Изобретение относится к способу лечения ретровирусных инфекций млекопитающих, например вируса иммунодефицита человека (ВИЧ), с использованием комбинаций ингибиторов ретровирусной протеазы, которые эффективны в предупреждении репликации ретровирусов млекопитающих, подобных ВИЧ, in vitro и in vivo. Это изобретение, в частности, относится к соединениям, являющимся ингибиторами протеазы (соединениям- ингибиторам), используемым в комбинационной терапии с другими соединениями-ингибиторами протеазы.
Во время цикла репликации ретровирусов продукты транскрипции гена gag и gag-pol транслируются как белки. Эти белки затем процессируются вирусно кодированной протеазой (или протеиназой), производя вирусные ферменты и структурные белки кора вируса. Чаще всего белки-предшественники gag процессируются в коровьи белки и белки-предшественники pol процессируются в вирусные ферменты, например обратную транскриптазу и ретровирусную протеазу. Было показано, что верный процессинг белков-предшественников ретровирусной протеазой необходим для сборки инфекционных вирионов. Например, было показано, что мутации со сдвигом в протеазной области гена pol ВИЧ предотвращают процессинг белка- предшественника gag. Также было показано посредством сайт- направленного мутагенеза остатка аспарагиновой кислоты в активном сайте протеазы ВИЧ, что предотвращает процессинг белка-предшественника gag. Поэтому были сделаны попытки ингибирования вирусной репликации ингибированием действия ретровирусных протеаз.
Ингибирование ретровирусной протеазы обычно включает миметик в переходном состоянии, посредством чего ретровирусная протеаза подвергается действию миметического соединения, которое связывается (обычно обратимым способом) с ферментом в конкуренции с белками gag и gag-pol, тем самым ингибируя специфический процессинг структурных белков и высвобождение самой ретровирусной протеазы. Таким способом можно эффективно ингибировать протеазы ретровирусной репликации.
Было предложено несколько классов миметических соединений, в частности, для ингибирования протеаз, например для ингибирования протеазы ВИЧ. Такие миметические соединения включают изостеры гидроксиэтиламина, изостеры восстановленных амидов и непептидные изостеры. Смотри, например, EP 0346847; ЕР 0342541; Roberts et al., "Rational Design of Peptide-Based Proteinase Inhibitors, and 2,8 A Crystal Structure of C2 Symmetric Inhibitor Complexed to HIV-I Protease", Science, 249, 527 (1990) and S. Thaisrivogs, "Structure-Based Design of Non-Peptide HIV Protease Inhibitors", 35th Annual Buffalo Medicinal Chemistry Meeting, State University of New York et Buffalo, NY, May 22 - 25, 1994.
Проблемой ингибиторов ретровирусной протеазы, подобных ингибиторам протеазы ВИЧ, было развитие штаммов вирусов, устойчивых к ингибитору. Например, ингибитор L-735524 протеазы ВИЧ Merck and Co. эффективен против инфекции ВИЧ у человека, но позже L-735524 развивает у пациентов устойчивые штаммы ВИЧ (Waldholz, The Wall Street Journal, February 25, 1994, page B3; and Condra et al., Nature 374: 569 - 571 (1995)). Другие примеры можно найти в Vacca et al., Proc. Natl. Acad. Sci. USA 91: 4096 - 4100 (1994); Но et al., J. Virol. 68: 2016 - 2020 (1994); and Sardana et al., Biochem. 33: 2004 - 2010 (1994).
Данное изобретение относится к способу лечения ретровирусных инфекций млекопитающих, например вируса иммунодефицита человека (ВИЧ), с использованием комбинаций ингибиторов ретровирусной протеазы, которые эффективны в предупреждении репликации ретровирусов in vitro или in vivo. Это изобретение, в частности, относится к соединениям-ингибиторам протеазы, используемым в комбинационной терапии с другими соединениями-ингибиторами протеазы. Кроме того, эту комбинацию можно использовать в комбинации с другими противовирусными средствами.
Ретровирусная протеаза является критическим ферментом в процессе ретровирусной репликации. Размножению ретровируса, например ВИЧ, можно препятствовать путем воздействия на вирус ингибитора ретровирусной протеазы. Однако, при пролонгированном воздействии ингибитора протеазы на ретровирус может происходить селекция вариантных ретровирусов, так что появляется новый доминирующий штамм ретровируса, устойчивый к ингибитору протеазы. Этот новый доминирующий штамм ретровируса может продуцировать протеазу, которая больше не ингибируется или, как бывает более часто, недостаточно ингибируется ингибитором протеазы и может свободно размножаться даже в присутствии ингибитора протеазы, если концентрацию этого ингибитора существенно не повышают. Настоящее изобретение предлагает способ преодоления развития ретровирусных штаммов, которые устойчивы к ингибитору ретровирусной протеазы.
Настоящий способ предусматривает введение млекопитающему, например человеку, обезьяне, кошке и т.д. эффективного количества по меньшей мере двух ингибиторов ретровирусной протеазы. Это введение можно выполнять совместным введением по меньшей мере двух ингибиторов ретровирусной протеазы, т.е. введением двух или более ингибиторов ретровирусной протеазы так, чтобы эффективное количество по меньшей мере двух ингибиторов присутствовало у этого млекопитающего в любое время. Альтернативно, введение можно выполнять последовательным или чередующимся введением по меньшей мере двух ингибиторов ретровирусной протеазы, т.е. введением двух или более ингибиторов ретровирусной протеазы так, чтобы у этого млекопитающего в любое время присутствовало эффективное количество только одного ингибитора. При правильном выборе ингибиторов ретровирусной протеазы этот способ может эффективно регулировать размножение ретровирусов, даже в присутствии штаммов, устойчивых к любому одному из ингибиторов.
Ингибиторы ретровирусной протеазы выбирают на основе профиля устойчивого штамма (штаммов) ретровируса, который появляется in vivo или in vitro при воздействии ингибитора на размножающуюся культуру ретровирусов. Ингибиторы ретровирусной протеазы выбирают по отсутствию перекрестной устойчивости по меньшей мере у одного ретровирусного устойчивого штамма. Считается, что ретровирусный штамм является перекрестно устойчивым к двум ингибиторам протеазы, когда ретровирусный штамм устойчив к обоим ингибиторам. Хотя некоторая перекрестная устойчивость может допускаться, предпочтительно, чтобы не существовало никакой перекрестной устойчивости между выбранными ингибиторами ретровирусной протеазы, когда их берут как группу. Таким образом, вариант (или мутантный штамм) ретровируса, который может развиваться в результате воздействия первого ингибитора ретровирусной протеазы, будет все же ингибироваться вторым ингибитором ретровирусной протеазы, или который может развиваться в результате воздействия как первого, так и второго ингибиторов ретровирусной протеазы, будет все же ингибироваться третьим ингибитором ретровирусной протеазы или четвертым ингибитором ретровирусной протеазы и т.д.
Проводят сравнение профилей перекрестной устойчивости между разными ингибиторами протеазы и для комбинационной терапии отбирают соединения, которые предпочтительно проявляют слабую или не проявляют никакой перекрестной устойчивости. Фенотип устойчивости к лекарственному средству можно разделить на следующие: фенотип без устойчивости, фенотип с низким уровнем устойчивости (менее чем около 10-кратный сдвиг в EC50 или EC90), фенотип со средним уровнем устойчивости (от около 10- до около 100-кратного сдвига в EC10 или EC90) или фенотип с высоким уровнем устойчивости (более чем около 100-кратный сдвиг в EC50 или EC90). Ожидается, что устойчивость к лекарственному средству будет коррелировать с ослабленным действием на вирусную нагрузку пациента, когда достижимые in vivo концентрации ингибиторов имеют уменьшенное ингибирующее протеазу действие на устойчивый вирус. Таким образом, более предпочтительными комбинациями ингибиторов протеазы будут те, которые обладают профилями минимальной перекрестной устойчивости (т.е., предпочтительно, устойчивостью не более чем промежуточного уровня; более предпочтительно, устойчивостью не более чем низкого уровня; и наиболее предпочтительно, никакой устойчивостью) и максимальной, свойственной им активностью для вирусов дикого типа и/или устойчивых вирусов, выбранных против другого ингибитора. Например, предпочтительные соединения для использования в комбинации с первым соединением будут предпочтительно эффективны против штаммов вируса, которые обладают промежуточным уровнем, более предпочтительно высоким уровнем устойчивости к первому соединению. Фармакология и токсикология каждого ингибитора и комбинации также являются факторами при отборе ингибиторов для комбинационной терапии.
Более предпочтительно, выбирают ингибиторы ретровирусной протеазы, когда по меньшей мере один вирусный штамм, устойчивый к первому ингибитору ретровирусной протеазы, и по меньшей мере один вирусный штамм, устойчивый ко второму ингибитору ретровирусной протеазы, имеющие разные аминокислотные замещения в пептидной последовательности протеазы, действуют на ту же самую область сайта связывания субстрата протеазы и содействуют наблюдаемой устойчивости к ингибитору. Таким образом, число возможных аминокислотных замещений, которые могут иметь место в том же сайте в протеазе, ограничено. Это, в частности, справедливо, когда сайт критический к активности, эффективности и/или стабильности фермента.
Этот случай наблюдали в отношении к ингибиторам протеазы ВИЧ примеров 1 и 2. Ретровирусная устойчивость к соединению примера 1 являлась результатом мутации сайта у аминокислоты 88 протеазы ВИЧ (замещение аспарагина 88 аспарагиновой кислотой 88). Ретровирусная устойчивость к соединению Примера 2 также вытекает из мутации сайта у аминокислоты 88 протеазы ВИЧ (замещение аспарагина 88 серином 88). Известно, что некоторые замещения у аминокислоты 88 вызывают потерю ферментативной активности (Loeb et al. Nature 340: 387 - 400 (1989)). Таким образом, введение обоих ингибиторов протеазы ВИЧ примеров 1 и 2 значительно снижает вероятность дальнейшего успешного продуцирования устойчивого штамма вируса, перекрестно устойчивого к обоим ингибиторам. Никакой устойчивости к обоим ингибиторам, используемым в комбинации, не обнаруживали в течение 6 недель лечения по сравнению с появлением фенотипа, устойчивого к единственному ингибитору, в то же самое временное ограничение. В дополнение к мутациям сайтов, которые влияют на ферментативную активность, в том же самом варианте могут появляться также другие мутации сайтов, которые не влияют существенно на ферментативную активность и/или устойчивость.
Альтернативно, более предпочтительно выбирают ингибиторы ретровирусной протеазы, когда по меньшей мере один вирусный штамм, устойчивый к первому ингибитору ретровирусной протеазы, имеет повышенную чувствительность к этому второму ингибитору протеазы, или когда по меньшей мере один вирусный штамм, устойчивый ко второму ингибитору вирусной протеазы, имеет повышенную чувствительность к этому первому ингибитору протеазы.
Репрезентативные ингибиторы ретровирусной протеазы, которые пригодны для использования в настоящем способе, включают, но не ограничиваются ими, ингибиторы протеазы, обнаруженные и описанные в находящихся в совместном владении и совместно рассматриваемых родовых заявках США с номерами 08/152934 (подана 15 ноября 1993), 08/253531 (подана 3 июня 1994), 08/109787 (подана 20 августа 1993), 08/110911 (подана 24 августа 1993), 08/110913 (подана 24 августа 1993), 08/110912 (подана 24 августа 1993), 08/204827 (подана 2 марта 1994), 07/886556 (подана 20 мая 1992), 07/886663 (подана 20 мая 1992), 07/886531 (подана 20 мая 1992), 08/148817 (подана 8 ноября 1993), 08/886700 (подана 21 мая 1992) и 07/998187 (подана 29 декабря 1992) и заявках на патент PCT N PСT/US 93/10552 (подана 29 октября 1993), PСT/US 93/10460 (подана 29 октября 1993) и PСT/US 93/10461 (подана 29 октября 1993), каждая из которых включена здесь как ссылка во всей полноте. Дополнительные ингибиторы ретровирусной протеазы, которые пригодны для использования в настоящем способе, включают, но не ограничиваются ими, ингибиторы протеазы, предложенные и описанные в патенте США 5157041; EP 346847; родовой заявке США N 07/883825 (подана 15 мая 1992); WO 93/09096; Tet. Lett. 35: 673 - 676 (1994); Proc. Natl. Acad. Sci. USA, 91: 4096 - 4100 (1994); Y.M. Wong et al., Biopharm. and Drug Dispos. 15: 535 - 544 (1994); M.L. West and D.P. Fairlie. Trends Pharmacol. Sci. 16: 67-75 (1995) и S. Thaisrivongs, "HIV Protease Inhibitors", Ann. Reports Med. Chem., Vol. 29, Chap. 14, pp. 133 - 144 (1994) (Academic Press, J. Bristol, Ed.), каждый из которых включен здесь как ссылка во всей полноте.
Считается, что без дальнейшей разработки любой специалист в данной области может, применяя предшествующее описание, использовать настоящее изобретение в наиболее полной мере. Следующие предпочтительные конкретные воплощения изобретения не предназначены для обеспечения исчерпывающего описания всех возможных комбинаций соединений, а только для обеспечения примеров комбинаций лекарственных средств, которые, как ожидается, эффективны. Аналогичное испытание этих и других ингибиторов протеазы с использованием устойчивых вирусных изолятов, не ограниченных теми, которые перечислены ниже, может помочь установлению пригодных комбинаций лекарственных средств. Следовательно, следующие предпочтительные конкретные воплощения изобретения должны толковаться только как иллюстративные и никоим образом не ограничительные для остальной части описания изобретения.
Пример 1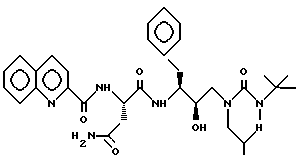
[1S-[1R*(R*), 2S*] ] -N1-[3-[[[(1,1-диметилэтил)амино]карбонил] (2-метилпропил)амино] -2-гидрокси-1-(фенилметил)пропил] - 2-[(2-хинолинилкарбонил)амино]бутандиамид можно получить в соответствии со способами, описанными в находящихся в совместном владении и совместно рассматриваемых заявках на патент США с номерами 08/152934 (подана 15 ноября 1993) и 08/156498 (подана 23 ноября 1993), которые включены здесь как ссылка во всей полноте.
Пример 2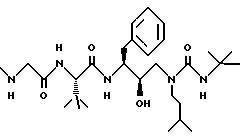
(2R, 3S)-3-(N-Метиламиноацетил-L-трет-бутилглицинил) амино-1-(N-изоамил-N-(трет-бутилкарбамоил)амино-4-фенил-2-бутанол можно получить в соответствии со способами, описанными в находящихся в совместном владении и совместно рассматриваемых заявках на патент США с номерами 08/109787 (подана 20 августа 1993), Attorney docket N 27766/1, поданными совместно с данной заявкой, и 08/156498 (подана 23 ноября 1993), все три включены здесь ссылкой во всей полноте.
Пример 3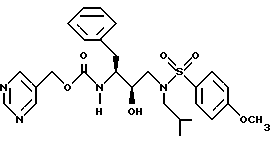
5-Пиримидилметиловый эфир [2R-гидрокси-3-[[(4-метоксифенил)-сульфонил] (2-метилпропил] амино] -1S-(фенилметил) пропил] карбаминовой кислоты можно получить в соответствии со способами, описанными в находящихся в совместном владении и совместно рассматриваемых заявках на патент США с номерами 08/110911 (подана 24 августа 1993) и 08/156498 (подана 23 ноября 1993), обе включены здесь ссылкой во всей полноте.
Пример 4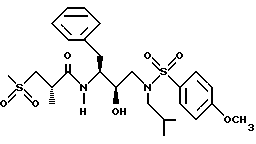
[1S-[1R*(R*),2S*]]-N-[2-Гидрокси-3-[N1-(2-метилпропил)-N1-(4-метоксифенилсульфонил)амино]-1- (фенилметил) пропил]-2-метил-3-(метилсульфонил)пропанамид можно получить в соответствии со способами, описанными в находящихся в совместном владении и совместно рассматриваемых заявках на патент США с номерами 08/110913 (подана 24 августа 1993) и 08/156498 (подана 23 ноября 1993), обе включены здесь ссылкой во всей полноте.
Пример 5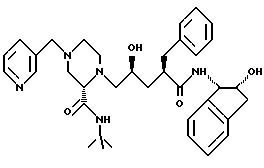
N-(2-(R)-Гидрокси-1(S)-инданил)-2-(R)-фенилметил-4(S)-гидрокси-5- (1-(4-(3-пиридилметил)-2(S)-N'-(трет-бутилкарбоксамидо)пиперазинил)) пентанамид (L-735524) можно получить в соответствии со способами, описанными в заявке на патент США N 07/888825 (подана 15 мая 1992), WO 93/09096, Tet. Lett. 35: 673 - 676 (1994) и Proc. Natl. Acad. Sci. USA, 91: 4096-4100 (1994), каждая из которых включена здесь ссылкой во всей полноте.
Пример 6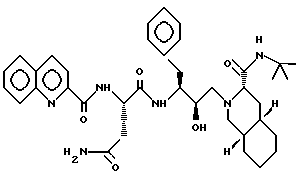
N-трет-бутилдекагидро-2-[2(R)-гидрокси-4-фенил-3(S)-[[N- (2-хинолилкарбонил)-L-аспарагинил] амино] бутил] -(4aR, 8aS) - изохинолин-3(S)-карбоксамид (Ro 31-8959) можно получить в соответствии со способами, описанными в патенте США 5157041, включенным здесь ссылкой во всей полноте.
Пример 7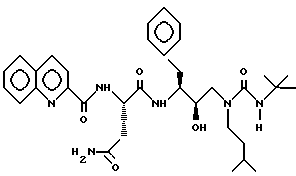
[1S-[1R*(R*), 2S] ] -N1-[3-[[[(1,1-Диметилэтил)амино]карбонил]-(3-метилбутил)амино] -2-гидрокси-1- (фенилметил) пропил] -2-[(2-хинолинилкарбонил)амино]бутандиамид можно получить в соответствии со способами, описанными в находящихся в совместном владении и совместно рассматриваемых заявках на патент США с номерами 08/152934 (подана 15 ноября 1993) и 08/156498 (подана 23 ноября 1993), обе включены здесь как ссылка во всей полноте.
Пример 8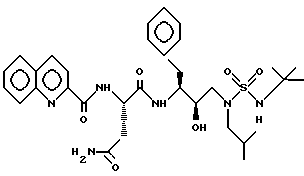
N-[3-[N2-[N1-(1,1-Диметилэтил)аминосульфонил]-N2- (2-метил-пpoпил)амино] -2R-гидpoкcи-1S-(фенилметил)пропил] -2S- [(2-хинолинилкарбонил)амино] бутанамид можно получить в соответствии со способами, описанными в находящихся в совместном владении и совместно рассматриваемых заявках PCT/US 93/10552 (от 29.10.93) и в заявке США 08/156498 от 23.11.93, включенных здесь в качестве ссылок.
Пример 9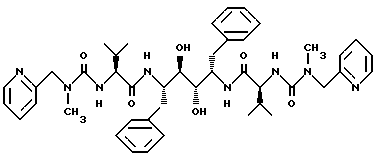
(2S,3R,4S,5S)-2,5-бис-[N-[N-[N-Метил-N-(2-пиридинилметил)амино]карбонил] валинил] амино] -3,4-дигидрокси-1,6-дифенилгексан (А-77003) можно получить в соответствии со способами, описанными в J. Med. Chem. 36: 320 - 330 (1993), которая включена здесь как ссылка во всей полноте.
Пример 10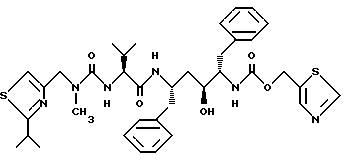
(2S, 3R, 4S,5S)-5-[N-[N-[N-Метил-N-(2-изопропил-4-тиазолил) метил)амино] карбонил] валинил] амино] -3-гидрокси-1,6-дифенилгексан (А-84538, АВТ-538) можно получить в соответствии со способами, описанными в заявке на патент PCT N WO 94/14436 (подана 16 декабря 1993), которая включена здесь как ссылка во всей полноте.
Пример 11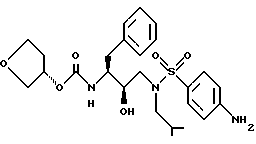
3S-тетрагидрофураниловый эфир [2R-гидрокси-3-[[(4-аминофенил)сульфонил] (2-метилпропил)амино] -1S-(фенилметил)пропил] -карбаминовой кислоты (VX-478) можно получить в соответствии со способами, описанными в заявке на патент PСT N WO 94/05639 (подана 7 сентября 1993), которая включена здесь как ссылка во всей полноте.
Пример 12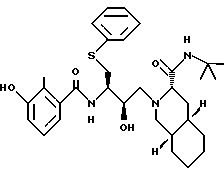
N-трет-Бутилдекагидро-2-[2-(R)-гидрокси-4-(фенилтио)-3(S)- [[N-[(2-метил-3-гидроксифенил)карбонил] амино] бутил] -(4аR,8aS)- изохинолин-3(S)-карбоксамид (AG-1343, AG-1350) можно получить в соответствии со способами, описанными в Bioorg. and Med. Chem. Let. 5:715-720, 5:721-726 и 5:727-732 (1995), каждая из которых включена здесь как ссылка во всей полноте. В частности, НОВТ- активный эфир 3-гидрокси-2-метил-бензойной кислоты (Bioorg. and Med. Chem. Let. 5:727-732 (1995)) можно соединить с N-трет- бутилдекагидро-2-[2(R)-гидрокси-4-(фенилтио)-3(S)-аминобутил] - (4aR, 8aS)-изохинолин-3(S)-карбоксамидом (Bioorg. and Med. Chem. Let. 5:715-720 (1995)).
Пример 13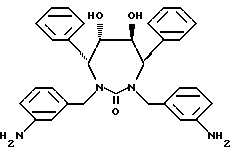
[4R-(4α, 5α, 6β, 7β ]-1,3-бис(3- Аминофенил)метил]гексагидро-5,6-дигидрокси-4,7-бис(фенилметил)- 2H-1,3-диазепин-2-он (DMP-450, ХМ-412) можно получить в соответствии со способами, описанными в заявке на патент PCT WO 93/07128, которая включена здесь как ссылка во всей полноте. В частности, 3-нитрофенилметилгалогенид, например 3-нитрофенилметилхлорид или -бромид, обрабатывают гидроксизащищенным производным [4R-((4 α , 5 α , 6 β , 7 β )] -гексагидро-5,6-дигидрокси-4,7-бис (фенилметил)-2H-1,3-диазепин-2-оном, затем удаляют защитные группы (освобождают) гидроксигруппы (смотри WO 93/07128) и восстанавливают нитрогруппы в аминогруппы. Такие восстановления можно проводить, применяя стандартные методики, хорошо известные специалистам данной области.
Пример 14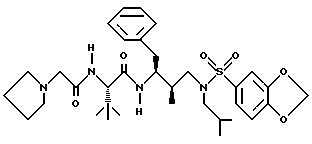
Получение N-[2R-гидрокси-3-[[(1,3-бензодиоксол-5-ил) сульфонил]-(2-метилпропил)амино]-1S-(фенилметил)пропил]-2S- [[(пирролидин-1-ил)ацетил]амино] -3,3-диметилбутанамида
Часть А: Получение 1,3-бензодиоксол-5-сульфонилхлорида
В раствор 4,25 г безводного N,N-диметилформамида при 0oC в атмосфере азота добавляли 7,84 г сульфурилхлорида, после чего образовалось твердое вещество. После перемешивания в течение 15 мин добавляли 6,45 г 1,3-бензодиоксола и смесь нагревали при 100oC в течение 2 ч. Реакционную смесь охлаждали, выливали в ледяную воду, экстрагировали хлористым метиленом, сушили над сульфатом магния, фильтровали и концентрировали, получая 7,32 г неочищенного материала в виде черного масла. Его хроматографировали на силикагеле с использованием смеси 20% хлористый метилен/гексан, получая 1,9 г (1,3-бензодиоксол-5-ил)сульфонилхлорида.
Альтернативно, в круглодонную колбу на 22 л, снабженную механической мешалкой, холодильником, нагревающим кожухом и капельной воронкой с уравновешенным давлением, заливали комплекс триоксид серы- диметилформамид (ДМФ) (2778 г, 18,1 моль). Затем добавляли дихлорэтан (4 л) и начинали перемешивание. Через капельную воронку затем в течение пяти минут добавляли 1,3-бензодиоксол (1905 г, 15,6 моль). Температуру затем повышали до 75oC и поддерживали ее в течение 22 ч (ЯМР показал, что реакция заканчивалась после 9 ч). Реакционную смесь охлаждали до 26oC и добавляли оксалилхлорид (2290 г, 18,1 моль) с такой скоростью, чтобы поддерживать температуру ниже 40oC (1,5 ч). Смесь нагревали до 67oC в течение 5 ч, после чего охладили до 16oC ледяной баней. Реакцию тушили водой (5 л) со скоростью, которая позволяла поддерживать температуру ниже 20oC. После завершения добавления воды смесь перемешивали в течение 10 мин. Слои разделяли и органический слой промывали снова дважды водой (5 л). Органический слой сушили сульфатом магния (500 г) и фильтровали для удаления осушителя. Растворитель удаляли в вакууме при 50oC. Полученную теплую жидкость оставляли для охлаждения, во время которого начиналось образование твердого вещества. Через 1 ч это твердое вещество промывали гексаном (400 мл), фильтровали и сушили, получая целевой сульфонилхлорид (2823 г). Промывочный гексан концентрировали и образованное твердое вещество промывали 400 мл гексана, получая дополнительный сульфонилхлорид (464 г). Общий выход был 3287 г (95,5%, считая на 1,3-бензодиоксол).
Часть В: Получение 2S-[бис(фенилметил)амино]- 3-фенилпропанола
СПОСОБ 1: Получение 2S-[бис(фенилметил)амино]-3-фенилпропанола восстановлением DIBAL фенилметилового эфира
Стадия 1: Раствор L-фенилаланина (50,0 г, 0,302 моль), едкого натра (24,2 г, 0,605 моль) и карбоната калия (83,6 г, 0,605 моль) в воде (500 мл) нагревали до 97oC. Затем медленно добавляли бензилбромид (108,5 мл, 0,605 моль) (время добавления - 25 мин). Смесь перемешивали при 97oC в течение 30 мин в атмосфере азота. Раствор охлаждали до комнатной температуры и экстрагировали толуолом (2 х 250 мл). Комбинированный органический слой промывали водой и соляным раствором, сушили над сульфатом магния, фильтровали и концентрировали до образования масла. Фенилметиловый эфир N,N-бис(фенилметил)-L-фенилаланина можно очищать колоночной хроматографией (силикагель, 15% этилацетат/гексан). Обычно продукт достаточно чистый для использования непосредственно в следующей стадии без дальнейшей очистки. EIMS: m/z 434.
Стадия 2: Бензилированный фенилметиловый эфир фенилаланина (0,302 моль) из предыдущей реакции растворяли в толуоле (750 мл) и охлаждали до -55oC. 1,5 М раствор DIBAL в толуоле (443,9 мл, 0,666 моль) добавляли со скоростью, позволяющей поддерживать температуру между -55 и -50oC (время добавления - 1 ч). Смесь перемешивали в течение 20 мин в атмосфере азота и затем гасили при -55oC медленным добавлением метанола (37 мл). Холодный раствор затем выливали в холодный (5oC) 1,5 н. раствор HCl (1,8 л). Осажденный твердый материал (приблизительно 138 г) отделяли фильтрованием и промывали толуолом. Твердый материал суспендировали в смеси толуола (400 мл) и воды (100 мл). Смесь охлаждали до 5oC и обрабатывали 2,5 н. NaOH (168 мл) и затем перемешивали при комнатной температуре до растворения твердого материала. Слой толуола отделяли от водной фазы и промывали водой и соляным раствором, сушили над сульфатом магния, фильтровали и концентрировали до объема 75 мл (89 г). В остаток добавляли этилацетат (25 мл) и гексан (25 мл), после чего начал кристаллизоваться целевой спиртовый продукт. Через 30 мин для промотирования дальнейшей кристаллизации добавляли дополнительные 50 мл гексана. Твердый материал отделяли фильтрованием и промывали 50 мл гексана, получая 34,9 г продукта первой порции. Вторую порцию продукта (5,6 г) выделяли повторным фильтрованием маточной жидкости. Две порции объединяли и кристаллизовали из этилацетата (20 мл) и гексана (30 мл), получая 40 г 2S-[бис(фенилметил)амино]-3-фенилпропанола, выход 40%, считая на L-фенилаланин. Анализ. Рассчитано для C23H25ON: C 83,34; H 7,60; N 4,23. Найдено C 83,43; H 7,59; N 4,22.
СПОСОБ 2: Получение 2S-[бис(фенилметил)амино]бензопропанола N,N-дибензилированием L-фенилаланилола
L-Фенилаланилол (176,6 г, 1,168 моль) добавляли в перемешиваемый раствор карбоната калия (484,6 г, 3,506 моль) в 710 мл воды. Смесь нагревали до 65oC в атмосфере азота. Раствор бензилбромида (400 г, 2,339 моль) в этаноле 3A (305 мл) добавляли с такой скоростью, чтобы поддерживалась температура между 60-68oC. Двухфазный раствор перемешивали при 65oC в течение 55 мин и затем оставляли для охлаждения до 10oC при энергичном перемешивании. Масляный продукт отверждали в небольшие гранулы. Продукт разбавляли 2,0 л водопроводной воды и перемешивали в течение 5 минут для растворения неорганических побочных продуктов. Продукт отделяли фильтрованием при пониженном давлении и промывали водой до pH 7. Полученный неочищенный продукт перекристаллизовывали из 1,1 л смеси этилацетат/гептан (1:10). Продукт отделяли фильтрованием (при -8oC), промывали 1,6 л охлажденной (-10oC) смесью этилацетат/гексан (1: 10) и сушили на воздухе, получая 339 г (выход 88%) 2S-[бис(фенилметил)амино] -3-фенилпропанола. Т.пл. = 71,5-73,0oC.
Часть C: Получение 2S-[бис(фенилметил)амино] -3-фенилпропанальдегид (пропановый альдегид)
СПОСОБ 1: 2S-[бис(Фенилметил)амино]-3-фенилпропанол (200 г, 0,604 моль) растворяли в триэтиламине (300 мл, 2,15 г, 0,604 моль). Смесь охлаждали до 12oC и добавляли раствор комплекса триоксид серы/пиридин (380 г, 2,39 моль) в диметилсульфоксиде (ДМСО) (1,6 л) со скоростью, позволяющей поддерживать температуру между 8 - 17oC. Раствор перемешивали при температуре окружающей среды в атмосфере азота в течение 1,5 ч. Реакционную смесь охлаждали ледяной водой и гасили 1,6 л холодной воды (10-15oC) в течение 45 мин. Образованный раствор экстрагировали этилацетатом (2,0 л), промывали 5%-ной лимонной кислотой (2,0 л) и соляным раствором (2,2 л), сушили над MgSO4 (280 г) и фильтровали. Растворитель удаляли в вакууме и остаток затем сушили в вакууме, получая 198,8 г 2S-[бис(фенилметил)амино]-3-фенилпропанальдегида в виде бледно- желтого масла (99,9%). Полученный неочищенный продукт был достаточно чистым для использования непосредственно в следующей стадии без очистки.
СПОСОБ 2: Раствор оксалилхлорида (8,4 мл, 0,096 моль) в дихлорметане (240 мл) охлаждали до -74oC. Затем медленно добавляли раствор МДСО (12,0 мл, 0,155 моль) в дихлорметане (50 мл) со скоростью, позволяющей поддерживать температуру при -74oC (время добавления - 1,25 ч). Смесь перемешивали в течение 5 мин, затем добавляли раствор 2S-[бис(фенилметил)амино]-3-фенилпропанола (0,074 моль) в 100 мл дихлорметана (время добавления - 20 мин, температура от -75oC до -68oC). Раствор перемешивали при -78oC в атмосфере азота в течение 35 мин. Затем в течение 10 мин (температура от -78oC до -68oC) добавляли триэтиламин (41,2 мл, 0,295 моль), после чего осаждалась аммониевая соль. Холодную смесь перемешивали в течение 30 мин и затем добавляли воду (225 мл). Слой дихлорметана отделяли от водной фазы и промывали водой, соляным раствором, сушили над сульфатом магния, фильтровали и концентрировали. Остаток разбавляли этилацетатом и гексаном и затем фильтровали для дальнейшего удаления аммониевой соли. Фильтрат концентрировали, получая 2S-[бис(фенилметил)амино] -3- фенилпропанальдегид. Этот альдегид переносили в следующую стадию без очистки.
СПОСОБ 3: В смесь 1,0 г (3,0 ммоль) 2S-[бис(фенилметил)амино]-3- фенилпропанола, 0,531 г (4,53 моль) N-метилморфолина, 2,27 г молекулярных сит (4А) и 9,1 мл ацетонитрила добавляли 53 мг (0,15 моль) перрутената тетрапропиламмония (ТРАР). Смесь перемешивали в течение 40 мин при комнатной температуре и концентрировали при пониженном давлении. Остаток суспендировали в 15 мл этилацетата, фильтровали через подушку силикагеля. Фильтрат концентрировали при пониженном давлении, получая продукт, содержащий приблизительно 50% 2S-[бис(фенилметил) амино]-3-фенилпропанальдегида в виде бледно-желтого масла.
СПОСОБ 4: В раствор 1,0 г (3,02 моль) 2S-[бис(фенилметил) амино]-3-фенилпропанола в 9,0 мл толуола добавляли 4,69 мг (0,03 ммоль) 2,2,6,6-тетраметил-1-пиперидинилокси, свободного радикала (TEMPO), 0,32 г, (3,11 ммоль) бромида натрия, 9,0 мл этилацетата и 1,5 мл воды. Смесь охлаждали до 0oC и медленно, в течение 25 мин, добавляли водный раствор 2,87 мл 5% бытового отбеливателя, содержащего 0,735 г (8,75 ммоль) бикарбоната натрия, и 8,53 мл воды. Смесь перемешивали при 0oC в течение 60 мин. После еще двух добавок отбеливателя (1,44 мл каждого) смесь перемешивали в течение 10 мин. Водный слой экстрагировали дважды 20 мл этилацетата. Объединенный органический слой промывали 4,0 мл раствора, содержащего 25 мг иодида калия и воду (4,0 мл), 20 мл 10%-ного водного раствора тиосульфата натрия и затем соляным раствором. Органический раствор сушили над сульфатом магния, фильтровали и концентрировали при пониженном давлении, получая 1,34 г неочищенного масла, содержащего небольшое количество целевого альдегидного продукта, 2S-[бис(фенилметил)амино]-3- фенилпропанальдегида.
Часть D: Получение N,N-дибензил-3(S)-амино-1,2-(S)-эпокси-4-фенилбутана
СПОСОБ 1: Раствор 2S-[бис(фенилметил)амино] -3- фенилпропанальдегида (191,7 г, 0,58 моль) и хлориодметана (56,4 мл, 0,77 моль) в тетрагидрофуране (1,8 л) охлаждали до температуры от -30 до -35oC в реакторе из нержавеющей стали в атмосфере азота. Затем добавляли раствор н-бутиллития в гексане (1,6 М, 365 мл, 0,58 моль) со скоростью, которая позволяет поддерживать температуру ниже -25oC. После добавления смесь перемешивали при температуре от -30 до -35oC в течение 10 мин. Другие добавления реагентов проводили следующим образом: (1) добавляли дополнительный хлориодметан (17 мл), затем н-бутиллитий (110 мл) при < -25oC. После добавления смесь перемешивали при температуре от -30 до -35oC в течение 10 мин. Это повторяли один раз. (2) Добавляли дополнительный хлориодметан (8,5 мл, 0,11 моль), затем н- бутиллитий (55 мл, 0,88 моль) при < -25oC. После добавления смесь перемешивали при температуре от -30 до -35oC в течение 10 мин. Это повторяли 5 раз. (3) Добавляли дополнительный хлориодметан (8,5 мл, 0,11 моль), затем н-бутиллитий (37 мл, 0,059 моль) при < -25oC. После добавления смесь перемешивали при температуре от -30 до -35oC в течение 10 мин. Это повторяли один раз. Наружное охлаждение прекращали и смесь нагревали до температуры окружающей среды в течение 4 - 16 часов, когда ТСХ (силикагель, 20% этилацетат/гексан) показала, что реакция закончилась. Реакционную смесь охлаждали до 10oC и гасили 1452 г 16%-ного раствора хлорида аммония, поддерживая температуру ниже 23oC. Смесь перемешивали в течение 10 мин и органический и водный слои разделяли. Водную фазу экстрагировали этилацетатом (2х500 мл). Слой этилацетата объединяли со слоем тетрагидрофурана. Объединенный раствор сушили над сульфатом магния (220 г), фильтровали и концентрировали в вакууме. Остаток в виде коричневого масла сушили при 70oC в вакууме (0,79 атм) в течение 1 ч, получая 222,8 г неочищенного материала. Неочищенный продукт обычно используют непосредственно в следующей стадии без очистки.
СПОСОБ 2: Раствор неочищенного альдегида (0,074 моль) и хлориодметана (7,0 мл, 0,096 моль) в тетрагидрофуране (285 мл) охлаждали до -78oC в атмосфере азота. Затем добавляли 1,6 М раствор н-бутиллития в гексане (25 мл, 0,040 моль) со скоростью, позволяющей поддерживать температуру при -75oC. После первого добавления снова добавляли дополнительный хлориодметан (1,6 мл, 0,022 моль) и затем н-бутиллитий (23 мл, 0,037 моль), поддерживая температуру -75oC. Смесь перемешивали в течение 15 мин. Каждый из реагентов: хлориодметан (0,70 мл, 0,010 моль) и н-бутиллитий (5 мл, 0,008 моль) добавляли еще 4 раза в течение 45 мин при -75oC. Охлаждающую баню затем удаляли и раствор нагревали до 22oC в течение 1,5 часа. Смесь выливали в 300 мл насыщенного водного раствора хлорида аммония. Слой тетрагидрофурана отделяли. Водную фазу экстрагировали этилацетатом (1х300 мл). Объединенный органический слой промывали соляным раствором, сушили над сульфатом магния, фильтровали и концентрировали, получая коричневое масло (27,4 г). Продукт можно использовать в следующей стадии без очистки.
СПОСОБ 3: Раствор 2S-[бис(фенилметил)амино] -3-фенилпропанальдегида (178,84 г, 0,54 моль) и бромхлорметана (46 мл, 0,71 моль) в тетрагидрофуране (1,8 л) охлаждали до температуры от -30 до -35oC в реакторе из нержавеющей стали в атмосфере азота. Затем добавляли раствор н-бутиллития в гексане (1,6 М, 340 мл, 0,54 моль) со скоростью, позволяющей поддерживать температуру ниже -25oC. После добавления смесь перемешивали при температуре между -30 и -35oC в течение 10 мин. Другие добавления реагентов проводили следующим образом: (1) добавляли дополнительный бромхлорметан (14 мл), затем н-бутиллитий (102 мл) при < -25oC. После добавления смесь перемешивали при температуре между -30 и -35oC в течение 10 мин. Это повторяли один раз. (2) Добавляли дополнительный бромхлорметан (7 мл, 0,11 моль), затем н-бутиллитий (51 мл, 0,082 моль) при < -25oC. После добавления смесь перемешивали при температуре между -30 и -35oC в течение 10 мин. Это повторяли 5 раз. (3) Добавляли дополнительный бромхлорметан (7 мл, 0,11 моль), затем н-бутиллитий (51 мл, 0,082 моль) при < -25oC. После добавления смесь перемешивали при температуре между -30 и -35oC в течение 10 мин. Это повторяли один раз. Наружное охлаждение прекращали и смесь нагревали до температуры окружающей среды в течение 4 - 16 ч, когда ТСХ (силикагель, 20% этилацетат/гексан) показала, что реакция закончилась. Реакционную смесь охлаждали до 10oC и гасили 1452 г 16%-ного раствора хлорида аммония, поддерживая температуру ниже 23oC. Смесь перемешивали в течение 10 мин и органический и водный слои разделяли. Водную фазу экстрагировали этилацетатом (2х500 мл). Слой этилацетата объединяли со слоем тетрагидрофурана. Объединенный раствор сушили над сульфатом магния (220 г), фильтровали и концентрировали в роторном испарителе при 65oC. Остаток в виде коричневого масла сушили при 70oC в вакууме (0,79 атм) в течение 1 ч, получая 22.2,8 г неочищенного материала.
Часть E: Получение соли N-[3(S)-[N,N-бис(фенилметил)амино]-2(R)- гидрокси-4-фенилбутил]-N-изобутиламин щавелевая кислота
Стадия 1: В раствор неочищенного N,N-дибензил-3(S)-амино -1,2(S)-эпокси-4-фенилбутана (388,5 г 1,13 моль) в изопропаноле (2,7 л) или этилацетате добавляли изобутиламин (1,7 кг, 23,1 моль) в течение 2 мин. Температура повышалась от 25oC до 30oC. Раствор нагревали до 82oC и перемешивали при этой температуре в течение 1,5 ч. Теплый раствор концентрировали в вакууме. Остаток в виде коричневого масла сушили в вакууме (0,8 мм рт.ст.) в течение 16 ч, получая 450 г продукта в виде неочищенного масла.
Стадия 2: В раствор щавелевой кислоты (8,08 г, 89,72 ммоль) в метаноле (76 мл) добавляли раствор неочищенного 3(S)-[N,N-бис(фенилметил)амино]-1-(2-метилпропил)амино-4-фенилбутан-2(R)-ола в этилацетате (90 мл) в течение 15 мин. Смесь перемешивали при комнатной температуре в течение около 2 ч. Твердый материал выделяли фильтрованием, промывали этилацетатом (2х20 мл) и сушили в вакууме в течение около 1 ч, получая 21,86 г 97% диастереомерно чистой соли, т.пл.=174,99oC; микроанализ: Рассчитано: C 71,05%, H 7,50%, N 5,53%; Найдено: C 71,71%, H 7,75%, N 5,39%.
Альтернативно, неочищенный 3(S)-[N, N-бис(фенилметил) амино]- 1-(2-метилпропил)амино-4-фенилбутан-2(R)-ол (5 г) растворяли в метил-трет-бутиловом эфире (МТБЭ) (10 мл) и добавляли щавелевую кислоту (1 г) в метаноле (4 мл). Смесь перемешивали в течение около 2 ч. Полученную твердую часть отделяли фильтрованием, промывали холодным МТБЭ и сушили, получая 2,1 г белого твердого продукта с 98,9% диастереомерной чистоты (на основе площади пика ВЭЖХ).
Часть F. Получение 1-[N-[(1,3-бензодиоксол-5-ил] сульфонил] -N-(2-метилпропил)амино]-3(S)-[N,N-бис(фенилметил)амино]-фенил-2 (R)-бутанола
В соль N-[3(S)-[N,N-бис(фенилметил)амино]-2(R)-гидрокси-4-фенилбутил]- N-изобутиламин щавелевая кислота (354,7 г, 0,7 моль) в 1,4-диоксане (2000 мл) добавляли раствор карбоната калия (2.41,9 г, 1,75 моль) в воде (250 мл). Смесь перемешивали в течение 2 ч при комнатной температуре, затем в течение 15 мин добавляли 1,3-бензодиоксол-5-сульфонилхлорид (162,2 г, 0,735 моль) в 1,4-диоксане (250 мл). Реакционную смесь перемешивали при комнатной температуре в течение 18 ч. Добавляли этилацетат (1000 мл) и воду (500 мл) и перемешивание продолжали в течение еще 1 ч. Водный слой отделяли и далее экстрагировали этилацетатом (200 мл). Объединенный слой этилацетата промывали 25%-ным соляным раствором (500 мл) и сушили над безводным сульфатом магния. После фильтрования и промывания сульфата магния этилацетатом (200 мл) растворитель удаляли в вакууме, получая целевой сульфамид в виде вязкого желтого пенящегося масла (440,2 г, выход 105%). ВЭЖХ/МС (электрораспыление) (m/z 601 [М+H]+).
Альтернативно, соль N-[3(S)-[N,N-бис(фенилметил)амино]-2(R)- гидрокси-4-фенилбутил] -N-изобутиламин щавелевая кислота (2800 г, 5,53 моль) и ТГФ (4 л) добавляли в круглодонную колбу на 22 л, снабженную механической мешалкой. Карбонат калия (1921 г, 13,9 моль) растворяли в воде (2,8 л) и добавляли в суспензию в ТГФ. Смесь затем перемешивали в течение 1 ч. 1,3-бензодиоксол-5-сульфонилхлорид (1281 г, 5,8 моль) растворяли в ТГФ (1,4 л) и добавляли в реакционную смесь в течение 25 мин. Дополнительные 200 мл ТГФ применяли для промывания капельной воронки. Реакционную смесь перемешивали в течение 14 ч и затем добавляли воду (4 л). Смесь перемешивали в течение 30 мин и оставляли для разделения слоев. Слои удаляли и водный слой промывали дважды ТГФ (500 мл). Объединенный слой ТГФ сушили над сульфатом магния (500 г) в течение 1 ч. Этот раствор затем фильтровали для удаления осушителя и применяли в последующих реакциях.
Часть G: Получение соли 1-[N-[(1,3-бензодиоксол-5-сульфонил]-N- (2-метилпропил)амино]-3(S)-амино-4-фенил-2(R)-бутанол метансульфокислота
Неочищенный 1-[N-[(1,3-бензодиоксол-5-ил)сульфонил] -N-(2-метилпропил) амино] -3(S)-[бис(фенилметил)амино]-4-фенил-2(R)-бутанол (6,2 г, 0,010 моль) растворяли в метаноле (40 мл). В раствор затем добавляли метансульфокислоту (0,969 г, 0,010 моль) и воду (5 мл). Смесь помещали в склянку Парра на 500 мл для гидрирования, содержащую 20% Pd(ОН)2 на угле (255 мг, содержание воды 50%). Склянку помещали в гидрогенизатор и продували 5 раз азотом и 5 раз водородом. Реакцию проводили при 35oC под давлением водорода 4,430 атм в течение 18 ч. Добавляли дополнительный катализатор (125 мг) и после продувания гидрирование продолжали в течение дополнительных 20 ч. Смесь фильтровали через целит, который промывали метанолом (2 х 10 мл). Приблизительно одну треть метанола удаляли при пониженном давлении. Оставшееся количество метанола удаляли азеотропной перегонкой с толуолом при 80 мм рт.ст. Добавляли толуол в виде порций по 15, 10, 10 и 10 мл. Продукт кристаллизовали из смеси и фильтровали и промывали дважды толуолом порциями по 10 мл. Твердый продукт сушили при комнатной температуре при 1 мм рт.ст. в течение 6 ч, получая соль амина (4,5 г, 84%): m/z 421 [М+H]+.
Альтернативно, в раствор неочищенного 1-[N-[(1,3- бензодиоксол-5- ил)сульфонил] -N-(2-метилпропил)амино] -3(S)-[бис(фенилметил)амино]-4-фенил- 2(R)-бутанола в ТТФ добавляли воду (500 мл), затем метансульфокислоту (531 г, 5,5 моль). Раствор перемешивали для обеспечения полного смешивания и добавляли в автоклав на 18,9 л. При помощи ТГФ (500 мл) в автоклав добавляли катализатор Pearlman (200 г 20% Pd(OH)2 на C/50% воды). Реактор продували четыре раза азотом и четыре раза водородом. В реактор загружали водород под давлением 4,2 атм и перемешивали при начальной скорости 450 об/мин. Через 16 ч анализ ВЭЖХ показал, что все еще присутствовало небольшое количество монобензилсодержащего промежуточного продукта. Добавляли дополнительный катализатор (50 г) и реакцию проводили в течение ночи. Раствор затем фильтровали
через целит (500 г) для удаления катализатора и концентрировали в вакууме в виде пяти порций. К каждой порции добавляли толуол (500 мл) и удаляли в вакууме для азеотропного удаления остаточной воды. Полученный твердый материал разделяли на три части и каждую промывали метил-трет-бутиловым эфиром (2 л) и фильтровали. Остаточный растворитель удаляли при комнатной температуре в вакуумном сушильном шкафу при менее чем 1 мм рт.ст., получая 2714 г ожидаемой соли.
Часть H: Получение N-[2R-гидрокси-3-[[(1,3-бензодиоксол-5-ил)сульфонил] (2-метилпропил)амино]-1S-(фенилметил)-пропил]-2S-[(фенилметоксикарбонил)амино]- 3,3-диметилбутанамида
В раствор 118,8 г (0,776 моль) N-гидроксибензотриазола и 137,1 г (0,52 моль) N-карбобензилоксикарбонил-L-трет-лейцина в 750 мл безводного ДМФ при 0oC в атмосфере азота добавляли 109,1 г (0,57 моль) EDC. После перемешивания при 0oC в течение 2 ч добавляли раствор 273 г (0,53 моль) метансульфоната 2R-гидрокси-3-[[(1,3- бензодиоксол-5-ил)сульфонил] (2-метилпропил)амино] -1S-(фенилметил)пропиламина, предварительно нейтрализованный 228 мл (210 г, 2,08 моль) 4-метилморфолина, в 250 мл безводного ДМФ. После перемешивания при 0oC в течение 30 мин смесь перемешивали 18 ч при комнатной температуре. Растворители удаляли при пониженном давлении при 45oC, добавляли 1,5 л этилацетата, промывали 5%-ной лимонной кислотой, насыщенным раствором бикарбоната натрия, соляным раствором, сушили над безводным сульфатом магния, фильтровали и концентрировали, получая 400 г неочищенного материала. Его хроматографировали в виде 3 порций на хроматографе Prep 2000 на силикагеле с использованием в качестве элюента 20-50% этилацетат/гексан, получая 320 г очищенного материала, m/e = 674 (M+Li), 98% по ВЭЖХ.
Часть I: Получение N-[2R-гидрокси-3-[[(1,3-бензодиоксол-5- ил) сульфонил] (2-метилпропил)амино] -1S-(фенилметил)пропил] - 2S-амино-3,3-диметилбутанамида
Раствор 312 г N-[2R-гидрокси-3-[[1,3-бензодиоксол-5-ил) сульфонил](2-метилпропил)амино] -1S-(фенилметил)пропил]-2S- [(фенилметоксикарбонил)амино] -3,3-диметилбутанамида в 1 л тетрагидрофурана гидрировали в присутствии 100 г катализатора, 4% палладия на угле, под давлением водорода 4,2 атм в течение 6 часов при комнатной температуре. Катализатор удаляли фильтрованием и растворитель удаляли при пониженном давлении, получая 240 г целевого соединения.
Часть J: Получение N-[2R-гидрокси-3-[[(1,3-бензодиоксол-5-ил)сульфонил] (2-метилпропил)амино] -1S-(фенилметил)пропил] -2S-[(хлорацетил) амино]-3,3-диметилбутанамида
В раствор 234,3 г (0,439 моль) N[2R-гидрокси-3-[[(1,3-бензодиоксол-5-ил) сульфонил] (2-метилпропил)амино] - 1S-(фенилметил)пропил]-2S-амино-3,3-диметилбутанамида в 1 л хлористого метилена добавляли 80 мл (59,5 г, 0,46 моль) диизопропилэтиламина, затем медленно при комнатной температуре добавляли 78,8 г (0,46 моль) хлоруксусного ангидрида, поддерживая температуру ниже 35oC. После перемешивания в течение дополнительного 1 ч анализ ВЭЖХ показывал, что все еще присутствовало небольшое количество исходного соединения, и добавляли 1,5 г хлоруксусного ангидрида. Через 10 мин растворители удаляли при пониженном давлении, добавляли 1 л этилацетата, промывали 5%-ной лимонной кислотой, насыщенным раствором бикарбоната натрия, соляным раствором, сушили над безводным сульфатом магния, фильтровали и концентрировали, получая 314 г неочищенного материала. Его хроматографировали в виде 3 порций на хроматографе Prep 2000 на силикагеле с использованием 20-50% этилацетат/гексан, получая 165 г целевого соединения, m/e = 616, (M+Li), 98% по ВЭЖХ.
Часть K: Получение N-[2R-гидрокси-3-[[(1,3-бензодиоксол-5-ил)сульфонил] (2-метилпропил)амино]-1S-(фенилметил) пропил]-2S-[[(пирролидин-1-ил) ацетил] амино]-3,3-диметилбутанамида
В 164,2 г (0,27 моль) N-[2R-гидрокси-3-[[(1,3-бензодиоксол-5-ил) сульфонил] (2-метилпропил)амино]-1S-(фенилметил)-пропил]-2S- [(хлорацетил)амино] -3,3-диметилбутанамида добавляли 500 мл тетрагидрофурана, растворитель удаляли при пониженном давлении для удаления любого количества этилацетата и затем добавляли 350 мл тетрагидрофурана. В этот раствор при 10oC затем добавляли 130 мл (1,56 моль) пирролидина. Через 1 ч растворители удаляли при пониженном давлении, добавляли 1 л этилацетата, промывали насыщенным раствором бикарбоната натрия, соляным раствором, сушили над безводным сульфатом магния, фильтровали и концентрировали, получая 185 г неочищенного материала, анализ которого ВЭЖХ показал его 98,9% чистоту. Его разделяли на 3 порции и хроматографировали на хроматографе Prep 2000 с использованием сначала 50% этилацетат/гексан, затем 5% метанол/этилацетат, получая 160 г очищенного материала (99% по ВЭЖХ). Его затем перекристаллизовали из 460 мл диэтилового эфира и 70 мл гексана, получая 121 г целевого продукта (> 99% по ВЭЖХ), m/e 651 (M+Li), т.пл. = 112-114oC.
Пример 15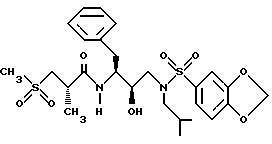
Получение N-[2R-гидрокси-3-[(2-метилпропил)[1,3-бензодиоксол-5-ил) сульфонил] амино] -1S-(фенилметил)пропил]-25-метил-3-(метилсульфонил) пропанамида
Часть А: Получение 2(S)-метил-3-(метилсульфонил)пропионовой кислоты
Стадия 1: В раствор 200 г (1,23 моль) D-(-)-3-ацетил-- β-меркаптоизомасляной кислоты в 1,0 л метанола добавляли 161,0 г (2,47 моль) едкого кади, растворенного в 500 мл метанола, поддерживая температуру ниже 10oC при охлаждении ледяной баней. После перемешивания дополнительно 20 мин добавляли 117 мл (156 г, 1,23 моль) диметилсульфата, поддерживая температуру ниже 20oC. Ледяную баню удаляли и смесь перемешивали в течение дополнительных 60 мин. Соль удаляли фильтрованием, растворители удаляли при пониженном давлении и добавляли этилацетат. После отделения водяной слой подкисляли концентрированной соляной кислотой, экстрагировали этилацетатом, сушили над безводным сульфатом магния, фильтровали и концентрировали, получая 164 г (99%) целевой 2S-метил-3-(метилтио)пропионовой кислоты, m/e 133 (M-H).
Стадия 2: В раствор 10,0 г (74,6 ммоль) 2S-метил-3-(метилтио)пропионовой кислоты в 150 мл ацетона и 30 мл воды, охлажденный до 18oC в ледяной бане, добавляли 161,8 г (263 ммоль) оксона. После добавления приблизительно половины материала температура поднималась до 24oC, добавление прекращали, температуру снижали до 18oC, затем добавление продолжали. После перемешивания при 15-20oC в течение 15 мин баню удаляли и реакционную смесь перемешивали при комнатной температуре в течение 1 ч. Твердую часть отделяли фильтрованием и промывали ацетоном, фильтрат концентрировали приблизительно до 40 мл и остаток растворяли в 200 мл этилацетата. Слой этилацетата сушили над безводным сульфатом магния, фильтровали и концентрировали, получая 11,4 г масла. Его растворяли в минимальном количестве этилацетата и добавляли гексан для образования осадка. Его собирали, получая 6,95 г целевого продукта, m/z = 167 (М+Н).
Часть В: Получение N-[2R-гидрокси-3-[(2-метилпропил)-[1,3-бензодиоксол- 5-ил)сульфонил] амино] -1S-(фенилметил) пропил]-2S-метил-3-(метилсульфонил) пропанамида
В раствор 5,0 г (30 ммоль) 2S-метил-3-(метилсульфонил) пропионовой кислоты и 6,90 г (45 ммоль) N-гидроксибензотриазола в 30 мл безводного ДМФ при 0oC в атмосфере азота добавляли 6,34 г (33 ммоль) EDC. Приблизительно через 10 мин весь EDC растворялся. Через 60 мин при 0oC добавляли раствор 15,5 г (30 ммоль) метансульфоната 2R-гидрокси-3-[[(1,3-бензодиоксол-5- ил)сульфонил] (2-метилпропил)амино] -1S-(фенилметил)пропиламина в 30 мл безводного ДМФ, предварительно нейтрализованный 3,4 мл (31,6 ммоль) 4-метилморфолина. После выдерживания 3 часа при 0oC смесь затем перемешивали ночью в течение 17 ч. ДМФ удаляли при пониженном давлении, добавляли этилацетат, промывали 5%-ной лимонной кислотой, насыщенным раствором бикарбоната натрия, водой, соляным раствором, сушили над безводным сульфатом магния, фильтровали и концентрировали, получая 16 г неочищенного материала, который имел чистоту 88% по ВЭЖХ. Продукт хроматографировали на силикагеле с использованием 20-80% этилацетат/гексан, получая чистый продукт, который перекристаллизовали из смеси этилацетат/гексан, получая 8,84 г чистого продукта, т.пл. 131,8-133,8oC.
Альтернативно, в раствор 35,0 г (211 ммоль) 2S-метил-3-(метилсульфонил) пропионовой кислоты и 48,3 г (315 ммоль) N-гидроксибензотриазола в 210 мл безводного ДМФ при 0oC в атмосфере азота добавляли 44,4 г (231 ммоль) EDC. Приблизительно через 30 мин весь EDC растворялся. Через дополнительные 60 мин при 0oC добавляли раствор 108,8 г (211 ммоль) метансульфоната 2R-гидрокси-3-[[(1,3-бензодиоксол-5-ил)сульфонил] (2-метилпропил) амино] -1S-(фенилметил)пропиламина в 350 мл безводного ДМФ, предварительно нейтрализованный 24 мл (22,3 г) 4-метилморфолина. После выдерживания 2 ч при 0oC смесь затем перемешивали ночью в течение 18 ч. ДМФ удаляли при пониженном давлении, добавляли 1 л этилацетата, промывали 5%-ной лимонной кислотой, насыщенным раствором бикарбоната натрия, водой, соляным раствором, сушили над безводным сульфатом магния, фильтровали и концентрировали, получая 120,4 г неочищенного материала, который имел чистоту 90% по ВЭЖХ. Продукт перекристаллизовали дважды из 750-1000 мл абсолютного этанола, получая 82,6 г целевого продукта.
Пример 16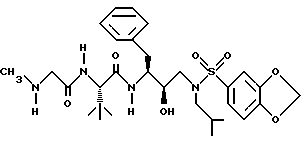
Получение 2S-[[(N-метиламино)ацетил] амино] -N-[2R-гидрокси-3- [[(1,3-бензодиоксол-5-ил)сульфонил] -(2-метилпропил)-амино]-1S- (фенилметил)пропил] -3,3-диметилбутанамида
В 6,55 г (10,7 ммоль) N-[2R-гидрокси-3-[[(1,3-бензодиоксол-5-ил)сульфонил]- (2-метилпропил)амино]-1S-(фенилметил)-пропил]-2S- [(хлорацетил)амино] -3,3-диметилбутанамида добавляли 25 мл тетрагидрофурана, растворитель удаляли при пониженном давлении для удаления любого количества этилацетата и затем добавляли 25 мл тетрагидрофурана. В этот раствор при 10oC добавляли 19 мл (214 ммоль) 40%-ного водного метиламина. Через 2 ч растворители удаляли при пониженном давлении, добавляли 1 л этилацетата, промывали насыщенным раствором бикарбоната натрия, соляным раствором, сушили над безводным сульфатом магния, фильтровали и концентрировали, получая 6,0 г продукта (чистота 98%).
Пример 17
Соединения-ингибиторы ретровирусной протеазы данного изобретения являются эффективными ингибиторами протеазы ВИЧ. Ферментативный анализ, описанный ниже, можно использовать в отборе ингибиторов ретровирусной протеазы для применения в комбинационной терапии. При помощи этого способа можно рассчитать IC50 (концентрация, при которой соединение-ингибитор снижает активность фермента на 50%) для таких соединений.
Ферментативный способ следующий. Субстратом является 2-аминобензоил-Ile-Nle-Phe(n-NO2)-Gln-Arg-NH2. Положительным контролем является MVT-101 (Miller, М. et al., Science 246, 1149 (1989)). Буфером для анализа является 20 мМ фосфат натрия, pH 6,4, 20% глицерин, 1 мМ этилендиаминтетрауксусная кислота, 1 мМ дитиотреитол (DTT) и 0,1% CHAPS. Субстрат растворяют в ДМСО, затем разбавляют в 10 раз в буфере для анализа. Конечная концентрация субстрата в анализе около 80 мкМ. Протеазу ВИЧ разбавляют в буфере для анализа до конечной концентрации фермента около 12,3 нМ, считая на молекулярную массу 10780.
Конечная концентрация ДМСО около 14% и конечная концентрация глицерина около 18%. Испытуемое соединение растворяют в ДМСО и разбавляют в ДМСО приблизительно в десять раз (10 х), затем добавляют 10 мкл субстрата. Повышение флюоресценции регистрируют в 4 временных точках (0, 8, 16 и 24 минуты) при комнатной температуре. Каждый анализ проводят в дупликатных лунках.
Пример 18
Эффективность выбранных соединений-ингибиторов протеазы ВИЧ данного изобретения можно определить при помощи описанного выше ферментативного анализа и следующего анализа CD4+ - клеточной линии. Антивирусную активность ингибиторов протеазы выражают как величины эффективной концентрации 50 (EC50) и/или эффективной концентрации 90 (EC90). Они являются концентрациями ингибиторов, которые требуются для ингибирования вирусной репликации на 50% или 90%, соответственно.
Способ анализа ингибирования ВИЧ остро инфицированных клеток является по существу автоматизированным колориметрическим анализом на основе тетразолия, описанным Pauwels et al. , J. Virol. Methods 20, 309-321 (1988). Анализы проводят в 96-луночных тканевых культуральных планшетах. CD+ - клеточную линию, например CEM, MT-2, MT-4 и подобные клеточные линии, выращивают в среде RPMI-1640 (Gibco), дополненной 10% фетальной телячьей сывороткой, и затем обрабатывают полибреном (2 мкг/мл). Среду объемом 80 мкл, содержащую 1 • 104 клеток, помещают диспенсером в каждую лунку тканевого культурального планшета. В каждую лунку добавляют 100 мкл испытуемого соединения, растворенного в тканевой культуральной среде (или среду без испытуемого соединения в качестве контроля) для достижения желаемой конечной концентрации и клетки инкубируют при 37oC в течение 1 часа. Замороженную культуру ВИЧ-1 разбавляют в культуральной среде до концентрации 5 • 104 TCID50 на мл (TCID50 = доза вируса, которая инфицирует 50% клеток в тканевой культуре) и 20 мкл вирусной пробы (содержащей 1000 TXID50 вируса) добавляют в лунки, содержащие испытуемое соединение, и лунки, содержащие только среду (инфицированные контрольные клетки). Некоторые лунки получают культуральную среду без вируса (неинфицированные контрольные клетки). Подобным образом, присущую токсичность испытуемого соединения определяют добавлением в несколько лунок, содержащих испытуемое соединение, среды без вируса. В сумме тканевые культуральные планшеты содержат следующие эксперименты (см. табл. A в конце описания).
В экспериментах 2 и 4 конечные концентрации испытуемых соединений составляют 1, 10, 100 и 500 мкг/мл. В качестве положительного лекарственного контроля включают азидотимидин (AZT) или дезоксиинозин (ddI). Испытуемые соединения растворяют в ДМСО и разбавляют тканевой культуральной средой так, чтобы конечная концентрация ДМСО не превышала 1,5% в любом случае. ДМСО добавляют во все контрольные лунки в соответствующей концентрации.
После добавления вируса клетки инкубируют при 37oC в увлажненной, содержащей 5% CO2 атмосфере в течение 7 дней. Испытуемые соединения можно добавлять в дни 0, 2 и 5, если желательно. На 7-й день после инфицирования клетки в каждой лунке ресуспендируют и 100 мкл пробы каждой клеточной суспензии удаляют для анализа. В каждый мкл клеточной суспензии добавляют 20 мкл раствора 5 мг/мл бромида 3-(4,5-диметилтиазол-2-ил)-2,5-дифенилтетразолия (МТТ) и клетки инкубируют в течение 4 ч при 37oC в среде с 5% CO2. Во время этого инкубирования содержание МТТ метаболически снижается живыми клетками, что приводит к продуцированию в клетке окрашенного формазанового продукта. В каждую пробу добавляют 100 мкл 10% додецилсульфат натрия в 0,01 н. HCl для лизирования клеток и пробы инкубируют всю ночь. Поглощение у 590 нм для каждой пробы определяют при помощи микропланшет-ридера Molecular Devices. Цитотоксичность и антивирусную эффективность испытуемого соединения определяют путем сравнения величин поглощения, полученных в лунках, содержащих инфицированные или неинфицированные клетки, инкубированные с соединениями, и неинфицированных, необработанных контрольных лунках.
ПРОЦЕДУРЫ С КУЛЬТУРОЙ ВИЧ СТИМУЛИРОВАНИЕ ЛИМФОЦИТОВ ДОНОРА
Лейкоцитные пленки получали из American Red Cross or Blood Bank et Washington University School of Medicine. Эти препараты предварительно тестируют на антитела на ВИЧ и CMV (цитомегавирусы) и поверхностный антиген HBV (вирус гепатита В) и ALT (активность аланинтрансферазы) как маркер для "не-А, не-В" гепатита. Обогащенную лейкоцитами кровь (30 мл) удаляют из пластикового контейнера и 15 мл переносят диспенсером в две центрифужные пробирки на 50 мл с навинчивающимся колпачком. Каждую пробу разбавляют равным объемом стерильного PBS (забуференный фосфатом физиологический раствор, ЗФР) и смешивают пипетированием. Фиколл-пак (15 мл) или LSM помещают ниже разбавленных проб крови при помощи пипетки Пастера и путем предоставления возможности раствору опускаться на дно пробирки. Каждую из пробирок затем центрифугируют при 1300 об/мин (400 g) в течение 45 мин при 20oC. После центрифугирования лимфоцитную зону у границы раздела удаляют и переносят в пробирку на 50 мл. Добавляют ЗФР для разбавления отделенных лимфоцитов и затем центрифугируют при 1300 об/мин в течение 8 мин. Дебрис промывают два раза ресуспендированием в ЗФР и снова центрифугируют. Конечный дебрис снова суспендируют в 20 мл ЗФР пипетированием и общее число жизнеспособных клеток определяют по исключению Trypan Blue.
АНАЛИЗЫ ОСТРОЙ ИНФЕКТИВНОСТИ С ИСПОЛЬЗОВАНИЕМ КЛИНИЧЕСКИХ ИЗОЛЯТОРОВ
Приблизительно 3•107 клеток активируют в течение 48 ч приблизительно с 3-5 мкг/мл РНА (фитогемагглютин, ФГА) в среде RPMI, содержащей 10% фетальную телячью сыворотку и IL-2 (10 Е/мл). Количественные вирусные растворы добавляют в суспензию активированных лимфоцитов при множественности инфекции около 0,001 - 0,01. Суспензию клетка-вирус инкубируют при 37oC в течение 2 ч для проведения вирусной абсорбции. Остаточный вирусный инокулум удаляют центрифугированием и клетки ресуспендируют в среде RPMI, содержащей 10% FBS и 10 Е/мл IL-2. Эти инфицированные клетки добавляют в тест-соединение, разбавленное в полной тканевой культуральной среде из исходного раствора (10 мг/мл) в ДМСО в титрационных микропланшетах с 96 лунками, получая около 5•105 клеток на лунку (на 200 мкл). Инфицированные, необработанные клетки и клетки, обработанные только ДМСО (0,1%) или AZT или DDI, применяли в качестве контроля. Культуры проверяли на образование синцитий на 7 и 11 день после инфицирования или супернатанты испытывали на активность обратной транскриптазы или антиген р24.
АНАЛИЗЫ ХРОНИЧЕСКОЙ ИНФЕКТИВНОСТИ
СЕМ-клетки, хронически инфицированные HXB2 (лабораторный штамм ВИЧ-1), добавляют в 6 лунок титрационного микропланшета с 12 лунками, получая 5•104 клеток на лунку. Половину лунок обрабатывают испытуемым соединением при разных концентрациях и то же самое число неинфицированных СЕМ-клеток сохраняют без добавления соединения. Свежую среду с испытуемым соединением или без него добавляют каждый день в течение трех последующих дней. Культуры затем инкубируют в течение 48 часов без изменения в среде. Клетки собирают центрифугированием, промывают 2 раза в PBS и ресуспендируют в 50 мкл буфера 2х Laemmli, содержащего 0,125 М Трис, pH 6,8, 4% SDS (додецилсульфат натрия), 20% глицерин, 10% бета-меркаптоэтанола и 0,02% Bromophenol blue. Супернатанты культур пропускают через фильтр 0,22 мкм для удаления дебриса и центрифугируют при 50000 об/мин в течение 90 мин для концентрирования вирусных частиц. Вирусный осадок ресуспендируют в 50 мкл буфера 2х Laemmli. Клеточные или вирусные суспензии кипятят в течение 5 мин и затем подвергают электрофоретическому разделению в 10 - 20% SDS-полиакриламидном градиентном геле. Содержимое геля затем переносят в нитроцеллюлозу электроблоттингом. ВИЧ-специфичные белки обнаруживают, используя моноклональные антитела на р24 и р17, затем антимышиный козлиный IgG, соединенный с биотином, и авидин, соединенный с HRP. Ферментативное превращение 4-хлор-1-нафтола применяли для визуализации специфических белков, распознаваемых моноклональными антителами. В дополнение, исследовалась инфективность вируса, продуцированная хронически инфицированными СЕМ-клетками в присутствии или в отсутствие испытуемого соединения. Фильтрованные супернатанты серийно разбавляли и применяли для инфицирования неинфицированных СЕМ-клеток (около 1•104/лунку). Культуры исследовали на образование синцитий в дни 7 и 11 после инфицирования или супернатанты испытывали на активность обратной транскриптазы или антиген p24.
МИКРОАНАЛИ3 ОБРАТНОЙ ТРАНСКРИПТА3Ы (RT).
Микроанализ RT является адаптацией нескольких стандартных анализов RT. Он был разработан для проведения количественного измерения активности RT ВИЧ в небольшом объеме и облегчения обработки различных проб (см. табл. В).
СПОСОБ:
1. Добавляют 50 мкл коктейля RT на лунку в микротитровальный планшет на 96 лунок с U-образным дном.
2. Добавляют 10 - 20 мкл на лунку раствора, не содержащего клетки супернатанта.
3. Перемешивают лунку механическим ротором.
4. Инкубируют при 37oC в течение 2 ч.
5. Отсасывают на фильтровальную бумагу DE81 или эквивалент при помощи ТОМТЕК.
6. Промывают при помощи 2X SSC четыре раза.
7. Промывают 95%-ным этанолом один раз.
8. Сушат фильтр.
9. Подготавливаются для подсчета счетчиком Beta-plate (Pharmacia).
Пример 19 (схему I см. в конце описания).
Многократные циклы ARE, повторяемые с повышаемой концентрацией соединения до наблюдения сдвига в EC50
Следующим является культуральный способ, применяемый для отбора мутантов, устойчивых к ингибитору протеазы ВИЧ. Инфицированные клетки выращивали непрерывно в присутствии ингибитора протеазы. Некоторые культуры подвергали по чередующимся неделям действию высоких и низких концентраций ингибиторов. Другие пересевали при постоянной концентрации. Концентрации лекарственных средств повышали периодически до наблюдения постоянного сдвига в EC50. Сдвиг в кривой зависимости от дозы обычно выявляли у концентрации лекарственного средства от 0,5 до 1 мкл/мл или выше (5-10•EC50) и он зависел от обрабатываемого вирусного изолята. Применяли как лабораторные адаптированные, так и первичные клинические изоляты ВИЧ. Те же вирусные изоляты пересевали таким же образом в отсутствие лекарственных средств, так что можно было сделать прямое сравнение нуклеозидных последовательностей обработанных и необработанных изолятов. Обычно варианты ВИЧ-1, устойчивые к ингибиторам протеазы, отбирали серийным пересеванием (выращиванием) в присутствии нескольких ингибирующих концентраций указанных ингибиторов протеазы (смотри Markowitz et al. , Journal of Virology 69: 701 - 706 (1995)). Варианты ВИЧ-1, перечисленные ниже, показывают мутации, которые присутствуют в выбранных вирусных изолятах и не присутствуют в контрольных, необработанных вирусных изолятах.
RF представляет штамм ВИЧ-1RF ВИЧа-1 и RFR представляет смесь устойчивых штаммов, полученных отбором RF против соединения Примера 1. RFR содержит смесь вирусных штаммов, имеющих генотипы протеазы G48V (14/40 клоны), G48V, V82A (18/40 клоны), G48V, L90S (2/40 клоны), G48V, 154T, V82A (1/40 клоны), G48M (1/40 клоны), G48V, Q61H (1/40 клоны), V131, G48V (1/40 клоны), G48V, F53L, V82A (1/40 клоны) и G48V, V82A, C95Y (1/40 клоны). RFR2 представляет смесь устойчивых штаммов, полученных клонированием RFR тремя циклами роста при ограничивающем разведении. RFR2 содержит смесь вирусных штаммов, имеющих генотипы протеазы G48V, V82A (13/15 клоны), G17E, G48V, V82A (1/15 клоны) и G48V, V82A, N88D, N88D (1/15 клоны). RFRR представляет смесь устойчивых штаммов, полученных отбором RF против соединений Примеров 1 и 2 и затем клонированием тремя циклами роста при ограничивающем разведении. RFRR содержит смесь вирусных штаммов, имеющих генотипы протеазы G48V, L63P, V82A (7/9 клоны), G48V, 154T, L63P, V82A, N88S (1/9 клоны) и G48V, 154T, L63P, G73M, V82A (1/9 клоны). SF162 представляет штамм SF-162 ВИЧа-1 и SF162R представляет смесь устойчивых штаммов, полученных отбором SF162 против соединения Примера 1. SF162R содержит смесь вирусных штаммов, имеющих генотипы протеазы M46I, F53L, L63P, A71V, N88D (2/3 клоны) и M461, F53L, L63P, A71V, N88D, Q92R (1/3 клоны). 89-959 представляет штамм 89-959 ВИЧа-1 и 89-959R представляет смесь устойчивых штаммов, полученных отбором 89-959 против соединения Примера 2. 89-959R содержит смесь вирусных штаммов, имеющих генотипы протеазы N88S (4/5 клоны) и D25N, T26A, D30N, D37N, R41K, G73D, R87K, N88S (1/4 клоны). NL4 представляет штамм ВИЧ-1NL4 ВИЧа-1. NL4 (G48V) представляет штамм, имеющий синтетически генерированную сайт-направленную мутацию в протеазе глицина в валин в аминокислотном положении номер 48. NL4 (184V) представляет штамм, имеющий синтетически генерированную сайт-направленную мутацию в протеазе изолейцина в валин в аминокислотном положении 84. NL4 (R8Q, M46I) представляет штамм, имеющий синтетически генерированные сайт-направленные мутации в протеазе аргинина в глутамин в аминокислотном положении номер 8 и метионина в изолейцин в аминокислотном положении номер 46. NL4 (P22-538) представляет смесь устойчивых штаммов, полученных отбором ВИЧ-1NL4-3 против соединения примера 10 после 22 пассажей, содержащих генотипы протеазы M46I, L63P, A71V, V82F, 184V, (4/10); M46I, L63P, V82F, 184V (3/10) и M46I, A71V, V82F, 184V (3/10). NL4(P37- 538) представляет смесь устойчивых штаммов, полученных отбором ВИЧ-1NL4-3 против соединения Примера 10 после 37 пассажей, содержащих генотип протеазы M46I, L63P, A71V, 184A. NL(538/524) представляет устойчивые штаммы, полученные селекцией NL4 (P22-538) против соединения Примера 5 после 24 пассажей, содержащие генотип протеазы M46I, L63P, A71V, 184A. NL4(538/P7-AG) представляет смесь устойчивых штаммов, полученных отбором NL4(P22-538) против соединения Примера 12 после 7 пассажей, содержащих генотипы протеазы M46I, L631, A71V, 184A и V32I, V82I. NL4(538/P24-AG) представляет смесь устойчивых штаммов, полученных отбором NL4(P22- 538) против соединения Примера 12 после 24 пассажей, содержащих генотип протеазы M46I, L63P, A71V, 184A. NLA(pI9-003) представляет устойчивые штаммы, полученные отбором ВИЧ-1NL4-3 против соединения Примера 9 после 19 пассажей, содержащие генотип протеазы R8K, M46I. NL4 (Р34-003) представляет устойчивые штаммы, полученные отбором BИЧ- 1NL4-3 против соединения Примера 9 после 34 пассажей, содержащие генотип протеазы R8K, M46I, L63P, A71V, L90M. Результаты устойчивости вирусных изолятов суммированы в Таблицах 1 - 11.
Пример 20
Результаты устойчивости вирусных изолятов, суммированные в Таблицах 1 - 3, получали в соответствии со следующей методикой анализа или ее небольшими модификациями. Приблизительно 3•107 клеток активируют в течение 46 часов приблизительно с 3-5 мкг/мл РНА в среде RPMI, содержащей 10% фетальную телячью сыворотку и IL-2 (10 Е/мл). В суспензию активированных лимфоцитов добавляют количественные вирусные исходные растворы при множественности инфекции около 0,001-0,01. Суспензию клетка-вирус инкубируют при 37oC в течение 2 ч для проведения абсорбции вируса. Остаточный вирусный инокулум удаляют центрифугированием и клетки ресуспендируют в RPMI, содержащей 10% FBS и 10 Е/мл IL-2. Эти инфицированные клетки добавляют в испытуемое соединение, разведенное в полной тканевой культуральной среде из исходного раствора (10 мг/мл) в ДМСО в планшетах для микротитрования с 96 лунками, получая около 5 • 105 клеток на лунку в 200 мкл. В качестве контроля применяли инфицированные, необработанные клетки и клетки, обработанные только ДМСО (0,1%) или AZT или DDI. Культуры обследовали на образование синцитий в дни 7 и 11 после инфицирования или супернатанты испытывали на активность обратной транскриптазы или антиген р24.
Пример 21
Результаты устойчивости вирусных изолятов, суммированные в таблицах 4-6, были получены в соответствии со следующей методикой анализа или ее небольших модификаций. Анализы проводили в тканевых культуральных планшетах на 96 лунок. Клетки СЕМ-Т4 суспендируют в 90% RPMI-среде (Gibco BRL Life Technologies, Inc. , Gaithsburg, MD), 10% термообработанной фетальной телячьей сыворотке (Gibco BRL Life Technologies, Inc., Gaithsburg, MD) до конечной концентрации 5 • 105 жизнеспособных клеток на мл. Замороженную аликвотную пробу культуры ВИЧ (штамм ВИЧ-1RF) быстро оттаивают (в водяной бане с температурой 37oC) и добавляют в СЕМ-Т4-клетки, получая конечную концентрацию около 0,001-0,01 инфекционных единиц на клетку. Суспензию вирус-клетка быстро смешивают образованием завихрения и 100 мкл сразу добавляют в 100 мкл разбавления каждого тест-соединения (получен как 2х концентрат в 90% RPMI, 10% FBS) каждой лунки тканевого культурального планшета. Каждый планшет содержит контрольные лунки, которые содержат клетки и вирус, но не содержат испытуемое соединение. 3'-Азидо-3'дезокситимидин (AZT) включают в качестве положительного контроля во всех анализах.
Тканевые культуральные планшеты инкубируют при 37oC в увлажненной, содержащей 5% CO2 атмосфере в течение 7 дней. Уровень вирусной репликации затем определяют измерением активности обратной транскриптазы в супернатантах, используя стандартные методики (как описано ранее и смотри, например, Techniques in HIV Research, Aldovini and Walker, eds., 1990, Stockton Press, NY).
Пример 22
Результаты устойчивости вирусных изолятов, суммированные в таблицах 7 - 11, получали в соответствии с методикой анализа, описанной Markowitz et al., Journal of Virology vol. 69, 701 - 706 (1995), которая включена здесь ссылкой во всей полноте.
Пример 23
Ингибиторы протеазы примеров 1 и 2, которые содержат необычный изостер гидроксиэтилкарбамида, применяли для отбора устойчивых к лекарственному средству вариантов ВИЧ-1 in vitro. Клинические и лабораторные штаммы ВИЧ-1 пересевали в Т-клеточных линиях или периферических кровяных мононуклеарных клетках (PBMCs) в присутствии повышаемых концентраций лекарственного средства. Устойчивые варианты постоянно показывали величины EC50 по меньшей мере, в 10 раз выше, чем у контрольного вируса, пересеянного в течение идентичного периода, но в отсутствие ингибитора. Вирусную ДНК амплифицировали PCR и нуклеотидную последовательность гена, кодирующего протеазу, определяли с использованием стандартных методик. В вирусах, устойчивых к ингибиторам протеазы примеров 1 и 2, соответственно, постоянно наблюдали аминокислотное изменение в положении 88 во многих из выбранных вариантах. Остаток Asn у 88 находится в пределах структурно сохраняемого спирального домена, присутствующего как в мономерной, так и в димерной аспарагиновых протеиназах. Соответствующая карбоксиконцевая последовательность Gly-Arg-Asp/Asn (остатки 86-88) необычна для ретровирусных аспарагиновых протеиназ. Хотя любое объяснение этих результатов является только размышлением, моделирующие исследования, основанные на темплатах, полученных на основании рентгеновских спектров высокого разрешения структур прототипичных гидроксиэтилкарбамидных ингибиторов, соединенных с рекомбинантной протеазой ВИЧ-1, по-видимому, подтверждают, что Asn88-мутации могут изменять конформацию протеазы.
Соединения-ингибиторы ретровирусной протеазы данного изобретения являются благоприятно эффективными антивирусными соединениями и, в частности, являются эффективными ингибиторами ретровирусов, в частности, лентивирусов, как показано выше. Таким образом, соединения - предмет данного изобретения являются эффективными ингибиторами ВИЧ. Предполагается, что соединения - предмет изобретения будут также ингибировать другие штаммы ВИЧ, например ВИЧ-2, и другие вирусы, такие как, например, вирус VISNA и вирус иммунодефицита Simian (SIV), HTLV-1 и HTLV-2. Таким образом, соединения - предмет данного изобретения эффективны в лечении и/или профилактике ретровирусных инфекций.
Подразумевается также, что данное изобретение включает, когда возможно, сольват или гидраты соединений-ингибиторов ретровирусной протеазы, которые получают и выделяют способами, известными в данной области.
Соединения-ингибиторы ретровирусной протеазы можно применять в форме солей, полученных из неорганических или органических кислот. Эти соли включают, но не ограничиваются ими, следующие соли: ацетат, адипат, альгинат, цитрат, аспартат, бензоат, бензолсульфонат, бисульфат, бутират, камфорат, камфорасульфонат, диглюконат, циклопентанпропионат, додецилсульфат, этансульфонат, глюкогептаноат, глицерофосфат, полусульфат, гептаноат, гексаноат, фумарат, гидрохлорид, гидробромид, гидроиодид, 2- гидроксиэтансульфонат, лактат, малеат, метансульфонат, никотинат, 2-нафталинсульфонат, оксалат, палмоат, лектинат, персульфат, 3- фенилпропионат, пикрат, пивалат, пропионат, сукцинат, тартрат, тиоцианат, тозилат, мезилат и ундеканоат.
Примеры кислот, которые можно применять для образования фармацевтически приемлемых солей с кислотами, включают такие неорганические кислоты, как соляная кислота, серная кислота и фосфорная кислота, и такие органические кислоты, как щавелевая кислота, малеиновая кислота, янтарная кислота и лимонная кислота, предпочтительна гидрохлоридная соль. Другие примеры включают соли с щелочными металлами или щелочноземельными металлами, например натрием, калием, кальцием или магнием, или с органическими основаниями.
Общая суточная доза, вводимая пациенту в виде одной или разделенных доз, может составлять, например, от 0,01 до 50 мг/кг массы тела ежедневно и, более обычно, от 0,1 до 30 мг. Дозированные унифицированные композиции могут содержать такие количества субчастей их, чтобы составить суточную дозу.
Количество активного ингредиента, которое можно комбинировать с материалами-носителями для получения разовой лекарственной формы, будет изменяться в зависимости от пациента, которому его вводят, и конкретного способа введения.
Схему приема лекарственного средства для лечения болезненного состояния соединениями-ингибиторами ретровирусной протеазы и/или композиции выбирают в соответствии со множеством факторов, включая тип, возраст, массу, пол, диету и медицинское состояние пациента, тяжесть болезни, способ введения, фармакологическую ценность, например активность, эффективность, фармакокинетические и токсикологические профили конкретного применяемого соединения, применяют ли систему для доставки в организм лекарственного средства, вводят ли соединение как часть комбинации лекарственных средств. Таким образом, реально применяемая схема приема лекарственного средства может широко изменяться и, следовательно, может отклоняться от предпочтительной схемы приема лекарственного средства, предложенной выше.
Соединения данного изобретения можно вводить перорально, парентерально, ингаляцией распылением, ректально или местно в унифицированных лекарственных готовых препаративных формах, содержащих, если требуется, обычные нетоксичные фармацевтически приемлемые носители и вспомогательные средства. Местное введение может включать также использование чрескожного введения, например чрескожных пластырей или устройств для ионофореза. Термин парентеральный, как использованный здесь, включает подкожные инъекции, внутривенную, внутримышечную, интрастериальную (внутригрудинную) инъекцию или способы инфузии.
Инъецируемые препараты, например стерильные инъецируемые водные или маслянистые суспензии, можно приготовить в соответствии с известными способами, использующими пригодные диспергирующие или смачивающие средства или суспендирующие средства. Стерильный инъецируемый препарат может также быть стерильным инъецируемым раствором или суспензией в нетоксичном парентерально приемлемом разбавителе или растворителе, например как раствор в 1,3-бутандиоле. Среди приемлемых разбавителей и растворителей, которые можно применять, имеется вода, раствор Рингера и изотонический раствор хлористого натрия. В дополнение, в качестве растворителя или суспендирующей среды обычно применяют стерильные жирные масла. Для этой цели можно применять любое жирное масло мягкого действия, включая синтетические моно- или диглицериды. В дополнение, жирные кислоты, например олеиновая кислота, находят применение в инъецируемых препаратах.
Суппозитории для ректального введения лекарственного средства можно получить смешиванием лекарственного средства с пригодным нераздражающим наполнителем, например какао-масло и полиэтиленгликолями, которые твердые при обычной температуре, но жидкие при ректальной температуре и, следовательно, будут плавиться в прямой кишке и высвобождать лекарственное средство.
Твердые лекарственные формы для перорального введения могут включать капсулы, таблетки, пилюли, порошки и гранулы. В таких твердых лекарственных формах активное соединение можно смешивать по меньшей мере с одним инертным разбавителем, например сахарозой, лактозой или крахмалом. Такие лекарственные формы могут содержать также, как в обычной практике, дополнительные вещества, другие чем инертные разбавители, например смазывающие вещества, такие как стеарат магния. В случае капсул, таблеток и пилюль лекарственные формы могут содержать также буферы. Таблетки и пилюли можно дополнительно получать с энтеросолюбильными покрытиями.
Жидкие лекарственные формы для перорального введения могут включать фармацевтически приемлемые эмульсии, растворы, суспензии, сиропы и элексиры, содержащие инертные разбавители, обычно используемые в этой области, например воду. Такие композиции могут содержать также вспомогательные средства, например смачивающие средства, эмульгирующие и суспендирующие средства, и подслащивающие средства, корригенты и отдушки.
Хотя соединения-ингибиторы ретровирусной протеазы данного изобретения можно вводить в качестве единственных активных фармацевтических средств, их также можно использовать в комбинации с другими противовирусными средствами, которые эффективны против ретровирусов, например ВИЧ-1. Такие соединения включают, но не ограничиваются ими, другие ингибиторы протеазы ВИЧ-1, различные аналоги нуклеозидов, ненуклеозидные ингибиторы обратной транскриптазы, антагонисты тирозина-минотрансферазы (tat) и ингибиторы глюкозидазы.
Примеры ингибиторов протеазы ВИЧ-1 включают, но не ограничиваются ими, Po 31-859 (Roberts, N.A. et al. Science 1990, 248, 258 - 261, and Drugs of Future 1991, 16(3), 210 - 212), KNI-272, (Kagayama, S., et al. Antimicrobial Agens and Chemotherapy 1993, 610 - 817), ряд циклических карбамидов (Lam, P. , et al., "De Novo Design and Discovery of Potent, Nonpeptidal HIV-1 Protease Inhibitors", paper 96 at the 205th American Ghemical Society National Meeting, Medicinal Chemistry Division, Denver, CO, March 28-April 2, 1993), L-735 524 (Dorsey, В. D., et al., "L-735 524: The Rational Design of Potent and Orally Bioavailable HIV Protease Inhibitor", paper 6 et the 206th American Chemical Society National Meeting, Medicinal Chemistry Division, Chicago, IL, August 22 - 27, 1993) и их аналоги.
Примеры конкурирующих нуклеозидных аналогов включают, но не ограничиваются ими, азидотимидин (AZT), дидеоксиинозин (DDI), DDC, ЗTC, 4DT и РМЕА. Примеры ненуклеозидных неконкурирующих ингибиторов обратной транскриптазы включают, но не ограничиваются ими, класс пиридонов (Wei, J. S., et ai., J. Med. Chem. 1993, 36, 249 - 255; Hoffrnan, J. M., et al., J. Med. Chem. 1992, 35, 3784 - 3791; Saari et al., J. Med. Chem. 1992, 35, 3792 - 3802; Drugs of the Future 1992 17(4), 283 - 285) и их аналоги; класс бис(гетероарил)пиперазинов (Romero, D. L., et al., J. Med. Chem. 1993, 36, 1505 - 1508; Romero, D. L., et al., Proc. Natl. Acad. Sci. USA 1991, 34, 746 - 751 and 3178 - 3198) и их аналоги и трициклические пиридобензо- и депиридодиазепиноны (Hargrave, К. D. , J. Med. Chem. 1991, 34, 2231 - 2241; Merluzzi, M. J. Science 1990, 250, 1411 - 1413) и их аналоги и 5-хлор-3-(фенилсульфонил)индол-2-карбоксамид и его аналоги (Williams, Т.М. et al., J. Med. Chem. 1993, 36, 1291 - 1294). Примеры антагонистов tat включают, но не ограничиваются ими, Rо 5-3335 и Ro 24-7429 (Hsu, M.C. et al., Proc. Natl. Acad. Sci. USA 1993, 909, 6395 - 6399; Tam, S. et al., "TAT INHIBITORS: A NEW CLASS OF ANT I-HIV AGENTS", paper 372, et the 204th American Chemical Society National Meeting, Organic Chemistry Division, Washington, DC, August 23 - 28, 1992) и их антагонисты. Примеры ингибиторов гликозидазы включают, но не ограничиваются ими, кастаноспермин, 6-бутриловый эфир кастаноспермина, бутил- 1-деоксинойримицин, пер-бутриловый N-бутил-1-деоксинойримицина и их аналоги и пролекарства.
Терапевтические средства можно приготовить как отдельные композиции, которые дают (вводят) по существу в одно и то же время, или терапевтические средства можно давать в виде единственных композиций, так чтобы все активные агенты были в организме пациента в терапевтически эффективном количестве. Альтернативно, терапевтические агенты можно вводить пациенту в разное время, так чтобы только один или два активных агента в какое-либо время были в организме пациента в терапевтически эффективном количестве.
Соединения и способы данного изобретения являются эффективными противовирусными соединениями и, в частности, эффективными ретровирусными ингибиторами, как показано выше. Таким образом, соединения - предмет данного изобретения являются эффективными ингибиторами протеазы ВИЧ. Предполагается, что соединения - предмет данного изобретения будут также ингибировать другие ретровирусы, такие как другие лентивирусы, в частности, другие штаммы ВИЧ, например ВИЧ- 2, вирус Т-клеточной лейкемии человека, вирус саркомы Рауса, вирус иммунодефицита simian, вирус лейкемии кошек, вирус иммунодефицита кошек и подобные вирусы. Таким образом, соединения - предмет данного изобретения эффективны в лечении и/или профилактике ретровирусных инфекций.
Являющиеся предметом изобретения соединения и способы эффективны также в предотвращении роста ретровирусов в растворе. Культуры как человеческих, так и животных клеток, например культуры Т-лимфоцитов, используют для ряда хорошо известных целей, например исследовательских и диагностических способов, включающих буж-измерители и стандарты. До и во время роста и хранения клеточной культуры соединения данного изобретения можно добавлять в клеточную культуральную среду в эффективной концентрации для предотвращения неожидаемой и нежелательной репликации ретровируса, который может нечаянно или неведомо присутствовать в клеточной культуре. Вирус может присутствовать в клеточной культуре первоначально, например известно, что ВИЧ присутствует в Т-лимфоцитах человека задолго до того, как становится возможным обнаружение его в крови, или из-за подвергания культуры действию вируса. Это использование соединений и способов данного изобретения предотвращает нежелательное или неожидаемое действие потенциально летальных ретровирусов на исследователя или клинициста.
Предшествующее описание только иллюстрирует данное изобретение и не предназначено для ограничения изобретения описанными соединениями. Подразумевается, что варианты и изменения, которые очевидны специалисту в данной области, входят в объем и сущность данного изобретения, которые определяются в прилагаемой формуле изобретения.
Из предшествующего описания специалист данной области может легко установить основные характеристики этого изобретения и без отклонения от существа и объема его может сделать разные изменения и модификации изобретения, чтобы приспособить его к различным употреблениям и условиям.
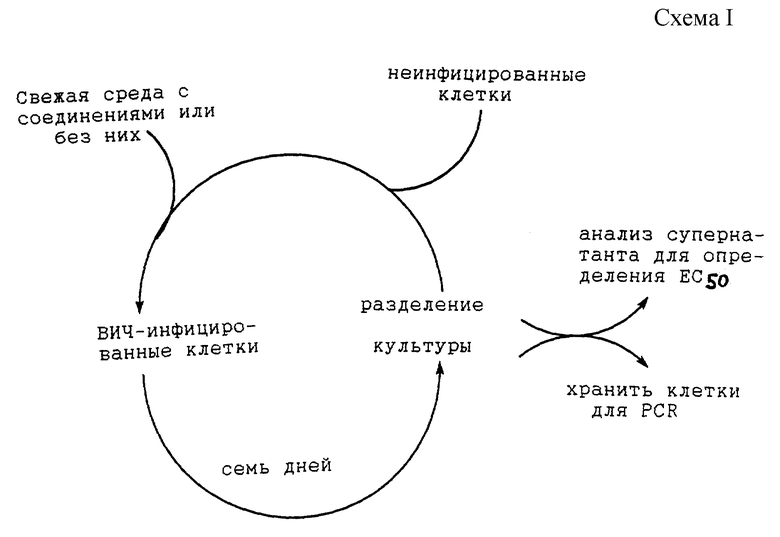

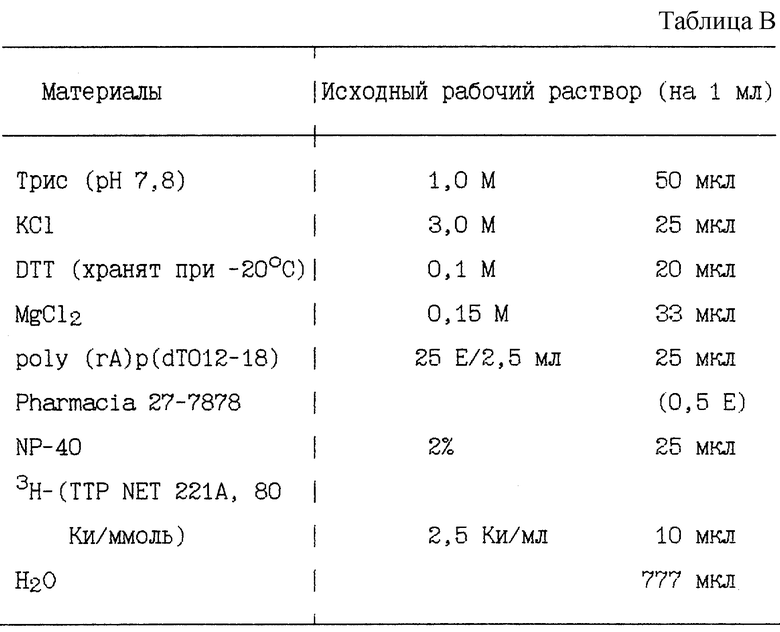

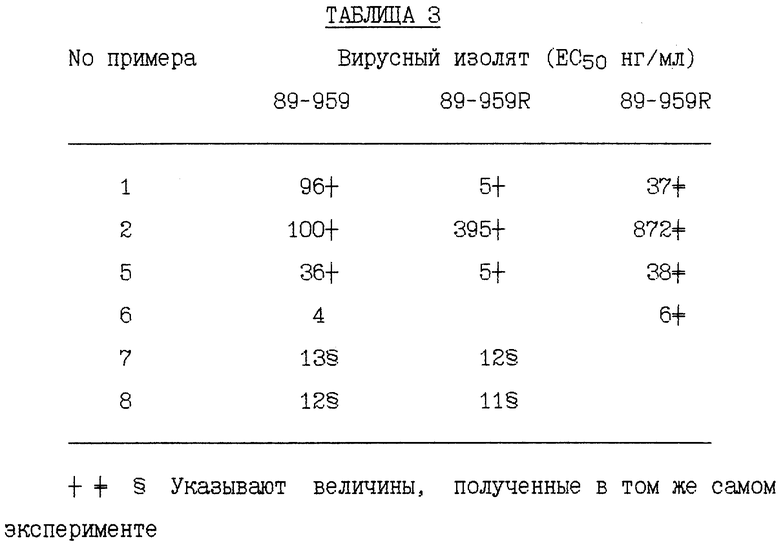

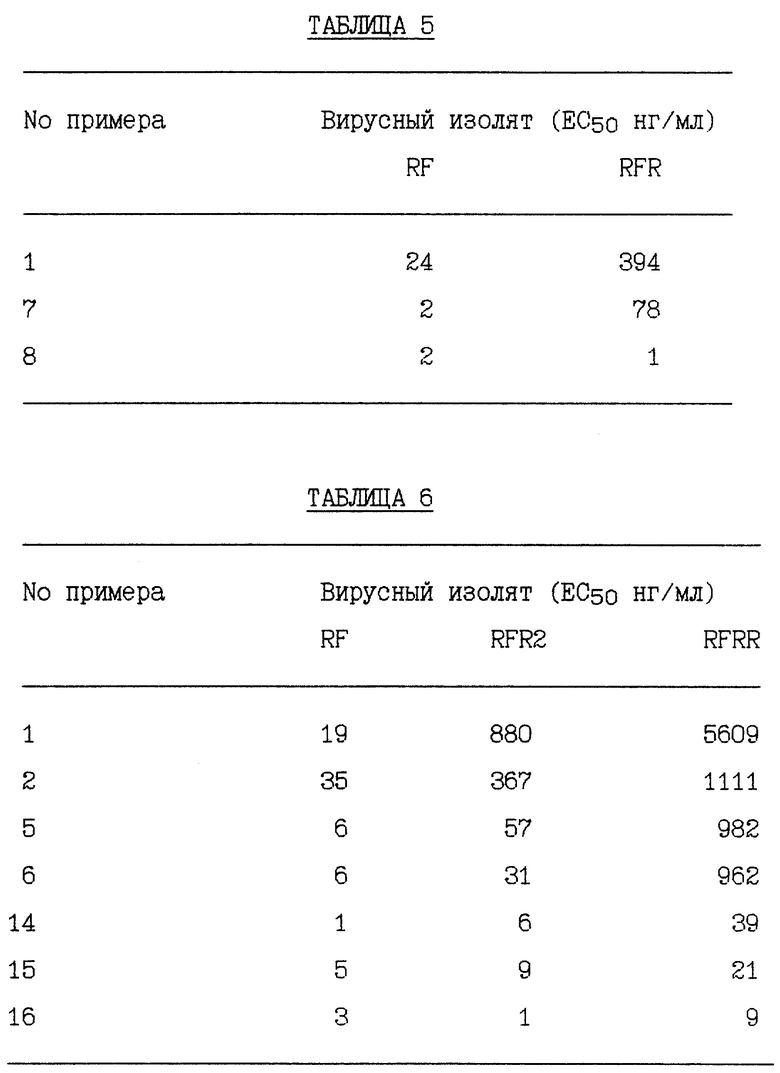
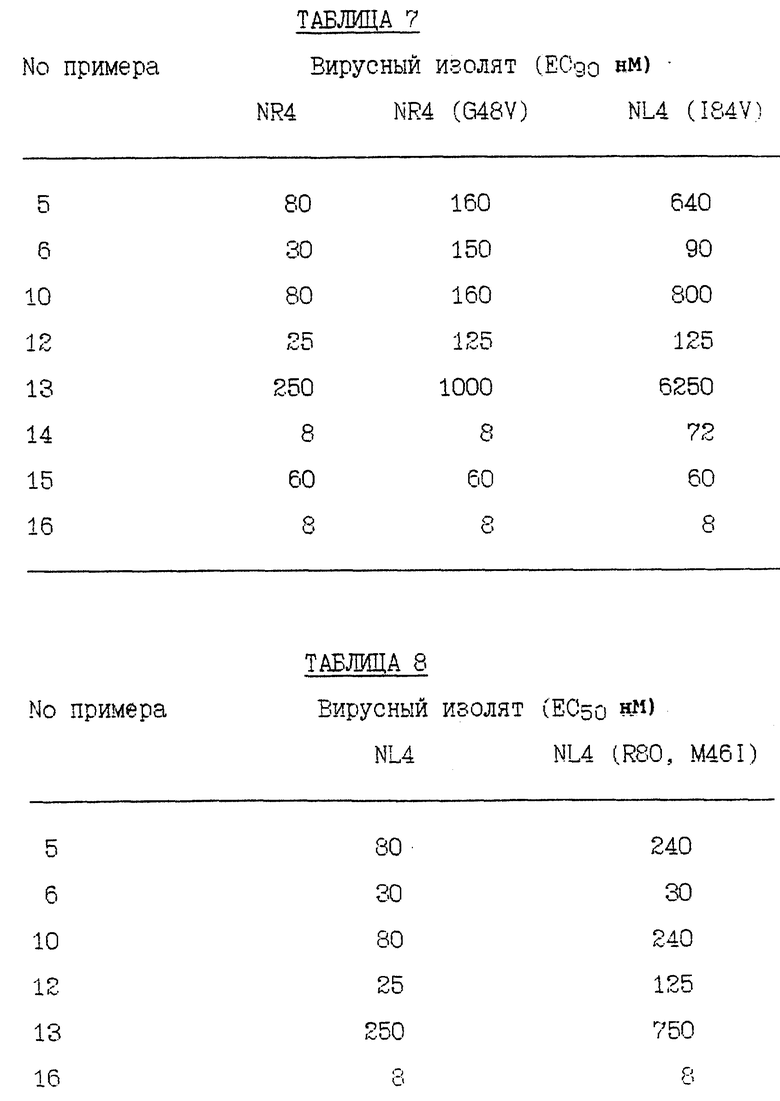
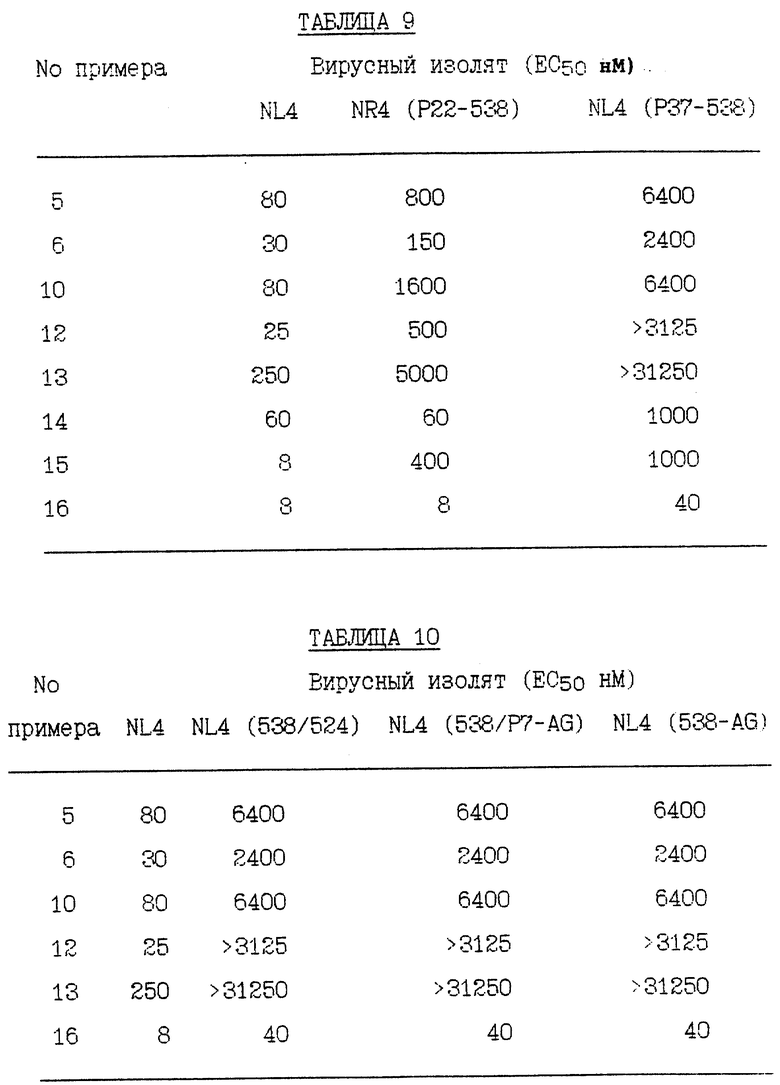
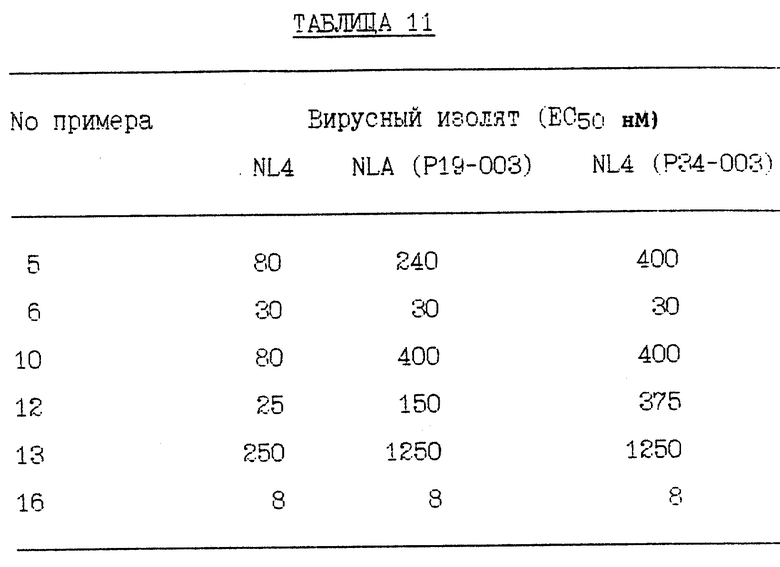
Изобретение относится к области биотехнологии. Способ предусматривает использование комбинаций ингибиторов ретровирусной протеазы, которые эффективны в предотвращении репликации ретровирусов in vitro или in vivo. Соединения ингибиторы протеазы используются также в комбинационной терапии с другими соединениями - ингибиторами протеазы, а также в комбинационной терапии с противовирусными средствами, другими, чем ингибиторы протеазы. Способ позволяет преодолеть развитие ретровирусных штаммов, которые устойчивы к ингибитору ретровирусной протеазы. 2 с. и 18 з.п. ф-лы, 13 табл.
| Nature, 6 April 1995, vol | |||
| Устройство для телефонирования по проводам токами высокой частоты | 1921 |
|
SU374A1 |

