Настоящее изобретение относится к получению класса 5-гетероциклических замещенных триптаминов, например 5-(1,2,4-триазол-1-ил)триптаминовых соединений, терапевтически активных как противомигреневые агенты. Изобретение относится к усовершенствованному способу получения этих производных 5-гетероциклического замещенного триптамина, который включает катализируемое палладием связывание и замыкание кольца.
Краткое описание литературных данных
Сложные физиологические и патофизиологические процессы нейротрансмиттера серотонина (5-НТ) становятся все более понятными.1 (Ссылки, обозначаемые верхними индексами, перечислены в конце). В соответствии с одной из своих ролей серотонин действует как вазоконстриктор в головном мозге и тем самым проявляет благоприятные свойства при лечении мигрени. Его потенциал в качестве фармацевтического агента, однако, ограничен из-за его быстрого метаболизма in vivo. В течение последних нескольких лет множество попыток было посвящено разработке N, N-диалкилтриптаминов в качестве агонистов рецептора 5-HT1D для достижения желаемой активности и селективности при лечении мигрени. Суматриптан является первым из данного класса лекарств, одобренных для данной цели. 2 МК-0462 (разработанный фирмой Merck & Со.) описан в патенте США 5298520 и является также сильнодействующим агонистом рецептора 5-HT1D, который проходит клинические испытания.
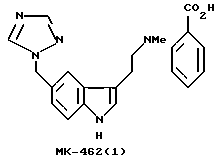
Обычно данный класс соединений получают с помощью индольной реакции Фишера для получения структуры N, N-диметилтриптамина. Применение данной методики для синтеза МК-0462, однако, является неэффективным и дает низкий выход из-за нестабильности бензилтриазольного остатка к условиям реакции, которая обычно ведет к полимеризации триазольного остатка, давая олигомеры. Желаемым в данной области является высоко эффективный способ получения N, N-диметилтриптамина, МК-0462(1), который устраняет нежелательную тенденцию к полимеризации триазолила.
Larock et al. показали, что циклизация йоданилинового фрагмента с имеющимся в структуре ацетиленом с использованием катализа палладием дает 2,3-дизамещенные индолы с выходами от хороших к абсолютным.3 Smith et al. также продемонстрировали это для 4-пиримидинильных и пиридинильных производных индол-3-ил-алкилпиперазинов, как это показано в публикации ЕР 0548831 A1. Два других применения данной методики продемонстрирированы при синтезе гетероконденсированных пирролов4a и триптофанов4b. Однако все эти методы требуют трифенилфосфина в качестве части системы катализатора, что является опасным для окружающей среды.
Применение методики катализируемого палладием связывания к конкретному синтезу 5-триазолил N, N-диметилтриптаминовой кольцевой системы не было описано ранее.
Краткое описание изобретения
Мы обнаружили, что МК-0462 может быть синтезирован с высоким выходом с помощью катализируемого палладием связывания/замыкания кольца 3-йод-4-аминобензилтриазола с защищенным подходящим образом производным бутинола и соответствующего триптофола с последующим превращением гидроксиэтильного остатка в диметиламиноэтил. Преимущество данного нового способа заключается в том, что он не требует использования трифенилфосфина, а также хлорида тетрабутиламмония и хлорида лития и устраняет тенденцию триазолильной полимеризации, имеющей место в индольном синтезе по Фишеру. В целом, способ может быть использован для получения 5-замещенных триптаминов, в которых 5-заместитель представляет собой триазолил, триазолилметил, имидазолил, имидазолилметил, тетразолил или тетразолилметил.
Согласно настоящему изобретению предоставляется способ, включающий стадию приведения в контакт соединения Структуры I с соединением Структуры II для образования соединения Структуры III: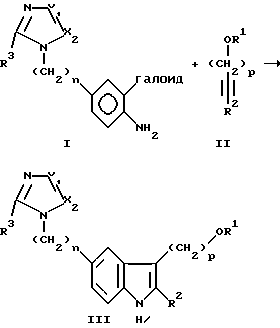
указанный процесс проводят в сухом инертном органическом растворителе для Структур I и II при температуре в пределах от около 70o до 120oC в присутствии палладиевого катализатора, растворимого в упомянутом растворителе, присутствующего в количестве от 0,5 до 5 молярных процентов относительно I, и в присутствии неорганического или органического соединения амина, которое выполняет функцию акцептора протона, то есть улавливателя кислоты, который не реагирует химически с упомянутым катализатором,
где X1 и X2 являются, независимо, кольцевыми атомами азота или углерода; галоид представляет Br или I; n является целым числом 0-1; p является целым числом 1-4; R3 является H или линейным или разветвленным C1-C4 алкилом; R1 является H или радикалом, который функционирует как гидрокси защитная группа, которая может быть удалена при гидролизе слабой кислотой, например, с помощью контакта со смесью HCI/MeOH, например 1:1 2 н. HCl/MeOH, при 0-30oC; R2 является радикалом, который функционирует как конечная защитная группа концевого углерода ацетилена, которая может быть удалена при гидролизе слабой кислотой, например, при контактировании со смесью HCI/MeOH, например 1:1 2 н. HCI/MeOH, при 0-30oC.
ОПИСАНИЕ ИЗОБРЕТЕНИЯ И ПРЕДПОЧТИТЕЛЬНЫХ ВОПЛОЩЕНИЙ
Синтез МК-462(1) иллюстрируется на следующей Схеме 1 ниже.
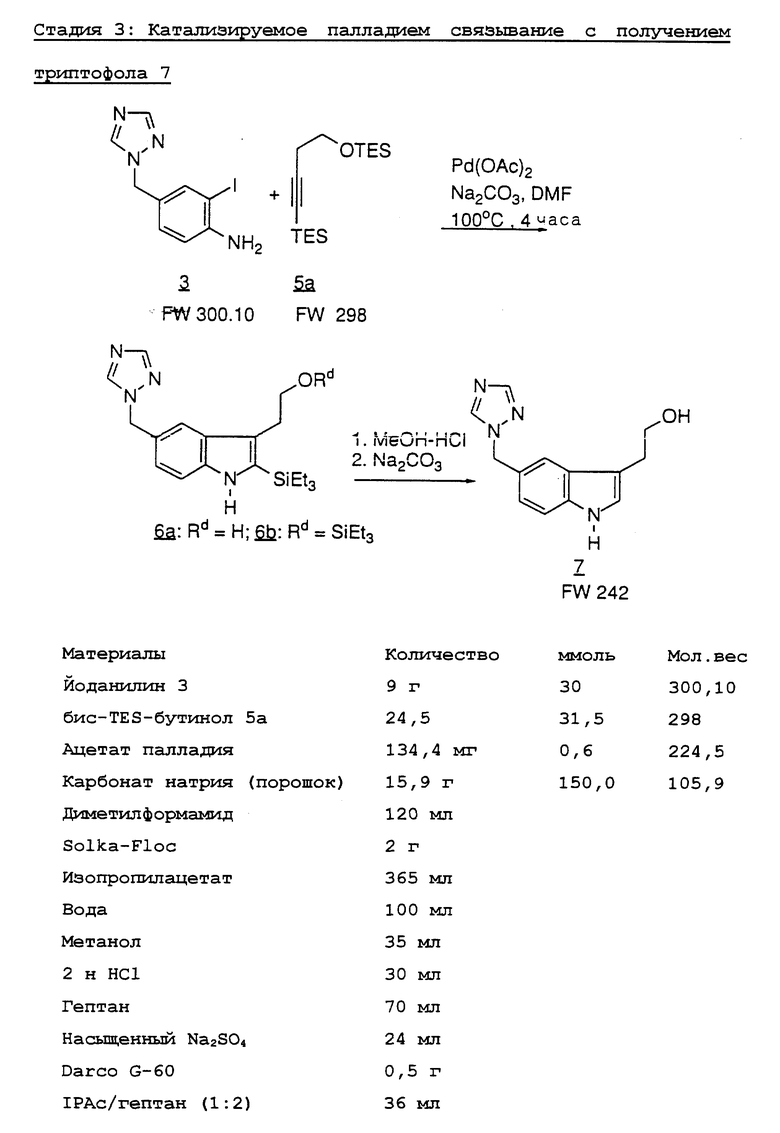
Ключевым элементом синтеза является получение предшественника триптофола 7, который может быть получен путем связывания 3-йод-4-аминобензилтриазола 3 с соответственно защищенным производным бутинола 5.
СХЕМА 1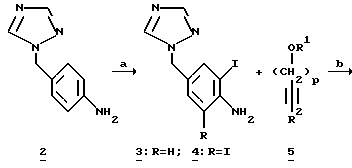
a Условия реакции: a) ICl, CaCO3, MeOH-H2O; b) 2 молярных процента Pd(OAc)2, Na2CO3, ДМФ, 100oC; с) MeOH-HCI; d)1. MsCl, Et3N, ТГФ; 2. 40% HNMe2; e) Бензойная кислота, изопропанол, комн. темп.
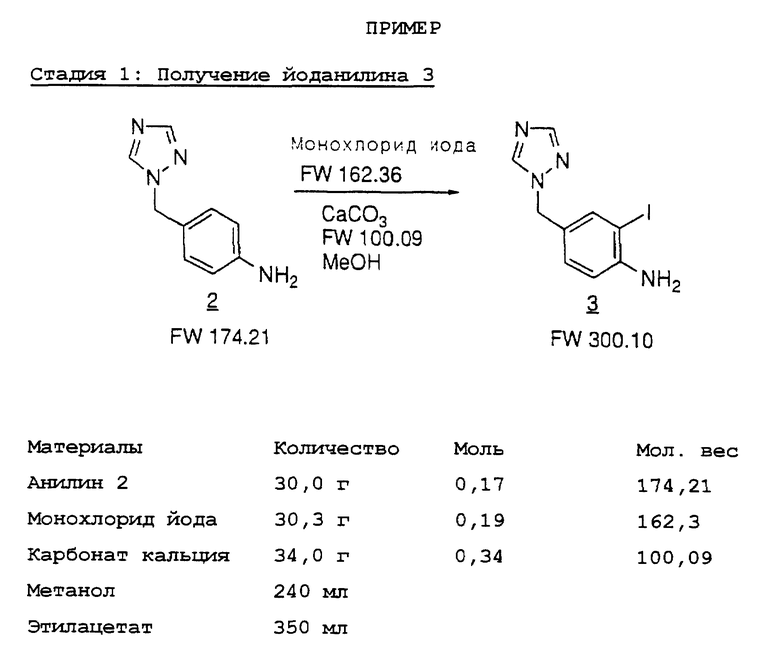
Синтез МК-462(1) начинают с получения йоданилина 3. 4- Аминобензилтриазол 2 доступен с помощью 3-стадийного способа с общим выходом >90% из 4-нитробензилбромида и 4-амино-1,2,4- триазола с использованием модифицированной процедуры, известной из литературы.5 Реакция 2 с монохлоридом йода в присутствии CaCO3 в водном метаноле дает 4-триазолилйоданилин 3 с выходом 91%; происходит некоторое дополнительное присоединение йода, давая 1-3% дийоданилина 4. Дополнительное присоединение йода нетрудно контролировать, поскольку это происходит гораздо более медленно.
Было найдено, что катализируемое палладием связывание/замыкание кольца между йоданилином 3 и бутинолом 5 протекает гладко с неожиданно высоким выходом в отсутствие стандартных требуемых реагентов трифенилфосфина, тетрабутиламмоний хлорида и литийхлорида, а также в отсутствии какой-либо индуцируемой триазолилом полимеризации.
Реакция связывания между йоданилином 3 и различными производными 3-бутин-1-ола подробно изучена в деталях (таблица). Было найдено, что для предотвращения связывания концевого углерода ацетилена необходима силильная защита. 3 Силильные группы вводят с помощью образования дианиона с BuLi с последующим гашением 2 эквивалентами силилхлорида. В случае TBDMS-защищенного (третичный бутилдиметилсилил) алкина бис-силилирование неожиданно не проходит до конца; а получается смесь 1:1 5d и 5e. Обнаружено, что альтернативная О-защита может быть произведена с помощью селективного гидролиза О-силильной группы; например 5a превращают в 5b с количественным выходом, используя разбавленную HCl в водном метаноле. Гидроксигруппа 5c может затем защищаться TBDMS или ТНР группой, давая алкины 5f и 5g соответственно с количественными выходами.
Самое простое производное 5c связывают с йоданилином 3, получая 2-ТМЗ-индол 6c с выходом 56%.6 Также образуется нежелательный региоизомер 9 (5%) (смотри ниже на Диаграмме 1). Образуются также другие нежелательные примеси. Считается, что ответственной за эти побочные продукты и низкий выход является ТМЗ-группа. Мы нашли неожиданно, что более стабильные TES и TBDMS группы на алкине дают индолы 6а и 6d8 соответственно с более высокими выходами (номера 1 и 4). Хотя более стабильная C-защита дает лучшие результаты, громоздкий TBDMS бутин связывается значительно медленнее; поэтому мы нашли, что особенно полезной защитной группой в этом конкретном синтезе является TES, поскольку она дает приемлемую скорость связывания и стабильность.
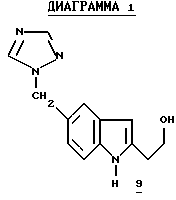
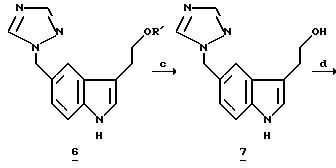
Десилилирование объединенных индолов 6a и 6b в MeOH-HCI дает триптофол 78 с общим выходом 70-80% после обработки и кристаллизации (Схема 1). Десилилирование 2-силилированных индолов может также осуществляться с другими кислотами, такими как алкановые кислоты, AlCl3, метансульфоновая кислота и другие сульфоновые кислоты. Однако мы обнаружили, что система MeOH-HCI является решительно более полезной и удобной для использования, особенно с точки зрения защиты окружающей среды. Превращение 3 и 5a в 7 производится непосредственно без выделения 6. Региоизомер 9 (6%) при кристаллизации 7 удаляется из маточной жидкости.
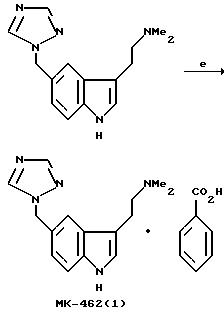
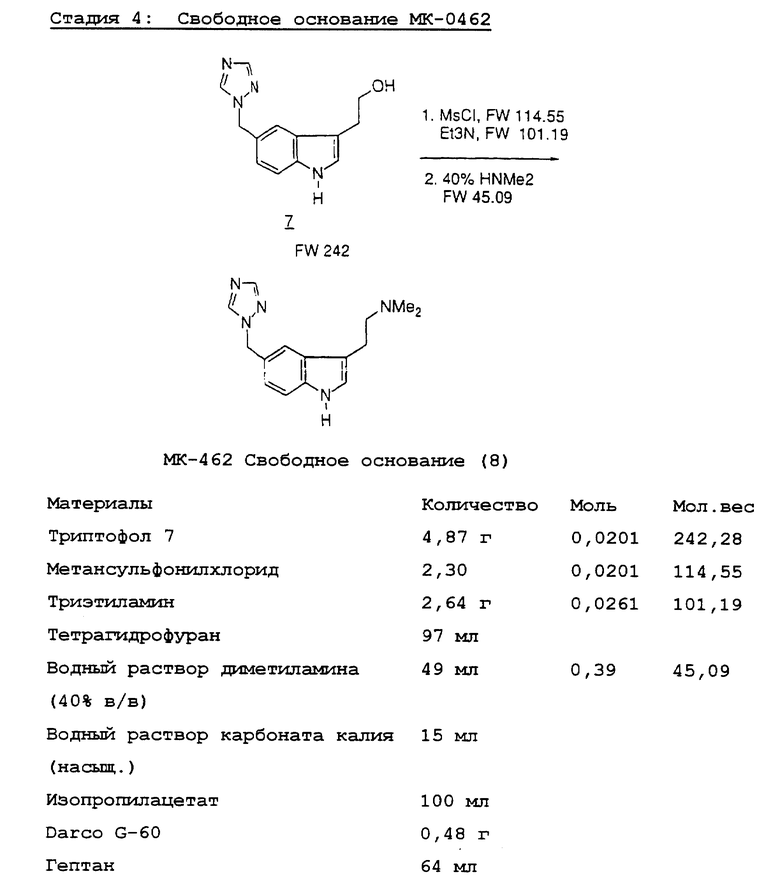
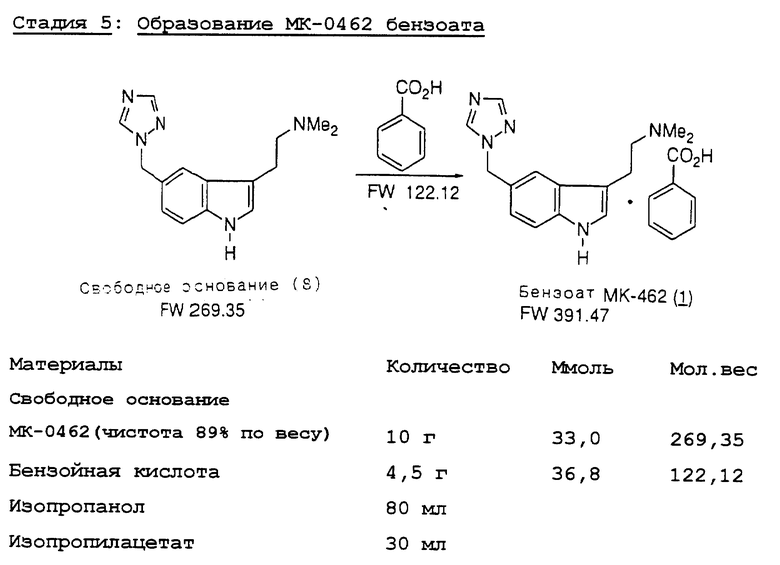
Превращение триптофола 7 в МК-0462 (1) включает образование мезилата из триптофола 7 с последующим замещением диметиламина, давая свободное основание МК-0462 88 с выходом 79%. Мы неожиданно обнаружили, что мезилат является склонным к полимеризации из-за внутримолекулярного алкилирования триазолом; поэтому мезилат обрабатывают непосредственно 40% диметиламином. Выделенный триптамин затем очищают путем добавления раствора бензойной кислоты к свободному основанию, получая МК-0462 в виде бензоатной соли с выходом 95%.
Этот новый синтез МК-0462(1), характеризуемый катализируемым палладием связыванием йоданилина 3 и бис-TES-бутинола 5a с образованием индольного кольца, является эффективным процессом, поддающимся увеличению масштаба, так что несмотря на образование нескольких примесей, неожиданно не требуется хроматографических очисток в противоположность стандартному индольному синтезу Фишера.
ССЫЛКИ И ПРИМЕЧАНИЯ
1. Glennon Т.A.; Darmani N.A; Martin B.R. Life Sciences 1991, 48,2493.
2. (a) Feniuk W.; Humpherey P. P. A. Drug Dev. Res. 1992, 26, 235; (b) Hopkins S.J. Drug of Today 1992, 28, 155.
3. Lorack R.C.; Yum E.K. J. Am.Chem.Soc. 1991, 113, 6689.
4. (a) Wensbo D.; Eriksson Jeschke T.; Annby U.; Gronowitz.; Cohen L.A. Tetrahedron Lett. 1993, 34, 2823. (b) Wensbo D.; Eriksson Jeschke Т.; Annby U.; Gronowitz.; Cohen L.A. Tetrahedron Lett. 1993, 34, 6471.
5. Astleford В. А., Goe G. L.; Keay J.G.; Scriven E. F.V. J.Org.Chem. 1989, 54, 731-732.
6. (а) 5c был куплен у Farchan Laboratories. (b) Pd (OAc)2 покупали у Johnson-Matthey.
7. Все новые соединения характеризуются с помощью 1H-ЯМР, 13C-ЯМР и элементного анализа. Избирательные данные (1H-ЯМР при 250 МГц, 13C-ЯМР при 62,5 МГц);
Индол 6b: 1H-ЯМР(CDCl3) δ: 0,90 (м, 15H), 1,60 (т, J=5,2 Гц, 1H), 3,09 (т, J=7,9 Гц, 2H), 3,85 (дт, J=7,9, 5,2 Гц, 2H), 5,40 (с, 2H), 7,10 (дд, J= 8,3, 1,4 Гц, 1H), 7,35 (д, J=8,3 Гц, 1H), 7,60 (д, J=1,4 Гц, 1H), 7,92 (с, 1H), 7,98 (с, 1H), 8,10 (с, 1H); 13C-ЯМР (MeOH-d4) 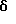 : 152,1, 144,5, 140,5, 134,0, 130,3, 126,2, 123,0, 122,3, 119,9, 112,7, 64,5, 55,3, 30,9, 7,9, 4,6; Вычислено: C19H27N5OSi: C 64,18; H 7,66; N 15,76, Hайдено: C 63,81; H 7,87; N 16,15.
: 152,1, 144,5, 140,5, 134,0, 130,3, 126,2, 123,0, 122,3, 119,9, 112,7, 64,5, 55,3, 30,9, 7,9, 4,6; Вычислено: C19H27N5OSi: C 64,18; H 7,66; N 15,76, Hайдено: C 63,81; H 7,87; N 16,15.
Триптофол 7: т. пл. 131-132oC; 1H-ЯМР (DMSO-d6) δ: 2,81 (т, J=7,4 Гц, 2H), 3,63 (дт, J= 7,4, 5,3 Гц, 2H), 4,65 (т, J=5,3 Гц, 1H), 5,43 (с, 2H), 7,00 (дд, J=8,4, 1,4 Гц, 1H), 7,15 (д, J=2,0 Гц, 1H), 7,51 (с, 1H), 7,94 (с, 1H), 8,62 (с, 1H), 10,85 (с, 1H); 13C-ЯМР (DMSO-d6) δ 151,3, 143,6, 135,7, 127,3, 125,8, 123,6, 121,1, 118,3, 111,7, 111,4, 61,5, 53,0, 28,7; Вычислено: C13H14N4O: C 64,44; H 5,82; N 23,12, Hайдено: C 64,38; H 5,85; N 23,28.
Триптамин 8: т. пл. 120-121oC; 1H-(DMSO-d6) δ: 2,34 (с, 6H), 2,63 (м, 2H), 2,93 (м, 2H), 5,43 (с, 2H), 7,05 (м, 2H), 7,31 (д, J=8,3 Гц, 1H), 7,56 (с, 1H), 7,97 (с, 1H), 7,99 (с, 1H), 8,49 (с, 1H); 13C-ЯМР (CDCl6) δ: 151,7, 142,8, 136,4, 127,7, 124,5, 123,1, 121,9, 119,1, 113,9, 112,0, 60,2, 54,6, 45,3, 23,5; Вычислено: C15H19N5: C 66,89; H 7,11; N 26,00, Hайдено: C 66,89; H 7,20; N 26,04.
Описанный выше специфический синтез МК-462 может также быть распространен на другие активные аналоги, содержащие имидазол, триазол или тетразол в 5 позиции индольного кольца, присоединенный через концевой атом азота, через метиленовую группу или присоединенный непосредственно к 5 положению индольного кольца как проиллюстрировано на следующей схеме: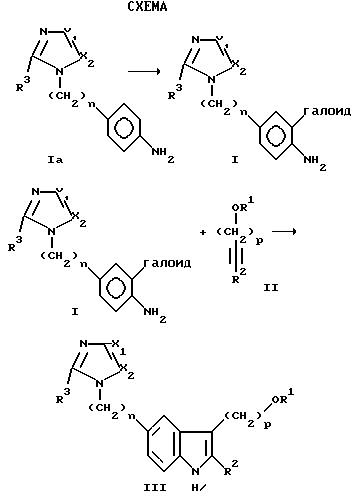
На начальной стадии процесса Cтруктура Ia подвергается реакции с галоидирующим агентом, образуя Cтруктуру I, при температуре примерно от -10 до 10oC в подходящем растворителе и в присутствии подходящего акцептора протона.
Галоидирующим агентом может быть, например, монохлорид йода, N-йодсукцинимид, N-бромсукцинимид и тому подобное.
Термином "галоид", как он здесь используется, обозначается Br или I.
Значениями для "n" являются 0, 1, а значениями для "p" являются 1, 2, 3 и 4.
Растворителем на данной стадии может быть MeOH, MeOH-H2O, EtOH, THF-H2O, CH2Cl2 и аналогичные, и полезным растворителем является 95% MeOH-H2O.
Полезные акцепторы протона, которые могут использоваться, включают: CaCO3, K2CO3, Na2CO3, Li2CO3, LiOH, KOH, NaOH, NaHCO3 и аналогичные. Когда используют N-бромсукцинимид или N- йодсукцинимид, отдельный акцептор протона не требуется.
Полезным набором условий реакции для стадии галоидирования является MeOH-H2O (95%) в качестве растворителя, температура около 0oC, посредством чего реакция осуществляется при атмосферном давлении в инертной атмосфере, например в атмосфере сухого N2, в присутствии карбоната кальция.
Структуру II, являющуюся защищенным 1-алкинолом, получают с помощью реакции исходного 1-алкинола IIa, который может быть выбран из 2-пропин-1-ола (пропаргиловый спирт), 3-бутин-1-ола, 4-пентин- 1-ола и 5-гексин-1-ола: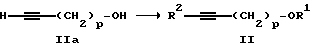
где p= 1-4, в подходящем инертном органическом эфирном растворителе, например тетрагидрофуране, диоксане, диэтиловом эфире, 1,2- диметоксиэтане и тому подобных, в сухой атмосфере, например в атмосфере сухого N2, при температуре от -50oC до -10oC при небольшом избытке н-бутиллития, составляющем около 2,1 молей на моль алкинола, в течение достаточного времени, например 2-8 часов, для полного генерирования дилитийаниона алкинола. Затем защитная группа присоединяется с помощью добавления предшественника, например хлортриметилсилана, также с небольшим избытком, около 2,1 моля на моль литийдианиона алкинола, и обеспечивается возможность перемешивания в течение 1-4 часов для завершения реакции. Реакционная смесь обрабатывается с помощью обычных процедур, давая дизащищенный алкинол II.
R1 защитная группа может быть селективно удалена с помощью гидролиза слабой кислотой, например, при перемешивании в смеси около 1:1 по объему 2 н. HCl/MeOH при температуре ниже 30oC, например 0-30 oC, и выделении продукта. Полученный в результате спирт может селективно защищаться еще одной защитной группой, R1, следуя описанной выше процедуре защиты, давая II, где R1 и R2 являются различными защитными группами.
Силилирующими агентами, которые могут использоваться, являются обычно галоидированные тригидрокарбилсиланы, например хлортриэтилсилан.
Тетрагидропиранильная, ТГФ, защитная группа может применяться с использованием исходного соединения дигидропирана в качестве предшественника в присутствии кислотного катализатора, например п-CH3PhSO2OH, для преобразования алкинола в ТНР эфир.
Структуру I затем связывают со Структурой II для получения Структуры III с помощью реакции, катализируемой палладием, в сухом инертном органическом растворителе, содержащем растворимый палладиевый катализатор, и в присутствии акцептора протонов, представляющего ароматический амин, алкиламин или неорганическое основание, который не является ядом катализатора, при температуре примерно от 70 до 120oC.
В Структуре III R3 является H или C1-C4 линейным или разветвленным алкилом, включающим метил, этил, н-пропил, изопропил, н-бутил, втор-бутил, изобутил и трет-бутил.
R1 является H или гидрокси защитной группой, выбранной из: силильного лиганда SiR3 a, где каждый Ra независимо выбран из линейного или разветвленного C1-C4алкила (как описано выше) или фенила; и тетрагидропиранила.
Представительные примеры радикалов SiR3 a включают триметилсилил, триэтилсилил, трибутилсилил, трифенилсилил, диметил-трет-бутилсилил, диметилфенилсилил, дифенилметилсилил, триизопропилсилил и тому подобные.
R2 ведет себя как защитная группа конечного атома углерода ацетилена и имеет такую же структуру, как описанный выше SiR3 a.
Как R1, так и R2 являются удаляемыми путем гидролиза с помощью слабой кислоты, например путем контактирования со смесью растворителей 2 н. HCI/MeOH около 1: 1 по объему при температуре около 0-30 oC в течение 1-24 часов для полного удаления R1, R2 радикалов.
Органическим растворителем, пригодным на данных стадиях связывания/замыкания кольца, должен быть растворитель, в котором Структура I, Структура II и палладий-катализатор являются растворимыми и совместимыми и который химически инертен в условиях реакции.
Классами растворителей, пригодными в этих реакциях, являются N,N-ди(C1-C4) C1-C2-алканоамиды, C4-C8 линейные эфиры, C4-C6 циклические моно- или диэфиры, ди C1-C4 алкоксиэтаны, C6-C10 ароматические углеводороды, моно- или дихлор C1-C4 алканы, алкилнитрилы и аналогичные или их смеси.
Характерные представители растворителей включают: диметилформамид, диметилацетамид, диэтилэфир, дипропилэфир, тетрагидрофуран, диоксан, 1,2-диметоксиэтан, бензол, толуол, о- ксилол, м- ксилол, п- ксилол, ацетонитрил, пропионитрил и аналогичные или их смеси.
Температуру поддерживают в пределах от 70 до 120oC. Подходящей является температура в пределах примерно 90-100oC. Обычно реакцию проводят в сухом N2 при атмосферном давлении.
Палладиевый катализатор, полезный в реакции, может быть выбран, например, из следующих классов: Pd алканоаты, Pd ацетонаты, Pd галогениды, Pd галогенидные комплексы, Pd- бензилиденацетоновые комплексы и аналогичные. Представительные примеры включают: Pd (II) ацетат, Pd(II) ацетилацетонат, Pd(O)бис-дибензилиден ацетон, Pd(II)бромид, Pd (II) хлорид, Pd (II) йодид, Pd (II) сульфат, Pd (II) трифторацетат, Pd(II)Cl2(CH3CN)2 и аналогичные. Подходящим катализатором является палладий ацетат.
Палладиевый катализатор используют в количестве от около 0,5 до 5 молярных процента по отношению к йоданилину 1, и полезный интервал составляет примерно от 2 до 3 молярных процентов растворимого палладиевого катализатора по отношению к йоданилину 1.
Акцептором протона, пригодным на данной стадии, является основное соединение, которое может быть органическим или неорганическим, действует как акцептор протона и не является "ядом катализатора". С помощью термина "яд катализатора" обозначают взаимодействие с катализатором, которое подавляет его каталитическую активность и предотвращает из-за данного связывание/замыкание кольца между Структурами I и II.
Подходящие классы акцепторов протона включают алкиламины, ароматические амины, гетероциклические амины, карбонаты, бикарбонаты, фосфаты, бифосфаты щелочных металлов I Группы и щелочноземельных металлов II Группы и аналогичные.
Представительные соединения включают литий карбонат, натрий карбонат, калий карбонат, натрий бикарбонат, калий бикарбонат, кальций карбонат, триэтиламин, диизопропилэтиламин, пиридин, N,N- диметиланилин, 4-диметиламинопиридин и тому подобное.
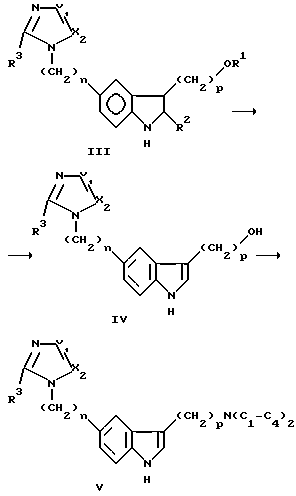
Удаление защитных групп R1 и R2 из III обычно выполняется с помощью гидролиза слабой кислотой без отдельного выделения III. Когда связывание/замыкание кольца заканчивается, растворители обычно удаляются при пониженном давлении. Смесь 2 н. HCl/MeOH около 1:1 по объему добавляют к концентрату III при комнатной температуре, и смеси позволяют перемешиваться при температуре ниже 30oC, например при 0-30oC, в течение 2-4 часов для полного удаления обеих R1 и R2 защитных групп с получением IV.
Замещение гидроксила в IV диалкиламином с получением V обычно проводится в две стадии в одном реакционном сосуде.
Спирт IV может подвергаться реакции с мезилхлоридом, (CF3SO2)2O и аналогичными в сухом инертном органическом растворителе, например тетрагидрофуране, диоксане, диэтилэфире, 1,2- диметоксиэтане, дихлорметане и аналогичных, при температуре примерно от -30 до -10oC в атмосфере сухого N2 в присутствии акцептора протона, представляющего растворимый алифатический или ароматический амин, например триэтиламин, пиридин, диэтилметиламин, диизопропилэтиламин, трибутиламин, 4- диметиламинопиридин и аналогичные, с образованием промежуточного мезилата или сульфоната in situ.
Диалкиламиновый аналог V может затем получаться с помощью простого добавления диалкиламина к мезилатному содержимому в реакционном сосуде и последующего перемешивания при комнатной температуре в течение 1 часа.
Полученная Структура V может отделяться как она есть или подвергаться реакции с соответствующими фармацевтически приемлемыми кислотами, например HCl, H2SO4, бензойной кислотой, янтарной кислотой, молочной кислотой, малеиновой кислотой и аналогичными, с получением соответствующей аддитивной соли кислоты.
Характерными примерами Структуры V, которые могут быть получены с помощью данного процесса, являются:
N,N-Диметил-2- [5- (1,2,4-триазол-1-илметил) -1H-индол-3-ил] -этиламин;
N,N-Диметил-2- [5- (1,3-имидазол-1-илметил) -1H-индол-3-ил] - этиламин;
N, N-Диметил-2- [5- (5-метил-1,2, 3, 4-триазол-1-илметил) - 1H-индол-3-ил] этиламин;
N,N-Диметил-2- [5- (1,3,4-триазол-1-илметил) -1H-индол-3-ил] - этиламин;
N,N-Диметил-2- [5- (I, 3,4-триазол-1-ил) -1H-индол-3-ил] этиламин;
N,N-Диэтил-2- [5- (1,2,4-триазол-1-илметил) -1H-индол-3- ил] -этиламин;
N,N-Диэтил-2- [5- (1,3-имидазол-1-илметил) -1H-индол-3-ил] - этиламин;
N, N-Диэтил-2- [5- (5-метил-1,2,3,4-тетразол-1-илметил) -1H- индол-3-ил] этиламин;
N,N-Диэтил-2- [5- (1,3,4-триазол-1- илметил) -1H-индол-3-ил] -этиламин;
N,N-Диэтил-2- [5- (1,3,4-триазол-1-ил) -1H-индол-3-ил] этиламин;
N,N-Диметил-[5- (1,2,4-триазол-1-илметил) -1H-индол-3-ил] - метиламин;
N,N-Диметил- [5- (1,3-имидазол-1-илметил) -1H-индол-3-ил] - метиламин;
N, N-Диметил- [5- (5-метил-1,2,3,4-тетразол-1-илметил) -1H- индол-3 -ил] метиламин;
N,N-Диметил- [5- (1,3,4-триазол-1- илметил) -1H-индол-3-ил] -метиламин;
N,N-Диметил- [5- (1,3,4-триазол-1-ил) -1H-индол-3-ил] метиламин;
N, N-Диэтил-3- [5- (1,2,4-триазол-1-илметил) -1H-индол-3- ил] -пропиламин;
N,N-Диметил-3- [5- (1,3-имидазол-1-ил) -1H-индол-3-ил] - пропиламин;
N, N-Диэтил-3- [5- (5-метил-1,2,3,4-тетразол-1-илметил) -1H- индол-3-ил] пропиламин;
N, N-Диметил-3- [5- (1,3,4-триазол-1-илметил) -1H-индол-3-ил] -пропиламин;
N,N-Диэтил-3- [5- (1,3,4-триазол-1-ил) -1H-индол-3-ил] пропиламин;
N,N-Диметил-4- [5- (3-метил-1,2,4,5-тетразол-1- илметил) -1H-индол-3-ил] бутиламин;
N,N-Диметил-4- [5- (2-этил-1,3-этил-имидазол-1-илметил) -1H- индол-3-ил] бутиламин;
N, N-Диметил-4- [5- (5-этил-1,2,3,4-триазол-1-илметил) -1H- индол-3-ил] бутиламин;
N, N-Диметил-4- [5- (2-метил-1,3,4-триазол-1-илметил) -1H- индол-3 -ил] бутиламин;
N, N-Диметил-4- [5- (2-этил-1,3,4-триазол-1-ил) -1H-индол-3-ил] бутиламин.
Включены также спиртовые аналоги описанных выше аминов, включающие, например,
2- [5- (1,2,4-триазол-1-илметил)-1H-индол-3-ил] этиловый спирт;
2- [5- (1,3-имидазол-1-илметил)-1H-индол-3-ил] этиловый спирт;
2-[5- (5-метил-1,2,3,4 -тетразол-1-илметил) -1H-индол-3-ил] этиловый спирт;
2- [5- (1,3,4-триазол-1-илметил) -1H-индол-3-ил] этиловый спирт;
2-[5-(1,3,4-триазол-1-ил)-1H-индол-3-ил]этиловый спирт;
[5-(1,2,4-триазол-1-илметил)-1H-индол-3-ил]метиловый спирт;
3-[5- (1,3-имидазол-1-илметил)-1H-индол-3-ил]пропиловый спирт;
4 -[5- (5- метил-1,2,3,4 -тетразол-1-илметил) -1H-индол-3-ил] бутиловый спирт;
2-[5- (2-метил-1,3,4-триазол-1-илметил) -1H-индол-3-ил] - этиловый спирт;
2- [5- (5-метил-1,3,4-триазол-1-ил) -1H-индол- 3-ил] этиловый спирт.
Следующий пример иллюстрируют изобретение как оно представлено изобретателями и не предназначен для ограничения объема или сути настоящего изобретения.
Стадия 1. К смеси истолченного в порошок карбоната кальция (34 г, 0,34 моль) и анилина 2 (30,0 г, 0,17 моль) в метаноле (240 мл) и воде (12 мл) добавляют при 0oC в атмосфере азота раствор монохлорида йода (30,3 г, 0,19 моль) в метаноле (120 мл) на протяжении 0,5 часа.
Смесь подогревают до комнатной температуры и гасят полунасыщенным раствором тиосульфата натрия (5 мл). Смесь перемешивают в течение 30 минут. Твердый продукт отфильтровывают и промывают этилацетатом (100 мл).
Фильтрат концентрируют в вакууме до 100 мл, разбавляют этилацетатом (250 мл), промывают полунасыщенным тиосульфатом натрия (200 мл), сушат над сульфатом магния и концентрируют до 100 мл. Добавляют гексаны для осаждения йоданилина 3 в виде бледно-рыжевато-коричневого твердого продукта (48,5 г, 91%).
Перекристаллизация йоданилина 3 (24 г) из этанола дает йоданилин 3 (14,5 г, 60% извлечение) в виде белого порошка; т.пл. 114-115oC.
Стадия 2. Сухой тетрагидрофуран (15,9 л) загружают в колбу, снабженную механической мешалкой и термопарой в атмосфере азота, и загружают в колбу 3-бутин-1-ол (958,5 г, 13,68 моль). Смесь охлаждают до -30oC и добавляют по каплям на протяжении 4 часов н-BuLi (17,1 л, 27,36 моль), поддерживая температуру ниже - 20oC.
Смесь выдерживается при -20oC в течение 1,2 часа. Добавляют по каплям на протяжении 55-60 минут хлортриэтилсилан (4,218 кг, 28,04 моль), сохраняя реакционную смесь при температуре ниже - 10oC. Затем смеси позволяют подогреться до комнатной температуры. Реакция завершается через 1,5 часа при приблизительно 22oC.
Раствор охлаждают до -10oC и добавляют 1% (вес/вес) Na2CO3 (8,4 л) на протяжении 25 минут при температуре <0oC. Добавляют гептан (10 л) и слои разделяют. Водный слой экстрагируют гептаном (10 л). Органические слои объединяют и промывают водой (22 л), и концентрируют до бледного оранжево-желтого масла, получая продукт 5a (выход 98,1%, чистота 93,8 вес.%).
Стадия 3. К сухому диметилформамиду (12 мл) добавляют раствор бис-TES-бутинола 5a в гептане (24,5 г, 31,5 ммоль, 40% по весу). Смесь концентрируют в вакууме до объема 22 мл. К концентрату добавляют диметилформамид (78 мл), йоданилин 3 (9 г, 30 ммоль) и карбонат натрия (15,9 г, 0,15 моль) в виде порошка. Смесь дегазируют с вакуум/азотной откачкой.
Добавляют ацетат палладия (134,4 мг, 0,6 ммоль), и смесь подогревают до 100oC в течение 4 часов.
Смесь продуктов охлаждают до комнатной температуры и отфильтровывают через Solka-Floc. Лепешку промывают диметилформамидом (30 мл). Объединенный фильтрат и промывочную жидкость перегоняют при 26 мм рт.ст. (т. кип. ДМФ: 67oC) до ≈ 25 мл, удаляя ≈ 100 мл дистиллята. Добавляют изопропилацетат (IPAC) (150 мл) и воду (50 мл) к дистилляционному остатку. Полученную в результате смесь отфильтровывают через 2 г Solka-Folc, и лепешку промывают изопропилацетатом (15 мл). Объединенные фильтраты промывают водой (50 мл) и концентрируют до 50 мл.
Вышеуказанный концентрат разбавляют метанолом (35 мл), и через 20 минут добавляют 2 н. HCl (30 мл, 2 экв), поддерживая температуру реакционной смеси ниже 30oC. Смесь выдерживают при комнатной температуре в течение 2 часов или до тех пор, пока реакция не будет полной.
Добавляют гептан (36 мл) и гептан-изопропилацетатный слой отделяют. Метанольно-водный слой, содержащий продукт 7, концентрируют в вакууме до 65 мл с удалением метанола (20 мл).
Добавляют к смеси изопропилацетат (50 мл). Смесь охлаждают до 18oC с последующим добавлением насыщенного водного карбоната натрия (24 мл) на протяжении 10 минут. Добавляют к смеси изопропилацетат (50 мл). Водный слой отделяют и экстрагируют изопропилацетатом (100 мл). Объединенный органический раствор (200 мл) обрабатывают Darco G-60 (0,5 г). Смесь перемешивают в течение 5 часов и отфильтровывают. Фильтрат концентрируют до 100 мл, получая мелкодисперсную суспензию, с последующим добавлением гептана (34 мл). Суспензию выдерживают при комнатной температуре в течение 1 часа. Твердый продукт отфильтровывают и промывают гептаном/изопропилацетатом (2:1; 36 мл). Продукт сушат, получая триптофол 7 (5,5 г, 75%). ЯМР данные и аналитические данные по C, H, N представлены выше в Ссылках и примечаниях".
Стадия 4. Триптофол 7 (4,87 г) суспендируют в сухом тетрагидрофуране (97 мл) и добавляют высушенный на молекулярном сите триэтиламин (2,64 г, 26,1 ммоль). Суспензию охлаждают до -20oC и добавляют метансульфонилхлорид (2,30 г, 20,1 ммоль) при температуре <-15oC на протяжении 45 минут. Реакционную смесь выдерживают в течение 30 минут при -20oC.
Суспензию отфильтровывают при температуре <-15oC и отфильтрованную лепешку промывают холодным безводным тетрагидрофураном (25 мл).
Водный раствор диметиламина (40%, в/в, 49 мл, 0,39 моль) добавляют к объединенным фильтратам. Реакционной смеси дают возможность подогреться до комнатной температуры.
Большую часть ТГФ удаляют с помощью дистилляции в вакууме при температуре <30oC (окончательный объем 60 мл). Добавляют изопропилацетат (50 мл) и насыщенный водный раствор карбоната калия (5 мл). Слои хорошо перемешивают и разделяют. Водный слой экстрагируют изопропилацетатом (50 мл).
Объединенные органические слои промывают насыщенным водным раствором карбоната калия (10 мл). К разбавленному органическому слою добавляют изопропилацетат (20 мл), и раствор продукта сушат с помощью нагревания с обратным холодильником с использованием ловушки Дина-Старка. Раствор охлаждают и обрабатывают Darco G-60 (0,5 г) в течение 60 минут и смесь фильтруют. Фильтраты концентрируют до 20 мл с помощью перегонки в вакууме, вносят затравку и оставляют кристаллизоваться в течение > 1 часа. К осажденному слою на протяжении 1 часа добавляют гептан (64 мл) и суспензию охлаждают до 0oC. После 1 часа выдержки суспензию отфильтровывают. Продукт промывают холодным 4:1 гептаном-изопропилацетатом (2 x 10 мл) и сушат в вакууме при 40oC. Свободное основание МК-0462 (8) получают в виде кремовоокрашенного твердого вещества (4,30 г, выход 73%). ЯМР данные и C, H, N аналитические данные представлены выше в "Ссылках и примечаниях".
Стадия 5. К раствору свободного основания МК-0462 (10 г, чистота 89% по весу) в изопропиловом спирте (80 мл) при комнатной температуре на протяжении 10 минут добавляют раствор бензойной кислоты (4,5 г, 36,8 ммоль) в изопропилацетате (20 мл). Смесь выдерживают при комнатной температуре в течение 0,5 часа, охлаждают до 0-5oC и фильтруют. Лепешку промывают изопропилацетатом (10 мл) и сушат, получая сырую бензоатную соль МК-0462 (13,1 г, чистота 95% по весу, выход согласно анализу 96%). Перекристаллизация из EtOH дает чистую твердую соль МК-0462 (13,1 г, чистота 95% по весу, выход согласно анализу 96%). Перекристаллизация из EtОH дает чистый твердый продукт. Элементный анализ и аналитические спектры согласуются с предложенной структурой.


N,N-Диметил-2-[5-(1,2,4-триазол-1-ил-метил)-1Н-индол-3-ил]-этиламин получают взаимодействием 2-[5-(1,2,4-триазол-1-ил-метил)-1H-индол-3-ил] этилового спирта с мезилхлоридом при -20° в сухом ТГФ с последующей обработкой полученного in situ промежуточного мезилата 40%-ным водным диметиламином. Способ не требует использования трифенилфосфина и хлорида лития и устраняет тенденцию триазолильной полимеризации. 2 с. и 9 з.п. ф-лы, 1 табл.

отличающийся тем, что он включает стадию обработки 2-[5-(1,2,4-триазол-1-илметил)-1Н-индол-3-ил]- этилового спирта формулы IV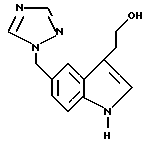
мезилхлоридом при -20°С в сухом тетрагидрофуране, в течение времени, достаточного для образования in situ промежуточного мезилата и последующую стадию обработки промежуточного мезилата 40%-ным водным диметиламином в течение времени, достаточного для образования соединения формулы V.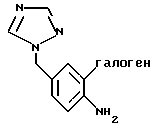
где галоген представляет собой Вг или J с защищенным бутинольным производным формулы II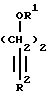
где R1 представляет Н, SiR3 a;
R2 представляет SiR3 a;
R3 a представляет линейный или разветвленный С1-С4 алкил или тетрагидропиранил, причем процесс осуществляют в ДМФ при 70 - 120°C в присутствии ацетата палладия и в присутствии неорганического соединения, выполняющего функцию акцептора протона и невзаимодействующего химически с катализатором, с последующим снятием защитных групп обработкой слабой кислотой до образования соединения формулы IV.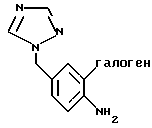
получают взаимодействием 4-амино-бензилтриазола формулы Ia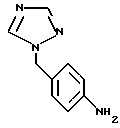
с галогенирующим агентом в смеси 95%-ным МеОН/Н2O в присутствии карбоната кальция.
| US 5298520 A, 1994 | |||
| Устройство для измерения намагниченности | 1974 |
|
SU497512A1 |
| Машковский М.Д | |||
| Лекарственные средства | |||
| -Москва.: Медицина, 1986, ч.1, с | |||
| КОПИРОВАЛЬНЫЙ СТАНОК ДЛЯ ДЕРЕВА | 1921 |
|
SU447A1 |

