 мл пиридина, и к этому раствору добавляли 220 мг уксусного ангидрида. Полученную смесь перемешивали в течение 2 часов при комнатной температуре. После отгонки растворителя при пониженном давлении, остаток экстрагировали хлороформом. Органический слой промывали водным раствором карбоната калия, а затем осушали безводным сульфатом натрия. После этого растворитель отгоняли при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (элюент: хлороформ/метанол=9:1), а затем кристаллизовали из диэтилового эфира, в результате чего получали 340 мг идентифицированного выше соединения в виде белых кристаллов.
ЯМР: δ 11,84 (шир. с, 1Н), 8,24 (шир. с., 1Н), 6,63-7,52 (м, 8Н), 4,52-5,30 (м, 6Н), 3,30-3,92 (м, 9Н), 2,0-2,62 (м, 7Н), 1,25 (д, 6Н).
МС (m/z): 613 (М+ +Н), 466.
Пример получения 10
4-Бромо-5-[3-{2-(4-(4-хлоробензил)-пиперазин-1-ил)-этокси} -4-метоксибензиламино]-3(2Н)-пиридазинона гидрохлорид (Соединение N 7)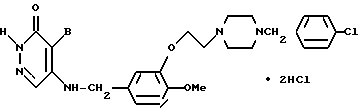
К размешанному раствору, состоящему из 440 мг 4-бромо-5-[3-{2-(4-хлорофобензил)пиперазин-1-ил)этокси} -4- метоксибензиламино]-3(2Н)-пиридазинона и 5 мл хлороформа, добавляли раствор метанола в 10% соляной кислоты до тех пор, пока pH не становился равным 2-3, и эту смесь перемешивали в течение 2 часов при комнатной температуре. К реакционному раствору добавляли диэтиловый эфир для кристаллизации, и получали 465 мг идентифицированного выше соединения в виде белых кристаллов, имеющего температуру плавления 176-183oC.
МС (m/z): 562 (М+-2HCl), 482, 238, 223 (100%), 203, 125, 91.
Пример получения 11
4-Бромо-5-3-{ 2-(4-(4-хлоробензил)-пиперазин-1-ил)этокси} - 4-метоксибензиламино]-3(2Н)-пиридазинона фумарат (соединение N 8)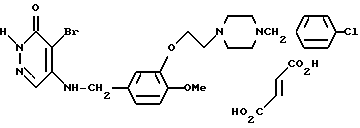
Смесь, содержащую 163 мг 4-бромо-5-[3-{2-(4-(4-хлоробензил)- пиперазин-1-ил)-этокси} -4-метоксибензиламино]-3(2Н)-пиридазинона, 33 мг фумаровой кислоты и 4 мл хлороформа, перемешивали при комнатной температуре в течение 3 часов. К реакционному раствору добавляли диэтиловый эфир для кристаллизации, в результате чего получали 120 мг идентифицированного выше соединения в виде белых кристаллов, имеющего т.пл. 178-185oC.
MC (m/z): 562 (M+-(CHCO2H)2), 482, 237, 223, 125 (100%), 91.
Пример получения 12
4-Бромо-5-[3-{ 2-(4-(4-хлоробензил)-пиперазин-1-ил)этокси] -4-метоксибензиламино}-3(2Н)-пиридазинона сульфат (соединение N 9)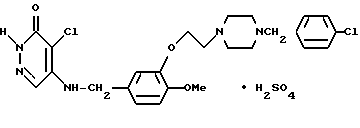
Смесь, содержащую 700 мг 4-бромо-5-[3-{2-(4- (4-хлоробензил)- пиперазин-1-ил)-этокси}-4-метоксибензиламино]-3(2Н)-пиридазинона, 5 мл метанола, 5 мл хлороформа, и 140 мг серной кислоты, перемешивали в течение трех часов при комнатной температуре. Реакционный раствор отгоняли при пониженном давлении. Полученный остаток кристаллизовали из изопропилового эфира/диэтилового эфира, и получали 800 мг идентифицированного выше соединения в виде белых кристаллов, имеющего т.пл. 158-162oC.
МС (m/z): 482 (М+ -Br-H2SO4), 238, 223 (100%), 125.
Соединения, полученные в соответствии с вышеприведенными примерами получений, представлены в табл. II. Структуры этих соединений, номера которых указаны в данной таблице, описаны в табл. I. В последней колонке табл. II указан номер прилагаемого примера получений.
Пример 1 получения композиции (таблетки)
Соединение N 39 - 10 г
Лактоза - 20 г
Крахмал - 4 г
Крахмал для получения пасты - 1 г
Стеарат магния - 0,1 г
Кальцийкарбоксиметилцеллюлоза - 7 г
Всего - 42,1 г
Вышеуказанные компоненты смешивали обычным способом, и эту смесь формовали в покрытые сахаром таблетки, каждая из которых содержала 50 мг активного ингредиента.
Пример 2 получения композиции (капсулы)
Соединение N 43 - 10 г
Лактоза - 20 г
Микрокристаллическая целлюлоза - 10 г
Стеарат магния - 1 г
Всего - 41 г
Вышеуказанные компоненты смешивали обычным способом, и полученной смесью наполняли желатиновые капсулы, каждая из которых содержала 50 мг активного ингредиента.
Пример 3 получения композиции (мягкие капсулы)
Соединение N 7 - 10 г
Кукурузное масло - 35 г
Всего - 45 г
Вышеуказанные компоненты смешивали и формовали обычным способом с получением мягких капсул.
Пример 4 получения композиции (мази)
Соединение N 25 - 1,0 г
Оливковое масло - 20 г
Белый вазелин - 79 г
Всего - 100 г
Вышеуказанные компоненты смешивали обычным способом и получали 1% мазь.
Пример 5 получения композиции (аэрозольная суспензия)
А) Соединение N 37 - 0,25%
Изопропилмиристат - 0,10%
Этанол - 26,40%
В) 60-40% смеси 1,2-дихлоротетрафтороэтана и 1-хлоропентафтороэтана - 73,25%
Компоненты вышеуказанной композиции (А) смешивали. Полученную смесь раствора загружали в контейнер, снабженный клапаном, а пропеллент (В) инжектировали через сопло с клапаном до монометрического давления, составляющего около 2,46-2,81 мг/см2, в результате чего получали аэрозольную суспензию.
Примеры испытаний
I. Бронхорасширяющее действие
1. In. vitro - анализ
Лекарственное средство:
Образец испытуемого лекарственного средства растворяли в 100% диметилсульфоксида (ДМСО, Wako. Junguaku) и разводили для дальнейшего использования. Лейкотриен D4 (LTD4, Ultrafine) и изопротеренол (Isoproterenol, Sigma) разводили дистиллированной водой. Индометацин (Indo Sigma) растворяли в 100% этанола (Et OH, Komune Kagaku). Аминофилин (AP,Sigma) и дигидрохлорид гистамина (His. Wako Junyaku) растворяли в дистиллированной воде. Конечные концентрации ДМСО и Et OH в бане составляли не более чем 0,25% (об./об), и не более чем 0,1% (об./об.), соответственно.
Метод 1:1
Морскую свинку (300-450 г) умерщвляли путем обескровливания и вынимали трахею. После удаления жира и соединительных тканей трахею разрезали и делили на 2-3 спиральные полоски, каждая из которых имела ширину около 2 мм и содержала ткани 4 гладких мышц. Каждый образец, полученный таким образом, был суспендирован в 8-мл бане для органов, содержащей модифицированный раствор Тироде, аэрированный 95% O2 + 5 % CO2 при 37oC, после чего к нему была приложена нагрузка в 1 г. Релаксацию мышцы регистрировали на самописце (Yokogawa Hokushin Electric, type 3066) с использованием изотонического преобразователя (Nihon Kohden, TD -112S).
Указанный модифицированный раствор Тироде имел следующий состав (мМ):
NaCl 137; KCl 2,5; CaCl2 1,8; MoCl2 1,0; NaHCO3 20; NaH2PO4 0,32; Глюкоза 11.
Затем образец оставляли на 50-60 минут и подвергали сокращению с помощью дигидрохлорида гистамина (100 мкМ). После установления реакции на постоянном уровне образцы промывали и оставляли на 20-30 минут. Затем добавляли индометацин (5 мкМ) и после инкубирования в течение 30 минут образцы подвергали сокращению путем добавления LTD4 (30 нМ). После стабилизации реакции целиком добавляли образец испытуемого лекарственного средства. И наконец, для достижения реакции максимальной релаксации добавляли АР(1 мМ). Полученный результат выражали в процентах релаксации по отношению к АР-индуцированной релаксации, которую принимали за 100%; после чего, измеряли концентрацию, при которой достигается 50%-ная релаксация (EC50, мкМ). В качестве контрольного лекарственного средства использовали АР. Результаты представлены в табл. III-I.
Метод 1-2;
Измерения проводили методом, аналогичным методу 1-1. Образец выдерживали в течение 60-90 минут, а затем подвергали релаксации путем добавления 1 мкМ изопретеренола. После этого образец промывали, и эту операцию повторяли с интервалом 30-40 минут до тех пор, пока реакция релаксации не достигала стабильного уровня. Затем для релаксации образца целиком добавляли испытуемое лекарственное средство. И наконец, для достижения максимума реакции релаксации добавляли 1 мМ АР. Полученный результат выражали в процентах релаксации по отношению к АР-индуцированной релаксации, которую принимали за 100%; и, кроме того, определяли концентрацию, при которой достигается 50%-ная релаксация (EC50, мкМ). Конечную концентрацию ДМСО в бане доводили до 0,2 об. /об.%. В качестве контрольного лекарственного средства использовали АР. Результаты представлены в табл. III-2.
In vivo-испытания
Воздействие на анафилактический бронхостеноз, опосредованный эндогенно высвобождающейся МРС-А (медленно реагирующей субстанцией А) у пассивно сенсибилизированных морских свинок
За 1-2 дня до начала эксперимента самцов морских свинок (350-450 г) пассивно сенсибилизировали путем внутривенной (i.v.) инъекции 0,125 мл кроличьей сыворотки против EA (EA=яичный альбумин) (Capple Laboratories). Антиген-индуцированный анафилактический бронхостеноз, опосредованный эндогенно высвобождающейся MPC-A, измеряли с помощью модифицированного метода Konzett u Rossler (Arch. Exp. Path.Pharmak., 195, 71, 1940). Сенсибилизированных морских свинок анестезировали путем внутрибрюшинной инъекции уретана (1,5 г/кг). Правую яремную вену канюлировали для введения всех агентов, а трахею канюлировали для регистрации полной легочной резистентности. Морским свинкам проводили искусственную вентиляцию легких с помощью небольшого респиратора для животных (Shinato, Model SN-480-7), установленного на ударный объем сердца 4,5 мл, и на частоту дыхания 50 вдохов в мин. Изменения в легочной резистентности измеряли с помощью датчика давления (Nihon Kohden, Model ТР-602Т), подсоединенного к Т-трубке трахеотомической канюли. Процент максимального бронхостеноза получали посредством зажима трахеи. Затем после описанной хирургической подготовки, животных предварительно обрабатывали индометацином (2 мг/кг, 10 мин), пириламином (2 мг/кг, 6 мин), и пропранололом (0,1 мг/кг, 5 мин), а затем проводили антигенную стимуляцию EA (0,2 мг/кг). Все испытуемые соединения вводили перорально за 2 часа до введения провокационной пробы EA. Ингибирование (%) бронхостеноза определяли по следующей формуле: Ингибирование (%) = 1,0 - % максимального бронхостеноза в условиях испытания/ % максимального бронхостеноза в условиях контроля • 100. Максимальный уровень бронхостеноза составлял 62±6% (среднее значение ± ср. кв. ош.; n = 6), а число испытуемых животных составляло 5-6.
В табл. III-3 приводится % ингибирования при дозе испытуемого соединения, составляющей 30 мг/кг.
II. Противоаллергическое действие
Тест на связывание с использованием 3-Н-пириламина (тест на связывание с рецептором гистамина H1 )
Этот тест проводили по методу Chang и др. (J.Neurochem 32, 1653 (1979)).
Меченный тритием пириламин добавляли к суспензии бычьего мозжечка и 50 мМ фосфатно-буферного раствора (pH 7,5), после чего смесь оставляли на 30 минут при 25oC. Затем смесь быстро фильтровали при отсасывании через фильтровальную бумагу из стекловолокна, и на этой фильтровальной бумаге определяли радиоактивность. Степень ингибирования по отношению к H1-рецептору при концентрации испытуемого соединения 10 мкМ, определяли по следующему уравнению:
Степень ингибирования (%) =
{1-(Величина связывания в присутствии лекарственного средства - величина неспецифического связывания)/(суммарная величина связывания - величина неспецифического связывания)} • 100, где суммарная величина связывания представляет собой 3H-пириламин-связывающую радиоактивность в отсутствие испытуемого соединения, а величина неспецифического связывания представляет собой 3H-пириламин-связывающую радиоактивность в присутствии 10 мкМ трипролизина. Результаты показаны в табл. IV.
III. Действие, ингибирующее агрегацию тромбоцитов
Эффект ингибирования агрегации тромбоцитов у кроликов
Кровь из брюшной артерии самцов японских белых кроликов (вес: 1,8 - 2,5 кг) собирали в шприц, содержащий 1/10 объем 3,8% цитрата натрия. Полученную таким образом кровь центрифугировали при 200 х г в течение 7 минут при комнатной температуре, и получали обогащенную тромбоцитами плазму (PRP). Кроме того, остаток подвергали центрифугированию при 2000 х г в течение 10 минут и получали обедненную тромбоцитами плазму (РРР). Измерения осуществляли путем разведения плазмы, обогащенной тромбоцитами, плазмой, обедненной тромбоцитами, до 300000/мм3. PRP и РРР помещали в кювету, и пределы измерений коэффициента пропускания доводили до 0% в случае PRP, и до 100% в случае РРР. После этого испытуемое лекарственное средство, растворенное в 100 % диметилсульфоксиде (ДМСО), добавляли к PRP (конечная концентрация ДМСО составляла 0,25%). После инкубирования в течение 2 минут при 37oC и при 900 об/мин, добавляли агент, стимулирующий агрегацию тромбоцитов, и регистрировали кривую агрегации. Действие испытуемого лекарственного средства, направленное против агрегации тромбоцитов, выражали как концентрацию (1C50, мкМ), при которой наблюдается 50%-ное ингибирование агрегации контрольного образца. Агент, стимулирующий агрегацию, а именно ADP был использован при минимальной концентрации (5 - 10 мкМ), которая вызывает максимальную агрегацию. Измерения агрегации тромбоцитов проводили с использованием следящего устройства NBS HEMA TPACER 601. Результаты представлены в табл. V.
Промышленное применение
Как свидетельствуют описанные выше результаты, соединения настоящего изобретения обладают прекрасной бронхорасширяющей активностью, противоаллергической активностью, и активностью, ингибирующей агрегацию тромбоцитов. Соединения настоящего изобретения обладают сильным фармакологическим действием даже при пероральном введении. Поэтому эти соединения могут быть использованы в качестве профилактических и терапевтических лекарственных средств для лечения аллергических заболеваний немедленного типа, таких, как бронхиальная астма, аллергический ринит, крапивница, и сенная лихорадка; различных воспалительных заболеваний, таких, как ревматический артрит, артрит позвоночника; ишемических болезней, таких, как стенокардия, и инфаркт миокарда; а также различных тромботических заболеваний.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ 3(2Н)-ПИРИДАЗИНОНА ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 1991 |
|
RU2054004C1 |
| Способ получения производных 3(2Н)-пиридазинона | 1988 |
|
SU1584750A3 |
| АЗОЛЬНЫЕ СОЕДИНЕНИЯ, СПОСОБ ИХ ПОЛУЧЕНИЯ, ПРОТИВОГРИБКОВАЯ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБ ЛЕЧЕНИЯ ГРИБКОВОЙ ИНФЕКЦИИ | 1998 |
|
RU2189982C2 |
| Способ получения производных 3(2Н)пиридазинона или их фармацевтически приемлемых солей | 1986 |
|
SU1468415A3 |
| ПРОИЗВОДНЫЕ 3(2Н) - ПИРИДАЗИНОНА | 1992 |
|
RU2054422C1 |
| БЕНЗОПИРАНОВОЕ СОЕДИНЕНИЕ | 2005 |
|
RU2366658C2 |
| ГЕТЕРОЦИКЛИЧЕСКОЕ СОЕДИНЕНИЕ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СПОСОБ АНТАГОНИЗИРОВАНИЯ АНГИОТЕНЗИНА II | 1992 |
|
RU2168510C2 |
| СОЕДИНЕНИЕ ТИАЗОЛИДИНА ПИРИДИНОВОГО ТИПА ИЛИ ЕГО СОЛЬ, ГИПОГЛИКЕМИЧЕСКОЕ СРЕДСТВО, АНТИГЛИКАЦИОННОЕ СРЕДСТВО И ФАРМАЦЕВТИЧЕСКОЕ СРЕДСТВО, ИНГИБИРУЮЩЕЕ ГИПЕРГЛИКЕМИЮ, НЕФЕРМЕНТАТИВНУЮ ГЛИКАЦИЮ И АЛЬДОЗОРЕДУКТАЗУ, ДЛЯ ПРЕДУПРЕЖДЕНИЯ И ЛЕЧЕНИЯ САХАРНОГО ДИАБЕТА И ДИАБЕТИЧЕСКИХ ОСЛОЖНЕНИЙ | 1995 |
|
RU2125053C1 |
| ЗАМЕЩЕННЫЕ ПРОИЗВОДНЫЕ БЕНЗОПИРАНА ПРОТИВ АРИТМИИ | 2002 |
|
RU2291867C2 |
| ПРОИЗВОДНЫЕ 4-АМИНОБЕНЗОПИРАНА | 2002 |
|
RU2288226C2 |
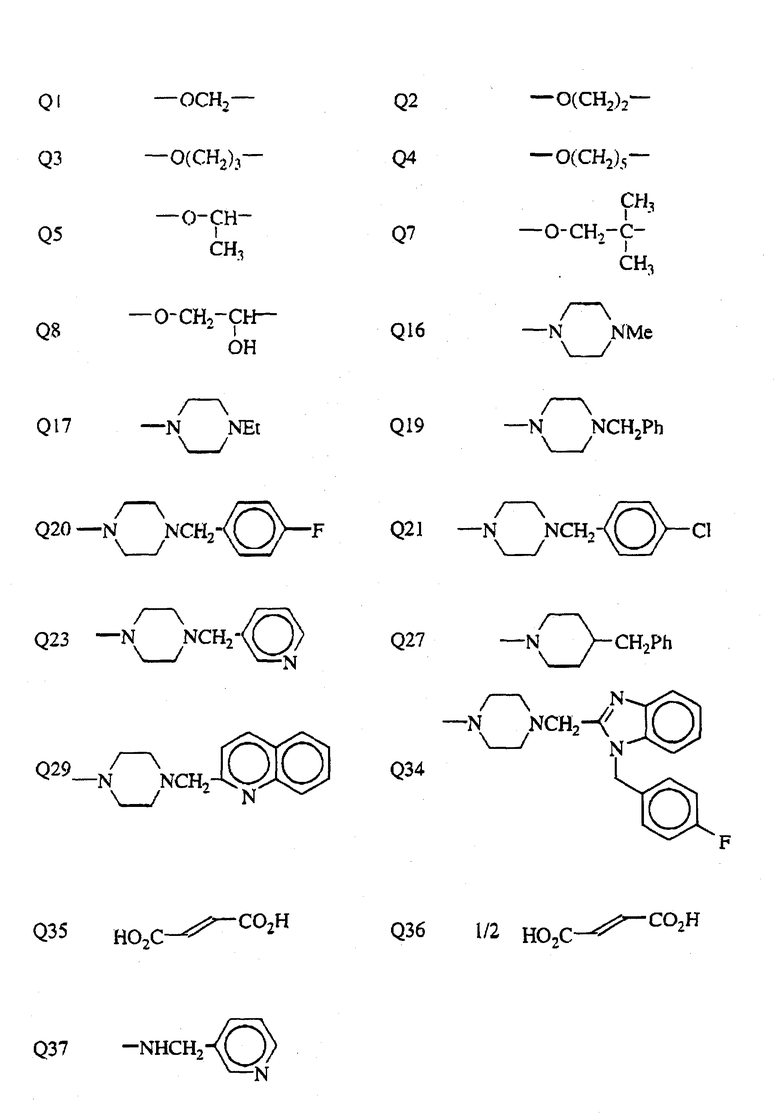
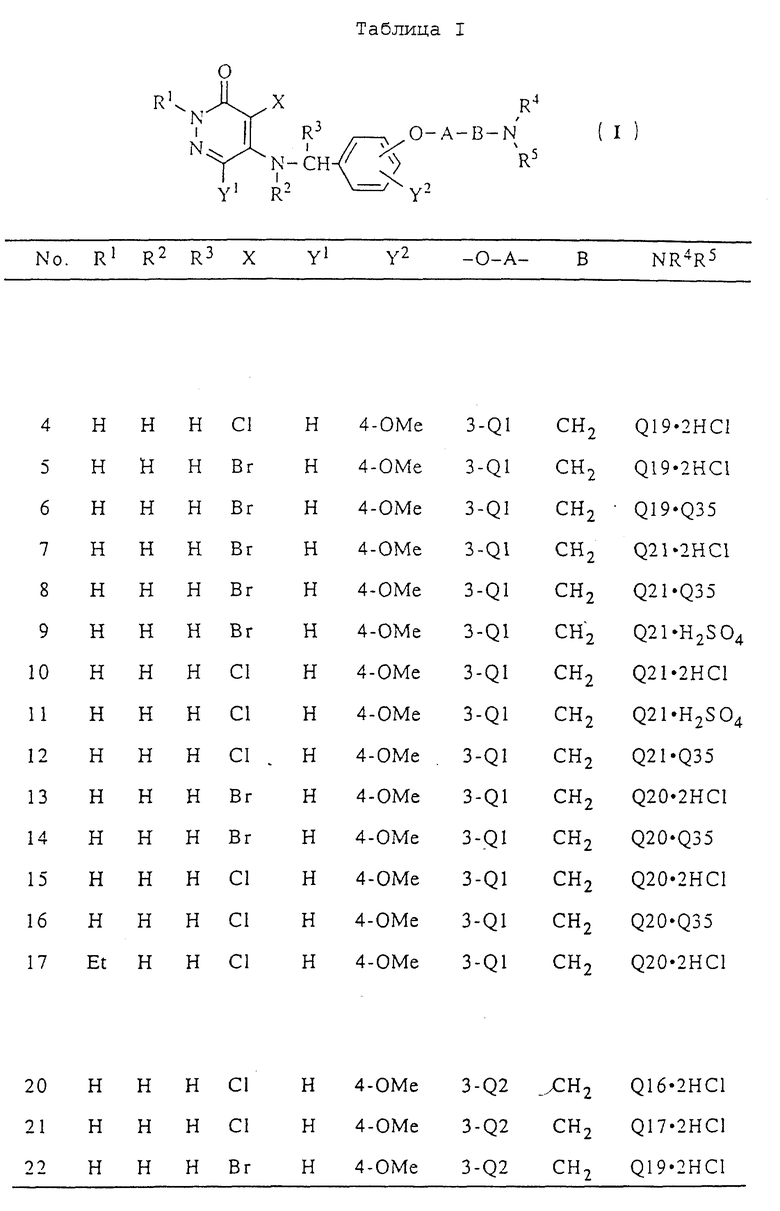
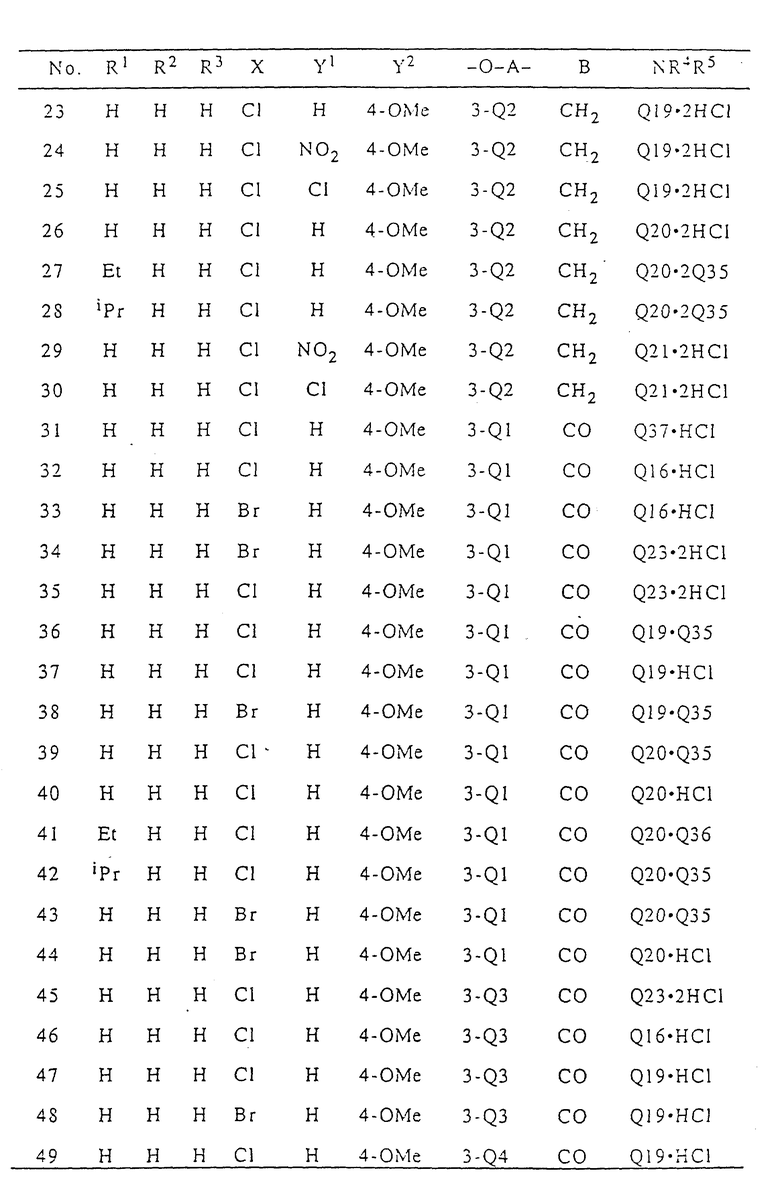
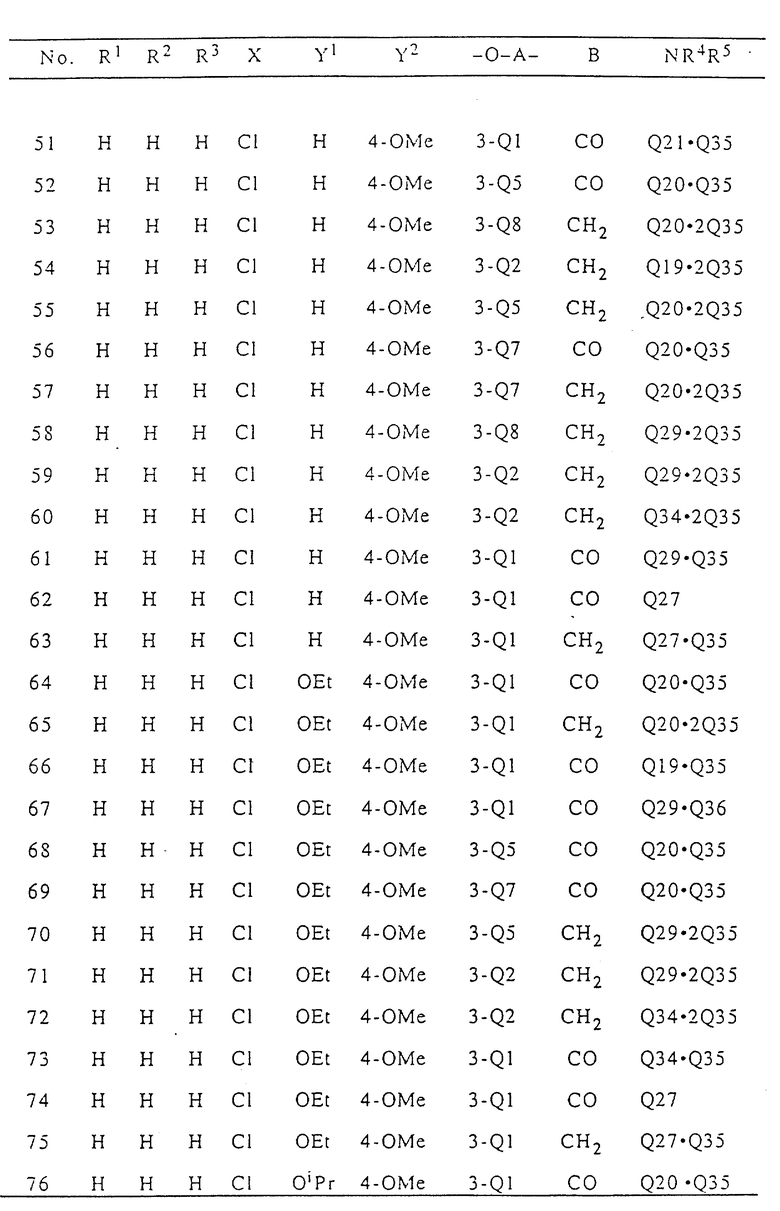
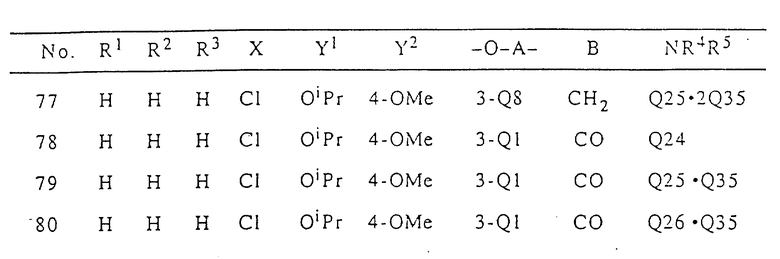
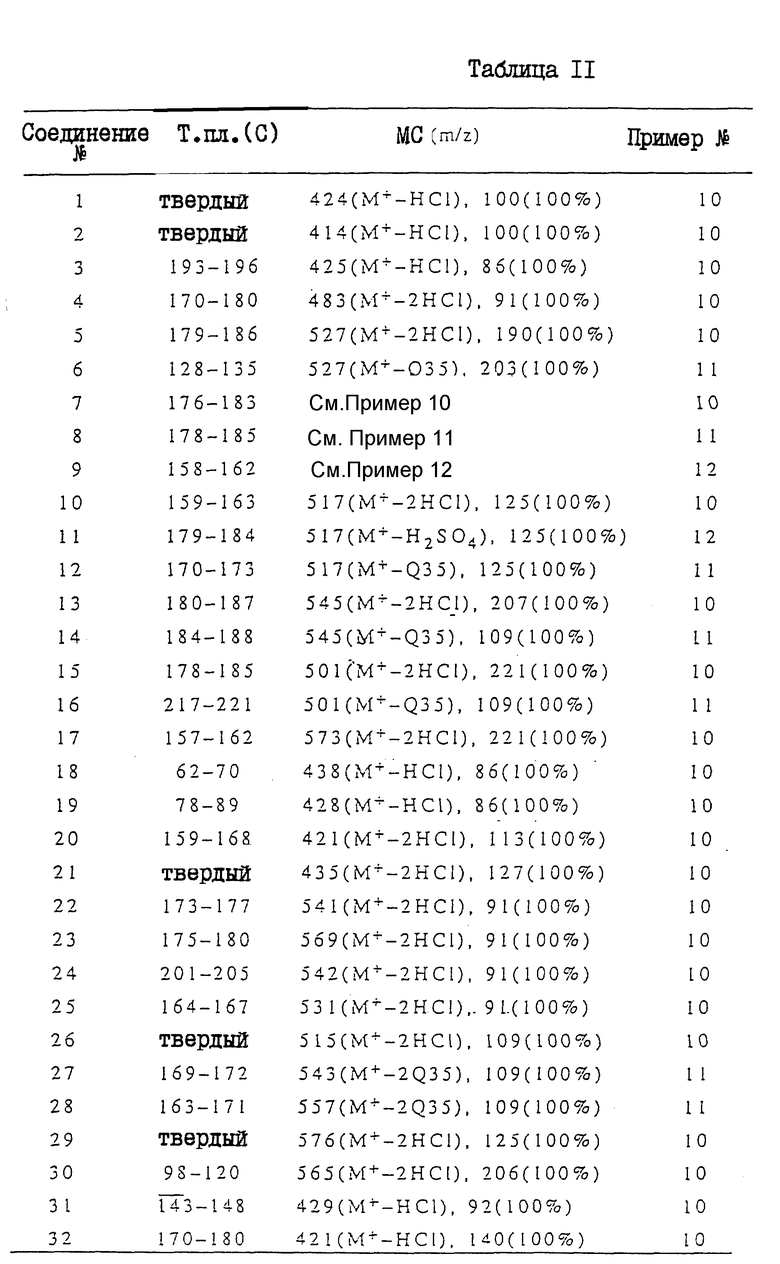

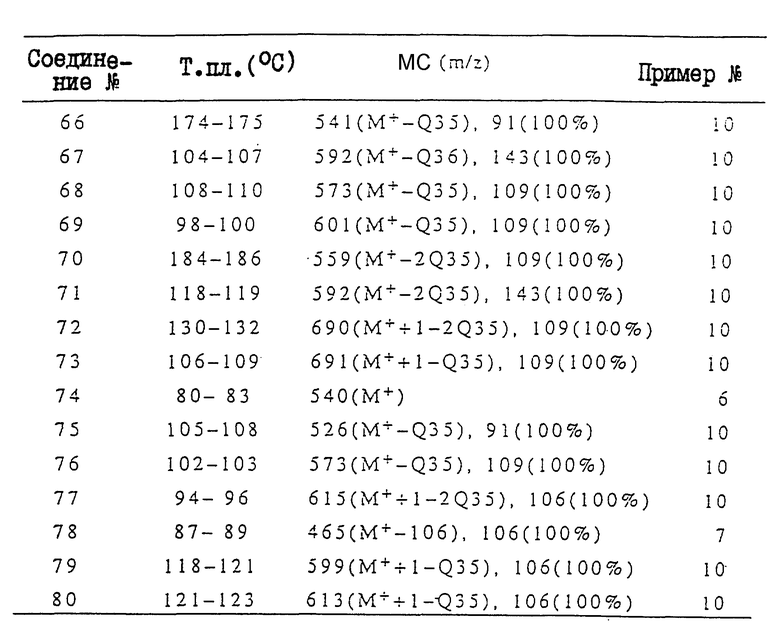
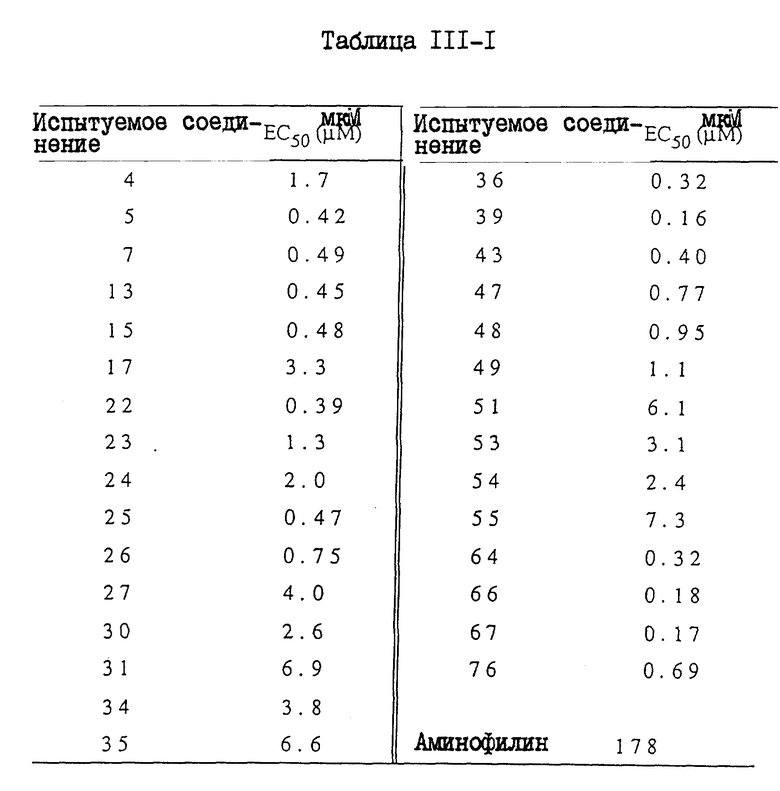
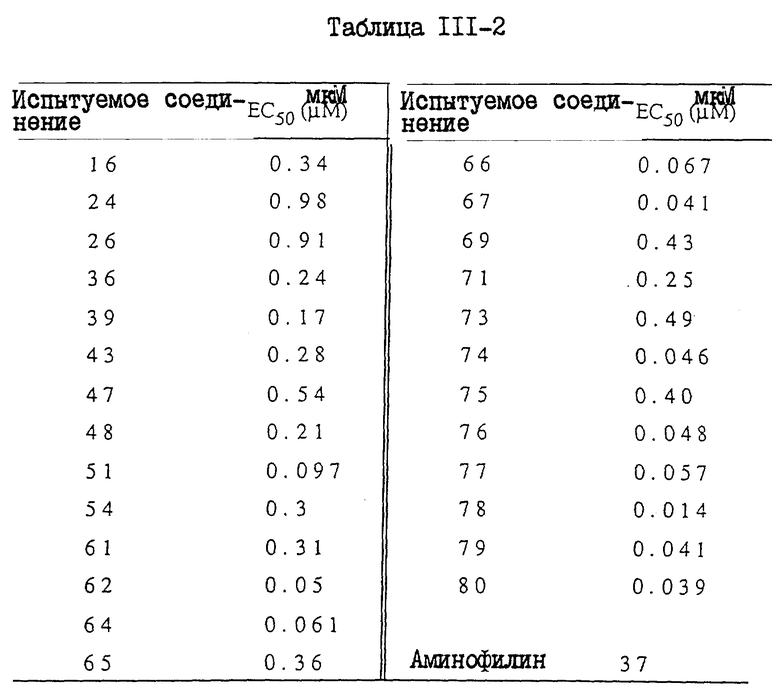

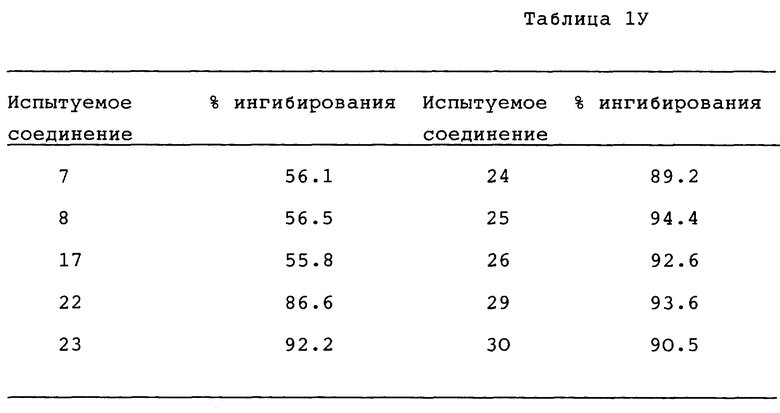
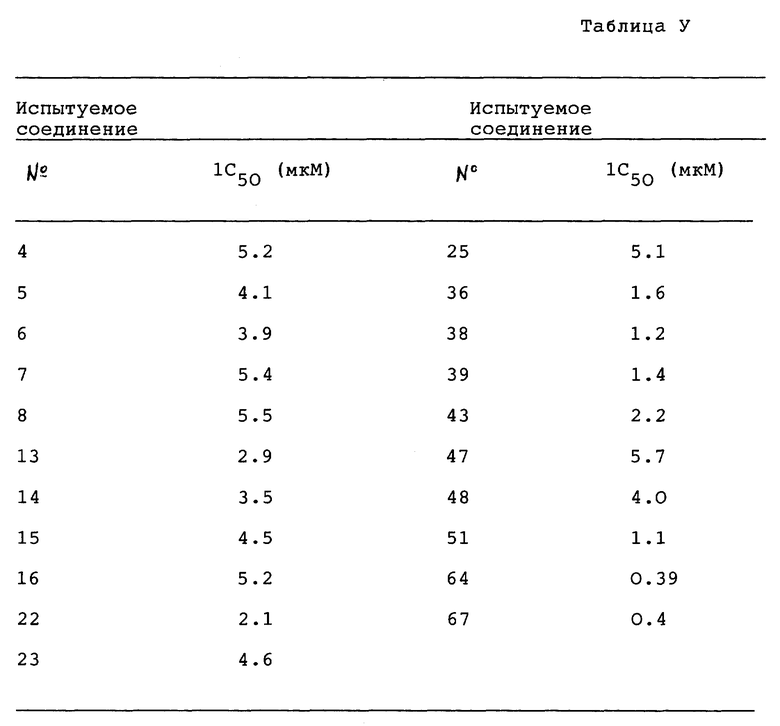
Представлено новое 3(2Н) -пиридазиноновое производное формулы I, где R1 представляет атом водорода или С1-4-алкил; каждый R2 и R3 представляет атом водорода; Х представляет атом хлора или атом брома; Y1 представляет атом водорода, атом галогена, нитрогруппу или С1-4-алкоксигруппу; Y2 представляет С1-4- алкоксигруппу; А представляет С1-5-алкиленовую цепь, которая может быть замещена гидроксильной группой; В представляет карбонильную группу или метиленовую цепь, которая может быть замещена С1-4-алкильной группой; а) R4 представляет атом водорода, R5 представляет Z-Ar, где Z представляет собой С1-5-алкиленовую цепь, а Ar представляет пиридил, либо в) R4 и R5, взятые вместе со смежным атомом азота, образуют 4-замещенное пиперазиновое кольцо формулы II, где R6 представляет собой С1-4-алкильную группу, которая может быть замещена фенильной группой, которая может быть замещена Y3, где Y3 является атомом галогена, аминогруппой, N-формиламиногруппой или С1-4-алкилкарбониламиногруппой; пиридилом, хинолином или группой формулы III, где R9 является С1-4-алкилом или бензилом, который может быть замещен атомом галогена, или с) R4 и R5 вместе со смежным атомом азота образуют 4-замещенное пиперидиновое кольцо формулы IV, где R11 - С1-4-алкил, который может быть замещен фенильной группой. Соединения обладают бронхорасширяющей активностью, противоаллергической и/или антитромботической активностью, описываются приемлемые аддитивные соли кислоты, а также фармацевтическая композиция на основе соединения формулы I. 2 с. и 1 з.п.ф-лы, 5 табл.
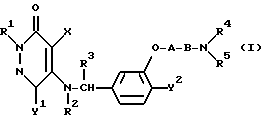
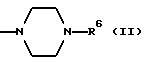
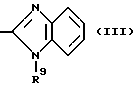
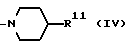
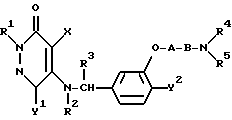
где R1 представляет атом водорода или С1-4-алкил;
каждый из R2 и R3 представляют атом водорода;
X представляет собой атом хлора или атом брома;
Y1 представляет собой атом водорода, атом галогена, нитрогруппу или С1-4-алкоксигруппу;
Y2 представляет собой С1-4-алкоксигруппу;
А представляет собой С1-5-алкиленовую цепь, которая может быть замещена гидроксильной группой;
В представляет собой карбонильную группу или метиленовую цепь, которая может быть замещена С1-4-алкильной группой;
а) R4 представляет собой атом водорода, а R5 представляет собой -Z-Ar, где Z представляет собой C1-5-алкиленовую цепь, а Ar представляет пиридил, либо в) R4 и R5, взятые со смежным атомом азота, образуют 4-замещенное пиперазиновое кольцо формулы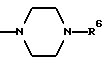
где R6 представляет собой С1-4-алкильную группу, причем эта алкильная группа может быть замещена фенильной группой, которая может быть замещена Y3, где Y3 является атомом галогена, аминогруппой, N-формиламиногруппой или С1-4-алкилкарбониламиногруплой; пиридилом; хинолилом или группой формулы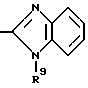
где R9 является С1-4-алкильной группой, или бензильной группой, которая может быть замещена атомом галогена, или с) R4 и R5 вместе со смежным атомом азота образуют 4-замещенное пиперидиновое кольцо формулы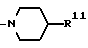
где R11 представляет собой С1-4-алкильную группу, которая может быть замещена фенильной группой,
или их фармацевтически приемлемые аддитивные соли кислоты.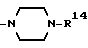
где R14 представляет собой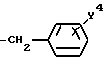
где Y4 является атомом водорода, атомом галогена, аминогруппой, N-формиламиногруппой или С1-4-алкоксикарбониламиногруппой, или
или
или
где R15 представляет собой бензильную группу, которая может быть замещена атомом галогена,
или их фармацевтически приемлемые аддитивные соли кислоты.
Приоритет по п.1 и п.3,
где R1, R2 и R3 имеют значения, определенные в п.1, Y1 представляет собой атом водорода, атом галогена, нитрогруппу, Y2 представляет собой С1-4-алкоксигруппу, А представляет собой С1-5-алкиленовую цепь, В представляет собой карбонильную группу или метиленовую цепь и R4 и R5 имеют значения, указанные для а) либо б) когда R4 и R5, взятые вместе со смежным атомом азота, образуют указанное 4-замещенное пиперазиновое кольцо, то R6 является С1-4 алкильной группой, которая может быть замещена фенильной группой, которая может быть замещена Y3, где Y3 является атомом галогена; или пиридилом или их фармацевтически приемлемые аддитивные соли кислоты - 29.06.93.
где R1, R2, R3, X, Y1 и Y2 имеют значения, определенные в п.1, А представляет собой С1-5алкиленовую цепь, которая может быть замещена гидроксильной группой, В представляет собой карбонильную группу или метиленовую цепь, которая может быть замещена С1-4алкильной группой, и R4 и R5 имеют значения, указанные для а), либо б) когда R4 и R5, взятые вместе со смежным атомом азота, образуют указанное 4-замещенное пиперазиновое кольцо, то R6 является С1-4 алкильной группой, причем эта алкильная группа может быть замещена фенильной группой, которая может быть замещена Y3, где Y3 является атомом галогена, аминогруппой, N-формиламиногруппой или С1-4 алкил карбониламиногруппой; пиридилом; хинолилом или группой формулы
где R9 является С1-4алкильной группой или бензильной группой, которая может быть замещена атомом галогена, или с) R4 и R5 вместе со смежным атомом азота образуют вышеуказанное 4-замещенное пиперидиновое кольцо, или их фармацевтически приемлемые аддитивные соли кислоты - 26.05.94.
| АВТОМАТИЧЕСКИЙ КЛАПАН ДЛЯ ПОЛИВНЫХ ТРУБОПРОВОДОВ | 0 |
|
SU186817A1 |
| Пневматический вибратор | 1973 |
|
SU482208A1 |
| Способ получения производных 3(2Н)-пиридазинона | 1988 |
|
SU1584750A3 |
| ПНЕВМАТИЧЕСКИЙ ГАЗОАНАЛИЗАТОР | 0 |
|
SU201765A1 |
| Мамковский М.Д | |||
| Лекарственные средства | |||
| -М.: Медицина, 1985, т | |||
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| Парный рычажный домкрат | 1919 |
|
SU209A1 |

