Предпосылки создания изобретения
Изобретение относится к фармацевтической липосомальной композиции, содержащей 2-{[5-{3-хлор-2-метил-4-[2-(4-метилпиперазин-1-ил)этокси]фенил}-6-(4-фторфенил)тиено[2,3-d]пиримидин-4-ил]окси}-3-(2-{[2-(2-метоксифенил)пиримидин-4-ил]метокси}фенил) пропановую кислоту, обозначаемую в настоящей заявке как 'Соединение А', или ее фармацевтически приемлемую соль. Более конкретно, изобретение относится к липосомальному носителю, композиции органического концентрата, содержащей Соединение А, и фармацевтической композиции для парентерального введения, содержащей липосомы и Соединение А. Кроме того, изобретение относится к применению таких композиций для лечения злокачественного новообразования. 'Соединение А' в контексте настоящей заявки включает все его энантиомеры, диастереоизомеры и атропизомеры, или их смеси, а также необязательно включает его фармацевтически приемлемые соли.
Структура Соединения А представляет собой
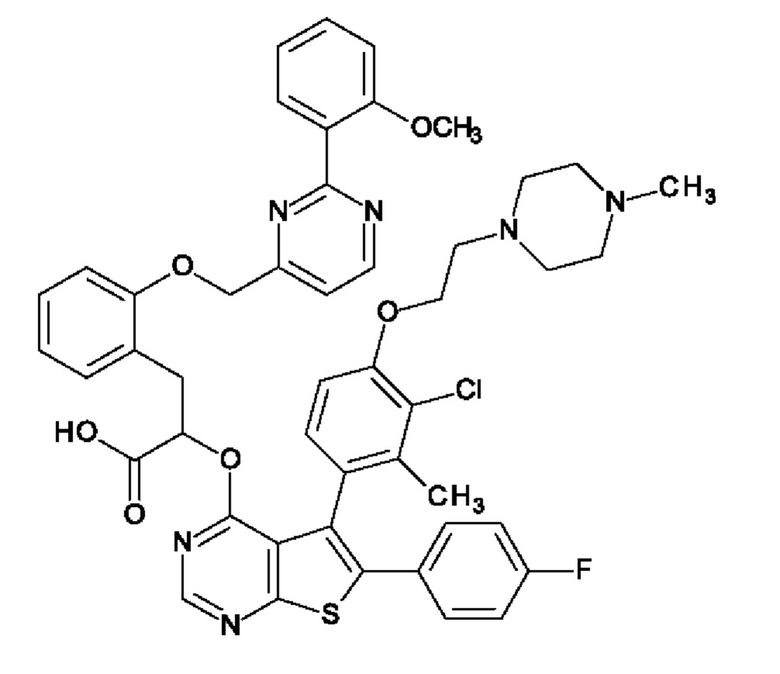
2-{[5-{3-хлор-2-метил-4-[2-(4-метилпиперазин-1-ил)этокси]фенил}-6-(4-фторфенил)тиено[2,3-d]пиримидин-4-ил]окси}-3-(2-{[2-(2-метоксифенил)пиримидин-4-ил]метокси}фенил)пропановую кислоту.
В предпочтительном варианте осуществления изобретения, Соединение А представляет собой:
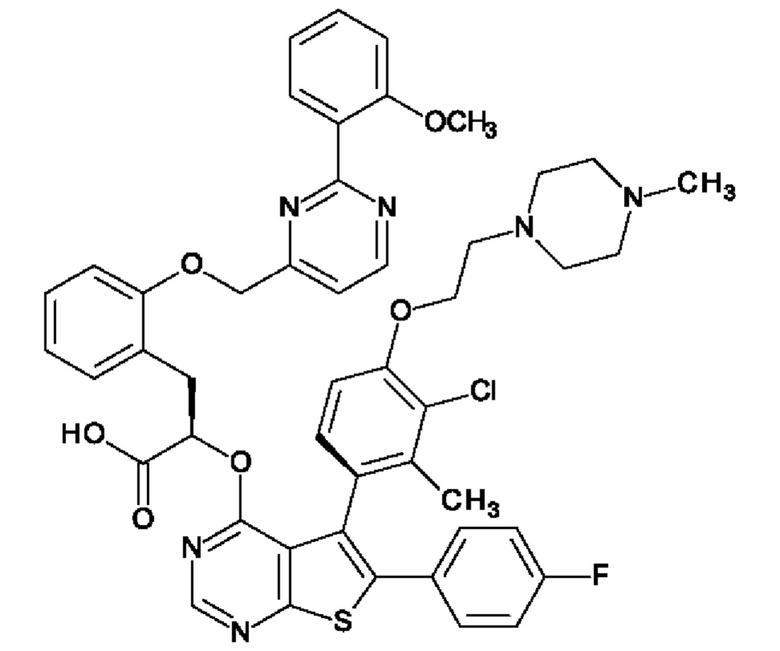
(2R)-2-{[(5Sa)-5-{3-хлор-2-метил-4-[2-(4-метилпиперазин-1-ил)этокси]фенил}-6-(4-фторфенил)тиено[2,3-d]пиримидин-4-ил]окси}-3-(2-{[2-(2-метоксифенил)пиримидин-4-ил]метокси}фенил)пропановую кислоту. В другом варианте осуществления изобретения. Соединение А, используемое в композиции, описанной в настоящей заявке, представляет собой свободную молекулу (не его соль).
Приготовление Соединения А, его применение в качестве ингибитора Mcl-1 для лечения злокачественного новообразования и его фармацевтические препараты описаны в WO 2015/097123, содержание которого включено в качестве ссылки. Приготовление, в частности, описано в Примере 30 в WO 2015/097123.
Соединение А является оптически активным, имеет один хиральный центр и хиральную ось. Оно имеет ограниченную водную растворимость для всех значений рН, включая физиологически релевантные значения рН. Для обеспечения безопасного и эффективного введения Соединения А, и для получения необходимых терапевтических эффектов. Соединение А необходимо солюбилизировать.
Существуют различные пути солюбилизации плохо растворимых соединений для парентерального введения. Типичными подходами являются оптимизация рН или применение со-растворителя (например, PEG300, PEG400, пропиленгликоля или этанола). Если эти подходы, по какой-либо причине, не осуществимы, то можно рассматривать применение поверхностно-активных веществ (например, Tween® 80 или Cremophor EL®). Тем не менее, эти виды поверхностно-активных веществ часто связаны с побочными эффектами. Циклодекстрины проявили себя в качестве безопасных солюбилизирующих агентов, но с ограничениями, что они не являются эффективными солюбилизаторами для всех соединений. Кроме того, соединения с высокой растворимостью в природных маслах (например, Propofol), могут солюбилизироваться в парентеральных жировых эмульсиях.
Другой возможностью солюбилизации плохо растворимых соединений является применение фосфолипидов (van Hoogevest P., Xiangli L., и Alfred F. "Drug delivery strategies for poorly water-soluble drugs: the industrial perspective" Expert Opinion on Drug Delivery 2011, 8(11), 1481-1500). Таким образом, фосфолипиды сами представляют собой одно из дополнительных средств для солюбилизации плохо растворимых соединений, вдобавок к обычным подходам. Тем не менее, солюбилизация определенного плохо растворимого соединения с помощью фосфолипидов не может быть предсказана.
Задачей настоящего изобретения является обеспечение композиции, которая успешно может использоваться для солюбилизации и парентеральной доставки Соединения А. В особенности, существует необходимость обеспечения безопасной и эффективной фармацевтической композиции для Соединения А. Другими задачами является обеспечение композиции, которая является стабильной в релевантных условиях и контейнерах, и которая позволяет осуществить возможность введения подходящей дозы Соединения А в течение целесообразных временных сроков. В соответствии с другой задачей, композиция должна иметь возможность приготавливаться с помощью надежного и функционального способа.
Сущность изобретения
Настоящее изобретение обеспечивает композицию, содержащую Соединение А, подходящую для парентерального введения пациентам. В особенности, такое введение осуществляют путем внутривенной инъекции или инфузии. Изобретение дополнительно обеспечивает две раздельные композиции, которые могут быть смешаны совместно незадолго до введения пациенту, для обеспечения композиции, подходящей для введения. Одна из композиций для смешивания представляет собой композицию органического концентрата, содержащую Соединение А, а вторая композиция для смешивания представляет собой липосомальный носитель. Если две раздельные композиции смешаны совместно, то Соединение А является загруженным на липосомы в липосомальном носителе, предоставляющем возможность солюбилизации Соединения А, что приводит к получению фармацевтической липосомальной композиции, подходящей для применения в клинике.
Предпочтительно, изобретение обеспечивает композицию, содержащую Соединение А, которая поддерживает химическую стабильность Соединения А. Например, ограничено образование нежелательного атропизомера и/или продуктов окисления и/или продуктов разложения.
Предпочтительно, изобретение обеспечивает композицию, содержащую Соединение А, которая имеет оптимальную химическую стабильность, например, удается избежать образования геля и/или удается избежать осаждения компонентов. Неожиданно было обнаружено, что композиции, содержащие Соединение А, устойчивые к гелеобразованию, при котором композиция образует гель. Такое гелеобразование может быть обратимым или необратимым. Гелеобразование осложняет или препятствует манипуляциям с композицией, содержащей Соединение А, для ее предложенного применения, и его следует избегать.
Композиция (органический концентрат), содержащая Соединение А, должна предоставлять возможность эффективной и оптимальной загрузки Соединения А в липосомы в липосомальном носителе путем простого смешивания 2 композиций совместно, незадолго до введения пациенту.
Предпочтительно; изобретение обеспечивает фармацевтическую липосомальную композицию, которая позволяет осуществить быстрое высвобождение Соединения А из липосом после внутривенного введения.
Таким образом, изобретение, раскрытое в настоящей заявке, позволяет осуществить эффективное введение Соединения А пациентам, несмотря на сложные химические характеристики Соединения А и сложные физические характеристики препаратов, содержащих Соединение А.
Краткое описание фигур
На Фигуре 1 представлены эффективность и переносимость липосомально приготовленного Соединения А после введения раз в неделю в дозе 10 мг/кг самкам бестимусных крыс, несущим MV4; 11 AML ксенотрансплантаты.
На Фигуре 2 представлены эффективность и переносимость липосомально приготовленного Соединения А после введения раз в неделю в дозе 30 мг/кг самкам бестимусных крыс, несущим MV4; 11 AML ксенотрансплантаты.
На Фигуре 3 представлено схематическое изображение принципов анализа высвобождения на основании MLV, которое предоставляет возможность мониторинга высвобождения соединения А из липосом в MLV.
На Фигуре 4 представлены профиль высвобождения Соединения А из SUV в 100% ePL (яичные фосфолипиды) при разнообразных различных SUV : MLV соотношениях (об./об.).
Подробное описание изобретения
Липосома представляет собой сферический пузырек, имеющий по меньшей мере один липидный бислой. Липосома может использоваться в качестве носителя для доставки питательных веществ и лекарственных средств. Липосомы наиболее часто состоят из фосфолипидов, в частности фосфатидилхолина, но также могут включать другие липиды, такие как яичный фосфатидилэтаноламин, при условии, что они совместимы с липидной бислойной структурой.
'Липосомальный носитель' в контексте настоящей заявки обозначает жидкость, содержащую липосомы, указанные липосомы содержат фосфолипиды. Указанный липосомальный носитель является подходящим для солюбилизации Соединения А в водной окружающей среде после смешивания с Соединением А, в особенности, если Соединение А обеспечивается в виде органического концентрата, описанного в настоящей заявке. Указанный липосомальный носитель является подходящим для загрузки с Соединением А перед введением пациенту.
Липосомальный размер, как раскрыто в настоящей заявке, относится к размеру, как определено с помощью фотонно-корреляционной спектроскопии (PCS). Средний размер, выраженный в виде среднего диаметра Z и коэффициента полидисперсности, определяют с использованием фотонно-корреляционной спектроскопии в соответствии ISO 13321.
Фармацевтическая композиция, описанная в настоящей заявке, в особенности, представляет собой фармацевтическую липосомальную композицию. 'Фармацевтическая липосомальная композиция' обозначает композицию, содержащую липосомы, которые пригодны для фармацевтического введения.
'Гелеобразование', в контексте настоящей заявки, обозначает образование геля. При образовании геля, композиция становится более вязкой, менее свободно текучей. Это значительно усложняет или делает невозможным обработку композиции для предназначенной цели.
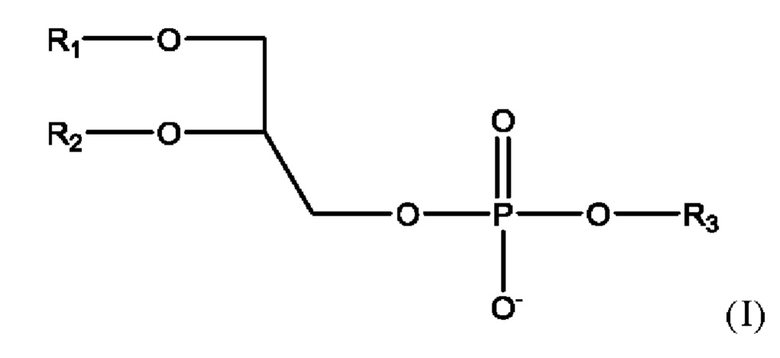
Фосфолипид, используемый в липосомальном носителе в настоящей заявке, содержит по меньшей мере один фосфолипид формулы
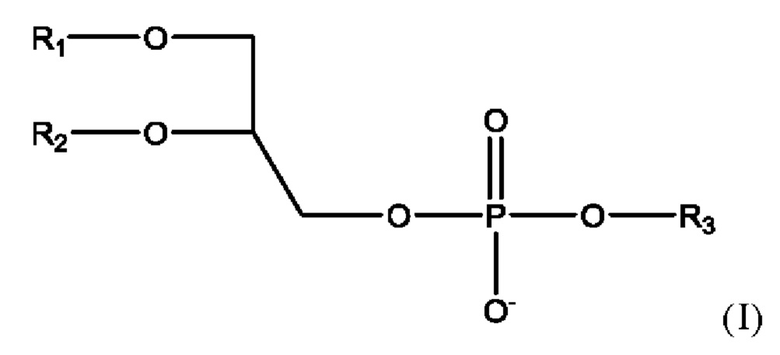
где
• R1 представляет собой С10-С24ацил;
• R2 представляет собой С10-С24ацил, или альтернативно R2 представляет собой водород или С10-С20ацил;
• R3 представляет собой водород, 2-триметиламино-1-этил, 2-амино-1-этил, С1-С4алкил, С1-С5алкил, замещенный карбокси, С2-С5алкил, замещенный карбокси и гидрокси, С2-С5алкил, замещенный карбокси и амино, инозитоловую группу или глицериловую группу;
или соль такого соединения.
Термин ацил, используемый в настоящей заявке ранее, представляет собой следующую группу:
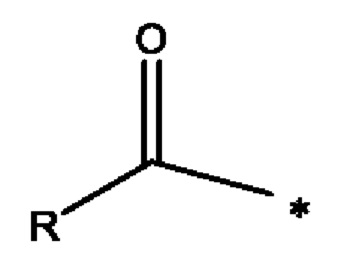
где * представляет собой точку присоединения R1 или R2 к остальной молекуле, и, например, R представляет собой неразветвленную алкильную цепь, которая может быть насыщенной или частично насыщенной.
Фосфолипид может быть нейтральным или может быть заряженным. Он может быть двухцепочечным или одноцепочечным амфифильным. Примерами нейтральных фосфолипидов с двойными цепями являются фосфатидилхолин (PC), фосфатидилэтаноламин (РЕ) и сфингомиелин. Примерами заряженных фосфолипидов являются фосфатидная кислота (РА), фосфатидилинозитол (PI), и фосфатидилсерин (PS) и фосфатидилглицерин (PG). Углеводородная цепь может быть либо ненасыщенной или насыщенной. В одном варианте осуществления, она имеет от 14 до 18 атомов углерода.
Для парентеральных препаратов, фосфолипид может быть двухцепочечным и может содержать фракцию менее чем 20% одноцепочечного амфифильного. Для парентеральных препаратов, фосфолипид может представлять собой фосфатидилхолин (PC) и может содержать фракцию менее чем 40% заряженного фосфолипида.
Одноцепочечный липид может представлять собой моноацильное производное нейтрального или заряженного фосфолипида, но он также может представлять собой моноацильное(ые) производное(ые) гликолипидов и сфинголипидов. Тем не менее, для парентерального применения, моноацильное производное не является предпочтительным. Деацилирование можно осуществлять путем ферментативного гидролиза с помощью фосфолипазы А2 или химическими средствами. Углеводородная цепь может быть либо ненасыщенной или насыщенной и может иметь, в особенности, от 14 до 18, или от 14 до 24 атомов углерода. Липиды могут иметь происхождение из природных растений или животных или микробиологических источников, синтезированных или частично синтезированных, включая производные полиэтиленгликоля (PEG) моноацильные фосфолипиды, например, пэгилированный моноацил фосфатидил этаноламин.
В предпочтительном варианте осуществления, R2 не представляет собой водород.
В особенности, фосфолипид, используемый в липосомальном носителе, описанном в настоящей заявке, выбирают из яичного лецитина, соевого лецитина или синтетических фосфолипидов. Примерами синтетических фосфолипидов для применения в настоящей заявке являются РОРС (пальмитоил олеоил фосфатидилхолин), DOPC (1,2-Диолеоил-sn-глицеро-3-фосфохолин), DMPC (1,2-Димиристоил-sn-глицеро-3-фосфохолин) и DPPC (1,2-Дипальмитоил-sn-глицеро-3-фосфохолин), в частности синтетические фосфолипиды РОРС (пальмитоил олеоил фосфатидилхолин), DOPC (1,2-Диолеоил-sn-глицеро-3-фосфохолин), и DMPC (1,2-Димиристоил-sn-глицеро-3-фосфохолин), в особенности РОРС. Более предпочтительно, фосфолипид, используемый в липосомальном носителе, выбирают из яичного лецитина или соевого лецитина, содержащего по меньшей мере 75% фосфатидилхолина или по меньшей мере 70% фосфатидилхолина. В другом варианте осуществления, можно использовать яичный лецитин с 71,5% мас./мас. (±7,5% мас./мас.) яичного PC (фосфатидилхолина) и 15% мае./мае. (±3% w/w) яичного РЕ (фосфатидилэтаноламина). В предпочтительном варианте осуществления изобретения, указанный фосфолипид выбирают из Lipoid E 80 S и Lipoid E 80. В предпочтительном варианте осуществления, указанный фосфолипид представляет собой Lipoid E 80 S. Если используют Е 80, то олеат натрия необязательно включают в препарат в качестве стабилизатора коллоидной стабильности.
В предпочтительных вариантах осуществления изобретения, фосфолипиды выбирают таким образом, чтобы способствовать более быстрому высвобождению Соединения А в кровообращение.
'Органический концентрат' в контексте настоящей заявки обозначает композицию, которая представляет собой раствор, содержащий органический, смешивающийся с водой растворитель, содержащий Соединение А, который является пригодным для смешивания с липосомальным носителем для обеспечения возможности загрузки в липосомы. В особенности, указанный органический концентрат является пригодным для смешивания с и загрузкой липосомальным носителем, как описано в настоящей заявке. Полученная смесь является подходящей для введения пациенту.
'Загрузка' обозначает инкорпорирование или перенос Соединения А в липосомы из органического концентрата.
'Отрицательно заряженный или полярный фосфолипидный стабилизатор' обозначает фосфолипид, содержащие по меньшей мере некоторые отрицательно заряженные липиды или полярный липид. В особенности, отрицательно заряженный или полярный фосфолипидный стабилизатор выбирают из натриевой или аммониевой соли DMPG (1,2-Димиристоил-sn-глицеро-3-фосфо-рац-глицерин или димиристоил фосфатидилглицерин), натриевой соли POPG (1-Пальмитоил-2-олеоил-sn-глицеро-3[Фосфо-рац-(1-глицерин)]), натриевой соли DOPS (1,2-Диолеоил-sn-глицеро-3-фосфосерин), натриевой соли DOPG (1,2-Диолеоил-sn-глицеро-3[Фосфо-рац-(1-глицерин)]), натриевой или аммониевой соли DPPG (1,2-Дипальмитоил-sn-глицеро-3[Фосфо-рац-(1-глицерин)]), натриевой или аммониевой соли DSPG (1,2-Дистеароил-sn-глицеро-3[Фосфо-рац-(1-глицерин)}), натриевой соли соевой фосфатидной кислоты (РА), натриевой соли яичной фосфатидной кислоты (РА), натриевой соли соевого фосфатидилсерина (PS), натриевой соли яичного фосфатидилглицерина (PG), натриевой соли соевого фосфатидилглицерина (PG), натриевой соли фосфатидилинозитола (PI), Lipoid S 75, Lipoid E 80 S и олеата натрия. В особенности, отрицательно заряженный или полярный фосфолипидный стабилизатор представляет собой 1,2-Димиристоил-sn-глицеро-3-фосфо-рац-глицерин, натриевую или аммониевую соль, или Lipoid E 80 S, предпочтительно 1,2-Димиристоил-sn-глицеро-3-фосфо-рац-глицерин, натриевую соль. Указанный 'отрицательно заряженный или полярный фосфолипидный стабилизатор' облегчает загрузку Соединения А в липосомы.
'Отрицательно заряженный или полярный фосфолипидный стабилизатор' может представлять собой яичный лецитин или соевый лецитин.
'Агент, корригирующий тоничность' обозначает фармацевтически приемлемое соединение, которое можно добавлять к препарату для придания ему изотоничности с плазмой крови человека. Агенты, корригирующие тоничность, включают, например, декстрозу, глюкозу, маннит, сахарозу, лактозу, трегалозу, глицерин и NaCl, в особенности сахарозу или глицерин, более предпочтительно сахарозу. Тоничность представляет собой 'эффективную осмоляльность' и равна сумме концентраций растворенных веществ, которые имеют способность проявлять осмотический потенциал в пределах мембраны. Парентеральные препараты должны быть изотоничными с плазмой крови. Агенты, корригирующие тоничность, хорошо известны квалифицированному специалисту в данной области техники.
Подходящий растворитель может быть выбран квалифицированным специалистом, занимающимся составлением рецептуры препаратов. В органическом концентрате в настоящей заявке, растворитель или растворители могут быть выбраны из пропиленгликоля, этанола, PEG300, Super-Refined® PEG300, PEG400 и Super-Refined® PEG400, в особенности пропиленгликоля и этанола.
Super-Refined® относится к сверхчистым PEG300 или PEG400.
'Буфер' используется для предотвращения изменений рН раствора и подходящие примеры хорошо известны квалифицированному специалисту, занимающемуся составлением рецептуры препаратов.
'Контейнер' обозначает ампулу или флакон с резиновой пробкой и крышечкой, одно- или двухкамерный шприц, инфузионный мешок или сосуд, изготовленный из полимерных материалов или стекла, подходящий для содержания композиций для парентерального введения. Он также включает любой сосуд для удерживания жидкостей.
'Свободная молекула' в контексте настоящей заявки обозначает соединение, которое не используется в форме соли, образованное наружными противоионами. Соединение А может присутствовать в цвиттер-ионной форме, и термин 'свободная молекула' в контексте настоящей заявки включает цвиттер-ионную форму.
В контексте настоящей заявки, термин 'содержащий' обозначает 'включающий' и не предназначен для исключения присутствия любого дополнительного компонента, если из контекста не следует другое, например, когда компоненты вместе составляют сумму до 100%.
В контексте настоящей заявки, термин 'лечить', 'лечение' или 'терапия' любого заболевания или нарушения относится в одном варианте осуществления, к облегчению заболевания или нарушения (то есть, замедлению или остановке или уменьшению развития заболевания или по меньшей мере одного из его клинических симптомов). В другом варианте осуществления, 'лечить', 'лечение' или 'терапия' относится к облегчению или улучшению по меньшей мере одного физического параметра, включая те, которые еще могут не ощущаться пациентом. В еще другом варианте осуществления, 'лечить', 'лечение' или 'терапия' относится к модуляции заболевания или нарушения, либо физически, (например, стабилизация распознаваемого симптома), физиологически, (например, стабилизация физического параметра), или в обоих аспектах.
'Стабилизатор от гелеобразования' в контексте настоящей заявки обозначает стабилизатор от гелеобразования композиции, содержащей Соединение А. Это определение стабилизаторов включает, например, электролиты или полимеры. Примерами подходящих электролитов являются хлорид натрия, хлорид калия, фосфат натрия двух- и одноосновный, сульфат аммония и аргинин, предпочтительно хлорид натрия. При необходимости, к препарату можно добавлять воду с электролитом. Примерами подходящих полимеров являются PEG300 и PEG400, в особенности PEG300. Стабильность от гелеобразования можно исследовать с помощью методик, хорошо известных квалифицированному специалисту в данной области техники, и, в особенности, можно использовать методики, описанные в настоящей заявке. Пример 12 в настоящей заявке обеспечивает методики, которые можно применять для тестирования гелеобразования. Когда понимают склонность к гелеобразованию для выбранной композиции, то квалифицированный специалист, занимающийся составлением рецептуры препаратов, может применить информацию для выбора оптимальных условий хранения для композиций, содержащих Соединение А.
В одном необязательном варианте осуществления, PEG300 представляет собой Super-Refined® PEG300, который представляет собой сверхчистый PEG300.
В одном предпочтительном варианте осуществления, выбирают стабилизатор, который предотвращает гелеобразование композиции, содержащей Соединение А, и который также уменьшает или предотвращает осаждение компонентов композиции, содержащей Соединение А. Например, PEG300 представляет собой особый интерес в этом отношении. В одном необязательном варианте осуществления, PEG300 представляет собой Super-Refined® PEG300, и который также может поддержать химическую стабильность Соединения А.
Смешивание 'незадолго до введения пациенту' обозначает вплоть до трех дней до введения, в особенности вплоть до 24 часов до введения, и, например, вплоть до 6 часов перед введением пациенту.
'Гистидин', в контексте настоящей заявки, обозначает, в особенности, 'L-гистидин'.
Благоприятно, в определенных вариантах осуществления изобретения, благодаря очень небольшому размеру частиц липосом, препарат является прозрачным, что предоставляет возможность визуального контроля завершенности растворения Соединения А перед введением. В предпочтительном варианте осуществления изобретения, композицию вводят пациентам внутривенно.
Варианты осуществления изобретения
Ниже описаны различные варианты осуществления изобретения.
Е1. Композиция органического концентрата, содержащая Соединение А, которое представляет собой 2-{[5-{3-хлор-2-метил-4-[2-(4-метилпиперазин-1-ил)этокси]фенил}-6-(4-фторфенил)тиено[2,3-d]пиримидин-4-ил]окси}-3-(2-{[2-(2-метоксифенил)пиримидин-4-ил]метокси}фенил)пропановую кислоту или ее фармацевтически приемлемую соль и отрицательно заряженный или полярный фосфолипидный стабилизатор.
Е2. Композиция органического концентрата в соответствии с вариантом осуществления Е1, содержащая Соединение А, которое представляет собой (2R)-2-{[(5Sa)-5-{3-хлор-2-метил-4-[2-(4-метилпиперазин-1-ил)этокси]фенил}-6-(4-фторфенил)тиено[2,3-d]пиримидин-4-ил]окси}-3-(2-{[2-(2-метоксифенил)пиримидин-4-ил]метокси}фенил)пропановую кислоту или ее фармацевтически приемлемую соль и отрицательно заряженный или полярный фосфолипидный стабилизатор.
Е3. Композиция органического концентрата в соответствии с вариантами осуществления Е1 или Е2, где Соединение А представляет собой свободную молекулу.
Е4. Композиция органического концентрата в соответствии с любым из вариантов осуществления Е1 - Е3, где отрицательно заряженный или полярный фосфолипидный стабилизатор выбирают из натриевой или аммониевой соли DMPG (1,2-Димиристоил-sn-глицеро-3-фосфо-рац-глицерин или димиристоил фосфатидилглицерин), натриевой соли POPG (1-Пальмитоил-2-олеоил-sn-глицеро-3[Фосфо-рац-(1-глицерин)]), натриевой соли DOPS (1,2-Диолеоил-sn-глицеро-3-фосфосерин), натриевой соли DOPG (1,2-Диолеоил-sn-глицеро-3[Фосфо-рац-(1-глицерин)]), натриевой или аммониевой соли DPPG (1,2-Дипальмитоил-sn-глицеро-3[Фосфо-рац-(1-глицерин)]), натриевой или аммониевой соли DSPG (1,2-Дистеароил-sn-глицеро-3[Фосфо-рац-(1-глицерин)]), натриевой соли соевой фосфатидной кислоты (РА), натриевой соли яичной фосфатидной кислоты (РА), натриевой соли соевого фосфатидилсерина (PS), натриевой соли яичного фосфатидилглицерина (PG), натриевой соли соевого фосфатидилглицерина (PG), натриевой соли фосфатидилинозитола (PI), Lipoid E 80 S и олеата натрия.
Е5. Композиция органического концентрата в соответствии с любым из вариантов осуществления Е1 - Е4, где отрицательно заряженный или полярный фосфолипидный стабилизатор представляет собой натриевую соль димиристоил фосфатидилглицерина (DMPG) или Lipoid Е 80 S.
Е6. Композиция органического концентрата в соответствии с вариантом осуществления Е5, где отрицательно заряженный или полярный фосфолипидный стабилизатор представляет собой 1,2-Димиристоил-sn-глицеро-3-фосфо-рац-глицерин, натриевую соль.
Е7. Композиция органического концентрата в соответствии с любым из вариантов осуществления Е1 - Е6, дополнительно содержащая один или несколько растворителей.
Е8. Композиция органического концентрата в соответствии с любым из вариантов осуществления Е1 - Е7, содержащая по меньшей мере один растворитель, выбранный из пропиленгликоля, этанола, PEG300 и PEG400.
Е9. Композиция органического концентрата в соответствии с любым из вариантов осуществления Е1 - Е8, содержащая по меньшей мере один растворитель, выбранный из:
• пропиленгликоля и этанола, или
• пропиленгликоля, PEG300 и этанола.
Е10. Композиция органического концентрата в соответствии с любым из вариантов осуществления Е1 - Е9, содержащая оба компонента, пропиленгликоль и этанол.
Е11. Композиция органического концентрата в соответствии с любым из вариантов осуществления E1 - Е10, дополнительно содержащая стабилизатор, выбранный из хлорида натрия, хлорида калия, фосфата натрия двух- и одноосновного, сульфата аммония и аргинина, предпочтительно хлорида натрия.
Е12. Композиция органического концентрата в соответствии с любым из вариантов осуществления Е1 - Е11, дополнительно содержащая воду.
Е13. Липосомальный носитель, содержащий фосфолипид и агент, корригирующий тоничность.
Е14. Липосомальный носитель в соответствии с вариантом осуществления Е13, содержащий от 5% мас./мас. до 20% мас./мас. фосфолипида, в особенности от 7% мас./мас. до 13% мас./мас. фосфолипида.
Е15. Липосомальный носитель в соответствии с вариантами осуществления Е13 или Е14, где фосфолипид выбирают из:
где
• R1 представляет собой С10-С24ацил;
• R2 представляет собой С10-С24ацил, или альтернативно R2 представляет собой водород или С10-С20ацил;
• R3 представляет собой водород, 2-триметиламино-1-этил, 2-амино-1-этил, С1-С4алкил,
С1-С5алкил, замещенный карбокси, С2-С5алкил, замещенный карбокси и гидрокси, С2-С5алкил, замещенный карбокси и амино, инозитоловую группу или глицериловую группу;
или соли такого соединения.
Е16. Липосомальный носитель в соответствии с вариантами осуществления Е13 или Е14, где фосфолипид выбирают из яичного лецитина, соевого лецитина или синтетических фосфолипидов.
Е17. Липосомальный носитель в соответствии с любым из вариантов осуществления Е13 - Е16, где фосфолипид выбирают из яичного лецитина, соевого лецитина, РОРС (пальмитоил олеоил фосфатидилхолин), DOPC (1,2-Диолеоил-sn-глицеро-3-фосфохолин), и DMPC (1,2-Димиристоил-sn-глицеро-3-фосфохолин), в частности РОРС (пальмитоил олеоил фосфатидилхолин), DOPC (1,2-Диолеоил-sn-глицеро-3-фосфохолин), и DMPC (1,2-Димиристоил-sn-глицеро-3-фосфохолин), в особенности РОРС.
Е18. Липосомальный носитель в соответствии с любым из вариантов осуществления Е13, Е14, Е16 или Е17, где фосфолипид выбирают из яичного лецитина или соевого лецитина, содержащего по меньшей мере 75% фосфатидилхолина, или по меньшей мере 70% фосфатидилхолина.
E19. Липосомальный носитель в соответствии с любым из вариантов осуществления Е13, Е14, Е16, Е17 или Е18, где фосфолипид представляет собой Lipoid Е 80 S.
Е20. Липосомальный носитель в соответствии с любым из вариантов осуществления Е13 - Е19, где агент, корригирующий тоничность, выбирают из декстрозы, глюкозы, маннита, сахарозы, лактозы, трегалозы, глицерина и NaCl.
Е21. Липосомальный носитель в соответствии с вариантом осуществления Е20, где агент, корригирующий тоничность, представляет собой сахарозу или глицерин, в особенности сахарозу.
Е22. Липосомальный носитель в соответствии с любым из вариантов осуществления Е13 - Е21, дополнительно содержащий буфер.
Е23. Липосомальный носитель в соответствии с вариантом осуществления Е22, где буфер представляет собой гистидин.
Е24. Липосомальный носитель в соответствии с любым из вариантов осуществления Е13 - Е23, дополнительно содержащий воду.
Е25. Липосомальный носитель в соответствии с любым из вариантов осуществления Е13 - Е24, где липосомы, присутствующие в липосомальном носителе, имеют средний размер от 20 нм до 300 нм, предпочтительно от 20 нм до 100 нм.
Е26. Липосомальный носитель в соответствии с вариантом осуществления Е25, где липосомы имеют средний размер от 20 нм до 80 нм, предпочтительно от 40 нм до 70 нм.
Е27. Фармацевтическая композиция, содержащая фосфолипид и Соединение А, которое представляет собой 2-{[5-{3-хлор-2-метил-4-[2-(4-метилпиперазин-1-ил)этокси]фенил}-6-(4-фторфенил)тиено[2,3-d]пиримидин-4-ил]окси}-3-(2-{[2-(2-метоксифенил)пиримидин-4-ил]метокси}фенил)пропановую кислоту, или ее фармацевтически приемлемую соль.
Е28. Фармацевтическая липосомальная композиция, содержащая:
а. фосфолипид, как описано в любом из вариантов осуществления Е15 - Е19, и
б. Соединение А, которое представляет собой 2-{[5-{3-хлор-2-метил-4-[2-(4-метилпиперазин-1-ил)этокси]фенил}-6-(4-фторфенил)тиено[2,3-d]пиримидин-4-ил]окси}-3-(2-{[2-(2-метоксифенил)пиримидин-4-ил]метокси}фенил) пропановую кислоту или ее фармацевтически приемлемую соль.
Е29. Фармацевтическая композиция в соответствии с вариантом осуществления Е27, где Соединение А представляет собой (2R)-2-{[(5Sa)-5-{3-хлор-2-метил-4-[2-(4-метилпиперазин-1-ил)этокси]фенил}-6-(4-фторфенил)тиено[2,3-d]пиримидин-4-ил]окси}-3-(2-{[2-(2-метоксифенил)пиримидин-4-ил]метокси}фенил)пропановую кислоту или ее соль, предпочтительно свободную молекулу.
Е30. Фармацевтическая липосомальная композиция в соответствии с вариантом осуществления Е28, где Соединение А представляет собой (2R)-2-{[(5Sa)-5-{3-хлор-2-метил-4-[2-(4-метилпиперазин-1-ил)этокси]фенил}-6-(4-фторфенил)тиено[2,3-d]пиримидин-4-ил]окси}-3-(2-{[2-(2-метоксифенил)пиримидин-4-ил]метокси}фенил) пропановую кислоту или ее соль, предпочтительно свободную молекулу.
Е31. Фармацевтическая композиция в соответствии с любым из вариантов осуществления Е27 - Е30, для инфузии или внутривенной инъекции.
Е32. Фармацевтическая композиция в соответствии с любым из вариантов осуществления Е27 - Е31, содержащая от 5% мас./мас. до 20% мас./мас. фосфолипида, в особенности от 7% мас./мас. до 13% мас./мас. фосфолипида.
Е33. Фармацевтическая композиция в соответствии с любым из вариантов осуществления Е27 - Е32, где фосфолипид выбирают из тех, которые описаны в вариантах осуществления Е15 - Е19, в особенности Е18 или Е19.
Е34. Фармацевтическая композиция в соответствии с любым из вариантов осуществления Е27 - Е33, дополнительно содержащая отрицательно заряженный или полярный фосфолипидный стабилизатор.
Е35. Фармацевтическая композиция в соответствии с вариантом осуществления Е34, где отрицательно заряженный или полярный фосфолипидный стабилизатор выбирают из натриевой или аммониевой соли DMPG (1,2-Димиристоил-sn-глицеро-3-фосфо-рац-глицерин или димиристоил фосфатидилглицерин), натриевой соли POPG (1-Пальмитоил-2-олеоил-sn-глицеро-3[Фосфо-рац-(1-глицерин)]), натриевой соли DOPS (1,2-Диолеоил-sn-глицеро-3-фосфосерин), натриевой соли DOPG (1,2-Диолеоил-sn-глицеро-3[Фосфо-рац-(1-глицерин)]), натриевой или аммониевой соли DPPG (1,2-Дипальмитоил-sn-глицеро-3[Фосфо-рац-(1-глицерин)]), натриевой или аммониевой соли DSPG (1,2-Дистеароил-sn-глицеро-3[Фосфо-рац-(1-глицерин)]), натриевой соли соевой фосфатидной кислоты (РА), натриевой соли яичной фосфатидной кислоты (РА), натриевой соли соевого фосфатидилсерина (PS), натриевой соли яичного фосфатидилглицерина (PG), натриевой соли соевого фосфатидилглицерина (PG), натриевой соли фосфатидилинозитола (PI), Lipoid Е 80 S, и олеата натрия, в особенности натриевой соли димиристоил фосфатидилглицерина (DMPG), натриевой соли 1,2-Димиристоил-sn-глицеро-3-фосфо-рац-глицерина, и Lipoid Е 80 S.
Е36. Фармацевтическая композиция в соответствии с любым из вариантов осуществления Е27 - Е35, дополнительно содержащая один или несколько растворителей, в особенности те растворители, которые описаны в любом из вариантов осуществления Е8, Е9 или Е10.
Е37. Фармацевтическая композиция в соответствии с любым из вариантов осуществления Е27 - Е36, дополнительно содержащая стабилизатор, выбранный из хлорида натрия, хлорида калия, фосфата натрия двух- и одноосновного, сульфата аммония и аргинина, предпочтительно хлорида натрия.
Е38. Фармацевтическая композиция в соответствии с любым из вариантов осуществления Е27 - Е37, дополнительно содержащая агент, корригирующий тоничность.
Е39. Фармацевтическая композиция в соответствии с вариантом осуществления Е38, где агент, корригирующий тоничность, выбирают из декстрозы, глюкозы, маннита, сахарозы, лактозы, трегалозы, глицерина и NaCl, в особенности сахарозы и/или глицерина, более предпочтительно сахарозы.
Е40. Фармацевтическая композиция в соответствии с любым из вариантов осуществления Е27 - Е39, дополнительно содержащая буфер, в особенности гистидин.
Е41. Фармацевтическая композиция в соответствии с любым из вариантов осуществления Е27 - Е40, содержащая липосомы.
Е42. Фармацевтическая композиция в соответствии с любым из вариантов осуществления Е27 - Е41, где соотношение фосфолипида (из липосомального носителя) к Соединению А в виде свободной молекулы составляет от 100:1 до 4,5:1 частей по весу.
Е43. Фармацевтическая композиция в соответствии с вариантом осуществления Е42, где соотношение фосфолипида из липосомального носителя к Соединению А в виде свободной молекулы составляет от 50:1 до 6:1 частей по весу, в особенности от 20:1 до 9:1 частей по весу.
Е44. Фармацевтическая композиция в соответствии с любым из вариантов осуществления Е27 - Е43, где загруженные липосомы имеют средний размер от 20 нм до 100 нм, в особенности от 50 нм до 90 нм, более предпочтительно от 50 нм до 80 нм.
Е45. Фармацевтическая композиция для инфузии или внутривенной инъекции содержащая смесь липосомального носителя в соответствии с любым из вариантов осуществления Е13 - Е26, и композиции органического концентрата в соответствии с любым из вариантов осуществления Е1 - Е12.
Е46. Фармацевтическая композиция для инфузии в соответствии с вариантом осуществления Е45, дополнительно содержащая глюкозу для инфузии.
Е47. Композиция органического концентрата, содержащая:
а. Соединение А, которое представляет собой (2R)-2-{[(5Sa)-5-{3-хлор-2-метил-4-[2-(4-метилпиперазин-1-ил)этокси]фенил}-6-(4-фторфенил)тиено[2,3-d]пиримидин-4-ил]окси}-3-(2-{[2-(2-метоксифенил)пиримидин-4-ил]метокси}фенил)пропановую кислоту,
б. отрицательно заряженный или полярный фосфолипидный стабилизатор, и
в. по меньшей мере один растворитель, выбранный из пропиленгликоля, этанола, PEG300 и PEG400.
Е48. Композиция органического концентрата в соответствии с вариантом осуществления Е47, содержащая пропиленгликоль, этанол и PEG300, в особенности оба компонента, пропиленгликоль и этанол.
Е49. Композиция органического концентрата в соответствии с вариантами осуществления Е47 или Е48, дополнительно содержащая стабилизатор, выбранный из хлорида натрия, хлорида калия, фосфата натрия двух- и одноосновного, сульфата аммония и аргинина, в особенности хлорида натрия.
Е50. Фармацевтическая композиция для инфузии или внутривенной инъекции содержащая:
а. фосфолипид, выбранный из тех, которые описаны в любом из вариантов осуществления Е15 - Е19,
б. Соединение А, которое представляет собой (2R)-2-{[(5Sa)-5-{3-хлор-2-метил-4-[2-(4-метилпиперазин-1-ил)этокси]фенил}-6-(4-фторфенил)тиено[2,3-d]пиримидин-4-ил]окси}-3-(2-{[2-(2-метоксифенил)пиримидин-4-ил]метокси}фенил)пропановую кислоту, и
в. отрицательно заряженный или полярный фосфолипидный стабилизатор, выбранный из тех; которые описаны в любом из вариантов осуществления Е4 - Е6.
Е51. Фармацевтическая композиция в соответствии с вариантом осуществления Е50, дополнительно содержащая стабилизатор, выбранный из хлорида натрия, хлорида калия, фосфата натрия двух- и одноосновного, сульфата аммония и аргинина, в особенности хлорида натрия.
Е52. Фармацевтическая композиция в соответствии с вариантами осуществления Е50 или Е51, дополнительно содержащая оба компонента, пропиленгликоль и этанол.
Е53. Набор, содержащий:
а. липосомальный носитель в соответствии с любым из вариантов осуществления Е13 - Е26, и
б. композицию органического концентрата в соответствии с любым из вариантов осуществления Е1 - Е12, или Е47 - Е49, или Е55 - Е66, Е81 или Е82.
Е54. Набор в соответствии с вариантом осуществления Е53, где а. и б. предназначены для смешивания друг с другом непосредственно перед введением пациенту.
Е55. Композиция органического концентрата, содержащая:
а. Соединение А, которое представляет собой 2-{[5-{3-хлор-2-метил-4-[2-(4-метилпиперазин-1-ил)этокси]фенил}-6-(4-фторфенил)тиено[2,3-d]пиримидин-4-ил]окси}-3-(2-{[2-(2-метоксифенил)пиримидин-4-ил]метокси}фенил)пропановую кислоту или ее фармацевтически приемлемую соль,
б. отрицательно заряженный или полярный фосфолипидный стабилизатор, и
в. стабилизатор от гелеобразования.
Е56. Композиция органического концентрата в соответствии с вариантом осуществления Е55, содержащая Соединение А, которое представляет собой (2R)-2-{[(5Sa)-5-{3-хлор-2-метил-4-[2-(4-метилпиперазин-1-ил)этокси]фенил}-6-(4-фторфенил)тиено[2,3-d]пиримидин-4-ил]окси}-3-(2-{[2-(2-метоксифенил)пиримидин-4-ил]метокси}фенил)пропановую кислоту или ее фармацевтически приемлемую соль.
Е57. Композиция органического концентрата в соответствии с вариантом осуществления Е55 или Е56, где Соединение А представляет собой свободную молекулу.
Е58. Композиция органического концентрата в соответствии с любым из вариантов осуществления Е55 - Е57, где отрицательно заряженный или полярный фосфолипидный стабилизатор выбирают из натриевой или аммониевой соли DMPG (1,2-Димиристоил-sn-глицеро-3-фосфо-рац-глицерин или димиристоил фосфатидилглицерин), натриевой соли POPG (1-Пальмитоил-2-олеоил-sn-глицеро-3[Фосфо-рац-(1-глицерин)]), натриевой соли DOPS (1,2-Диолеоил-sn-глицеро-3-фосфосерин), натриевой соли DOPG (1,2-Диолеоил-sn-глицеро-3[Фосфо-рац-(1-глицерин)]), натриевой или аммониевой соли DPPG (1,2-Дипальмитоил-sn-глицеро-3[Фосфо-рац-(1-глицерин)]), натриевой или аммониевой соли DSPG (1,2-Дистеароил-sn-глицеро-3[Фосфо-рац-(1-глицерин)]), натриевой соли соевой фосфатидной кислоты (РА), натриевой соли яичной фосфатидной кислоты (РА), натриевой соли соевого фосфатидилсерина (PS), натриевой соли яичного фосфатидилглицерина (PG), натриевой соли соевого фосфатидилглицерина (PG), натриевой соли фосфатидилинозитола (PI), яичного лецитина (например, Lipoid Е 80 S), соевого лецитина (например, Lipoid S 75), и олеата натрия, предпочтительно натриевой соли DMPG.
Е59. Композиция органического концентрата в соответствии с вариантом осуществления Е58, где отрицательно заряженный или полярный фосфолипидный стабилизатор представляет собой 1,2-Димиристоил-sn-глицеро-3-фосфо-рац-глицерин, натриевую или аммониевую соль, яичный лецитин (например, Lipoid Е 80 S), соевый лецитин (например, Lipoid S 75), или, предпочтительно 1,2-Димиристоил-sn-глицеро-3-фосфо-рац-глицерин, натриевую соль.
Е60. Композиция органического концентрата в соответствии с любым из вариантов осуществления Е55 - Е59, где стабилизатор от гелеобразования представляет собой полимер или электролит.
Е61. Органический концентрат в соответствии с вариантом осуществления Е60, где стабилизатор от гелеобразования представляет собой электролит, выбранный из хлорида натрия, хлорида калия, фосфата натрия двух- и одноосновного, сульфата аммония и аргинина, в особенности хлорида натрия, хлорида калия, фосфата натрия двух- и одноосновного, и сульфата аммония.
Е62. Композиция органического концентрата в соответствии с вариантом осуществления Е61, где стабилизатор от гелеобразования представляет собой хлорид натрия.
Е63. Композиция органического концентрата в соответствии с вариантом осуществления Е60, где стабилизатор от гелеобразования представляет собой полимер, выбранный из PEG300 и PEG400, предпочтительно PEG300.
Е64. Композиция органического концентрата в соответствии с вариантом осуществления Е63, где PEG300 присутствует в количестве от 5% мас./мас. до 30% мас./мас. композиции концентрата, в особенности от 15% мас./мас. до 20% мас./мас. более предпочтительно 15% мас./мас.
Е65. Композиция органического концентрата в соответствии с любым из вариантов осуществления Е55 - Е64, дополнительно содержащая растворитель.
Е66. Композиция органического концентрата в соответствии с вариантом осуществления Е65, дополнительно содержащая растворитель, выбранный из пропиленгликоль и этанол, и в особенности содержащая оба компонента, пропиленгликоль и этанол.
Е67. Фармацевтическая композиция, полученная в результате смешивания композиции органического концентрата в соответствии с любым из вариантов осуществления Е1 - E12, или Е47 - Е49, или Е55 - Е66, и липосомального носителя, где указанный липосомальный носитель содержит фосфолипид и агент, корригирующий тоничность, в особенности где указанный агент, корригирующий тоничность, выбирают из декстрозы, глюкозы, маннита, сахарозы, лактозы, трегалозы, глицерина и NaCl.
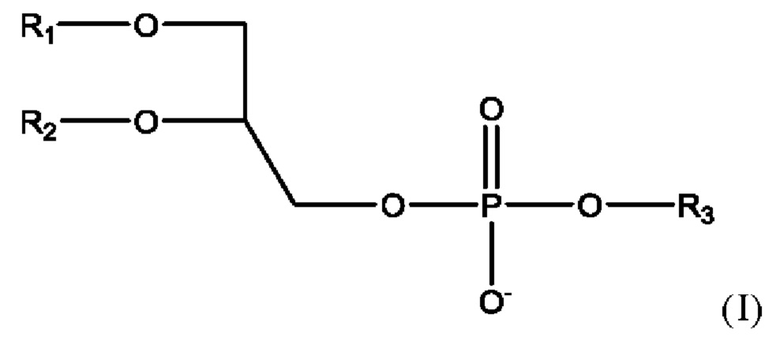
Е68. Фармацевтическая композиция в соответствии с вариантом осуществления Е67, где фосфолипид представлен формулой
где
• R1 представляет собой С10-С24ацил;
• R2 представляет собой С10-С24ацил, или альтернативно, R2 представляет собой водород или С10-С20ацил;
• R3 представляет собой водород, 2-триметиламино-1-этил, 2-амино-1-этил, С1-С4алкил,
С1-С5алкил, замещенный карбокси, С2-С5алкил, замещенный карбокси и гидрокси, С2-С5алкил, замещенный карбокси и амино, инозитоловую группу или глицериловую группу;
или соль такого соединения.
Е69. Фармацевтическая композиция в соответствии с вариантом осуществления Е67, где фосфолипид выбирают из яичного лецитина, соевого лецитина или синтетических фосфолипидов.
Е70. Фармацевтическая композиция в соответствии с вариантами осуществления Е67 - Е69, где фосфолипид выбирают из яичного лецитина, соевого лецитина, РОРС (пальмитоил олеоил фосфатидилхолин), DOPC (1,2-Диолеоил-sn-глицеро-3-фосфохолин), DMPC (1,2-Димиристоил-sn-глицеро-3-фосфохолин) и DPPC (1,2-Дипальмитоил-sn-глицеро-3-фосфохолин), в частности РОРС (пальмитоил олеоил фосфатидилхолин), DOPC (1,2-Диолеоил-sn-глицеро-3-фосфохолин), и DMPC (1,2-Димиристоил-sn-глицеро-3-фосфохолин), в особенности РОРС.
Е71. Фармацевтическая композиция в соответствии с любым из вариантов осуществления Е67, Е69 или Е70, где фосфолипид выбирают из яичного лецитина или соевого лецитина, содержащего по меньшей мере 75% фосфатидилхолина, или по меньшей мере 70% фосфатидилхолина, и, в особенности, выбирают из Lipoid Е 80 S.
Е72. Фармацевтическая композиция в соответствии с любым из вариантов осуществления Е67 - Е71, где агент, корригирующий тоничность, выбирают из декстрозы, глюкозы, маннита, сахарозы, лактозы, трегалозы, глицерина и NaCl, в особенности сахарозы или глицерина.
Е73. Липосомальная фармацевтическая композиция, содержащая, дополнительно к липосомам:
а. Соединение А, которое представляет собой 2-{[5-{3-хлор-2-метил-4-[2-(4-метилпиперазин-1-ил)этокси]фенил}-6-(4-фторфенил)тиено[2,3-d]пиримидин-4-ил]окси}-3-(2-{[2-(2-метоксифенил)пиримидин-4-ил]метокси}фенил)пропановую кислоту или ее фармацевтически приемлемую соль; и
б. стабилизатор от гелеобразования.
Е74. Липосомальная фармацевтическая композиция в соответствии с вариантом осуществления Е73, содержащая:
а. Соединение А, как описано в любом из вариантов осуществления Е2 - Е3, и
б. стабилизатор от гелеобразования, как описано в любом из вариантов осуществления Е60 - Е63.
Е75. Липосомальная фармацевтическая композиция в соответствии с вариантом осуществления Е74, дополнительно содержащая отрицательно заряженный или полярный фосфолипидный стабилизатор.
Е76. Липосомальная фармацевтическая композиция в соответствии с вариантом осуществления Е75, где указанный отрицательно заряженный или полярный фосфолипидный стабилизатор представляет собой структуру, как описано в Е4 - Е6 или Е58 или Е59.
Е77. Липосомальная фармацевтическая композиция в соответствии с любым из вариантов осуществления Е73 - Е76, дополнительно содержащая растворитель.
Е78. Липосомальная фармацевтическая композиция в соответствии с вариантом осуществления Е77, где указанный растворитель выбирают из пропиленгликоля и этанола, или в особенности содержащая оба компонента, пропиленгликоль и этанол.
Е79. Липосомальная фармацевтическая композиция в соответствии с любым из вариантов осуществления Е73 - Е78, содержащая фосфолипид, как описано в любом из вариантов осуществления Е15 - Е19, или любым из вариантов осуществления Е68 - Е71.
Е80. Липосомальная фармацевтическая композиция в соответствии с любым из вариантов осуществления Е73 - Е79, дополнительно содержащая агент, корригирующий тоничность, в особенности где указанный агент, корригирующий тоничность, выбирают из декстрозы, глюкозы, маннита, сахарозы, лактозы, трегалозы, глицерина и NaCl.
Е81. Композиция органического концентрата, содержащая;
а. Соединение А, которое представляет собой (2R)-2-{[(5Sa)-5-{3-хлор-2-метил-4-[2-(4-метилпиперазин-1-ил)этокси]фенил}-6-(4-фторфенил)тиено[2,3-d]пиримидин-4-ил]окси}-3-(2-{[2-(2-метоксифенил)пиримидин-4-ил]метокси}фенил)пропановую кислоту или ее фармацевтически приемлемую соль, предпочтительно свободную молекулу,
б. отрицательно заряженный или полярный фосфолипидный стабилизатор, например, выбранный из натриевой или аммониевой соли DMPG (1,2-Димиристоил-sn-глицеро-3-фосфо-рац-глицерин или димиристоил фосфатидилглицерин), натриевой соли POPG (1-Пальмитоил-2-олеоил-sn-глицеро-3[Фосфо-рац-(1-глицерин)]), натриевой соли DOPS (1,2-Диолеоил-sn-глицеро-3-фосфосерин), натриевой соли DOPG (1,2-Диолеоил-sn-глицеро-3[Фосфо-рац-(1-глицерин)]), натриевой или аммониевой соли DPPG (1,2-Дипальмитоил-sn-глицеро-3[Фосфо-рац-(1-глицерин)]), натриевой или аммониевой соли DSPG (1,2-Дистеароил-sn-глицеро-3[Фосфо-рац-(1-глицерин)]), натриевой соли соевой фосфатидной кислоты (РА), натриевой соли яичной фосфатидной кислоты (РА), натриевой соли соевого фосфатидилсерина (PS), натриевой соли яичного фосфатидилглицерина (PG), натриевой соли соевого фосфатидилглицерина (PG), натриевой соли фосфатидилинозитола (PI), Lipoid E 80 S, и олеата натрия, и
в. стабилизатор, выбранный из хлорида натрия, хлорида калия, фосфата натрия двух- и одноосновного, сульфата аммония и аргинина, предпочтительно хлорида натрия.
Е82. Композиция органического концентрата, содержащая:
а. Соединение А, которое представляет собой (2R)-2-{[(5Sa)-5-{3-хлор-2-метил-4-[2-(4-метилпиперазин-1-ил)этокси]фенил}-6-(4-фторфенил)тиено[2,3-d]пиримидин-4-ил]окси}-3-(2-{[2-(2-метоксифенил)пиримидин-4-ил]метокси}фенил)пропановую кислоту,
б. отрицательно заряженный или полярный фосфолипидный стабилизатор, например, выбранный из натриевой или аммониевой соли DMPG (1,2-Димиристоил-sn-глицеро-3-фосфо-рац-глицерин или димиристоил фосфатидилглицерин), натриевой соли POPG (1-Пальмитоил-2-олеоил-sn-глицеро-3[Фосфо-рац-(1-глицерин)]), натриевой соли DOPS (1,2-Диолеоил-sn-глицеро-3-фосфосерин), натриевой соли DOPG (1,2-Диолеоил-sn-глицеро-3[Фосфо-рац-(1-глицерин)]), натриевой или аммониевой соли DPPG (1,2-Дипальмитоил-sn-глицеро-3[Фосфо-рац-(1-глицерин)]), натриевой или аммониевой соли DSPG (1,2-Дистеароил-sn-глицеро-3[Фосфо-рац-(1-глицерин)]), натриевой соли соевой фосфатидной кислоты (РА), натриевой соли яичной фосфатидной кислоты (РА), натриевой соли соевого фосфатидилсерина (PS), натриевой соли яичного фосфатидилглицерина (PG), натриевой соли соевого фосфатидилглицерина (PG), натриевой соли фосфатидилинозитола (PI), яичного лецитина (например, Lipoid Е 80 S), соевого лецитина (например, Lipoid E75), и олеата натрия, в частности DMPG натрия; и
в. стабилизатор от гелеобразования, например, выбранный из
• полимера, где указанный полимер представляет собой в особенности PEG300 и PEG400, и
• электролита, где указанный электролит представляет собой в особенности хлорид натрия, хлорид калия, фосфат натрия двух- и одноосновный, сульфат аммония и аргинин, более предпочтительно хлорид натрия, хлорид калия, фосфат натрия двух- и одноосновный, сульфат аммония, в частности хлорид натрия
Е83. Фармацевтическая композиция, содержащая:
а. фосфолипид, выбранный из яичного лецитина, соевого лецитина, РОРС (пальмитоил олеоил фосфатидилхолин), DOPC (1,2-Диолеоил-sn-глицеро-3-фосфохолин), DMPC (1,2-Димиристоил-sn-глицеро-3-фосфохолин) и DPPC (1,2-Дипальмитоил-sn-глицеро-3-фосфохолин), в частности РОРС (пальмитоил олеоил фосфатидилхолин), DOPC (1,2-Диолеоил-sn-глицеро-3-фосфохолин), и DMPC (1,2-Димиристоил-sn-глицеро-3-фосфохолин), в особенности РОРС,
или где фосфолипид выбирают из яичного лецитина или соевого лецитина, содержащего по меньшей мере 70% фосфатидилхолина;
б. Соединение А, которое представляет собой (2R)-2-{[(5Sa)-5-{3-хлор-2-метил-4-[2-(4-метилпиперазин-1-ил)этокси]фенил}-6-(4-фторфенил)тиено[2,3-d]пиримидин-4-ил]окси}-3-(2-{[2-(2-метоксифенил)пиримидин-4-ил]метокси}фенил)пропановую кислоту или ее соль, предпочтительно свободную молекулу,
в. отрицательно заряженный или полярный фосфолипидный стабилизатор, например, натриевую или аммониевую соль DMPG (1,2-Димиристоил-sn-глицеро-3-фосфо-рац-глицерин или димиристоил фосфатидилглицерин), натриевую соль POPG (1-Пальмитоил-2-олеоил-sn-глицеро-3[Фосфо-рац-(1-глицерин)]), натриевую соль DOPS (1,2-Диолеоил-sn-глицеро-3-фосфосерин), натриевую соль DOPG (1,2-Диолеоил-sn-глицеро-3[Фосфо-рац-(1-глицерин)]), натриевую или аммониевую соль DPPG (1,2-Дипальмитоил-sn-глицеро-3[Фосфо-рац-(1-глицерин)]), натриевую или аммониевую соль DSPG (1,2-Дистеароил-sn-глицеро-3[Фосфо-рац-(1-глицерин)]), натриевую соль соевой фосфатидной кислоты (РА), натриевую соль яичной фосфатидной кислоты (РА), натриевую соль соевого фосфатидилсерина (PS), натриевую соль яичного фосфатидилглицерина (PG), натриевую соль соевого фосфатидилглицерина (PG), натриевую соль фосфатидилинозитола (PI), Lipoid E 80 S, и олеат натрия, в частности 1,2-Димиристоил-sn-глицеро-3-фосфо-рац-глицерин, натриевую или аммониевую соль, яичный лецитин или Lipoid Е 80 S, предпочтительно 1,2-Димиристоил-sn-глицеро-3-фосфо-рац-глицерин, натриевую соль; и
г. стабилизатор от гелеобразования, например, выбранный из
• полимера, где указанный полимер представляет собой в особенности PEG300 и PEG400, и
• электролита, где указанный электролит представляет собой в особенности хлорид натрия, хлорид калия, фосфат натрия двух- и одноосновный, сульфат аммония и аргинин, более предпочтительно хлорид натрия, хлорид калия, фосфат натрия двух- и одноосновный, сульфат аммония, в частности хлорид натрия.
Е84. Способ солюбилизации Соединения А, как описано в любом из вариантов осуществления E1, E2 или Е3, в водной окружающей среде, включающий смешивание:
а. липосомального носителя в соответствии с любым из вариантов осуществления Е13 - Е26, и
б. композиции органического концентрата в соответствии с любым из вариантов осуществления Е1 - Е12, или Е47 - Е49, или Е55 - Е66, Е81 или Е82.
Е85. Способ получения композиции Соединения А, пригодной для парентерального введения, включающий смешивание:
а. липосомального носителя в соответствии с любым из вариантов осуществления Е13 - Е26, и
б. композиция органического концентрата в соответствии с любым из вариантов осуществления Е1 - Е12 или Е47 - Е49, или Е55 - Е66, Е81 или Е82.
Е86. Способ в соответствии с вариантом осуществления Е84 или Е85, где смешивание осуществляют непосредственно перед введением пациенту.
Е87. Способ в соответствии с любым из вариантов осуществления Е84, Е85 или Е86, где смешивание и липосомную загрузку осуществляют путем повторной инверсии, в особенности встряхивая контейнер интенсивно путем инверсии.
Е88. Способ в соответствии с любым из вариантов осуществления Е84 - Е87, где смешивание и липосомную загрузку осуществляют путем инверсии контейнера, содержащего оба компонента а. и 6, в особенности многократно, или встряхивая интенсивно путем инверсии, до тех пор, пока в смеси не будет видимых частиц.
Е89. Способ модуляции активности Mcl-1 рецептора у субъекта, где способ включает введение субъекту терапевтически эффективного количества композиции в соответствии с любым из вариантов осуществления Е27 - Е46, или Е50 - Е52, или Е67 - Е80, или Е83.
Е90. Способ лечения злокачественного новообразования, включающий введение субъекту терапевтически эффективного количества композиции в соответствии с любым из вариантов осуществления Е27 - Е46, или Е50 - Е52, или Е67 - Е80, или Е83.
Е91. Способ в соответствии с вариантом осуществления Е90, где злокачественное новообразование выбирают из злокачественных новообразований мочевого пузыря, головного мозга, молочной железы и матки, хронических лимфоидных лейкозов, злокачественного новообразования толстой кишки, пищевода и печени, лимфобластных лейкозов, острого миелоидного лейкоза, лимфом, например, неходжкинской В-клеточной лимфомы и диффузной В-крупноклеточной лимфомы, меланом, злокачественных болезней крови, например, миелодиспластического синдрома, миелом, например, множественной миеломы, рака яичников, немелкоклеточного рака легкого, рака предстательной железы, рака поджелудочной железы и мелко клеточного рака легкого.
Е92. Способ в соответствии с вариантом осуществления Е91, где злокачественное новообразование выбирают из неходжкинской В-клеточной лимфомы, диффузной В-крупноклеточной лимфомы, множественной миеломы, миелодиспластического синдрома, хронических лимфоидных лейкозов и острого миелоидного лейкоза.
Е93. Фармацевтическая композиция в соответствии с любым из вариантов осуществления Е27 - Е46, или Е50 - Е52, или Е67 - Е80, или Е83, для применения в качестве лекарственного средства.
Е94. Фармацевтическая композиция для применения в соответствии с вариантом осуществления Е93, где указанное применение предназначено для лечения злокачественного новообразования, в особенности, где злокачественное новообразование выбирают из злокачественных новообразований мочевого пузыря, головного мозга, молочной железы и матки, хронических лимфоидных лейкозов, злокачественного новообразования толстой кишки, пищевода и печени, лимфобластных лейкозов, острого миелоидного лейкоза, лимфом, например, неходжкинской В-клеточной лимфомы и диффузной В-крупноклеточной лимфомы, меланом, злокачественных болезней крови, например, миелодиспластического синдрома, миелом, например, множественной миеломы, рака яичников, немелкоклеточного рака легкого, рака предстательной железы, рака поджелудочной железы и мелко клеточного рака легкого.
Е95. Фармацевтическая композиция для применения в соответствии с вариантом осуществления Е94, где указанное злокачественное новообразование выбирают из неходжкинской В-клеточной лимфомы, диффузной В-крупноклеточной лимфомы, множественной миеломы, миелодиспластического синдрома, хронических лимфоидных лейкозов и острого миелоидного лейкоза.
Е96. Применение композиции органического концентрата в соответствии с любым из вариантов осуществления Е1 - Е12 или Е47 - Е49, или Е55 - Е66, Е81 или Е82, для приготовления лекарственного средства для лечения злокачественного новообразования.
Е97. Применение в соответствии с вариантом осуществления Е96, для приготовления липосомального лекарственного средства для лечения злокачественного новообразования.
Е98. Применение в соответствии с вариантом осуществления Е96 или Е97, где злокачественное новообразование выбирают из злокачественных новообразований мочевого пузыря, головного мозга, молочной железы и матки, хронических лимфоидных лейкозов, злокачественного новообразования толстой кишки, пищевода и печени, лимфобластных лейкозов, острого миелоидного лейкоза, лимфом, например, неходжкинской В-клеточной лимфомы и диффузной В-крупноклеточной лимфомы, меланом, злокачественных болезней крови, например, миелодиспластического синдрома, миелом, например, множественной миеломы, рака яичников, немелкоклеточного рака легкого, рака предстательной железы, рака поджелудочной железы и мелкоклеточного рака легкого, в особенности неходжкинской В-клеточной лимфомы, диффузной В-крупноклеточной лимфомы, множественной миеломы, миелодиспластического синдрома, хронических лимфоидных лейкозов и острого миелоидного лейкоза.
Е99. Комбинация, содержащая:
• фармацевтическую композицию в соответствии с любым из вариантов осуществления Е27 - Е46, или Е50 - Е52, или Е67 - Е80, или Е83, и
• один или несколько терапевтически активных средств, для одновременного, последовательного или раздельного применения.
Е100. Композиция содержащая Соединение А, как определено в E1, E2 или Е3, и стабилизатор от гелеобразования.
В предпочтительном варианте осуществления изобретения, пациентов выбирали, например, в соответствии со следующими критериями:
Основные критерии включения:
 Возраст ≥ 18 лет
Возраст ≥ 18 лет
Для пациентов с множественной миеломой:
 Гистологически подтвержденная лимфома или подтвержденная множественная миелома. В предпочтительном варианте осуществления изобретения для пациентов с множественной миеломой, множественная миелома является рецидивирующей и/или трудно поддающейся стандартному лечению
Гистологически подтвержденная лимфома или подтвержденная множественная миелома. В предпочтительном варианте осуществления изобретения для пациентов с множественной миеломой, множественная миелома является рецидивирующей и/или трудно поддающейся стандартному лечению
 Характеризуется хромосомной аберрацией
Характеризуется хромосомной аберрацией
 Измеряемые проявления болезни:
Измеряемые проявления болезни:
- М-белок в сыворотке ≥ 0,5 г/дл
- М-белок в моче ≥ 200 мг/24 часа или
- Уровень свободной легкой цепи (FLC) в сыворотке ≥ 100 мг/л вовлеченного FLC.
Для пациентов с гистологически подтвержденной DLBCL:
 Документально подтвержденная положительная МУС, определенная с помощью флуоресцентной гибридизаций in situ (FISH) или иммуногистохимии (IHC)
Документально подтвержденная положительная МУС, определенная с помощью флуоресцентной гибридизаций in situ (FISH) или иммуногистохимии (IHC)
 Измеряемые проявления болезни (в соответствии с Рекомендациями относительно первичной оценки, определения стадий и оценки ответной реакции ходжкинской и не-ходжкинской лимфомы)
Измеряемые проявления болезни (в соответствии с Рекомендациями относительно первичной оценки, определения стадий и оценки ответной реакции ходжкинской и не-ходжкинской лимфомы)
Основные критерии исключения
1. Данные в анамнезе относительно хронического заболевания почек, включая
 активную инфекцию гепатита В или С.
активную инфекцию гепатита В или С.
 первичный билиарный цирроз
первичный билиарный цирроз
 цирроз печени, или
цирроз печени, или
 портальную гипертензию
портальную гипертензию
2. Хронический панкреатит в анамнезе
3. Пациенты с высоким риском синдрома лизиса опухоли (TLS)
4. Наличие в анамнезе или диагностированные пневмония, интерстициальное заболевание легких или воспалительное заболевание кишечника.
5. Нарушение сердечной деятельности или клинически значимые сердечные заболевания, включая любые из следующих параметров:
 Фракция выброса левого желудочка (LVEF) < 50%, как определено с помощью многопроекционного радиоизотопного сканирования (MUGA) или эхокардиограммы (ECHO)
Фракция выброса левого желудочка (LVEF) < 50%, как определено с помощью многопроекционного радиоизотопного сканирования (MUGA) или эхокардиограммы (ECHO)
 QT, скорректированные с поправкой Фредеричиа (QTcF) > 450 мс (пациенты мужского пола), >460 мс (пациенты женского пола)
QT, скорректированные с поправкой Фредеричиа (QTcF) > 450 мс (пациенты мужского пола), >460 мс (пациенты женского пола)
6. Тяжелые и/или неконторолируемые медицинские состояния, которые, по мнению исследователя, могут оказывать влияние на безопасность индивидуума или нарушать оценку результатов исследования.
7. Предшествующее лечение с применением ингибитора Mcl-1.
8. Предшествующее лечение с применением ингибитора Bcl-2 (для пациентов, включенных в исследование с применением максимально переносимой дозы, определенной в ходе этапа повышения доз).
9. Пациенты с первичной опухолью ЦНС или вовлеченной опухолью ЦНС.
Липосомальный носитель, описанная в настоящей заявке, может быть получен с помощью любого метода получения, который приводит к получению липосом со средним размером частиц вплоть до 1000 нм, предпочтительно вплоть до 300 нм, в особенности вплоть до 100 нм. Размер липосомы, как описано в настоящей заявке, относится к среднему диаметру Z с использованием фотонно-корреляционной спектроскопии в соответствии с ISO 13321, который описывает полученный средний размер частиц и коэффициент полидисперсности. Типичный способ получения включает диспергирование, смешивание с высокой скоростью сдвига и последующую гомогенизацию под высоким давлением, и такие способы хорошо известны квалифицированному специалисту в данной области техники.
Органический концентрат образовывается путем смешивания компонентов до тех пор, пока не образуется гомогенный раствор, в котором растворено Соединение А.
Для образования композиции для введения, концентрат добавляют к липосомальному носителю, после этого сразу смешивают, например, путем повторной инверсии, или интенсивного встряхивания. Альтернативно, концентрат можно добавлять к носителю во время вращения.
Благоприятно, в предпочтительном варианте осуществления изобретения, обеспечивается липосомальный носитель, который может быть смешан с Соединением А незадолго до введения для получения прозрачной композиции. В особенности, это достигается путем смешивания липосомального носителя с органическим концентратом, содержащим Соединение А, как описано в настоящей заявке. Прозрачность является желательной для проверки того, что Соединение А солюбилизировалось перед введением. Это может быть оценено с помощью визуального осмотра или прозрачность определяют при наиболее подходящей длине волны, в зависимости от концентрации липидов, например, 660 нм или используя трансмиссионную ячейку или кювету 1 см. В аспекте изобретения, фармацевтическая композиция для введения (липосомы, загруженные Соединением А) не должна снижать трансмиссию по сравнению с липосомальным носителем не более, чем на 30%, предпочтительно не более, чем на 20%, наиболее предпочтительно не более, чем на 10%.
В предпочтительном варианте осуществления изобретения, липосомный носитель, при смешивании с Соединением А, предоставляет возможность максимальной загрузки Соединения А на липосомы, в особенности, если Соединение А представлено в виде органического концентрата, как описано в настоящей заявке.
В Примерах в настоящей заявке, 'Соединение А' обозначает (2R)-2-{[(5Sa)-5-{3-хлор-2-метил-4-[2-(4-метилпиперазин-1-ил)этокси]фенил}-6-(4-фторфенил)тиено[2,3-d]пиримидин-4-ил]окси}-3-(2-{[2-(2-метоксифенил)пиримидин-4-ил]метокси}фенил)пропановую кислоту (без соли).
ПРИМЕР 1: Органический концентрат, содержащий Соединение А и хлорид натрия


Приготовление нерасфасованного растворителя:
Воду для инъекций, затем безводный этанол, после этого DMPG-Na загружали в сосуд и перемешивали до увлажнения DMPG-Na порошка. После этого добавляли приблизительно четверть пропиленгликоля и смесь перемешивали и нагревали. Продолжали перемешивать и температуру поддерживали в течение 90 минут или до полного растворения DMPG-Na. После завершения растворения, смесь впоследствии охлаждали до комнатной температуры, добавляли оставшийся пропиленгликоль и смесь перемешивали до тех пор, пока она не становилась гомогенной. После этого добавляли хлорид натрия и смесь нагревали в течение 90 минут или до полного растворения хлорида натрия. Полученный раствор охлаждали до комнатной температуры.
Приготовление органического концентрата лекарственного средства:
Нерасфасованный растворитель, приготовленный, как описано выше, загружали в сосуд, добавляли Соединение А и смесь перемешивали до увлажнения порошка Соединения А. Продолжали перемешивать в течение 60 минут или до полного растворения Соединения А. Концентрат фильтровали через стерильную 0,2 мкм Pall Fluorodyne® II DFL мембрану в Kleenpak™ капсулах KA3DFLP1G или KA3DFLP1. После фильтрации, полученный органический концентрат лекарственного продукта (61,75 мг/мл) переносили в стерильные, апирогенные флаконы.
ПРИМЕР 2: Липосомальный носитель для разведения
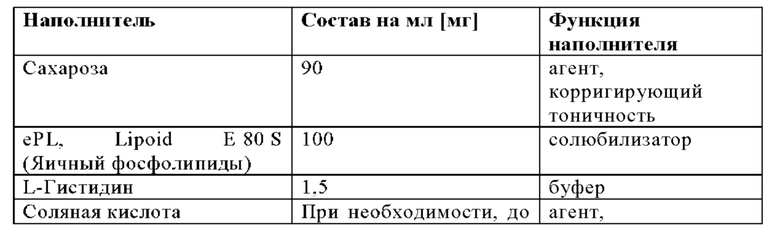

Приготовление липосомального носителя для разведения
Поскольку Лецитин (ePL) подвержен окислению, то кислород удаляли во всех технически возможных случаях и материалы предпочтительно защищали путем подачи слоя азота.
Воду для инъекций, после этого L-Гистидин, загружали в сосуд и перемешивали до тех пор, пока L-Гистидин полностью не растворялся. Загружали сахарозу и смесь перемешивали до тех пор, пока сахароза полностью не растворялась. Проверяли значение рН и доводили до 6,5±0,2 путем добавления раствора 1 н. соляной кислоты или 1 н. гидроксида натрия, после этого раствор перемешивали до тех пор, пока он не становился гомогенным. Затем в сосуд добавляли Lipoid Е 80 S. Смесь перемешивали до тех пор, пока не получали гомогенную дисперсию.
Линейное смешивание
Дисперсию, полученные выше, перемешивали и переносили в другой сосуд с помощью линейного гомогенизатора (Kinematica MEGATRON MT-SV 1-45 М), регулируемого при потоке 2-5 л/мин. Линейное смешивание продолжали до тех пор, пока сосуд с источником не ставился пустым.
Гомогенизация под высоким давлением
Сосуд, содержащий дисперсию лецитина, как описано выше, соединяли с гомогенизатором высокого давления (Avestin Emulsiflex С160) в рециркуляционной петле, и дисперсию охлаждали до 2-8°С, перемешивая при этом при 300 об. в мин. После повышения давления в сосуде до 0,5 бар, насос устанавливали на максимальную скорость и гомогенизатор высокого давления накачивали с помощью открытого клапана для удаления воздуха в системе. После этого гомогенизатор высокого давления устанавливали на 60 Гц, поток на 130-140 л/ч и давление на 950-1050 бар, и осуществляли гомогенизацию под высоким давлением в течение 19 проходов (около 7,5-8 часов), затем на 20-ом проходе, содержимое сосуда переносили в другой сосуд с помощью гомогенизатора высокого давления.
Предварительную фильтрацию осуществляли с помощью PVDF (поливинилидендифторидная мембрана) фильтра 0,2 мкм, и отфильтрованный раствор хранили при комнатной температуре. После этого раствор стерильно фильтровали через PVDF стерилизующий фильтр и затем переносили в стерильные флаконы, депирогенизированные путем обработки сухим жаром.
Этот водный препарат можно инъецировать или инфузировать как таковой или разводить с помощью инфузионных растворов (например, 5% Глюкоза) перед инфузией.
ПРИМЕР 3А: Препарат для инфузии
Органический концентрат из Примера 1 смешивали с липосомальным носителем из Примера 2 следующим образом. Липосомальному носителю из Примера 2 предоставляли возможность нагреться до комнатной температуры (то есть, до 20-25°С) путем вынимания из холодильника по меньшей мере за 30 минут до смешивания. Общий объем активного липосомального носителя, который будут вводить пациенту, можно определить, как указано в Таблице 3А.1 ниже. Липосомальный носитель содержит небольшие липосомы и используется в качестве солюбилизирующего носителя, способного растворять Соединение А в водной окружающей среде после смешивания с органическим концентратом, содержащим соединение А.

Одни флакон органического концентрата Соединения А (185,25 мг [включая излишек] Соединения А в 3 мл) комбинировали с одним флаконом липосомального носителя (2,1 грамм Lipoid Е 80 S в 21 мл), для получения извлекаемого объема 24 мл активного липосомального носителя, содержащего Соединение А, содержащего в целом 180 мг Соединения А). Для одного пациента может быть необходимым приготовить больше флаконов. Необходимо приготовить достаточное количество флаконов активного липосомального носителя, достаточного для введения дозы пациенту.
Органический концентрат Соединения А (3 мл) асептически и точно отбирали быстро в один флакон липосомального носителя и сразу смешивали путем инвертирования флакона ≥ 40 раз. Начальная мутность исчезает. Полученный препарат активного липосомального носителя будет немного более мутным по сравнению с липосомальным носителем для разведения. Он не должен содержать видимых частиц после смешивания. Если пациенту необходимо вводить более, чем 180 мг Соединения А, то стадии добавления и смешивания повторяют, при необходимости.
После этого рассчитывают общий объем раствора глюкозы, которые необходимо удалить из инфузионного мешка (например, используя Ecobag Глюкоза 5%, или Ecoflac Глюкоза 5%). Это объем является идентичным объему активного липосомального носителя, как представлено в Таблице 3А.1. Рассчитанный объем раствора глюкозы асептически и точно отбирают из инфузионного мешка (и отбрасывают).
Рассчитанный объем активного липосомального носителя (содержащего Соединение А) асептически и точно отбирают из одного или нескольких флаконов и инъецируют в глюкозный мешок, приготовленный выше, и смешивают хорошо путем переворачивания мешка по меньшей мере 10 раз. Если необходимо меньше, чем кратно 180 мг Соединения А, то остаток из флаконов необходимо выбрасывать.
Приготовленный инфузионный мешок, готовый для инфузии, следует анализировать визуально относительно наличия частиц. Содержимое мешка может проявлять незначительно опалесценцию или незначительную мутность в зависимости от концентрации Соединения А. Если видны частицы, то мешок отбраковывают и готовят свежий инфузионный мешок.
Соединение А немного чувствительно к свету, поэтому инфузионный мешок помещают под светонепроницаемое покрывало.
Один из примеров процедуры представлен ниже. Свежеприготовленные инфузионные мешки хранили в течение вплоть до 5 часов при комнатной температуре (то есть, при 20-25°С) плюс вплоть до 8 часов в условиях холодильника (2-8°С), которое включало фактическое время инфузии. Если выдерживали при комнатной температуре 20-25°С, то можно приготовить разведенный раствор для инфузии в инфузионном мешке и инфузию начинать в течение двух часов после приготовления и вводить при контролированной комнатной температуре. Если внутривенный раствор не вводят сразу же, то раствор необходимо поместить в холодильник при 2-8°С только не более 8 часов. Если приготовленный раствор охлаждают в холодильнике перед введением, то мешок можно довести до комнатной температуры перед введением. Инфузионный препарат, содержащий необходимую концентрацию лекарственного продукта Соединения А, можно инфузировать с помощью инфузионной системы и встроенного фильтра 1, 2 мкм после инфузионного мешка, расположенного максимально близко, насколько это возможно, к катетеру.
ПРИМЕР 3В: Препарат для инфузии
Используя такую же процедуру, как описано в Примере 3А, липосомальный носитель из Примера 2 смешивали с органическим раствором следующего состава:

Можно использовать следующие соотношения компонентов для смешивания:
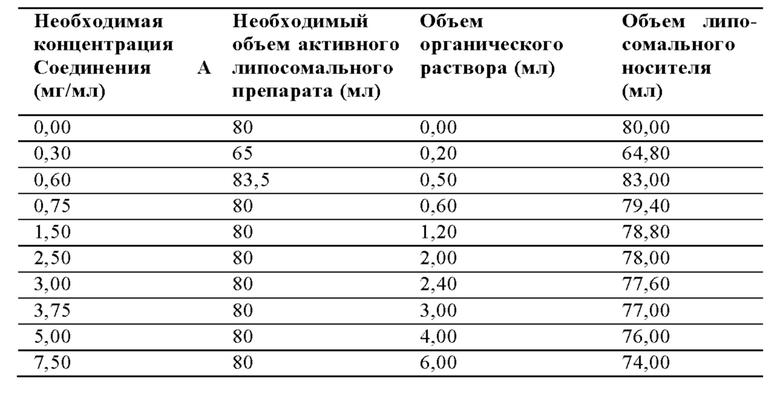
ПРИМЕР 4: Эффективность липосомального препарата Соединения А на модели ксенотрансплантат MV4; 11 острого миелоидного лейкоза у бестимусных крыс, используя схему внутривенного введения раз в неделю
Методы
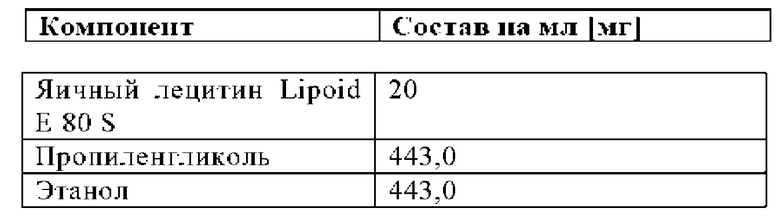
Таблица 4.1: Композиция органического растворителя, используемая для приготовления дозированного раствора для группы с носителем:
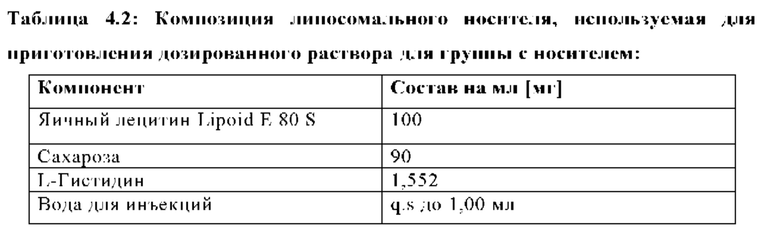
Липосомальный носитель получали, используя компоненты, указанные в Таблице 4.2, и методы, описанные в настоящей заявке в Пример 2. Органический раствор (Таблица 4.1) смешивали с липосомальным носителем в соотношении 1:32, 33 (об./об.) и использовали для дозирования группе с носителем в дозе 10 мл/кг.
В этом исследовании оценивали противоопухолевую активность и переносимость Соединения А в препарате на основе липосом после в/в инфузии бестимусным крысам, несущим MV4; 11 опухоль. Эксперименты осуществляли на самках бестимусных крыс Crl:NIH-Foxn1rnu (Charles River).
Клеточную линию MV4; 11 luc острого миелоидного лейкоза (AML) человека получали от родительских MV4; 11 клеток, которые имели происхождение от пациента. Родительские клетки MV4; 11 получали из DSMZ банка клеток. MV4; 11 luc клетки использовали для всех исследований in vivo и культивировали в RPMI-1640 среде (BioConcept Ltd. Amimed, №1-41F01-I), дополненной 10% FCS (GE health care life sciences, № H30071,03) 4 мМ L-глутамином (BioConcept Ltd. Amimed, №5-10K00-H), 0,6 мг/мл генетицина (Life Technologies, №10131-027) при 37°C в атмосфере 5% CO2 на воздухе. Клетки поддерживали в диапазоне от 0,2 до 1,1×106 клеток/мл. Для получения MV4; 11 luc ксенотрансплантатов, клетки собирали и ресупендировали в HBSS (Gibco, №14175) и смешивали с Matrigel (BD Bioscience, №354234) (1:1 об./об.) перед инъецированием 200 мкл, содержащих 1×107 клеток, подкожно в правую заднюю лапу животным, которых анестезировали с помощью изофлурана. За двадцать четыре часа перед инокуляцией клеток, всех животных облучали с применением 5 Гр в течение 2 минут, используя γ-излучатель.
За ростом опухоли наблюдали регулярно после инокуляции клеток и животных рандомизировали на леченные группы, когда их опухоли достигали подходящего объема. Размер опухоли, в мм, рассчитывали с помощью формулы: (L×W2×π/6), где W = ширина и L = длина опухоли.
Животных, несущих опухоли, разделяли на леченные группы (n=5), со средним объем опухоли приблизительно 450 мм3. После этого животных лечили с применением носителя или препарата Соединения А в дозе 10 и 30 мг/кг.
Соединение А приготавливали в виде препарат на основе липосом, используя органический концентрат лекарственного средства (описанный в Примере 1) и носитель (описанный в Примере 2 в настоящей заявке). Дозированные растворы приготавливали путем смешивания продукта из Примеров 1 и 2 при соотношении 1:7,236 (об./об.), затем разведения этой смеси путем добавления 5% глюкозы при соотношении 1:6,50 (об./об.) для дозы 10 мг/кг и 1:1,50 (об./об.) для дозы 30 мг/кг.
Носитель и Соединение А вводили один раз в неделю (QW) путем в/в инфузии 15 минут в латеральную хвостовую вену в дозе 10 мл/кг. Животных анестезировали с помощью изофлурана/О2 приблизительно в течение 20 минут для осуществления инфузии. Объемы опухолей измеряли, используя штангенциркули 2-3 раза в неделю. Животных взвешивали 2-3 раза в неделю и часто исследовали для выявления признаков любых побочных эффектов.
Данные изменения опухоли и веса тела анализировали статистически, используя GraphPad Prism 7.00 (программное обеспечение GraphPad). Если колебания в данных были нормально распределены, то данные анализировали, используя однофакторный ANOVA с ретроспективным критерием Даннетта для сравнения леченной группы относительно контрольной группы. Апостериорный критерий Тьюки использовали для внутригруппового сравнения. В других случаях, использовали тест Краскела-Уоллиса с ретроспективным критерием Даннетта. Где применимо, результаты представляли в виде среднего значения ± СПС.
В качестве критерия определения эффективности, рассчитывали значение %Т/С после окончания эксперимента в соответствии со следующей формулой:
(Δобъема опухолилеченные/Δобъема опухоликонтроль)*100
Регрессию опухоли рассчитывали в соответствии со следующей формулой:
-(Δобъема опухолилеченные/объема опухолилеченные в начале)*100
где Δобъема опухоли представляют собой средний объем опухоли в день оценки минус средний объем опухоли в начале эксперимента.
Результаты: Эффективность и переносимость
Препарат Соединения А на основе липосом проявляет существенную дозозависимую эффективность после инфузионного введения. В инфузионной дозе 10 мг/кг QW, индуцировалась максимальная противоопухолевая ответная реакция после первой дозы (61% регрессия в день 3 после начала лечения) и ответ ослаблялся при каждой последующей дозе, что приводило к постепенному увеличению объема опухоли в динамике. В день 24 (последний день измерения объема опухоли в группе с носителем) регрессия составила 6% после введения дозы 10 мг/кг QW (р<0,05 по сравнению с группой с носителем). После инфузии липосомального препарата Соединения А в дозе 30 мг/кг QW в течение 2 недель, достигали полной регрессии (CR) во всех MV4; 11 ксенотрансплантатах в день 13 после начала лечения. Полная регрессия поддерживалась у всех животных вплоть до дня 21, после чего наблюдали повторный рост опухоли у 3 животные и 2 животные проявило отсутствие роста опухоли в течение дальнейших 60 дней после достижения CR. В день 25 после начала лечения с применением 30 мг/кг, средний объем опухоли проявил 92% регрессию (р<0,05 по сравнению с группой с носителем). На основании изменений веса тела обе схемы дозирования липосомального препарата хорошо переносились. Результаты представлены на Фигурах 1 и 2.
ПРИМЕР 5: Данные стабильности для композиции из Примера 1
Исследовали стабильность композиции из Примера 1, Соединение А 185,25 мг в 3,0 мл, путем тестирования продолжительностей хранения вплоть до 3 месяцев, и светостабильности.
5.1. Тестирование при 5°С/ ОВ окружающей среды (относительная влажность)
В течение 3 месяцев хранения во флаконах в условиях 5°С/ОВ окружающей среды, наблюдали за композицией из Примера 1 Соединения А 185,25 мг/ 3,0 мл, и она была физически и химически стабильной. Не наблюдали существенных изменений в химических или физических свойствах.
5.2. Тестирование при 25°С/60% ОВ
В течение 3 месяцев хранения во флаконах в условиях 25°С/60% ОВ, наблюдали за композицией из Примера 1 Соединения А 185,25 мг/ 3,0 мл, и она была физически стабильной. Не наблюдали существенных изменений в течение 3 месяцев. Наблюдали повышенные уровни нежелательного атропизомера. Все результаты оставались в допустимых пределах.
5.3. Тестирование при 40°С/75% ОВ
В течение 3 месяцев хранения во флаконах в условиях 40°С/75% ОВ, наблюдали за Композицией из Примера 1 Соединения А 185,25 мг/ 3,0 мл, и она была физически стабильной. Не наблюдали существенных изменений в течение 3 месяцев, за исключением повышенных уровней нежелательного атропизомера в динамике. Все результаты находились в допустимых пределах через 1,5 месяца.
5.4. Светостабильность
Во время тестирования светостабильности во флаконах наблюдали, что композиция из Примера 1 Соединения А 185,25 мг/ 3,0 мл не была ни химически, ни физически стабильной. Наблюдали, что все результаты для образца, защищенного от воздействия света, находились в допустимых пределах.
Выводы
Данные удовлетворительной стабильности получали в течение 3 месяцев при 5°С/ОВ окружающей среды и 25°С/60% ОВ и 1,5 месяца при 40°С/75% ОВ. Доступные данные стабильности указывают на удовлетворительную стабильность композиции из Примера 1 Соединения А 185,25 мг/ 3,0 мл. Необходима защита от воздействия света.
ПРИМЕР 6: Данные стабильности для композиции из Примера 2
Исследовали стабильность композиции носителя из Примера 2 (21,7 мл) путем тестирования продолжительности хранения вплоть до 3 месяцев и светостабильности.
6.1. Тестирование при 5°С/ОВ окружающей среды
Не наблюдали изменений внешнего вида контейнера, внешнего вида содержимого и количественных показателей фосфолипидов вплоть до 3 месяцев хранения при 5°С, по сравнению с исходными результатами. Не наблюдали существенных изменений ни для значения рН, ни для размера частиц.
6.2. Тестирование при 25°С/60% ОВ
Не наблюдали изменений внешнего вида контейнера и внешнего вида содержимого вплоть до 3 месяцев хранения при 25°С, по сравнению с исходными результатами. Исходное рН 6,6 снижалось до рН 6,3 в течение 3 месяцев соответственно. Не наблюдали существенных изменений для размера частиц согласно фотонно-корреляционной спектроскопии и количественных показателей фосфолипидов вплоть до 3 месяцев. Невидимые невооруженным глазом твердые частицы повышались, однако значения оставались в диапазоне приемлемых значений.
6.3. Тестирование при 40°С/75% ОВ
Не наблюдали изменений внешнего вида контейнера и внешнего вида содержимого вплоть до 3 месяцев хранения при 40°С, по сравнению с исходными результатами. Наблюдали незначительное изменение значения рН, которое незначительно снижалось от рН 6,6 изначально, до 6,3 после хранения в течение 3 месяцев при 40°С.
Не наблюдали существенных изменений в течение размер частиц согласно фотонно-корреляционной спектроскопии и количественных показателей фосфолипидов вплоть до 3 месяцев хранения до 40°С. Достоверные отличия наблюдали при тестировании невидимых невооруженным глазом твердых частиц. Тем не менее, эти условия рассматриваются как стрессовые условия по сравнению с долгосрочным хранением при 5°С.
6.4. Светостабильность
Тестирование светостабильности осуществляли в продуктом в стеклянном флаконе и сравнивали с флаконом, защищенном от действия света в картонной коробке. В исследовании светостабильности не наблюдали существенных изменений для параметров внешнего вида контейнера, внешнего вида содержимого, рН раствора, видимых твердых частиц и количественных показателей фосфолипидов после хранения.
Выводы
Удовлетворительные данные стабильности были продемонстрированы для условий долгосрочного хранения при 5°С и условий «ускоренного старения» при 25°С/60% ОВ вплоть до 3 месяцев. Удовлетворительные данные стабильности были продемонстрированы для условий «ускоренного старения» при 40°С/75% ОВ вплоть до 1,5 месяцев, ограниченные результатами невидимых невооруженным взглядом твердых частиц. Для внешнего вида контейнера, внешнего вида содержимого и размера частиц не наблюдали изменений для всех условий хранения вплоть до 3 месяцев. Значения рН были стабильными и в нормативных пределах для всех условий хранения. Удовлетворительные данные стабильности были показаны для долгосрочного хранения при 5°С/ОВ окружающей среды. Носитель рассматривается как светостабильный.
ПРИМЕР 7: Исследование высвобождения in vitro. Соединение А: высвобождение Соединения А из небольших липосом на акцептор, содержащий избыток липидов, присутствующих в виде больших многослойных везикул (MLV)
Задачей этого эксперимента являлось исследование скорости высвобождения Соединения А из небольших однослойных везикул (SUV; используемых в настоящей заявке в качестве системы носителя лекарственного средства, см. Пример 3А) в большие многослойные везикулы (MLV), состоящие из яичных фосфолипидов, и используемые в качестве липофильного акцептора для высвобожденного лекарственного средства (см. Shabbits J.A., Chiu G.N. and Mayer L.D. "Development of an in vitro drug release assay that accurately predicts in vivo drug retention for liposome-based delivery systems" Journal of Controlled Release 2002, 84(3), 161-170).
Приготовление и осуществление исследования высвобождения, используя многослойные везикулы в качестве акцептора
Приготовление и характеристики MLV
Многослойные везикулы приготавливали с помощью метода пленочной гидратации. Вкратце, яичные фосфолипиды (Lipoid Е 80 S) взвешивали и полностью растворяли в органическом растворителе (EtOH/DCM при 4:1 об./об.) для обеспечения гомогенной смеси всех компонентов.
После этого, органический растворитель удаляли, используя ротационное выпаривание (первая стадия: 200 мбар, 50°С, 2 часа; вторая стадия: 30 мбар, 50°С, 45 минут) для получения гомогенной липидной пленки. Для обеспечения полного удаления органического растворителя, колбу продували азотом. После этого, пленку регидратировали, используя забуференный фосфатом солевой раствор (PBS, рН 7,4), получая MLV и, после завершения регидратирования, MLV подвергали воздействию ультразвука в течение 15 минут.
Для того, чтобы MLV были ресуспендированы в той же самой системе буферов, что и SUV, суспензию центрифугировали в течение 10 минут при 14000 g, супернатант (PBS) удаляли, и заменяли на сахарозно-гистидиновый буфер. В завершение, смесь тщательно перемешивали вихревым способом для обеспечения полного повторного диспергирования MLV в новой среде перед началом эксперимента высвобождения.
MLV препараты характеризовали на основании их размера с помощью лазерной дифракции, используя систему HELOS KR (SympaTec) после разведения 1:5000, используя PBS, получая х10, x50, и x90 значения.
х10 = величина частиц, соответствующая 10% кумулятивного распределению меньше номинального размера, x50 = средняя величина частиц (то есть, 50% частиц являются меньшими и 50% частиц являются большими), и х90 = величина частиц, соответствующая 90% кумулятивного распределению меньше номинального размера.

Исследование высвобождения, используя MLV
Для исследования высвобождения, используя MLV, использовали следующую установку для всех экспериментов:
SUV (донор) смешивали с MLV (акцептор) в колбе Эрленмейера объемом 25 мл с пробкой при перемешивании (500 об./мин.) при различных соотношениях (1:40, 1:100, 1:200, 1:500; SUV:MLV об./об.). 1 мл образцов отбирали в различные временные точки и сразу же центрифугировали (14000 об./мин., 10 минут) для отделения MLV от SUV для дальнейшей обработки. На Фигуре 3 в настоящей заявке представлено схематическое изображение принципов анализа высвобождения на основании MLV.
После этого, MLV порцию удаляли путем отсасывания и 50 мкл SUV порции смешивали с 450 мкл ACN:H2O для приготовления образца ВЭЖХ. После этого образец центрифугировали (14000 об./мин., 10 минут), супернатант фильтровали, используя фильтр 0,22 мкл и вводили в ВЭЖХ для анализа.
Метод высокоэффективной жидкостной хроматографии для Соединения А
Применяли систему высокоэффективной жидкостной хроматографии (ВЭЖХ) с УФ-обнаружением, включая следующие настройки оборудования и системы растворителей:


Исследование высвобождения Соединения А, используя MLV
Высвобождение Соединения А определяли, используя MLV в качестве акцепторного компартмента. В данном случае, Соединение А-загруженные SUV (7,5 мг/мл) добавляли к этим MLV при различных соотношениях (об./об.). На Фигуре 4 представлен профиль высвобождения Соединения А из SUV к 100% ePL при разнообразных различных SUV:MLV соотношениях (об./об.).
Примечание: Для всех экспериментов MLV высвобождения, максимальную концентрацию (maxC = 100%) получали путем разведения 7,5 мг/мл загруженных SUV 1 к 40, 1 к 100, 1 к 200, или 1 к 500 (об./об.) в сахарозно-гистидиновом буфере вместо MLV.
Используя этот тип экспериментальной модели, наблюдали очень быстрое высвобождение Соединения А из SUV в акцептор MLV. В данном случае, для разведения 1 к 40 только приблизительно 44% используемого исходного количества Соединения А было обнаружено в SUV порции в образце только через 2 минуты. Таким образом, ~60% было высвобождено или уже перенесено на акцепторную MLV порцию в эту временную точку. Более того, увеличение коэффициента разведения (1:100, 1:200, 1:500 SUV:MLV об./об.) только ускоряет и увеличивает высвобождение лекарственного средства, так как при t=2 минуты разведение 1 к 100 демонстрирует высвобождение ~70%, разведение 1 к 200 ~80%, и для разведения 1 к 500 почти 90% всего количества лекарственного средства не было обнаружено нигде в SUV при t=2 минуты и, следовательно, было высвобождено в акцепторный компартмент.
Вывод
Был продемонстрирован быстрый профиль высвобождения Соединения А с высвобождением вплоть до 90% лекарственного вещества только через 2 минуты. Данные показывают, что липосомы физически не удерживают Соединение А, инкапсулированное в присутствии избытка липидов, но существует быстрое перераспределение Соединения А между всеми липидами, присутствующими во время инкубации. В условиях in vivo предполагают сопоставимое поведение. Препарат обеспечивает чрезвычайно благоприятный баланс эффективной солюбилизации Соединения А, с желательным быстрым профилем высвобождения Соединения А.
ПРИМЕР 8: Композиция органического концентрата, содержащая Соединение А и PEG300, вариант 1


Композиция может быть приготовлена, используя способ, аналогичный описанному в Примере 1, или используя способы, хорошо известные квалифицированному специалисту в данной области техники.
ПРИМЕР 9: Композиция органического концентрата, содержащая Соединение А и PEG300, вариант 2

Композиция может быть приготовлена, используя способ, аналогичный описанному в Примере 1, или используя способы, хорошо известные квалифицированному специалисту в данной области техники.
ПРИМЕР 10: Композиция органического концентрата, содержащая Соединение А и PEG300, вариант 3
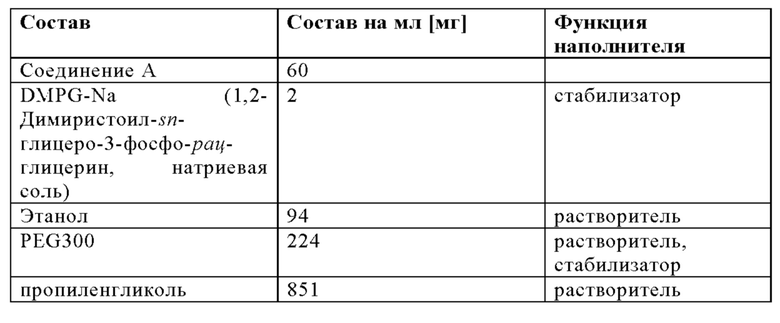
Композиция может быть приготовлена, используя способ, аналогичный описанному в Примере 1, или используя способы, хорошо известные квалифицированному специалисту в данной области техники.
Сравнительный Пример 11: Органический концентрат, содержащий Соединение А, без стабилизатора от гелеобразования

Композиция может быть приготовлена, используя способ, аналогичный описанному в Примере 1, или используя способы, хорошо известные квалифицированному специалисту в данной области техники.
ПРИМЕР 12: Исследование физической стабильности органических концентратов из Примеров 1, 8-10
Исследование физической стабильности осуществляли с помощью двух раздельных методов: а) устойчивость к затравливанию и б) диагностическая стабильность при интенсивном перемешивании при 250 об./мин. при 2°С.
а. Эксперимент затравливания осуществляли путем добавления ~ 1 мкл желированного препарата (Соединение А: 60 мг/мл, DMPG: 4,0 мг/мл, пропиленгликоль 851 мг/мл, этанол: 94 мг/мл: этот раствор застывал при хранении при перемешивании при 2°С) в прозрачные концентраты из Примеров 1, 8 и 9. Образцы перемешивали вихревым способом в течение 10 секунд и хранили в холодильнике в течение ночи при 2-8°С. Примеры 1, 8 и 9 не проявляли гелеобразования Соединения А при затравливании.
б. Исследования диагностической стабильности в условиях перемешивания осуществляли путем помещения 2 мл каждого органического концентрата в стеклянную пробирку и гомогенного перемешивания при 250 об./мин. при 2°С. Как показано в Таблице ниже, гелеобразования не происходило для препаратов, описанных в Примерах 1, 8-10, в течение 10 дней. Через 13 дней композиция из Примера 1 затвердевала, тогда как препараты, описанные в Примерах 8-10, оставались стабильными, то есть прозрачными растворами.

Благоприятно, препараты, содержащие PEG300, устойчивые к гелеобразованию в эксперименте перемешивания, описанном выше, в сочетании с резистентностью к затравливанию. Гелеобразованием Примера 1 можно управлять путем выбора подходящей температуры хранения, например, путем хранения при комнатной температуре.
ПРИМЕР 13: Липосомальная загрузка, используя концентрат композиции из Примеров 1,8-10
Загрузку липосом тестировали при шкале 23,7 мл, используя такой же самый подход, как и описанный в Примере 3А.
Органический концентрат из Примеров 8-10 проявлял сопоставимые характеристики загрузки липосом по сравнению с Примером 1. Примеры 8-10 приводили к получению липосомальных препаратов с сопоставимой мутностью по сравнению с препаратами, полученными из Примера 1.


Для облегчения липосомальной загрузки Примера 8 и Примера 9, исследовали загрузку с применением 19 G игл и сравнивали с иглой 21 G. Применение 19 G иглы приводило к незначительно меньшему размеру частиц и незначительно меньшей мутности по сравнению с инъекцией с применением 21 G иглы.
ПРИМЕР 14: Физическая стабильность препаратов органических концентратов плацебо
Препараты плацебо (без Соединения А) тестировали при -20°С; экстремальное условие, которое может ускорять возможное осаждение компонентов препарата. Физическую стабильность органических концентратов плацебо, описанных ниже, исследовали вплоть до 17 дней.
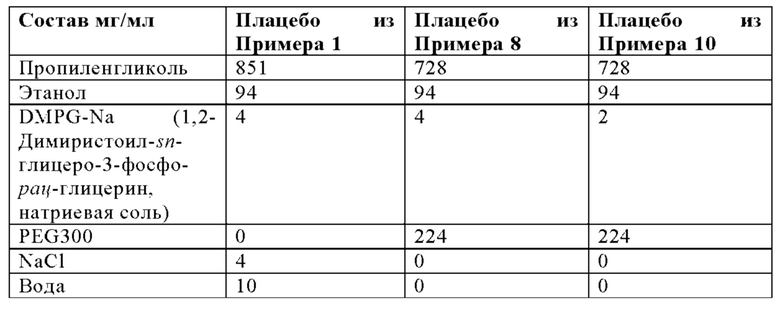
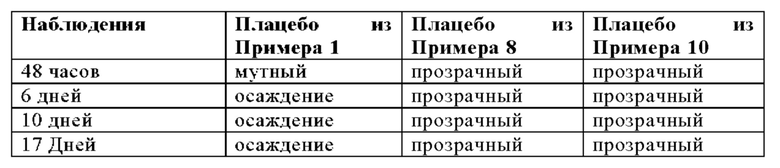
Осаждение наблюдали в композиции плацебо из Примера 1. Композиция плацебо из Примеров 8 и 10 оставалась прозрачной и не наблюдали осаждение во время осуществления эксперимента.
ПРИМЕР 15: Химическая стабильность активных органических концентратов из Примеров 1, 8 и 9
Тестировали химическую стабильность активного соединения в препаратах из Примеров 1, 8 и 9, в особенности для исследования уровней примесей, возникающих вследствие окислительного разложения, и образования нежелательного атропизомера.
Идентификация, исследование и продукты разложения с помощью ВЭЖХ



Продукты окислительного разложения были низкими для всех вариантов препаратов, указанных выше. Обнаруженное повышение нежелательного атропизомера было связано с температурой и временем, но не изменялось для тестируемых вариантов препаратов.
При визуальном контроле не обнаруживали изменения цвета концентрата в любых тестируемых препаратах, указанных выше.
| название | год | авторы | номер документа |
|---|---|---|---|
| ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ ДЛЯ ПРИМЕНЕНИЯ ПРИ ЛЕЧЕНИИ БОЛИ | 2020 |
|
RU2812902C2 |
| АНЕСТЕТИЧЕСКИЕ КОМПОЗИЦИИ С ЗАМЕДЛЕННЫМ ВЫСВОБОЖДЕНИЕМ И СПОСОБЫ ИХ ПОЛУЧЕНИЯ | 2019 |
|
RU2820649C2 |
| АНЕСТЕТИЧЕСКИЕ КОМПОЗИЦИИ С ЗАМЕДЛЕННЫМ ВЫСВОБОЖДЕНИЕМ И СПОСОБЫ ИХ ПОЛУЧЕНИЯ | 2018 |
|
RU2791481C2 |
| КОМПОЗИЦИИ НА ОСНОВЕ МЕЛАТОНИНА ДЛЯ ПАРЕНТЕРАЛЬНОГО ВВЕДЕНИЯ | 2015 |
|
RU2685698C2 |
| КОНТРАСТИРУЮЩИЙ АГЕНТ, СОДЕРЖАЩИЙ ПОЛУФТОРОУГЛЕВОДОРОДНОЕ ВЕЩЕСТВО | 2014 |
|
RU2672588C2 |
| ФОСФОЛИПИДНЫЙ ДЕПО-ПРЕПАРАТ | 2010 |
|
RU2595865C2 |
| ПРИМЕНЕНИЕ СТАБИЛЬНЫХ ПРИ ХРАНЕНИИ ВЯЗКИХ ДЕПО-ФОСФОЛИПИДОВ ДЛЯ ЛЕЧЕНИЯ РАН | 2011 |
|
RU2578437C2 |
| ЛИОФИЛИЗИРОВАННЫЙ ПРОДУКТ И СУСПЕНЗИЯ НАПОЛНЕННЫХ ГАЗОМ МИКРОВЕЗИКУЛ | 2020 |
|
RU2827409C2 |
| СПОСОБЫ УМЕНЬШЕНИЯ ОСЛОЖНЕНИЙ ОТ ВНУТРИСУСТАВНОГО СТЕРОИДА | 2019 |
|
RU2806023C2 |
| ЛИПОСОМАЛЬНАЯ КОМПОЗИЦИЯ ДЛЯ ИСПОЛЬЗОВАНИЯ В ПЕРИТОНЕАЛЬНОМ ДИАЛИЗЕ | 2013 |
|
RU2609860C2 |
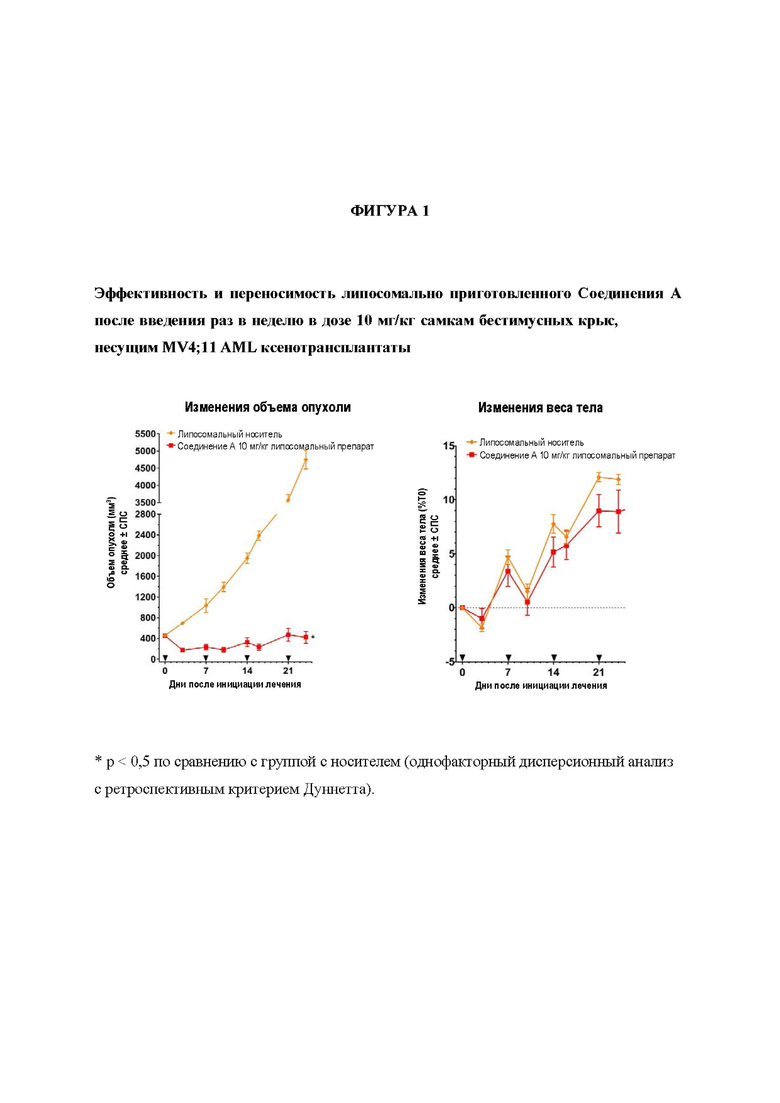
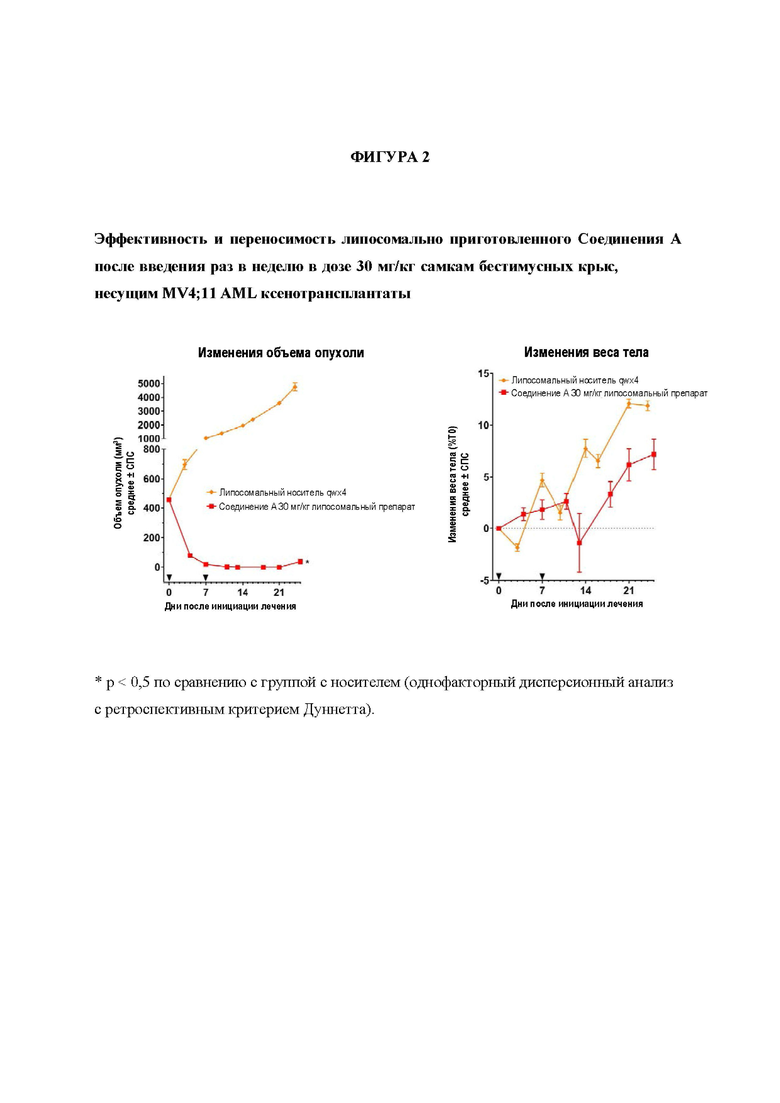
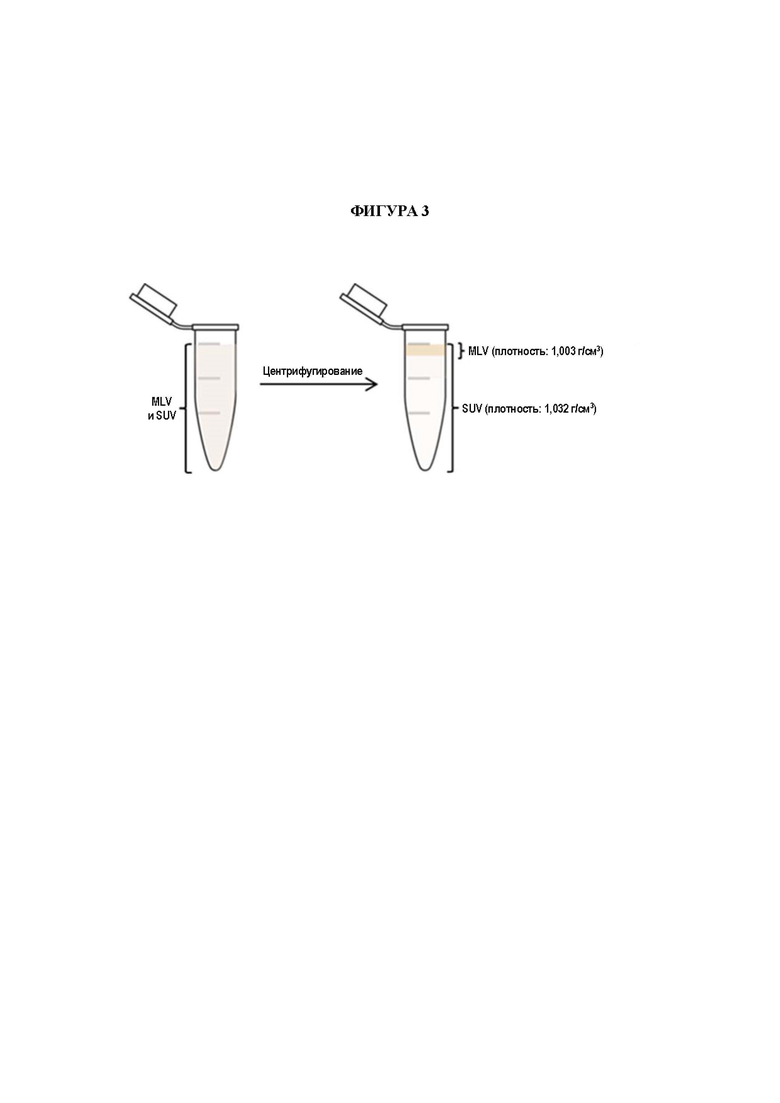
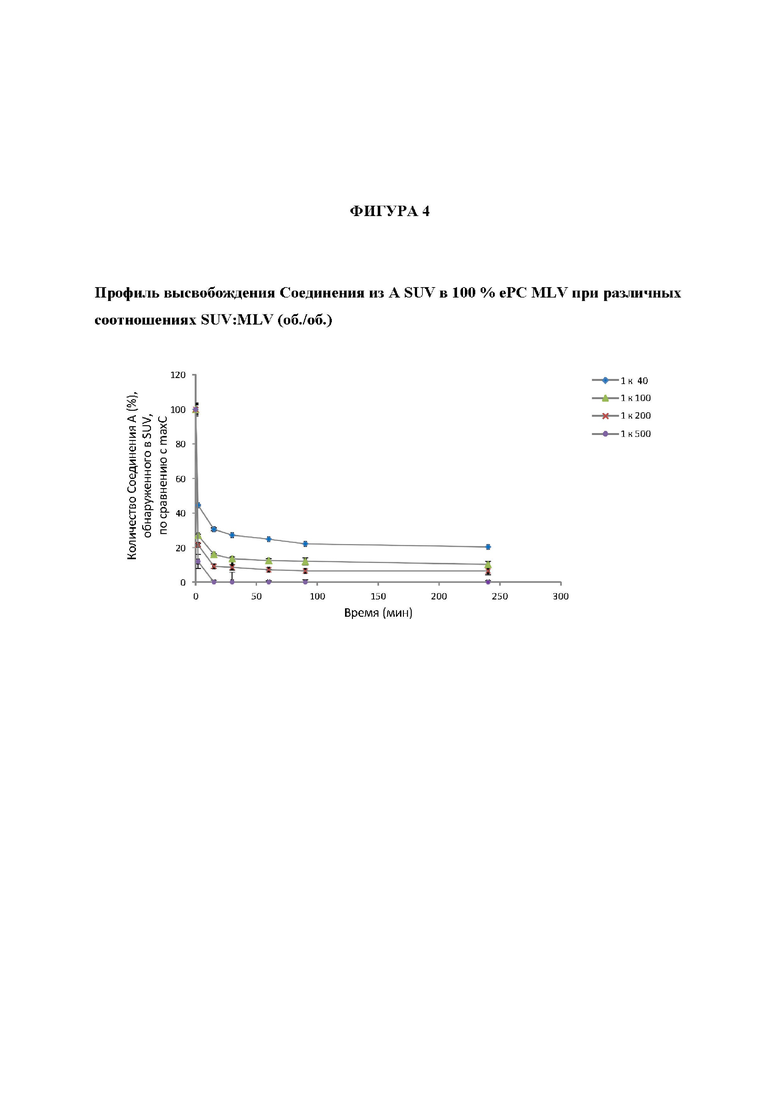
Изобретение относится к фармацевтической липосомальной композиции, содержащей 2-{[5-{3-хлор-2-метил-4-[2-(4-метилпиперазин-1-ил)этокси]фенил}-6-(4-фторфенил)тиено[2,3-d]пиримидин-4-ил]окси}-3-(2-{[2-(2-метоксифенил)пиримидин-4-ил]метокси}фенил)пропановую кислоту (Соединение А) или ее фармацевтически приемлемую соль. Более конкретно, изобретение относится к липосомальному носителю, композиции органического концентрата, содержащей Соединение А, и фармацевтической композиции для парентерального введения, содержащей липосомы и Соединение А. Кроме того, изобретение относится к применению таких композиций для лечения злокачественного новообразования. 7 н. и 20 з.п. ф-лы, 4 ил., 18 табл., 15 пр.
1. Композиция органического концентрата для приготовления фармацевтической композиции для парентерального введения, содержащая:
а) Соединение А, которое представляет собой 2-{[5-{3-хлор-2-метил-4-[2-(4-метилпиперазин-1-ил)этокси]фенил}-6-(4-фторфенил)тиено[2,3-d]пиримидин-4-ил]окси}-3-(2-{[2-(2-метоксифенил)пиримидин-4-ил]метокси}фенил)пропановую кислоту или ее фармацевтически приемлемую соль,
б) отрицательно заряженный или полярный фосфолипидный стабилизатор, где этот отрицательно заряженный или полярный фосфолипидный стабилизатор выбирают из натриевой или аммониевой соли DMPG (1,2-Димиристоил-sn-глицеро-3-фосфо-рац-глицерин или димиристоил фосфатидилглицерин), натриевой соли POPG (1-Пальмитоил-2-олеоил-sn-глицеро-3[Фосфо-рац-(1-глицерин)]), натриевой соли DOPS (1,2-Диолеоил-sn-глицеро-3-фосфосерин), натриевой соли DOPG (1,2-Диолеоил-sn-глицеро-3[Фосфо-рац-(1-глицерин)]), натриевой или аммониевой соли DPPG (1,2-Дипальмитоил-sn-глицеро-3[Фосфо-рац-(1-глицерин)]), натриевой или аммониевой соли DSPG (1,2-Дистеароил-sn-глицеро-3[Фосфо-рац-(1-глицерин)]), натриевой соли соевой фосфатидной кислоты (РА), натриевой соли яичной фосфатидной кислоты (РА), натриевой соли соевого фосфатидилсерина (PS), натриевой соли яичного фосфатидилглицерина (PG), натриевой соли соевого фосфатидилглицерина (PG), натриевой соли фосфатидилинозитола (PI), яичного лецитина (например, Lipoid Е 80 S), соевого лецитина (например, Lipoid S 75), и олеата натрия, и
в) стабилизатор от гелеобразования, где этот стабилизатор от гелеобразования представляет собой полимер, выбранный из PEG300 и PEG400, или электролит, выбранный из хлорида натрия, хлорида калия, фосфата натрия двух- и одноосновного и сульфата аммония.
2. Композиция органического концентрата по п. 1, содержащая Соединение А, которое представляет собой (2R)-2-{[(5Sa)-5-{3-хлор-2-метил-4-[2-(4-метилпиперазин-1-ил)этокси]фенил}-6-(4-фторфенил)тиено[2,3-d]пиримидин-4-ил]окси}-3-(2-{[2-(2-метоксифенил)пиримидин-4-ил]метокси}фенил)пропановую кислоту или ее фармацевтически приемлемую соль.
3. Композиция органического концентрата по п. 1 или 2, где Соединение А представляет собой свободную молекулу.
4. Композиция органического концентрата по любому из пп. 1-3, где отрицательно заряженный или полярный фосфолипидный стабилизатор представляет собой 1,2-Димиристоил-sn-глицеро-3-фосфо-рац-глицерин, натриевую или аммониевую соль, яичный лецитин (например, Lipoid Е 80 S), соевый лецитин (например, Lipoid S 75), предпочтительно 1,2-Димиристоил-sn-глицеро-3-фосфо-рац-глицерин, натриевую соль.
5. Композиция органического концентрата по любому из пп. 1-4, где стабилизатор от гелеобразования представляет собой электролит, выбранный из хлорида натрия, хлорида калия, фосфата натрия двух- и одноосновного, и сульфата аммония.
6. Композиция органического концентрата по любому из пп. 1-4, где стабилизатор от гелеобразования представляет собой хлорид натрия.
7. Композиция органического концентрата по любому из пп. 1-4, где стабилизатор от гелеобразования представляет собой полимер, выбранный из PEG300 и PEG400, предпочтительно PEG300.
8. Композиция органического концентрата по любому из пп. 1-7, дополнительно содержащая растворитель.
9. Композиция органического концентрата по любому из пп. 1-8, дополнительно содержащая растворитель, выбранный из пропиленгликоля и этанола, или в особенности содержащая оба компонента, пропиленгликоль и этанол.
10. Фармацевтическая композиция, полученная в результате смешивания композиции органического концентрата по любому из пп. 1-9 и липосомального носителя, где указанный липосомальный носитель содержит фосфолипид и агент, корригирующий тоничность.
11. Фармацевтическая композиция по п. 10, где фосфолипид выбирают из яичного лецитина, соевого лецитина или синтетических фосфолипидов.
12. Фармацевтическая композиция по п. 10 или 11, где фосфолипид представлен формулой
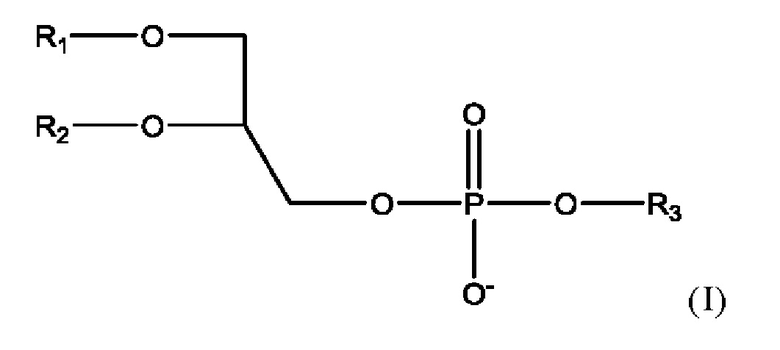
где
• R1 представляет собой С10-С24ацил;
• R2 представляет собой С10-С24ацил;
• R3 представляет собой водород, 2-триметиламино-1-этил, 2-амино-1-этил, С1-С4алкил, С1-С5алкил, замещенный карбокси, С2-С5алкил, замещенный карбокси и гидрокси, С2-С5алкил, замещенный карбокси и амино, инозитоловую группу или глицериловую группу;
или соль такого соединения.
13. Фармацевтическая композиция по любому из пп. 10, 11, где фосфолипид выбирают из яичного лецитина, соевого лецитина, РОРС (пальмитоил олеоил фосфатидилхолин), DOPC (1,2-Диолеоил-sn-глицеро-3-фосфохолин) и DMPC (1,2-Димиристоил-sn-глицеро-3-фосфохолин), в частности РОРС (пальмитоил олеоил фосфатидилхолин), DOPC (1,2-Диолеоил-sn-глицеро-3-фосфохолин) и DMPC (1,2-Димиристоил-sn-глицеро-3-фосфохолин), в особенности РОРС.
14. Фармацевтическая композиция по любому из пп. 10, 11 или 13, где фосфолипид выбирают из яичного лецитина или соевого лецитина, содержащего по меньшей мере 70% фосфатидилхолина, и в особенности выбирают из Lipoid Е 80 S.
15. Фармацевтическая композиция по любому из пп. 10-14, где агент, корригирующий тоничность, выбирают из декстрозы, глюкозы, маннита, сахарозы, лактозы, трегалозы, глицерина и NaCl, в особенности сахарозы или глицерина.
16. Липосомальная фармацевтическая композиция, содержащая липосомальный носитель, который содержит фосфолипид и агент, корригирующий тоничность, и дополнительно:
а) Соединение А, которое представляет собой 2-{[5-{3-хлор-2-метил-4-[2-(4-метилпиперазин-1-ил)этокси]фенил}-6-(4-фторфенил)тиено[2,3-d]пиримидин-4-ил]окси}-3-(2-{[2-(2-метоксифенил)пиримидин-4-ил]метокси}фенил)пропановую кислоту или ее фармацевтически приемлемую соль;
б) стабилизатор от гелеобразования, где этот стабилизатор от гелеобразования представляет собой полимер, выбранный из PEG300 и PEG400, или электролит, выбранный из хлорида натрия, хлорида калия, фосфата натрия двух- и одноосновного и сульфата аммония; и
в) отрицательно заряженный или полярный фосфолипидный стабилизатор, где этот отрицательно заряженный или полярный фосфолипидный стабилизатор выбирают из натриевой или аммониевой соли DMPG (1,2-Димиристоил-sn-глицеро-3-фосфо-рац-глицерин или димиристоил фосфатидилглицерин), натриевой соли POPG (1-Пальмитоил-2-олеоил-sn-глицеро-3[Фосфо-рац-(1-глицерин)]), натриевой соли DOPS (1,2-Диолеоил-sn-глицеро-3-фосфосерин), натриевой соли DOPG (1,2-Диолеоил-sn-глицеро-3[Фосфо-рац-(1-глицерин)]), натриевой или аммониевой соли DPPG (1,2-Дипальмитоил-sn-глицеро-3[Фосфо-рац-(1-глицерин)]), натриевой или аммониевой соли DSPG (1,2-Дистеароил-sn-глицеро-3[Фосфо-рац-(1-глицерин)]), натриевой соли соевой фосфатидной кислоты (РА), натриевой соли яичной фосфатидной кислоты (РА), натриевой соли соевого фосфатидилсерина (PS), натриевой соли яичного фосфатидилглицерина (PG), натриевой соли соевого фосфатидилглицерина (PG), натриевой соли фосфатидилинозитола (PI), яичного лецитина (например, Lipoid Е 80 S), соевого лецитина (например, Lipoid S 75), и олеата натрия.
17. Липосомальная фармацевтическая композиция по п. 16, содержащая:
а) Соединение А, как описано в п. 2 или 3, и
б) стабилизатор от гелеобразования, как описано в любом из пп. 5, 6 или 7.
18. Липосомальная фармацевтическая композиция по п. 16, где указанный отрицательно заряженный или полярный фосфолипидный стабилизатор является таким, как описано в п. 4.
19. Липосомальная фармацевтическая композиция по любому из пп. 16-18, дополнительно содержащая растворитель.
20. Липосомальная фармацевтическая композиция по п. 19, где указанный растворитель выбирают из пропиленгликоля и этанола, или, в особенности, содержащая оба компонента, пропиленгликоль и этанол.
21. Липосомальная фармацевтическая композиция по любому из пп. 16-20, содержащая фосфолипид, как описано в любом из пп. 11-14.
22. Липосомальная фармацевтическая композиция по любому из пп. 16-21, где агент, корригирующий тоничность, выбирают из декстрозы, глюкозы, маннита, сахарозы, лактозы, трегалозы, глицерина и NaCl.
23. Применение фармацевтической композиции по любому из пп. 10-22 в качестве лекарственного средства для лечения злокачественного новообразования.
24. Применение фармацевтической композиции по п. 23, где злокачественное новообразование выбирают из злокачественных новообразований мочевого пузыря, головного мозга, молочной железы и матки, хронических лимфоидных лейкозов, злокачественного новообразования толстой кишки, пищевода и печени, лимфобластных лейкозов, острого миелоидного лейкоза, лимфом, например неходжкинской В-клеточной лимфомы и диффузной В-крупноклеточной лимфомы, меланом, злокачественных болезней крови, например миелодиспластического синдрома, миелом, например множественной миеломы, рака яичников, немелкоклеточного рака легкого, рака предстательной железы, рака поджелудочной железы и мелкоклеточного рака легкого.
25. Применение композиции органического концентрата по любому из пп. 1-9 для приготовления лекарственного средства для лечения злокачественного новообразования.
26. Комбинация для лечения злокачественного новообразования, содержащая:
а) фармацевтическую композицию по любому из пп. 10-22, и
б) один или несколько терапевтически активных средств для одновременного, последовательного или раздельного применения.
27. Набор для лечения злокачественного новообразования, содержащий:
а) липосомальный носитель, и
б) композицию органического концентрата по любому из пп. 1-9.
| EA 201401292 A2, 2015.06.30 | |||
| ЛИПОСОМАЛЬНАЯ КОМПОЗИЦИЯ | 2010 |
|
RU2476216C1 |
| Пьезоэлектрический микрофон | 1926 |
|
SU5923A1 |
| SHABBITS J A et al., "Development of an in vitro drug release assay that accurately predicts in vivo drug retention for liposome-based delivery systems", JOURNAL OF CONTROLLED RELEASE, ELSEVIER, AMSTERDAM, NL, 84 (3), 05 December 2002, page 161-170, найдено онлайн, | |||

