Изобретение относится к соединениям, выполняющим новые функции ингибиторов костной резобции/промоторов остеогенеза.
Нормальная регенерация костей характеризуется равновесием между костной резорбцией и остеогенезом, и в случае преобладания костной резорбции происходит растворение и уменьшение костных компонентов, что ведет к костным заболеваниям, например, остеопорозу. Известно, что половые гормоны, например, экстроген, способны подавлять костную резорбцию, вследствие чего применяются в Европе и Америке для профилактики и лечения остеопороза. Тем не менее, до настоящего времени не подтверждено, что такие гормоны концентрируются в костях, и не следует недооценивать возможность онкогеннеза в результате введения одних только таких гормонов.
С другой стороны, антибиотики тетрациклинового типа обладают способностью концентрироваться в костях, но при этом не способны ни ингибировать костную резорбцию, ни способствовать образованию костного вещества. И только в описании патента США N 4925833 указано, что тетрациклин промотирует синтез костных белков в опытах на клеточном уровне. Хотя синтез костных белков и необходим для остеогенеза, одного только синтеза костных белков мало для остеогенеза.
И до настоящего времени не известны вещества, способные промотировать остеогенез, и которые могут быть использованы в качестве профилактических и лечебных средств при костных заболеваниях.
Соответственно, целью настоящего изобретения является создание средства для лечения костных заболеваний, ингибирующего костную резорбцию и способствующего остеогенезу, предпочтительно синергически, и способного концентрироваться в костях.
Создателями настоящего изобретения проведены разнообразные исследования, направленные на решение вышеуказанных проблем, и в ходе этих исследований было обнаружено, что соединения, полученные ковалентным связыванием антибиотиков тетрациклинового типа и гормонов стероидного типа, например, экстрогена, с применением связующего соединения, промотируют остеогенез, помимо способности ингибировать костную резорбцию, а способны накапливаться в костях, которые являются предметом настоящего изобретения.
Соответственно, в настоящем изобретении предлагаются промоторы остеогенеза/ингибиторы костной резорбции, имеющие общую формулу (I):
X-Y-Z
(где X представляет одновалентную группу следующей формулы (II):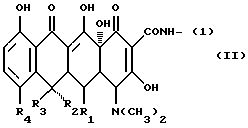
(где R1 представляет водород или гидроксил, R2 представляет водород или гидроксил, R3 представляет водород или метил и R4 представляет водород, галоген или диметиламиногруппу);
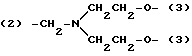
Y представляет двухвалентную или трехвалентную группу следующих формул (III), (IV) или (V):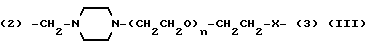
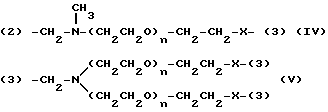
(где n = 0 - 4 и X представляет прямую связь -O- или -NH-);
Z представляет одновалентную группу, образованную удалением водорода или гидроксила из соединения следующей формулы (VI):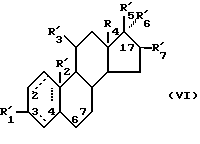
(где R'1 представляет HO- или O=; R'2 представляет атом водорода или метил; R'3 представляет атом водорода, фенил или замещенный фенил; R'4 представляет метил или этил; R'5 представляет гидроксил, кетогруппу или ацетил; R'6 представляет водород, гидроксил, метил, этинил или пропинил, или R'5 и R'6 совместно образуют группу =O; R'7 представляет водород, гидроксил или =O, или R'6 и R'7 вместе присоединены к кислородам 2,2-диоксипропильной группы и символом 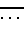 обозначена простая или двойная связь), причем присоединение этой группы происходит в 2-положение, 3-положение, 4-положение, 6-положение, 7-положение или 17-положение, или по фенильной группе, присоединенной в 11-положении, связь (1) в формуле (II) и связь (2) в формулах (III) - (V) непосредственно соединены, связь (3) в формулах (III) - (V) и любая связь в вышеуказанных положениях формулы (VI) непосредственно соединены).
обозначена простая или двойная связь), причем присоединение этой группы происходит в 2-положение, 3-положение, 4-положение, 6-положение, 7-положение или 17-положение, или по фенильной группе, присоединенной в 11-положении, связь (1) в формуле (II) и связь (2) в формулах (III) - (V) непосредственно соединены, связь (3) в формулах (III) - (V) и любая связь в вышеуказанных положениях формулы (VI) непосредственно соединены).
В вышеуказанной формуле (II) галоген представлен, например: хлором, фтором, бромом или йодом, предпочтительно хлором.
Краткие пояснения к диаграммам
На фиг. 1 приведена фотография, иллюстрирующая результаты эксперимента N 3.
На фиг. 2 приведена фотография, иллюстрирующая результаты эксперимента N 3.
На фиг. 3 приведена фотография, иллюстрирующая результаты эксперимента N 3.
На фиг. 1-3 приведены результаты одного и того же эксперимента, повторенного трижды.
Предпочтительными примерами одновалентной группы формулы (II), являющейся фрагментом соединения формулы (I) в качестве активного компонента фармацевтического препарата изобретения, включают:
Формулу (II-1):
Одновалентная группа тетрациклина, представленная формулой (II-I) (где (в формуле (II)) R1 - водород, R2 - гидроксил, R3 - метил и R4 - водород):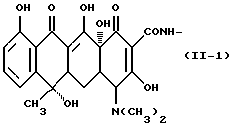
Формулу (II-2):
Одновалентная группа террамицина, представленная формулой (II-2) (где (в формуле (II)) R1 - гидроксил, R2 - гидроксил, R3 - метил и R4 - метил):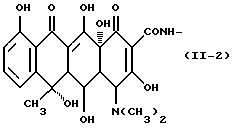
Формулу (II-3):
Одновалентная группа хлортетрациклина, представленная формулой (II-3) (где (в формуле (II)) R1 - водород, R2 - гидроксил, R3 - метил и R4 - хлор):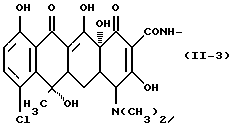
Формулу (II-4):
Одновалентная группа дезокситетрациклина, представленная формулой (II-4) (где (в формуле (II)) R1 - гидроксил, R2 - водород, R3 - метил и R4 - водород):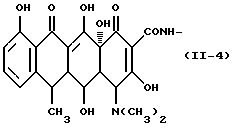
Формулу (II-5):
Одновалентная группа аминотетрациклина, представленная формулой (II-5) (где (в формуле (II)) R1 - водород, R2 - водород, R3 - водород и R4 - диметиламиногруппа):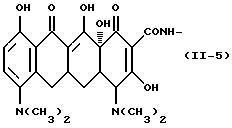
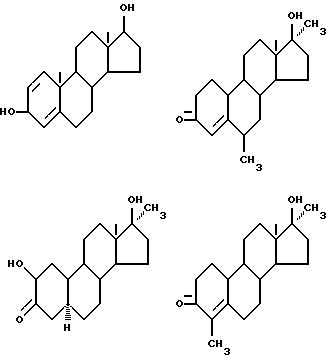
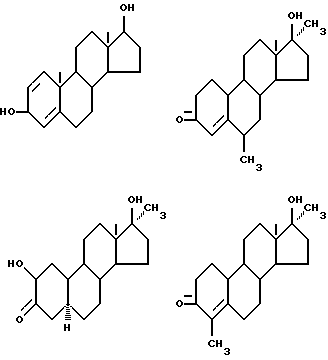
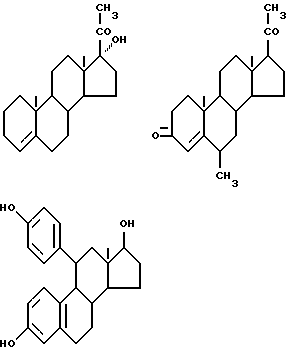
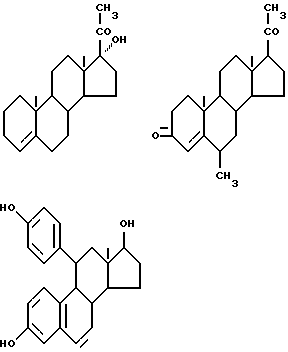
Предпочтительные примеры одновалентных групп соединения формулы (VI), являющихся фрагментами соединений формулы (I) в качестве активных компонентов фармацевтических препаратов настоящего изобретения, включают следующее.
Формулу (VI-1):
Одновалентная группа эстрона представлена формулой (VI-1) (где (в формуле (VI)) R'5 и R'6 совместно образуют группу =O и R'7 - водород):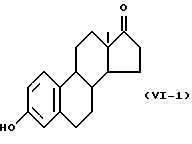
Формулу (VI-2):
Одновалентная группа эстрадиола представлена формулой (VI-2) (где (в формуле (VI)) R'5 - гидроксил, R'6 - водород и R'7 - водород):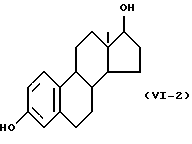
Формулу (VI-3):
Одновалентная группа эстроалкинола представлена формулой (VI-3) (где (в формуле (VI)) R'5 - гидроксил, R'6 - этинил и R'7 - водород):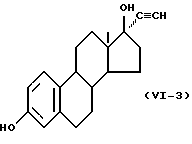
Формулу (VI-4):
Одновалентная группа эстриола представлена формулой (VI-4) (где (в формуле (VI)) R'5 - гидроксил, R'6 - водород и R'7 - гидроксил):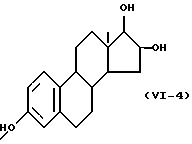
Соединительная группа в соединениях вышеприведенных формул (VI) - (VI-4) может находиться в 3-положении, 6-положении или 17-положении.
Соединения формулы (VI) могут быть также представлены одновалентными группами, полученными удалением водорода или гидроксильной группы в следующих соединениях: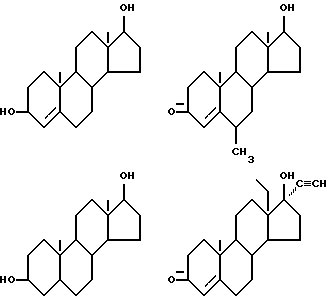
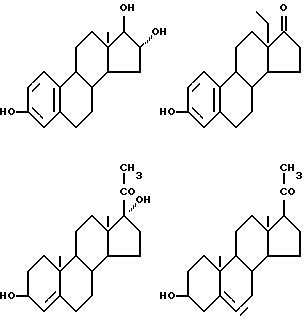
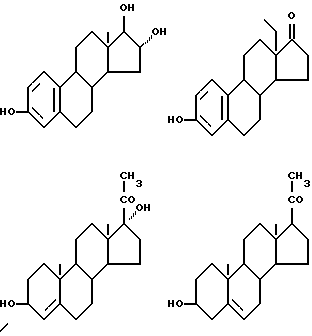
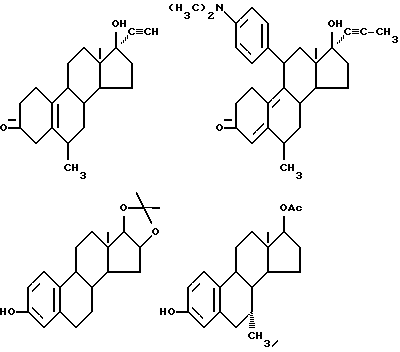
Соответственно активный компонент фармацевтического препарата настоящего изобретения может быть обозначен следующим шифром; например:
/II-1/-/III/-(3) /V-1/ (цифрой в скобках () указано положение связующей группы в группе формулы /VI-1/. Возможные варианты таких сочетаний представлены таблицей А (см.в конце описания).
Сами соединения, охарактеризованные выше, могут быть получены известными способами. К примеру, связующее соединение формул (III-V) вначале соединяют со стероидным соединением формулы (VI) и полученный продукт присоединения соединяют с соединением тетрациклинового ряда.
Присоединение связующего соединения формул (III) - (V) в 3-положении стероидного соединения формулы (VI) осуществляют, например, согласно следующей схеме реакции: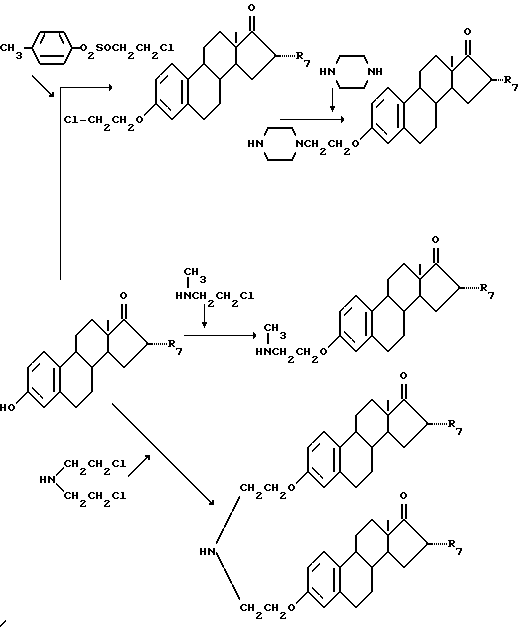
Кроме того, соединение, в котором R'5 - гидроксил и R'6 - водород или этинил, может быть получено, например, следующей реакцией: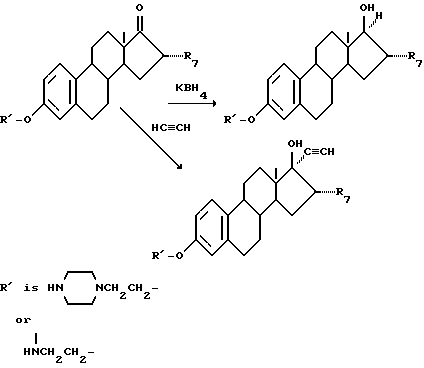
Для присоединения связующего соединения формул (III) - (V) в 6-положении стероидного соединения формулы (VI) вначале в 6-положение стероидного соединения вводят группу =O, после чего проводят следующую реакцию: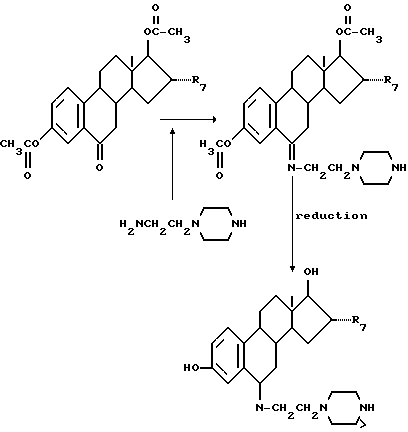
Затем к продукту реакции связующего соединения со стероидным соединением может быть присоединено тетрациклиновое соединение путем связывания N-атома связующего соединения с N-атомом амидной группы тетрациклинового соединения с помощью формальдегида.
Фармацевтические препараты настоящего изобретения могут быть приготовлены в дозированной форме для перорального или парентерального введения, например: внутривенным вливанием, чрезкожной инъекцией, внутримышечной инъекцией, внутрибрюшинной инъекцией и т.д. Эффективная ежедневная доза для человека равна 0.2 - 200 мг при пероральном введении и 0.1 - 100 мг при парентеральном введении. Соединения настоящего изобретения характеризуются чрезвычайно низкой токсичностью, и значение ЛД50 соединения I, полученного в примере 1, например, для мышей составляет 143 мг/кг.
Фармацевтические препараты настоящего изобретения могут быть получены в виде обычных форм, определяемых путем введения. В случае перорального введения фармацевтический препарат может иметь вид, например: капсулы, таблетки, гранулы, порошка, жидкого препарата и т.д. Такие формы получают обычным путем. Жидкие препараты могут быть получены, например, растворением или суспендированием активного компонента настоящего изобретения в приемлемой среде, например, водном буферном растворе и т.п. Порошковый препарат может быть приготовлен смешиванием активного компонента с порошкообразным наполнителем таким, как крахмал, например, кукурузный крахмал и/или сахарид, например, лактоза.
Таблетки могут быть получены смешиванием активного компонента с наполнителем, например, вышеуказанным наполнителем и связующим средством, например, крахмальной пастой и прессованием смеси в таблетирующей машине. Гранулы могут быть приготовлены смешиванием активного компонента с наполнителем, связующим средством и т.д., разбавлением смеси с жидкостью, например: водой и/или глицерином, просеиванием полученного продукта через сито с его гранулированием и высушиванием полученных гранул. Капсулы могут быть приготовлены капсулированием вышеописанных порошка или гранул в капсулы соответствующего размера.
Парентеральные дозированные формы могут быть получены растворением или суспендированием активного компонента в физиологическом солевом растворе или буферном солевом, таком, как, например, фосфатный буферный раствор. Парентеральные дозированные формы могут представлять высушенные вымораживанием продукты, которые перед употреблением необходимо растворить или суспендировать, и добавками для сушки вымораживанием могут служить сахарид, например, лактоза или обычные добавки для сушки вымораживанием.
Примеры
Далее приводятся конкретные примеры соединений настоящего изобретения. Однако, объем изобретения в целом не ограничен только этими примерами.
Пример синтеза 1
Синтез 1-1
3-хлорэтокси-17-оксиэстра-1,3,5(10)-триена (соединение 1-1)
К толуольному раствору, полученному смешиванием 27.1 г эстрона, 22.2 г 3-хлорэтокси-17-оксиэстра-1,3,5(10)-триена и небольшого количества триэтиланилинхлорида, добавляют раствор NaOH. После установления pH 10 реакцию продолжают 4 часа и растворитель испаряют. Перекристаллизацией твердого остатка из спирта получают соединение (1-1, R7 = H), выход 79%
Т.пл. 86 - 88oC.
Элементный анализ: C 72.4, H 4.3, Cl 10.71.
Синтез 1-2
N-[17-оксиэстра-1,3,5(10)-триен-3-оксиэтил]-пиперазина (соединение 1-2)
Смесь 7.8 г вышеописанного соединения (1-1), 46.6 г безводного пиперазина и 120 мл диметилформамида (ДМФА) нагревают 5 часов при 80 - 100oC. После удаления ДМФА испарением и перекристаллизации полученного твердого остатка из спирта и ацетона получают белое кристаллическое соединение (VI), выход 85%.
Т.пл. 140 - 142oC.
Элементный анализ: C 75.2, H 9.2, N 7.4.
Синтез 1-3
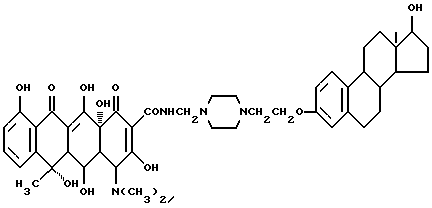
N-4-[17-оксиэстра-1,3,5(10)-триен-3-оксиэтил] -пиперазин-1- метилтетрациклина (соединение 1-3)
Смесь 3.8 г вышеописанного соединения (1-2), 0.03 г метаформальдегида и 15 мл изопропанола нагревают 2 часа при 40oC. После добавления 3.5 г тетрациклина смесь перемешивают 5 часов. По окончании реакции продукт реакции фильтруют и промывают изопропанолом и этиловом эфиром. В результате получают соединение (1-3) в виде твердого желтого вещества (R1 = R4 = H, R2 = OH, R3 = CH3), выход 95%.
Т.пл. 160oC (разл.).
Элементный анализ: C 67.21, H 7.12, N 6.67.
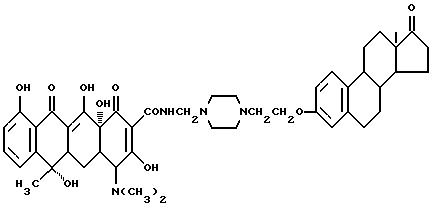
Пример синтеза 2
Синтез 2-1
N-[17 β -Гидроксиэстра-1,3,5(10)-триен-3-оксиэтил]пиперазин (соединение 2-1)
В метиловом спирте растворяют 3.8 г соединения (1-2) примера 1. После добавления 0.5 г боргидрата калия в щелочных условиях реакционную смесь нагревают с перемешиванием 3 часа. Реакционную смесь нейтрализуют кислотой и метиловый спирт испаряют. Перекристаллизацией полученного твердого остатка из спирта получают белые кристаллы (соединение 2-1, R7 = H), выход 91%.
Т.пл. 141 - 142oC.
Элементный анализ: C 75.21, H 9.23, N 7.14.
2-2
Синтез N-4-[17 β -гидроксиэстра-1,3,5(10)-триен-3- оксиэтил]пиперазин-1-метилентетрациклина (соединение 2-2)
Смесь 3.84 г вышеописанного соединения (2-1), 0.03 г метаформальдегида и 20 мл изопропанола нагревают 2 часа при 40oC. После добавления 3.5 г тетрациклина реакционную смесь перемешивают 5 часов. По окончании реакции продукт реакции фильтруют и промывают изопропанолом и этиловым эфиром. В результате получают бледно-желтое твердое вещество (соединение 2-2) (R1 = R4 = H, R2 = OH, R3 = CH3), выход 95%.
Т.пл. 165oC (разл.).
Элементный анализ: C 67.3, H 7.34, N 6.54.
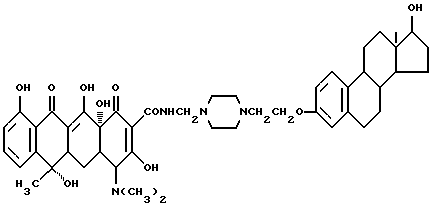
Пример синтеза 3
Синтез N-4-[17 β -гидроксиэстра-1,3,5(10)-триен-3-оксиэтил]-1- пиперазин-1-метилен(окситетрациклина) (соединение 3-1)
Смесь 3.8 г соединения примера 2 (2-1), 0.03 г метаформальдегида и 20 мл изопропанола нагревают 2 часа при 40oC. После добавления 3.5 г тетрамицина реакционную смесь перемешивают 5 часов. По окончании реакции и обработки по методике примера 1-3 получают бледно-желтое твердое вещество (соединение 3) (R1 = R2 = OH, R3 = CH3, R4 = H), выход 93%.
Т.пл. 171oC (разл.).
Элементный анализ: C 65.62, H 7.1, N 6.67.
Пример синтеза 4
4-1
Синтез бис-N,N-[17-оксиэстра-1,3,5(10)-триен-3-оксиэтил]амина (соединение 4-1)
К смеси 3.6 г мастагенхлорида, 12 г эстрона, 4 г триэтиланилина, воды и толуола добавляют при перемешивании раствор NaOH. После перемешивания смеси 5 часов растворитель испаряют. Перекристаллизацией твердого остатка из спирта получают целевое соединение, выход 72%.
Т.пл. 256 - 259oC.
Элементный анализ: C 78.5, H 8.6, N 2.31.
4-2
Синтез бис-N, N-[17-оксиэстра-1,3,5(10)-триен-3- оксиэтил]аминометилентетрациклина (соединение 4-2)
Смесь 6.1 г вышеописанного соединения (4-1), 0.03 г метаформальдегида и 20 мл изопропанола нагревают 2 часа при 40oC. После добавления 3.5 г тетрациклина реакционную смесь перемешивают 8 часов. По окончании реакции получают бледно-желтое твердое вещество (соединение 4-2) (R1 = R4 = H, R2 = OH, R3 = CH3), выход 68%.
Т.пл. 183oC (разл.).
Элементный анализ: C 71.1, H 7.21, N 3.89.
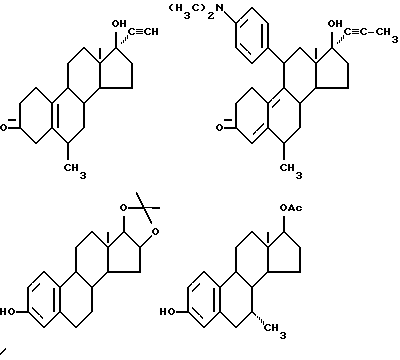
Строение соединения представлено следующими молекулярными формулами: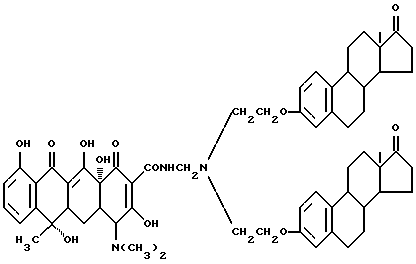
Пример синтеза 5
5-1
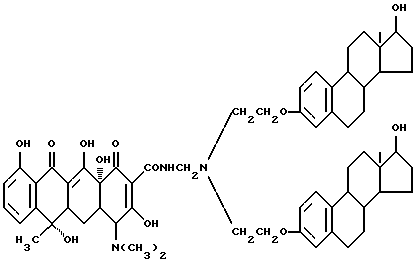
Синтез бис-N, N-[17 β -гидроксиэстра-1,3,5(10)-триен-3-оксиэтил]амина (соединение 5-1)
К 6.1 г вышеописанного соединения (4-1) добавляют метиловый спирт и после прибавления в щелочных условиях 0.5 г боргидрата калия реакционную смесь перемешивают 5 часов. Затем реакционную смесь нейтрализуют кислотой и метиловый спирт испаряют. Твердый остаток очищают в растворе ацетона и в растворе спирта. Эстрон-17-кетогруппу в соединении (4-1) восстанавливают в -17 β -гидроксигруппу. С выходом 82% получено белое вещество.
Т.пл. 193 - 197oC.
Элементный анализ: C 78.41, H 8.51, N 2.33.
5-2.
Синтез бис-N, N-[17 β -гидроксиэстра-1,3,5(10)триен-3- оксиэтил]аминометилентетрациклина (соединение 5-2)
Смесь 5.4 г соединения (5-1), описанного выше, 0.03 г метаформальдегида и 20 мл изопропанола перемешивают 2 часа при 40oC. После добавления 3.5 г тетрациклина реакционную смесь перемешивают 8 часов. По окончании реакции получено бледно-желтое твердое вещество (соединение 5-2) (R1 = R4 = H, R2 = OH, R3 = CH3), выделенное в тех же условиях, что и в примере 1-3, выход 94%
Т.пл. 171oC (разл.).
Элементный анализ: C 71.02, H 7.02, N 3.98.
Строение соединения представлено следующей молекулярной формулой: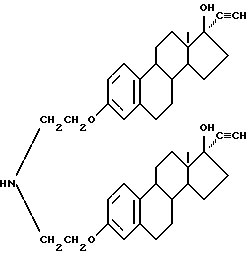
Пример синтеза 6
6-1.
Синтез бис-N, N-[17 β -гидрокси-17 α/ -этинилэстра-1,3,5(10)-триен-3-оксиэтил]амина (соединение 6-1)
В смеси 100 мл тетрагидрофурана и 1 г гидроксида калия растворяют 6.1 г соединения (4-1) примера 4 и через смесь при интенсивном перемешивании при 0oC пропускают газообразный ацетилен до полного завершения реакции. Добавлением кислоты реакционную смесь нейтрализуют до pH 4 и растворитель испаряют. Продукт реакции промывают водой и сушат. Последующей перекристаллизацией из спирта и хлороформа получено белое твердое соединение (6-1), выход 78%.
Т.пл. 201 - 205oC.
Элементный анализ: C 79.21, H 8.58, N 2.18.
Строение соединения соответствует следующей молекулярной формуле: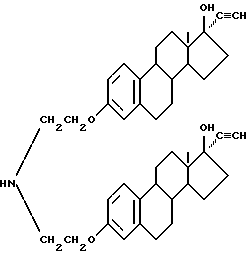
6-2.
Смесь 6.6 г вышеописанного соединения (6-1), и 0.03 г метаформальдегида и 20 мл изопропанола нагревают 2 часа при 60oC. Затем добавляют 3.4 г тетрациклина и реакционную смесь перемешивают 8 часов. По окончании реакции обработкой по методике примера 1-3 с выходом 93% получают бледно-желтое твердое вещество (соединение 6-2), т.е. бис-N,N-[17 β -гидрокси-17 α -этинилэстра-1,3,5(10)триен-3-оксиэтил] аминометилентетрациклин (R1 = R4 = H, R2 = OH, R3 = CH3).
Т.пл. 178oC (разл.).
Элементный анализ: C 72.1, H 7.12, N 3.9.
Строение соединения соответствует следующей молекулярной формуле: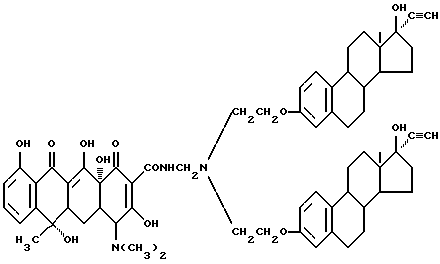
Пример синтеза 7
7-1.
Синтез N-[17-оксиэстра-1,3,5(10)-триен-3-оксиэтил] -N-метиламина (соединение 7-1)
К толуольному раствору 2.7 г эстрона, 1 г хлорэтилметиламина и небольшого количества триэтиланилина добавляют раствор гидроксида натрия. После установления pH 10 реакционную смесь выдерживают 4 часа. Затем растворитель испаряют и перекристаллизацией твердого остатка из спирта получают соединение (7-1, R7 = H), выход 71%.
Т.пл. 262 - 266oC.
Элементный анализ: C 75.24, H 9.41, N 4.28.
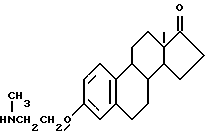
7-2.
Смесь 3.3 г вышеописанного соединения (7-1), 0.03 г метаформальдегида и 20 мл изопропанола нагревают 2 часа при 60oC и затем добавляют 3.5 г тетрациклина. Реакционную смесь перемешивают 8 часов. По окончании реакции обработкой по методике примера 1-3 с выходом 90% получают бледно-желтое твердое вещество (соединение 7-2), т.е. N-[17-оксиэстра-1,3,5(10)-триен-3-оксиэтил] -N-метиламинометилентетрациклин (R1 = R4 = H, R2 = OH, R3 = CH3).
Т.пл. 190oC (разл.).
Элементный анализ: C 68.8, H 7.72, N 3.62.
Строение соединения соответствует следующей молекулярной формуле: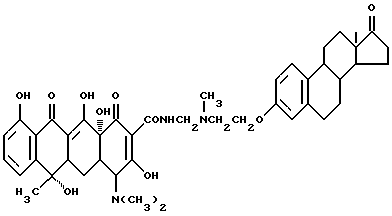
Пример синтеза 8
8-1.
Синтез N-[3,17 β -дигидроксиэстра-1,3,5(10)триен-6- аминоэтил]пиперазина (соединение 8-1)
120 мл тетрагидрофурана растворяют 5.2 г соединения (8-0) и к раствору добавляют 3.2 г аминоэтилпиперазина. Реакционную смесь перемешивают 2 часа, ТГФ удаляют испарением и к остатку добавляют 100 метилового спирта и 2.8 г муравьиной кислоты. Перемешивание реакционной смеси продолжают еще 3 часа. Метиловый спирт удаляют испарением и перекристаллизацией из спирта получают соединение 8-1.
Т.пл. 172 - 177oC.
8-2.
Синтез N-4-(3,17 β -дигидроксиэстра-1,3,5(10)-триен-6- аминоэтил)пиперазин-1-метилентетрациклина (соединение 8-2)
Смесь 4.1 г вышеописанного соединения (8-1), 0.03 г метаформальдегида и 20 мл изопропанола нагревают 2 часа при 50oC. После добавления 3.5 г тетрациклина реакционную смесь перемешивают 5 часов. По окончании реакции продукт реакции фильтруют, промывают изопропанолом и этиловым эфиром и после сушки в вакууме с выходом 81.2% получают бледно-желтое твердое вещество (соединение 8-2) с температурой плавления 167oC (разл.).
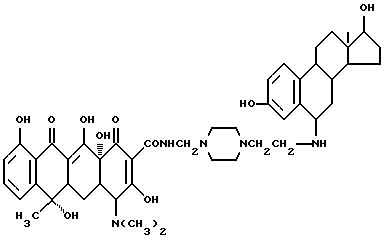
Соединение (8-0) получено следующим образом:
1. Получение 17 β -эстроалкинола
В метиловом спирте растворяют 4 г эстрона и к раствору при 30oC по каплям прибавляют смесь 0.8 г боргидрата калия, 1.76 г гидроксида натрия и 8.8 мл воды. Реакционную смесь выдерживают 2 часа, нейтрализуют до нейтральной реакции разбавленной уксусной кислотой и затем разбавляют водой. Образовавшееся твердое вещество отфильтровывают, промывают водой и сушат. Перекристаллизацией продукта из водного спирта с выходом 97.2% получают белые кристаллы с температурой плавления 173 - 174oC.
2. Получение диацетата 17 β -эстроалкинола
Раствор 10 г 17 β -эстроалкинола в пиритеине смешивают с 35 мл уксусной кислоты. Реакционную смесь перемешивают один час и переносят в ледяную воду с образованием твердого вещества. Образовавшийся осадок фильтруют, сушат и перекристаллизацией продукта реакции из абсолютного спирта с выходом 97% получают белые кристаллы с температурой плавления 126 - 128oC.
3. Получение диацетата 6-карбонил-17 β -эстроалкинола (Соединение (XII))
В бензоле растворяют 5 г диацетата 17 β -эстроалкинола, при охлаждении по каплям прибавляют 0.45 трехокиси хрома и полученную смесь растворяют в 30 мл ледяной уксусной кислоты, 20 мл уксусной кислоты и 30 мл бензола. По окончании реакции смесь перемешивают еще некоторое время и затем переносят в воду. Продукт реакции экстрагируют этиловым эфиром, промывают насыщенным раствором гидрокарбоната натрия и водой, сушат, концентрируют и последующей очисткой на силикагеле получают целевое соединение, выход 40%, температура плавления 173 - 175oC.
Пример синтеза 9
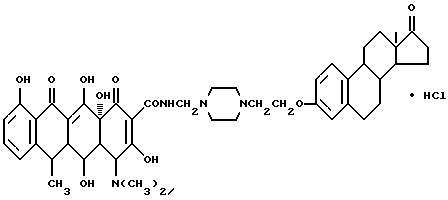
Синтез N-4-[17-оксиэстра-1,3,5(10)-триен-3-оксиэтил] -1-пиперазин-1-метилендоксициклина HCl (соединение 9)
Смесь 2.2 г соединения (1-2), 0.2 г полиформальдегида и 100 мл изопропанола перемешивают 1.5 часа с нагреванием при 60oC, после чего добавляют 3 г гидрохлорида доксициклина. Затем реакционную смесь перемешивают еще 2.5 часа при 60oC. По окончании реакции продукт реакции отфильтровывают, промывают изопропанолом и этиловым эфиром и после сушки получают бледно-желтое вещество (соединение 9) с температурой плавления 172oC (разл.), выход 87%.
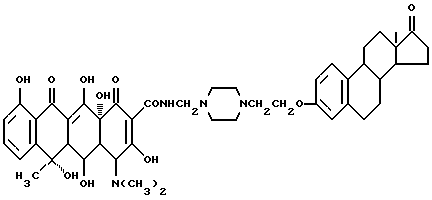
Пример синтеза 10
Синтез N-4-[17-оксиэстра-1,3,5(10)-триен-3-оксиэтил] пиперазин-1- метилен(окситетрациклина) (соединение 10)
Смесь 0.91 г соединения (1-2), 80 мг полиформальдегида и 50 мл изопропанола нагревают с перемешиванием 2 часа при 60oC. После добавления 1 г террамицина реакционную смесь перемешивают еще 3 часа при 60oC. Продукт реакции отфильтровывают, промывают изопропанолом и этиловым эфиром и после сушки с выходом 89% получают бледно-желтое твердое вещество с температурой плавления 175oC (разл.).
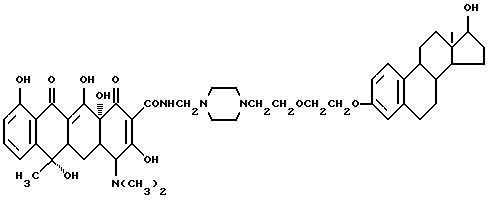
Пример синтеза 11
Синтез N-4-[17-гидроксиэстра-1,3,5(10)-триен-3-этоксиэтил]- пиперазин-1-метилентетрациклина (соединение 11)
Смесь 4.1 г N-(17-гидроксиэстрон-1,3,5(10)-триен-3-оксиэтил)пиперазина, 0.5 г полиформальдегида и 50 мл изопропанола нагревают 2 часа при 60oC с перемешиванием. После добавления 4 г тетрациклина реакционную смесь выдерживают 3 часа при 40 - 45oC. Продукт реакции отфильтровывают и промыванием изопропанолом и этиловым эфиром с выходом 68.6% получают бледно-желтое твердое вещество (соединение 11) с температурой плавления 154oC (разл.).
Пример синтеза 12
Синтез 17-гидроксиандрост-4-ен-3-оксиэтилметилентетрациклина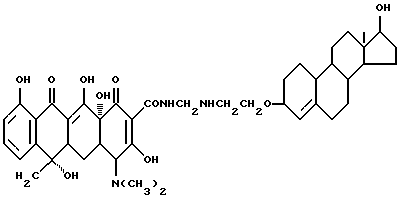
Смесь 3.3 г 3-аминоэтокси-17-гидроксиандрост-4-ена, 0.3 г метаформальдегида и 40 мл изопропанола нагревают 4 часа при 60oC. После добавления 4.5 г тетрациклина реакционную смесь перемешивают 5 часов. Продукт реакции отфильтровывают и после промывания изопропанолом и этиловым эфиром с выходом 92% получают желтое твердое вещество.
Элементный анализ: C 66.78, H 7.41, N 5.38.
Пример синтеза 13
Синтез 17-гидроксиандрост-4-ен-3-он-6-метилен(оксиэтиламинометилен) тетрациклина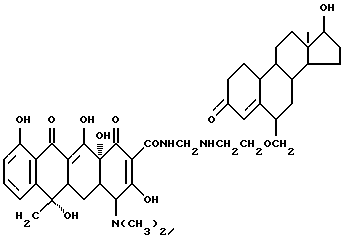
Смесь 3.6 г 6-аминоэтоксиметилен-17-гидроксиандрост-4-ен-3-она, 0.3 г метаформальдегида, 4.5 г тетрациклина и 30 мл ацетона перемешивают 24 часа в темноте при комнатной температуре. По окончании реакции продукт реакции отфильтровывают и после промывания ацетоном и этиловым эфиром с выходом 86% получают желтое твердое вещество.
Элементный анализ: C 67.9, H 7.67, N 5.18.
Пример синтеза 14
Синтез 17-гидроксиандростан-3-(оксиэтиламинометилен)тетрациклина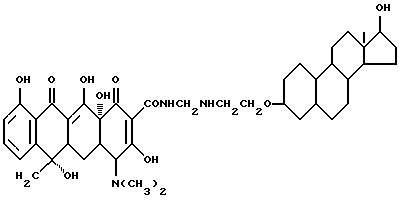
Смесь 3.35 г 3-аминоэтокси-17-гидроксиандростана, 0.3 г метаформальдегида, 4.5 г тетрациклина и 30 мл ацетона перемешивают 30 часов в темноте при комнатной температуре. По окончании реакции продукт реакции отфильтровывают и после промывания ацетоном и этиловым эфиром с выходом 85% получают желтое твердое вещество.
Элементный анализ: C 66.61, H 7.69, N 5.4.
Пример синтеза 15
Синтез 17 β -гидрокси-18-метил-19-норандрост-4-ен-3-он-17 α -(бутиниленаминометилен)тетрациклина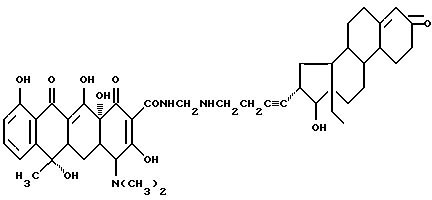
Смесь 3.7 г 17 α -аминоэтилэтинил-17 β -гидрокси-18-метил-19-норандрост-4-ен-3-она, 0.3 г метаформальдегида и 30 мл изопропанола нагревают 4 часа при 60oC. После добавления 4.5 г тетрациклина реакционную смесь перемешивают 6 часов. По окончании реакции продукт реакции отфильтровывают и после промывания изопропанолом и этиловым эфиром с выходом 85% получают желтое твердое вещество.
Элементный анализ: C 68.51, H 7.11, N 5.17.
Пример синтеза 16
Синтез 16 α , 17 β -дигидроксиэстра-1,3,5(10)-триен-3-(оксиэтиламинометилен)тетрациклина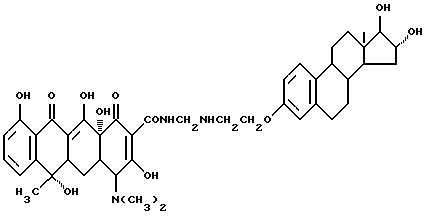
Смесь 3.3 г 3-аминоэтокси-16 α , 17 β/ -дигидроксиэстра-1,3,5(10)-триена, 0.3 г метаформальдегида и 50 мл изопропанола нагревают 2 часа при 80oC и затем охлаждают до 40oC. После добавления 4.5 г тетрациклина реакционную смесь выдерживают 6 часов. По окончании реакции продукт реакции отфильтровывают и после промывания изопропанолом и этиловым эфиром с выходом 85% получают желтое твердое вещество.
Элементный анализ: C 65.48, H 6.82, N 5.13.
Пример синтеза 17
Синтез 18-метил-17-оксиэстра-1,3,5(10)-триен-3-(оксиэтиламинометилен)тетрациклина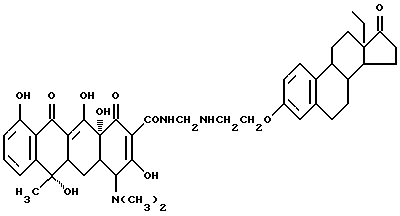
Смесь 3.3 г 3-аминоэтокси-18-метилэстра-1,3,5(10)-триена, 0.3 г метаформальдегида и 50 мл изопропанола выдерживают 48 часов в темноте при 30oC. По окончании реакции продукт реакции отфильтровывают и после промывания изопропанолом и этиловым эфиром с выходом 82% получают желтое твердое вещество.
Элементный анализ: C 67.45, H 6.86, N 5.27.
Пример синтеза 18
Синтез 17 α -гидроксипрегна-4-ен-20-он-3-(оксиэтиламинометилен) тетрациклина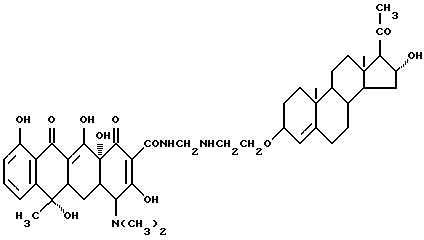
Смесь 3.8 г 3-аминоэтокси-17 α -гидроксипрегн-4-ен-20-она, 0.3 г метаформальдегида и 50 мл изопропанола нагревают 2 часа при 60oC. Реакционную смесь охлаждают до 40oC и добавляют 4.5 г тетрациклина. Затем реакционную смесь выдерживают 5 часов при 60oC. По окончании реакции продукт реакции отфильтровывают и после промывания ацетоном и этиловым эфиром с выходом 87% получают желтое твердое вещество.
Элементный анализ: C 66.52, H 7.37, N 5.01.
Пример синтеза 19
Синтез прегна-5-ен-20-он-3-(оксиэтиламинометилен)тетрациклина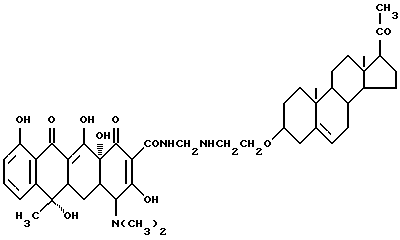
Смесь 3.6 г 3-аминоэтоксипрегна-5-ен-20-она, 0.6 г метаформальдегида и 40 мл изопропанола нагревают 2 часа при 80oC. Реакционную смесь охлаждают до 40oC, добавляют 4.5 г тетрациклина и выдерживают 6 часов при 40oC. По окончании реакции продукт реакции отфильтровывают и после промывания изопропанолом и этиловым эфиром с выходом 93% получают твердое вещество.
Элементный анализ: C 67.7, H 7.36, N 5.05.
Пример синтеза 20
17-Гидроксиандрост-1,4-диен-3-(оксиэтиламинометилен)доксициклин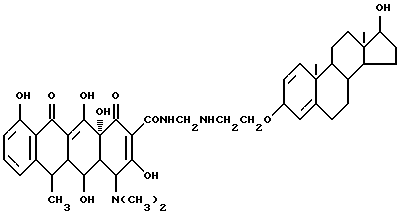
Смесь 3.3 г 3-аминоэтокси-17 β -гидроксиандрост-1,4-диена, 0.3 г метаформальдегида и 50 мл изопропанола нагревают 2 часа при 80oC. Реакционную смесь охлаждают до 40oC, добавляют 4.5 г гидрохлорида доксициклина и выдерживают 4 часа. По окончании реакции промыванием продукта реакции изопропанолом и этиловым эфиром с выходом 89% получают желтое твердое вещество.
Элементный анализ: C 67.23, H 7.25, N 5.28.
Пример синтеза 21
Синтез 17 α -метил-17 β -гидроксиандрост-4-ен-3-он-6-(метиленоксиэтиламинометилен)доксициклина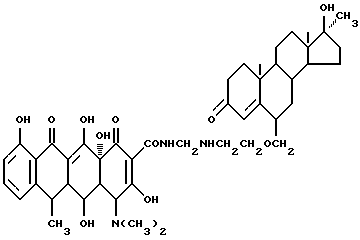
Смесь 3.8 г 6-аминоэтоксиметилен-17 α -метил-17 β -гидроксиандрост-4-ен-3-она, 0.3 г метаформальдегида и 25 мл изопропанола нагревают 4 часа при 60oC. Затем реакционную смесь охлаждают до 40oC, добавляют 4.5 г гидрохлорида доксициклина и выдерживают 8 часов. По окончании реакции промыванием продукта реакции изопропанолом и этиловым эфиром с выходом 86% получают желтое твердое вещество.
Элементный анализ: C 63.72, H 7.02, N 5.13.
Пример синтеза 22
Синтез 17 α -метил-17 β -гидроксиандрост-3-он-2-(оксиэтиламинометилен)доксициклина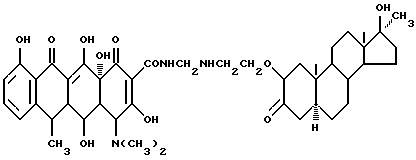
Смесь 3.6 г 2-аминоэтокси-17 α -метил-17 β -гидроксиандростан-3-она, 0.3 г метаформальдегида и 30 мл изопропанола нагревают 2 часа при 60oC, а затем добавляют 4.5 г гидрохлорида доксициклина и реакционную смесь дополнительно выдерживают. По окончании реакции промыванием продукта реакции изопропанолом и этиловым эфиром с выходом 91% получают желтое твердое вещество.
Элементный анализ: C 65.91, H 7.51, N 5.07.
Пример синтеза 23
Синтез 17 α -метил-17  -гидрокси-19-норандрост-4-ен-3-он-6-(метиленоксиэтиламино)доксициклина
-гидрокси-19-норандрост-4-ен-3-он-6-(метиленоксиэтиламино)доксициклина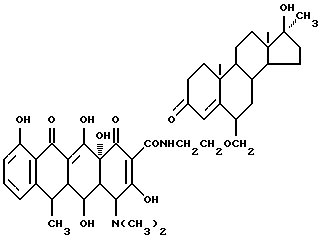
Смесь 3.7 г 6-аминоэтоксиметилен-17 α -метил-17 β -гидрокси-19-норандрост-4-ен-3-она, 0,3 г метаформальдегида и 50 мл ацетона выдерживают 2 часа при 30oC, затем добавляют 4.5 г гидрохлорида доксициклина и реакционную смесь выдерживают еще 30 часов. По окончании реакции продукт реакции фильтруют и промыванием изопропанолом и этиловым эфиром с выходом 87% и получают желтое твердое вещество.
Элементный анализ: C 63.64, H 7.03, N 5.18.
Пример синтеза 24
Синтез 17 α -этинил-17 β -гидроксиандрост-5(10)-ен-3-он-6-(метиленоксиэтиламинометилен)доксициклина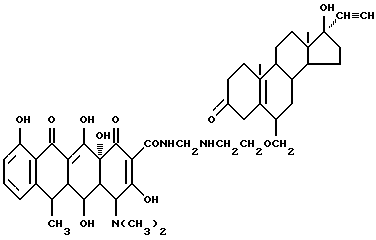
Смесь 3.7 г 6-аминоэтоксиметилен-17 α -этинил-17 β -гидроксиандрост-5(10)-ен-3-она, 0.3 г метаформальдегида и 50 мл изопропанола нагревают 2 часа при 60oC, затем добавляют 4.5 г гидрохлорида доксициклина и реакционную смесь нагревают еще 8 часов при 40oC. По окончании реакции продукт реакции фильтруют и промыванием изопропанолом и этиловым эфиром с выходом 87% получают желтое твердое вещество.
Элементный анализ: C 66.81, H 7.06, N 5.01.
Пример синтеза 25
17 α -Пропилен-17 β -гидрокси-11-диметиламинофениландрост- 4,9-диен-3-он-6-(метиленоксиэтиламинометилен)доксициклин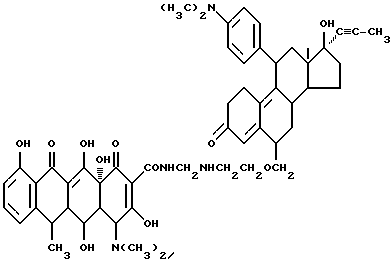
Смесь 5 г 6-аминоэтоксиметилен-11-(4'-диметиламинофенил)-17 α -пропилен-17 β -гидроксиандрост-4,9-диен-3-она, 0.3 г метаформальдегида и 40 мл изопропанола нагревают 2 часа при 80oC. После охлаждения реакционной смеси до 40oC добавляют 4.5 г гидрохлорида доксициклина и реакционную смесь выдерживают 6 часов. По окончании реакции продукт реакции фильтруют и после промывания изопропанолом и этиловым эфиром с выходом 90% получают желтое твердое вещество.
Элементный анализ: C 68.76, H 6.88, N 5.72.
Пример синтеза 26
Синтез 16,17-изопропилиден-16,17-диоксиэстра-1,3,5(10)- триен-3-(оксиэтиламинометилен)доксициклина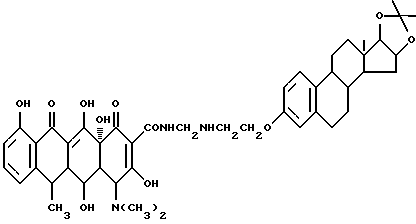
Смесь 3.7 г 16,17-изопропилиден-16,17-диоксиэстра-1,3,5(10)-триен-3-аминоэтилового простого эфира, 0.3 г метаформальдегида и 50 мл изопропанола нагревают 2 часа при 60oC, затем добавляют 4.5 г гидрохлорида доксициклина и реакционную смесь дополнительно выдерживают 8 часов. По окончании реакции продукт реакции фильтруют и после промывания изопропанолом и этиловым эфиром с выходом 95% получают желтое твердое вещество.
Элементный анализ: C 66.71, H 6.9, N 5.18.
Пример синтеза 27
Синтез 3,17-дигидроксиэстра-1,3,5(10)-триен-17-ацетат- 7-(метиленоксиэтиламинометилен)доксициклина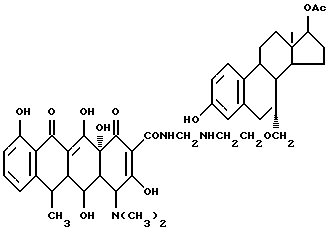
Смесь 3.8 г 7-аминоэтилоксиметиленэстра-3,17-диен-17-ацетата, 0.3 г метаформальдегида и 50 мл изопропанола нагревают 4 часа при 60oC, затем добавляют 4.5 г гидрохлорида доксициклина и реакционную смесь дополнительно выдерживают 4 часа. По окончании реакции продукт реакции фильтруют и после промывания изопропанолом с выходом 88% получают желтое твердое вещество.
Элементный анализ: C 65.34, H 6.68, N 4.95.
Пример синтеза 28
Синтез 17-гидроксипрегна-4-ен-20-он-3-(оксиэтиламинометилен)доксициклина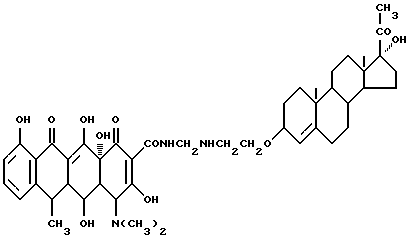
Смесь 3.7 г 3-аминоэтокси-17  -гидроксипрегна-4-ен-20-она, 0.3 г метаформальдегида и 40 мл изопропанола нагревают 2 часа при 60oC, затем добавляют 4.5 г гидрохлорида доксициклина и реакционную смесь дополнительно выдерживают 4 часа. По окончании реакции продукт реакции фильтруют и после промывания изопропанолом и этиловым эфиром с выходом 92% получают желтое твердое вещество.
-гидроксипрегна-4-ен-20-она, 0.3 г метаформальдегида и 40 мл изопропанола нагревают 2 часа при 60oC, затем добавляют 4.5 г гидрохлорида доксициклина и реакционную смесь дополнительно выдерживают 4 часа. По окончании реакции продукт реакции фильтруют и после промывания изопропанолом и этиловым эфиром с выходом 92% получают желтое твердое вещество.
Элементный анализ: C 66.41, H 7.4, N 5.14.
Пример синтеза 29
Синтез прегна-4-ен-3,20-дион-6-(метиленоксиэтиламинометилен)доксициклина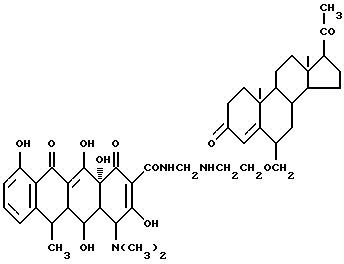
Смесь 3.9 г 6-аминоэтоксиметиленпрегна-4-ен-3,20-диона, 0.3 г метаформальдегида и 50 мл изопропанола нагревают 3 часа при 60oC, затем добавляют 4.5 г гидрохлорида доксициклина и реакционную смесь дополнительно выдерживают 4 часа. По окончании реакции продукт реакции фильтруют и после промывания изопропанолом и этиловым эфиром с выходом 93% получают желтое твердое вещество.
Элементный анализ: C 67.77, H 7.36, N 4.59.
Пример синтеза 30
Синтез 3,17-дигидроксиэстра-1,3,5(10)-триен-11-(4- феноксиэтиламино)метилендоксициклина \\6 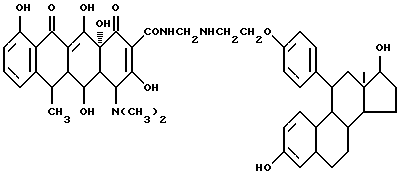
Смесь 4 г 11-(4'-аминоэтоксифенил)-3,17-дигидроксиэстра- 1,3,5(10)-триена, 0.3 г метаформальдегида и 50 мл изопропанола нагревают 2 часа при 60oC, затем добавляют 4.5 г гидрохлорида доксициклина и реакционную смесь дополнительно выдерживают 8 часов. По окончании реакции продукт реакции отфильтровывают и после промывания изопропанолом и этиловым эфиром с выходом 89% получают желтое твердое вещество.
Элементный анализ: C 68.13, H 6.69, N 4.73.
Пример синтеза 31
Синтез 17 β -гидрокси-17 α -метиландростано-(3,2-c)пиразол-N-метилентетрациклина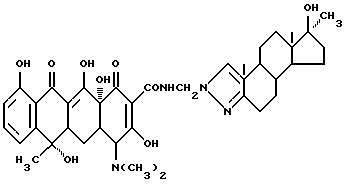
Смесь 3.29 г 17 β -гидрокси-17 α - метиландростано(3,2-c)пиразола, 0.3 г метаформальдегида и 30 мл изопропанола нагревают 2 часа при 40oC, добавляют 4.5 г тетрациклина и реакционную смесь перемешивают 6 часов. По окончании реакции продукт реакции отфильтровывают и после промывания изопропанолом и этиловым эфиром с выходом 89% получают желтое твердое вещество.
Элементный анализ: C 67.48, H 7.07, N 7.30.
Опыт 1.
Распределение соединения в организме
В соединение 1-3, полученное в примере синтеза 1-3, вводят радиоактивную метку 3H (0.34 мCi/мг) и соединение инъектируют в вену хвоста мыши в дозе 20 μ Ci/20 г. Мышей в группах по пять мышей в каждой умерщвляют через 1 минуту, 5 минут, 15 минут, 30 минут, 1 час, 4 часа, 6 часов, 24 часа, 48 часов и 72 часа после инъекции и из глазницы каждой мыши отбирают 50 мкл крови. Кроме того, у каждой мыши удаляют сердце, матку, малый кишечник, кости (бедренная кость) и т.д. и 50 мг ткани (50 мкл крови) помещают в пластиковые пробирки для анализа. В пробирку кроме того помещают 0.2 мл перхлорной кислоты, 0.4 мл перекиси водорода и каплю н-октилового спирта и нагревают на водяной бане 45 минут при 75oC. Затем отбирают 0.1 мл этой обработанной жидкости и помещают в колбу для хранения сцинцилляционного раствора, где смешивают с 5 мл 0.5% сцинцилляционного раствора. После того, как раствор становится прозрачным, его для определения cpm в образце и его радиоактивности помещают в FJ 2105 жидкостной сцинцилляционный счетчик. Отдельно определяют dpm 52 образцов, полученных в четырех группах животных (метод внешнего стандарта).
Количество лекарственного средства внутри ткани определяют следующим образом.
Количество лекарственного средства в ткани (cpm/мг) = (cpm образца) + (количество обработанной ткани (мг)).
Количество лекарственного средства в ткани (мкг/мг) = (cpm/мг образца • 6) + (эффективность счетчика (E) • 2.22 • 10 • 0.34 mCi/мг (удельная радиоактивность)).
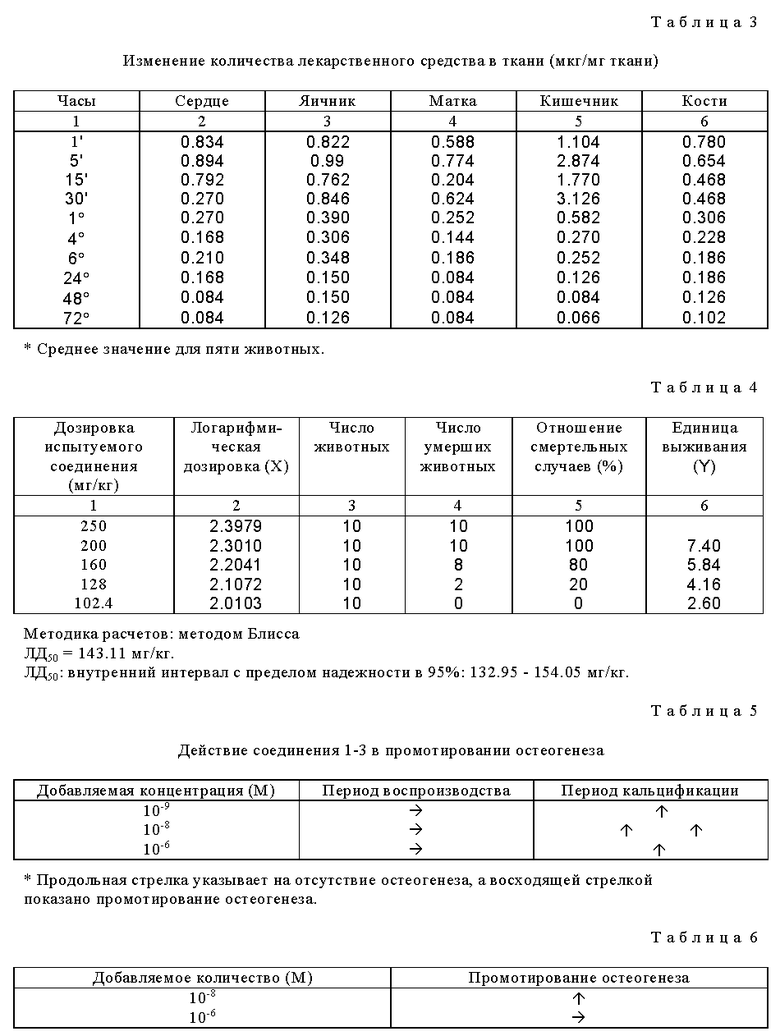
Получены результаты, представленные в табл. 1-4 (см. в конце описания).
Опыт 2.
Анализ на острую токсичность
(1) Образец
Соединение 1-3 представляет собой бледно-желтый кристаллический порошок, серийный номер соединения 930 113. Раствор соединения в концентрации 50 мг/мл имеет pH 5 и представляет собой прозрачную бледно-желтую жидкость. Жидкость может быть получена в лаборатории исследования остеопороза, кафедра фармации, Уэст Чайна Юниверсити Медикал Сайэнс Скул (УЧЮМСС).
(2) Животные
В экспериментах используются здоровые мыши породы Канминг, первого класса, весом от 18 до 21 г, половина самцов и половина самок. Мыши выращиваются Экспериментальным центром при УЧЮМСС для выращивания животных.
(3) Определение полулетальной дозы (ЛД50)
Подготовлено четыре-пять групп мышей, получающих дозу в равных отношениях (1 : 0.7 - 0.8) в пределах интервала ЛД 0 - 100%, выявленного в предварительных испытаниях. Лекарственное средство для перорального приема готовят суспендированием твердого соединения 1-3 в 1% CMCNa2 с образованием суспензии. Для инъекций растворы лекарственного средства приготовлены методом разбавления с низким удельным весом путем растворения соединения 1-3 в физиологическом солевом растворе и получения растворов с различной концентрацией. Животных подвергают голоданию (но при свободном доступе к воде) и через 20 часов наугад независимо от пола и веса набирают группы по десять мышей. Лекарственное средство дают один раз в день в дозе 0.2 мл/10 г и затем животных осматривают. Мертвых животных подвергают вскрытию и любые патологические изменения отмечают невооруженным глазом.
(4) Результаты испытаний
(a) Определение на мышах максимально терпимой дозы соединения 1-3.
Предварительными испытаниями выявлена максимально терпимая доза, не ведущая к смерти. После того, как 20 мышей (10 самцов и 10 самок) получают одной дозой химическое соединение в максимальной концентрации и в максимально возможном для перорального введения количестве, животных наблюдают в течение 7 дней. В результате никаких нарушений у мышей не отмечено, как не отмечено смертельных случаев. Максимально терпимая доза (МТД) составляет >6 г/кг.
(b) После инъектирования соединения 1-3 в вену хвоста мышей получены результаты, представленные в табл. 4 (см. в конце описания).
(5) Выводы
Максимально терпимая доза (МТД) соединения 1-3 при разовом введении мыши составляет, по меньшей мере, 6 г/кг, и токсичность соединения чрезвычайно мала. Значение ЛД50 при внутривенной инъекции в вену хвоста мыши составляет 143.11 мг/кг. После внутривенной инъекции активность мыши падает, затем мышь начинает прыгать, наблюдаются спазмы. Глазное яблоко выступает из глазницы и белеет, наблюдается недержание мочи и кала. Хотя основная часть отравленных мышей погибает мгновенно, та небольшая часть выживших мышей все равно погибает в течение 24 часов. Мыши, выжившие в течение 24 часов, не погибают и в течение последующих 7 дней. Никакой зависимости смертельных случаев от пола животных не отмечено, как не отмечено никаких патологических изменений после вскрытия при наблюдении невооруженным глазом. Испытания проводились при температуре 17oC.
Опыт 3.
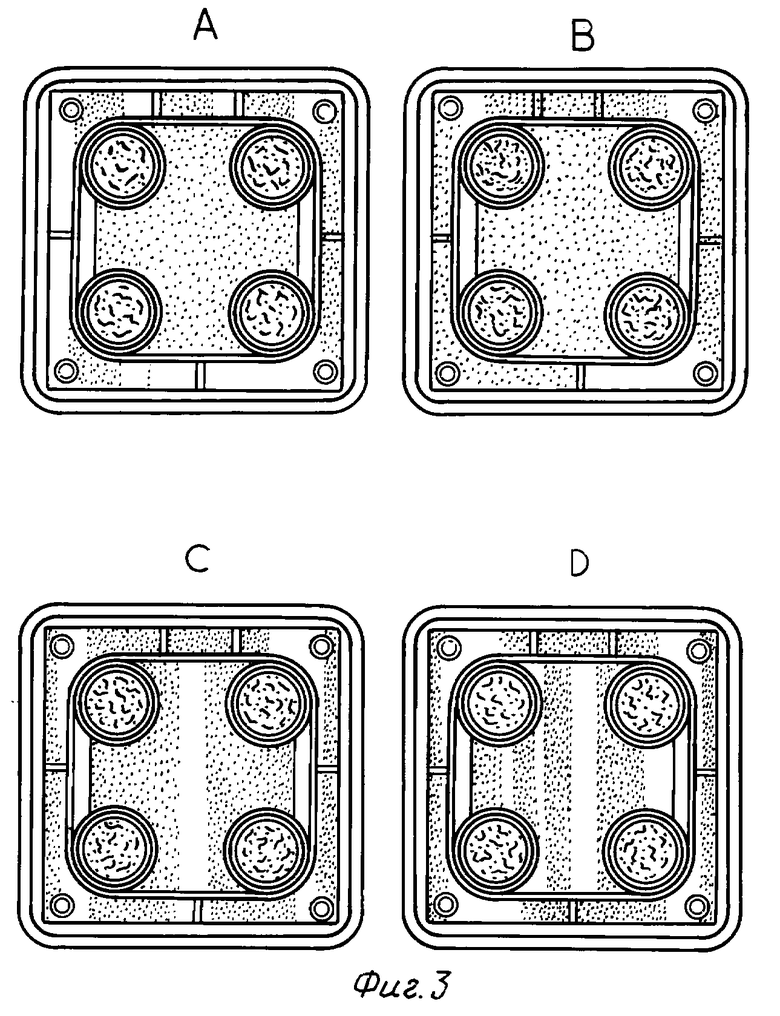
Исследование остеогенеза (1)
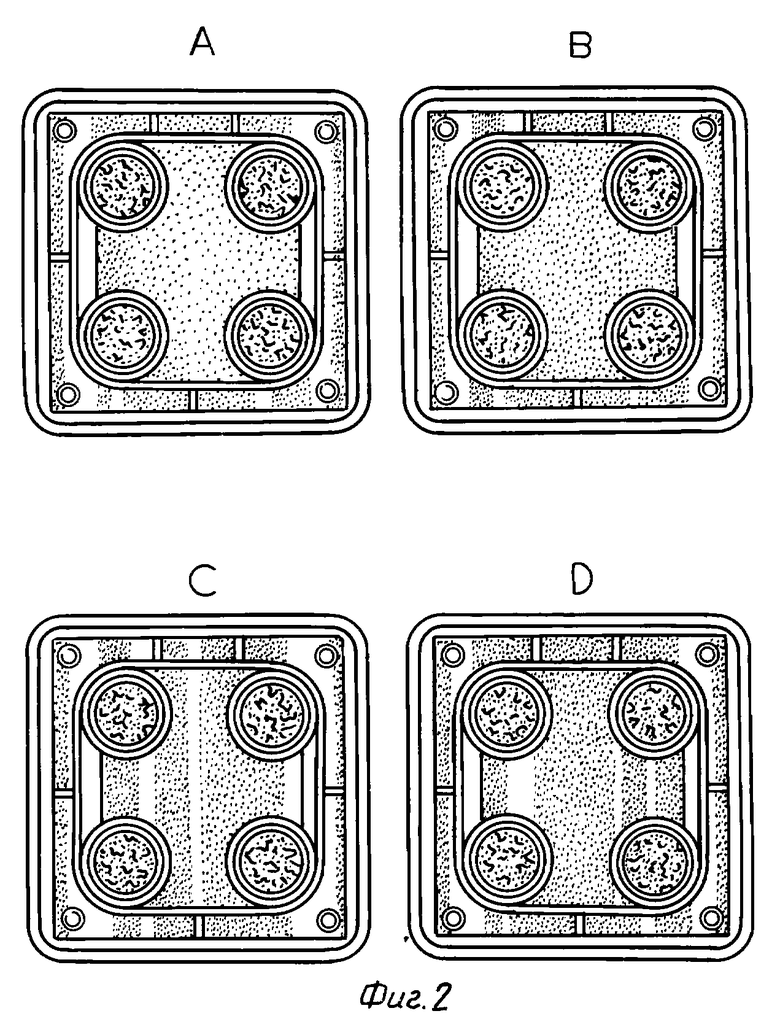
В качестве испытуемых клеток применяется инкубационная система первого поколения остеобластов, происходящих из свода черепа крыс Уистер (самки, возраст 6 месяцев). После начала инкубирования в среду один раз в день добавляют образцы лекарственного средства в дозе 10-6 М, 10-8 М или 10-9 М на второй и третий день (период воспроизводства). Или же соединение 1-3 в вышеуказанном количестве добавляют в среду один раз в день в течение 4 дней (период кальцификации), начиная с седьмого дня от начала инкубирования. На четырнадцатый день от начала инкубирования клетки окрашивают по фон Коссу и проводят определение фосфатов. Области костных узлов, окрашенные в коричневый цвет, видны невооруженным глазом, и используются в качестве показателя остеогенеза. Получены результаты, представленные в табл. 5 (см. в конце описания).
На фиг. 1 - 3 показаны результаты, полученные при трехкратном повторении одной и той же вышеприведенной методики. На фотографиях буквой A указано отсутствие лекарственного средства, буква B указывает на добавление соединения в концентрации 10-9 М, буква C - в концентрации 10-8 М и буква D - в концентрации 10-6 М.
Из вышеприведенных результатов очевидно, что соединение настоящего изобретения обладает промотирующим остеогенез действием.
Опыт 4.
Исследование остеогенеза (2)
В качестве испытуемых клеток использована инкубационная система первого поколения клеток костного мозга из бедренной кости крыс Уистер (самки, возраст 6 месяцев), и соединение 1-3 добавляли один раз в день в количестве 10-8 М или 10-6 М к среде на седьмой, девятый и одиннадцатый день от начала инкубирования (период кальцификации). Исследования проводят по методике опыта 3. Полученные результаты приведены в табл. 6 (см. в конце описания).
Как следует из приведенных в таблице результатов, соединение настоящего изобретения обладает промотирующим остеогенез действием.




Описываются новые производные тетрациклина общей формулы I, где значения R1, R2, R3, R4 указаны в п.1 формулы изобретения. Соединения способны концентрироваться в костной ткани и обладают ингибирующим резорбцию и промотирующим окостенение действием. 7 з.п. ф-лы., 3 ил., 7 табл.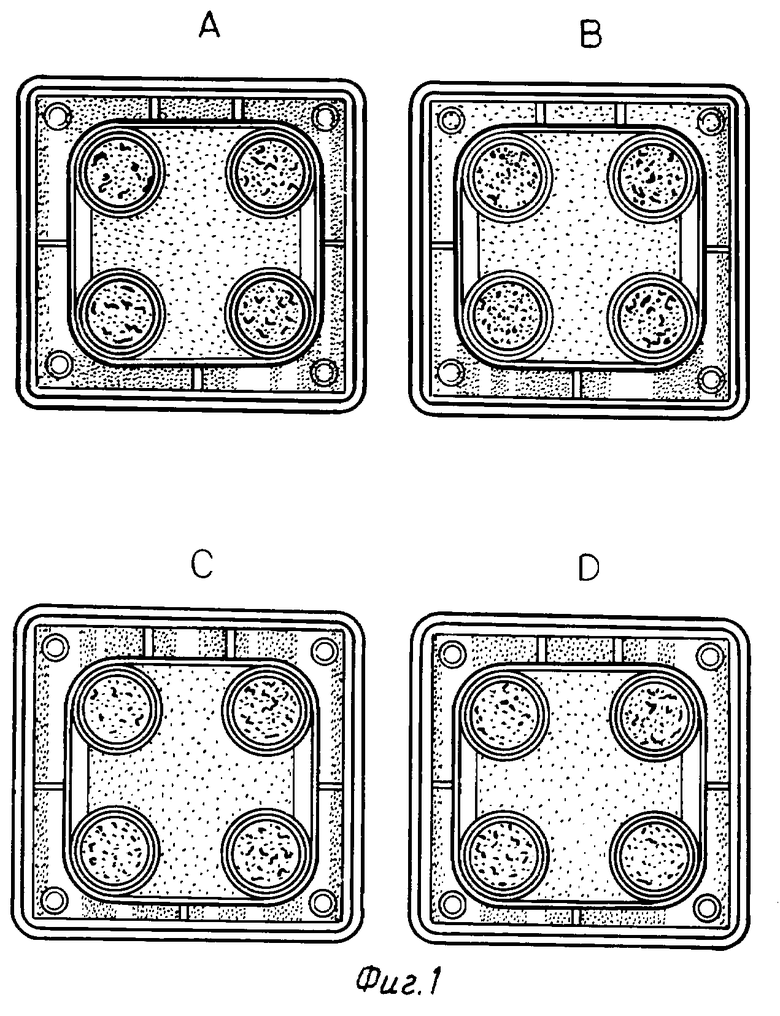
X-Y-Z,
где X представляет одновалентную группу общей формулы II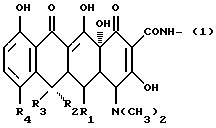
где R1 представляет водород или гидроксил;
R2 представляет водород или гидроксил;
R3 представляет метил;
R4 представляет водород;
Y представляет двухвалентную или трехвалентную группу общей формулы III, IV, V или VI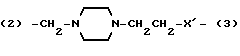
где Х' представляет собой простую связь, -O - или NН;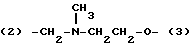
(2)-(CH2NH)mCH2CH2-W- (3)
где m равно 0 или 1;
W представляет -О-, -ОСН2-, простую связь или -C≡C-; причем, когда m равно 0, W представляет -OCH2-;
Z представляет одновалентную группу, образованную удалением атома водорода или гидроксильной группы в соединении общей формулы VII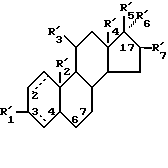
где  представляет НО- или O=;
представляет НО- или O=;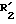 представляет атом водорода или метил;
представляет атом водорода или метил;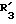 представляет атом водорода, фенил или замещенный фенил;
представляет атом водорода, фенил или замещенный фенил;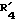 представляет метил или этил;
представляет метил или этил;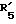 представляет гидроксил, кетогруппу или ацетил;
представляет гидроксил, кетогруппу или ацетил;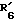 представляет водород, гидроксил, метил, этинил или пропинил или
представляет водород, гидроксил, метил, этинил или пропинил или 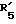 и
и 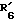 совместно образуют группу =O;
совместно образуют группу =O;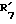 представляет водород, гидроксил, или
представляет водород, гидроксил, или 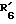 и
и 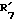 вместе соединяются с атомом кислорода 2,2-диоксипропильной группы;
вместе соединяются с атомом кислорода 2,2-диоксипропильной группы;
символ 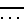 обозначает простую или двойную связь,
обозначает простую или двойную связь,
причем группы формул III - IV присоединяются во 2 положение, 3 положение, 6 положение, 7 положение или 17 положение, или по фенильной группе, присоединенной в II положении группы Z, причем (1) формулы II и (2) формул III - VI связаны непосpeдcтвeннo и (3) группы формул III-VI связаны непосредственно c любой группой формулы VII.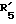 и
и 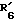 совместно образуют группу =O и
совместно образуют группу =O и 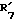 представляет водород.
представляет водород.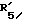 представляет гидроксил,
представляет гидроксил, 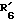 представляет водород и
представляет водород и 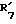 представляет водород.
представляет водород. представляет гидроксил,
представляет гидроксил, 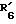 представляет этинил,
представляет этинил, 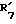 представляет водород.
представляет водород.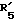 представляет гидроксил,
представляет гидроксил, 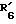 представляет водород и
представляет водород и 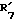 представляет гидроксил.
представляет гидроксил.
Приоритет по пунктам:
25.03.93 по пп. 1-8;
29.12.93 по пп. 1-8 уточнение признаков.
| Устройство для охлаждения водою паров жидкостей, кипящих выше воды, в применении к разделению смесей жидкостей при перегонке с дефлегматором | 1915 |
|
SU59A1 |
| Тринус Ф.П | |||
| Фармако-терапевтический справочник | |||
| - Киев, Здоровья, 1989, с | |||
| Одновальный, снабженный дробителем, торфяной пресс | 1919 |
|
SU261A1 |
