Область техники
Данное изобретение относится к ингибиторам фермента 11-бэта-гидроксистероидной дегидрогеназы I типа (11β-HSD-1 или HSD-1) и способам лечения, использующим такие соединения. Соединения полезны для лечения сахарного диабета, такого как инсулиннезависимый сахарный диабет 2 типа (NIDDM), инсулинорезистентности, ожирения, липидных расстройств и других заболеваний и состояний.
ПРЕДПОСЫЛКИ ИЗОБРЕТЕНИЯ
Сахарный диабет обусловлен множеством факторов и наиболее просто характеризуется повышенными уровнями содержания глюкозы в плазме (гипергликемия) натощак. Обычно различают две формы сахарного диабета: сахарный диабет 1 типа или инсулинзависимый диабет (IDDM), при котором у пациентов вырабатывается небольшое количество инсулина или совсем не вырабатывается инсулин - гормон, регулирующий потребление глюкозы, и сахарный диабет 2 типа или инсулиннезависимый сахарный диабет (NIDDM), при котором инсулин у пациентов вырабатывается и даже проявляется гиперинсулинемия (т.е. уровни содержания инсулина в плазме равны и даже являются повышенными по сравнению с субъектами, не страдающими диабетом), и в то же время наблюдается гипергликемия. Сахарный диабет 1 типа обычно лечится экзогенным введением инсулина посредством инъекции. Однако у пациентов с сахарным диабетом 2 типа зачастую развивается "инсулинорезистентность", так что действие инсулина в стимулировании метаболизма глюкозы и липидов в основных чувствительных к инсулину тканях, а именно мышечных тканях, тканях печени и жировых тканях, ослабевает. У пациентов, которые являются инсулинорезистентными, но не больны сахарным диабетом, наблюдаются повышенные уровни содержания инсулина, которые компенсируют их инсулинорезистентность, поэтому уровни содержания глюкозы в плазме не повышены. У пациентов с NIDDM уровни содержания инсулина в плазме, даже когда они повышены, являются недостаточными для преодоления выраженной инсулинорезистентности, что приводит к гипергликемии.
Инсулинорезистентность обусловлена, прежде всего, нарушением рецепторного связывания, которое еще не полностью изучено. Невосприимчивость к инсулину приводит к недостаточной активации поглощения глюкозы, сниженному окислению глюкозы и накоплению гликогена в мышце, неадекватной инсулиновой репрессии липолиза в жировой ткани и неадекватному продуцированию и секреции глюкозы печенью.
Продолжительная или неконтролируемая гипергликемия, которая имеет место у больных сахарным диабетом, связана с повышенной заболеваемостью и преждевременной смертностью. Аномальный гомеостаз глюкозы также связан как прямо, так и опосредованно, с ожирением, гипертензией и нарушениями в липидном, липопротеиновом и аполипопротеиновом метаболизме. Больные сахарным диабетом 2 типа являются пациентами с повышенным риском сердечно-сосудистых осложнений, таких как атеросклероз, коронарная болезнь сердца, удар, заболевание периферических сосудов, гипертензия, нефропатия, нейропатия и ретинопатия. Следовательно, терапевтический контроль гомеостаза глюкозы, метаболизма липидов, ожирения и гипертензии является крайне важным для клинического контроля и лечения сахарного диабета.
Большое количество пациентов с инсулинорезистентностью, у которых однако не наблюдается развития сахарного диабета 2 типа, также имеют риск развития симптомов, называемых "синдромом Х" или "метаболическим синдромом". Синдром Х характеризуется инсулинорезистентностью в сочетании с патологическим ожирением, гиперинсулинемией, высоким кровяным давлением, низким уровнем HDL и высоким уровнем VLDL. Такие пациенты, не зависимо от наличия или отсутствия развития явного сахарного диабета, являются пациентами с повышенным риском развития сердечно-сосудистых осложнений, перечисленных выше.
Лечение сахарного диабета 2 типа обычно включает физическую нагрузку и диету. Повышение уровня содержания инсулина в плазме введением сульфонилмочевин (например, толбутамида и глипизида) или меглитинида, которые стимулируют секрецию панкреатическими β-клетками большего количества инсулина, и/или инъекцией инсулина, когда сульфонилмочевины или меглитинид неэффективны, может приводить к тому, что концентрации инсулина становятся достаточно высокими, чтобы стимулировать инсулинрезистентные ткани. Однако в этом случае могут быть получены опасно низкие уровни содержания глюкозы в плазме и в конечном итоге может иметь место повышенный уровень инсулинорезистентности.
Бигуаниды повышают чувствительность к инсулину, приводя к некоторой коррекции гипергликемии. Однако многие бигуаниды, например фенформин и метформин, вызывают молочный ацидоз, тошноту и диарею.
Глитазоны (то есть 5-бензилтиазолидин-2,4-дионы) составляют более новый класс соединений, которые могут облегчать гипергликемию и другие симптомы диабета 2 типа. Данные фармацевтические средства по существу повышают чувствительность к инсулину в мышечной, печеночной и жировой ткани, приводя к частичной или полной коррекции повышенных уровней содержания глюкозы в плазме по существу не вызывая гипергликемии. Глитазоны, которые в настоящее время поступают в продажу, являются агонистами рецептора, активируемого пролифератором пироксисомы (PPAR) гамма-подтипа. Считается, что PPAR-γ агонизм ответственен за повышение сенсибилизации к инсулину, которая наблюдается при использовании глитазонов. Более новые PPAR агонисты, которые разработаны для лечения диабета 2 типа и/или дислипидемии, являются агонистами одного или нескольких PPAR альфа-, гамма- и дельта-подтипов.
В настоящее время сохраняется потребность в новых способах лечения сахарного диабета и связанных с ним состояний. Данное изобретение удовлетворяет этим и другим потребностям.
КРАТКОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Данное изобретение относится к соединению, представленному формулой I
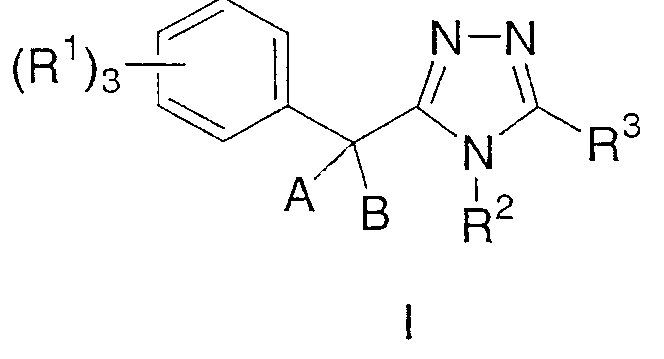
или его фармацевтически приемлемой соли или сольватам, где:
А и В могут быть взяты отдельно или вместе,
когда взяты отдельно,
А представляет собой галоген, С1-6-алкил, ОС1-6-алкил или фенил, причем указанные алкил, фенил или алкильная часть ОС1-6-алкила являются необязательно замещенными 1-3 галогеновыми группами; и
B представляет собой Н, галоген, С1-6-алкил, -ОС1-6-алкил, -SC1-6-алкил, С2-6-алкенил, фенил или нафтил, причем указанные алкил, алкенил, фенил, нафтил и алкильные части -ОС1-6-алкила и -SC1-6-алкила являются необязательно замещенными 1-3 группами, выбранными из галогена, ОН, СН3О, CF3 и OCF3;
когда взяты вместе,
А и В вместе представляют собой (а) С1-4-алкилен, необязательно замещенный 1-3 галогеновыми группами и 1-2 Ra группами, где Ra представляет собой С1-3-алкил, ОС1-3-алкил, С6-10-арилС1-6-алкилен или фенил, необязательно замещенный 1-3 галогеновыми группами, или (b) С2-5-алкандиил, так что они образуют с атомом углерода, к которому присоединены, 3-6-членный цикл, причем указанный цикл необязательно содержит 1 двойную связь или 1-2 гетероатома, выбранных из О, S и N, и указанный 3-6-членный цикл является необязательно замещенным С1-4-алкиленом, оксо, этилендиокси или пропилендиокси и необязательно дополнительно замещенным 1-4 группами, выбранными из галогена, С1-4-алкила, галоген-С1-4-алкила, С1-3-ацила, С1-3-ацилокси, С1-3-алкокси, С1-6-алкил-ОС(О)-, С2-4-алкенила,С2-4-алкинила, С1-3-алкокси-С1-3-алкила, С1-3-алкокси-С1-3алкокси, фенила, CN, OH, D, NH2, NHRa и N(Ra)2, где Ra принимает значения, определенные выше;
каждый R1 представляет собой Н или независимо выбран из группы, включающей ОН, галоген, С1-10-алкил, С1-6-алкокси и С6-10-арил, причем указанные С1-10-алкил, С6-10-арил и алкильная часть С1-6-алкокси являются необязательно замещенными 1-3 галогенами, ОН, ОС1-3-алкильной, фенильной или нафтильной группами, и указанные фенил и нафтил являются необязательно замещенными 1-3 заместителями, независимо выбранными из галогена, ОСН3,OCF3, CH3, CF3 и фенила, где указанный фенил необязательно замещен 1-3 галогеновыми группами,
или две R1 группы вместе представляют собой конденсированный С5-6-алкильный или арильный цикл, который может быть необязательно замещенным 1-2 ОН или Ra группами, где Ra принимает значения, определенный выше;
R2 и R3 взяты вместе или отдельно;
когда взяты вместе, R2 и R3 представляют собой (а) С3-8 алкандиил, образующий конденсированный 5-10-членный неароматический цикл, необязательно содержащий 1-2 двойные связи и необязательно содержащий 1-2 гетероатома, выбранных из О, S и N; или (b) конденсированную 6-10-членную ароматическую моноциклическую или бициклическую группу, причем указанные алкандиил и ароматическая моноциклическая или бициклическая группа являются необязательно замещенными 1-6 атомами галогенов и 1-4 группами, выбранными из ОН, С1-3-алкила, ОС1-3-алкила, галоген-С1-3алкила, галоген-С1-3-алкокси и фенила, и указанный фенил является необязательно замещенным 1-4 группами, независимо выбранными из галогена, С1-3-алкила, ОС1-3-алкила, и указанный С1-3-алкил и С1-3-алкильная часть ОС1-3-алкила являются необязательно замещенными 1-3 галогеновыми группами;
когда взяты отдельно,
R2 выбран из группы, включающей (а) С1-14-алкил, необязательно замещенный 1-6 галогеновыми группами и 1-3 заместителями, выбранными из ОН, ОС1-3-алкила и фенила, причем указанный фенил является необязательно замещенным 1-4 группами, независимо выбранными из галогена, ОСН3, OCF3, CH3 и CF3, и указанная С1-3-алкильная часть ОС1-3-алкила является необязательно замещенной 1-3 галогеновыми группами; (b) фенил или пиридил, необязательно замещенный 1-3 галогенами, ОН или Ra группами, где Ra принимает значения, определенные выше; (с) С2-10-алкенил, необязательно замещенный 1-3 заместителями, независимо выбранными из галогена, ОН и ОС1-3-алкила, причем указанная С1-3-алкильная часть ОС1-3-алкила является необязательно замещенной 1-3 галогеновыми группами; (d) СН2СО2Н; (е) СН2СО2-С1-6-алкил; (f) CH2C(O)NHRa, где Ra принимает значения, определенные выше; (g) NH2, NHRa и N(Ra)2, где Ra принимает значения, определенные выше;
и R3 выбран из группы, включающей С1-14-алкил, С2-10-алкенил, SC1-6-алкил, С6-10-арил, гетероциклил и гетероарил, причем указанные алкил, алкенил, арил, гетероциклил, гетероарил и алкильная часть SC1-6-алкила являются необязательно замещенными (а) R; (b) 1-6 галогеновыми группами и (с) 1-3 группами, выбранными из ОН, NH2, NHC1-4-алкила, группы N(C1-4-алкил)2, С1-4-алкила, ОС1-4-алкила, CN, C1-4-алкил-S(O)x-, где х равен 0, 1 или 2, С1-4-алкил-SO2NH-, H2NSO2-, C1-4-алкил-NHSO2- и (С1-4-алкил)2NSO2-, причем указанный С1-4-алкил и С1-4-алкильные части указанных групп являются необязательно замещенными фенилом и 1-3 галогеновыми группами, и
R выбран из гетероциклила, гетероарила и арила, причем указанная группа является необязательно замещенной 1-4 группами, выбранными из галогена, С1-4-алкила, С1-4-алкил-S(O)x-, где х принимает значения, определенные выше, С1-4-алкил-SO2NH-, H2NSO2-, C1-4-алкил-NHSO2-, (C1-4-алкил)2NSO2-, CN, OH, OC1-4-алкила, и указанный С1-4-алкил и С1-4-алкильные части указанных групп являются необязательно замещенными 1-5 галогенами и 1 группой, выбранной из ОН и ОС1-3-алкила.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Данное изобретение относится к соединению, представленному формулой I:
или его фармацевтически приемлемой соли или сольвату, где:
А и В могут быть взяты отдельно или вместе;
когда взяты отдельно,
А представляет собой галоген, С1-6-алкил, ОС1-6-алкил или фенил, причем указанные алкил, фенил и алкильная часть ОС1-6-алкила являются необязательно замещенными 1-3 галогеновыми группами; и
В представляет собой Н, галоген, С1-6-алкил, -ОС1-6-алкил, -SC1-6-алкил, C2-6-алкенил, фенил или нафтил, причем указанные алкил, алкенил, фенил, нафтил и алкильные части -ОС1-6-алкила и -SC1-6-алкила являются необязательно замещенными 1-3 группами, выбранными из галогена, ОН, СН3О, CF3 и OCF3; и
когда взяты вместе,
А и В вместе представляют собой (а) С1-4-алкилен, необязательно замещенный 1-3 галогеновыми группами и 1-2 Ra группами, где Ra представляет собой С1-3-алкил, ОС1-3-алкил, С6-10-ар-С1-6-алкилен или фенил, необязательно замещенный 1-3 галогеновыми группами, или (b) С2-5-алкандиил, так что вместе с атомом углерода, к которому присоединены, они образуют 3-6-членный цикл, причем указанный цикл необязательно содержит 1 двойную связь или 1-2 гетероатома, выбранных из О, S и N, и указанный 3-6-членный цикл является необязательно замещенным С1-4-алкиленом, оксо, этилендиокси или пропилендиокси и необязательно дополнительно замещенным 1-4 группами, выбранными из галогена, С1-4-алкила, галоген-С1-4-алкила, С1-3-ацила, С1-3-ацилокси, С1-3-алкокси, С1-6-алкил-ОС(О)-, С2-4-алкенила, С2-4-алкинила, С1-3-алкокси-С1-3-алкила, С1-3-алкокси-С1-3-алкокси, фенила, CN, OH, D, NH2, NHRa и N(Ra)2, где Ra принимает значения, определенные выше;
каждый R1 представляет собой Н или независимо выбран из группы, включающей ОН, галоген, С1-10-алкил, С1-6-алкокси и С6-10-арил, причем указанные С1-10-алкил, С6-10-арил и алкильная часть С1-6-алкокси являются необязательно замещенными 1-3 галогенами, ОН, ОС1-3-алкильными, фенильными или нафтильными группами, и указанные фенил и нафтил являются необязательно замещенными 1-3 заместителями, независимо выбранными из галогена, ОСН3,OCF3, CH3, CF3 и фенила, где указанный фенил является необязательно замещенным 1-3 галогеновыми группами,
или две R1 группы вместе образуют конденсированный С5-6-алкильный или арильный цикл, который может быть необязательно замещенным 1-2 ОН или Ra группами, где Ra принимает значения, определенные выше;
R2 и R3 взяты вместе или отдельно;
когда взяты вместе, R2 и R3 представляют собой (а) С3-8-алкандиил, образующий конденсированный 5-10-членный неароматический цикл, необязательно содержащий 1-2 двойные связи и необязательно содержащий 1-2 гетероатома, выбранных из О, S и N; или (b) конденсированную 6-10-членную ароматическую моноциклическую или бициклическую группу, причем указанные алкандиил и ароматическая моноциклическая или бициклическая группа являются необязательно замещенными 1-6 атомами галогенов и 1-4 группами, выбранными из ОН, С1-3-алкила, ОС1-3-алкила, галоген-С1-3-алкила, галоген-С1-3-алкокси и фенила, и указанный фенил является необязательно замещенным 1-4 группами, независимо выбранными из галогена, С1-3-алкила, ОС1-3-алкила, и указанный С1-3-алкил и С1-3-алкильная часть ОС1-3-алкила являются необязательно замещенными 1-3 галогеновыми группами;
когда взяты отдельно,
R2 выбран из группы, включающей: (а) С1-14-алкил, необязательно замещенный 1-6 галогеновыми группами и 1-3 заместителями, выбранными из ОН, ОС1-3-алкила и фенила, причем указанный фенил необязательно замещен 1-4 группами, независимо выбранными из галогена, ОСН3, OCF3, CH3 и CF3, и указанная С1-3-алкильная часть ОС1-3-алкила является необязательно замещенной 1-3 галогеновыми группами; (b) фенил или пиридил, необязательно замещенный 1-3 галогенами, ОН или Ra группами, где Ra принимает значения, определенные выше; (с) С2-10-алкенил, необязательно замещенный 1-3 заместителями, независимо выбранными из галогена, ОН и ОС1-3-алкила, причем указанная С1-3-алкильная часть ОС1-3-алкила необязательно замещена 1-3 галогеновыми группами; (d) СН2СО2Н; (е) СН2СО2С1-6-алкил; (f) CH2C(O)NHRa, где Ra принимает значения, определенные выше; (g) NH2, NHRa и N(Ra)2, где Ra принимает значения, определенные выше;
и R3 выбран из группы, включающей С1-14-алкил, С2-10-алкенил, SC1-6-алкил, С6-10-арил, гетероциклил и гетероарил, причем указанные алкил, алкенил, арил, гетероциклил, гетероарил и алкильная часть SC1-6-алкила являются необязательно замещенными (а) R; (b) 1-6 галогеновыми группами и (с) 1-3 группами, выбранными из ОН, NH2, NHC1-4-алкила, N(C1-4-алкил)2, С1-4-алкила, ОС1-4-алкила, CN, C1-4-алкил-S(O)x-, где х равен 0, 1 или 2, С1-4-алкил-SO2NH-, H2NSO2-, C1-4-алкил-NHSO2- и (С1-4-алкил)2NSO2-, причем указанный С1-4-алкил и С1-4-алкильные части указанных групп необязательно замещены фенилом и 1-3 галогеновыми группами, и
R выбран из гетероциклила, гетероарила и арила, причем указанная группа необязательно замещена 1-4 группами, выбранными из галогена, С1-4-алкила, С1-4-алкил-S(O)x-, где х принимает значения, определенные выше, С1-4-алкил-SO2NH-, H2NSO2-, C1-4-алкил-NHSO2-, (C1-4-алкил)2NSO2-, CN, OH, OC1-4-алкила, и указанный С1-4-алкил и С1-4-алкильные части указанных групп необязательно замещены 1-5 галогеновыми группами и 1 группой, выбранной из ОН и ОС1-3-алкила.
В данном описании применимы следующие определения.
Термин "Ac" означает ацетил, то есть группу СН3С(О)-.
Термин "алкил", а также префикс "алк" в других группах, таких как алкокси и алканоил, означает углеродные цепи, которые могут быть линейными или разветвленными, или их сочетания, если углеродная цепь не определена иначе. Примеры алкильных групп включают метил, этил, пропил, изопропил, бутил, втор- и трет-бутил, пентил, гексил, гептил, октил, нонил и т.п. Когда позволяет указанное количество атомов углерода, например для С3-С10, термин "алкил" включает также циклоалкильные группы и сочетания линейных или разветвленных алкильных цепей с циклоалкильными структурами. Когда количество атомов углерода не указано, подразумевается С1-6.
Термин "алкенил" означает углеродные цепи, которые содержат, по меньшей мере, одну углерод-углеродную двойную связь и которые могут быть линейными или разветвленными, или их сочетания, если углеродная цепь не определена иначе. Примеры алкенила включают винил, аллил, изопропенил, пентенил, гексенил, гептенил, 1-пропенил, 2-бутенил, 2-метил-2-бутенил и т.п. Когда указанное количество атомов углерода позволяет, например для С5-С10, термин "алкенил" включает также циклоалкенильные группы и сочетания линейных, разветвленных и циклических структур. Когда количество атомов углерода не указано, подразумевается С2-6.
Термин "алкинил" означает углеродные цепи, которые содержат, по меньшей мере, одну углерод-углеродную тройную связь и которые могут быть линейными или разветвленными или представлять собой их сочетания. Примеры алкинила включают этинил, пропаргил, 3-метил-1-пентинил, 2-гептинил и т.п.
Термин "алкандиил" относится к углеродным цепям, которые являются бифункциональными, таким как -СН2-, -(СН2)2-, -(СН2)3- и т.п. Алкандиильные группы являются линейными или разветвленными, если не указано другое. Для сравнения, алкильные группы являются монофункциональными.
Термин "алкилен" в данном описании относится к атому углерода или углеродной цепи, который(ая) присоединен(а) через двойную связь.
Термин "циклоалкил" относится к подгруппе алкила и означает насыщенный карбоцикл, содержащий указанное количество атомов углерода. Примеры циклоалкила включают циклопропил, циклобутил, циклопентил, циклогексил, циклогептил и т.п. Циклоалкильная группа обычно является моноциклической, если не указано другое. Циклоалкильные группы являются насыщенными, если не указано другое.
Термин "арил" означает ароматическую моно- или полициклическую систему, содержащую в кольце атомы углерода. Предпочтительными арилами являются ароматические моноциклические или бициклические 6-10-членные циклические системы. Фенил и нафтил являются предпочтительными арилами. Наиболее предпочтительным арилом является фенил.
Термины "гетероцикл" и "гетероциклил" относятся к насыщенным или ненасыщенным неароматическим циклам или циклическим системам, содержащим, по меньшей мере, один гетероатом, выбранный из О, S и N, и дополнительно включающим атом серы в окисленных формах, SO и SO2. Примеры гетероциклов включают тетрагидрофуран (ТГФ), дигидрофуран, 1,4-диоксан, морфолин, 1,4-дитиан, пиперазин, пиперидин, 1,3-диоксолан, имидазолидин, имидазолин, пирролин, пирролидин, тетрагидропиран, дигидропиран, оксатиолан, дитиолан, 1,3-диоксан, 1,3-дитиан, оксатиан, тиоморфолин и т.п.
Термин "гетероарил" означает ароматический или частично ароматический гетероцикл, который содержит в цикле, по меньшей, один гетероатом, выбранный из О, S и N (включая SO). Таким образом, гетероарилы включают гетероарилы, конденсированные с другими видами циклов, такими как арилы, циклоалкилы и гетероциклы, которые являются неароматическими. Примеры гетероарильных групп включают пирролил, изоксазолил, изотиазолил, пиразолил, пиридил, оксазолил, оксадиазолил, тиадиазолил, тиазолил, имидазолил, триазолил, тетразолил, фурил, триазинил, тиенил, пиримидил, бензизоксазолил, бензоксазолил, бензотиазолил, бензотиадиазолил, дигидробензофуранил, индолинил, пиридазинил, индазолил, изоиндолил, дигидробензотиенил, индолизинил, хинолизинил, циннолинил, фталазинил, хиназолинил, нафтиридинил, карбазолил, бензодиоксолил, хиноксалинил, пуринил, фуразанил, изобензилфуранил, бензимидазолил, бензофуранил, бензотиенил (включая S-оксид), хинолил, индолил, изохинолил, дибензофуранил, нафтиридил и т.п. Гетероциклильные и гетероарильные группы включают циклы и циклические системы, содержащие от 3 до 15 атомов, образующих 1-3 цикла.
Термин "галоген" относится к фтору, хлору, брому и йоду. Хлор и фтор обычно являются предпочтительными. Фтор является наиболее предпочтительным, когда галогены замещают алкильную или алкоксигруппу (например, CF3O и CF3CH2O).
Термин "фармацевтическая композиция" означает препарат, включающий активный(е) ингредиент(ы) и носитель, а также любой продукт, который образуется в результате, непосредственно или опосредованно, сочетания, комплексообразования или агрегации любых двух или нескольких ингредиентов или в результате реакции диссоциации или реакции другого типа одного или нескольких ингредиентов. Соответственно, фармацевтические композиции данного изобретения включают композиции, полученные смешением соединения или соединений данного изобретения и фармацевтически приемлемого носителя.
Соединения формулы I могут содержать один или несколько асимметрических центров и, следовательно, могут существовать в виде рацематов или рацемических смесей, индивидуальных энантиомеров, диастереомерных смесей и индивидуальных диастереомеров. Все эти изомерные формы включены в область данного изобретения.
Некоторые соединения, описанные в данном изобретении, содержат двойные олефиновые связи. В область данного изобретения включены как Е, так и Z геометрические изомеры в чистой форме, а также в виде смеси.
Некоторые соединения, описанные в данном изобретении, могут существовать в виде таутомеров, которые имеют различные точки присоединения водорода, что сопровождается смещением одной или нескольких двойных связей. Например, кетон и его енольная форма являются кето-енольными таутомерами. Изобретение включает как отдельные таутомеры, так и их смеси.
Если необходимо, рацемические смеси соединений формулы I могут быть разделены с выделением отдельных энантиомеров. Разделение может проводиться способами, хорошо известными в данной области техники, такими как сочетание рацемической смеси соединений формулы I с энантиомерно чистым соединением с образованием диастереомерной смеси, которая затем подвергается разделению на отдельные диастереомеры стандартными методами, такими как фракционная кристаллизация или хроматография. Реакция сочетания зачастую является реакцией получения солей с использованием энантиомерной чистой кислоты или основания. Диастереомерные производные после этого могут быть превращены в, по существу, чистые энантиомеры отщеплением присоединенного хирального остатка от диастереомерного соединения.
Рацемическая смесь соединений формулы I также может разделяться непосредственно хроматографическими методами с использованием хиральных неподвижных фаз, и эти методы хорошо известны в данной области техники.
Альтернативно, энантиомеры соединений общей формулы I могут быть получены стереоселективным синтезом с использованием оптически чистых исходных веществ или реагентов.
Один аспект данного изобретения, который представляет особый интерес, относится к соединению формулы I, где А и В взяты вместе и представляют собой С2-5-алкандиил, так что вместе с атомом углерода, к которому присоединены, они образуют 3-6-членный цикл, причем указанный цикл необязательно содержит 1 двойную связь или 1-2 гетероатома, выбранных из О, S и N, и указанный 3-6-членный цикл является необязательно замещенным С1-4-алкиленом, оксо, этилендиокси или пропилендиокси и дополнительно необязательно замещенным 1-4 группами, выбранными из галогена, С1-4-алкила, галоген-С1-4-алкила, С1-3-ацила, С1-3-ацилокси, С1-3-алкокси, С1-6-алкил-ОС(О)-, С2-4-алкенила, С2-4-алкинила, С1-3-алкокси-С1-3-алкила, С1-3-алкокси-С1-3-алкокси, фенила, CN, OH, D, NH2, NHRa и N(Ra)2, где Ra представляет собой С1-3-алкил, ОС1-3-алкил, С6-10-ар-С1-6-алкилен или фенил, необязательно замещенный 1-3 атомами галогена. В данный аспект изобретения включены все другие переменные, которые первоначально определены согласно формуле I.
Другим аспектом данного изобретения, который представляет еще больший интерес, является соединение, описанное выше, где А и В взяты вместе и представляют собой С2-4-членную алкандиильную группу, так что вместе с атомом углерода, к которому присоединены, они образуют 3-5-членный цикл, необязательно замещенный 1-2 группами, выбранными из галогена, С1-4-алкила, галоген-С1-4-алкила, С1-3-алкокси, С1-3-алкокси-С1-3-алкила, С1-3-алкокси-С1-3-алкокси и фенила. В данный аспект включены все другие переменные, которые первоначально определены в соответствии с формулой I.
Еще более точно, аспект изобретения, который представляет интерес, относится к соединению, описанному выше, где А и В взяты вместе и представляют собой С2-4 алкандиильную группу, так что вместе с атомом углерода, к которому присоединены, они образуют 3-5-членный цикл, причем указанный цикл является незамещенным или замещен 1-2 галогеновыми группами. В данный аспект включены все переменные, которые определены в соответствии с формулой I.
Еще более точно, аспект данного изобретения, который представляет интерес, относится к соединению, описанному выше, где 1-2 галогеновые группы представляют собой атомы фтора. В данный аспект включены все переменные, которые определены в соответствии с формулой I.
В другом аспекте данного изобретения, который представляет интерес, раскрыто соединение формулы I, где две группы R1 представляют собой Н и одна группа R1 выбрана из группы, включающей ОН, галоген, С1-10-алкил, С1-6-алкокси и С6-10-арил, причем указанные С1-10-алкил, С6-10-арил и алкильная часть С1-6-алкокси являются необязательно замещенными 1-3 галогенами, ОН, ОС1-3алкильной, фенильной или нафтильной группами, и указанный фенил и нафтил являются необязательно замещенными 1-3 заместителями, выбранными из галогена, ОСН3, OCF3, CH3, CF3 и фенила, где указанный фенил необязательно замещен 1-3 галогеновыми группами. В данный аспект включены все другие переменные, которые первоначально определены согласно формуле I.
Более точно, аспект данного изобретения, который представляет интерес, относится к соединению формулы I, где одна группа R1 представляет собой Н и две другие группы R1 выбраны из группы, включающей ОН, галоген, С1-10-алкил и С1-6-алкокси, причем указанный С1-10-алкил и алкильная часть С1-6-алкокси являются необязательно замещенными 1-3 галогеновыми группами. В данный аспект включены все другие переменные, которые первоначально определены согласно формуле I.
Еще более точно, аспект данного изобретения, который представляет интерес, относится к соединению формулы I, где две группы R1 представляют собой галоген или метил. Данный аспект включает все другие переменные, которые первоначально определены согласно формуле I.
В другом аспекте данного изобретения раскрыто соединение формулы I, где R2 взят отдельно от R3 и выбран из группы, включающей (а) С1-14-алкил, необязательно замещенный 1-6 галогеновыми группами и 1-3 заместителями, выбранными из ОН, ОС1-3-алкила и фенила, причем указанный фенил является необязательно замещенным 1-4 группами, независимо выбранными из галогена, ОСН3, OCF3, CH3 и CF3, и указанная С1-3-алкильная часть ОС1-3-алкила является необязательно замещенной 1-3 галогеновыми группами; (b) фенил или пиридил, необязательно замещенный 1-3 галогеновыми, ОН или Ra группами; (с) С2-10-алкенил, необязательно замещенный 1-3 заместителями, независимо выбранными из галогена, ОН и ОС1-3алкила, причем указанная С1-3-алкильная часть ОС1-3-алкила является необязательно замещенной 1-3 галогеновыми группами; (d) CH2CO2H; (e) CH2CO2C1-6-алкил; (f) СН2С(О)NHRa и (g) NH2, NHRa и N(Ra)2 и
Ra представляет собой С1-3-алкил, ОС1-3-алкил, С6-10-арил-С1-6-алкилен или фенил, необязательно замещенный 1-3 галогеновыми группами. Данный аспект изобретения включает все другие переменные, которые первоначально определены в соответствии с формулой I.
Более точно, в данном аспекте настоящего изобретения раскрыто соединение формулы I, где R2 взят отдельно от R3 и представляет собой С1-14-алкил, необязательно замещенный 1-6 галогеновыми группами и 1-3 заместителями, выбранными из ОН, ОС1-3-алкила и фенила, причем указанный фенил является необязательно замещенным 1-4 группами, независимо выбранными из галогена, ОСН3, OCF3, CH3 и CF3, и алкильная часть ОС1-3-алкила является необязательно замещенной 1-3 галогеновыми группами. Данный аспект включает все другие переменные, которые первоначально определены в соответствии с формулой I.
Еще более точно, аспект данного изобретения, который представляет особый интерес, относится к соединению формулы I, где R2 взят отдельно от R3 и представляет собой метил или циклопропил. Данный аспект включает все другие переменные, которые первоначально определены в соответствии с формулой I.
В другом аспекте данного изобретения, соединение, которое представляет интерес, определено в соответствии с формулой I, где R3 взят отдельно от R2 и выбран из группы, включающей С1-14-алкил, С2-10-алкенил, SC1-6-алкил, С6-10-арил, гетероциклил и геретоарил, причем указанный алкил, алкенил, арил, гетероциклил, гетероарил и алкильная часть SC1-6-алкила являются необязательно замещенными (а) R; (b) 1-6 галогеновыми группами и (с) 1-3 группами, выбранными из ОН, NH2, NHC1-4-алкила, N(С1-4-алкил)2, C1-4-алкила, OC1-4-алкила, CN, C1-4-алкил-S(O)x-, где х равен 0, 1 или 2, С1-4-алкил-SO2NH-, H2NSO2-, C1-4-алкил-NHSO2- и (С1-4-алкил)2NSO2-,причем указанный С1-4-алкил и С1-4-алкильные части указанных групп являются необязательно замещенными фенилом и 1-3 галогеновыми группами, и R выбран из гетероциклила, гетероарила и арила, причем указанная группа является необязательно замещенной 1-4 группами, выбранными из галогена, С1-4-алкила, С1-4-алкил-S(O)x-, где х принимает значения, определенные выше, С1-4-алкил-SO2NH-, H2NSO2-, C1-4-алкил-NHSO2-, (C1-4-алкил)2NSO2-, CN, OH, OC1-4-алкила, причем указанный С1-4-алкил и С1-4-алькильные части указанных групп являются необязательно замещенными 1-5 атомами галогенов и 1 группой, выбранной из ОН и ОС1-3-алкила. Данный аспект включает все другие переменные, которые первоначально определены согласно формуле I.
Более точно, соединение, которое представляет интерес, определено в соответствии с формулой I, где R3 взят отдельно от R2 и выбран из группы, включающей С1-14-алкил, С6-10-арил, гетероциклил и гетероарил, причем указанные группы являются необязательно замещенными (а) R; (b) 1-6 галогеновыми группами и (с) 1-3 группами, выбранными из ОН, NH2, NHC1-4-алкила, N(C1-4-алкил)2, С1-4-алкила, ОС1-4-алкила, CN, С1-4-алкил-S(O)x-, где х равен 0, 1 или 2, С1-4-алкил-SO2NH-, H2NSO2-, C1-4-алкил-NHSO2-, (C1-4-алкил)2NSO2-, причем указанный С1-4-алкил и С1-4-алкильные части указанных групп являются необязательно замещенными фенилом и 1-3 атомами галогенов. Данный аспект изобретения включает все другие переменные, которые первоначально определены согласно формуле I.
Еще более точно, соединение, которое представляет интерес, определено в соответствии с формулой I, где R3 взят отдельно и выбран из группы, включающей циклопропил, необязательно замещенный метилом или фенилом; фенил, необязательно замещенный галогеном, ОН, ОСН3 или OCF3; гетероарил, выбранный из бензимидазолила, индолила, бензофуранила и дигидробензофуранила, причем указанные гетероарильные группы являются необязательно замещенными (а) R; (b) 1-6 галогеновыми группами или (с) 1-3 группами, выбранными из OH, NH2, NHC1-4-алкила, группы N(C1-4-алкил)2, С1-4-алкила, ОС1-4-алкила, CN, C1-4-алкил-S(O)x-, где х равен 0, 1 или 2, С1-4-алкил-SO2NH-, H2NSO2-, C1-4-алкил-NHSO2-, (C1-4-алкил)2NSO2-, причем указанный С1-4-алкил и С1-4-алкильные части указанных групп являются необязательно замещенными фенилом и 1-3 галогеновыми группами, и R выбран из гетероциклила, гетероарила и арила, причем указанная группа является необязательно замещенной 1-4 группами, выбранными из галогена, С1-4-алкила, ОН, ОС1-4-алкила, и указанный С1-4-алкил и С1-4-алкильные части указанных групп являются необязательно замещенными 1-5 галогеновыми группами и 1 группой, выбранной из ОН и ОС1-3-алкила. Данный аспект изобретения включает все другие переменные, которые первоначально определены согласно формуле I.
В другом аспекте данного изобретения, который представляет интерес, описано соединение формулы I, где R2 и R3 взяты вместе и представляют собой (а) С3-8-алкандиил, образующий конденсированный 5-10-членный неароматический цикл, необязательно содержащий 1 двойную связь и необязательно содержащий 1 гетероатом, выбранный из О, S и N; или (b) конденсированную 6-10-членную ароматическую моноциклическую или бициклическую группу,
причем указанные алкандиил и ароматическая моноциклическая или бициклическая группа являются необязательно замещенными 1-3 атомами галогенов и 1-2 группами, выбранными из ОН, C1-3-алкила, ОС1-3-алкила, галоген-С1-3-алкила, галоген-С1-3-алкокси и фенила, причем указанный фенил является необязательно замещенным 1-2 группами, независимо выбранными из галогена, С1-3-алкила, ОС1-3-алкила, и указанный С1-3-алкил и С1-3-алкильная часть ОС1-3-алкила являются необязательно замещенными 1-3 галогеновыми группами. Данный аспект изобретения включает все другие переменные, которые первоначально определены в соответствии с формулой I.
Более точно, аспект данного изобретения, который представляет интерес, относится к соединению формулы I, где R выбран из гетероциклила, гетероарила и арила, причем указанная группа является необязательно замещенной 1-4 галогеновыми группами и 1-2 группами, выбранными из С1-4-алкила, С1-4-алкил-S(O)x-, где х равен 0, 1 или 2, С1-4-алкил-SO2NH-, H2NSO2-, C1-4-алкил-NHSO2-, (C1-4-алкил)2NSO2-, CN, OH и ОС1-4-алкила, причем указанный С1-4-алкил и С1-4-алкильные части указанных групп являются необязательно замещенными 1-3 галогеновыми группами и 1 группой, выбранной из ОН и ОС1-3-алкила. Данный аспект включает все другие переменные, которые первоначально определены в соответствии с формулой I.
Соединения, которые входят в объем данного изобретения, включают соединения, описанные в примерах. Иллюстративными, но не ограничивающими, примерами соединений данного изобретения являются ингибиторы 11β-HSD-1, представляющие соединения следующих структурных формул
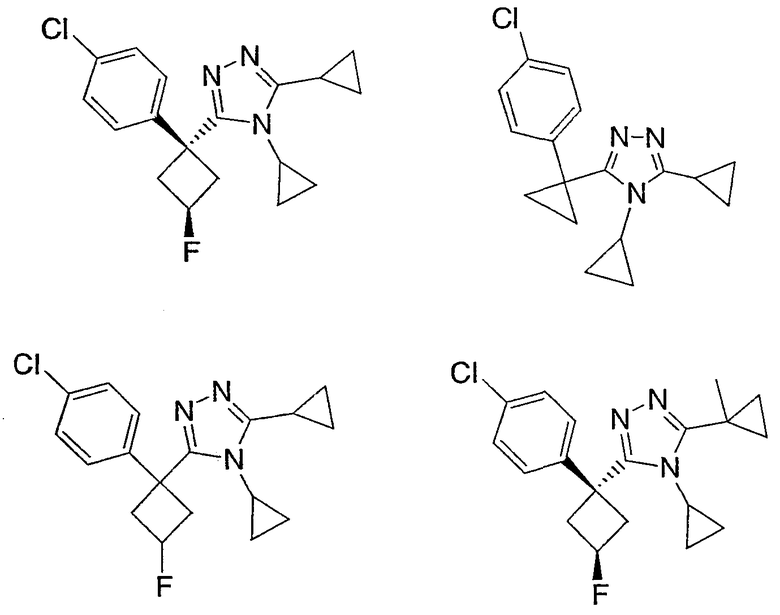
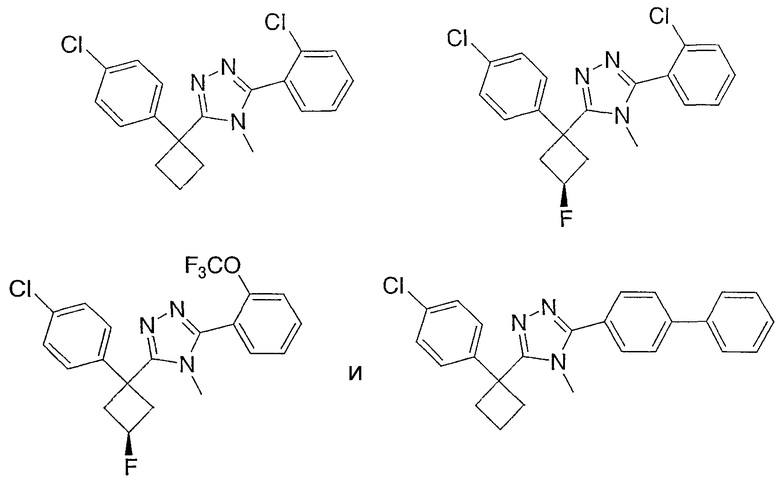
и их фармацевтически приемлемые соли и сольваты.
Еще один аспект данного изобретения относится к фармацевтической композиции, включающей соединение согласно формуле I или его соль или гидрат в сочетании с фармацевтически приемлемым носителем.
Еще один аспект данного изобретения относится к способу лечения гипергликемии, сахарного диабета или инсулинорезистентности у пациента-млекопитающего, нуждающегося в таком лечении, который включает введение указанному пациенту эффективного количества соединения согласно формуле I или его соли или сольвата.
В еще одном аспекте данного изобретения раскрыт способ лечения инсулиннезависимого сахарного диабета у пациента-млекопитающего, нуждающегося в таком лечении, который включает введение указанному пациенту соединения согласно формуле I в количестве, эффективном для лечения сахарного диабета.
В еще одном аспекте данного изобретения описан способ лечение ожирения у пациента-млекопитающего, нуждающегося в таком лечении, который включает введение указанному пациенту соединения согласно формуле I в количестве, которое эффективно для лечения ожирения.
В еще одном аспекте данного изобретения описан способ лечения синдрома Х у пациента-млекопитающего, нуждающегося в таком лечении, который включает введение указанному пациенту соединения согласно формуле I в количестве, которое эффективно для лечения синдрома Х.
В еще одном аспекте данного изобретения описан способ лечения липидного расстройства, выбранного из группы, включающей дислипидемию, гиперлипидемию, гипертриглицеридемию, гиперхолестеринемию, низкое содержание HDL и высокое содержание LDL, у пациента-млекопитающего, нуждающегося в таком лечении, который включает введение указанному пациенту соединения согласно формуле I в количестве, эффективном для лечения указанного липидного расстройства.
В еще одном аспекте данного изобретения описан способ лечения атеросклероза у пациента-млекопитающего, нуждающегося в таком лечении, который включает введение указанному пациенту соединения согласно формуле I в количестве, эффективном для лечения атеросклероза.
В еще одном аспекте данного изобретения раскрыт способ лечения состояния, выбранного из группы, включающей (1) гипергликемию, (2) низкую толерантность к глюкозе, (3) инсулинорезистентность, (4) ожирение, (5) липидные расстройства, (6) дислипидемию, (7) гиперлипидемию, (8) гипертриглицеридемию, (9) гиперхолестеринемию, (10) низкие уровни содержания HDL, (11) высокие уровни содержания LDL, (12) атеросклероз и его последствия, (13) сосудистый рестеноз, (14) панкреатит, (15) отложение жировой ткани в области брюшины, (16) нейродегенеративное заболевание, (17) ретинопатию, (18) нефропатию, (19) нейропатию, (20) синдром Х и другие состояния и расстройства, компонентом которых является инсулинорезистентность, у пациента-млекопитающего, нуждающегося в таком лечении, который включает введение пациенту соединения согласно формуле I в количестве, эффективном для лечения указанного состояния.
В другом аспекте данного изобретения раскрыт способ лечения состояния, выбранного из группы, включающей (1) гипергликемию, (2) низкую толерантность к глюкозе, (3) инсулинорезистентность, (4) ожирение, (5) липидные расстройства, (6) дислипидемию, (7) гиперлипидемию, (8) гипертриглицеридемию, (9) гиперхолестеринемию, (10) низкие уровни содержания HDL, (11) высокие уровни содержания LDL, (12) атеросклероз и его последствия, (13) сосудистый рестеноз, (14) панкреатит, (15) отложение жировой ткани в области брюшины, (16) нейродегенеративное заболевание, (17) ретинопатию, (18) нефропатию, (19) нейропатию, (20) синдром Х и другие состояния и расстройства, компонентом которых является инсулинорезистентность, у пациента-млекопитающего, нуждающегося в таком лечении, который включает введение пациенту эффективного количества соединения согласно формуле I и соединения, выбранного из группы включающей:
(a) DP-IV ингибиторы;
(b) сенсибилизаторы инсулина, выбранные из группы, включающей (i) PPAR агонисты и (ii) бигуаниды;
(c) инсулин и имитаторы инсулина;
(d) сульфонилмочевины и другие средства, усиливающие секрецию инсулина;
(е) ингибиторы α-глюкозидазы;
(f) антагонисты глюкогонового рецептора;
(g) GLP-1, имитаторы GLP-1 и агонисты GLP-1 рецептора;
(h) GIP, имитаторы GIP и агонисты GIP рецептора;
(i) PACAP, имитаторы РАСАР и агонисты РАСАР рецептора 3;
(j) средства, снижающие содержание холестерина и выбранные из группы, включающей (i) ингибиторы HMG-CoA редуктазы, (ii) вещества, усиливающие экскрецию, (iii) никотиниловый спирт, никотиновую кислоту и их соли, (iv) PPARα агонисты, (v) двойные PPAR α/γ агонисты, (vi) ингибиторы абсорбции холестерина, (vii) ингибиторы ацил-СоА:холестерин ацилтрансферазы и (viii) антиоксиданты;
(k) PPAR δ агонисты;
(l) соединения для лечения ожирения;
(m) ингибитор транспортера подвздошной желчной кислоты;
(n) противовоспалительные средства, отличные от глюкокортикоидов;
(о) ингибиторы протеин-тирозин-фосфатазы-1В (РТР-1В),
причем указанные соединения вводятся пациенту в количестве, эффективном для лечения указанного состояния.
В другом аспекте данного изобретения раскрыт способ лечения состояния, выбранного из группы, включающей гиперхолестеринемию, атеросклероз, низкие уровни содержания HDL, высокие уровни содержания LDL, гиперлипидемию, гипертриглицеридемию и дислипидемию, у пациента-млекопитающего, нуждающегося в таком лечении, который включает введение пациенту терапевтически эффективного количества соединения согласно формуле I и ингибитора HMG-CoA редуктазы.
Более точно, в другом аспекте данного изобретения раскрыт способ лечения состояния, выбранного из группы, включающей гиперхолестеринемию, атеросклероз, низкие уровни содержания HDL, высокие уровни содержания LDL, гиперлипидемию, гипертриглицеридемию и дислипидемию, у пациента-млекопитающего, нуждающегося в таком лечении, где ингибитор HMG-CoA редуктазы представляет собой статин.
Еще более точно, в другом аспекте изобретения описан способ лечения состояния, выбранного из группы, включающей гиперхолестеринемию, атеросклероз, низкие уровни содержания HDL, высокие уровни содержания LDL, гиперлипидемию, гипертриглицеридемию и дислипидемию, у пациента-млекопитающего, нуждающегося в таком лечении, где ингибитор HMG-CoA редуктазы представляет собой статин, выбранный из группы, включающей ловастатин, симвастатин, правастатин, флувастатин, аторвастатин, итавастатин, ZD-4522 и ривастатин.
В другом аспекте данного изобретения раскрыт способ лечения атеросклероза у пациента - человека, нуждающегося в таком лечении, где ингибитор HMG-CoA редуктазы представляет собой статин, который дополнительно включает введение ингибитора абсорбции холестерина.
Более точно, в другом аспекте данного изобретения раскрыт способ лечения атеросклероза у пациента - человека, нуждающегося в таком лечении, где ингибитор HMG-CoA редуктазы представляет собой статин и ингибитор абсорбции холестерина представляет собой эзетимиб.
В другом аспекте данного изобретения раскрыта фармацевтическая композиция, которая включает
(1) соединение согласно формуле I,
(2) соединение, выбранное из группы, включающей
(a) DP-IV ингибиторы;
(b) сенсибилизаторы инсулина, выбранные из группы, включающей (i) PPAR агонисты и (ii) бигуаниды;
(c) инсулин и имитаторы инсулина;
(d) сульфонилмочевины и другие средства, усиливающие секрецию инсулина;
(е) ингибиторы α-глюкозидазы;
(f) антагонисты глюкагонового рецептора;
(g) GLP-1, имитаторы GLP-1 и агонисты GLP-1 рецептора;
(h) GIP, имитаторы GIP и агонисты GIP рецептора;
(i) PACAP, имитаторы РАСАР и агонисты РАСАР рецептора 3;
(j) средства, снижающие содержание холестерина и выбранные из группы, включающей (i) ингибиторы HMG-CoA редуктазы, (ii) вещества, усиливающие экскрецию, (iii) никотиниловый спирт, никотиновую кислоту и их соли, (iv) PPARα агонисты, (v) двойные PPAR α/γ агонисты, (vi) ингибиторы абсорбции холестерина, (vii) ингибиторы ацил-СоА:холестерин ацилтрансферазы и (viii) антиоксиданты;
(k) PPAR δ агонисты;
(l) соединения для лечения ожирения;
(m) ингибитор транспортера подвздошной желчной кислоты;
(n) противовоспалительные средства, отличные от глюкокортикоидов;
(о) ингибиторы протеин-тирозин-фосфатазы-1В (РТР-1В);
и
(3) фармацевтически приемлемый носитель.
Термин "фармацевтически приемлемая соль" относится к солям, полученным из фармацевтически приемлемых оснований или кислот, включая неорганические или органические основания и неорганические или органические кислоты. Соли, полученные из неорганических оснований, включают соли алюминия, аммония, кальция, меди, трехвалентного железа, двухвалентного железа, лития, магния, трехвалентного марганца, двухвалентного марганца, калия, натрия, цинка и т.п. Особенно предпочтительными являются соли аммония, кальция, магния, калия и натрия. Соли в твердой форме могут иметь несколько кристаллических структур, а также могут существовать в форме гидратов и полигидратов.
Соли, полученные из фармацевтически приемлемых органических оснований, включают соли первичных, вторичных и третичных аминов, замещенных аминов, в том числе существующих в природе замещенных аминов, циклических аминов, и основных ионно-обменных смол, таких как аргинин, бетаин, кофеин, холин, N,N'-дибензилэтилендиамин, диэтиламин, 2-диэтиламиноэтанол, 2-диметиламиноэтанол, этаноламин, этилендиамин, N-этилморфолин, N-этилпиперидин, глюкамин, глюкозамин, гистидин, гидрабамин, изопропиламин, лизин, метилглюкамин, морфолин, пиперазин, пиперидин, полиаминовые смолы, прокаин, пурины, теобромин, триэтиламин, триметиламин, трипропиламин, трометамин и т.п.
Когда соединение данного изобретения является основным, соли могут быть получены из фармацевтически приемлемых кислот, включая неорганические и органические кислоты. Такие кислоты включают уксусную, бензолсульфоновую, бензойную, камфорсульфоновую, лимонную, этансульфоновую, фумаровую, глюконовую, глутаминовую, бромистоводородную, хлористоводородную, изетионовую, молочную, малеиновую, яблочную, миндальную, метансульфоновую, слизевую, азотную, памовую, пантотеновую, фосфорную, янтарную, серную, винную, п-толуолсульфоновую кислоту и т.п. Особенно предпочтительные фармацевтически приемлемые кислоты включают лимонную, бромистоводородную, хлористоводородную, малеиновую, фосфорную, серную и винную кислоты. В большинстве случаев соединения данного изобретения являются основными, поскольку триазольный цикл является основным. Производные триазола данного изобретения также могут быть получены и обрабатываться в виде фармацевтически не приемлемых солей (например, трифторацетатных солей) в процессе синтеза их перед применением для получения фармацевтических средств.
Будет понятно, что ссылки на соединения формулы I в данном изобретении включают фармацевтически приемлемые соли, а также фармацевтически неприемлемые соли, когда они используются в качестве предшественников свободных соединений или их фармацевтически приемлемых солей или в других синтетических манипуляциях.
Сольваты и, в частности, гидраты соединений формулы I включены в область данного изобретения.
Метаболиты соединений данного изобретения, которые являются терапевтически активными и которые определены формулой I, также включены в область данного изобретения. Пролекарства представляют собой соединения, которые превращаются в терапевтически активные соединения при введении пациенту или после введения пациенту. Соединения, которые сами по себе не обладают структурами, заявленными в данном изобретении, но которые превращаются в активные соединения, определенные формулой I в процессе или после введения млекопитающему пациенту, являются пролекарствами и соединениями данного изобретения, поскольку являются их активными метаболитами, которые определены формулой I.
Соединения, описанные в данном изобретении, являются селективными ингибиторами 11β-HSD1 фермента. Следовательно, данное изобретение относится к применению ингибитора 11β-HSD1 для ингибирования редуктазной активности 11β-гироксистероидной дегидрогеназы, которая ответственна за превращение кортизона в кортизол. Избыточный кортизол связан с рядом расстройств, включая NIDDM, ожирение, дислипидемию, инсулинорезистентность и гипертензию. Введение соединений снижает уровень содержания кортизола и других 11β-гидроксистероидов в тканях-мишенях, снижая, таким образом, эффекты избыточных количеств кортизола и других 11β-гидроксистероидов. Ингибирование 11β-HSD1 может применяться для лечения и контроля заболеваний, проводимых патологически высокими уровнями содержания кортизола и других 11β-гидроксистероидов, таких как NIDDM, ожирение, гипертензия и дислипидемия.
Данное изобретение включает применение 11β-HSD1 ингибитора для лечения, контроля, облегчения, профилактики, задержки атаки или снижения риска развития заболеваний и состояний, которые описаны выше и которые проводятся избыточными или неконтролируемыми количествами кортизола и/или других кортикостероидов у пациента-млекопитающего, в частности у человека, введением эффективного количества соединения формулы I или его фармацевтически приемлемой соли или сольвата. Ингибирование 11β-HSD1 фермента ограничивает превращение кортизона, который в нормальном состоянии является инертным, в кортизол, который может вызывать симптомы или вносить вклад в симптомы данных заболеваний и состояний, если присутствует в избыточных количествах.
NIDDM и гипертензия
Соединения данного изобретения являются селективными ингибиторами 11β-HSD1 и не воздействуют на 11β-HSD2. В то время как ингибирование 11β-HSD1 полезно для снижения уровней содержания кортизола и лечения состояний, связанных с ним, ингибирование 11β-HSD2 связано с серьезными побочными эффектами, такими как гипертензия.
Кортизол является важным и широко известным противовоспалительным гормоном, который также выступает в качестве антагониста действия инсулина в печени, так что чувствительность к инсулину снижается, приводя к повышенному глюконеогенезу и повышенным уровням содержания глюкозы в печени. Для пациентов с уже ослабленной толерантностью к глюкозе характерна большая вероятность развития сахарного диабета 2 типа в присутствии патологически высоких уровней содержания кортизола.
Высокие уровни содержания кортизола в тканях, где присутствует минералокортикоидный рецептор, зачастую приводит к гипертензии. Ингибирование 11β-HSD1 смещает соотношение кортизола и кортизона в специфических тканях в сторону кортизона.
Введение терапевтически эффективного количества 11β-HSD1 ингибитора эффективно для лечения, контроля и облегчения симптом NIDDM, и регулярное введение терапевтически эффективного количества 11β-HSD1 ингибитора задерживает или предотвращает атаку NIDDM, особенно у людей.
Ожирение, метаболический синдром, дислипидемия
Избыточные уровни содержания кортизола связывали с ожирением, возможно, вследствие повышенного печеночного глюконеогенеза. Патологическое ожирение тесно связано с интолерантностью к глюкозе, гиперинсулинемией, гипертриглицеридемией и другими факторами синдрома Х, такими как высокое кровяное давление, повышенное содержание VLDL и пониженное содержание HDL (Montague et al., Diabetes, 2000, 49: 883-888). Таким образом, введение эффективного количества 11β-HSD1 ингибитора полезно для лечения или контроля ожирения. Длительное лечение 11β-HSD1 ингибитором также полезно для задержки или профилактики начала ожирения, особенно если пациент использует ингибитор 11β-HSD1 в сочетании с контролируемой диетой и физической нагрузкой.
Посредством снижения инсулинорезистентности и поддержания содержания глюкозы в плазме на уровне нормальных концентраций соединения данного изобретения могут применяться для лечения и профилактики состояний, которые сопровождают сахарный диабет II типа и инсулинорезистентность, включая метаболический синдром ("синдром Х"), ожирение, реактивную гипогликемию и диабетическую дислипидемию.
Атеросклероз
Как описано выше, ингибирование активности 11β-HSD1 и снижение количества кортизола благоприятны для лечения и контроля гипертензии. Поскольку гипертензия и дислипидемия способствуют развитию атеросклероза, введение терапевтически эффективного количества ингибитора 11β-HSD1 данного изобретения может быть особенно полезно для лечения, контроля, задержки начала развития или профилактики атеросклероза.
Другие применения
При использовании для лечения соединений данного изобретения можно лечить, контролировать, предотвращать заболевания, расстройства или состояния, перечисленные ниже, или можно замедлять их развитие: (1) гиперликемия, (2) низкая толерантность к глюкозе, (3) инсулинорезистентность, (4) ожирение, (5) липидные расстройства, (6) дислипидемия, (7) гиперлипидемия, (8) гипертриглицеридемия, (9) гиперхолестеринемия, (10) низкие уровни содержания HDL, (11) высокие уровни содержания LDL, (12) атеросклероз и его последствия, (13) сосудистый рестеноз, (14) панкреатит, (15) патологическое ожирение, (16) нейродегенеративное заболевание, (17) ретинопатия, (18) нефтопатия, (19) нейропатия, (20) синдром Х и другие расстройства, компонентом которых является инсулинорезистентность.
Перечисленные выше заболевания и состояния можно лечить с использованием соединений формулы I, или соединение может вводиться для профилактики или снижения риска развития заболеваний и состояний, описанных выше. Поскольку конкурентное ингибирование 11β-HSD2 может иметь вредные побочные эффекты или может фактически повышать количество кортизола в ткани-мишени, где желательно снижение содержания кортизола, селективные ингибиторы 11β-HSD1 с небольшим ингибированием 11β-HSD2 или без такого ингибирования являются желательными.
Ингибиторы 11β-HSD1 формулы I обычно имеют значение константы ингибирования IC50 менее примерно 500 нМ, предпочтительно менее примерно 100 нМ. Обычно отношение значения IC50 11β-HSD2 к значению IC50 11β-HSD1 равно, по меньшей мере, примерно двум или более, предпочтительно примерно десяти или более. Еще более предпочтительными являются соединения с соотношением IC50 для 11β-HDS1 и IC50 для 11β-HDS2, равным примерно 100 или более. Например, для соединений, имеющих IC50, соединения идеально показывают константу ингибирования 11β-HSD2 более примерно 500 нМ, предпочтительно более 1000 нМ.
Соединения формулы I могут использоваться в сочетании с одним или несколькими другими лекарственными средствами для лечения, профилактики, подавления или облегчения течения заболеваний или состояний, для которых применимы соединения формулы I или другие лекарственные средства. Обычно сочетание лекарственных средств является более надежным или более эффективным, чем применение одного лекарственного средства, или сочетание является более надежным или более эффективным, чем должно было ожидаться на основании сложения свойств отдельных лекарственных средств. Такое(ие) другое(ие) лекарственное(ые) средство(а) могут вводиться способами и в количестве, которые обычно применяются, одновременно или последовательно с соединением формулы I. Когда соединение формулы I используется одновременно с одним или несколькими другими лекарственными средствами, предпочтительным является сложный препарат, содержащий такое(ие) другое(ие) лекарственное(ые) средство(а) и соединение формулы I. Однако комбинированная терапия также включает терапевтические лечения, при которых соединение формулы I и одно или несколько других лекарственных средств вводятся по различным перекрывающимся схемам лечения. Это подразумевает, что при использовании в сочетании с другими активными ингредиентами соединение данного изобретения или другой активный ингредиент или оба лекарственных средства могут применяться эффективно в более низких дозах, чем при применении каждого отдельно. Соответственно, фармацевтические композиции данного изобретения включают композиции, которые в дополнение к соединению формулы I содержат одно или несколько других активных ингредиентов.
Примеры других активных ингредиентов, которые могут вводиться в сочетании с соединением формулы I и могут вводиться раздельно или вместе в одной фармацевтической композиции, включают, но без ограничения только ими:
(a) ингибиторы дипептидилпептидазы IV (DP-IV);
(b) сенсибилизаторы инсулина, включая (i) PPAR γ агонисты, такие как глитазоны (например, троглитазон, пиоглитазон, энглитазон, МСС-555, розиглитазон и т.п.) и другие PPAR лиганды, включая двойные агонисты PPAR α/γ, такие как KRP-297, и PPARα агонисты, такие как гемфиброзил, клофибрат, фенофибрат и безафибрат, и (ii) бигуаниды, такие как метформин и фенформин;
(c) инсулин и имитаторы инсулина;
(d) сульфонилмочевины и другие средства, усиливающие секрецию инсулина, такие как толбутамид и глипизид, меглитинид и аналогичные средства;
(е) ингибиторы α-глюкозидазы (такие как акарбоза);
(f) антагонисты глюкогонового рецептора, такие как средства, описанные в WO 98/04528, WO 99/01423, WO 00/39088 и WO 00/69810;
(g) GLP-1, имитаторы GLP-1 и агонисты GLP-1 рецептора, такие как средства, описанные в WO 00/42026 и WO 00/59887;
(h) GIP, имитаторы GIP, такие как средства, описанные в WO 00/58360, и агонисты GIP рецептора;
(i) PACAP, имитаторы РАСАР и агонисты РАСАР рецептора 3, такие как средства, описанные в WO 01/23420;
(j) средства, снижающие содержание холестерина, такие как (i) ингибиторы HMG-CoA редуктазы (ловастатин, симвастатин, правастатин, флувастатин, аторвастатин, ривастатин, итавастатин, росувастатин и другие статины), (ii) вещества, усиливающие экскрецию (холестирамин, колестипол и диалкиламиноалкильные производные сшитого декстрана), (iii) никотиниловый спирт, никотиновая кислота и их соли, (iv) ингибиторы абсорбции холестерина, такие как, например, эзетимиб и бэта-ситостерол, (v) ингибиторы ацил-СоА:холестерин ацилтрансферазы, такие как, например, авасимиб, (vi) антиоксиданты, такие как пробукол;
(k) PPAR δ агонисты, такие как средства, описанные в WO 97/28149;
(l) соединения для лечения ожирения, такие как фенфлурамин, дексфенфлурамин, фентермин, сибутрамин, орлистат, нейропептидные Y5 ингибиторы, СВ1 рецепторные инверсные агонисты и антагонисты и β3 адренергические рецепторные агонисты;
(m) ингибитор транспортера подвздошной желчной кислоты;
(n) средства, предназначенные для применения при воспалительных состояниях и отличные от глюкокортикоидов, такие как аспирин, нестероидные противовоспалительные лекарственные средства, азулфидин и селективные ингибиторы циклооксигеназы 2;
(о) ингибиторы протеин-тирозин-фосфатазы-1В (РТР-1В).
Описанные выше сочетания включают соединение формулы I или его фармацевтически приемлемую соль, гидрат или сольват, не только с одним или несколькими другими активными соединениями. Примеры, не ограничивающиеся приведенным перечнем, включают сочетания соединений формулы I с двумя или несколькими активными соединениями, выбранными из бигуанидов, сульфонилмочевин, ингибиторов HMG-CoA редуктазы, PPAR агонисты, ингибиторы РТР-1В, ингибиторы DP-IV и соединения для лечения ожирения.
Для обеспечения млекопитающего, особенно человека, эффективной дозой соединения данного изобретения может использоваться любой подходящий способ введения. Например, могут использоваться пероральный, ректальный, местный, парентеральный, глазной, легочный, назальный способы введения. Лекарственные формы включают таблетки, пастилки, дисперсии, суспензии, растворы, капсулы, кремы, мази, аэрозоли и т.п. Предпочтительно, соединение формулы вводится перорально.
Эффективная доза активного ингредиента изменяется в зависимости от конкретного применяемого соединения, способа введения, состояния, подлежащего лечению, и тяжести состояния. Такие дозы могут без труда определяться квалифицированным специалистом.
При лечении или для профилактики заболеваний и состояний, описанных в данном изобретении, для которых показаны соединения формулы I, удовлетворительные результаты получены при введении соединений данного изобретения в суточной дозе от примерно 0,1 до примерно 100 миллиграммов на килограмм (мг/кг) массы тела, предпочтительно при приеме в виде разовой суточной дозы или в виде дозы, поделенной примерно на прием от двух до шести раз в день. Таким образом, общая суточная доза находится в интервале от примерно 0,1 мг до примерно 1000 мг, предпочтительно от примерно 1 мг до примерно 50 мг. Для типичного взрослого пациента с массой тела 70 кг общая суточная доза будет находиться в интервале от примерно 7 мг до примерно 350 мг. Данная дозировка может корректироваться для обеспечения оптимального терапевтического результата.
Другой аспект данного изобретения относится к фармацевтической композиции, которая включает соединение формулы I или его фармацевтически приемлемую соль, гидрат или сольват в сочетании с фармацевтически приемлемым носителем.
Композиции включают композиции, подходящие для перорального, ректального, местного, парентерального (включая подкожное, внутримышечное или внутривенное), глазного (офтальмологическое), чрескожного, легочного (назальную или трансбукальную ингаляцию) или назального введения, хотя наиболее подходящий способ в любом случае будет зависеть от природы и тяжести состояния, подлежащего лечению, и активного ингредиента. Они могут традиционно поставляться в стандартной лекарственной форме и могут быть получены любыми способами, хорошо известными в фармацевтической области.
Соединение формулы I может объединяться с фармацевтическим носителем в соответствии с традиционными фармацевтическими способами получения смесей. Носители принимают самые различные формы. В частности, носители для жидких композиций для перорального введения включают, например, воду, гликоли, масла, спирты, вкусовые агенты, консерванты, красители и другие компоненты, используемые при получении жидких суспензий, эликсиров и растворов для перорального введения. Для приготовления твердых лекарственных форм для перорального введения, например, порошков, твердых и мягких капсул и таблеток, используются такие носители как крахмал, сахара и микрокристаллическая целлюлоза, разбавители, гранулирующие агенты, смазывающие агенты, связующие вещества, дезинтегрирующие агенты и т.п. Твердые препараты для перорального введения предпочтительны по сравнению с жидкими препаратами для перорального введения.
Твердые лекарственные формы для перорального введения могут также содержать связующее вещество, такое как трагакант, аравийская камедь, кукурузный крахмал или желатин; наполнители, такие как гидрофосфат кальция; дезинтегрирующий агент, такой как кукурузный крахмал, картофельный крахмал, альгиновая кислота; смазывающее вещество, такое как стеарат магния; и подслащивающее вещество, такое как сахароза, лактоза или сахарин. Капсулы могут также содержать жидкий носитель, такой как жирное масло.
Могут присутствовать и другие вещества для того, чтобы выступать в качестве покрытий или для модификации физической формы стандартной дозы. Например, таблетки могут быть покрыты шеллаком, сахаром или тем и другим.
Таблетки могут быть покрыты стандартными водными или неводными методами. Обычно процентное содержание активного ингредиента, конечно, может изменяться от примерно 2 процентов до примерно 60 процентов (мас./мас.). Таким образом, таблетки содержат соединение формулы I или его соль или гидрат в количестве в интервале от примерно 0,1 мг до примерно 1,5 г, предпочтительно в интервале от примерно 1,0 мг до примерно 500 мг, и наиболее предпочтительно, в интервале от примерно 10 мг до примерно 100 мг.
Жидкие препараты для перорального введения, такие как сиропы или эликсиры, могут содержать помимо активного ингредиента сахарозу в качестве подслащивающего вещества, метил- и пропилпарабены в качестве консервантов, краситель и вкусовую добавку, такую как вишневая или апельсиновая вкусовая добавка.
Препараты для парентерального введения, обычно в форме раствора или суспензии, как правило получают с водой, и они необязательно включают поверхностно-активное вещество, такое как гидроксипропилцеллюлоза. Дисперсии могут быть получены в глицерине, жидких полиэтиленгликолях и их смесях в маслах. Обычно препараты в разбавленной форме также содержат консервант.
Фармацевтические лекарственные формы для инъекций, включая водные растворы и дисперсии и порошки для быстрого получения растворов или дисперсий для инъекций, также являются стерильными и должны быть достаточно жидкими для возможности введения их с помощью шприца; они должны быть стабильными в условиях производства и хранения и обычно являются консервированными. Следовательно, носитель включает растворитель или дисперсионную среду, содержащую, например, воду, этанол, многоатомный спирт (например, глицерин, пропиленгликоль и жидкий полиэтиленгликоль), их подходящие смеси и растительные масла.
БИОЛОГИЧЕСКИЕ ИСПЫТАНИЯ: ОПРЕДЕЛЕНИЕ КОНСТАНТ ИНГИБИРОВАНИЯ
В условиях in vitro ферментативную активность испытуемых соединений оценивают посредством опыта сцинтилляционного приближения (Scintillation Proximity Assay - SPA). Вкратце, субстрат тритированного кортизона, NADPH кофактор и титрованное соединение выдерживают с 11β-HSD1 ферментом при 37°С для создания условий протекания превращения кортизола. После инкубации препарат SPA гранул, покрытых белком А, предварительно смешанными с антикортизольным моноклональным антителом и неспецифическим 11β-HSD ингибитором, добавляют в каждую ячейку. Смесь встряхивают при 15°С и затем считывают с помощью жидкостного сцинтилляционного счетчика, подходящего для применения на 96-луночных планшетах. Определяют процент ингибирования по сравнению с неингибированной контрольной лункой и строят графики IC50. Аналогичное испытание проводят с использованием 11β-HSD2, где тритированный кортизол и NAD используют в качестве субстрата и кофактора соответственно. Для начала опыта 40 мкл субстрата (25 нМ 3Н-кортизон + 1,25 мМ NADPH в 50 мМ HEPES буфера, рН 7,4) добавляют в подготовленные лунки на 96-луночном планшете. Твердое соединение растворяют в ДМСО с концентрацией 10 мМ с последующим 50-кратным разбавлением в ДМСО. Разбавленный раствор затем титруют 4-кратно 7 раз. 1 мкл каждого титрованного соединения затем дважды добавляют к субстрату. Для начала реакции 10 мкл 11β-HSD1 микросом из СНО трансфектантов добавляют в каждую лунку при подходящей концентрации для достижения примерно 10% конверсии исходного вещества. Для окончательного подсчета процента ингибирования добавляют ряд лунок, которые представляют минимум и максимум опыта: одна серия лунок содержит субстрат без соединения или фермента (фоновый сигнал), другая серия лунок содержит субстрат и фермент без какого бы то ни было соединения (максимальный сигнал). Планшеты в течение непродолжительного периода вращают с низкой скоростью в центрифуге для объединения реагентов, герметично закрывают клейкой лентой, осторожно перемешивают и выдерживают при 37°С в течение 2 часов. После инкубирования 45 мкл SPA гранул, предварительно суспендированных с антикортизольным моноклональным антителом и неспецифическим 11β-HSD ингибитором, добавляют в каждую лунку. Планшеты снова герметично закрывают и осторожно встряхивают в течение более 1,5 часов при 15°С. Данные планшета считывают с помощью жидкостного сцинтилляционного счетчика, такого как Topcount. Для контроля для ингибирования связывания антитела против кортизола с кортизолом субстрат с 1,25 нМ [3]H кортизола добавляют в указанные одиночные лунки. 1 мкл 200 мкМ соединения добавляют в каждую из этих лунок в месте с 10 мкл буфера вместо фермента. Любое вычисленное ингибирование обусловлено тем, что соединение вмешивается в процесс связывания кортизола с антителом на SPA гранулах.
БИОЛОГИЧЕСКИЕ ИСПЫТАНИЯ: ОПРЕДЕЛЕНИЕ ИНГИБИРОВАНИЯ IN VIVO
В общих словах, испытываемое соединение дозированно перорально вводят млекопитающему и оставляют его на некоторое время, обычно от 1 до 24 часов. Тритированный кортизон вводят внутривенно и спустя несколько минут производят забор крови. Из отделенной плазмы экстрагируют стероиды и анализируют с помощью ВЭЖХ. Относительные уровни содержания 3Н-кортизона и продукта его восстановления, 3Н-кортизола, определяют для группы, принимающей соединения, и контрольной группы, принимающей дозировки носителя. Из полученных значений вычисляют абсолютную конверсию, а также процент ингибирования.
Более точно, соединения приготавливают для перорального введения растворением их в носителе (5% (об./об.) гидроксипропил-бэта-циклодекстрин в Н2О или эквивалент) при нужной концентрации для получения дозы обычно 10 миллиграммов на килограмм. Утром натощак растворы вводят ICR мышам (поставляемым Charles River) через зонд питания в дозе 0,5 мл на животное трем животным в опытной группе.
По истечении нужного времени, обычно 1 часа или 4 часов, 0,2 мл 3 мкМ 3Н-кортизона в dPBS вводят инъекцией в хвостовую вену. Животное отсаживают в клетку на 2 минуты и затем безболезненно умерщвляют в камере с СО2. После экспирации мышь удаляют и кровь собирают сердечной пункцией. Кровь помещают в пробирку для отделения сыворотки и выдерживают при комнатной температуре не менее 30 минут для соответствующей коагуляции. По истечении периода инкубации кровь разделяют на сыворотку центрифугированием при 3000 g (4°С) в течение 10 минут.
Для анализа стероидов в сыворотке их сначала экстрагируют органическим растворителем. 0,2 Мл сыворотки переносят в чистую пробирку микроцентрифуги. Для этого добавляют 1,0 мл этилацетата с последующим энергичным взбалтыванием в течение 1 минуты. Быстрое вращение на центрифуге приводит к образованию слоя водных белков сыворотки, и органический супернатант становится прозрачным. 0,85 мл верхней органической фазы переносят в чистую пробирку микроцентрифуги и сушат. Высушенный образец снова суспендируют в 0,250 мл ДМСО с высокой концентрацией кортизона и кортизола для анализа методом ВЭЖХ.
0,200 мл образца впрыскивают с хроматографическую колонку Metachem Inertsil C-18, уравновешенную 30% метанолом. Разделение целевых стероидов проводят с небольшим линейным градиентом до 50% метанола; одновременный контроль с помощью УФ при 254 нМ охлажденных стандартных растворов во вновь суспендированном растворе служит в качестве внутреннего стандарта. Тритиевый сигнал получают с помощью радиохроматографического детектора, который загружает данные в программу для анализа. Процент конверсии 3Н-кортизона в 3Н-кортизол вычисляют в виде отношения AUC для кортизола к объединенному AUC для кортизона и кортизола.
Приведенные далее примеры иллюстрируют данное изобретение и не предназначены для ограничения его области.
ПРИМЕР 1
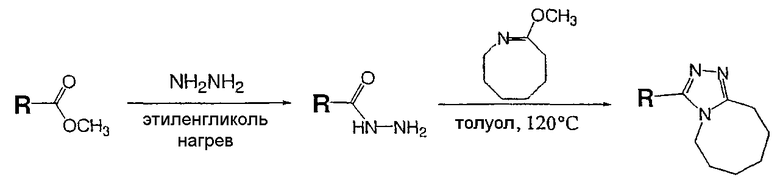
Общая схема
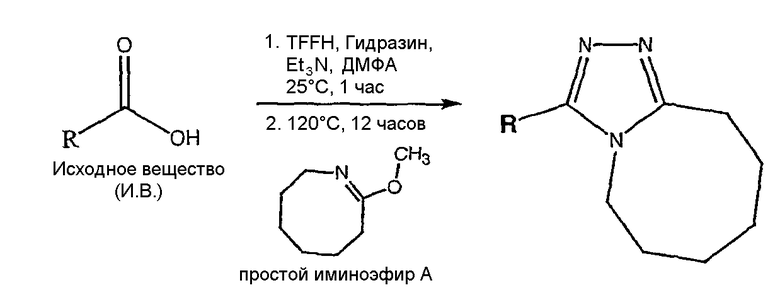
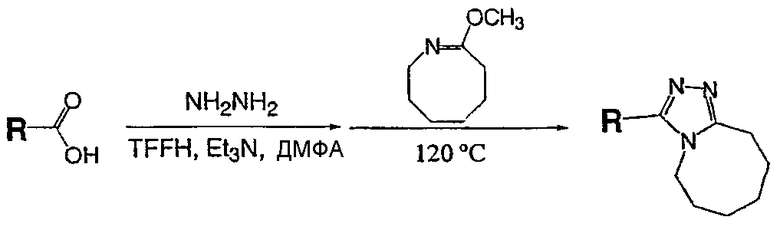
Описанный далее синтез одномерного единичного массива чистых соединений проводят на системе марки Myriad Core System. Все реакторы перед применением сушат в потоке азота при 120°С в течение 12 часов. Растворители сушат над ситами в течение, по меньшей мере, 12 часов перед применением. Реагенты и вспомогательные соединения (карбоновые кислоты и 8-метокси-2,3,4,5,6,7-гексагидроазоцин (простой иминоэфир А)) растворяют в подходящих растворителях непосредственно перед применением.
Синтез
Карбоновую кислоту, показанную в таблице ниже в качестве исходного вещества, добавляют в сухой реактор Myriad объемом 10 мл со стеклянной пористой перегородкой в атмосфере азота (714 мл, 0,1 ммоль, 0,14 М в N,N-диметилформамиде (ДМФА)). Гексафторфосфатфтор-N,N,N',N'-тетраметилформамидия (TFFH) (200 мкл, 0,1 ммоль, 0,5 М в ДМФА), затем триэтиламин (400 мкл, 0,2 ммоль, 0,5 М в ДМФА) и гидразин (240 мкл, 0,12 ммоль, 0,5 М в ДМФА) добавляют в реактор в атмосфере азота. Реакционную смесь выдерживают в течение 1 часа при 25°С с перемешиванием газом (1-секундный импульс каждые 5 минут). 8-Метокси-2,3,4,5,6,7-гексагидроазоцин (простой иминоэфир А, 600 мкл, 0,15 ммоль, 0,25 М в ДМФА) добавляют в реактор в атмосфере азота. Реакционную смесь выдерживают в течение 12 часов при 120°C с перемешиванием газом (1-секундный импульс каждые 5 минут) и затем охлаждают до комнатной температуры. После охлаждения сырую реакционную смесь анализируют ЖХ-МС (метод 1). Сырую реакционную смесь очищают препаративной ВЭЖХ с использованием масс-спектрометрического обнаружения (метод 2). Чистоту собранных фракций анализируют ЖХ-МС (метод 1); фракции, которые, как показывает анализ, имеют чистоту более 90%, объединяют во взвешенных EPA пузырьках объемом 40 мл и лиофилизуют.
Условия ВЭЖХ
МС: API-ES режим ионизации, область сканирования массы (100-600 атомных единиц массы)
ELSD: детектор рассеяния света.
Параметры лиофилизации
Установленная исходная температура замерзания: 1 час при -70°С
Установленная температура сушки фазового конденсатора: -50°С
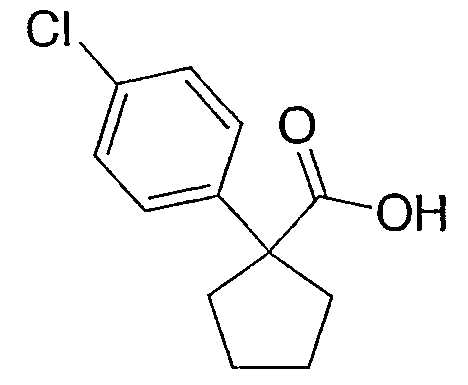
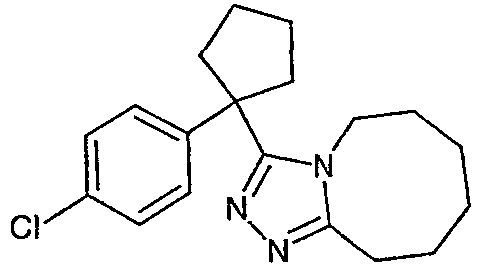
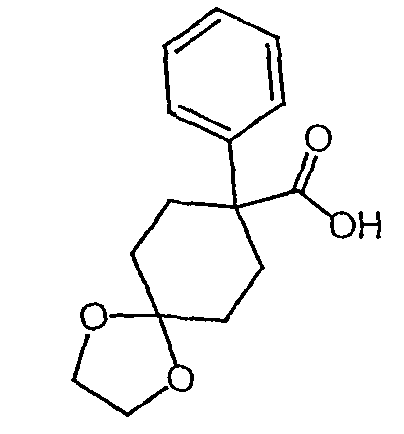
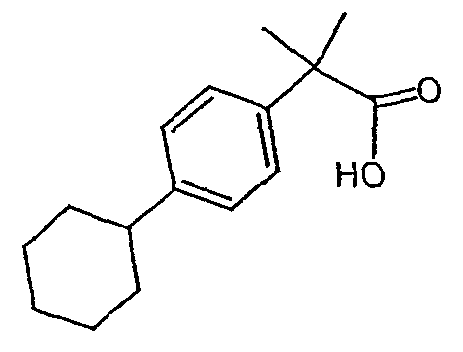
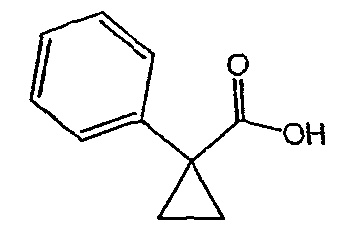
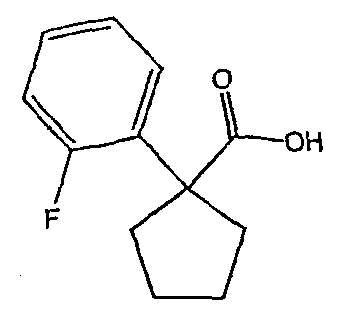
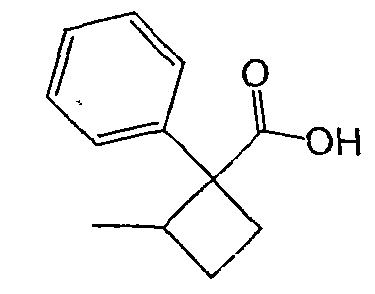
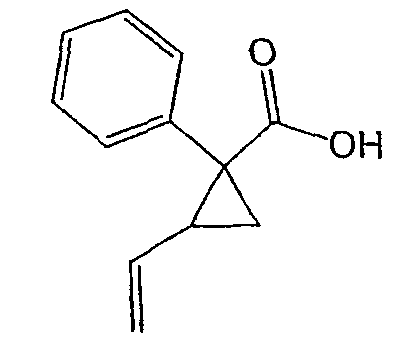
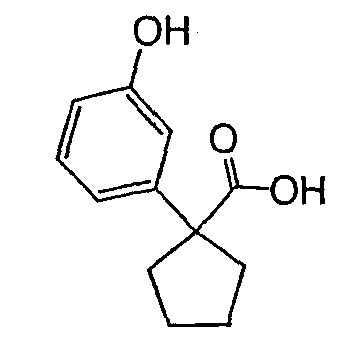
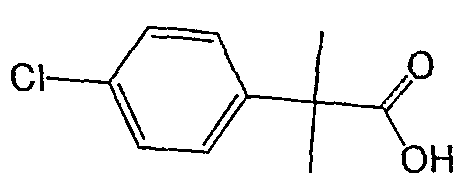
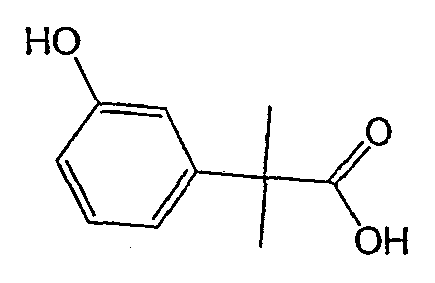
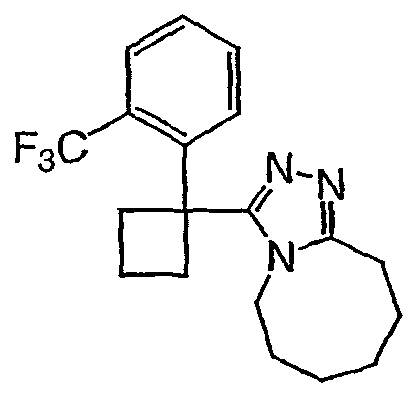
Исходным веществом для примера 1-1 является
ПРИМЕР 2
Методика 2А
Общая схема
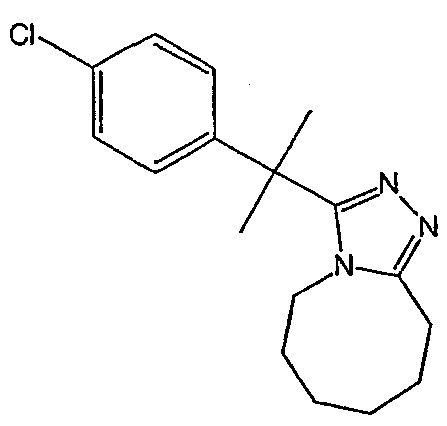
Получение 3-(1,1-дифенилпропил)-5,6,7,8,9,10-гексагидро[1,2,4]триазоло[4,3-a]азоцина (2-1)
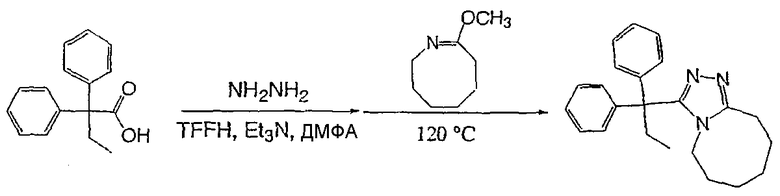
2,2-Дифенилбутановую кислоту (39,6 мг, 0,166 ммоль) растворяют в ДМФА (0,33 мл). К раствору добавляют гексафторфосфат фтор-N,N,N',N'-тетраметилформамидия (TFFH, 43,6 мг) и безводный триэтиламин (46,4 мкл) и раствор охлаждают до 0°С. Спустя 10 минут, добавляют моногидрат гидразина (6,5 мкл). Полученную реакционную смесь перемешивают при комнатной температуре в течение 30 минут, после чего ВЭЖХ/МС показывает полное превращение в 2,2-дифенилбутангидразид. Добавляют 8-метокси-2,3,4,5,6,7-гексагидроазоцин (38 мл, 0,249 ммоль) и раствор перемешивают при 120°С в течение ночи. Смесь нагревают до комнатной температуры, продукт очищают препаративной ВЭЖХ и выделяют в виде соли трифторуксусной кислоты. Соль добавляют в насыщенный раствор гидрокарбоната натрия и экстрагируют этилацетатом с получением свободного основания. Экстракт сушат над сульфатом магния, фильтруют и упаривают с получением очищенного триазола (2-1) в виде твердого белого вещества; МС ESI (m/z) 346,3.
Соединения 2-2 - 2-23 получают по существу в соответствии с этой же методикой с использованием подходящей карбоновой кислоты (И.В.). Образование продукта контролируют ВЭЖХ/МС.
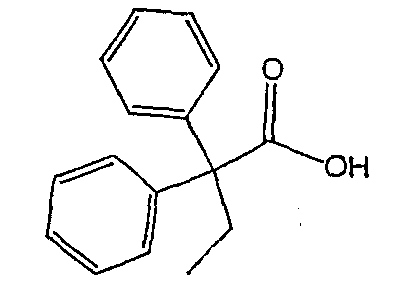
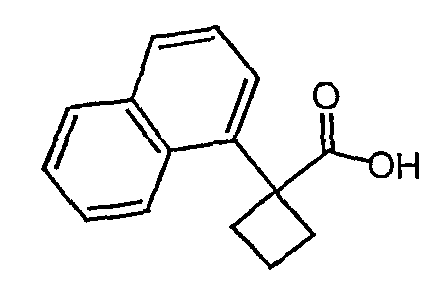
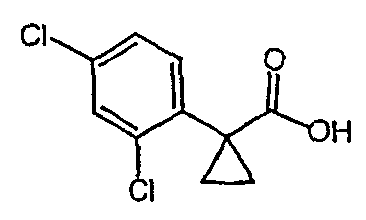
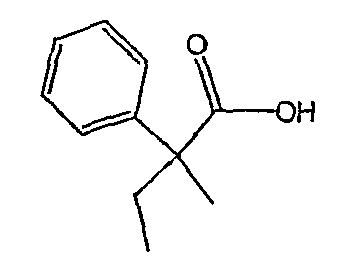
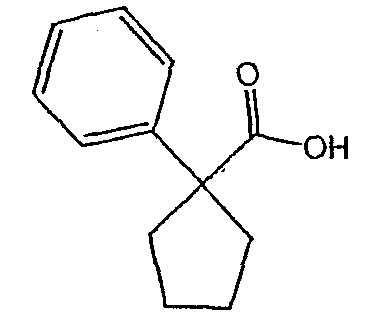
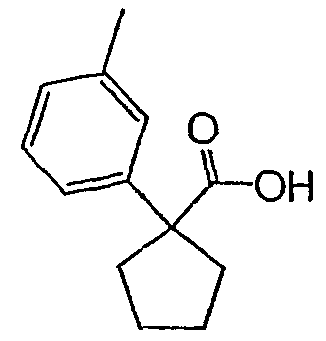
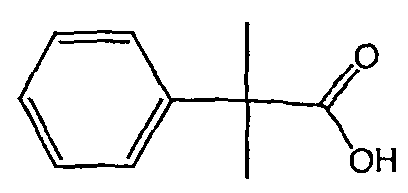
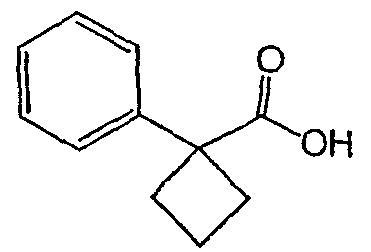
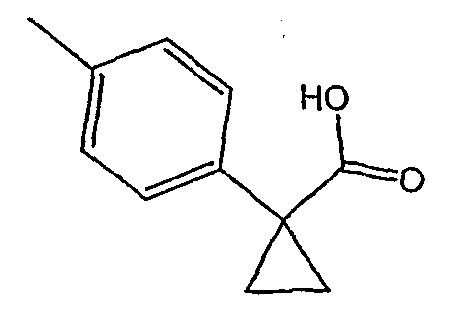
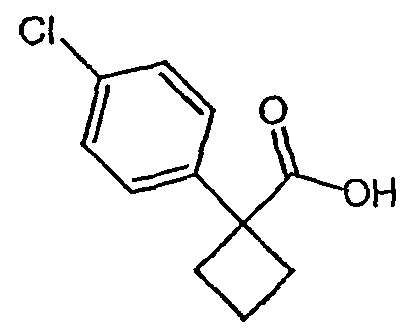
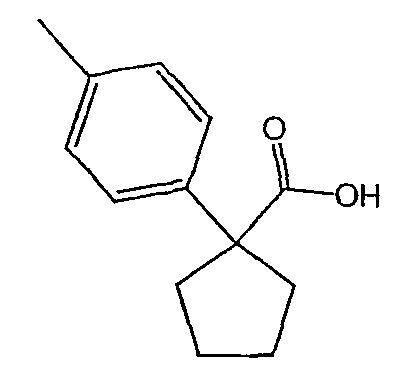
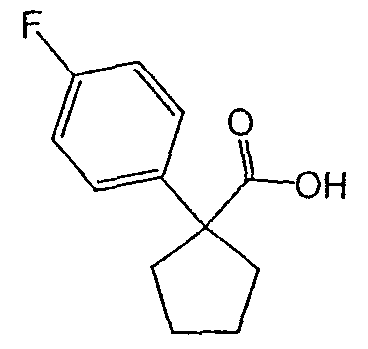
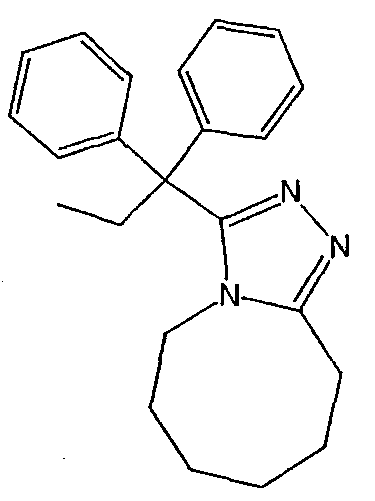
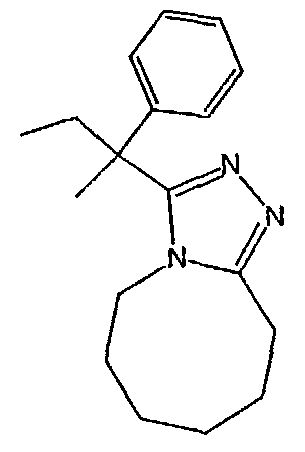
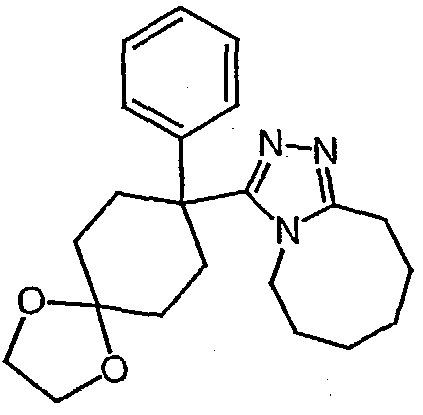
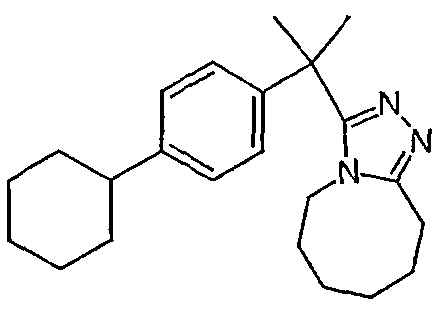
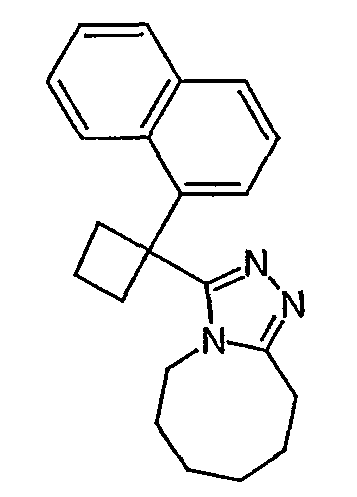
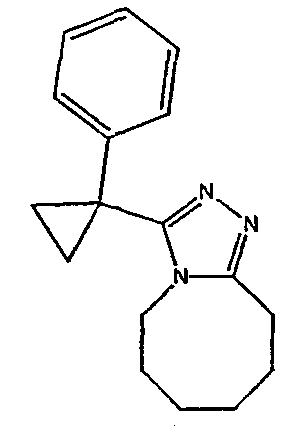
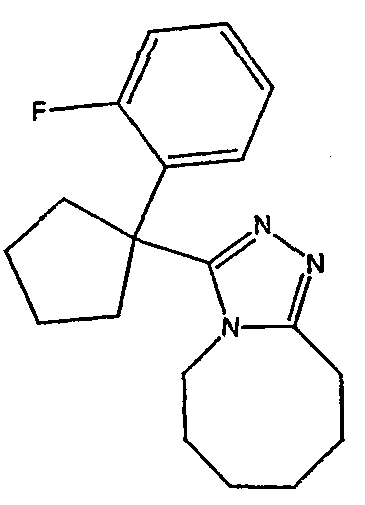
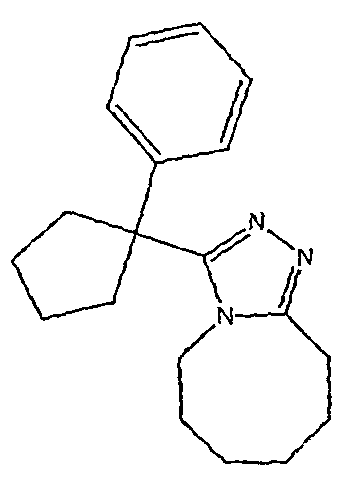
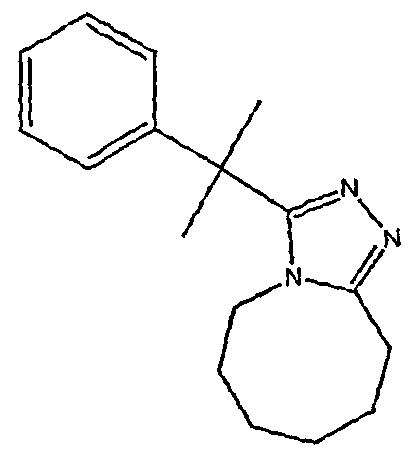
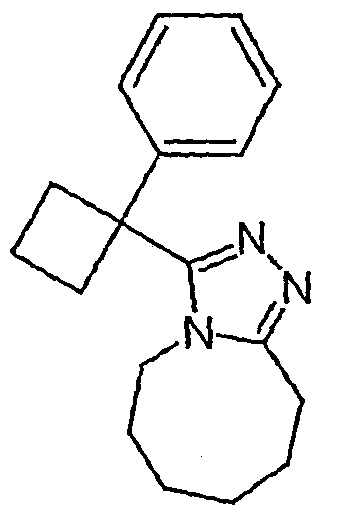
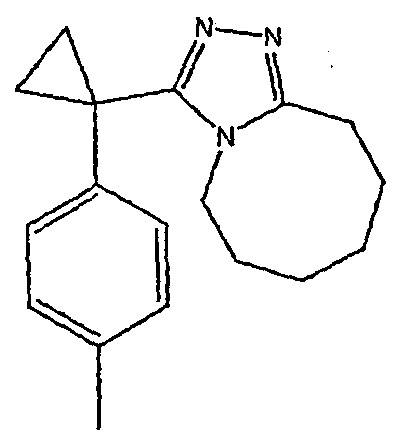
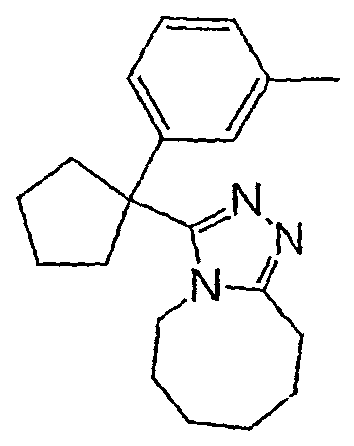
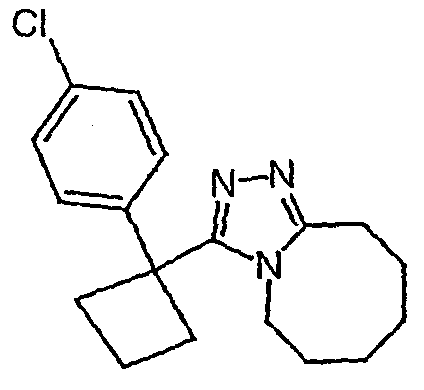
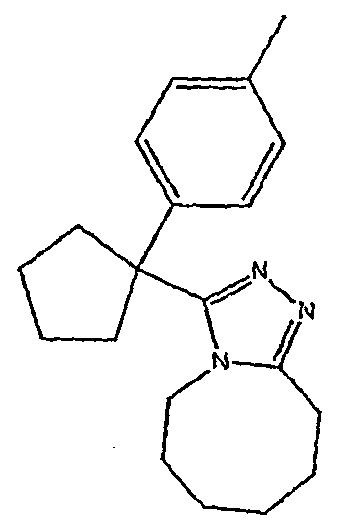
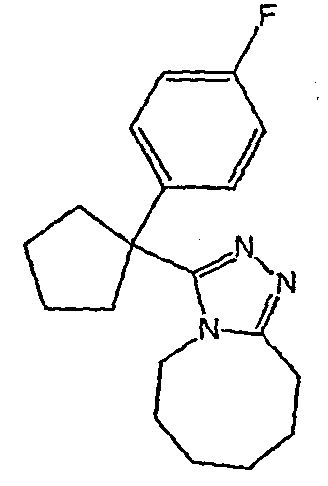
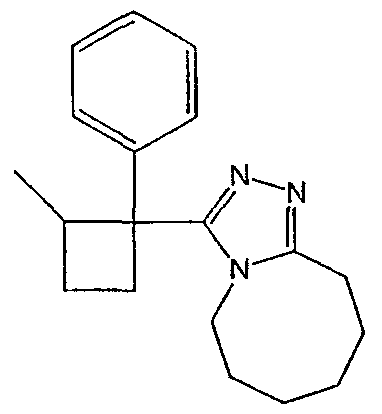
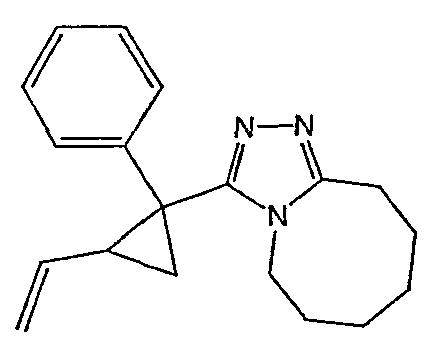
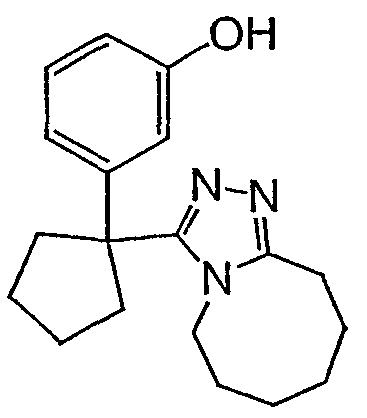
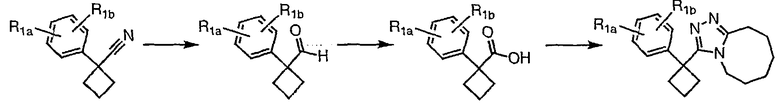
Методика 2В
Общая схема
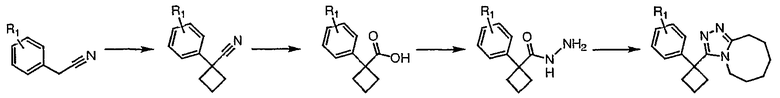
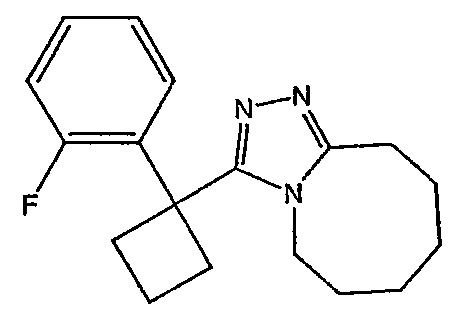
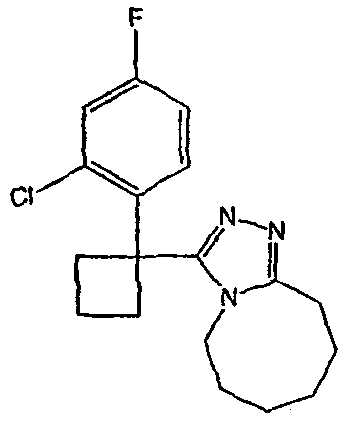
Получение 3-[1-(2-фторфенил)циклобутил]-5,6,7,8,9,10-гексагидро[1,2,4]триазоло[4,3-a]азоцина (2-24)
Стадия 1
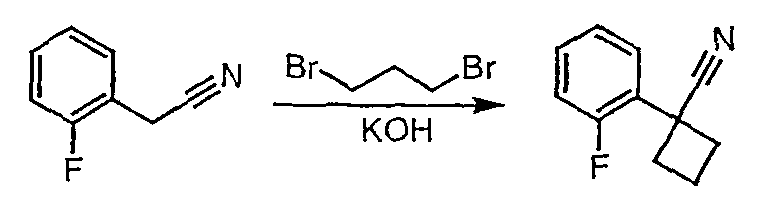
Гидроксид калия (1,78 г) растворяют в диметилсульфоксиде (5,8 мл) [1]. (2-Фторфенил)ацетонитрил (0,97 г, 7,2 ммоль) и 1,3-дибромпропан (0,95 мл, 9,3 ммоль) растворяют в этиловом эфире (2 мл), и полученную смесь по каплям при энергичном перемешивании добавляют к раствору гидроксида калия при комнатной температуре. Смесь перемешивают при комнатной температуре в течение одного часа, затем реакцию гасят добавлением воды (3,8 мл), охлажденной льдом. Смесь фильтруют через рыхлый слой целита, который промывают эфиром (20 мл). Фильтрат переносят в делительную воронку и водный слой экстрагируют эфиром (3×10 мл). Органические слои объединяют, сушат над сульфатом магния, фильтруют и упаривают до светло-желтого масла (1,0 г). Чистый 1-(2-фторфенил)циклобутанкарбонитрил (0,45 г) получают после хроматографии на силикагеле.
Стадия 2
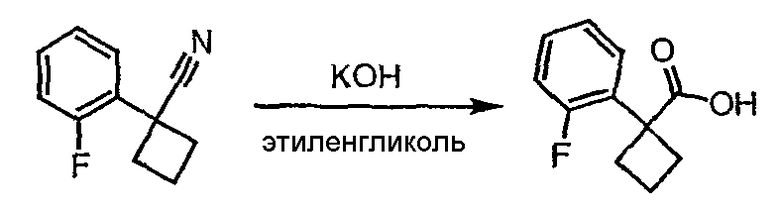
1-(2-Фторфенил)циклобутанкарбонитрил (0,21 г, 1,15 ммоль) и гидроксид калия (0,194 г) растворяют в этиленгликоле (2 мл). Полученную смесь кипятят с обратным холодильником в течение 3 часов при 198°С, затем выливают в воду (5 мл) и экстрагируют эфиром (2×5 мл). Водный раствор подкисляют HCl и экстрагируют эфиром (3×5 мл). Экстракты объединяют, сушат над сульфатом магния, фильтруют и упаривают с получением неочищенной карбоновой кислоты.
Стадия 3
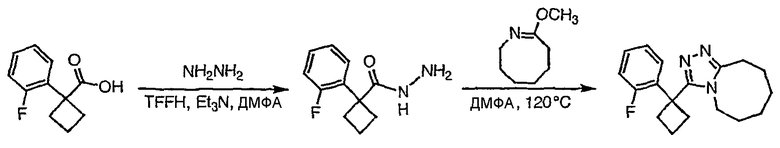
1-(2-фторфенил)циклобутанкарбоновую кислоту (51,3 мг, 0,264 ммоль) растворяют в ДМФА (0,52 мл). К полученному раствору при комнатной температуре добавляют гексафторфосфат фтор-N,N,N',N'-тетраметилформамидия (TFFH, 74,6 мг, 0,282 ммоль) и безводный триэтиламин (71,0 мкл, 0,509 ммоль). Спустя 5 минут добавляют безводный гидразин (10 мкл, 0,319 ммоль). Смесь перемешивают при комнатной температуре в течение 30 минут, после чего ВЭЖХ-МС показывает образование 1-(2-фторфенил)циклобутанкарбогидразида с хорошим выходом.
8-Метокси-2,3,4,5,6,7-гексагидроазоцин (47 мкл, 0,412 ммол) добавляют к раствору 1-(2-фторфенил)циклобутанкарбогидразида и реакционную смесь перемешивают при 120°С в течение ночи. Раствор охлаждают, концентрируют и продукт очищают препаративной ВЭЖХ с получением его в виде соли трифторуксусной кислоты. Соль добавляют к насыщенному раствору гидрокарбоната натрия и экстрагируют этилацетатом с получением свободного основания. Экстракт сушат над сульфатом магния, фильтруют и упаривают с получением очищенного триазола (2-24) в виде твердого вещества.
Соединения 2-25 - 2-32 получают, по существу, в соответствии с этой же методикой с использованием соответствующего фенилацетонитрила. Образование продукта контролируют с помощью ВЭЖХ/МС.
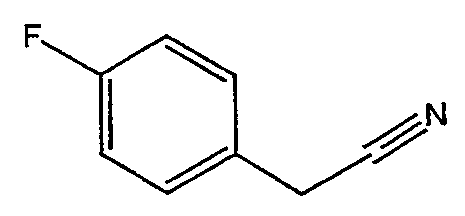
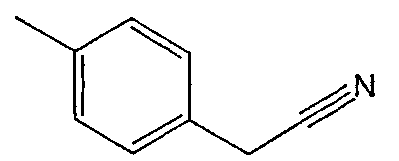
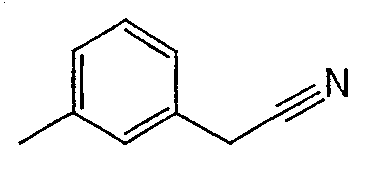
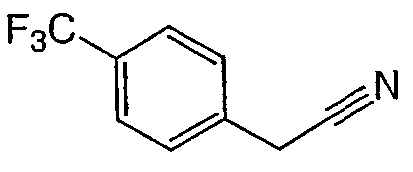
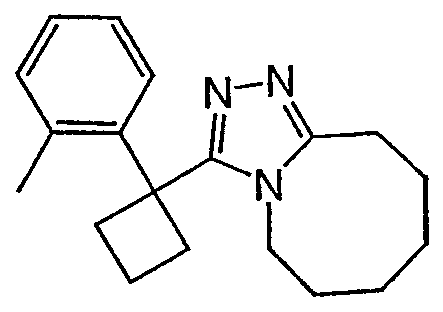
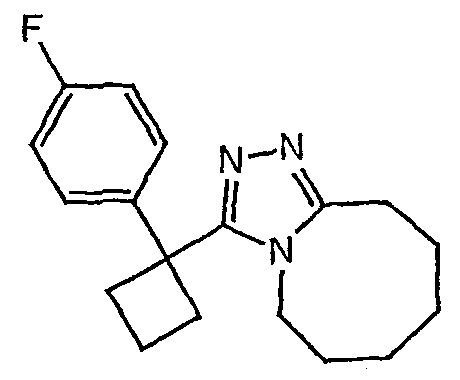
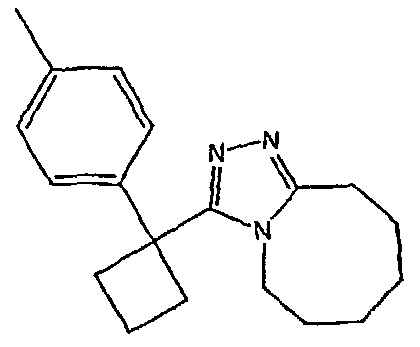
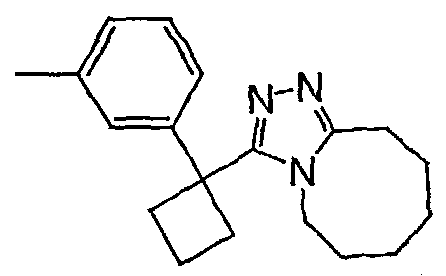

Методика 2С
Общая схема
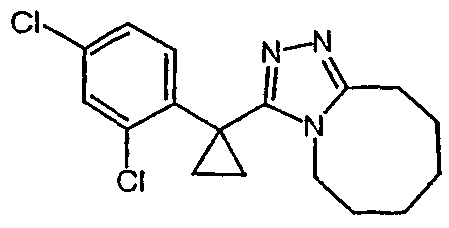
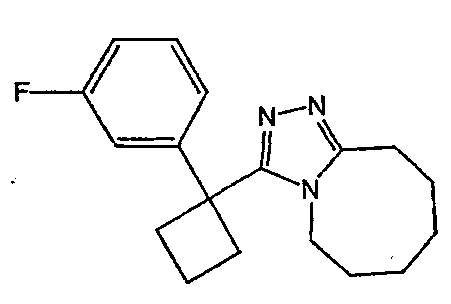
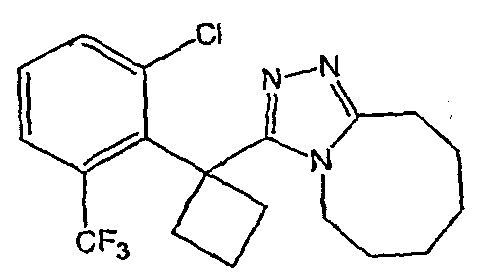
Получение 3-[1-(3,4-дифторфенил)циклобутил]-5,6,7,8,9,10-гексагидро[1,2,4]триазоло[4,3-a]азоцина (2-33)
Стадия 1
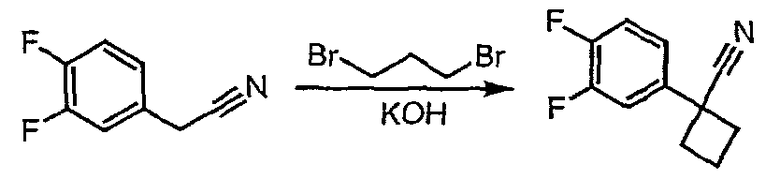
(3,4-Дифторфенил)ацетонитрил превращают в 1-(3,4-дифторфенил)циклобутанкарбонитрил в соответствии со способом, описанным в методике 2В, стадия 1.
Стадия 2
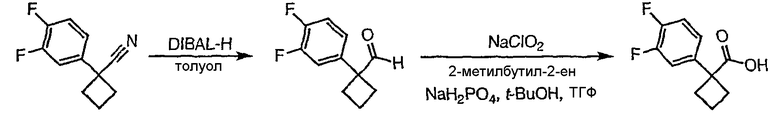
1-(3,4-Дифторфенил)циклобутанкарбонитрил (384,5 мг, 1,99 ммоль) растворяют в толуоле (30 мл) и охлаждают до -78°С. К полученному раствору по каплям добавляют гидрид диизобутилалюминия (DIBAL-H) (1,0 М раствор в гексанах) (3,98 мл, 3,98 ммоль). Смесь перемешивают при -78°С в течение 30 минут, затем добавляют 5% серную кислоту (2 мл). Реакционную смесь нагревают до комнатной температуры, перемешивают в течение 20 минут и фильтруют через рыхлый слой целита. Целит промывают этилацетатом, весь фильтрат переносят в делительную воронку и промывают водой. Органический слой сушат над сульфатом натрия и упаривают с получением целевого альдегида.
1-(3,4-Дифторфенил)циклобутанкарбальдегид (240,0 мг, 1,22 ммоль) растворяют в смеси трет-бутанол/тетрагидрофуран/2-метилбут-2-ен (3,0 мл/1,0 мл/1,0 мл) и энергично перемешивают при комнатной температуре. Хлорит натрия (243,4 мг, 2,69 ммоль) и дигидрофосфат натрия (370,4 мг, 2,68 ммол) растворяют в воде (1,2 мл) и по каплям добавляют к полученному ранее раствору. Смесь перемешивают в течение часа, после чего ТСХ показывает, что реакция завершена. Летучие растворители удаляют в вакууме и продукт разбавляют водой и промывают гексаном (3 мл). Водный раствор подкисляют 6 N водной хлористоводородной кислотой до рН 2. Раствор экстрагируют этилацетатом (3×20 мл), объединенные органические слои промывают раствором соли (5 мл), сушат над сульфатом магния, фильтруют и упаривают с получением целевой карбоновой кислоты (125 мг).
Стадия 3
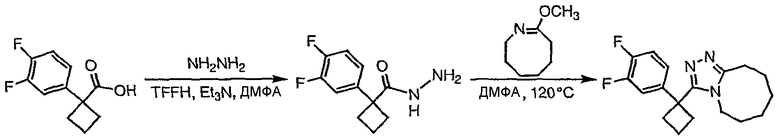
1-(3,4-Дифторфенил)циклобутанкарбоновую кислоту подвергают превращению в 3-[1-(3,4-дифторфенил)циклобутил]-5,6,7,8,9,10-гексагидро[1,2,4]триазоло[4,3-a]азоцин (2-33) в соответствии со способом, описанным в методике 2В, стадия 3; МС ESI (m/z) 318,2.
Соединения 2-34 - 2-38 получают по существу в соответствии с этой же методикой, используя соответствующий дизамещенный фенилацетонитрил. Образование продукта контролируют ВЭЖХ/МС.
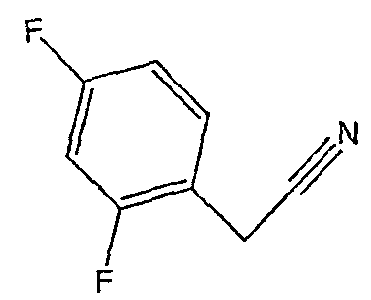
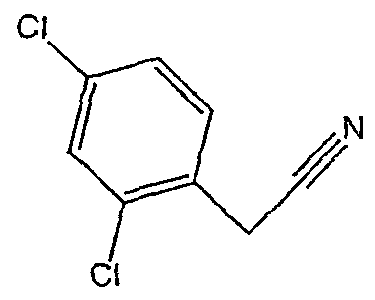
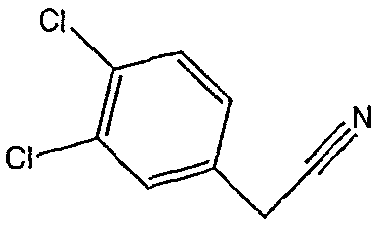
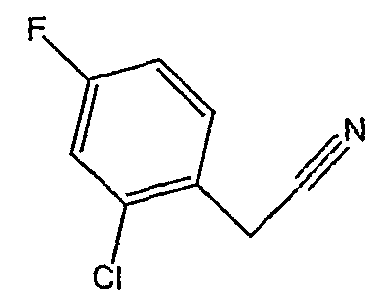
Методика 2D
Общая схема
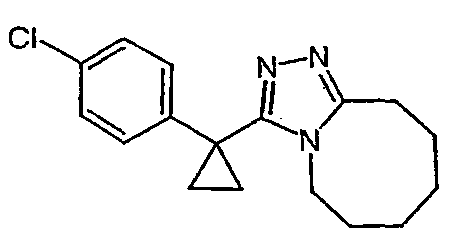
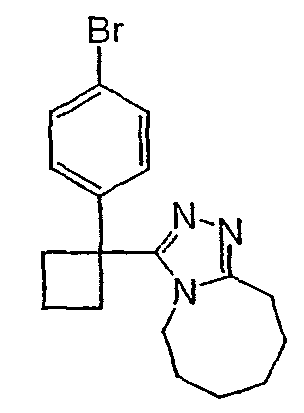
Получение 3-(1-(4-хлорфенил)циклогексил)-5,6,7,8,9,10-гексагидро[1,2,4]триазоло[4,3-a]азоцина (2-39)
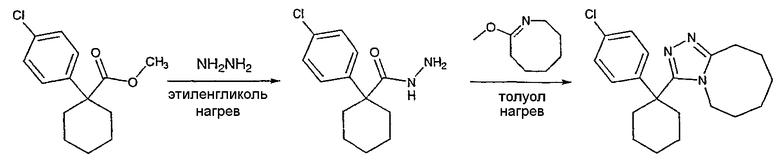
Метил 1-(4-хлорфенил)циклогексанкарбоксилат (277 мг) и гидразингидрат (0,30 мл) растворяют в этиленгликоле (5 мл), нагревают до 150°С и выдерживают при этой температуре в течение 15 часов. Раствор охлаждают и добавляют воду (5 мл). Образующийся осадок отфильтровывают и сушат в вакууме, получая ацилгидразид (108 мл) в виде твердого белого вещества.
Безводный толуол добавляют к смеси 1-(4-хлорфенил)циклогексанкарбогидразида (62 мг) и 8-метокси-2,3,4,5,6,7-гексагидроазоцина (40,1 мг). Реактор нагревают до 120°С и выдерживают при этой температуре в течение ночи, после чего охлаждают до комнатной температуры и растворитель выпаривают. Неочищенный продукт очищают колоночной хроматографией, получая 3-(1-(4-хлорфенил)циклогексил)-5,6,7,8,9,10-гексагидро[1,2,4]триазоло[4,3-a]азоцин (2-39) в виде твердого белого вещества.
Пример 3
Методика 3А
Получение 1-(4-хлорфенил)циклобутанкарбогидразида
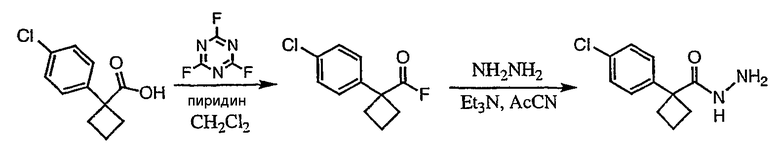
1-(4-Хлорфенил)циклобутанкарбоновую кислоту (10,0 г) растворяют в дихлорметане (150 мл) и охлаждают до -10°С на бане со смесью лед/раствор соли. К полученному раствору добавляют пиридин (3,84 мл) и затем цианурфторид (8,9 мл в 25 мл дихлорметана). Смесь перемешивают при комнатной температуре в течение часа, после чего ТСХ показывает, что реакция завершена. Раствор переносят в делительную воронку, содержащую лед (150 мл). После энергичного встряхивания органический слой удаляют, сушат над сульфатом магния, фильтруют и упаривают с получением карбонилфторида.
Безводный гидразин (2,02 мл) растворяют в ацетонитриле (100 мл) и охлаждают до 0°С. Добавляют триэтиламин (12,8 мл) и затем 1-(4-хлорфенил)циклобутанкарбонилхлорид (10 г) в ацетонитриле (25 мл). Раствор перемешивают при комнатной температуре в течение часа, после чего ацетонитрил удаляют выпариванием. Продукт получают после хроматографии на силикагеле.
Методика 3В
Получение 1-(4-хлорфенил)циклопропанкарбогидразида
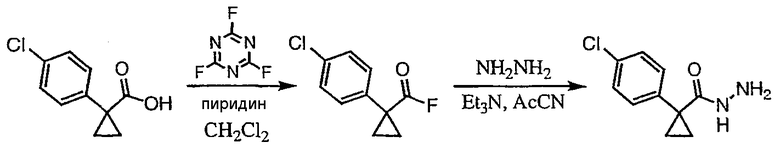
1-(4-Хлорфенил)циклопропанкарбогидразид получают в соответствии с методикой 3А, используя 1-(4-хлорфенил)циклопропанкарбоновую кислоту.
Методика 3С
Получение 1-(4-фторфенил)циклобутанкарбогидразида
Стадия 1
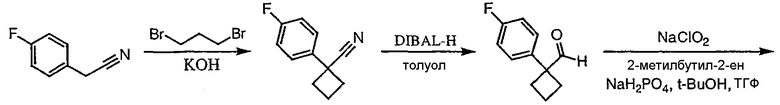
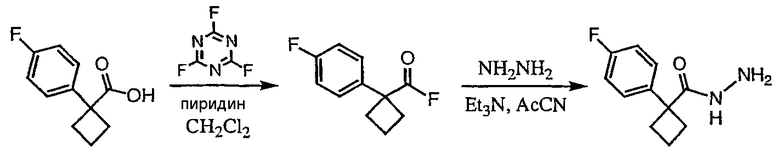
Гидроксид калия (8,2 г, 146,1 ммоль) растворяют в диметилсульфоксиде (100 мл) [1]. (4-Фторфенил)ацетонитрил (6,87 г, 50,8 ммоль) и 1,3-дибромпропан (56,4 ммоль) растворяют в этиловом эфире (10 мл) и полученную смесь при энергичном перемешивании по каплям добавляют к раствору гидроксида калия при комнатной температуре. Смесь перемешивают в течение двух часов, после чего реакцию гасят добавлением воды (100 мл), охлажденной льдом. Смесь фильтруют через рыхлый слой целита, который промывают эфиром (10 мл). Фильтрат переносят в делительную воронку и водный слой экстрагируют эфиром (3×100 мл). Органические слои объединяют, сушат над сульфатом магния, фильтруют и упаривают, получая продукт (8,85 г) в виде бледно-желтого масла.
Стадия 2
Неочищенный 1-(4-фторфенил)циклобутанкарбонитрил (8,85 г, 50,5 ммоль) растворяют в безводном толуоле (100 мл) и охлаждают до -78°С. К полученному раствору по каплям добавляют гидрид диизобутилалюминия (DIBAL-H) (1,0 М раствор в гексанах, 60,6 мл). Реакцию контролируют ТСХ (гексан: этилацетат 9:1). Смесь перемешивают при -78°С в течение часа, после чего добавляют 5% серную кислоту (20 мл). Реакционную смесь нагревают до комнатной температуры, перемешивают в течение 20 минут и фильтруют через рыхлый слой целита. Целит промывают этилацетатом, весь фильтрат переносят в делительную воронку и промывают водой. Органический слой сушат над сульфатом натрия, фильтруют и упаривают досуха, получая целевой альдегид.
1-(4-Дифторфенил)циклобутанкарбальдегид (8,8 г, 49,4 ммоль) растворяют в трет-бутаноле (90 мл), тетрагидрофуране (30 мл) и 2-метилбут-2-ене (30 мл) и энергично перемешивают при комнатной температуре. Хлорит натрия (9,8 г, 108,7 ммоль) и дигидрофосфат натрия (15,0 г, 108,7 ммоль) растворяют в воде (54 мл) и по каплям добавляют к полученному ранее раствору. Смесь перемешивают в течение часа, после чего ТСХ показывает, что реакция завершена. Летучие растворители удаляют в вакууме, продукт разбавляют водой и затем промывают гексаном (3 мл). Водный раствор подкисляют 6 N водной хлористоводородной кислотой до рН 2. Раствор экстрагируют этилацетатом (3×150 мл), объединенные органические слои промывают раствором соли (20 мл), сушат над сульфатом магния, фильтруют и упаривают, получая 1-(4-фторфенил)циклобутанкарбоновую кислоту (8,0 г).
Полученную карбоновую кислоту подвергают превращению в 1-(4-фторфенил)циклобутанкарбогидразид в соответствии со способом, описанным в методике 3А.
Методика 3D
Получение 1-(4-фторфенил)циклопропанкарбогидразида
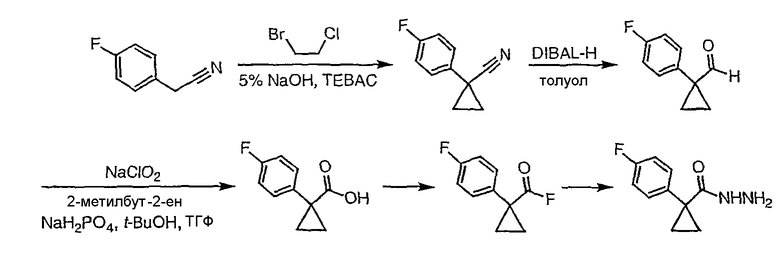
1-(4-Фторфенил)ацетонитрил (3,77 г, 27,9 ммоль), 1-бром-2-хлорэтан (5,0 г, 34,9 ммоль) и хлорид бензилтриэтиламмония (ТЕВАС, 127,6 мг, 0,56 ммоль) добавляют в колбу и энергично перемешивают [2]. К полученному раствору по каплям добавляют гидроксид калия (50% в воде, 195 ммоль). Смесь перемешивают при 40°С в течение 5 часов и в течение ночи при комнатной температуре, реакционную смесь разбавляют водой и экстрагируют дихлорметаном. Органический слой отделяют, промывают 1 N водной хлористоводородной кислотой, водой и сушат над сульфатом магния. Полученный раствор фильтруют и дихлорметан выпаривают, получая неочищенный продукт (4,5 г).
1-(4-Фторфенил)циклопропанкарбоновую кислоту получают из неочищенного 1-(4-фторфенил)циклопропилкарбонитрила в соответствии со способом, описанным в методике 3С, стадия 2. Полученную карбоновую кислоту подвергают превращению в 1-(4-фторфенил)циклопропанкарбогидразид в соответствии с методикой 3А.
Методика 3Е
Общая схема
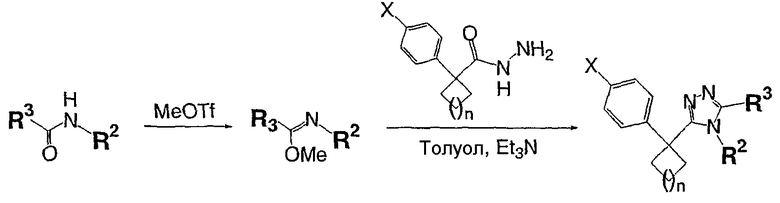
Получение 3,4-дициклопропил-5-(1-фенилциклобутил)-4Н-1,2,4-триазола (3-1)
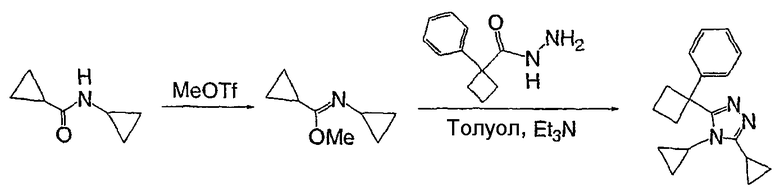
Метилтрифторметансульфонат (89,1 мкл) добавляют к N-циклопропилциклопропанкарбоксамиду (98,6 мг, 0,788 ммоль). Полученную смесь перемешивают в течение 30 минут при 60°С, после чего ЯМР четко показывает превращение в метил N-циклопропилциклопропанкарбоксимидоат.
Толуол (2 мл), триэтиламин (223 мкл) и 1-фенилциклобутанкарбогидразид (90 мг) добавляют к метил N-циклопропилциклопропанкарбоксимидоату и перемешивают при 60°С в течение 3 часов, затем при 110°С в течение 1 часа. Смесь охлаждают и упаривают, остаток очищают препаративной ВЭЖХ, получая продукт в виде соли трифторуксусной кислоты. Соль добавляют к насыщенному раствору гидрокарбоната натрия и экстрагируют этилацетатом, получая свободное основание. Органический экстракт сушат над сульфатом магния, фильтруют и упаривают с получением 3,4-дициклопропил-5-(1-фенилциклобутил)-4Н-1,2,4-триазола (3-1); МС ESI (m/z) 280,2.
Другое соединение примера 3 получают по существу в соответствии с этой же методикой, используя соответствующие карбоксамид и ацилгидразид. В качестве растворителя при получении 3-2 используют ацетонитрил. Соединение 3-19 выделяют в качестве побочного продукта в синтезе 3-18. Метиламиды получают из их соответствующих сложных метиловых эфиров и метиламина с использованием хорошо известных методик. Другие амиды стандартно получают из коммерчески доступных карбоновых кислот и аминов, используя 1-(диметиламинопропил)-3-этилкарбодиимид гидрохлорид в качестве реагента и методики, описанные в литературе. Получение ацилгидразидов описано в методиках 3А, 3В, 3С и 3D.
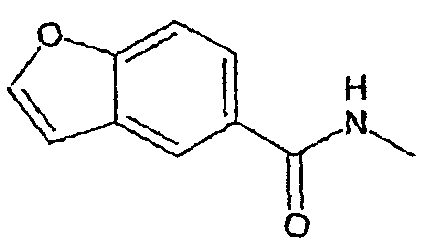
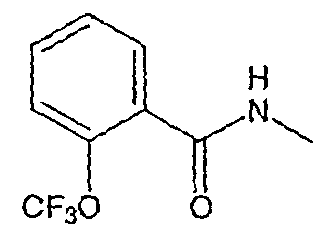
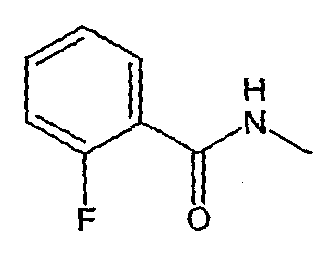
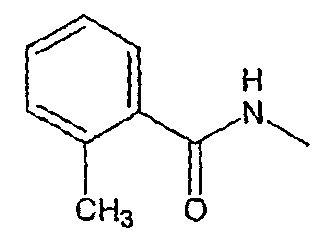
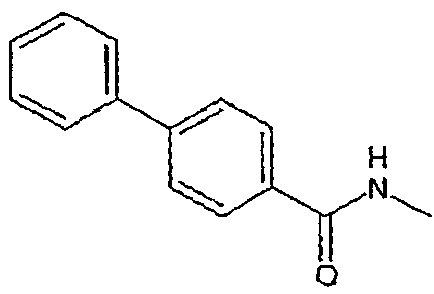
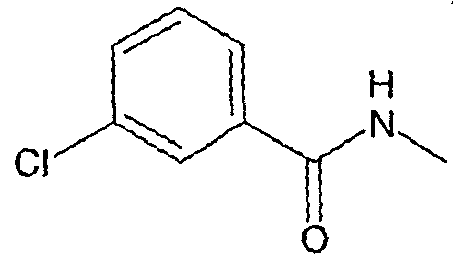
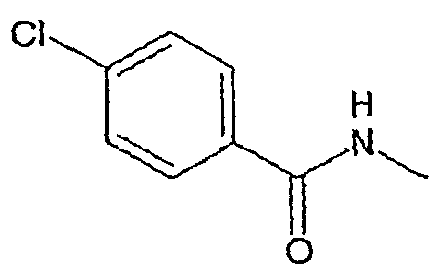
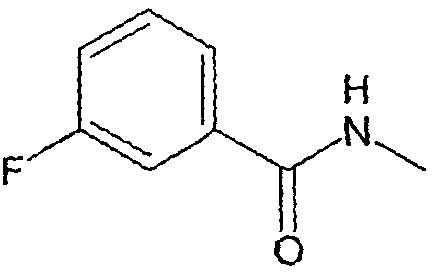
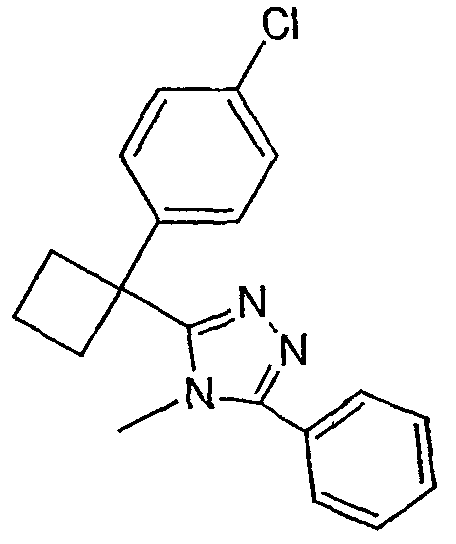
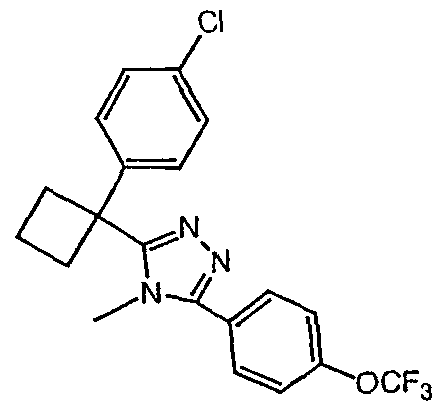
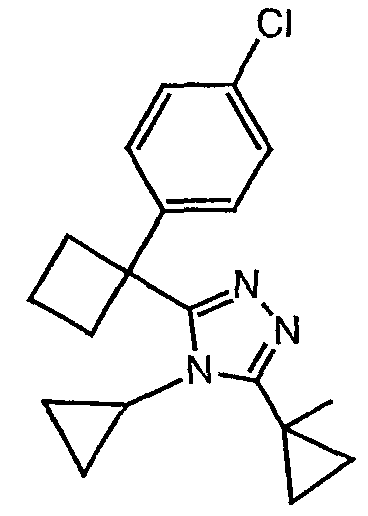
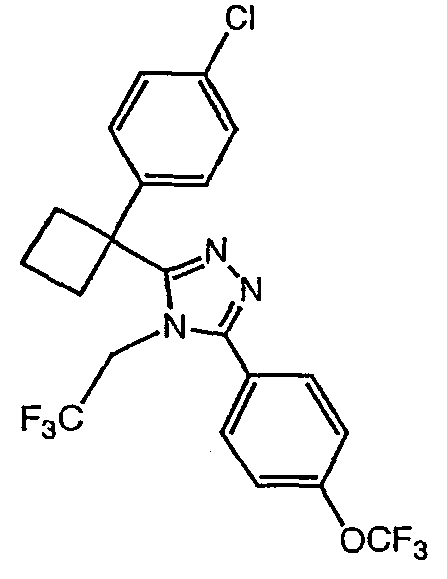
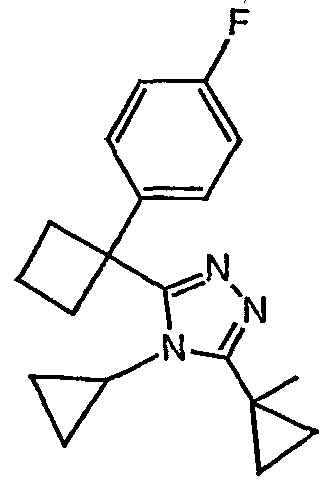
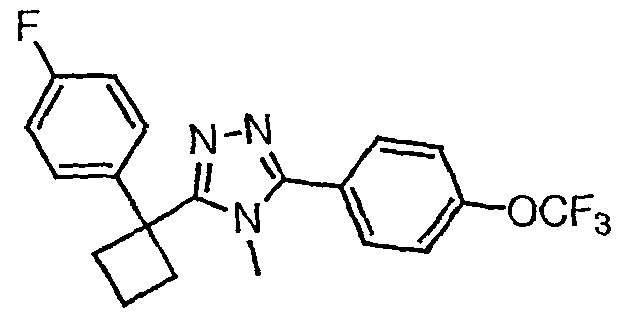
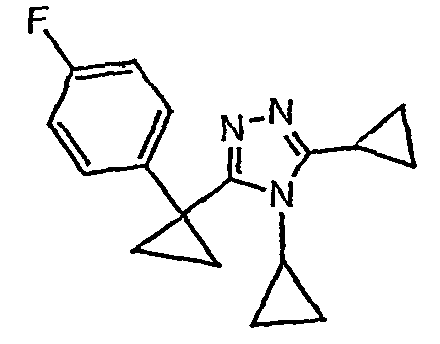
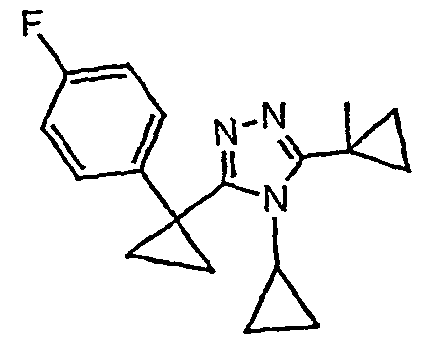
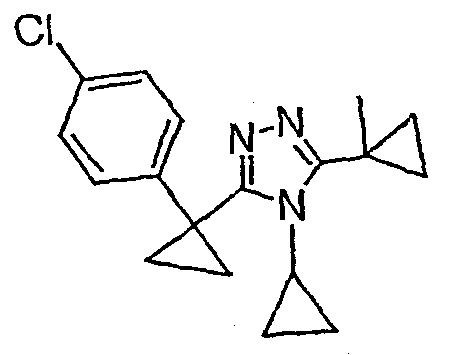
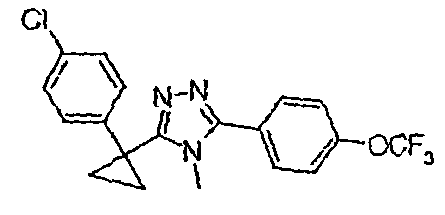
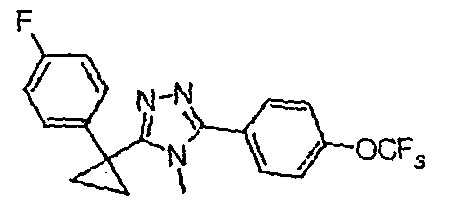
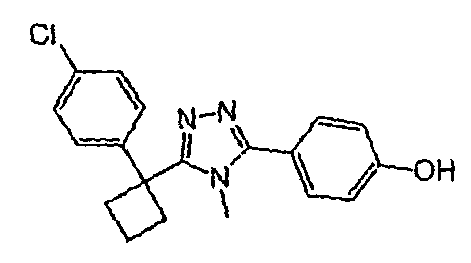
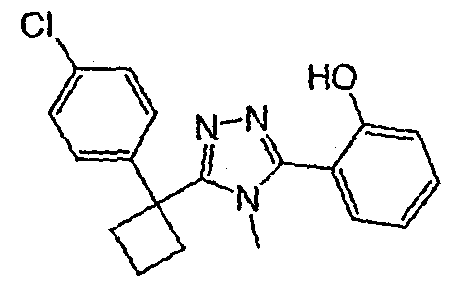
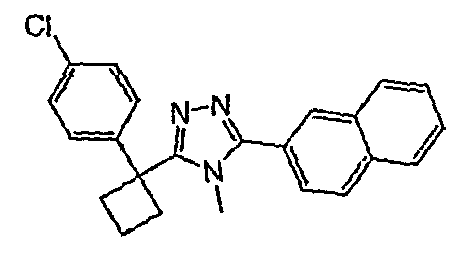
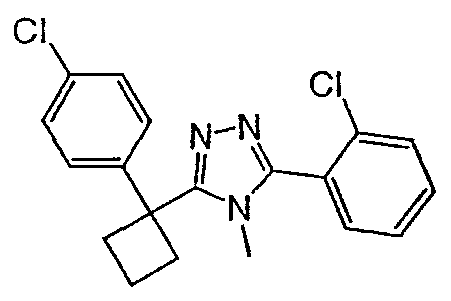
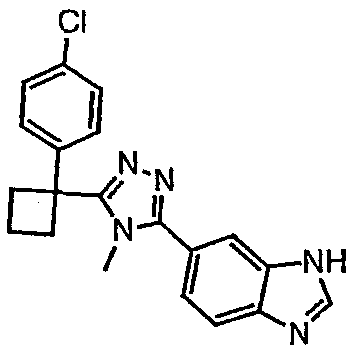
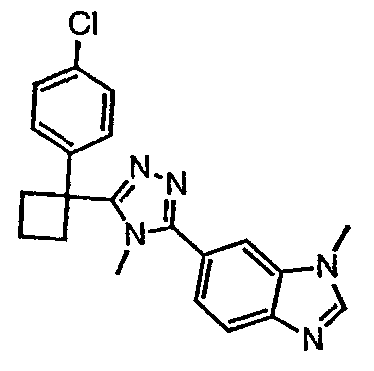
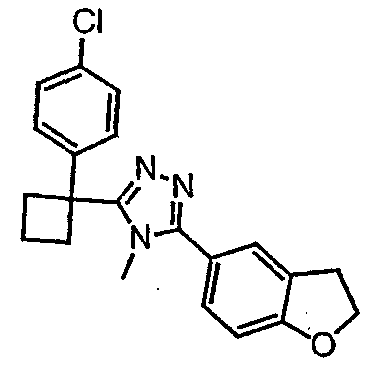
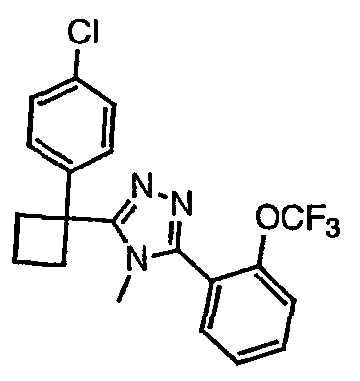
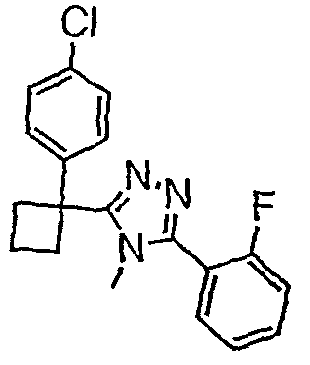
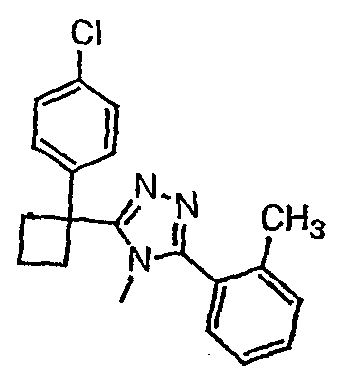
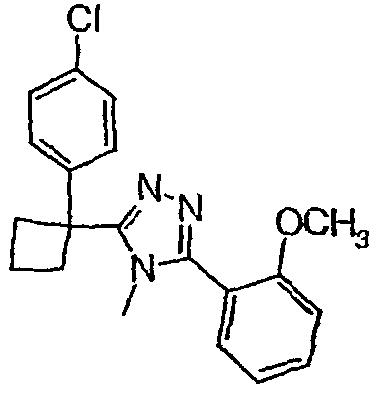
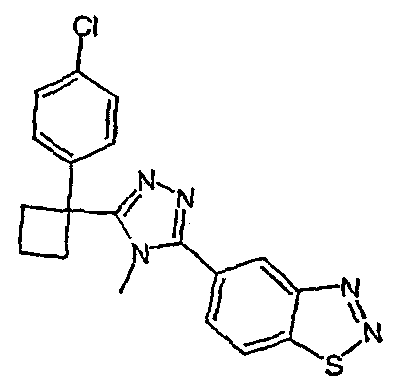
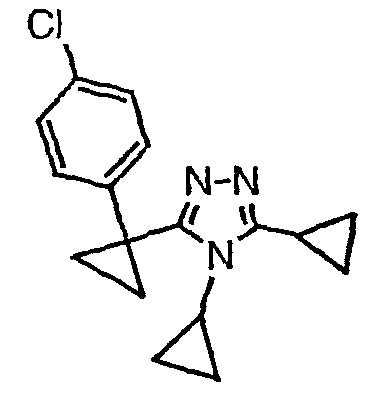
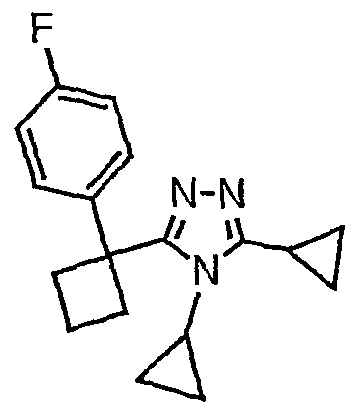
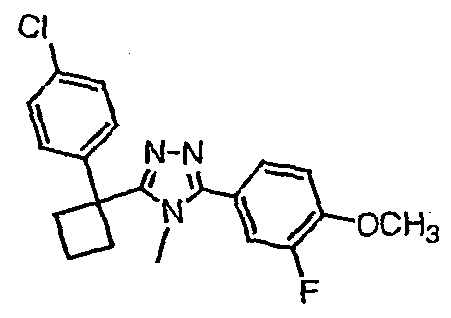
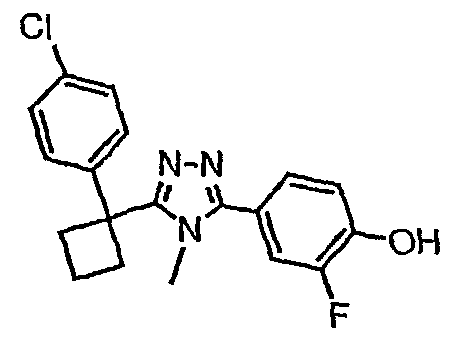
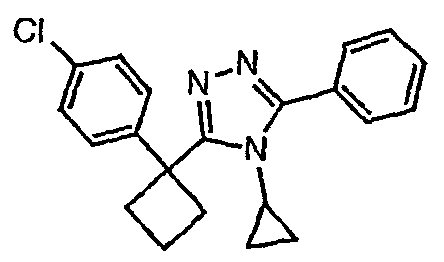
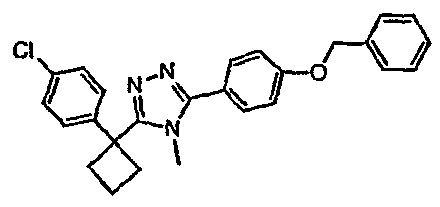
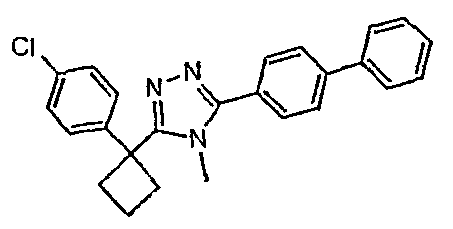
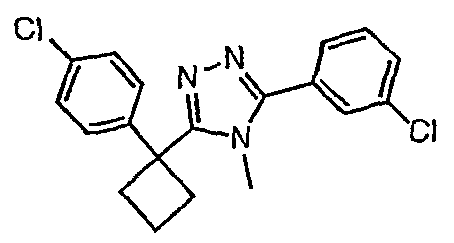
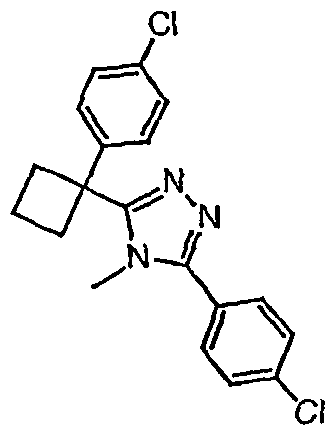
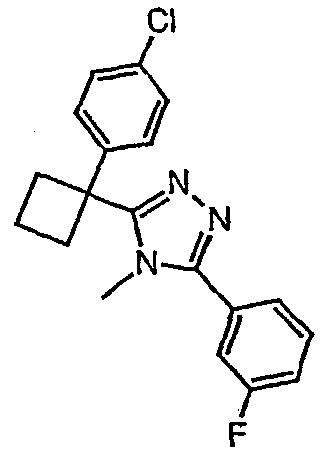
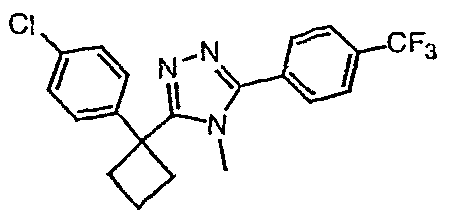
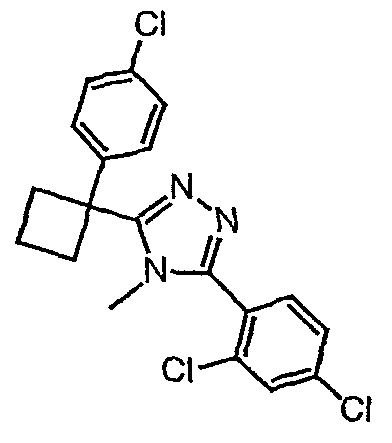
Метод препаративной ВЭЖХ для примера 3:
Используют методику, описанную в примере 2.
Метод аналитической ЖХ для примера 3:
Используют методики, идентичные описанным в примере 2.
Пример 4
Методика 4А
Получение 3-[1-(4-хлорфенил)-(Z)-3-(метоксиметокси)циклобутил]-5,6,7,8,9,10-гексагидро[1,2,4]триазоло[4,3-a]азоцина (4-1) и 3-[1-(4-хлорфенил)-(Е)-3-(метоксиметокси)циклобутил]-5,6,7,8,9,10-гексагидро[1,2,4]триазоло[4,3-a]азоцина (4-2)
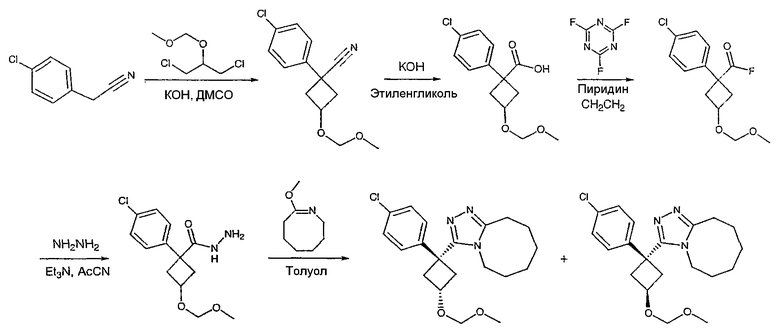
Гидроксид калия (2,57 г) растворяют в диметилсульфоксиде (8,0 мл). (4-Хлорфенил)ацетонитрил (1,58 г, 10,4 ммоль) и 1,3 дихлор-2-(метоксиметокси)пропан (1,993 г) растворяют в диэтиловом эфире (3 мл) и полученную смесь добавляют по каплям к раствору гидроксида калия с энергичным перемешиванием при комнатной температуре. Смесь перемешивают при комнатной температуре в течение часа, после чего реакцию гасят добавлением воды (5,5 мл), охлажденной льдом. Смесь фильтруют через рыхлый слой целита, который промывают эфиром (30 мл). Фильтрат переносят в делительную воронку и водный слой экстрагируют эфиром (3×15 мл). Органические слои объединяют, сушат над сульфатом магния, фильтруют и упаривают. Продукт очищают хроматографией на силикагеле, получая 1-(4-хлорфенил)-3-(метоксиметокси)циклобутанкарбонитрил (1,28 г) в виде смеси изомеров (˜2:1).
Нитрил (1,28 г) и гидроксид калия (2,2 г) растворяют в этиленгликоле (13 мл). Смесь выдерживают в течение шести часов при 198°С, после чего охлаждают до комнатной температуры, выливают в воду (15 мл) и промывают эфиром (2×20 мл). Водный раствор осторожно подкисляют водной хлористоводородной кислотой и экстрагируют эфиром (2×20 мл). Органические слои объединяют, сушат над сульфатом магния, фильтруют и упаривают, получая продукт в виде коричневого масла (0,9068 г).
1-(4-Хлорфенил)-3-(метоксиметокси)циклобутанкарбоновую кислоту (0,9068 г) и пиридин (0,40 мл) растворяют в дихлорметане (12 мл) и охлаждают до -10°С. Цианурфторид (1,0 мл) растворяют в дихлорметане (2 мл) и по каплям добавляют к реакционной смеси. Спустя 30 минут, реакционную смесь переносят в делительную воронку, содержащую лед (10 мл). После энергичного встряхивания дихлорметановый слой удаляют, сушат над сульфатом магния, фильтруют и упаривают.
Неочищенный фторангидрид растворяют в ацетонитриле (3 мл) и добавляют при перемешивании к раствору безводного гидразина (140 мкл), триэтиламина (1,0 мл) и ацетонитрила (15 мл) при 0°С. Спустя 10 минут, реакцию завершают с помощью ВЭЖХ/МС и сушат в вакууме.
Часть неочищенного 1-(4-хлорфенил)-3-(метоксиметокси)циклобутанкарбогидразида (456,1 мг) растворяют в безводном толуоле (7 мл) и смешивают с 8-метокси-2,3,4,5,6,7-гексагидроазоцином (228 мкл). Раствор нагревают до 120°С и выдерживают при этой температуре в течение 3 часов, затем медленной охлаждают до комнатной температуры. Продукт частично очищают хроматографией на силикагеле (100% этилацетат → 5% метанол в этилацетате → 10% метанол в этилацетате), получая смесь 4-1 и 4-2 в соотношении 62:38, соответственно. Изомеры разделяют препаративной ВЭЖХ и выделяют в виде их солей трифторуксусной кислоты. Каждую соль отдельно добавляют к насыщенному раствору гидрокарбоната натрия и экстрагируют этилацетатом. Очищенные свободные основания 3-[1-(4-хлорфенил)-цис-3-(метоксиметокси)циклобутил]-r-5,6,7,8,9,10-гексагидро[1,2,4]триазоло[4,3-a]азоцин (4-1) и 3-[1-(4-хлорфенил)-транс-3-(метоксиметокси)циклобутил]-r-5,6,7,8,9,10-гексагидро[1,2,4]триазоло[4,3-a]азоцин (4-2) сушат над сульфатом магния, фильтруют и концентрируют. Изомеры, 4-1 и 4-2, более эффективно разделяют хиральной препаративной ВЭЖХ (ChiralPak OD (Daicel Chemical Industries), колонка 2 см × 25 см, 20% изопропанол/гептан, 6 мл/мин), МС ESI (m/z) 376,2.
Методика 4В
Получение 3-(4-хлорфенил)-цис-3-(5,6,7,8,9,10-гексагидро[1,2,4]триазоло[4,3-a]азоцин-3-ил)циклобутан-r-ол (4-3)
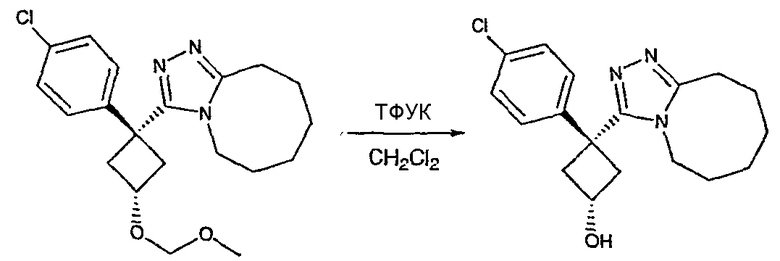
3-[1-(4-Хлорфенил)-цис-3-(метоксиметокси)циклобутил]-r-5,6,7,8,9,10-гексагидро[1,2,4]триазоло[4,3-a]азоцин (4-1) (53 мг) растворяют в дихлорметане (1 мл) и перемешивают при комнатной температуре. К полученному раствору добавляют трифторуксусную кислоту (0,2 мл) и раствор перемешивают в течение ночи при комнатной температуре. Летучие растворители удаляют в вакууме, и остаток очищают хроматографией на силикагеле, получая 3-(4-хлорфенил)-цис-3-(5,6,7,8,8,9,10-гексагидро[1,2,4]триазоло[4,3-a]азоцин-3-ил)циклобутан-r-ол (4-3) в виде твердого белого вещества.
3-(4-Хлорфенил)-транс-3-(5,6,7,8,9,10-гексагидро[1,2,4]триазоло[4,3-a]азоцин-3-ил)циклобутан-r-ол (4-4) получают по существу в соответствии с этой же методикой, используя эпимерное исходное вещество (4-2).
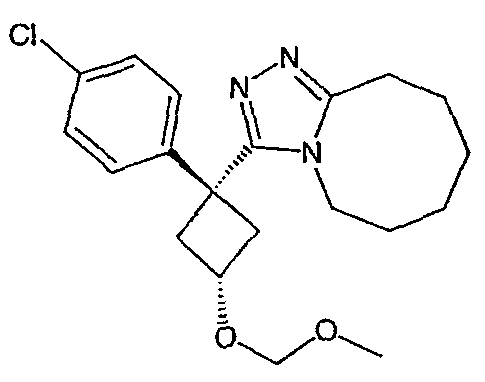
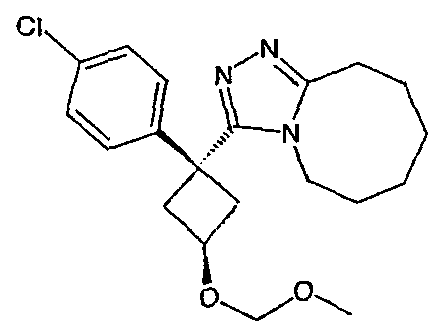
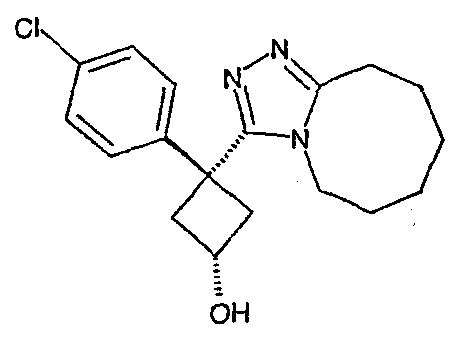
Методика 4С
Получение 3-(4-хлорфенил)-3-(5,6,7,8,9,10-гексагидро[1,2,4]триазоло[4,3-a]азоцин-3-ил)циклобутанона (4-5)
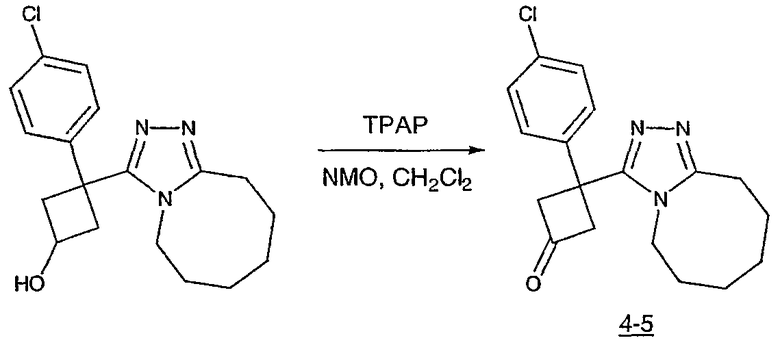
Смесь спиртов (4-3 и 4-4) (114,1 мг) растворяют в дихлорметане (5 мл) и охлаждают до 0°С. К полученному раствору добавляют перрутенат тетрапропиламмония (ТРАР, 12,1 мг) и 4-метилморфолин N-оксид (60,4 мг) и реакционную смесь перемешивают при комнатной температуре в течение трех часов. После этого неочищенную реакционную смесь загружают непосредственно на колонку с силикагелем и очищают (100% дихлорметан → 5% метанол в дихлорметане → 10% метанол в дихлорметане), получая 3-(4-хлорфенил)-3-(5,6,7,8,9,10-гексагидро[1,2,4]триазоло[4,3-a]азоцин-3-ил)циклобутанон (4-5); МС ESI (m/z) 330,1.
Методика 4D
Получение 3-[1-(4-хлорфенил)-3-метиленциклобутил]-5,6,7,8,9,10-гексагидро[1,2,4]триазоло[4,3-a]азоцина (4-6)
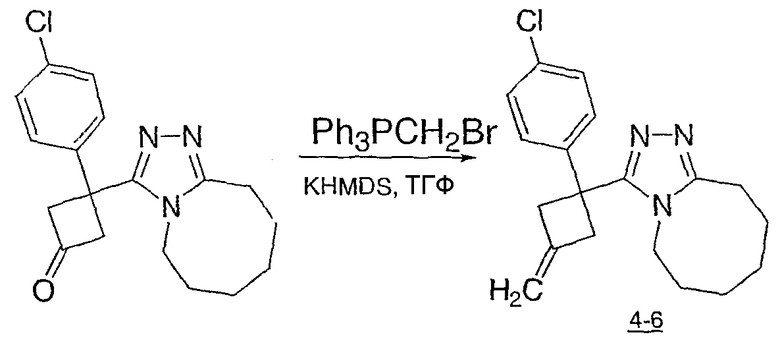
3-(4-Хлорфенил)-3-(5,6,7,8,9,10-гексагидро[1,2,4]триазоло[4,3-a]азоцин-3-ил)циклобутанон (4-5) (52 мг) растворяют во свежеперегнанном тетрагидрофуране (2 мл). К полученному раствору добавляют бромид метилтрифенилфосфония (281 мг) и затем бис(триметилсилил)амид калия (KHMDS, 0,5 M в толуоле, 1,25 мл). Реакционную смесь перемешивают в течение 24 часов при комнатной температуре, после чего неочищенный продукт добавляют к насыщенному раствору гидрокарбоната натрия и экстрагируют этилацетатом. Органический слой собирают, сушат над сульфатом магния, фильтруют и концентрируют. Продукт очищают колоночной хроматографией на силикагеле, получая 3-[1-(4-хлорфенил)-3-метиленциклобутил]-5,6,7,8,9,10-гексагидро[1,2,4]триазоло[4,3-a]азоцин (4-6); МС ES (m/z) 328,2.
Методика 4Е
Получение 3-[1-(4-хлорфенил)-3,3-дифторциклобутил]-5,6,7,8,9,10-гексагидро[1,2,4]триазоло[4,3-a]азоцина (4-7)
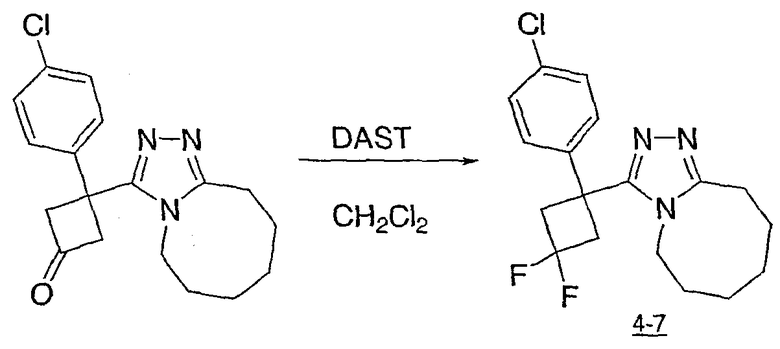
3-(4-Хлорфенил)-3-(5,6,7,8,9,10-гексагидро[1,2,4]триазоло[4,3-a]азоцин-3-ил)циклобутанон (4-5) (11,4 мг) растворяют в дихлорметане (1 мл). К полученному раствору добавляют трифторид (диэтиламино)серы (DAST, 73 мкл) и раствор перемешивают при комнатной температуре в течение 24 часов. Раствор выливают в насыщенный водный раствор гидрокарбоната натрия и экстрагируют дихлорметаном. Органический слой сушат над сульфатом магния, фильтруют и концентрируют. Остаток очищают хроматографией на силикагеле (100% дихлорметан → 1% метанол в дихлорметане → 5% метанол в дихлорметане), получая 3-[1-(4-хлорфенил)-3,3-дифторциклобутил]-5,6,7,8,9,10-гексагидро[1,2,4]триазоло[4,3-a]азоцин (4-7); МС ESI (m/z): 352,1.
Методика 4F
Получение 3-[1-(4-хлорфенил)-транс-3-фторциклобутил]-r-5,6,7,8,9,10-гексагидро[1,2,4]триазоло[4,3-a]азоцина (4-8)
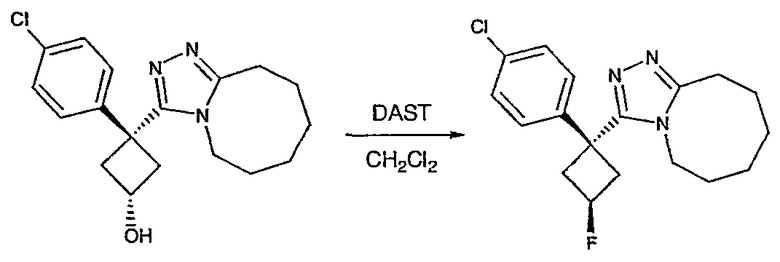
3-(4-Хлорфенил)-цис-3-(5,6,7,8,9,10-гексагидро[1,2,4]триазоло[4,3-a]азоцин-3-ил)циклобутан-r-ол (4-3) (21,3 мг) растворяют в безводном дихлорметане (1,5 мл) и охлаждают до 0°С. К полученному раствору добавляют трифторид (диэтиламино)серы (DAST, 80 мкл). Раствор нагревают до комнатной температуры и перемешивают в течение ночи. Продукт выливают в насыщенный водный раствор бикарбоната натрия и экстрагируют дихлорметаном. Органический слой сушат над сульфатом магния, фильтруют и концентрируют. Остаток очищают хроматографией на силикагеле (100% дихлорметан → 1% метанол в дихлорметане → 5% метанол в дихлорметане), получая 3-[1-(4-хлорфенил)-транс-3-фторциклобутил]-r-(5,6,7,8,9,10-гексагидро[1,2,4]триазоло[4,3-a]азоцин (4-8).
3-[1-(4-Хлорфенил)-транс-3-фторциклобутил]-r-(5,6,7,8,9,10-гексагидро[1,2,4]триазоло[4,3-a]азоцин (4-9) получают, по существу, в соответствии с этой же методикой, используя эпимерное исходное вещество (4-4).
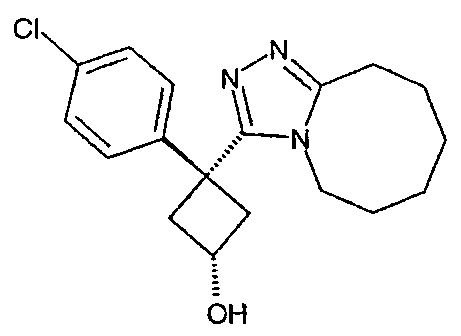
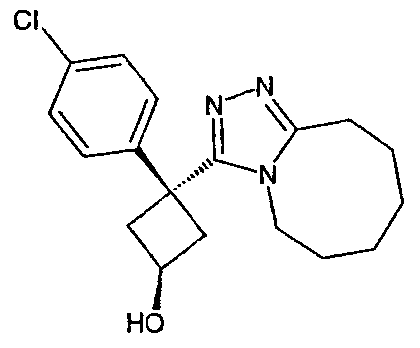
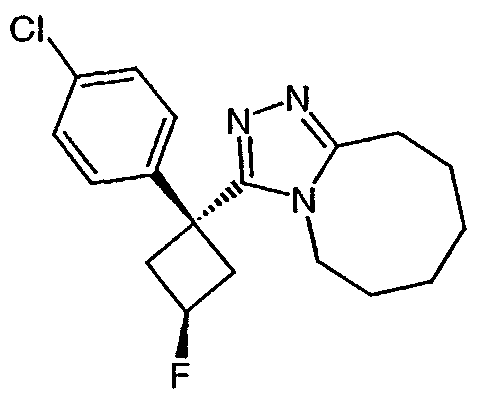
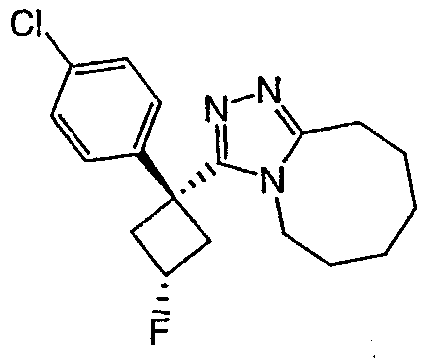
Методика 4G
Получение 3-(3-метил-1-фенилциклобутил)-5,6,7,8,9,10-гексагидро[1,2,4]триазоло[4,3-a]азоцина (4-10)
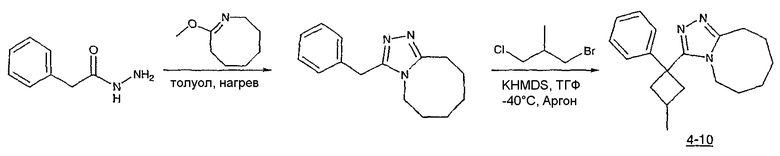
2-Фенилацетогидразин (1,01 г) добавляют к раствору безводного толуола (11 мл) и 8-метокси-2,3,4,5,6,7-гексагидроазоцина (0,96 мл). Смесь выдерживают при 60°С в течение 3 часов, затем при 110°С в течение ночи. Раствор охлаждают до комнатной температуры и концентрируют. Остаток очищают хроматографией на силикагеле, получая 3-бензил-5,6,7,8,9,10-гексагидро[1,2,4]триазоло[4,3-a]азоцин в виде твердого белого вещества.
3-Бензил-5,6,7,8,9,10-гексагидро[1,2,4]триазоло[4,3-a]азоцин (287,6 мг) и 1-бром-3-хлор-2-метилпропан (140 мкл) растворяют в безводном тетрагидрофуране, освобожденном от кислорода, и раствор охлаждают до -40°С в атмосфере аргона. К полученному раствору по каплям добавляют бис(триметилсилил)амид калия (KHMDS, 0,5 М в толуоле, 2,5 мл). Спустя 30 минут, добавляют вторую аликвоту KHMDS (2,5 мл). Еще через 30 минут снова добавляют KHMDS (2,15 мл) и раствору дают возможность медленно нагреться до комнатной температуры. Спустя один час, реакцию гасят водой и реакционную смесь добавляют в раствор соли. Смесь экстрагируют этилацетатом, органический слой сушат сульфатом магния, фильтруют, упаривают и очищают хроматографией на силикагеле, получая 3-[1-(4-хлорфенил)-(Z)-3-(метоксиметокси)циклобутил]-5,6,7,8,9,10-гексагидро[1,2,4]триазоло[4,3-a]азоцин (4-10) в виде смеси изомеров (˜1,2:1); МС ESI (m/z): 296,2.
Методика 4Н
Получение 1-(4-хлорфенил)-транс-3-фторциклобутан-r-карбогидразида
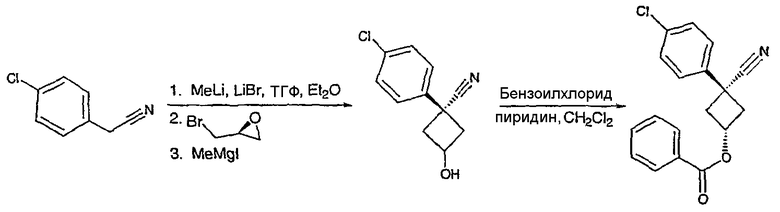
(4-Хлорфенил)ацетонитрил (14,04 г) растворяют в свежеперегнанном тетрагидрофуране (250 мл) и перемешивают при -78°С в атмосфере аргона [1]. К полученному раствору по каплям добавляют метиллитий (LiBr-комплекс, 1,5 М в диэтиловом эфире, 62 мл, 1 экв.) так, чтобы температура реакции сохранялась на уровне ниже -66°С. Раствор перемешивают в течение часа при -78°С, при этом цвет раствора изменяется от желтого до глубокого красного. К раствору по каплям добавляют эпибромгидрин и раствор перемешивают в течение дополнительных 90 минут. В раствор добавляют йодид метилмагния (3,0 М в диэтиловом эфире, 31 мл), и раствор, который становится светло-коричневым при медленном нагревании до комнатной температуры, перемешивают в течение ночи. Реакцию гасят водой (75 мл) и подкисляют до рН 2 5 N водной хлористоводородной кислотой (˜30 мл). Раствор соли добавляют до тех пор, пока слои не разделятся. Органический слой собирают, водный слой повторно экстрагируют диэтиловым эфиром (2×50 мл). Органические слои объединяют, сушат сульфатом магния, фильтруют и концентрируют.
Неочищенный 1-(4-хлорфенил)-3-гидроксициклобутан-1-карбонитрил (смесь цис:транс изомеров с соотношением примерно 4,2:1) растворяют в дихлорметане (150 мл) и перемешивают при 0°С. К раствору добавляют пиридин (11,3 мл) и затем бензоилхлорид (10,8 мл), раствор нагревают до комнатной температуры и перемешивают в течение 2,5 часов. Затем добавляют дополнительное количество пиридина (2 мл) и бензоилхлорида (2 мл) и реакционную смесь перемешивают при 30°С в течение ночи. Реакционный раствор добавляют к насыщенному раствору гидрокарбоната натрия и экстрагируют дихлорметаном. Органический слой промывают насыщенным хлоридом аммония, сушат над хлоридом магния, фильтруют и концентрируют, получая масло красноватого цвета. Два изомера разделяют хроматографией на силикагеле (25% дихлорметан/гексаны → 33% дихлорметан/гексаны → 50% дихлорметан/гексаны → 100% дихлорметан), получая целевой 3-(4-хлорфенил)-цис-3-цианоциклобутилбензоат (18,63 г).
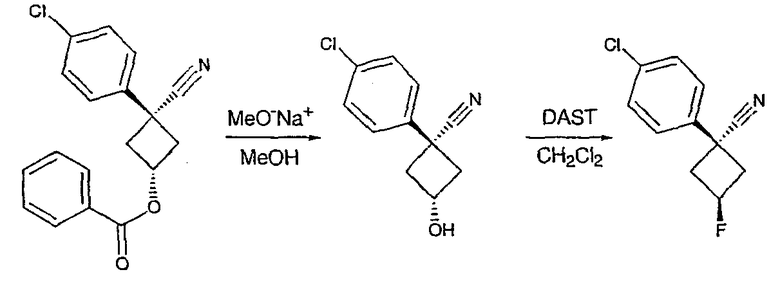
3-(4-Хлорфенил)-цис-3-цианоциклобутилбензоат (6,42 г) растворяют в смеси метанол/тетрагидрофуран (10 мл/20 мл) и перемешивают при комнатной температуре. Гидроксид лития моногидрат (1,1 г) растворяют в воде (10 мл) и добавляют в раствор бензоата. Спустя 10 минут, добавляют твердый хлорид аммония (˜2 г) и летучие растворители выпаривают. Оставшуюся водную смесь экстрагируют диэтиловым эфиром, органический слой сушат сульфатом магния, фильтруют и концентрируют, получая целевой циклобутанол.
Навеску 1-(4-хлорфенил)-цис-3-гидроксициклобутан-r-карбонитрила (1,13 г) растворяют в безводном дихлорметане и перемешивают при 0°С. В раствор добавляют трифторид (диэтиламино)серы (DAST, 1,43 г), раствор нагревают до 40°С и выдерживают при этой температуре в течение 10 часов. Добавляют дополнительное количество DAST (0,5 мл) и реакционный раствор перемешивают при 40°С в течение ночи. Раствор охлаждают, добавляют насыщенный водный раствор гидрокарбоната натрия и дважды экстрагируют дихлорметаном. Органические экстракты объединяют, сушат сульфатом магния, фильтруют и концентрируют. Неочищенный остаток осторожно хроматографируют на силикагеле (10% этилацетат/гексаны → 20% этилацетат/гексаны → 25% этилацетат/гексаны), получая 1-(4-хлорфенил)-транс-3-фторциклобутан-r-карбонитрил (1,024 г).
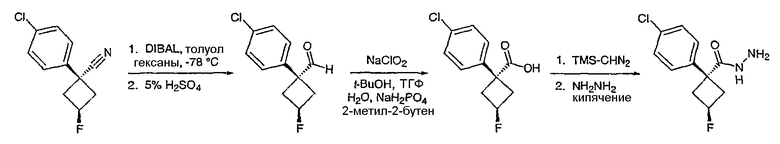
1-(4-Хлорфенил)-транс-3-фторциклобутан-r-карбонитрил (1,65 г) растворяют в безводном толуоле (30 мл) и охлаждают до -78°С. Раствор гидрида диизобутилалюминия (DIBAL, 1 М в гексанах, 9,4 мл) добавляют в течение 10 минут, и полученный раствор перемешивают в течение 30 минут. Реакцию гасят добавлением 5% серной кислоты (2,5 мл), и смесь нагревают до комнатной температуры. Спустя 1 час, смесь фильтруют через рыхлый слой целита. Целит промывают этилацетатом и весь фильтрат выливают в воду (20 мл). Слои разделяют, и водный раствор экстрагируют этилацетатом. Органические слои объединяют, сушат над сульфатом магния, фильтруют и концентрируют.
Неочищенный альдегид растворяют в смеси трет-бутанол/тетрагидрофуран/2-метилбут-2-ен (15 мл/5 мл/5 мл) и полученный раствор перемешивают при комнатной температуре. Хлорид натрия (1,56 г) и дигидрофосфат натрия (2,39 г) растворяют в воде (7 мл) и добавляют к раствору при энергичном перемешивании. Спустя 80 минут, летучие растворители удаляют в вакууме и смесь подкисляют до рН 2 водной 1 N хлористоводородной кислотой. Продукт экстрагируют этилацетатом (3×30 мл). Экстракты объединяют, сушат над сульфатом магния, фильтруют и упаривают, получая целевую карбоновую кислоту.
1-(4-Хлорфенил)-транс-3-фторциклобутан-r-карбоновую кислоту (5,68 г) растворяют в смеси дихлорметан/метанол (40 мл/10 мл). К полученному раствору добавляют (триметилсилил)диазометан (15 мл, 2,0 М в гексанах) до получения неисчезающего желтого окрашивания. Раствор перемешивают при комнатной температуре в течение 1 часа, после чего ТСХ показывает, что реакция завершена. Для гашения (триметилсилил)диазометана добавляют уксусную кислоту (2 мл) и раствор концентрируют, получая 1-(4-хлорфенил)-транс-3-фторциклобутан-r-карбоксилат.
Неочищенный сложный метиловый эфир (5,8 г) растворяют в толуоле (15 мл). Добавляют безводный гидразин (3,1 мл, 98,8 ммоль) и реакционную смесь кипятят с обратным холодильником в течение двух дней. Раствор охлаждают до комнатной температуры и толуол удаляют в вакууме, продукт очищают хроматографией на силикагеле (100% этилацетат), получая 1-(4-хлорфенил)-транс-3-фторциклобутан-r-карбогидразид в виде твердого белого вещества (4,82 г).
Методика 4I
Общая схема
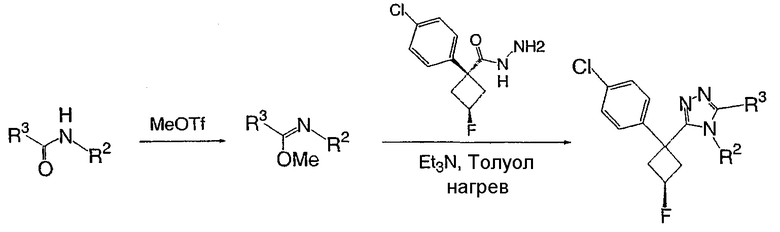
Получение 3-[1-(4-хлорфенил)-транс-3-фторциклобутил]-4,5-дициклопропил-r-4Н-1,2,3-триазола (4-11)
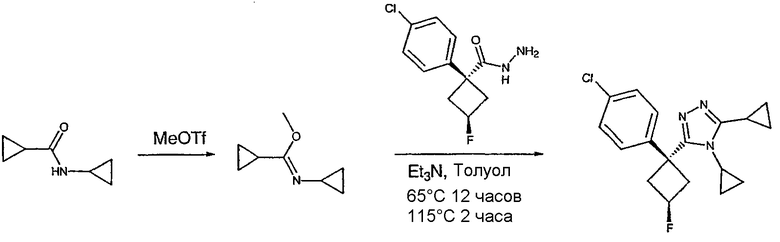
Метил трифторметансульфонат (84,1 мкл) добавляют к N-циклопропилциклопропанкарбоксамиду (93,0 мг). Смесь нагревают до 65°С, выдерживают при этой температуре в течение 2 минут, затем охлаждают до комнатной температуры. Толуол (1 мл), триэтиламин (207 мкл) и 1-(4-хлорфенил)-транс-3-фторциклобутан-r-карбогидразид (108 мг) добавляют к метил N-циклопропилциклопропанкарбоксимидоату и полученную смесь перемешивают при 60°С в течение ночи и при 115°С в течение 2 часов. Раствор охлаждают, затем концентрируют и остаток очищают хроматографией на силикагеле (100% этилацетат → 1% метанол в этилацетате → 3% метанол в этилацетате → 5% метанол в этилацетате), получая очищенный 3-[1-(4-хлорфенил)-транс-3-фторциклобутил]-4,5-дициклопропил-r-4Н-1,2,4-триазол (4-11).
Соединения 4-12 - 4-15 получают, по существу, в соответствии с этой же методикой, используя соответствующий карбоксамид в качестве исходного вещества и 1-(4-хлорфенил)-транс-3-фторциклобутан-r-карбогидразид.
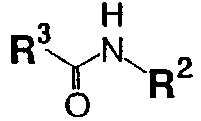
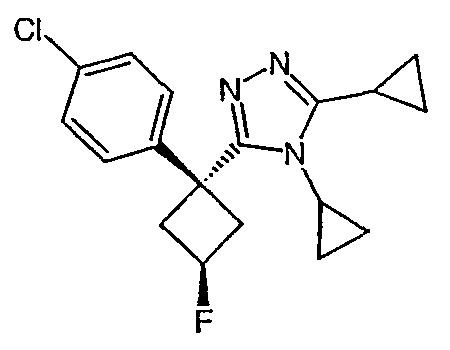
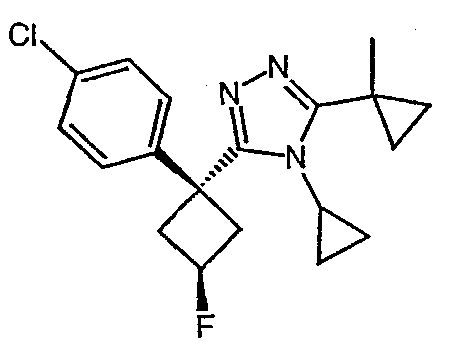
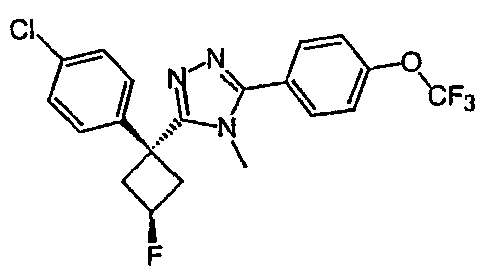
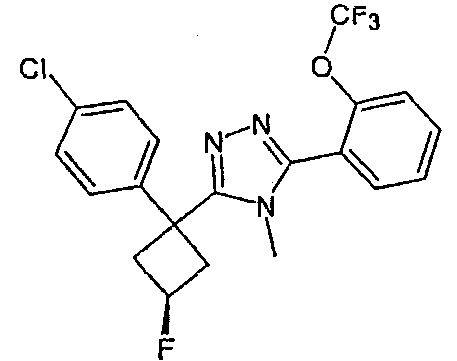
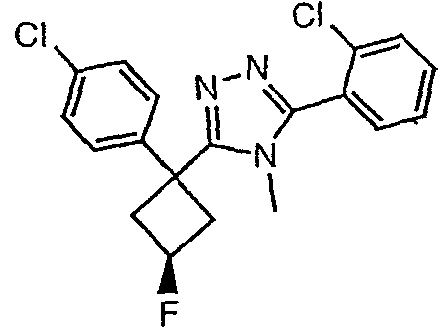
Метод препаративной ВЭЖХ для примера 4:
Используют метод препаративной ВЭЖХ, описанный в примере 2.
Аналитический метод ЖХ идентичен методу, описанному в примере 2.
Ссылки
1. Jeffery, J.E.; Kerrigan, F.; Miller, T.K.; Smith, G.J; Tometzki, G.B.; J. Chem. Soc., Perkin Trans 1, 1996, (21), 2583-2589.
2. Fedorynski, M.; Jonczyk, A. Org. Prep. Proced Int., 1995, 27 (3), 355-359.
3. Suzuki, H.; Tsutsui, H; Kano, A.; Katoh, S.; Morita, T.; Matsuda, K.; Iibuchi, N.; Ogawa, M. Heterocycles, 1997, 45 (9), 1657-61.
В то время, как в описании представлены предпочтительные воплощения данного изобретения, подразумевается, что множество альтернативных воплощений также входят в объем, определяемый формулой изобретения. Следовательно, изобретение определено шире, чем частные воплощения, раскрытые в описании.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ 1,2,4-ТРИАЗОЛО[4,3-B]ПИРИДО[3,2-D]ПИРИДАЗИНА, СПОСОБ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБ ИНГИБИРОВАНИЯ ФОСФОДИЭСТЕРАЗЫ-4 | 1998 |
|
RU2202552C2 |
| ПРОИЗВОДНЫЕ 5-АРИЛ-1Н-1,2,4-ТРИАЗОЛА И СОДЕРЖАЩАЯ ИХ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2000 |
|
RU2249588C2 |
| ИНГИБИТОР, ПРЕДСТАВЛЯЮЩИЙ СОБОЙ ПРОИЗВОДНОЕ ПИРИДАЗИНА, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ЕГО ПРИМЕНЕНИЕ | 2020 |
|
RU2807611C2 |
| НОВЫЕ ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ КАК ИНГИБИТОРЫ МОНОАЦИЛГЛИЦЕРИНЛИПАЗЫ | 2019 |
|
RU2769507C1 |
| АМИНОТЕТРАГИДРОПИРАНЫ В КАЧЕСТВЕ ИНГИБИТОРОВ ДИПЕПТИДИЛПЕПТИДАЗЫ-IV ДЛЯ ЛЕЧЕНИЯ ИЛИ ПРЕДУПРЕЖДЕНИЯ ДИАБЕТА | 2010 |
|
RU2550508C2 |
| ЗАМЕЩЕННЫЕ [1,2,4]ТРИАЗОЛО[4,3-a]ПИРИДИНЫ, ПРОЯВЛЯЮЩИЕ СВОЙСТВА АНТАГОНИСТОВ АДЕНОЗИНОВЫХ А2А РЕЦЕПТОРОВ, И ИХ ПРИМЕНЕНИЕ | 2013 |
|
RU2534804C1 |
| ПРОИЗВОДНЫЕ ТРИАЗОЛА КАК ИНГИБИТОРЫ 11-БЕТА-ГИДРОКСИСТЕРОИДДЕГИДРОГЕНАЗЫ-1 | 2003 |
|
RU2360910C2 |
| ИНГИБИТОРЫ ОКСАЗИНМОНОАЦИЛГЛИЦЕРИНЛИПАЗЫ (MAGL) | 2019 |
|
RU2794334C2 |
| СПОСОБЫ И КОМПОЗИЦИИ ДЛЯ НАПРАВЛЕННОЙ ДЕГРАДАЦИИ БЕЛКА | 2020 |
|
RU2820673C2 |
| ПРОИЗВОДНЫЕ ТРИАЗОЛОПИРИДИНА В КАЧЕСТВЕ МОДУЛЯТОРОВ АКТИВНОСТИ TNF | 2014 |
|
RU2677697C1 |
Описываются новые производные 1,2,4-триазола общей формулы I
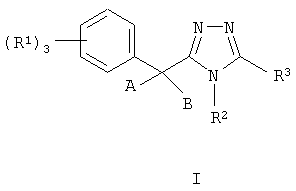
где А и В могут быть взяты отдельно или вместе, причем, когда последние взяты отдельно, А означает C1-6 алкил или фенил, а В означает C1-6 алкил; А и В, взятые вместе, означают С2-5 алкандиил, так что они образуют с С-атомом 3-6-членный цикл, необязательно замещенный C1-4-алкиленом, оксо, этилендиокси, С1-4-алкилом, 1-2 галогенами, С1-3-алкокси-С1-3-алкокси или гидрокси; каждый R1 независимо означает водород, ОН, галоген, С3-6 циклоалкил, С1-6-алкил, возможно замещенный 1-3 галоидами; или две R1 группы, стоящие у соседних углеродных атомов, образуют 6-членный арильный цикл; R2 и R3 могут быть взяты вместе или отдельно; когда они взяты вместе, то представляют собой С3-8-алкандиил, образующий конденсированный 5-10-членный неароматический цикл; когда R2 и R3 взяты отдельно, то R2 означает С1-6-алкил, возможно замещенный 1-3 галогенами, или циклопропил, и R3 означает циклопропил, возможно замещенный С1-4-алкилом, нафтил, фенил, возможно замещенный галогеном, ОН, C1-6алкилом, где указанный С1-6-алкил необязательно замещен 1-3 галогеновыми группами, OC1-6алкилом, где указанный OC1-6алкил необязательно замещен 1-3 галогеновыми группами, фенилом или бензилоксигруппой, дигидробензофуранил, бензотиадиазолил или бензимидазолил, возможно замещенный С1-6алкилом их фармацевтически приемлемые соли или сольваты и фармацевтическая композиция на их основе. Указанные соединения являются ингибиторами 11-β-гидроксистероидной дегидрогеназы I и могут быть использованы в медицине для лечения сахарного диабета, ожирения и дислипидемии. 3 н. и 16 з.п.ф-лы, 17 табл.
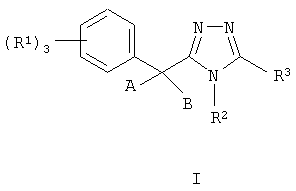
или их фармацевтически приемлемые соли или сольваты,
где
А и В могут быть взяты отдельно или вместе, причем
когда А и В взяты отдельно, А представляет собой C1-6 алкил или фенил и В представляет собой C1-6 алкил, и
когда А и В взяты вместе, они представляют собой С2-5 алкандиил, так что они образуют с атомом углерода, к которому присоединены, 3-6-членный цикл, необязательно замещенный C1-4-алкиленом, оксо, этилендиокси, С1-4алкилом, 1-2 галогенами, С1-3-алкокси-С1-3-алкокси или ОН;
каждый R1 представляет собой Н, С3-6 циклоалкил, ОН, галоген, C1-6-алкил, необязательно замещенный 1-3 галогенами,
или две R1 группы, стоящие у соседних атомов углерода, образуют 6-членный арильный цикл;
R2 и R3 могут быть взяты вместе или отдельно, причем
когда R2 и R3 взяты вместе, они представляют собой С3-8-алкандиил, образующий конденсированный 5-10-членный неароматический цикл, и
когда R2 и R3 взяты отдельно, R2 представляет собой С1-6-алкил, необязательно замещенный 1-3 галогенами, или циклопропил, и R3 представляет собой циклопропил, необязательно замещенный C1-4 алкилом, нафтил, фенил, необязательно замещенный галогеном, ОН, C1-6 алкилом, где указанный C1-6 алкил необязательно замещен 1-3 галогенами, OC1-6 алкилом, где указанный OC1-6 алкил необязательно замещен 1-3 галогенами, фенилом или бензилоксигруппой, дигидробензофуранил, бензотиадиазолил или бензимидазолил, необязательно замещенный C1-6 алкилом.
или его фармацевтически приемлемую соль или сольват.
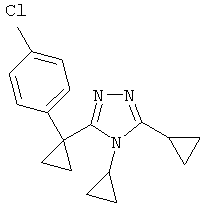
или его фармацевтически приемлемая соль или сольват.
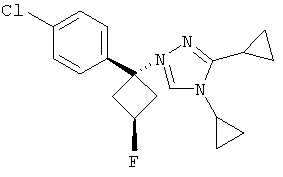
или его фармацевтически приемлемая соль или сольват.
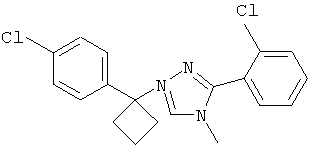
или его фармацевтически приемлемая соль или сольват.
или его фармацевтически приемлемая соль или сольват.
| ПРЕОБРАЗОВАТЕЛЬ СИГНАЛОВ | 0 |
|
SU190094A1 |
| H.REIMLINGER et al | |||
| CHEMISCHE BERICHTE | |||
| Клапанный регулятор для паровозов | 1919 |
|
SU103A1 |
| ПРОИЗВОДНЫЕ ТРИАЗОЛА, СПОСОБ ИХ ПОЛУЧЕНИЯ И ИНСЕКТОАКАРИЦИДНАЯ КОМПОЗИЦИЯ | 1993 |
|
RU2114107C1 |

