Изобретение относится к ингибиторам ретровирусной протеазы и более конкретно к новым соединениям, композициям и способы ингибирования ретровирусных протеаз. В частности, данное изобретение относится к содержащим сульфонамид соединениям гидроксиэтиламина, ингибирующим протеазы, композициям и способу ингибирования ретровирусных протеаз, таких как протеаза вируса иммунодефицита человека /ВИЧ/, и лечению ретровирусной инфекции, например инфекции ВИЧ. Данное изобретение относится также к способам получения таких соединений, а также промежуточных продуктов, применимых в таких способах.
Во время цикла репликации ретровирусов в качестве протеинов выступают продукты gag и gag-pol генов. Эти белки затем процессируются кодируемой вирусом протеазой /или протеиназой/, давая вирусные ферменты и структурные протеины вирусного ядра. В наиболее типичном случае белки-предшественники gag процессируются с образованием белков ядра, а белки-предшественники pol процессируются в вирусные ферменты, например, обратную транскриптазу и ретровирусную протеазу. Было показано, что точный процессинг белков-предшественников ретровирусной протеазой необходим для сборки инфекционных вирионов. Например, было показано, что мутации со сдвигом рамки в протеазной области pol гена ВИЧ предотвращает процессинг gag белка-предшественника. Также было показано при помощи сайт-направленного мутагенеза остатка аспарагиновой кислоты в протеазе ВИЧ, что процессинг gag белка-предшественника не мог осуществляться. Поэтому были сделаны попытки ингибирования репликации вируса путем ингибирования действия ретровирусных протеаз.
В ингибировании ретровирусных протеаз может участвовать вещество-имитатор переходного состояния. Ретровирусную протеазу приводят в контакт с таким соединением, которое связывается с ферментом, конкурируя с gag и gag-pol белками и ингибируя вследствие этого репликацию структурных белков и, что более важно, саму ретровирусную протеазу. Таким способом можно эффективно ингибировать протеазы ретровируса.
Были предложены несколько классов соединений, в частности, для ингибирования таких протеаз, как протеаза ВИЧ. К таким соединениям относятся изостеры гидроксиэтиламина и изостеры восстановленного амида. См., например, EP 0346847; EP 0342541; Roberts et al., "Rational Design of Peptide-Based Proteinase Inhibitors", Science 248, 358 /1990/; и Erickson et al., "Design Activity and 2,8  Crystal Structure of a C2 Symmetric Inhibitor Complexed to HIV-I Protease", Science 249, 527 /1990/.
Crystal Structure of a C2 Symmetric Inhibitor Complexed to HIV-I Protease", Science 249, 527 /1990/.
Известны несколько классов соединений, применимых в качестве ингибиторов протеолитического фермента ренина. См., например, US N 4599198; ИК, 2184730; GB 2209752; EP 0264795; GB 2200115 и US SIR H725. Из них GB 2200115, GB 2209752, EP 0264795, US SIR H725 и US 4599198 описывают содержащие мочевину гидроксиэтиламины в качестве ингибиторов ренина. GB 2200115 описывает также сульфамоилсодержащие гидроксиэтиламины в качестве ингибиторов ренина, а EP 0264795 описывает некоторые сульфонамидсодержащие гидроксиэтиламины в качестве ингибиторов ренина. Однако известно, что хотя ренин и протеазы ВИЧ классифицируются как аспартилпротеазы, нельзя заранее предсказать, будут ли эффективные ингибиторы ренина эффективными ингибиторами протеазы ВИЧ.
Данное изобретение относится к соединениям и композициям, ингибирующим вирус. Более конкретно, изобретение относится к соединениям и композициям, ингибирующим ретровирусные протеазы, способу ингибирования ретровирусных протеаз, способам получения соединений и промежуточных продуктов, применимых в этих способах. Соединения данного изобретения представляют собой обладающие ингибирующей активностью сульфонамидсодержащие соединения гидроксиэтиламина.
В соответствии с данным изобретением предлагается ингибирующее ретровирусные протеазы соединение формулы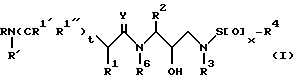
или его фармацевтически приемлемые соли, пролекарство или эфир, где
R обозначает водород, алкоксикарбонил, аралкоксикарбонил, алкилкарбонил, циклоалкилкарбонил, циклоалкилалкоксикарбонил, циклоалкилалканоил, алканоил, аралканоил, ароил, арилоксикарбонил, арилоксикарбонилалкил, арилоксиалканоил, гетероциклилкарбонил, гетероциклилоксикарбонил, гетероциклилалканоил, гетероциклилалкоксикарбонил, гетероаралканоил, гетероаралкоксикарбонил, гетероарилоксикарбонил, гетероароил, алкил, алкенил, алкинил, циклоалкил, арил, аралкил, арилоксиалкил, гетероарилоксиалкил, гидроксиалкил, аминокарбонил, аминоалканоил и моно- и дизамещенные аминокарбонил и моно- и дизамещенные аминоалканоилрадикалы, в которых заместители выбраны из алкила, арила, аралкила, циклоалкила, циклоалкилалкила, гетероарила, гетероаралкила, гетероциклоалкила, гетероциклоалкилалкила, или, если радикалы аминокарбонил и аминоалканоил являются дизамещенными, заместители вместе с атомом азота, к которому они присоединены, образуют радикал гетероциклоалкил или гетероарил;
R' обозначает водород, радикалы, описанные для R3, или R''SO2-, где R'' обозначает радикалы, описанные для R3; или R и R' вместе с азотом, к которому они присоединены, образуют радикалы гетероциклоалкил и гетероарил;
R1 обозначает водород; -CH2SO2NH2, -CH2CO2CH3, -CO2CH3, -CONH2, -CH2C(O)NHCH3, -C(OH3)2(SH), -C(CH3)2(SCH3), -C(CH3)2(S[O]CH3), -C(CH3)2(S[O] 2CH3), алкил, галогеналкил, алкенил, алкинил, циклоалкил и аминокислотные боковые цепи, выбранные из таких боковых цепей, как аспарагин, S-метилцистеин и его сульфоксидные /SO/ и сульфоновые /SO2/ производные, изолейцин, аллоизолейцин, аланин, лейцин, трет-лейцин, фенилаланин, орнитин, гистидин, норлейцин, глутамин, треонин, глицин, аллотреонин, серин, O-алкилсерин, аспарагиновая кислота, β-цианоаланин и валин;
R1' и R1'' независимо друг от друга обозначают водород и радикал, описанный для R1, или R1' и R1'', вместе с R1 и атомами углерода, с которыми соединены R1, R1' и R1'', обозначают циклоалкил;
R2 обозначает алкил, арил, циклоалкил, циклоалкилалкил и аралкил, иногда замещенные группой, выбранной из радикалов, таких как алкил и галоген, -NO2, -CN, -CF3, -OR9 и -SR9, где R9 обозначает водород и алкил и галоген;
R3 обозначает водород, алкил, галогеналкил, алкенил, алкинил, гидроксиалкил, алкоксиалкил, циклоалкил, циклоалкилалкил, гетероциклоалкил, гетероарил, гетероциклоалкилалкил, арил, аралкил, гетероаралкил, аминоалкил и моно- и дизамещенный аминоалкил, где заместители выбраны из таких радикалов, как алкил, арил, аралкил, циклоалкил, циклоалкилалкил, гетероарил, гетероаралкил, гетероциклоалкил и гетероциклоалкилалкил, или в случае дизамещенного аминоалкила заместители вместе с атомом азота, к которому они присоединены, образуют гетероциклоалкил или гетероарил;
R4 обозначает радикалы, описанные для R3, за исключением водорода;
R6 обозначает водород и алкил;
x = 0, 1 или 2;
t = 0 или 1;
Y обозначает O, S и NR15, где R15 обозначает водород и радикалы, описанные для R3.
Группа наиболее предпочтительных соединений, описанных формулой I, представлены формулой II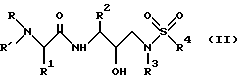
где R обозначает водород, алкоксикарбонил, аралкоксикарбонил, алкилкарбонил, циклоалкилкарбонил, циклоалкилалкоксикарбонил, циклоалкилалканоил, алканоил, аралканоил, ароил, арилоксикарбонил, арилоксикарбонилалкил, арилоксиалканоил, гетероциклилкарбонил, гетероциклилоксикарбонил, гетероциклилалканоил, гетероциклилалкоксикарбонил, гетероаралканоил, гетероаралкоксикарбонил, гетероарилоксикарбонил, гетероароил, алкил, алкенил, циклоалкил, арил, аралкил, арилоксиалкил, гетероарилоксиалкил, гидроксиалкил, аминокарбонил и моно- и дизамещенные аминокарбонил и моно- и дизамещенные аминоалканоилрадикалы, в которых заместители выбраны из таких радикалов, как алкил, арил, аралкил, циклоалкил, циклоалкилалкил, гетероарил, гетероаралкил, гетероциклоалкил, гетероциклоалкилалкил, или в случае дизамещенного аминоалканоила заместители вместе с атомом азота, с которым они соединены, могут образовывать гетероциклоалкил или гетероарил;
R' обозначает водород и радикалы, описанные для R3, или R и R' вместе с атомом азота, к которому они присоединены, обозначают гетероциклоалкил и гетероарил;
R1 обозначает водород; -CH2SO2NH2, -CH2CO2CH3, -CO2CH3, -CONH2, -CH2C(O)NHCH3, -C(CH3)2(SH), -C(CH3)2(SCH3), -C(CH3)2(S[O]CH3), -C(CH3)2(S[O] 2CH3), алкил, галогеналкил, алкенил, алкинил, циклоалкил и аминокислотные боковые цепи, выбранные из таких боковых цепей, как аспарагин, S-метилцистеин и его сульфоксидные /SO/ и сульфоновые /SO2/ производные, изолейцин, аллоизолейцин, аланин, лейцин, трет-лейцин, фенилаланин, орнитин, гистидин, норлейцин, глутамин, треонин, глицин, аллотреонин, серин, O-метилсерин, аспарагиновая кислота, β-цианоаланин и валин;
R2 обозначает алкил, арил, циклоалкил, циклоалкилалкил и аралкил, иногда замещенные группой, выбранной из таких радикалов, как алкил и галоген, -NO2, -C≡N, CF3, -OR9, -SR9, где R9 обозначает водород и алкил;
R3 обозначает алкил, галогеналкил, алкенил, алкинил, гидроксиалкил, алкоксиалкил, циклоалкил, циклоалкилалкил, гетероциклоалкил, гетероарил, гетероциклоалкилалкил, арил, аралкил, гетероаралкил, аминоалкил и моно- и дизамещенный аминоалкил, в которых заместители выбраны из таких радикалов, как алкил, арил, аралкил, циклоалкил, циклоалкилалкил, гетероарил, гетероаралкил, гетероциклоалкил и гетероциклоалкилалкил, или в случае дизамещенного аминоалкила заместители вместе с атомом азота, к которому они присоединены, образуют гетероциклоалкил или гетероарил;
R4 обозначает радикалы, описанные для R3.
Более предпочтительная группа соединений формулы II состоит из соединений, в которых:
R обозначает водород, алкоксикарбонил, аралкоксикарбонил, алкинилкарбонил, циклоалкилкарбонил, циклоалкилалкоксикарбонил, циклоалкилалканоил, алканоил, аралканоил, ароил, арилоксикарбонил, арилоксикарбонилалкил, арилоксиалканоил, гетероциклилкарбонил, гетероциклилоксикарбонил, гетероциклилалканоил, гетероциклилалкоксикарбонил, гетероаралканоил, гетероаралкоксикарбонил, гетероарилоксикарбонил, гетероароил, алкил, алкенил, циклоалкил, арил, аралкил, арилоксиалкил, гетероарилоксиалкил, гидроксиалкил, аминокарбонил, аминоалканоил и моно- и дизамещенные аминокарбонил, и аминоалканоил, в которых заместители выбраны из радикалов алкил, арил, аралкил, циклоалкил, циклоалкилалкил, гетероарил, гетероаралкил, гетероциклоалкил, гетероциклоалкилалкил, или в случае дизамещенного аминоалканоила заместители вместе с атомом азота, с которым они соединены, образуют гетероциклоалкил или гетероарил;
R' обозначает водород и радикалы, описанные для R3, или R и R' вместе с атомом азота, с которым они соединены, обозначают гетероциклоалкил и гетероарил;
R1 обозначает CH2C(O)NHCH3, C(CH3)2(SCH3), C(CH3)2(S[O]CH3), C(CH3)2(S[O] 2CH3), алкил, алкенил и алкинил и аминокислотные боковые цепи, выбранные из таких боковых цепей, как аспарагин, валин, треонин, аллотреонин, изолейцин, трет-лейцин, S-метилцистеин и его сульфоновые и сульфоксидные производные, аланин, аллоизолейцин;
R2 обозначает алкил, циклоалкилалкил и аралкил, иногда замещенные галогеном и радикалами формул -OR9 и -SR9, где R9 обозначает алкил; и
R3 и R4 независимо друг от друга обозначают алкил, алкенил, алкоксиалкил, циклоалкил, циклоалкилалкил, гетероциклоалкил, гетероциклоалкилалкил, арил, аралкил и гетероалкил.
Наибольший интерес представляют собой соединения формулы II, в которой:
R обозначает алкоксикарбонил, аралкоксикарбонил, алкилкарбонил, циклоалкилкарбонил, циклоалкилалкоксикарбонил, циклоалкилалканоил, алканоил, аралканоил, ароил, арилоксикарбонил, арилоксикарбонилалкил, арилоксиалканоил, гетероциклилкарбонил, гетероциклилоксикарбонил, гетероциклилалканоил, гетероциклилалкоксикарбонил, гетероаралканоил, гетероаралкоксикарбонил, гетероарилоксикарбонил, гетероароил, аминокарбонил, аминоалканоил и моно- и дизамещенные аминокарбонил и аминоалканоил, в которых заместители выбраны из таких радикалов, как алкил, арил, аралкил, циклоалкил, циклоалкилалкил, гетероарил, гетероаралкил, гетероциклоалкил, гетероциклоалкилалкил, или в случае дизамещенного аминоалканоила эти заместители вместе с атомом азота, с которым они соединены, образуют гетероциклоалкил или гетероарил;
R' обозначает водород и радикалы, описанные для R3, или R и R' вместе с атомом азота, к которому они присоединены, обозначают гетероциклоалкил или гетероарил;
R1 обозначает CH2C(O)NHCH3, C(CH3)2(SCH3), C(CH3)2(S[O]CH3), C(CH3)2(S[O] 2CH3), метил, пропаргил, т-бутил, изопропил и втор-бутил и аминокислотные боковые цепи, выбранные из группы, состоящей из таких боковых цепей, как аспарагин, валин, S-метилцистеин, аллоизолейцин, изолейцин и β-цианоаланин;
R2 обозначает CH3SCH2CH2-, изобутил, н-бутил, бензил, 4-фторбензил, 2-нафтилметил и циклогексилметил;
R3 обозначает изоамил, н-бутил, изобутил и циклогексил; и
R4 обозначает фенил, замещенный фенил и метил.
Другая группа предпочтительных соединений формулы I представляет собой соединения формулы III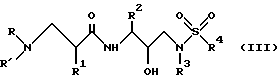
в которой R обозначает водород, алкоксикарбонил, аралкоксикарбонил, алкилкарбонил, циклоалкилкарбонил, циклоалкилалкоксикарбонил, циклоалкилалканоил, алканоил, аралканоил, ароил, арилоксикарбонил, арилоксикарбонилалкил, арилоксиалканоил, гетероциклилкарбонил, гетероциклилоксикарбонил, гетероциклилалканоил, гетероциклилалкоксикарбонил, гетероаралканоил, гетероаралоксикарбонил, гетероарилоксикарбонил, гетероароил, алкил, алкенил, циклоалкил, арил, аралкил, арилоксиалкил, гетероарилоксиалкил, гидроксиалкил, аминокарбонил, аминоалканоил и моно- и дизамещенные аминокарбонил и аминоалканоил, в которых заместители выбраны из таких радикалов, как алкил, арил, аралкил, циклоалкил, циклоалкилалкил, гетероарил, гетероаралкил, гетероциклоалкил, гетероциклоалкилалкил, или в случае дизамещенного аминоалканоила эти заместители вместе с атомом азота, к которому они присоединены, образуют гетероциклоалкил или гетероарил;
R' обозначает водород и радикалы, описанные для R3, или R и R' вместе с атомом азота, к которому они присоединены, обозначают гетероциклоалкил и гетероарил;
R1 обозначает водород, - CH2SO2NH2, -CH2CO2CH3, -CO2CH3, -CONH2, -CH2C(O)NHCH3, -C(CH3)2(SH), -C(CH3)2(SCH3), -C(CH3)2(S[O]CH3), -C(CH3)2(S[O] 2CH3), алкил, галогеналкил, алкенил, алкинил, циклоалкил и аминокислотные боковые цепи, выбранные из таких боковых цепей, как аспарагин, S-метилцистеин и его сульфоксидные /SO/ и сульфонамидные /SO2/ производные, изолейцин, аллоизолейцин, аланин, лейцин, трет-лейцин, фенилаланин, орнитин, гистидин, норлейцин, глутамин, треонин, глицин, аллотреонин, серин, аспарагиновая кислота, β-цианоаланин и валин;
R2 обозначает алкил, арил, циклоалкил, циклоалкилалкил и аралкил, иногда замещенные группой, выбранной из радикалов алкила и водорода, -NO2, -C≡N, CF3, -OR9, -SR9, где R9 обозначает водород и алкил;
R3 обозначает алкил, галогеналкил, алкенил, алкинил, гидроксиалкил, алкоксиалкил, циклоалкил, циклоалкилалкил, гетероциклоалкил, гетероарил, гетероциклоалкилалкил, арил, аралкил, гетероаралкил, аминоалкил и моно- и дизамещенный аминоалкил, в которых заместители выбраны из таких радикалов, как алкил, арил, аралкил, циклоалкил, циклоалкилалкил, гетероарил, гетероаралкил, гетероциклоалкил и гетероциклоалкилалкил, или в случае дизамещенного аминоалкила эти заместители вместе с атомом азота, к которому они присоединены, образуют гетероциклоалкил или гетероарил;
R4 обозначает радикалы, описанные для R3.
Более предпочтительная группа соединений формулы III состоит из соединений, в которых
R обозначает водород, алкоксикарбонил, аралкоксикарбонил, алкилкарбонил, циклоалкилкарбонил, циклоалкилалкоксикарбонил, циклоалкилалканоил, алканоил, аралканоил, ароил, арилоксикарбонилалкил, арилоксиалканоил, гетероциклилкарбонил, гетероциклилоксикарбонил, гетероциклилалканоил, гетероциклилалкоксикарбонил, гетероаралканоил, гетероаралкоксикарбонил, гетероарилоксикарбонил, гетероароил, алкил, алкенил, циклоалкил, арил, аралкил, арилоксиалкил, гетероарилоксиалкил, гидроксиалкил, аминокарбонил, аминоалканоил и моно- и дизамещенные аминокарбонил и аминоалканоил, в которых заместители выбраны из таких радикалов, как алкил, арил, аралкил, циклоалкил, циклоалкилалкил, гетероарил, гетероаралкил, гетероциклоалкил, гетероциклоалкилалкил, или в случае дизамещенного аминоалканоила эти заместители вместе с атомом азота, с которым они соединены, образуют гетероциклоалкил или гетероарил;
R' обозначает водород и радикалы, описанные для R3, или R и R' вместе с атомом азота, с которым они соединены, обозначают гетероциклоалкил и гетероарил;
R1 обозначает водород, алкил и алкенил и аминокислотные боковые цепи, выбранные из группы, состоящей из таких аминокислот, как аспарагин, валин, треонин, аллотреонин, изолейцин, трет-лейцин, S-метилцистеин и его сульфоновые и сульфоксидные производные, аланин и аллоизолейцин;
R2 обозначает алкил, циклоалкил и аралкил, иногда замещенные галогеном и радикалами формулы -OR9 и -SR9, где R9 обозначает водород и радикалы алкил и галоген;
R3 и R4 независимо друг от друга обозначают алкил, алкенил, алкоксиалкил, циклоалкил, циклоалкилалкил, гетероциклоалкил, гетероциклоалкилакил, арил, аралкил, гетероарил и гетероаралкил.
Наиболее предпочтительны соединения формулы III, в которых
R обозначает водород, алкоксикарбонил, аралкоксикарбонил, алкилкарбонил, циклоалкилкарбонил, циклоалкилалкоксикарбонил, циклоалкилалканоил, алканоил, аралканоил, ароил, арилоксикарбонил, арилоксикарбонилалкил, арилоксиалканоил, гетероциклилкарбонил, гетероциклилоксикарбонил, гетероциклилалканоил, гетероциклилалкоксикарбонил, гетероаралканоил, гетероаралканоилкарбонил, гетероарилоксикарбонил, гетероароил, аминокарбонил, аминоалканоил и моно- и дизамещенные аминокарбонил и аминоалканоил, в которых заместители выбраны из таких радикалов, как алкил, арил, аралкил, циклоалкил, циклоалкилалкил, гетероарил, гетероаралкил, гетероциклоалкил, гетероциклоалкилалкил, или в случае дизамещенного аминоалканоила эти заместители вместе с атомом азота, к которому они присоединены, образуют гетероциклоалкил или гетероарил;
R' обозначает водород и радикалы, описанные для R3, или R и R' вместе с атомом азота, к которому они присоединены, обозначают гетероциклоалкил и гетероарил;
R1 обозначает водород, метил, пропаргил, т-бутил, изопропил, и втор-бутил и аминокислотные боковые цепи, выбранные из группы, состоящей из таких аминокислот, как аспарагин, валин, S-метилцистеин, аллоизолейцин, треонин, серин, аспарагиновая кислота, β-цианоаланин и аллотреонин;
R2 обозначает CH3SCH2CH2-, изобутил, н-бутил, бензил, 4-фторбензил, 2-нафтилметил и циклогексилметил; и
R3 обозначает алкил, циклогексил, изобутил, изоамил и н-бутил;
R4 обозначает метил, фенил и замещенный фенил, в котором заместители выбраны из таких заместителей, как галоген, алкоксигидрокси-, нитро- и аминогруппа.
Еще одной предпочтительной группой соединений формулы I являются соединения формулы IV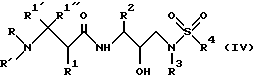
в которой R обозначает водород, алкоксикарбонил, аралкоксикарбонил, алкилкарбонил, циклоалкилкарбонил, циклоалкилалкоксикарбонил, циклоалкилалканоил, алканоил, аралканоил, ароил, арилоксикарбонил, арилоксикарбонилалкил, арилоксиалканоил, гетероциклилкарбонил, гетероциклилоксикарбонил, гетероциклилалканоил, гетероциклилалкоксикарбонил, гетероаралканоил, гетероаралкоксикарбонил, гетероарилоксикарбонил, гетероароил, алкил, алкенил, циклоалкил, арил, аралкил, арилоксиалкил, гетероарилоксиалкил, гидроксиалкил, аминокарбонил, аминоалканоил и моно- и дизамещенные аминокарбонил аминоалканоил, в которых заместители выбраны из таких радикалов, как алкил, арил, аралкил, циклоалкил, циклоалкилалкил, гетероарил, гетероаралкил, гетероциклоалкил, гетероциклоалкилалкил, или в случае дизамещенного аминоалканоила эти заместители вместе с атомом азота, с которым они соединены, образуют гетероциклоалкил или гетероарил;
R' обозначает водород и радикалы, описанные для R3, или R и R' вместе с азотом, к которому они присоединены, обозначают гетероциклоалкил и гетероарил;
R1 обозначает водород, -CH2SO2NH2, -CH2CO2CH3, -CO2CH3, -CONH2, -CH2C(O)NHCH3, -C(CH3)2(SH), -C(CH3)2(SCH3), -C(CH3)2(S[O]CH3), -C(CH3)2(S[O] 2CH3), алкил, галогеналкил, алкенил, алкинил, циклоалкил и аминокислотные боковые цепи, выбранные из таких аминокислот, как аспарагин, S-метилцистеин и его сульфоксидные /SO/ и сульфоновые /SO2/ производные, изолейцин, аллоизолейцин, аланин, лейцин, трет-лейцин, фенилаланин, орнитин, гистидин, норлейцин, глутамин, треонин, глицин, аллотреонин, серин, аспарагиновая кислота, β-цианоаланин и валин;
R1' и R1'' независимо друг от друга обозначают водород и радикалы, описанные для R1, или одни из R1' и R1'', вместе с R1 и атомами углерода, с которыми R1, R1' и R1'' соединены, обозначают циклоалкил;
R2 обозначает алкил, арил, циклоалкил, циклоалкилалкил и аралкил, иногда замещенные группой, выбранной из радикалов, таких алкил и галоген, -NO2, -C≡N, CF3, -OR9 и SR9, где R9 обозначает водород или алкил;
R3 обозначает алкил, галогеналкил, алкенил, алкинил, гидроксиалкил, алкоксиалкил, циклоалкил, циклоалкилалкил, гетероциклоалкил, гетероарил, гетероциклоалкилалкил, арил, аралкил, аминоалкил и моно- и дизамещенный аминоалкилы, в которых заместители выбраны из таких радикалов, как алкил, арил, аралкил, циклоалкил, циклоалкилалкил, гетероарил, гетероаралкил, гетероциклоалкил и гетероциклоалкилалкил, или в случае дизамещенного аминоалкила эти заместители вместе с атомом азота, к которому они присоединены, образуют гетероциклоалкил или гетероарил; и
R4 обозначает радикалы, описанные для R3.
Более предпочтительная группа соединений формулы IV состоит из соединений, в которых
R обозначает арилалканоил, гетероароил, арилоксиалканоил, арилоксикарбонил, алканоил, аминокарбонил, монозамещенный аминоалканоил или дизамещенный аминоалканоил или моно- или диалкиламинокарбонил;
R' обозначает водород и радикалы, описанные для R3, или R и R' вместе с азотом, к которому они присоединены, обозначают гетероциклоалкил или гетероарил;
R1, R1' и R1'' независимо друг от друга обозначают водород и алкилы, имеющий ~ 1 - 4 атомов углерода, алкенил, алкинил, аралкил и радикалы, выраженные формулой -CH2C(O)R'' или -C(O)R'', в которых R'' обозначает R38, -NR38R39 и OR38, где R38 и R39 независимо друг от друга обозначают водород или алкил, имеющий ~ 1 - 4 атомов углерода;
R2 обозначает алкил, циклоалкилалкил и аралкил, иногда замещенные галогеном и радикалами, выраженными формулой -OR9 и -SR9, где R9 обозначает водород и алкил;
R3 и R4 независимо друг от друга обозначают алкил, алкенил, алкоксиалкил, циклоалкил, циклоалкилалкил, гетероциклоалкил, гетероциклоалкилалкил, арил, аралкил, гетероарил и гетероаралкил.
Наибольший интерес представляют собой соединения формулы IV, в которых:
R обозначает арилалканоил, арилоксикарбонил, арилоксиалканоил, алканоил, аминокарбонил, монозамещенный аминоалканоил или дизамещенный аминоалканоил или моно- или диалкиламинокарбонил;
R' обозначает водород и радикалы, описанные для R3, или R и R' вместе с азотом, к которому они присоединены, обозначают гетероциклоалкил или гетероарил;
R1, R1', R1'' независимо друг от друга обозначают водород, метил, этил, бензил, фенилпропил и пропаргил;
R2 обозначает CH3SCH2CH2-, изобутил, н-бутил, бензил, 4-фторбензил, 2-нафтилметил и циклогексилметил;
R3 обозначает алкил, циклогексил, изобутил, изоамил и н-бутил;
R4 обозначает метил, фенил и замещенный фенил, в котором заместители выбраны из галогена, алкокси-, амино-, и нитрогруппы.
В данном описании термин "алкил", один или в сочетании, обозначает алкил с прямой или разветвленной цепью, содержащий от 1 до 10, предпочтительно от 1 до 8, атомов углерода. Примерами таких радикалов являются метил, этил, н-пропил, изопропил, н-бутил, изобутил, втор-бутил, трет-бутил, пентил, изоамил, гексил, октил и т.п. Термин "алкенил", один или в сочетании, обозначает углеводородный радикал с прямой или разветвленной цепью, имеющий одну или несколько двойных связей и содержащий от 2 до приблизительно 18 атомов углерода, предпочтительно от 2 до приблизительно 8 атомов углерода. Примерами подходящих алкенилов являются этенил, пропенил, алкил, 1,4-бутадиенил и т. п. Термин "алкинил", один или в сочетании, обозначает углеводородный радикал с прямой цепью, имеющий одну или несколько тройных связей и содержащий от 2 до приблизительно 10 атомов углерода. Примерами алкинилов являются этинил, пропинил /пропаргил/, бутинил и т.п. Термин "алкокси", один или в сочетании, обозначает радикал алкилового простого эфира, в котором термин алкил имеет описанное выше значение. Примерами подходящих радикалов алкиловых эфиров являются метокси-, этокси-, н-пропокси-, изопропокси-, н-бутокси-, изобутокси-, втор-бутокси-, трет-бутоксигруппа и т.п. Термин "циклоалкил", один или в сочетании, обозначает насыщенный или частично насыщенный моноциклический, бициклический или трициклический алкил, в котором каждая циклическая часть молекулы содержит приблизительно от 3 до 8 атомов углерода и является циклической. Термин "циклоалкилалкил" обозначает описанный выше алкил, который замещен циклоалкильным радикалом, содержащим приблизительно от 3 до 8, предпочтительно от 3 до приблизительно 6, атомов углерода. Примерами таких циклоалкильных радикалов являются циклопропил, циклобутил, циклопентил, циклогексил и т.п. Термин "арил", один или в сочетании, обозначает фенильный или нафтильный радикал, который иногда несет один или несколько заместителей, выбранных из таких радикалов, как алкил, алкоксигруппа, галоген, гидрокси-, амино-, нитро-, цианогруппа, галогеналкил и др., такой как фенил, п-толил, 4-метоксифенил, 4-(трет-бутокси)фенил, 4-фторфенил, 4-хлорфенил, 4-гидроксифенил, 2-нафтил, 2-нафтил и др. Термин "аралкил", один или в сочетании, обозначает алкил, описанный выше, в котором один атом водорода заменен арилом, описанным выше, таким как бензил, 2-фенилэтил, и т.п. Термин "аралкоксикарбонил", один или в сочетании, обозначает радикал формулы -C(O)-O-аралкил, в которой термин "аралкил" имеет данное выше значение. Примером аралкоксикарбонильного радикала является бензилоксикарбонил. Термин "арилокси" обозначает радикал формулы арил-O-, в которой термин "арил" имеет данное выше значение. Термин "алканоил", один или в сочетании, обозначает ацильный радикал, производный от алканкарбоновой кислоты. Примерами алканоила являются ацетил, пропионил, бутирил, валерил, 4-метилвалерил и т.п. Термин "циклоалкилкарбонил" обозначает ацильную группу, производную моноциклической или содержащей мостики циклоалканкарбоновой кислоты, например циклопропанкарбонил, циклогексанкарбонил, адамантанкарбонил и т.п., или из конденсированной с бензольным кольцом моноциклической циклоалканкарбоновой кислоты, которая необязательно замещена, например, алкансиламиногруппой, такой как 1,2,3,4-тетрагидро-2-нафтоил-1,2-ацетамидо-1,2,3,4-тетрагидро-2-нафтоилом. Термин "аралканоил" обозначает ацильный радикал, производный от арилзамещенной алканкарбоновой кислоты, такой как фенилацетил, 3-фенилпропионил (гидроциннамоил), 4-фенилбутирил, (2-нафтил)ацетил, 4-хлоргидроциннамоил, 4-аминогидроциннамоил, 4-метоксигидроциннамоил и т.п. Термин "ароил" обозначает ацильный радикал, производный от ароматической карбоновой кислоты. Примерами таких радикалов являются ароматические карбоновые кислоты, необязательно замещенные бензойная или нафтойная кислота, например бензоил, 4-хлорбензоил, 4-карбоксибензоил, 4-(бензилоксикарбонил)бензоил, 1-нафтоил, 2-нафтоил, 6-карбокси-2-нафтоил, 6-(бензилоксикарбонил)-2-нафтоил, 3-бензилокси-2-нафтоил, 3-гидрокси-2-нафтоил, 3-(бензилоксиформамидо)-2-нафтоил и т. п. Гетероциклил- или гетероциклоалкилчасти, гетероциклилкарбонила, гетероциклилоксикарбонила, гетероциклилалкоксикарбонила или гетероциклилалкила и т.п. представляют собой насыщенный или частично ненасыщенный моноциклический, бициклический или трициклический гетероцикл, который содержит один или несколько гетероатомов, выбранных из группы, состоящей из азота, кислорода и серы, необязательно замещенный на одном или нескольких атомах углерода галогеном, алкилом, алкоксигруппой, оксогруппой и т.п. и /или/ на вторичном атоме азота (т.е. = NH-) алкилом, аралкоксикарбонилом, алканоилом, фенилом или фенилалкилом или на третичном атоме азота /т.е. =N-/ оксидогруппой, и который присоединен через атом углерода. Гетероарильная часть гетероароила, гетероароилоксикарбонила или гетероаралкоксикарбонила или подобных групп представляет собой ароматический моноциклический, бициклический или трициклический гетероцикл, который содержит гетероатом и иногда замещен, как описано выше, в зависимости от определения гетероциклила. Примерами таких гетероциклильных и гетероарильных групп являются пирролидинил, пиперидинил, пиперазинил, морфолинил, тиаморфолинил, пирролил, имидазолил (например, имидазол-4-ил, 1-бензилоксикарбонилимидазол-4-ил и т. д.), пиразолил, пиридил, пиразинил, пиримидинил, фурил, тиенил, триазолил, оксазолил, тиазолил, индолил (например, 2-индолил и др.), хинолинил (например, 2-хинолинил, 3-хинолинил, 1-оксидо-2-хинолинил и т.д.), изохинолинил (например, 1-изохинолинил, 3-изохинолинил и т.д.), тетрагидрохинолинил (например, 1,2,3,4-тетрагидро-2-хинолил и т.д.), 1,2,3,4-тетрагидроизохинолинил (например, 1,2,3,4-тетрагидро-1-оксо-изохинолинил и т.д.), хиноксалинил, β-карболинил, 2-бензофуранкарбонил, 1-, 2-, 4- или 5-бензимидазолил и т.п. Термин "циклоалкилалкоксикарбонил" обозначает ацильную группу, производную от циклоалкилалкоксикарбоновой кислоты формулы циклоалкилалкил-O-COOH, в которой циклоалкилалкил имеет данное выше значение. Термин "арилоксиалканоил" обозначает ацильный радикал формулы арил-O-алканоил, в которой арил и алканоил имеют данное выше значение. Термин "гетероциклилоксикарбонил" обозначает ацильную группу, производную от гетероциклил-O-COOH, где гетероциклил имеет данное выше значение. Термин "гетероциклилалканоил" представляет собой ацильный радикал, производный от гетероциклилзамещенной алканкарбоновой кислоты, где гетероциклил имеет данное выше значение. Термин "гетероциклилалкоксикарбонил" обозначает ацильный радикал, произведенный от гетероциклилзамещенной алкан-O-COOH, где гетероциклил имеет данное выше значение. Термин "гетероарилоксикарбонил" обозначает ацильный радикал, производный от карбоновой кислоты, представленной формулой гетероарил-O-COOH, где гетероарил имеет данное выше значение. Термин "аминокарбонил", один или в сочетании, обозначает аминозамещенную карбонильную (карбамоильную) группу, производную от аминозамещенной карбоновой кислоты, в которой аминогруппа может быть первичной, вторичной или третичной аминогруппой, содержащей такие заместители, как водород, алкил, арил, аралкил, циклоалкил, циклоалкилалкил и др. Термин "аминоалканоил" обозначает ацильную группу, производную от аминозамещенной алканкарбоновой кислоты, в которой аминогруппа может быть первичной, вторичной или третичной аминогруппой, содержащей такие заместители, как водород, алкил, арил, аралкил, циклоалкил, циклоалкилалкил и т.п. Термин "галоген" означает фтор, хлор, бром и йод. Термин "галогеналкил" означает алкильный радикал, имеющий данное выше значение, в котором один или несколько водородов заменены галогеном. Примерами таких галогеналкильных радикалов являются хлорметил, 1-бромэтил, фторметил, дифторметил, трифторметил, 1,1,1-трифторэтил и др. Термин "уходящая (отщепляемая) группа" обычно относится к группам, легко замещаемым нуклеофилом, таким как аминный, тиоловый или спиртовой нуклеофил. Подобные отщепляемые группы хорошо известны специалистам. Примерами таких отщепляемых групп являются (но не ограничены ими) N-гидроксисукцинимид, N-гидроксибензотриазол, галогениды, трифлаты, тозилаты и т.п. Предпочтительные уходящие группы будут указаны при необходимости.
Способы получения соединений формулы I изложены ниже. Следует отметить, что основной способ относится к получению соединений, имеющих определенную стереохимию, например абсолютная стереохимия в отношении гидроксильной группы обозначена как /R/. Однако такие способы, как правило, применимы для тех же соединений противоположной конфигурации, например, имеющих стереохимию в отношении гидроксильной группы /S/. Кроме того, соединения, имеющие /R/ стереохимию, можно применять для получения соединений, имеющих /S/ стереохимию. Например, /R/ соединение можно превратить в /S/ соединение при помощи хорошо известных способов.
Получение соединений формулы I
Соединения данного изобретения, представленные формулой I, можно получить при помощи следующего основного способа. Этот способ схематически показан в схемах 1 и 2 (см. в конце описания).
N-защищенное производное хлоркетона аминокислоты, имеющее формулу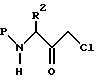
в которой P обозначает аминозащищающую группу, а R2 имеет описанное выше значение, восстанавливают до соответствующего спирта при помощи подходящего восстанавливающего агента. Пригодные аминозащищающие группы хорошо известны специалистам и могут представлять собой карбобензоксигруппу, т-бутоксикарбонил и др. Предпочтительной аминозащищающей группой является карбобензоксигруппа. Предпочтительным N-замещенным хлоркетоном является N-бензилоксикарбонил-L-фенилаланинхлорметилкетон. Предпочтительным восстанавливающим агентом является боргидрид. Реакцию восстановления проводят при температуре от -10oC до приблизительно 25oC, предпочтительно при приблизительно 0oC, в подходящей системе растворителей, такой как, например, тетрагидрофуран и т.п. N-защищенные хлоркетоны коммерчески доступны, например в Bachem. Inc., Torrance, California. В другом случае, хлоркетоны можно получить способом, изложенным в S.J. Fittkau. J. Pract. Chem., 315, 1037 /1973/, с последующей N-защитой при помощи хорошо известных специалистам способов.
Галогенпроизводное спирта может применяться непосредственно, как описано ниже, или предпочтительно оно затем реагирует, предпочтительно при комнатной температуре, с подходящим основанием в подходящей системе растворителей с образованием N-защищенного аминоэпоксида формулы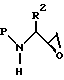
в которой P и R2 имеют описанные выше значения. Подходящими системами растворителей для получения аминоэпоксида являются этанол, метанол, изопропанол, тетрагидрофуран, диоксан и др. в том числе их смеси. Подходящими основаниями для получения эпоксида из восстановленного хлоркетона являются гидроксид калия, гидроксид, натрия, т-бутоксид калия, DBU и т.п. Предпочтительным из них является гидроксид калия.
Альтернативно, защищенный аминоэпоксид можно получить, как описано в совместной и дополняющей заявке PCT Patent Application Serial N PCT/US 93/04804, включенной здесь в качестве ссылки, исходя из L-аминокислоты, которая реагирует с подходящей аминозащищающей группой, в подходящем растворителе с образованием аминозащищенного сложного эфира L-аминокислоты формулы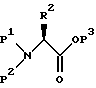
в которой P3 обозначает карбоксилзащищающую группу, например метил, этил, бензил, трет-бутил и т.п.; R2 имеет описанное выше значение; и P1 и P2 независимо друг от друга выбраны из таких аминозащищающих групп /но не только из них/, как арилалкил, замещенный арилалкил, циклоалкенилалкил и замещенный циклоалкенилалкил, аллил, замещенный аллил, ацил, алкоксикарбонил, аралкоксикарбонил и силил. Примерами арилалкила являются /но не ограничены ими/ бензил, о-метилбензил, тритил и бензгидрил, которые необязательно могут быть замещены галогеном, C1-C8-алкилом, алкокси-, гидрокси-, нитро-, амино-, алкиламино-, ациламиногруппой, алкиленом и ацилом или их солями, такими как соли фосфония или аммония. Примерами арильных групп являются фенил, нафталенил, инданил, антраценил, дуренил, 9-(9-фенилфторенил) и фенантренил, циклоалкенилалкил или замещенный циклоалкиленилалкил, содержащие C6-C10-циклоалкилы. Подходящие ацильные группы представляют собой карбобензоксигруппу, т-бутоксикарбонил, изобутоксикарбонил, бензоил, замещенный бензоил, бутирил, ацетил, трифторацетил, трихлорацетил, фталоил и т.п.
Кроме того, P1 и /или/ P2-защищающие группы могут образовывать гетероциклическое кольцо с азотом, к которому они присоединены, например, 1,2-бис(метилен)бензол, фталимидил, сукцинимидил, малеимидил и т.п. Эти гетероциклические группы могут далее включать смежные арильные и циклоалкильные кольца. Кроме того, гетероциклические группы могут быть моно-, ди- или тризамещенными, например нитрофталимидил. Термин силил относится к атому кремния, необязательно замещенному одним или несколькими алкилами, арилами и аралкилами.
Подходящими силилзащитными группами являются /но не ограничены ими/ триметилсилил, триэтилсилил, триизопропилсилил, трет-бутилдиметилсилил, диметилфенилсилил, 1,2-бис(диметилсилил)бензол, 1,2-бис(диметилсилил)этан и дифенилметилсилил. Силилирование аминогрупп для получения моно- или бисдисилиламиногрупп может дать производные аминоспирта, аминокислоты, эфиров аминокислот и амидов. В случае аминокислот, эфиров аминокислот и амидов восстановление карбонильной группы обеспечивает получение требуемого моно- или биссилиламиноспирта. Силилирование аминоспирта может дать N,N,O-трисилилпроизводное. Удаление силила из силилэфирной группы легко достигается обработкой, например, гидроксидом металла или фторидом аммония, либо в виде отдельной стадии реакции, либо in situ во время получения аминоальдегидного реагента. Подходящими силилирующими агентами являются, например, триметилсилилхлорид, трет-бутилдиметилсилилхлорид, фенилдиметилсилилхлорид, дифенилметилсилилхлорид или продукты их сочетания с имидазолом или DMF. Способы силилирования аминов и удаления силилзащитных групп хорошо известны специалистам. Способы получения этих содержащих аминогруппу производных из соответствующих аминокислот, амидов или сложных эфиров аминокислот также хорошо известны специалистам в области органической химии, в частности в области химии аминокислот/сложных эфиров аминокислот или аминоспиртов.
Предпочтительно P1 и P2 независимо друг от друга представляют собой аралкил или замещенный аралкил. Более предпочтительно, каждый из P1 и P2 является бензилом.
Аминозащищенный сложный эфир L-аминокислоты затем восстанавливают до соответствующего спирта. Например, амино-защищенный сложный эфир L-аминокислоты может быть восстановлен диизобутилалюминийгидридом при -78oC в подходящем растворителе, таком как толуол. Предпочтительными восстанавливающими агентами являются алюмогидрид лития, боргидрид лития, боргидрид натрия, боран, литий-три-трет-бутоксиалюминийгидрид, комплекс боран/THF (тетрагидрофуран). Наиболее предпочтительным восстанавливающим агентом является диизобутилалюминийгидрид /DiBAL-H/ в толуоле. Полученный спирт превращают затем, например по способу окисления Swern, в соответствующий альдегид формулы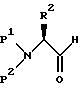
в которой P1, P2 и R2 имеют описанное выше значение. Так, раствор спирта в дихлорметане добавляют к охлажденному /-75o ... -68oC/ раствору оксалилхлорида в дихлорметане и DMSO в дихлорметане и перемешивают в течение 35 минут.
Приемлемыми окисляющими реагентами являются, например, комплекс триоксид серы/пиридин и DMSO, оксалилхлорид и DMSO, ацетилхлорид или ангидрид и DMSO, трифторацетилхлорид или ангидрид и DMSO, метансульфонилхлорид и DMSO или тетрагидротиафен-S-оксид, толуолсульфонилбромид и DMSO, трифторметансульфонилангидрид /ангидрид/ и DMSO, пентахлорид фосфора и DMSO диметилфосфорилхлорид и DMSO и изобутилхлорформиат и DMSO. Условия окисления описаны Rutz et al. [Angew. Chem. 99, p. 1186, /1987/, Angew. Chem. Int. Ed. Eng. 26, p. 1141, 1987/, применявшими оксалилхлорид и DMSO при -78oC.
Предпочтительным способом окисления, описанным в данном изобретении, является применение комплекса триоксида серы с пиридином, триэтиламина и DMSO при комнатной температуре. Эта система обеспечивает очень хорошие выходы целевого хирально защищенного аминоальдегида, применимого без очистки, т.е. необходимость очистки килограммов промежуточных продуктов при помощи хроматографии исключается и широкомасштабные операции становятся менее опасными. Реакция при комнатной температуре также исключает необходимость применения низкотемпературного реактора, что делает способ более пригодным для коммерческого производства.
Реакцию можно проводить в инертной атмосфере, например в атмосфере азота или аргона, или в нормальном или сухом воздухе, при атмосферном давлении или в запаянном реакционном сосуде при повышенном давлении. Предпочтительной является атмосфера азота. Примерами аминовых оснований являются, например, трибутиламин, триизопропиламин, N-метилпиперидин, N-метилморфолин, азабициклононан, диизопропилэтиламин, 2,2,6,6-тетраметилпиперидин, N,N-диметиламинопиридин или смеси этих оснований. Предпочтительным является триэтиламин. Кроме чистого DMSO в качестве растворителя может быть использована смесь DMSO с непротонными или галогенированными растворителями, такими как тетрагидрофуран, этилацетат, толуол, ксилол, дихлорметан, этилендихлорид и т. п. Биполярные апротонные сорастворители включают в себя ацетонитрил, диметилформамид, диметилацетамид, ацетамид, тетраметилмочевину и ее циклический аналог, N-метилпирролидон, сульфолан и т.п. Кроме N,N-дибензилфенилаланинола в качестве предшественника альдегида могут быть использованы обсуждаемые выше производные фенилаланинола, для получения соответствующего N-монозамещенного (либо P1, либо P2 = H) или N,N-дизамещенного альдегида.
Кроме того, для получения таких альдегидов можно проводить восстановление гидридом амидного или эфирного производного фенилаланина с азотом, защищенным алкилом, бензилом или циклоалкенилом, замещенного фенилаланина или содержащего циклоалкил аналога производного фенилаланина. Гидридный перенос представляет собой дополнительный способ синтеза альдегида в условиях, при которых можно избежать конденсации альдегида /например, окисление по Оппенгауэру/.
Альдегиды данного способа могут быть также получены способами восстановления защищенного фенилаланина и аналогов фенилаланина или их амидных или сложноэфирных производных, например амальгамой натрия с HCl в этаноле, или лития, или натрия, или калия, или кальция в аммиаке. Реакцию проводят при температуре от приблизительно -20oC до приблизительно 45oC, предпочтительно от 5oC до приблизительно 25oC. Два дополнительных способа получения альдегида с защищенным азотом предусматривают окисление соответствующего спирта отбеливателем в присутствии каталитического количества свободного радикала 2,2,6,6-тетраметил-1-пиридилокси, а во втором способе окисления спирта до альдегида выполняют с применением каталитического количества перрутената тетрапропиламмония в присутствии N-метилморфолин-N-оксида.
Альтернативно, кислое хлоридное производное защищенного фенилаланина или производного фенилаланина, описанное выше, может быть восстановлено водородом и катализатором, таким как Pd на карбонате бария или сульфате бария, с дополнительным замедлителем катализатора, таким как сера или тиол, или без него /восстановление по Розенмунду/.
Альдегид, полученный при окислении по Swern, реагирует затем с галогенметиллитием реагентом, генерируемым in situ реакцией алкиллитиевого или ариллитиевого соединения с дигалогенметаном формулы X1CH2X2, в которой X1 и X2, независимо друг от друга являются I, Br или Cl. Например, раствор альдегида и хлорметана в THF охлаждают до -78oC и добавляют раствор н-бутиллития в гексане. Образующийся продукт представляют собой смесь диастереомеров соответствующих аминозащищенных эпоксидов формул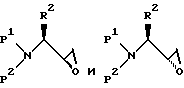
Эти диастереомеры можно разделить, например, при помощи хроматографии, или, альтернативно, они могут разделиться после реакции в последующих стадиях. Для соединений, имеющих /S/-стереохимию, вместо D-аминокислоты можно использовать L-аминокислоту.
Добавление хлорметиллития или бромметиллития к хиральному аминоальдегиду является высокоизбирательным в отношении диастереомеров. Предпочтительно, хлорметиллитий или бромметиллитий получают in situ реакцией дигалогенметана и н-бутиллития. Приемлемыми метиленирующими галогенметанами являются хлорйодметан, дибромметан, дийодметан, бромфторметан и т.п. Сульфонатный эфир продукта присоединения, например, бромисто-водородной кислоты к формальдегиду является также метиленирующим агентом. Тетрагидрофуран является предпочтительным растворителем, но можно применять и другие растворители, такие как толуол, диметоксиэтан, этилендихлорид, метиленхлорид, в виде чистых растворителей или в виде смеси. Биполярные апротонные растворители, такие как ацетонитрил, DMF, N-метилпирролидон, применимы в качестве растворителей или в качестве части смеси растворителей. Реакцию можно проводить в инертной атмосфере, такой как азот или аргон. N-бутиллитий может быть заменен другими органометаллическими реагентами, такими как метиллитий, трет-бутиллитий, втор-бутиллитий, фениллитий, фенилнатрий и т.п. Реакцию можно проводить при температуре между приблизительно -80oC и 0oC, но предпочтительно между приблизительно -80oC и -20oC. Наиболее предпочтительна температура между -40oC и -15oC. Реагенты можно добавлять только один раз, но в определенных условиях предпочтительны многократные добавления. Предпочтительно проведение реакции при атмосферном давлении, однако при определенных условиях, например в среде с высокой влажностью, предпочтительно повышенное давление.
Альтернативные способы превращения в эпоксиды согласно данному изобретению включают в себя замещение других видов заряженных предшественников метиленирования с последующий обработкой их основанием для образования аналогичного аниона. Примерами таких видов молекул являются тозилат или трифлат триметилсульфоксония, галогенид тетраметиламмония, галогенид метилдифенилсульфоксония, где галогенид представляет собой хлорид, бромид или йодид.
Превращение альдегидов данного изобретения в их эпоксидное производное можно также проводить в несколько стадий. Например, добавление аниона тиоанизола, полученного, например, из бутил- или ариллитиевого реагента, к защищенному аминоальдегиду, окисление образующегося защищенного аминосульфоспирта хорошо известными окислителями, такими как пероксид водорода, трет-бутилгипохлорид, переоксид или перйодат натрия, с образованием сульфоксида. Алкилирование сульфоксида, например, метилйодидом или бромидом, метилтозилатом, метилмезилатом, метилтрифлатом, этилбромидом, изопропилбромидом, бензилхлоридом и т.п., в присутствии органического или неорганического основания. Альтернативно, защищенный аминосульфоспирт может быть алкилирован, например, приведенными выше алкилирующими агентами с образованием солей сульфония, которые затем превращают в целевые эпоксиды при помощи трет-амина или минеральных оснований.
Требуемые эпоксиды при наиболее предпочтительных условиях образовывались диастереоселективно в количественном соотношении 85:15 /S:R/. Этот продукт можно очистить хроматографически с образованием диастереомерно и энантиомерно чистого продукта, но более удобно применять его без очистки для получения ингибиторов ретровирусных протеаз. Вышеупомянутый способ применим для смесей оптических изомеров так же, как и для разделенных соединений. Если желателен определенный оптический изомер, то он может быть получен путем выбора исходного материала, например L-фенилаланина, D-фенилаланина, L-фенилаланинола, D-фенилаланинола, D-гексагидрофенилаланинола и т.п., или разделение может происходить на промежуточных или конечных стадиях. Хиральные вспомогательные вещества, такие как один или два эквивалента камфорсульфоновой кислоты, лимонной кислоты, камфорной кислоты, 2-метоксифенилуксусной кислоты и т. п. , могут быть использованы для образования солей, сложных эфиров или амидов соединений данного изобретения. Эти соединения или производные могут быть кристаллизованы или разделены хроматографически с применением хиральной или ахиральной колонки, как это известно специалистам в данной области.
Затем аминоэпоксид реагирует в подходящей системе растворителей с равным количеством, или предпочтительно с избытком, желаемого амина формулы: R3NH2, в которой R3 представляет собой водород или имеет описанное выше значение. Реакцию можно проводить в широком диапазоне температур, например приблизительно от 10oC до приблизительно 100oC, но предпочтительно, хотя и необязательно, проводить эту реакцию при температуре, при которой растворитель начинает кипеть. Подходящими системами растворителей являются протонные, непротонные и биполярные апротонные органические растворители, такие как, например, системы, в которых растворителем является спирт, например метанол, этанол, изопропанол и др., простые эфиры, такие как тетрагидрофуран, диоксан и т.п., и толуол, N,N-диметилформамид, диметилсульфоксид, и их смеси. Предпочтительным растворителем является изопропанол. Примерами аминов, соответствующих формуле R3NH2, являются бензиламин, изоамиламин, циклогексанметиламин, нафтиленметиламин и т. п. Образующийся продукт представляет собой производное 3-(N-замещенная амино)-3-(R2)-1-(NHR3)-пропан-2-ола (далее называемое аминоспиртом) и может быть представлен формулами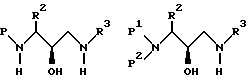
в которых P, P1, P2, R2 и R3 имеют описанное выше значение. Альтернативно, вместо аминоэпоксида может быть использован галоспирт.
Затем аминоспирт, описанный выше, реагирует в подходящем растворителе с сульфонилхлоридом /R4SO2Cl/ или сульфонилангидридом в присутствии акцептора кислоты. Растворителями, в которых может проходить эта реакция, являются метиленхлорид, тетрагидрофуран. Подходящими акцепторами кислоты являются триэтиламин, пиридин. Предпочтительными сульфонилхлоридами являются метансульфонилхлорид и бензолсульфонилхлорид. Образующееся производное сульфонамида /сульфамида/ может быть представлено в зависимости от применяемого эпоксида формулами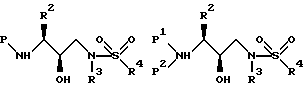
в которых P, P1, P2, R2, R3 и R4 имеют описанное выше значение. Эти промежуточные продукты применимы для получения ингибиторных соединений данного изобретения и сами являются также активными ингибиторами ретровирусных протеаз.
Сульфонилгалогениды формулы R4SO2X могут быть получены реакцией подходящего реактива Гриньяра или алкиллитиевого реагента с сульфурилхлоридом или диоксидом серы с последующим окислением галогеном, предпочтительно хлором. Также тиолы могут быть окислены до сульфонилхлоридов при помощи хлора в присутствии воды при тщательно контролируемых условиях. Кроме того, сульфокислоты могут быть превращены в сульфонилгалогениды при помощи таких реагентов, как PCl5, и также в ангидриды при помощи подходящих дегидратирующих агентов. Сульфокислоты, в свою очередь, могут быть получены при помощи хорошо известных специалистам способов. Такие сульфокислоты также коммерчески доступны. Для получения соединений, в которых -SO2- заменена на -SO- или -S-, соответственно, вместо сульфонилгалогенидов можно использовать сульфинилгалогениды /R4SOX/ или сульфенилгалогениды /R4SX/ соответственно.
После получения производного сульфонамида аминозащищающую группу P или аминозащищающие группы P1 и P2 удаляют при условиях, которые не влияют на оставшуюся часть молекулы. Эти способы хорошо известны специалистам в данной области и включают в себя гидролиз, гидрогенолиз и т.п. Предпочтительным способом является удаление защитной группы, например, карбобензоксигруппы, гидрогенолизом с применением палладия на угле в подходящей системе растворителей, таких как спирт, уксусная кислота и т.п. или их смеси. Если защитной группой является т-бутоксикарбонил, ее можно удалить с применением неорганической или органической кислоты, например HCl или трифторуксусной кислоты, в подходящей системе растворителей, например, в диоксане или метиленхлориде. Образующийся продукт представляет собой соль амина. После нейтрализации соли амин реагирует с аминокислотой или соответствующим производным аминокислоты, представленным формулой (PN[CR1'R1'']tCH(R1)COOH), в которой t, R1, R1' и R1'' имеют описанные выше значения, с образованием антивирусных соединений данного изобретения, имеющих формулу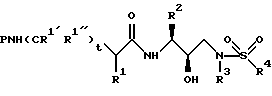
в которой t, P, R1, R1', R1'', R2, R3 и R4 имеют описанные выше значения. Предпочтительными защитными группами в этом случае являются бензилоксикарбонил или т-бутоксикарбонил. Если амин реагирует с производным аминокислоты, например, когда t = 1 и R1' и R1'' оба представляют собой H, так что аминокислота является β-аминокислотой, такие β-аминокислоты можно получить в соответствии со способом, изложенным в дополнительной заявке US Serial N 07/345808. Если t = 1 один из R1' и R1'' представляет собой H и R1 представляет собой H, так что аминокислота представляет собой гомо-β-аминокислоту, такие гомо-β-аминокислоты можно получить по способу, изложенному в дополнительной заявке US Serial N 07/853561. Если t = 0 и R1 является алкилом, алкенилом, алкинилом, циклоалкилом, -CH2SO2NH2, -CH2CO2CH3, -CO2CH3, -CONH2, -CH2C(O)NHCH3, -C(CH3)2(SH), -C(CH3)2(SCH3), -C(CH3)2(S[O] CH3), -C(CH3)2(S[O2]CH3), или аминокислотной боковой цепью, такие материалы хорошо известны и многие из них коммерчески доступны от Sigma-Aldrich.
N-защищающая группа может быть затем удалена, если требуется, с применением описанных выше способов и затем может реагировать с карбоксилатом формулы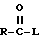
где R имеет описанное выше значение, а L представляет собой подходящую уходящую /отщепляемую/ группу, такую как галоген. Предпочтительно, если R1 является боковой цепью, природной α-аминокислоты, R представляет собой 2-хинолинкарбонил, производный от N-гидроксисукцинимид-2-хинолинкарбоксилата, т.е. L является гидроксисукцинимидом. Раствор свободного амина /или его соли с уксусной кислотой/ и приблизительно 1,0 эквивалент карбоксилата смешивают в подходящей системе растворителей и иногда обрабатывают (до 5 эквивалентов) основанием, таким как N-метилморфолин, при комнатной температуре. Подходящей системой растворителей являются тетрагидрофуран, метиленхлорид или N,N-диметилформамид и др., в том числе их смеси.
Альтернативно аминоспирт, защищенный от разрыва эпоксидной связи, может быть защищен далее по месту вновь введенной аминогруппы защитной группой P1, которая не удаляется при удалении первой защитной группы P. Специалисты могут выбрать подходящие комбинации P и P1. Одним из таких вариантов является вариант, когда P представляет собой Cbz, а P1 представляет собой Boc. Образующееся соединение, представленное формулой
может быть введено в дальнейшие стадии синтеза с образованием соединения формулы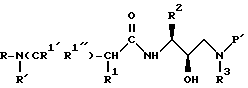
и новую защитную группу P1 избирательно удаляют. После снятия защиты образующийся амин реагирует с образованием производного сульфонамида, описанного выше. Это избирательное снятие защиты и дальнейшее превращение в сульфонамид можно выполнять либо в конце синтеза, либо на какой-либо промежуточной стадии, если желательно.
Вместо сульфонилгалогенидов можно применять сульфинилгалогениды /RSOCl/ и сульфенилгалогениды /RSCl/ для получения соединений, в которых часть молекулы -SO2- заменена на -SO- или -S- соответственно.
Предполагается, что для получения соединений формул, имеющих R6, эти соединения можно получать после изложенного выше способа и перед взаимодействием производного сульфонамида или его аналога, например, взаимодействием с аминокислотой PNH(CH2)tCH(R1)COOH, проводимым по способу, известному специалистам как восстановительное аминирование. Так, цианоборгидрид натрия и подходящий альдегид или кетон могут реагировать с производным сульфонамида или его аналогом при комнатной температуре для восстановительного аминирования какого-либо из соединений формул I-IV. Также предлагается, что если R3 промежуточного продукта аминоспирта является водородом, ингибирующие соединения данного изобретения, в которых R3 представляет собой алкил или другие заместители, в которых α-C содержит по меньшей мере один водород, могут быть получены при помощи восстановительного аминирования конечного продукта реакции между аминоспиртом и амином или на любой другой стадии синтеза для получения таких соединений.
Предлагаемые эквиваленты основных формул, изложенных выше, для антивирусных соединений и производных, а также промежуточные соединения представляют собой соединение, в других отношениях соответствующие им и имеющие те же самые основные свойства, например их таутомеры, так же как и соединения, в которых одна или несколько из R-групп представляют собой простые вариации описанных выше заместителей, например, в которых R представляет собой алкил с большим количеством атомов углерода, чем указанный алкил. Кроме того, в случае, если заместитель обозначает водород, то точная химическая природа заместителя, иного, чем водород, в этом положении, например, радикал гидрокарбил или галоген, гидроксигруппа, аминогруппа и т.п., не является критической до тех пор, пока она не действует неблагоприятно на общую активность и /или/ на методику синтеза.
Приведенные выше химические реакции описаны, как правило, в виде их самого широкого применения в получении соединений данного изобретения. Иногда эти реакции могут оказаться неприменимыми в таком именно виде для каждого соединения внутри описанного диапазона соединений. Специалисты легко поймут, для каких соединений это может иметь место. Во всех таких случаях такие реакции можно проводить при помощи общепринятых модификаций, известных специалистам в этой области, например, с применением подходящей защиты мешающих групп, изменением реагентов, обычной модификацией условий реакции и т.п., или путем применения других реакций, описанных здесь или общепринятых, для получения соответствующих соединений данного изобретения. Во всех препаративных способах все исходные материалы известны или могут быть легко получены из известных исходных материалов.
Без дальнейшей детализации специалисты в данной области смогут, используя предшествующее описание, использовать данное изобретение в его самой полной степени. Поэтому следующие далее отдельные предпочтительные варианты следует понимать лишь как иллюстративные, но не ограничивающие данное изобретение в какой бы то ни было степени.
Все реагенты использовали без очистки в таком виде, в каком они были получены. Все H-ЯМР и C-ЯМР-спектры получали либо на Varian VXR-300, либо на VXR-400 спектрометре ядерного магнитного резонанса.
Примеры 1-9 иллюстрируют получение промежуточных продуктов. Эти промежуточные продукты использованы для получения ингибирующих соединений данного изобретения, иллюстрированном в примерах 10-16. Кроме того, промежуточные продукты примеров 2-6 также представляют собой ингибиторы ретровирусных протеаз и, в частности, ингибируют протеазу ВИЧ.
Пример 1A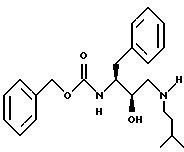
Получение N[3(S)-бензилоксикарбониламино-2(R)-гидрокси-4-фенилбутил] -N-изоамиламина
Часть A:
К раствору 75,0 г /0,226 моля/ N-бензилоксикарбонил-L-фенилаланинхлорметилкетона в смеси 807 мл метанола и 807 мл тетрагидрофурана при -2oC добавляли 13,17 г /0,348 моля, 1,54 экв./ твердого боргидрида натрия в течение 100 минут. Растворители удаляли при пониженном давлении при 40oC и остаток растворяли в этилацетате /приблизительно 1 л/. Раствор промывали последовательно 1 М кислым сульфатом натрия, насыщенным раствором бикарбоната натрия и насыщенным раствором хлорида натрия. После высушивания над безводным сульфатом магния и фильтрования раствор удаляли при пониженном давлении. К полученному маслу добавляли гексан /приблизительно 1 л/ и смесь нагревали до 60oC при интенсивном перемешивании. После охлаждения до комнатной температуры твердый материал собирали и промывали 2 л гексана. Полученный твердый материал перекристаллизовывали из горячего этилацетата и гексана, получая 32,3 г /выход 43%/ N-бензилоксикарбонил-3(S)-амино-1-хлор-4-фенил-2-(S)-бутанола, т.пл. 150-151oC и M+Li+ = 340.
Часть B:
К раствору 6,52 г /0,116 моля, 1,2 экв./ гидроксида калия в 968 мл абсолютного спирта при комнатной температуре добавляли 32,3 г /0,097 моля/ N-CBZ-3(S)-амино-1-хлор-4-фенил-2(S)-бутанола. После перемешивания в течение 15 минут растворитель удаляли при пониженном давлении и твердый материал растворяли в метиленхлориде. После промывания водой, высушивания над сульфатом магния, фильтрования и упаривания получали 27,9 г белого твердого вещества. Перекристаллизация из горячего этилацетата и гексана дала 22,3 г /выход 77%/ N-бензилоксикарбонил-3(S)-амино-1,2(S)-эпокси-4-фенилбутана, т.пл. 102-103oC и MH+ 298.
Часть C:
Раствор N-бензилоксикарбонил-3(S)-амино-1,2(S)-эпокси-4-фенилбутана /1,0 г, 3,36 ммоля/ и изоамиламина /4,90 г, 67,2 ммоля, 20 экв./ в 10 мл изопропанола нагревали для дефлегмации в течение 1,5 часов. Раствор охлаждали до комнатной температуры, концентрировали в вакууме и затем выливали в 100 мл перемешиваемого гексана, в результате чего продукт кристаллизовался из раствора. Продукт выделяли фильтрованием и сушили на воздухе, получая 1,18 г, 95% N-[[3(S)-фенилметилкарбамоил)амино-2(R)-гидрокси-4-фенилбутил] N-[(3- метилбутил)]амина, т.пл. 108,0-109,5oC, MH+ m/z = 371.
Пример 1B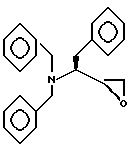
Получение N,N-дибензил-3(S)-амино-1,2-(S)-эпокси-4-фенилбутана
Стадия A:
Раствор L-фенилаланина /50,0 г, 0,302 моля/, гидроксида натрия /24,2 г, 0,605 моля/ и карбоната калия /83,6 г, 0,605 моля/ в воде /500 мл/ нагревали до 90oC. Затем медленно добавляли бензилбромид /108,5 мл, 0,912 моля/ /время добавления приблизительно 25 минут/. Затем смесь перемешивали при 97oC в течение 30 минут. Раствор охлаждали до комнатной температуры и экстрагировали толуолом /2 х 250 мл/. Объединенные органические слои промывали водой, солевым раствором, сушили над сульфатом магния, фильтровали и концентрировали, получая масляный продукт. Этот неочищенный продукт использовали в следующей стадии без очистки.
Стадия B:
Неочищенный бензилированный продукт предыдущей стадии растворяли в толуоле /750 мл/ и охлаждали до -55oC. 1,5 М раствор DIBAL-H в толуоле /443,9 мл, 0,666 моля/ добавляли со скоростью, позволяющей поддерживать температуру между -55o и -50oC /время добавления - 1 час/. Смесь перемешивали в течение 20 минут при -55oC. Реакцию останавливали при -55oC медленным добавлением метанола /37 мл/. Затем холодный раствор выливали в холодный раствор /5oC/ 1,5 N HCl /1,8 л/. Осажденное твердое вещество /прибл. 138 г/ отфильтровывали и промывали толуолом. Твердый материал суспендировали в смеси толуола /400 мл/ и воды /100 мл/. Смесь охлаждали до 5oC, обрабатывали 2,5 N NaOH /186 мл/ и затем перемешивали при комнатной температуре до растворения твердого материала. Слой толуола отделяли от водной фазы и промывали водой и солевым раствором, сушили над сульфатом магния, фильтровали и концентрировали до объема 75 мл /89 г/. Затем к остатку добавляли этилацетат /25 мл/ и гексан /25 мл/, после чего спиртовой продукт начинал кристаллизоваться. После 30 минут добавляли дополнительно 50 мл гексана для ускорения дальнейшей кристаллизации. Твердое вещество отфильтровывали и промывали 50 мл гексана, получая приблизительно 35 г материала. Вторая часть материала могла быть выделена повторным фильтрованием маточной жидкости. Твердые вещества объединяли и перекристаллизовывали из этилацетата /20 мл/ и гексана /30 мл/, получая в целом, в двух частях, приблизительно 40 г /40% от L-фенилаланина/ аналитически чистого спиртового продукта. Маточные жидкости объединяли и концентрировали /34 г/. Остаток обрабатывали этилацетатом и гексаном, что дало дополнительно 7 г /выход прибл. 7%/ слегка загрязненного твердого продукта. Возможна дальнейшая оптимизация извлечения продукта из маточной жидкости.
Альтернативно, спирт получали из L-фенилаланинола. L-фенилаланинол /176,6 г, 1,168 моля/ добавляли к перемешиваемому раствору карбоната калия /484,6 г, 3,506 моля/ в 710 мл воды. Смесь нагревали до 65oC в атмосфере азота. Раствор бензилбромида /400 г, 2,339 моля/ в 3A этаноле /305 мл/ добавляли при скорости, позволяющей поддерживать температуру между 60o и 68oC. Двухфазный раствор перемешивали при 65oC в течение 55 минут и затем охлаждали до 10oC при интенсивном перемешивании. Маслянистый продукт отверждали до небольших гранул. Продукт разводили 2,0 л водопроводной воды и перемешивали 5 минут для растворения неорганических побочных продуктов. Продукт выделяли фильтрованием при пониженном давлении и промывали водой до pH 7. Полученный неочищенный продукт сушили на воздухе в течение ночи, получая полусухое твердое вещество /407 г/, которое перекристаллизовывали из 1,1 л смеси этилацетат/гептан /1:10 по объему/. Продукт выделяли фильтрованием /при -8oC/, промывали 1,6 л холодной /-10oC/ смеси этилацетат/гептан /1:10 по объему/ и сушили на воздухе, получая 339 г /88% выход/ β S-2-[бис(фенилметил)амино] бензолпропанола, т.пл. 71,5-73,0oC. Если требуется, можно получить дополнительный продукт из маточного раствора. Другие аналитические свойства были идентичны свойствам соединения, полученного, как описано выше.
Стадия C:
Раствор оксалилхлорида /8,4 мл, 0,095 моля/ в дихлорметане /240 мл/ охлаждали до -74oC. Затем раствор DMSO /12,0 мл, 0,155 моля/ в дихлорметане /50 мл/ медленно добавляли со скоростью, позволяющей поддерживать температуру при -74oC /время добавления - прибл. 1,25 часа/. Смесь перемешивали 5 минут и добавляли раствор спирта /0,074 моля/ в 100 мл дихлорметана /время добавления - 20 минут, температура -75 ... -68oC/. Раствор перемешивали при -78oC в течение 35 минут. Затем добавляли триэтиламин /41,2 мл, 0,295 моля/ в течение 10 минут /температура -78 ... -68oC/, после чего осаждалась соль аммония. Холодную смесь перемешивали в течение 30 минут и затем добавляли воду /225 мл/. Слой дихлорметана отделяли от водной фазы и промывали водой, солевым раствором, сушили над сульфатом магния, фильтровали и концентрировали. Остаток разводили этилацетатом и гексаном и затем отфильтровывали для дальнейшего удаления соли аммония. Фильтрат концентрировали, получая целевой альдегидный продукт. Альдегид использовали на следующей стадии без очистки.
В литературе сообщались температуры более высокие, чем -70oC, для окисления по Сверну (Swern). Другие модификации Сверна и альтернативы окисления по Сверну также возможны.
Альтернативно альдегид получали следующим образом. /200 г, 0,604 моля/ растворяли в триэтиламине /300 мл, 2,15 моля/. Смесь охлаждали до 12oC и раствор комплекса триоксид серы/пиридин /380 г, 2,39 моля/ в DMSO /1,6 л/ добавляли при скорости, позволяющей поддерживать температуру между 8o и 17oC /время добавления - 1,0 ч/. Раствор перемешивали при температуре окружающей среды в атмосфере азота в течение 1,5 часов, после чего реакцию завершали ТСХ-анализом /33% этилацетат/гексан, силикагель/. Реакционную смесь охлаждали охлажденной льдом, водой и погашениями 1,6 л холодной воды /10-15oC/ через 45 минут. Образующийся раствор экстрагировали этилацетатом /2,0 л/, промывали 5% лимонной кислотой /2,0 л/ и солевым раствором /2,2 л/, сушили над MgSO4 /280 г/ и фильтровали. Растворитель удаляли на роторном испарителе при 35-40oC и затем сушили под вакуумом, получая 198,8 г α S-[бис-(фенилметил)амино] -бензолпропанальдегида в виде бледно-желтого масла /99,9%/. Полученный неочищенный продукт был достаточно чистым для использования его сразу же в следующей стадии без дополнительной очистки. Аналитические данные для этого соединения совпадают с опубликованными в литературе. [α]
Стадия D:
Раствор α S-[бис(фенилметил)амино]бензолпропанальдегида /191,7 г, 0,58 моля/ и хлорйодметана /56,4 мл, 0,77 моля/ в тетрагидрофуране /1,8 л/ охлаждали до -30 ... -35oC /более низкая температура, такая как -70oC, также работала хорошо, но более высокие температуры легче получить в широкомасштабных операциях/ в реакторе из нержавеющей стали в атмосфере азота. Затем раствор н-бутиллития в гексане /1,6 М, 365 мл, 0,58 моля/ добавляли при скорости, позволяющей поддерживать температуру ниже -25oC. После добавления смесь перемешивали при -30 ... -35oC 10 минут. Следующие добавления реагентов проводили таким образом:
/1/ Дополнительно добавляли хлорйодметан /17 мл/, с последующим добавлением н-бутиллития /110 мл/ при температуре ниже -25oC. После добавления смесь перемешивали при -30 ... -35oC 10 минут. Это повторяли один раз.
/2/ Дополнительно добавляли хлорйодметан /8,5 мл, 0,11 моля/ с последующим добавлением н-бутиллития /55 мл, 0,088 моля/ при температуре ниже -25oC. После добавления смесь перемешивали при -30 ... -35oC 10 минут. Это повторяли 5 раз.
/3/ Дополнительно добавляли хлорйодметан /8,5 мл, 0,11 моля/ с последующим добавлением н-бутиллития /37 мл, 0,059 моля/ при температуре ниже -25oC. После добавления смесь перемешивали при -30 ... -35oC 10 минут. Это повторяли один раз. Наружное охлаждение прекращали и смесь нагревали до температуры окружающей среды в течение 4-16 часов, когда ТСХ-анализ (силикагель, 20% этилацетат/гексан) показывал, что реакция завершена. Реакционную смесь охлаждали до 10oC и реакцию гасили при помощи 1452 г 16% раствора хлорида аммония /полученного растворением 232 г хлорида аммония в 1220 мл воды/, поддерживая температуру ниже 23oC. Смесь перемешивали 10 минут и разделяли органический и водный слой. Водную фазу экстрагировали этилацетатом /2 х 500 мл/. Слой этилацетата соединяли со слоем тетрагидрофурана. Объединенный раствор сушили над сульфатом магния /220 г/, фильтровали и концентрировали на роторном испарителе при 65oC. Коричневый масляный остаток сушили при 70oC в вакууме /0,8 бар/ в течение 1 ч, получая 222,8 г неочищенного материала. /Вес неочищенного продукта был более 100%. Вследствие относительной нестабильности продукта на силикагеле продукт обычно использовали в следующей стадии без очистки/. Отношение диастереомеров неочищенной смеси было определено при помощи протонного ЯМР: (2S)/(2R): 86:14. Минорные и основные эпоксидные диастереомеры характеризовались в этой смеси при помощи ТСХ-анализа (силикагель, 10% этилацетат/гексан), Rf = 0,29 и 0,32 соответственно. Аналитическую пробу каждого из диастеромеров получали очисткой на ТСХ-силикагеле (3% этилацетат/гексан) и характеризовали следующим образом:
N,N α S-Трис(фенилметил)-2S-оксиранметанамин
1H-ЯМР /400 МГц, CDCl3/ δ 2,49 (AB-System, 1H, JAB = 2,82), 2,76 и 2,77 (AB-System, 1H, JAB = 4,03 Гц), 2,83 (м, 2H), 2,99 и 3,03 (AB-System, 1H, JAB = 10,1 Гц), 3,15 (м, 1H), 3,73 и 3,84 (AB-System, 4H, JAB = 14,00 Гц), 7,21 (м, 15H); 13C-ЯМР (400 МГц, CDCl3) δ 139,55, 129,45, 128,42, 128,14, 129,09, 126,94, 125,97, 60,32, 54,23, 52,13, 45,99, 33,76; HRMS рассчит. для C24H26NO /M+1/ 344,477, найдено 344,2003.
N,N, α S-Трис(фенилметил)-2R-оксиранметанамин
1H-ЯМР /300 МГц, CDCl3/ δ 2,20 (м, 1H), 2,59 (м, 1H), 2,75 (м, 2H), 2,97 (м, 1H), 3,14 (м, 1H), 3,85 (AB-System, 4H), 7,25 (м, 15H). ЖХВР на хиральной стационарной фазе: Pirkle-Whelk-01 Column /250 х 4,6 мм 1.D./, подвижная фаза: гексан/изопропанол (99,5:0,5, об./об.), скорость потока: 1,5 мл/мин, детектирование УФ-детектором при 210 нм. Время удерживания /8/ 9,38 мин, время удерживания энантиомера /4/: 13,75 мин.
Альтернативно, раствор неочищенного альдегида 0,074 моля и хлорйодметана /7,0 мл, 0,096 моля/ в тетрагидрофуране /285 мл/ охлаждали до -78oC в атмосфере азота. 1,6 М раствор н-бутиллития в гексане /25 мл, 0,040 моля/ добавляли затем при скорости, позволяющей поддерживать температуру при -75oC /время добавления - 15 минут/. После первого добавления добавляли дополнительно хлорйодметан /1,6 мл, 0,022 моля/ с последующим добавлением н-бутиллития /23 мл, 0,037 моля/, поддерживая температуру при -75oC. Смесь перемешивали 15 минут. Каждый из реагентов, хлорйодметан /0,70 мл, 0,010 моля/ и н-бутиллитий /5 мл, 0,008 моля/ добавляли еще 4 раза в течение 45 минут при -75oC. Охлаждающую баню затем удаляли и раствор подогревали до 22oC в течение 1,5 часа. Смесь выливали в 300 мл насыщенного раствора хлорида аммония в воде. Тетрагидрофурановый слой отделяли. Водную фазу экстрагировали этилацетатом /1 х 300 мл/. Объединенные органические слои промывали солевым раствором, сушили над сульфатом магния, фильтровали и концентрировали, получая коричневое масло /27,4 г/. Продукт можно было использовать в следующей стадии без очистки. Целевой диастереоизомер мог быть очищен перекристаллизацией на последующей стадии. Продукт мог быть также очищен хроматографически.
Альтернативно, раствор α S-[Бис(фенилметил)амино]бензолпропанальдегида /178,84 г, 0,54 моля/ и бромхлорметана /46 мл, 0,71 моля/ в тетрагидрофуране /1,8 л/ охлаждали до -30 ... -35oC /более низкая температура, такая как -70oC, также работала хорошо, но более высокие температуры легче получить в широкомасштабных операциях/ в реакторе из нержавеющей стали в атмосфере азота. Затем раствор н-бутиллития в гексане /1,6 М, 340 мл, 0,54 моля/ добавляли при скорости, позволяющей поддерживать температуру ниже -25oC. После добавления смесь перемешивали при -30 ... -35oC 10 минут. Дальнейшие добавления реагентов проводили следующим образом:
/1/ дополнительно добавляли бромхлорметан /14 мл/ с последующим добавлением н-бутиллития /102 мл/ при температуре ниже -25oC. После добавления смесь перемешивали при -30 ... -35oC 10 минут. Это повторяли 1 раз.
/2/ Дополнительно добавляли бромхлорметан /7 мл, 0,11 моля/ с последующим добавлением н-бутиллития /51 мл, 0,082 моля/ при температуре ниже -25oC. После добавления смесь перемешивали при -30 ... -35oC 10 минут. Это повторяли 5 раз.
/3/ Дополнительно добавляли бромхлорметан /7 мл, 0,11 моля/ с последующим добавлением н-бутиллития /51 мл, 0,082 моля/ при температуре ниже -25oC. После добавления смесь перемешивания при -30 ... -35oC 10 минут. Это повторяли 1 раз. Наружное охлаждение прекращали и смесь нагревали до температуры окружающей среды в течение 4-16 часов, когда ТСХ-анализ (силикагель, 20% этилацетат/гексан) показывал, что реакция завершена. Реакционную смесь охлаждали до 10oC и реакцию останавливали при помощи 1452 г 16% раствора хлорида аммония /полученного растворением 232 г хлорида аммония в 1220 мл воды/, поддерживая температуру ниже 23oC. Смесь перемешивали 10 минут и органическую и водную фазы разделяли. Водную фазу экстрагировали этилацетатом /2 х 1500 мл/. Этилацетатный слой соединяли с тетрагидрофурановым слоем. Объединенный раствор сушили над сульфатом магния /220 г/, фильтровали и концентрировали на роторном испарителе при 65oC. Коричневый масляный остаток сушили при 70oC в вакууме /0,8 бар/ в течение 1 часа, получая 222,8 г неочищенного материала.
Пример 2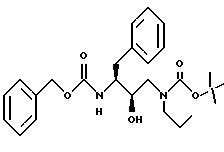
Получение N-[[3-(фенилметилкарбамоил)амино] -2R-гидрокси-4- фенил]-1-[(2-метилпропил)амино-2-(1,1-диметилэтоксил)карбонил]бутана
К раствору 7,51 г /20,3 ммоля/ N-[[3S-(фенилметилкарбамоил)амино]-2R- гидрокси-4-фенилбутил] -N-(2-метилпропил)] амина в 67 мл безводного тетрагидрофурана добавляли 2,25 г /22,3 ммоля/ триэтиламина. После охлаждения до 0oC добавляли 4,4 г /20,3 ммоля/ ди-трет-бутилкарбоната и перемешивание продолжали при комнатной температуре в течение 21 часа. Летучие соединения удаляли в вакууме, добавляли этилацетат, затем промывали 5% лимонной кислотой, насыщенным раствором бикарбоната натрия, солевым раствором, сушили над сульфатом магния, фильтровали и концентрировали, получая 9,6 г неочищенного продукта. Хроматографирование на силикагеле с применением смеси 30% этилацетат/гексан дало 8,2 г чистого N-[[3S-фенилметилкарбамоил)амино]-2R-гидрокси-4-фенил]-1-[(2- метилпропил)амино-2-(1,1-диметилэтоксил)карбонил]бутана, масс-спектр m/e = 477 /M+Li/.
Пример 3A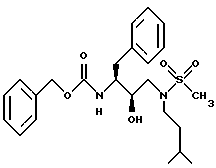
Получение фенилметил[2R-гидрокси-3-[(3-метилбутил)метилсульфонил) амино] -1S-(фенилметил)пропил]карбамата
К раствору N[3(S)-бензилоксикарбониламинно-2(R)-гидрокси-4- фенилбутил] -N-изоамиламина /2,0 г, 5,2 ммоля/ и триэтиламина /723 мкл, 5,5 ммоля/ в дихлорметане /20 мл/ добавляли по каплям метансульфониллхлорид /метансульфохлорид/ /400 мкл, 5,2 ммоля/. Реакционную смесь перемешивали 2 часа при комнатной температуре, затем раствор дихлорметана концентрировали прибл. до 5 мл и вносили в колонку с силикагелем /100 г/. Эту колонку элюировали хлороформом, содержащим 1% этанол и 1% метанол. Фенилметил[2R-гидрокси-3-[(3-метилбутил)(метилсульфонил)амино] - 1S-(фенилметил)пропил]карбамат получали в виде белого твердого вещества. Анал. рассчит. для C24H34N2O5S: C 62,31; H 7,41; N 6,06. Найдено: C 62,17; H 7,55; N 5,97.
Пример 3B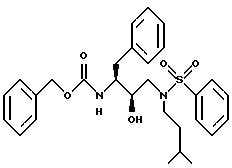
Получение фенилметил[2R-гидрокси-3-[(3-метилбутил)(фенилсульфонил) амино]-1S-(фенилметил)пропил]карбамата
Реакцией N[3(S)-бензилоксикарбониламино-2(R)-гидрокси-4- фенилбутил]-N-изоамиламина /1,47 г, 3,8 ммоля/, триэтиламина /528 мкл, 3,8 ммоля/ и бензолсульфохлорида /483 мкл, 3,8 ммоля/ получают фенилметил[2R-гидрокси-3-[(3-метилбутил)фенилсульфонил)амино]- 1S-(фенилметил)пропил]-карбамат. Колоночная хроматография на силикагеле с элюированием хлороформом, содержащим 1% этанол, дала чистый продукт. Анал. рассчит. для C29H36N2O5: C 66,39; H 6,92; N 5,34. Найдено: C 66,37; H 6,93; N 5,26.
Пример 4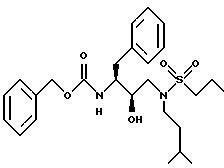
Получение фенилметил[2R-гидрокси-3-/(3-метилбутил)(н-пропансульфонил) амино]-1S-(фенилметил)пропил/карбамата
К раствору N[3(S)-бензилоксикарбониламино-2(R)-гидрокси-4- фенилбутил] -N-изоамиламина /192 мг, 0,5 ммоля/ и триэтиламина /139 мкл, 1,0 ммоль/ в дихлорметане /10 мл/ добавляли по каплям триметилсилилхлорид /63 мкл, 0,5 ммоля/. Смесь перемешивали в течение 1 часа при комнатной температуре, охлаждали до 0oC на бане со льдом и затем добавляли каплями н-пропансульфохлорид /56 мкл, 0,5 ммоля/. Реакционную смесь перемешивали в течение 1,5 часов при комнатной температуре, затем разбавляли этилацетатом /50 мл/ и промывали последовательно 1 N HCl, водой, насыщенным раствором бикарбоната натрия и насыщенным раствором хлорида натрия /по 25 мл каждого/. Органический раствор сушили над сульфатом магния, фильтровали и концентрировали до масла. Масло перемешивали с метанолом /10 мл/ в течение 16 часов, концентрировали и остаток хроматографировали на силикагеле /50 г/, элюируя колонку 10% этилацетатом с гексаном. Фенилметил[2R-гидрокси-3-[(3-метилбутил)-(н-пропансульфонил)амино] - 1S-(фенилметил)пропил] карбамат перекристаллизовывали из смеси этиловый эфир/гексан, получая белое твердое вещество. Аналит. рассчитано: для C26H38N2O5S: C 63,64; H 7,81; N 5,71. Найдено: C 63,09; H 7,74; N 5,64.
Пример 5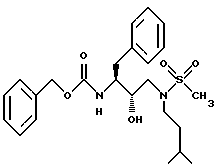
Способ, описанный в примере 2, применяли для получения фенилметил[2S-гидрокси-3-[(3-метилбутил)метилсульфонил)амино] - 1S-(фенилметил)пропил] карбамата.
К раствору N[3(S)-бензилоксикарбониламино-2(S)-гидрокси-4- фенилбутил] -N-изоамиламина /192 мг, 0,5 ммоля/ и триэтиламина /139 мкл, 0,55 ммоля/ в дихлорметане /8 мл/ добавляли по каплям метансульфохлорид /39 мкл, 0,55 ммоля/. Реакционную смесь перемешивали в течение 16 часов при комнатной температуре, затем дихлорметановый раствор вносили в колонку с силикагелем /50 г/. Колонку элюировали дихлорметаном, содержащим 2,5% метанола. Фенилметил[2S-гидрокси-3-[(3-метилбутил)-(метилсульфонил)амино]- 1S-(фенилметил)пропил] карбамат получали в виде белого твердого вещества. Аналит. рассчитано для C24H34N2O5S•0,2H2O: C 61,83; H 7,44; N 6,01. Найдено: C 61,62; H 7,40; N 5,99.
Пример 6
Согласно способам предшествующих примеров 1-5 были получены промежуточные соединения, представленные в таблицах 1A и 1B.
Следующие примеры 7-9 иллюстрируют получение промежуточных продуктов β-аминокислот. Эти промежуточные продукты могут взаимодействовать с промежуточными соединениями примеров 1-6 с образованием ингибиторных соединений данного изобретения, содержащих β-аминокислоты.
Пример 7
A. Получение 4-(4-метоксибензил))итаконата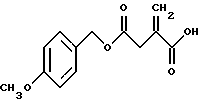
5-литровую трехгорлую круглодонную колбу, снабженную капельной воронкой с постоянным давлением, обратным холодильником, входным отверстием для азота и механической мешалкой, загружали итаконовым ангидридом /660,8 г, 5,88 моля/ и толуолом /2300 мл/. Раствор нагревали с обратным холодильником и обрабатывали 4-метоксибензиловым спиртом /812,4 г, 5,88 моля/, добавляемым по каплям в течение 2,6 часов. Этот раствор кипятили с обратным холодильником еще 1,5 часа и затем содержимое колбы выливали в три 2-литровые колбы Эрленмейера для кристаллизации. Раствору давали остыть до комнатной температуры, после чего происходила кристаллизация целевого мономера. Продукт выделяли фильтрованием на воронке Бюхнера и сушили на воздухе, получая 850,2 г, 58% материала с т.пл. 83-85oC. Второй сбор, 17%, был выделен после охлаждения фильтрата в бане со льдом. 1H-ЯМР /CDCl3/ 300 МГц, 7,32 (д, J = 8,7 Гц, 2H), 6,91 (д, J = 8,7 Гц, 2H), 6,49 (с, 1H), 5,85 (с, 1H), 5,12 (с, 2H), 3,83 (с, 3H), 3,40 (с, 2H).
B. Получение метил-4(4-метоксибензил)итаконата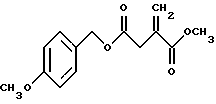
5-литровую круглодонную трехгорлую колбу, снабженную обратным холодильником, входным отверстием для азота, капельной воронкой с постоянным давлением и механической мешалкой, загружали 4(4-метоксибензил)итаконатом /453,4 г, 1,81 моля/ и обрабатывали 1,5-диазобицикло[4.3.0]нон-5-еном /275,6 г, 1,81 моля/, /DBN/ добавляемым по каплям так, чтобы температура не повышалась выше 15oC. К этой перемешиваемой смеси добавляли раствор метилйодида /256,9 г, 1,81 моля/ в 250 мл толуола из капельной воронки в течение 45 минут. Раствору давали нагреться до комнатной температуры и перемешивали дополнительно 3,25 часа.
Осажденный DBN гидройодид удаляли фильтрованием, промывали толуолом и фильтрат сливали в делительную воронку. Раствор промывали насыщенным водным раствором NaHCO3 /2 х 500 мл/, 0,2 N HCl /1 х 500 мл/ и солевым раствором /2 х 500 мл/, сушили над безводным сульфатом магния, фильтровали и растворитель удаляли в вакууме. Это дало прозрачное бесцветное масло, 450,2 г, 94%, ЯМР которого соответствовал предлагаемой структуре. 1H-ЯМР /CDCl3/ 300 МГц 7,30 (д, J = 8,7 Гц, 2H), 6,90 (д, J = 8,7 Гц, 2H), 6,34 (с, 1H), 5,71 (с, 1H), 5,09 (с, 2H), 3,82 (с, 3H), 3,73 (с, 3H), 3,38 (с, 2H), 13C-ЯМР /CDCl3/ 170,46, 166,47, 159,51, 133,55, 129,97, 128,45, 127,72, 113,77, 66,36, 55,12, 51,94, 37,64.
C. Получение метил-4(4-метоксибензил)-2(R)-метилсукцината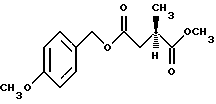
500-миллилитровую склянку Фишера-Портера загружали метил-4(4-метоксибензил)итаконатом /71,1 г, 0,269 моля/, катализатором родий (R,R) DiPAMP /204 кг, 0,269 ммоля, 0,1 мол.%/ и дегазированным метанолом /215 мл/. Через склянку пропускали 5 раз азот и 5 раз водород до конечного давления 40 Psig. Гидрогенизация начиналась сразу же и приблизительно через 1 час поглощение начинало уменьшаться, после 3 часов поглощение водорода прекращалось, колбу продували азотом, открывали и содержимое склянки концентрировали на роторном испарителе, получая коричневое масло, которое поглощалось в кипящем изооктане /прибл. 200 мл, это повторяли дважды/, раствор фильтровали через слой целита /броунмиллерита/ и фильтрат концентрировали в вакууме, получая 66,6 г, 93% прозрачного бесцветного масла. 1H-ЯМР /CDCl3/ 300 МГц 7,30 (д, J = 8,7 Гц, 2H), 6,91 (д, J = 8,7 Гц, 2H), 5,08 (с, 2H), 3,82 (с, 3H), 3,67 (с, 3H), 2,95 (ддк, J = 5,7, 7,5, 8,7 Гц, 1H), 2,79 (дд, J = 8,1, 16,5 Гц, 1H), 2,45 (дд, J = 5,7, 16,5 Гц, 1H), 1,23 (д, J = 7,5 Гц, 3H).
D. Получение метил-2(R)-метилсукцината
3-литровую трехгорлую круглодонную колбу, снабженную входным отверстием для азота, механической мешалкой, обратным холодильником и капельной воронкой с постоянным давлением, нагружали метил-4(4-метоксибензил)-(2R)-метилсукцинатом /432,6 г, 1,65 моля/ и толуолом /1200 мл/. Раствор при перемешивании обрабатывали трифторуксусной кислотой /600 мл/ из капельной воронки в течение 0,25 часа. Раствор приобретал темно-пурпурный цвет и внутренняя температура повышалась до 45oC. После перемешивания в течение 2,25 часа температура была 27oC и раствор становился розовым. Раствор концентрировали на роторном испарителе. Остаток разбавляли водой /2200 мл/ и насыщенным водным NaHCO3 /1000 мл/. Дополнительное количество NaHCO3 добавляли, пока кислота не была нейтрализована. Водную фазу экстрагировали этилацетатом /2 х 1000 мл/ для удаления побочных продуктов и водный слой подкисляли до pH = 1,8 концентрированной HCl. Этот раствор экстрагировали этилацетатом /4 х 1000 мл/, промывали солевым раствором, сушили над безводным сульфатом магния, фильтровали и концентрировали на роторном испарителе, получая бесцветную жидкость /251 г/, более 100%, которую перегоняли в вакууме через прибор с коротким путем жидкости, фракция 1: температура бани 120oC, @ более 1 мм, т. кип. 25-29oC; фракция 2: температура бани 140oC, @ 0,5 мм, т.кип. 95-108oC, 151 г, [α]d @ 25oC = +1,38oC /c = 15,475, MeOH/, [α]d = 8,48oC /чистая/; фракция 3: температура бани 140oC, т.кип. 108oC, 36 г, [α]d @ 25oC = 11,49oC /c = 15,00, MeOH/, [α]d = +8,98oC /чистая/. Фракции 2 и 3 объединяли, получая 189 г, 78% продукта. 1H-ЯМР /CDCl3/ 300 МГц 11,6 (шир. с, 1H), 3,72 (с, 3H), 2,92 (ддк, J = 8,7, 6,9, 8,0 Гц, 1H), 2,81 (дд, J = 8,0, 16,8 Гц, 1H), 2,47 (дд, J = 5,7, 16,8 Гц, 1H), 1,26 (д, J = 6,9 Гц, 3H).
E. Получение метилитаконата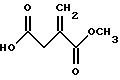
50-миллилитровую круглодонную колбу, снабженную обратным холодильником, входным отверстием для азота и магнитной мешалкой загружали метил-4-(4-метоксибензил)итаконатом /4,00 г, 16 ммолей/, 12 мл толуола и 6 мл трифторуксусной кислоты. Раствор выдерживали при комнатной температуре 18 часов и затем летучие вещества удаляли в вакууме. Остаток разбавляли этилацетатом и экстрагировали три раза насыщенным водным раствором бикарбоната натрия. Объединенный водный экстракт подкисляли до pH 1 водным бисульфатом калия и затем экстрагировали три раза этилацетатом. Объединенный раствор в этилацетате промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния, фильтровали и концентрировали в вакууме. Остаток подвергали вакуумной перегонке, получая 1,23 г, 75% чистого продукта, т.кип. 85-87oC, @ 0,1 мм. 1H-ЯМР /CDCl3/ 300 МГц 6,34 (с, 1H), 5,73 (с, 2H), 3,76 (с, 3H), 3,38 (с, 2H), 13C-ЯМР /CDCl3/ 177,03, 166,65, 129,220, 132,99, 52,27, 37,46.
F. Перегруппировка Куруиуса метил-2(R)-метилсукцината: получение метил-N-Moz-α-метил-β-аланина
5-литровую круглодонную четырехгорлую колбу, снабженную входным отверстием для азота, обратным холодильником, механической мешалкой, капельной воронкой с постоянным давлением и отверстие для термометра, загружали метил-2(R)-метилсукцинатом /184,1 г, 1,26 моля/, триэтиламином /165,6 г, 218 мл, 1,64 моля, 1,3 экв/ и толуолом /1063 мл/. Раствор нагревали до 85oC и затем обрабатывали по каплям раствором дифенилфосфорилазида /346,8 г, 1,26 моля/ в течение 1,2 часа. Раствор поддерживали при этой температуре еще в течение 1 часа и затем смесь обрабатывали 4-метоксибензиловым спиртом /174,1 г, 1,26 моля/ в течение 0,33 часа из капельной воронки. Раствор перемешивали при 88oC еще в течение 2,25 часа и затем охлаждали до комнатной температуры. Содержимое колбы выливали в разделительную воронку и промывали насыщенным водным раствором NaHCO3 /2 х 500 мл/, 0,2 N HCl /2 х 500 мл/, солевым раствором /1 х 500 мл/, сушили над безводным сульфатом магния, фильтровали и концентрировали в вакууме, получая 302,3 г, 85% целевого продукта в виде слегка коричневого масла. 1H-ЯМР /CDCl3/ 300 МГц 7,32 (д, J = 8,4 Гц, 2H), 6,91 (д, J = 8,4 Гц, 2H), 5,2 (шир. м, 1H), 5,05 (с, 2H), 3,83 (с, 3H), 3,70 (с, 3H), 3,35 (м, 2H), 2,70 (м, 2H), 1,20 (д, J = 7,2 Гц, 3H).
G.Гидролизметил-N-Moz-α-метил-β-аланина:получениеα-метил-β-аланин-гидрохлорида
5-литровую трехгорлую круглодонную колбу, снабженную обратным холодильником, входным отверстием для азота, механической мешалкой, загружали метил-N-Moz-α-метил β-аланином /218,6 г, 0,78 моля/, ледяной уксусной кислотой /975 мл/ и 12 N HCl /1960 мл/. Затем раствор нагревали с обратным холодильником в течение 3 часов. После того, как раствор охлаждали до комнатной температуры /прибл. 1 час/, водную фазу декантировали из органического остатка /полимера/ и концентрировали водную фазу на роторном испарителе. После добавления ацетона к концентрированному остатку образовалось слегка желтое твердое вещество, которое суспендировали ацетоном, и белое твердое вещество выделяли фильтрованием на воронке Бюхнера. Последние следы ацетона удаляли под вакуумом, получая 97,7 г, 90% чистого продукта, т.пл. 128,5-130,5oC, @ 25oC = 9,0oC /c = 2,535, метанол/. 1H-ЯМР /D2O/ 300 МГц 3,29 (дд, J = 8,6, 13,0 Гц, 1H), 3,16 (дд, J = 5,0, 13,0 Гц, 1H), 2,94 (ддк, J = 7,2, 5,0, 8,6 Гц, 1H), 1,30 (д, J = 7,2 Гц, 3H); 13C-ЯМР /D2O/ 180,84, 44,56, 40,27, 17,49.
H. Получение N-Boc-α-метил-β-аланина
Раствор α-метил-β аланин-гидрохлорида /97,7 г, 0,70 моля/ в воде /1050 мл/ и диоксан /1050 мл/ доводили до pH 8,9 раствором 2,9 N NaOH. Перемешиваемый раствор обрабатывали дитретбутилпирокарбонатом /183,3 г, 0,84 моля, 1,2 экв./, добавляя сразу все его количество. pH раствора поддерживали между 8,7 и 9,0 путем периодического добавления 2,5 N NaOH. После 2,5 часов pH стабилизировался и реакция считалась завершенной. Раствор концентрировали на роторном испарителе /температуру поддерживали менее 40oC/. Избыток ди-трет-бутилпирокарбоната удаляли экстрагированием дихлорметаном и водный раствор затем подкисляли холодной 1 N HCl и сразу же экстрагировали этилацетатом /4 х 1000 мл/. Объединенный этилацетатный экстракт промывали солевым раствором, сушили над безводным сульфатом магния, фильтровали и концентрировали на роторном испарителе, получая густое масло /127,3 г, 90% выход неочищенного продукта/, которое перемешивали с н-гексаном, после чего образовались кристаллы чистого продукта /95,6 г, 68%/, т.пл. 76-78oC, [α]d @ 25oC = -11,8oC /c = 2,4, EtOH/. Вторую часть получали концентрированием фильтрата и разбавлением гексаном, 15,4 г, объединенный выход 111,05, 78%. 1H-ЯМР /ацетон D6/ 300 МГц 11,7 (шир. с, 1H), 6,05 (шир. с, 1H), 3,35 (м, 1H), 3,22 (м, 1H), 2,50 (м, 1H), 1,45 (с, 9H), 1,19 (д, J = 7,3 Гц, 3H); 13C-ЯМР /ацетон D6/ 177,01, 79,28, 44,44, 40,92, 29,08, 15,50. Элементный анализ, рассчитанный для C9H17NO4: C 53,19; H 8,42; N 6,89. Найдено: C 53,36; H 8,46; N 6,99.
I. Получение N-4-метоксибензилоксикарбонил-α-метил -β-аланина
Раствор метилового эфира N-4-метоксибензилкарбонил-α- метил-β-аланина /2,81 г, 10,0 ммолей/ в 30 мл 25% водного метанола обрабатывали гидроксидом лития /1,3 экв./ при комнатной температуре в течение 2 часов. Раствор концентрировали в вакууме и остаток поглощали в смеси воды и эфира, фазы разделяли и органическую фазу отбрасывали. Водную фазу подкисляли водным кислым сульфатом калия до pH 1,5 и затем экстрагировали трижды эфиром. Объединенную эфирную фазу промывали водным раствором хлорида натрия, сушили над безводным сульфатом магния, фильтровали и концентрировали в вакууме, получая 2,60 г, 97% N-4-метоксибензилоксикарбонил-α -метил-β-аланина /N-Moz-AMBA/, который очищали перекристаллизацией из смеси этилацетата и гексана, получая 2,44 г, 91% чистого продукта, т.пл. 96-97oC, MH+ = 268. 1H-ЯМР /D6-ацетон/ 300 МГц 1,16 (3H, д, J = 7,2 Гц, 2,70 (1H, м), 3,31 (2H, м), 3,31 (3H, с), 4,99 (2H, с), 6,92 (2H, д, J = 8,7 Гц), 7,13 (2H, д, J = 8,7 Гц).
Пример 8
По способу примера 7 были получены представленные в таблице 2 β-аминокислоты.
Пример 9
По способу, изложенному в примере 7, были получены следующие соединения β-аминокислот.
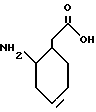
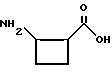
Пример 10A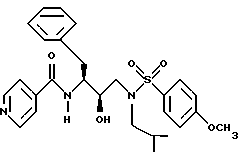
Получение N-[2R-гидрокси-3-[[(4-метоксифенил)сульфонил] (2- метилпропил)амино]-1S-(фенилметил)пропил]-4-пиридинкарбоксамида
К раствору 231 мг /0,57 ммоля/ 2R-гидрокси-3-[(2-метилпропил-(4-метоксифенил)сульфонил] амино- 1S-(фенилметил)пропиламина в 3 мл метиленхлорида при 0oC добавляли 288 мг /2,85 ммоля/ триэтиламина и затем 112 мг /0,63 ммоля/ гидрохлорида изоникотиноилхлорида. После 19 часов при комнатной температуре растворитель удалялся, добавляли этилацетат, затем промывали насыщенным раствором бикарбоната натрия, солевым раствором, сушили над сульфатом магния, фильтровали и концентрировали, получая 290 мг неочищенного продукта. Его хроматографировали на силикагеле с применением смеси 3-5% изопропанол/метиленхлорид в качестве элюента, получая 190 мг целевого соединения; масс-спектр, рассчит. для C27H34N3O5S /M+H/ 512,2219; обнаруженный 512,2280.
Пример 10B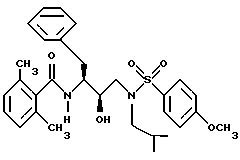
Получение N-[2R-гидрокси-3-[[(4-метоксифенил)сульфонил] (2- метилпропил)амино]-1S-(фенилметил)пропил]-2,6-диметилбензамида
К раствору 83 мг /0,55 ммоля/ 2,6-диметилбензойной кислоты и 125 мг /0,82 ммоля/ N-гидроксибензотриазола в 3 мл безводного DMF при 0oC добавляли 117 мг /0,61 ммоля/ 1-(3-диметиламинопропил)-3-этилкарболиимидгидрохлорида. После 2 часов при 0oC добавляли 203 мг /0,50 ммоля/ 2R-гидрокси-3-[(2-метилпропил)(4-метоксифенил)сульфонил]амино- 1S-(фенилметил)пропиламина. После 22 часов при комнатной температуре растворитель удаляли в вакууме, добавляли этилацетат, затем промывали насыщенным раствором бикарбоната натрия, солевым раствором, сушили над сульфатом магния, фильтровали и концентрировали, получая 300 мг неочищенного продукта. Хроматография его на силикагеле с применением смеси 20-50% этилацетат/гексан в качестве элюента дала 37 мг целевого продукта; масс-спектр, рассчитанный для C30H38N2O5S /M+H/, 539,2580, обнаружено 539,2632.
Пример 11A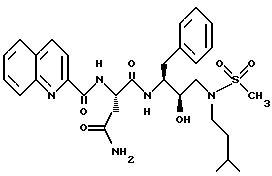
Получение N1-[2R-гидрокси-3-[(3-метилбутил)(метилсульфонил)амино]- 1S-(фенилметил)пропил]-2S-[(2-хинолинилкарбонил)амино]-бутандиамида
Часть A:
Раствор фенилметил[2R-гидрокси-3-[(3-метилбутил)(метилсульфонил) амино] -1S-(фенилметил)пропил] карбамата, полученный, как описано в примере 3, /100 мг/ в метаноле /10 мл/ гидрировали над 10% палладием на угле в течение 2 часов, фильтровали через диатомовую землю и концентрировали, получая продукт в виде масла.
Часть B:
Раствор N-CBZ-L-аспарагина /61 мг, 0,23 ммоля/ и N-гидроксибензотриазола /33 мг, 0,22 ммоля/ в DMF /2 мл/ охлаждали до 0oC на бане со льдом и затем добавляли EDC /42 мг, 0,22 ммоля/. Раствор перемешивали 30 минут при 0oC и затем добавляли продукт части A /69 мг, 0,21 ммоля/ в DMF /2 мл/. После 30 минут при 0oC реакционную смесь нагревали до комнатной температуры и перемешивали в течение 16 часов. Затем реакционную смесь выливали в 50%-насыщенный раствор бикарбоната натря /100 мл/ и образующийся белый осадок собирали вакуумным фильтрованием, промывали водой и сушили в вакууме. Фенилметил[3-амино-1S-[[2R-гидрокси-3-[(3-метилбутил)(метилсульфонил) амино] -1S-(фенилметил)амино] карбонил] -3-оксопропилкарбамат получали в виде белого твердого вещества. Аналитически рассчитано для C28H40N4O7S•0,5H2O: C 57,42; H 7,06; N 9,57. Найдено: C 57,72; H 7,21; N 9,24.
Часть C:
Раствор фенилметил[3-амино-1S-[[2R-гидрокси-3-[(3-метилбутил) (метилсульфонил)амино]-1S-(фенилметил)амино]карбонил]-3-оксопропил карбамата /135 мг, 0,23 ммоля/ в метаноле /15 мл/ гидрировали над 10% палладием на угле в течение 6 часов, фильтровали через диатомову землю и концентрировали, получая продукт в виде масла.
Часть D:
К раствору продукта части C /101 мг, 0,23 ммоля/ в DMF /5 мл/ добавляли сложный эфир N-гидроксисукцинимида и 2-хинолинкарбоновой кислоты /67 мг, 0,25 ммоля/. Реакцию проводили с перемешиванием при комнатной температуре в течение 16 часов, затем реакционную смесь выливали в 50%-насыщенный раствор бикарбоната натрия /60 мл/. Полученное твердое вещество собирали вакуумным фильтрованием, промывали водой и сушили в вакууме. N1-[2R-гидрокси-3-[(3-метилбутил)(метилсульфонил)амино] - 1S-(фенилметил)пропил] -2S-[(2-хинолинилкарбонил)амино] бутандиамид получали в виде белого твердого вещества. Аналитически рассчит. для C30H39N5O6S•0,1H2O: C 58,52; H 6,71; N 11,37. Найдено: C 58,34; H 6,35; N 11,13.
Пример 11B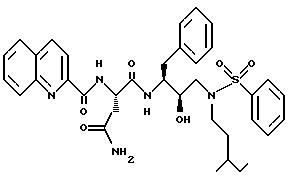
Получение N1-[2R-гидрокси-3-[(3-метилбутил(фенилсульфонил)амино] - 1S-(фенилметил)пропил]-2S-[(2-хинолинилкарбонил) амино]бутандиамида
Часть A:
CBZ-защищенное соединение фенилметил[2R-гидрокси-3-[(3- метилбутил)(фенилсульфонил)амино] -1S- (фенилметил)пропил] карбамат /200 мг, 0,38 ммоля/ подвергали снятию защиты гидрированием над 10% палладием на угле и получали продукт в виде масла.
Часть B:
Свободный амин из части A взаимодействовал с N-CBZ-L-аспарагином /109 мг, 0,41 ммоля/ в присутствии N-гидроксибензотриазола /63 мг, 0,41 ммоля/ в EDC /77 мг, 0,40 ммоля/, давая фенилметил[3-амино-1S-[[2R-гидрокси-3-[(3- метилбутил)(фенилсульфонил)амино]-1S-(фенилметил)амино]карбонил]-3- оксопропил]карбамат в виде белого твердого вещества. Аналит. рассчит. для C23H42N4O7S: C 62,05; H 6,63; N 8,77. Найдено: C 61,86; H 6,60; N 8,64.
Часть C:
С продукта части B (110 мг, 0,17 ммоля) защиту снимали гидрированием над 10% палладием на угле, получая продукт в виде масла.
Часть D:
Полученный свободный амин взаимодействовал со сложным эфиром N-гидроксисукцинимида и 2-хинолинкарбоновой кислоты /45 мг, 0,17 ммоля/, давая N1-[2R-гидрокси-3-[(3-метилбутил)(фенилсульфонил)амино]- 1S-(фенилметил)пропил] -2S-[(2-хинолинилкарбонил)амино]бутандиамид в виде белого твердого вещества. Аналит. рассчит. для C35H41N5O6S: C 63,71; H 6,26; N 10,61. Найдено: C 63,69; H 6,42; N 10,42.
Пример 12A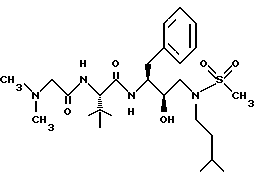
Получение 2S-[[(диметиламино)ацетил] амино] -N-[2R-гидрокси-3-[(3- метилбутил)метилсульфонил) амино]-1S-(фенилметил)пропил]-3,3-диметилбутанамида
Часть A:
К раствору N-CBZ-L-трет-лейцина /100 мг, 0,38 ммоля/ и N-гидроксибензотриазола /52 мг, 0,34 ммоля/ в DMF /3 мл/ добавляли EDC /65 мг, 0,34 ммоля/. Раствор перемешивали 60 минут при комнатной температуре и затем добавляли продукт примера 10, части A /105 мг, 0,32 ммоля/ в DMF /2 мл/. Реакционную смесь перемешивали в течение 16 часов при комнатной температуре, затем выливали в 50%-насыщенный раствор бикарбоната натря /50 мл/. Водную смесь экстрагировали дважды этилацетатом /25 мл/. Объединенные этилацетатные слои промывали водой /25 мл/ и сушили над сульфатом магния. Фильтрование и концентрирование дали масло, которое хроматографировали на силикагеле /50 г/ с элюированием 2,5% метанолом в дихлорметане. Фенилметил[1S-[[[2R-гидрокси-3-[(3-метилбутил)-(метилсульфонил)амино] - 1S-(фенилметил)пропил]амино]-карбонил]-2,2-диметилпропил]карбамат получали в виде липкого твердого вещества. Аналит. рассчит. для C30H45N3O6•2H2O: C 58,55; H 8,09; N 6,83. Найдено: C 58,38; H 7,77; N 7,10.
Часть B:
Раствор фенилметил[1S-[[[2R-гидрокси -3-[(3-метилбутил)- (метилсульфонил) амино]-1S-(фенилметил)пропил] амино]карбонил]-2,2- диметилпропил] карбамата /100 мг, 0,17 ммоля/ в метаноле /10 мл/ гидрировали над 10% палладием на угле в течение 2 часов. Реакционную смесь фильтровали через диатомову землю и концентрировали до масла.
Часть C:
N, N-диметилглицин /20 мг, 0,19 ммоля/, N-гидроксибензотриазол /28 мг, 0,18 ммоля/ и EDC /35 мг, 0,18 ммоля/ перемешивали в DMF /4 мл/ при комнатной температуре 40 минут. Продукт из части B в DMF /4 мл/ добавляли и реакционную смесь перемешивали в течение 16 часов, затем выливали в 50%-насыщенный бикарбонат натрия /50 мл/. Водную смесь экстрагировали трижды дихлорметаном /30 мл/, который в свою очередь промывали водой /30 мл/ и сушили над сульфатом магния. Фильтрование и концентрирование дали масло. Его хроматографировали на силикагеле /50 г/, элюируя сначала 2,5% метанолом в дихлорметане /400 мл/, а затем 5% метанолом в дихлорметане. 2S-[[(диметиламино)ацетил] амино]-N-[2R-гидрокси-3-[(3- метилбутил)метилсульфонил) амино] -1S(фенилметил)пропил]- 3,3-диметилбутанамид получали в виде белого твердого вещества. Аналит. рассчит. для C26H46N4O5S•CH2Cl2: C 56,04; H 8,34; N 9,87. Найдено: C 56,06; H 8,36; N 9,70.
Пример 12B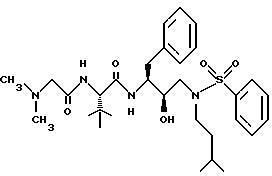
Получение 2S-[[(диметиламино)ацетил] амино] -N-[2R-гидрокси-3-[(3- метилбутил)(фенилсульфонил)амино] -1S-(фенилметил)пропил] -3,3- диметилбутанамида
Часть A:
К раствору N-CBZ-L-трет-лейцина /450 мг, 1,7 ммоля/ и N-гидроксибензотриазола /260 мг, 1,7 ммоля/ в DMF /10 мл/ добавляли EDC /дихлорэтан/ /307 мг, 1,6 ммоля/. Раствор перемешивали 60 минут при комнатной температуре и затем добавляли продукт примера 11, части A /585 мг, 1,5 ммоля/ в DMF /2 мл/. Реакционную смесь перемешивали в течение 16 часов при комнатной температуре, затем выливали в 50%-насыщенный раствор бикарбоната натрия /200 мл/. Водную смесь экстрагировали трижды этилацетатом /50 мл/. Объединенные этилацетатные слои промывали водой /50 мл/ и насыщенным раствором NaCl /50 мл/, затем сушили над сульфатом магния. Фильтрование и концентрирование дали масло, которое хроматографировали на силикагеле /50 г/ с элюцией 20% этилацетатом в гексане. Феннилметил[1S-[[[2R-гидрокси -3-[(3-метилбутил) (фенилсульфонил)амино]- 1S-(фенилметил)пропил] амино]карбонил]-2,2 -диметилпропил] карбамат получали в виде твердого вещества. Аналит. рассчитано для C35H47N3O6S: C 65,91; H 7,43; N 6,59. Определено: C 65,42; H 7,24; N 6,55.
Часть B:
Раствор фенилметил[1S-[[[2R-гидрокси-3- [(3-метилбутил) (фенилсульфонил) аминно] -1S-(фенилметил)пропил] амино]карбонил]-2,2- диметилпропил]карбамата /200 мг, 0,31 ммоля/ в метаноле /15 мл/ гидрировали над 10% палладием на угле 2 часа. Реакционную смесь фильтровали через диатомову землю и концентрировали до масла.
Часть C:
Полученный свободный амин из части B /150 мг, 0,3 ммоля/ соединяли с диизопропилэтиламином /114 мкл, 0,33 ммоля/ в дихлорметане /5 мл/. К смеси добавляли бромацетилхлорид /27 мкл, 0,33 ммоля/ (по каплям). Реакционную смесь перемешивали в течение 30 минут при комнатной температуре, затем разбавляли дихлорметаном /30 мл/ и экстрагировали 1 N HCl и затем насыщенным раствором NaCl /по 25 мл каждого/. Органический раствора сушили над сульфатом магния и концентрировали до твердого вещества. 2S-[[бромацетил]амино] -N-[2R- гидрокси-3-[(3-метилбутил) (фенилсульфонил)аминно]-1S-(фенилметил) пропил] -3,3-диметилбутанамид был достаточно чистым для применения в следующей стадии. Этот материал можно также получить с заменой бромуксусного ангидрида на бромацетилхлорид, также можно использовать хлорацетилхлорид или хлоруксусный ангидрид.
Часть D:
Продукт части C растворяли в дихлорметане /5 мл/ и добавляли диизопропилэтиламин /114 мкл, 0,66 ммоля/ и диметиламингидрохлорид /53 мг, 0,66 ммоля/. Реакцию перемешивали в течение 18 часов, затем концентрировали под током азота до приблизительно 1 мл. Остаток хроматографировали на силикагеле /50 г/ с применением 2% метанола в дихлорметане. 2S-[(диметиламино)ацетил] амино] -N-[2R-гидрокси-3-[(3- метилбутил)(фенилсульфонил) амино] -1S-(фенилметил)пропил] -3,3- диметилбутанамид получали в виде твердого вещества. Анал. рассчит. для C31H48N4O5S: C 63,24; H 8,22; N 9,52. Определено: C 63,03; H 8,01; N 9,40.
Пример 12C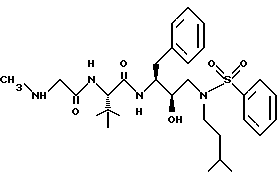
Получение 2S-[[(метиламино)ацетил] амино]-N-[2R-гидрокси-3-[(3- метилбутил)(фенилсульфонил) амино]-1S-(фенилметил) пропил]-3,3-метилбутанамида
2S-[[Бромацетил]амино]-N-[2R-гидрокси -3-[(3-метилбутил) (фенилсульфонил)амино] -1S-(фенилметил)пропил]-3,3-диметилбутанамид /103 мг, 0,16 ммоля/ и 40% водный метиламин /42 мкл, 0,49 ммоля/ соединяли в этаноле /2 мл/ и перемешивали при комнатной температуре в течение 24 часов. Реакционную смесь концентрировали досуха и растирали с эфиром. Твердый материал удаляли фильтрованием, а фильтрат концентрировали до масла. Масло хроматографировали на силикагеле /50 г/ с применением 4% метанола в дихлорметане. 2S-[[(метиламино)ацетил] амино]-N-[2R-гидрокси-3-[(3-метилбутил) (фенилсульфонил) амино]-1S-(фенилметил) пропил]-3,3-диметилбутанамид получали в виде твердого вещества. Анал. рассчит. для C30H46N4O5S: C 62,69; H 8,07; N 9,75. Определено: C 62,38; H 8,14; N 9,60.
Пример 12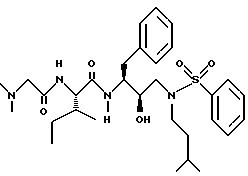
Получение 2S-[[(диметиламино)ацетил] амино] -N-[2R-гидрокси-3-[(3- метилбутил)(фенилсульфонил) амино]-1S-(фенилметил)пропил] -3S-метилпентанамида
Часть A:
К раствору амина /продукта примера 11, части A/ /2,79 г, 7,1 ммоля/ в 27 мл диоксана добавляли сложный эфир N-t-бутилкарбонил-L-изолейцин-N-гидроксисукцинимида и реакцию перемешивали в атмосфере азота в течение 16 часов. Содержимое концентрировали в вакууме и остаток растворяли в этилацетате, промывали 5% водным кислым сульфатом калия, насыщенным раствором бикарбоната натрия и насыщенным раствором хлорида натрия. Органический слой сушили над сульфатом магния, фильтровали и концентрировали, получая 4,3 г неочищенного материала, который хроматографировали с применением смеси этилацетат:гексан /3: 1/, получая 3,05 г, 72% 2S-[[(1,1-диметилэтокси)карбонил] амино]-N-[2R- гидрокси-3-[(3-метилбутил)(фенилсульфонил) амино] -1S-(фенилметил) пропил]- 3S-метилпентанамида.
Часть B:
/3,05 г, 5,0 ммолей/ продукта из части A растворяли в 20 мл 4 N HCl в диоксане и перемешивали в атмосфере азота 1,5 часа. Содержимое концентрировали в вакууме и растирали с диэтиловым эфиром. Неочищенную гидрохлоридную соль откачивали при 1 мм Hg досуха, получая 2,54 г продукта в виде гидрохлорида.
Часть C:
(2,54, 5,0 ммолей) гидрохлорида амина растворяли в 50 мл тетрагидрофурана и добавляли 4-метилморфолин /1,01 г, 10 ммолей/, получая осадок. К этой суспензии добавляли хлоруксусный ангидрид /0,865 г, 5,0 ммолей/ и перемешивали в течение 40 минут. Содержимое концентрировали в вакууме и остаток разделяли в этилацетате /200 мл/ и 5% KHSO4. Органический слой промывали раствором насыщенного бикарбоната натрия и насыщенного хлорида натрия, сушили над сульфатом магния, фильтровали и концентрировали, получая неочищенный продукт. Очистка на силикагеле с применением в качестве элюента смеси этилацетат:гексан /1:1/ дала 1,89 г чистого хлорацетамида.
Часть D:
К раствору хлорацетамида /1,89 г, 3,2 ммоля/ из части C в 25 мл тетрагидрофурана добавляли 4,0 мл 50% водного диметиламина и раствор перемешивали в течение 1 часа. Раствор концентрировали в вакууме и остаток растворяли в этилацетате и промывали водой. Органический слой сушили над сульфатом магния, фильтровали и концентрировали, получая неочищенный продукт, который очищали кристаллизацией из этилацетата и изооктана, получая 1,80 г /88% выход/, т. пл. 121-122oC, H-ЯМР, масс-спектр: рассчит. 589,3424, определено 589,3405.
Пример 12E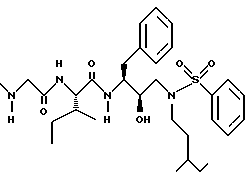
Получение 2S-[[(метиламино)ацетил] амино]-N-[2R-гидрокси-3-[(3- метилбутил)(фенилсульфонил) амино]-1S-(фенилметил) пропил-3S-метилпентанамида
К раствору хлорацетамида примера 12D, части C /2,36 г, 4,0 ммоля/ в тетрагидрофуране /25 мл/ добавляли 3 мл водного метиламина /40 вес.%/ и реакцию перемешивали в течение 1 часа. Содержимое концентрировали и остаток разделяли между этилацетатом /100 мл/ и водой /100 мл/. Органический слой сушили над сульфатом магния, фильтровали и концентрировали, получая неочищенный продукт, который очищали перекристаллизацией из смеси этилацетат/гептан; /M + H/ 575, ЯМР: определено 575,3267.
Пример 12F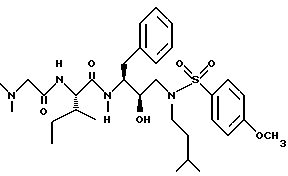
Получение 2S-[[(диметиламино)ацетил] амино] -N-[2R-гидрокси-3-[(3- метилпропил)(4-метоксифенилсульфонил) амино] -1S-(фенилметил)пропил] -3S- метилпентанамида
Часть A:
К раствору 2R-гидрокси-3- [(2-метилпропил)(4-метоксифенилсульфонил) амино] -1S-пропиламина /1,70 г, 4,18 ммоля/ в 40 мл дихлорметана добавляли эфир N-карбобензилокси-L- изолейцин-N-гидроксисукцинамида /1,51 г, 4,18 ммоля/ и раствор перемешивали в атмосфере азота в течение 16 часов. Содержимое концентрировали в вакууме и остаток перерастворяли в этилацетате. Раствор в этилацетате промывали водным раствором 5% KHSO4, насыщенным раствором бикарбоната натрия и насыщенным раствором хлорида натрия, сушили над сульфатом магния, фильтровали и концентрировали, получая 2,47 г неочищенного продукта. Продукт очищали хроматографически на силикагеле с применением в качестве элюента смеси гексан/этилацетат /12: 1/, получая 2,3 г /выход 84%/ 2-[(карбобензилокси)амино] -N-[2-гидрокси-3-[(3-метилпропил)(4- метоксифенилсульфонил)амино]-1-(фенилметил)пропил] -3-метилпентанамид, [4-(R*,S*,S*,)].
1,18 г /1,8 ммоля/ продукта части A растворяли в 50 мл метанола и к раствору добавляли 250 мг 10% палладия на угле под током азота. Суспензию гидрировали при 50 пси /50 • 6894, 757 Па/ водорода в течение 20 часов. Содержимое продували азотом и фильтровали через целит /броунмеллерит/ и концентрировали в вакууме, получая 935 мг 2S-(амино)-N-[2R-гидрокси-3-[(3 -метилпропил)(4-метоксифенилсульфонил) амино]-1S-(фенилметил) пропил]-3S-метилпентанамида, который использовали без дальнейшей очистки.
Часть C:
0,935 г /1,8 ммоля/ амина из части B растворяли в 15 мл диоксана и к раствору добавляли 4-метилморфолин /190 мг, 1,85 ммоля/ и затем хлоруксусный ангидрид /0,315 г, 1,8 ммоля/. Реакционную смесь перемешивали в атмосфере азота в течение 3 часов, концентрировали в вакууме и вновь растворяли в этилацетате. Раствор в этилацетате промывали 50 мл 5% водного KHSO4, насыщенным раствором NaHCO3 и насыщенным раствором NaCl, сушили над сульфатом магния, фильтровали и концентрировали, получая 613 мг /выход 68%/ 2S-[(хлорацетил)амино] -N-[2R- гидрокси-3-[(3-метилпропил)(4- метоксифенилсульфонил) амино] -1S-(фенилметил)пропил]-3S-метилпентанамида после очистки хроматографией на силикагеле с применением смеси гексан:этилацетат /1:1/.
Часть D:
К раствору хлорацетамида из части C /673 мг, 1,10 ммоля/ в 20 мл тетрагидрофурана добавляли 5 мл 50 вес.% водного диметиламина и раствор перемешивали в течение 1 часа. Раствор концентрировали и остаток вновь растворяли в 50 мл этилацетата и промывали 25 мл воды. Слой этилацетата сушили над сульфатом магния, фильтровали и концентрировали, получая неочищенное твердое вещество, которое очищали на колонке силикагеля с применением элюента 97:3 /дихлорметан: метанол/, получая 400 мг 2S-[[(диметиламино)ацетил] амино]-N-[2R-гидрокси-3-[(3-метилпропил)(4- метоксифенилсульфонил) амино] -1-(фенилметил)пропил] -3-метилпентанамида.
Пример 13A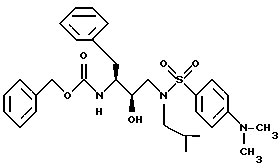
Получение фенилметилового эфира [2R-гидрокси-3-[[(4-диметиламинофенил)сульфонил] (2-метилпропил)амино] - 1S-(фенилметил)пропил] карбаминовой кислоты
К раствору 100 мг /0,19 ммоля/ фенилметилового эфира [2R-гидрокси-3-[[(4-фторфенил)сульфонил](2-метилпропил)амино]- 1S-(фенилметил)пропил]карбаминовой кислоты в 1 мл пиридина добавляли 53 мл триэтиламина и 120 мкл /0,95 ммоля/ 40% водного диметиламина. После нагревания в течение 24 часов при 100oC раствор охлаждали, добавляли этилацетат, промывали 5% лимонной кислотой, насыщенным раствором бикарбоната натрия, сушили над сульфатом магния, фильтровали и концентрировали. Оставшееся твердое вещество перекристаллизовывали из этилацетата/гексана, получая 10 мг целевого продукта; масс-спектр m/e = 540 /M+H/.
Пример 13B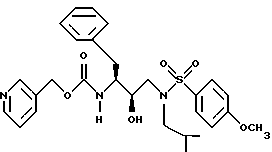
Получение 3-пиридилметилового эфира [2R-гидрокси-3-[[(4-метоксифенил)сульфонил] (2 -метилпропил)амино]- 1S-(фенилметил) пропил]-карбаминовой кислоты
Часть A:
Раствор N-бензилоксикарбонил-3S-амино-1,2-S-эпокси-4-фенилбутана /50 г, 0,168 моля/ и изобутиламина /246 г, 3,24 моля/ в 650 мл изопропанола нагревали с обратным холодильником в течение 1,25 часов. Раствор охлаждали до комнатной температуры, концентрировали в вакууме и затем выливали в 1 л перемешиваемого гексана, после чего продукт кристаллизовался из раствора, собирали его и сушили на воздухе, получая 57,6 г N-[3S-бензилоксикарбониламино-2R-гидрокси-4-фенил] -N-изобутиламина, т.пл. 108-109,5oC, масс-спектр m/e = 371 /M + H/.
Часть B:
Амин из части A /1,11 г, 3,0 ммоля/ и триэтиламин /324 мг, 3,20 ммоля/ в 20 мл метиленхлорида обрабатывали 715 мг /3,46 ммоля/ 4-метоксибензолсульфохлорида. Раствор перемешивали при комнатной температуре в течение 6 часов, концентрировали, растворяли в этилацетате, затем промывали 1 N кислым сульфатом калия, насыщенным раствором бикарбоната натрия, солевым раствором, сушили над сульфатом магния, фильтровали и концентрировали, получая прозрачное масло. Его перекристаллизовывали из диэтилового эфира, получая 1,27 г фенилметилового эфира [2R-гидрокси-3-[[(4-метоксифенил)сульфонил] (2- метилпропил)амино]-1S-(фенилметил)пропил]-карбаминовой кислоты.
Часть C:
Раствор 930 мг /3,20 ммоля/ продукта части B в 30 мл метанола гидрировали в присутствии 70 мг 10% палладия на угольном катализаторе при 40 psig /40 • 6894, 757 Па/ в течение 17 часов, катализатор удаляли фильтрованием и раствор концентрировали, получая 704 мг [2R-гидрокси-3-[[(4-метоксифениил)сульфонил] (2-метилпропил)амино]- 1S-(фенилметил)пропиламина, масс-спектр m/e = 407 /M + H/, который был использован на следующей стадии без очистки.
Часть D:
К раствору 2,5 г /22,9 ммоля/ 3-пиридилкарбинола в 100 мл безводного ацетонитрила добавляли 8,8 г /34,4 ммоля/ N,N-дисукцинимидилкарбоната и 5,55 мл /68,7 ммоля/ пиридина. Раствор перемешивали в течение 1 часа и затем концентрировали в вакууме. Остаток растворяли в этилацетате, затем промывали насыщенным раствором бикарбоната натрия, солевым раствором сушили над сульфатом магния, фильтровали и концентрировали, получая 5,3 г N-гидроксисукцинимид-3-пиридилметилкарбоната, масс-спектр m/e = 251 /M + H/, который использовали сразу же в следующей стадии без очистки.
Часть E:
К раствору амина из части C /2,87 г, 7,0 ммолей/ и 1,38 мл триэтиламина в 24 мл безводного метиленхлорида добавляли раствор 1,65 г /6,6 ммоля/ N-гидроксисукцинимид-3-пиридилкарбоната из части D в 24 мл метиленхлорида. Раствор перемешивали в течение 1 часа, добавляли 100 мл метиленхлорида, затем промывали насыщенным раствором бикарбоната натрия, солевым раствором, сушили над сульфатом магния, фильтровали и концентрировали, получая 3,69 г неочищенного продукта. Хроматография на силикагеле с применением смеси 2% метанол/метиленхлорид давала 3,27 г 3-пиридилметилового эфира [2R-гидрокси-3-[[4- метоксифенилсульфонил] (2-метилпропил)амино]- 1S-(фенилметил)пропил]-карбаминовой кислоты, масс-спектр m/e = 548 /M + Li/.
Пример 13C
Получение 3-пиридилметилового эфира [2R-гидрокси-3- [(фенилсульфонил)(2-метилпропил)амино] - 1S-(фенилметил)пропил]-карбаминовой кислоты
Часть A:
Раствор N-бензилоксикарбонил-3S-амино-1,2 -S-эпокси-4-фенилбутана /50 г, 0,168 моля/ и изобутиламина /246 г, 3,24 моля/ в 650 мл изопропилового спирта нагревали с обратным холодильником в течение 1,25 часов. Раствор охлаждали до комнатной температуры, концентрировали в вакууме и затем выливали в 1 л перемешиваемого гексана, после чего продукт кристаллизовали из раствора, собирали и сушили на воздухе, получая 57,6 г N-[3S-бензилоксикарбониламино-2R -гидрокси-4-фенил]-N-изобутиламина, т.пл. 108-109,5oC, масс-спектр m/e = 371 /M + H/.
Часть B:
Амин из части A /0,94 г, 2,5 ммоля/ и триэтиламин /288 мг, 2,85 ммоля/ в 20 мл метиленхлорида обрабатывали 461 мг /2,61 ммоля/ бензолсульфохлоридом. Раствор перемешивали при комнатной температуре в течение 16 часов, концентрировали, растворяли в этилацетате, затем промывали 1 N кислым сульфатом калия, насыщенным раствором бикарбоната натрия, солевым раствором, сушили над сульфатом магния, фильтровали и концентрировали, получая прозрачное масло. Его перекристаллизовывали из диэтилового эфира и гексана, получая 0,73 г фенилметилового эфира [2R-гидрокси-3-[(фенилсульфонил)(2- метилпропил)амино] -1S-(фенилметил)пропил] -карбаминовой кислоты, т.пл. 95-99oC масс-спектр m/e = 511 /M + H/.
Часть C:
Раствор 500 мг фенилметилового эфира [2R-гидрокси-3-[(фенилсульфонил)(2-метилпропил)амино] - 1S-(фенилметил)пропил]-карбаминовой кислоты в 20 мл метанола гидрировали в присутствии 250 мг 10% палладия на угольном катализаторе при 40 пси в течение 3 часов, катализатор удаляли фильтрованием и раствор концентрировали, получая 352 г [2R-гидрокси-3-[(фенилсульфонил)(2 -метилпропил)амино]- 1S-(фенилметил)пропиламина, масс-спектр m/e = 377 /M + H/, который использовали сразу без очистки в следующей стадии.
Часть D:
К раствору 1,24 ммоля 5-норборнен-2,3-дикарбоксиимидокарбонохлоридата /Henklein P., et al., Synthesis 1987, 166-167/ в 1 мл безводного метиленхлорида добавляли раствор 43 мкл /2,44 ммоля/ 3-пииридилкарбинола в 129 мкл /1,6 ммоля/ пиридина в 1 мл метиленхлорида при 0oC под азотом. После 4 часов при комнатной температуре добавляли 150 мг /0,4 ммоля/ [2R-гидрокси-3-[(фенилсульфонил)(2 -метилпропил)амино]- 1S-(фенилметил)пропиламина из части C и 100 мкл пиридина. После перемешивания в течение 15 часов при комнатной температуре добавляли этилацетат, затем промывали 1 N HCl, насыщенным раствором бикарбоната натрия, солевым раствором, сушили над сульфатом магния, фильтровали и концентрировали, получая 175 мг неочищенного продукта. Хроматография на силикагеле с применением смеси 1% метанол/метиленхлорид дала 69 мг чистого 3-пиридилметилового эфира [2R-гидрокси-3-[(фенилсульфонил)(2- метилпропил)амино] -1S-(фенилметил)пропил]-карбаминовой кислоты, масс-спектр m/e = 512,2267 /M + H/; рассчит. для C27H33N3O5S: 512,2219.
Пример 13D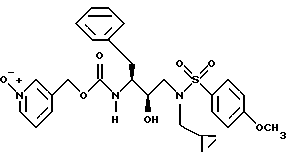
Получение 3-пиридилметилового эфира [2R-гидрокси-3-[[(4-метоксифенил) сульфонил] (2-метилпиридил)амино] - 1S-(фенилметил)пропил]-карбаминовой кислоты, N-оксида.
К раствору 211 мг /0,39 ммоля/ 3-пиридилметилового эфира [2R-гидрокси-3-[[(4- метоксифенил)сульфонил](2-метилпропил)амино]- 1S-(фенилметил)пропил] -карбаминовой кислоты в 5 мл метиленхлорида при 0oC добавляли 500 мг 50% 3-хлорпербензойной кислоты. После перемешивания при комнатной температуре в течение 1 часа добавляли этилацетат, раствор промывали насыщенным бикарбонатом натрия, 0,2 N раствором гидроксида аммония и солевым раствором, сушили над сульфатом магния, фильтровали и концентрировали, получая 200 мг неочищенного продукта. Его хроматографировали на C18 материале (колонке) с обращенной фазой с применением смеси 20-40% ацетонитрил/вода, затем 100% ацетонитрила, получая 90 мг целевого продукта, который затем перекристаллизовывали из смеси этилацетат/изооктан, получая 34 мг названного в заглавии примера соединения в чистом виде. Масс-спектр m/e = 564 /M + Li/.
Пример 13E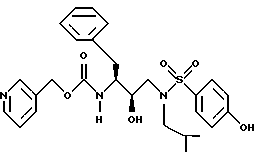
Получение 3-пиридилметилового эфира [2R-гидрокси-3-[[(4-гидроксифенил)сульфонил] (2-метилпропил)амино] - 1S-(фенилметил)пропил] -карбаминовой кислоты
Часть A:
Раствор 0,98 г (1,85 ммоля) фенилметилового эфира
[2R-гидрокси-3-[[(4-фторфенил)сульфонил] (2-метилпропил)амино] - 1S-(фенилметил)пропил]-карбаминовой кислоты в 3,8 мл безводного DMF добавляли к 22 мг /7,4 ммоля/ 80% гидрида натрия в 2 мл DMF. К этой смеси добавляли 0,40 г /3,7 ммоля/ бензилового спирта. После 2 часов раствор охлаждали до 0oC, добавляли воду и затем этилацетат. Органический слой промывали 5% лимонной кислотой, насыщенным раствором бикарбоната натрия и солевым раствором и сушили над сульфатом магния. Фильтровали и концентрировали, получая 0,90 г неочищенного материала. Его хроматографировали на основном оксиде алюминия с применением смеси 3% метанол/метиленхлорид, получая 0,70 г циклического карбамата [2R-гидрокси-3-[(2-метилпропил)(4- гидроксифенил)сульфонил] амино-1S-(фенилметил)пропиламина. Масс-спектр m/e = 509 /M + H/.
Часть B:
К раствору 0,66 г /1,28 ммоля/ циклического карбамата из части A в 15 мл этанола добавляли 2,6 мл /6,4 ммоля/ 2,5 гидроксида натрия. После 1 часа нагревания с обратным холодильником добавляли 4 мл воды и раствор кипятили с обратным холодильником еще 8 часов. Летучие вещества удаляли, добавляли этилацетат и промывали водой, солевым раствором, сушили над сульфатом магния. Затем фильтровали и концентрировали, получая 550 мг неочищенного 2R-гидрокси-3-[(2-метилпропил)(4-гидроксифенил) сульфонил]амино- 1S-(фенилметил)пропиламина.
Часть C:
Раствор неочищенного 2R-гидрокси-3-[(2-метилпропил)(4- бензилоксифенил)сульфонил] амино-1S-(фенилметил)пропиламина в 10 мл этанола гидрировали в присутствии 500 мг 10% палладия на угольном катализаторе при 50 psig /50 • 6894, 757 Па/ водорода в течение 2 часов. Катализатор удаляли фильтрованием и растворитель удаляли в вакууме, получая 330 мг 2R-гидрокси-3-[(2-метилпропил)(4- гидроксифенил)сульфонил]амино- 1S-(фенилметил)пропиламина, масс-спектр m/e = 393 /M + H/.
Часть D:
К раствору 320 мг /0,82 ммоля/ амина из части C в 6 мл DMF добавляли 192 мг /0,76 ммоля/ N-гидроксисукцинимид-3-пиридилметилкарбоната. После 15 часов при комнатной температуре DMF удаляли в вакууме, добавляя этилацетат, промывали водой, солевым раствором, сушили над сульфатом магния, фильтровали и концентрировали, получая 390 мг неочищенного материала, хроматография на силикагеле с применением смеси 50-80% этилацетат/гексан дала 180 мг 3-пиридилметилового эфира [2R-гидрокси-3-[[(4-гидроксифенил)сульфонил] (2 -метилпропил)амино] - 1S-(фенилметил)пропил]-карбаминовой кислоты, масс-спектр m/e = 528 /M + H/.
Пример 13F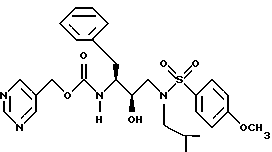
Получение 5-пиримидилметилового эфира [2R-гидрокси-3-[[(4-метоксифенил)сульфонил] (2-метилпропил)амино] - 1S-(фенилметил)пропил] -карбаминовой кислоты
К раствору 9,5 мг /0,09 ммоля/ 5-пиримидилкарбинола в 1 мл безводного ацетонитрила при комнатной температуре добавляли 24 мг /0,09 ммоля/ N,N-дисукцинимидилкарбоната и 19,1 мкл /0,24 ммоля/ пиридина. После перемешивания в течение 5 часов добавляли 32 мг /0,08 ммоля/ 2R-гидрокси-3-[(2-метилпропил)(4-метоксифенил)сульфонил] амино- 1S-(фенилметил)пропиламина и раствор перемешивали в течение 48 часов. После концентрирования в вакууме добавляли метиленхлорид, затем промывали смесью насыщенного раствора бикарбоната с солевым раствором /1:1/, сушили над сульфатом магния, фильтровали и концентрировали, получая 27 мг неочищенного продукта. Хроматография на силикагеле с применением смеси 2% метанол/метиленхлорид дала 22 мг целевого продукта, масс-спектр m/e = 543 /M + H/.
Пример 14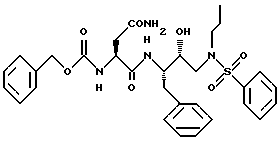
Получение фенилметил[3-амино-1S-[[2R-гидрокси-3-[(3- пропил)(фенилсульфонил)амино]-1S-(фенилметил)амино]карбонил]-3- оксопропил]карбамата
Фенилметил[2R-гидрокси-3-[(3-пропил)(фенилсульфонил)амино] - 1S-(фенилметил)пропил] карбамат /200 мг, 0,40 ммоля/ подвергали реакции снятия защитной группы гидрированием над 10% палладием на угле и образующийся свободный амин соединяли с N-CBZ-L-аспарагином /157 мг, 0,42 ммоля/ в присутствии N-гидроксибензотриазола /114 мг, 0,84 ммоля/ и EDC /130 мг, 0,67 ммоля/, получая фенилметил[3-амино-1S-[[2R-гидрокси-3-[(3-пропил)(фенилсульфонил)амино] - 1S-(фенилметиламино] карбонил] -3-оксопропил] карбамат в виде твердого вещества. Анал. рассчит. для C31H38N4O7S•0,2H2O: C 60,61; H 6,30; N 9,12. Определено: C 60,27; H 6,16; N 8,93.
Пример 15A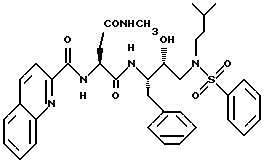
Получение N1[2R-гидрокси-3-[(3 -метилбутил)(фенилсульфонил) амино]-N4- метил-1S-(фенилметил)пропил]-2S-[(2- хинолинилкарбонил)амино]бутандиамида
Часть A:
N2-[(1,1-диметилэтокси) карбонил] -N-метил-L-аспарагин получали из α-бензилового эфира Boc-L-аспарагиновой кислоты /1,0 г, 3,09 ммоля/, метиламина•HCl /209 мг, 3,09 ммоля/, EDC /711 мг, 3,7 ммоля/, 1-гидроксибензотриазола /627 мг, 4,63 ммоля/ и N-метилморфолина /0,7 мл, 6,3 ммоля/ в DMF /20 мл/. После перемешивания в течение ночи при ком. температуре реакционную смесь разбавляли этилацетатом, промывали водой, насыщенным раствором бикарбоната натрия, 5% лимонной кислотой, солевым раствором, сушили над сульфатом магния и концентрировали до масла, масло растворяли в 20 мл сухого этанола и гидрировали в присутствии 10% (вес/вес) палладия на угле при атмосферном давлении и комнатной температуре в течение ночи. Смесь фильтровали через целит и концентрировали до белой твердой пены, 670 мг.
Часть B:
Раствор фенилметил[2R-гидрокси-3-[(3- метилбутил)(фенилсульфонил)амино]- 1S-(фенилметил)пропил] карбамата /310 мг, 0,59 ммоля/ в метаноле /10 мл/ гидрировали над 10% палладием на угле в течение 3 часов, фильтровали через диатомову землю и концентрировали, получая продукт в виде масла /214 мг/. Этот свободный амин /208 мг, 0,53 ммоля/ соединяли с N2-[(1,1-диметилэтокси) карбонил] -N-метил-L-аспарагином /137 мг, 0,76 ммоля/ и EDC /130 мг, 0,67 ммоля/, получая 290 мг N1(2R-гидрокси-3-[(3-метилбутил) (фенилсульфонил)амино] -N4-метил- 1S-(фенилметил) пропил]-2S-[(1,1-диметилэтоксикарбонил)амино]бутандиамида.
Часть C:
N1[2R-гидрокси-3-[(3-метилбутил)(фенилсульфонил) амино] -N4-метил- 1S-(фенилметил)пропил] -2S-[1,1-диметилэтоксикарбонил) амино] бутандиамид /270 мг, 0,43 ммоля/ перемешивали в 4 N HCl в диоксане /5 мл/ при комнатной температуре в течение 0,5 часов. Растворитель и избыток реагента упаривали досуха. Продукт сушили в вакууме. Этот материал /125 мг, 0,225 ммоля/ реагировали затем с эфиром N-гидроксисукцинимида и 2-хинолинкарбоновой кислоты /61 мг, 0,225 ммоля/, N-метилморфолином /50 мкл, 0,45 ммоля/ в метиленхлориде /2 мл/ в течение 3 часов. Продукт N1[2R-гидрокси-3-[(3-метилбутил) (фенилсульфонил)амино] -N4-метил- 1S-(фенилметил)пропил]-2S-[(2- хинолинилкарбонил)амино]бутандиамид очищали хроматографией на силикагеле. Анал. расчит. для C36H43N5O6S•0,2H2O: C 63,83; H 6,45; N 10,34. Определено: C 63,64; H 6,40; N 10,34.
Пример 15B
Согласно описанным выше способам было получено также следующее соединение: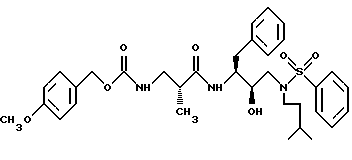
Получение (4-метоксифенил)метилового эфира [3-[[2-гидрокси-3-[(3-метилбутил)(фенилсульфонил) амино]- 1-(фенилметил)пропил] амино]-2-метил-3-оксопропил]-карбаминовой кислоты, [1S-[1R*(S*),2S*]]
4,10 г /7,8 ммоля/ фенилметилового эфира [2R-гидрокси-3-[(3-метилбутил) (фенилсульфонил)амино]- 1S-(фенилметил)пропил]-карбаминовой кислоты, [R-(R*, S*)] гидрогенировали в растворе метанола и этанола с применением каталитического 20% Pd/C при избыточном давлении 50 пси водорода в течение 3 часов. Катализатор отфильтровывали и растворители удаляли в вакууме, получая 3,0 г свободного амина.
В отдельной колбе 2,09 г /7,8 ммоля/ N-Moz-AMBA добавляли к 10 мл диметилформамида и 1,58 г /1,5 экв./ N-гидроксибензотриазола и раствор перемешивали в течение 30 минут. К нему добавляли свободный амин в 10 мл диметилформамида и реакционную смесь перемешивали в течение 20 часов. Растворитель удаляли выпариванием и неочищенный материал разделяли между этилацетатом и насыщенным водным раствором бикарбоната натрия. Этилацетатный слой промывали 5% кислым сульфатом калия и солевым раствором, сушили над сульфатом магния, фильтровали и концентрировали, получая 2,58 г чистого продукта после перекристаллизации из этилацетата, эфира и гексана, выход 52%.
Пример 16
Согласно способам примеров 1-15 были получены соединения, представленные в таблице 3.
Пример 17
Соединения данного изобретения являются эффективными ингибиторами протеазы ВИЧ. При помощи ферментного текста, описанного ниже, соединения, представленные в описанных здесь примерах, ингибировали фермент ВИЧ. Предпочтительные соединения данного изобретения и их рассчитанные величины IC50 /ингибирующие концентрации 50%, т.е. концентрации, при которых ингибиторующее соединение уменьшает активность фермента на 50%/ представлены в таблице 16. Способ определения активности фермента описан ниже. Субстратом является 2-Ile-Nle-Phe(p-NO2)-Gln-Arg-NH2. Положительный контроль - MVT-101 /Miller M., et al., Science, 246, 1149 /1989//. Условия теста следующие:
Тест-буфер: 20 мМ фосфат натрия, pH 6,4
20% глицерин
1 мМ ЭДТА
1 мМ ДТГ
0,1% CHAPS
Указанный выше субстрат растворяли в DMSO, затем разбавляли 10 раз тест-буфером. Конечная концентрация субстрата в тесте составляла 80 мкМ.
Протеазу ВИЧ разбавляли тест-буфером до конечной концентрации фермента 12,3 нМ на основе мол. массы 10780.
Конечная концентрация DMSO равна 14% и конечная концентрация глицерина равна 18%. Тестируемое соединение растворяли в DMSO и разводили DMSO до 10 х тест-концентрации; добавляли 10 мкл препарата фермента, материалы смешивали и затем выдерживали эту смесь при температуре окружающей среды в течение 15 минут. Ферментную реакцию инициировали добавлением 40 мкл субстрата. Увеличение флуоресценции наблюдали в 4 временных точках /0,8, 16 и 24 минуты/ при температуре окружающей среды. Каждое определение проводили дважды /в двух ячейках/.
Предшествующие примеры можно повторить с тем же успехом с заменой описанных в общем виде или специфических реагентом и /или/ применяемых условий на реагенты и условий, использованных в предшествующих примерах.
Пример 18
Эффективность соединений, приведенных в таблице 15 определяли согласно описанному выше ферментному тесту и CEM-клеточному тесту.
Способ определения ингибирования ВИЧ инфицированных клеток представляет собой автоматизированный колориметрический тест на основе тетразолия, описанный Pauwles et al., J. Virol. Methods, 20, 309-321 /1988/. Тесты проводили в планшетах с 96-ячейками для культуры ткани. CEM-клетки, CD4+-клеточная линия, выращенная в RPM1-1640 среде /Gibco/ с добавлением 10% плодной сыворотки теленка и затем обработанная полибреном /2 мкг/мл/. По 80 мкл среды, содержащей 1•104 клеток, заливали в каждую ячейку планшета для культуры ткани. В каждую ячейку добавляли 100 мкл тестируемого соединения, растворенного в культуральной среде /или среды без тестируемого соединения в качестве контроля/, получая желаемую конечную концентрацию, и клетки инкубировали при 37oC в течение 1 часа. Замороженную культуру ВИЧ-1 разводили культуральной средой до концентрации 5•104 TCID50 на мл /TCID50 = дозе вируса, которая инфицирует 50% клеток в культуре ткани/ и 20 мкл этой пробы вируса /содержащих 1000 TCID50 вируса/ добавляли в ячейки, содержащие тестируемое соединение, и в ячейки, содержащие только среду /инфицированные контрольные клетки/. Несколько ячеек получали культуральную среду без вируса /неинфицированные контрольные клетки/. Подобным образом собственную внутреннюю токсичность тестируемого соединения определяли путем добавления среды без вируса в несколько ячеек, содержащих тестируемое соединение. В целом планшеты для культуры ткани содержали следующие варианты опытов (см. таблицу А в конце описания).
В экспериментах 2 и 4 конечные концентрации тестируемых соединений были 1, 10, 100 и 500 мкг/мл. В положительный лекарственный контроль включали либо азидотимидин /AZT/, либо дидезоксиинозин /ddI/. Тестируемые соединения растворяли в DMSO и разводили средой для культуры ткани таким образом, что конечная концентрация DMSO не превышала 1,5% в любом из случаев. DMSO добавляли к контрольным ячейкам в подходящей концентрации.
После добавления вируса клетки инкубировали при 37oC в увлажненной атмосфере 5% CO2 в течение 7 дней. Тестируемые соединения могли добавляться в дни 0, 2 и 5, если желательно. На 7-ой день после инфицирования клетки в каждой ячейке ресуспендировали и пробы по 100 мкл каждой клеточной суспензии брали для анализа. 20 мкл раствора 5 мг/мл 3-(4,5-диметилтиазол-2 -ил)-2,5-дифенилтетразолийбромида /MTT/ добавляли к каждой пробе 100 мкл клеточной суспензии и клетки инкубировали в течение 4 часов при 27oC в атмосфере 5% CO2. Во время этой инкубации MTT метаболически восстанавливался живыми клетками, приводя к образованию в клетке окрашенного продукта формазана. К каждой пробе добавляли 100 мкл 10% додецилсульфата натрия в 0,01 N HCl для лизиса клеток и пробы инкубировали в течение ночи. Поглощение при 590 нм определяли для каждой пробы при помощи считывающего устройства Molecular Device microplate. Величины поглощения для каждой серии ячеек сравнивали для оценки контрольной вирусной инфекции, ответной реакции неинфицированных контрольных клеток, а также оценки тестируемого соединения на его цитотоксичность и антивирусную эффективность.
Соединения данного изобретения являются эффективными антивирусными соединениями и, в частности, эффективными ингибиторами ретровирусов, как показано выше. Так, соединения данного изобретения являются эффективными ингибиторами протеазы ВИЧ. Предполагается, что эти соединения будут также ингибировать другие ретровирусы, такие как лентивирусы, в частности другие штаммы ВИЧ, например ВИЧ-2, вирус T-клеточного лейкоза человека, респираторно-синцитиальный вирус, вирус иммунодефицита обезьяноподобных, вирус лейкоза собак, вирус иммунодефицита собак, гепаднавирус, цитомегаловирус и пикорнавирус. Таким образом, соединения данного изобретения эффективны в лечении и /или/ профилактике ретровирусных инфекций.
Соединения данного изобретения могут иметь один или несколько асимметричных атомов углерода и поэтому способны существовать в форме оптических изомеров, так же как в форме их рацемических или нерацемических смесей. Оптические изомеры можно получить разделением рацемических смесей согласно обычным способам, например путем образования диастереоизомерных солей обработкой оптически активными кислотой или основанием. Примерами пригодных для этого кислот являются винная, дибензоилвинная, дитолуолвинная и камфорсульфоновая кислота. Затем разделяют смесь диастереоизомеров кристаллизацией с последующим выделением оптических оснований из этих солей. Другой способ разделения оптических изомеров предусматривает применение хиральной хроматографической колонки, улучшающей разделение энантиомеров. Еще один доступный способ предусматривает синтез ковалентных молекул диастереомеров реакцией соединений формулы I с оптически чистой кислотой в активированной форме или с оптически чистым изоцианатом. Синтезированные диастереоизомеры могут быть разделены общепринятыми способами, такими как хроматография, перегонка, кристаллизация или сублимация, с последующим гидролизом для получения энантиомеров чистого соединения. Оптически активные соединения формулы I могут быть получены известными способами с применением оптически активных исходных материалов. Эти изомеры могут быть в форме свободной кислоты, свободного основания, сложного эфира или соли.
Соединения данного изобретения могут быть использованы в форме солей, производных из неорганических или органических кислот. Эти соли включают /но не ограничены ими/ следующие соли: ацетат, адипат, альгинат, цитрат, аспартат, бензоат, бензолсульфонат, бусульфат, бутират, камфорат, камфорсульфонат, диглюконат, циклопентанпропионат, додецилсульфат, этансульфонат, глюкогептаноат, глицерофосфат, гемисульфат, гептаноат, гексаноат, фумарат, гидрохлорид, гидробромид, гидройодид, 2-гидроксиэтансульфонат, лактат, малеат, метансульфонат, никотинат, 2-нафталенсульфонат, оксалат, пальмоат, пектинат, персульфат, 3-фенилпропионат, пикрат, пивалат, пропионат, сукцинат, тартрат, тиоцианат, тозилат, мезилат и ундеканоат. Также основные азотсодержащие группы могут быть кватернизованы такими агентами, как низшие алкилгалогениды, такие как метил-, этил-, пропил- и бутилхлориды, бромиды и йодиды; диалкилсулфатами, такими как диметил-, диэтил-, дибутил и диамилсульфаты, галогенидами с длинной цепью, такими как децил-, лаурил-, миристил и стеарилхлориды, бромиды и йодиды, аралкилгалогенидами, такими как бензил- и фенэтилбромиды, и др. В результате получают растворимые или диспергируемые в воде или в масле продукты.
Примерами кислот, которые можно применять для образования фармацевтически приемлемых кислотно-аддитивных солей являются такие неорганические кислоты, как соляная кислота, серная кислота и фосфорная кислота, и такие органические кислоты, как щавелевая кислота, малеиновая кислота, янтарная кислота и лимонная кислота. Другие примеры включают в себя соли со щелочными металлами или щелочно-земельными металлами, такими как натрий, калий, кальций или магний, или с органическими основаниями.
Полная суточная доза, вводимая хозяину в виде однократной дозы или в виде разделенных доз, составляет, например, 0,01-10 мг/кг веса тела в день и более обычно 0,01-1 мг. Композиции стандартных доз могут содержать такие количества в отдельных дозировках, чтобы можно было получить дневную дозу.
Количество активного ингредиента, которое может быть соединено с материалами носителя для образования единичной дозировочной формы будет варьировать в зависимости от хозяина, проходящего лечение, и от способа введения.
Схему приема лекарственного средства для лечения заболевания соединениями и /или/ композициями данного изобретения выбирают в соответствии со множеством факторов, в том числе таких факторов, как тип, возраст, вес, пол, условия диеты и лечения больного, тяжесть заболевания, способ введения, фармакологические свойства, такие как активность, эффективность, фармакокинетические и токсилогические профили применяемого соединения, а также в зависимости от того, применяют ли систему доставки лекарства или соединение вводят в виде части комбинированного лекарственного препарата. Поэтому в действительности схему приема можно широко варьировать и, следовательно, отклоняться от предпочтительной схемы приема, описанной выше.
Соединения данного изобретения можно вводить перорально, парентерально, при помощи ингалятора, ректально или местно в виде препаратов в стандартных дозах, содержащих обычные нетоксичные фармацевтически приемлемые носители, адъюванты и наполнители, если желательно. Местное введение может предусматривать также применение трансдермальных пластырей и ионтофорезных устройств для чрескожного введения. Термин парентеральный охватывает здесь подкожные инъекции, внутривенные, внутримышечные, внутригрудинные инъекции или инфузионные способы.
Препараты для инъекций, например, пригодные для инъекций водные или маслянистые суспензии, могут быть приготовлены при помощи подходящих диспергирующих или увлажняющих и суспендирующих средств. Такие стерильные препараты для инъекций могут также быть стерильным инъецируемым раствором или суспензией в нетоксичном парентерально приемлемом разбавителе или растворителе, например в виде раствора в 1,3-бутандиоле. Среди приемлемых наполнителей и растворителей могут быть применены вода, раствор Рингера и изотонический раствор хлорида натрия. Кроме того, в качестве растворителя или суспендирующей среды применяют стерильные нелетучие масла. Для этой цели может быть применено любое легкое нелетучее масло, в том числе синтетические моно- или диглицериды. Кроме того, для приготовления инъецируемых препаратов можно применять жирные кислоты, такие как олеиновая кислота.
Суппозитории для ректального введения можно приготовлять смешиванием лекарственного средства с подходящим нераздражающим наполнителем, таким как какао-масло и полиэтиленгликоли, которые являются твердыми при обычных температурах и расплавляются при температуре в прямой кишке с выделением лекарственного средства.
Твердыми лекарственными формами для перорального введения могут быть капсулы, таблетки, пилюли, порошки и гранулы. В таких твердых лекарственных формах активное соединение может быть смешано по меньшей мере с одним инертным разбавителем, таким как сахароза, лактоза, или крахмал. Такие лекарственные формы могут также содержать, как и в обычной практике, дополнительные вещества, иные, чем инертные разбавители, например смазывающие средства, такие как стеарат магния. В случае капсул, таблеток и пилюль лекарственные формы могут также содержать буферные агенты. Таблетки и пилюли могут иметь наружное покрытие.
Жидкими лекарственными формами для перорального введения могут быть фармацевтически приемлемые эмульсии, растворы, суспензии, сиропы и эликсиры, содержащие инертные разбавители, применяемые обычно в фармакологии, такие как вода. Такие композиции могут также содержать адъюванты, такие как увлажняющие агенты, эмульгаторы и суспендирующие агенты, а также подслащивающие и ароматизирующие средства.
Хотя соединения данного изобретения можно вводить в виде единственного активного фармацевтического агента, их можно применять также в сочетании с одним или несколькими иммуномодуляторами, антивирусными агентами или иными антиинфекционными агентами. Например, соединения данного изобретения можно вводить в комбинации с AZT, DDI, DDC или с ингибиторами глюкозидазы, такими как N-бутил-1-деоксиджирамицин или его пролекарствами, для профилактики и /или/ лечения СПИДа. При введении в виде комбинации эти терапевтические агенты могут быть приготовлены в виде отдельных композиций, которые даются одновременно или в другое время, или эти терапевтические агенты могут даваться в виде единой композиции с соединениями данного изобретения.
Вышеизложенное представляет собой лишь иллюстрацию данного изобретения и не должно ограничивать изобретение только описанными соединениями. В сфере действия и природе данного изобретения предполагаются вариации и изменения, которые очевидны специалистам и которые определены в прилагаемой формуле изобретения.
Из вышеизложенного описания специалисты могут легко определить основные характеристики данного изобретения и без отхода от объема и сферы действия изобретения могут внести различные изменения и модификации в изобретение для приспособления его к различным условиям и областям применения.
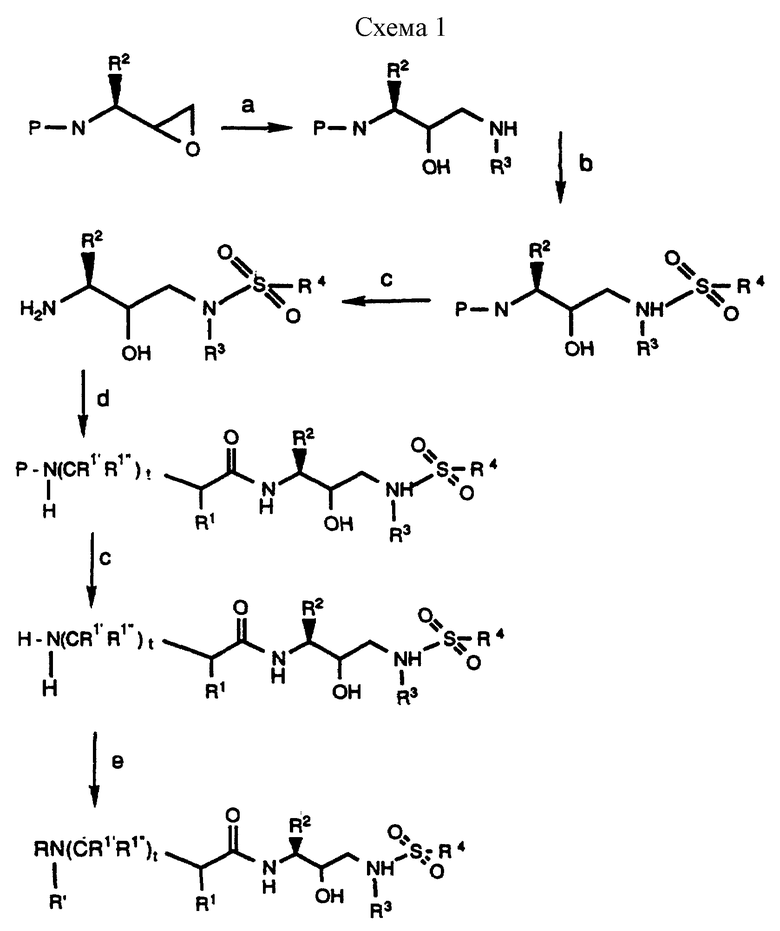
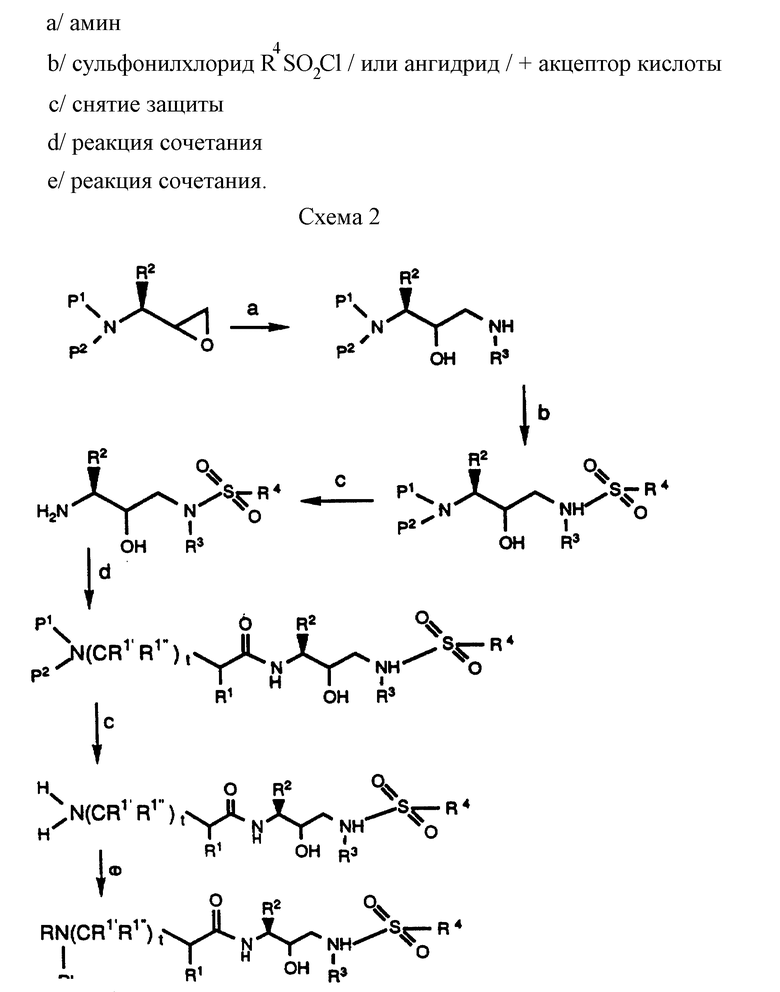

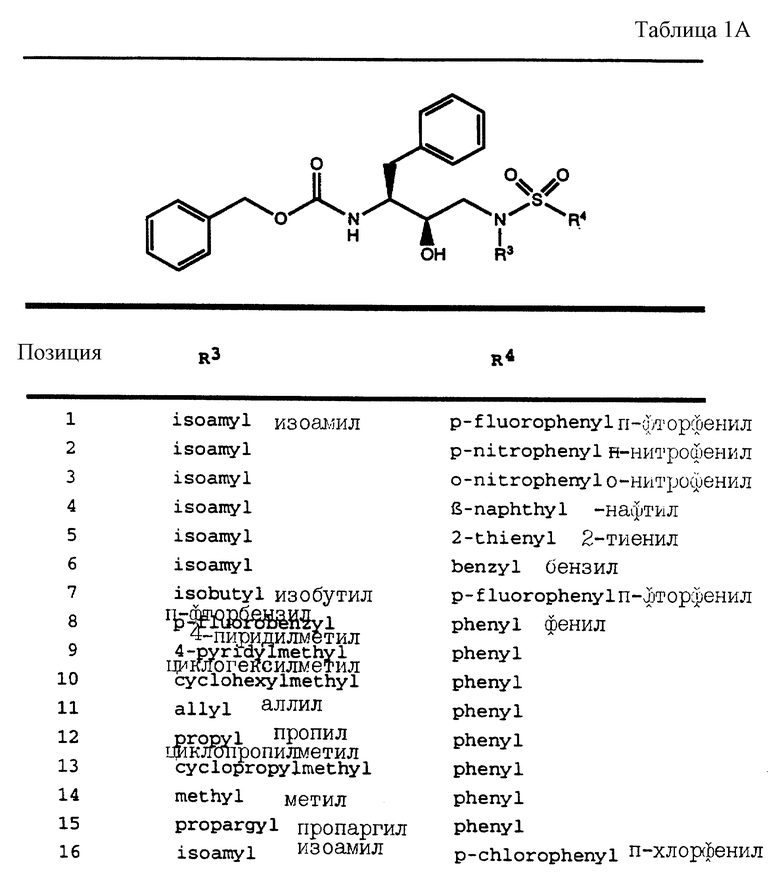

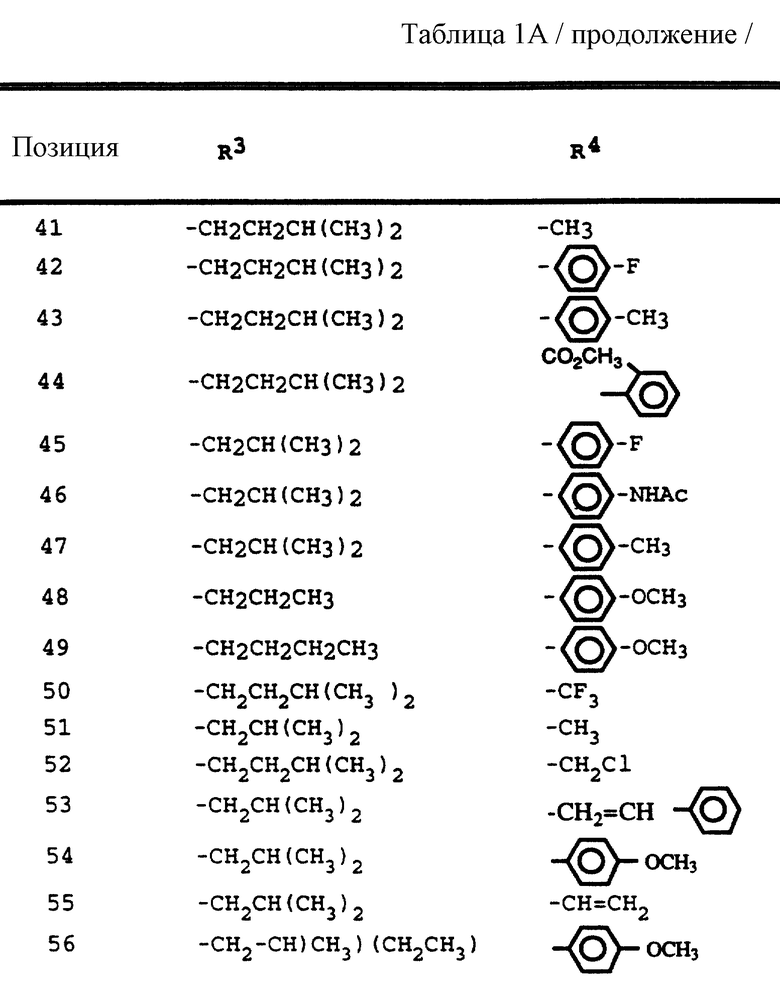
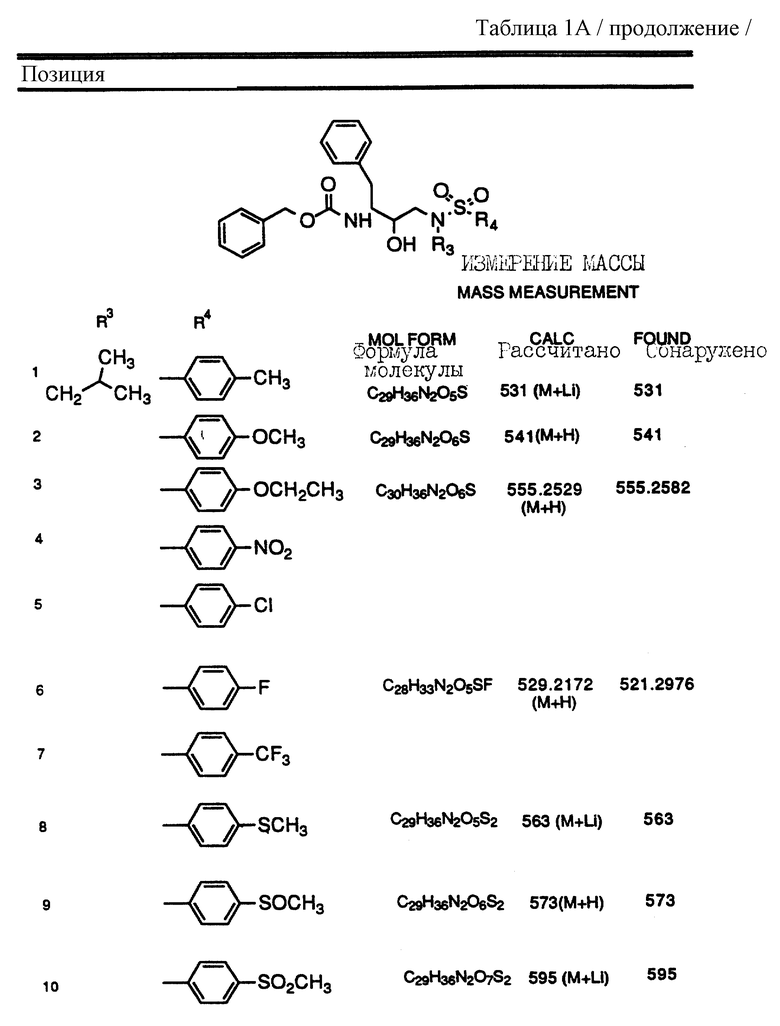
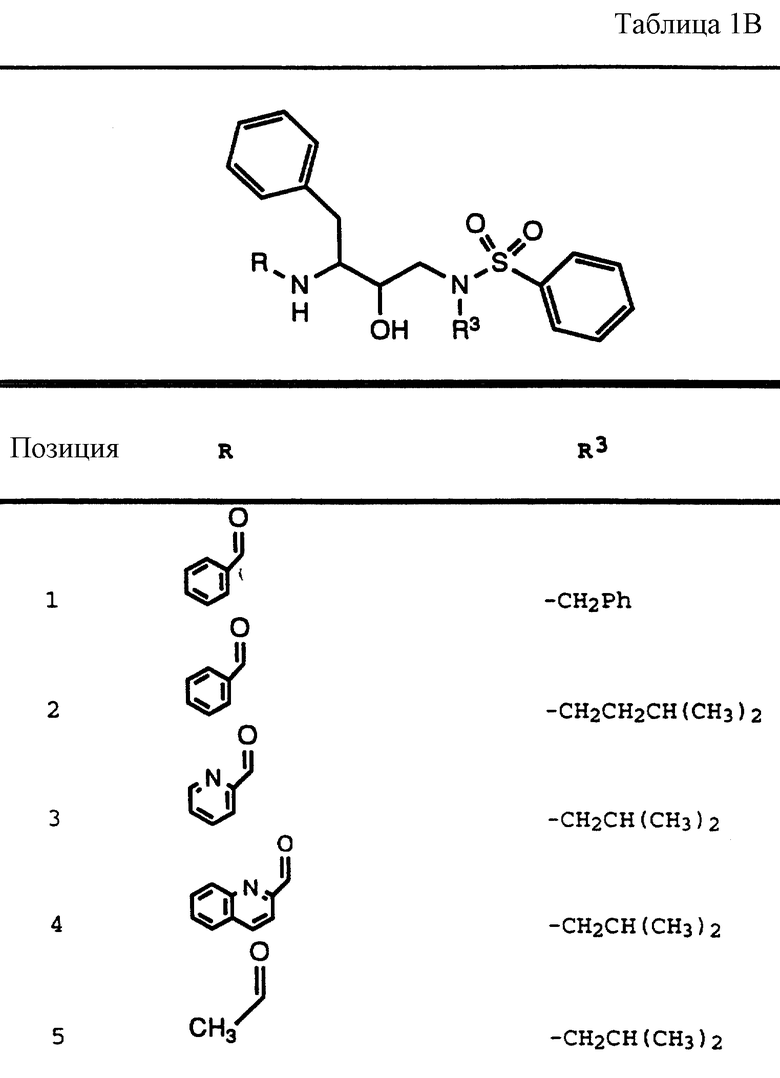
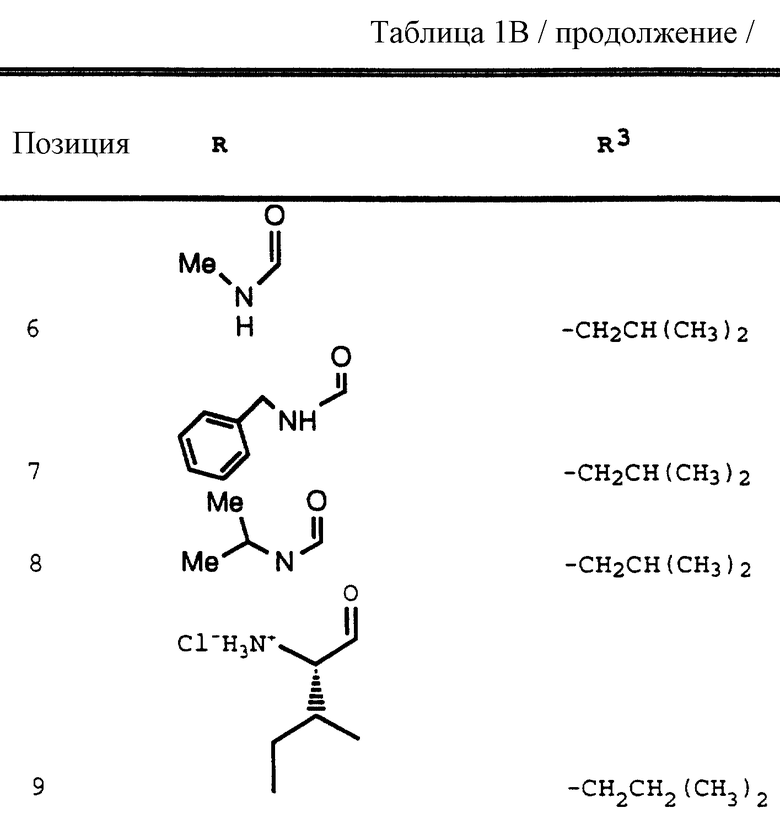
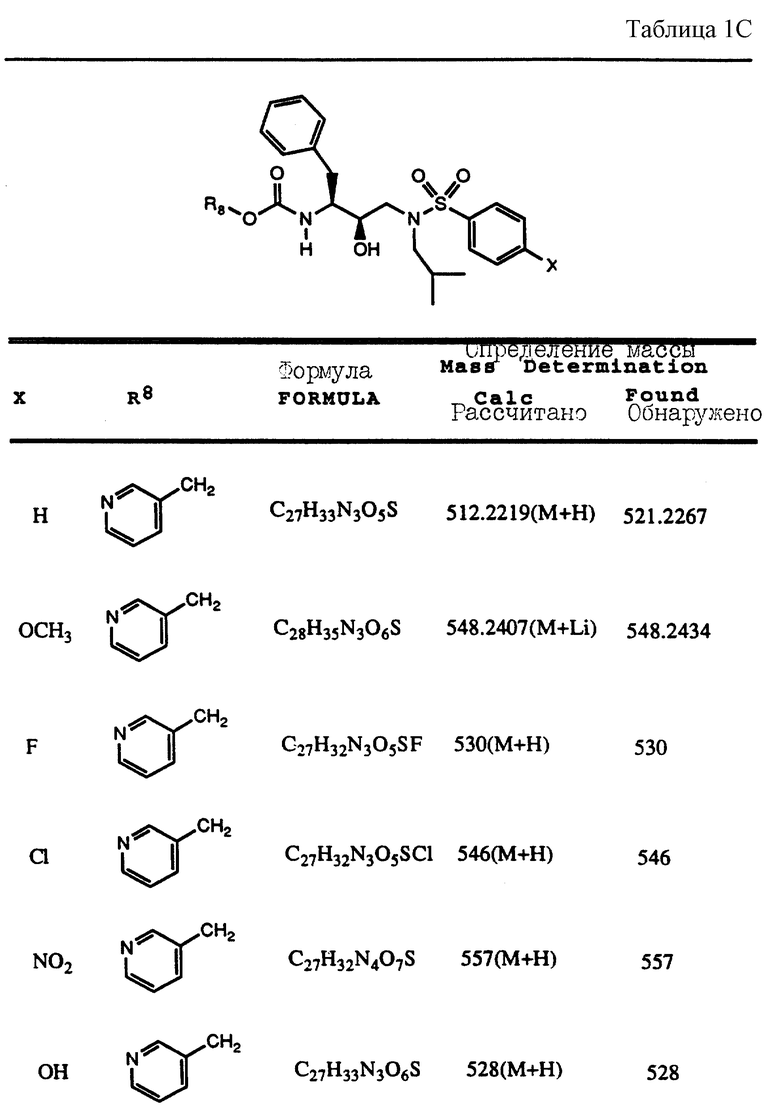
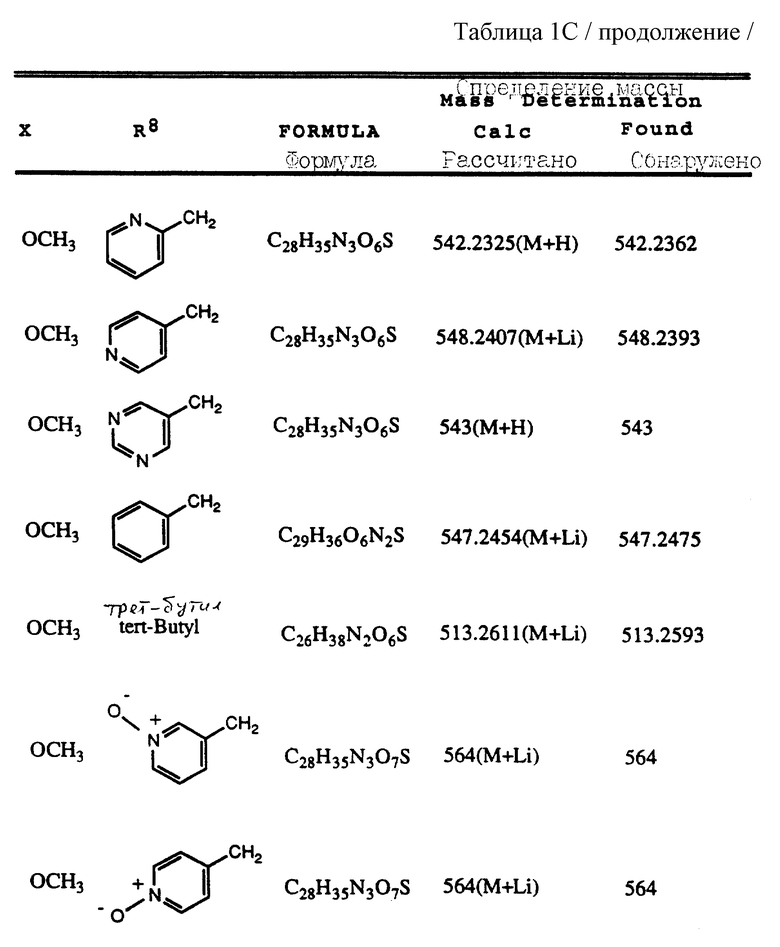
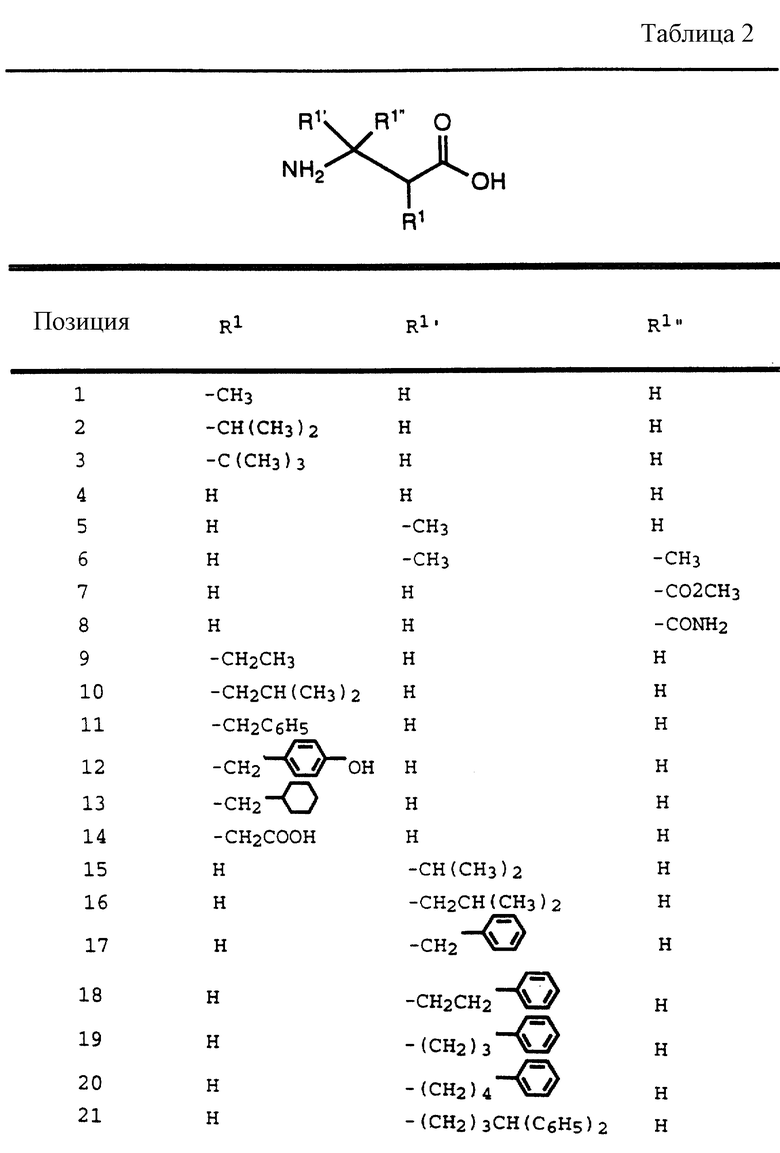
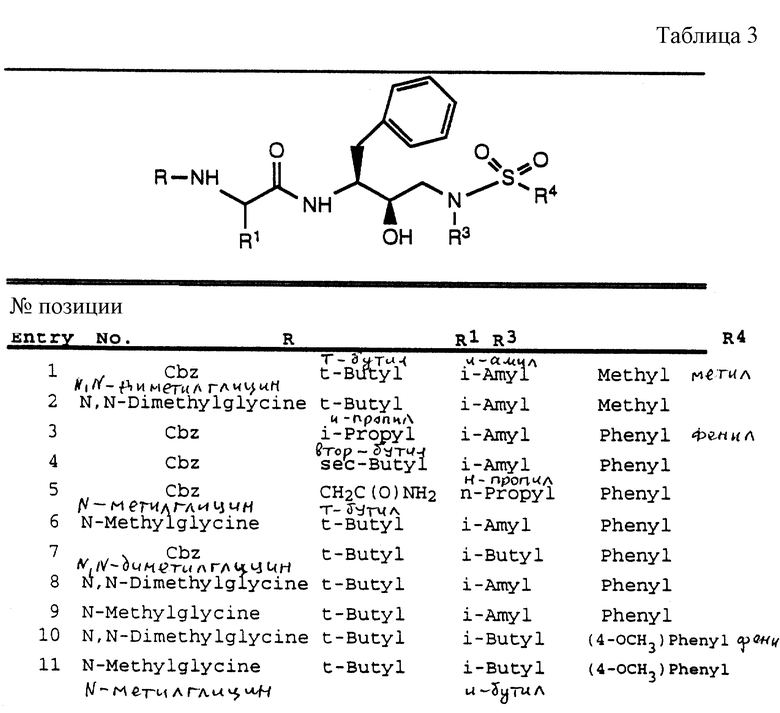
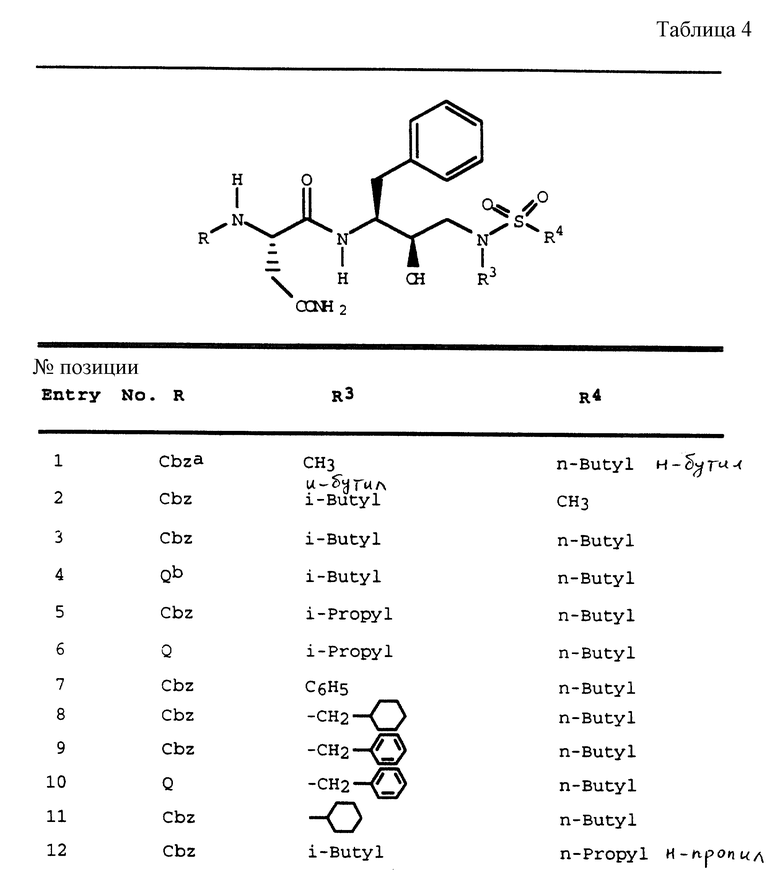

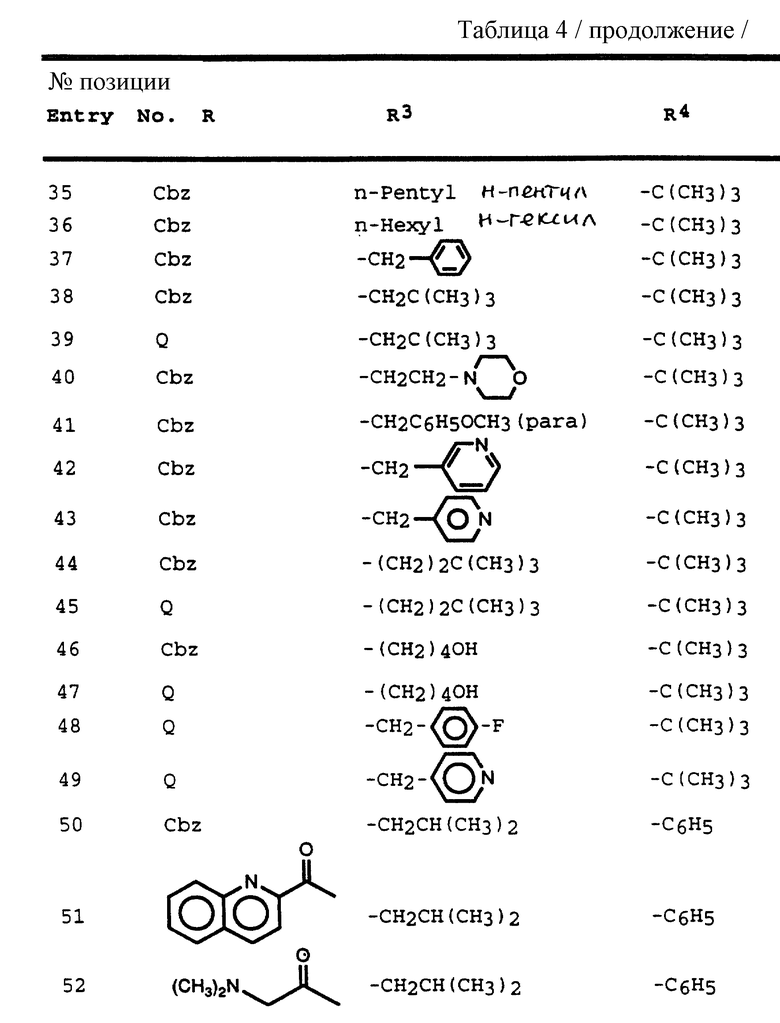
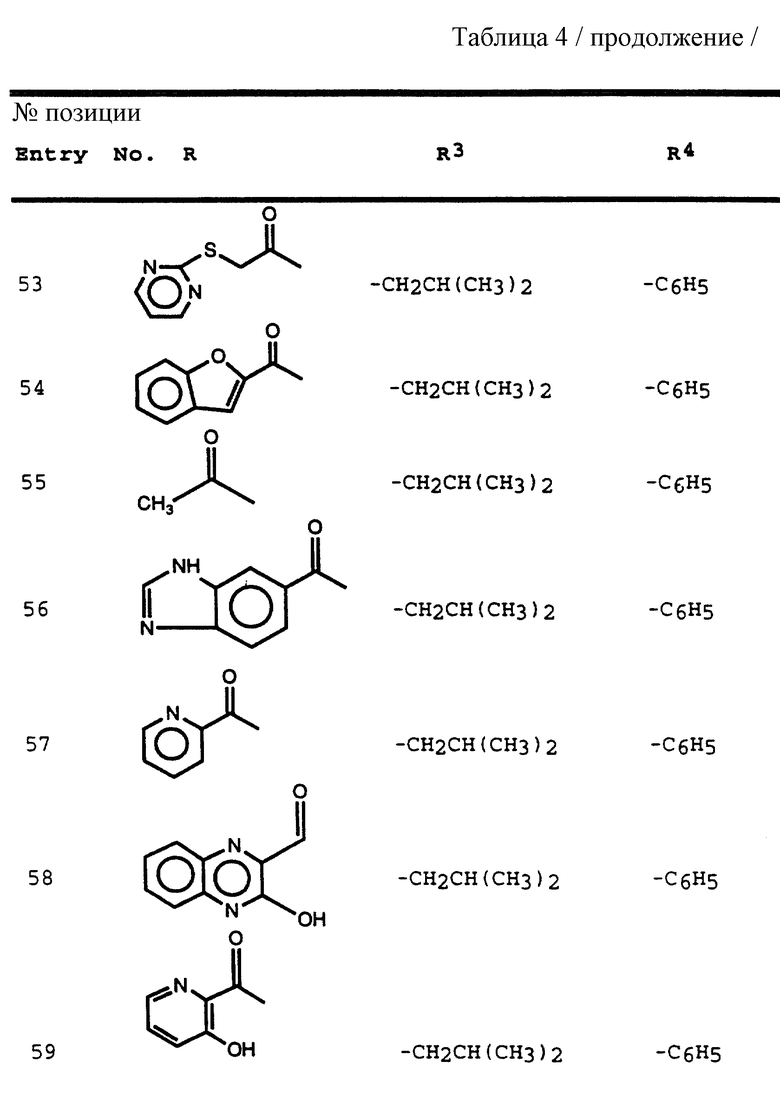
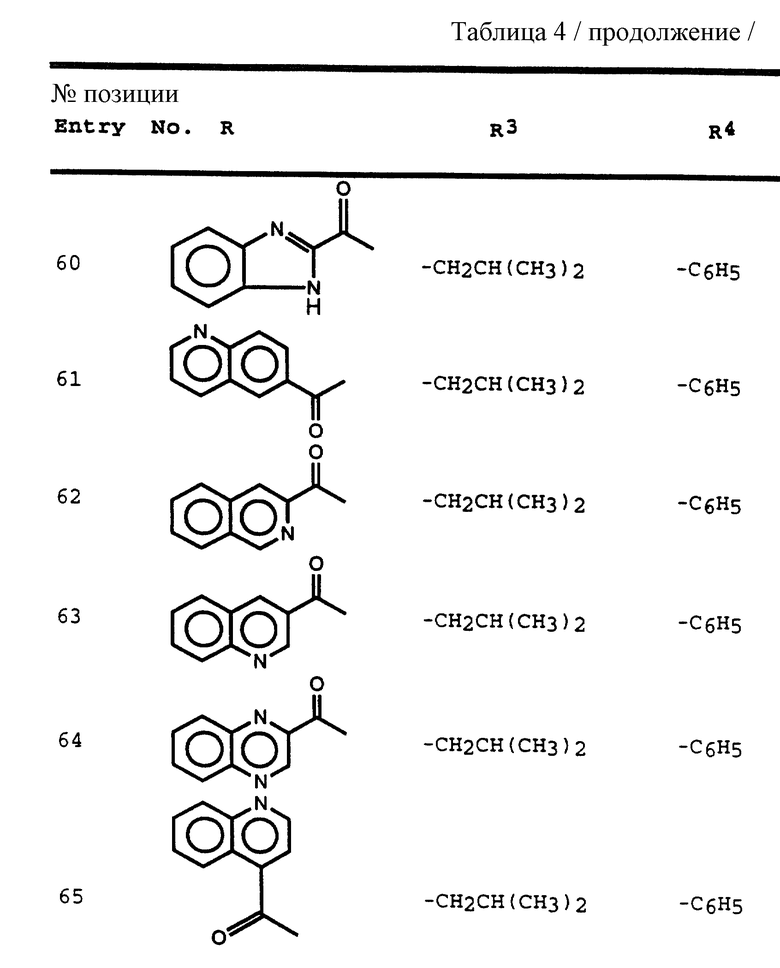
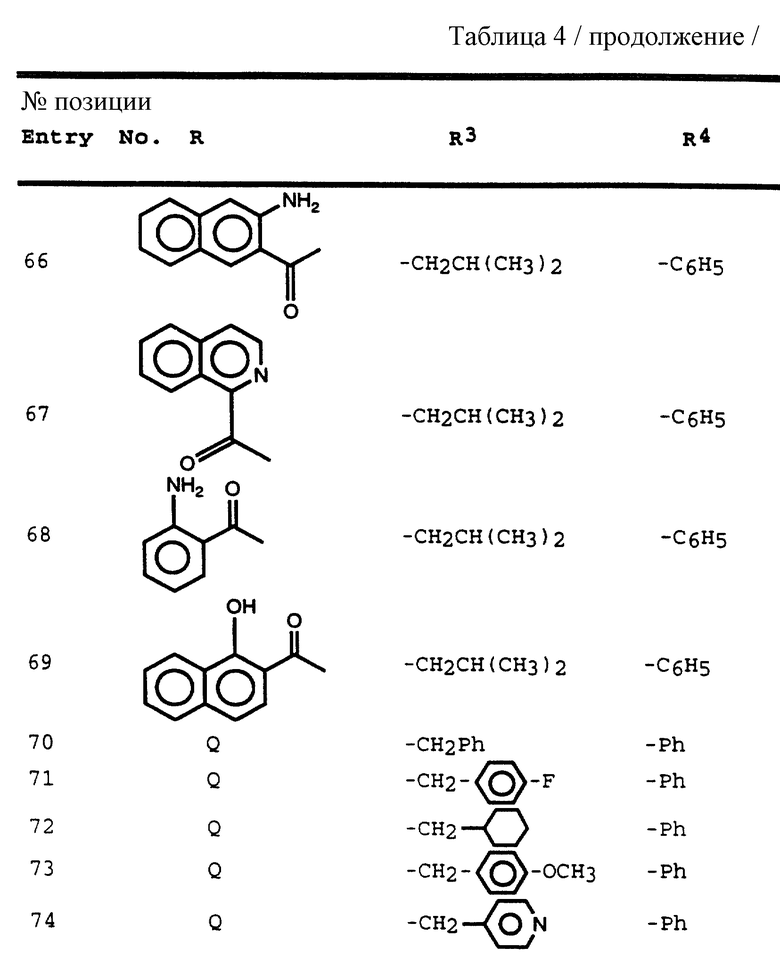
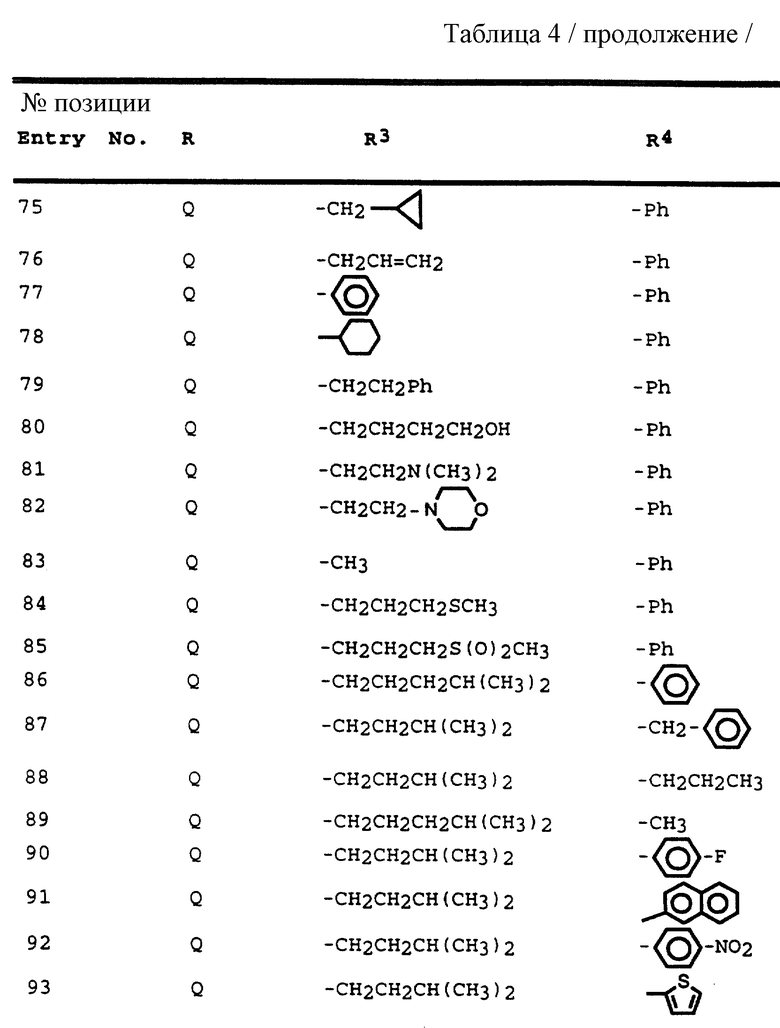
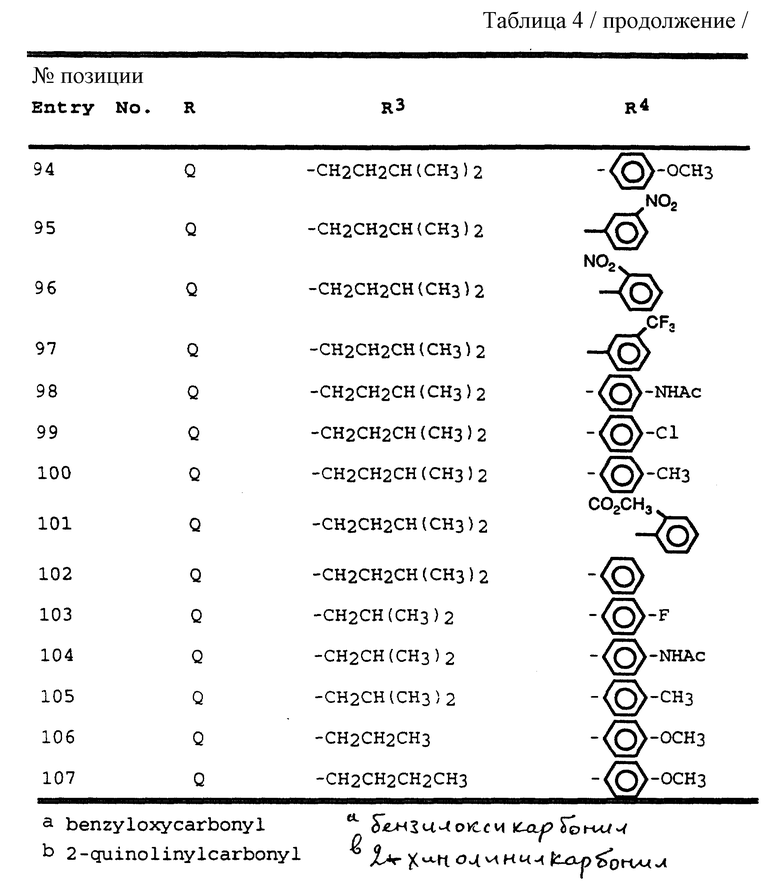
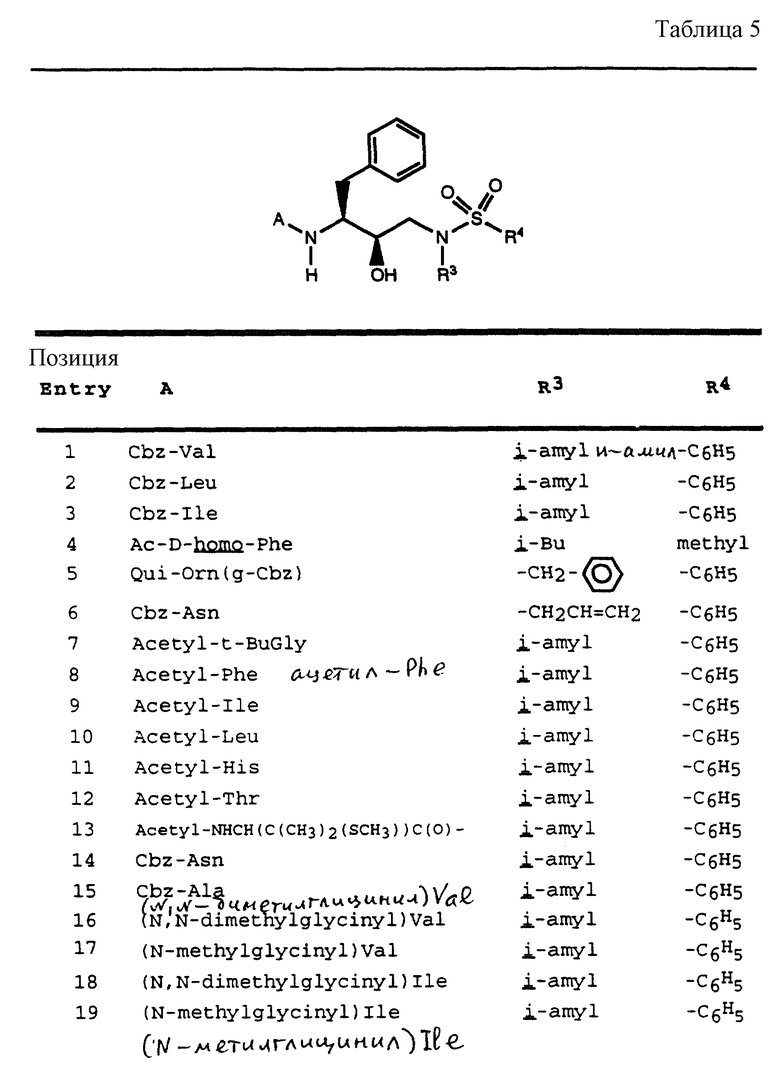
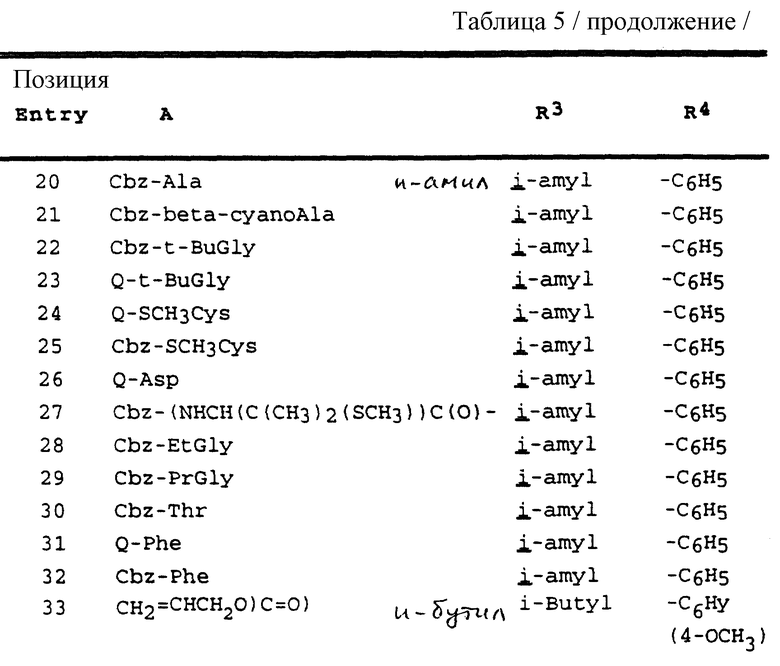
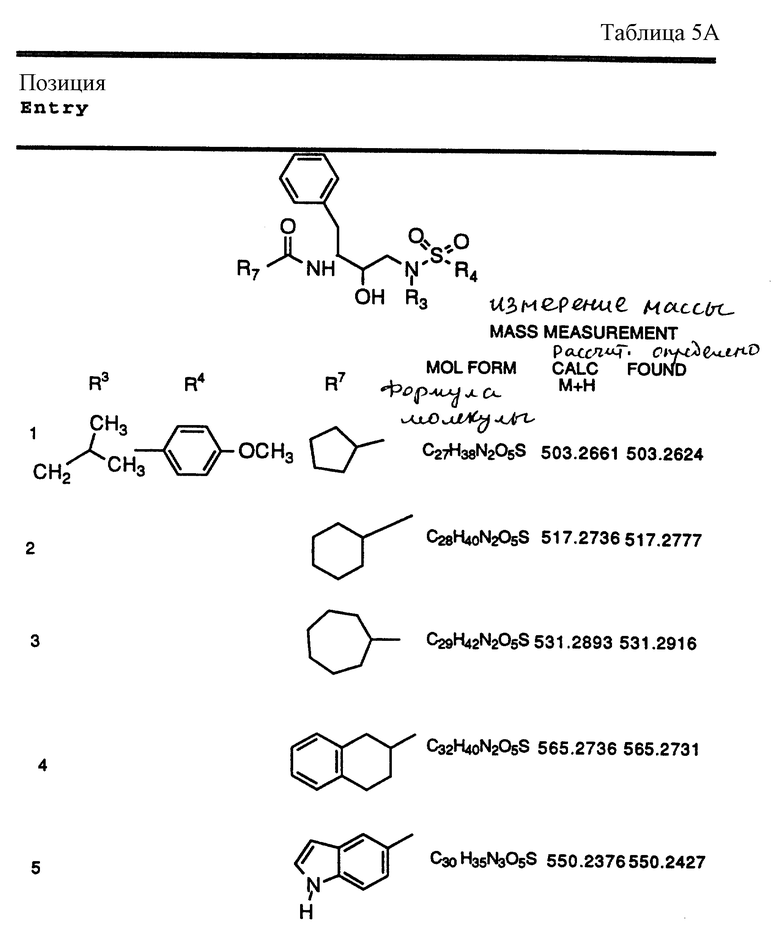
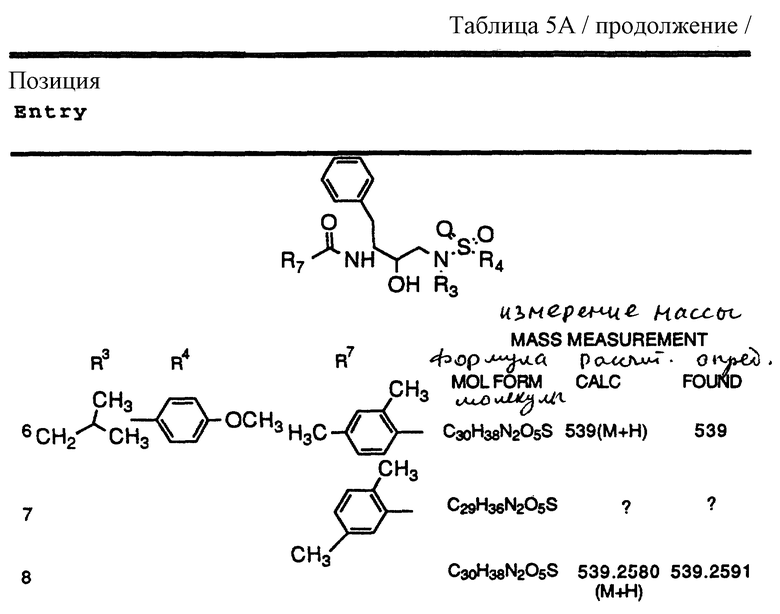
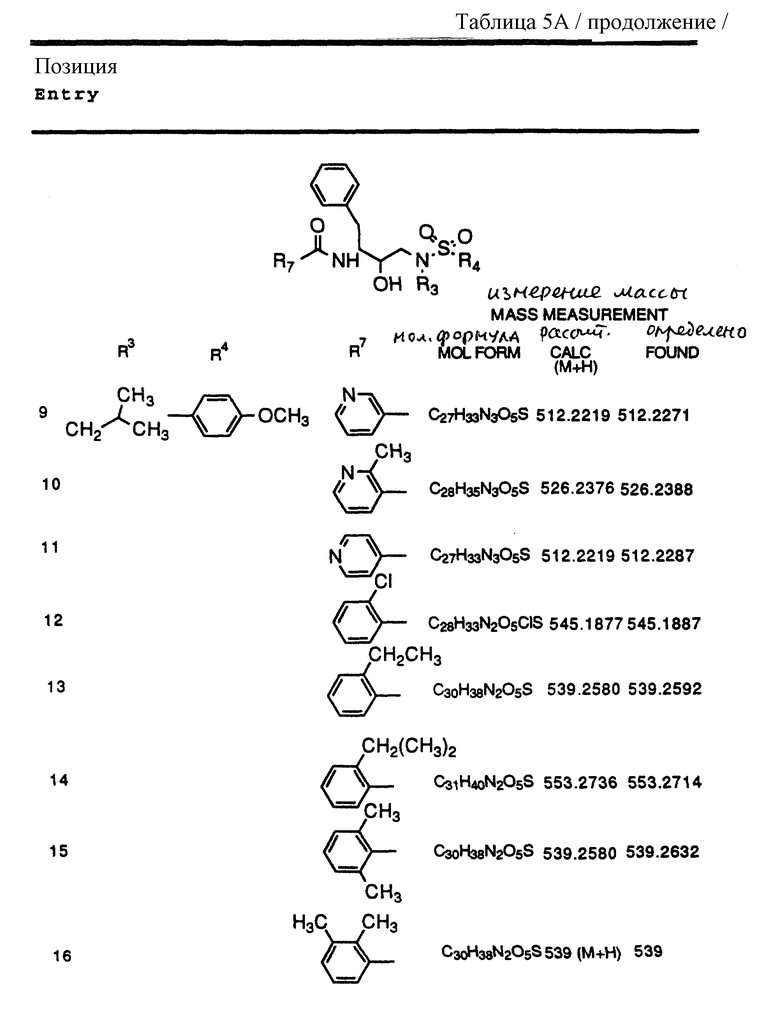
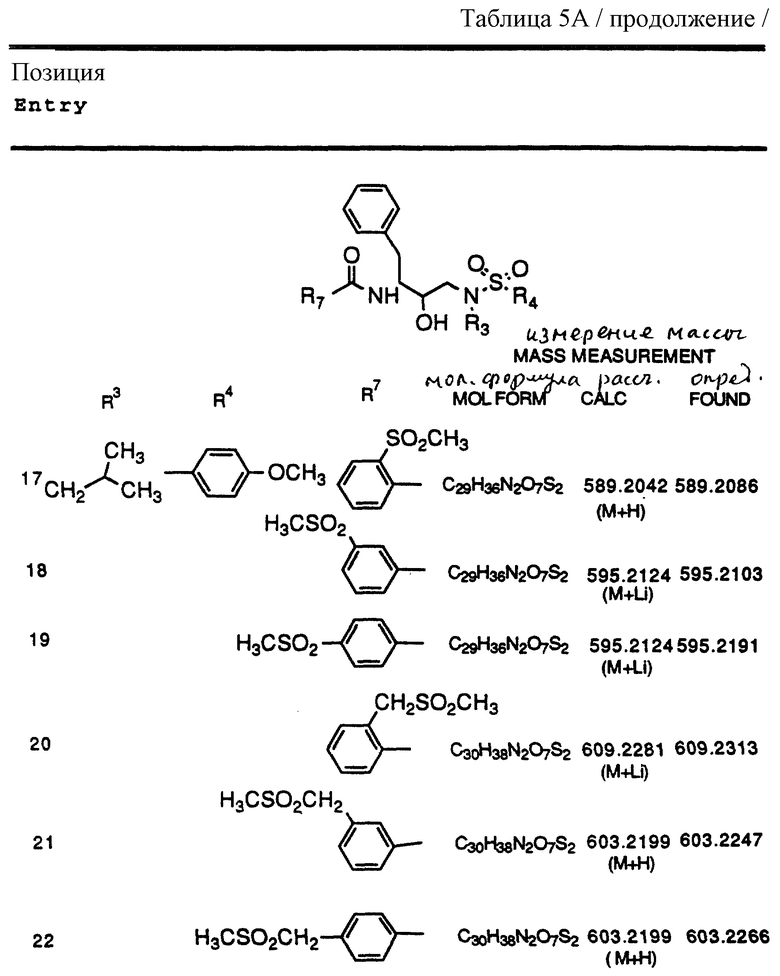
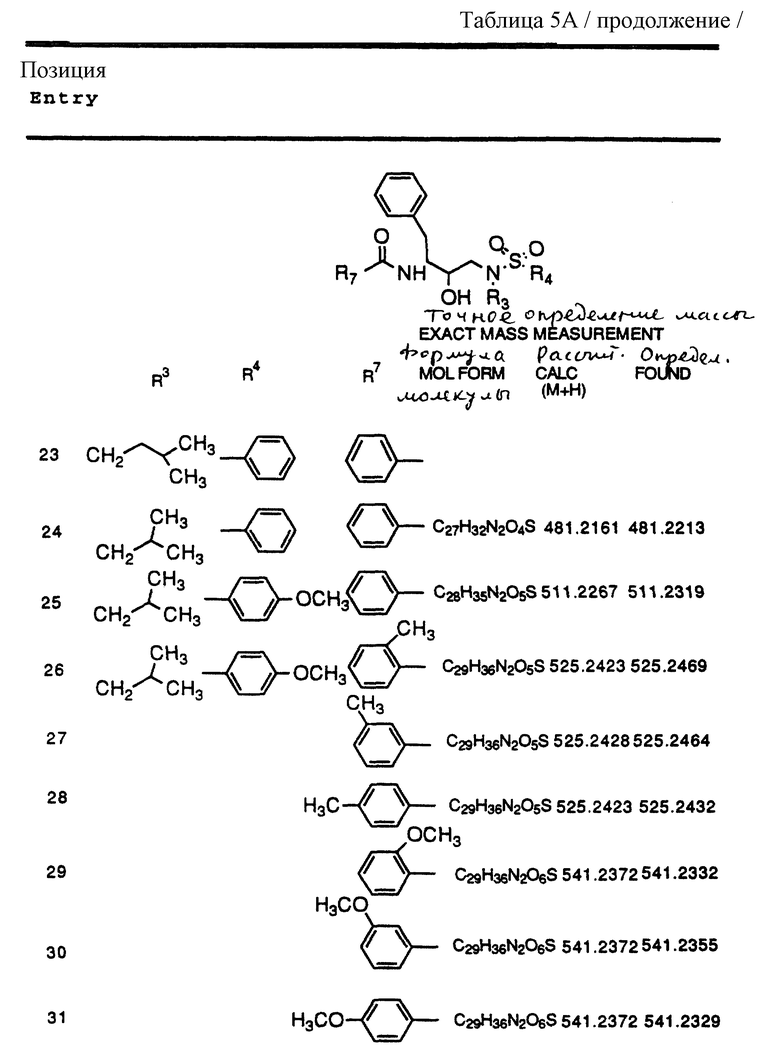
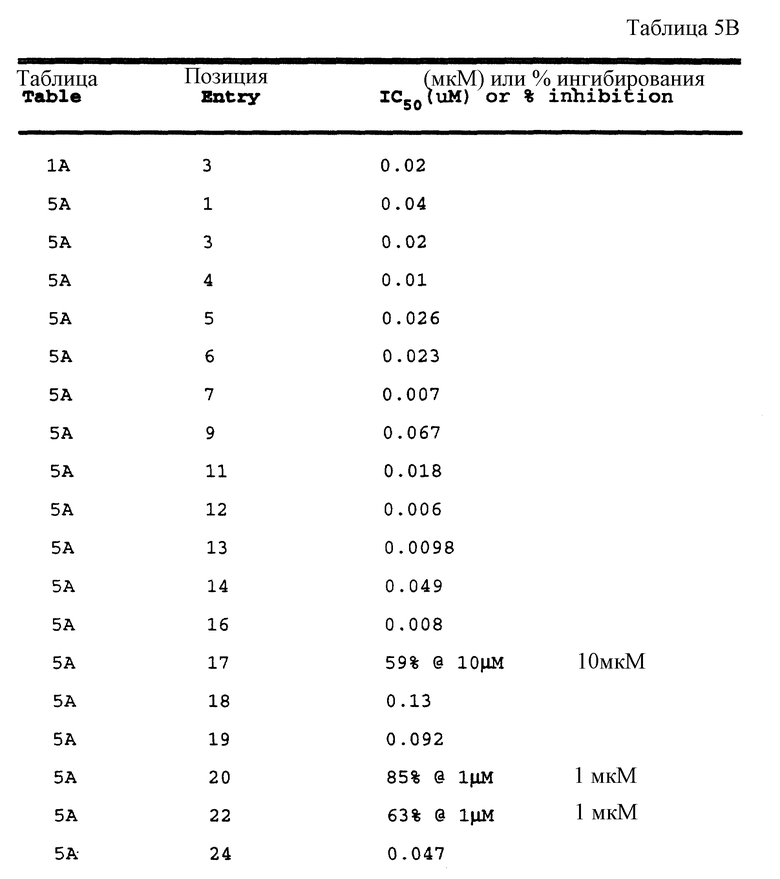
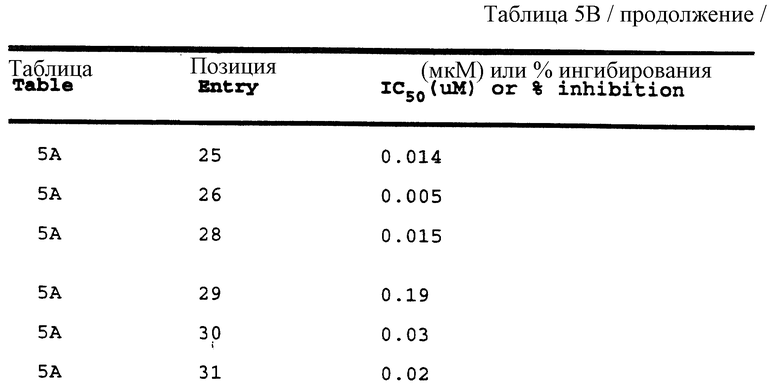
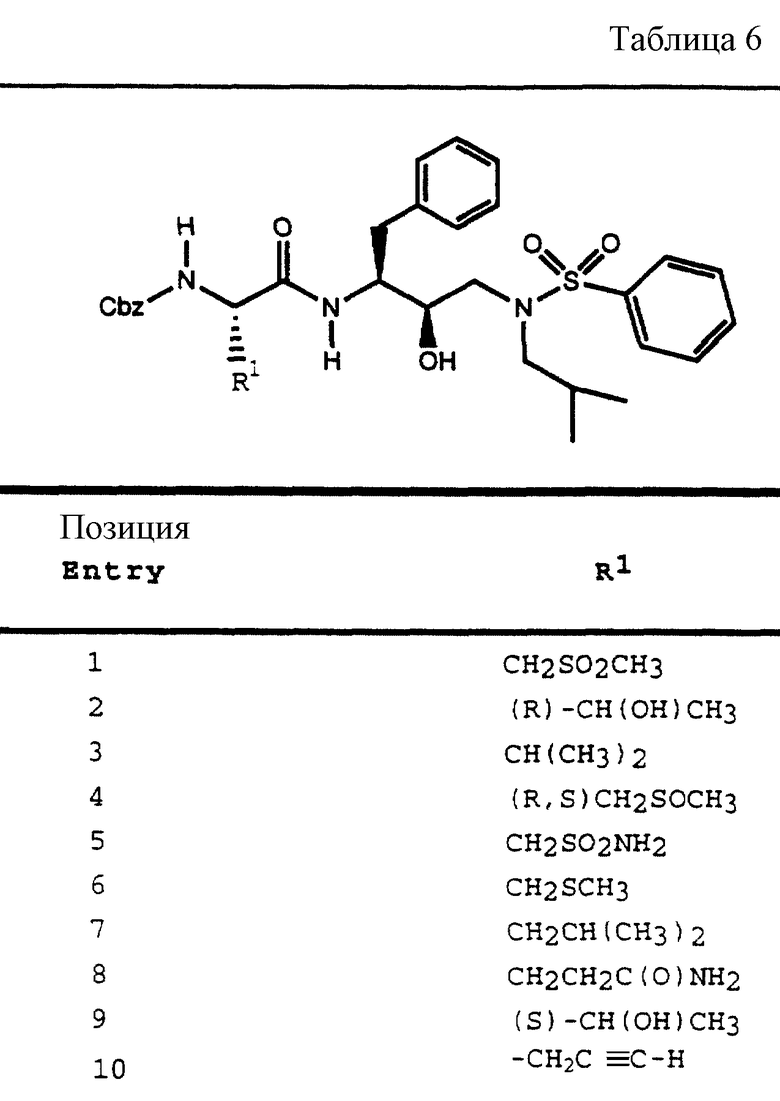
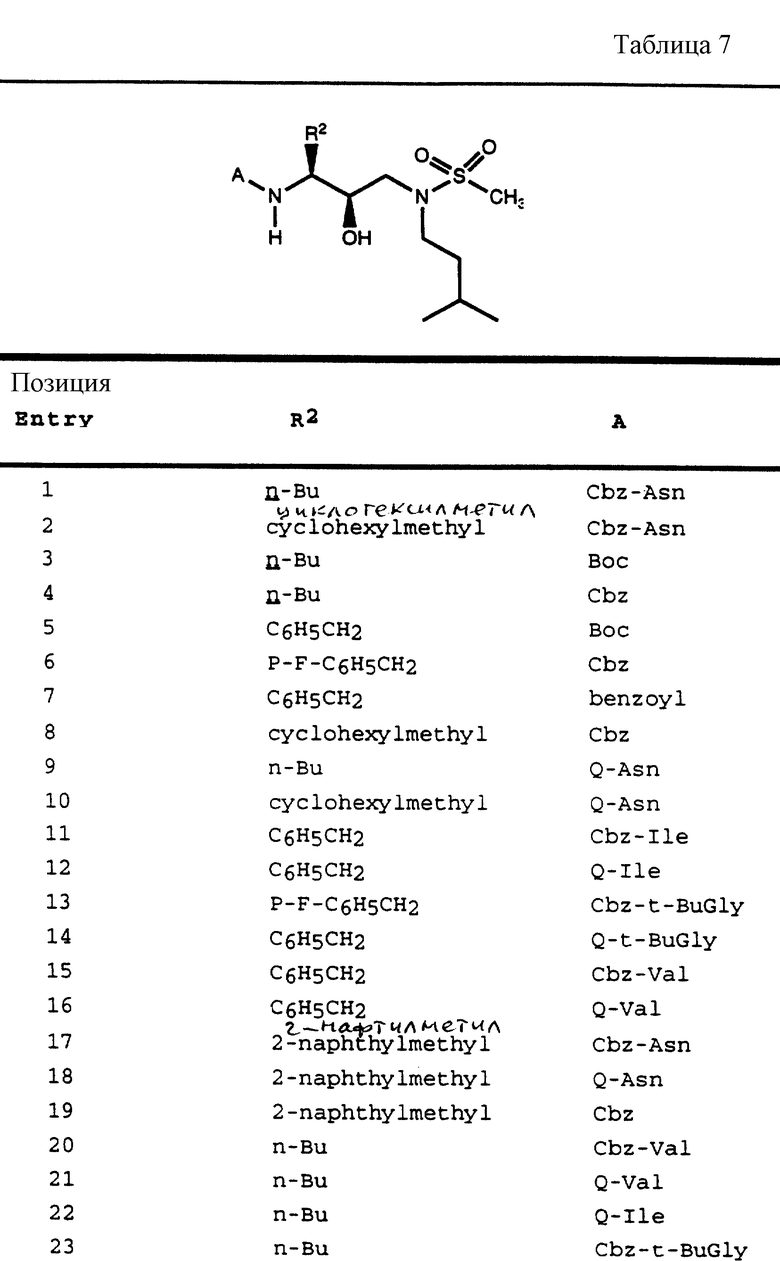

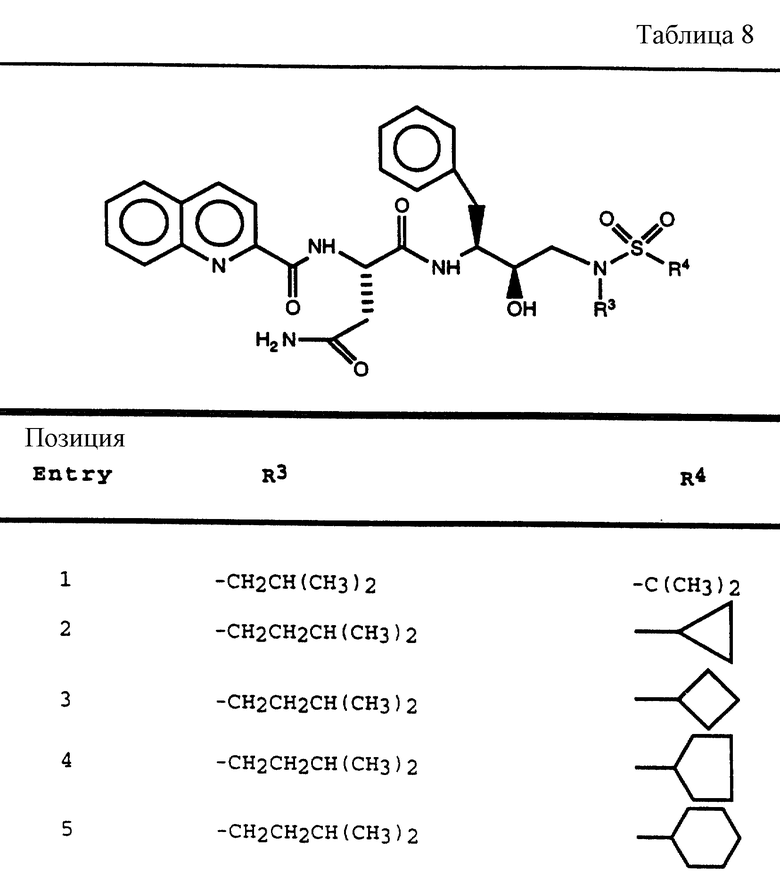
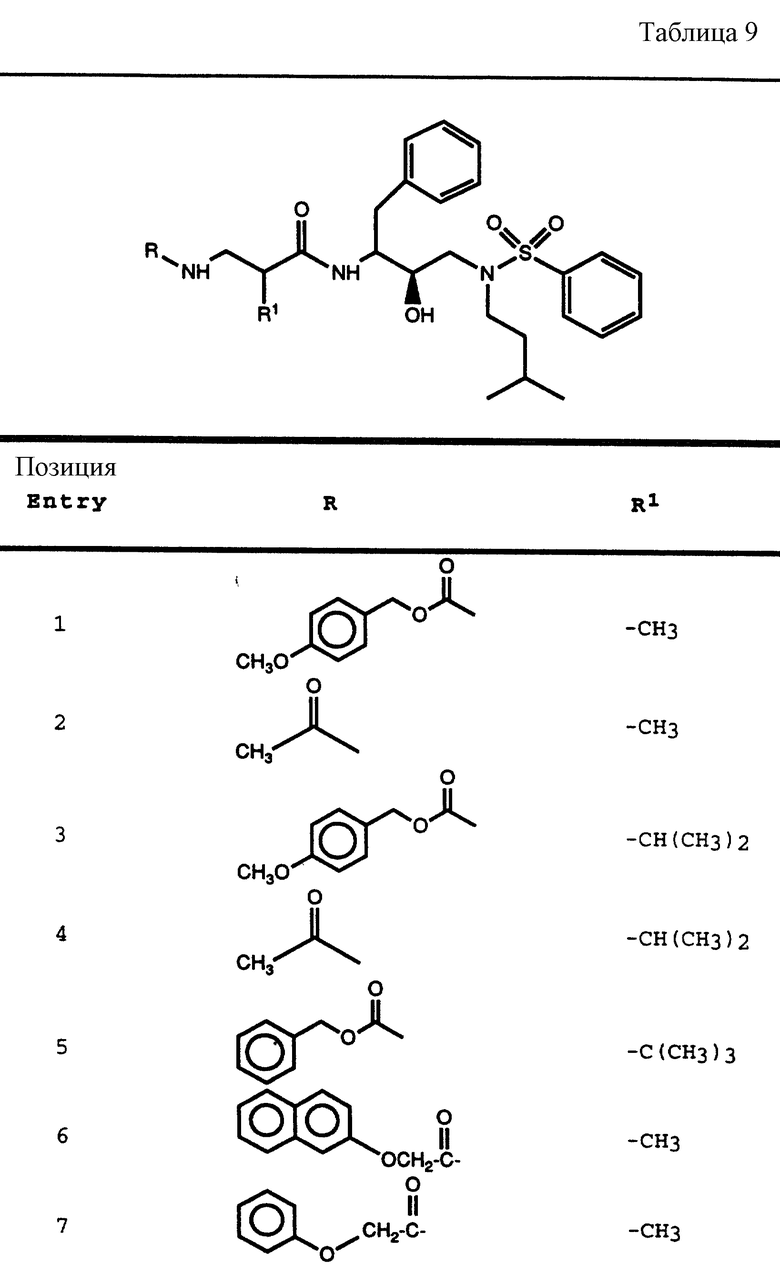
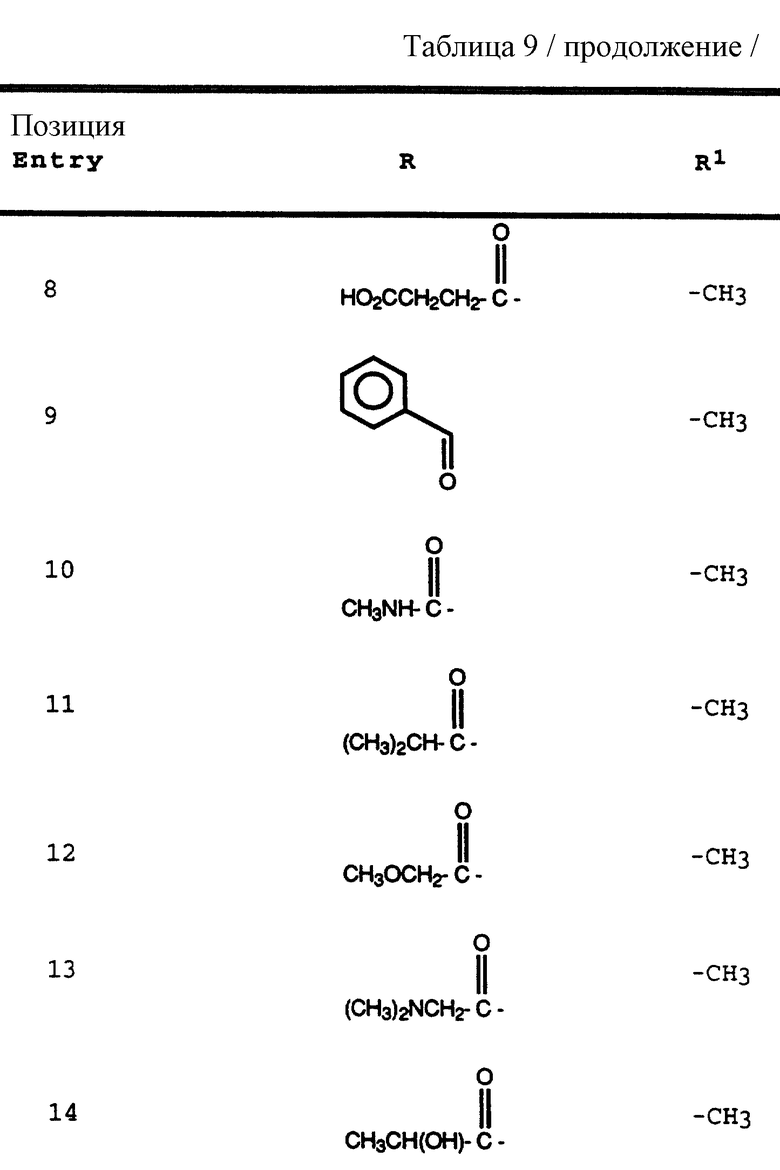
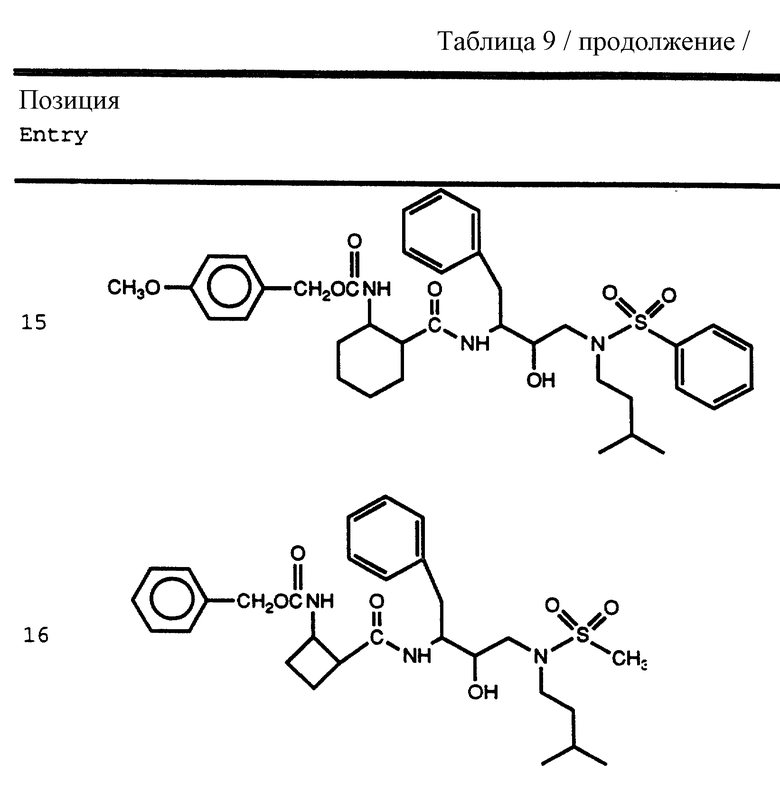
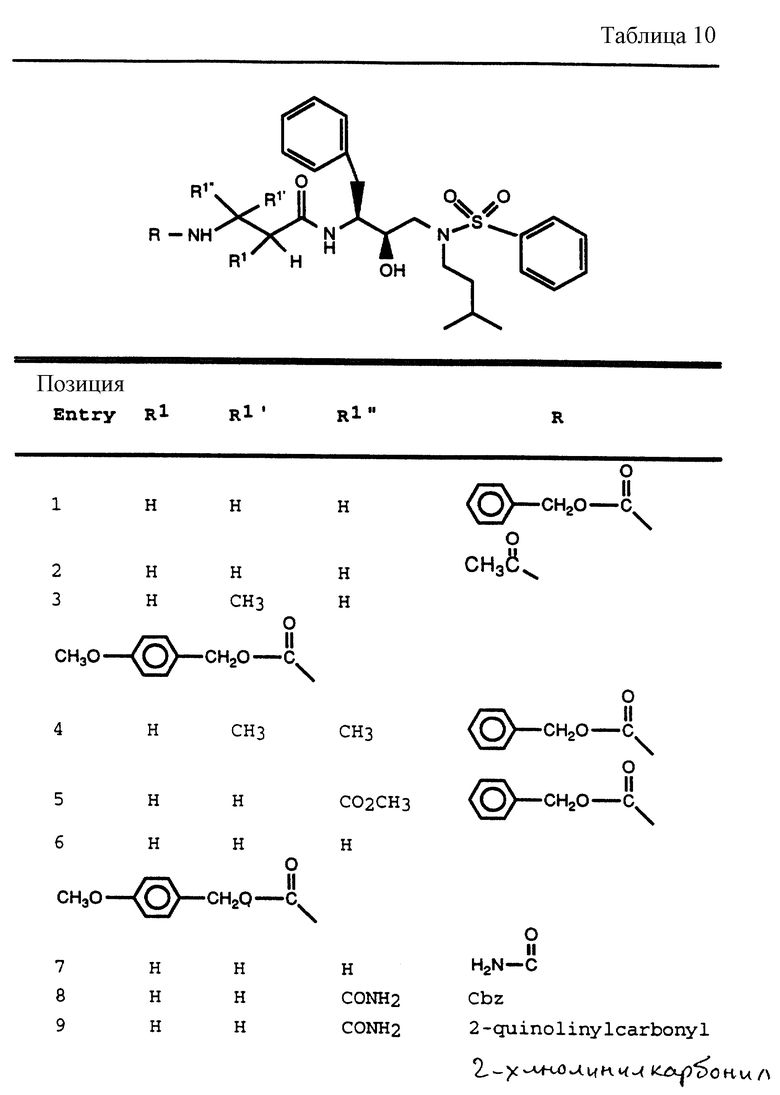
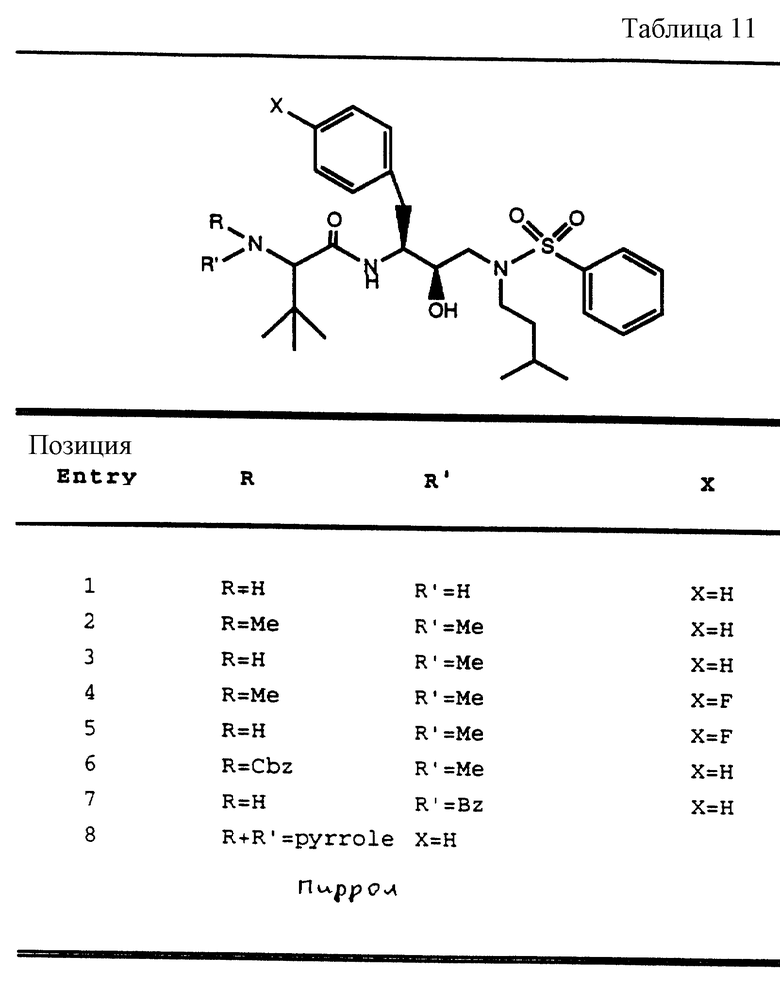
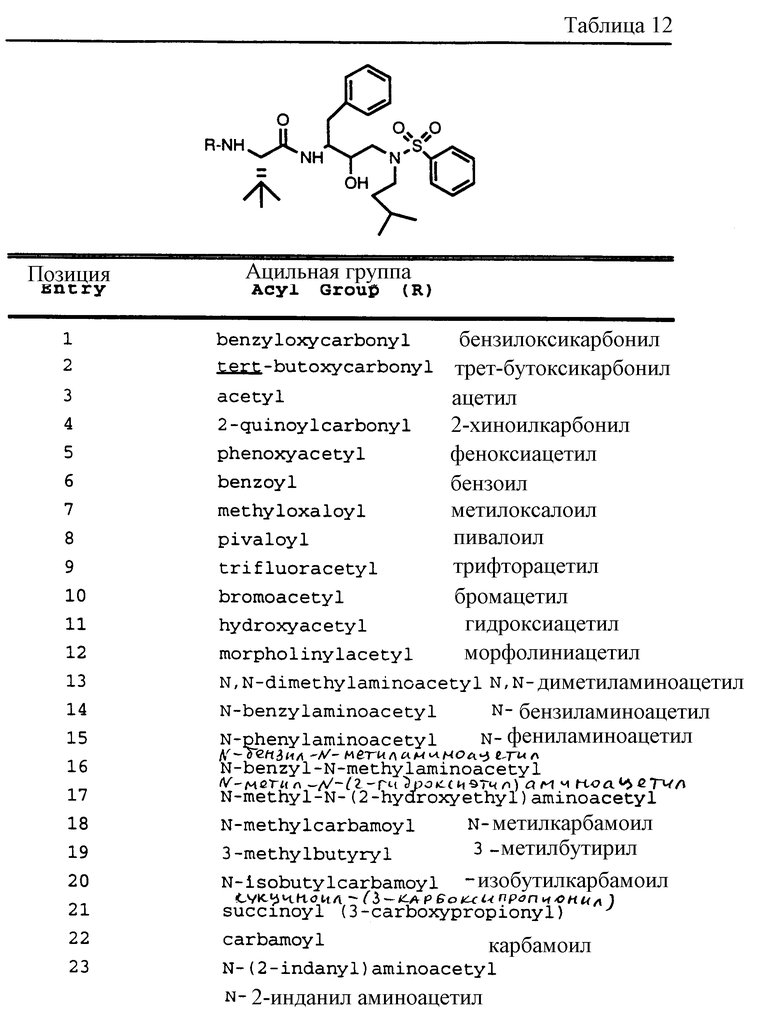
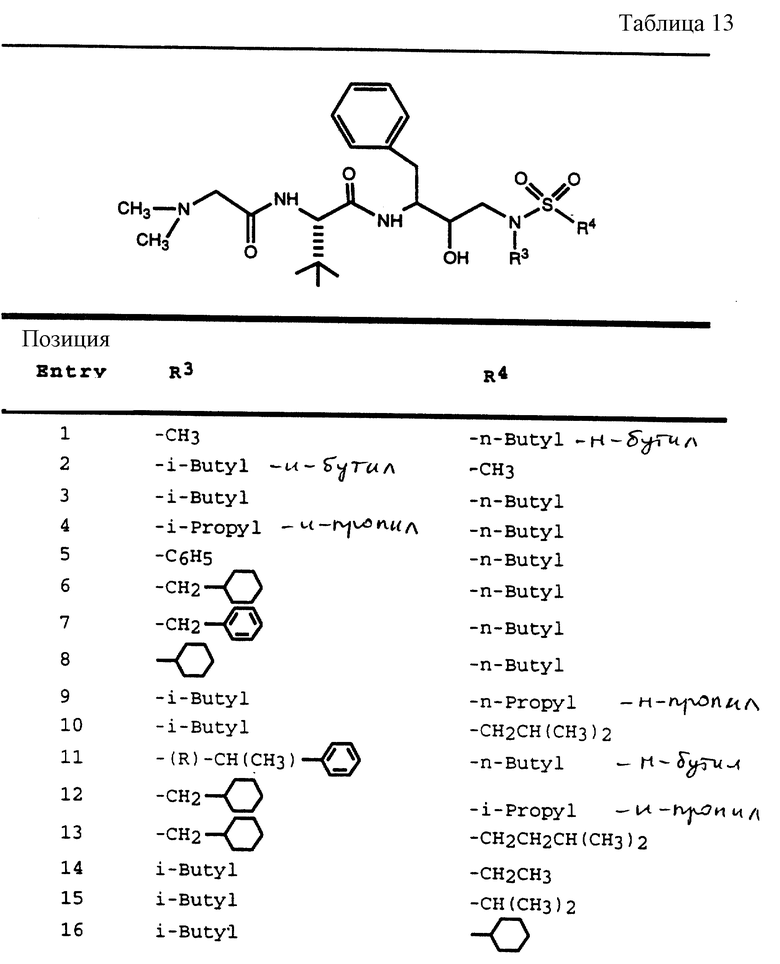
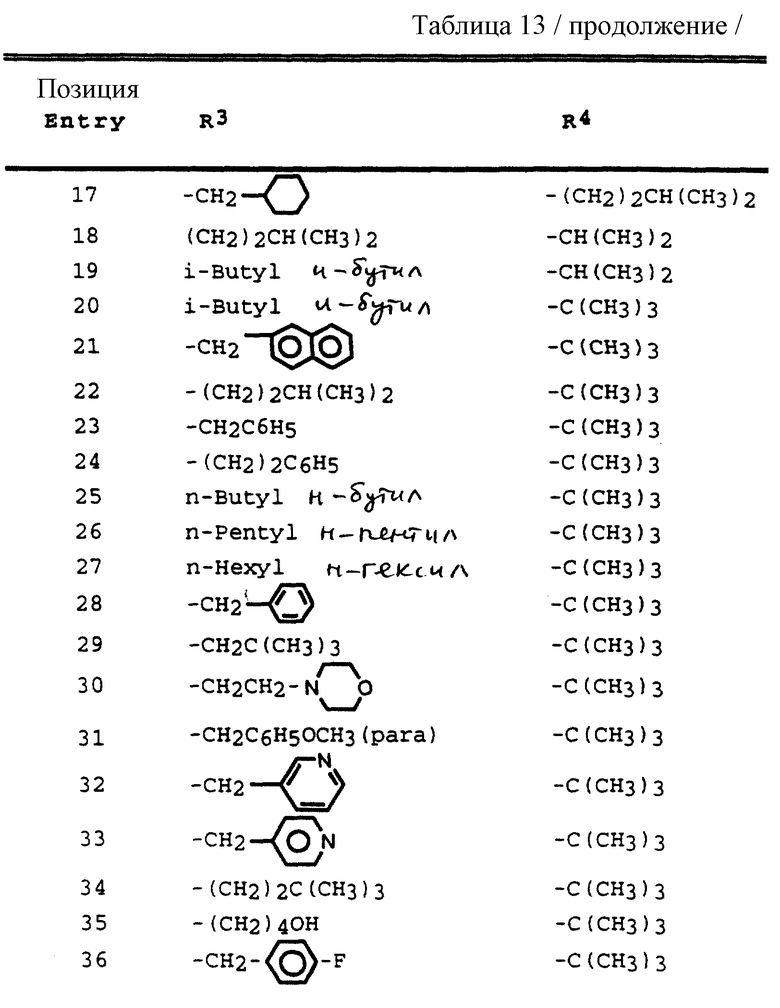
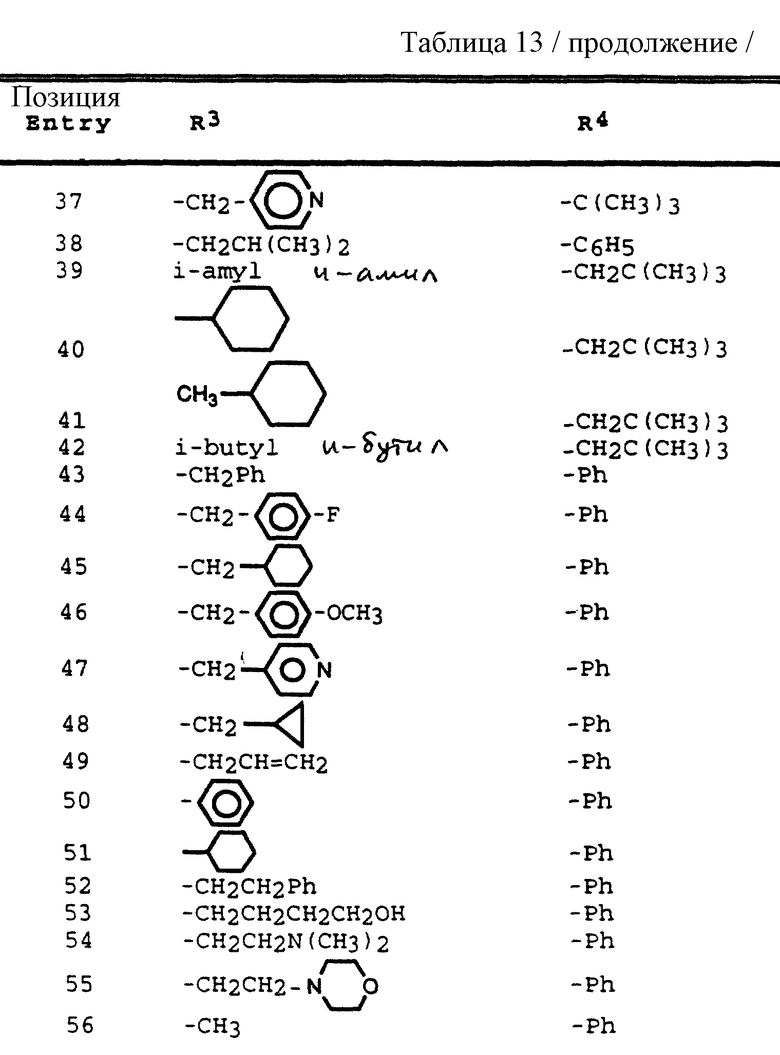
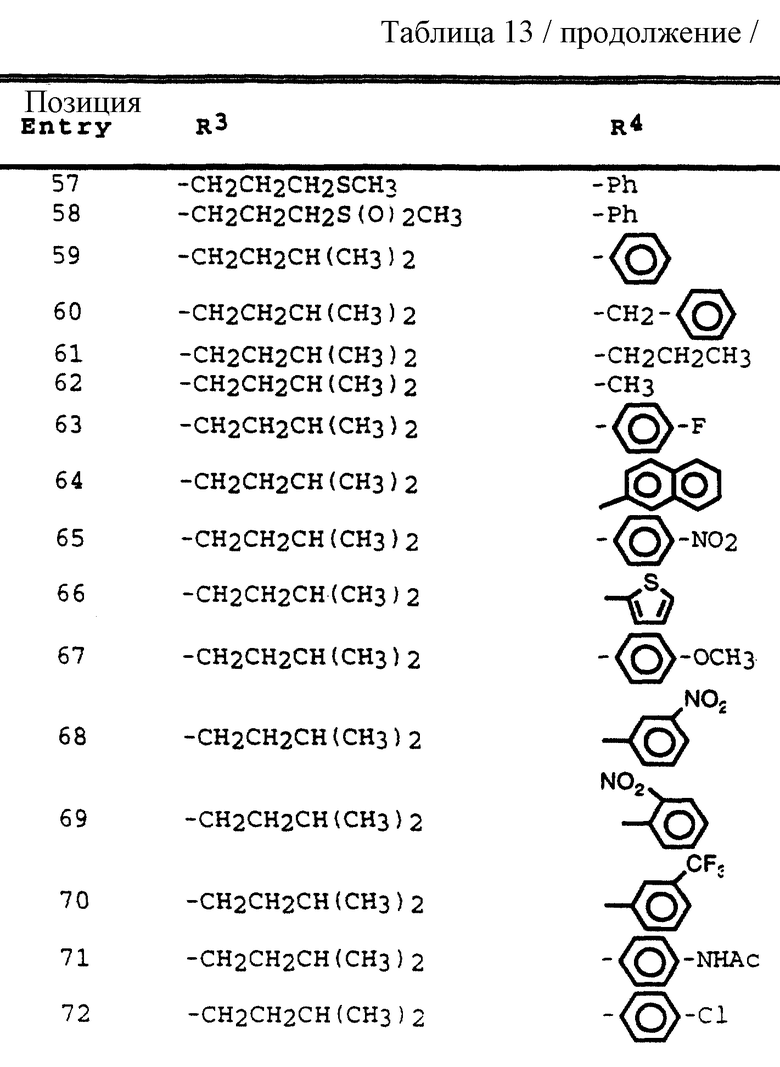
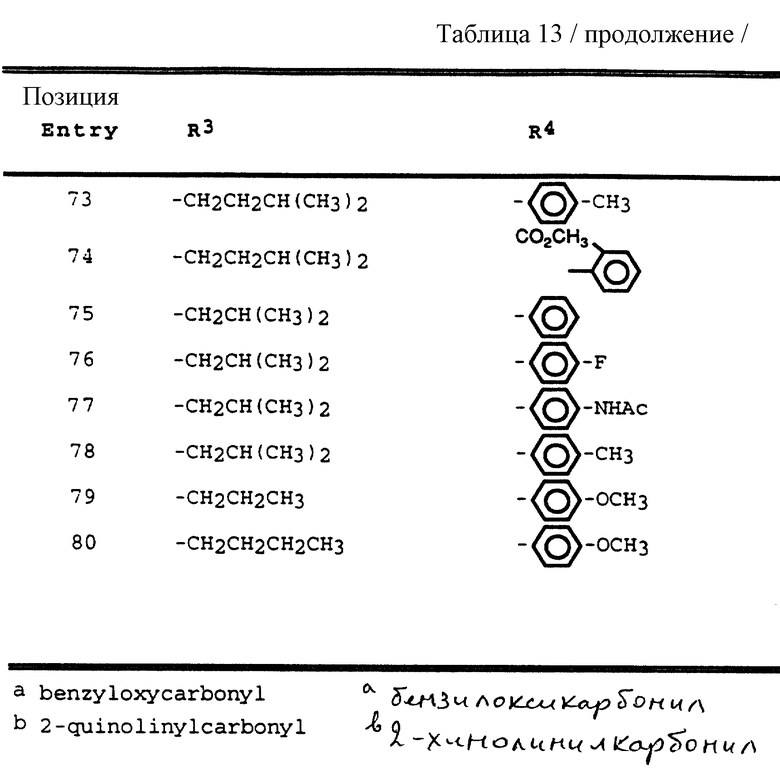
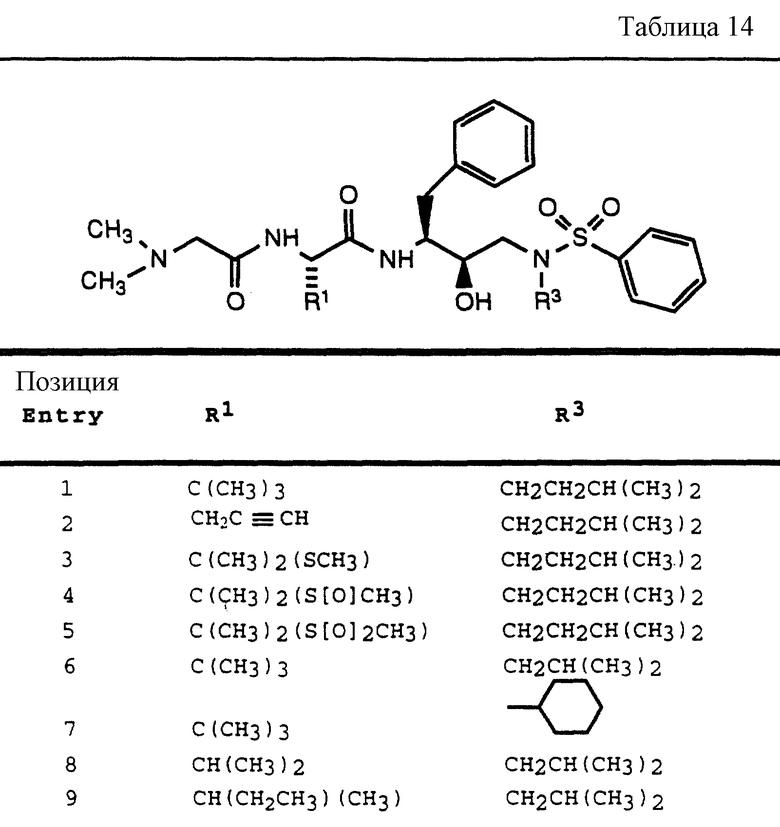
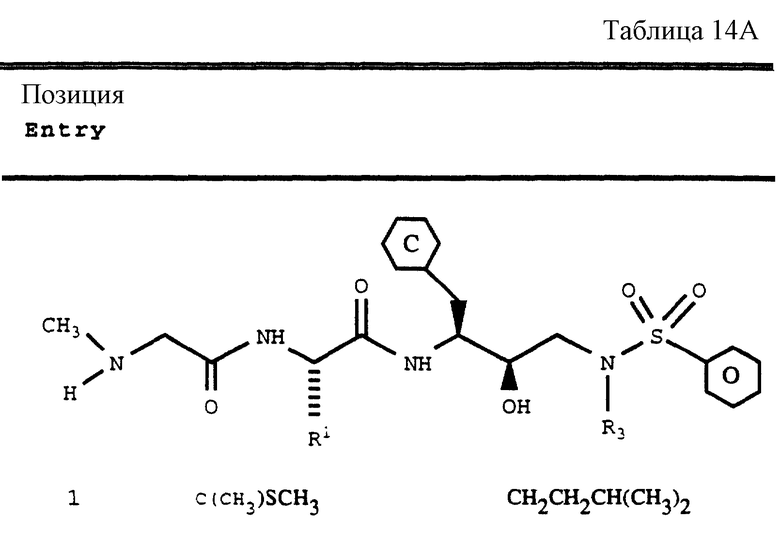
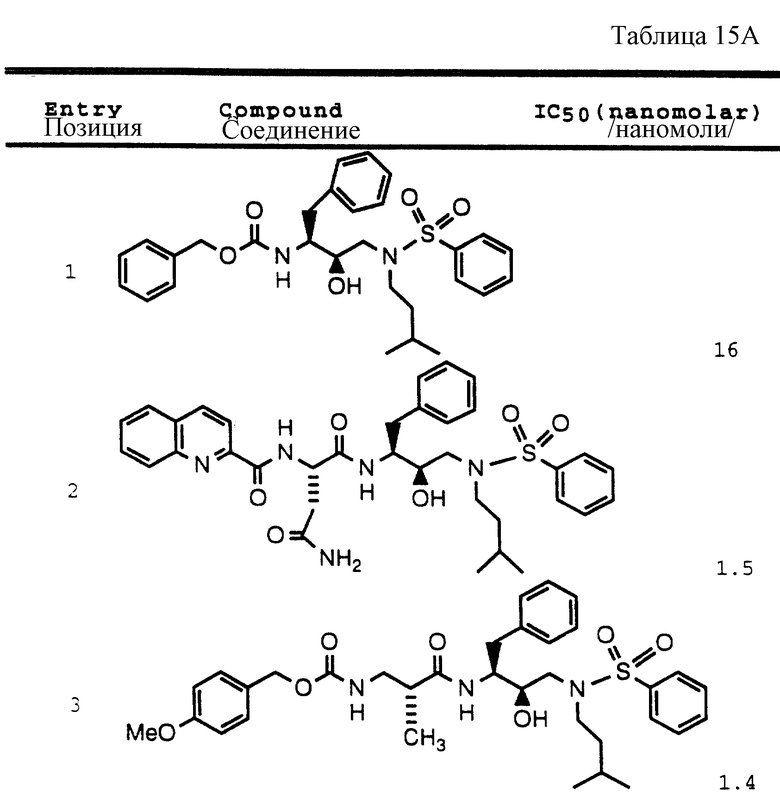
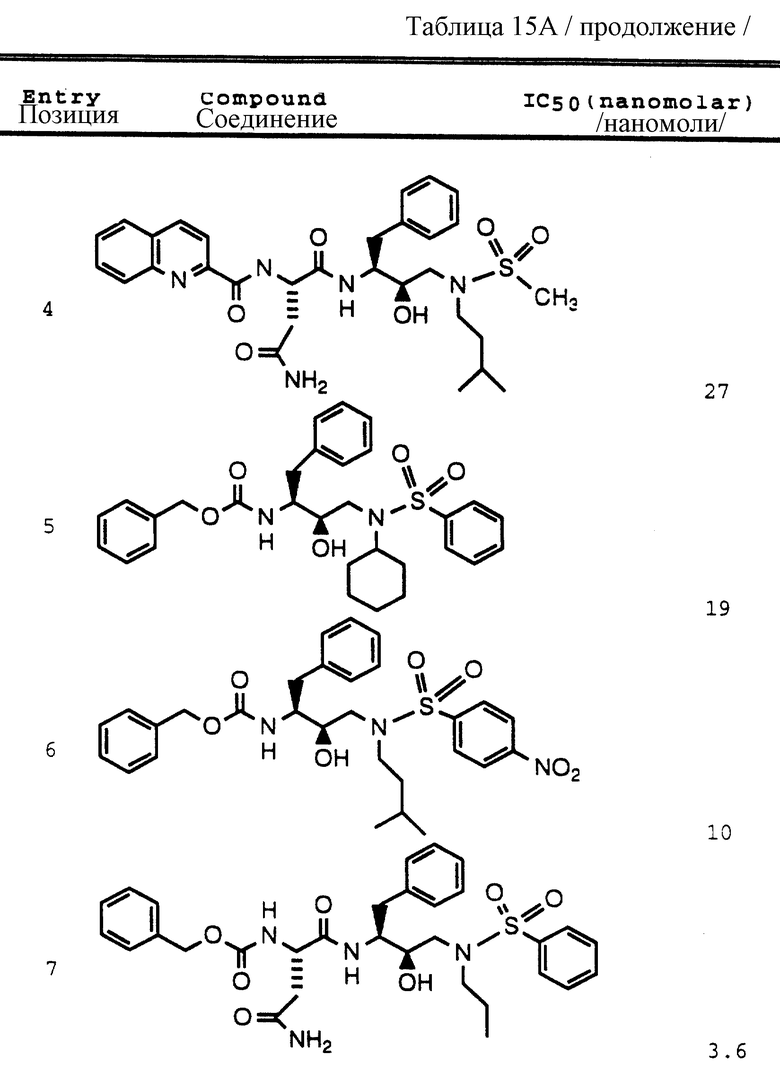
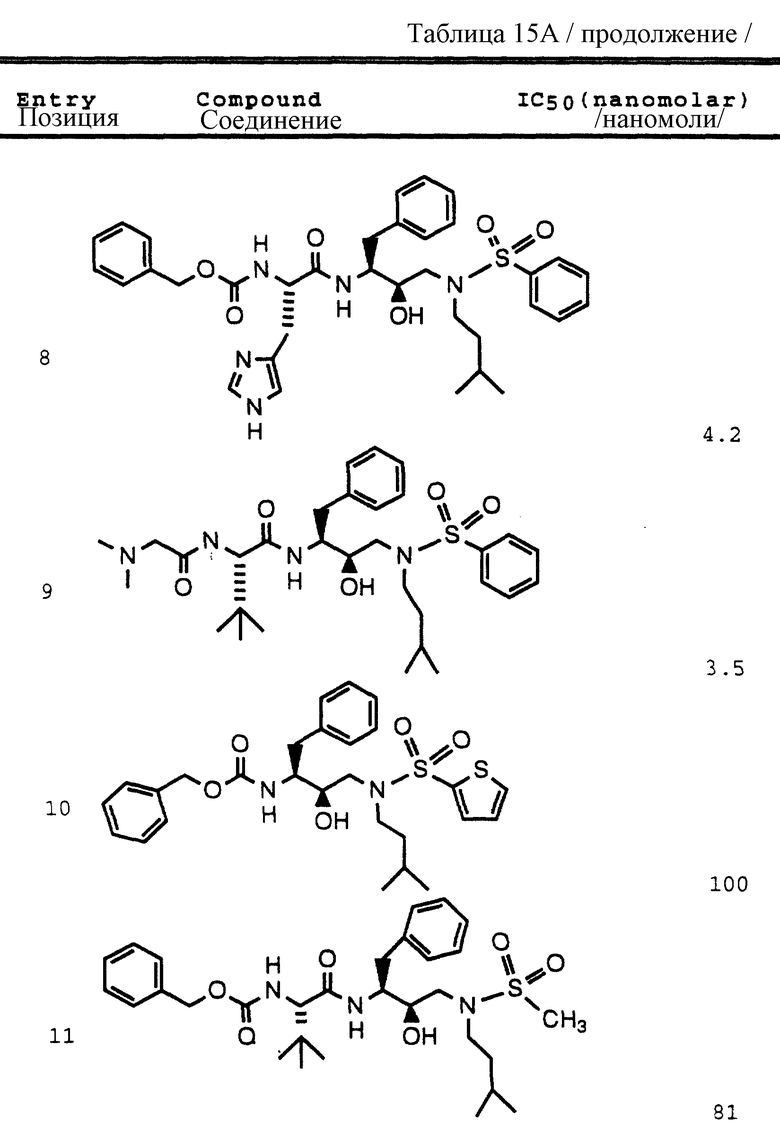
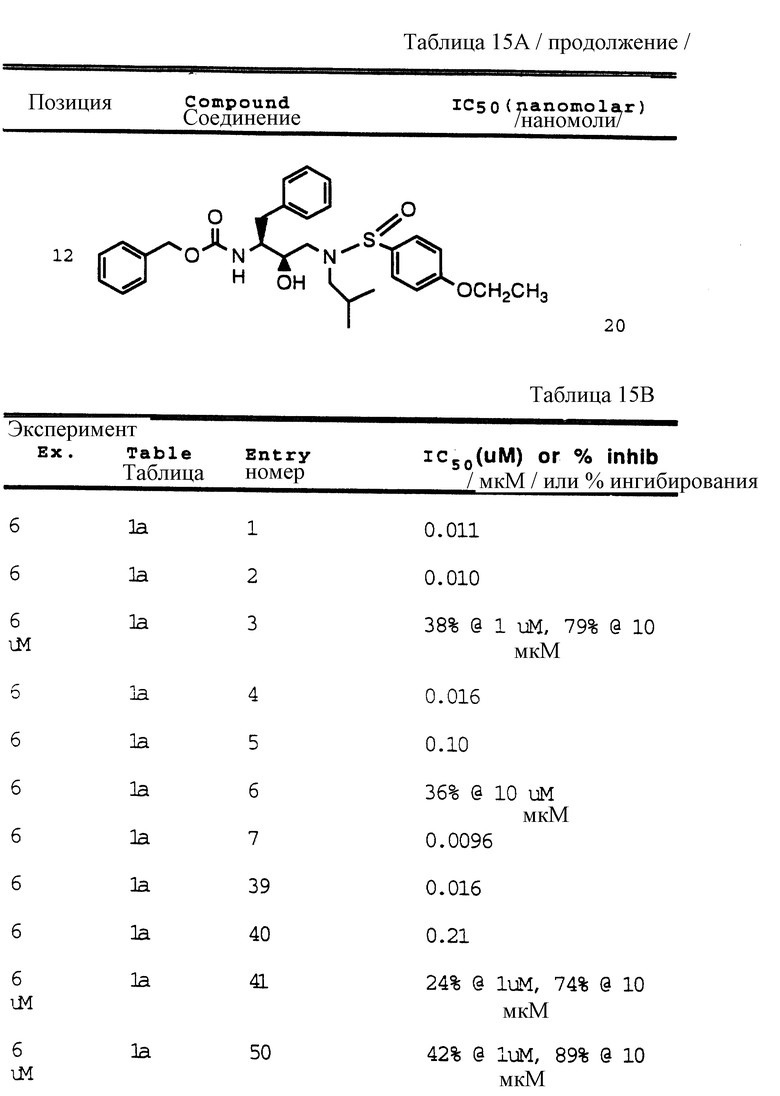

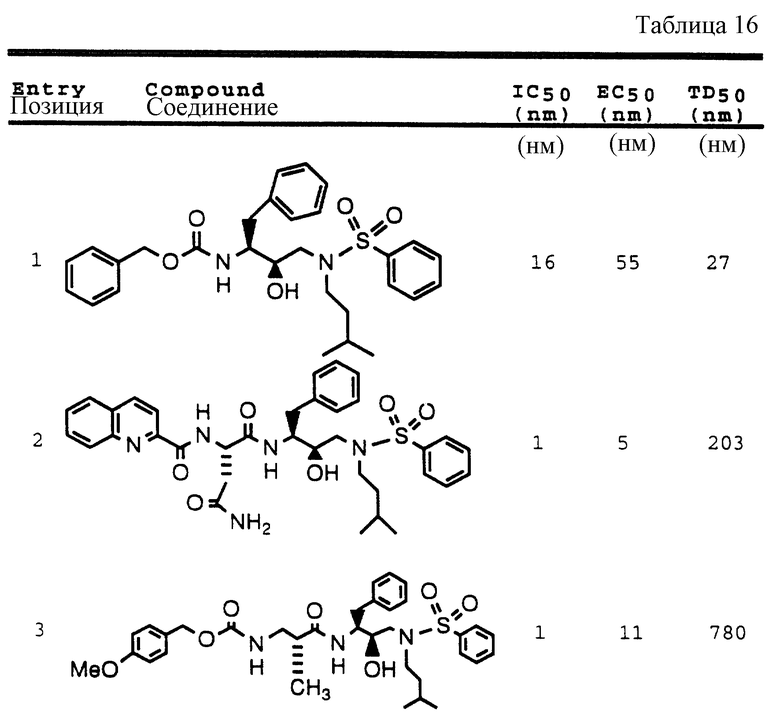
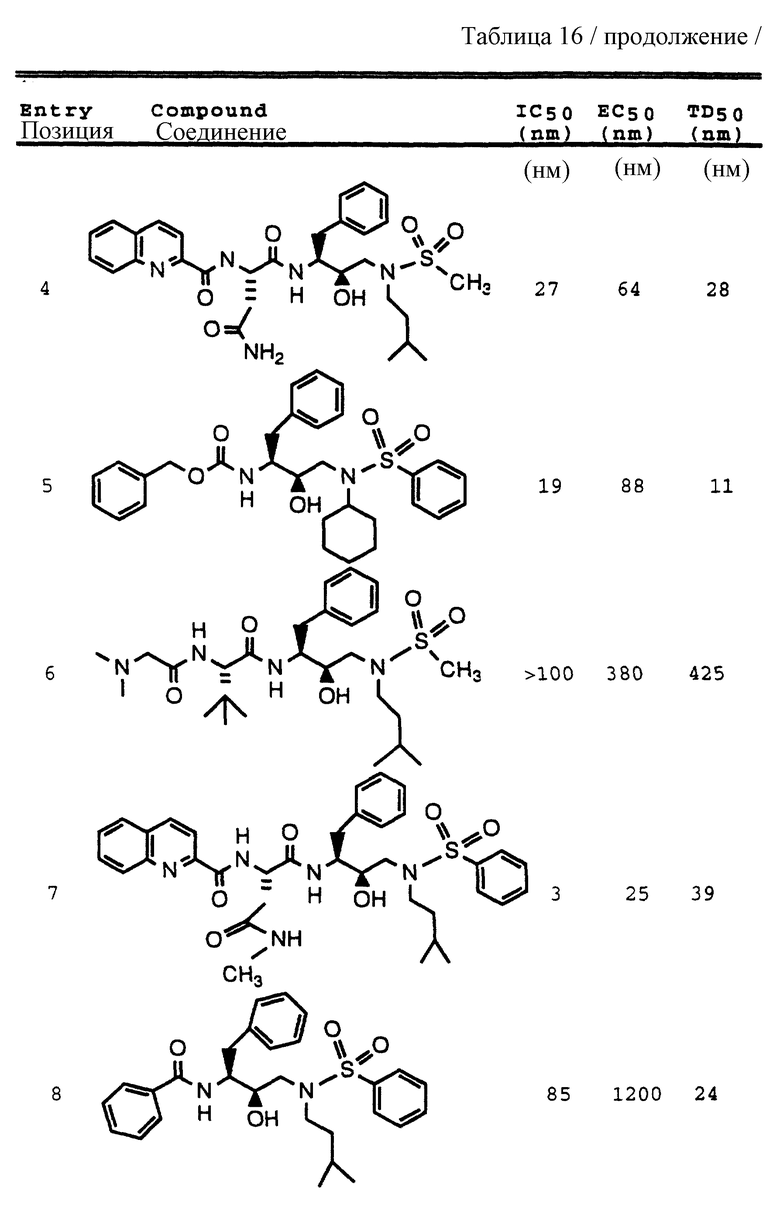
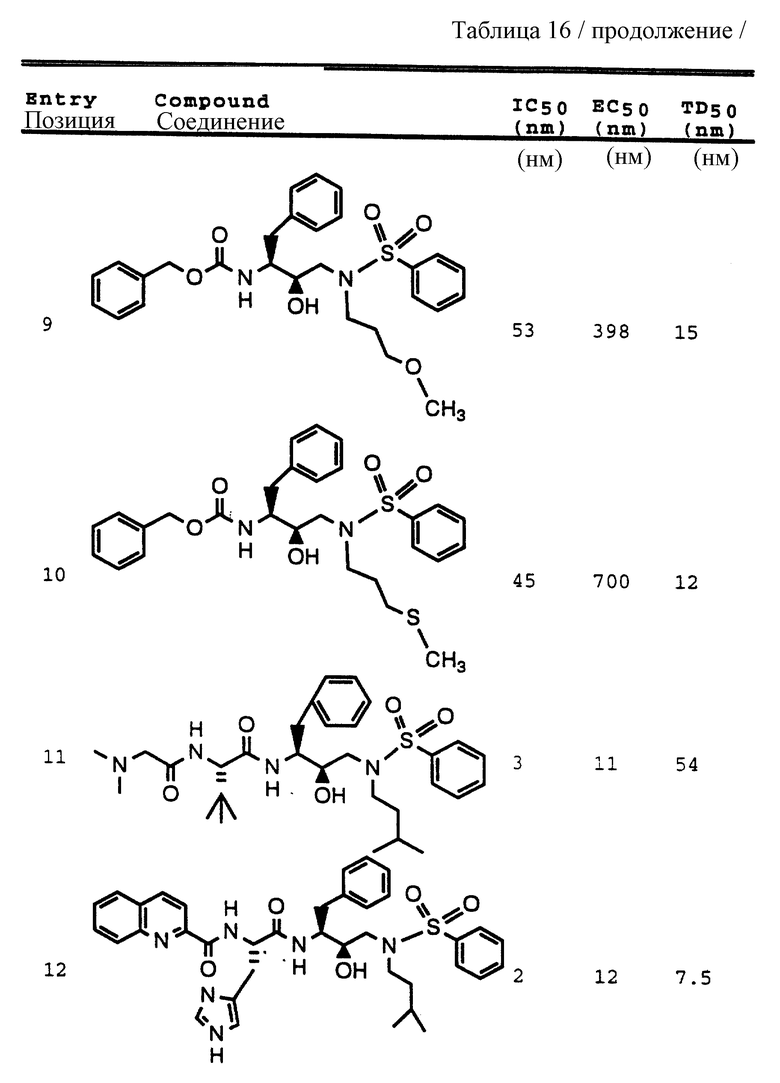
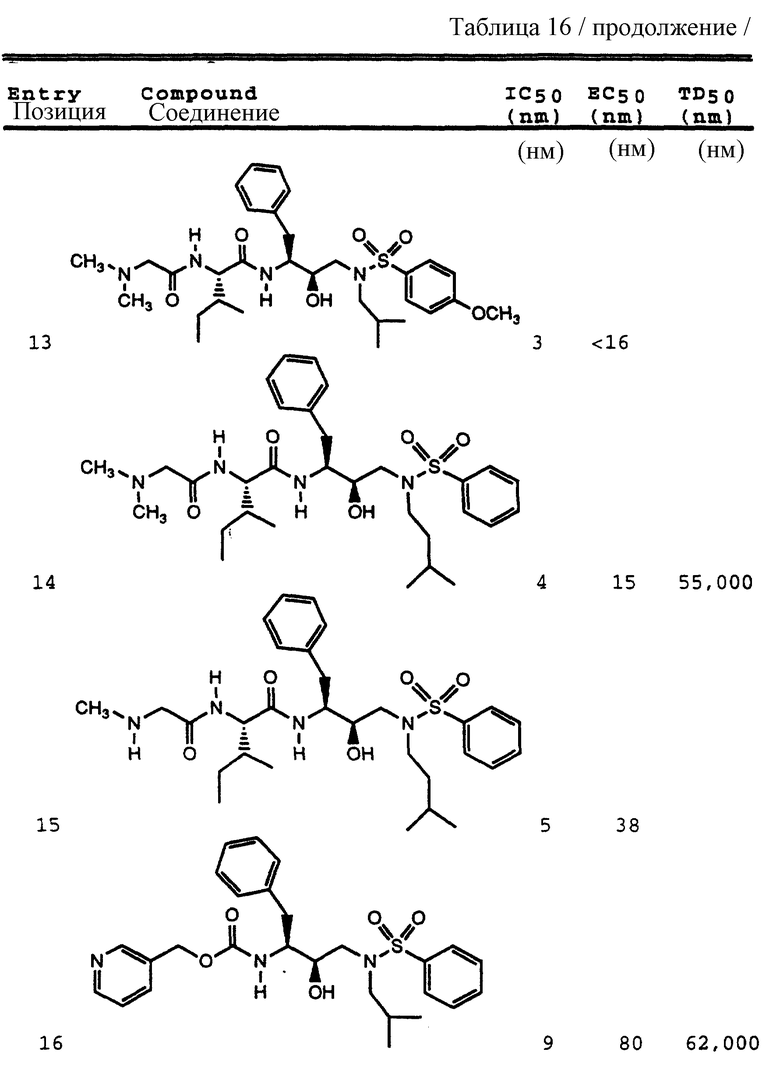
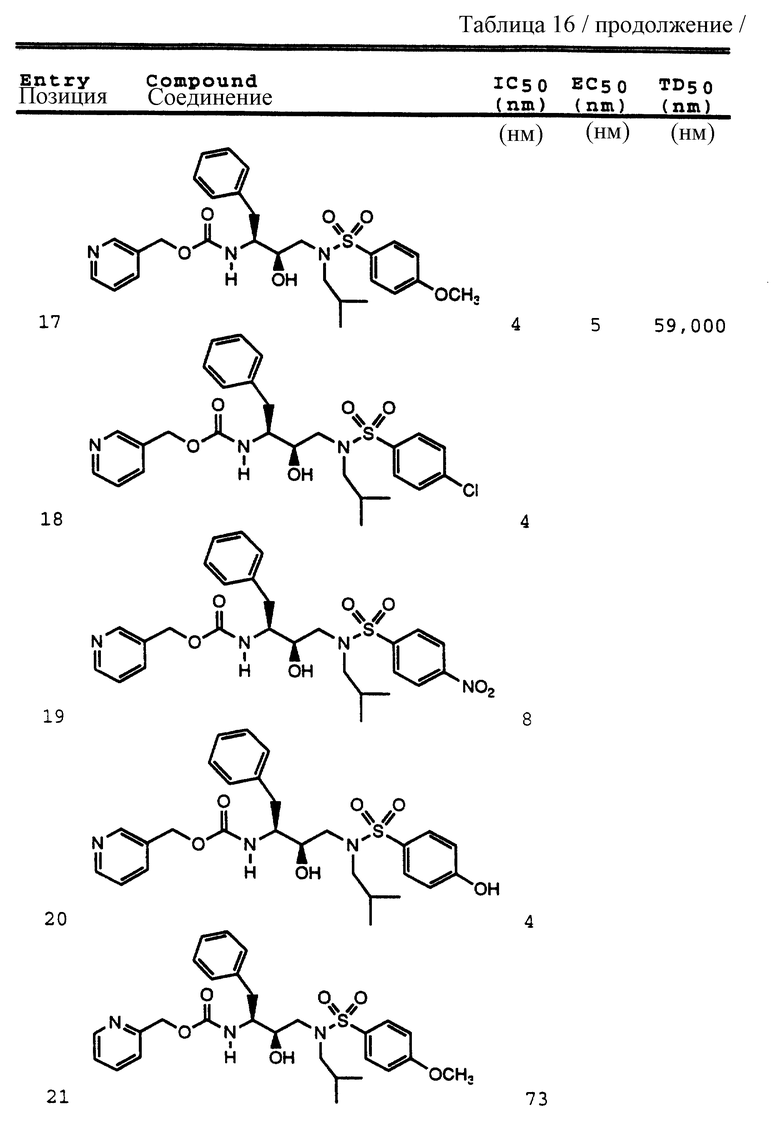
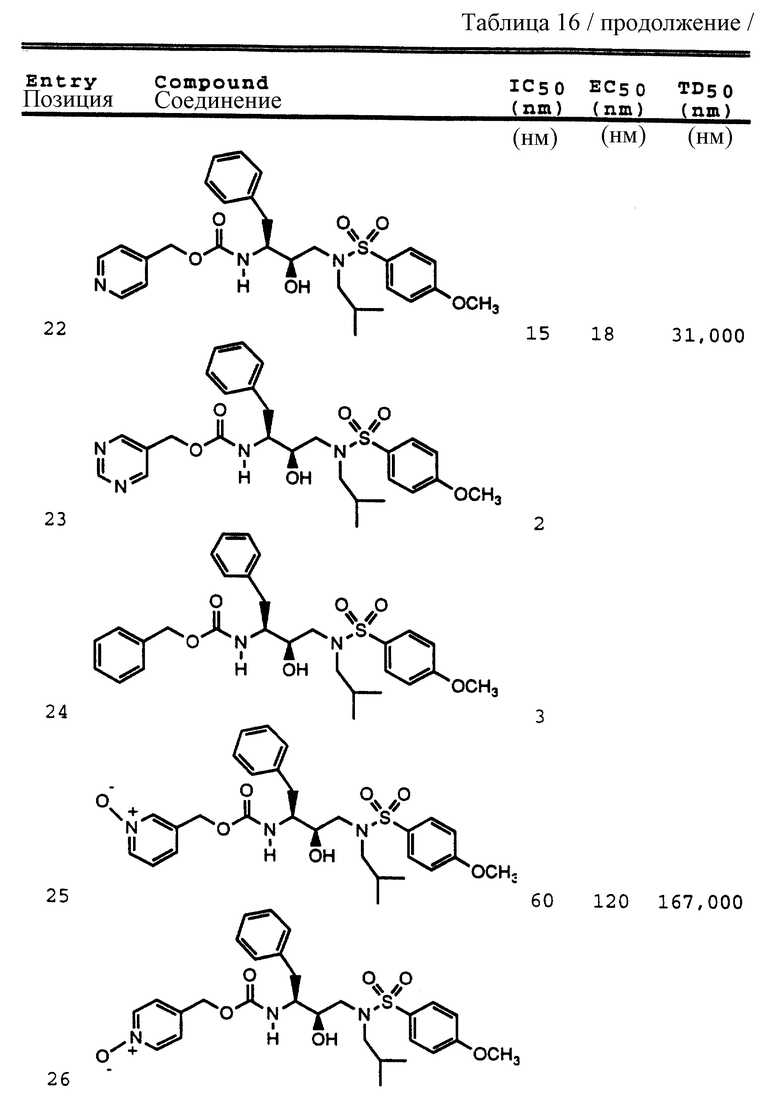

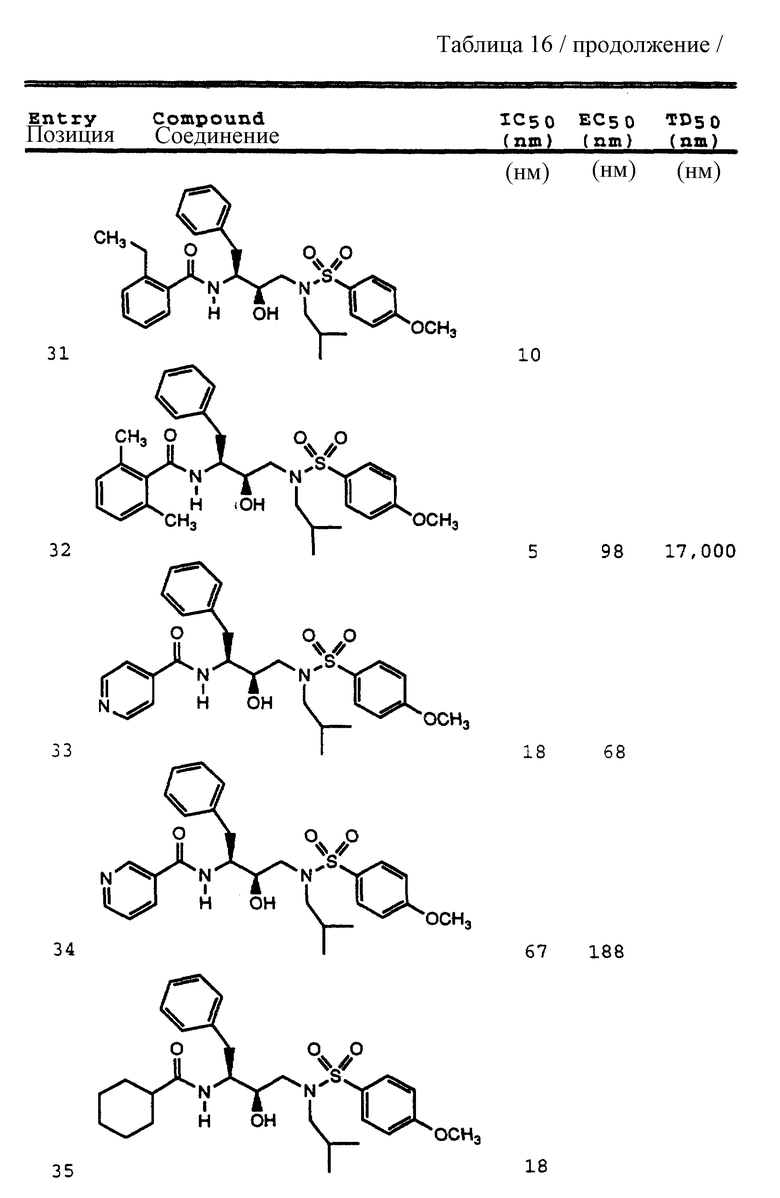
Изобретение относится к производным гидроксиэтиламиносульфонамидам формул I, II, III, где R - фенил-C1-C8алкоксикарбонил, где фенил может быть замещен C1-C8алкокси; хинолинилкарбонил, моно- или ди-C1-C8алкиламино-C1-C8алканоил; R' - H, C1-C8алкил; R1 - H, C1-C8алкил, C2-C8алкенил, -C(O)NH2, CH2C(O)NH2, -CH2C(O)NHCH3, C(CH3)2(SCH3), аминокислотная боковая цепь, такая как глицин; R1' и R1'' оба - H; R2 - фенил-C1-C8алкил; R3 - H, C1-C8алкил, C1-C8алкокси-C1-C8алкил, C2-C8алкенил; R4 - C1-C8алкил, фенил, метоксифенил; R6 - H; Y - O; x = 1, 2; t = 0 или 1. Соединения I, II, III относятся к ингибиторам ретровирусной протеазы и могут быть использованы в фармацевтической композиции для ингибирования ВИЧ-протеазы, а также в способе ингибирования ВИЧ-инфекций. 16 с. и 45 з.п.ф-лы, 23 табл.
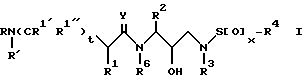
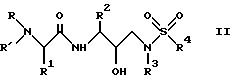
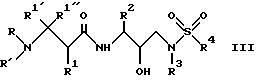
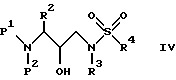
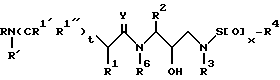
или их фармацевтически приемлемые соли,
где R - фенил-C1-C8алкоксикарбонил, где фенил может быть замещен C1-C8алкокси; хинолинилкарбонил, моно- или ди-C1-C8алкиламино-C1-C8алканоил; R' - водород, C1-C8алкил;
R1 - водород, C1-C8алкил, C2-C8алкенил, -C(O)NH2, CH2C(O)NH2, -CH2C(O)NHCH3, C(CH3)2(SCH3), аминокислотная боковая цепь, такая, как глицин;
R1' и R1'' оба - водород;
R2 - фенил-C1-C8алкил;
R3 - водород, C1-C8алкил, C1-C8алкокси-C1-C8алкил, C2-C8алкенил;
R4 - C1-C8алкил, фенил, метоксифенил;
R6 - водород;
Y - кислород;
x = 1 или 2;
t = 0 или 1.
фенилметил[2R-гидрокси-3-[(3-метилбутил)(метилсульфонил)амино] -1S-(фенилметил)пропил]карбамат;
фенилметил[2R-гидрокси-3-[(3-метилбутил)(фенилсульфонил)амино] -1S-(фенилметил)пропил]карбамат;
N1-[2R-гидрокси-3-[(3-метилбутил)(метилсульфонил)амино]-1S-(фенилметил)пропил]-2S-[(2-хинолинилкарбонил)амино]бутандиамид;
N1-[2R-гидрокси-3-[(3-метилбутил)(метилсульфонил)амино]-1S-(фенилметил)пропил]-2S-[(фенилметилоксикарбонил)амино]бутандиамид;
N1-[2R-гидрокси-3-[(3-метилбутил)(фенилсульфонил)амино]-1S-(фенилметил)пропил]-2S-[(2-хинолинилкарбонил)амино]бутандиамид;
N1-[2R-гидрокси-3-[(3-метилбутил)(фенилсульфонил)амино]-1S-(фенилметил)пропил]-2S-[(фенилметилоксикарбонил)амино]бутандиамид;
2S-[[(диметиламино)ацетил]амино]-N-[2R-гидрокси-3-[(3-метилбутил)(фенилсульфонил)амино]-1S-(фенилметил)пропил]-3,3-диметилбутанамид;
2S-[[(метиламино)ацетил]амино]-N-[2R-гидрокси-3-[(3-метилбутил)(фенилсульфонил)амино]-1S-(фенилметил)пропил]-3,3-диметилбутанамид;
N1-[2R-гидрокси-3-[(3-метилбутил)(фенилсульфонил)амино] -N4-метил-1S-(фенилметил)пропил]-2S-[(2-хинолинилкарбонил)амино]бутандиамид;
[3-[[2-гидрокси-3-[(3-метилбутил)(фенилсульфонил)амино]-1-(фенилметил)пропил] амино] -2-метил-3-оксопропил] -(4-метоксифенил)метиловый эфир, [1S-(1R*(S*),2S*]]-.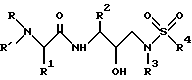
или их фармацевтически приемлемые соли,
где R - фенил-C1-C8алкоксикарбонил, где фенил может быть замещен C1-C8алкокси, хинолинилкарбонил, моно- или ди-C1-C8алкиламино-C1-C8алканоил; R' - водород, C1-C8алкил;
R1 - водород, C1-C8алкил, C2-C8алкенил, -C(O)NH2, CH2C(O)NH2, -CH2C(O)NHCH3, C(CH3)2(SCH3), аминокислотная боковая цепь, такая, как глицин;
R2 - фенил-C1-C8алкил;
R3 - C1-C8алкил, C1-C8алкокси-C1-C8алкил, C2-C8алкенил;
R4 - C1-C8алкил, фенил, метоксифенил.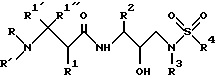
или их фармацевтически приемлемые соли,
где R - фенил-C1-C8алкоксикарбонил, где фенил может быть замещен C1-C8алкокси, хинолинилкарбонил, моно- или ди-C1-C8алкиламино-C1-C8алканоил; R' - водород, C1-C8алкил;
R1 - водород, C1-C8алкил, C2-C8алкенил, -C(O)NH2, CH2C(O)NH2, -CH2C(O)NHCH3, C(CH3)2(SCH3), аминокислотная боковая цепь, такая, как глицин;
R1' и R1'' оба - водород;
R2 - фенил-C1-C8алкил;
R3 - C1-C8алкил, C1-C8алкокси-C1-C8алкил, C2-C8алкенил;
R4 - C1-C8алкил, фенил, метоксифенил.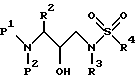
или их фармацевтически приемлемые соли,
где P1 - фенил-C1-C8алкоксикарбонил, фенилкарбонил, где фенил может быть замещен одним или двумя заместителями, выбранными из галогена, C1-C8алкила, C1-C8алкокси, групп -SO2CY3, -CH2SO2CH3, гетероарил-C1-C8алкоксикарбонил, гетероарилкарбонил, где гетероарил может быть замещен C1-C8алкилом и в качестве гетероатомов содержит один или два атома азота, C1-C8алкоксикарбонил, C3-C8циклоалкилкарбонил;
R2 - водород;
R2 - фенил-C1-C8алкил;
R3 - C1-C8алкил, C1-C8алкокси-C1-C8алкил, C3-C8циклоалкил;
R4 - C1-C8алкил, фенил, необязательно замещенный галогеном, C1-C8алкилом, гидрокси, NO2, C1-C8алкокси, диметиламиногруппой.
фенилметил[2R-гидрокси-3-[(2-метилпропил)(фенилсульфонил)амино] -1S-(фенилметил)пропил]карбамат;
фенилметил[2R-гидрокси-3-[(2-метилпропил)(4-метоксифенилсульфонил)амино] -1S-(фенилметил)пропил]карбамат;
фенилметил[2R-гидрокси-3-[(2-метилпропил)(4-фторфенилсульфонил)амино] -1S-(фенилметил)пропил]карбамат;
фенилметил[2R-гидрокси-3-[(2-метилпропил)(4-нитрофенилсульфонил)амино] -1S-(фенилметил)пропил]карбамат;
фенилметил[2R-гидрокси-3-[(2-метилпропил)(4-хлорфенилсульфонил)амино] -1S-(фенилметил)пропил]карбамат;
фенилметил[2R-гидрокси-3-[(2-метилпропил)(4-ацетамидофенилсульфонил)амино]-1S-(фенилметил)пропил]карбамат;
фенилметил[2R-гидрокси-3-[(2-метилпропил)(4-аминофенилсульфонил)амино] -1S-(фенилметил)пропил]карбамат;
фенилметил[2R-гидрокси-3-[(3-метилбутил)(4-метоксифенилсульфонил)амино] -1S-(фенилметил)пропил]карбамат;
фенилметил[2R-гидрокси-3-[(3-метилбутил)(4-фторфенилсульфонил)амино] -1S-(фенилметил)пропил]карбамат;
фенилметил[2R-гидрокси-3-[(3-метилбутил)(4-нитрофенилсульфонил)амино] -1S-(фенилметил)пропил]карбамат;
фенилметил[2R-гидрокси-3-[(3-метилбутил)(4-хлорфенилсульфонил)амино] -1S-(фенилметил)пропил]карбамат;
фенилметил[2R-гидрокси-3-[(2-метилпропил)(4-метоксифенилсульфонил)амино] -1S-(4-фторфенилметил)пропил]карбамат;
фенилметил[2R-гидрокси-3-[(2-метилпропил)(4-фторфенилсульфонил)амино] -1S-(4-фторфенилметил)пропил]карбамат;
фенилметил[2R-гидрокси-3-[(бутил)(фенилсульфонил)амино]-1S-(фенилметил)пропил]карбамат;
фенилметил[2R-гидрокси-3-[(циклогексилметил)(фенилсульфонил)амино] -1S-(фенилметил)пропил]карбамат;
фенилметил[2R-гидрокси-3-[(циклогексил)(фенилсульфонил)амино] -1S-(фенилметил)пропил]карбамат;
фенилметил[2R-гидрокси-3-[(пропил)(фенилсульфонил)амино]-1S-(фенилметил)пропил]карбамат;
пентамид 2S-[[(диметиламино)ацетил] амино] -N-2R-гидрокси-3-[(3-метилпропил)(4-метоксифенилсульфонил)амино]-1S-(фенилметил)пропил]-3S-метила;
пентамид 2S-[[(метиламино)ацетил] амино] -N-2R-гидрокси-3-[(4-метилбутил)(фенилсульфонил)амино]-1S-(фенилметил)пропил]-3S-метила;
пентамид 2S-[[(диметиламино)ацетил] амино]-N-2R-гидрокси-3-[(4-метилбутил)(фенилсульфонил)амино]-1S-(фенилметил)пропил]-3S-метила;
[2R-гидрокси-3-[[(4-метоксифенил)сульфонил] (2-метилпропил)амино] -1S-(фенилметил)пропиламин;
2R-гидрокси-3-[(2-метилпропил)(4-гидроксифенил)сульфонил] амино-1S-(фенилметил)пропиламин;
[2R-гидрокси-3-[(фенилсульфонил)(3-метилбутил)амино]-1S-(фенилметил)пропиламин;
[2R-гидрокси-3-[(фенилсульфонил)(2-метилпропил)амино]-1S-(фенилметил)пропиламин;
[2R-гидрокси-3-[(фенилсульфонил)(циклогексилметил)амино]-1S-(фенилметил)пропиламин;
[2R-гидрокси-3-[(фенилсульфонил)(циклогексил)амино]-1S-(фенилметил)пропиламин;
4-пиридинкарбоксамид N-[2R-гидрокси-3-[[(4-метоксифенил)сульфонил](2-метилпропил)амино]-1S-(фенилметил)пропил];
бензамид N-[2R-гидрокси-3-[[(4-метоксифенил)сульфонил] (2-метилпропил)амино]-1S-(фенилметил)пропил]-2,6-диметил;
бензамид N-[2R-гидрокси-3-[[(4-метоксифенил)сульфонил] (2-метилпропил)амино]-1S-(фенилметил)пропил]-2-метил;
бензамид N-[2R-гидрокси-3-[[(4-метоксифенил)сульфонил] (2-метилпропил)амино]-1S-(фенилметил)пропил]-2-этил;
бензамид N-[2R-гидрокси-3-[[(4-метоксифенил)сульфонил] (2-метилпропил)амино]-1S-(фенилметил)пропил]-2-хлор;
3-пиридилметиловый эфир [2R-гидрокси-3-[[(4-метоксифенил)сульфонил](2-метилпропил)амино]-1S-(фенилметил)пропил] карбаминовой кислоты;
N-оксид 3-пиридилметилового эфира [2R-гидрокси-3-[[(4-метоксифенил)сульфонил] (2-метилпропил)амино] -1S-(фенилметил)пропил] карбаминовой кислоты;
3-пиридилметиловый эфир [2R-гидрокси-3-[[фенилсульфонил] (2-метилпропил)амино]-1S-(фенилметил)пропил] карбаминовой кислоты;
4-пиридилметиловый эфир [2R-гидрокси-3-[[(4-метоксифенил)сульфонил](2-метилпропил)амино]-1S-(фенилметил)пропил] карбаминовой кислоты;
N-оксид 4-пиридилметилового эфира [2R-гидрокси-3-[[(4-метоксифенил)сульфонил] (2-метилпропил)амино] -1S-(фенилметил)пропил] карбаминовой кислоты;
3-пиридилметиловый эфир [2R-гидрокси-3-[[(4-хлорфенил)сульфонил](2-метилпропил)амино]-1S-(фенилметил)пропил] карбаминовой кислоты;
3-пиридилметиловый эфир [2R-гидрокси-3-[[(4-нитрофенил)сульфонил](2-метилпропил)амино]-1S-(фенилметил)пропил] карбаминовой кислоты;
3-пиридилметиловый эфир [2R-гидрокси-3-[[(4-фторфенил)сульфонил](2-метилпропил)амино]-1S-(фенилметил)пропил] карбаминовой кислоты;
3-пиридилметиловый эфир [2R-гидрокси-3-[[(4-гидроксифенил)сульфонил] (2-метилпропил)амино]-1S-(фенилметил)пропил] карбаминовой кислоты; или
5-пиридилметиловый эфир [2R-гидрокси-3-[[(4-метоксифенил)сульфонил](2-метилпропил)амино]-1S-(фенилметил)пропил] карбаминовой кислоты.
| УСТРОЙСТВО ДЛЯ КОМПЕНСАЦИИ СИСТЕМАТИЧЕСКИХ | 0 |
|
SU264795A1 |
| Массообменный аппарат | 1972 |
|
SU468641A1 |
| Автоматический огнетушитель | 0 |
|
SU92A1 |
| Способ щитовой проходки в сложных горногидрогеологических условиях | 1969 |
|
SU289204A1 |
| Шувалова Е.П | |||
| Инфекционные болезни | |||
| - М.: Медицина, 1990, с | |||
| 532 - 543 | |||
| Краткий курс молекулярной фармакологии/ Под редакцией В.П.Сергеева | |||
| - М., 1975, с | |||
| Печь-кухня, могущая работать, как самостоятельно, так и в комбинации с разного рода нагревательными приборами | 1921 |
|
SU10A1 |
