Тахикинины представляют собой семейство пептидов, которым присуща обычная амидированная концевая последовательность
Phe-Xaa-Gly-Leu-Met-NH2,
далее называемая последовательностью N 1 (SEQ ID NO: 1). Вещество P являлось первым пептидом этого семейства, которое было выделено, хотя его очистка и определение его основной последовательности не были осуществлены до начала 1970-х годов. Вещество P имеет следующую аминокислотную последовательность:
Arg-Pro-Lys-Pro-Gln-Gln-Phe-Phe-Gly-Leu-Met-NH2,
которая далее здесь называется последовательностью N 2 (SEQ ID NO : 2).
На рубеже 1983-1984 годов появилось несколько сообщений о выделении двух новых тахикининов млекопитающих, называемых теперь нейрокинином A (который также известен как вещество K, нейромедин L и нейрокинина d) и нейрокинином B (известным также как нейромедин K и нейрокинин β) См. обзор об этих открытиях в J.E.Maggio, Peptides, 6 (Supplement 3): 237-243 (1985). Нейрокинин A имеет аминокислотную последовательность
His-Lys-Thr-Asp-Ser-Phe-Val-Gly-Leu-Met-NH2,
называемую далее последовательностью N 3 (SEQ ID NO:3). Структура нейрокинина B представляет собой аминокислотную последовательность
Asp-Met-His-Asp-Phe-Phe-Val-Gly-Leu-Met-NH2,
называемую далее последовательностью N 4 (SEQ ID NO:4).
Тахикинины широко распространены как в центральной, так и в периферической нервных системах, выделяются из нервов и влияют на многие биологические действия, которые в большинстве случаев зависят от активации специфических рецепторов, выраженных на мембране клеток-мишеней. Тахикинины также продуцируются рядом тканей, не являющихся нервными тканями.
Тахикинины млекопитающих - вещество P, нейрокинин A и нейрокинин B - действуют через три основных подтипа рецепторов, обозначаемых как NK-1, NK-2 и NK-3 соответственно. Эти рецепторы присутствуют во многих органах.
Полагают, что вещество P, inter alia, вовлекается в трансмиссию болевых ощущений, в том числе боли, ассоциируемой с головными болями при мигрени, и боли при артрите. Эти пептиды также причастны к желудочно-кишечным расстройствам и болезням желудочно-кишечного тракта, таким как воспалительные заболевания кишечника. Тахикинины также играют некую роль во многих других заболеваниях, как обсуждается ниже.
Принимая во внимание многочисленность заболеваний, ассоциируемых с избытком тахикининов, разработка антагонистов тахикининовых рецепторов послужит для устранения таких клинических состояний. Первые антагонисты тахикининовых рецепторов представляли собой пептидные производные. Такие антагонисты обеспечивали ограниченное фармацевтическое применение ввиду их метаболической неустойчивости.
Недавние публикации описывают новые классы непептидных антагонистов тахикининовых рецепторов, которые, как правило, обладают большей пероральной биологической доступностью и большей метаболической устойчивостью, чем ранее разработанные классы антагонистов рецепторов тахикининов. Примеры таких непептидных тахикининовых рецепторов имеются в публикации европейского патента 591040 A1, опубликованной 6 апреля 1994; в публикации по договору о патентной кооперации ВОИС 94/01402, опубликованной 3 марта 1994; в публикации по договору о патентной кооперации ВОИС 93/011609, опубликованной 21 января 1993.
Настоящее изобретение, по существу, предлагает класс непептидных антагонистов тахикининовых рецепторов. Благодаря своей непептидной природе соединения настоящего изобретения свободны от недостатков, в смысле метаболической устойчивости, присущих известным антагонистам рецепторов тахикининов на основе пептидов.
Настоящее изобретение включает в себя способы лечения или предупреждения физиологического нарушения, ассоциируемого с избытком тахикининов, и упомянутые способы включают введение млекопитающему, нуждающемуся в упомянутом лечении, эффективного количества соединения формулы I
в которой m равен 0, 1, 2 или 3; n равен 0 или 1; o представляет собой 0, 1 или 2; p равен 0 или 1;
R представляет собой фенил, 2- или 3-индолил, 2- или 3-индолинил, бензотиенил, бензофуранил или нафтил; и эти группы R могут быть замещены одним или двумя галогенами, (C1-C3)-алкокси-группами, трифторметилами, (C1-C4)-алкилами, фенил-(C1-C3)- алкоксигруппами или (C1-C4)-алканоильными группами;
R1 представляет собой тритил, фенил, дифенилметил, фенокси-группу, фенилтиогруппу, пиперазинил, пиперидинил, пирролидинил, морфолинил, индолинил, индолил, бензотиенил, гексаметилениминил, бензофуранил, тетрагидропиридинил, хинолинил, изохинолинил, восстановленный хинолинил, восстановленный изохинолинил, фенил-((C1-C4)-алкил)-, фенил-((C1-C4)-алкокси)-, хинолин- (C1-C4)-алкил)-, изохинолинил-((C1-C4)- -алкил)-, восстановленный хинолинил-((C1-C4)-алкил)-, восстановленный изохинолил-((C1-C4)-алкил)-, бензоил-((C1-C3)-алкил)-, (C1-C4)-алкил или -NH-CH2-R5;
при этом любая из групп R1 может быть замещена галогеном, (C1-C4)-алкилом, (C1-C4)-алкоксигруппой, трифторметилом, аминогруппой, (C1-C4)-алкиламиногруппой, ди((C1-C4)-алкил)аминогруппой или (C2-C4)-алканоиламиногруппой;
или любая из групп R1 может быть замещена фенилом, пиперазинилом, (C3-C8)-циклоалкилом, бензилом, (C1-C4)-алкилом, пиперидинилом, пиридинилом, пиримидинилом, (C2-C6)-алкиламиногруппой, пирролидинилом, (C2-C6)-алканоилом или (C1-C4)-алкоксикарбонилом,
и любая из этих групп может быть замещена галогеном, (C1-C4)-алкилом, (C1-C4)-алкоксигруппой, трифторметилом, аминогруппой, (C1-C4)-алкиламиногруппой, ди(C1-C4)-алкил)аминогруппой или (C2-C4)-алканоиламиногруппой;
или R1 представляет собой аминогруппу, отщепляющуюся группу, водород, (C1-C4)-алкиламиногруппу или ди((C1-C4)-алкил)аминогруппу;
R5 представляет собой пиридил, анилино-((C1-C3)-алкил)- или анилинокарбонил;
R2 представляет собой водород, (C1-C4)- алкил, (C1-C4)-алкилсульфонил, карбокси-(C1-C3)-алкил)-, (C1-C3)- алкоксикарбонил-((C1-C3)-алкил)- или -CO-R6;
R6 представляет собой водород, (C1-C4)-алкил, (C1-C3)-галоидалкил, фенил, (C1-C3)-алкоксигруппу, (C1-C3)-гидроксиалкил, аминогруппу, (C1-C4)-алкиламиногруппу, ди((C1-C4)- алкил)аминогруппу или -(CH2)q-R7; q равен 0-3;
R7 представляет собой карбоксигруппу, (C1-C4)-алкоксикарбонил, (C1-C4)- алкилкарбонилоксигруппу, аминогруппу, (C1-C4)- алкиламиногруппу, ди((C1-C4)-алкил)аминогруппу, (C1-C6)-алкоксикарбониламиногруппу или
феноксигруппу, фенилтиогруппу, пиперазинил, пиперидинил, пирролидинил, морфолинил, индолинил, индолил, бензотиенил, бензофуранил, хинолинил, изохинолинил, восстановленный хинолинил, восстановленный изохинолинил, фенил-((C1-C4)-алкил)-, хинолинил-((C1-C4)-алкил-, изохинолинил- ((C1-C4)-алкил)-, восстановленный хинолинил-((C1-C4)-алкил)-, восстановленный изохинолинил-((C1-C4)-алкил)-, бензоил-(C1-C3)-алкил;
из которых любая арильная или гетероциклическая R7-группа может быть замещена галогеном, трифторметилом, (C1-C4)-алкоксигруппой, (C1-C4)-алкилом, аминогруппой, (C1-C4)-алкиламиногруппой, ди((C1-C4)-алкил)аминогруппой или (C2-C4)-алканоиламиногруппой;
или любая из групп R7 может быть замещена фенилом, пиперазинилом, (C3-C8)-циклоалкилом, бензилом, пиперидинилом, пиридинилом, пиримидинилом, пирролидинилом, (C2-C6)-алканоилом или (C1-C4)-алкоксикарбонилом;
из которых любая группа может быть замещена галогеном, трифторметилом, аминогруппой, (C1-C4)-алкоксигруппой, (C1-C4)-алкилом, (C1-C4)-алкиламиногруппой, ди((C1-C4)- алкил)аминогруппой или (C2-C4)-алканоиламиногруппой;
R8 представляет собой водород или (C1-C6)-алкил;
R3 представляет собой фенил, фенил-((C1-C6)-алкил)-, (C3-C8)-циклоалкил, (C5-C8)-циклоалкенил, (C1-C8)-алкил, нафтил, (C2-C8)-алкенил или водород;
и любая из этих групп, за исключением водорода, может быть замещена одним или двумя галогенами, (C1-C3)- алкоксигруппами, (C1-C3)-алкилтиогруппами, нитрогруппами, трифторметилами или (C1-C3)-алкильными группами; и
R4 представляет собой водород или (C1-C3)- алкил;
при условии, что если R1 представляет собой водород или галоген, R3 представляет собой фенил, фенил- ((C1-C6)-алкил)-, (C3-C8)-циклоалкил, (C5-C8)-циклоалкенил или нафтил;
или его фармацевтически приемлемой соли.
В другом варианте своего осуществления настоящее изобретение включает в себя новые соединения формулы I и их фармацевтически приемлемые соли, их сольваты и пролекарства, а также фармацевтические формулировки, содержащие в качестве активного ингредиента соединение формулы I в сочетании с фармацевтически приемлемым носителем, разбавителем или эксципиентом. Настоящее изобретение также включает в себя новые способы синтеза соединений формулы I.
Температура, упоминаемая здесь, дается в градусах по Цельсию (oC). Все используемые здесь единицы являются единицами массы, за исключением жидкостей, для которых приводятся единицы объема.
Используемый здесь термин "(C1-C6)-алкил" относится к линейным или разветвленным одновалентным насыщенным алифатическим цепям с 1-6 атомами углерода и включает, но не ограничивается ими, метил, этил, пропил, изопропил, бутил, изобутил, трет-бутил, пентил, изопентил и гексил. Термин "(C1-C6)-алкил" включает в свое определение термин "(C1-C4)-алкил",
"Двухвалентный (C1-C4)-алкил" означает линейную или разветвленную двухвалентную насыщенную алифатическую цепь, содержащую от одного до четырех атомов углерода. Типичными двухвалентными (C1-C4)-алкильными группами являются метилен, этилен, пропилен, 2-метилпропилен, бутилен и подобные группы.
"Галоген" обозначает хлор, фтор, бром или йод.
"Галоид(C1-C4)алкил" означает линейную или разветвленную алкильную цепь, содержащую от одного до четырех атомов углерода, к которой присоединены 1, 2 или 3 атома галогена. Типичными галоид(C1-C4)алкильными группами являются хлорметил, 2-бромэтил, 1-хлоризопропил, 3-фторпропил, 2,3-дибромбутил, 3-хлоризобутил, йод-трет-бутил, трифторметил и подобные группы.
"Гидрокси(C1-C4)алкил" представляет собой линейную или разветвленную алкильную цепь, содержащую от одного до четырех атомов углеродов, с присоединенной к ней гидроксильной группой. Типичными гидрокси(C1-C4)алкильными группами являются гидроксиметил, 2-гидроксиэтил, 1-гидроксиизопропил, 2-гидроксипропил, 2-гидроксибутил, 3-гидроксиизобутил, гидрокси-трет-бутил и подобные группы.
"(C1-C6)-Алкилтиогруппа" представляет собой линейную или разветвленную алкильную цепь, содержащую от одного до шести атомов углерода, с присоединенным к ней атомом серы. Типичными (C1-C6)-алкилтиогруппами являются метилтиогруппа, этилтиогруппа, пропилтиогруппа, изопропилтиогруппа, бутилтиогруппа и подобные группы. Термин "(C1-C6)-алкилтиогруппа" включает в свое определение термин "(C1-C4)-алкилтиогруппа".
Используемый здесь термин "(C2-C8)-алкенил" означает линейную или разветвленную одновалентную ненасыщенную алифатическую цепь, содержащую от двух до восьми атомов углерода. Типичными (C2-C6)-алкенильными группами являются этенил (известный также как винил), 1-метилэтенил, 1-метил-1- пропенил, 1-бутенил, 1-гексенил, 2-метил-2-пропенил, 1-пропенил, 2-пропенил, 2-бутенил, 2-пентенил и подобные группы.
"(C5-C8)-Циклоалкенил представляет собой углеводородное кольцо, содержащее от пяти до восьми атомов углерода и имеющее по крайней мере одну двойную связь в пределах кольца, которое является незамещенным или замещенным 1, 2 или 3 заместителями, выбираемыми, независимо, среди галогена, галоид (C1-C4)алкила, (C1-C4)-алкила, (C1-C4)-алкоксигруппы, карбоксигруппы, (C1-C4)-алкоксикарбонила, карбамоила, N-(C1-C4)-алкилкарбамоила, аминогруппы, (C1-C4)-алкиламиногруппы или -(CH2)a-Rc, где a равно 1, 2, 3 или 4 и Rc представляет собой гидроксильную группу, (C1-C4)-алкоксигруппу, карбоксигруппу, (C1-C4)-алкоксикарбонил, аминогруппу, карбамоил, (C1-C4)-алкиламиногруппу или ди((C1-C4)- алкил)аминогруппу.
"(C1-C4)-Алкиламиногруппа" представляет собой линейную или разветвленную аминоалкильную цепь, содержащую от одного до четырех атомов углерода, присоединенных к аминогруппе. Типичными (C1-C4)-алкиламиногруппами являются метиламиногруппа, этиламиногруппа, пропиламиногруппа, изопропиламиногруппа, бутиламиногруппа, втор-бутиламиногруппа и подобные группы.
"Ди((C1-C4)-алкил) аминогруппа" представляет собой диалкиламиноцепь, содержащую две алкильные цепи, причем каждая содержит, независимо от другой, от одного до четырех атомов углерода, соединенных с обычной аминогруппой. Типичными ди((C1-C4)-алкил)-аминогруппами являются диметиламиногруппа, этилметиламиногруппа, метилизопропиламиногруппа, трет-бутилизопропиламиногруппа, ди-трет-бутиламиногруппа и подобные группы.
"Арилсульфонил" представляет собой арильную группу, присоединенную к сульфонильной группе. Используемый здесь термин "арил" означает фенил, нафтил, гетероциклическую группу или ненасыщенную гетероциклическую группу, которая, необязательно, замещается 1, 2 или 3 заместителями, выбираемыми среди галогена, галоид(C1-C4)-алкила, (C1-C4)-алкила, (C1-C4)-алкоксигруппы, карбоксигруппы, (C1-C4)-алкоксикарбонила, карбамоила, N-(C1-C4)алкилкарбамоила, аминогруппы, (C1-C4)-алкиламиногруппы, ди(C1-C4)алкиламиногруппы или -(CH2)a-Rb, где a равно 1, 2, 3 или 4, и Rb представляет собой гидроксильную группу, (C1-C4)-алкоксигруппу, карбоксигруппу, (C1-C4)-алкоксикарбонил, аминогруппу, карбамоил, (C1-C4)-алкиламиногруппу или ди(C1-C4)алкиламиногруппу.
Термин "гетероцикл" относится к незамещенному или замещенному устойчивому 5-7-членному моноциклическому или 7-10-членному бициклическому гетероциклическому кольцу, которое является насыщенным и которое состоит из атомов углерода и одного-трех гетероатомов, выбираемых из группы, состоящей из атомов азота, кислорода и серы, и при этом гетероатомы азота и серы могут быть, необязательно, окисленными, и гетероатом азота, необязательно, может быть кватернизован и включает бициклическую группу, в которой любое из гетероциклических колец, определение которым дается выше, слито с бензольным кольцом. Гетероциклическое кольцо может быть присоединено к любому гетероатому или атому углерода, которые образуют устойчивую структуру. Гетероцикл является незамещенным или замешенным 1, 2 или 3 заместителями, выбираемыми, независимо, среди галогена, галоид(C1-C4)алкила, (C1-C4)- алкила, (C1-C4)-алкоксигруппы, карбоксигруппы, (C1-C4)-алкоксикарбонила, карбамоила,
N-(C1-C4)-алкилкарбамоила, аминогруппы, (C1-C4)-алкиламиногруппы, ди(C1-C4)-алкиламиногруппы или -(CH2)a-Rd, где a равно 1, 2, 3 или 4, и Rd представляет собой гидроксильную группу, (C1-C4)-алкоксигруппу, карбоксигруппу, (C1-C4)-алкоксикарбонил, аминогруппу, карбамоил, (C1-C4)-алкиламиногруппу или ди(C1-C4)алкиламиногруппу.
Термин "ненасыщенный гетероцикл" относится к незамещенному или замещенному устойчивому 5-7-членному моноциклическому или 7-10-членному бициклическому гетероциклическому кольцу, которое содержит одну или несколько двойных связей и которое состоит из атомов углерода и одного-трех гетероатомов, выбираемых из группы, состоящей из атомов азота, кислорода или серы, и при этом гетероатомы азота и серы могут быть, необязательно, окисленными, и гетероатом азота может быть, необязательно, кватернизован, и включает бициклическую группу, в которой любое из гетероциклических колец, определение которым дается выше, слито с бензольным кольцом. Ненасыщенное гетероциклическое кольцо может быть присоединено по любому гетероатому или атому углерода, которые дают устойчивую структуру. Ненасыщенный гетероцикл может быть незамещенным или замещенным 1, 2 или 3 заместителями, выбираемыми, независимо, среди галогена, галоид(C1-C4)алкила, (C1-C4)-алкила, (C1-C4)-алкоксигруппы, карбоксигруппы, (C1-C4)-алкоксикарбонила, карбамоила,
N-(C1-C4)алкилкарбамоила, аминогруппы, (C1-C4)алкиламиногруппы, ди(C1-C4)- алкиламиногруппы или -(CH2)a-Re, где a равно 1, 2, 3 или 4; и Re представляет собой гидроксильную группу, (C1-C4)-алкоксигруппу, карбоксигруппу, (C1-C4)-алкоксикарбонил, аминогруппу, карбамоил, (C1-C4)-алкиламиногруппу или ди(C1-C4)алкиламиногруппу.
Примерами таких гетероциклов и ненасыщенных гетероциклов являются пиперидинил, пиперазинил, ацепинил, пирролил, 4-пиперидинил, пирролидинил, пиразолил, пиразолидинил, имидазолил, имидазолинил, имидазолидинил, пиридил, пиразинил, пиримидинил, пиридазинил, оксазолил, оксазолидинил, изоксазолил, изоксазолидинил, морфолинил, тиазолил, тиазолидинил, изотиазолил, хинуклидинил, изотиазолидинил, индолил, хинолинил, изохинолинил, бензимидазолил, тиадиазолил, бензопиранил, бензотиазолил, бензазолил, фурил, тетрагидрофурил, тетрагидропиранил, тиенил, бензотиенил, тиаморфолинилсульфоксид, тиаморфолинилсульфон, оксадиазолил, триазолил, тетрагидрохинолинил, тетрагидроизохинолинил, 3-метилимидазолил, 3-метоксипиридил, 4-хлорхинолинил, 4-аминотиазолил, 8-метилхинолинил, 6-хлорхиноксалинил, 3-этилпиридил, 6-метоксибензимидазолил, 4-гидроксифурил, 4-метилизохинолинил, 6,8-дибромхинолинил, 4,8-диметилнафтил, 2-метил-1,2,3,4- тетрагидроизохинолинил, N-метилхинолил-2-ил, 2-трет-бутоксикарбонил-1,2,3,4-изохинолин-7-ил и подобные группы.
"(C1-C6)-Алкоксигруппа" представляет собой линейную или разветвленную алкильную цепь, содержащую от одного до шести атомов углерода, присоединенную к атому кислорода. Типичными (C1-C6)-алкоксигруппами являются метоксигруппа, этоксигруппа, пропоксигруппа, изопропоксигруппа, бутоксигруппа, трет-бутоксигруппа, пентоксигруппа и подобные группы.
Термин "(C1-C6)-алкоксигруппа" включает в свое определение термин "(C1-C6)-алкоксигруппа".
"(C2-C6)-Алканоил" представляет собой линейную или разветвленную алкильную цепь, содержащую от одного до пяти атомов углерода, присоединенную к карбонильной группе. Типичными (C2-C6)-алканоильными группами являются этаноил, пропаноил, изопропаноил, бутаноил, трет-бутаноил, пентаноил, гексаноил, 3-метилпентаноил и подобные группы.
"(C1-C4)-Алкоксикарбонил" представляет собой линейную или разветвленную алкоксицепь, содержащую от одного до четырех атомов углерода, связанных с карбонильной группой. Типичными (C1-C4)-алкоксикарбонильными группами являются метоксикарбонил, этоксикарбонил, пропоксикарбонил, изопропоксикарбонил, бутоксикарбонил, трет-бутоксикарбонил и подобные группы.
"(C3-C8)-Циклоалкил" представляет собой насыщенную углеводородную кольцевую структуру, содержащую от трех до восьми атомов углерода, которая является незамещенной или замещенной 1, 2 или тремя заместителями, выбираемыми, независимо, среди галогена, галоид(C1-C4)-алкила, (C1-C4)-алкила, (C1-C4)-алкоксигруппы, карбоксигруппы, (C1-C4)-алкоксикарбонила, карбамоила,
N-(C1-C4)-алкилкарбамоила, аминогруппы, (C1-C4)-алкиламиногруппы, ди(C1-C4)алкиламиногруппы или -(CH2)a-Rf, где a равно 1, 2, 3 или 4, и Rf представляет собой гидроксильную группу, (C1-C4)-алкоксигруппу, карбоксигруппу, (C1-C4)-алкоксикарбонил, аминогруппу, карбамоил, (C1-C4)алкиламиногруппу или ди(C1-C4)алкиламиногруппу. Типичными (C3-C8)-циклоалкильными группами являются циклопропил, циклопентил, циклогексил, циклогептил, 3-метилциклопентил, 4-этоксициклогексил, 4-карбоксициклогептил, 2-xлopциклoгексил, циклобутил, циклооктил и подобные группы.
Термин "аминозащищающая группа", который используется в настоящем описании, относится к заместителям аминогруппы, которые обычно используют для блокирования или защиты функциональной аминогруппы во время реагирования других функциональных групп соединения. Примерами таких аминозащищающих групп являются формил, тритил, фталимидогруппа, трихлорацетил, хлорацетил, бромацетил, йодацетил и блокирующие группы уретанового типа, такие как бензилоксикарбонил, 4-фенилбензилоксикарбонил, 2-метилбензилоксикарбонил, 4-метоксибензилоксикарбонил, 4-фторбензилоксикарбонил, 4-хлорбензилоксикарбонил, 3-хлорбензилоксикарбонил, 2-хлорбензилоксикарбонил, 2,4-дихлорбензилоксикарбонил, 4-бромбензилоксикарбонил, 3-бромбензилоксикарбонил, 4-нитробензилоксикарбонил, 4-цианобензилоксикарбонил, трет-бутоксикарбонил, 1,1-дифенилэт-1- илоксикарбонил, 1,1-дифенилпроп-1-илоксикарбонил, 2-фенилпроп-2- илоксикарбонил, 2-(п-толуил)-проп-2-илоксикарбонил, циклопентанилоксикарбонил, 1-метилциклопентанилоксикарбонил, циклогексанилоксикарбонил, 1-метилциклогексаноилоксикарбонил, 2-метилциклогексанилоксикарбонил, 2-(4-толуилсульфонил)- этоксикарбонил, 2-(метилсульфонил)этоксикарбонил, 2-(трифенилфосфино)-этоксикарбонил, фторенилметоксикарбонил ("FMOC"), 2-(триметилсилил)этоксикарбонил, аллилоксикарбонил, 1-(триметилсилилметил)проп-1-енил-оксикарбонил, 5-бензизоксалилметоксикарбонил, 4-ацетоксибензилоксикарбонил, 2,2,2-трихлорэтоксикарбонил, 2-этинил-2-пропоксикарбонил, циклопропилметоксикарбонил, 4-(децилокси)бензилоксикарбонил, изоборнилоксикарбонил, 1-пиперидилоксикарбонил и подобные группы; бензоилметилсульфонильная группа, 2-нитрофенилсульфенильная группа, дифенилфосфиноксидная и подобные аминозащищающие группы. Вид используемой аминозащищающей группы обычно не является критическим обстоятельством, пока образовавшаяся аминогруппа является устойчивой в условиях последующих реакций в других положениях молекулы промежуточного соединения, и может быть селективно удалена в соответствующий момент без разрушения остальной части молекулы, включая любую другую аминозащищающую группу. Предпочтительными аминозащищающими группами являются тритил, трет-бутоксикарбонил (трет-БОК), аллилоксикарбонил и бензилоксикарбонил. Другие примеры групп, подходящих под вышеупомянутые термины, описываются в E.Haslam, "Protective Groups in Organic Chemistry", (J.G.W.McOmie, ed., 1973), Chapter 2; и в T. W.Greene and P.G.M.Wuts, "Protective Groups in Organic Synthesis" (1991), Chapter 7.
Термин "карбоксизащищающая группа", который используется в настоящем описании, относится к заместителям карбоксигруппы, которые обычно используют для блокирования или защиты функциональной карбоксигруппы во время реакций других функциональных групп соединения. Примерами таких карбоксизащищающих групп являются метил, п-нитробензил, п-метилбензил, п-метоксибензил, 3,4-диметоксибензил, 2,4-диметоксибензил, 2,4,6-триметоксибензил, 2,4,6-триметилбензил, пентаметилбензил, 3,4-метилендиоксибензил, бензгидрил, 4,4'-диметоксибензгидрил, 2,2', 4,4'-тетраметоксибензгидрил, трет-бутил, трет-амил, тритил, 4-метокситритил, 4,4'-диметокситритил, 4,4',4''-триметокситритил, 2-фенилпроп-2-ил, триметилсилил, трет-бутилдиметилсилил, фенацил, 2,2,2-трихлорэтил, 2-(ди- (н-бутил)метилсилил) этил, п-толуолсульфонэтил, 4-нитробензилсульфонилэтил, аллил, циннамил, 1-(триметилсилил-метил) проп-1-ен-3-ил и подобные группы. Предпочтительными карбоксизащищающими группами являются аллил, бензил и трет-бутил. Другие примеры таких групп можно найти в E.Haslam, цит. выше, Chapter 5, и в T.W.Greene et.al., цит. выше, Chapter 5.
Используемый здесь термин "отщепляющаяся группа" относится к группе атомов, которая перемещается от атома углерода под действием нуклеофила в реакции нуклеофильного замещения. Термин "отщепляющаяся группа", используемый в настоящем описании, включает в себя, но не ограничивается, активирующие группы.
Используемый здесь термин "ативирующая группа" относится к отщепляющейся группе, которая, когда берется с карбонильной группой (-C=O), к которой она присоединена, с большей вероятностью принимает участие в реакции ацилирования, чем в случае, если такая группа не присутствует, как в свободной кислоте. Такие активирующие группы хорошо известны специалистам в этой области техники и могут представлять собой, например, сукцинимидоксигруппу, фталимидоксигруппу, бензотриазолилоксигруппу, бензолсульфонилоксигруппу, метансульфонилоксигруппу, толуолсульфонилоксигруппу, азидогруппу или -O-CO-((C4-C7)-алкил).
Соединения, используемые в способе настоящего изобретения, имеют множество асимметрических центров. Вследствие наличия таких хиральных центров соединения настоящего изобретения встречаются в виде рацематов, смесей энантиомеров и отдельных энантиомеров, а также в виде диастереомеров и смесей диастереомеров. Все асимметрические формы - отдельные изомеры и их сочетания - входят в объем настоящего изобретения.
Термины "R" и "S" используют здесь так, как они обычно используются в органической химии - для обозначения специфической конфигурации хирального центра. Термин "R" (rectus) относится к конфигурации хирального центра со взаимосвязью групповых приоритетов (от наивысшего к другому наинизшему) по часовой стрелке при взгляде вдоль связи по направлению к группе с наинизшим приоритетом. Термин "S" (sinister) относится к такой конфигурации хирального центра, когда групповые приоритеты (от наивысшего к другому наинизшему) взаимосвязаны в направлении против часовой стрелки при взгляде вдоль связи в направлении группы с наинизшим приоритетом. Приоритет групп основывается на их атомном номере (в порядке убывания атомного номера). Частичный список приоритетов и обсуждение стереохимии содержится в "Nomenclature of Organic Compounds: Principles and Practice", (J.H.Fletcher, et.al., eds., 1974) на pp. 103-120.
Кроме системы (R) - (S), в настоящей заявке используется также более старая система D-L для обозначения абсолютной конфигурации, особенно в отношении аминокислот. В этой системе формула проекции Фишера ориентируется таким образом, что атом углерода под номером 1 в главной цепи располагается наверху. Префикс "D" используется для обозначения абсолютной конфигурации изомера, в котором функциональная (определяющая) группа располагается справа от атома углерода в хиральном центре, и "L" - для изомера, в котором она располагается слева.
Как упоминалось выше, настоящее изобретение включает фармацевтически приемлемые соли соединения, определяемого формулой I. Соединение настоящего изобретения может обладать достаточно кислотной, достаточной основной или обеими функциональными группами и, соответственно, реагировать с рядом органических и неорганических оснований и неорганических и органических кислот с образованием фармацевтически приемлемых солей.
Употребляемый здесь термин "фармацевтически приемлемая соль" относится к солям соединений вышеупомянутой формулы, которые, по существу, являются нетоксичными для живых организмов. Типичные фармацевтически приемлемые соли включают соли, получаемые при взаимодействии соединений настоящего изобретения с фармацевтически приемлемыми минеральными или органическими кислотами или органическими и неорганическими основаниями. Такие соли известны как соли присоединения кислот и соли присоединения оснований.
Кислоты, которые обычно используют для образования солей присоединения кислот, представляют собой неорганические кислоты, такие как соляная кислота, бромистоводородная кислота, йодистоводородная кислота, серная кислота, фосфорная кислота и подобные кислоты, и органические кислоты, такие, как п-толуолсульфоновая и метансульфоновая кислота, щавелевая кислота, п-бромфенилсульфоновая кислота, угольная кислота, янтарная кислота, лимонная кислота, бензойная кислота, уксусная кислота и подобные кислоты. Примерами таких фармацевтически приемлемых солей являются сульфаты, пиросульфаты, бисульфаты, сульфиты, бисульфиты, фосфаты, моногидрофосфаты, дигидрофосфаты, метафосфаты, пирофосфаты, бромиды, йодиды, ацетаты, пропионаты, дигидрохлориды, изобутираты, капроаты, гептаноаты, пропиолаты, оксалаты, малонаты, сукцинаты, субераты, себацаты, фумараты, малеаты, бутин-1,4-диоаты, гексин-1,6-диоаты, бензоаты, хлорбензоаты, метилбензоаты, гидроксибензоаты, метоксибензоаты, фталаты, ксилолсульфонаты, фенилацетаты, фенилпропионаты, фенилбутираты, цитраты, лактаты, γ -гидроксибутираты, гликоляты, тартраты, метансульфонаты, пропансульфонаты, нафталин-1- сульфонаты, нафталин-2-сульфонаты, манделаты и подобные соли. Предпочтительными фармацевтически приемлемыми солями присоединения кислот являются соли, образованные с такими минеральными кислотами, как хлористоводородная кислота и бромистоводородная кислота, и соли, образованные с такими органическими кислотами, как малеиновая кислота и метансульфоновая кислота.
Соли присоединения оснований включают соли, образованные с неорганическими основаниями, такими как гидроксиды аммония и щелочных или щелочно-земельных металлов, карбонаты, бикарбонаты и т.п. Такие основания, пригодные для получения солей по настоящему изобретению, включают, таким образом, гидроксид натрия, гидроксид калия, гидроксид аммония, карбонат калия, карбонат натрия, бикарбонат натрия, бикарбонат калия, гидроксид калия, карбонат кальция и подобные основания. Особенно предпочтительными являются формы калиевых и натриевых солей.
Следует представлять, что конкретный контрион, образующий часть любой соли настоящего изобретения, обычно не является критическим обстоятельством, пока соль в целом является фармацевтически приемлемой и пока контрион не способствует появлению нежелательных качественных свойств соли в целом.
Настоящее изобретение также включает в себя фармацевтически приемлемые сольваты соединений формулы I. Многие соединения формулы I могут соединяться с растворителями, такими как вода, метанол, этанол и ацетонитрил, с образованием фармацевтически приемлемых сольватов, таких как соответствующие гидраты, метанолаты, этанолаты и ацетонитрилаты.
Особенно предпочтительными соединениями, применяемыми по способу настоящего изобретения, являются соединения формулы I, в которых
а) R представляет собой замещенный или незамещенный 2- или 3-индолил, фенил или нафтил;
b) n равен 1;
c) R1 представляет собой фенил, замещенный фенил, пиперидинил, замещенный пиперидинил, пиперазинил, замещенный пиперазинил, пирролидинил, пиридил, бензоил или морфолинил;
d) R2 представляет собой -CO-R6, (C1-C4)-алкилсульфонил или (C1-C3)- алкоксикарбонил-((C1-C3)алкил)-;
е) R3 представляет собой фенил, замещенный фенил, (C3-C8)-циклоалкил, замещенный (C3-C8)-циклоалкил, нафтил или замещенный нафтил;
f) R8 представляет собой водород или метил.
Наиболее предпочтительной группой соединений, применяемых в способах настоящего изобретения, являются соединения формулы I, в которых R представляет собой необязательно замещенный индолил, R1 представляет собой замещенный пиперидинил или замещенный пиперазинил, R8 представляет собой водород, и R2 представляет собой ацетил или метилсульфонил. Другой предпочтительной группой соединений, применяемых в способах настоящего изобретения, являются соединения формулы I, в которых R представляет собой нафтил, R1 представляет собой необязательно замещенный фенил, замещенный пиперидинил или замещенный пиперазинил, R2 представляет собой ацетил или метилсульфонил, и R3 представляет собой фенил или замещенный фенил.
Особенно предпочтительными соединениями настоящего изобретения являются соединения формулы I, в которых
a) R представляет собой замещенный или незамещенный 2- или 3-индолил, фенил или нафтил;
b) n равен 1;
c) R1 представляет собой тритил, фенил, замещенный фенил, пиперидинил, замещенный пиперидинил, пиперазинил, замещенный пиперазинил, пирролидинил, пиридил, бензоил или морфолинил;
d) R2 представляет собой -CO-R6, (C1-C4)-алкилсульфонил или (C1-C3)- алкоксикарбонил-((C1-C3)алкил)-;
e) R3 представляет собой фенил, замещенный фенил, (C3-C8)- циклоалкил, замещенный (C3-C8)-циклоалкил, нафтил или замещенный нафтил; и
f) R8 представляет собой водород или метил.
Соединения настоящего изобретения могут быть получены множеством способов, хорошо известных специалистам в этой области техники. Конкретный порядок стадий, требуемых для получения соединений формулы I, зависит от конкретного соединения, которое синтезируют, исходного соединения и относительной лабильности замещаемых групп.
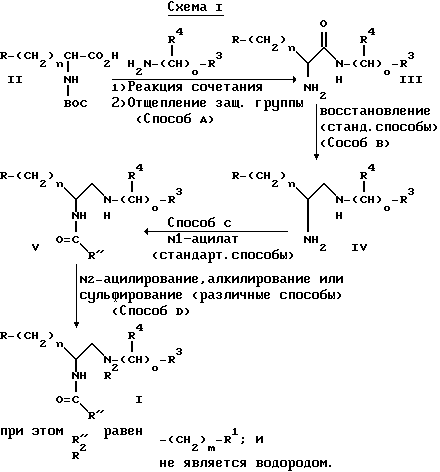
Примеры таких последовательностей реакций изображаются на схемах I-IV. Сочетание замещенного амина с соединением формулы II (способ A) может быть осуществлено многими способами, известными в технике, причем конкретные используемые способы зависят от конкретного соединения формулы II, которое применяют в качестве исходного вещества, и типа замещенного амина, используемого в реакции сочетания. В таких реакциях сочетания часто используют обычно применяемые реагенты связывания, такие как 1,1-карбонил-диимидазол, дициклогексилкарбодиимид, диэтилазодикарбоксилат, 1-гидроксибензотриазол, алкилхлорформиат и триэтиламин, фенил-дихлорфосфат и хлорсульфонилизоцианат. Примеры таких способов описываются ниже. После отщепления защитной группы от аминогруппы получают соединения формулы III.
Соединение формулы III затем восстанавливают, превращая амид в амин (способ B). Амиды могут быть восстановлены до аминов с использованием процедур, хорошо известных в технике. Такое восстановление можно осуществить с применением алюмогидрида лития, так же как и с применением многих других алюмогидридов. С другой стороны, амиды могут быть восстановлены каталитическим гидрированием, хотя для этого обычно требуются высокие температуры и давление. Для восстановления амида может быть использован борогидрид натрия в сочетании с другими реагентами. В такой реакции восстановления особенно применимыми являются комплексы борана, такие как комплекс борана с диметилсульфидом.
Следующей стадией на схеме 1 (способ C) является селективное ацилирование первичного амина с применением стандартных методов, типичным примером которых является способ C. Первичный амин является легко доступным для селективного замещения по сравнению со вторичным амином в силу более высоких пространственных требований вторичного амина.
Такое ацилирование может быть осуществлено с применением любого из большого числа технических приемов, систематически используемых специалистами в органической химии. Одной из таких реакционных схем является замещение с применением ангидрида, такого как ацетангидрид. Другая реакционная схема, часто применяемая для ацилирования первичного амина, использует карбоновую кислоту, предпочтительно, с активирующим агентом, как описано выше в способе A. В реакции типа аминодеалкоксилирования применяют сложные эфиры как средство ацилирования первичного амина. Активированные эфиры, которые аттенюируют для обеспечения усиленной селективности, являются очень эффективными ацилирующими агентами.

Первичные амины также можно ацилировать с применением амидов, и осуществление этого является, по существу, реакцией обмена. Такую реакцию обычно осуществляют с солью амина. При этой реакции часто добавляют трифторид бора, обычно в форме комплекса трифторида бора с диэтиловым эфиром, чтобы образовать комплекс с отщепляющимся аммиаком.
Следующей процедурой является процедура замещения вторичного амина (способ D). Для большинства соединений формулы I такое замещение является алкилированием, ацилированием или сульфированием. Такое замещение обычно выполняют, используя хорошо известные способы. Обычно алкилирования можно достичь с применением галоидалкилов и т.п., а также хорошо известными способами гидроалкилирования, как показано в способе G, схема II, см. выше, при использовании альдегидов или кетонов. Прописи многих реакций ацилирования, обсужденных выше, являются достаточно эффективными также для ацилирования вторичных аминов. Для сульфирования вторичных аминов могут быть использованы алкил- и арилсульфонилхлориды.
Во многих случаях одной из последних стадий при синтезе соединений формулы I является удаление амино- или карбоксизащищающей группы. Такие процедуры, которые изменяются в зависимости от типа защищающей группы, так же как и для случая относительной неустойчивости других групп соединения, подробно описываются во многих справочниках, таких как T.W.Greene, et. al., Protective Groups in Organic Synthesis (1991).
Схемы II и III изображают альтернативные прописи и стратегию синтеза соединений формулы I. Многие отдельные реакции подобны реакциям, описанным на схеме I, но реакции по схемам II и III осуществляются другим, но еще хорошо известным специалистам в этой области техники, рядом стадий.

На завершающем этапе R2a сочетается с карбонильной группой, к которой она присоединяется, что эквивалентно R2.
Чтобы получить один их оптических изомеров в количестве, предпочтительном по сравнению с энантиомером, практики могут пойти по одному из следующих путей. Во-первых, можно получить смесь энантиомеров, и затем разделить два энантиомера. Обычно применяемым способом расщепления рацемической смеси (или смеси энантиомеров) на отдельные энантиомеры является превращение сначала энантиомеров в диастереомеры посредством соли с оптически активной кислотой или основанием. Затем эти диастереомеры можно разделить, используя различную растворимость, фракционную кристаллизацию, хроматографию или подобные методы. Другие подробности расщепления энантиомерных смесей можно найти в J. Jacques, et. al., "Enantiomers, Racemates, and Resolutions" (1991).
Кроме схем, описанных выше, при практическом осуществлении настоящего изобретения можно также выбрать энантиоспецифическую схему получения соединений формулы I. Приведенная ниже схема IV изображает такое типичное решение реакции синтеза, которое сохраняет хиральный центр, присутствующий в исходном веществе в нужной ориентации, в данном случае - "R"-конфигурацию. По таким реакционным схемам обычно получают соединения, в которых более 95% целевого продукта представляет собой нужный энантиомер.
Многие из стадий синтеза, используемых по схеме IV, являются такими же, какие используются по другим схемам, особенно, по схема III.

Далее следует описание характерных примеров условий реакций, используемых при получении соединений формулы I.
Способ A
Взаимодействие карбоновой кислоты и первичного амина с образованием амида
Получение 2-трет-бутоксикарбониламино-3-(1H-индол-3-ил)- N-(2-метоксибензил)пропанамида
К раствору N-(трет-бутоксикарбонил)триптофана (46,4 г, 152,6 ммоль) в 500 мл диоксана добавляют порциями карбонилдиимидазол (25,4 г, 156 ммоль). Получающуюся в результате смесь перемешивают в течение 2,5 часов при комнатной температуре и затем перемешивают при 45oC в течение 30 минут. Затем добавляют 2-метоксибензиламин (20,7 мл, 158,7 ммоль), и реакционную смесь затем перемешивают в течение 16 часов при комнатной температуре.
Удаляют диоксан при пониженном давлении. Продукт обрабатывают этилацетатом и водой, и органический слой последовательно промывают IN соляной кислотой, насыщенным раствором бикарбоната натрия, водой и соляным раствором, затем сушат над сульфатом натрия, и удаляют растворитель. Заключительная кристаллизация из метанола дает 52,2 г однородного продукта в виде желтых кристаллов.
Выход 80,8%, т.пл. 157-160oC.
Отщепление защитной группы первичного амина
Синтез 2-амино-3-(1H-индол-3-ил)-N-(2-метоксибензил)-пропанамида
К смеси 2-трет-бутоксикарбониламино-3-(1H-индол-3-ил)-N- (2-метоксибензил)пропанамида, полученного выше (25,1 г, 59,2 ммоль), и анизола (12 мл, 110,4 ммоль) при 0oC добавляют по каплям водный раствор трифторуксусной кислоты (118 мл, 1,53 ммоль) в 50 мл воды. Эту смесь перемешивают при 0oC в течение одного часа, и затем продолжают перемешивание в течение 2,5 часов при температуре окружающей среды. Затем смесь сильно охлаждают в течение 16 часов.
Удаляют летучие при пониженном давлении. Продукт обрабатывают этилацетатом и насыщенным раствором бикарбоната натрия, и затем органический слой промывают соляным раствором и сушат его над сульфатом натрия. Растворители удаляют под вакуумом. Перекристаллизация из раствора в диэтиловом эфире с циклогексаном (1:1) дает 18,0 г (94,2%) однородного продукта в виде не совсем белого порошка. Т.пл. 104-108oC.
Способ B
Восстановление амидкарбонила
Синтез 2-амино-3-(1H-индол-3-ил)-1-[N-(2-метоксибензил)-амино]- пропана
К кипящему, с обратным холодильником, раствору 2-амино-3- (1H-индол-3-ил)-N-(2-метоксибензил)пропанамида (9,81 г, 30,3 ммоль), полученного так, как описано выше, в 100 мл безводного тетрагидрофурана добавляют по каплям 10 М комплекс боран-метил-сульфид (9,1 мл, 91,0 ммоль). Получающуюся в результате смесь кипятят с обратным холодильником в течение 2 часов. Затем смесь охлаждают до комнатной температуры, и избыток борана гасят добавлением по каплям 160 мл метанола. Получающуюся в результате смесь кипятят с обратным холодильником в течение 15 минут, и удаляют метанол при пониженном давлении.
Остаток растворяют в насыщенном метанольном растворе хлористоводородной кислоты (250 мл), и раствор кипятят с обратным холодильником в течение 1 часа. Удаляют метанол под вакуумом, и продукт выделяют добавлением 5N раствора гидроксида натрия с последующей экстракцией диэтиловым эфиром. Затем продукт сушат над сульфатом натрия. Растворители удаляют под вакуумом. Флэш-хроматография (силикагель, элюирование смесью метанол:метилен:хлорид:гидроксид аммония - 10: 100: 0,5) дает 7,1 г смеси названного в заголовке соединения (75%) и индолинового производного названного в заголовке соединения (25%) в виде янтарного масла.
Способ C
Ацилирование первичного амина
Получение 3-(1H-индол-3-ил)-1-[N-(2-метоксибензил)амино]-2- [N-(2-((4-фенил)пиперазин-1-ил)ацетил)амино]пропана [соединение примера 17]
Смесь натриевой соли 2-((4-фенил)пиперазин-1-ил)уксусной кислоты (1,64 г, 6,8 ммоль) и гидробромида триэтиламина (1,24 г, 6,8 ммоль) в 35 мл безводного диметилформамида нагревают до 50oC и оставляют при этой температуре на 35 минут. Дают возможность смеси остыть до комнатной температуры. К смеси добавляют 1,1-карбонилдиимидазол (1,05 г, 6,5 ммоль). Получающуюся в результате смесь перемешивают в течение 3 часов при комнатной температуре.
К этой реакционной смеси добавляют 2-амино-3-(1H-индол-3- ил)-[N-(2-метоксибензил)амино]пропан (75%) и индолиновое производное (25%), полученные выше, растворенные в 10 мл безводного диметилформамида. Получающуюся в результате смесь перемешивают в течение 16 часов при комнатной температуре. Удаляют диметилформамид при пониженном давлении.
Названное в заголовке соединение и его индолиновое производное обрабатывают этилацетатом и водой и затем промывают соляным раствором и сушат над сульфатом натрия. Растворители удаляют под вакуумом. Этот процесс дает 3,2 г смеси названного в заголовке соединения и его индолинового производного в виде желтого масла. Эти два соединения затем разделяют высокоэффективной жидкостной хроматографией, используя колонку для хроматографии с обращенной фазой и затем колонку с силикагелем, и получают названный в заголовке продукт (выход 5,2%) в виде желтой пены.
Способ D
Методика ацилирования вторичного амина
Получение 1-[N-этоксикарбонил-N-(2-метоксибензил)амино] - 3-(1H-индол-3-ил)-2-[N-(2-((4-фенил)пиперазин-1-ил)ацетил)aмино] - пропана [соединение примера 28]
К раствору 3-(1H-индол-3-ил)-1-[N-(2-метоксибензил)амино] - 2-[N-(2-((4-фенил)пиперазин-1-ил)ацетил)амино] пропана (0,43 г, 0,85 ммоль) и триэтиламина (130 мкл, 0,93 ммоль) в 5 мл безводного тетрагидрофурана добавляют по каплям этилхлорформиат (89 мкл, 0,93 ммоль). Получающуюся в результате смесь перемешивают в течение 16 часов при комнатной температуре. Тетрагидрофуран удаляют при пониженном давлении.
Ацилированный продукт распределяют между этилацетатом и 0,2N раствором гидроксида натрия, и затем органическую фазу промывают последовательно водой и соляным раствором и сушат над сульфатом натрия. Флэш-хроматография (силикагель, метанол:метиленхлорид - 2,5:97,5) дает 390 мг однородного, названного в заголовке продукта в виде белой пены.
Получение 3-(1H-индол-3-ил)-1-[N-(2-метоксибензил)-N- (метиламинокарбонил)амино-2-[N-(2-((4-фенил)пиперазин-1-ил)- ацетил)-амино] пропана [соединение примера 29]
К раствору 3-(1H-индол-3-ил)-1-[N-(2-метоксибензил)амино] - 2-[N-(2-((4-фенил)пиперазин-1-ил)ацетил)амино] пропана (0,40 г, 0,78 ммоль) в 10 мл безводного тетрагидрофурана при комнатной температуре добавляют по каплям метилизоцианат (140 мкл, 2,3 ммоль). Получающуюся в результате смесь затем перемешивают в течение 16 часов при комнатной температуре. Удаляют тетрагидрофуран под вакуумом. Названный в заголовке продукт выделяют посредством последовательного промывания этилацетатом, водой и соляным раствором и последующей сушки над сульфатом натрия. Флэш-хроматография с применением силикагеля и метанола с метиленхлоридом (5/95) в качестве элюента дает 396 мг однородного продукта в виде желтого масла.
Алкилирование вторичного амина
Получение 1-[N-этил-N-(2-метоксибензил)амино]-3-(1H- индол-3-ил)-2-[N-(2-((4-фенил)пиперазин-1-ил)ацетил)амино]- пропана [соединение примера 9]
К раствору 3-(1H-индол-3-ил)-1-[N-(2-метоксибензил)амино] - 2-[N-(2-((4-фенил)пиперазин-1-ил)ацетил)амино] пропана (0,41 г, 0,80 ммоль) в 5 мл безводного N, N-диметилформамида при комнатной температуре добавляют этилйодид (120 мкл, 1,5 ммоль) и карбонат калия (120 мг, 0,87 ммоль). Эту смесь затем нагревают до 50oC и выдерживают при этой температуре в течение 4 часов, после чего ее перемешивают при комнатной температуре в течение 16 часов. Затем удаляют N,N-диметилформамид при пониженном давлении. Продукт распределяют между этилацетатом и водой, и затем промывают органическую фазу соляным раствором и сушат над сульфатом натрия. Растворители удаляют под вакуумом. Препаративной тонкослойной хроматографией получают 360 мг названного в заголовке продукта в виде желтой пены.
Способ E
Восстановление карбонила амида
Получение 1,2-диамино-3-(1H-индол-3-ил)пропана
К раствору триптофанамида (20,3 г, 0,1 моль) в тетрагидрофуране (24,4 мл) при комнатной температуре и при перемешивании добавляют эфират трифторида бора (12,3 мл, 0,1 ммоль). При кипячении с обратным холодильником и постоянном перемешивании добавляют по каплям боран-метилсульфид (32,25 мл, 0,34 моль). Реакционную смесь кипятят с обратным холодильником при перемешивании в течение пяти часов. Осторожно, по каплям, добавляют смесь тетрагидрофурана с водой (26 мл, 1:1). Добавляют раствор гидроксида натрия (160 мл, 5N), и смесь кипятят с обратным холодильником и при перемешивании в течение 16 часов.
Разделяют слои охлажденной смеси, и водный слой экстрагируют тетрагидрофураном два раза, каждый раз по 40 мл. Эти тетрагидрофурановые экстракты объединяют и упаривают. Добавляют этилацетат (800 мл), и этот раствор промывают насыщенным раствором хлорида натрия три раза по 80 мл. Этилацетатный экстракт сушат над сульфатом натрия, фильтруют и упаривают, получают 18,4 г (97%) названного в заголовке соединения.
Защита первичного амина
Получение 2-амино-1-трет-бутоксикарбониламино-3-(1H-индол-3-ил) пропана
К полученному выше 1,2-диамино-3-(1H-индол-3-ил) пропану (1,06 г, 5,6 ммоль), который растворяют в 28 мл тетрагидрофурана, при комнатной температуре добавляют по каплям ди-трет-бутилдикарбонат (0,90 мл, 3,9 ммоль) в 10 мл тетрагидрофурана. Это добавление по каплям осуществляют в течение 5 часов. Упаривают растворитель. Флэш-хроматография с применением этанола/гидроксида аммония/этилацетата дает 0,51 г (1,76 ммоль, 31%) нужного карбамата.
Ацилирование вторичного амина
Получение 1-трет-бутоксикарбониламино-3-(1H-индол-3-ил)- 2-[N-(2-((4-фенил)пиперазин-1-ил)ацетил)амино]пропана [соединение примера 151]
Суспензию 2-((4-фенил)пиперазин-1-ил)уксусной кислоты (2,47 г, 11,2 ммоль) и триэтиламина (3,13 мл, 22,5 ммоль) в ацетонитриле (1200 мл) при перемешивании кипятят с обратным холодильником непродолжительное время. Пока получающийся в результате раствор все еще остается теплым, добавляют карбонилдиимидазол (1,82 г, 11,2 ммоль), и смесь кипятят с обратным холодильником в течение 10 минут. Затем к реакционной смеси добавляют 2-амино-1-трет-бутоксикарбониламино-3-(1H-индол-3-ил)-пропан (3,25 г, 11,2 ммоль) в 50 мл ацетонитрила. Получающуюся в результате смесь кипятят с обратным холодильником при перемешивании в течение 30 минут, и затем в течение ночи перемешивают при комнатной температуре.
Реакционную смесь затем кипятят с обратным холодильником при перемешивании в течение 5 часов, и затем удаляют растворитель под вакуумом. Остающееся масло промывают раствором карбоната натрия, а затем водой шесть раз, после чего промывают насыщенным раствором хлорида натрия. Получающуюся в результате жидкость сушат над сульфатом натрия и фильтруют. Оставшийся остаток затем сушат под вакуумом. Фильтрат уменьшают в объеме, и затем частично очищают хроматографией. Образец с хроматографии объединяют с остатком на фильтре, получая в общем 3,94 г (выход 72%) названного в заголовке продукта.
Способ F
Отщепление защитной группы первичного амина
Синтез 1-амино-3-(1H-индол-3-ил)-2-[N-(2-((4-фенил)- пиперазин-1-ил)ацетил)амино]пропана [соединение примера 150]
К охлажденному льдом 70% водному раствору трифторуксусной кислоты (2,8 мл трифторуксусной кислоты в общем объеме 4 мл) добавляют 1-трет-бутоксикарбониламино-3-(1H-индол-3-ил)-2-[N- (2-((4-фенил)пиперазин-1-ил)ацетил)амино] пропан (0,80 г, 1,63 ммоль) и анизол (4 мл). Эту смесь перемешивают в течение 35 минут, получая в результате прозрачный раствор. Раствор перемешивают еще в течение одного часа и затем упаривают.
Затем к остающейся в результате жидкости добавляют этилацетат, после чего промывают раствором карбоната натрия. После этой промывки осуществляют еще три промывки насыщенным раствором хлорида натрия. Образующийся в результате раствор затем сушат над сульфатом натрия, фильтруют и упаривают, получают 0,576 г (выход 90%) названного в заголовке продукта реакции.
Способ G
Гидроалкилирование первичного амина
Получение 1-[N-(2-хлорбензил)амино] -3-(1H-индол-3-ил)-2- [N-(2-((4-фенил)пиперазин-1-ил)ацетил)амино]пропана [соединение примера 2]
Соединяют в толуоле 2-хлорбензальдегид (0,112 г, 0,8 ммоль) и 1-амино-3-(1H-индол-3-ил)-2-[N-(2-((4-фенил)пиперазин-1- ил)ацетил)амино] пропан (0,156 г, 0,398 ммоль). Получающуюся в результате смесь затем перемешивают и нагревают и затем упаривают. К остатку добавляют толуол, и эту смесь снова упаривают. К остатку добавляют тетрагидрофуран, и смесь затем охлаждают на ледяной бане.
Затем к реакционной смеси добавляют цианоборогидрид натрия (0,025 г, 0,4 ммоль). К вышеупомянутой жидкой смеси периодически добавляют газообразный хлористый водород. Смесь перемешивают при комнатной температуре в течение 16 часов, и затем уменьшают ее объем под вакуумом.
К остатку затем добавляют разбавленную соляную кислоту, и раствор дважды экстрагируют эфиром. Кислый водный экстракт подщелачивают, добавляя по каплям 5N раствор гидроксида натрия. Этот подщелаченный раствор затем экстрагируют этилацетатом. Объединенные этилацетатные экстракты промывают насыщенным раствором хлорида натрия, сушат над сульфатом натрия, фильтруют и упаривают. Этот процесс с последующей хроматографией дает на выходе 0,163 г (выход 79%) названного в заголовке продукта реакции.
Способ H
Тритилирование
Получение 3-(1H-индол-3-ил)-2-(N-трифенилметиламино)пропанамида
Триптофанамид (26,43 г, 0,130 моль) суспендируют в 260 мл метиленхлорида, и эту смесь промывают азотом, а затем создают атмосферу аргона. Растворяют тритилхлорид (38,06 г, 0,136 моль) в 75 мл метиленхлорида. Раствор тритилхлорида медленно добавляют к раствору триптофанамида, который помещен на ледяную баню, причем добавление продолжается в течение 25 минут. Затем реакционную смесь перемешивают в течение ночи.
Реакционную смесь затем выливают в делительную воронку и промывают 250 мл воды, а затем 250 мл соляного раствора. Когда органический слой фильтруют через сульфат натрия для осушки, твердое вещество выпадает в осадок. Фильтрат собирают, и удаляют растворитель.
Затем к собранному твердому веществу добавляют этилацетат, и эту смесь перемешивают и затем сильно охлаждают в течение ночи. На следующий день получающиеся в результате твердое вещество несколько раз промывают холодным этилацетатом и затем сушат под вакуумом. Выход 49,76 г (85,9%).
Восстановление карбонила
Получение 1-амино-3-(1H-индол-3-ил)-2-(N-трифенилметиламино)- пропана
В атмосфере аргона суспендируют 3-(1H-индол-3-ил)-2-(N- трифенилметиламино)пропанамид (48,46 г, 0,108 моль) в 270 мл тетрагидрофурана. Эту смесь затем кипятят с обратным холодильником. Затем к реакционной смеси медленно добавляют комплекс боран-метилсульфид (41,3 г, 0,543 моль). Во время добавления боран-метилсульфидного комплекса растворяется весь исходный амид. Этот раствор затем перемешивают в течение ночи при 83oC на масляной бане.
После охлаждения раствора к нему добавляют смесь тетрагидрофурана с водой (1: 1, всего 75 мл). Затем к смеси добавляют раствор гидроксида натрия (5N, 230 мл), и смесь затем кипятят с обратным холодильником в течение 30 минут.
После разделения водного и органического слоев собирают органический слой. Водный слой затем экстрагируют тетрагидрофураном. Органические слои объединяют, и затем удаляют растворители упариванием. Получающуюся в результате жидкость обрабатывают этилацетатом и соляным раствором, и второй раз промывают соляным раствором. Затем раствор сушат над сульфатом натрия, и растворители удаляют под вакуумом, получают 48,68 г нужного промежуточного соединения.
Замещение первичного амина
Получение 1-[N-(2-метоксибензил)амино]-3-(1H-индол-3-ил)- 2-(N-трифенилметиламино)пропана
К 1-амино-3-(1H-индол-3-ил)-2-(N-трифенилметиламино)пропану (48,68 г, 0,109 моль), растворенному в толуоле (1,13 л), добавляют 2-метоксибензальдегид (23,12 г, 0,169 моль), причем 2-метоксибензальдегид предварительно очищают путем промывания основанием. Реакционную смесь перемешивают в течение ночи. Растворители удаляют под вакуумом.
Извлечение твердое вещество растворяют в 376 мл смеси тетрагидрофурана с метанолом (1:1). К этому раствору добавляют борогидрид натрия (6,83 г, 0,180 моль). Эту смесь перемешивают на ледяной бане в течение 4 часов. Растворители удаляют упариванием. Оставшуюся жидкость обрабатывают 1200 мл этилацетата и 1000 мл смеси соляного раствора и 20N раствора гидроксида натрия (1:1). Эту часть экстрагируют дважды этилацетатом, каждый раз по 500 мл, и затем экстракты сушат над сульфатом натрия. Затем растворители удаляют путем упаривания в течение ночи, и получают 67,60 г (выход > 99%) нужного продукта.
Способ J
Тритилирование
Получение 3-(1H-индол-3-ил)-2-(N-трифенилметиламино)-пропановой кислоты [N-тритилтриптофан]
К суспензии триптофана (100,0 г, 0,490 моль) в безводном метиленхлориде (800 мл) в атмосфере азота при перемешивании добавляют с умеренной скоростью хлортриметилсилан (70,0 мл, 0,527 моль). Эту смесь непрерывно перемешивают в течение 4,25 часов. Добавляют триэтиламин (147,0 мл, 1,055 моль), а затем добавляют раствор трифенилметилхлорида (147,0 г, 0,552 моль) в метиленхлориде (400 мл), используя капельную воронку. Смесь перемешивают при комнатной температуре в атмосфере азота по крайней мере в течение 20 часов. Реакцию гасят путем добавления метанола (500 мл).
Раствор концентрируют на роторном испарителе почти досуха, и смесь снова растворяют в метиленхлориде и этилацетате. Затем осуществляют водную обработку, включающую обработку 5% лимонной кислотой (2x) и соляным раствором (2x). Органический слой сушат над сульфатом натрия, фильтруют и концентрируют досуха на роторном испарителе. Твердое вещество растворяют в горячем диэтиловом эфире, и затем добавляют гексан для промотирования кристаллизации. Таким способом выделяют 173,6 г (0,389 моль) чистой для анализа 3-(1H-индол-3-ил)-2-(N-трифенилметиламино)пропановой кислоты в виде светлого желтовато-коричневого твердого вещества, причем за два сбора получают общий выход 79%.
Сочетание
Получение 3-(1H-индол-3-ил)-N-(2-метоксибензил)-2-(N- трифенилметиламино)пропанамид
К раствору 3-(1H-индол-3-ил)-2-(N-трифенилметиламино) пропановой кислоты (179,8 г, 0,403 моль), 2-метоксибензиламина (56,0 мл, 0,429 моль) и гидрата гидроксибензотриазола (57,97 г, 0,429 моль) в безводном тетрагидрофуране (1,7 л) и в безводном N,N-диметилформамиде (500 мл) при перемешивании в атмосфере азота при 0oC добавляют триэтиламин (60,0 мл, 0,430 моль) и гидрохлорид 1-(3-диметиламинопропил)-3-этоксикарбодиимида (82,25 г, 0,429 моль). Смеси дают возможность нагреваться до комнатной температуры в атмосфере азота по крайней мере в течение 20 часов. Смесь концентрируют на роторном испарителе и затем снова растворяют в метиленхлориде, и осуществляют водную обработку 5% раствором лимонной кислоты (2x), насыщенным раствором бикарбоната натрия (2x) и соляным раствором (2x). Органический слой сушат над безводным сульфатом натрия, фильтруют и концентрируют досуха на роторном испарителе. Названный в заголовке продукт затем отфильтровывают в виде розового твердого вещества в двух партиях. Выделяют 215,8 г (0,381 моль) вещества, чистого для анализа (выход 95%).
Восстановление
Получение 3-(1H-индол-3-ил)-1-[N-(2-метоксибензил)амино] -2- (N-трифенилметиламино)пропана
Red-Al® [3,4M, раствор бис(2-метоксиэтокси)алюмогидрида натрия в толуоле] (535 мл, 1,819 моль), растворенный в безводном тетрагидрофуране (400 мл), постепенно добавляют, используя капельную воронку, к кипящему с обратным холодильником раствору полученного выше продукта ацилирования - 3-(1H-индол-3-ил)-N- (2-метоксибензил)-2-(N-трифенилметиламино)пропанамида (228,6 г, 0,404 моль) в безводном тетрагидрофуране (1,0 л) в атмосфере азота. Реакционная смесь становится раствором пурпурного цвета. Реакцию гасят по крайней мере через 20 часов путем постепенного добавления избытка насыщенного раствора сегнетовой соли (тетрагидрат виннокислого калия-натрия). Органический слой отделяют, промывают соляным раствором (2x), сушат над безводным сульфатом натрия, фильтруют и концентрируют до масла на роторном испарителе. Дальнейшую очистку не проводят, и продукт используют непосредственно на следующей стадии.
Способ K
Ацилирование
Получение 3-(1H-индол-3-ил)-1-[N-(2-метоксибензил)- ацетиламино]-2-[N-трифенилметиламино)пропана
К раствору 3-(1H-индол-3-ил)-1-[N-(2-метоксибензил)- амино]-2-(N-трифенилметиламино)пропана (0,404 моль) в безводном тетрагидрофуране (1,2 л) при перемешивании, в атмосфере азота, при 0oC добавляют триэтиламин (66,5 мл, 0,477 моль) и уксусный ангидрид (45,0 мл, 0,477 моль). Через 4 часа смесь концентрируют на роторном испарителе, снова растворяют в метиленхлориде и этилацетате, промывают водой (2x) и соляным раствором (2x), сушат над безводным сульфатом натрия, фильтруют и концентрируют до твердого вещества на роторном испарителе. Получающееся в результате твердое вещество растворяют в хлороформе, и раствор загружают на силикагель 60 (230-400 меш) и элюируют смесью этилацетата с гексаном (1:1). Продукт затем кристаллизуют из смеси этилацетата с гексаном. Получающийся в результате продукт-3-(1H-индол-3-ил)-1-[N- (2-метоксибензоил)ацетиламино] -2-(N-трифенилметиламино)пропан - кристаллизуют и выделяют в три приема, получая 208,97 г (выход 87%) вещества, чистого для анализа.
Способ L
Детритилирование
Получение 2-амино-3-(1H-индол-3-ил)-1-[N-(2-метоксибензил)- ацетиламино] пропана
К раствору 3-(1H-индол-3-ил)-1-[N-(2-метоксибензил)- ацетиламино]-2-(N-трифенилметиламино)пропана (14,11 г, 23,763 ммоль) в безводном метиленхлориде при перемешивании, в атмосфере азота, при 0oC добавляют муравьиную кислоту (9,0 мл, 238,540 ммоль). Через 4 часа реакционную смесь концентрируют до масла на роторном испарителе и снова растворяют в диэтиловом эфире и 1,0N соляной кислоте. Водный слой дважды промывают диэтиловым эфиром и подщелачивают до pH свыше 12 гидроксидом натрия. Продукт экстрагируют метиленхлоридом (4x). Органические экстракты объединяют, сушат над безводным сульфатом натрия и концентрируют на роторном испарителе до белой пены. Выделяют соединение 2-амино- 3-(1H-индол-3-ил)-1-[N-(2-метоксибензил)ацетиламино] пропан (7,52 г, 21,397 ммоль), получая выход 90%. В дальнейшей очистке нет необходимости.
Способ M
Бромацетилирование
Получение 2-[(2-бром)ацетил] амино-3-(1H-индол-3-ил)-1-[N- (2-метоксибензил)ацетиламино]пропана
К раствору 2-амино-3-(1H-индол-3-ил)-1-[N-(2-метоксибензил)- ацетиламино] пропана (7,51 г, 21,369 ммоль) в безводном тетрагидрофуране (100 мл) при перемешивании, в атмосфере азота, при 0oC добавляют диизопропилэтиламин (4,1 мл, 23,537 ммоль) и бромацетилбромид (2,05 мл, 23,530 ммоль). Через 2 часа добавляют этилацетат, и реакционную смесь промывают дважды водой, 1,0N соляной кислотой (2x), насыщенным раствором бикарбоната натрия (2x) и соляным раствором. Органический слой сушат над безводным сульфатом натрия, фильтруют и концентрируют на роторном испарителе до желто-коричневой пены. Таким способом получают 2-[(2-бром)ацетил] -амино-3-(1H- индол-3-ил)-1-[N-(2-метоксибензил)ацетиламино] пропан с количественным выходом. В дальнейшей очистке нет необходимости.
Способ N
Нуклеофильное замещение
Получение 1-[N-(2-метоксибензил)ацетиламино] -3-(1H-индол- 3-ил)-2-[N-(2-((4-циклогексил)пиперазин-1-ил)ацетил)амино] пропана [соединение примера 74]
К раствору 2-[(2-бром)ацетил] амино-3-(1H-индол-3-ил)-1- [N-2-метоксибензол)ацетиламино]пропана (21,369 ммоль) и порошка карбоната калия (3,56 г, 25,758 ммоль) в метиленхлориде при перемешивании, в атмосфере азота, добавляют 1-циклогексилпиперазин (3,65 г, 22,492 ммоль). Реакционную смесь перемешивают в течение ночи при комнатной температуре. Соли отфильтровывают, и раствор концентрируют на роторном испарителе до коричневой пены. Нужный продукт очищают на колонке Prep 500, используя 10 л элюента с градиентом, начиная от 100% метиленхлорида и заканчивая смесью 5% метанола, 94,5% метиленхлорида и 0,5% гидроксида аммония. Загрязненные фракции объединяют и очищают далее препаративной высокоэффективной жидкостной хроматографией с обращенной фазой (метанол/ ацетонитрил/вода/ацетат аммония). После объединения вещества с обеих хроматографических очисток выделяют 10,43 г (18,663 ммоль) названного в заголовке соединения (выход 87%).
Альтернативным способом ацилирования первичного амина, как показывает конечная стадия прописи синтеза по схеме IV, является взаимодействие соединения формулы
с карбоксилатом калия формулы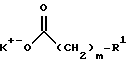
в присутствии изобутилхлорформиата и N-метилморфолина. Эту реакцию обычно осуществляют в присутствии растворителя, не принимающего участия в реакции, такого как метиленхлорид, при пониженных температурах, обычно от -30oC до 10oC, предпочтительнее - при температуре от -20oC до 0oC. В этой реакции, как правило, используют эквимолярные количества обоих реагентов, хотя рабочими являются и другие соотношения. Пример такого способа ацилирования первичного амина приводится в следующем далее примере.
Способ P
Получение (R)-1-[N-(2-метоксибензил)ацетиламино-3-(1H- индол-3-ил)-2-[N-(2-((4-циклогексил)пиперазин-1-ил)ацетил)- амино] -пропана [соединение примера 75]
Названное в заголовке соединение получают, охлаждая сначала калиевую соль 2-((4-циклогексил) пиперазин-1-ил) уксусной кислоты до температуры от -8oC до -15oC в 5 объемах безводного метиленхлорида. Затем к этой смеси добавляют изобутилхлорформиат с такой скоростью, чтобы температура не превышала -8oC. Эту реакционную смесь затем перемешивают в течение 1 часа, причем поддерживают температуру в интервале от -8oC до -15oC.
Затем к этой смеси добавляют дигидрохлорид (R)-2-амино-3- (1H-индол-3-ил)-1-[N-(2-метоксибензил)ацетиламино] пропана с такой скоростью, чтобы температура не поднималась выше 0oC. Далее к этой смеси добавляют N-метилморфолин с такой скоростью, чтобы температура не превышала 0oC. Эту смесь затем перемешивают в течение 1 часа при температуре от -15oC до -8oC.
Реакцию гасят путем добавления 5 объемов воды. Органический слой один раз промывают насыщенным раствором бикарбоната натрия. Затем органическую фазу сушат над безводным карбонатом калия и фильтруют для удаления осушающего агента. К фильтрату затем добавляют 2 эквивалента концентрированной соляной кислоты, и после этого добавляют 1 объем изопропилового спирта. Затем заменяют метиленхлорид изопропиловым спиртом под вакуумом путем перегонки.
Конечный объем изопропилового спирта затем под вакуумом снижают до трех объемов. Затем реакционную смесь охлаждают до 20-25oC, и дают возможность продукту реакции кристаллизоваться по крайней мере в течение одного часа. Затем нужный продукт извлекают фильтрацией, и промывают его достаточным количеством изопропилового спирта, чтобы получить бесцветный фильтрат. Затем кристаллический остаток на фильтре сушат под вакуумом при 50oC.
Табл. 1 показывает множество соединений, полученных, по существу, с применением стадий, описанных на схемах I-IV. Специалист в этой области техники может легко понять, что определенный порядок стадий должен использоваться во многих случаях, чтобы избежать реакций, иных, чем те, к которым стремятся. Например, как в вышеупомянутых способах, часто является необходимостью использование защитной группы для блокирования реакции в определенной части молекулы.
Аббревиатуры, которые используются в табл.1, являются обычно употребляемыми в этой области техники и должны быть без труда понятными для практика, работающего в этой области техники. Например, аббревиатура "Ph" означает фенильную группу, "i-Pr" означает изопропильную группу, "Me" обозначает метильную группу, "Et" относится к этильной группе, "t-Вu" описывает трет-бутильную группу и т.п.
В табл.1 в первой колонке указывается номер примера соединения. В следующих колонках (это могут быть одна, две или три колонки) описываются образцы заместителей конкретного примера соединения. В колонке, озаглавленной "MpoC" (Т. пл.oC), указывается температура плавления соединения, если оно находится в твердом состоянии, или отмечается форма вещества при температуре окружающей среды. В следующей колонке, озаглавленной "MS" (м.-с.), указывается масса соединения, которая определяется масс-спектроскопией. В следующей колонке указывается профиль ядерного магнитного резонанса синтезированного соединения-примера. В последних колонках приводится молекулярная формула соединения- примера, а также его элементный анализ.
В табл.1 в данных о спектрах ЯМР употребляются следующие обозначения: s - синглет, d - дублет, t - триплет, q - квартет, m - мультиплет, dd - двойной дублет, brs - широкий синглет, brd - широкий дублет, brt - широкий триплет и Hz - герц.
Биологическую активность соединений настоящего изобретения оценивают, используя начальный скрининг-анализ, который быстро и точно измеряет связывание испытуемого соединения с известными сайтами рецепторов NK-1 и NK-2. Испытания, пригодные для оценки антагонистов тахикининов, хорошо известных в технике. См. , например, J.Jukic et al. Life Sciences, 49:1463-1469 (1991); N. Kucharczyk, et al., Journal of Medicinal Chemistry, 36: 1654-1661 (1993); N. Rouissi, et al. , Biochemical and Biophysical Research Communications, 176:894-901 (1991).
Анализ связывания рецептора NK-1
Радиорецепторный анализ связывания осуществляют, используя в качестве основы опубликованную схему - D.G. Payan, et. al., Journal of Immunology, 133: 3260-3265 (1984). При этом анализе аликвоту клеток 1M9 (1 • 106 клеток/пробирку в среде RPMI 1604 с добавлением 10% фетальной телячьей сыворотки) инкубируют с 20 pM вещества P, меченного 125I, в присутствии конкурента в возрастающих концентрациях, в течение 45 минут при 4oC.
Клеточная линия 1M9 является хорошо охарактеризованной и легко доступной линией человеческих клеток. См., например, Annals of the New York Academy of Science, 190: 221-234 (1972); Nature (London), 251:443-444 (1974); Proceedings of the National Academy of Sciences (USA), 71:84-88 (1974). Эти клетки выращивают обычным способом в RPMI 1640 с добавлением 50 мгк/мл гентамицинсульфата и 10% фетальной телячьей сыворотки.
Реакцию завершают фильтрацией через фильтр из стекловолокна в системе для сбора выросших в культуре клеток, применяя фильтры, предварительно замоченные в течение 20 минут в 0,1% полиэтиленимине. Специфическое связывание меченого вещества P определяют в присутствии 20 нМ немеченого лиганда.
Анализ связывания рецептора NK-2
Клетки CHO-hNK-2R, CHO-производные от клеточной линии, трансформированной человеческим NK-2-рецептором, экспрессирующие около 400000 таких рецепторов на клетку, выращивают в 75-см3 колбах или роллер-флаконах в минимальной поддерживающей среде (альфа-модификации) с 10% фетальной бычьей сыворотки. Генная последовательность человеческого NK-2-рецептора дается в N.P. Gerard, et al. Journal of Biological Chemistry, 265:20455-20462 (1990).
Для получения мембран 30 конфлюентных роллерных культур разобщают путем промывания каждого роллер-флакона 10 мл физиологического раствора, забуференного фосфатом Дульбекко (ЗФР), без кальция и магния, с последующим добавлением 10 мл бесферментного раствора для разъединения клеток (на основе ЗФР, от Speciality Media, Inc.). Спустя 15 минут, диссоциированные клетки собирают и центрифугируют при 1000 об/мин в течение 10 минут на клинической центрифуге. Мембраны получают гомогенизацией клеточных осадков в 300 мл 50 мМ трисбуфера, pH 7,4, на гомогенизаторе Tekmar® в течение 10- 15 секунд с последующим центрифугированием при 12000 об/мин (20000 • q), при применении ротора Beckman JA-14®. Осадки в пробирках промывают один раз, используя вышеописанную процедуру. Осадки ресуспендируют в 100-120 мл 50 мМ трис-буфера, pH 7,4 и 4 мл аликвоты хранят в замороженном состоянии при -70oC. Концентрация белка в таком препарате составляет 2 мг/мл.
Для анализа рецепторного связывания одну 4-мл аликвоту препарата мембран CHO-hNK-2R суспендируют в 40 мл проверочного буфера, содержащего 50 мМ трис, pH 7,4, 3 мМ хлорида марганца, 0,02% альбумина бычьей сыворотки (BSA) и 4 мкг/мл химостатина. Для образца используют 200 мкл гомогената (40 мкг белка). Радиоактивный лиганд представляет собой [125I] йодогистидил-нейрокинин A (New England Nuclear, NEX- 252), 2200 Ки/ммоль. Лиганд готовят в проверочном буфере с 20 нКи на 100 мкл; конечная концентрация при анализе составляет 20 pM. Неспецифическое связывание определяют с применением 1 мкМ эледоизина. Для построения стандартной кривой "концентрация-отклик" используют десять концентраций эледоизина от 0,1 до 1000 нМ.
Все образцы и эталоны для инкубации добавляют в 10 мкл диметилсульфоксида (ДМСО) для скрининга (единичная доза) или 5 мкл ДМСО для определения IC50. Порядок добавления для инкубации: 190 или 195 мкл проверочного буфера, 200 мкл гомогената, 10 или 5 мкл образца в ДМСО, 100 мкл радиоактивного лиганда. Образцы инкубируют в течение 1 часа при комнатной температуре и затем фильтруют на 48-луночном харвестере клеток Brandel через фильтры GF/B, которые предварительно замачивают в течение двух часов в 50 мМ трис-буфере, pH 7,7, содержащем 0,5 % BSA. Фильтр промывают 3 раза приблизительно 3 мл холодного 50 мМ трис-буфера, pH 7,7. Эти круглые фильтры затем помещают в полистирольные пробирки 12х75 мм, и проводят подсчет радиоактивности на счетчике гамма-квантов.
В приведенной ниже табл. 2 даются результаты некоторых таких анализов связывания нейрокинина. В первой колонке дается номер примера соединения, которое проверяют как антагониста, из числа соединений, которые подробно описаны в представленной выше табл. 1. В следующих колонках указывается концентрация проверяемого соединения (в наномолях), при которой ингибируются пятьдесят процентов связывания соответствующего нейрокинина, как указывается в заголовке, или процент ингибирования такого связывания при указанной концентрации. Некоторые величины являются средними результатами нескольких экспериментов.
Поскольку соединения формулы I являются эффективными антагонистами тахикининовых рецепторов, эти соединения имеют значение при лечении широкого ряда клинических состояний, которые характеризуются наличием избытка тахикинина. Таким образом, настоящее изобретение предлагает способ лечения или предупреждения физиологического нарушения, ассоциируемого с избытком тахикининов, и этот способ включает введение млекопитающему, нуждающемуся в упомянутом лечении, эффективного количества соединения формулы I или его фармацевтически приемлемой соли. Термин "физиологическое нарушение, ассоциируемое с избытком тахикининов", включает в себя нарушения, ассоциируемые с несоответствующей стимуляцией тахикининовых рецепторов, не считающейся с фактическим количеством тахикинина, присутствующим в месте действия.
Такие физиологические нарушения могут включать расстройства центральной нервной системы, такие как страх, депрессия, психоз и шизофрения; нейродегенеративные нарушения, такие как деменция, включая сенильную деменцию типа Альцгеймера, болезнь Альцгеймера, деменцию, ассоциируемую со СПИДом, и синдром Дауна; демиелинирующие болезни, такие как рассеянный склероз и боковой амиотрофический склероз, и другие нейропатологические расстройства, такие как периферическая невропатия, такая как диабетическая и вызванная химиотерапией невропатия, и пост-герпетическая и другие невралгии; острые и хронические заболевания с непроходимостью дыхательных путей, такие как респираторный дистресс-синдром взрослых, бронхопневмония, бронхоспазм, хронический бронхит, позывы к кашлю, и астма; воспалительные заболевания, такие как воспаление кишечника, псориаз, фиброз, остеоартрит и ревматоидный артрит; болезни скелетно-мышечной системы, такие как остеопороз, аллергии, такие как экзема и ринит; аллергические расстройства, такие как при действии сумаха; глазные болезни, такие как конъюнктивит, весенний конъюнктивит и подобные; кожные болезни, такие как контактный дерматит, атопический дерматит, крапивница и другие экзематозные дерматиты; расстройства вследствие вредных привычек, такие как алкоголизм; связанные со стрессом соматические нарушения; рефлекторную симпатическую дистрофию, такую как плечевой синдром; дистимические нарушения; побочные иммунологические реакции, такие как отторжение трансплантированных тканей, и нарушения, связанные с иммунным усилением или подавлением, такие как красная системная волчанка; желудочно-кишечные расстройства или болезни, ассоциируемые с невральной регуляцией внутренних органов, такие как неспецифический язвенный колит, болезнь Крона и синдром раздраженной толстой кишки; нарушения функции мочевого пузыря, такие как гиперрефлексия мышц мочевого пузыря и недержание; артериосклероз; болезни соединительной и коллагеновой ткани, такие как склеродермия и эозифильный фасциолиз; раздражающие симптомы доброкачественной гипертрофии простаты; нарушения кровотока, вызванные болезнями с расширением и сужением сосудов, такими как стенокардия, мигрень и болезнь Рейно; и боль или ноцицепцию, например, которые свойственны или ассоциируются с любым из вышеупомянутых состояний, в особенности, передачу боли при мигрени. Например, соединения формулы I могут подойти для применения при лечении расстройств центральной нервной системы, таких как страх, психоз и шизофрения, нейродегенеративных нарушений, таких как болезнь Альцгеймера и синдром Дауна; респираторных заболеваний, таких как бронхоспазм и астма; воспалительных заболеваний, таких как воспаление кишечника, остеоартрит и ревматоидный артрит; побочных иммунологических нарушений, таких как отторжение пересаженных тканей; желудочно-кишечных расстройств и заболеваний, таких как расстройства, ассоциируемые с невральной регуляцией внутренних органов, таких как неспецифический язвенный колит, болезнь Крона и синдром раздраженной толстой кишки; недержания; нарушений кровотока, вызванных вазодилатацией; и боли или ноцицепции, например, такой, какая присуща или которая ассоциируется с любым из вышеупомянутых состояний, или передачи боли при мигрени.
Результаты нескольких экспериментов показывают, что многие соединения формулы I являются селективными антагонистами тахикининовых рецепторов. Эти соединения связываются преимущественно с одним подтипом рецептора тахикинина, по сравнению с другими такими рецепторами. Такие соединения являются особенно предпочтительными.
Например, антагонисты NK-1 являются особенно предпочтительными при лечении боли, особенно-хронической боли, такой как невропатическая боль, послеоперационная боль и мигрень, боли, ассоциируемой с артритом, боли, связанной с раковым заболеванием, хронической боли в нижней части спины, "гистаминовой" головной боли, боли при герпесной невралгии, фантомной боли, центральной боли, зубной боли, невропатической боли, опио-резистантной боли, висцеральной боли, боли при операции, боли при повреждении кости, боли при родах и родоразрешении, боли, являющейся результатом ожога, послеродовой боли, боли при ангине и боли в мочеполовой системе, включая боль при цистите.
Кроме лечения боли, антагонисты NK-1 особенно предпочтительны при лечении и предупреждении недержания мочи; раздражающих симптомов доброкачественной гипертрофии простаты; нарушений подвижности желудочно-кишечного тракта, таких как синдром раздраженной толстой кишки; болезней с острой и хронической непроходимостью дыхательных путей, таких как бронхоспазм, бронхопневмония, астма и респираторный дистресс-синдром взрослых; артериосклероза; воспалительных состояний, таких как воспаление кишечника, неспецифический язвенный колит и болезнь Крона, ревматоидный артрит, остеоартрит, нейрогенное воспаление, аллергия, ринит, кашель, дерматит, крапивница, псориаз, конъюнктивит, вызванный раздражением миоз; отторжения пересаженных тканей; транссудации плазмы, являющейся результатом цитокиновой химиотерапии и подобного; травмы спинного мозга; удара; церебрального удара (ишемии); болезни Альцгеймера; болезни Паркинсона; рассеянного склероза; бокового амиотрофического склероза; шизофрении; страха и депрессии.
Антагонисты NK-2 являются особенно предпочтительными при лечении недержания мочи, бронхоспазма, астмы, респираторного дистресс-синдрома взрослых, нарушений подвижности желудочно-кишечного тракта, таких как синдром раздраженной толстой кишки, и боли.
Кроме описанных выше испытаний на связывание in vitro, многие соединения настоящего изобретения проверены in vivo на модельных системах для состояний, ассоциируемых с избытком тахикининов. Из таких соединений, проверенных in vivo, многие показывают эффективность для упомянутых состояний.
Соединения формулы I вводят, как правило, в форме фармацевтических композиций. Эти соединения можно вводить множеством способов, включая пероральный, ректальный, трансдермальный, подкожный, внутривенный, внутримышечный и интраназальный. Эти соединения являются эффективными как в виде композиций для инъекций, так и в виде композиций для перорального введения. Такие композиции готовят способами, хорошо известными в фармацевтической технике, и такие композиции содержат по крайней мере одно активное соединение.
Настоящее изобретение включает фармацевтические композиции, которые в качестве активного ингредиента содержат соединения формулы I в сочетании с фармацевтически приемлемыми носителями. При изготовлении композиций настоящего изобретения активный ингредиент обычным образом смешивают с эксципиентом, разбавляют эксципиентом или заключают в эксципиент, который может находиться в форме капсулы, саше, пакетика или иной емкости. Когда эксципиент служит в качестве разбавителя, он может представлять собой твердое вещество, полутвердое вещество или жидкость, которые действуют как разбавители, носители или среды для активного ингредиента. Так, композиции могут иметь форму таблеток, пилюль, порошков, лепешек, саше, крахмальных капсул, эликсиров, суспензий, эмульсий, растворов, сиропов, аэрозолей (как в твердой, так и в жидкой среде), мазей, содержащих, например, до 10 мас.% активного соединения, мягких и жестких желатиновых капсул, суппозиториев, стерильных растворов для инъекций и стерильно упакованных порошков.
При приготовлении формулировки может иметься необходимость размолоть активное соединение, чтобы получить соответствующий размер частиц перед тем, как соединять это вещество с другими ингредиентами. Если активное вещество является, по существу, нерастворимым, его обычно измельчают до частиц размером менее 200 меш. Если активное вещество в значительной степени растворяется в воде, обычно размер частиц путем измельчения доводят, например, до 40 меш, чтобы обеспечить по существу равномерное распределение в формулировке.
Некоторыми примерами подходящих эксципиентов являются лактоза, декстроза, сахароза, сорбит, маннит, крахмалы, аравийская камедь, фосфат кальция, альгинаты, трагакант, желатин, силикат кальция, микрокристаллическая целлюлоза, поливинилпирролидон, целлюлоза, вода, сироп и метилцеллюлоза. Формулировки могут включать, дополнительно, смазки, такие как тальк, стеарат магния и вазелиновое масло; смачиватели; эмульгаторы и суспендирующие агенты; консерванты, такие как метил- и пропилгидроксибензоаты; подслащивающие вещества; и корригенты. Композиции настоящего изобретения могут быть составлены с помощью процедур, известных в технике, таким образом, чтобы обеспечить быстрое, длительное или замедленное выделение активного ингредиента после введения пациенту.
Композиции настоящего изобретения формулируют, предпочтительно, в единицах лекарственной формы, причем каждая доза содержит от 5 до 100 мг, обычнее - от 10 до 30 мг активного ингредиента. Термин "единица лекарственной формы" относится к физически отдельным единицам, применимым в качестве стандартных доз для людей и других млекопитающих, причем каждая единица содержит предварительно установленное количество активного вещества, вычисленное таким образом, чтобы получить желаемое лечебное действие, в сочетании с подходящим фармацевтически приемлемым эксципиентом.
Активное соединение является эффективным в широком интервале дозировки. Например, суточная дозировка обычно попадает в интервал от 0,5 до 30 мг на кг массы тела. При лечении взрослых людей особенно предпочтительным является интервал от 1 до 15 мг/кг/сутки в однократной или разделенной дозе. Однако следует понимать, что фактически вводимое количество соединения будет определять лечащий врач, с учетом относящихся к делу обстоятельств, в том числе, состояния, которое лечат, выбранного способа введения лекарственного препарата, конкретного применяемого соединения, возраста, массы и реакции отдельного пациента и тяжести симптомов у него, и, следовательно, упомянутые выше дозировочные интервалы не подразумевают какого-либо ограничения объема изобретения. В отдельных случаях уровень дозировки ниже низшего предела вышеупомянутого интервала может являться более чем адекватным, в то время как в других случаях могут применяться дозы, превышающие верхний предел - без появления какого-либо вредного побочного действия, при условии, что такие большие дозы сначала разделяются на несколько меньших доз для введения в течение суток.
Для приготовления твердых композиций, таких как таблетки, основной активный ингредиент смешивают с фармацевтически приемлемым эксципиентом, чтобы образовать твердую преформулировочную композицию, содержащую однородную смесь соединения настоящего изобретения. Когда такие преформулировочные композиции упоминаются как однородные, это означает, что активный ингредиент равномерно диспергирован в композиции, таким образом, что композицию можно легко разделить на равно эффективные единицы лекарственной формы, такой как таблетки, пилюли и капсулы. Такую твердую преформулировку затем разделяют на единичные лекарственные формы описанного выше типа, содержащие от 0,1 до 500 мг активного ингредиента настоящего изобретения.
На таблетки или пилюли по настоящему изобретению затем может быть нанесено покрытие, или они могут быть составлены иным способом, для того, чтобы получить лекарственную форму, имеющую преимущество пролонгированного действия. Например, таблетка или пилюля могут содержать внутренний и наружный компонент лекарственной формы, причем последний является формой оболочки для первого. Оба компонента могут быть разделены энтеросолюбильным слоем, который служит для сопротивления разрушению в желудке и дает возможность внутреннему компоненту проходить неповрежденным в двенадцатиперстную кишку или задерживаться при выделении. Ряд материалов может быть использован для таких энтеросолюбильных слоев или покрытий, причем такие материалы включают некоторые полимерные кислоты и смеси полимерных кислот с такими материалами, как шеллак, цетиловый спирт и ацетат целлюлозы.
Жидкие формы, в которые новые композиции настоящего изобретения могут быть включены для введения их перорально или путем инъекции, включают водные растворы, сиропы с соответствующим корригентом, водные или масляные суспензии, и эмульсии с корригентом с пищевыми маслами, такими как хлопковое масло, сезамное масло, кокосовое масло или арахисовое масло, а также элексиры и подобные фармацевтические носители.
Композиции для ингаляции или инсуффляции включают растворы и суспензии в фармацевтически приемлемых водных или органических растворителях или в их смесях и порошки. Жидкие или твердые композиции могут содержать подходящие фармацевтически приемлемые эксципиенты, которые описаны выше. Предпочтительно, композиции вводят респираторным путем через рот или нос для местного или системного действия. Композиции, предпочтительно, в фармацевтически приемлемых растворителях, могут быть распылены с применением инертных газов. Распыляемые растворы могут вдыхаться непосредственно из распыляющего устройства, или распыляющее устройство может быть присоединено к маске для лица, палатке или аппарату с периодическим избыточным давлением. Композиции в виде растворов, суспензий или порошков могут вводиться, предпочтительно, через рот или нос, из устройств, которые доставляют формулировку соответствующим способом.
Пример препарата 1
Готовят твердые желатиновые капсулы, содержащие следующие ингредиенты.
Ингредиент - Количество (мг/капсулу)
Соединение примера 51 - 30,0
Крахмал - 305,0
Стеарат магния - 5,0
Перечисленные выше ингредиенты смешивают, и заполняют смесью желатиновые капсулы - по 340 мг в капсулу.
Пример препарата 2
Готовят таблетки, используя перечисленные ниже ингредиенты.
Ингредиент - Количество (мг/таблетку)
Соединение примера 66 - 25,0
Микрокристаллическая целлюлоза - 200,0
Коллоидный диоксид кремния - 10,0
Стеариновая кислота - 5,0
Компоненты смешивают и прессуют в таблетки, масса каждой из которых составляет 240 мг.
Пример препарата 3
Готовят сухой порошок для ингаляции, содержащий перечисленные далее компоненты.
Ингредиент - Масса, %
Соединение примера 17 - 5
Лактоза - 95
Активное соединение смешивают с лактозой, и смесь загружают в ингалятор для сухого порошка.
Пример препарата 4
Готовят таблетки, каждая из которых содержит 30 мг активного ингредиента, как описано ниже.
Ингредиент - Количество (мг/таблетку)
Соединение примера 14 - 30,0 мг
Крахмал - 45,0 мг
Микрокристаллическая целлюлоза - 35,0 мг
Поливинилпирролидон (в виде 10% раствора в воде) - 4,0 мг
Натрийкарбоксиметилкрахмал - 4,5 мг
Стеарат магния - 0,5 мг
Тальк - 1,0 мг
Всего - 120 мг
Активный ингредиент, крахмал и целлюлозу просеивают через сито N 20 меш (США) и тщательно смешивают. К полученному в результате порошку подмешивают раствор поливинилпирролидона, и полученную смесь просеивают через сито 16 меш (США). Полученные таким образом гранулы сушат при 50-60oC и просеивают через сито 16 меш (США). Затем к гранулам добавляют натрийкарбоксиметилкрахмал, стеарат магния и тальк, предварительно просеянные через сито N 30 меш (США), и все это, после перемешивания, прессуют на таблетирующей машине, и получают таблетки массой по 120 мг.
Пример препарата 5
Капсулы, каждая из которых содержит по 40 мг лекарственного препарата, изготовляют следующим образом.
Ингредиент - Количество (мг/капсулу)
Соединение примера 13 - 40,0 мг
Крахмал - 109,0 мг
Стеарат магния - 1,0 мг
Всего - 150,0 мг
Активный ингредиент, целлюлозу, крахмал и стеарат магния смешивают, просеивают через сито N 20 меш (США), и заполняют смесью твердые желатиновые капсулы, по 150 мг в каждую капсулу.
Пример препарата 6
Суппозитории, каждый из которых содержит 25 мг активного ингредиента, готовят следующим образом.
Ингредиент - Количество
Соединение примера 18 - 25 мг
Глицериды насыщенных жирных кислот до - 2000 мг
Активный ингредиент просеивают через сито N 60 меш (США) и суспендируют в глицеридах насыщенных жирных кислот, предварительно расплавленных при минимальном необходимом для этого нагрева.
Смесь затем выливают в форму для суппозиториев емкостью 2,0 г, и оставляют охлаждаться.
Пример препарата 7
Суспензии, которые содержат 50 мг лекарственного средства в 5 мл дозе, готовят следующим образом.
Ингредиент - Количество
Соединение примера 43 - 50,0 мг
Ксантановая смола - 4,0 мг
Натрийкарбоксиметилцеллюлоза (11%) и микрокристаллическая целлюлоза (89%) - 50,0 мг
Сахароза - 1,75 г
Бензоат натрия - 10,0 мг
Отдушка и краситель - g.v.
Очищенная вода - До 5,0 мл
Лекарственное средство, сахарозу и ксантановую смолу смешивают, пропускают через сито N 10 меш (США) и затем смешивают с предварительно подготовленным раствором микрокристаллической целлюлозы и натрийкарбоксиметилцеллюлозы в воде. Разводят в некотором количестве воды бензоат натрия, отдушку и краситель, и добавляют при перемешивании. Затем добавляют воду в количестве, достаточном для получения требуемого объема.
Пример препарата 8
Капсулы, каждая из которых содержит 15 мг лекарственного средства, готовят следующим образом.
Ингредиент - Количество (мг/капсулу)
Соединение примера 58 - 15,0 мг
Крахмал - 407,0 мг
Стеарат магния - 3,0 мг
Всего - 425,0 мг
Активный ингредиент, целлюлозу, крахмал и стеарат магния смешивают, пропускают через сито N 20 меш (США) и заполняют смесью твердые желатиновые капсулы - по 425 мг в каждую капсулу.
Пример препарата 9
Форму для внутривенного введения готовят из следующих ингредиентов.
Ингредиент - Количество
Соединение примера 91 - 250,0 мг
Изотонический физиологический раствор - 1000 мл
Пример препарата 10
Препарат для местного применения готовят так, как описано ниже.
Ингредиент - Количество
Соединение примера 67 - 1 - 10 г
Эмульгирующий воск - 30 г
Вазелиновое масло - 20 г
Бесцветный мягкий парафин - До 100 г
Бесцветный мягкий парафин нагревают до расплавления. Вводят в него вазелиновое масло и эмульгирующий воск, и перемешивают до растворения. Добавляют соединение примера 67, и продолжают перемешивание до образования дисперсии. Затем смесь охлаждают до твердого состояния.
Пример препарата 11
Подъязычные или буккальные таблетки, каждая из которых содержит 10 мг активного ингредиента, можно приготовить следующим образом.
Ингредиент - Количество на таблетку
Активный ингредиент - 10,0 мг
Глицерин - 210,5 мг
Вода - 143,0 мг
Цитрат натрия - 4,5 мг
Поливиниловый спирт - 26,5 мг
Поливинилпирролидон - 15,5 мг
Всего - 410,0 мг
Глицерин, воду, цитрат натрия, поливиниловый спирт и поливинилпирролидон смешивают вместе при непрерывном перемешивании и поддержании температуры около 90oC. Когда полимеры перейдут в раствор, раствор охлаждают до 50-55oC, и постепенно подмешивают лекарственное средство. Однородную смесь разливают в формы, изготовленные из инертного материала, и получают содержащую лекарственное средство диффузионную матрицу, имеющую толщину 2-4 мм. Эту диффузионную матрицу затем разрезают, и формируют отдельные таблетки, имеющие соответствующий размер.
Другая предпочтительная форма, используемая в способах настоящего изобретения, использует кожный пластырь. Его можно применять для непрерывной или прерывистой инфузии соединений настоящего изобретения в регулируемых количествах. Конструкции и применение пластыря для доставки фармацевтических средств достаточно известны в литературе (См., например, патент США 5023252 от 11 июня 1991). Такой пластырь изготавливается для непрерывной, пульсирующей или другой доставки фармацевтических агентов.
Бывает необходимо ввести фармацевтическую композицию прямо или косвенно в мозг. Приемы непосредственного введения обычно включают введение катетера для доставки лекарственного средства в желудочковую систему, чтобы обойти гематоэнцефалический барьер. Одна из таких имплантируемых систем доставки, применяемая для переноса биологического действующего начала в специфические анатомические области организма, описывается в патенте США 5011472 от 30 апреля 1991.
Приемы непрямой доставки, которые обычно являются предпочтительными, включают, как правило, приготовление композиций, обеспечивающих латентность лекарственного препарата путем превращения гидрофильных лекарственных препаратов в жирорастворимые лекарственные препараты или пролекарства. Латентность обычно достигается через блокирование гидроксильных, карбонильных, сульфатных и первичных аминных групп, присутствующих в лекарственном средстве, которое делает лекарственное средство более жирорастворимым и поддающимся транспортировке через гематоэнцефалический барьер. С другой стороны, может быть усилена доставка гидрофильных лекарственных средств посредством внутриартериального вливания гипертонических растворов, которые могут временно открыть гематоэнцефалический барьер.
По результатам испытаний заявленные соединения можно отнести к категории нетоксичных продуктов.
| название | год | авторы | номер документа |
|---|---|---|---|
| ТРИГИДРАТ ДИГИДРОХЛОРИДА (R)-3-(1Н-ИНДОЛ-3-ИЛ)-1-[N-(2-МЕТОКСИБЕНЗИЛ)АЦЕТИЛАМИНО]-2-[N-(2-(4-(ПИПЕРИДИН-1-ИЛ)ПИПЕРИДИН-1-ИЛ)АЦЕТИЛ)АМИНО]ПРОПАНА И ФАРМАЦЕВТИЧЕСКИЙ СОСТАВ | 1995 |
|
RU2151769C1 |
| ПРИМЕНЕНИЕ 6-ГЕТЕРОЦИКЛО-4-АМИНО-1,3,4,5-ТЕТРАГИДРОБЕНЗ(CD) ИНДОЛА ДЛЯ ЛЕЧЕНИЯ РВОТЫ ИЛИ ТОШНОТЫ ПРИ ДВИЖЕНИИ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1993 |
|
RU2138262C1 |
| ПРОИЗВОДНЫЕ ЦИКЛИЧЕСКИХ ПЕПТИДОВ ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ, СПОСОБ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1993 |
|
RU2129562C1 |
| ПРОИЗВОДНЫЕ 1-КАРБА-(1-ДЕТИА)-ЦЕФЕМА | 1993 |
|
RU2107067C1 |
| НОВЫЕ 1Н-ИНДОЛ-3-АЦЕТАМИДЫ КАК ИНГИБИТОРЫ SPLA И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 1994 |
|
RU2162463C2 |
| СПОСОБ ПОЛУЧЕНИЯ 5-ЗАМЕЩЕННЫХ ПИРРОЛО (2,3-α)ПИРИМИДИНОВ | 1993 |
|
RU2127274C1 |
| СПОСОБ ПОЛУЧЕНИЯ СЛОЖНОГО ЭФИРА СУЛЬФОКСИДА 3-ЭКЗОМЕТИЛЕНЦЕФАМА | 1992 |
|
RU2010795C1 |
| ПРОИЗВОДНЫЕ ГЛИКОПЕПТИДА ИЛИ ИХ СОЛИ, СПОСОБ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1995 |
|
RU2145609C1 |
| ПРОИЗВОДНЫЕ СУЛЬФОНИЛМОЧЕВИНЫ ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ОСНОВЕ ПРОИЗВОДНЫХ СУЛЬФОНИЛМОЧЕВИНЫ, ОБЛАДАЮЩАЯ ПРОТИВООПУХОЛЕВОЙ АКТИВНОСТЬЮ | 1994 |
|
RU2127259C1 |
| ПРОИЗВОДНЫЕ ТРИПЕПТИДОВ В ВИДЕ R- ИЛИ RS-ФОРМЫ ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ НЕТОКСИЧНЫЕ СОЛИ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1991 |
|
RU2077538C1 |


















































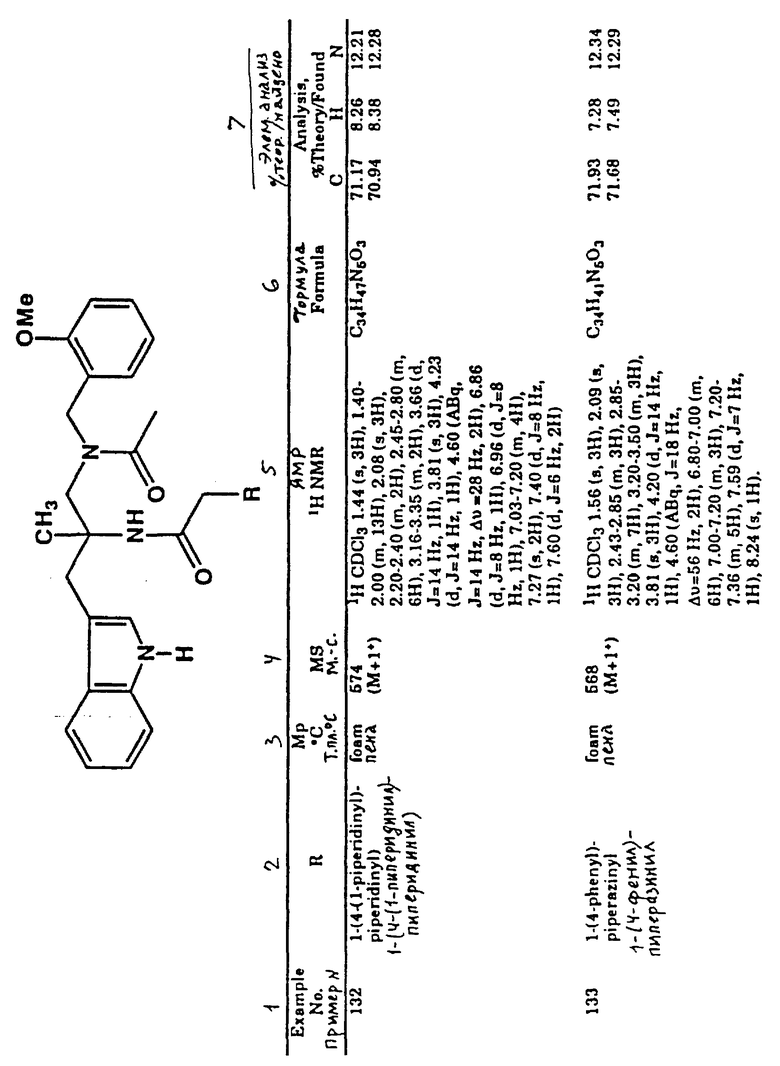


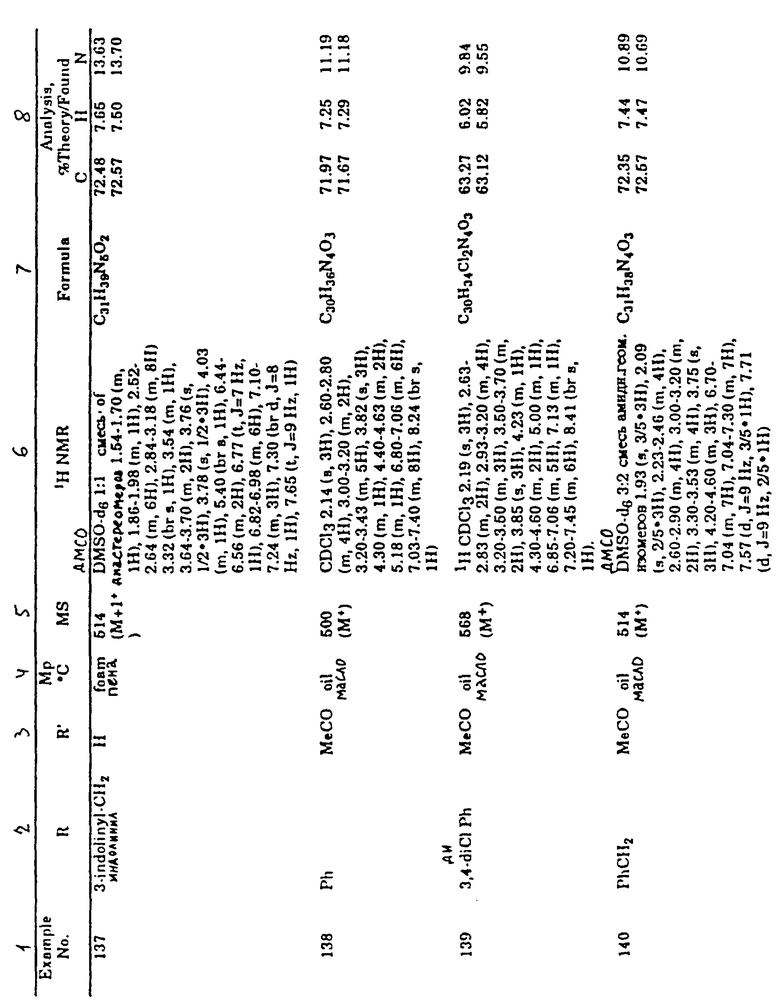


























Описываются новые непептидные антагонисты рецепторов тахикинина общей формулы (I)
где значения R, R1, R2, R3, R4, R8, n, m, указаны в п.1 формулы изобретения. Описывается также фармацевтическая композиция, являющаяся антагонистом рецепторов NK-1 и NК-2, на основе соединений общей формулы I. 2 с. и 10 з.п. ф-лы, 2 табл.
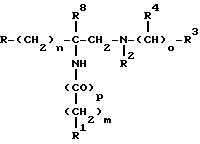
в которой m равен 0 или 1;
n равен 0 или 1;
o представляет собой 0,1 или 2;
p равен 1;
R представляет собой фенил, 2- или 3-индолил, 2- или 3-индолинил, бензотиенил, бензофуранил или нафтил, и любая из групп R может быть замещена одним или двумя галогенами, (C1 - C3)-алкоксигруппами, трифторметилами, (C1 - C4)-алкилами, фенил-(C1 - C3)-алкоксигруппами или (C1 - C4)-алканоильными группами;
R1 представляет собой фенил, фенокси - группу, фенилтиогруппу, гексаметилениминил, пиперазинил, пиперидинил, пирролидинил, морфолинил, индолинил, 1,2,3,4-тетрагидроизохинолил, (C1 - C4)-алкил или -NH-CH2-R5; при этом любая из группы R1 может быть замещена фенилом, (C3 - C8)-циклоалкилом, бензилом, (C1 - C4)-алкилом, пиперидинилом, пиримидинилом, (C2 - C6)-алканоиламиногруппой, (C1 - C4)-алкоксикарбонилом, и фенил может быть замещен галогеном, трифторметилом, или R1 представляет собой галоген, R5 представляет собой пиридил, анилино-(C1 - C3-алкил)- или анилинокарбонил;
R2 представляет собой водород, (C1 - C4)-алкил, (C1 - C4)-алкилсульфонил, карбокси-((C1 - C3)-алкил)-, (C1 - C3)-алкоксикарбонил-((C1 - C3)-алкил)- или -CO-R6, R6 представляет собой водород, (C1 - C4)-алкил, (C1 - C3)-галоидалкил, фенил, (C1 - C3)-алкоксигруппу, (C1 - C4)-алкиламиногруппу, ди((C1 - C4)-алкил)аминогруппу;
R8 представляет собой водород или (C1 - C6)-алкил;
R3 представляет собой фенил, фенил-((C1 - C6)-алкил)-, (C3 - C8)-циклоалкил, (C1 - C8)-алкил, и любая из этих групп - фенил, фенил-((C1 - C6)-алкил)-, (C3 - C8)-циклоалкил, (C1 - C8)-алкил, может быть замещена одним или двумя галогенами, (C1 - C3)-алкоксигруппами, (C1 - C3)-алкилтиогруппами, трифторметилами или (C1 - C3)-алкильными группами; и
R4 представляет собой водород, при условии, что если R1 представляет собой галоген, R3 представляет собой фенил, фенил-((C1 - C6)-алкил)-, (C3 - C8)-циклоалкил;
или его фармацевтически приемлемая соль или сольват.
где Ra представляет собой галоген, (C1 - C3)-алкоксигруппу, (C1 - C3)-алкилтиогруппу, нетрогруппу, трифторметил или (C1 - C3)-алкил.
| WO 9310073 A, 27.05.93 | |||
| WO 9322279 A, 11.11.93 | |||
| Машковский М.Д | |||
| Лекарственные средства | |||
| - М.: Медицина, 1972, ч | |||
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| Приспособление с иглой для прочистки кухонь типа "Примус" | 1923 |
|
SU40A1 |
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| Выбрасывающий ячеистый аппарат для рядовых сеялок | 1922 |
|
SU21A1 |
