Настоящее изобретение относится к новым синтетическим пептидам. Более конкретно оно относится к синтетическому пептиду, который в сочетании со смесью пептидов проявляет сильную поверхностную активность. Настоящее изобретение относится также к промежуточным соединениям, используемым для получения указанных синтетических пептидов, способу получения указанных синтетических пептидов, легочным поверхностно-активным веществам (ПАВ), содержащим указанные пептиды и смесь липидов, а также к лекарственному препарату для лечения респираторного дистресс-синдрома, содержащему указанные легочные ПАВ в качестве активного ингредиента.
Респираторный дистресс-синдром представляет собой заболевание, при котором поверхностная активность альвеол снижена вследствие недостатка легочного ПАВ. Это приводит к коллапсу альвеол, что в свою очередь ведет к тяжелым нарушениям дыхания. Этот синдром часто встречается у недоношенных младенцев и является причиной высокого уровня смертности. Известно, что композиции легочных ПАВ высокоэффективны для лечения неонатального респираторного дистресс-синдрома.
Взрослые также подвержены гипоксии, вызванной различными причинами, и известно много примеров, когда диффузные затемнения, похожие на матовое стекло, наблюдаются в обоих легких на рентгеновских снимках грудной клетки, и происходит нарушение дыхания, несмотря на применение аппарата искусственной вентиляции легких и т.п. В работе автора Ueda и др. (см. Hiromoto Yasuda, "Biosurfactants. Chapter 3. Medical Practices Using Surfactants. Section 1. Clinical Applications of Surfactants. V.Aspiration Pneumonia and Surfactants", стр. 184, 1990 г. Science Forum. Co. Ltd.) описано 2 случая пневмонии у взрослых (т.е. 1 случай: пневмония в результате вдыхания газообразного нитрата, и 2 случай: рецидивирующая аспирационная пневмония в результате опухоли головного мозга, оба эти случая вызывали гипоксию и вели к ухудшению общего состояния пациентов и к нарушениям дыхания), в которых удалось добиться значительного улучшения и спасти жизни пациентов применив инъекции легочного ПАВ в дыхательные пути. Послеоперационные нарушения дыхания могут происходить после операций на сердце, поскольку в процессе операции дыхание останавливается. Влияние легочных ПАВ на такие нарушения дыхания также описано в литературе (см. Shuichi Nosaka et al., Journal of Japanese Medical Society for Biological Interface, Vol. 22, p. 66, 1991).
Таким образом, заместительная терапия, заключающаяся во введении легочных ПАВ извне и через дыхательные пути, дает значительный терапевтический эффект при лечении респираторного дистресс-синдрома.
В последнее время было обнаружено 4 типа апопротеинов, которые являются уникальными легочными ПАВ для млекопитающих. Это - ПАВ-апопротеин А и ПАВ-апопротеин D, которые являются гидрофильными, ПАВ-апопротеин B (далее обозначаемый SP-B) и ПАВ-апопротеин C (далее обозначаемый SP-C), которые являются гидрофобными (см. Toyoaki Akino and Yoshio Kuroki, Respiration and Circulation, Vol. 38, N 18, p. 722, 1990; Hiromoto Yasuda et al., Biosurfactants: Chapter 2. The Biochemistry of Surfactants-Surfactants and Apoprotens, p. 131, 1990, Science Forum, Co. Ltd.).
SP-C (последовательность N 1), полученный из легких человека, состоит из 35 аминокислот и является высокогидрофобным апопротеином, который богат валином и имеет фенилаланин в качестве П-концевой аминокислоты, SP-C, выделенные из легких быков (последовательно N 2), свиней (последовательность N 3), крыс и т. п. Также состоит из 34 - 35 аминокислот и, хотя последовательности аминокислот в N-концевой позиции различаются в зависимости от видов животных, они почти полностью соответствуют человеческому SP-C.
В японском патенте N Hei-3-502095 также указано на то, что синтетический пептид (последовательность N 4) с указанными ниже 32 аминокислотами, которые являются частью структуры SP-C, представляет собой минимальный фрагмент, обладающий высокой поверхностной активностью, что смеси этого пептида с липидами эффективно применяются для лечения респираторного дистресс-синдрома, и что сравнение поверхностной активности этого минимального фрагмента пептида с поверхностной активностью других фрагментов синтетических пептидов, которые имеют более короткие последовательности аминокислот, показывает, что снижение поверхностной активности происходит не из-за отсутствия той или иной аминокислоты, а вследствие уменьшения длины пептидной цепочки.
Ранее некоторые из авторов настоящего изобретения провели работу, результаты которой показали, что смеси синтетического пептида (далее обозначаемого TP-C), структура которого является частью структуры SP-C, с липидами эффективны при лечении респираторного дистресс-синдрома и эти смеси применили при лечении нескольких пациентов (см. японский патент N Hei-5-518188).
Что касается получения синтетических пептидов, то известно, что по мере удлинения аминокислотной последовательности пептида, все чаще образуются дефектные пептиды, затрудняется выделение и очистка пептидов, увеличивается время, необходимое для получения пептидов, все труднее становится организовать получение пептидов в больших количествах и т.п.
Также из соображений сохранности, композиции легочных ПАВ часто поставляются в виде сухих порошков, которые перед использованием разводят физиологическим раствором и вводят в виде суспензии. Для того чтобы повысить дисперсность суспензии, содержащей легочные ПАВ, применяют добавку суспендирующих агентов, таких как маннитол (см. опубликованный японский патент N Hei-1-60451) и липофилизацию при температуре первичного замораживания от -1 до -10oC. Однако эти способы трудноосуществимы на практике и желательно было разработать более простые способы получения препаратов.
Композиция легочных ПАВ (далее называемая S-35), полученная путем соединения SP-C со смесью липидов, в которую входят холинфосфоглицерид, кислый фосфолипид и жирнокислый аналог, обладает очень плохой диспергируемостью в физиологическом растворе, что затрудняет приготовление суспензии, которая должна быть достаточно однородной, чтобы ее можно было использовать в качестве композиции. Причина этого - образование дисульфидных связей цистеиновыми остатками в пептиде, из-за чего повышается когезионная способность пептида и гидрофобность самого легочного ПАВ.
Поскольку TP-C обладает низкой растворимостью в большинстве растворителей, необходимо было использовать трифторуксусную кислоту (ТФК) при приготовлении композиции легочных ПАВ. Проблемы с TP-C заключались в том, что требовалось много времени на концентрирование и высушивание для удаления как можно большего количества ТФК, а также в том, что из-за остатков ТФК суспензия легочных ПАВ становилась подкисленной в процессе приготовления композиции легочных ПАВ.
Авторы настоящего изобретения тщательно изучили синтетические пептиды с точки зрения вышеописанных проблем и разработали настоящее изобретение, открыв, что новые синтетические пептиды (далее называемые "синтетические пептиды по изобретению"), которые содержат приведенную ниже аминокислотную последовательность и гидрофильной пептидной частью специфической последовательностью с N-концевой части и гидрофобную пептидную часть, образованную, в основном, последовательностью Лей и/или Нле в C-концевой части, легко выделять и очищать, их можно производить в больших количествах, они хорошо растворимы в муравьиной кислоте, ТФК, трифторэтане, диметилсульфоксиде (ДМСО), хлороформе, смесях хлороформа с метанолом, в метаноле, этиленхлоргидрине и тетрагидрофуране и, в частности, обладают значительно более высокой растворимостью в метаноле по сравнению с синтетическим SP-C и TP-C. Авторы настоящего изобретения обнаружили также, что легочные ПАВ, приготовленные из синтетических пептидов по изобретению и смесей липидов, даже в тех случаях, когда их получают обычными способами с применением лиофилизации при температуре -20oC или ниже и без добавки суспендирующих агентов, образуют достаточно однородные суспензии, более однородные по сравнению с S-35 или синтетическими легочными ПАВ (далее называемые "SF-3"; см. опубликованный японский патент N Hei-2-87685), включающими в себя только смесь липидов, состоящую из холинфосфоглицерида, кислого фосфолипида и жирнокислого аналога или по сравнению с веществом (далее называемым "S-TA"; см. опубликованный японский патент # Sho-61-9924), содержащим помимо жирной кислоты, вещество, состоящее из фосфолипида, нейтрального липида, холестерина, углеводов и минимальных количеств протеинов, присутствующих в легких млекопитающих, и вместе с тем, синтетические пептиды по изобретению обладают сильной поверхностной активностью, которая эквивалентна поверхностной активности S-35, S-TA или легочных ПАВ, состоящих из TP-C и смеси липидов.
Xaa-Про-Вал-Xbb-Xcc-Лиз-Арг-W
(Xaa может не присутствовать или может представлять собой Цис или Сер, Xbb представляет собой Гис или Асн, Xcc представляет собой Лей или Иле, а W представляет собой гидрофобную часть).
Синтетический пептид по изобретению содержит гидрофильную пептидную часть, описанную указанной ниже специфической последовательностью в N-концевой части, и пептидную часть, имеющую гидрофобную пептидную часть, содержащую в основном Лей и/или Нле в С-концевой части, и представляет собой синтетический пептид, обладающий сильной поверхностной активностью в смеси с липидами.
Xaa-Про-Вал-Xbb-Xcc-Лиз-Арг
(Xaa может не присутствовать или может представлять собой Цис или Сер, Xbb представляет собой Гис или Асн, а XCC представляет собой Лей или Иле).
Несмотря на то, что гидрофобная пептидная часть состоит из таких гидрофобных аминокислот, как Лей, Нле, Иле, Вал, Фен, Нва и Три, она обычно включат 12 или более, предпочтительно 12 - 20 молекул Лей и/или Нле. Несмотря на то, что с точки зрения упрощения синтеза и т.п., предпочтительно, чтобы эта гидрофобная пептидная часть состояла из тех же гидрофобных аминокислот, она может состоять из подходящей последовательности молекул Лей и Нле или может содержать в своей последовательности от 1 до 5 молекул Иле, Вал, Нва, Три и других гидрофобных аминокислот.
Синтетические пептиды по изобретению включают также синтетические пептиды, в которых аминокислота пли пептид добавлен к N-концевой части и/или C-концевой части вышеуказанного синтетического пептида. Аминокислота, добавляемая к N-концевой части, может представлять собой Цис или Сер. Далее, к N-концевой части может быть добавлен пептид с последовательностью Фен-Гли-Иле-Про. Тиольную группу или гидроксильную группу, присутствующую в вышеуказанном синтетическом пептиде, можно ацилировать жирной кислотой с 14 - 18 атомами углерода (предпочтительно пальмитиновой кислотой), или же можно провести реакцию ацетоамидометилирования. Пептид, добавляемый к C-концу, может иметь последовательность Гли-Ала-Лей-Лей или Гли-Ала-Лей-Лей-Мет-Гли-Лей.
Далее, синтетические пептиды по изобретению включают также такие синтетические пептиды (за исключением пептидов, частично имеющих структуру природных SP-C), которые содержат пептидную группу, обладающую хорошей гидрофильностью, и которые проявляют сильную поверхностную активность, если их соединить со смесью липидов, даже после добавки, удаления и замещения одной или нескольких аминокислот, входящих в состав этих пептидов.
Синтетические пептиды по изобретению можно получить химическими способами или методами генной инженерии, хотя химические способы являются предпочтительными с точки зрения выделения и очистки.
Химические способы получения синтетических пептидов по изобретению включают способы поэтапного удлинения цепочки и конденсации фрагментов с применением жидкофазного синтеза или твердофазного синтеза, таких как способы, основанные на применении азидов, хлорангидридов кислот, ангидридов кислот, смешанных ангидридов кислот, N-N'-дициклогексилкарбодиимида (способ DCC), активированного эфира (p-нитрофенольного эфира, p-гидроксисукцинимидэфира, карбоимидазола и т.п.), окислительно-восстановительными способами активации N-N'-дициклогексилкарбодиимида.
Изобретение предлагает также способ конденсации фрагмента для получения синтетических пептидов по изобретению, в котором гидрофильная пептидная группа, содержащая защищенный N-конец и защищенные функциональные боковые цепи, служит промежуточным соединением.
По сравнению со способом поэтапного удлинения цепочки способ конденсации фрагмента позволяет легче производить очистку целевого вещества, он лучше подходит для синтеза больших количеств пептидов и позволяет предотвращать потери вследствие неожиданных ошибок. Синтетические пептиды по изобретению можно получить путем конденсации гидрофобной части с предварительно полученной гидрофильной пептидной частью, имеющей защищенный N-конец и защищенные функциональные боковые цепочки, путем жидкофазного или твердофазного синтеза. Выбор защитных групп для N-конца и функциональных боковых цепочек не ограничен какими-либо особыми требованиями, можно использовать любые защитные группы, применяемые в обычном синтезе пептидов. 9-Флуоренилметилоксикарбонил (Fmoc), 2-хлорбензилоксикарбонил (2-ClZ) или трет-бутилоксикрабонил (Boc) могут использоваться в качестве защитных групп для концевой аминокислотной группы, 9-флуоренилметилоксикарбонил (Fmoc), трет-бутилоксикарбонил (Boc) или карбобензокси (Z) или тозил (Tos) можно использовать в качестве защитной группы для лизина, Трт, Fmoc, Boc, динитрофенола (Dпр)Bom, Bz1 или тозил можно использовать в качестве защитной группы для гистидина, а 4-метокси-2,3,6-триметилбензолсульфонил (Mtr), Pmc, Mts или тозил можно использовать в качестве защитной группы для аргинина. Таким образом, пептиды, которые можно использовать в качестве промежуточных соединений для получения синтетических пептидов по изобретению, включают Fmoc-Про-Вал-Гис(Трт)-Лей-Лиз(Boc)-Арг(Mtr)
Fmoc-Про-Вал-Асн-Лей-Лиз(Вос)-Арг(Mtr) и
Fmoc-Про-Вал-Асн-Иле-Лиз(Вос)-Арг(Mtr).
Легочные ПАВ (далее называемые "ПАВ по изобретению") можно получить путем соединения пептида по изобретению со смесью липидов, состоящей из холинфосфоглицерида, кислого липида и жирнокислого аналога
Соотношение компонентов композиции следует подобрать так, чтобы весовое соотношение каждого из этих компонентов по отношению к суммарному сухому весу готового продукта составляло: синтетический пептид - 0,1 - 5,0 мас.%, холинфосфоглицерид - 50,6 - 80,5 мас.%, кислый фосфолипид - 4,5 - 37,6 мас. %, аналог жирной кислоты - 4,6 - 24,6 мас.%.
Примеры холинфосфоглицеридов, которые могут быть использованы в ПАВ по изобретению, включают 1,2-диацилглицеро-(3)-фосфохолины, такие как 1,2-дипальмитоилглицероло-(3)-фосфохолин (дипальмитоилфосфатидилхолин), 1,2-дистеароилглицеро-(3)-фосфохолин, 1-пальмитоил-2-стеароилглицеро-(3)-фосфохолин и 1-стеароил-2-пальмитоилглицеро-(3)-фосфохолин и т.п.; 1-алкил-2-ацилглицеро-(3)-фосфохолины, такие как 1-гексадицел-2-пальмитоилглицеро-(3)-фосфохолин и 1-октадецил-2-пальмитоилглицеро-(3)-фосфохолин и т.п.; а также 1-2-диалкилглицеро-(3)-фосфохолины, такие как 1,2-дигексадецилглицеро-(3)-фосфохолин и т.п. Несмотря на то, что вышеуказанные соединения имеют оптические изомеры на основе второго атома углерода глицеринового остатка, любой изомер (D-, L- и DL-) можно использовать в качества ПАВ по изобретению. Помимо одиночных соединений из числа вышеуказанных холинфосфоглицеридов в настоящем изобретении могут быть использованы смеси, состоящие из двух или более различных 1,2-диацилглицеро-(3)-фосфохолинов, имеющих ацильные группы, предпочтительно две насыщенные ацильные группы с 12 - 24 атомами углерода, или смеси, состоящие из вышеуказанных смесей и вышеуказанных одиночных соединений.
В числе подходящих кислых фосфолипидов, например: 1,2-диацил-sn-глицеро-(3)-фосфорная кислота (L -α- фосфатидная кислота), 1,2-диацил-sn-глицеро-(3)фосфо-L-серин (фосфатидилсерин), 1,2-диацил-sn-глицеро-(3)-фосфо-sn-глицерин (фосфатидилглицерин) и 1,2-диацил-sn-глицеро-(3)-фосфо-(1)-L-мио-инозит (фосфатидилинозит). Первая и вторая позиции в этих соединениях могут быть замещены одной и той же или разными ацильными группами. Предпочтительно, чтобы указанная ацильная группа имела 12 - 24 атома углерода.
Примеры подходящих жирнокислых аналогов включают свободные жирные кислоты, соли щелочных металлов жирных кислот, сложные жирнокислые алкилэфиры, жирнокислые глицеринэфиры и амиды жирных кислот, а также смеси, включающие два или более из указанных соединений, и спирты жирных кислот и амины жирных кислот.
В настоящем описании термин "аналоги жирной кислоты" охватывает вышеуказанные спирты жирных кислот и алифатические амины.
В качестве свободной жирной кислоты можно использовать миристиновую кислоту, пальмитиновую кислоту или стеариновую кислоту, хотя предпочтительной является пальмитиновая кислота.
Натриевые, калиевые, магниевые и кальциевые соли вышеуказанных свободных жирных кислот можно использовать в качестве соли щелочного металла жирной кислоты, хотя предпочтительным является пальмитат натрия. Низшие алкилэфиры с 1 - 4 атомами углерода можно использовать в качестве жирнокислых алкилэфиров, хотя предпочтительным является этилпальмитат. Сложные эфиры моноглицерина можно использовать в качестве сложного эфира глицерина жирной кислоты, хотя предпочтительным является монопальмитин.
Спирты с 14 - 18 атомами углерода можно использовать в качестве спирта жирной кислоты, хотя предпочтительным является гексадециловый спирт. Амины с 14 - 18 атомами углерода можно использовать в качестве алифатического амина, хотя предпочтительным является гексадециламин.
Вышеуказанные холинфосфоглицериды, кислые фосфолипиды и жирнокислые аналоги могут представлять собой продукты, выделенные из растений или животных, полусинтетические продукты или синтетические продукты, можно применять такие продукты, выпускаемые промышленностью.
ПАВ по изобретению можно получить путем высушивания и отверждения при пониженном давлении смеси растворов пептида по изобретению с вышеуказанной смесью липидов, с последующим суспендированием полученного остатка в подходящей суспендирующей жидкости, с последующей лиофилизацией.
Примеры растворителей, которые можно использовать для получения раствора пептида по изобретению, включают муравьиную кислоту, ТФК, трифторэтанол, ДМСО, хлороформ-метанол, хлороформ, метанол, этиленхлоргидрин и тетрагидрофуран.
Примеры растворителей, которые можно использовать для получения растворов смеси липидов, включают хлороформ и хлороформ-метанол /1:2 - 5:1 (o/o)/.
Примеры суспендирующей жидкости включают воду и смеси воды с этанолом /4: 1 - 20:1 (o/o)/, хотя предпочтительными являются смеси воды с этанолом. Суспендирование проводят в течение 5 - 60 минут, предпочтительно в течение 15 - 30 минут, при 30 - 60oC, предпочтительно при 40 - 50oC.
ПАВ по изобретению, полученное таким способом, неизбежно содержит небольшое количество остаточной воды. Однако желательно высушивать ПАВ до содержания воды 5,0 мас.% или ниже по отношению к суммарной массе продукта. Если ПАВ высушен до такой степени, то остаток этанола становится незаметным в тех случаях, когда используются смеси воды с этанолом.
Сухие порошковые композиции ПАВ по изобретению можно суспендировать и диспергировать до однородного состояния в растворе, содержащем фазиологически-пригодные концентрации соли одновалентного или двухвалентного металла, например 0,9% хлорида натрия или 1,5 мМ хлорида кальция, или в растворе физиологического буфера, содержащем такие соли. Для приготовления раствора используют ручные миксеры или миксеры с переменной скоростью, или ультразвуковой аппарат.
Переходим к описанию поверхностной активности, способности к образованию суспензии и фармакологических свойств ПАВ по изобретению, полученных вышеуказанными способами.
(1) Поверхностная активность
1) Снижение поверхностного натяжения
Степень снижения поверхностного натяжения измеряли способом, описанным в работе Tanaka et al. (Journal of Japanese Medical Society for Biological Interface, Vol. 13, N 2, p. 87, 1982).
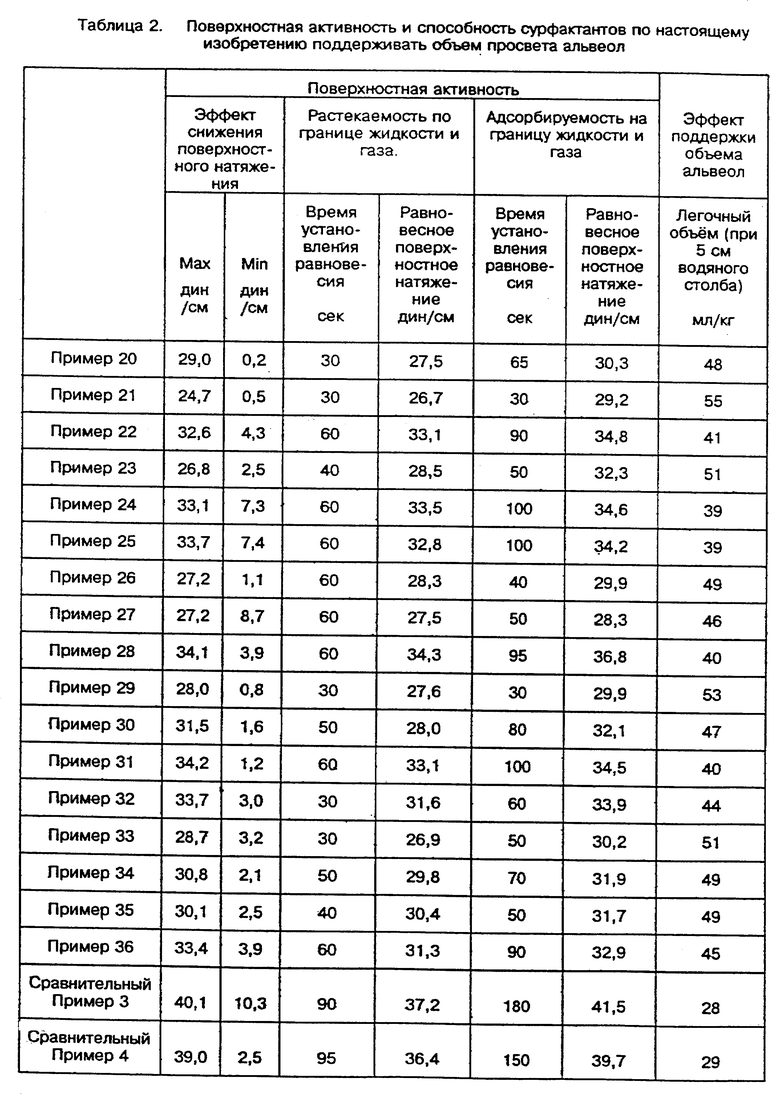
Суспензии ПАВ по изобретению капали на физиологический раствор (с поверхностным натяжением 54,0 см2) так, что на 1 см2 площади приходилось 1,0 - 2,0 мкг ПАВ по изобретению. Указанную площадь поверхности сжимали и расширяли в пределах 54,0 - 21,6 см2 в течение 2 - 5 минут и непрерывно измеряли поверхностное натяжение при 37oC с помощью измерительного прибора Wilhelmy (фирмы Kyowa Interface Science Co. Ltd.). Максимальное поверхностное натяжение составляло 24,7 - 34,1 дин/см, а минимальное поверхностное натяжение составляло 0,2 - 8,7 дин/см, свидетельствует о том, что благодаря действию ПАВ по изобретению, поверхностное натяжение физиологического раствора снизилось.
С помощью этого же способа измерили влияние SF-3 на снижение поверхностного натяжения и получили следующие результаты: максимальные значения поверхностного натяжения - 26,8 - 50,3 дин/см и минимальные значения поверхностного натяжения - 1,0 - 13,5 дин/см.
Поверхностное натяжение самого физиологического раствора при 37oC составляло 70,5 дин/см.
2) Способность распределяться по поверхности раздела газ-жидкость.
Суспензию ПАВ по изобретению капали на поверхность физиологического раствора так, чтобы приходилось 0,8 - 1,5 мкг ПАВ на 1 см2 поверхности физиологического раствора, и с помощью вертикальной пластины измеряли изменение поверхностного натяжения, начиная с того момента, когда капля суспензии попала на раствор. Во время измерения температура составляла 37oC.
Период уравновешивания - это время, в течение которого поверхностное натяжение достигает постоянного значения, начиная с момента, когда капля суспензии попадает на физиологический раствор, а значение после достижения этого равновесия называется равновесным поверхностным натяжением.
ПАВ по изобретению образовывали пленку на поверхности раздела газ-жидкость в течение короткого периода времени (30 - 60 секунд) и снижали поверхностное натяжение до 26,7 - 34,3 дин/см.
Тем же способом измеряли способность SF-3 распределяться по поверхности, эти измерения показали, что поверхностное натяжение через 120 секунд снизилось до 38,1 - 52,9 дин/см.
Этот факт свидетельствует о том, что ПАВ по изобретению способен быстро распределяться по поверхности раздела газ-жидкость и вызывать снижение поверхностного натяжения.
3) Способность к адсорбции на поверхности раздела газ-жидкость
Приготовили суспензии физиологического раствора, содержащие 0,2 - 1,0 мг ПАВ по изобретению на 1 мл при 37oC и измерили скорость адсорбции суспендированных ПАВ по изобретению на поверхности раздела газ-жидкость.
Способность к адсорбции измеряли способом, описанным в работе King et al. (American Journal of Physiology, N 223, p. 715, 1972).
Т.е. суспензию впрыскивали на дно сосуда из тефлона диаметром 5 см, содержащего физиологический раствор, а затем перемешивали магнитной мешалкой. Способность к адсорбции определяли по изменению поверхностного натяжения после прекращения перемешивания.
ПАВ по изобретению снизили поверхностное натяжение до значений от 28,3 до 36,8 дин/см за период от 30 до 100 секунд после прекращения перемешивания, после чего поверхностное натяжение оставалось неизменным.
Этот факт свидетельствует о том, что ПАВ по изобретению в суспендированном состоянии плавали и адсорбировались на поверхности раздела газ-жидкость в течение 30-100 секунд и образовали пленку, обладающую сильной поверхностной активностью.
Те же измерения, проведенные для SF-3, показали, что постоянное поверхностное натяжение в пределах от 42,2 до 58,3 дин/см было достигнуто через 150 секунд или более.
Этот факт свидетельствует о том, что адсорбционное действие SF-3 ниже, чем ПАВ по изобретению, и что ПАВ по изобретению обладают выраженной способностью стимулировать адсорбцию на поверхности.
(2) Способность переходить в суспензию
Испытания на способность легочных ПАВ переходить в суспензию проводили способом, описанным в японском патенте Hei-4-76965.
Способность переходить в суспензию определяли исходя из диспергируемости в конкретные моменты времени после начала суспендирования, а также из максимального размера диспергированных частиц через 2 минуты после начала суспендирования.
Тесты на диспергируемость проводили следующим образом. 60 мг каждого из легочных ПАВ разлили по 20 мл флаконам. Затем в каждый из указанных флаконов добавили по 2 мл физиологического раствора и поместили флаконы в шейкер типа Iwaki КМ Shaker-V-S (изготовленный фирмой Iwaki Sangyo Co Ltd.) и встряхивали с интенсивностью 270 движений в минуту. Степень дисперсии каждого образца наблюдали визуально с помощью увеличительного стекла каждые 30 секунд в течение первых 2 минут после начала встряхивания, каждую минуту в течение с 2 по 4 минуты после начала встряхивания и каждые 10 минут после 4-й минуты от начала встряхивания.
Степень перехода в суспензию определяли через определенные промежутки времени 2 человека, причем каждый проводил оценку 10 образцов. Образцы считались суспендированными, если в сосуде не наблюдаются скопления вещества и если композиция равномерно диспергирована в физиологическом растворе до образования белой плотной суспензии.
Эффективность диспергирования каждый сотрудник определял в установленные моменты времени в качестве процентного отношения образцов, где суспендирование завершилось, к суммарному количеству образцов (10 флаконов), а затем выводился средний показатель для обоих сотрудников.
Максимальные размеры диспергированных частиц измеряли следующим способом. 60 мг каждого легочного ПАВ разливали по 20 мл флаконам. Затем в каждый флакон добавили по 2 мл физиологического раствора и флаконы встряхивали в течение 2 минут так, как описано выше. После этого под микроскопом находили самую крупную частицу и измеряли ее диаметр микрометром. Таким образом было обнаружено, что ПАВ по изобретению в основном были суспендированы в течение 2 минут, и что максимальные размеры частиц этих ПАВ составляли 0,8 мм и менее, что свидетельствует о высокой способности к суспендированию.
(3) Фармакологические свойства
1) Острая токсичность
Острую токсичность ПАВ по изобретению тестировали на мужских особях мышей 1CR и крыс вистеровских в возрасте 5 недель. ЛД50 при оральном введении в ЛД50 при перитонеальном введении для мышей составляли 2,4 - 10,0 г/кг и 1,0 - 5,0 г/кг соответственно, а для крыс эти дозы составляли 1,5 - 5,0 г/кг и 1,5 - 2,5 г/кг соответственно.
2) Подострая токсичность.
ПАВ по изобретению назначали внутрибрюшинно взрослым вистеровским крысам в суточных дозах 300 - 600 мг/кг в течение 1 месяца. В отношении массы крыс отклонений не наблюдалось, не выявлено также аномальных явлений при гистологических наблюдениях невооруженным глазом.
3) Воздействие, заключающееся в сохранении объема альвеол
Недоношенные детеныши кролика в гестационном периоде 27 дней практически не вырабатывают легочных ПАВ и находятся в состоянии нехватки легочных ПАВ. Поэтому их использовали в качестве моделей для изучения неонатального респираторного дистресс-синдрома.
У 5 детенышей кролика в гестационном периоде 27 дней измеряли объемы альвеол (далее называемые объемом легких) под различным давлением в дыхательных путях при 37oC.
На шее детеныша делали надрез и с помощью водяного манометра, прикрепленного к трахее, проводили непрерывные измерения, начиная с 5 минуты после введения ПАВ по изобретению через дыхательные пути. С помощью двухканального шприцевого насоса с независимым приводом N 940 (изготовленного фирмой Harvard Co. , USA). давление в дыхательных путях поднимали до 30 см H2O, чтобы заставить альвеолы расшириться. Затем давление в дыхательных путях снизили до 0 см H2O для того, чтобы вызвать коллапс альвеол, и при этом измеряли объем легких при различных давлениях H2O. Затем объемы легких указывали в миллилитрах на 1 кг массы (мл/кг).
ПАВ по изобретению (60 мг/кг) вводили путем прямой инъекции в дыхательные пути 0,05 - 0,5 мл суспензии физиологического раствора с концентрациями ПАВ в 1,0 - 6,0 (в/о).
Объем легких в тот момент, когда давление снижается до 5 см H2O указывает на функциональную остаточную способность и, чем выше этот объем, тем выше активность легочного ПАВ.
В качестве контроля вводили физиологический раствор без ПАВ по изобретению. Объемы легких (при 5 см H2O) у недоношенных детенышей кролика в контрольной группе составляли 1 - 5 мл/кг, указывая на то, что альвеолы едва расширялись.
Доношенные детеныши в гестационном периоде 30 дней обладают нормальными уровнями легочных ПАВ. Объемы легких таких детенышей (при 5 см H2O) составляют 35 - 53 мл/кг, указывая на то, что альвеолы достаточно расширяются и что можно вызвать нормальное дыхание.
В тех случаях, когда вводили SF-3, объемы легких (при 5 см H2O) недоношенных детенышей составляли 15 - 25 млг/кг, указывая на неадекватное расширение альвеол.
В тех случаях, когда вводили ПАВ по изобретению, объемы легких (при 5 см H2O) составляли 39 - 55 мл/кг, указывая на то, что ПАВ по изобретению повышают объем легких у недоношенных детенышей до нормальных уровней.
Как описано выше, синтетические пептиды по изобретению обладают способностью сильно повышать поверхностную активность липидных смесей. Это позволяет изготавливать лечебные препараты для лечения респираторного дистресс-синдрома, которые эффективны в отношении поверхностной активности, способности к суспендированию и фармакологических свойств, используя ПАВ по изобретению, которые состоят из синтетических пептидов по изобретению и смеси липидов.
Композиции, содержащие ПАВ по изобретению в качестве активного ингредиента, можно использовать также для лечения других заболеваний, для лечения которых применяются легочные ПАВ, включая послеоперационные нарушения дыхания, астму, бронхит, неонатальный некротический энтерит, язвы желудка и двенадцатиперстной кишки, заболевания органов дыхания, вызванные вирусами и трубной непроходимостью, для предотвращения спайки маточных труб и послеоперационных спект, а также в качестве отхаркивающего.
Лекарственные препараты по изобретению для лечения респираторного дистресс-синдрома содержат 50 - 100 мг ПАВ по изобретению на одну дозу для детей и 50 - 5000 мг ПАВ на одну дозу для взрослых. Такие дозы получают путем суспендирования в воде, физиологическом растворе или буферах и т.п., которые являются физиологически переносимыми, в концентрациях 1,0 - 10,0% (в/о). Эти суспензии вводят в течение 72 часов после наступления нарушения дыхания путем инъекций или распыления в дыхательных путях, производимого от 1 до 10 раз. Композиции можно вдыхать и в несуспендированном виде, т.е. в виде порошка. Дозировки, способ применения и частоту введения можно изменять в зависимости от симптомов и состояния пациентов, а также от сопутствующего курса лечения.
Лекарственные препараты по изобретению могут содержать, при необходимости, такие фактические вспомогательные вещества, как стабилизаторы, консерванты, изотонирующие агенты, буферы, суспендирующие агенты, антиокислители и поверхностно-активные вещества, или такие лекарства, как бронхолитические средства, антиаллергические препараты, карценостатические средства, противовирусные агенты, противовоспалительные препараты и противогрибковые препараты.
Лекарственный препарат по изобретению может быть в виде жидкости или порошка. Лекарственные препараты по изобретению можно расфасовать в запечатанные сосуды, такие как флаконы и ампулы, и хранить в виде стерильных композиций.
Теперь переходим к более подробному описанию настоящего изобретения с помощью приведенных ниже примеров.
(1) Получение пептидов
В приведенных ниже примерах молекулярную массу синтезированных пептидов измеряли методом масс-спектрометрии с бомбардировкой быстрыми атомами (МСББА). В качестве масс-спектрометра использовали JMS-S102A (JEOL. Ltd.), а в качестве источника ионов - цезиевую пушку (10 Кее).
Пример 1
Пептид (Пептид A) с последовательностью 5 синтезировали на поверхности фенилацетамидометилового полимера (ФАМ) методом твердофазного синтеза по способу, описанному в работе "The Peptides" (Gross E. and Meinenhofe J. издатели., Barany G. and Merrifield R. авторы, том. 2, стр. 1 - 284, Academie Press, New York, 1980).
Лейциновый остаток на C-конце трансформировали в трет-бутилоксикарбониллейцин (Вос-Лей) и связали его с полимером ФАМ через оксиметилфенилацетамидметил. После связывания C-конца, Вос-Лей-ФАМ-полимер (0,70 моль/г, 0,35 г) перенесли в реакционный контейнер пептидного синтезатора (модель 99ОЕ, производитель Beekman Instruments, Inc.). Затем в направлении N-конца на поверхности полимера ввели аминокислоты (к которым были присоединены соответствующие защитные группы) с помощью симметричных ангидридов, чтобы синтезировать полностью защищенный пептид-O-полимер. Однако при конденсации аргинина произвели двойное присоединение, используя N,N-дициклогексилкарбордиимид /гидроксибензотриазол [Connic et al. Chem. Ber., 103, 788-798 (1970)] .
N-концевые аминогруппы всех аминокислот были защищены Вос-группами, а функциональные боковые цепочки были защищены следующими группами (которые ввели до того, как аминокислоты использовали в реакциях):
Арг-Тоз : (тозил)
Лиз-2-ClZ : (2-хлорбензилоксикарбонил)
Цис-4MeBz1 : (4-метилбензил)
Гис-Тоз : (тозил)
Завершение реакции конденсации этих соединений подтвердили с помощью кайзеровского теста с использованием нингидрина. Полностью защищенный пептид-O-полимер (155 мг) в течение 5 минут набухал в дихлорметане. Затем защитную группу N-α- Вос удаляли с помощью ТФК, содержащей 1% (о/о) индола и 0,1% (о/о) этандитиола. После этого пептид отщепляли от полимера, обрабатывая незащищенный пептид-O-полимер безводным фтористым водородом (11 мл), в который добавили p-крезол (1 мл), p-тиокрезол (0,2 г) и ДМСО (1 мл); обработку производили в течение 60 минут при 0oC.
Фтористый водород и ДМСО отгоняли под вакуумом при 0oC. Отделенный пептид и полимер промывали три раза холодным диэтиловым эфиром (15 мл) и экстрагировали отделенный пептид, промывая четыре раза в холодной ТФК (5 мл). Экстрагированную жидкость немедленно фильтровали и добавляли ледяную воду (150 мл) до выпадания в осадок неочищенного пептида. Неочищенный пептид затем центрифугировали при 1000 xo и 0oC в течение 30 минут и выделяли в виде осадка. Этот осадок промывали диэтиловым эфиром (15 мл). После повторной промывки с использованием диэтилового эфира, этилацетата и дистиллированной воды, получали 84 мг пептида A.
Затем этот неочищенный пептид растворяли в 50% водном растворе ДМСО и очищали высокоэффективной жидкостной хроматографией с обращенной фазой (ВЭЖХ) с использованием "μ Bondasheres" и колонки C8-300 для отбора очищенного пептида A.
Элюирование производили в течение 5 минут с использованием 50% водного раствора ацетонитрила, содержащего 0,1% ТФК, в качестве элюента. После этого элюирование производили в течение 30 минут при линейном градиенте концентрации, образованной вышеуказанным элюентом, и с использованием 80% водного раствора ацетонитрила, содержащего 0,1% ТФК.
Присутствие пептида в элюате наблюдали при 245 нм (спектрофотометр фирмы Japan Spectroscopic Co. Ltd., модель 870-UV) и с помощью дифференциального рефрактометра (фирмы Shimadzu Manufacturing corporation, модель RID-6A).
МСББА (М+Н+) : 3837,1 (вычисленная молекулярная масса; 3835,9)
Пример 2
Пептид (пептид В) с последовательностью 6 синтезировали методом твердофазного синтеза с использованием многопептидной твердофазной системы синтеза "Kokku - San" (торговое наименование; производитель Kokusan Chemical Works Co. , Ltd.) с применением способов, описанных в работе "Solid Phase Peptide Synthesis-A Practical Approach" авторы E.Atherton and R.S.Sheppard (стр. 25-189, Oxford University Press, Oxford) и в работе Kenichi Akagi et al. (Chem. Pharm. Bull., 37(10), pp. 2661-2664, 1989).
В качестве исходного полимера использовали N-α-9- флуоренилметилоксикарбонил-лейцин-O-полимер (Fmoc-Лей-O-полимер) (0,20 ммоль/0,5 г), в котором N-α-9- флуоренилметилоксикарбонил-лейцин (Fmoc-Лей) связан с сополимером 4-(гидроксиметил) и феноксиметила (сополимер 1% стирола с дивинилбензолом). Указанный полимер набухал в течение 20 минут в N,N-диметилформамиде (ДМФ), затем его промывали четыре раза ДМФ. После этого добавили 20% пиперидин в ДМФ и смесь перемешивали встряхиванием до удаления защитных групп. Эту операцию повторили три раза, чтобы полностью удалить защитные группы. После этого трижды промывали ДМФ, три раза промыли N-метил-2-пирролидоном и три раза снова ДМФ до удаления избытка пиперидина в полимере. Наличие пиперидина проверяли с помощью лакмусовой бумаги.
После этого добавляли ДМФ (6 мл), Fmос-Лей (0,5 ммоль), N-гидроксибензотриазол (0,5 ммоль) и N,N'-диизопропилкарбодиимид (0,5 ммоль) и смесь перемешивали встряхиванием в течение 90 минут для того, чтобы прошла реакция конденсации. Затем полимер промыли четыре раза ДМФ до удаления избытка реактивов. Завершение реакции конденсации с помощью кайзер-теста с использованием нингидрина.
По этой схеме проводили синтез и поэтапно добавляли аминокислоты в направлении N-конца на поверхности полимера до получения пептид-O-полимера с полностью защищенными N-концом и функциональными группами.
Реакции конденсации для введения аргинина, лизина, гистидина, пролина и цистеина проводили дважды, каждый раз в течение 120 минут.
После этого к защищенному пептид-O-полимеру добавили 20% пиридин в ДМФ для того, чтобы удалить с N-конца защитную группу Fmoc. Затем пептид-O-полимер промыли шесть раз ДМФ и шесть раз метанолом и высушили при пониженном давлении. Затем к высушенному пептид-O-полимеру (100 мг) добавляли m-крезол (0,2 мл), 1,2-этандитиол (0,5 мл), тиоанизол (1,2 мл), ТФК (7,5 мл) и триметилсилилоромид (1,4 мл), добавку производили при перемешивании на ледяной бане. После этого смесь перемешивали в течение 120 минут на ледяной бане до удаления защитных групп с функциональных боковых цепочек и до удаления пептида с полимера, а затем фильтровали через стеклянный фильтр (С3). Фильтрат концентрировали при пониженном давлении приблизительно до 5 мл с помощью испарителя. После этого добавили диэтиловый эфир, чтобы пептид выпал в осадок. Осадок собирали на стеклянный фильтр (C3), пять раз промывали диэтиловым эфиром и, после высушивания под пониженным давлением получили 60 мг пептида B.
N-концевые аминогруппы всех аминокислот были защищены группами Fmoc, а функциональные боковые цепочки были защищены следующими (которые ввели до того, как аминокислоты использовали в реакциях):
Арг-Mtr : (4-метокси-2,3,6-триметилбензолсульфонил)
Лиз-Вос : (трет-бутилоксикарбонил)
Цис-Трт : (тритил)
Гис-Трт : (тритил)
Приблизительно 100 мг неочищенного пептида растворяли в ТФК (1 мл) и добавили в четыре раза больше растворителя подвижной фазы, т.е. 10 мМ β- меркаптоэтанола в ТФК-дихлорметане (5:95, о/о), получив раствор 20 мг/мл, который очищали ВЭЖХ с использованием колонки Asahipak GS-510 (диаметр 7,5 х 500 мм) (торговое наименование; производитель Asahi Chemical Industry Co., Ltd.), получив чистый пептид B.
В качестве элюента использовали 10 мМ β- меркаптоэтанол в ТФК-дихлорметане (5: 95, о/о), элюирование производили при скорости потока в 0,8 мл/мин, в течение 80 минут. Присутствие пептида в элюате наблюдали при 245 нм (спектрофотометр фирмы Japan Spectroscopic Co., Ltd., модель 870-UV) и с помощью дифференциального рефтактометра (фирмы Shimadzu Manufacturing Сorporation, модель RID-6A).
МСББА (М+Н+): 3017,9 (вычисленная молекулярная масса; 3016,9).
Пример 3
Пептид (пептид C) с последовательностью 7 получали способом, который описан в Примере 2.
МСББА (М+Н+): 3116,0 (вычисленная молекулярная масса: 3115,1).
Пример 4
Пептид (пептид D) с последовательностью 8 получали способом, который описан в Примере 2.
МСББА (М+Н+): 2663,7 (вычисленная молекулярная масса; 2662,5).
Пример 5
Пептид (пептид E) с последовательностью 9 получали способом, который описан в Примере 2.
МСББА (М+Н+): 2211,2 (вычисленная молекулярная масса; 2209,9).
Пример 6
Пептид (пептид F) с последовательностью 10 получали способом, который описан в Примере 2.
МСББА (М+Н+): 2647,5 (вычисленная молекулярная масса; 2646,4).
Пример 7
Пептид (пептид G) с последовательностью 11 получали способом, который описан в Примере 2.
МСББА (М+Н+): 3018,1 (вычисленная молекулярная масса; 3016,9).
Пример 8
Пептид (пептид H) с последовательностью 12 синтезировали методом твердофазного синтеза с использованием многопептидной твердофазной системы синтеза, описанной в Примере 2. В качестве исходного полимера использовали N-N-α-9-флуоренилметилоксикарбонил-норлейцин-O-полимер (Fmoc-Nle-O-полимер) (0,20 ммоль/0,5 г). Указанный полимер набухал в течение 20 минут в N,N-диметилформамиде (ДМФ), затем его промывали четыре раза ДМФ. После этого добавляли 20% пиперидин в ДМФ и смесь перемешивали встряхиванием до удаления защитных групп. Эту операцию повторили три раза, чтобы полностью удалить защитные группы. После этого девять раз промывали ДМФ, чтобы удалить избыток пиперидина в полимере. Присутствие остатка пиперидина контролировали с помощью лакмусовой бумаги.
После этого добавили ДМФ (6 мл), Fmoc-Нле (0,5 ммоль), N-гидроксибензотриазол (0,5 ммоль) и N,N'-диизопропилкарбодиимид (0,5 ммоль), и смесь перемешивали встряхиванием в течение 90 минут для того, чтобы прошла реакция конденсации. Затем полимер промыли четыре раза ДМФ до удаления избытка реактивов. Завершение реакции конденсации проверяли с помощью кайзер-теста с использованием нингидрина.
По этой схеме проводили синтез и поэтапно добавляли аминокислоты в направлении N-конца на поверхности полимера до получения пептид-O-полимера с полностью защищенными N-концом и функциональными группами.
Реакции конденсации для введения аргинина, лизина, гистидина, пролина и цистеина проводили дважды, каждый раз в течение 120 минут.
После этого к защищенному пептид-O-полимеру добавили 20% пиридин в ДМФ для того, чтобы удалить с N-конца защитную группу Fmoc. Затем пептид-O-полимер промыли шесть раз ДМФ и шесть раз метанолом и высушили при пониженном давлении. Затем к высушенному пептид-O-полимеру (100 мг) добавили m-крезол (0,2 мл), 1,2-этандитиол (0,5 мл), тиоанизол (1,2 мл), ТФК (7,5 мл) и триметилсилилбромид (1,4 мл), добавку производили при перемешивании на ледяной бане. После этого смесь перемешивали в течение 120 минут на ледяной бане до удаления защитных групп с функциональных боковых цепочек и до удаления пептида с полимера, а затем фильтровали через стеклянный фильтр (C3). Фильтрат концентрировали при пониженном давлении приблизительно до 5 мл с помощью испарителя. После этого добавили диэтиловый эфир, чтобы пептид выпал в осадок. Осадок собирали на стеклянный фильтр (C3), пять раз промывали диэтиловым эфиром и после высушивания под пониженным давлением получали 65 мг пептида H.
N-концевые аминогруппы всех аминокислот были защищены группами Fmoc, а функциональные боковые цепочки были защищены следующими группами (которые ввели до того, как аминокислоты использовали в реакциях):
Арг-Mtr : (4-метокси-2,3,6-триметилбензолсульфонил)
Лиз-Вос : (трет-бутилоксикарбонил)
Цис-трт : (тритил)
Гис-Вос : (трет-бутилоксикарбонил)
Приблизительно 10 мг неочищенного пептида растворяли в 3,0 мл смешанного растворителя (хлороформ-метанол, 2:1, о/о). Затем образец очищали на колонке Sephadex 1H-60 (диаметр 2,5 см x 90 см), уравновешенной смешанным растворителем (хлороформ-метанол, 2:1, о/о) до получения чистого пептида H.
Присутствие пептида в элюате наблюдали при 245 нм (спектрофотометр фирмы Japan Spectroscopic Co., Ltd., модель 870-UV) и с помощью дифференциального рефрактометра (фирмы Shimadzu Manufacturing corporation, модель RID-6A).
МСББА (М+Н1): 2663,6 (вычисленная молекулярная масса; 2662,5).
Пример 9
Пептид (пептид I) с последовательностью 13 получали способом, который описан в Примере 8.
МСББА (М+Н+): 2560,4 (вычисленная молекулярная масса; 2553,3).
Пример 10
Пептид (пептид Q) с последовательностью 14 получали способом, который описан в Примере 8.
МСББА (М+Н+): 2663,8 (вычисленная молекулярная масса; 2662,5).
Пример 11
Пептид (пептид K) с последовательностью 15 получали способом, который описан в Примере 2.
МСББА (М+Н+): 2663,5 (вычисленная молекулярная масса; 2662,5).
Пример 12
Пептид (пептид L) с последовательностью 16 получали способом, который описан в Примере 2.
МСББА (М+Н+): 2503,6 (вычисленная молекулярная масса; 2502,4).
Пример 13
Пептид (пептид M) с последовательностью 17 получали способом, который описан в Примере 2.
МСББА (М+Н+): 2736,7 (вычисленная молекулярная масса; 2735,5).
Пример 14
Пептид (пептид N) с последовательностью 18 получали способом, который описан в Примере 2.
МСББА (М+Н+): 2640,4 (вычисленная молекулярная масса; 2639,4).
Пример 15
Пептид (пептид O) с последовательностью 19 получали способом, который описан в Примере 8.
МСББА (М+Н+): 2640,3 (вычисленная молекулярная масса; 2639,4).
Пример 16
Fmoc-Про-Вал-Гис(Трт)-Лей-Лиз(Вoc)-Арг(Mtr)
Названный пептид (пептид P) синтезировали методом твердофазного синтеза с использованием Peptide Synthesizer System 9050 (производитель Millipore Corp.)
В качестве исходного полимера использовали N-α-9- флуоренилметилкарбонил N-ω-4- метокси-2,3,6- триметилбензолсульфонил-аргинин-O-полимер (Fmoc-Арг(Mtr)-O-полимер) (0,20 ммоль), в котором N-α-9- флуоренилметилкарбонил N-ω-4- метокси-2,3,6-триметилбензолсульфонил-аргинин связан с 2-метокси-4-алкоксибензилалкоголь-полимером (полимер "Sasrin" торговое наименование фирмы Bachem Co., Ltd). К этому полимеру поэтапно добавляли аминокислоты в направлении N-конца на поверхности полимера в соответствии с протоколом "Peptide Synthesizer System 9050" с целью синтезировать пептид-O-полимер с полностью защищенными N-концом и функциональными группами.
Затем полностью защищенный пептид-O-полимер пять раз промывали метанолом и высушивали при пониженном давлении. После этого к высушенному пептид-O-полимеру (330 мг) добавили раствор ТФК-дихлорметана (1:9, о/о); добавку производили при перемешивании на ледяной бане. Затем смесь перемешивали на ледяной бане в течение 30 минут, после чего перемешивали в течение 90 минут при комнатной температуре до удаления пептида из полимера, причем защитные группы все еще были присоединены к полимеру. После этого смесь фильтровали через стеклянный фильтр (C3) и фильтрат концентрировали при пониженном давлении до около 5 мл, используя испаритель. Затем добавили диэтиловый эфир, чтобы пептид выпал в осадок. Полученный осадок собирали на стеклянный фильтр (C3), промывали пять раз диэтиловым эфиром и высушивали при пониженном давлении. Таким образом получали 180 мг пептида P.
N-концевые аминогруппы всех аминокислот были защищены группами Fmoc, а функциональные боковые цепочки были защищены следующими группами (которые ввели до того, как аминокислоты использовали в реакциях):
Арг-Mtr: (4-метокси-2,3-триметилбензолсульфонил)
Лиз-Вос: (трет-бутилоксикарбонил)
Гис-Трт: (тритил)
Затем раствор ТФР-дихлорметана (1:99, о/о) добавили в неочищенный пептид до получения образца раствора 10 мг/мл, который очищали ВЭЖХ с использованием колонки Asahipak CS-510 (диаметр 21,5 х 500 мм) (торговое наименование; фирмы Asahi Chemical Industry Co., Ltd.), получая чистый пептид P.
Раствор ТФК-дихлорметана (1:99, о/о) использовали в качестве элюата, элюирование проводили при скорости потока в 8,1 мл/мин в течение 120 минут. Присутствие пептида в элюате наблюдали при 245 нм (спектрофотометр фирмы Japan Spectroscopic Co., Ltd, модель 870-UV) и с помощью дифференциального рефрактометра (фирмы Shimadzu Manufacturing Сorporation модель RID-6A).
МСББА (М+Н+): 1405,0 (вычисленная молекулярная масса; 1403,8).
Пример 17
H-Нле-(Нле)14-Нле-O-полимер синтезировали с твердофазной многопептидной системой как описано в Примере 8.
Далее, после добавки ДМФ в синтезированный H-Нле-(Нле)14-Нле-O-полимер вместо Fmoc-Арг(Mtr) добавили пептид P. После этого добавили N-гидроксибензотриазол и N,N'-диизопропилкарбодиимид и смесь перемешивали встряхиванием в течение 8 часов. Эту реакцию конденсации провели дважды. Завершение реакции конденсации проверяли с помощью кайзер-теста с использованием нингидрина.
После этого способом, описанным в Пример 8, защитные группы удаляли с функциональных групп, и пептид отделяли от полимера и очищали ВЭЖХ до получения пептида с последовательностью 13 (пептида I).
МСББА (М+Н+): 2560,2 (вычисленная молекулярная масса; 2559,3).
Пример 18
Пептид (пептид O), в котором тиольные группы в пептиде находились в ацетамидометилированном (АСМ) виде получали способом, описанным в Примере 4, за исключением того, что вместо Fmoc-Цис-(Трт) использовали Fmoc-Цис-(АСМ).
МСББА (М+Н+): 2734,7 (вычисленная молекулярная масса; 2733,6).
Пример 19
Пептид R получали путем этерификации тиольных групп пептида E пальмитиновой кислотой по способу, описанному в патенте EP 0458167 A1 на имена
МСББА (М+Н+): 2448,5 (вычисленная молекулярная масса; 2448,3).
Сравнительный пример 1
Пептид с последовательностью 20 получали способом, описанным в Примере 2.
МСББА (М+Н+): 2793,8 (вычисленная молекулярная масса; 2792,6).
Сравнительный пример 2
Пептид T с последовательностью 21 получали способом, описанным в Примере 2.
МСББА (М+Н+): 3018,3 (вычисленная молекулярная масса; 3016,9).
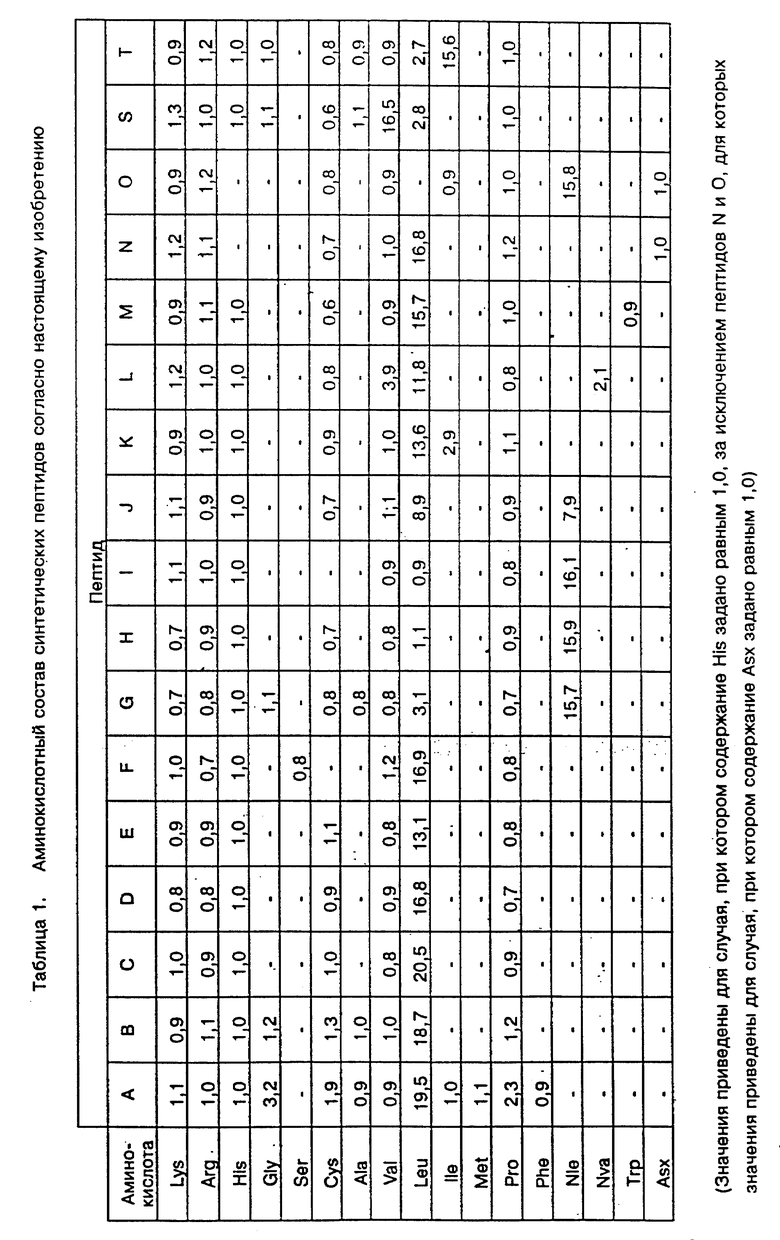
Анализ аминокислотного состава синтетических пептидов по изобретению
Синтетические пептиды по изобретению были подвергнуты кислотному гидролизу с использованием раствора 12 н. HCl-ТФК (2:1, о/о), содержащего 5% (м/о) фенола; реакцию проводили под вакуумом при 150oC в течение 1, 2, 4, 6, 12, 24, 48 и 72 часов, и продукты гидролиза после удаления их из кислоты подвергли анализу с использованием "Системы автоматического анализа аминокислот Shimadzu" (1C-9A). Триптофан (Три) пептида M подвергли щелочному гидролизу, используя 4,2 н. водный гидроксид натрия; реакцию проводили в течение 16, 24 и 32 часов, после чего смесь нейтрализовали HCl, а затем провели анализ с помощью "Системы автоматического анализа аминокислот". Аминокислотный состав, вычисленный на основе коэффициентов кислотности аминокислот, выход которых был наибольшим при гидролизе в течение от 1 до 72 часов, в значительной степени совпадал с показателями, рассчитанными из химических формул.
Результаты приведены в Таблице 1.
(2) Получение ПАВ по изобретению
ПАВ по изобретению получали посредством смешивания пептидов по изобретению с тремя липидными компонентами - холинфосфоглицеридом, кислым фосфолипидом и жирнокислым аналогом.
Пример 20
Стерилизованный 1,2-дипальмитоилглицеро-(3)-фосфохолин (1350 мг), 1,2-диацил-sn-глицеро-(3)-фосфо-sn-глицерол (с ацильной группой, имеющий 14 - 24 атома углерода; производства фирмы Sigma Chemical Co., Ltd.) (450 мг) и миристиновую кислоту (200 мг) растворяли при комнатной температуре в смеси хлороформ-метанол (2: 1, о/о) (100 мл), а 25 мг пептида A растворяли в ТФК (1,0 мл). Полученные растворы смешивали вместе, а затем высушивали и отверждали при пониженном давлении. Полученный остаток суспендировали в смеси вода-этанол (9: 1, о/о) (100 мл) в течение 15 минут при 40oC. После замораживания полученной суспензии при -50oC и последующего высушивания в течение 36 часов под вакуумом в 85 - 100 мкНо, получали ПАВ (2070 мг) в виде белого порошка.
Полученный порошок не содержал поддающихся обнаружению количеств этанола, а содержание каждого из компонентов по отношению к суммарной массе ПАВ составляло: 65,2 мас.% 1,2-дипальмитоилглицеро-(3)-фосфохолина, 21,7 мас.% 1,2-диацил-sn-глицеро-(3)-фосфо-sn-глицерола, 9,7 мас. % миристиновой кислоты, 1,2 мас.% пептида A и 2,2 мас.% воды.
Пример 21
1,2-Дипальмитоилглицеро-(3)-фосфохолин (300 мг), 1,2-диацил-sn-глицеро-(3)-фосфо-sn-глицерол (с ацильной группой, имеющей 14-24 атома углерода; производства фирмы Sigma Chemical Co. , Ltd.) (100,0 мг) и пальмитиновую кислоту (40,0 мг) растворяли в смеси хлороформ-метанол (2:1, о/о) (300 мл), а 10,0 мг пептида B растворяли в смеси хлороформ:метанол) (2:1, о/о) (2,0 мл). Полученные растворы смешивали вместе, а затем высушивали и отверждали при пониженном давлении. Полученный остаток суспендировали в смеси вода-этанол (9:1, о/о) (100 мл) в течение 20 минут при 45oC. После замораживания полученной суспензии при -60oC и последующего высушивания в течение 40 часов под вакуумом в 60 - 110 мкНо, получали 459,1 мг ПАВ в виде белого порошка.
Полученный порошок не содержал поддающихся обнаружению количеств этанола, а содержание каждого из компонентов по отношению к суммарной массе ПАВ составляло : 65,3 мас.% 1,2 - дипальмитоилглицеро-(3)-фосфохолина, 21,8 мас. % 1,2-диацил-sn-глицеро-(3)-фосфо-sn-глицерола, 8,7 мас.% пальмитиновой кислоты, 2,2 мас.% пептида B и 2,0 мас.% воды.
Пример 22
1,2-Дипальмитоилглицеро-(3)-фосфохолин (280,0 мг), 1,2-дилауроил-sn-глицеро-(3)-фосфо-sn-глицерол (120,0 мг) и пальмитиновую кислоту (27,0 мг) растворяли в смеси хлороформ-метанол (2:1, о/о) (150 мл), а 2,8 мг пептида C растворяли в смеси хлороформ-метанол (1:2, о/о) (0,5 мл). Полученные растворы смешивали вместе, а затем высушивали и отверждали при пониженном давлении. Полученный остаток суспендировали в смеси вода-этанол (8:2, о/о) (100 мл) в течение 45 минут при 40oC. После замораживания полученной суспензии при -65oC и последующего высушивания в течение 36 часов под вакуумом в 50 - 80 мкНо, получали 437,6 мг ПАВ в виде белого порошка.
Полученный порошок не содержал поддающихся обнаружению количеств этанола, а содержание каждого из компонентов по отношению к суммарной массе ПАВ составляло: 64,0 мас.% 1,2-дипальмитоилглицеро-(3)-фосфохолина, 27,4 мас.% 1,2-дилаурил-sn-глицеро-(3)-фосфо-sn-глицерола, 6,2 мас.% пальмитиновой кислоты, 0,6 мас.% пептида С и 1,8 мас.% воды.
Пример 23
Выполнив те же операции, которые описаны в Примере 21, но при использовании пептида D вместо пептида B, и 1-пальмитоил-2-олеил-sn-глицеро-(3)-фосфо-sn-глицерола вместо 1,2-диацил-sn-глицеро-(3)-фосфо-sn-глицерола (с ацильной группой, имеющей 14 - 24 атома углерода; производства фирмы Sigma Chemical Co., Ltd.), получали 451,9 мг ПАВ в виде белого порошка.
Полученный порошок не содержал поддающихся обнаружению количеств этанола, а содержание каждого из компонентов по отношению к суммарной массе ПАВ составляло: 66,4 мас.% 1,2-дипальмитоилглицеро-(3)-фосфохолина, 22,1 мас.% 1-пальмитоил-2-олеоил-sn-глицеро-(3)-фосфо-sn-глицерола, 8,9 мас.% пальмитиновой кислоты, 2,2 мас.% пептида и 0,4 мас.% воды.
Пример 24
1,2-Дипальмитоилглицеро-(3)-фосфохолин (320,0 мг), 1,2-димиристоил-sn-глицеро-(3)-фосфо-sn-глицерол (80,0 мг) пальмитиновую кислоту (60,0 мг) растворяли в смеси хлороформ-метанол (1:1, о/о) (200 мл), а 14,0 мг пептида E растворяли в ТФК (0,3 мл). Полученные растворы смешивали вместе, а затем высушивали и отверждали при пониженном давлении. Полученный остаток суспендировали в смеси вода-этанол (10: 1, о/о) (50 мл) в течение 60 минут при 45oC. После замораживания полученной суспензии при -45oC и последующего высушивания в течение 24 часов под вакуумом в 50 - 110 мкНо, получали 479,2 мг ПАВ в виде белого порошка.
Полученный порошок не содержал поддающихся обнаружению количеств этанола, а содержание каждого из компонентов по отношению к суммарной массе ПАВ составляло: 66,8 мас.% 1,2-диаальмитоилглицеро-(3)-фосфохолина, 16,7 мас.% 1,2-димиристоил-sn-глицеро-(3)-фосфо-sn-глицерола, 12,5 мас.% пальмитиновой кислоты, 2,9 мас.% пептида E и 1,1 мас.% воды.
Пример 25
Выполнив те же операции, которые описаны в Примере 21, но при использовании пептида F (22,0 мг) вместо пептида B (10,0 мг) и 1,2-дистеароил-sn-глицеро-(3)-фосфо-sn-глицерола вместо 1,2-диацил-sn-глицеро-(3)-фосфо-sn-глицерола (с ацильной группой, имеющей 14 - 24 амома углерода; производства фирмы Sigma Chemical Co., Ltd.) получали 463,9 мг ПАВ в виде белого порошка.
Полученный порошок не содержал поддающихся обнаружению количеств этанола, а содержание каждого из компонентов по отношению к суммарной массе ПАВ составляло: 64,7 мас. % 1-2-пальмитоилглицеро-(3)-фосфохолина, 21,6 мас.% 1,2-дистеароил-sn-глицеро-(3)-фосфо-sn-глицерола, 8,6 мас. % пальмитиновой кислоты, 4,7 мас.% пептида F и 0,4 мас.% воды.
Пример 26
Выполнив те же операции, которые описаны в Примере 21, но при использовании пептида C вместо пептида B, получали 454,1 мг ПАВ в виде белого порошка.
Полученный порошок не содержал поддающихся обнаружению количеств этанола, а содержание каждого из компонентов по отношению к суммарной массе ПАВ составляло: 66,1 мас.% 1,2-дипальмитоилглицеро-(3)-фосфохолина, 22,0 мас.% 1,2-ацил-глицеро-(3)-фосфо-sn-глицерола (с ацильной группой, имеющей 14 - 24 атома углерода; производства фирмы Sigma Chemical Co., Ltd.), 8,8 мас.% пальмитиновой кислоты, 2,2 мас.% пептида C и 0,9 мас.% воды.
Пример 27
1,2-Дипальмитоилглицеро-(3)-фосфохолин (210 мг), 1,2-диацил-sn-глицеро-(3)-фосфо-sn-глицерол (с ацильной группой, имеющей 14 - 24 атома углерода; производства фирмы Sigma Chemical Co., Ltd.) (90,0 мг) и стеариновую кислоту (33,0 мг) растворяли в смеси хлороформ-метанол (3:1, о/о) (100 мл), а 1,9 мг пептида H растворяли в метаноле (0,5 мл). Полученные растворы смешивали вместе, а затем высушивали и отверждали при пониженном давлении. Полученный остаток суспендировали в смеси вода-этанол (9:1, о/о) (90 мл) в течение 15 минут при 50oC. После замораживания полученной суспензии при -55oC и последующего высушивания в течение 28 часов под вакуумом в 100 - 120 мкНо получали 340,2 мг ПАВ в виде белого порошка.
Полученный порошок не содержал поддающихся обнаружению количеств этанола, а содержание каждого из компонентов по отношению к суммарной массе ПАВ составляло: 61,7 мас.% 1,2-дипальмитоилглицеро-(3)-фосфохолина, 26,5 мас.% 1,2-диацил-sn-глицеро-(3)-фосфо-sn-глицерола, 9,7 мас.% стеариновой кислоты, 0,5 мас.% пептида H и 1,6 мас.% воды.
Пример 28
1,2-Дипальмитоилглицеро-(3)-фосфохолин (210,0 мг), 1-пальмитоил-2-олеоил-sn-глицеро-(3)-фосфо-L-серин (90,0 мг) и пальмитиновую кислоту (33,0 мг) растворяли в смеси хлороформ-метанол (4:1, о/о) (100 мл), а 11,0 мг пептида I растворяли в ТФК (0,5 мл). Полученные растворы смешивали вместе, а затем высушивали и отверждали при пониженном давлении. Полученный остаток суспендировали в смеси вода-этанол (9:1, о/о) (110 мл) в течение 25 минут при 45oC. После замораживания полученной суспензии при -55oC и последующего высушивания в течение 28 часов под вакуумом в 100 - 120 мкНо получали 348,7 мг ПАВ в виде белого порошка.
Полученный порошок не содержал поддающихся обнаружению количеств этанола, а содержание каждого из компонентов по отношению к суммарной массе ПАВ составляло: 60,2 мас.% 1,2-дипальмитоилглицеро-(3)-фосфохолина, 25,8 мас.% 1-пальмитоил-2-олеоил-sn-глицеро-(3)-фосфо-L-серина, 9,5 мас.% пальмитиновой кислоты, 3,2 мас.% пептида I и 1,3 мас.% воды.
Пример 29
Выполнив те же операции, которые описаны в Пример 21, но при использовании пептида Q вместо пептида B, получали 459,3 мг ПАВ в виде белого порошка.
Пример 30
Выполнив те же операции, которые описаны в Пример 21, но при использовании пептида K вместо пептида B, получали 452,5 мг ПАВ в виде белого порошка.
Пример 31
Выполнив те же операции, которые описаны в Примере 21, но при использовании пептида L вместо пептида B, получали 456,6 мг ПАВ в виде белого порошка.
Пример 32
Выполнив те же операции, которые описаны в Примере 21, но при использовании пептида M вместо пептида B, получали 453,9 мг ПАВ в виде белого порошка.
Пример 33
Выполнив те же операции, которые описаны в Примере 21, но при использовании пептида N вместо пептида B, получали 452,5 мг ПАВ в виде белого порошка.
Пример 34
Выполнив те же операции, которые описаны в Примере 21, но при использовании пептида O вместо пептида B, получали 458,1 мг ПАВ в виде белого порошка.
Пример 35
1,2-Дипальмитоилглицеро-(3)-фосфохолин (30,0 мг), 1,2-диацил-sn-глицеро-(3)-фосфо-sn-глицерол (с ацильной группой, имеющей 14 - 24 атома углерода; производства фирмы Sigma Chemical Co., Ltd.) (10,0 мг) и пальмитиновую кислоту (4,0 мг) растворяли в смеси хлороформ-метанол (2:1. о/о) (30 мл), а 1,0 мг пептида Q растворяли в смеси хлороформ-метанол (2:1, о/о) (2,0 мл). Полученные растворы смешивали вместе, а затем высушивали и отверждали при пониженном давлении. Полученный остаток суспендировали в смеси вода-этанол (9:1, о/о) (10 мл) в течение 20 минут при 45oC. После замораживания полученной суспензии при -60oC и последующего высушивания в течение 36 часов под вакуумом в 60 - 120 мкНо получали 45,4 мг ПАВ в виде белого порошка.
Полученный порошок не содержал поддающихся обнаружению количеств этанола, а содержание каждого из компонентов по отношению к суммарной массе ПАВ составляло: 66,1 мас.% 1,2-дипальмитоилглицеро-(3)-фосфохолина, 22,0 мас.% 1,2-диацил-sn-глицеро-(3)-фосфо-sn-глицерола, 8,8 мас.% пальмитиновой кислоты, 2,2 мас.% пептида Q и 0,9 мас.% воды.
Пример 36
Выполнив те же операции, которые описаны в Пример 35, но при использовании пептида R вместо пептида Q, получали 45,7 мг ПАВ в виде белого порошка.
Сравнительный пример 3
Выполнив те же операции, которые описаны в Примере 21, за исключением того, что вместо раствора пептида B (10,0 мг) в смеси хлороформ-метанол (2: 1, о/о) (2,0 мл), использовали раствор пептида S (10,0 мг) в ТФК (0,3 мл) и получали 455,2 мг ПАВ в виде белого порошка.
Полученный порошок не содержал поддающихся обнаружению количеств этанола, а содержание каждого из компонентов по отношению к суммарной массе ПАВ составляло: 65,9 мас.% 1,2-дипальмитоилглицеро-(3)-фосфохолина, 22,0 мас.% 1,2-ацил-sn-глицеро-(3)-фосфо-sn-глицерола (с ацильной группой, имеющей 14-24 атома углерода; производства фирмы Sigma Chemical Co., Ltd.), 8,8 мас. % пальмитиновой кислоты, 2,2 мас.% пептида S и 1,1 мас.% воды.
Сравнительный пример 4
Выполнив те же операции, которые описаны в Примере 21, за исключением того, что вместо раствора пептида B (10,0 мг) в смеси хлороформ-метанол (2: 1, о/о) (2,0 мл), использовали раствор пептида T (10,0 мг) в ТФК (0,3 мл) и получали 456,0 мг ПАВ в виде белого порошка.
Полученный порошок не содержал поддающихся обнаружению количеств этанола, а содержание каждого из компонентов по отношению к суммарной массе ПАВ составляло: 65,8 мас.% 1,2-дипальмитоилглицеро-(3)-фосфохолина, 21,9 мас.% 1,2-ацил-sn-глицеро-(3)-фосфо-sn-глицеро (с ацильной группой имеющей 14 - 24 атома углерода; производства фирмы Sigma Chemical Co., Ltd.), 8,8 мас.% пальмитиновой кислоты, 2,2 мас.% пептида T и 1,3 мас.% воды.
В Таблице 2 даны результаты тестов на поверхностную активность ПАВ по изобретению и на их способность поддерживать объем альвеол.
Возможное применение
Как указано выше, новые синтетические пептиды по изобретению легко выделять и очищать, их можно получать способами, приемлемыми для массового производства, эти пептиды обладают высокой растворимостью в обычных растворителях и их проще суспендировать до получения однородной суспензии; эти пептиды обладают поверхностной активностью, эквивалентной поверхностной активности обычных композиций.
Таким образом, настоящее изобретение описывает лекарственные препараты для лечения респираторного дистресс-синдрома, который представляет собой заболевание, дающее тяжелые нарушения дыхания.







Описываются новые синтетические пептиды, имеющие следующую специфическую последовательность общей формулы I
Xaa-Pro-Val-Xbb-Xcc-Lys-Arg-W,
Xaa может не присутствовать или может представлять собой Cys или Ser; Хbb представляет собой His или Asn; Хсс представляет собой Leu или Ile; W представляет собой гидрофобную часть пептида, содержащую 12-20 молекул Leu и/или Nle, причем тиольная или гидроксильная группа может быть ацилирована жирной кислотой C14-C18 или подвергнута реакции ацетамидометилирования. Описываются также композиции и лекарственный препарат на основе соединений формулы I для лечения респираторного дистресс-синдрома. 5 с. и 2 з.п.ф-лы, 2 табл.
Xaa-Pro-Val-Xbb-Xсс-Lys-Arg-W
Xaa может не присутствовать или может представлять собой Cys или Ser;
Xbb представляет собой His или Asn;
Xсс = Leu, Ile;
W представляет собой гидрофобную часть пептида, содержащую 12-20 молекул Leu и/или Nle,
причем тиольная или гидроксильная группа может быть ацилирована жирной кислотой C14-C18 или подвергнута реакции ацетамидометилирования.
Xaa- Pro-Val-Xbb-Xсс-Lys-Arg,
значения Xaa, Xbb, Xсс, W - см. п.1 формулы изобретения.
Синтетические пептиды по п.1 или 2 - 0,1-5,0
Холинфосфоглицерид - 50,6-85,0
Кислый фосфолипид - 4,5-37,6
Аналог жирной кислоты - 4,6-24,6
7. Лекарственный препарат для лечения респираторного дистресс-синдрома, содержащий в качестве активного ингредиента поверхностно-активное вещество, включающее в себя синтетический пептид по п.1 или 2, холинфосфоглицерид, кислый фосфолипид и аналог жирной кислоты.
| Способ получения органорастворимых производных хитозана | 1989 |
|
SU1823878A3 |
| Автоматический огнетушитель | 0 |
|
SU92A1 |
| Домовый номерной фонарь, служащий одновременно для указания названия улицы и номера дома и для освещения прилежащего участка улицы | 1917 |
|
SU93A1 |
| ЭЛЕКТРОННЫЙ КОММУТАТОР | 0 |
|
SU307513A1 |
| ЧЕТЫРЕХКОМПОНЕНТНЫЙ ОБЪЕКТИВI:•. г-.---- V.---;M :.-..?!S ,:^'-^'-... ., - -••.,•; •• ; i ilK ;;:^':'з;г.-";, Л;':': '':' :';:''iiБИ5ЛИО'-«ПлА I | 0 |
|
SU348967A1 |
