Изобретение относится к ингибиторам ретровирусных протеаз и, более конкретно, относится к новым соединениям и к композиции и способу ингибирования ретровирусных протеаз. Данное изобретение, в частности, относится к сульфонамидсодержащим гидроксиэтиламиновым соединениям, ингибиторам протеаз, к композиции и способу ингибирования ретровирусных протеаз таких, как протеаза вируса иммунодефицита человека (ВИЧ), и к лечению ретровирусных инфекций, например ВИЧ-инфекции. Настоящее изобретение относится также к способам получения таких соединений, так же как и к промежуточным соединениям, полезным в таких процессах.
Во время цикла репликации ретровирусов продукты gag и gag-pol гена транслируются в виде протеинов. Эти протеины или белки впоследствии перерабатываются с помощью вирусно кодированной протеазы (или протеиназы), давая вирусные ферменты и структурные протеины вирусного ядра (сердцевины). Наиболее обычно gag предшествующие протеины перерабатываются в сердцевинные белки, а pol предшествующие белки перерабатываются в вирусные ферменты, например обратную транскриптазу и ретровирусную протеазу. Было показано, что для сборки инфекционных виронов необходима правильная переработка предшествующих белков с помощью ретровирусной протеазы. Например, было показано, что мутации сдвига рамки генетического кода в протеазной области pol гена ВИЧ предотвращает переработку gag предшествующего белка. Было показано, что благодаря сайт-направленному мутагенезу остатка аспарагиновой кислоты в ВИЧ протеазе эта переработка gag предшествующего белка предотвращается. Таким образом, делались попытки ингибировать вирусную репликацию путем ингибирования действия ретровирусных протеаз.
Ингибирование ретровирусных протеаз может вовлекать миметики переходного состояния, посредством чего ретровирусная протеаза подвергается воздействию соединения миметика, которое связывается с ферментом конкурентно с gag и gag-pol белками, тем самым ингибируя репликацию структурных белков, и что более важно, самой ретровирусной протеазы. Таким образом протеазы ретровирусной репликации могут эффективно ингибироваться.
Предлагалось несколько классов соединений, в частности, для ингибирования протеаз, таких, как для ингибирования ВИЧ-протеазы. Такие соединения включают гидроксиэтиламиновые изостеры и восстановленные амидные изостеры. См., например EP 0346847; EP 0342541; Roberts и др., "Rational Desing of Peptide-Baset Proteinase Inhibitors", Science, 248, 358 (1990); и Erickson и др. , "Desing Activity, and 2.8. 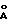 Crystal Structure of a C2 Symmetric Inhibitor Complexed to HIV-I Protease", Science, 249, 527 (1990).
Crystal Structure of a C2 Symmetric Inhibitor Complexed to HIV-I Protease", Science, 249, 527 (1990).
Известно, что несколько классов миметических соединений полезны в качестве ингибиторов протеолитического фермента ренина. Смотри, например, патент США 4599198, патент Объед. Корол-ва 2184730; GB 2209752; EP 0264795; GB 2200115 и США SIR H725. Из них GB 2200115, GB 2209752, EP 0264795, США SIR H725 и США 4599198 раскрывают мочевиносодержащие гидроксиэтиламиновые ингибиторы ренина, GB 2200115 раскрывают также сульфамоилсодержащие гидроксиэтиламиновые ингибиторы ренина, а EP 0264795 раскрывают некоторые сульфонамидсодержащие гидроксиэтиламиновые ингибиторы ренина. Однако, известно, что, хотя рениновые и ВИЧ-протеазы обе классифицируются как аспартил-протеазы, обычно нельзя предсказать, что соединения, которые являются эффективными ингибиторами ренина, будут эффективными ингибиторами ВИЧ-протеазы.
Настоящее изобретение направлено на соединение и композиции, ингибирующие вирусы. Более конкретно, настоящее изобретение направлено на соединение и композиции, ингибирующие ретровирусные протеазы, к способу ингибирования ретровирусных протеаз, к процессам получения соединений и к промежуточным соединениям, полезным в таких процессах. Соединения, о которых идет речь, характеризуются как сульфонилалканоиламино-гидроксиэтиламино-сульфонамидные ингибиторные соединения.
В соответствии с настоящим изобретением представляются соединения, ингибирующие ретровирусные протеазы, формулы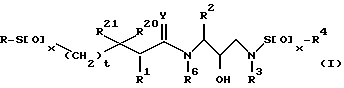
или их фармацевтически приемлемые соли, пролекарства или сложные эфиры,
в которой R - водород, алкил, алкенил, алкинил, гидроксиалкил, алкоксиалкил, циклоалкил, циклоалкилалкил, гетероциклоалкил, гетероарил, гетероциклоалкилалкил, арил, аралкил, гетероаралкил, аминокарбонилалкил, аминоалкилкарбонилалкил, аминоалкил, алкилкарбонилалкил, арилоксиалкилкарбонилалкил, аралкоксикарбонилалкил радикалы и моно- и ди-замещенные аминокарбонилалкильные аминоалкилкарбонилалкильные и аминоалкильные радикалы, в которых указанные заместители выбраны из алкила, арила, аралкила, циклоалкила, циклоалкила, гетероарила, гетероаралкила, гетероциклоалкила и гетероциклоалкилалкильного радикалов, или в случае дизамещенного радикала, указанные заместители вместе с атомов азота, к которому они присоединены, образуют гетероциклоалкильный или гетероарильный радикал;
каждый x независимо представляют 0, 1 или 2;
t = 0 или 1;
R1, R20 и R21 независимо - водород, -CH2SO2NH2, -CH2CO2CH3, -CO2CH3, -CONH2, -CH2C(O)NHCH3, -C(CH3)2(SH), -C(CH3)2(SCH3), -C(CH3)2(S[O]CH3),
-C(CH3)2(S[O]2CH3) алкил, галоидалкил, алкенил, алкинил и циклалкильный радикалы и аминокислотные боковые цепи выбраны из аспарагина, S-метилцистеина и их сульфоксидных (SO) и сульфоновых (SO2) производных, изолейцина, алло-изолейцина, аланина, лейцина, трет-лейцина, фенилаланина, орнитина, гистидина, норлейцина, глютамина, треонина, глицина, алло-треонина, серина, O-алкил-серина, аспарагиновой кислоты, бетациано-аланина и валина;
R2 - алкил, арил, циклоалкил, циклоалкилалкил и аралкильный радикалы, причем эти радикалы необязательно замещены группой, выбранной из -NO2, CH, -C≡N, CF3, -OR9, -SR9, галоидалкила и галогена и алкильных радикалов, в которых R9 - водород или алкильный радикал;
R3 - водород, алкил, галоидалкил, алкенил, алкинил, гидроксиалкил, алкоксиалкил, циклоалкил, циклоалкилалкил, гетероциклоалкил, гетероарил, гетероциклоалкил-алкил, арил, аралкил, гетероаралкил, аминоалкил и моно и ди-замещенные аминоалкильные радикалы, в которых указанные заместители выбраны из алкила, арила, аралкила, циклоалкила, циклоалкилалкила, гетероарила, гетероаралкила, гетероциклоалкила и гетероциклоалкилалкильного радикалов, или в случае, ди-замещенных аминоалкильных радикалов, указанные заместители наряду с атомом азота, к которому они присоединены, образуют гетероциклоалкильный или гетероарильный радикал;
R4 - радикалы, определенные для R3, исключая водород;
Y - 0, S или NR15, где R15 - водород, и радикалы, определенные для R3; и
R6 - водород или алкильные радикалы.
Предпочтительным классом ретровирусных ингибирующих соединений настоящего изобретения являются соединения, представленные формулой: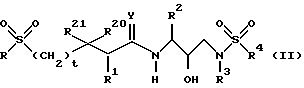
или их фармацевтически приемлемые соли, пролекарства или сложные эфиры, предпочтительно, в которых абсолютная стереохимия у гидрокси группы обозначается как (R);
R - алкил, алкенил, алкинил, циклоалкил, гидроксиалкил, циклоалкилалкил, гетероциклоалкил, гетероциклоалкилалкил, алкоксиалкил, арил, гетероарил, аралкил, гетероалкил, гетероаралкил, аминокарбонилалкил, аминоалкилкарбонилалкил, алкилкарбонилалкил, арилоксиалкилкарбонил и аралкоксикарбонилалкильный радикалы;
R1, R20 и R21 независимо - водород, -CH2SO2NH2, -CH2CO2CH3, -CO2CH3, -CONH2, -CH2C(O)NHCH3, -C(CH3)2(SCH3), -C(CH3)2(S[O] CH3), -C(CH3)2(S[O] 2CH3), алкил, галоидалкил, алкенил, алкинил и циклоалкильный радикалы, и аминокислотные боковые цепи, выбранные из аспарагина, S-метил-цистеина, и их сульфоксидных (SO) и сульфоновых (SO2) производных, изолейцина, алло-изолейцина, аланина, лейцина, трет-лейцина, фенилаланина, орнитина, гистидина, норлейцина, глютамина, треонина, глицина, алло-треонина, серина, O-метил-серина, аспарагиновой кислоты, бета-циано-аланина и валиновых боковых цепей;
R2-алкил, арил, циклоалкил, циклоалкилалкил, и аралкильный радикалы, эти радикалы являются необязательно замещенными группой, выбранной из алкильного и галогенового радикалов, NO2, CN, -C≡N, CF3, OR9 и SR9, где R9 - водород, или алкил, или галогеновый радикал;
R3 - алкил, галоидалкил, алкенил, гидроксиалкил, алкоксиалкил, циклоалкил, циклоалкилалкил, гетероциклоалкил, гетероциклоалкилалкил, арил, гетероарил, аралкил или гетероаралкил;
R4 - радикалы, определенные для R3, за исключением водорода;
t=0 или 1;
Y - O, S или NR15, где R15 - водород или радикал, определенный для R3; предпочтительно Y представляет O.
Предпочтительным классом соединений из соединений формулы I являются соединения, представленные формулой: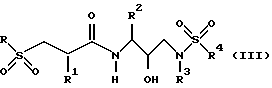 или их фармацевтически приемлемые соли, пролекарства или сложные эфиры, в которых R, R1, R2, R3 и R4 имеют значения, определенные для формулы (II).
или их фармацевтически приемлемые соли, пролекарства или сложные эфиры, в которых R, R1, R2, R3 и R4 имеют значения, определенные для формулы (II).
В том смысле, как он используется здесь, термин "алкил", один или в сочетании, означает алкильный радикал с прямой или разветвленной цепью, содержащий от 1 до примерно 10, предпочтительно 1 - 8 атомов углерода. Примеры таких радикалов включают метил, этил, н-пропил, изопропил, н-бутил, изобутил, втор-бутил, трет-бутил, пентил, изоамил, гексил, октил и аналогичные. Термин "алкенил" один или в сочетании, означает углеводородный радикал с прямой или разветвленной цепью, имеющий одну или более двойных связей и содержащий от 2 до примерно 18 атомов углерода, предпочтительно от 2 до примерно 8 атомов углерода. Примеры подходящих алкенильных радикалов включают этенил, пропенил, 1,4-бутадиенил и аналогичные. Термин "алкинил", один или в сочетании, означает углеводородный радикал с прямой цепью, имеющий одну или более тройных связей и содержащий от 2 до примерно 10 атомов углерода. Примеры алкинильных радикалов включают этинил, пропинил, пропаргил и аналогичные. Термин "алкокси", один или в сочетании, обозначает алкиловый эфирный радикал, в котором термин "алкиловый" имеет значения, указанные выше. Примеры подходящих алкиловых эфирных радикалов включают метокси, этокси, н-пропокси, изопропокси, н-бутокси, изобутокси, втор-бутокси, трет-бутокси и аналогичные. Термин "циклоалкил", один или в сочетании, обозначает насыщенный или частично насыщенный моноциклический, бициклический или трициклический алкильный радикал, в котором каждый циклический фрагмент содержит от примерно 3 до примерно 8 атомов углерода. Термин "циклоалкилалкил" означает алкильный радикал, определенный выше, который замещен циклоалкильным радикалом, содержащим примерно 3 - 8, предпочтительно 3 - 6, атомов углерода. Примеры таких циклоалкильных радикалов включают циклопропил, циклобутил, циклопентил, циклогексил и аналогичные. Термин "арил", один или в сочетании, означает фенильный или нафтильный радикал, который необязательно несет один или более заместителей, выбранных из алкила, алкокси, галогена, гидрокси, амино, нитро, циано, галоидалкила и аналогичных, такой как фенил, n-толил, 4-метоксифенил, 4-(трет-бутокси)фенил, 4-фторфенил, 4-хлорфенил, 4-гидроксифенил, 1-нафтил, 2-нафтил и аналогичные. Термин "аралкил", один или в сочетании, означает алкильный радикал, определенный выше, в котором один атом водорода замещен арильным радикалом, определенным выше, такой, как бензил, 2-фенилэтил и аналогичные. Термин "аралкоксикарбонил", один или в сочетании, означает радикал формулы -C(O)-C-аралкил, в котором термин "аралкил" имеет значения, данные выше. Примером аралкоксикарбонильного радикала является бензилоксикарбонил. Термин "арилокси" означает радикал формулы арил-O-, в которой термин "арил" имеет значения, данные выше. Термин "алканоил", один или в сочетании, означает ацильный радикал, происходящий из алканкарбоновой кислоты, примеры которого включают ацетил, пропионил, бутирил, валерил, 4-метилвалерил и аналогичные. Термин "циклоалкилкарбонил" означает ацильную группу, являющуюся производной моноциклической или мостиковой циклоалканкарбоновой кислоты, такую, как циклопропанкарбонил, циклогексанкарбонил, адамантанкарбонил и аналогичные, или бенз-сконденсированной моноциклической циклоалканкарбоновой кислоты, которая необязательно замещена группой, например, алканоиламино, такая, как 1,2,3,4-тетрагидро-2-нафтоил, 2-ацетамидо-1,2,3,4-тетрагидро-2-нафтоил. Термин "аралканоил" означает ацильный радикал, происходящий из арил-замещенной алканкарбоновой кислоты такой, как фенилацетил, 3-фенилпропионил (гидроксициннамоил), 4-фенил-бутирил, (2-нафтил)ацетил, 4-хлоргидроциннамоил, 4-аминогидроциннамоил, 4-метоксигидроциннамоил и аналогичные. Термин "ароил" означает ацильный радикал, происходящий из ароматической карбоновой кислоты. Примеры таких радикалов включают радикалы ароматических карбоновых кислот, необязательно замещенной бензойной или нафтойной кислоты такие, как бензоил, 4-хлорбензоил, 4-карбоксибензоил, 4-(бензил-оксикарбонил)бензоил, 1-нафтоил, 2-нафтоил, 6-карбокси-2-нафтоил, 6-(бензилоксикарбонил)-2-нафтоил, 3-бензилокси-2-нафтоил, 3-гидрокси-2-нафтоил, 3-(бензилоксиформамидо)-2-нафтоил и аналогичные. Гетероциклильная или гетероциклоалкильная часть гетероциклилкарбонила, гетероциклилоксикарбонила, гетероциклилалкоксикарбонила или гетероциклилалкильной группы или аналогичных, представляет насыщенный или частично ненасыщенный моноциклический, бициклический или трициклический гетероцикл, который содержит один или более гетеро-атомов, выбранных из азота, кислорода и серы, который является необязательно замещенным у одного или более атомов углерода галогеном, алкилом, алкокси, оксо и аналогичными, и/или вторичного атома азота (т.е. -NH-)алкилом, аралкоксикарбаноилом, алканоилом, фенилом или фенилалкилом, или у третичного атома азота (т. е. =N-) группой оксидо, и который присоединен через атом углерода. Гетероарильная часть или фрагмент гетероароильной, гетероарилоксикарбонильной или гетероаралкоксикарбонильной группы или аналогичных является ароматическим моноциклическим, бициклическим или трициклическим гетероциклом, который содержит гетеро-атомы и необязательно замещен, как определено выше в отношении определения гетероциклил. Примерами таких гетероциклильных и гетероарильных групп являются пирролидинил, пиперидинил, пиперазинил, морфолинил, тиаморфолинил, пирролил, имидазолил (например, имидазол-4-ил, 1-бензилоксикарбонилимидазол-4-ил, и др.), пиразолил, пиридил, пиразинил, пиримидинил, фурил, тиенил, триазолил, оксазолил, тиазолил, индолил (например, 2-индолил и др.), хинолинил (например, 2-хинолинил, 3-хинолинил, 1-оксидо-2-хинолинил и др.), изохинолинил (например, 1-изохинолинил, 3-изохинолинил и др.), тетрагидрохинолинил (например, 1,2,3,4-тетрагидро-2-хинолил и др.), 1,2,3,4-тетрагидроизохинолинил (например, 1,2,3,4-тетрагидро-1-оксо-изохинолинил и др.), хиноксадинил, β- карболинил, 2-бензофуранкарбонил, 1-, 2-, 4- или 5-бензимидазолил и аналогичные. Термин "циклоалкилалкоксикарбонил" означает ацильную группу, происходящую из циклоалкилалкоксикарбоновой кислоты формулы циклоалкилалкил-O-COOH, в которой циклоалкилалкил имеет значения, данные выше. Термин "арилоксиалканоил" означает ацильный радикал формулы арил-O-алканоил, в которой арил и алканоил имеют значения, приведенные выше. Термин "гетероциклилоксикарбонил" означает ацильную группу, происходящую из гетероциклил-O-COOH, где гетероциклил имеет значения, определенные выше. Термин "гетероциклилалканоил" представляет ацильный радикал, происходящий из гетероциклил-замещенной алканкарбоновой кислоты, в которой гетероциклил имеет значения, приведенные выше. Термин "гетероциклилалкоксикарбонил" означает ацильный радикал, происходящий из гетероциклил-замещенной алкан-O-COOH, где гетероциклил имеет значения, приведенные выше. Термин "гетероарилоксикарбонил" означает ацильный радикал, происходящий из карбоновой кислоты, представленной формулой гетероарил-O-COOH, в которой гетероарил имеет значения, приведенные выше. Термин "аминокарбонил", один или в сочетании, означает аминозамещенную карбонильную (карбамоильную) группу, происходящую из аминозамещенной карбоновой кислоты, в которой аминогруппа может быть первичной, вторичной или третичной аминогруппой, содержащей заместители, выбранные из водорода и алкильного, арильного, аралкильного, циклоалкильного, циклоалкилалкильного радикалов и аналогичных. Термин "аминоалканоил" означает ацильную группу, происходящую из аминозамещенной алканкарбоновой кислоты, в которой амино группа может быть первичной, вторичной или третичной аминогруппой, содержащей заместителя, выбранные из водорода, и алкильной, арильной, аралкильной, циклоалкильной, циклоалкилалкильной групп и аналогичных. Термин "галоген" означает фтор, хлор, бром или йод. Термин "уходящая или удаляемая группа" обычно относится к группам, легко заменяемым нуклеофилом, таким, как амин, тиол или спиртовый нуклеофил. Такие удаляемые группы хорошо известны в технике. Примеры таких удаляемых групп включают, но не ограничиваются ими, N-гидроксисукцинимид, N-гидроксибензотриазол, галогениды, трифлаты, тозилаты и аналогичные. Предпочтительные удаляемые группы указаны здесь в соответствующих разделах.
Процедуры получения соединений формулы I представлены ниже. Следует понимать, что показана общая процедура, которая относится к получению соединений, имеющих определенную стереохимию, абсолютная стереохимия у гидроксильной группы обозначается как (R). Однако, такие процедуры обычно применимы и к соответствующим соединениям противоположной конфигурации, например, когда стереохимия у гидроксильной группы является (S). В дополнение к сказанному, соединения, имеющие (R) стереохимию, могут использоваться для получения соединений, имеющих (S) стереохимию, и наоборот. Например, соединение, имеющее (R) стереохимию, может превращаться в соединение (S) стереохимии с использованием хорошо известных методов.
Получение соединений формулы I
Соединения II настоящего изобретения могут получаться с использованием следующей общей процедуры; N-защищенное хлоркетоновое производное аминокислоты формулы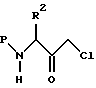
в которой P представляет аминозащищающую группу, и R2 имеет значения, определенные выше,
восстанавливается в соответствующий спирт с использованием соответствующего восстанавливающего агента. Подходящие аминозащищающие группы хорошо известны в технике и включают карбобензокси, бутирил, трет-бутоксикарбонил, ацетил, бензоил и аналогичные. Предпочтительной группой, защищающей аминогруппу, является карбобензокси. Предпочтительным N-защищенным хлоркетоном является N-бензилоксикарбонил-L-фенилаланин-хлорметилкетон. Предпочтительным восстанавливающим агентом является боргидрид натрия. Реакция восстановления проводится при температуре от -10oC до примерно 25oC, предпочтительно примерно при 0oC, в подходящей системе растворителя, такой, как, например, тетрагидрофуран и аналогичные. N-Защищенные хлоркетоны являются промышленно доступным, например, такими, как поставляются фирмой Bachem, Inc., Торранс, Калифорния. Альтернативно, хлоркетоны могут получаться с помощью процедуры, представленной в работе S. J. Fittkau, J. Prakt. Chem., 315, 1037 (1973), и впоследствии N-защищаются с использованием процедур, которые хорошо известны в технике.
Галоид-спирт может использоваться непосредственно, как описано ниже, или, предпочтительно, подвергается затем реакции, предпочтительно при комнатной температуре, с подходящим основанием в подходящей системе растворителя с получением N-защищенного аминоэпоксида формулы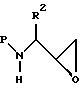
где P и R2 имеют значения, определенные выше.
Подходящие системы растворителей для получения аминоэпоксида включают этанол, метанол, изопропанол, тетрагидрофуран, диоксан и аналогичные, включая смеси их. Подходящие основания для получения эпоксида из восстановленного хлоркетона включают гидроокись калия, гидроокись натрия, трет-бутилат калия, DBU и аналогичные. Предпочтительным основанием является гидроокись калия.
Альтернативно защищенный аминоэпоксид может получаться исходя из L-аминокислоты, которая вводится в реакцию с подходящей аминозащищающей группой в подходящем растворителе, давая эфир аминозащищенной L-аминокислоты формулы: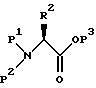
где P1 и P2 независимо - водород, бензильная и аминозащищающая группы (определенные выше в отношении P), при условии, что P1 и P2 оба не являются водородами; P3 представляет защитную группу карбоксила (такую, как метил, этил, третичный-бутил, бензил и аналогичные); и R2 имеет значения, определенные выше.
Сложный эфир аминозащищенной L-аминокислоты затем восстанавливается в соответствующий спирт. Например, эфир аминозащищенной L-аминокислоты может восстанавливаться диизобутилалюминийгидридом при -78oC в подходящем растворителе, таком, как толуол. Получающийся в результате спирт затем превращается, например, по способу окисления Сверна, в соответствующий альдегид формулы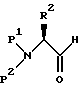
где P1, P2 и R2 имеют значения, определенные выше.
Так, раствор спирта в дихлорметане добавляется к охлажденному (от -78 до -68oC) раствору оксалилхлорида в дихлорметане и DMCO в дихлорметане и перемешивается в течение 35 минут.
Альдегид, получающийся в результате окисления Сверна, затем подвергается взаимодействию с галоидметиллитиевым реагентом, который генерируется на месте с помощью реакции алкиллитиевого или ариллитиевого соединения с дигалоидметаном, представленным формулой X1CH2X2, где X1 и X2 представляют независимо I, Br или C1. Например, раствор альдегида и хлориодметана в ТГФ охлаждается до -78oC, и добавляется раствор н-бутиллития в гексане. Получающийся продукт представляет смесь диастереомеров соответствующих аминозащищенных эпоксидов формул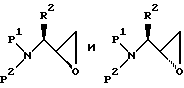
Диастереомеры могут разделяться, например, с помощью хроматографии, или, альтернативно, сразу подвергаться реакции на последующих стадиях, после чего диастереомерные продукты могут разделяться. Для соединений, имеющих (S) стереохимию, вместо L-аминокислоты может использоваться D-аминокислота.
Аминоэпоксид затем вводится в реакцию в подходящей системе растворителя с равным количеством или предпочтительно с избытком желаемого амина формулы
R3NH2,
где R3 представляет водород или имеет значения, определенные выше.
Реакция может проводиться в широком интервале температур примерно 10-100oC и предпочтительно, но не обязательно, проводится при температуре, при которой начинает дефлегмировать растворитель. Подходящие системы растворителей включают протонные, непротонные и диполярные апротонные органические растворители такие, как, например, спирт такой, как метанол, этанол, изопропанол и аналогичные, простые эфиры такие, как тетрагидрофуран, диоксан и аналогичные, и толуол, N,N-диметилформамид, диметилсульфоксид, и смеси их. Предпочтительным растворителем является изопропанол. Примеры аминов, соответствующих формуле R3NH2, включают бензиламин, изобутиламин, н-бутиламин, изопентиламин, изоамиламин, циклогексанметиламин, нафталинметиламин и аналогичные. Получающийся продукт представляет 3-(N-защищенный амино)-3-(R2)-1-(NHR3)-пропан-2-ольное производное (называемое здесь ниже аминоспиртом), представленное формулами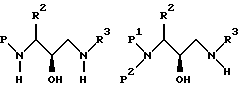
где P, P1, P2, R2 и R3 имеют значения, описанные выше.
Альтернативно галоидспирт может использоваться вместо аминоэпоксида.
Аминоспирт, определенный выше, затем подвергается реакции в подходящем растворителе с сульфонилхлоридом (R4SO2Cl) или сульфонилангидридом в присутствии акцептора кислоты. Подходящие растворители, в которых может проводиться реакция, включают метиленхлорид, тетрагидрофуран и аналогичные. Подходящие акцепторы кислоты включают триэтиламин, пиридин и аналогичные. Предпочтительными сульфонилхлоридами являются метансульфонилхлорид и бензолсульфонилхлорид. Получающееся сульфонамидное производное может быть представлено, в зависимости от используемого эпоксида, формулами: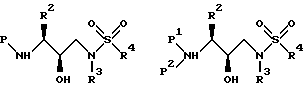
где P, P1, P2, R2, R3 и R4 имеют значения, определенные выше.
Эти промежуточные соединения полезны для получения ингибиторных соединений настоящего изобретения и являются также активными ингибиторами ретровирусных протеаз.
Сульфонилгалогениды формулы R4SO2X могут получаться с помощью реакции подходящего Гриньяровского или алкиллитиевого реагента с сульфурилхлоридом, или двухокисью серы с последующим окислением галогеном, предпочтительно хлором. Тиолы также могут окисляться в сульфонилхлориды с использованием хлора в присутствии воды при тщательно регулируемых условиях. Дополнительно, сульфоновые кислоты могут превращаться в сульфонилгалогениды с использованием таких реагентов, как PCl5, а также в ангидриды с использованием подходящих дегидратирующих реагентов. Сульфоновые кислоты могут в свою очередь получаться с использованием процедур, хорошо известных в технике. Такие сульфоновые кислоты также промышленно доступны.
Вместо сульфонилгалогенидов для получения соединений, в которых фрагмент -SO2- заменен соответственно группой -SO- и -S-, могут использоваться сульфинилгалогениды (R4SOCl) и сульфенилгалогениды (R4SCl).
После получения сульфонамидного производного аминозащищающая группа P удаляется, или удаляются группы P1 и P2, в условиях, которые не оказывают отрицательного воздействия на остальную часть молекулы. Эти методы хорошо известны в технике и включают кислотный гидролиз, гидрогенолиз и аналогичные. Предпочтительный метод включает удаление защитной группы, например, удаление карбобензокси группы, с помощью гидрогенолиза с использованием палладия на угле в подходящей системе растворителя, такой, как спирт, уксусная кислота и аналогичные, или их смеси. Когда защитной группой является трет-бутоксикарбонильная группа, она может удаляться с использованием неорганической или органической кислоты, например HCl или трифторуксусной кислоты, в подходящей системе растворителя, например диоксане или метиленхлориде. Получающийся в результате продукт является аминово-солевым производным. Когда защитной группой является бензильная группа, она может удаляться с помощью гидрогенолиза. После нейтрализации соли амин затем подвергается реакции с сульфоном формулы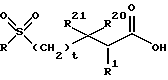
где R, R1, R20, R21 и t имеют значения, определенные выше.
Сульфон получается согласно следующей процедуре.
Меркаптан формулы RSH подвергается реакции с замещенным метакрилатом формулы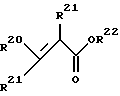
путем реакции присоединения Микаэля. Присоединение Микаэля проводится в подходящем растворителе и в присутствии подходящего основания, с получением соответствующего тиольного производного, представленного формулой: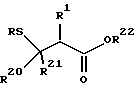
где R и R1 - радикалы, определенные выше; R20 и R21 - водород или радикалы, определенные для R1; и R22 представляет защитную группу карбоксила, такую, как метил, этил, бензил, трет-бутил или аналогичные.
Подходящие растворители, в которых может проводиться присоединение Микаэля, включают протонные, непротонные и диполярные апротонные органические растворители, например спирты, такие, как, например, метанол, этанол, бутанол и аналогичные, а также простые эфиры, например ТГФ, и ацетонитрил, ДМФ, ДМСО, и аналогичные, включая их смеси. Подходящие основания включают алкокиси металлов 1 группы такие, как метилат натрия, этилат натрия, бутилат натрия и аналогичные, а также гидриды металлов 1 группы такие, как гидрид натрия, включая их смеси.
Тиольное производное превращается в соответствующий сульфон или сульфоксид формулы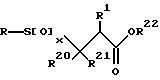
с помощью окисления тиольного производного подходящим агентом окисления в подходящем растворителе. Подходящие агенты окисления включают, например, перекись водорода, метаперборат натрия, оксон (пероксимоносульфат калия), метахлорпероксибензойную кислоту, периодную кислоту и аналогичные, включая их смеси. Подходящие растворители включают уксусную кислоту (для метапербората натрия), а для других перкислот - простые эфиры, такие, как ТГФ и диоксан, и ацетонитрил, ДМФ и аналогичные, включая их смеси.
Сульфон затем превращается в соответствующую свободную кислоту формулы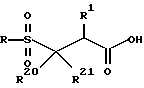
Один метод включает использование подходящего основания, например гидроокиси лития, гидроокиси натрия и аналогичного, включая их смеси, в подходящем растворителе таком, как, например, ТГФ, вода, ацетонитрил, ДМФ, ДМСО, метиленхлорид и аналогичные, включая их смеси. Другие методы, которые могут использоваться для снятия защиты, зависят от природы R22. Например, когда R22 представляет трет-бутильную группу, может использоваться сильная кислота, такая, как соляная кислота, или трифторуксусная кислота. Когда R22 представляет бензильную группу, она может удаляться с помощью гидрогенолиза.
Свободная кислота может затем сочетаться с использованием хорошо известных в технике процедур с сульфонамидным производным аминоспирта или его аналогом, который описан выше. Получающийся продукт является соединением, представленным формулой I.
Альтернативно, можно сочетать сульфонамидный изостер с промышленно доступной кислотой.
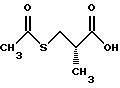
удалять тиоцетильную группу подходящим основанием таким, как гидроокись, или амин такой, как аммиак, а затем подвергать получающийся тиол реакции с алкилирующим агентом таким, как алкилгалогенид, тозилат или мезилат, с получением соединений следующей структуры: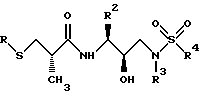
Сера затем может окисляться в соответствующий сульфон или сульфоксид с использованием подходящих окисляющих агентов, как описано выше, давая желаемые соединения следующей структуры: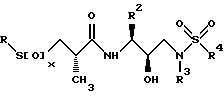
Альтернативно для получения соединений формулы I, замещенный метакрилат формулы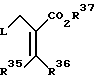
где L представляет удаляемую группу, определенную ранее, R35 и R36 - водород или радикалы, определенные для R1; R37 - алкильные, аралкильные, циклоалкильные и циклоалкилалкильные радикалы,
подвергается взаимодействию с подходящим сульфонирующим агентом таким, как, например, сульфиновая кислота, представленная формулой RSO2M, где R - радикалы, определенные выше, и M - металл, способный образовывать соль кислоты, например натрий, давая соответствующий сульфон, представленный формулой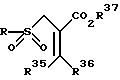
где R, R35, R36 и R37 имеют значения, определенные выше.
Сульфон затем гидролизуется в присутствии подходящего основания такого, как гидроокись лития, гидроокись натрия и аналогичные, в соединение, представленное формулой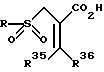
где R, R35 и R36 - радикалы, определенные выше.
Получающееся в результате соединение затем асимметрически гидрируется с использованием катализатора асимметрического гидрирования, такого, как, например, комплекс рутений-BINAP, давая восстановленный продукт, существенно обогащенный более активным изомером, представленный формулой: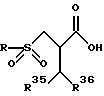
где R, R35 и R36 - радикалы, определенные выше.
Когда более активный изомер имеет R-стереохимию, может использоваться катализатор асимметрического гидрирования Ru(R - BINAP). Соответственно, когда более активный изомер имеет S-стереохимию, может использоваться катализатор Ru(R - BINAP). Когда оба изомера являются активными, или когда желательно иметь смесь двух диастереомеров, для восстановления указанного выше соединения, может использоваться такой катализатор гидрирования, как платина или палладий на угле. Восстановленное соединение затем присоединяется к сульфонамидному изостеру, как описано выше, давая соединения формулы II.
Альтернативно, кислота или производное кислоты, соответственно замещенной удаляемой группой (обсуждаемой выше), может обрабатываться меркаптаном и основанием (см. выше), давая органический сульфид. Производные кислоты определяются выше. Получающийся в результате сульфид может окисляться в соответствующий сульфоксид или сульфон с помощью методов, обсуждаемых ранее.
Предпочтительный способ получения 2(S)-метил-3-(метилсульфонил)пропионовой кислоты заключается в следующем. Исходят из промышленно доступных соединений следующей структуры: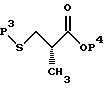
где P3 представляет защитную группу серы, предпочтительно бензоил или ацетил, и P4 представляет или водород, или защитную группу карбоновой кислоты, такую, как метил, этил, трет-бутил, бензил и аналогичные. Предпочтительно P4 представляет трет-бутил. Защитная группа серы P3 может селективно удаляться с использованием методов, известных специалистам в данной области техники. Например, когда P3 представляет или бензоил, или ацетил, она может удаляться с помощью обработки неорганическим основанием или амином, предпочтительно аммиаком, в соответствующем растворителе, таком, как метанол, этанол, изопропанол, толуол или тетрагидрофуран. Предпочтительным растворителем является метанол. Данный метод дает соединение следующей структуры.
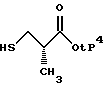
которое может алкилироваться у атома серы соединением структуры
RX,
где R имеет значения, определенные выше, и X представляет подходящую удаляемую группу, такую, как галогенидная (хлорид, бромид, иодид), мезилатная, тозилатная или трифлатная. Реакция выполняется в присутствии подходящего основания такого, как триэтиламин, диизопропилэтиламин, 1,8-диазабицикло[5.4.0] ундец-7-ен (DBU) и аналогичные, в подходящем растворителе таком, как толуол, тетрагидрофуран или метиленхлорид. Предпочтительным основанием является DBU, а предпочтительным растворителем является толуол. Когда R представляет метильную группу, RX может быть метилхлоридом, метилбромидом, метилиодидом, или диметилсульфатом. Предпочтительно RX представляет метилиодид. Продукт реакции представляет соединение структуры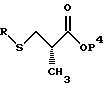
Сера может затем окисляться или в сульфоксид, или сульфон с использованием методов, известных специалистам в данной области. Подходящими окисляющими агентами являются метахлорпербензойная кислота, перекись водорода, перборат натрия и аналогичные. Соответствующими растворителями являются метиленхлорид, толуол, уксусная кислота, пропионовая кислота и аналогичные. Предпочтительный способ предусматривает использование перекиси водорода или пербората натрия в уксусной кислоте. Сульфоновый продукт имеет структуру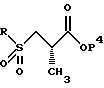
Защитная группа карбоновой кислоты P4 может затем удаляться с использованием методов, хорошо известных специалистам в данной области техники. Например, когда P4 представляет трет-бутильную группу, она может удаляться с помощью обработки кислотой такой, как соляная кислота или трифторуксусная кислота. Согласно предпочтительному способу используется 4 норм. соляная кислота в диоксане. Данная процедура дает желаемое конечное соединение структуры: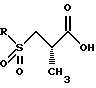
Вполне понятно, что специалисты в данной области могут использовать вариации синтетических приемов и последовательностей таких, как использование иных защитных групп для серы (P3) или для карбоновой кислоты (P4), и иных реагентов для осуществления тех же преобразований.
Предусматривается, что для получения соединений формул, имеющих R6, эти соединения могут приготавливаться после процедуры, представленной выше, и перед сочетанием сульфонамидного производного или его аналога с сульфоном, осуществляемым с помощью процедуры, называемой в технике восстановительным аминированием. Так, цианоборгидрид натрия и соответствующий альдегид или кетон могут подвергаться реакции с сульфонамидным производным соединением или соответствующим аналогом при комнатной температуре для того, чтобы восстановительно аминировать любое из соединений формул I - III. Предусматривается также, что, когда R3 аминоспиртового промежуточного соединения представляет водород, ингибиторные соединения могут получаться с помощью восстановительного аминирования конечного продукта реакции между аминоспиртом и амином, или на любой другой стадии синтеза для получения ингибиторных соединений.
Эквивалентами соединений общих формул, представленных выше, для антивирусных соединений и производных, а также промежуточных соединений являются соединения, в иных отношениях соответствующие им и имеющие те же общие свойства, в которых одна или более из различных R групп представляют простые вариации заместителей, определенных здесь, например, в которых R представляет высшую алкильную группу, в отличие от указанной. Кроме того, когда заместитель обозначен как водород, или может быть водородом, точная химическая природа заместителя, который является отличным от водорода в данном положении, например, углеводородный радикал или галоген, гидрокси, амино и аналогичная функциональная группа, не является критической, если он не оказывает отрицательного воздействия на общую активность и/или процедуру синтеза.
Химические реакции, описанные выше, раскрыты в их самом широком смысле применительно к получению соединений данного изобретения. Иногда реакции не могут быть применимы, как они описаны, к каждому соединению, включенному в описываемый объем изобретения. Соединения, в отношении которых это имеет место, легко могут быть определены специалистами в данной области. Во всех таких случаях или могут быть успешно проведены реакции с помощью обычных модификаций, известных специалистам в данной области техники, например, с помощью соответствующей защиты препятствующих групп, или путем замены реагентов на альтернативные общепринятые реагенты, или с помощью обычных модификаций условий реакции и аналогичными методами, или для получения соответствующих соединений данного изобретения могут быть применены другие реакции, описанные здесь, или общепринятые реакции. Во всех методиках получения все исходные материалы являются или известными, или могут легко получаться из известных исходных материалов.
Следует полагать, что специалисты в данной области без дополнительных разъяснений смогут с использованием представленного выше описания использовать настоящее изобретение во всех отношениях полностью. Следующие ниже предпочтительные конкретные воплощения поэтому должны пониматься лишь как иллюстративные и не ограничивающие никоим образом объем описываемого изобретения.
Все реагенты использовались в том виде, как они получались без очистки. Все протонные и углеродные ЯМР спектры были получены на ЯМР спектрометре или Вариан VXR-300, или VXR-400.
Пример 1A
Получение N[3(S)-бензоилоксикарбониламино-2(R)-гидрокси-4- фенилбутил] -N-изоамиламина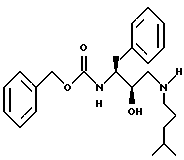
Часть A.
К раствору N-бензоилоксикарбонил-L-фенилаланинхлорметилкетона (75 г, 0,2 моля) в смеси 800 мл метанола и 800 мл тетрагидрофурана добавлялся боргидрид натрия (13,17 г, 0,348 моля, 1,54 эквив.) на протяжении 100 минут. Раствор перемешивался при комнатной температуре в течение 2 часов, а затем концентрировался в вакууме. Остаток растворялся в 1000 мл этилацетата и промывался 1 норм. KHSO4, насыщенным водным NaHCO3, насыщенным водным NaCl, сушился над безводным сульфатом магния, фильтровался и концентрировался в вакууме, давая масло. Неочищенный продукт растворялся в 1000 мл гексанов при 60oC и оставлялся охлаждаться до комнатной температуры, после чего образовавшиеся кристаллы отделялись фильтрованием и промывались равными количествами гексанов. Данное твердое вещество затем перекристаллизовывалось из горячего этилацетата и гексанов, давая 32,3 г 43% N-бензилоксикарбонил-3(S)-амино-1-хлор-4-фенил-2(S)-бутанола, т. пл. 150 - 151oC, FAB MS: MLi = 340.
Часть B.
Раствор гидроокиси калия (6,52 г, 0,116 моля, 1,2 эквив.) в 970 мл абсолютного этанола обрабатывался N-бензилоксикарбонил-3(S)-амино-1-хлор-4-фенил-2(S)-бутанолом (32,3 г, 0,097 моля). Данный раствор перемешивался при комнатной температуре в течение 15 минут, а затем концентрировался в вакууме, давая белое твердое вещество. Твердое вещество растворялось в дихлорметане и промывалось водой, сушилось над безводным сульфатом магния, фильтровалось и концентрировалось в вакууме, давая белое твердое вещество. Данное твердое вещество кристаллизовалось из гексанов и этилацетата, давая 22,3 г. 77% N-бензилоксикарбонил-3(S)-амино-1,2(S)-эпокси-4-фенилбутана, т. пл. 102 - 103oC, FAB MC: MH+ = 298.
Часть C.
Раствор N-бензилоксикарбонил-3(S)-амино-1,2-(S)-эпокси-4- фенилбутана (11,54 г, 38,81 ммоля) и изоамиламина (66,90 г. 0,767 моля, 19,9 эквивалентов) в 90 мл изопропилового спирта нагревался до температуры дефлегмации в течение 3,1 часа. Раствор охлаждался до комнатной температуры и частично концентрировался в вакууме, а остальной раствор выливался в 200 мл перемешиваемых гексанов, после чего продукт кристаллизовался из раствора. Продукт отделялся с помощью фильтрования и сушился на воздухе, давая 11,76 г, 79% N-[[3(S)-фенилметилкарбамоил)амино-2(R)-гидрокси-4-фенилбутил] -N- [(3-метилбутил)]амина, т.пл. 118 - 122oC, FAB MC: MH+ = 385.
Пример 1B
Получение N-[[3(S)-фенилметилкарбамоил)амино-2R-гидрокси-4- фенил]-1-[(2-метилпропил)амино-2-(1,1-диметилэтоксил)карбонил]бутана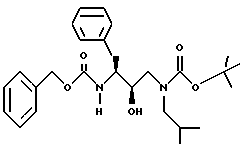
К раствору 7,51 г (20,3 ммоля) N-[[3S-(фенилметилкарбамолил)амино]-2R-гидрокси-4-фенилбутил] -N- (2-метилпропил)] амина в 67 мл безводного тетрагидрофурана добавлялось 2,25 г (22,3 ммоля) триэтиламина. После охлаждения до 0oC добавлялось 4,4 г (20,3 ммоля) ди-трет-бутилдикарбоната и перемешивание продолжалось при комнатной температуре в течение 21 часа. Летучие вещества удалялись в вакууме, добавлялся этилацетат, затем смесь промывалась 5%-ной лимонной кислотой, насыщенным бикарбонатом натрия, раствором поваренной соли, сушилась над сульфатом магния, фильтровалась и концентрировалась, давая 9,6 г неочищенного продукта. Хроматография на силикагеле с использованием смеси 30%-ный этилацетат/гексан давала 8,2 г чистого N-[[3S-(фенилметилкарбамоил)амино] -2R-гидрокси-4-фенил]-1-[(2- метилпропил)амино-2-(1,1-диметилэтоксил)карбонил]бутана, масс спектр m/e = 477 (M + Li).
Пример 2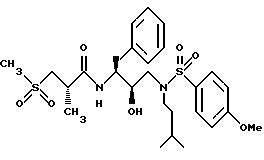
Получение [1S-[1R*(R*), 2S*] ]-N[2- гидрокси-3-[(3-метилбутил)(4-метоксифенилсульфонил)амино]-1- (фенилметил)пропил]-2-метил-3-(метилсульфонил)пропанамида
Часть A.
Раствор аминоспирта из примера 1, часть C (1,1515 г, 2,99 ммоля) и триэтиламина (313,5 мг, 3,10 ммоля) в 15 мл дихлорметана обрабатывался 4-метоксибензолсульфонилхлоридом (630,6 мг, 3,05 ммоля) с помощью шприца. Раствор перемешивался при комнатной температуре в течение 40 минут и затем концентрировался в вакууме. Остаток растворялся в этилацетате и промывался 1 норм. KHSO4 насыщенным водным раствором бикарбоната натрия, солевым раствором, сушился над безводным сульфатом магния, фильтровался и концентрировался, давая 1,5622 г белой пены. Неочищенный продукт очищался с помощью перекристаллизации из смеси гексанов и этилацетата, давая 1,1047 г, 67% чистого продукта, т. пл. 95 - 98oC. FAB Масс спектр высокого разрешения, вычислено для C30H38N2O6S: 555,2529. Найдено: 555,2559.
Часть B.
Раствор продукта из части A (970 мг, 1,68 ммоля) в 30 мл метанола обрабатывался 70 мг катализатора 10% палладия на угле и гидрировался при 41 фунт./кв.дюйм (2,883 кг/кв.см) в течение 16 часов при комнатной температуре. Катализатор удалялся с помощью фильтрования, и фильтрат концентрировался в вакууме, давая прозрачное масло, которое затвердевало при стоянии, т.пл. 81 - 85oC, FAB MC: MH+ = 421, 764,1 мг, которое использовалось непосредственно на следующей стадии.
Стадия C.
Смесь 2(S)-метил-3-метилсульфонилпропионовой кислоты (194 мг, 1,17 ммоля), N-гидроксибензотриазола (276 мг, 1,34 ммоля) и гидрохлорида 1-(3-диметиламинопропил)-3-этилкарбодиимида (EDC) (256 мг, 1,34 ммоля) растворялась в 3,5 мл диметилформамида (ДМФ) и оставлялась взаимодействовать в течение 30 минут при 0oC. Амин со Стадии B (451,1 мг, 1,07 ммоля) растворенный в 1,5 мл ДМФ, добавлялся к вышеуказанной смеси, и все перемешивалось при комнатной температуре в течение 16 часов. Раствор затем выливался в 20 мл насыщенного водного раствора бикарбоната натрия и экстрагировался 4 раза этилацетатом. Объединенные этилацетатные экстракты промывались 5% водной лимонной кислотой, насыщенным водным бикарбонатом натрия, солевым раствором, сушились над безводным сульфатом магния, фильтровались и концентрировались, давая прозрачное масло, которое кристаллизовалось при стоянии. Данное вещество перекристаллизовывалось из гексанов и этилацетата, давая 517,6 мг, 85% чистого продукта с т. пл. 125 - 129oC. BP FAB MC: вычислено для C27H40N2O7S2: 569,2355. Найдено: 569,2397.
Пример 3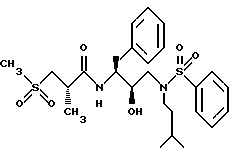
Получение [1S-[1R*(R*),2S*]]-N-[2- гидрокси-3-[(2-метилпропил)(фенилсульфонил)амино]-1-(фенилметил) пропил]-2-метил-3-(метилсульфонил)-пропанамида
Стадия A.
Раствор N-бензилоксикарбонил-3(S)-амино-1,2(S)-эпокси-4-фенилбутана (50,0 г, 0,168 моля) и изобутиламина (246 мг, 3,24 моля, 20 эквивалентов) в 650 мл изопропилового спирта нагревался до условий дефлегмации в течение 1,25 часов. Раствор охлаждался до комнатной температуры, концентрировался в вакууме, а затем выливался в 1 л перемешиваемого гексана, после чего продукт кристаллизовался из раствора. Продукт отделялся фильтрованием и сушился на воздухе, давая 57,56 г, 92% N-[3-(S)-бензилоксикарбониламино-2(R)-гидрокси-4-фенил]-N- изобутиламина, т.пл. 108,0 - 109,5oC, MH+ m/z = 371.
Стадия B.
Амин со стадии A (936,5 мг, 2,53 ммоля) и триэтиламин (288,5 мг, 2,85 ммоля) растворялся в 20 мл дихлорметана и обрабатывался бензолсульфонилхлоридом (461 мг, 2,61 ммоля). Раствор перемешивался при комнатной температуре в течение 16 часов, а затем концентрировался в вакууме. Остаток растворялся в этилацетате, и данный раствор промывался 1 норм. бисульфатом калия, насыщенным водным бикарбонатом натрия, солевым раствором, сушился над безводным сульфатом магния, фильтровался и концентрировался, давая прозрачное масло 1,234 г. Данное масло кристаллизовалось из смеси эфира и гексанов, 729,3 мг, 56,5%, т.пл. 95 - 99oC, FAB MC: MH+ = 511.
Стадия C.
Раствор фенилметил [2(R)-гидрокси-2-[2-метилпропил]- (бензолсульфонил)амино] -1-S-(фенилметил)пропилкарбамата (671,1 мг, 1,31 ммоля) со стадии B в 10 мл метанола гидрировался над 50 мг 10%-ного палладия на угле при 40 фунт/кв.дюйм (2,812 кг/см2) при комнатной температуре в течение 15 часов. Катализатор удалялся фильтрованием через диатомовую землю и фильтрат концентрировался, давая белую пену, 474,5 мг, 96%, FAB MC: MH+ = 377, которая использовалась непосредственно на следующей стадии без дальнейшей очистки.
Стадия D
Смесь 2(S)-метил-3-(метилсульфонил)пропионовой кислоты (210,6 мг, 1,27 ммоля), N-гидроксибензотриазола (260,4 мг, 1,70 ммоля) и EDC (259 мг, 1,35 ммоля) в 3,5 мл ДМФ перемешивалась при 0oC в течение получаса. Амин со стадии C (474 мг, 1,15 ммоля), растворенный в 2 мл ДМФ, добавлялся к указанному выше раствору и перемешивался при комнатной температуре в течение 16 часов, а затем выливался в 100 мл 50% насыщенного водного бикарбоната натрия. Водный раствор экстрагировался этилацетатом. Этилацетатный раствор промывался 5%-ной водной лимонной кислотой, насыщенным водным бикарбонатом натрия, солевым раствором, сушился над безводным сульфатом магния, фильтровался и концентрировался, давая белую пену 560,5 мг, которая кристаллизовалась из этилацетата и гексанов, давая 440,3 мг чистого продукта, т.пл. 112 - 116,5oC, HR FAB MC.
Вычислено для C25H36N2O6S2: 525,2093. Найдено: 525,2077.
Пример 4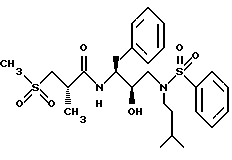
Получение [1S-[1R*(R*),2S*]]-N-[2-гидрокси- 3-[(3-метилбутил)(фенилсульфонил)амино]-1-(фенилметил)пропил]-2- метил-3-(метилсульфонил)-пропанамида
Стадия A.
Смесь N-[3(S)-бензилоксикарбониламино-2(R)-гидрокси-4-фенилбутил] - N-[(3-метилбутил)]амина (Пример 1, стадия C) (3,89 г, 10,1 ммоля) и триэтиламина (1,02 г, 10,1 ммоля) растворялась в 25 мл тетрагидрофурана (ТГФ) и обрабатывалась раствором ди-трет-бутилпирокарбоната (2,21 г, 10,1 ммоля), растворенного в 10 мл ТГФ. Раствор перемешивался при комнатной температуре в течение полутора часов и затем концентрировался в вакууме. Остаток растворялся в этилацетате и промывался 1 норм. бисульфатом калия, насыщенным водным бикарбонатом натрия, солевым раствором, сушился над безводным сульфатом магния, фильтровался и концентрировался, давая густое прозрачное масло, 4,66 г, 98,5%, Rf = 0,23 на силикагеле при элюировании смесь. 5/1 гексаны:этилацетат. Данное вещество использовалось непосредственно на следующей стадии без дальнейшей очистки.
Стадия B.
Продукт со стадии A (4,66 г, 10,1 ммоля) растворялся в 40 мл безводного этанола и обрабатывался 30 мг 15%-ного палладиевого катализатора на угле. Смесь затем гидрировалась в течение 18 часов при комнатной температуре и 40 фунт/кв.дюйм (2,812 кг/кв.см). Катализатор удалялся фильтрованием через диатомовую землю, и фильтрат концентрировался, давая масло, которое использовалось непосредственно на последующей стадии без очистки.
Стадия C.
Смесь 2(S)-метил-3-метилсульфонилпропионовой кислоты (1,39 г, 8,3 ммоля), N-гидроксибензотриазола (1,84 г, 12,0 ммолей) и гидрохлорида 1-(3-диметиламинопропил)-3-этилкарбодиимида (EDC) (1,77 г, 9,2 ммоля) растворялась в 10 мл диметилформамида и оставлялась реагировать в течение 30 минут при 0oC. Амин со стадии B (2,80 г, 8,0 ммолей), растворенный в 10 мл ДМФ, добавлялся к вышеуказанной смеси и перемешивался при комнатной температуре в течение 24 часов. Раствор концентрировался в вакууме, и остаток брался в этилацетат. Этилацетатный раствор промывался 5%-ной водной лимонной кислотой, насыщенным водным бикарбонатом натрия, солевым раствором, сушился над безводным сульфатом магния, фильтровался и концентрировался, давая прозрачное масло, которое очищалось с помощью флэш хроматографии, давая 3,00 г, 75%, данное вещество использовалось непосредственно на следующей стадии.
Стадия D.
Продукт со стадии C (3,00 г, 6,02 ммоля) обрабатывался 30 мл 4 норм. HCl в диоксане при комнатной температуре в течение 24 часов. Раствор концентрировался в вакууме, и полутвердый остаток растирался с эфиром и сушился в вакууме, давая белое аморфное твердое вещество, т.пл. более 250oC, становится желтым при 221oC, FAB MC, MH+ = 436.
Стадия E.
Продукт со стадии D растворялся в дихлорметане и обрабатывался насыщенным водным бикарбонатом натрия, давая раствор свободного амина. Органическая фаза сушилась над безводным сульфатом магния, фильтровалась и концентрировалась в вакууме, давая вещество (610 мг, 1,75 ммоля). Данный амин суспендировался в 50 мл ТГФ и обрабатывался последовательно триэтиламином (1,01 г, 10 ммолей) и бензолсульфонилхлоридом (283 мг, 1,75 ммоля). Раствор перемешивался при комнатной температуре в течение 19,5 часов. Твердые вещества удалялись фильтрованием, фильтрат концентрировался и растворялся в дихлорметане. Дихлорметановый раствор промывался 1 норм. бисульфатом калия, насыщенным водным бикарбонатом натрия, солевым раствором, сушился над безводным сульфатом магния, фильтровался и концентрировался, давая масло, которое растиралось с метанолом, давая белое твердое вещество, которое отделялось фильтрованием. Неочищенное твердое вещество затем кристаллизовалось из этилацетата и гексанов, давая 200 мг, 21% материала с т.пл. 112 - 115oC, HR FAB MC, вычислено для C26H38N2O6S2: 538,2171. Найдено: 533,2180.
Пример 5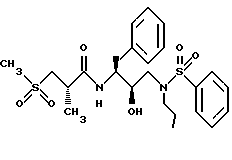
Получение [1S-[1R*(R*),2S*]]-N-[2- гидрокси-3-(пропил)(фенилсульфонил)амино]-1-(фенилметил)пропил]-2- метил-3-(метилсульфонил)-пропанамида
Стадия A
Раствор N-бензилоксикарбонил 3(S)-амино-1,2(S)-эпокси-4-фенилбутана (6,06 г, 21,4 ммоля) и н-пропиламина (20,9 г, 0,35 ммоля) в 100 мл изопропилового спирта нагревался до дефлегмации в течение 3 часов. Раствор затем концентрировался в вакууме, давая твердое вещество, которое кристаллизовалось из гексанов и этилацетата, давая 6,53 г, 90% желаемого продукта, т.пл. 120-123oC, FAB MC: MH+=357.
Стадия B
Амин со стадии A вводился в реакцию с бензолсульфонилхлоридом по способу, аналогичному примеру 3, стадии B. Получающееся соединение (1,426 г, 2,87 ммоля), растворенное в 25 мл метанола, гидрировалось над 40 мг 10% палладия на угле при 40 фунт/кв.дюйм (2,812 кг/кв.см) в течение 16 часов при комнатной температуре. Раствор затем фильтровался через диатомовую землю, и фильтрат концентрировался, давая 1,04 г, 100%, прозрачного масла, которое использовалось непосредственно на следующей стадии без дополнительной очистки, HR FAB MC вычислено для C15H24N2O3S: 363,1742. Найдено: 363,1763.
Стадия C
Смесь 2(S)-метил-3-(метилсульфонил)пропионовой кислоты (243,7 мг, 1,47 ммоля), N-гидроксибензотриазола (332,0 мг, 2,16 ммоля) и EDC (304,8 мг, 1,59 ммоля) в 2,5 мл ДМФ перемешивалась при 0oC в течение получаса, а затем обрабатывалась раствором свободного амина со стадии B (513,3 мг, 1,42 ммоля) в 1,5 мл ДМФ. Раствор перемешивался при комнатной температуре в течение 16 часов, а затем выливался в 80 мл 50% насыщенного водного бикарбоната натрия. Раствор экстрагировался этилацетатом, и этилацетатный раствор промывался 5% водной лимонной кислотой, насыщенным водным бикарбонатом натрия, солевым раствором, сушился над безводным сульфатом магния, фильтровался и концентрировался, давая белую пену, 576,8 мг, которая очищалась с помощью кристаллизации из смеси этилацетат/гексаны, давая 441,1 мг, 61% продукта с т.пл. 134-136,5oC, HR FAB MC, вычислено для C24H34N2O6S2+Li : 517,2019. Найдено: 517,1973.
Пример 6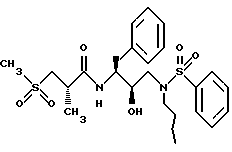
Получение [IS-[IR*(R*), 2S*]]-N-[2-гидрокси-3-(бутил)(фенилсульфонил)амино]-1-(фенилметил) пропил]-2-метил-3-(метилсульфонил)-пропанамида
Стадия A
В результате реакции N-бензилоксикарбонил 3(S)-амино-1,2(S)-эпокси-4-фенилбутана (1,48 г, 5,0 ммолей) и н-бутиламина (7,314 г, 100,0 ммолей) получают 1,50 г (80%) N-[3(S)-бензилоксикарбониламино-2(R)-гидрокси-4-фенилбутил]-N-бутиламина, т.пл. 125-128oC, FAB MC, Спектр: MH+=371.
Стадия B.
Амин со стадии A (1,67 г, 4,5 ммолей) и триэтиламин (859,4 мг) растворялись в 60 мл дихлорметана и обрабатывались бензолсульфонилхлоридом (822,3 мг, 4,66 ммоля) при комнатной температуре. После перемешивания в течение 15 минут раствор концентрировался в вакууме, и остаток растворялся в этилацетате. Этилацетатный раствор промывался 1 норм. бисульфатом калия, насыщенным водным бикарбонатом натрия, солевым раствором, сушился над безводным сульфатом магния, фильтровался и концентрировался, давая масло. Масло кристаллизовалось из гексанов и эфира, давая 2,04 г, 89% чистого продукта, т.пл. 68 - 77oC, FAB MC: MH+=511.
Стадия C.
Раствор фенилметил [2(R)-гидрокси-3-[н-бутил](бензолсульфонил)амино]-IS-(фенилметил)пропилкарбамата со стадии B (1,86 г, 3,64 ммоля) в 40 мл метанола гидрировался над 110 мг 10% палладия на угле при 40 фунт/кв.см (2,812 кг/кв. см) в течение 4 часов. Раствор фильтровался через диатомовую землю и концентрировался в вакууме, давая твердое вещество, т.пл. 68-88oC, FAB MC: MH+=377, которое использовалось на следующей стадии без дальнейшей очистки.
Стадия D.
Смесь 2(S)-метил-3-(метилсульфонил)пропионовой кислоты (288,4 мг, 1,74 ммоля) EDC (369,6 мг, 1,93 ммоля) и N-гидроксибензотриазола (368,1 мг, 2,41 ммоля) растворялась в 3,5 мл ДМФ и перемешивалась при 0oC в течение 30 минут. Данный раствор затем обрабатывался амином со стадии C (621,9 мг, 1,65 ммоля), растворенным в 2 мл ДМФ. Смесь оставлялась перемешиваться при комнатной температуре в течение 30 часов, а затем концентрировалась в вакууме. Остаток растворялся в этилацетате, промывался 1 норм. бисульфатом калия, насыщенным водным бикарбонатом натрия, солевым раствором, сушился над безводным сульфатом магния, фильтровался и концентрировался в вакууме, давая масло. Сырой продукт очищался с помощью флэш хроматографии на силикагеле при элюировании смесями гексаны/этилацетат, давая желаемый продукт в виде белого твердого вещества, 353 мг, 41%, т.пл. 99 - 103oC, HR FAB MC: вычислено для C25H36N2O6S2: 531,2175. Найдено: 513,2176.
Пример 7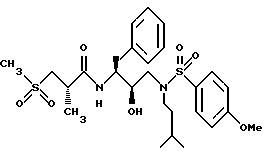
Получение [IS-[IR*(R*), 2S*] ]-N-[2-гидрокси-3-[(2-метилпропил)(4-метоксифенилсульфонил) амино]-1-(фенилметил)пропил]-2-метил-3-(метилсульфонил)-пропанамида
Стадия A
Амин из примера 3, стадии A, N-[3(S)-бензилоксикарбониламино-2(R)-гидрокси-4-фенил] -N- изобутиламин (1,1131 г, 3,00 ммоля) и триэтиламин (324,0 мг, 3,20 ммоля) в 20 мл дихлорметана обрабатывался 4-метокси-бензолсульфонилхлоридом (715,4 мг, 3,46 ммоля). Раствор перемешивался при комнатной температуре в течение 6 часов, а затем концентрировался в вакууме. Остаток растворялся в этилацетате и промывался 1 норм. бисульфатом калия, насыщенным водным бикарбонатом натрия, солевым раствором, сушился над безводным сульфатом магния, фильтровался и концентрировался, давая прозрачное масло. Масло кристаллизовалось из эфира, давая белое твердое вещество 1,273 г, 78%, т.пл. 97 - 101oC, чистого продукта, FAB MC: MH+ = 541.
Стадия B
Продукт со стадии A (930 мг, 1,68 ммоля) растворялся в 30 мл метанола и гидрировался при 40 фунт/кв. дюйм (2,812 кг/кв. см) над 70 мг 10% палладия на угле при комнатной температуре в течение 17 часов. Катализатор удалялся фильтрованием через диатомовую землю, и фильтрат концентрировался в вакууме, давая 704 мг прозрачного масла, которое затвердевало при стоянии, т.пл. 105 - 110oC, FAB MC, MH+= 407, и использовалось непосредственно на следующей стадии без дальнейшей очистки.
Стадия C
Смесь 2-метил-3-(метилсульфонил)пропиновой кислоты (174,9 мг, 1,05 ммоля), N-гидроксибензотриазола (230 мг, 1,50 ммоля) и EDC (220,5 мг, 1,15 ммоля) в 2 мл ДМФ перемешивалась при 0oC в течение 0,5 часа, а затем обрабатывалась амином со стадии B (401,2 мг, 0,99 ммоля) в 1 мл ДМФ. Раствор перемешивался при комнатной температуре в течение 16 часов, а затем выливался в 20 мл насыщенного водного бикарбоната натрия. Водный раствор экстрагировался этилацетатом, а затем этилацетатный раствор промывался 5% водной лимонной кислотой, насыщенным водным бикарбонатом натрия, солевым раствором, сушился над безводным сульфатом магния, фильтровался и концентрировался в вакууме, давая прозрачное масло, 260 мг, которое очищалось с помощью флэш хроматографии на силикагеле при элюировании гексанами и этилацетатом, давая 52,7 мг, 9,6%, т.пл. 87 - 92oC, HR FAB MC, вычислено для C26H38N2O7S2: 555,2199. Найдено: 555,2234.
Пример 8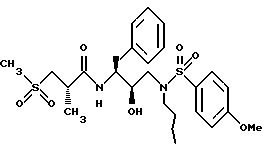
Получение [IS-(1R*(R*), 2S*] ]-N-[2-гидрокси-3-[(бутил)-(4-метоксифенилсульфонил)-амино] -1- (фенилметил)пропил] -2-метил-3-(метилсульфонил)-пропанамида
Стадия A
Амин со стадии A примера 6 (1,52 мг, 4,10 ммоля) и триэтиламин (488 мг, 4,82 ммоля) в 30 мл дихлорметана обрабатывался 4-метоксибензолсульфонилхлоридом (869 мг, 4,20 ммоля) при комнатной температуре в течение 3 часов. Раствор удалялся в вакууме, и остаток брался в этилацетат. Этилацетатный раствор промывался 1 норм. бисульфатом калия, насыщенным водным бикарбонатом натрия, солевым раствором, сушился над безводным сульфатом магния, фильтровался и концентрировался, давая белое твердое вещество, которое промывалось эфиром и сушилось на воздухе, давая 1,71 г, 77%, т.пл. 118 - 120oC, FAB MC; M + Li = 547, чистого продукта.
Стадия B
Продукт со стадии A (1,514 г, 2,80 ммолей) в 30 мл метанола гидрировался при 40 фунт/кв.дюйм (2,812 кг/кв.см) над 110 мг 10% палладия на угле в течение 16 часов при комнатной температуре. Катализатор удалялся фильтрованием через диатомовую землю, и фильтрат концентрировался, давая белое твердое вещество, 1,20 г, 100%, т.пл. 103-108oC, HR FAB MC: вычислено для C21H30N2O4S: 413,2086. Найдено: 413,2121, которое использовалось непосредственно на следующей стадии без дальнейшей очистки.
Стадия C
Смесь 2(S)-метил-3-(метилсульфонил)пропионовой кислоты (354,4 мг, 2,13 ммоля), N-гидроксибензотриазола (473,4 мг, 3,09 ммоля) и EDC (445,3 мг, 2,33 ммоля) в 1,5 мл ДМФ перемешивалась при 0oC в течение 25 минут, а затем подвергалась обработка амином со стадии B (815 мг, 2,0 ммоля) в 2 мл ДМФ. Смесь перемешивалась при комнатной температуре в течение 16 часов, а затем выливалась в 50 мл насыщенного водного бикарбоната натрия, а затем экстрагировалась этилацетатом. Этилацетатный раствор промывали 5% водной лимонной кислотой, насыщенным водным бикарбонатом натрия, солевым раствором, сушился над безводным сульфатом магния, фильтровался и концентрировался в вакууме, давая 905 мг белой пены. Продукт очищался с помощью флэш хроматографии на силикагеле при элюировании смесью этилацетат/гексаны, давая 711,6 мг, 65%, чистого продукта, т. пл. 87 - 92oC, HR FAB MC, M + Li; вычислено для C26H38N2O7S2: 561,2281. Найдено: 561,2346.
Пример 9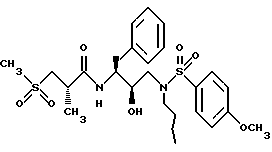
Получение [1S-(1R*(R*), 2S*]]-N-[2-гидрокси-3-[(пропил)-(4-метоксифенилсульфонил)-амино] -1- (фенилметил)пропил] -2-метил-3-(метилсульфонил)-пропанамида
Стадия A
Раствор продукта со стадии A примера 5 (620 мг, 1,74 ммоля) и триэтиламина (250 мг, 2,47 ммоля) в 15 мл дихлорметана обрабатывался 4-метоксибензолсульфонилхлоридом (371 мг, 1,79 ммоля) при комнатной температуре в течение 2,33 часов. Растворитель удалялся в вакууме, и остаток брался в этилацетат, а затем промывался 1 норм. бисульфатом калия, насыщенным водным бикарбонатом натрия, солевым раствором, сушился над безводным сульфатом магния, фильтровался и концентрировался, давая 1,0622 г, белой пены. Неочищенный продукт очищался с помощью флэш хроматографии на силикагеле при элюировании гексанами и этилацетатом, давая 615 мг, 67% чистого продукта с т.пл. 88 - 92oC, HR FAB MC; вычислено для C28H34N2O6S: 5533, 2298. Найдено: 533,2329.
Стадия B.
Раствор карбаминовой кислоты, продукта со стадии A (519 мг, 0,98 ммоля) в 30 мл метанола обрабатывался 70 мг 10% палладиевого катализатора на угле и гидрировался при 46 фунт/кв. дюйм (3,234 кг/кв. см) в течение 22 часов при комнатной температуре. Катализатор удалялся фильтрованием через диатомовую землю, и фильтрат концентрировался в вакууме, давая прозрачное масло, которое затвердевало при стоянии, т.пл. 124-127oC, FAB MC; M + Li = 399,387 мг, 100%, которое использовалось непосредственно на следующей стадии.
Стадия C.
Смесь 2(S)-метил-3-метилсульфонилпропионовой кислоты (138,5 мг, 0,83 ммоля), N-гидроксибензотриазола (174,6 мг, 1,14 ммоля) и 1-(3-диметиламинопропил)-3-этилкарбодиимида гидрохлорида (EDC) (171,8 мг, 0,90 ммоля) растворялась в 2,5 мл диметилформамида и оставлялась реагировать в течение 30 минут при 0oC. Амин со стадии B (304,9 мг, 0,78 ммоля), растворенный в 1,5 мл ДМФ, добавлялся в указанной выше смеси и перемешивался при комнатной температуре в течение 14,5 часов. Раствор затем выливался в 20 мл насыщенного водного бикарбоната натрия и экстрагировался этилацетатом. Этилацетатные экстракты промывались 5% водной лимонной кислотой, насыщенным водным бикарбонатом натрия, солевым раствором, сушились над безводным сульфатом магния, фильтровались и концентрировались, давая белое твердое вещество. Данное вещество перекристаллизовывалось из гексанов и этилацетата, давая 228 мг, 54% чистого продукта с т. пл. 115 - 118oC, HR FAB MC; вычислено для C27H40N2O7S2: 541,2042. Найдено: 541,2064.
Пример 10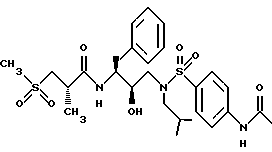
Получение [1S-[1R*(R*), 2S*]]-N-[2-гидрокси-3-[(2-метилпропил)-(4-ацетамидо)фенилсульфонил) амино]-1-(фенилметил)пропил]-2-метил-3-(метилсульфонил)-пропанамида
Стадия A.
Раствор продукта из примера 3, стадии A (1,1082 г, 2,99 ммоля) и триэтиламина (713 мг, 3,05 ммоля) в 20 мл дихлорметана обрабатывался N-ацетилсульфанилилхлоридом (713,2 мг, 3,05 ммоля) при комнатной температуре в течение 3,67 часов. Растворитель удалялся в вакууме, и остаток брался в этилацетат, и затем промывался 1 норм. бисульфатом калия, насыщенным водным бикарбонатом натрия, солевым раствором, сушился над безводным сульфатом магния, фильтровался и концентрировался, давая 1,398 г, белого твердого вещества, т.пл. 155 - 158oC, FAB MC; M + Li = 574.
Стадия B.
Раствор продукта со стадии A (900 мг, 1,58 ммоля) в 30 мл метанола обрабатывался 90 мг 10% палладиевого катализатора на угле и гидрировался при 32 фунт/кв. дюйм (2,250 кг/кв. см) в течение 15 часов при комнатной температуре. Катализатор удалялся фильтрованием через диатомовую землю, и фильтрат концентрировался в вакууме, давая белую пену, FAB MC; M + H+=334, 680 мг, 99%, которая использовалась непосредственно на следующей стадии без дальнейшей очистки.
Стадия C.
Смесь 2(S)-метил-3-метилсульфонилпропионовой кислоты (159,7 мг, 0,96 ммоля), N-гидроксибензотриазола (210,8 мг, 1,38 ммоля), и гидрохлорида 1-(3-диметиламинопропил)- 3-этилкарбодиимида (EDC) (203,9 мг, 1,06 ммоля) растворялась в 1,5 мл диметилформамида и оставлялась реагировать в течение 30 минут при 0oC. Амин со стадии B (401,9 мг, 1,06 ммоля), растворенный в 0,5 мл ДМФ, добавлялся к указанной выше смеси и перемешивался при комнатной температуре в течение 16,5 часов. Раствор затем выливался в 75 мл насыщенного водного бикарбоната натрия и экстрагировался этилацетатом. Этилацетатные экстракты промывались 5% водной лимонной кислотой, насыщенным водным бикарбонатом натрия, солевым раствором, сушились над безводным сульфатом магния, фильтровались и концентрировались, давая белую пену, 490 мг. Данное вещество кристаллизовалось из гексанов и этилацетата, давая 428 мг, 80% чистого продукта с т.пл. 123 - 127oC, HR FAB MC; вычислено для C27H39N3O7S2: 588,2398. Найдено: 588,2395.
Пример 11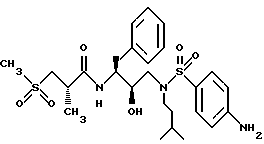
Получение [1S-[1R*(R*), 2S*]]-N-[2-гидрокси-3-[(3-метилбутил)(4-аминофенилсульфонил)амино] -1- (фенилметил)пропил] -2-метил-3-(метилсульфонил)-пропанамида
Стадия A.
Раствор продукта из примера 1, стадии C (1,1812 г, 3,07 ммоля) и триэтиламина (325,7 мг, 3,22 ммоля) в 20 мл дихлорметана обрабатывался 4-нитробензолсульфонилхлоридом (767 мг, 90% чистоты, 3,11 ммоля) при комнатной температуре в течение 10 минут. Растворитель удалялся в вакууме, а остаток брался в этилацетат и затем промывался 1 норм. бисульфатом калия, насыщенным водным бикарбонатом натрия, солевым раствором, сушился над безводным сульфатом магния, фильтровался и концентрировался, давая 2,3230 г рыжевато-коричневого твердого вещества, которое кристаллизовалось из смеси этилацетата и петролейного эфира, давая 870 мг, 50%, т.пл. 130 - 132oC чистого продукта, HR FAB MC; M + Li. Вычислено для C29H35N3O7SLi: 576,2316. Найдено: 576,2350.
Стадия B.
Раствор продукта со стадии A (574 мг, 1,01 ммоля) в 40 мл метанола (раствор не был полностью гомогенным) обрабатывался 70 мг катализатора 10% палладий на угле и гидрировался при 42 фунт/кв. дюйм (2,953 кг/кв. см) в течение 15 часов при комнатной температуре. Катализатор удалялся фильтрованием через диатомовую землю, и фильтрат концентрировался в вакууме, давая белое твердое вещество, которое кристаллизовалось из хлороформа, т.пл. 123 - 127oC, FAB MC: M + Li = 412, 400 мг, 91%, которое использовалось непосредственно на следующей стадии без дальнейшей очистки.
Стадия C.
Смесь 2(S)-метил-3-метилсульфонилпропионовой кислоты (112,3 мг, 0,675 ммоля), N-гидроксибензотриазола (159,1 мг, 1,04 ммоля) и гидрохлорида 1-(3-диметиламинопропил)-3-этилкарбодиимида (EDC) (147,8 мг, 0,77 ммоля) растворялась в 1,0 мл диметилформамида и оставлялась реагировать в течение 30 минут при 0oC. К указанной выше смеси добавлялся амино со стадии B (261,9 мг, 0,646 ммоля), растворенный в 0,5 мл ДМФ и смесь перемешивалась при комнатной температуре в течение 16,5 часов. Раствор затем выливался в 75 мл насыщенного водного бикарбоната натрия и экстрагировался этилацетатом. Этилацетатные экстракты промывались 5% водной лимонной кислотой, насыщенным водным бикарбонатом натрия, солевым раствором, сушились над безводным сульфатом магния, фильтровались и концентрировались, давая белую пену, 326,3 мг. Данное вещество очищалось с помощью флэш хроматографии на силикагеле при элюировании этилацетатом, давая 213,6 мг, 64% чистого продукта в виде белой пены, FAB MC; MH+=554.
Пример 12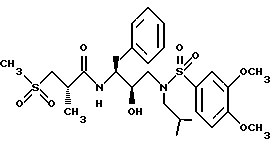
Получение [1S-[1R*(R*), 2S*]]-N-[2-гидрокси-3-[(2-метилпропил)(3,4-диметоксифенилсульфонил) амино]-1-(фенилметил)пропил]-2-метил-3-(метилсульфонил)-пропанамида
Стадия A.
Раствор продукта из примера 3, стадии A (1,5356 г, 4,14 ммоля) и триэтиламина (522 мг, 5,17 ммоля) в 15 мл дихлорметана обрабатывался 3,4-диметоксибензолсульфонилхлоридом (1,0087 г, 4,26 ммоля) при комнатной температуре в течение 14 часов. Растворитель удалялся в вакууме, и остаток брался в этилацетат, а затем промывался 1 норм. бисульфатом калия, насыщенным водным бикарбонатом натрия, солевым раствором, сушился над безводным сульфатом магния, фильтровался и концентрировался, давая 2,147 г, 90,5%, белого твердого вещества, т.пл. 124 - 127oC, HR FAB MC; M + Li; вычислено для C30H38N2O7S+Li: 577,2560. Найдено: 577,2604.
Стадия B
Раствор карбаминовой кислоты, продукта со стадии A (513 мг, 0,90 ммоля) в 30 мл метанола перемешивался с 20 мг катализатора палладиевой черни и 10 мл муравьиной кислоты в течение 15 часов при комнатной температуре. Катализатор удалялся фильтрованием через диатомовую землю, и фильтрат концентрировался в вакууме, а остаток брался в этилацетат. Этилацетатный раствор промывался насыщенным водным бикарбонатом натрия, солевым раствором и сушился над безводным сульфатом магния, фильтровался и концентрировался, давая прозрачное масло, 220 мг. Данное вещество кристаллизовалось из гексанов и этилацетата, давая 178 мг, 40% чистого продукта с т.пл. 130-133oC, HR FAB MC; M + Li; вычислено для C27H40N2O8S2Li: 591,2386. Найдено: 591,2396.
Пример 13
Получение (1S-[1R*(R*), 2S*]]-N-[2-гидрокси-3-[(2-метилпропил)(4-гидроксифенилсульфонил)амино]- 1-(фенилметил)пропил]-пропанамида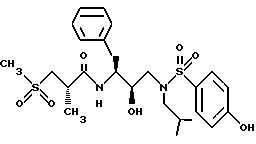
Стадия A.
Раствор 0,98 г (1,85 ммоля) [2R-гидрокси-3-[[(4-фторфенил)сульфонил] (2-метилпропил)амино] -1S- (фенилметил)пропил]-фенилметилового эфира карбаминовой кислоты в 3,8 мл безводного ДМФ добавлялся к 22 мг (7,4 ммоля) 80% гидрида натрия в 2 мл ДМФ. К данной смеси добавлялось 0,40 г (3,7 ммоля) бензилового спирта. Спустя 2 часа раствор охлаждался до 0oC, добавлялась вода, а затем этилацетат. Органический слой промывался 5% лимонной кислотой, насыщенным бикарбонатом натрия и солевым раствором, сушился над сульфатом магния, фильтровался и концентрировался, давая 0,90 г сырого вещества. Данное вещество хроматографировалась на основной окиси алюминия с использованием смеси 3% метанол/метиленхлоридом, давая 0,70 г 2R-гидрокси-3-[(2-метилпропил)-(4-бензилоксифенил)сульфонил] амино-1- (фенилметил)пропиламина, циклического карбамата; масс спектр m/e = 509 (M+H).
Стадия B.
К раствору 0,65 г (1,28 ммоля) циклического карбамата со стадии A в 15 мл этанола добавлялось 2,6 мл (6,4 ммоля) 2,5 норм. раствора гидроокиси натрия. После 1 часа нагревания с обратным холодильником добавлялось 4 мл воды, и раствор нагревался в условиях дефлегмации в течение дополнительных восьми часов. Летучие вещества удалялись, добавлялся этилацетат, и смесь промывалась водой, солевым раствором, сушилась над сульфатом магния, фильтровалась и концентрировалась, давая 550 мг неочищенного 2R-гидрокси-3-[(2-метилпропил)(4-бензилоксифенил)сульфонил]амино-1S- (фенилметил)пропиламина.
Стадия C.
Раствор неочищенного 2R-гидрокси-3-[(2-метилпропил)-(4-бензилоксифенил)сульфонил]амино-1S- (фенилметил)пропиламина в 10 мл этанола гидрировался в присутствии 500 мг катализатора 10% палладия на угле под давлением водорода 50 фунт/кв. дюйм (3,515 кг/кв.см) в течение 2 часов. Катализатор удалялся фильтрованием, и растворитель удалялся в вакууме, давая 330 мг 2R-гидрокси-3-[(2-метилпропил)(4-гидроксифенил)сульфонил]амино-1S- (фенилметил)пропиламина, масс спектр m/e=393 (M+H).
Стадия D.
К раствору 337 мг (2,03 ммоля) 2(S)-метил-3-(метилсульфонил)пропионовой кислоты и 423 мг (2,21 ммоля) N-гидроксибензотриазола в 4 мл безводного ДМФ при 0oC добавлялось 423 мг (2,76 ммоля) гидрохлорида 1-(3-диметиламинопропил)-3-этилкарбодиимида. После перемешивания в течение 2 часов добавлялся амин со стадии C выше, и раствор перемешивался при комнатной температуре в течение 17 часов. Растворитель удалялся в вакууме, добавлялся этилацетат, а затем смесь промывалась насыщенным водным бикарбонатом натрия, солевым раствором, сушилась над сульфатом магния, фильтровалась и концентрировалась, давая 939 мг неочищенного продукта. Хроматография на силикагеле с использованием смеси 2-5% метанол/метиленхлорид давала 533 мг пропанамида, N-[2-гидрокси-3-[(2-метилпропил)(4-гидроксифенилсульфонил)амино]-1- (фенилметил)пропил] -2-метил-3-(метилсульфонил)-, [1S-[1R*-(R*), 2S*]], масс спектр m/e = 547 (M+Li).
Пример 14.
Следующие общие приемы могут использоваться для получения дополнительных соединений, охватываемых объемом настоящего изобретения. Общая процедура синтеза аминоэпоксидов.
К раствору 0,226 моля N-бензиоксикарбонил-L-фенилаланинхлорметилкетона в смеси 807 мл метанола и 807 мл тетрагидрофурана при -2oC добавляется 1,54 эквив. твердого боргидрида натрия на протяжении ста минут. Растворители затем удаляются при пониженном давлении при 40oC, и остаток растворяется в этилацетате (приблизительно 1 л). Раствор промывается последовательно 1 М бисульфатом калия, насыщенным бикарбонатом натрия и затем насыщенными растворами хлористого натрия. После сушки над безводным сульфатом магния и фильтрования, раствор удаляется при пониженном давлении. К получающемуся в результате маслу добавляется гексан (приблизительно 1 л), и смесь подогревается до 60oC при вихревом перемешивании. После охлаждения до комнатной температуры твердые вещества собираются и промываются 2 литрами гексана. Получающееся в результате твердое вещество перекристаллизуется из горячего этилацетата и гексана, давая 32,3 г (43% выход) N-бензилоксикарбонил-3(S)-амино-1-хлор-4-фенил-2(S)-бутанола, т.пл. 150-151oC, и M + Li*=340.
Стадия B.
К раствору 1,2 эквивалента гидроокиси калия в 968 мл абсолютного этанола при комнатной температуре добавляется 0,097 моля N-CBZ-3(S)-амино-1-хлор-4-фенил-2(S)-бутанола. После перемешивания в течение пятнадцати минут растворитель удаляется при пониженном давлении, и твердые вещества растворяются в метиленхлориде. После промывания водой, сушки над сульфатом магния, фильтрования и отгонки с паром получают белое твердое вещество. Перекристаллизация из горячего этилацетата и гексана дает N-бензилоксикарбонил-3(S)-амино-1,2(S)-эпокси-4-фенилбутан.
Альтернативная процедура синтеза аминоэпоксидов.
Стадия A.
Раствор L-фенилаланина (50,0 г, 0,302 моля), гидроокиси натрия (24,2 г, 0,605 моля) и карбоната калия (83,6 г, 0,605 моля) в воде (500 мл) нагревается до 97oC. Затем медленно добавляется бензилбромид (108,5 мл, 0,912 моля) (время добавления = примерно 25 минут). Смесь затем перемешивается при 97oC в течение 30 минут. Раствор охлаждается до комнатной температуры и экстрагируется толуолом (2х250 мл). Объединенные органические слои затем промываются водой, солевым раствором, сушатся над сульфатом магния, фильтруются и концентрируются, давая продукт в виде масла. Неочищенный продукт затем используется на следующей стадии без очистки.
Стадия B.
Сырой бензилированный продукт описанной выше стадии растворяется в толуоле (750 мл) и охлаждается до -55oC. Затем добавляется 1,5 М раствор DIBAL-H в толуоле (443,9 мл, 0,666 моля) со скоростью поддержания температуры между -55oC и -50oC (время добавления - 1 час). Смесь перемешивается в течение 20 минут при -55oC. Реакционная смесь гасится при -55oC медленным добавлением метанола (37 мл). Холодный раствор затем выливается в холодный (5oC) 1,5 норм. раствор HCl (1,8 л). Выпавшее в осадок твердое вещество (приблизительно 138 г) отфильтровывается и промывается толуолом. Твердый материал суспендируется в смеси толуола (400 мл) и воды (100 мл). Смесь охлаждается до 5oC, обрабатывается 2,5 норм. гидроокисью натрия (186 мл), а затем перемешивается при комнатной температуре до тех пор, пока твердое вещество не растворится. Толуольный слой отделяется от водной фазы и промывается водой и солевым раствором, сушится над сульфатом магния, фильтруется и концентрируется до объема 75 мл (89 г). Затем к остатку добавляются этилацетат (25 мл) и гексан (25 мл), после чего спиртовый продукт начинает кристаллизоваться. Спустя 30 минут для промотирования дальнейшей кристаллизации добавляется дополнительно 50 мл гексана. Твердое вещество отфильтровывается и промывается 50 мл гексана, давая приблизительно 35 г вещества. Еще одна порция вещества может быть отделена с помощью повторного отфильтровывания маточной жидкости. Твердые вещества объединяются и перекристаллизуются из этилацетата (20 мл) и гексана (30 мл), давая в 2 сборах примерно 40 г (40% из L-фенилаланина) аналитически чистого спиртового продукта. Маточные жидкости объединяются и концентрируются (34 г). Остаток обрабатывается этилацетатом и гексаном, что дает дополнительно 7 г (-7% выход) слегка нечистого твердого продукта. Возможно осуществить дальнейшую оптимизацию при выделении из маточной жидкости.
Стадия C.
Раствор оксалилхлорида (8,4 мл, 0,096 моля) в дихлорметане (240 мл) охлаждается до -74oC. Затем медленно добавляется раствор ДМСО (12,0 мл, 0,155 моля) в дихлорметане (50 мл) со скоростью, поддерживающей температуру на уровне -74oC (время добавления примерно 1,25 часа). Смесь перемешивается в течение 5 минут с последующим добавлением раствора спирта (0,074 моля) в 100 мл дихлорметана (время добавления примерно 20 минут, температура -75oC до -68oC). Раствор перемешивается при -78oC в течение 35 минут. Затем на протяжении 10 минут добавляется триэтиламин (41,2 мл, 0,295 моля) (температура от -78 до -68oC), после чего выпадает в осадок аммониевая соль. Холодная смесь перемешивается в течение 30 минут, а затем добавляется вода (225 мл). Дихлорметановый слой отделяется от водной фазы и промывается водой, солевым раствором, сушится над сульфатом магния, фильтруется и концентрируется. Остаток разбавляется этилацетатом и гексаном, а затем фильтруется для дополнительного удаления аммониевой соли. Фильтрат концентрируется, давая желаемый альдегидный продукт. Альдегид используется на следующей стадии без очистки.
В литературе для окисления по Сверну сообщались температуры выше, чем -70oC. Возможны также другие модификации Сверна и альтернативные приемы окисления по Сверну.
Раствор сырого альдегида 0,074 моля и хлориодметана (7,0 мл, 0,096 моля) в тетрагидрофуране (285 мл) охлаждается до -78oC, 1,6 М раствор н-бутиллития в гексане (25 мл, 0,040 моля) затем добавляется со скоростью, обеспечивающей поддержание температуры при -75oC (время добавления - 15 минут). После первого добавления снова добавляется дополнительно хлориодметан (1,6 мл, 0,022 моля), с последующим добавлением н-бутиллития (23 мл, 0,037 моля) при сохранении температуры на уровне -75oC. Смесь перемешивается в течение 15 минут. Каждый из реагентов, хлориодметан (0,70 мл, 0,010 моля) и н-бутиллитий (5 мл, 0,008 моля) добавляются еще 4 раза на протяжении 45 минут при -75oC. Охлаждающая баня затем удаляется и раствор подогревается до 22oC на протяжении 1,5 часов. Смесь выливается в 300 мл насыщенного водного раствора хлористого аммония. Тетрагидрофурановый слой отделяется. Водная фаза экстрагируется этилацетатом (1х300 мл). Объединенные органические слои промываются солевым раствором, сушатся над сульфатом магния, фильтруются и концентрируются, давая коричневое масло (27,4 г). Продукт можно использовать на следующей стадии без очистки. Желаемый диастереомер может очищаться с помощью перекристаллизации на последующей стадии образования сульфонамида. Альтернативно продукт может очищаться с помощью хроматографии.
Общая процедура синтеза производных 1,3-диамино-4-фенил-бутан-2-ола (Аминоспирты).
Смесь амина R3NH2 (20 эквивалентов) в сухом изопропиловом спирте (20 мл/ммоль эпоксида, подвергаемого превращению) нагревается до температуры дефлегмации и затем обрабатывается N-Cbz аминоэпоксидом формулы: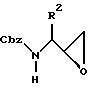
из воронки для добавления твердого вещества на протяжении периода 10-15 минут. После того, как добавление завершается, раствор поддерживается в условиях нагревания с обратным холодильником в течение дополнительных 15 минут, и за ходом реакции следят по данным ТСХ. Реакционная смесь затем концентрируется в вакууме, давая масло, а затем оно обрабатывается н-гексаном при быстром перемешивании, после чего из раствора выпадает в осадок вещество с раскрытым кольцом. Осаждение обычно завершается в пределах 1 часа, и продукт затем отделяется с помощью фильтрования на воронке Бюхнера, а затем сушится воздухом. Продукт дополнительно сушится в вакууме. Данный способ дает аминоспирты достаточной чистоты для большинства целей.
Таблица 1 показывает характерные представители аминоспиртов, полученных согласно описанным выше общим приемам.
Общая процедура для осуществления реакции аминоспиртов с сульфонилгалогенидами или сульфонилангидридами. Получение сульфонамидов.
К раствору N[3(S)-бензилоксикарбониламино-2-(R)-гидрокси-4-фенилбутил] -N-изоамиламина (2,0 г, 5,2 ммоля) и триэтиламина (723 мкл, 5,5 ммолей) в дихлорметане (20 мл) добавляется по каплям метансульфонилхлорид (400 мкл, 5,2 ммоля). Реакционная смесь перемешивается в течение 2 часов при комнатной температуре, затем дихлорметановый раствор концентрируется до приблизительно 5 мл и наносится на силикагельную колонку (100 г). Колонка элюируется хлороформом, содержащим 1% этанол и 1% метанол.
Альтернативно в результате реакции N-[3(S)-бензилоксикарбониламино-2(R)-гидрокси-4-фенилбутил]-N- изоамиламина (1,47 г, 3,8 ммоля), и триэтиламина (528 мкл, 3,8 ммоля) и бензолсульфонилхлорида (483 мкл, 3,8 ммоля) можно получить соответствующее (фенилсульфонил)амино производное.
В Таблице 2 показаны характерные представители сульфонамидов, полученных согласно описанной выше процедуре.
Общая процедура удаления защитных групп с помощью гидрогенолиза с использованием палладия на угле
A. Спиртовый растворитель.
Cbz - Защищенное пептидное производное растворяется в метаноле (приблиз. 20 мл/ммоль), и в атмосфере азота добавляется 10% палладиевый катализатор на угле. Реакционный сосуд герметизируется и продувается 5 раз азотом, а затем 5 раз водородом. Давление поддерживается при 50 фунт/кв. дюйм (3,515 кг/кв. см) в течение 1-16 часов, а затем водород заменяется азотом, и раствор фильтруется через средство из целита для удаления катализатора. Растворитель удаляется в вакууме, давая свободное аминопроизводное подходящей чистоты для применения непосредственно на следующей стадии.
B. Растворитель уксусная кислота
Cbz- Защищенное пептидное производное растворяется в ледяной уксусной кислоте (20 мл/ммоль) и в атмосфере азота добавляется катализатор 10% палладий на угле. Реакционный сосуд продувается 5 раз азотом и 5 раз водородом и затем поддерживается при давлении 40 фунт/кв. дюйм (2,812 кг/кв. см) в течение примерно 2 часов. Водород затем заменяется азотом, и реакционная смесь фильтруется через фильтровальное средство из целита для удаления катализатора. Фильтрат концентрируется, и получающийся в результате продукт берется в безводный эфир и выпаривается досуха 3 раза. Конечный продукт, ацетатная соль, сушится в вакууме и имеет подходящую чистоту для последующего превращения.
Общая процедура удаления Boc-защитной группы с помощью 4 норм. соляной кислоты в диоксане
Boc-Защищенная аминокислота или пептидное производное обрабатывается раствором 4 норм. HCl в диоксане при перемешивании при комнатной температуре. Обычно реакция снятия защиты завершается за 15 минут, за ходом реакции следят по данным тонкослойной хроматографии (ТСХ). После завершения реакции избыток диоксана и HCl удаляется с помощью выпаривания в вакууме. Последние следы диоксана и HCl наилучшим образом удаляются с помощью повторного выпаривания из безводного эфира или ацетона. Гидрохлоридная соль, полученная таким образом, тщательно сушится в вакууме и является подходящей для дальнейшей реакции.
Процедуры получения сульфонильных соединений
Процедуры, описанные ниже в примерах 13A, 13B и 13C иллюстрируют приемы получения сульфонилалканоильных соединений, которые могут присоединяться к сульфонамидам, полученным выше.
Пример 14A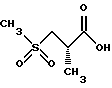
Получение 2(S)-метил-3-(метилсульфонил)-пропионовой кислоты
К раствору 10 г трет-бутилового эфира D-(-)-S-бензоил-b-меркаптоизомасляной кислоты в 20 мл метанола добавлялся барботированием через раствор газообразный аммиак при 0oC. Реакционной смеси затем давали возможность подогреваться до комнатной температуры, смесь перемешивалась на протяжении ночи и концентрировалась при пониженном давлении. Получающаяся в результате смесь твердого вещества (бензамида) и жидкости фильтровалась, давая 5,21 г бледного масла, которое затем затвердевало. Данное вещество идентифицировалось как трет-бутиловый эфир 2(S)-метил-3-меркаптопропионовой кислоты.
К раствору 5,21 г трет-бутилового эфира 2(S)-метил-3-меркаптопропионовой кислоты в 75 мл толуола при 0oC добавлялось 4,50 г 1,8-диазабицикло [5.4.0] ундец-7-ена и 1,94 мл метилиодида. После перемешивания при комнатной температуре в течение 2,5 часов, летучие вещества удалялись, добавлялся этилацетат, осуществлялась промывка разбавленной соляной кислотой, водой, солевым раствором, сушка и концентрирование, давая 2,82 г бледного масла, идентифицированного как трет-бутиловый эфир 2(S)-метил-3-(тиометил)пропионовой кислоты.
К раствору 2,82 г трет-бутилового эфира 2(S)-метил-3-(тиометил)пропионовой кислоты в 50 мл уксусной кислоты добавлялось 5,58 г пербората натрия, и смесь нагревалась до 55oC в течение 17 часов. Реакционная смесь выливалась в воду, экстрагировалась метиленхлоридом, промывалась водным бикарбонатом натрия, сушилась и концентрировалась, давая 2,68 г трет-бутилового эфира 2(S)-метил-3-(метилсульфонил)-пропионовой кислоты в виде белого твердого вещества.
К 2,68 г трет-бутилового эфира 2(S)-метил-3-(метилсульфонил) пропионовой кислоты добавлялось 20 мл смеси 4 норм. соляной кислоты/диоксана, и смесь перемешивалась при комнатной температуре в течение 19 часов. Растворитель удалялся при пониженном давлении, давая 2,18 г сырого продукта, который перекристаллизовывался из смеси этилацетат/гексан, давая 1,44 г 2(S)-метил-3-(метилсульфонил)пропионовой кислоты в виде белых кристаллов.
Пример 14B
Стадия A.
Раствор метилметакрилата (7,25 г, 72,5 ммолей) и фенэтилмеркаптана (10,0 г, 72,5 ммоля) в 100 мл метанола охлаждался на ледяной бане и обрабатывался метилатом натрия (100 г, 1,85 ммоля). Раствор перемешивался в атмосфере азота в течение 3 часов, а затем концентрировался в вакууме, давая масло, которое бралось в эфир и промывалось 1 норм. водным бисульфатом калия, насыщенным водным хлоридом натрия, сушился над безводным сульфатом магния, фильтровался и концентрировался, давая 16,83 г, 97,5% метил 2-(R,S)-метил-4-тиа-6-фенилгексаноата в виде масла. ТСХ на SiO2 с элюированием смесью 20:1 гексан:этилацетат (объем/объем) Rf = 0,41. Альтернативно, можно использовать вместо метилметакрилата 3-бром-2-метилпропионат.
Стадия B.
Раствор метил 2-(R,S)-метил-4-тио-6-фенилгексаноата (4,00 г, 16,8 ммолей) в 100 мл дихлорметана перемешивался при комнатной температуре и обрабатывался порциями мета-хлорпероксибензойной кислотой (7,38 г, 39,2 ммолей) на протяжении приблизительно 40 минут. Раствор перемешивался при комнатной температуре в течение 16 часов, а затем фильтровался, и фильтрат промывался насыщенным водным бикарбонатом натрия, 1 норм. гидроокисью натрия, насыщенным водным хлористым натрием, сушился над безводным сульфатом магния, фильтровался и концентрировался, давая 4,50 г, 99% желаемого сульфона. Неочищенный сульфон растворялся в 100 мл тетрагидрофурана и обрабатывался раствором гидроокиси лития (1,04 г, 24,5 ммоля) в 40 мл воды. Раствор перемешивался при комнатной температуре в течение 2 минут, а затем концентрировался в вакууме. Остаток затем подкислялся 1 норм. водным бисульфатом калия до pH 1, а затем экстрагировался три раза этилацетатом. Объединенный этилацетатный раствор промывался насыщенным водным хлористым натрием, сушился над безводным сульфатом магния, фильтровался и концентрировался, давая белое твердое вещество. Твердое вещество бралось в кипящую смесь этилацетат/гексан и оставлялось стоять спокойно, после чего образовывались белые иглы, которые отделялись фильтрованием и сушилась воздухом, давая 3,38 г, 79% 2-(R,S)-метил-3- β- фенэтилсульфонил)-пропионовой кислоты, т.пл. 91 - 93oC.
Стадия C
Раствор 2-(R, S)-метил-3- ( β- фенэтилсульфонил)пропионовой кислоты (166,1 мг, 0,65 ммоля), N-гидроксибензотриазола (HOBT) (146,9 мг, 0,97 ммоля) и гидрохлорида 1-(3-диметиламинопропил)-3-этилкарбодиимида (EDC) (145,8 мг, 0,75 ммоля) в 4 мл безводного диметилформамида (ДМФ) охлаждался до 0oC и перемешивался в атмосфере азота в течение получаса. Данный раствор затем обрабатывался желаемым изостером сульфонамида и перемешивался при комнатной температуре в течение 16 часов. Раствор выливался в 30 мл 60% насыщенного водного раствора бикарбоната натрия. Водный раствор затем декантировался от органического остатка. Органический остаток брался в дихлорметан и промывался 10% водной лимонной кислотой, солевым раствором, сушился над безводным сульфатом магния, фильтровался и концентрировался. Флэш хроматография смеси на силикагеле с элюированием смесью 1:1 гексан/этилацетат может использоваться и давать раздельные диастереомеры.
Пример 14C
Стадия A
Раствор метил 2-(бромметил)-акрилата (26,4 г, 0,148 моля) в 100 мл метанола обрабатывался метансульфинатом натрия (15,1 г, 0,148 моля) порциями на протяжении 10 минут при комнатной температуре. Раствор затем перемешивался при комнатной температуре в течение периода 1,25 часа, и раствор концентрировался в вакууме. Остаток затем брался в воду, экстрагировался четыре раза этилацетатом. Объединенный этилацетатный раствор промывался насыщенным хлоридом натрия, сушился над безводным сульфатом магния, фильтровался и концентрировался, давая белое твердое вещество, 20,7 г, которое бралось в кипящую смесь ацетон/метил-трет-бутиловый эфир и оставлялось стоять, после чего образовывались кристаллы чистого метил 2-(метилсульфонилметил)акрилата (18,0 г, 68%), т.пл. 65 - 68oC.
Стадия B.
Раствор метил 2-(метилсульфонилметил)акрилата (970 мг, 5,44 ммоля) в 15 мл тетрагидрофурана обрабатывался раствором гидроокиси лития (270 мг, 6,4 ммоля) в 7 мл воды. Раствор перемешивался при комнатной температуре в течение 5 минут, а затем подкислялся до pH1 1 норм. водным бисульфатом калия, и раствор экстрагировался три раза этилацетатом. Объединенный этилацетатный раствор сушился над безводным сульфатом магния, фильтровался и концентрировался, давая 793 мг, 89% 2-(метилсульфонилметил)акриловой кислоты, т.пл. 147 - 149oC.
Стадия C.
Раствор 2-(метилсульфонилметил)акриловой кислоты (700 мг. 4,26 ммоля) в 20 мл метанола загружался в бутыль Фишера-Портера наряду с 10% палладиевым катализатором на угле в атмосфере азота. Реакционный сосуд запечатывался и продувался пять раз азотом, а затем пять раз водородом. Давление поддерживалось при 50 фунт/кв. дюйм (3,515 кг/кв. см) в течение 16 часов, а затем водород заменялся азотом, и раствор фильтровался через фильтровальное средство из целита для удаления катализатора, и фильтрат концентрировался в вакууме, давая 682 мг, 96% 2-(R,S)-метил-3-метилсульфонил-пропионовой кислоты.
Стадия D
Раствор 2-(R, S)-метил-3-(метилсульфонил)пропионовой кислоты (263,5 мг, 1,585 ммоля), N-гидроксибензотриазола (НОВТ) (322,2 мг, 2,13 ммоля) и гидрохлорида 1-(3-диметиламинопропил)-3-этилкарбодиимида (EDC) (339,1 мг, 1,74 ммоля) в 4 мл безводного диметилформамида (ДМФ) охлаждался до 0oC и перемешивался в атмосфере азота в течение 0,5 часа. Данный раствор затем обрабатывался желаемым сульфонамидом и перемешивался при комнатной температуре в течение 16 часов. Раствор выливался в 60 мл 60% насыщенного водного раствора бикарбоната натрия. Водный раствор затем декантировался с органического остатка. Органический остаток брался в дихлорметан и промывался 10% водной лимонной кислотой, солевым раствором, сушился над безводным сульфатом магния, фильтровался и концентрировался, давая желаемый продукт.
Пример 14D
Получение сульфоновых ингибиторов из L-(+)-S-ацетил β- меркаптоизомасляной кислоты
Стадия A
Круглодонная колба загружалась желаемым изостером сульфонамида (2,575 ммоля), например, амин со стадии C примера 3 может присоединяться к L-(+)-S-ацетил β- меркаптомасляной кислоте в присутствии гидрохлорида 1-(3-диметиламино-пропил)-3-этилкарбодиимида (EDC) (339,1 мг, 1,74 ммоля) в 10 мл метиленхлорида, и реакционная смесь оставляется перемешиваться при комнатной температуре в течение 16 часов. Раствор концентрируется в вакууме, и остаток берется в этилацетат, промывается 1 норм. бисульфатом калия, насыщенным водным бикарбонатом натрия, солевым раствором, сушится над безводным сульфатом магния, фильтруется и концентрируется, давая масло, которое может очищаться с помощью радиальной хроматографии на SiO2 при элюировании этилацетатом, давая чистый продукт.
Стадия B
Раствор продукта стадии A (0,85 ммоля) в 10 мл метанола обрабатывается безводным аммиаком в течение примерно 1 минуты при 0oC. Раствор перемешивается при данной температуре в течение 16 часов, и затем концентрируется в вакууме, давая желаемый продукт, который может использоваться непосредственно на следующей стадии без дальнейшей очистки.
Стадия C
Раствор продукта стадии B (0,841 ммоля) в 10 мл сухого толуола в атмосфере азота обрабатывается сразу же последовательно 1,8-диазабицикло[5.4.0]ундец-7-еном (DBU), (128,1 мг, 0,841 ммоля) и иодметаном (119,0 мг, 0,841 ммоля). После нахождения в течение получаса при комнатной температуре реакционная смесь разбавляется этилацетатом, промывается 1 норм. бисульфатом калия, насыщенным водным бикарбонатом натрия, солевым раствором. После того как раствор сушится над безводным сульфатом магния, фильтруется и концентрируется в вакууме, получается желаемый продукт и может использоваться непосредственно на следующей стадии.
Стадия D:
Раствор продукта стадии C (0,73 ммоля) и пербората натрия (500 мг, 3,25 ммоля) в 30 мл ледяной уксусной кислоты подогревается до 55oC в течение 16 часов. Раствор концентрируется в вакууме, а затем остаток берется в этилацетат, промывается водой, насыщенным водным бикарбонатом натрия, солевым раствором, сушится над безводным сульфатом магния, фильтруется и концентрируется, давая желаемый продукт.
Представители сульфонов, полученные согласно описанным выше общим процедурам, показаны в Таблице 3.
Общая процедура сочетания сульфонильных соединений с сульфонамидами.
Смесь сульфонилалканоильного соединения (приблизительно 1 ммоль), N-гидроксибензотриазола (1,5 ммоля) и гидрохлорида 1-(3-диметиламинопропил)-3-этилкарбодиимида (EDC) (1,2 ммоля) растворяется в подходящем растворителе таком, как ДМФ, и оставляется реагировать в течение примерно 30 минут при 0oC. Сульфонамид (1,05 ммоля) растворяется в ДМФ, добавляется к указанной выше смеси и перемешивается при комнатной температуре в течение периода времени, достаточного для того, чтобы произошла реакция. Раствор затем выливается в насыщенный водный бикарбонат натрия и экстрагируется, например, этилацетатом. Экстракты промываются, сушатся, фильтруются и концентрируются. Получающийся материал затем кристаллизуется из подходящего растворителя или смеси растворителей, таком, как гексан и этилацетат, давая продукт.
Характерные представители соединений, полученных согласно этим общим процедурам, показаны в Таблице 3a.
Пример 15
При использовании общих и конкретных процедур, описанных в примерах 1-14, могут быть получены соединения, показанные в Таблицах 4 - 8.
Пример 16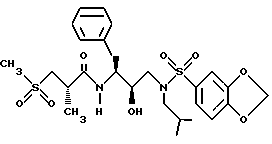
Получение N-[2R-гидрокси-3-[(2-метилпропил)[(1,3-бензодиоксол-5-ил)сульфонил)амино]- 1S-(фенилметил]пропил]-2S-метил-3-(метилсульфонил)пропанамида.
К раствору 5,0 г (30 ммоль) 2S-метил-3-(метилсульфонил)пропионовой кислоты и 6,90 г (45 ммоль) N-гидроксибензотриазола в 30 мл безводного ДМФ при 0oC в атмосфере азота добавлялось 6,34 г (33 ммоль) EDC. Спустя приблизительно 10 минут весь EDC растворялся. Спустя 60 минут при 0oC добавлялся раствор 15,5 г (30 ммоль) метансульфоната 2R-гидрокси-3-[[1,3-бензодиоксол-5-ил)-сульфонил] (2-метилпропил)амино] -1S- (фенилметил)пропиламина в 30 мл безводного ДМФ, предварительно нейтрализованный 3,4 миллилитрами (31,6 ммоль) 4-метилморфолина. После 3 часов при 0oC смесь затем перемешивалась на протяжении ночи в течение 17 часов. ДМФ удалялся при пониженном давлении, добавлялся этилацетат, промывался 5% лимонной кислотой, насыщенным бикарбонатом натрия, водой, солевым раствором, сушился над безводным сульфатом магния, фильтровался и концентрировался, давая 16 г сырого или неочищенного вещества, которое по данным ВЭЖХ было 88% чистоты. Продукт хроматографировался на силикагеле с использованием 20%-80% смеси этилацетат/гексан, давая чистый продукт, который перекристаллизовывается из смеси этилацетат/гексан, давая 8.84 г чистого продукта т.пл. 131,8-133,8oC.
Альтернативно к раствору 35,0 г (211 ммоль) 2S-метил-3-(метилсульфонил)пропионовый кислоты и 48,3 г (315 ммоль) N-гидроксибензотриазола в 210 мл безводного ДМФ при 0oC в атмосфере азота добавлялось 44,4 г (231 ммоль) EDC. Спустя приблизительно 30 минут весь EDC растворялся. Спустя 60 минут при 0oC добавлялся раствор 108,8 г (211 ммоль) метансульфоната 2R-гидрокси-3-[[(1,3-бензодиоксол-5-ил)сульфонил] (2-метилпропил)амино] -1S-(фенилметил)пропиламина в 350 мл безводного ДМФ, предварительно нейтрализованный 24 мл-ми (22,3 г) 4-метилформолина. После 2 часов при 0oC смесь затем перемешивалась на протяжении ночи в течение 18 часов. ДМФ удалялся при пониженном давлении, добавлялся 1 л этилацетата, смесь промывалась 5% лимонной кислотой, насыщенным бикарбонатом натрия, водой, солевым раствором, сушилась над безводным сульфатом магния, фильтровалась и концентрировалась, давая 120,4 г сырого или неочищенного вещества, которое по данным ВЭЖХ было 90% чистоты. Продукт кристаллизовался дважды из 750-1000 мл абсолютного этанола, давая 82,6 г желаемого продукта, по данным ВЭЖХ вещество было 99% чистоты.
Пример 17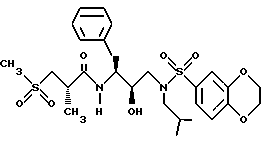
Получение N-(2R-гидрокси-3-[(2-метилпропил)[(1,4-бензодиоксан-6-ил)сульфонил] амино]-1S-(фенилметил)пропил]-2S-метил-3-(метилсульфонил)пропанамида.
Часть A: Раствор 1-[(2-метилпропил)[1,4-бензодиоксан-6-ил) сульфонил] амино] -3S-[(фенилметоксикарбонил)амино] -4-фенилбутан-2R-ола (0,6 г, 1,06 ммоль) в ТГФ (10 мл) гидрировался при 50 фунт./кв. дюйм (3,515 кг/кв.см) в присутствии 10% Pd/C (0,4 г) в течение 12 часов, при комнатной температуре. Катализатор удалялся с помощью фильтрования, фильтрат концентрировался при пониженном давлении.
Часть B: Получающийся в результате остаток из части A растворялся в метиленхлориде (4,0 мл) и добавлялся к охлажденной (0oC) смеси 2S-метил-3-(метилсульфонил)пропионовой кислоты (0,2 г, 1,2 ммоль), HOBt (0,25 г, 1,6 ммоль) и EDC (0,24 г, 1,25 ммоль) в смеси растворителей ДМФ (2 мл) и метиленхлорида (2 мл) и перемешивался при 0oC в течение 2 часов. После перемешивания в течение 3 часов при комнатной температуре реакционная смесь разбавлялась метиленхлоридом (15 мл), промывалась холодной 0,5 норм. NaOH (2 х 10 мл), водой (3 х 15 мл), сушилась (Na2SO4) и концентрировалась в вакууме. Остаток очищался с помощью флэш хроматографии на силикагеле с использованием EtOAc в качестве элюента, давая желаемый сульфонамид в виде белого аморфного порошка (0,5 г, 82%). Rt=19,9 мин. FABMS m/z 589 (M+ Li)+; HRMS-FAB вычислено для C27H39N2O3S2 583,2148 (MH)+, найдено 583,2115.
Пример 18.
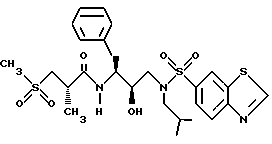
Получение N-[2R-гидрокси-3-[(2-метилпропил)[(бензотиазол-5-ил)сульфонил] амино] -1S-(фенилметил)пропил]-2S-метил-3-(метилсульфонил)пропанамида
Смесь 2-(S)-метил-3-метилсульфонилпропионовой кислоты (0.220 г, 1,325 ммоль), гидроксибензотриазола (0,178 г, 1,325 ммоль), EDC (0,253 г, 1,325 ммоль) в ДМФ (20 мл) перемешивалась в течение 1 часа при комнатной температуре. Добавлялся хлоргидрат [2R-гидрокси-3-[(бензотиазол-6-сульфонил)-(2-метилпропил)амино] - 1S-(фенилметил)пропиламина (0,620 г, 2,66 ммоль) и перемешивался в течение 18 часов. Реакционная смесь концентрировалась в вакууме, и остаток распределялся между этилацетатом (200 мл) и лимонной кислотой (5%, 100 мл). Органический слой промывался насыщенным бикарбонатом натрия (100 мл), солевым раствором (100 мл), сушился (сульфатом магния) и концентрировался. Хроматография с использованием смеси этилацетат:гексан 3: 1) давала 0,330 г (43%) желаемого продукта в виде порошка. Вычислено: M=581; Найдено: M+Li=588.
Пример 19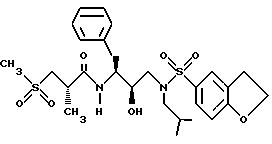
Получение N-[2R-гидрокси-3-[(2-метилпропил)[(2,3-бензофуран- 5-ил)сульфонил]амино]-1S-(фенилметил)пропил]-2S-метил-3- (метилсульфонил)пропанамида
К раствору 0,170 г (1 ммоль) 2-(S)-метил-3-метилсульфонилпропионовой кислоты, растворенной в 5 мл сухого диметилформамида, добавлялось 1,5 эквивалента N-гидроксибензотриазола, и раствор охлаждался в ледяной ванне. К данному охлажденному раствору добавлялось 0,19 г (1,0 ммоль) EDC, и раствор перемешивался в течение 30 минут. К данному раствору добавлялся 2R-гидрокси-3-[[(2,3-дигидробензофуран-5-ил) сульфонил] (2-метилпропил)амино] -1S-(фенилметил)пропиламин (0,418 г, 1,0 ммоль), и реакционная смесь перемешивалась в течение 16 часов. Содержимое концентрировалось на вращательном испарителе, и остаток растворялся в этилацетате, промывался 5% бисульфатом калия, насыщенным бикарбонатом натрия и солевым раствором, сушился над сульфатом магния, фильтровался и концентрировался, давая сырое масло. Очистка с помощью флэш хроматографии (SiO2) с использованием смеси 1:1 этилацетат: гексан в качестве элюента давала очищенный продукт: масс спектр, m/z = 573,5.
Пример 20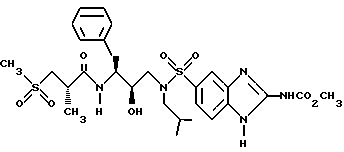
Получение N-[2R-гидрокси-3-[(2-метилпропил)[(2-карбометоксиамино) бензимидазол-5-ил)сульфонил]амино]-1S-(фенилметил)пропил]-2S-метил-3- (метилсульфонил)пропанамида
Смесь 2-(S)-метил-3-метилсульфонилпропионовой кислоты (157,0 мг, 0,94 ммоль), гидрата 1-гидроксибензотриазола (144,0 мг, 0,94 ммоль) и хлоргидрата 1-(3-диметиламино-пропил)-3-этилкарбодиимида (EDC) (180,0 мг, 0,94 ммоль) растворялась в диметилформамиде (5,0 мл), и раствор перемешивался при комнатной температуре в течение 45 минут. Затем добавлялись 2R-гидрокси-3-[[(2-(карбометоксиамино)бензимидазол- 5-ил)сульфонил] (2-метилпропил)амино] -1S-(фенилметил)пропиламин (459 мг, 0,94 ммоль) и N-метилморфолин (202,0 мг, 2,0 ммоль), и реакционная смесь перемешивалась при комнатной температуре в течение 16 часов. Раствор выливался в этилацетат (75 мл), и этилацетатный слой промывался 10% водной уксусной кислотой (3 х 25 мл), насыщенным водным бикарбонатом натрия (3 х 25 мл) и насыщенным водным хлоридом натрия (25 мл). Органический слой сушился над безводным сульфатом натрия, и растворитель удалялся в вакууме. Получающийся в результате остаток растворялся в горячем этилацетате (25 мл). Раствор охлаждался до комнатной температуры, и начинал образовываться осадок. Добавлялись гексаны (25 мл), и раствор перемешивался при комнатной температуре в течение 2 часов. Получающийся продукт собирался с помощью вакуумного фильтрования, давая чистый N-[2R-гидрокси-3-[(2-метилпропил)[2-(карбометоксиамино)бензимидазол -5-ил)сульфонил] амино] -1S-(фенилметил)-пропил] -2S-метил-3- (метилсульфонил)пропанамид в виде белого твердого вещества (395 мг, 65%); FAB-MS вычислено для C28H39N5O8S2 m/z = 637 (M+H), найдено m/z = 644 (M+Li).
Пример 21.
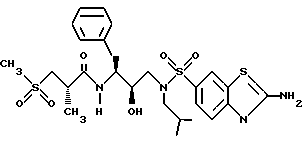
Получение N-[2R-гидрокси-3-[(2-метилпропил)[(2-аминобензотиазол-6-ил)сульфонил] амино]-1-S-(фенилметил)пропил]-2S-метил-3-(метилсульфонил)пропанамида
К раствору 2-(S)-метил-3-метилсульфонилпропионовой кислоты (0,249 г, 1,5 ммоль) в 5 мл сухого диметилформамида, добавлялось 1,5 эквивалента N-гидроксибензотриазола, и раствор охлаждался в ледяной ванне. К данному охлажденному раствору добавлялся EDC (0,200 г, 1,5 ммоль), и раствор перемешивался в течение 30 минут. К данному раствору добавлялся 2R-гидрокси-3-[[(2-аминобезотиазол-6-ил)сульфонил](2-метилпропил)амино]-1S- (фенилметил)пропиламин (0,673 г, 1,5 ммоль), и реакционная смесь перемешивалась в течение 16 часов. Содержимое концентрировалось в вакууме, и остаток растворялся в этилацетате, промывался 5% бисульфатом калия, насыщенным бикарбонатом натрия и солевым раствором, сушился над сульфатом магния, фильтровался и концентрировался, давая сырое масло. Очистка с помощью колоночной флэш хроматографии на силикагеле с использованием смеси 1:1:0,1 этилацетат: гексан: метанол в качестве элюента давала чистый N-[2R-гидрокси-3-[(2-метилпропил)-[(2- аминобензотиазол-6-ил)сульфонил]амино]-1S-(фенилметил)пропил] - 2S-метил-3-(метилсульфонил)пропанамид: FAB-MS: m/z - 598,6 (M+H).
Пример 22
Соединения настоящего изобретения являются эффективными ингибиторами ВИЧ протеазы. При использовании ферментного анализа, описанного ниже, соединения, представленные в примерах, описанных здесь, ингибировали ВИЧ фермент. Предпочтительные соединения настоящего изобретения и их вычисленные величины ИК50 или IC50 (концентрация, ингибирующая 50%, т.е., концентрация, при которой ингибиторное соединение уменьшает активность фермента на 50%) показаны в Таблице 9. Ферментный метод описывается ниже. Субстратом является 2-аминобензоил-Ile-Nle-Phe-(p-NO2)-Cln-ArgNH2. Положительным контролем является MVT-101 (Miller, M. и др., Science, 246, 1149 (1989)). Условия анализа являются следующими:
Буфер для анализа: 20 мМ фосфат натрия, pH 6,4; 20% глицерин; 1 мМ ЕДТА; 1 мМ ДТТ; 0,1% CHAPS.
Описанный выше субстрат растворяется в ДМСО, затем разбавляется 10-кратно буфером для анализа. Конечная концентрация субстрата в опыте составляет 80 мкМ.
ВИЧ протеаза разбавляется в буфере для анализа до конечной концентрации фермента 12,3 наномолярной, в расчете на молекулярный вес 10780.
Конечная концентрация ДМСО составляет 14%, а конечная концентрация глицерина - 18%. Испытываемое соединение растворяется в ДМСО и разбавляется до 10-кратной опытной концентрации; добавляется 10 мкл ферментного препарата, вещества смешиваются, и затем смесь инкубируется при температуре окружающей среды в течение 15 минут. Ферментная реакция инициируется добавлением 10 мкл субстрата. Увеличение флуоресценции регистрируется в 4 временных точках (0, 8, 16 и 24 минуты) при температуре окружающей среды. Каждый анализ осуществляется двукратно.
Пример 23
Эффективность соединений настоящего изобретения определялась в опыте на CEM клетках.
Метод анализа ингибирования ВИЧ остро инфицированных клеток является автоматизированным колориметрическим анализом на основе тетразолия, по существу таким, как описан авторами Pauwles и др., J. Virol. Methods, 20, 308-321 (1988). Опыты проводились на культуре ткани на 96-луночных пластинах. CEM клетки, линия клеток CD4+, выращивались в RPMI-1640 среде (Gibco) с добавлением 10% плодной бычьей сыворотки, а затем обрабатывались полибреном (2 мкг/мл). 80 мкл объем среды, содержащей 1 • 104 клеток, помещался в каждую лунку пластины для тканевой культуры. В каждую лунку добавлялся 100 мкл объем испытываемого соединения, растворенного в среде культуры ткани (или среда без испытываемого соединения в качестве контроля) для достижения желаемой конечной концентрации, и клетки инкубировались при -37oC в течение 1 часа. Замороженная культура ВИЧ-1 разбавлялась в культуральной среде до концентрации 5 • 104 TCID50 на мл (TCID50 = доза вируса, которая инфицирует 50% клеток в культуре ткани), и в лунки, содержащие испытываемое соединение и в лунки, содержащие только среду (инфицированные контрольные клетки) добавлялся 20 мкл объем пробы вируса (содержащий 1000 TCID50 вируса). Несколько лунок получали культуральную среду без вируса (неинфицированные контрольные клетки). Аналогичным образом определялась токсичность испытываемого соединения путем добавления среды без вируса в несколько лунок, содержащих испытываемое соединение. Вкратце пластины с культурой ткани имели следующие экспериментальные показатели (см. табл. A).
В экспериментах 2 и 4 конечные концентрации испытываемых соединений были 1, 10, 100 и 500 мкг/мл. В качестве положительного лекарственного контроля был включен или азидотимидин (AZT), или дидеоксиинозин (ddI). Испытываемые соединения растворялись в ДМСО и разбавлялись в тканевой культуральной среде так, чтобы конечная концентрация ДМСО не превышала в любом случае 1,5%. В соответствующей концентрации добавлялся ДМСО во все контрольные лунки.
После добавления вируса клетки инкубировались при 37oC в увлажненной атмосфере 5% CO2 в течение 7 дней. Испытываемые соединения можно было добавлять в дни 0, 2 и 5 при желании. На 7 день, после инфицирования, клетки в каждой лунке повторно суспендировались и 100 мкл пробы каждой суспензии клеток удалялись для анализа. В каждые 100 мкл суспензии клеток добавлялся 20 мкл объем 5 мг/мл раствора бромида 3-(4,5-диметилтиазол-2-ил)-2,5-дифенилтетразолия (MTT), и клетки инкубировались в течение 4 часов при 27oC в 5% CO2 окружающей среде. Во время инкубирования MTT метаболически восстанавливался живыми клетками; приводя в результате к получению в клетках окрашенного формазанового продукта. В каждую пробу добавлялся 100 мкл 10% додецилсульфата натрия в 0,01% норм. HCl для лизирования клеток, и пробы инкубировались на протяжении ночи. Для каждой пробы определялась абсорбция при 590 нм с использованием молекулярного микроплатного считывающего устройства. Величины абсорбции для каждого комплекта лунок сравнивались для оценки ответной реакции вирусной контрольной инфекции, неинфицированных контрольных клеток, а также испытываемого соединения по цитотоксичности противовирусной эффективности. Результаты представлены в Таблице 10.
Пример 24
С использованием описанного соответственно в примерах 22 и 23, приведенных в описании, ферментного анализа и анализа CEM клеток соединения, представленные в описанных выше примерах, испытывались на ингибирование ВИЧ фермента. Результаты испытаний представлены в Таблице 11.
Предполагается, что с использованием процедур, представленных в приведенных выше примерах, с учетом общего описания, могут быть получены другие соединения настоящего изобретения и что такие соединения будут проявлять активность в качестве ингибиторов ВИЧ протеазы, по существу, аналогичную активности соединений, представленных в примерах.
Соединения настоящего изобретения являются эффективными противовирусными соединениями и, в особенности, являются эффективными ретровирусными ингибиторами, как показано выше. Так, описываемые изобретения являются эффективными ингибиторами ВИЧ протеазы. Следует понимать, что описываемые соединения также будут ингибировать другие ретровирусы такие, как иные лентивирусы, в частности, иные штаммы ВИЧ, например ВИЧ-2, вирус лейкемии T-клеток человека, респираторный синцитиальный вирус, вирус иммунодефицита обезьяны, кошачий вирус лейкемии кошачий вирус иммунодефицита, гепаданвирус, цитомегаловирус и пикорнавирус. Таким образом, описываемые соединения являются эффективными при лечении и/или профилактике ретровирусных инфекций.
Соединения настоящего изобретения могут иметь один или более асимметрических атомов углерода и, таким образом, способны существовать в форме оптических изомеров так же, как и в форме рацемических и нерацемических смесей их. Оптические изомеры могут получаться с помощью разделения рацемических смесей в соответствии с общепринятыми процессами, например с помощью образования диастереоизомерных солей путем обработки оптически активной кислотой или основанием. Примерами соответствующих кислот являются винная, диацетилвинная, дибензоилвинная, дитолуилвинная и камфорсульфоновая кислота, а затем разделения смеси диастереизомеров с помощью кристаллизации с последующим освобождением оптически активных оснований из этих солей. Иной способ разделения оптических изомеров включает использование хиральной хроматографической колонки, оптимально выбранной для максимилизации разделения энантиомеров. Еще один доступный метод включает синтез корвалентных диастереоизомерных молекул с помощью реакции соединений формулы I с оптически чистой кислотой в активированной форме, или оптически чистым изоцианатом. Синтезированные диастереиозомеры могут разделяться с помощью общепринятых средств таких, как хроматография, перегонка, кристаллизация или сублимирование, а затем гидролизоваться для получения энантиомерно чистого соединения. Оптически активные соединения формулы I аналогично могут получаться с использованием оптически активных исходным материалом. Эти изомеры могут быть в форме свободной кислоты, свободного основания, сложного эфира или соли.
Соединения настоящего изобретения могут использоваться в форме солей, получаемых из неорганических или органических кислот. Эти соли включаются, но не ограничиваются следующими: ацетат, адипинат, альгинат, цитрат, аспарагинат, бензоат, бензолсульфонат, бисульфат, бутират, камфорат, камфорсульфонат, диглюконат, циклопентанпропионат, додецилсульфат, этансульфонат, глюкогептаноат, глицерофосфат, гемисульфат, гептаноат гексаноат, фумарат, гидрохлорид, гидробромид, гидроиодид, 2-гидркоси-этансульфонат, лактат, малеат, метансульфонат, никотинат, 2-нафталинсульфонат, оксалат, пальмоат, пектинат, персульфат, 3-фенилпропионат, пикрат, пивалат, пропионат, сукцинат, тартрат, тиоцианат, тозилат, мезилат и ундеканоат. Основные азотсодержащие группы могут также кватернизоваться такими агентами, как низшие алкилгалогениды такие, как метил-, этил-, пропил- и бутил-хлориды, -бромиды и -иодиды; диалкилсульфаты такие, как диметил-, диэтил-, дибутил- и диамил-сульфаты, галогениды с длинной цепью такие, как децил-, лаурил-, миристил- и стеарил-хлориды, -бромиды и -иодиды, аралкилгалогениды такие, как бензил- и фенотил-бромиды, и другие. Таким образом могут получаться продукты, растворимые или диспергируемые в воде или масле.
Примеры кислот, которые могут применяться для образования фармацевтически приемлемых кислотно-аддитивных солей, включают такие неорганические кислоты, как соляная, серная и фосфорная кислоты, и такие органические кислоты, как щавелевая, малеиновая, янтарная и лимонная кислоты. Другие примеры включают соли с щелочными металлами или щелочноземельными металлами такими, как натрий, калий, кальций или магний, или с другими основаниями.
Общая суточная доза, назначаемая субъекту-хозяину в виде одной или в виде раздельных доз, может составлять количества, например, от 0.001 до 10 мг/кг веса тела в день и более обычно 0.01 - 1 мг. Композиции в виде единичных доз могут содержать такие количества множественных доз, чтобы в целом составить дневную дозу.
Количество активного ингредиента, которое может сочетаться с материалами носителя для получения единичной дозы, варьирует в зависимости от субъекта, подвергаемого лечению, и от конкретного способа назначения.
Дозировочный режим для лечения болезненных состояний с помощью соединений и/или композиций данного изобретения выюирается в соответствии с разнообразными факторами, включающими тип, возраст, вес, пол, диету и медицинское состояние пациента, тяжесть заболевания, способ назначения, фармакологические факторы такие, как активность, эффективность, фармакокинетический и токсикологический профиль конкретно применяемого соединения, используется ли система доставки лекарства и назначается ли соединение в виде части комбинации лекарств. Таким образом, фактически применяемый дозировочный режим может широко варьироваться, и поэтому, он может отклоняться от предпочтительного режима дозировок, представленного выше.
Соединения настоящего изобретения могут назначаться орально, парентерально, с помощью ингаляционного спрея, ректально, или топически в виде препаративных форм единичных доз, содержащих общепринятые нетоксичные фармацевтические приемлемые носители, адьюванты и разбавители, в зависимости от необходимости или желания. Топическое или местное назначение может также предусматривать использование трансдермального назначения такого, как трансдермальные бляшки или приспособления для ионотерапии. Термин парентеральный, как он используется здесь, включает подкожные инъекции, внутривенные, внутримышечные, интрастернальные инъекции или приемы инфузии или вливания.
Инъецируемые препараты, например стерильные инъецируемые водные или маслянистые суспензии, могут формироваться в соответствии с известными приемами с использованием подходящих диспергирующих и/или смачивающих агентов и суспендирующих агентов. Стерильный инъецируемый препарат может также представлять стерильный инъекцируемый раствор или суспензию в нетоксичном парентерально приемлемом разбавителе или растворителе, например в виде растворов в 1,3-бутандиоле. В числе приемлемых разбавителей или носителей и растворителей, которые могут применяться, находятся вода, раствор Рингера, и изотоничный натрийхлоридный раствор. В дополнение к сказанному, в качестве растворителя или суспендирующей среды обычно применяются стерильные, фиксированные масла. Для этой цели может применяться любое смешанное фиксированное масло, включая синтетические моно- или диглицериды. В дополнение к изложенному, при получении инъецируемых препаратов находят применение жирные кислоты, такие, как олеиновая кислота.
Суппозитории или медицинские свечи для рентального назначения лекарств могут приготавливаться с помощью смешения лекарства с подходящим эксципиентом таким, как масло какао, и полиэтиленгликоли, которые при обычных температурах являются твердыми, но жидкими при ректальной температуре и, поэтому, способны плавиться в прямой кишке и высвобождать лекарство.
Твердые дозированные формы для орального назначения могут включать капсулы, таблетки, пилюли, порошки и гранулы. В таких твердых дозированных формах активное соединение может быть смешано с по крайней мере одним инертным разбавителем таким, как сахароза, лактоза или крахмал. Такие дозированные формы могут также включать, как и обычно в практике, дополнительные вещества, иные, чем инертные разбавители, например смазочные агенты такие, как стеарат магния. В случае капсул, таблеток и пилюль дозированные формы могут также включать буферные агенты. Таблетки и пилюли могут дополнительно приготавливаться с энтерическими покрытиями.
Жидкие дозировочные формы для орального назначения могут включать фармацевтически приемлемые эмульсии, растворы, суспензии, сиропы и эликсиры, содержащие инертные разбавители, обычно используемые в данной области техники, такие, как вода. Такие композиции могут также включать вспомогательные вещества или адъюванты такие, как смачивающие агенты, эмульгирующие и суспендирующие агенты, и послащивающие, вкусовые и ароматизирующие агенты.
Хотя соединения изобретения могут назначаться как один активный фармацевтический агент, они могут также использоваться в сочетании с одним или большим числом иммуномодуляторов, антивирусных агентов или других антиинфекционных агентов. Например, соединения данного изобретения могут назначаться в сочетании с AZT, DDI, DDC или с N-бутил-1-деоксинойиримицин для профилактики и/или лечение СПИДа. При назначении в сочетании терапевтические агенты могут преобразовываться в препаративные формы в виде отдельных композиций, которые даются для приема одновременно или в различное время, или терапевтические агенты могут даваться в виде единой композиции.
Предшествующее описание дается лишь для иллюстрации изобретения и оно не предназначено для ограничения и изобретения конкретно раскрытыми соединениями. Имеется в виду, что вариации и изменения, очевидные специалистам в данной области, находятся в рамках объема и природы изобретения, которые определяются прилагаемыми пунктами формулы изобретения или притязаний.
Из приведенного выше описания специалист в данной области сможет легко понять существенные признаки для характеристики данного изобретения, и не отклоняться от сути и объема его, сможет произвести различные изменения и модификации изобретения для приспособления его к различным видам применения и к условиям.
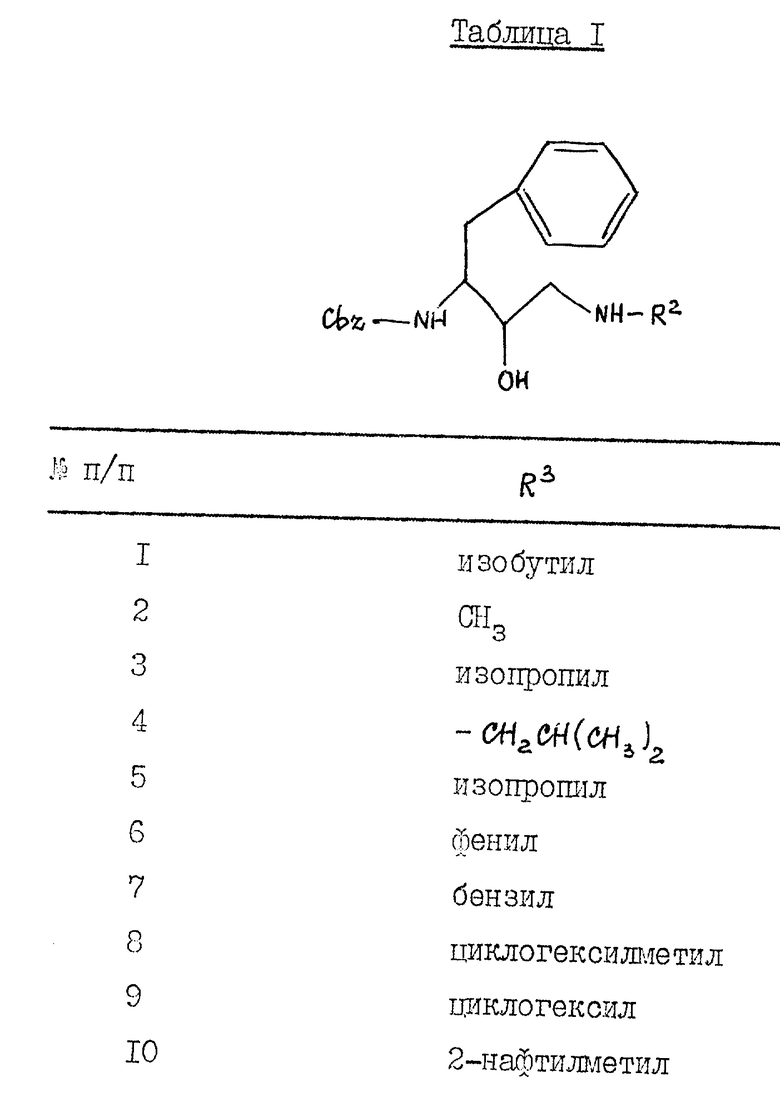

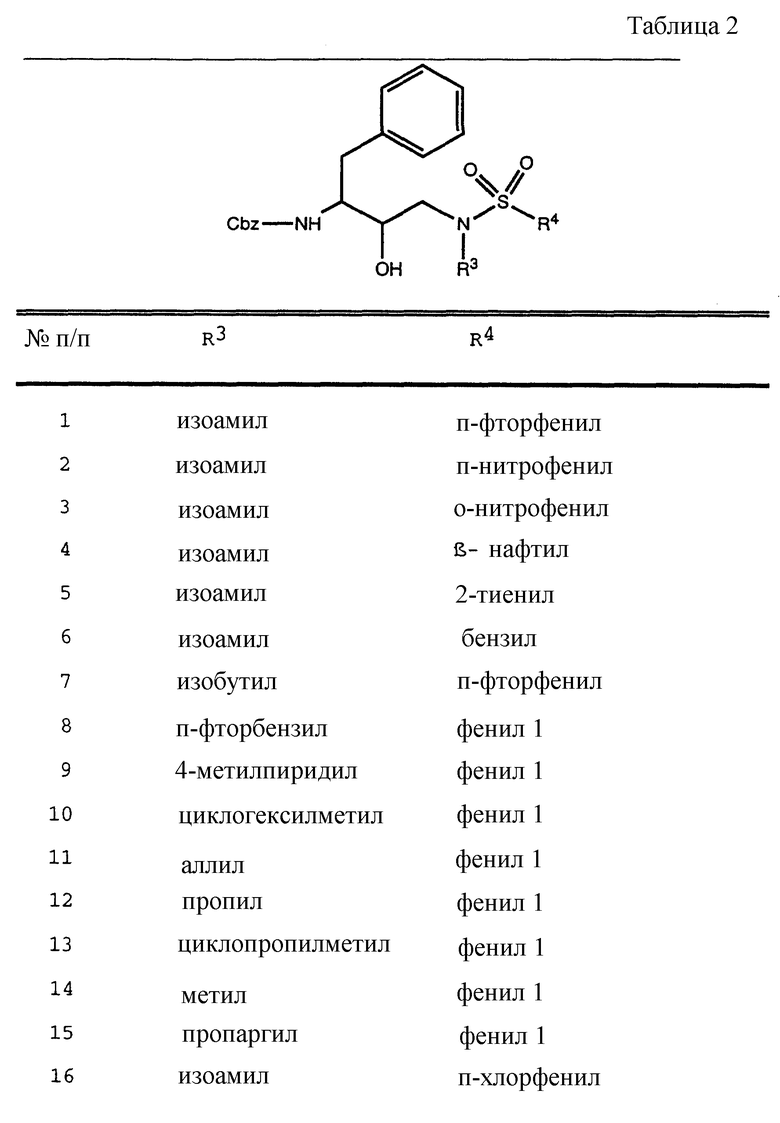

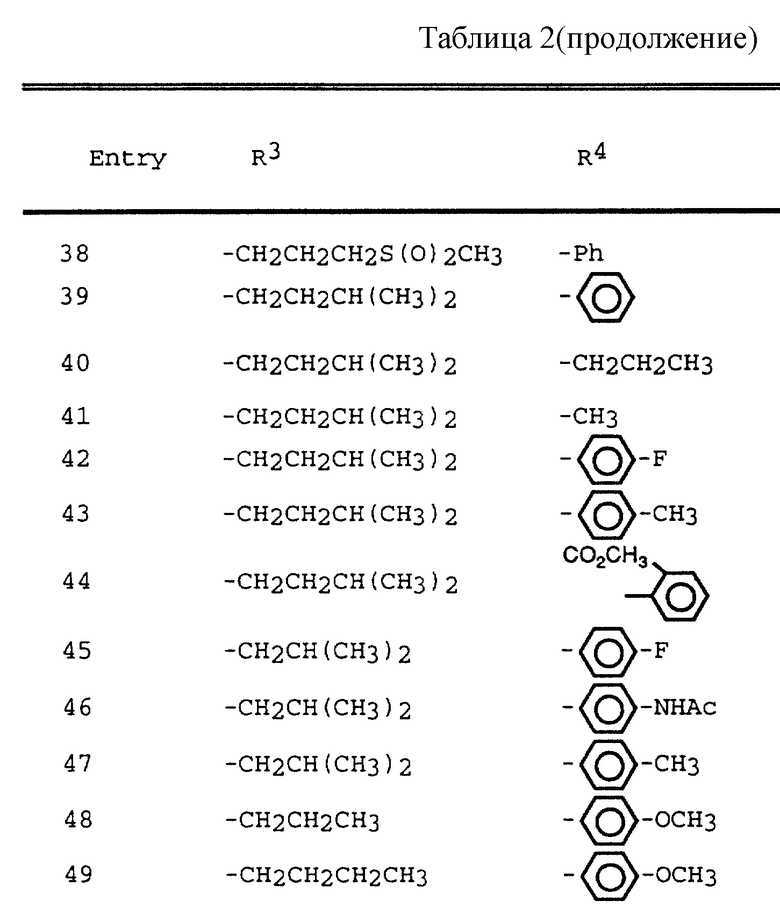
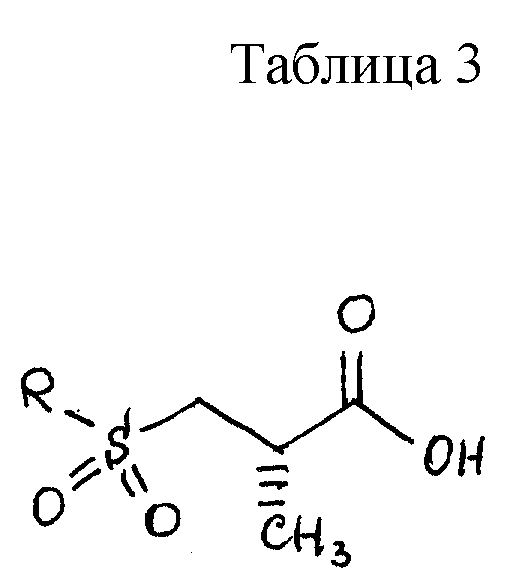
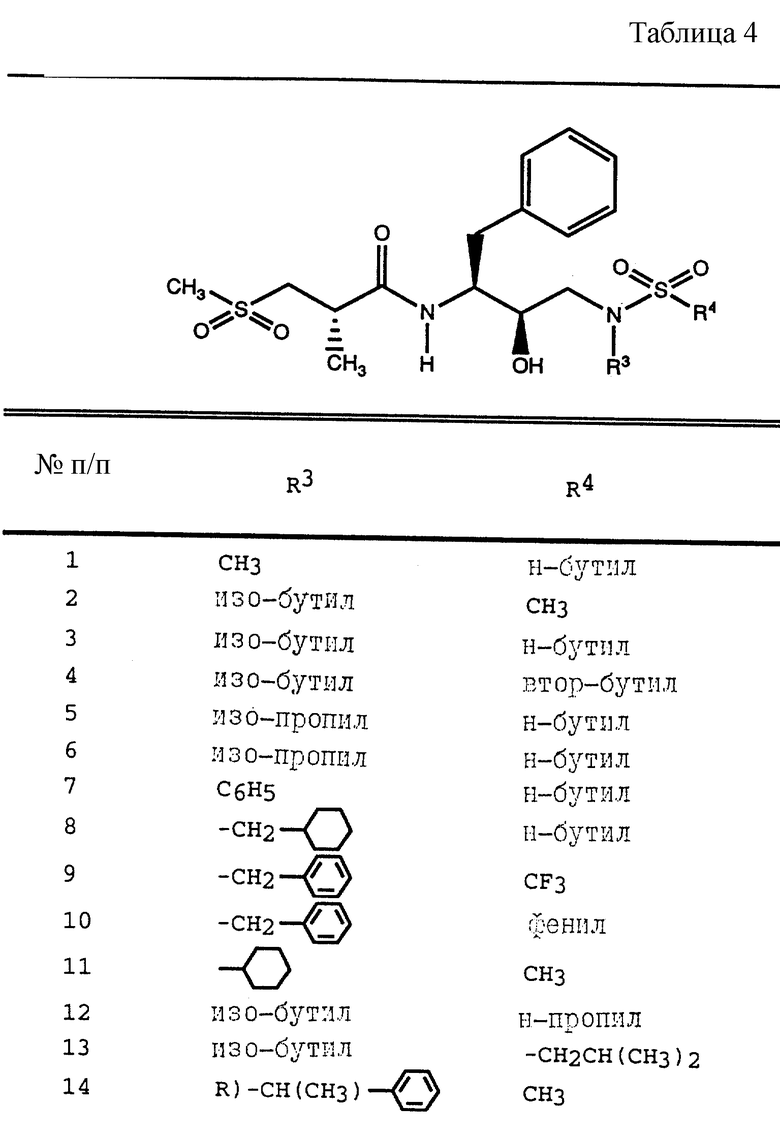
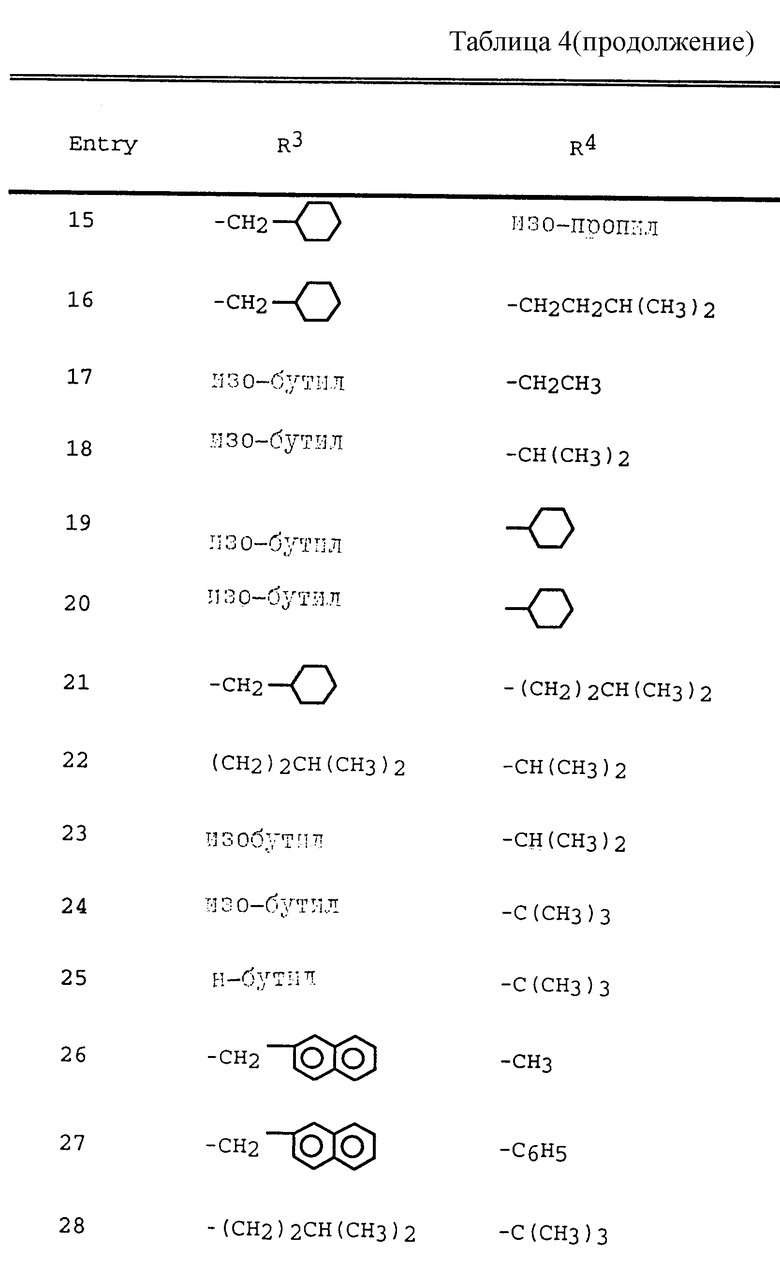

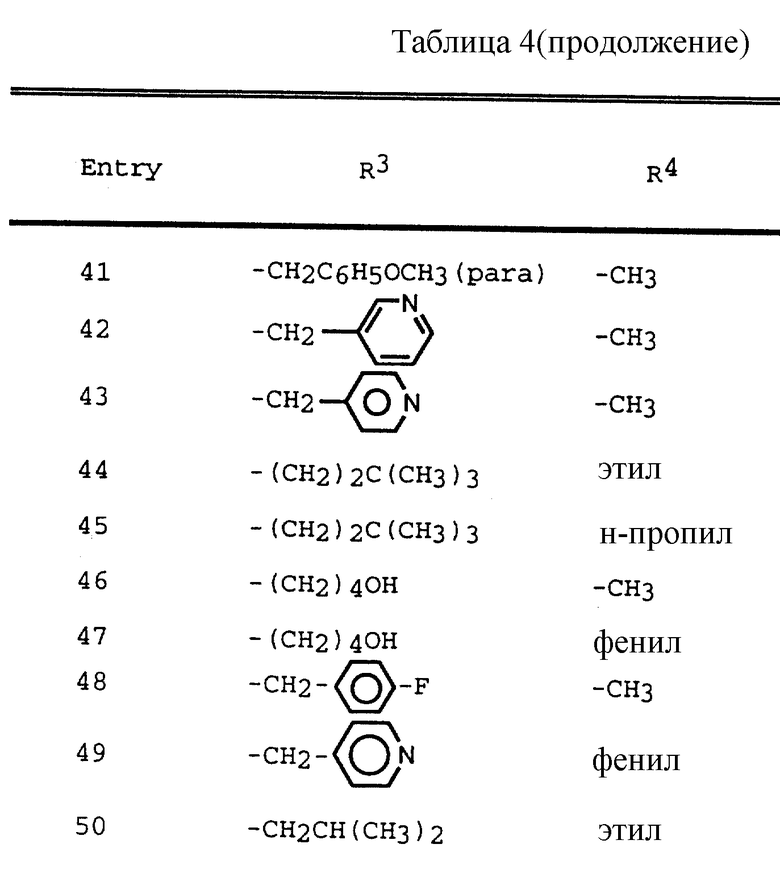
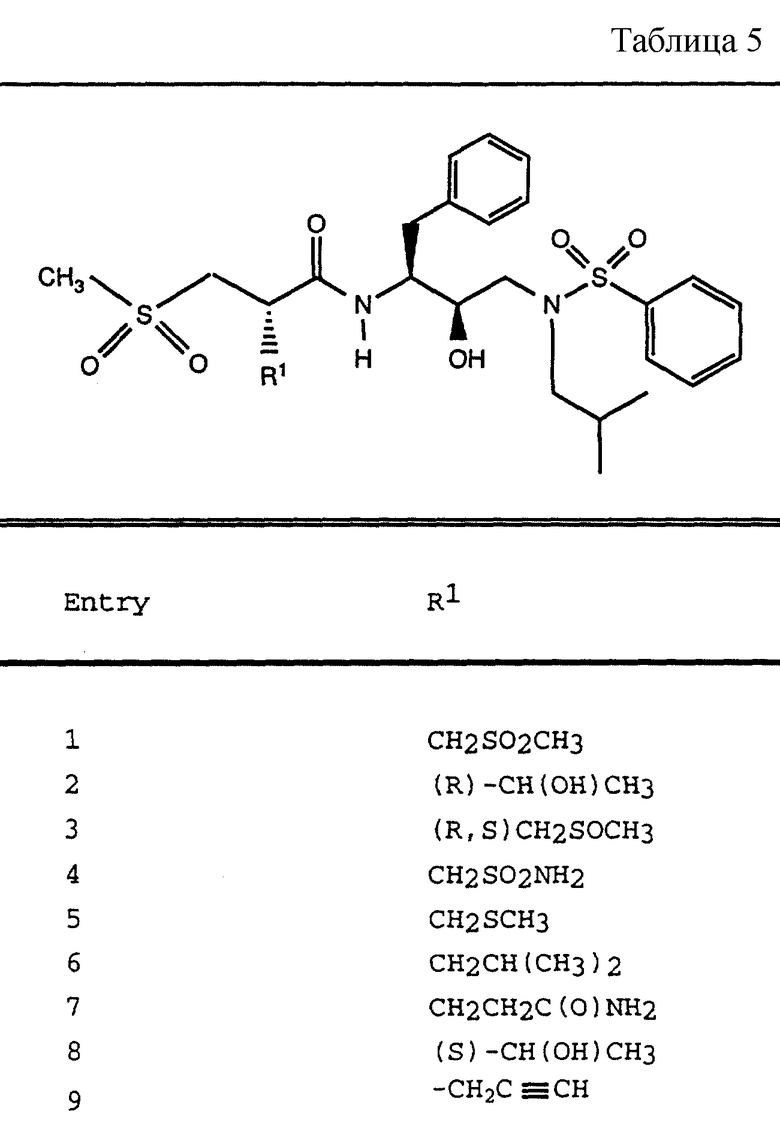
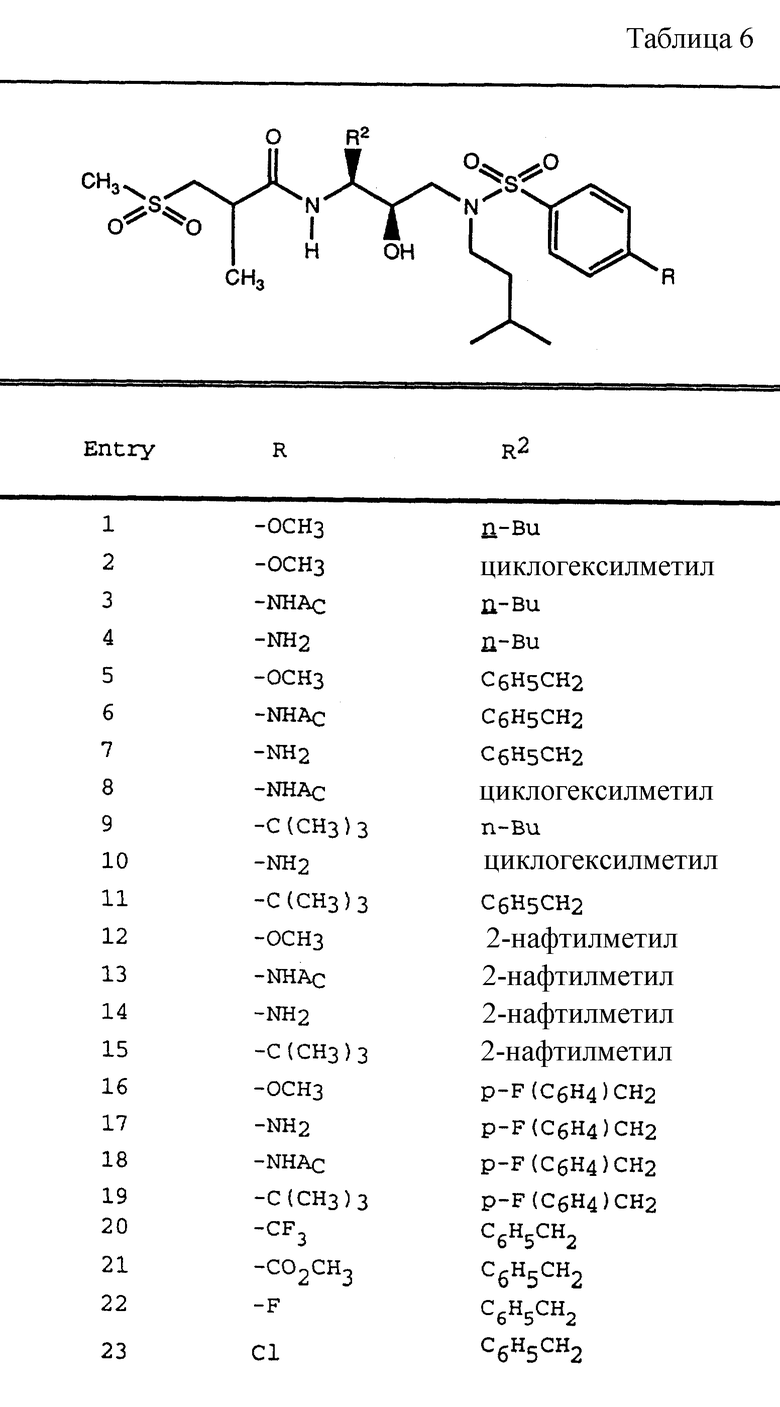
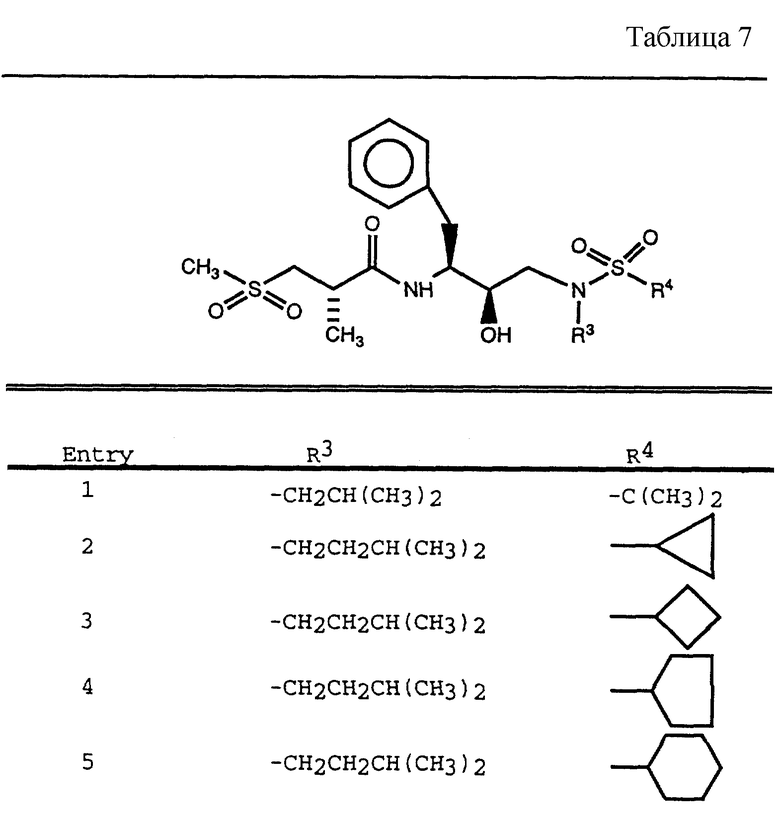
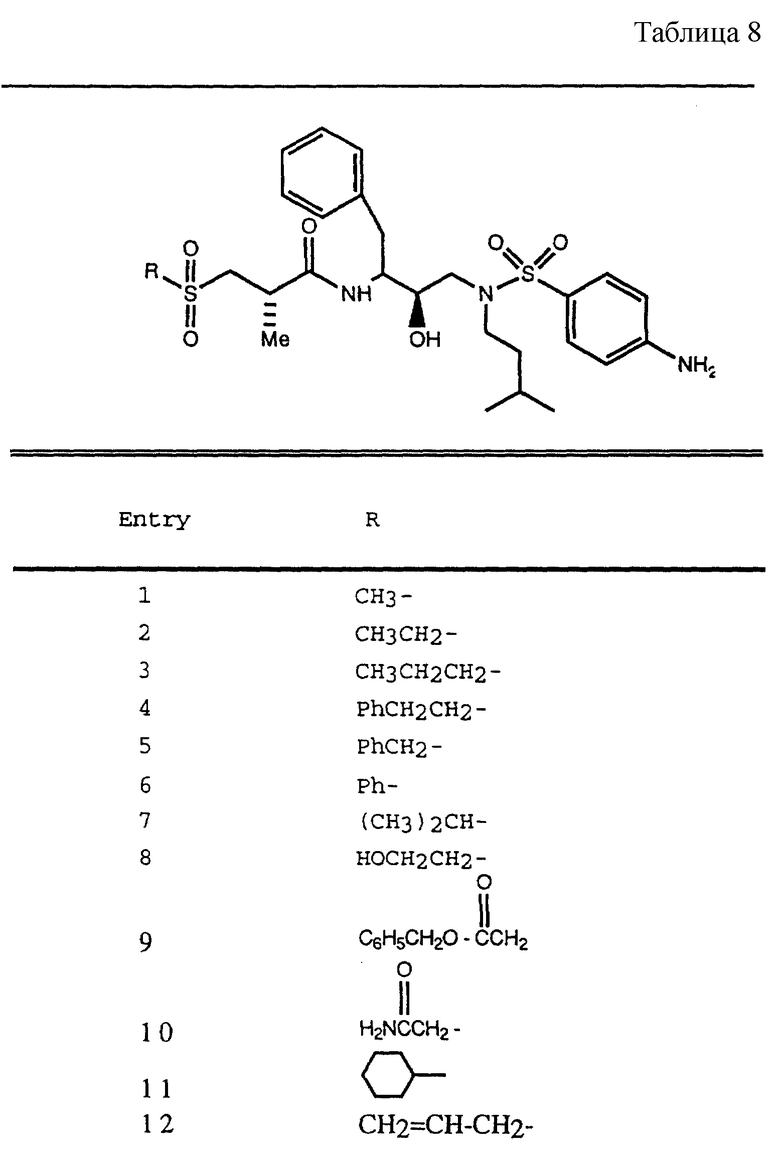


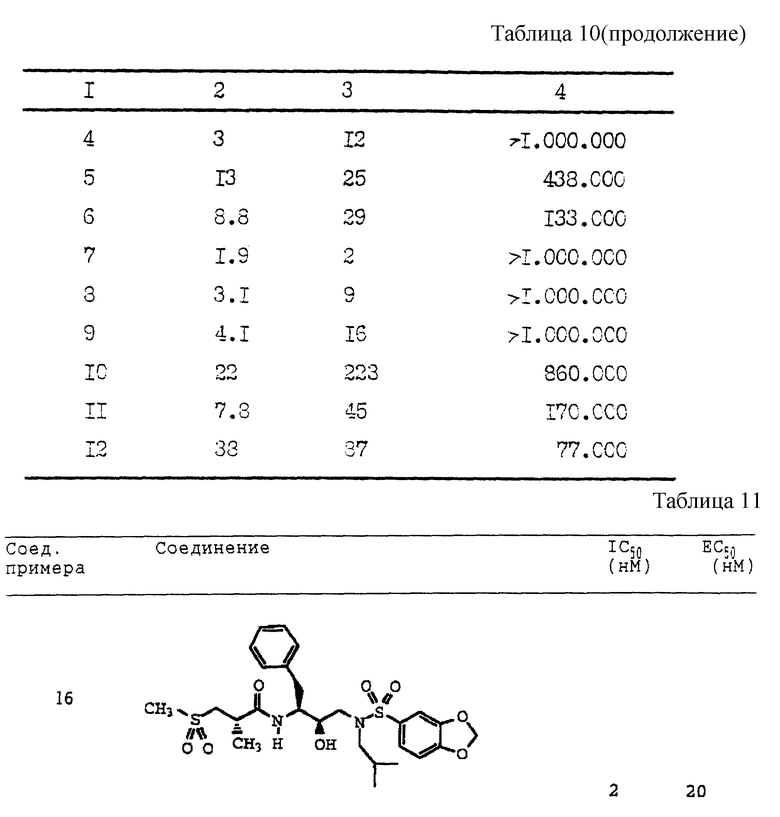
Описывается новое соединение - сульфонилалканоиламино-гидроксиэтиламино-сульфонамидное соединение общей формулы (I), где R-алкил; R1, R20, R21-каждый независимо водород или алкил; R2-алкил или аралкил, R3-алкил; R4-арил, который необязательно замещен одним или несколькими заместителями, выбранными из группы, включающей алкил, алкокси, гидрокси, амино, ацетамино; или бензотиазол, бензодиоксолил, бензофурил, бензимидазолил, бензодиоксанил; R6-водород; Y-кислород; x=2; t=0 или 1, или его фармацевтически приемлемые соли. Соединения настоящего изобретения являются эффективными противовирусными соединениями, в особенности эффективными ретровирусными ингибиторами. В частности, они являются эффективными ингибиторами ВИЧ-протеазы, а также будут ингибировать другие ретровирусы, такие, как иные лентивирусы, в частности, иные штаммы ВИЧ, вирус лейкемии Т-клеток человека и т.д. 3 с. и 31 з.п. ф-лы, 11 табл.
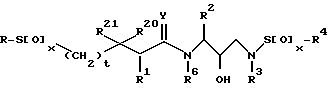
в которой R - алкил;
R1, R20, R21 - каждый независимо водород или алкил;
R2 - алкил или аралкил;
R3 - алкил;
R4 - арил, который необязательно замещен одним или несколькими заместителями, выбранными из группы, включающей алкил, алкокси, гидрокси, амино, ацетамино; или бензотиазол, бензодиоксолил, бензофурил, бензимидазолил, бензодиоксанил;
R6 - водород;
Y - кислород;
x = 2;
t = 0 или 1,
или его фармацевтически приемлемые соли.
[1S-[1R*(R*),2S*]]-N-[2-гидрокси-3-[(2-метилпропил)(фенилсульфонил)амино]-1-(фенилметил)пропил]-2-метил-3-(метилсульфонил)-пропанамид,
[1S-[1R*(R*),2S*]]-N-[2-гидрокси-3-[(3-метилбутил)(фенилсульфонил)амино] -1-(фенилметил)пропил]-2-метил-3-(метилсульфонил)-пропанамид,
[1S-[1R*(R*), 2S*] ]-N-[2-гидрокси-3-[(пропил)(фенилсульфонил)амино]-1-(фенилметил)пропил]-2-метил-3-(метилсульфонил)-пропанамид,
[1S-[1R*(R*), 2S*] ] -N-[2-гидрокси-3-[(бутил)(фенилсульфонил)амино]-1-(фенилметил)пропил]-2-метил-3-(метилсульфонил)-пропанамид,
[1S-[1R*(R*), 2S*] ] -N-[2-гидрокси-3-[(2-метилпропил)(4-метоксифенилсульфонил)амино] -1-(фенилметил)пропил] -2-метил-3-(метилсульфонил)-пропанамид,
[1S-[1R*(R*), 2S*] ] -N-[2-гидрокси-3-[(бутил)(4-метоксифенилсульфонил)амино]-1-(фенилметил)пропил]-2-метил-3-(метилсульфонил)-пропанамид,
[1S-[1R*(R*), 2S*] ] -N-[2-гидрокси-3-[(пропил)(4-метоксифенилсульфонил)амино]-1-(фенилметил)пропил]-2-метил-3-(метилсульфонил)-пропанамид,
[1S-[1R*(R*),2S*]]-N-[2-гидрокси-3-[(2-метилпропил)(4-ацетамидо)фенилсульфонил)амино] -1-(фенилметил)пропил]-2-метил-3-(метилсульфонил)-пропанамид,
[1S-[1R*(R*),2S*]]-N-[2-гидрокси-3-[(3-метилбутил)(4-амино)фенилсульфонил)амино] -1-(фенилметил)пропил]-2-метил-3-(метилсульфонил)-пропанамид,
[1S-[1R*(R*), 2S*] ] -N-[2-гидрокси-3-[(2-метилпропил)(3,4-диметокси)фенилсульфонил)амино] -1-(фенилметил)пропил]-2-метил-3-(метилсульфонил)-пропанамид, или
[1S-[1R*(R*), 2S*] ]-N-[2-гидрокси-3-[(3-метилбутил)(4-метоксифенилсульфонил)амино]-1-(фенилметил)пропил]-2-метил-3-(метилсульфонил)-пропанамид.
| WO 9208701 А, 1992 | |||
| ВСЕСОЮЗНАЯ | 0 |
|
SU377139A1 |
| Балаян М.С | |||
| Приобретенный иммунодефицитный синдром | |||
| Клиническая медицина | |||
| Колосниковая решетка с чередующимися неподвижными и движущимися возвратно-поступательно колосниками | 1917 |
|
SU1984A1 |
