Настоящее изобретение относится к новому соединению, в частности, к 1-метиловому эфиру N[N-(3,3-диметилбутил)-L-α- аспартил]-L-гексагидрофенилаланина, которое может быть использовано в качестве подслащивающего агента, а также к способу его получения.
Это новое соединение в особенности применимо для подслащивания различных продуктов, в частности напитков, пищевых продуктов, кондитерских изделий, жевательной резинки, предметов гигиены и туалета, а также косметических, фармацевтических и ветеринарных продуктов.
Известно, что для того, чтобы подслащивающий агент можно было бы использовать в промышленном масштабе, он должен обладать, во-первых, высоким подслащивающим потенциалом при низкой стоимости его использования, и, во-вторых, удовлетворительной стабильностью, то есть стабильностью, совместимой с условиями применения.
Из множества имеющихся на рынке подслащивающих агентов в настоящее время наиболее широко используется дипептидное производное-1-метиловый эфир N-L- -α- аспартил-L-фенилаланина (Пат. США 3492131), известный как аспартам и имеющий следующую формулу:
Подслащивающая способность аспартама относительно низка, так как она в 200 раз ниже, чем у сахарозы в расчете на вес. Несмотря на свои прекрасные органолептические свойства, основные недостатки этого соединения, которыми являются дороговизна вследствие относительно низкой подслащивающей способности и достаточно низкая стабильность в некоторых условиях использования подслащивающих агентов, особенно в нейтральной среде, ограничивают область его промышленного применения.
Поэтому пищевая промышленность испытывает явную необходимость в новом подслащивающем агенте, который имеет высокую подслащивающую способность для уменьшения стоимости и который по меньшей мере также стабилен как аспартам и даже более стабилен, чем аспартам, особенно в нейтральной среде.
Предметом настоящего изобретения является новый подслащивающий агент 1-метиловый эфир N-[N-(3,3-диметилбутил)-L- -α- аспартил]-L- гексагидрофенилаланина, имеющий формулу:
Это новое подслащивающее соединение, следовательно, представляет собой N-алкилированное производное гексагидрированного аспартама (гексагидроаспартама).
Гексагидроаспартам формулы
уже описан в литературе, в частности во Фр. патенте 2013158. Его подслащивающая способность аналогична аспартаму: он слаще сахарозы приблизительно в 225 раз из расчета на вес.
Совершенно неожиданно было обнаружено, что соединение настоящего изобретения приблизительно в 50 (пятьдесят) раз слаще аспартама и в 12000 (двенадцать тысяч) раз слаще сахарозы (столового сахара) из расчета на вес.
Более того, как было найдено, стабильность соединения настоящего изобретения на много выше таковой аспартама в обычных условиях, используемых при приготовлении пищевых продуктов. Это преимущество является наиболее важным, поскольку ограничения использования аспартама в некоторых пищевых продуктах обусловлены его очень низкой стабильностью в средах, близких к нейтральной, то есть для величин pH приблизительно 7, величин, которые часто встречаются в продуктах, таких как молочные продукты, кондитерские изделия или другие продукты, требующие при приготовлении высокой температуры, жевательные резинки и зубные пасты.
Соединение настоящего изобретения, хотя и получено непосредственно из аспартама, обладает тем преимуществом, что не содержит фрагмент L- фенилаланина, поскольку фенильная группа этой аминокислоты восстановлена в циклогексильную в соединении настоящего изобретения. Более того, благодаря своей очень высокой подслащивающей способности соединение настоящего изобретения будет использоваться в пищевых продуктах в концентрации в 60 раз ниже таковой для аспартама. Следовательно, часто обсуждаемое присутствие в пищевых продуктах некоторых составляющих аспартама, а именно присутствие метанола или L-фенилаланина, будет соответственно весьма существенно снижено или подавлено за счет использования соединения настоящего изобретения.
Целью настоящего изобретения является соединение настоящего изобретения в качестве подслащивающего агента, а также подслащивающие композиции, включающие соединение настоящего изобретения, также как и использование данного соединения для подслащивания различных продуктов, указанных в вводной части.
Подслащивающий агент настоящего изобретения может быть добавлен в любой пищевой продукт, которому требуется придание сладкого вкуса, при условии, что он добавляется в достаточных пропорциях для достижения требуемого уровня сладости. Оптимальная концентрация использования подслащивающего агента зависит от многих факторов, таких, например, как условия хранения и использования продуктов, особые составляющие продуктов и требуемый уровень сладости. Любой квалифицированный специалист может легко определить оптимальную пропорцию подслащивающего агента, которая должна быть использована для получения пищевого продукта, с помощью обычных чувствительных анализов. Подслащивающий агент настоящего изобретения в общем случае добавляется в пищевой продукт в пропорции от 0.5 до 50 мг подслащивающего агента на килограмм или литр пищевого продукта. Очевидно, что концентраты продуктов будут содержать большие количества подслащивающего агента, а затем перед использованием разбавляются.
Подслащивающий агент настоящего изобретения может быть добавлен в подслащиваемые продукты в чистом виде, но вследствие своей высокой подслащивающей способности он, как правило, смешивается с подходящим носителем или разбавителем.
Подходящие носители или разбавители предпочтительно выбираются из группы, включающей полидекстрозу, крахмал, мальтодекстрины, целлюлозу, метилцеллюлозу, карбоксиметилцеллюлозу и другие производные целлюлозы, альгинат натрия, пектины, камеди, лактозу, мальтозу, глюкозу, лейцин, глицерин, маннит, сорбит, бикарбонат натрия, фосфорную, лимонную, винную, фумаровую, бензойную, сорбиновую и пропионовую кислоты и их натриевые, калиевые и кальциевые соли, и их эквиваленты.
Подслащивающий агент настоящего изобретения в пищевом продукте может быть использован как единственный подслащивающий агент или в комбинации с другими подслащивающими агентами, такими как сахароза, зерновой сироп, фруктоза, сладкие дипептидные аналоги или производные (аспартам, алитам), неогесперидин дигидрохалькон, гидрированная изомальтулоза, стевиозид, L-сахара, солод, ксилит, сорбит, ацесульфам, сахарин и их натриевые, калиевые, аммониевые и кальциевые соли, цикламовая кислота и ее натриевая, калиевая и кальциевая соли, sucralose, монеллин, тауматин и их эквиваленты.
Второй объект изобретения относится к способу получения указанного выше соединения, заключающийся в том, что раствор аспартама и 3,3-диметилбутиральдегида при комнатной температуре обрабатывают водородом при относительном давлении 3 бар (0.3 МПа) в присутствии платинового катализатора.
В одном из предпочтительных вариантов осуществления этого способа указанный катализатор выбирают из группы, включающей платиновую чернь и оксид платины.
Оценка степени протекания реакции путем отбора пробы и определения количества образующегося продукта с помощью высокоэффективной жидкостной хроматографии (ВЭЖХ) позволяет специалисту легко определить время гидрирования, соответствующее используемым условиям.
В соответствии с одним из предпочтительных способов настоящего изобретения раствор аспартама и 3,3- диметилбутиральдегида представляет собой водно-спиртовой раствор с pH 4.5-5, полученный путем смешения 0.1 М раствора уксусной кислоты и метанола, причем концентрация аспартама в водно-спиртовом растворителе находится между 50 и 60 г/л, а концентрация 3,3-диметилбутиральдегида составляет между 20 и 30 г/л.
В предпочтительном варианте осуществления настоящего изобретения полученный продукт очищают путем осаждения и фильтрации после упаривания в вакууме спиртовой части растворителя.
Соединение настоящего изобретения может также быть получено другими методами. В частности, 1-метиловый эфир N-L -α- аспартил-L-гексагидрофенилаланина может быть подвергнут восстановительному N-алкилированию 3,3-диметилбутиральдегидом под действием цианборгидрида натрия или водорода в присутствии катализатора (см. , например, ohfune et al., Chem. Letters, 1984, pp. 441-444 и обзор R.N. Rylander, "Catalytic Hydrogenation in organic Synthesis", Academic Press, San Diego, 1993, pp. 165-174).
Однако способ настоящего изобретения является более предпочтительным, поскольку позволяет получать новое подслащивающее соединение непосредственно из аспартама в одну единственную стадию. Этот способ в сущности позволяет одновременно проводить восстановительное N-алкилирование 3,3- диметилбутиральдегидом и гидрирование фенильной группы аспартама с образованием циклогексильной группы.
Однако одновременное проведение восстановительного N-алкилирования и гидрирования фенильной группы возможно только с использованием ограниченного числа катализаторов и в очень специфических экспериментальных условиях, что единственно позволяет получать соединение настоящего изобретения высокой аналитической чистоты, необходимой для применения в пищевых продуктах.
Следует отметить, что использование аспартама в качестве исходного вещества при получении соединения настоящего изобретения неизбежно влечет за собой необходимость разрешения некоторых трудностей, связанных со свойствами аспартама.
В сущности, аспартам обладает относительно низкой растворимостью в большинстве органических растворителей, его растворимость в общем случае не превышает нескольких грамм на литр.
С другой стороны, если растворимость аспартама больше в водной среде, его стабильность в то же время в такой среде относительно низкая.
Более того, любые попытки увеличить температуру с целью улучшения растворимости аспартама приводят к усугублению процессов разложения.
Способ настоящего изобретения позволяет использовать аспартам в качестве исходного соединения с точки зрения обеспечения чистоты, необходимой для соединения, предназначенного для использования в продуктах питания.
Как уже отмечалось, качество соединения настоящего изобретения очень сильно зависит от экспериментальных условий, в которых осуществляют способ. Таким образом, тип катализатора, и в меньшей мере время и давление реакции гидрирования, природа реакционной среды и ее pH являются существенными параметрами.
Таким образом, настоящее изобретение описывает новое соединение, которое обладает, по сравнению с аспартамом, в 60 раз большей подслащивающей способностью и большей стабильностью как в нейтральной, так и в кислой среде. Более того, это новое соединение получают непосредственно и в одну единственную стадию из аспартама с высоким выходом и высокой степени чистоты.
Более полно настоящее изобретение иллюстрируется следующими примерами получения, которые не следует рассматривать как ограничивающие объем изобретения.
Пример 1
В реактор, снабженный мешалкой, обеспечивающей очень хороший перенос газообразного водорода в жидкую фазу, при перемешивании последовательно загружают 60 см3 0.1 М водного раствора уксусной кислоты, 1 г платиновой черни (Platinum black, Aldrich N 20,591-5), 2.55 г 3,3- диметилбутиральдегида, 30 см3 метанола и 5 г аспартама.
После продувки реактора азотом, смесь подвергают гидрированию при относительном давлении 3 бар (0.3 МПа) при комнатной температуре. За ходом реакции следят с помощью отбора проб и определения количества образующегося продукта методом высокоэффективной жидкостной хроматографии (ВЭЖХ). Концентрацию целевого продукта определяют с помощью предварительно полученной калибровочной кривой. Через 72 ч гидрирования наблюдается образование 97% целевого продукта.
После этого реакцию останавливают пропусканием тока азота и отделяют катализатор фильтрированием на тонком фильтре (0.5 мкм). При необходимости pH фильтрата доводят до 5 путем добавления нескольких капель 1 М раствора гидрооксида натрия. Затем метанол упаривают в вакууме при температуре не выше 40oC, при этом быстро осаждается белое твердое вещество. После этого смесь перемешивают несколько часов при комнатной температуре для полноты осаждения. Продукт отделяют фильтрацией, сушат и промывают приблизительно 50 см3 гексана. В итоге получают 4.5 г 1-метилового эфира N-[N-(3,3-диметилбутил)-L-α-аспартил] - L-гексагидрофенилаланина (выход 69%) в виде белого порошка высокой степени чистоты (более 98% по данным ВЭЖХ).
Пример 2
Реакцию проводили с использованием того же оборудования, растворителя и тех же реагентов при тех же концентрациях, которые описаны в Примере 1, но с использованием в качестве катализатора 1 г оксида платины (катализатор Адамса, оксид платины (IV), Aldrich N 20, 603-2), гидрирование осуществляют при относительном давлении 3 бар (0.3 МПа), при комнатной температуре, реакцию останавливали через 72 ч (образовалось 96% продукта). После очистки осаждением, как это описано в Примере 1, получали 4.5 г целевого продукта (выход 69%) в виде белого порошка очень высокой степени чистоты (более 98% по данным ВЭЖХ).
Чистоту соединения, полученного в соответствии со способом настоящего изобретения, контролируют классическими методами тонкослойной хроматографии, ИК-спектроскопии, УФ-спектроскопии, высокоэффективной жидкостной хроматографии (ВЭЖХ), с помощью термического анализа, измерением оптического вращения, ядерным магнитным резонансом и элементным анализом.
Физические характеристики полученного способом настоящего изобретения соединения приведены ниже.
Брутто-формула: C20H36NO5.
Молекулярная масса: 384.51.
Содержание воды (метод Карла Фишера): 3-5%.
Тонкослойная хроматография: Силикагель 60 F254 на алюминиевой подложке (Merck N 5554), элюент: бутанол- уксусная кислота-вода (8:2:2), проявление нингидрином: Rf 0.62.
Спектр ИК (KBr), см-1:3514(HOH), 3367, 3195(NH), 2957, 2920, 2850 (CH), 1755 (COOCH3), 1659 (CONH), 1620-1593 (COO), 1450, 1388, 1199, 1177, 773.
УФ-спектр: максимум при 222 нм.
Высокоэффективную жидкостную хроматографию проводят на колонке типа "Lichrospher 100 RP-18 endcapped." фирмы Merck, длиной 244 мм и диаметром 4 мм, элюент: 65 мМ раствор ацетата аммония- ацетонитрил (50:50), расход 1 мл/мин, детектор-рефрактометр, время удержания 4.9 мин.
Дифференциальный термический анализ проводят от 40 до 350oC со скоростью нагрева 10oC/мин, т.пл. 88oC, 200oC разложение отсутствует.
Оптическое вращение: [α]
Элементный анализ: Найдено (вычислено для содержания воды 4.5%), %: С 59.63 (59.78), H 9.15 (9.51), N 6.66 (6.97), O 23.35 (23.72).
Подслащивающая способность соединения настоящего изобретения оценивалась группой из восьми экспертов. Для этого сравнивали по вкусу водные растворы соединения настоящего изобретения различной концентрации с контрольными растворами сахарозы с концентрацией 2%, 5% и 10%. Подслащивающую способность испытываемого соединения сравнивали с сахарозой и затем выражали весовым отношением между соединением и сахарозой при равной степени сладости, то есть когда сладкий вкус раствора испытываемого соединения и контрольного раствора сахарозы, по мнению большинства экспертов, имели одинаковую степень сладости.
Подслащивающая способность соединения настоящего изобретения по весу приблизительно в 12000 раз выше, чем сахарозы, при сравнении с 2%-ным раствором сахарозы, в 10000 раз выше при сравнении с 5%- ным раствором сахарозы и в 5000 раз выше при сравнении с 10%-ным раствором сахарозы.
Стабильность соединения настоящего изобретения и аспартама измеряли путем определения количества продукта (с помощью высокоэффективной жидкостной хроматографии), остающегося после ускорения старения раствора при продолжительном нагревании при 70oC в нейтральной среде (фосфатный буфер с pH 7) или в кислой среде (фосфатный буфер с pH 3). Стабильность таким образом испытываемого соединения оценивалась временем полураспада (время, соответствующее 50% разложения).
Фиг. 1 представляет собой сравнительную диаграмму кривых стабильности аспартама (кривая а) и соединения настоящего изобретения (кривая б) в кислой среде при pH 3. Соединение настоящего изобретения имеет время полураспада приблизительно 60 ч, тогда как время полураспада аспартама в тех же условиях составляет только 24 ч. Это говорит о том, что стабильность соединения настоящего изобретения в 2.5 раза выше по сравнению с аспартамом.
Фиг. 2 представляет собой сравнительную диаграмму кривых стабильности аспартама (кривая а) и соединения настоящего изобретения (кривая б) в кислой среде при pH 7. Соединение настоящего изобретения имеет время полураспада приблизительно 11 ч, тогда как время полураспада аспартама в тех же условиях составляет только 10 мин. Это говорит о том, что стабильность соединения настоящего изобретения в 66 раз выше по сравнению с аспартамом.

1-Метиловый эфир N-[N-(3,3-диметилбутил)-L-α-аспартил] -L-гексагидрофенилаланина, который может быть использован в качестве подслащивающего агента, получают обработкой раствора аспартама и 3,3-диметилбутиральальдегида водородом при давлении 3 бар и комнатной температуре в присутствии платинового катализатора. 2 с. и 4 з.п. ф-лы, 2 ил.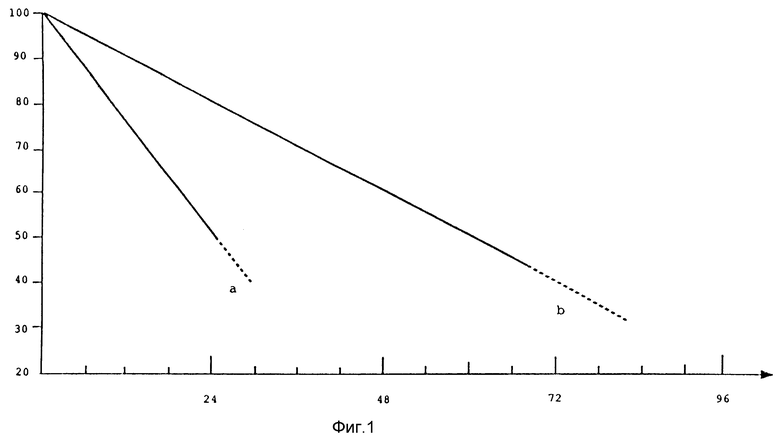

в качестве подслащивающего агента.
отличающийся тем, что раствор аспартама и 3,3-диметилбутиральдегида при комнатной температуре обрабатывают водородом при относительном давлении 3 бар в присутствии платинового катализатора.
| Способ получения N-формил- @ -Аспартилфенилаланина | 1985 |
|
SU1433414A3 |
| СТЕБЛЕПОДЪЕМНИКВСЕСОЮЗНЛЯП;Т:!1ТНО-ч;1[Ш^{ГНДЙБИБЛИОТЕКА | 0 |
|
SU334946A1 |
| СПОСОБ ФУНКЦИОНАЛЬНОГО ПРЕОБРАЗОВАНИЯ НЕСТАЦИОНАРНЫХ СВЕТОВЫХ ПОТОКОВ | 0 |
|
SU338946A1 |
| Чистовая форма к машине для выдувания стаканов | 1956 |
|
SU107593A1 |
| Patel et.al | |||
| J | |||
| Приспособление для изготовления в грунте бетонных свай с употреблением обсадных труб | 1915 |
|
SU1981A1 |
| Топочная решетка для многозольного топлива | 1923 |
|
SU133A1 |
| J | |||
| chem | |||
| Soc | |||
| Приспособление к индикатору для определения момента вспышки в двигателях | 1925 |
|
SU1969A1 |
| Кровля из глиняных обожженных плит с арматурой из проволочной сетки | 1921 |
|
SU120A1 |
