В патентной заявке США серии N 750647, поданной на рассмотрение 30 августа 1991 г., описываются антагонисты рецепторов фибриногена и способы получения антагонистов рецепторов фибриногена, которые получают в соответствии со способом настоящего изобретения. В частности, соединение формулы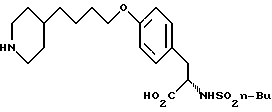
получают по 11-стадийной методике, при которой для получения эфира предполагается образование потенциально опасного NaH/ДМФА, при получении эфира, который требует хроматографической очистки.
В журнале J Heterocyclic Co., 3, 74 (1966) (Singerman et al.) описана методика получения 4-(4-пиридинил)бутилхлорида, состоящая из 6 стадий. По методике изобретения для получения названного соединения требуется только одна стадия.
В журнале J.Org. Chem., 31, 1996 (1966) (Solar et al.) описано O-алкилирование тирозина. Селективное O-алкилирование N-сульфонилированного тирозина, описанное в настоящем изобретении, является беспрецедентным.
Изобретение представляет собой высокоэффективный способ синтеза соединений формулы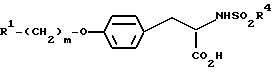
где заместитель R1 представляет собой шести-членное насыщенное или ненасыщенное гетероциклическое кольцо, содержащее один или два гетероатома, где гетероатомы представляют собой атом N, или NR6, где R6 представляет собой H или C1-10-алкил;
m принимает целые значения от двух до шести; и
заместитель R4 представляет собой арил, C1-10-алкил или C4-10-аралкил.
Изобретение представляет собой способ получения антагонистов рецепторов фибриногена формулы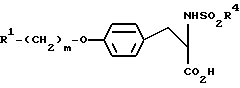
где заместитель R1 представляет собой шести-членное насыщенное или ненасыщенное гетероциклическое кольцо, содержащее один или два гетероатома, где гетероатомы представляют собой атом N, или NR6, где R6 представляет собой H или C1-10-алкил;
m принимает целые значения от двух до шести; и
заместитель R4 представляет собой арил C1-10-алкил или C4-10-аралкил
в соответствии с методикой по которой: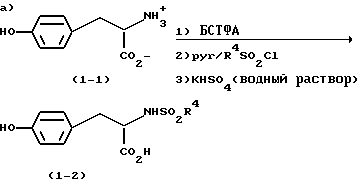
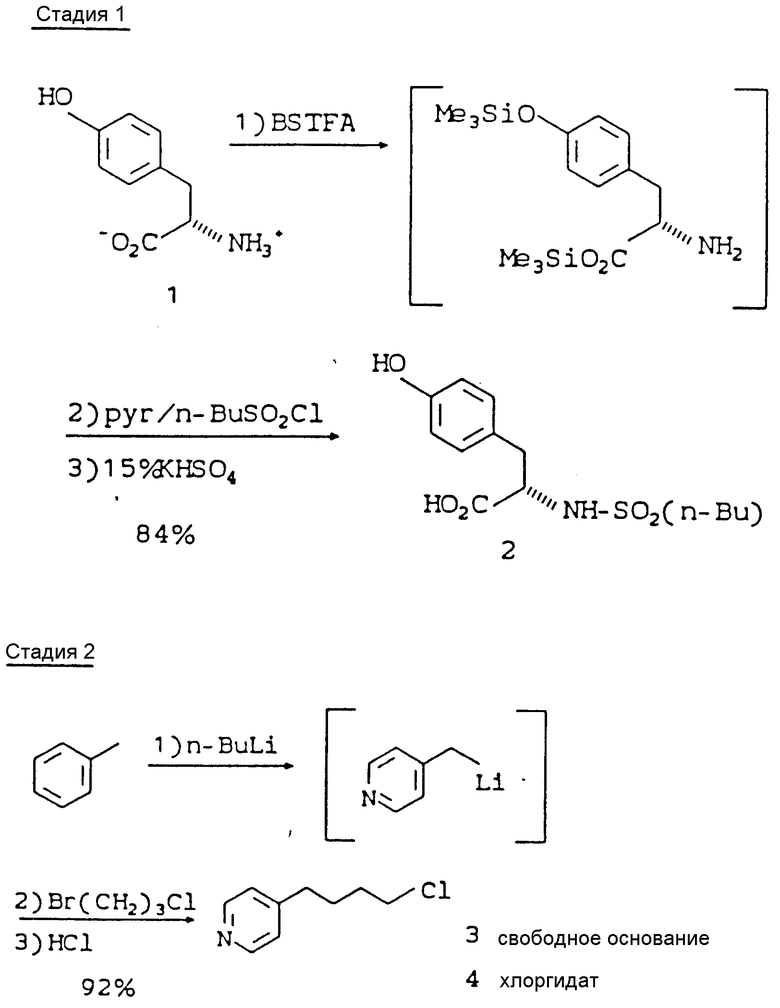
тирозин или производное тирозина (1-1) подвергают сульфонилированию с помощью R4SO2Cl, используя посредник бис-триметилсилил трифторацетамид (БСТФА), в ацетонитриле, с получением соответствующего сульфонамида (1-2);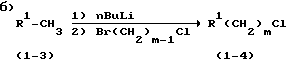
метилированный R1 (соединение 1-3) вначале реагирует с н.-Buli, а затем с линейной алкильной группой, содержащей атом Br на одном конце и Cl - на другом, с образованием R1(CH2)mCl (1-4);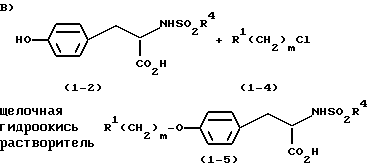
соединение (1-2) добавляют к соединению (1-4) и подвергают фенольному O-алкилированию в щелочной гидроокиси, предпочтительно в 3 н. КОН, в полярном апротонном растворителе, таком как, например, 1,3-диметил-3,4,5,6-тетрагидро-2(1Н)-пиримидинон (ДМП), метилсульфоксид (ДМСО), N-метил-пирролидинон (N-МП), 1,3-диметил-2-имидазолидинон (ДМИ), тетраметилмочевина (ТММ) или N, N-диметилацетамид (ДМАА), предпочтительно в апротонном растворителе, таком как ДМП или ДМСО, более предпочтительно в ДМСО, предпочтительно при приблизительно 65oC.
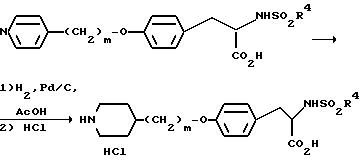
Когда заместитель R1 представляет собой пиридин, селективное гидрирование достигается при использовании Pd/C в уксусной кислоте.
Предпочтительно, настоящее изобретение представляет собой высокоэффективный способ синтеза соединения: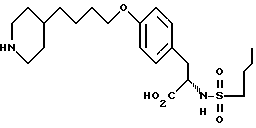
В синтезах настоящего изобретения триметилсилильные группы используются в качестве временной защиты, которая дает возможность осуществлять селективное сульфонилирование в одну стадию с использованием самого L-тирозина.
В рассматриваемом синтезе предпочтительно:
1) используется 4-пиколин в качестве латентной формы пиперидина, что исключает необходимость использовать защиту, и гомологизация атома углерода в положении 3 с помощью 3-бром-1-хлорпропана через 4-пиколиллитий;
2) временное бис-O,O'-силилирование (L)-тирозина с помощью БСТФА, которое обеспечивает селективное N-сульфонилирование с помощью н.-BuSO2Cl с образованием сульфонамида с высоким выходом и без рацемизации в одну стадию;
3) достигается с высоким выходом селективное образование фенилового эфира с использованием простого реагента - водного щелочного основания (NaOH или КОН, предпочтительно 3 н. раствор КОН) в ДМП (1,3-диметил-3,4,5,6-тетрагидро-2-(1Н)-пиримидинон) или в ДМСО; и
4) селективное гидрирование пиридинового кольца в присутствии тирозина достигается при использовании Pd/C в уксусной кислоте.
В синтезах настоящего изобретения используются недорогие исходные материалы и реагенты, а также исключается из настоящего процесса стадия использования при образовании простого эфира потенциально опасной смеси NaH/ДМФА, которая требует хроматографической очистки, как это описывается в работах предшествующего уровня. Способ настоящего изобретения не требует проведения такой хроматографической очистки.
Пример 1. В соответствии с методикой настоящего изобретения может быть получено следующее соединение: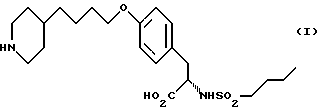
Ниже приведены четыре стадии способа, представленные в конце текста.
Стадия 1: N-н.-BuSO2-(L)-тирозин (2)
Круглодонную четырехгорлую колбу на 50 л, снабженную механической мешалкой, обратным холодильником, входным отверстием для азота, ловушкой хлористого водорода, нагревательным элементом и термопарой, продувают азотом и загружают (L)-тирозин (1040 г, 5,74 моль) 1, CH3CN (20,8 л), N,O-бис-(триметилсилил-трифторметил)ацетамид (3103 г, 12,054 моль). Суспензию нагревают до 85oC и выдерживают при слабом кипении в течение 2 ч. Полученный прозрачный раствор, который в соответствии с данными ПМР представляет собой преимущественно O,O'-бис-триметилсилил-(L)-тирозин, охлаждают до 40oC и медленно в течение 30 мин добавляют пиридин (544,84 г, 6,888 моль) и н.-BuSO2Cl (989,9 г, 6,314 моль). Затем полученную реакционную массу выдерживают при 70oC в течение 3 ч и далее при комнатной температуре в течение 14 ч. Упаривают почти весь растворитель в концентраторе периодического действия, и полученное масло обрабатывают 15%-ным раствором KHSO4 (20,8 л) и интенсивно перемешивают в течение 1 ч. Смесь экстрагируют изопропилацетатом (3 • 6,2 л). Объединенные органические слои обрабатывают с использованием EcosorbTM S-402 (3,12 кг) и перемешивают при комнатной температуре в течение ночи. Ecosorb отфильтровывают, и осадок на фильтре промывают изопропилацетатом (4,2 л). Фильтрат упаривают досуха, и полученное желтое масло растворяют в горячем этилацетате (45 - 50oC, 1,25 л). К полученному раствору при перемешивании добавляют гексан (3,74 л), и полученную суспензию перемешивают при комнатной температуре в течение ночи. Твердый осадок отфильтровывают, и остаток на фильтре промывают смесью этилацетат/гексан (0,2 л/1,89 л). После сушки в вакууме получают 1457 г (выход 84%) соединения 2 в виде белого твердого продукта.
Оценка с помощью ВЭЖХ: 99,6 A%; время удерживания - 7,55 мин; колонка - Zorbax RX-C8, 4,6 мм • 25 см вн.д; 220 нм; 1,5 мл/мин; линейный градиент от 10 до 90% A через 10 мин, A = CH3CN, B = 0,1% водная H3PO4; т. пл. 125 - 126,5oC, [α]
Спектр 1H-ЯМР (CD3OD), δ м.д.: 0,81 (т, J = 7,2 Гц, 3H); 1,24 (м, 2H); 1,45 (m, 2H); 2,61 (т, J = 7,9 Гц, 2H); 2,73 (A от ABX-системы, JAB = 13,8 Гц, JAX = 9,8 Гц, 1H); 3,07 (B от ABX-системы, JBA = 13,8 Гц, JBX = 4,7 Гц, 1H); 4,07 (X от ABX-системы JXA = 9,8 Гц, JXB = 4,7 Гц, 1H); 6,72 (д, J = 8,4 Гц, 2H); 7,10 (д, J = 8,4 Гц, 2H).
Спектр 13C-ЯМР (CD3OD), δ м. д.: 13,9, 22,5, 26,5, 39,1, 54,1, 59,5, 116,3, 129,2, 131,6, 157,5, 175,3.
Элементный анализ: вычислено для C13H19O5NS
C 51,81; H 6,35; N 4,65; S 10,64.
Найдено: C 51,73; H 6,28; N 4,60; S 10,82.
Стадия 2: 4-(4-Пиридинил)бутилхлорид, хлоргидрат (4)
При получении 4-(4-пиридинил)бутилхлорида необходимо нагревание 4-пиколина и н.-BuLi при 40oC в течение 2 ч для полного использования н.-BuLi. Без нагревания непрореагировавший н. -BuLi будет вступать в перекрестную реакцию металлирования с 3-бром-1-хлорпропаном с с образованием н.-бутилбромида, который затем будет взаимодействовать с 4-пиколиллитием с образованием 4-пентилпиридина. Обратное добавление 4-пиколиллития к 3-бром-1-хлорпропану при температуре -65oC имеет решающее значение для того, чтобы исключить образование продукта бис-алкилирования, 1,5-бис(4-пиридил)-пентана. Полное удаление ТГФ и воды имеет большое значение для мягкого образования хлоргидрата, так как ТГФ реагирует с HCl с образованием 4-хлорбутанола, который повышает растворимость хлоргидрата и снижает его выход, а присутствие воды значительно затрудняет фильтрование хлоргидрата из-за того, что последний становится липким. При соблюдении этих рекомендаций получают хлоргидрат 4-(4-пиридинил)бутилхлорида (4) с выходом 92% и со степенью чистоты 98%.
Четырехгорлую круглодонную колбу на 22 л, снабженную механической мешалкой, обратным холодильником, капельной воронкой с противодавлением и термопарой, продувают азотом в течение ночи. Затем в колбу загружают ТГФ (4,1 л) и 4-пиколин (838,2 г, 9,0 моль) и охлаждают до - 70oC. Затем медленно добавляют раствор н.-бутиллития (7,02 л, 1,41 М раствор) в гексане, поддерживая температуру реакционной массы не < -50oC.
Добавление продолжают в течение приблизительно 1 ч. При проведении реакции при температуре около 0oC значительно снижается выход и увеличивается образование примесей.
Баню с сухим льдом убирают и реакционной массе дают нагреться до комнатной температуры и затем выдерживают при температуре 40 - 45oC в течение 2 ч.
Температура 40 - 45oC является оптимальной для эффективного завершения образования литийпроизводного 4-пиколина без разложения. Без нагревания непрореагировавший н. -BuLi вступает в реакцию перекрестного металлирования с 3-бром-1-хлорпропаном с образованием 1-н.-бутилбромида. Последний реагирует с 4-пиколинлитием с образованием 4-пентилпиридина, который нельзя отделить от желаемого продукта. Нагревание до более высокой температуры приводит к значительному разложению реагентов.
Для растворения суспензии 4-пиколиллития добавляют ТГФ (4,1 л) и получают темно-оранжевый раствор. Полученный раствор охлаждают до 0oC, а затем с использованием пневматического насоса через полипропиленовую трубку осторожно добавляют к охлажденному до -75oC раствору 3-бром-1-хлорпропана в ТГФ (1,5 л), который находится в сухой трехгорлой круглодонной колбе на 50 л, снабженной механической мешалкой, входным/выходным отводом для азота и термопарой, при этом температуру реакционной массы поддерживают не выше -65oC.
Реакция 4-пиколиллития и 3-бром-1-хлорпропана является экзотермической. Решающее значение имеет сохранение температуры реакционной массы не более -65oC, что позволяет исключить взаимодействие требуемого продукта с 4-пиколиллитием с образованием 1,5-бис-(4-пиридинил)пентата. Время добавления составляет 2 ч.
Реакционной массе дают постепенно нагреться до температуры 0oC в течение ночи и затем обрабатывают 9 л воды, перемешивая в течение 10 мин, отделяя слои и экстрагируя водный слой изопропилацетатом (5 л). Объединенные органические слои концентрируют в вакууме при 40oC до одной трети первоначального объема в концентраторе периодического действия, оборудованном двумя дополнительными ловушками между приемной и подающей вакуумной линиями, а затем добавляют изопропилацетат (6 л) и снова концентрируют до одной трети исходного объема.
Полное удаление ТГФ и воды путем азеотропной отгонки с изопропилацетатом имеет большое значение для мягкого образования хлоргидрата. ТГФ реагирует с HCl с образованием 4-хлорбутанола, который повышает растворимость хлоргидрата и снижает его выход. В присутствии воды клейкий твердый хлоргидрат плохо фильтруется.
Реакционную массу охлаждают до -10oC и затем обрабатывают раствором 9,0 моль HCl в 3 л изопропилацетата.
Раствор HCl в изопропилацетате готовят днем раньше путем пропускания хлористого водорода и изопропилацетат при -10oC до тех пор, пока не поглотится 9,1 моль хлористого водорода (по весу) и хранят при комнатной температуре. Потери хлористого водорода составляют приблизительно 1%. Добавление HCl сопровождается повышением температуры, которая повышается до 35oC.
После перемешивания в течение 1 ч полученную суспензию переносят с помощью пневматического насоса через полипропиленовую трубку в заполненную азотом закрытую фильтрующую воронку, находящуюся в вакууме. Твердый остаток промывают несколько раз ТГФ (4,5 л общий объем) и сушат в вакууме в потоке азота при пониженном давлении. Получают 1710,4 г (92%) соединения 4 в виде белого твердого вещества.
Оценка с помощью ВЭЖХ: 98,0% площадь; время удерживания 2,40 мин; колонка - Zorbax RX-C8, 4,6 мм • 25 см вн.д; 220 нм; 1,5 мл/мин; изократность 50% /50% A/B, A = CH3CN, B = 0,01 М раствор натровая соль декансульфоновой кислоты в 0,1%-ном водном растворе H3PO4; время удерживания для 4-пиколина - 1,7 мин.
Tпл = 119 - 120,5oC; масс-спектр (химическая ионизация) m/z 169 (M+ - HCl).
Спектр 1H-ЯМР (CD3OD), δ м.д.: 1,79 - 2,00 (м, 4H); 3,01 (т, J = 7,3 Гц, 2H); 3,36 (т, J = 6,1 Гц, 2H); 8,0 (д, J = 6,7 Гц, 2H); 8,75 (д, J = 6,7 Гц, 2H).
Спектр 13C-ЯМР (CD3OD), δ м.д.: 28,1, 33,0, 36,1, 45,3, 128,6, 142,2, 166,1.
Элементный анализ: вычислено для C9H13NCl2 -
C 52,45; H 6,36; N 6,80; Cl 34,40.
Найдено: C 52,22; H 6,40; N 6,51; Cl 34,11.
Стадия 3: Образование фенилового эфира
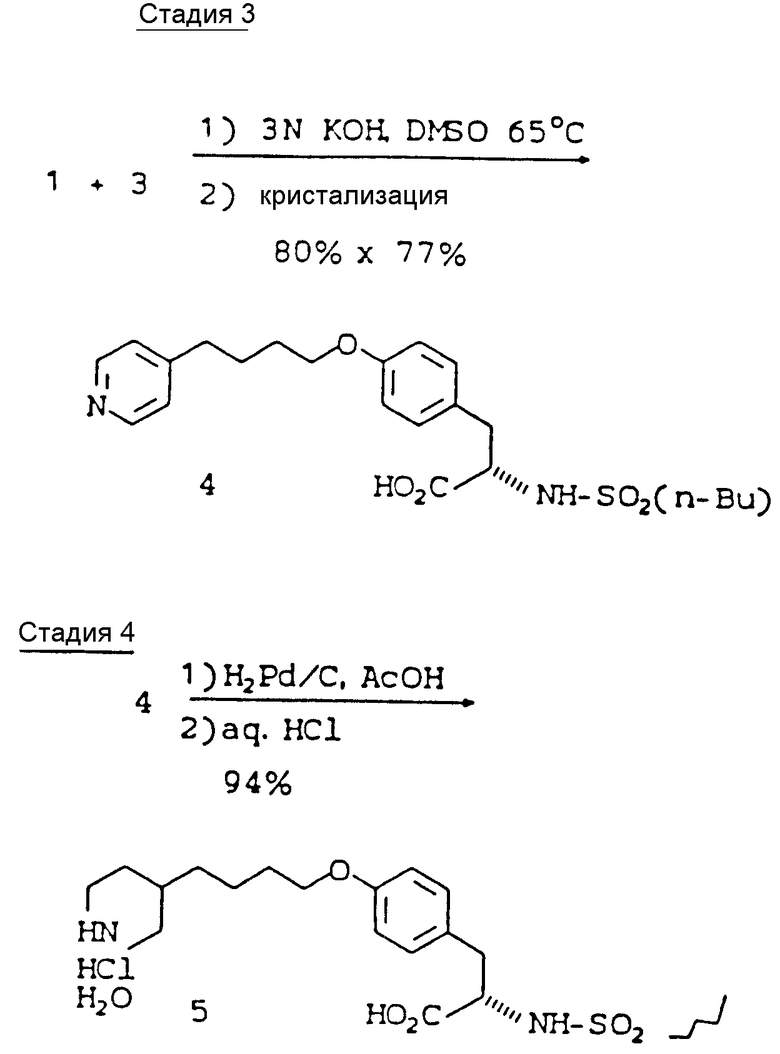
В реакции сопряжения с КОН в качестве посредника использование растворителей на основе мочевины (ДМП, ДМИ, ТММ) и ДМСО приводит к наиболее высокому выходу - 85 - 96%. Использование ДМАА и N-МП приводит к выходам менее 80%. ДМП, как установлено, представляет собой оптимальный растворитель, в котором минимизируется образованием продукта бис-алкилирования (1%), тогда как при использовании в качестве растворителя ДМСО количество образующегося бис-продукта является максимальным (2%).
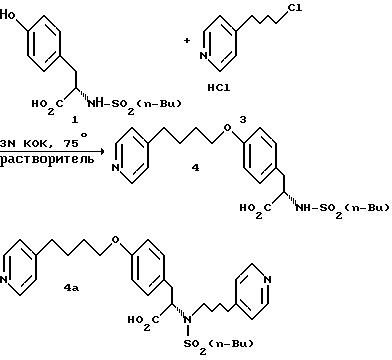
Разработан способ непосредственного выделения продукта сопряжения, при котором исключается использование больших объемов метиленхлорида для экстракции. Обработка с помощью Ecosorb водной разбавленной реакционной смеси и фильтрование с последующим доведением pH до изоэлектронной точки (pH 4,8) обеспечивает 80%-ный выход сырого продукта со степенью чистоты 93 - 95 A%. Последующее доведение pH до 5,5 и двухкратная обработка смесью 10% AcOH/вода приводит к удалению непрореагировавшего н.-BuLi-тирозина и бис-продукта и выделению с выходом 77% бежевого твердого продукта со степенью чистоты 99 A%. Если бис-продукт еще присутствует в недопустимых количествах (более 0,1%), следует провести еще одну обработку смесью 10% AcOH/вода. С другой стороны, можно получать величину pH 5,5, а не 4,8 после обработки с помощью Ecosorb и фильтрования, так что осаждение и удаление н.-BuLi-тирозина объединяется на одной стадии.
Стадия 3а: Образование эфира и очистка
В четырехгорлую круглодонную колбу на 50 л, снабженную механической мешалкой, обратным холодильником, входным отверстием для азота и термопарой, загружают N-н.-бутансульфонил-(L)-тирозин (1386,3 г, 4,60 моль), хлоргидрат 4-(4-пиридинил)-бутилхлорида (1137,8 г, 5,52 моль) и ДМСО (16,56 л). Затем при интенсивном перемешивании в течение 15 мин добавляют 3 н. водный раствор КОН (5,52 л, 16,56 моль).
Смешение 3 н. водного раствора КОН с остальными продуктами сопровождается небольшим повышением температуры. Температуру поддерживают в интервале 30 - 40oC с помощью водяной бани.
Добавляя иодид калия (7,64 г, 46,0 ммоль), и смесь выдерживают при 65oC в течение 24 ч и при 60oC в течение 12 ч (или до 95%-ного окончания реакции по данным ВЭЖХ). После охлаждения до комнатной температуры смесь разбавляют 0,25 н. рассолом NaOH (46 л) и экстрагируют один раз трет.-бутилметиловым эфиром (23 л). Водный слой обрабатывают с помощью Ecosorb S-402- (2 кг) и Nuchar SA (150 г), и полученную смесь (приблизительно 67 л) механически перемешивают в течение 1 ч. Смесь фильтруют через крупнопористый фильтр, и осадок на фильтре промывают 69 л воды. Объединенные фильтраты (около 136 л) помещают в сосуд на 200 л, снабженный pH-метром и механической мешалкой. При интенсивном перемешивании добавляют NaCl (2,5 кг), перемешивают в течение 30 мин, а затем добавляют 50%-ный водный раствор уксусной кислоты (около 4 л) до достижения pH 4,80 и продолжают перемешивание в течение 2 - 3 ч.
Начальная величина pH составляет 13,3. При pH около 4,8 наряду с твердым продуктом бежевого цвета образуется небольшое количество коричневого смолистого продукта. Продолжительное перемешивание необходимо для завершения образования кристаллического продукта. Если величина pH ниже 4,8, то следует добавить NaOH.
Полученную суспензию фильтруют через фильтрующую воронку с крупными порами, и осадок на фильтре промывают 23 л воды. Сырой продукт сушат при 40oC при остаточном вакууме в атмосфере азота в течение 20 ч и получают 1599 г (80%) смеси коричневого и бежевого твердого продукта, имеющего 95%-ную чистоту.
Основные примеси представляют собой исходный тирозин (0,75%) и продукт бис-алкилирования (2,75 A%). Маточные жидкости и объединенные промывные жидкости содержат согласно жидкостной хроматографии приблизительно 10% продукта.
Оценка с помощью ВЭЖХ: продукт 5, 96% площадь; время удерживания 6,76 мин; тирозин 1, время удерживания 7,66 мин; продукт бис-алкилирования, время удерживания 6,20 мин; колонка Zorbax RX-C8, 4,6 мм • 25 см вн.д; 220 нм; 1,5 мл/мин; линейный градиент от 10 до 90% A через 10 мин, A = CH3CN, B = 0,1% водная H3PO4.
Стадия 3б:
Твердый продукт по следующей методике подвергают дополнительной очистке до степени чистоты 99,4 A%.
Очистка продукта от примесей на этой стадии до проведения последующего гидрирования имеет решающее значение, так как продукт гидрирования бис-алкилированного продукта и тиразин 1 чрезвычайно трудно удалить.
В RB колбу на 50 л, снабженную термопарой и капельной воронкой, загружают сырой продукт 5 (1,5 кг, 3,45 моль) и 0,25 н. раствор NaOH (19,33 л, 4,83 моль). После завершения растворения твердого продукта при мягком нагревании до 60 - 70oC в течение нескольких мин добавляют 0,25 н. раствор NaHCO3 (4,83 л, 1,21 моль). Полученный раствор охлаждают до комнатной температуры и доводят величину pH до 7 при медленном добавлении 1 н. раствора HCl (приблизительно 2,65 л). Величину pH раствора дополнительно опускают до 5,5 при медленном добавлении 0,5 н. раствора HCl (приблизительно 5,10 л). Перемешивают в течение 1 ч, затем полученную суспензию фильтруют через грубую фильтрующую воронку со слоем фильтрующей бумаги и слоем полипропилена (10 мкм) и осадок на фильтре промывают водой (10 л). Твердый остаток сушат при остаточном вакууме с подачей азота. Получают 1,42 кг бежевого твердого продукта.
При такой обработке удаляется большая часть тирозина 1. Образец на этой стадии должен содержать не более 0,1% соединения 1. При последующей промывке в смеси 10% AcOH/вода удаляют примеси продукта бис-алкилирования. Фильтрование суспензии твердого продукта с использованием стеклянной фильтрующей воронки с пористостью M не рекомендуется, так как такое фильтрование имеет чрезвычайно небольшую скорость. Фильтрующая воронка с пористостью C также не должна использоваться из-за небольшого проскока продукта.
Твердый продукт суспендируют в 10%-ном растворе уксусной кислоты в воде (1 г/15 мл) и нагревают паром до 80oC в течение 5 мин, затем дают медленно охладиться до комнатной температуры в течение ночи. После перемешивания в течение 18 ч твердый продукт собирают с помощью грубого фильтра со слоем фильтровальной бумаги и слоем полипропилена (10 мкм), промывают водой (20 л) и частично сушат при остаточном вакууме при подпитке азотом в течение нескольких часов. Описанную выше процедуру повторяют, и твердый продукт промывают дистиллированной водой (20 л), метанолом (3 • 4 л) и сушат в вакууме в течение 2 дней при 35oC при подпитке азотом. Получают 1,16 кг (выход 77%, полный выход 62%) почти белого твердого соединения.
В дальнейшем показано, что промывка метанолом не является необходимой, но на этом этапе теряется около 5% продукта. Если содержание продукта бис-алкилирования составляет более 0,1%, следует провести еще одну обработку смесью 10%-ным раствором уксусной кислоты в воде.
Оценка с помощью ВЭЖХ: продукт 5, 99,8% площадь; время удерживания 6,76 мин; тирозин 1; время удерживания 7,66 мин; продукт бис-алкилирования, время удерживания 6,20 мин; колонка Zorbax RX-C8, 4,6 мм • 25 см вн.д; 220 нм; 1,5 мл/мин; линейный градиент от 10 до 90% A через 10 мин, A = CH3CN, B = 0,1% водная H3PO4.
Tпл = 137 - 138oC; [α]
Спектр 1H ЯМР (CD3OD), δ м.д.: 0,86 (т, J = 7,3 Гц, 3H); 1,33 (гекс, J = 7,3 Гц, 2H); 1,68 (m, 2H); 1,83 (m, 2H); 2,82 (m, 2H); 3,06 (A от ABX-системы, JAB = 13,9 Гц, JAX = 6,3 Гц, 1H); 3,16 (B от ABX-системы, JBA = 13,9 Гц, JBX = 5,0 Гц, 1H); 3,90 (т, J = 5,7 Гц, 2H); 4,32 (X от ABX-системы, JXA = 6,3 Гц, JXB = 5,0 Гц, 1H); 6,72 (д, J = 8,6 Гц, 2H); 7,17 (д, J = 8,6 Гц, 2H); 7,33 (д, J = 6,3 Гц, 2H); 8,49 (д, J = 6,3 Гц, 2H).
Спектр 13C ЯМР (CDCl3), δ м.д.: 13,5, 21,5, 25,4, 26,5, 28,6, 35,1, 38,9, 53,0, 57,9, 67,0, 114,3, 125,0, 128,7, 130,8, 145,9, 155,8, 157,7, 175,07.
Элементный анализ, вычислено для C22H30O5N2S -
C 60,81; H 6,96; N 6,45; S 7,38.
Найдено: C 60,53; H 6,88; N 6,26; S 7,65.
Стадия 4: Гидрирование
Селективное гидрирование пиридинового кольца до пиперидинового кольца проводят с использованием 10%-ного Pd/C в уксусной кислоте при 60oC, что приводит к образованию целевого продукта без восстановления фенольного кольца. Окончание реакции гидрирования устанавливают с помощью ВЭЖХ и спектроскопии ПМР в точке, близкой к точке теоретического поглощения водорода. Сразу же после исчезновения исходных продуктов гидрирование должно быть остановлено. После фильтрования реакционной смеси, упаривания уксусной кислоты с последующей кристаллизацией продукта из 6%-ной водной уксусной кислоты получают свободное основание 6. Следовые количества исходных продуктов удаляют путем промывки 6%-ной водной уксусной кислотой. Обработка свободного основания 2,5% об. концентрированной соляной кислоты (2,1 экв.) в изопропилацетате приводит к соединению 5 (хлоргидрат) моногидрат с 94%-ным выходом в виде бесцветного твердого продукта со степенью чистоты более 99,7 A% и с содержанием обеих примесных продуктов на уровне 0,1 A%.
Стадия 4a:
Фениловый эфир 5 (1,051 кг, 2,42 моль) и 10%-ный Pd/C (53 г, 5 мас.%) в уксусной кислоте (14 л) гидрируют в аппарате из нержавеющей стали на 5 галлонов при давлении 40 фунтов/кв.дюйм и при температуре 60oC. Когда реакция приближается к окончанию, из реакционной массы каждый час отбирают пробы, и реакцию останавливают сразу же после исчезновения исходных продуктов (приблизительно 5,5). Более продолжительное время реакции приводит к образованию примесей.
Образец в 1 мл фильтруют через тонкий слой Solka-Flock (промытый уксусной кислотой), промывают фильтрующий слой уксусной кислотой и упаривают досуха на роторном испарителе. Полученное масло обрабатывают несколькими мл воды до осаждения осадка, а затем снова упаривают в вакууме досуха. Полученный белый твердый продукт анализируют с помощью ПМР (CD3OD) и ВЭЖХ. Вся процедура занимает приблизительно 30 мин. В спектре ПМР (CD3OD) полное исчезновение пиридинового пика при 7,32 и 8,40 м.д. указывает на то, что исходный продукт полностью прореагировал. ВЭЖХ (с использованием условий линейного градиента) служит для контролирования примесей при времени удерживания около 8,0 мин. Количество этих примесей возрастает при более продолжительном гидрировании. Сигналы исходных соединений и продуктов реакции могут находиться очень близко друг к другу, поскольку их время удерживания составляет соответственно 6,76 и 6,80 мин.
Реакционную массу фильтруют через слой Solka-Flock (820 г, промытый 5 л уксусной кислоты) и промывают уксусной кислотой (14 л). Фильтрат концентрируют, получают густое масло, содержащее приблизительно 1 кг уксусной кислоты. Для концентрирования используют концентратор периодического действия, снабженный двумя дополнительными ловушками между подающей и остаточной вакуумными линиями при температуре 60oC (время операции приблизительно 5 ч). Добавляют дистиллированную воду (15 л) до получения концентрации 1 г/15 мл 6%-ной уксусной кислоты в воде, и полученную суспензию перемешивают при комнатной температуре в течение 18 ч, твердый продукт отфильтровывают на воронке Бюхнера с листом фильтрующей бумаги и со слоем полипропилена (10 мкм), промывают дистиллированной водой (10 л) и сушат в вакууме при подпитке азотом. Получают 1,03 кг (97%) белого твердого вещества.
Если количество соединения 5 все еще превышает количество, установленное в описании, то следует провести еще одну промывку 6%-ной уксусной кислотой (1 г/15 мл) (в течение по меньшей мере 6 ч). Типичным является выделение около 92% с 3-кратным уменьшением соединения 5.
Оценка с помощью ВЭЖХ: свободное основание 6, 99,5% площадь; время удерживания 6,94 мин; соединение 5; время удерживания 6,72 мин; соединение 5, время удерживания 7,39 мин; колонка - Zorbax RX-C8, 4,6 мм • 25 см вн.д; 220 нм; 1,5 мл/мин; линейный градиент от 20 до 70% A через 12 мин, A = CH3CN, B = 0,1% водная H3PO4.
Tпл = 223 - 225oC.
Спектр 1H ЯМР (CD3OD), δ м.д.: 0,88 (т, J = 7,3 Гц, 3H); 1,33 (м, 6H); 1,58 (m, 5H); 1,76 (m, 2H); 1,81 (m, 2H); 2,77 (т, J = 7,5 Гц, 2H); 2,80 (м, 2H); 2,88 (м, 2H); 3,03 (B от ABX-системы, JBA = 13,9 Гц, JBX = 4,6 Гц, 1H); 3,30 (м, 2H); 3,30 (м, 2H); 3,90 - 4,0 (м, 3H); 6,80 (д, J = 8,5 Гц, 2H); 7,18 (д, J = 8,6 Гц, 2H).
Элементный анализ: вычислено для C22H37O5N2S -
C 59,84; H 8,40; N 6,34; S 7,24.
Найдено: C 59,98; H 8,40; N 6,40; S 7,24.
Стадия 4б:
В трехгорлую RB колбу на 22 л, снабженную механической мешалкой, вводом для азота и капельной воронкой, загружают свободное основание 6 (316,0 г, 0,717 моль) и изопропилацетат (9,5 л). Смесь перемешивают при комнатной температуре (19oC) в течение 10 - 15 мин, затем по каплям добавляют концентрированную соляную кислоту (120 мл). Добавление продолжается около 40 мин, при этом температуру поддерживают около 19oC. Далее смесь перемешивают при комнатной температуре (10oC) еще 5 ч. Твердый продукт промывают изопропилацетатом (2 • 1 л) и сушат под азотом с отсасыванием в течение ночи. Выделяют соединение 6 в виде хлоргидрата моногидрата (348 г) с выходом 98%.
Оценка с помощью ВЭЖХ: свободное основание 6, 99,8% площадь; время удерживания 6,79 мин; колонка - Zorbax RX-C8, 4,6 мм • 25 см вн.д; 220 нм; 1,5 мл/мин; линейный градиент от 10 до 90% A через 10 мин, A = CH3CN, B = 0,1% водная H3PO4; или L - 700, 462, 99,8% площадь, время удерживания 6,94 мин; соединение 5, время удерживания 6,72 мин; соединение 1, время удерживания 7,39 мин; колонка - Zorbax RX-C8, 4,6 мм • 25 см вн.д; 220 нм; 1,5 мл/мин; линейный градиент от 20 до 70% A через 12 мин.
Хиральная ВЭЖХ: (L)-изомер - >99,9%, время удерживания 10 мин; (D)-изомер <0,1%. Время удерживания 8,5 мин. Колонка - ULTRON-ES-OVM, 4,6 мм • 25 мм вн. д., 5 м с защитной колонкой; 220 нм; 0,7 мл/мин; 90% буфер (6 г формиата аммония, с доведением до pH 4,1 с помощью муравьиной кислоты, 10% MeOH).
Tпл = 87 - 88oC, тпл = 131 - 132oC [α
Спектр 1H ЯМР (CD3OD), δ м.д.: 0,84 (т, J = 7,3 Гц, 3H); 1,23 (гекс, J = 7,3 Гц, 2H); 1,30 - 1,70 (m, 9H); 1,75 (m, 2H); 1,95 (m, 2H); 2,64 (т, J = 7,4 Гц, 2H); 2,77 (A от ABX-системы, JAB = 13,9 Гц, JAX = 9,8 Гц, 1H); 2,95 (м, 2H); 3,11 (B от ABX-системы, JBA = 13,9 Гц, JBX = 4,6 Гц, 1H); 3,47 (м, 2H); 3,95 (т, J = 6,2 Гц, 2H); 4,09 (X от ABX-системы JXA = 9,8 Гц, JXB = 4,6 Гц, 1H); 6,84 (д, J = 8,6 Гц, 2H); 7,18 (д, J = 8,6 Гц, 2H).
Спектр 13C ЯМР (CDCl3), δ м.д.: 14,0, 22,5, 24,0, 26,5, 30,0, 30,4, 34,8, 36,8, 39,0, 45,3, 54,1, 59,4, 68,7, 115,5, 130,4, 131,7, 159,6, 175,2.
ИК-спектр (вазелиновое масло, см-1): 3520, 3166, 2800 - 2300, 1727, 1610, 1595, 1324, 1256, 1141, 1119, 829.
Масс-спектр (HR) рассчитано для C22H37O5N2S - 441.2423;
найдено 441.2423 (M+ -H2O-HCl).
Элементный анализ: вычислено для C22H39O6ClN2S -
C 53,37; H 7,94; N 5,66; Cl 7,16; S 6,48.
Найдено: C 53,56; H 8,04; N 5,62; Cl 7,36; S 6,53.
Использование: в медицине в качестве антагонистов рецепторов фибриногена. Продукт: 4-(4-пиридил)бутиловый эфир N-сульфонилтирозина формулы I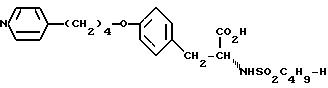
или его фармацевтически приемлемые соли. Реагент 1: тирозин. Реагент 2: H-C4H9SO2Cl. Реагент 3: метилированный пиридин. Реагент 4: Br(CH2)4Cl. Продукт взаимодействия реагентов 1 и 2 подвергают реакции с продуктом взаимодействия реагентов 3 и 4. Условия реакции: в присутствии щелочи. Продукт формулы I подвергают селективному гидрированию при использовании Pd/C в уксусной кислоте. Получают продукт формулы II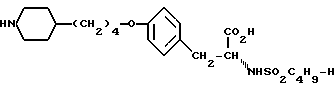
2 с. и 2 з.п.ф-лы.
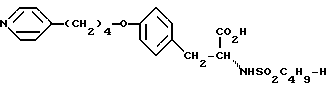
или его фармацевтически приемлемые соли.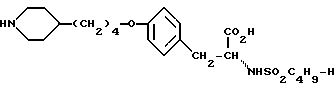
или его фармацевтически приемлемых солей, отличающийся тем, что осуществляют селективное гидрирование соединения формулы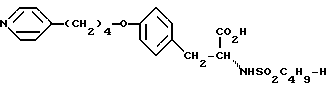
или его фармацевтически приемлемой соли.
| EP, заявка, 0004011A1, кл | |||
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
| EP, заявка, 0381033A1, к л | |||
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |

