Настоящее изобретение относится к производным 5-гетероцикло-1,5-бензодиазепина и их фармацевтически приемлемым солям, которые проявляют агонистическую активность в отношении ХЦК-А рецепторов, что позволяет им модулировать гормоны гастрин и холецистокинин (ХЦК) у млекопитающих.
Холецистокинины (ХЦК) и гастрин представляют собой пептиды с родственными структурами, которые присутствуют в желудочно-кишечной ткани и в центральной нервной системе. Холецистокинины включают в себя ХЦК-33, нейропептид, который первоначально был выделен в форме пептида, состоящего из тридцати трех аминокислот, его карбокси-концевой октапептид, ХЦК-8 (также встречающийся в природе нейропептид), и 39- и 12-аминокислотные формы. Гастрин существует в форме пептидов из 34, 17 или 14 аминокислот (34-, 17- и 14-аминокислотные формы), с минимальной активной последовательностью, представляющей собой C-концевой тетрапептид, Трп-Мет-Асп-Фен-NH2 (ХЦК-4), которая является общим структурным элементом, имеющимся как у ХЦК, так и у гастрина.
ХЦК и гастрин являются желудочно-кишечными гормонами и нейротрансмиттерами в невральной и периферической системах и выполняют свои соответствующие биологические функции посредством связывания со специфическими рецепторами, локализованными в различных участках тела. Имеются по меньшей мере два подтипа холецистокининовых рецепторов, именуемых ХЦК-А и ХЦК-Б, оба обнаруживаются в периферической и центральной нервной системе.
ХЦК-А рецептор, именуемый обычно как рецептор "периферического типа", обнаруживают главным образом в нервных волокнах поджелудочной железы, желчного пузыря, подвздошной кишки, пилорического сфинктера и на вагусных афферентных нервных волокнах. ХЦК рецепторы А-типа обнаруживают также в мозге в дискретных областях, где они служат для обеспечения многих функций ЦНС. Благодаря способности ХЦК-8 и ХЦК-селективных агонистов А-типа подавлять потребление пищи у некоторых видов животных возник значительный интерес к созданию новых веществ, функционирующих как ХЦК агонисты селективные по отношению к рецептору А-типа, которые могли бы служить средствами, снижающими аппетит.
ХЦК-Б или гастриновые рецепторы обнаруживают в периферических нейронах, в гладких желудочно-кишечных мускулах и слизистой оболочке желудочно-кишечного тракта, особенно в париетальных клетках, ECL клетках, D клетках и главных клетках. ХЦК-Б рецепторы преобладают также в мозге и они вовлечены в регуляцию тревоги, возбуждения и в действие нейролептических агентов.
Известна [патент США N 4988692] группа производных 3-ациламино-1-алкил-5-фенил-1,5-бензодиазепина, которые действуют как антагонисты холецистокинина, изменяя или блокируя действие эндогенного гормона на его рецепторы.
Известны [патент США N 4490304; заявка РСТ N W090/06937; заявка РСТ N W091/19733] пептидные производные, проявляющие ХЦК-А агонистическую активность. Было обнаружено, что такие соединения пригодны как для регуляции аппетита, так и для лечения и/или профилактики желудочно-кишечных расстройств или расстройств центральной нервной системы у животных, и в частности у человека.
Известно [патент США N 5187154] применение нейропептида холецистокинина (ХЦК) для контроля опорожнения желудка у пациентов в стадии раннего неинсулинзависимого диабета и страдающих быстрым опорожнением кишечника. Известно [патент США N 5187154] также, что соединения, которые подавляют опорожнение кишечника, могут быть использованы для облегчения или устранения симптомов, связанных с ранними или пред-диабетами. Конкретные симптомы включают повышенные уровни глюкозы и инсулина в крови, инсулиновую устойчивость, повышенную чувствительность к инфекции или гликурию при поддержании опорожнения кишечника в нормальных пределах.
В настоящее время мы открыли новую группу производных 5-гетероцикло-1,5-бензодиазепина, которые обладают агонистической активностью в отношении ХЦК-А рецептора, что дает им возможность модулировать гормоны гастрин и холецистокинин (ХЦК) у млекопитающих. Некоторые из этих соединений обладают также антагонистической активностью по отношению к ХЦК-Б рецепторам.
Таким образом, согласно настоящему изобретению предложены 5-гетероцикло-1,5-бензодиазепины общей формулы (I)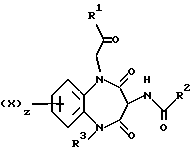
и их фармацевтически приемлемые соли,
где
X является водородом или галогеном;
R1 является группой -NR4R5,
где R4 обозначает C3-6-алкил, а
R5 обозначает фенил, замещенный в пара-положении метоксилом,
R2 является индолом, фенилом или группой -NHR11,
R3 является гетероциклической группой, присоединенной к азоту через атом углерода ее циклического кольца, выбранной из пиридила, пиримидинила или пиразолила, последний в свою очередь трижды замещен C1-4-алкилом, z равно 1.
Если R1 представляет собой группу NR4R5, то примеры приемлемых групп включают в себя группы, у которых R4 является C3-6-алкилом, таким как пропил или изопропил, а R5 представляет собой фенил, замещенный в пара-положении метоксилом.
Если R2 представляет собой группу NHR11, то R11 как правило является фенилом, возможно замещенным карбоксигруппой. Если R11 является однозамещенной фенильной группой, то заместитель как правило находится в мета-положении.
Примеры наиболее приемлемых R2 групп включают в себя индол или группу NHR11. Как правило R2 выбирают из группы индола, фенильной группы или группы NHR11. Более конкретно, R2 представляет собой индол, фенил или NHR11.
Если R3 представляет собой пиридил, то примеры приемлемых групп включают в себя 2-пиридил, 3-пиридил и 4-пиридил.
Если R3 представляет собой пиримидинил, то примеры приемлемых групп включают в себя 2-пиримидинил или 5-пиримидинил.
Если R3 представляет собой пиразолил, то примеры приемлемых групп включают 1,3,5-триметил-1Н-пиразол-4-ил.
Примеры наиболее приемлемых R3 групп включают пиридил, например 2-пиридил, 3-пиридил, 4-пиридил, пиримидинил, в частности 2-пиримидинил или 5-пиримидинил, либо 1,3,5-триметил-1Н-пиразол-4-ил.
Наиболее перспективная группа соединений согласно изобретению включает в себя те соединения, у которых R1 представляет собой группу NR4R5, в которой R4 является пропилом или изопропилом, a R5 является фенилом, возможно замещенным в пара-положении метоксилом; R2 представляет собой фенил или группу NHR11, где R11 представляет собой фенил или фенил, замещенный карбоксигруппой, причем заместитель предпочтительно находится в мета-положении, или индол; R34 представляет собой пиридил, например 2,3 или 4-пиридил, пиримидинил, например 2- или 5-пиримидинил, или 1,2,5-триметил-1Н-пиразол-4-ил, а X является водородом или фтором.
Наиболее интересен класс соединений по настоящему изобретению, который обладает очень высоким и селективным сродством к ХЦК-А рецептору, при этом исключительная эффективность наблюдается в тех случаях, когда R2 является индольной группой. В рамках этой группы наиболее перспективными соединениями являются те, у которых R4 представляет собой изопропил, R5 представляет собой п-метоксифенил, a R3 представляет собой пиридил, пиримидинил или 1,3,5-триметил-1Н-пиразол-4-ил или, более конкретно, R3 представляет собой 3-пиридил, а X представляет собой водород.
Предпочтительные соединения по изобретению включают в себя:
{ 1-[изопропил-(4-метокси-фенил)-карбамоилметил]-2,4-диоксо- 5-пиридин-2-ил-2,3,4,5-тетрагидро-1Н-бензо[b] [1,4] диазепин-3-ил} - амид 1Н-индол-2-карбоновой кислоты;
{ 1-[изопропил-(4-метокси-фенил)-карбамоилметил] - 2,4-диоксо-5-пиримидин-2-ил-2,3,4,5-тетрагидро-1Н-бензо[b] [1,4] диазепин-3-ил}-амид 1Н-индол-2-карбоновой кислоты;
2-[2,4-диоксо-3-(3-фенил-уреидо)-5-пиридин-2- ил-2,3,4,5-тетрагидро-бензо[b] [1,4] диазепин-1-ил]-N-изопропил-N- (4-метокси-фенил)-ацетамид и их энантиомеры.
Наиболее предпочтительным соединением по изобретению является
{ 1-[изопропил-(4-метокси-фенил)-карбамоилметил] -2,4-диоксо-5-пиридин- 3-ил-2,3,4,5-тетрагидро-1Н-бензо[b] [1,4]диазепин-3-ил}-амид 1Н-индол-2-карбоновой кислоты; и его энантиомеры.
В данном описании термин алкил, как правило, используют для обозначения алифатических изомеров соответствующего алкила как с прямой цепью, так и с разветвленной цепью. Так например, подразумевается, что C1-4-алкил включает в себя метил, этил, н-пропил, изопропил, н-бутил, изобутил, трет-бутил, н-фенил и т.д.
В данном описании термин циклоалкил обозначает все алициклические изомеры соответствующего алкила. Например, термин C3-6-алкил обозначает, в частности, такие группы, как циклопропил, циклопентил и циклогексил.
Подразумевается, что термин галоген обозначает F, Cl, Br или I.
Термин тетразол как группа или часть группы относится к (1Н)-тетразол-5-ильной группировке и ее таутомерам.
Специалистам следует иметь в виду, что в соединениях формулы (I) существуют стереоцентры. Соответственно, настоящее изобретение включает в себя все возможные стереоизомеры и геометрические изомеры формулы (I) и включает в себя не только рацемические соединения, но также и оптически активные изомеры. Если требуется соединение формулы (I) в виде отдельного энантиомера, то он может быть получен либо посредством разделения конечного продукта, либо путем стереоспецифического синтеза либо из изомерно чистого исходного материала, либо из любого приемлемого промежуточного соединения. Разделение конечного продукта, промежуточного соединения или исходного материала может быть осуществлено любым способом, известным из уровня техники [E.L.Eliel, Stereochemistry of Carbon Compounds (McGraw Hill, 1962); S.H.Wilen, Tables of Resoling Agents]. Кроме того, в ситуациях, когда возможны таутомеры соединений формулы (I), подразумевается, что настоящее изобретение включает в себя все таутомерные формы соединений.
Специалисту также следует иметь в виду, что соединения по настоящему изобретению также могут быть использованы в виде фармацевтически приемлемой соли. Физиологически приемлемые соли соединений формулы (I) включают в себя обычные соли, образованные из фармацевтически приемлемых неорганических и органических кислот, а также четвертично-аммониевые соли, полученные при добавлении кислот. Более конкретные примеры приемлемых солей включают в себя соляную, бромоводородную, серную, фосфорную, азотную, перхлорную, фумаровую, уксусную, пропионовую, янтарную, гликолевую, муравьиную, молочную, малеиновую, винную, лимонную, памоевую, малоновую, гидроксималеиновую, фенилуксусную, глутаровую, бензойную, салициловую, фумаровую, толуолсульфоновую, метансульфоновую, нафталин-2-сульфоновую, бензолсульфоновую и тому подобные кислоты. Другие кислоты, такие как щавелевая кислота, не являясь сами фармацевтически приемлемыми, могут быть использованы для получения солей, приемлемых в качестве промежуточных соединений при получении соединений по изобретению и их фармацевтически приемлемых солей. Приводимые здесь ссылки на соединение по изобретению включают в себя как соединения формулы (I), так и их фармацевтически приемлемые соли.
Соединения по настоящему изобретению обладают ХЦК-А агонистической активностью и могут рассматриваться как полные или частичные агонисты холецистокинина, поскольку они связываются с ХЦК-А рецепторами, полностью или частично стимулируя сжатие желчного пузыря и/или понижая потребление пищи у животных.
В качестве агонистов ХЦК-А рецепторов соединения по настоящему изобретению являются приемлемыми в качестве средств, понижающих аппетит при лечении ожирения, а также связанных с ним патологий, таких как диабеты или гипертензия. Более того, описанные здесь соединения предложены для новых способов индукции сытости, для регуляции аппетита и модификации всасывания пищи у млекопитающих, особенно у человека, для регуляции аппетита, лечения ожирения и поддержания потери веса. Соединения также приемлемы для лечения неинсулинзависимых диабетических состояний, ассоциированных с быстрым опорожнением кишечника.
Кроме того, определенные соединения по настоящему изобретению, могут также проявлять некоторую антагонистическую активность на особых сайт-специфических ХЦК-Б и гастриновых рецепторах, что демонстрируется их ингибированием ХЦК-4 стимулируемого сокращения изолированного продольного мускульно-мышечного сплетения подвздошной кишки морской свинки и стимулируемой пентагастрином секреции кислоты в изолированной крысиной слизистой оболочке желудка в известных [M.Patel and C.F.Spraggs in Br.J.Pharmac., (1992), 106, 275-282; J.J.Reeves and R.Stables in Br.J.Pharmac., (1985), 86,677-684] тестах.
Относительные величины сродства соединений по изобретению к ХЦК-А и ХЦК-Б рецепторам могут быть определены традиционными способами [Fomos et al. , J.Pharmacol.Exp.Ther., 1992, 261, 1056-1063].
Способность соединений по изобретению ингибировать секрецию желудочного сока, например стимулируемую пентагастрином секрецию желудочного сока, может быть определена у находящейся в сознании крысы с желудочной фистулой [Hedges and Parsons Journal of Physiology, 1977, 267, 191-194].
Соединения формулы (I) ингибируют или задерживают опорожнение кишечника, что может быть определено с помощью стандартных тестов. Так, например, крысы, лишенные пищи в течение 18 часов, могут быть предварительно обработаны тестируемым соединением, введенным внутрибрюшинно за определенный промежуток времени (20 минут) до подачи в качестве пищи метилцеллюлозы, которую вводят посредством желудочного зонда. Пища содержит маркерный элемент, фенол красный. Спустя определенные предварительно установленные интервалы времени крыс умерщвляют и определяют количество пищи в желудке путем измерения концентрации присутствующего маркерного вещества. Затем это значение сравнивают с контрольным животным, которое не было предварительно обработано тестируемым соединением.
Было установлено, что соединения по изобретению имеют наиболее благоприятный профиль активности в силу их высокой биологической доступности при пероральном введении в сочетании с относительно хорошей растворимостью в воде.
В частности, соединение формулы (I) или его фармацевтически приемлемая соль пригодна для применения в терапии, и в частности при лечении человека.
Специалисту следует иметь в виду, что ссылка на лечение распространяется как на профилактику, так и на лечение установившихся заболеваний или симптомов. Кроме того, следует иметь в виду, что количество соединения по изобретению, требуемое для лечения, варьирует в зависимости от природы подлежащего лечению состояния и возраста и состояния пациента и должно быть, в конечном счете, находиться на усмотрении лечащего врача или ветеринара. В общем случае, однако, дозы, применяемые для лечения взрослого человека, как правило лежат в интервале от 0,02 до 5000 мг в день, например 1-1500 мг в день. Требуемая доза для удобства может быть представлена в виде единичной дозы или разделенных доз, вводимых через определенные интервалы, например в виде двух, трех, четырех или более поддоз в день.
Хотя и возможно терапевтическое введение соединения по настоящему изобретению в виде сырого химического соединения, все же предпочтительно представлять активный ингредиент в виде фармацевтического препарата, содержащего соединение формулы (I) или его фармацевтически приемлемую соль совместно с одним или более чем одним фармацевтически приемлемым носителем и возможно другими терапевтическими и/или профилактическими ингредиентами. Носитель(и) должен быть "приемлемым" в том смысле, что он является совместимым с другими ингредиентами препарата и не опасен для самого реципиента.
Препараты включают в себя препараты, специально приготовленные для перорального, трансбуккального, парентерального, ректального введения или введения в виде имплантата, однако предпочтительным является пероральное введение. Для трансбуккального введения препарат может иметь форму таблеток или лепешек, изготовленных соответствующим образом. Таблетки и капсулы для перорального введения могут содержать обычные эксципиенты, такие как связующие агенты (например, сироп, гуммиарабик, желатин, сорбит, трагакант, крахмальный клейстер или поливинилпирролидон), наполнители (например, лактозу, сахар, микрокристаллическую целлюлозу, кукурузный крахмал, фосфат кальция или сорбит), смазочные материалы (например, стеарат магния, стеариновую кислоту, тальк, полиэтиленгликоль или диоксид кремния), разрыхлители (например, картофельный крахмал или крахмальный гликолят натрия) или смачивающие агенты, такие как лаурилсульфат натрия. Таблетки могут быть покрыты оболочкой с использованием известных из уровня техники способов. Приемлемые оболочки для таблеток включают обычные энтеросолюбильные оболочки.
С другой стороны, соединения по настоящему изобретению, могут быть включены в жидкие препараты для перорального введения, такие, например, как водные или масляные суспензии, растворы, эмульсии, сиропы или эликсиры. Кроме того, препараты, содержащие эти соединения, могут быть представлены в виде сухого продукта, предназначенного для смешивания перед употреблением с водой или другим приемлемым носителем. Такие жидкие препараты могут содержать обычные добавки, такие как суспендирующие агенты, например сироп сорбита, метилцеллюлозу, сироп из глюкозы/сахара, желатин, гидроксиэтилцеллюлозу, карбоксиметилцеллюлозу, гель стеарата алюминия или гидрогенизированные пищевые жиры; эмульгирующие агенты, такие как лецитин, моноолеат сорбитана или гуммиарабик; неводные носители (которые могут включать в себя пищевые масла), такие как миндальное масло, фракционированное кокосовое масло, масляные эфиры, пропиленгликоль или этиловый спирт; и консерванты, такие как метил- или пропил-п-гидроксибензоаты или сорбиновую кислоту. Такие препараты также могут быть изготовлены в виде суппозиториев, содержащих, например, обычные суппозиторные основы, такие как масло какао или другие глицериды.
При пероральном введении соединения по изобретению обычно вводят в виде таблеток в энтеросолюбильных оболочках или капсул, изготовленных из энтеросолюбильных материалов или покрытых энтеросолюбильной оболочкой.
Кроме того, препараты, содержащие соединения по настоящему изобретению могут быть изготовлены для парентерального введения посредством инъекции или непрерывного вливания. Препараты для инъекций могут иметь такие формы, как суспензии, растворы или эмульсии в масляных или водных носителях, и могут содержать препаративные агенты, такие как суспендирующие, стабилизирующие и/или диспергирующие агенты. С другой стороны, активный ингредиент может иметь форму порошка, предназначенного для смешивания перед применением с приемлемым носителем (например, стерильной апирогенной водой).
Препарат, содержащий соединение по изобретению, может быть изготовлен также в виде депо-препарата. Такие длительно действующие препараты могут вводиться путем имплантации (например, подкожно или внутримышечно) или посредством внутримышечной инъекции. Соответственно, могут быть приготовлены препараты соединений по изобретению с приемлемыми полимерными или гидрофобными материалами (например, в виде эмульсии в подходящем масле), ионообменными смолами или в виде слаборастворимых производных, таких например как слаборастворимая соль.
Препараты, содержащие соединения по изобретению могут содержать от 0,1 до 99% активного ингредиента, для таблеток и капсул это обычно 30-95% и 3-50% - для жидких препаратов. Соединения общей формулы (I) и их соли могут быть получены общими способами, описанными ниже. В следующем описании группы R1-R12 и X такие, как определено для соединений формулы (I), если это не оговорено особо, или могут являться группами, которые можно в них превратить.
Для любого из этих процессов может оказаться необходимым и/или желательным защитить чувствительные или реакционноспособные группы. Защитные группы применяют согласно стандартным методам органического синтеза [N.W. Green and P. G.M.Watts (1991) Protection Groups in Organic Synthesis, John Wiley & Sons] . Эти группы удаляют на соответствующей стадии синтеза, применяя методы, известные из уровня техники. Так, например, аминогруппы могут быть защищены группой, выбранной из арилметила (например, бензила), ацила или сульфонила, в частности аллилсульфонила, фталимида или тозила; если это желательно, то производят последующее удаление защитной группы путем гидролиза или гидрогенолиза, если это целесообразно, применяя стандартные условия. Гидроксильные или карбоксильные группы могут быть защищены с применением любой стандартной гидроксил- или карбоксил-защитной группы. Примерами приемлемых гидроксил- или карбоксил-защитных групп являются группы, выбранные из алкила, например метила, трет-бутила или метоксиметила, арилметила, например бензила, дифенилметила или трифенилметила, гетероциклических групп, таких как тетрагидропиранил, ацила, например ацетила или бензоила, и силильных групп, таких как триалкилсилил, например трет-бутилдиметилсилил. Гидроксил-защитные группы могут быть удалены соответствующими способами. Так, например, алкильные, силилацильные и гетероциклические группы могут быть удалены посредством гидролиза в кислых или щелочных условиях. Арилметильные группы, такие как трифенилметил, могут быть легко удалены посредством гидролиза в кислотных условиях. Аралиметильные группы, такие как бензил, могут быть отщеплены путем гидрогенолиза в присутствии катализатора из благородного металла, такого как палладий на угле. Силильные группы также могут быть легко удалены с помощью источника фторидных ионов, такого как тетра-н-бутиламмонийфторид.
Согласно первому общему способу А соединения формулы (I) могут быть получены посредством реакции взаимодействия амина формулы (III), где R1, R2, R3, X и z имеют значения, определенные в формуле (I)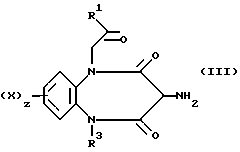
с соединением R11Y (IV), в котором Y является группой -NCO, HNCOCl или NHCORa, где Ra является феноксигруппой, замещенной нитрогруппой, или 1-имидазольнои группой.
Реакция обычно протекает в присутствии подходящего растворителя, такого как галогеноуглеводород (например, дихлорметана), эфир (например, тетрагидрофурана) или нитрил (например, ацетонитрила), либо их смеси при температуре в пределах от 0 до 80oC.
Соединения формулы (IV), в которой Y является -NCO, имеются в продаже или могут быть получены посредством взаимодействия аминов H2N-R11 с фосгеном или трифосгеном в подходящем растворителе, таком как метиленхлорид. Соединения формулы (IV), в которой Y является NHCOCl, также образуются в результате реакции взаимодействия аминов H2NR11 с фосгеном или трифосгеном в подходящем растворителе, таком как метиленхлорид. Соединения формулы (IV), в которой Y является NHCORa, а Ra является 1-имидазольной группой, получают путем обработки аминов HN2-R11 диимидазолкарбонилом в приемлемом растворителе (дихлорметане, эфире, тетрагидрофуране) при температуре в пределах от 0 до 80oC (обычно при комнатной температуре). Соединения формулы (IV), в которой Y является HNCORa, а Ra является феноксигруппой, замещенной нитрогруппой, получают в результате взаимодействия аминов H2N-R11 с соответствующим хлорформиатом RaCOCl в присутствии основания (пиридина, триэтиламина) в подходящем растворителе (дихлорметане) и при температуре от 0 до 50oC.
Согласно следующему общему способу Б соединения формулы (I) могут быть получены посредством реакции взаимодействия промежуточного соединения формулы (V)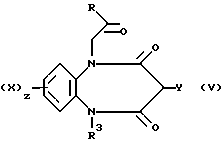
в которой Y является группой -NCO, -NHCOCl или NHCORa, где Ra является феноксигруппой, замещенной нитрогруппой, или 1-имидазольной группой, с амином (VI)
H2N-R11 (VI)
и возможно в присутствии основания, такого как третичный амин (например, триэтиламин).
Реакция обычно протекает в соответствующем растворителе, таком как галогенизированный углеводород (например, дихлорметан) или эфир (например, тетрагидрофуран), либо амид (например, N,N-диметилформамид), при температуре, как правило, варьирующей от комнатной температуры до температуры дефлегмации растворителя.
Как правило, соединения формулы (V) получают in situ из амина (III).
В частном случае способа (Б), когда Y является группой NHCORa, а Ra является 1-имидазольной группой, имидазолид (V) может быть образован in situ, и в этом случае амин формулы (VI) смешивают с соединением формулы (III)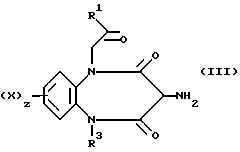
в присутствии карбонилдиимидазола при описанных выше условиях.
В способе Б, когда Y является группой NHCORa, а Ra является феноксигруппой, замещенной нитрогруппой, реакцию с первичным амином (VI) предпочтительно проводить в присутствии такого основания, как третичный амин, например триэтиламин.
В способе Б, когда Y является изоцианатной группой -N=C=O, реакцию с первичным амином (VI) предпочтительно проводят в апротонном растворителе, таком как галогенуглеводород, например метиленхлорид. Обычно изоцианат получают in situ перед добавлением первичного амина (VI).
Соединения формулы (V), в которой Ra является возможно замещенной феноксигруппой, могут быть получены из первичного амина (III) посредством реакции с соответствующим замещенным нитрогруппой фенилхлорформиатом в присутствии такого основания, как пиридин. Реакция может быть осуществлена в таком растворителе, как галогеноуглеводород, например дихлорметан, при температуре от 0 до 50oC.
Соединения формулы (V), в которой Ra является 1-имидазольной группой, могут быть получены посредством реакции соединения формулы (III) с карбонилдиимидазолом в присутствии подходящего растворителя, такого как галогенированный углеводород (например, дихлорметана) или эфир (например, тетрагидрофурана) при температуре в пределах от 0 до 80oC (обычно при комнатной температуре).
Соединения формулы (V), в которой Y является изоцианатной группировкой -N=C=O или карбамоилхлоридом -NHCOCl, могут быть получены из первичного амина (III) посредством реакции с фосгеном (COCl2) или трифосгеном в подходящем растворителе, таком как метиленхлорид.
Согласно следующему общему способу В соединения формулы (I) также могут быть получены посредством реакции взаимодействия соединения формулы (VII)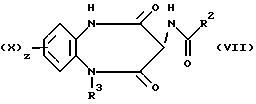
с галогенацетамидом, имеющим формулу (VIII)
R1COCH2Hal (VIII)
где Hal = Cl или Br.
Реакцию обычно проводят посредством обработки соединения формулы (VII) сильным основанием, таким как гидрид натрия, в полярном апротонном растворителе, таком как N,N-диметилформамид, с последующим взаимодействием с ацетилгалидом (VIII).
Ацетилгалид (VIII) получают путем взаимодействия амина R-1-H с соответствующим галоацетилбромидом в дихлорметане при 0oC в присутствии подходящего основания, такого как триэтиламин.
Амины R1-H, где R1 является группой -NR4R5 могут быть получены путем восстановительного алкилирования амина H2N-R5 соответствующим альдегидом или кетоном.
Согласно общему способу Г, соединения общей формулы (I) могут также быть получены в результате реакции взаимодействия промежуточного соединения формулы (III) с кислотами формулы (IX), приводимой ниже:
HOOC-R2 (IX)
Данная реакция взаимодействия промежуточного соединения формулы (III) с кислотой формулы (IX) может быть проведена в присутствии подходящего дегидратирующего агента, такого как, например, дициклогексикарбодиимид (ДЦК), 1-(3-диметиламинопропил)- 3-этилкарбодиимидгидрохлорид (ЭДХ) или 4-бензотриазол-1- илокситрис-(диметиламино)фосфонийгексафторфосфат (БОФ), в частности в присутствии подходящего спирта (N-гидроксилсукцинимида или N-гидроксибензотриазола).
С другой стороны, соединения общей формулы (I) могут быть получены посредством реакции взаимодействия промежуточных соединений формулы (III) с активированным производным кислоты (IX), таким как ее хлорангидрид или ангидрид, включая смешанные ангидриды.
Предпочтительные растворители для общего способа Г включают N,N-диметилформамид или дихлорметан. Предпочтительные температуры лежат между 0 и 60oC. Предпочтительные для этой реакции основания включают в себя триэтиламин, N-метилморфолин или N,N-диметиламинопирин (ДМАП).
Согласно следующему общему способу (Д) соединения формулы (I) могут быть получены в результате реакции взаимодействия соединения, соответствующего соединению формулы (I), в которой R3 представляет собой водород, с галогенидом R3Hal (где Hal является Cl или Br, a R3 является группой, определенной в формуле (I), более конкретно гетероарильной группой, например, пиридилом, пиримидинилом и т.д.). Реакцию обычно проводят в присутствии металлической меди и ацетата калия и в присутствии такого растворителя, как диметилсульфоксид или N,N-диметилформамид и предпочтительно реакцию проводят при температуре в пределах от 25 до 100oC.
Согласно следующему общему способу (Е) соединения по изобретению могут быть превращены в другие соединения по изобретению. Так, например, соединения формулы (I), в которой R8 является группой (CH2)bCO2H, могут быть получены в результате реакции взаимодействия соединения формулы (I), в которой R8 является водородом, с соединением Br(CH2)bCOOR*, где R* является C1-4-алкилом, в присутствии сильного основания, такого как гидрид натрия, с последующим удалением карбоксил-защитной группы обычными способами, например посредством кислотного или щелочного гидролиза.
Соединения формулы (I), в которой R11 является фенилом, замещенным алкоксикарбонильной группой, также могут быть гидролизованы обычными способами, например посредством кислотного гидролиза с образованием соединения формулы (I), в которой R11 является фенилом, замещенным карбоксилом.
Соединения формулы (III) могут быть получены путем восстановления соединений формулы (X)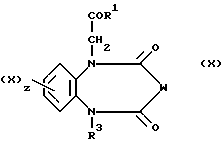
в которой W является CH-N3 или C=N-NHPh.
Соединения формулы (X), в которой W является CH-N3, могут быть восстановлены до соединения формулы (III) посредством гидрогенизации в присутствии подходящего катализатора, такого как 5-10%-ный палладий на носителе, таком как уголь или карбонат кальция, или оксида платины (IV).
Реакция обычно происходит в присутствии такого растворителя, как алканол (например, этанол), эфир (например, этилацетат) или уксусная кислота.
Соединения формулы (X), в которой W является C=N-NHPh, могут быть восстановлены до соединения формулы (III) в результате реакции с цинком и уксусной кислотой. Эта реакция может быть проведена при температуре в интервале 0-50oC.
Соединения формулы (X), в которой W является CHN3, могут быть получены из соединения формулы (X), в которой W является группой CH2, посредством обработки сильным основанием, таким как гидрид натрия или гексаметилдисилазид калия, либо трет-бутилат калия, а затем три-изопропилбензенсульфонилазидом или ди-третбутоксиазидодикарбоксилатом. Реакция обычно протекает в таком растворителе, как эфир (например, тетрагидрофуран), при температуре в интервале от -78oС до 20oC.
Соединения формулы (III) могут быть также получены в результате реакции взаимодействия соединения формулы (X), в которой W является CH2, с подходящим основанием, таким как бис(триметилсилил)амид натрия и O-(дифенил-фосфенил)гидроксиламин, в таком растворителе, как диметилформамид.
Соединения формулы (X), в которой W является группой C=NNHPh или CH2, могут быть получены посредством реакции взаимодействия ортофенилендиамина (XI) с двухкислотным хлоридом (XII), в котором Q является CH2 или C=NNHPh, в подходящем растворителе, таком как эфир, например тетрагидрофуран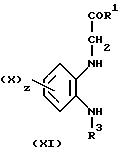
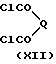
Соединение формулы (XII), в которой Q является группой C=NNHPh, может быть получено посредством реакции взаимодействия кетомалоновой кислоты с фенилгидразоном с последующим взаимодействием с пентахлоридом фосфора.
Соединения формулы (XI) являются либо известными соединениями, либо могут быть получены аналогичными способами. Так, например, соединения формулы (XI) могут быть получены посредством алкилирования амина (XIII)
Так амин (XIII) может быть подвергнут реакции взаимодействия с соединением R1COCH2Hal, в котором Hal является хлором или бромом, возможно в присутствии иодида натрия в таком растворителе, как N,N-диметилформамид, и такого основания, как карбонат калия.
Другой способ получения промежуточного соединения формулы (III), как указано ниже, включает в себя обработку промежуточного соединения формулы (XIV) гидридом натрия с последующим добавлением галогеноацетамида (VIII) в приемлемом растворителе, таком как N,N-диметилформамид, при 0oC с получением защищенного соединения формулы (XIV)
Промежуточное соединение (XVI) может быть превращено в амин (III) посредством обработки HBr в метиленхлориде.
Промежуточное соединение (XIV) получают из промежуточного соединения формулы (XVI) в результате реакции взаимодействия с бензилоксихлорфомиатом в дихлорметане, применяя триэтиламин в качестве основания. Эта реакция обычно протекает при комнатной температуре.
Промежуточное соединение (XVI) получают из фенилендиамина (XIII) в результате следующего процесса.
В результате реакции диамина (XII) с п-метоксибензоилхлоридом с последующим восстановлением образующегося амида с помощью литий-алюминийгидрида образуется N-защищенный диамин (XVII)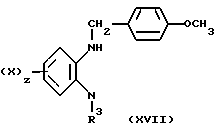
В результате реакции соединения (XVII) с двукислотным хлоридом (XII; Q= C= NNHPh) с последующим восстановлением с помощью цинка и уксусной кислоты получается амин (XVIII)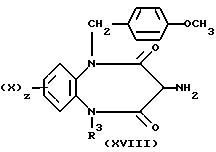
Соединение формулы (XVIII) может быть превращено в целевое соединение (XVI) посредством реакции с Ce(NO2)6NH4 (церий-аммонийнитрат).
Соединения формулы (VII) могут быть получены из соединения формулы (XVI) посредством общих способов А, Б и В, описанных выше.
Соединения, соответствующие соединениям формулы (I), но в которой R3 представляет собой водород, могут быть получены из соответствующего амина (III), в котором R3 представляет собой водород, применяя общие способы А, Б и В. Соединения формулы (III), в которой R3 является водородом, могут быть получены согласно общим способам, описанным выше для получения соединений формулы (III), в которой R3 является гетероциклической группой, но с использованием промежуточных соединений, в которых R3 является п-метоксибензильной группой, которая затем может быть удалена соответствующим способом.
Таким образом, в результате реакции диамина (XIX)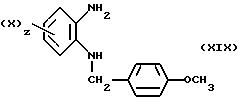
с галогенацетамидом (VIII) получают двузамещенный диамин (XX)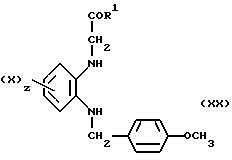
В результате реакции взаимодействия соединения (XX) с двукислотным хлоридом (XII), в котором Q является группой C=NNHPh, с последующим восстановлением с помощью цинка и уксусной кислоты получают бензодиазепин (XIX)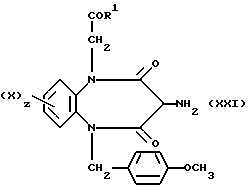
В результате реакции взаимодействия соединения формулы (XXI) с церий-аммонийнитратом получают соединение, соответствующее соединению формулы (III), в которой R3 является водородом.
Диамин (XIX) может быть получен в результате реакции нитрофторопроизводного (XXII)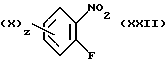
с п-метоксибензиламином с последующим восстановлением нитрогруппы.
Соединения формулы (X), в которой W является группой CH2, могут быть получены посредством реакции соединения формулы (XXIII), в которой X, z и R1 имеют значения, определенные в формуле (I),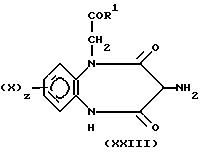
с бромидом R3Br (у которого R3 имеет значение, определенное выше) в присутствии порошка меди и ацетата калия. Реакцию предпочтительно проводить в полярном растворителе, таком как диметилформамид, и при нагревании.
Соединение (XXIII) может быть получено посредством реакции взаимодействия диамина (XXIV), у которого X1, z и R1 имеют значения, определенные выше,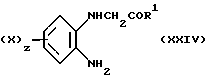
с малонилдихлоридом аналогично способу, описанному для получения соединения формулы (X), в которой W является CH2.
Соединения формулы (I) содержат по меньшей мере один асимметричный атом углерода, а именно атом углерода диазепинового кольца, к которому присоединена группировка замещенной мочевины. Конкретные энантиомеры соединений формулы (I) могут быть получены посредством разделения рацемической смеси соответствующими способами, такими как хиральная ЖХВР. В альтернативном случае требуемый энантиомер может быть получен из соответствующего энантиомерного амина формулы (III) посредством любого из способов, описанных выше для получения соединений формулы (I) из амина (III). Энантиомеры амина (III) могут быть получены из рацемического амина (II) с помощью соответствующих процедур, таких как солеобразование с приемлемой оптически активной кислотой или посредством препаративной хиральной ЖХВР.
Примеры
Следующие примеры приведены для иллюстрации синтеза некоторых конкретных соединений по настоящему изобретению и для иллюстрации частных вариантов применения общих способов А-Д. Соответственно приводимый ниже раздел Примеры никоим образом не служит для ограничения объема изобретения, рассматриваемого здесь.
Используемые здесь символы и условные обозначения, применяемые в данных процессах, схемах и примерах, идентичны применяемым в современной научной литературе, например в Journal of American Chemical Society. Если это не оговорено особо, все исходные материалы получали из коммерческих источников и применяли без дальнейшей очистки. В частности, в примерах и описании изобретения могут быть использованы следующие сокращения: г (граммы); мг (миллиграммы); л (литры); мл (миллилитры); фкд (фунты на квадратный дюйм); М (молярный); мМ (миллимолярный); в.в. (внутривенно); Гц (герц); моль (молей); КТ (комнатная температура); мин (минуты); ч (часы); Т.пл. (температура плавления); ТСХ (тонкослойная хроматография); MeOH (метанол); ТФУ (трифторуксусная кислота); ТГФ (тетрагидрофуран); диметилсульфоксид (ДМСО); ЭтОАц (этилацетат); дихлорметан (ДХМ); диметилформамид (ДМФ); 1,1-карбонилдиимидазол (КДИ); изобутилхлорфомиат (иБуХФ); N-гидроксисукцинимид (ГОСук); N-гидроксибензтиазол (ГОБТ); 1-(3-диметиламинопропил)-3-этилкарбодиимида гидрохлорид (ЭДХ); бис(2-оксо-3-оксазолидинил)фосфинхлорид (БОФ); трет-бутилоксикарбонил (БОК); дициклогексилкарбодиимид (ДЦК); бензилоксикарбонил (Кбз); 4-диметиламинопиридин (ДМАП). Все ссылки на эфир относятся к диэтиловому эфиру. Если не оговорено особо, все температуры выражены в oC (градусы Цельсия). Все реакции проходят при комнатной температуре, если не оговорено особо.
1ЯМР спектры снимали либо на Varian VXR-300, либо на Varian Unity-300 приборе. Химические сдвиги выражены в частях на миллион (млн-1 d единицы). Константы ассоциации выражены в единицах герц (Гц). Варианты расщепления обозначены как s, синглет; d, дуплет; t, триплет; q, квартет; m, мультиплет; b, широкий.
Масс-спектры низкого разрешения (МС) снимали на JOEL JMX-AX505HA, JOEL 8Х-102 и SCIEX-API спектрофотометрах. Все масс-спектры получали в виде положительных ионов под действием ионизации электронным облучением (ЭОИ), химической ионизации (ХИ), методов электронного удара (ЭУ) или бомбардировки быстрыми атомами (ББА). Инфракрасные спектры (ИК) получали на Nicolet 510FT-IR спектрометре, применяя 1-мм NaCl элемент. Вращения регистрировали на Perkin-Elmer 241 поляриметре. Все реакции контролировали посредством тонкослойной хроматографии на 0,25 мм E.Merck пластинах силикагеля (60F-254), визуализировали с помощью УФ-света, 7%-ной этанольной фосфомолибденовой кислоты или п-анисового альдегида. Колоночную хроматографию быстрого разделения проводили на силикагеле (230-400 меш, Merck).
Продукты очищали посредством препаративной жидкостной хроматографии высокого давления с обращенной фазой (ОФ-ЖХВД), используя Waters Model3000 Delta Prep, снабженный Delta-pak радиальным компрессионным элементом (C18, 300 А, 15 м, 47 мм х 300 мм). Система растворителей включала А, водную 0,1% трифторуксусной кислоты, Б, 60% ацетонитрила, 40% водной 0,1%-ной трифторуксусной кислоты и В, ацетонитрил. Все растворители содержали 0,1% ТФУ. Во всех случаях применяли линейные градиенты, скорость протока составляла 100 мл/минуту (tо= 5,0 мин). Аналитическую чистоту определяли посредством ОФ-ЖХВД, используя Waters 600Е систему, снабженную Waters 990 diode array спектрометром (I интервал 200-400 нМ). Стационарной фазой служила Vydac C18 колонка (5 м, 4,6 мм х 250 мм). Скорость протока составляла от 1,0 до 1,5 мл/мин (tо = 2,8 или 3,0 мин), а системы растворителей были такие же, как описано выше. Результаты представляли в виде в.у., времени удержания в минутах (% ацетонитрила через промежуток времени).
Пример 1
2-[2,4-Диоксо-3-(3-фенил-уреидо)-5-пиридин-2-ил-2,3,4,5- тетрагидробензо[b][1,4]диазепин-1-ил]-N-изопропил-N-(4-метокси-фенил)- ацетамид
К раствору 2-(3-амино-2,4-диоксо-5-пиридин-2-ил-2,3,4,5- тетрагидробензо[b] [1,4]диазепин-1-ил)-N-изопропил-N-(4-метокси-фенил)- ацетамида (100 мг) в метиленхлориде (1 мл) добавляли раствор фенилизоцианата (25,6 мг) в метиленхлориде (1 мл), полученную смесь перемешивали при к.т. в течение 4 часов. Растворители удаляли под вакуумом, а остаток перекристаллизовывали из этилацетата, получая в результате продукт, указанный в заголовке (44 мг) в виде беловатого вещества. 1H ЯМР (300 МГц, CDCl3): 1,03 (2xd, J=7 Гц, 6Н), 3,81 (s, 3H), 4,17 (d, J=16 Гц, 1Н), 4,37 (d, J=16 Гц, 1Н), 5,0 (sept, J=7,1 Гц, 1Н), 5,40 (d, J=6,3 Гц, 1Н), 6,40 (d, J=5,3 Гц, 1Н), 6,8- 7,3 (m,17H), 7,80 (br, 1Н), 8,42 (d, J=3,9 Гц, 1Н). ТСХ (10%-ный MeOH, CH2Cl2) Rf=0,53. m/z[MH]+= 593.
Пример 2
{ 1-[изопропил-(4-метокси-фенил)-карбамоилметил] -2,4-диоксо-5-пиридин- 2-ил-2,3,4,5-тетрагидро-1Н-бензо[1] [1,4] диазепин-3ил}-амид 1Н-индол-2-карбоновой кислоты
Раствор 2-(3-Амино-2,4-диоксо-5-пиридин-2-ил-2,3,4,5- тетрагидробензо[b] [1,4]диазепин-1-ил)-N-изопропил-N-(4-метокси-фенил)- ацетамида (276 мг), БОФ (244 мг), ГОБТ (78 мг), ДМАП (69 мг) и индол-2-карбоновой кислоты (98 мг) в ДМФ (1 мл) перемешивали при к. т. в течение 18 часов. Реакционную смесь разбавляли этилацетатом (15 мл), промывали 1 N водным раствором гидроксида натрия (2х15 мл), водой (20 мл), рассолом (20 мл), сушили (K2CO3) и концентрировали под вакуумом, получая в результате сырой продукт. После очистки посредством ЖХВД-ОФ с применением линейного градиента (от 20% А, 80% Б до 90% Б, 10% В, 30 мин) получали продукт, указанный в заголовке (26,6 мг), в виде белого лиофилизата; Тr=21,3 мин. 1H ЯМР (300 МГц, CDCl3): 0,98 (2xd, J= 7 Гц, 6Н), 3,68 (s, 3Н), 4,17 (d, J=16,6 Гц, 1Н), 4,37 (d, J=16,8 Гц, 1Н), 4,87 (sept, J=6,8 Гц, 1Н), 5,40 (d, J=6,8 Гц, 1Н), 6,40 (d, J=5,3 Гц, 1Н), 6,75-6,84 (m,5H), 6,92-6,99 (m,2H), 7,07 (t, J=7,3 Гц, 1Н), 7,09-7,2 (m,4H), 7,24 (t, J=9,2 Гц, 1H), 7,40 (t, J=9,1 Гц, 2H), 7,52 (d, J=7,9 Гц, 1Н), 7,72 (m,2H), 8,38 (d, J=4,4 Гц, 1Н), 9,19 (s,1H), m/z[MH]+=617.
Примеры 3 и 4
{ 1-[изопропил-(4-метокси-фенил)-карбамоилметил]-2,4-диоксо-5- пиридин-2-ил-2,3,4,5-тетрагидро-1Н-бензо[b] [1,4] диазепин-3-ил} -амид (+) или (-) 1Н-индол-2-карбоновой кислоты
Порцию весом 100 мг смеси из Примера 2 наносили на полупрепаративную Pirkle Д-Лейцин колонку и с помощью изократной системы растворителей, состоящей из гексана (77%), изопропилового спирта (20%) и ацетонитрила (3%), элюировали отдельные энантиомеры. Каждый из растворителей содержал 0,3% диэтиламина.
Соответствующие фракции собирали и объединяли. Растворитель удаляли под вакуумом, а остаток промывали водой. Полученный осадок отделяли фильтрацией и сушили под вакуумом.
Пример 3: (Pirkle Д-Лейцин, 2 мл/мин) tr=19,62 мин (100%); m/z[МН]+ = 617.
Пример 4: (Pirkle Д-Лейцин, 2 мл/мин) tr=22,797 мин (98,4%); m/z [MH]+ = 617.
Пример 5
{ 1-[изопропил-(4-метокси-фенил)-карбамоилметил]-2,4-диоксо-5-пиримидин- 2-ил-2,3,4,5-тетрагидро-1Н-бензо[b] [1,4] диазепин-3ил}-амид 1Н-индол-2-карбоновой кислоты
К раствору 2-(3-амино-2,4-диоксо-5-пиримидин-2-ил-2,3,4,5- тетрагидро-бензо[b] [1,4]диазепин-1-ил)-N-изопропил-N-(4-метокси- фенил)-ацетамида (933 мг, 1,97 ммоль) в ДМФ (20 мл) последовательно при перемешивании при температуре окружающей среды добавляли индол-2-карбоновую кислоту (333 мг, 2,07 ммоль), N-гидроксибензотриазол (266 мг, 1,97 ммоль) и гидрохлорид 1-(3-диметиламинопропил)-3-этилкарбодиимида (415 мг, 2,16 ммоль). Полученную смесь перемешивали при температуре окружающей среды в течение 18 часов. Растворитель выпаривали при пониженном давлении, получая в результате желтое масло, которое растворяли в этилацетате (100 мл), промывали водой (2х30 мл), сушили над MgSO4, фильтровали и концентрировали при пониженном давлении, получая в результате пену. Сырой продукт очищали посредством хроматографии быстрого разделения на силикагеле (30 г), элюируя этилацетатом (600 мл). Соответствующие фракции объединяли и концентрировали под вакуумом, получая в результате соединение, указанное в заголовке (902 мг, 1,46 ммоль), в виде белой пены: 1H ЯМР (CDCl3, 400 МГц): 9,44 (s,1H), 8,75 (d, 2H, J=4,4 Гц), 7,64-6,88 (m, 15H), 5,60 (d, 1H, J=6,8 Гц), 5,03 (m, 1H), 4,45 (d, 1H, J=16,6 Гц), 3,99 (d, 1H, J= 16,6 Гц), 3,81 (s, 3H), 1,07 (m,1H); TCX (CH2Cl2/CH3OH (19:1)): Rf=0,63; MC (ББА) m/z 618,2 (MH+).
Пример 6
[1-[изопропил-(4-метокси-фенил)- карбамоилметил] -2,4-диоксо-5-(1,3,5-триметил-1 -пиразол-4-ил)- 2,3,4,5-тетрагидро-1Н-бензо[b][1,4]диазепин-3-ил] -амид 1Н-индол-2-карбоновой кислоты
К раствору 2-(3-амино-2,4-диоксо-5-(1,3,5-триметил-1Н-пиразол- 4-ил)-2,3,4,5-тетрагидро-бензо[b] [1,4] диазепин-1-ил)-N-изопропил-N- (4-метокси-фенил)-ацетамида (385 мг, 0,76 ммоль) в ДХМ (5 мл) добавляли индол-2-карбоновую кислоту (148 мг, 0,92 ммоль), ГОБТ (125 мг, 0,92 ммоль), ЭДХ (176 мг, 0,92 ммоль) и TEA (2 d). Раствор перемешивали при КТ в течение 48 часов, затем вливали в ДХМ (100 мл). Смесь экстрагировали насыщенным бикарбонатом натрия (х2), рассолом, сушили над MgSO4 и концентрировали под вакуумом. Полученное вещество обрабатывали этанолом, получая в результате соединение, указанное в заголовке (159 мг): Tr=18,3 мин (30-55% С через 30 минут); 1H ЯMP (d6-ДMCO, 300 МГц) d 11,8 (s, 1Н), 8,45 (s, 1H), 7,7-6,9 (m, 12H), 5,36 (m, 1H), 4,78 (m, 1H), 4,23 (m, 2H), 3,79 (s, 3H), 3,64 (d, 3, J=26,6 Гц), 2,11 (d, 3, J=31 Гц), 1,5 (d, 3H, J=72 Гц); MC низкого разрешения (ББА) m/e 648 (МН+).
Пример 7
{ 1-[изопропил-(4-метокси-фенил)-карбамоилметил]- 2,4-диоксо-5-пиридин-3-ил-2,3,4,5-тетрагидро-1Н- бензо[b][1,4]диазепин-3-ил}-амид 1Н-индол-2-карбоновой кислоты
К раствору {1-изопропил-(4-метокси-фенил)-карбамоилметил]-2,4- диоксо-2,3,4,5-тетрагидро-1Н-бензо[b] [1,4] диазепин-3-ил} -амида 1Н-индол-2-карбоновой кислоты (0,14 г, 0,26 ммоль) и 3-бромпиридина (40 мкл, 0,42 ммоль) в ДМФ (1 мл) добавляли порошок меди (46 мг, 0,73 ммоль) и калиевую соль уксусной кислоты (38 мг, 0,73 ммоль). Гетерогенный раствор перемешивали при 100oC в течение 15 ч, а затем подвергали горячему фильтрованию через целит и промывали метанолом. Полученный осадок фильтровали и очищали посредством ЖХВД-ОФ (40-60% С через 30 мин), получая в результате соединение, указанное в заголовке (19 мг), в виде белого лиофилизата: Tr=8,7 мин (40-60% С через 30 мин); 1H ЯМР (d6-Ацетон, 300 МГц) d 10,98 (s, 1H), 9,02 (s, 1H), 8,76 (s, 1H), 7,85 (d, 1H, J=8,8 Гц), 7,5 (m, 15H), 5,68 (d, 1H, J=7,6 Гц), 5,02 (m, 1H), 4,65 (ABq, 2H, J=16,8, 135 Гц), 4,02 (s, 3H), 2,19 (d, 3H, J=2,0 Гц), 2,18 (d, 3H, J=2,0 Гц); MC низкого разрешения (ББА) m/e 617 (МН+).
Пример 8
2-[2,4-диоксо-3-(3-фенил-уреидо)-5-пиридин-3-ил-2,3,4,5- тетрагидробензо[b][1,4]диазепин-1-ил]-N-(метокси-фенил)ацетамид
К раствору 2-(3-амино-2,4-диоксо-5-пиридин-3-ил-2,3,4,5- тетрагидробензо[b] [1,4]диазепин-1-ил)-N-изопропил-N-(4-метокси- фенил)-ацетамида (140 мг, 0,295 ммоль) в метиленхлориде (1 мл) добавляли раствор фенилизоцианата (36,6 мг, 0,295 ммоль) в метиленхлориде (1 мл), полученный раствор перемешивали при КТ в течение 16 ч. Растворитель удаляли под вакуумом, а остаток перекристаллизовывали из 5%-ного метанола в этилацетате, получая в результате соединение, указанное в заголовке (49 мг), в виде белого порошка. 1H ЯМР (300 МГц, CDCl3): s 8,72 (s,1H), 8,56 (d, 1H, J=8,0 Гц), 7,92 (d, 1H, J=8,0 Гц), 6,91-7,43 (m, 16H), 6,43 (d, 1H, J=8,3 Гц), 5,43 (d, 1H, J=8,3 Гц), 4,95 (sept, 1H, J=6,8 Гц), 4,60 (d, 1H, J=6,8 Гц), 4,60 (d, 1H, J=11,8 Гц), 4,18 (d, 1H, J=11,8 Гц), 3,88 (s, 3H), 1,06 (m, 6H). MC низкого разрешения (ББА) m/e 593 (m+).
Пример 9
Трет-бутиловый эфир 3-(3-{1-[изопропил-(4-метокси-фенил)- карбамоилметил] -2,4-диоксо-5-пиридин-3-ил-2,3,4,5-тетрагидро-1Н- бенз[b] [1,4] диазепин-3-ил}-уреидо)-бензойной кислоты
К раствору трет-бутилового эфира м-аминобензоата (43,3 мг, 0,224 ммоль) в ТГФ (5 мл) и триэтиламине (0,068 мл) при 0oC добавляли трифосген (22,1 мг), полученную смесь перемешивали при 0oC в течение 1 ч перед добавлением 2-(3-амино-2,4-диоксо-5-пиридин-3-ил-2,3,4,5-тетрагидробензо[b] [1,4] диазепин-1-ил)-N-изопропил-N-(4-метокси-фенил)-ацетамида (106 мг, 0,224 ммоль), полученную смесь перемешивали при КТ в течение ночи. Растворитель удаляли под вакуумом, а остаток растворяли в этилацетате (50 мл) и промывали 0,5 N соляной кислотой (2х30 мл), водой (30 мл), рассолом (30 мл), сушили (MgSO4) и концентрировали под вакуумом, в результате чего получали соединение, указанное в заголовке (115 мг), в виде белого твердого вещества. 1H ЯМР (300 МГц, CDCl3): s 8,62 (s,1H), 8,59 (d,1H, J=8,0 Гц), 7,92 (d,1H, J=8,0 Гц), 6,91-7,83 (m, 15H), 6,43 (d, 1H, J=8,1 Гц), 5,43 (d,1H, J=8,1 Гц), 4,91 (sept, 1H, J= 6,8 Гц), 4,60 (d,1H, J=11,8 Гц), 4,18 (d,1H, J=11,8 Гц), 3,86 (s,3H),1,66 (s,9H).
Пример 10
3-(3-{ 1-[изопропил-(4-метокси-фенил)-карбамоилметил] -2,4-диоксо-5- пиридин-3-ил-2,3,4,5-тетрагидро-1Н-бензо[b] [1? 4]диазепин-3-ил}- уреидо)-бензойная кислота
Смесь трет-бутилового эфира 3-(3-[1- изопропил-(4-метокси-фенил)-карбамоилметил] -2,4-диоксо-5-пиридин- 3-ил-2,3,4,5-тетрагидро-1Н-бензо[b][1,4] диазепин-3-ил} -уреидо)- бензойной кислоты (115 мг, 0,224 ммоль) и 4N HCl в диоксане (1 мл) перемешивали при КТ в течение 1,75 ч, после чего добавляли еще 1 мл 4N HCl в диоксане и перемешивали реакционную смесь при КТ в течение ночи. Добавляли эфир (20 мл), образовавшийся осадок растирали в порошок с эфиром (3х30 мл), получая в результате соединение, указанное в заголовке, в виде белого порошка. 1H ЯМР (300 МГц, CDCl3): s 1H ЯМР (300 МГц, CDCl3): s 8,61 (s, 1H), 8,56 (d, 1H, J=8,2 Гц), 7,91 (d, 1H, J=8,2 Гц), 6,91-7,83 (m, 16H), 6,41 (d, 1H, J=7,9 Гц), 5,43 (d, 1H, J=7,9 Гц), 4,91 (sept, 1H, J=6,8 Гц), 4,60 (d, 1H, J=11,8 Гц), 4,18 (d, 1H, J=11,8 Гц), 3,86 (s, 3H), 1,06 (m, 6H).
MC низкого разрешения (ББА) m/e 637 (m+).
Пример 11
{1-[изопропил-(4-метокси-фенил)-карбамоилметил]-2,4-индолдиоксо-5- пиридин-4-ил-2,3,4,5-тетрагидро-1Н-бензо[b] [1,4] диазепин-3-ил}-амид 1Н-индол-2-карбоновой кислоты
К раствору 23,3 мг (0,049 ммоль) 2-(3-амино-2,4-диоксо-5-пиридин- 4-ил-2,3,4,5,5а, 9а-гексагидро-бензо[b][1,4]диазепин-1-ил)-N- изопропил-N-(4-метокси-фенил)-ацетамида в 0,5 мл ДМФ добавляли 8,5 мг (0,052 ммоль; 1,05 эквив) индол-2-карбоновой кислоты, 6,7 мг (0,049 ммоль; 1 эквив) N-гидроксибензотриазола, а затем при перемешивании при температуре окружающей среды - 10,4 мг (0,054 ммоль; 1,1 эквив) гидрохлорида 1-(3-диметиламинопропил)-3-этилкарбодиимида. Полученную смесь перемешивали при температуре окружающей среды в течение 18 часов. Растворитель выпаривали при пониженном давлении, после чего получали желтое масло, которое переносили в ДХМ (30 мл), промывали насыщ. NaHCO3, сушили над MgSO4, фильтровали и концентрировали при пониженном давлении с образованием желто-коричневой пены. Сырой продукт очищали флэш-хроматографией на силикагеле (4 г), элюируя последовательно смесью этилацетат/гексан (4:1; 100 мл), этилацетатом (100 мл). Соответствующие фракции объединяли и концентрировали под вакуумом, получая в результате 10,2 мг (0,017 ммоль) соединения, указанного в заголовке, в виде белой пены: 1H ЯМР (d6-Ацетон, 300 МГц) d 10,83 (s, 1H), 8,63 (s, 1H), 7,69 (m, 2H), 7,61 (d, 1H, J=8,2 Гц), 7,53 (m, 3H), 7,45 (m, 1H), 7,38-7,28 (m, 4H), 7,23 (t, 1H), 7,09 (m, 4H), 5,50 (d, 1H, J=7,6 Гц), 4,85 (m, 1H), 4,65 (d, 1H, J=16,6 Гц), 4,28 (d, 1H, J=16,7 Гц), 3,86 (s, 3H), 3,86 (s, 3H), 1,01 (m, 6H); TCX Rf= 0,36 (ЭтОН); MC (ББА) m/z 617,3.
Пример 12
Трет-бутиловый эфир 3-(3-{ 7-фтор-1-[изопропил-(4- метокси-фенил)-карбамоилметил] -2,4-диоксо-5-пиридин-3-ил-2,3,4,5- тетрагидро-1Н-бензо[b][1,4] диазепин-3-ил}-уреидо)-бензойной кислоты
Раствор 84 мг 2-(3-амино-7-фтор-2,4-диоксо-5-пиридин-3-ил- 2,3,4,5-тетрагидробензо[b] [1,4] диазепин-1-ил)-N-изопропил-N-(4- метокси-фенил)-ацетамида (0,165 ммоль) в 4 мл ацетонитрила объединяли с 59 мг трет-бутилового эфира 3-[(4-нитрофенил)оксикарбонил] -аминобензойной кислоты (0,165 ммоль, 1 экв) и нагревали при дефлегмации в атмосфере азота в течение 3 часов. Полученную взвесь охлаждали до 5oC, выдерживали при этой температуре в течение 30 минут, фильтровали и сушили при глубоком вакууме, получая в результате 93 мг соединения, указанного в заголовке, в виде кристаллического вещества. 1H ЯМР (300 МГц, d6-ДМСО) d 9,41 (s, 1H), 8,74 (d, 1H, J=2,3 Гц), 8,61 (dd, 1H, J=1,5, 4,9 Гц), 7,99 (m, 1H), 7,95 (m, 1H), 7,67 (dd, 1H, J= 5,6, 9,3 Гц), 7,57 (m, 2H), 7,48 (m, 1H), 7,31 (m, 4H), 7,10 (d, 2H, J=8,9 Гц), 6,97 (d, 1H, J=7,7 Гц), 6,87 (dd, 1Н, J=8,9 Гц), 5,16 (d, 1Н, J=7,6 Гц), 4,78 (m, 1H), 4,56 (d, 1H, J=16,5 Гц), 4,19 (d, 1H, J=16,5 Гц), 3,84 (s, 3H), 1,54 (s, 9H), 0,98 (m, 6H); MC низкого разрешения (ББА) m/e 710 (МН+).
Пример 13
3-(3-{ 7-фтор-1-[изопропил-(4-метокси-фенил)-карбамоилметил] -2,4-диоксо-5- пиридин-3-ил-2,3,4,5-тетрагидро-1Н-бензо[b][1,4]диазепин-3-ил}- уреидо)-бензойная кислота
Смесь 84 мг трет-бутилового эфира 3-(3- {7-фтор-1-[1-изопропил-(4-метокси-фенил)-карбамоилметил] -2,4- диоксо-5-пиридин-3-ил-2,3,4,5-тетрагидро-1Н- бензо[b][1,4]диазепин-3-ил}-уреидо)-бензойной кислоты (0,118 ммоль) и 4 мл трифторуксусной кислоты перемешивали в атмосфере азота в течение 1,5 часов. Трифторуксусную кислоту удаляли под вакуумом, а остаток измельчали в порошок с диэтиловым эфиром. Суспензию фильтровали, промывали диэтиловым эфиром и сушили при глубоком вакууме, получая в результате 85 мг соединения, указанного в заголовке, в виде белой кристаллической соли трифторуксусной кислоты. 1H ЯМР (300 МГц, d6-ДМСО) d 9,33 (s, 1H), 8,70 (b, 1H), 8,57 (d, 1H, J= 4,7 Гц), 8,00 (m, 1H), 7,97 (d, 1H, J=8,6 Гц), 7,61 (dd, 1H, J=5,5, 9,1 Гц), 7,55 (dd, 1H, J=4,88, 8,2 Гц), 7,47 (m, 2H), 7,26 (m, 4H), 7,05 (d, 2H, J=9,0 Гц), 6,92 (d,1H, J=7,8 Гц), 6,82 (dd, 1H, J=2,8, 9,6 Гц), 5,10 (d, 1H, J= 7,6 Гц), 4,72 (m, 1H), 4,51 (d, 1H, J=16,8 Гц), 4,14 (d, 1H, J=16,8 Гц), 3,78 (s, 3H), 0,92 (m, 6H); MC низкого разрешения (ББА) m/e 655 (МН+).
Примеры 14 и 15
[1-[изопропил-(4-метокси-фенил)-карбамоилметил] -2,4-диоксо-5- пиридин-3-ил-2,3,4,5-тетрагидро-1Н-бензо[b] [1,4] диазепин-3-ил] -амид (+) или (-) 1Н-индол-2-карбоновой кислоты
Посредством препаративной жидкостной хроматографии высокого давления (ЖХВД) с использованием waters Model 4000 Delta Prep, снабженных Daicel Chemical Industries Chiralpak-AD препаративной колонкой (20 мкм, 5 см х 50 см) в качестве стационарной фазы разделяли (> 99,9%) энантиомеры по Примеру 7. В качестве подвижной фазы наносили смесь из 72% гексана, 21% изопропилового спирта и 7% хлороформа. Применяли изократические условия при скорости протока 50 мл/мин (до = 16 мин). Соответствующие фракции объединяли, концентрировали под вакуумом и лиофилизировали из воды и ацетонитрила, получая целевые аналоги. Аналитическую чистоту определяли посредством ЖХВД с применением Hewlett Packard 1050 системы, снабженной спектрометром диодного типа (лямбда интервал 200-400). В качестве стационарной фазы применяли Daicel Chemical Industries Chiralpak-AD (10 мкм, 0,46 см х 25 см). Подвижные фазы были те же самые, что и описанные выше, а скорость протока составляла 1,0 мл/мин (до = 3 мин). Время удерживания, ВУ, выраженное в минутах, имело для двух изомеров следующие значения.
Энантиомер 1, Пример 14: ВУ = 25,06 мин.
Энантиомер 2, Пример 15: ВУ = 81,39 мин.
Промежуточное соединение 1
Изопропил-(4-метоксифенил)амин
К перемешивавшемуся раствору 4-метоксифениламина (1,24 г, 6,22 ммоль) в метаноле (15 мл) при температуре окружающей среды последовательно добавляли ледяную уксусную кислоту (415 мг, 6691 ммоль), ацетон (669 мг, 11,5 ммоль) и 1М цианоборгидрида натрия в ТГФ (12,7 мл, 12,6 ммоль). Реакционную смесь перемешивали в течение ночи при комнатной температуре. Доводили pH до 2 с помощью 6N HCl и перемешивали в течение 30 минут для удаления избытка цианоборгидрида натрия. Затем pH доводили до 8,5 с помощью 1N NaOH и полученный раствор экстрагировали диэтиловым эфиром (2 х 50 мл) и этилацетатом (50 мл). Органические экстракты объединяли, сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении, получая в результате соединение, указанное в заголовке (1,42 г, 5,91 ммоль), в виде желтого масла. 1Н ЯМР (300 МГц, CDCl3): d 6,78 (d, J=8,8 Гц, 2Н), 6,57 (d, J=9,1 Гц, 2Н), 3,75 (s, 3H), 3,55 (m, 1H), 2,92 (br s,1H), 1,18 (d, J=6,1 Гц, 6H); ТСХ (ЭтОАц/Гекс (2:3)): Rf= 0,72.
Промежуточное соединение 2
2-Бром-N-изопропил-N-(4-метоксифенил)ацетамид
К раствору изопропил-(4-метоксифенил)-амина (25,11 г, 152 ммоль) в дихлорметане (250 мл) при перемешивании при температуре окружающей среды добавляли триэтиламин (15,38 г, 152 ммоль). Раствор охлаждали на ледяной бане (< 3oC) и через 45 минут по каплям добавляли бромацетилбромид (30,68 г, 152 ммоль), растворенный в дихлорметане (100 мл). Реакционную смесь перемешивали в течение ночи при температуре окружающей среды, промывали 0,3N HCl (300 мл) и рассолом (300 мл), сушили над сульфатом натрия, фильтровали и упаривали при пониженном давлении, получая в результате темно-коричневое масло. Масло фильтровали через слой силикагеля (150 г), который элюировали смесью этилацетат/гексан (1:1, 90 мл), а фильтрат упаривали при пониженном давлении, получая в результате соединение, указанное в заголовке (41,05 г, 143 ммоль), в виде коричневого масла, которое кристаллизовалось при хранении.
1H ЯМР (300 МГц, CDCl3): d, 1,04 (d, J=6,8 Гц, 6Н), 3,53 (s, 2H), 3,84 (s, 3H), 4,93 (m, 1H), 6,93 (d, J=9,1 Гц, 2Н), 7,10 (d, J=9,1 Гц, 3Н);
ТСХ (ЭтОАц/Гексан (3:17)): Rf = 0,18.
Промежуточное соединение 3
2-(фенилгидразоно)-малоновая кислота
К интенсивно перемешиваемому раствору моногидрата кетомалоновой кислоты (29,33 г) в этаноле (140 мл) и воды (300 мл) при температуре окружающей среды в течение 40 минут по каплям добавляли фенилгидразин (23,3 г). Полученную суспензию перемешивали в течение ночи при температуре окружающей среды. Твердую фазу отделяли посредством фильтрации, промывали последовательно холодной водой (100 мл) и этанолом (25 мл) и сушили на воздухе. Затем проводили сушку при 75oC в течение ночи в вакууме, в результате чего получали соединение, указанное в заголовке, в виде желтого твердого вещества (42,38 г). 1H ЯМР (300 МГц, ДМСО-d6): d 7,12 (t,1H), 7,35-7,48 (m,4H); т.пл.: 155-157oC (разл.).
Промежуточное соединение 4
Двухлористый 2-(фенилгидразоно)-пропандиол
К перемешивавшейся суспензии 2-(фенилгидразоно)-малоновой кислоты (14,73 г) в хлороформе (90 мл) при 5oC по частям добавляли пентахлорид фосфора (36,84 г) в течение 20-минутного промежутка времени. После окончания добавления раствор нагревали до комнатной температуры и перемешивали в течение 1 часа, затем нагревали при дефлегмации в течение трех часов. Раствор охлаждали на ледяной бане, а образовавшийся осадок отделяли фильтрацией, промывали холодным гексаном (50 мл) и сушили в течение ночи под вакуумом, получая в результате соединение, указанное в заголовке (13,4 г), в виде светло-желтого твердого вещества. 1H (300 МГц, ДМСО-d6): d 7,12 (t, 1H), 7,20-7,56 (m, 4H); т.пл.: 135-138oC (разл).
Промежуточное соединение 5
2-(2-Аминопиридил)нитробензол
К раствору 2-аминопиридина (2,00 г) в ТГФ (10 мл) при 0oC в течение 1 часа добавляли гидрид натрия (60%-ный в масле, 1,26 г). По каплям добавляли раствор 2-фторнитробензола (2,21 мл) в ТГФ (8 мл), порученную смесь выдерживали при комнатной температуре в течение ночи. Добавляли насыщенный водный карбонат натрия (10 мл) и отделившуюся водную фазу экстрагировали в дихлорметан (3 х 10 мл). Объединенные органические экстракты промывали рассолом (20 мл), сушили (MgSO4) и концентрировали под вакуумом. После колоночной флэш-хроматографии (циклогексан:этилацетат; 3:1 в качестве элюента) получали соединение, указанное в заголовке (2,23 г), в виде красно-оранжевого твердого вещества. 1H ЯМР (300 МГц, CDCl3): 6,83 (m, 3Н), 7,42 (dt, J=1,7 Гц, 1Н), 7,40 (dt, J=1,7 Гц, 1Н), 8,08 (dd, J=1,7 Гц, 1Н), 8,21 (d, J=1,15 Гц, 1Н), 8,61 (dt, J=1,7 Гц, 1Н), 10,0 (s, 1Н). Т.пл. = 71oC.
Промежуточное соединение 6
N-(2-пиридил)фенилендиамин
2-(2-Аминопиридил)нитробензол (2,14 г) растворяли в ледяной уксусной кислоте (45 мл), добавляли железные опилки (5,57 г), полученную смесь перемешивали при КТ в течение 72 часов, после чего твердую фазу удаляли путем фильтрации через целит, а растворитель удаляли путем концентрирования под вакуумом. Добавляли водный карбонат натрия (2М), доводя pH до 8, эту смесь затем экстрагировали в дихлорметан (3 Х 30 мл). Объединенные органические фазы промывали рассолом (30 мл), сушили (MgSO4) и концентрировали под вакуумом, получая в результате соединение, указанное в заголовке (1,68 г), в виде бежевого твердого вещества. 1H ЯМР (300 МГц, CDCl3): 6,12 (br, 2H), 6,42 (d, J=6 Гц, 1Н), 6,68 (m, 1Н), 6,78 (m, 3H), 7,008 (dt, J=1,60 Гц, 1H), 7,15 (dd, 1,58 Гц, 1Н), 7,42 (m, 1H), 8,15 (br, 1H). TCX (циклогексан:этилацетат; 1:1) Rf = 0,2.
Промежуточное соединение 7
N-изопропил-N-(4-метокси-фенил)-2-[2-(пиридин-2-иламинофениламино]- ацетамид
Смесь N-(2-пиридил)фенилендиамина (2,00 г), изопропил-(4-метоксифенил)амина (3,08 г) и карбоната калия (1,50 г) в ДМФ (30 мл) перемешивали при КТ в течение 22 ч. Растворители удаляли под вакуумом, а остаток фракционировали между этилацетатом (50 мл) и водой (3х30 мл). Органическую фазу промывали рассолом (30 мл), сушили (MgSO4) и концентрировали под вакуумом, получая в результате сырой продукт, указанный в заголовке, в виде коричневого вещества. После двух перекристаллизаций из смеси этилацетатгексан (1:1) получали соединение, указанное в заголовке, в виде желтовато-коричневого вещества. 1H ЯМР (300 МГц, CDCl3): 1,01 (d, J=7 Гц, 6Н), 3,4 (s,2H), 3,83 (s, 3H), 4,89 (sept, J=7 Гц), 6,08 (s,1H), 6,31 (d, J=8,5 Гц, 1Н), 6,42 (d, J= 8,0 Гц, 1H), 6,66 (m,2H), 6,9-7,1 (m,6H), 7,22 (d, J=8,0 Гц, 1Н), 7,40 (t, J=6,5 Гц, 1Н), 8,18 (d, J=3,5 Гц, 1H). ТСХ (10%-ный MeOH, CH2Cl2) Rf=0,42.
Промежуточное соединение 8
2-[2,4-диоксо-3-(3-фенил-гидразоно)-5-пиридин-2-ил-2,3,4,5- тетрагидробензо[b][1,4]диазепин-1-ил]-N-изопропил-N- (метоксифенил)ацетамид
Раствор N-изопропил-N-(4-метоксифенил)-2-[2-(пиридин-2- иламинофениламино] ] -ацетамида (500 мг) в ТГФ (20 мл) и двухлористого 2-(фенилгидразоно)-пропандиола (317 мг) в ТГФ (20 мл) добавляли одновременно к объему ТГФ (20 мл) с температурой 0oC, полученную смесь оставляли нагреваться до КТ в течение ночи. Растворители удаляли под вакуумом, а остаток растворяли в этилацетате (50 мл) и промывали 2N водным бикарбонатом натрия (2х30 мл), водой (30 мл), рассолом (30 мл), сушили (MgSO4) и концентрировали под вакуумом, получая в результате сырой продукт. После колоночной флэш-хроматографии на силикагеле, элюируя 10%-ной смесью метанол/метиленхлорид, получали соединение, указанное в заголовке, в виде 3:2 смеси гидразонов (460 мг). Из-за того, что продукт представлял собой смесь диастереоизомеров, 1H ЯМР данные не диагностировали. ТСХ (10%-ный MeOH, CH2Cl2) Rf = 0,62.
Промежуточное соединение 9
2-(3-Амино-2,4-диоксо-5-пиридин-2-ил-2,3,4,5-тетрагидробензо[b] [1,4] диазепин-1-ил]-N-изопропил-N-(метокси-фенил)ацетамид
Смесь 2-[2,4-диоксо-3-(фенилгидразоно)-5- пиридин-2-ил-2,3,4,5-тетрагидробензо[b] [1,4] диазепин-1-ил)-N- изопропил-N-(4-метоксифенил)ацетамида (460 мг), цинковой пыли (430 мг) и уксусной кислоты (5,6 мл) перемешивали в течение 3 ч. Твердые частицы удаляли фильтрованием через целит, фильтрат концентрировали под вакуумом, а из остатка готовили азеотропную смесь с гексаном. Остаток растворяли в этилацетате (50 мл) и промывали 2N водным бикарбонатом натрия (2 х 30 мл), водой (30 мл), рассолом (30 мл), сушили (MgSO4) и концентрировали под вакуумом, получая в результате сырой продукт (270 мг), который использовали без дальнейшей очистки. 1H ЯМР (300 МГц, CDCl3): 1,03 (d, J=7 Гц, 6Н), 3,79 (s, 3H), 4,02 (d, J=10 Гц, 1Н), 4,40 (s, 1Н), 4,42 (d, J= 10 Гц, 1Н), 5,0 (sept, J=7,1 Гц, 1Н), 6,8-7,3 (m,10H), 7,44 (d, J=8,0 Гц, 1H), 7,68 (d, J= 8,0 Гц, 1Н), 7,80 (m,1H), 8,42 (d, J=3,9 Гц, 1Н). ТСХ (10%-ный MeOH, CH2Cl2) Rf=0,23.
Промежуточное соединение 10
(2-Нитро-фенил)-пиримидин-2-ил-амин
К раствору 2-аминопиримидина (10 г, 105 ммоль) в ДМФ (100 мл), охлажденному до 5oC, добавляли 60%-ный гидрид натрия в минеральном масле (5,47 г, 137 ммоль), полученную смесь перемешивали один час при охлаждении. Затем в течение 20 минут к перемешивавшемуся охлажденному раствору по каплям добавляли 1-фтор-2-нитробензол (14,83 г, 105 ммоль) в ДМФ (30 мл). Раствор оставляли для медленного нагрева до температуры окружающей среды при перемешивании в течение 3 часов. Продукт осаждали путем добавления воды (300 мл), отделяли фильтрованием и сушили, получая в результате соединение, указанное в заголовке (13,39 г, 61,9 ммоль), в виде оранжевого твердого вещества: 1H ЯМР (Ацетон-d6, 400 МГц): 10,34 (br s, 1Н), 9,00 (d, 1H, J=8,6 Гц), 8,60 (d, 2H, J= 4,8 Гц), 8,23 (d,1H, J=8,4 Гц), 7,74 (t, 1H), 7,17 (t, 1H), 7,06 (t, 1H); TCX (ЭтОАц/Гексан (3:17)): Rf = 0,27.
Промежуточное соединение 11
N-Пиримидин-2-ил-бензол-1,2-диамин
К раствору (2-нитрофенил)-пиримидин-2-ил-амина (13,2г, 61,1 ммоль) в смеси ЭтОАц (450 мл) и CH3OH (350 мл) добавляли катализатор - никель Ренея (16 г (увлажненный водой)), реакционную смесь гидрогенизировали при давлении водорода 1 атм при температуре окружающей среды в течение 3 часов. Катализатор отделяли фильтрованием, а фильтрат концентрировали под вакуумом до получения красно-коричневого вещества, после растирания в порошок которого с холодным CH3OH (250 мл) получали соединение, указанное в заголовке (8,21 г, 44,1 моль), в виде серого твердого вещества: 1H ЯМР (CDCl3, 400 МГц): 8,38 (d, 2H, J= 4,9 Гц), 7,37 (d, 1H, J=7,9 Гц), 7,08 (t, 1H), 7,00 (br s, 1Н), 6,83 (m, 2H), 6,67 (t, 1H), 3,60 (br s, 2H); ТСХ (ЭтОАц/Гексан (2:1)): Rf = 0,33.
Промежуточное соединение 12
N-Изопропил-N-(4-метокси-фенил)-2[2-(пиримидин-2-иламино)- фениламино] -ацетамид
К раствору N-пиримидин-2-ил-бензол-1,2-диамина (82 мг, 0,441 ммоль) в ДМФ (2 мл) добавляли карбонат калия (61 мг, 0,441 ммоль), иодид калия (7 мг, 0,044 ммоль) и 2-бром-N-изопропил-N-(4-метокси-фенил)-ацетамид (126 мг, 0,441 ммоль). Полученную реакционную смесь перемешивали в течение ночи при 60oC. Растворитель удаляли под вакуумом, а сырой материал фракционировали между CH2Cl2 (35 мл) и водой (15 мл). Органическую фазу отделяли, сушили над MgSO4, фильтровали и концентрировали до получения желтого масла, после растирания в порошок которого с ЭтОН (6 мл) и фильтрации получали соединение, указанное в заголовке (96,7 мг, 0,247 ммоль), в виде желтых кристаллов: 1H ЯМР (CDCl3, 300 МГц): 8,36 (d, 2H, J=4,9 Гц), 7,38 (dd, 1H, J=1,2, 7,8 Гц), 7,06-7,90 (m, 6H), 6,75 (t, 1H), 6,66 (t, 1H), 6,36 (dd, 1Н, J=1,2, 7,8 Гц), 4,97 (m, 1H), 3,87 (s, 3H), 3,43 (s, 2H), 1,04 (d, 6H, J=6,8 Гц), TCX (ЭтОАц): Rf = 0,62; МС (ББА) m/z 392,0 (MH+).
Промежуточное соединение 13
2-[2,4-диоксо-3-(фенил-гидразоно)-5-пиримидин-2-ил-2,3,4,5- тетрагидробензо[b][1,4]диазепин-1-ил]-N-изопропил-N-(4-метокси-фенил)- ацетамид
К взвеси N-изoпpoпил-N-(4-метoкcи-фeнил)-2-[2-(пиpимидин-2-илaминo)- фениламино] -ацетамида (500 мг, 1,28 ммоль) в ТГФ (12 мл), охлажденной на ледяной бане, по каплям в течение 5 минут добавляли двухлористый 2-(фенилгидразоно)пропандиол (344 мг, 0,141 ммоль) в ТГФ (6 мл). После завершения процедуры добавления раствор оставляли нагреваться до комнатной температуры в течение ночи при перемешивании. Растворитель упаривали при пониженном давлении, а полученное масло растворяли в этилацетате (80 мл), промывали насыщенным раствором бикарбоната натрия, сушили над MgSO4, фильтровали и концентрировали при пониженном давлении, получая в результате желтое масло. Сырой продукт очищали посредством флэш-хроматографии на силикагеле (15 г), элюируя смесью ЭтОА/Гексан (2:1, 250 мл). Соответствующие фракции объединяли и концентрировали, получая в результате соединение, указанное в заголовке (500 мг, 0,886 ммоль), в виде желтой пены: 1H ЯМР (CDCl3, 300 МГц): 11,22 (s, 1H), 8,58 (m, 2H), 7,70-6,90 (m, 14H), 5,05 (m, 1H), 4,46 (m, 1H), 3,82 (m, 4H), 1,12 (m, 6H); TCX (ЭтОА/Гексан (2:1)): Rf = 0,21; MC (ББА) m/z 564,1 (MH+).
Промежуточное соединение 14
2-(3-Амино-2,4-диоксо-5-пиримидин-2-ил-2,3,4,5-тетрагидробензо[b] [1,4] диазепин-1-ил]-N-изопропил-N-(4-метокси-фенил)-ацетамид
К перемешиваемому раствору 2-12,4- диоксо-3-(фенил-гидразоно)-5-пиримидин-2-ил-2,3,4,5-тетрагидробензо[b] [1,4] диазепин-1-ил]-N-изопропил-N-(4-метокси-фенил)-ацетамида (500 мг, 0,886 ммоль) в уксусной кислоте (12 мл) при температуре окружающей среды добавляли цинковую пыль (530 мг), полученную смесь перемешивали в течение 1 часа. Цинк отделяли фильтрованием, фильтрат концентрировали под вакуумом, а полученное масло фракционировали между водой (30 мл) и этилацетатом (80 мл). С помощью 6N гидроксида натрия доводили pH до 8 и фазы разделяли. Водную фазу экстрагировали этилацетатом (2х35 мл), органические экстракты объединяли, сушили над сульфатом магния, фильтровали и концентрировали под вакуумом, получая в результате желтую пену. Сырой продукт очищали посредством флэш-хроматографии на силикагеле (15 г), элюируя смесью метиленхлорид/метанол (19:1, 250 мл). Соответствующие фракции объединяли и концентрировали, получая соединение, указанное в заголовке (255 мг, 0,537 ммоль), в виде белого стекла; 1H ЯМР (CDCl3 400 МГц): 8,78 (d, 2H, 4,9 Гц), 7,59 (dd, 1h, J=1,1, 8,3 Гц), 7,33-7,25 (m, 3H), 7,16 (t, 1H), 7,05 (dd, 1H, J=1,4, 8,3 Гц), 6,96 (dd, 1H, J=2,7, 8,7 Гц), 6,89 (dd, 1H, J= 2,7, 8,5 Гц), 6,86 (dd, 1H, J=1,2, 8,2 Гц), 5,06 (m, 1H), 4,52 (d, 1H, J=16,6 Гц), 4,43 (s,1H), 3,85 (d, 1H, J=16,6 Гц), 3,82 (s, 3H), 2,60 (br s, 2H), 1,11 (d, 6H, J=1,0 Гц); TCX (CH2Cl2/CH3OH (9:1)): Rf = 0,48; MC (ББА) m/z 475,3 (MH+).
Промежуточное соединение 15
(2-Нитрофенил)-(1,3,5-триметил-1Н-пиразол-4-ил)-амин
К раствору 1-фтор-2-нитробензола (7,47 мл, 70,9 ммоль) в этаноле (35 мл) и H2O (105 мл) добавляли 4-амино-1,3,5-триметилпиразол (8,8 г, 70,9 ммоль). Раствор нагревали при дефлегмации в течение 15 часов, затем охлаждали до комнатной температуры. Осадок отделяли фильтрованием и промывали 25%-ным водным этанолом, получая в результате соединение, указанное в заголовке (8,6 г): 1H ЯМР (CDCl3, 300 МГц) d 8,81 (s, 1H), 8,18 (dd, 2H, J=1,6, 8,7 Гц), 6,69 (m, 1H), 6,61 (dd, 1H, J=1,2, 8,7 Гц), 3,77 (s, 3H), 2,10 (s, 3H), 2,06 (s, 3H); MC низкого разрешения (ББА) m/z 247 (MH+).
Промежуточное соединение 16
N-(1,3,5-Триметил-1Н-пиразол-4-ил)-бензол-1,2-диамин
К раствору (2-нитро-фенил)-(1,3,5-триметил-1Н-пиразол-4-ил)-амина (8,6 г, 35 ммоль) в этилацетате (175 мл) добавляли 10%-ный палладий на угле (1 г). Раствор перемешивали в атмосфере азота (50 фкд) в течение 15 ч, а затем фильтровали через слой целита, промывали этилацетатом и концентрировали под вакуумом, получая в результате соединение, указанное в заголовке (7,56 г): 1H ЯМР (CDCl3, 300 МГц) d 6,74 (m, 3H), 6,32 (d, 1H, J=1,9, 7,2 Гц), 4,6 (s, 1H), 3,75 (s, 3H), 4,1 (s, 2H), 2,05 (m, 6H); MC низкого разрешения (ББА) m/z 217 (MH+).
Промежуточное соединение 17
N-Изопропил-N-(4-метокси-фенил)-2-[2-(1,3,5-триметил-1Н-пиразолиламино)-фениламино]-ацетамид
К раствору N-(1,3,5-триметил- 1Н-пиразол-4-ил)-бензол-1,2-диамина (7,56 г, 38,5 ммоль) и 2-бром-N-изопропил-N-(4-метокси-фенил)-ацетамида (7,18 г, 38,5 ммоль) в ДМФ (70 мл) добавляли карбонат калия (14,5 г, 105 ммоль) и иодид калия (581 мг, 3,5 ммоль). Раствор нагревали при 80oC в течение 15 часов, а затем вливали в ДХМ (100 мл). Смесь экстрагировали H2O (Х4), 1 N НСl (Х2) и рассолом, сушили над MgSO4 и концентрировали под вакуумом. Полученную пену очищали путем растирания в порошок с Эт2O, получая в результате соединение, указанное в заголовке (11,9 г); 1H ЯМР (CDCl3, 300 МГц) d 8,08 (s, 1H), 7,07 (dd, 4H, J=2,2, 6,6 Гц), 6,63 (m, 2H), 6,31 (m, 2H), 5,03 (m, 1H), 4,76 (s, 1H), 3,87 (s, 3H), 3,74 (s, 3H), 3,46 (s, 2H), 2,02 (m, 6H), 1,07 (d, 6H, J=6,8 Гц); MC низкого разрешения (ББА) m/z 422 (МН+).
Промежуточное соединение 18
2-[2,4-Диоксо-3-(фенил-гидразоно)-5-(1,3,5-триметил-1Н-пиразол- 4-ил)-2,3,4,5-тетрагидро-бензо[b] [1,4] диазепин-1-ил]-N-изопропил-N- (4-метокси-фенил)-ацетамид
Раствор N-изопропил-N-(4-метокси-фенил)-2-[2-(1,3,5-триметил-1Н- пиразол-иламино)-фениламино] ацетамида (30 г, 7,1 ммоль) в ТГФ (70 мл) и раствор двухлористого 2-(фенил-гидразоно)-пропандиола (1,75 г, 7,1 ммоль) в ТГФ (70 мл) одновременно добавляли к ТГФ, охлажденному до 0oC. Раствор перемешивали при КТ в течение 15 часов, а затем концентрировали под вакуумом, получая в результате соединение, указанное в заголовке (8,1 г), которое применяли без дальнейшей очистки.
Промежуточное соединение 19
2-[3-Амино-2,4-Диоксо-5-(1,3,5-триметил-1Н-пиразол-4-ил)-2,3,4,5- тетрагидро-бензо[b] [1,4]диазепин-1-ил]-N-изопропил-N-(4-метокси-фенил)- ацетамид
К раствору 2-[2,4-диоксо-3-(фенил-гидразоно)-5-(1,3,5- триметил-1Н-пиразол-4-ил-2,3,4,5-тетрагидро-бензо[b] [1,4] диазепин-1-ил]- N-изопропил-N-(4-метокси-фенил)-ацетамида (4,6 г, 7,74 ммоль) в ледяной уксусной кислоте (45 мл) добавляли цинковую пыль (4,6 г). Гетерогенный раствор перемешивали при КТ в течение 15 часов, а затем фильтровали через слой целита, промывали этилацетатом и концентрировали до сухого состояния. Полученное масло растворяли в этилацетате (200 мл) и экстрагировали насыщенным бикарбонатом натрия (X3), рассолом, сушили с помощью MgSO4 и концентрировали под вакуумом. Полученное масло очищали посредством флэш-хроматографии на силикагеле (5% метанол/ДХМ), получая в результате соединение, указанное в заголовке (385 мг): 1H ЯМР (CDCl3, 300 МГц) d 7,60-7,80 (m, 10H), 5,05 (m, 1H), 4,4 (m, 1H), 3,82 (s, 3H), 3,74 (s, 3H), 3,62 (s, 2H), 2,24 (s, 3H), 1,20 (s, 3H), 1,09 (m, 6H); MC низкого разрешения (ББА) m/z 505 (МН+).
Промежуточное соединение 20
4-Метоксибензил-(2-нитрофенил)-амин
К 2-фторнитробензолу (80,65 мл, 0,77 ммоль), растворенному в этаноле (200 мл) и воде (600 мл), добавляли 4-метоксибензиламин (100 мл, 0,77 ммоль). Реакционную смесь нагревали при 92oC в течение 15 ч, охлаждали до комнатной температуры и отделяли фильтрованием оранжевый осадок. Осадок перекристаллизовывали из смеси этанол:вода 1/3, затем растворяли в этилацетате, сушили над MgSO4 и концентрировали под вакуумом, получая в результате соединение, указанное в заголовке (117,88 г): 1H ЯМР (300 МГц, CDCl3): d 8,35 (bs, 1H), 8,18 (d, J=8,5 Гц, 1H), 7,38 (dd, J=7,3, 7,8 Гц, 1), 7,27 (d, J=8,5 Гц, 2H), 6,85 (3H), 6,66 (dd, J=7,3, 7,8 Гц,1Н), 4,47 (коллапсированный ABq, J= 4,9 Гц, 2Н), 3,81 (s,3H); MC низкого разрешения (ББА) m/z 258,99 (МН+).
Промежуточное соединение 21
N-(4-метокси-бензил)-бензол-1,2-диамин
К (4-метоксибензил)-(2-нитрофенил)-амину (50 г, ммоль) в ледяной уксусной кислоте (500 мл), охлажденному до 15oC в трехгорлой круглодонной колбе, снабженной мешалкой, закрепленной в верхней части, добавляли цинковый порошок (50 г). Реакционную смесь нагревали до комнатной температуры и перемешивали в течение 15 ч. Реакционную смесь фильтровали через слой целита, и фильтрат концентрировали до сухого состояния. Полученное масло переносили в смесь этилацетат/вода, pH водного слоя доводили до 8 бикарбонатом натрия. Водный слой экстрагировали этилацетатом (Х4), сушили над MgSO4 и концентрировали под вакуумом.
Сырой продукт очищали посредством флэш-хроматографии на силикагеле (50% гексан/ДХМ), получая в результате соединение, указанное в заголовке (33,34 г): 1H ЯМР (300 МГц, CDCl3): d 7,32 (d, J=8,5 Гц, 2Н), 6,88 (d, J=8,5 Гц, 2Н), 6,97 (m, 4H), 4,25 (s, 2H), 3,81 (s, 3H); MC низкого разрешения (ББА) m/z 476 (МН+).
Промежуточное соединение 22
N-Изопропил-2[2-(4-метокси-бензиламино)-фениламино]-N-(4-метокси-фенил)- ацетамид
К раствору N-(4-метоксибензил)-бензол-1,2-диамина (2,42 г, 10,6 ммоль) в ДМФ (40 мл) добавляли карбонат калия (1,47 г, 10,6 ммоль), иодид калия (176 мг, 1,06 ммоль) и 2-бром-N-изопропил-N-(4-метокси-фенил)- ацетамид (3,03 г, 10,6 ммоль). Полученную реакционную смесь перемешивали в течение ночи при 60oC. Растворитель удаляли под вакуумом, а сырой материал растворяли в ЭтОАц (150 мл), промывали водой (2х50 мл) и рассолом (50 мл), сушили с помощью MgSO4, фильтровали и концентрировали до коричневого масла. Осадок, который образовывался при добавлении эфира (70 мл), отфильтровывали, получая в результате соединение, указанное в заголовке (1,39 г, 3,21 ммоль), в виде желтого вещества. Оставшийся фильтрат концентрировали при пониженном давлении до коричневого масла, которое очищали посредством флэш-хроматографии на силикагеле (40 г), элюируя смесью ЭтОАц/гексан (1:4, 900 мл). Соответствующие фракции объединяли и концентрировали при пониженном давлении, получая в результате вторую порцию соединения, указанного в заголовке (1,86 г, 4,29 ммоль), в виде коричневого масла; 1H ЯМР (300 МГц, CDCl3); d 7,30 (m, 2Н), 7,05 (m, 2Н), 6,95 (m, 2Н), 6,88 (m, 2Н), 6,73-6,61 (m, 3Н), 6,28 (m, 1Н), 5,01 (m, 1Н), 4,23 (s, 2H), 3,87 (s, 3H), 3,80 (s, 3H), 3,40 (s, 2H), 1,07 (m, 6H), ТСХ (ЭтОАц/гексан (1:4)): Rf = 0,14.
Промежуточное соединение 23
N-Изопропил-2-[5-(4-метокси-бензил)-2,4-диоксо-3-(фенил-гидразоно)- 2,3,4,5-тетрагидро-бензо[b][1,4]диазепин-1-ил]-N-(4-метокси-фенил)- ацетамид
К раствору N-изопропил-2-[2-(4-метоксибензиламино)-фениламино]-N-(4-метокси-фенил)- ацетамида (3,25 г, 7,51 ммоль) в ТГФ (50 мл), охлажденному на ледяной бане (< 5oC), по каплям в течение 20 минут добавляли двухлористый 2-(фенил-гидразоно)пропандиол (1,84 г, 7,51 ммоль) в ТГФ (50 мл). После завершения добавления раствор оставляли нагреваться до комнатной температуры и перемешивали в течение ночи. Растворитель упаривали при пониженном давлении, а полученное в результате масло растворяли в этилацетате (300 мл), промывали насыщенным раствором бикарбоната натрия (100 мл) и рассолом (50 мл), сушили над MgSO4, фильтровали и концентрировали при пониженном давлении, получая в результате соединение, указанное в заголовке (4,55 г, 7,51 ммоль), в виде желтого масла: 1H ЯМР (300 МГц, CDCl3): d 11,46 (s, 0,5H), 10,69 (s, 0,5H), 7,56-6,95 (m, 15H), 6,77-6,66 (m, 2H), 5,34 (m, 1H), 5,05 (m, 1H), 4,79 (m, 1H), 4,34-4,08 (m, 2H), 3,86 (s, 2H), 3,74 (s, 3H), 3,72 (s, 3H), 1,11 (m, 6H); TCX (ЭтОАц/гексан (2:3)): Rf = 0,30.
Промежуточное соединение 24
2-[3-Амино-5-(4-метокси-бензил)-2,4-диоксо-2,3,4,5-тетрагидро-бензо [b] [1,4]диазепин-1-ил]-N-изопропил-N-(4-метокси-фенил)- ацетамид
К перемешиваемому раствору N-изопропил-2-[5-(4-метокси- бензил)-2,4-диоксо-3-(фенил-гидразоно)-2,3,4,5- тетрагидробензо[b][1,4]диазепин-1-ил]-N-(4-метокси-фенил)-ацетамида (4,55 г, 7,51 ммоль) в уксусной кислоте (50 мл) добавляли цинковую пыль (4,50 г), полученную смесь перемешивали в течение 4 часов при температуре окружающей среды. Цинк отделяли фильтрованием, фильтрат концентрировали под вакуумом, полученное масло фракционировали между водой (150 мл) и этилацетатом (250 мл). Доводили pH до 8 с помощью 6N гидроксида натрия и разделяли фазы. Водную фазу экстрагировали этилацетатом (2х80 мл), органические фазы объединяли, сушили над сульфатом магния, фильтровали и концентрировали под вакуумом до коричневого масла. Сырой продукт очищали посредством флэш-хроматографии на силикагеле (70 г), элюируя последовательно смесями ЭтОАц/гексан (2:1, 500 мл) и метиленхлорид/метанол (19: 1, 500 мл). Соответствующие фракции объединяли и концентрировали, получая в результате соединение, указанное в заголовке (2,85 г, 5,52 ммоль), в виде коричневого масла; 1H ЯМР (300 МГц, CDCl3): d 7,43 (d, J=7,3 Гц, 1Н), 7,30-7,19 (m, 4H), 7,05-6,90 (m, 5H), 6,59 (d, J=8,8 Гц, 2Н), 5,13 (d, J= 15,1 Гц, 1Н), 5,00 (m, 1H), 4,86 (d, J=15,1 Гц, 1Н), 4,25 (s, 1H), 4,21 (d, J= 16,6 Гц, 1Н), 3,85 (s, 3H), 3,69 (s, 3H), 3,48 (d, J=16,6 Гц, 1Н), 1,09 (m, 6H); ТСХ (CH2Cl2/CH3OH (29:1): Rf = 0,11.
Промежуточное соединение 25
2-(3-Амино-2,4-диоксо-2,3,4,5-тетрагидро-бензо[b] [1,4] диазепин-1-ил] - N-изопропил-N-(4-метокси-фенил)-ацетамид
К перемешиваемому раствору 2-[3-амино-5-(4-метокси-бензил)-2,4- диоксо-2,3,4,5-тетрагидробензо[b] [1,4] диазепин-1-ил]-N-(4-метокси-фенил)- ацетамида (2,63 г, 5,09 ммоль) в смеси ацетонитрил/H2O (9:1, 70 мл) при температуре окружающей среды порциями в течение одного часа добавляли цериевый нитрат аммония (10,05 г, 18,3 ммоль). Раствор перемешивали в течение ночи при комнатной температуре. Раствор концентрировали под вакуумом, экстрагировали толуолом (2х50 мл), а остаток экстрагировали CH2Cl2 (3х50 мл), фильтровали и концентрировали до оранжевого стеклообразного вещества. Сырой продукт очищали посредством флэш-хроматографии на силикагеле (100 г), элюируя последовательно смесью CH2Cl2/CH3OH (15:1, 1,5 л) и CH2Cl2/CH3OH (10:1, 1,3 л). Соответствующие фракции объединяли и концентрировали при пониженном давлении, получая в результате соединение, указанное в заголовке (1,97 г, 4,97 ммоль), в виде коричневой пены: 1H ЯМР (300 МГц, ДМСО-d6): d 10,68 (br s, 1H), 7,39-6,98 (m,8H), 5,75 (s,1H), 4,76 (m,1H), 4,16-3,92 (m,4H), 3,77 (s,3H), 3,15 (m,2H), 0,97 (m,6H); TCX (CH2CL2/CH3OH (13:1): Rf = 0,21.
Промежуточное соединение 26
{ 1-[изопропил-(4-метокси-фенил)-карбамоилметил] -2,4-диоксо-2,3,4,5- тетрагидро-1Н-бензо[b][1,4]диазепин-3-ил}-амид 1Н-индол-2-карбоновой кислоты
К раствору 2-(3-амино-2,4-диоксо-2,3,4,5- тетpaгидpoбензo[b][1,4]диaзепин-1-ил]-N-изoпpoпил-N-(4-метoкcи- фенил)-ацетамида (350 мг, 0,883 ммоль) в ДМФ (10 мл) последовательно при перемешивании добавляли индол-2-карбоновую кислоту (142 мг, 0,883 ммоль), N-гидроксибензотриазол (119 мг, 0,883 ммоль) и гидрохлорид 1-(3-диметиламинопропил)-3-этилкарбодиимида (0,169 мг, 0,883 ммоль). Полученную смесь перемешивали при температуре окружающей среды в течение 18 часов. Растворитель выпаривали при пониженном давлении, получая желтое масло, которое растворяли в этилацетате (60 мл), промывали водой (20 мл) и рассолом (20 мл), сушили над MgSO4, фильтровали и концентрировали при пониженном давлении до желтого масла, которое затвердевало при стоянии. Сырой продукт растирали в порошок с кипящим этанолом (20 мл), охлаждали, фильтровали и сушили, получая в результате соединение, указанное в заголовке (169 мг, 0,313 ммоль), в виде желтого твердого вещества: 1H ЯМР (300 МГц, Ацетон-d6): d 10,85 (br s,1H), 9,83 (br s,1H), 7,67 (m, 2H), 7,53 (m, 2H), 7,37-7,20 (m, 7H), 7,04 (m, 3H), 5,28 (d, J=7,5 Гц, 1H), 4,87 (m, 1H), 4,31 (d, J=16,3 Гц, 1H), 4,13 (d, J=16,6 Гц, 1H), 3,83 (s, 3H), 1,02 (m, 6H); TCX (CH2Cl2/CH3OH (19:1): Rf = 0,24; MC (ББА): m/z = 540 (МН+).
Промежуточное соединение 27
N-Изопропил-N-(4-метокси-фенил)-2-фениламиноацетамид
Смесь N-изопропил-N-(4- метокси-фенил)бромацетамида (257,6 г, 924 ммоль), 1,2- фенилендиамина (100 г, 924 ммоль) и карбоната калия (128 г, 924 ммоль) в ДМФ (1200 мл) перемешивали при 0oC в течение 2 ч, а затем оставляли нагреваться до КТ в течение 20 часов. Реакционную смесь фильтровали через целит, и фильтрат концентрировали под вакуумом. Полученный остаток растворяли в ЭтОАц (1200 мл), промывали водой (3х200 мл), рассолом (200 мл), сушили (MgSO4) и концентрировали под вакуумом. После удаления около 70% растворителя образовывался осадок, который отделяли фильтрованием и промывали холодным ЭтОАц, сушили и получали соединение, указанное в заголовке (67,1 г), в виде бежевого вещества. Объединенные фильтраты концентрировали под вакуумом, получая темное масло (88 г). После двух перекристаллизаций из этанола получали вторую порцию соединения, указанного в заголовке, в виде бежевого вещества (29,6 г). 1H ЯМР (300 МГц, ДМСО-d6): s 7,22 (m, 2H), 7,05 (m, 2H), 6,47 (m, 1H), 6,34 (m, 2H), 5,95 (m, 1H), 4,81 (sept, 1H, J=6,8 Гц), 4,59 (dt, 1H, J=27,2, 6,1 Гц), 4,4 (s, 2H), 3,77 (s, 3H), 3,30 (s, 2H), 0,96 (d, 6H, J=6,8 Гц).
Промежуточное соединение 28
2-(2,4-диоксо-2,3,4,5-тетрагидро-бензо[b] [1,4]диазепин-1 ил]-N- изопропил-N-(4-метокси-фенил)-ацетамид
Раствор N-изопропил-N-(4-метокси-фенил)-2-фениламиноацетамида (20 г, 63,8 ммоль) в ТГФ (500 мл) и раствор малонилдихлорида (6,2 мл, 63,8 ммоль) в ТГФ (500 мл) добавляли одновременно в течение 40 минут к ТГФ (100 мл), полученный раствор перемешивали при КТ в течение 72 часов, после чего добавляли еще 3,0 мл малонилдихлорида. Спустя 5 ч растворители удаляли под вакуумом, а остаток растворяли в метиленхлориде (300 мл) и промывали 2N водным бикарбонатом натрия (2х200 мл). Объединенные органические фазы промывали водой (2х200 мл), рассолом (200 мл), сушили (MgSO4) и концентрировали под вакуумом, получая в результате сырой продукт (23,8 г). Затем его интенсивно растирали в порошок с эфиром, а полученное коричневое вещество затем очищали посредством колоночной флэш-хроматографии, элюируя 5%-ным метанолом в метиленхлориде, получая в результате соединение, указанное в заголовке (8,85 г) в виде бежевого вещества. 1H ЯМР (300 МГц, CDCl3): s 7,7 (s, 1H), 7,4 (m, 1H), 6,90-7,3 (m, 7H), 4,97 (sept., 1H, J=6,8 Гц), 4,4 (m, 1H), 3,81 (s, 3H), 3,78 (m, 1H), 3,40 (s, 2H), 1,06 (d, 6H, J=6,8 Гц).
Промежуточное соединение 29
2-(2,4-диоксо-5-пиридин-3-ил-2,3,4,5-тетрагидро-бензо[b] [1,4] диазепин-1-ил]-N-изопропил-N-(4-метокси-фенил)-ацетамид
Смесь 2-(2,4-диоксо-2,3,4,5-тетрагидробензо[b][1,4]диазепин-1-ил)- N-изопропил-N-(4-метокси-фенил)-ацетамида (3,5 г, 9,48 ммоль), медного порошка (1,09 г, 17,06 ммоль), 3-бромпиридина (0,91 мл, 9,48 ммоль) и ацетата калия (1,11 г, 11,37 ммоль) в ДМФ (80 мл) прогревали при 100oС в течение 7 ч. Добавляли еще 0,4 мл 3-бромпиридина и перемешивали полученную смесь при 100oС в течение еще 14 часов. Добавляли медный порошок (1,09 г, 17,06 ммоль), 3-бромпиридин (0,91 мл, 9,48 ммоль) и ацетат калия (1,11 г, 11,37 ммоль) и нагревали до 110oС в течение 6 ч. Твердые частицы удаляли фильтрованием, фильтрат концентрировали под вакуумом, а остаток фракционировали между этилацетатом (150 мл) и 10%-ным водным раствором гидроксида аммония (150 мл). Объединенные органические фазы промывали водой (2х100 мл), рассолом (50 мл), сушили (MgSO4) и концентрировали под вакуумом, получая в результате соединение, указанное в заголовке (2,56 г), в виде кремоподобного вещества. 1H ЯМР (300 МГц, CDCl3): s 7,86 (d, 1H, J=8,1 Гц), 7,01-7,43 (m, 10H), 6,92 (d, 1H, J= 8,3 Гц), 5,05 (sept, 1H, J=6,8 Гц), 4,22 (m, 2H), 3,88 (s, 3H), 3,58 (dd, 2H, J=32,2, 12,0 Гц), 1,06 (m, 6H).
Промежуточное соединение 30
2-(3-Азидо-2,4-диоксо-5-пиридин-3-ил-2,3,4,5-тетрагидро-бензо[b] [1,4] дазепин-1-ил)-N-изопропил-N-(4-метокси-фенил)-ацетамид
К раствору 2-(2,4-диоксо-5-пиридин-3-ил-2,3,4,5-тетрагидробензо[b][1,4] диазепин-1-ил)-N-изопропил-N-(4-метокси-фенил)-ацетамида (750 мг, 1,64 ммоль) в ТГФ (15 мл), охлажденному до -78oС в течение семи минут по каплям добавляли гексаметилдисилазид калия (0,5М в толуоле, 4,59 мл, 2,29 ммоль), полученный раствор перемешивали при -78oС в течение 17 мин. Добавляли 2,4,6-триизопропил-бензолсульфонилазид (632 мг, 2,05 ммоль) и перемешивали реакционную смесь при -78oС в течение 4 мин перед добавлением уксусной кислоты (0,235 мл), после чего оставляли реакционную смесь нагреваться до КТ. Растворители удаляли под вакуумом, получая в результате сырой продукт. Его очищали посредством колоночной флэш-хроматографии, элюируя 5%-ным метанолом в метиленхлориде, получая в результате соединение, указанное в заголовке (530 мг), в виде бесцветной пены. 1H ЯМР (300 МГц, CDCl3): s 7,96 (d,1H, J=8,0 Гц), 6,91-7,43 (m, 11H), 5,05 (sept, 1H, J=6,8 Гц), 4,42 (d, 1H, J=12,1 Гц), 4,38 (m, 1H), 4,22 (d, 1H, J=12,1 Гц), 3,88 (s, 3H), 1,06 (m, 6H).
Промежуточное соединение 31
2-(3-Амино-2,4-диоксо-5-пиридин-3-ил-2,3,4,5-тетрагидро-бензо[b] [1,4] диазепин-1-ил)-N-изопропил-N-(4-метокси-фенил)-ацетамид
Смесь 2-(3-азидо-2,4-диоксо-5-пиридин-3-ил- 2,3,4,5-тетрагидробензо[b] [1,4]диазепин-1-ил)-N-изопропил-N-(4- метокси-фенил)-ацетамида (530 мг, 1,06 ммоль) и 10%-ного палладия на угле (53 мг) перемешивали в этаноле (50 мл) в атмосфере азота в течение 9 ч. Добавляли следующую навеску палладия на угле (53 мг) и перемешивали полученную смесь еще в течение 16 ч. Твердые частицы удаляли фильтрованием через целит, фильтрат концентрировали под вакуумом, получая в результате соединение, указанное в заголовке (520 мг), которое применяли без дальнейшей очистки. 1H ЯМР (300 МГц, CDCl3): s 8,52 (m, 2H), 7,92 (d, 1H, J= 8,0 Гц), 6,91-7,43 (m, 11H), 5,05 (sept., 1H, J=6,8 Гц), 4,42-4,22 (m, 3H), 3,88 (s, 3H), 1,06 (m, 6H).
Промежуточное соединение 32
2-(2,4-Диоксо-5-пиридин-4-ил-2,3,4,5-тетрагидро-бензо[b] [1,4] диазепин-1-ил)-N-изопропил-N-(4-метокси-фенил)-ацетамид
К перемешивавшемуся раствору 400 мг 1 -[изопропил-(4-метокси-фенил)-амино]-1,5-дигидро-бензо[b][1,4] диазепин-2,4-диона в 20 мл ДМФ добавляли 212 мг (2,16 ммоль, 2,0 эквив) ацетата калия, 206 мг (3,25 ммоль, 3,0 эквив) медного порошка и 245 мг (2,62 ммоль, 2 эквив) 4-бромпиридина. Полученный раствор прогревали при 122oC в течение 7 ч. Реакционную смесь подвергали горячему фильтрованию через слой целита, слой промывали 10 мл метанола, а фильтрат концентрировали под вакуумом. Остаток разбавляли ЭтОАц (100 мл) и промывали 5%-ным гидроксидом аммония (5х25 мл). Органический слой отделяли, сушили (MgSO4), а растворители удаляли под вакуумом. После очистки материала посредством хроматографии быстрого разделения на силикагеле с использованием в качестве элюента смеси ЭтОАц/Гексан/NH4OH (80:20:1) с последующей перекристаллизацией из ЭтОН получали 80 мг (16%) соединения, указанного в заголовке: 1H ЯМР (Ацетон-d6, 300 МГц) d 8,57 (d, 1H, J=4,6 Гц), 7,46 (m, 3H), 7,31 (m, 4H), 6,97 (m, 3H), 4,86 (sept., 1Н), 4,48 (d, 1H, J=16,8 Гц), 4,21 (d, 1H, J=16,8 Гц), 3,86 (s, 3H), 3,67 (d, 1H, J=12,2 Гц), 3,22 (d, 1H, J= 12,2 Гц), 1,02 (m, 6H);
MC низкого разрешения (ББА) m/z 459,2 (МН+).
Промежуточное соединение 33
2-(3-Амино-2,4-Диоксо-5-пиридин-4-ил-2,3,4,5,5а, 9а-гексагидробензо[b] [1,4] диазепин-1-ил)-N-изопропил-N-(4-метокси-фенил)ацетамид
К перемешиваемому раствору 80 мг (0,175 ммоль) 2-(2,4-диоксо-5-пиридин-4-ил-2,3,4,5- тетрагидробензо[b] [1,4]диазепин-1-ил)-N-изопропил-N-(4-метокси-фенил)- ацетамида в 3 мл ДМФ при 0oC добавляли 0,210 мл (0,209 ммоль, 1,2 эквив) 1N бис(триметилсилил)амида натрия в ТГФ. После перемешивания в течение 0,5 ч добавляли 63 мг (0,262 ммоль; 1,5 эквив) O-(дифенил-фосфинил)гидроксиламина [Hager. J.C.S. Perkin I, 3284-3288] и перемешивали реакционную смесь в течение 16 часов при температуре окружающей среды. Реакционную смесь фильтровали, и фильтрат концентрировали под вакуумом. Сырой продукт очищали посредством флэш-хроматографии на 5 г силикагеля, элюируя последовательно ЭтОАц (100 мл), смесью ДХМ/CH3OH (19:1, 100 мл). Соответствующие фракции объединяли и концентрировали под вакуумом, получая в результате соединение, указанное в заголовке, в виде прозрачного стекла: МС низкого разрешения (ББА) m/z 474,2 (МН+); ТСХ Rf = 0,31 (ДХМ/CH3OH, 19:1)).
Промежуточное соединение 34
2-Амино-N-изопропил-N-(4-метокси-фенил)-ацетамид
Раствор 2,86 г 2-бром-N-изопропил-N-(4-метокси-фенил)ацетамида (10 ммоль) в 100 мл метанола насыщали аммонием при 0oC и оставляли на трое суток при температуре окружающей среды в закупоренной колбе. Метанол и аммоний удаляли под вакуумом, а остаток растворяли в 100 мл хлороформа и промывали водой (2х50 мл). Органический слой сушили над безводным MgSO4 фильтровали, концентрировали под вакуумом и сушили при глубоком вакууме, получая в результате 2,7 г соединения, указанного в заголовке, в виде масла. 1H ЯМР (300 МГц, CDCl3) d 6,96 (m, 4H), 4,99 (m, 1H), 3,84 (s, 3H), 2,97 (s, 2H), 1,58 (s, 2H), 1,05 (d,6H, J=6,6 Гц); МС низкого разрешения (ЭОИ) m/e 223 (МН+).
Промежуточное соединение 35
2-(4-фтор-2-нитро-фениламино)-N-изопропил-N-(4-метокси-фенил) ацетамид
Смесь 9,06 г 2,5-дифтор-нитробензола (60 ммоль) и 12,64 г 2-амино-N-изопропил-N-(4-метокси-фенил)-ацетамида (60 ммоль, 1,0 эквив) подвергали взаимодействию в 225 мл смеси 2:1 этанол/вода и нагревали с дефлегмацией в атмосфере азота при интенсивном перемешивании в течение ночи (приблизит. 16 часов). Полученный шлам охлаждали до температуры окружающей среды, фильтровали и промывали смесью 2:1 вода/этанол. Влажное вещество растворяли в метиленлхлориде, сушили над безводным сульфатом натрия и упаривали под вакуумом. Остаток растирали в порошок с гексаном, фильтровали и промывали гексаном. Продукт сушили в глубоком вакууме, получая при этом 9,32 г соединения, указанного в заголовке, в виде оранжевого вещества. 1H ЯМР (300 МГц, CDCl3) d 7,89 (dd, 1H, J=3,1, 9,3 Гц), 7,12 (m, 3H), 6,99 (m, 2H), 6,35 (dd, 1H, J=4,8, 9,2 Гц), 5,03 (m,1H), 3,89 (s, 3H), 3,57 (s, 2H), 1,09 (d, 6H, J= 6,8 Гц); МС низкого разрешения (ЭОИ) m/e 362 (МН+).
Промежуточное соединение 36
2-(2-Амино-4-фтор-фениламино)-N-изопропил-N-(4-метокси-фенил) ацетамид
Раствор 30 мл этилацетата, 175 мл этанола и 2,50 г 2-(4-фтор- 2-нитро-фениламино)-N-изопропил-N-(4-метокси-фенил)-ацетамида (6,92 ммоль) смешивали с 0,25 г палладия на угле (10%-ный по весу) и гидрогенизировали под водородным баллоном в течение 16 часов. Реакционную смесь фильтровали и упаривали под вакуумом, получая в результате 1,91 г соединения, указанного в заголовке, в виде твердого вещества. 1H ЯМР (300 МГц, CDCl3): d 7,03 (m, 2H), 6,94 (m, 2H), 6,36 (m, 3H), 4,99 (m, 1H), 4,37 (b, 3H), 3,86 (s, 3H), 3,39 (s, 2H), 1,07 (d, 6H, J=6,8 Гц); MC низкого разрешения (ББА) m/e 332 (МН+).
Промежуточное соединение 37
2-(7-Фтор-2,4-диоксо-2,3,4,5-тетрагидро-бензо[b] [1,4] диазепин-1-ил)- N-изопропил-N-(4-метокси-фенил)-ацетамид
Раствор 1,724 г 2-(2-амино-4-фтор-фениламино)-N- изопропил-N-(4-метокси-фенил)-ацетамида (5,20 ммоль) в 15 мл тетрагидрофурана переносили в воронку для подачи веществ. Раствор 0,506 мл малонилдихлорида (5,20 ммоль, 1,0 эквив) в 15 мл тетрагидрофурана переносили в отдельную воронку для подачи веществ. Растворы всех реагентов одновременно по каплям в течение 30 минут добавляли к 100 мл тетрагидрофурана при температуре окружающей среды в атмосфере азота при интенсивном перемешивании. После перемешивания в течение 20 минут при температуре окружающей среды в виде отдельной порции добавляли дополнительное количество малонилдихлорида (5,20 ммоль, 0,1 эквив). Реакционную смесь перемешивали дополнительно в течение 2,5 часов, а затем упаривали под вакуумом до получения остатка. Остаток очищали на силикагеле для быстрого разделения с элюцией сначала смесью 1:3 этилацетат/гексан, а затем 3: 1 этилацетат/гексан. Соответствующие фракции объединяли, упаривали под вакуумом до получения остатка и растирали в порошок с гексаном. Гексан удаляли под вакуумом, а оставшееся вещество сушили в глубоком вакууме, получая в результате 1,061 г соединения, указанного в заголовке, в виде дубильного вещества. 1H ЯМР (300 МГц, CDCl3): d 8,14 (b, 1H), 7,45 (dd, 1H, J=5,5, 9,2 Гц), 7,29 (m, 1H), 7,05 (m, 1H), 6,94 (m, 3H), 6,78 (m, 1H), 4,99 (m, 1H), 4,37 (d, 1H, J= 16,4 Гц), 3,82 (s, 3H), 3,69 (d, 1H, J=16,0 Гц), 3,40 (m, 2H), 1,09 (d, 6H, J=6,8 Гц); MC низкого разрешения (ББА) m/e 400 (МН+).
Промежуточное соединение 38
2-(7-Фтор-2,4-диоксо-5-пиридин-3-ил- 2,3,4,5-тетрагидро-бензо[b] [1,4] диазепин-1-ил)-N-изопропил-N-(4- метокси-фенил)-ацетамид
Смесь 0,880 г 2-(7-фтор-2,4-диоксо-2,3,4,5-тетрагидробензо[b][1,4]диазепин-1-ил)- N-изопропил-N-(4-метокси-фенил)-ацетамида (2,20 ммоль), 420 г меди (бронза, 6,61 ммоль, 3 эквив), 476 мг ацетата калия (4,85 ммоль, 2,2 эквив) и 0,290 мл 3-бромпиридина (4,85 ммоль, 2,2 эквив) в 10 мл диметилформамида прогревали при 100oC в атмосфере азота в течение 3 часов. Добавляли дополнительно 0,132 мл 3-бромпиридина (2,43 ммоль, 1,1 эквив) и продолжали реакцию еще в течение 2 часов. Реакционную смесь охлаждали до температуры окружающей среды, фильтровали через воронку из спеченного стекла, а затем упаривали до остатка под вакуумом. Остаток фракционировали между этилацетатом и водным гидроксидом аммония (5 мл конц., разведенного до 100 мл). После разделения слоев органический слой промывали водным гидроксидом аммония (5 мл конц., разведенного до 100 мл), а затем насыщенным водным рассолом. Органическую фазу затем трижды экстрагировали водной HCl (1N). Кислые слои объединяли и нейтрализовали водным гидроксидом натрия (1N). Нейтрализованную смесь дважды экстрагировали метиленхлоридом. Слои метиленхлорида объединяли, сушили над безводным сульфатом натрия, фильтровали и упаривали под вакуумом до остатка. Остаток растирали в порошок с гексаном, а затем концентрировали под вакуумом, получая в результате 0,705 г соединения, указанного в заголовке, в виде желто-коричневого твердого вещества. 1H ЯМР (300 МГц, CDCl3) d 8,60 (m, 2H), 8,08 (b, 1H), 7,54 (b, 1H), 7,39 (m, 1H), 7,15 (m, 2H), 7,00 (m, 3H), 6,57 (dd,1H, J=2,7, 9,2 Гц), 4,95 (m, 1H), 4,32 (d, 1H, J=17,9 Гц), 3,85 (s, 3H), 3,61 (d, 1H, J=12,1 Гц), 3,52 (d, 1H, J=12,1 Гц), 1,06 (d, 6H, J=6,8 Гц); MC низкого разрешения (ББА) m/e 477 (MH+).
Промежуточное соединение 39
Трет-бутиловый эфир 3-нитро-бензойной кислоты
Твердый трет-бутилат калия (3,82 г, 32,30 ммоль) добавляли к раствору 3-нитро-бензоилхлорида (5,00 г, 26,94 ммоль) в сухом тетрагидрофуране (70 мл) и перемешивали в течение 2 часов в атмосфере азота. Реакционную смесь концентрировали под вакуумом и фракционировали между дихлорметаном и водой. После разделения фаз водный слой снова экстрагировали этилацетатом. Органические слои объединяли, сушили над безводным сульфатом магния, фильтровали, а затем концентрировали под вакуумом. Сырой продукт очищали на силикагеле для быстрого разделения, применяя 0-5% градиент этилацетата в н-гексане. Фракции, содержавшие продукт, объединяли, концентрировали под вакуумом, а затем сушили в глубоком вакууме, получая в результате 3,82 г соединения, указанного в заголовке, в виде масла. 1H ЯМР (300 МГц, CDCl3) d 8,79 (m, 1H), 8,35 (m, 2H), 7,62 (m, 1H), 1,63 (s, 9H); MC низкого разрешения (ХИ) m/e 224 (МН+).
Промежуточное соединение 40
Трет-бутиловый эфир 3-амино-бензойной кислоты
Раствор трет-бутилового эфира 3-нитро-бензойной кислоты (3,77 г, 16,9 ммоль) в абсолютном этаноле (50 мл) смешивали с палладием на угле (10% по весу, 0,30 г) и гидрогенизировали под баллоном газообразного водорода приблизительно в течение 3 часов. Реакционную смесь фильтровали через слой кизельгура, а затем концентрировали под вакуумом до масла, которое кристаллизовалось при сушке в глубоком вакууме с получением в результате 3,26 г соединения, указанного в заголовке, в виде желто-коричневого твердого вещества. 1H ЯМР (300 МГц, CDCl3): d 7,38 (d, 1H, J=8,0 Гц), 7,29 (m, 1H), 7,19 (m, 1H), 6,83 (m, 1H), 1,58 (s, 9H); MC низкого разрешения (ХИ) m/e 194 (МН+).
Промежуточное соединение 41
Трет-бутиловый эфир 3-[(4-нитрофенил)оксикарбонил] -амино-бензойной кислоты
К раствору трет-бутилового эфира 3-амино-бензойной кислоты (3,15 г, 16,24 ммоль) в атмосфере азота при 0-5oC в течение 20 минут по каплям добавляли раствор 4-нитро-фенилхлорформиата в сухом дихлорметане (25 мл) и безводный пиридин (1,379 мл, 17,05 ммоль) в безводном дихлорметане (25 мл). Реакционную смесь оставляли при перемешивании нагреваться до температуры окружающей среды в течение ночи. После промывания водной HCl (1N) реакционный раствор сушили над безводным сульфатом магния и концентрировали под вакуумом до твердого состояния. Сырой продукт перемешивали в н-гексане в течение 30 мин, фильтровали и сушили в глубоком вакууме, получая в результате 4,460 г соединения, указанного в заголовке, в виде белого кристаллического вещества. 1H ЯМР (300 МГц, CDCl3) d 8,30 (m, 2H), 7,92 (m, 1H), 7,78 (d, 2Н, J=7,5 Гц), 7,43 (m, 3H), 7,11 (bs, 1H), 1,69 (s, 9H); MC низкого разрешения (L-SIMS) m/e 358 (MH+).
Промежуточное соединение 42
2-(3-Азидо-7-фтор-2,4-диоксо-5-пиридин-3-ил-2,3,4,5- тетрагидро-бензо[b] [1,4]диазепин-1-ил)-N-изопропил-N-(4-метокси-фенил) ацетамид
Раствор 262 мг 2-(7-фтор-2,4-диоксо-5-пиридин-2,3,4,5- тетрагидробензо[b] [1,4] диазепин-1-ил)-N-изопропил-N-(4-метоксифенил)- ацетамида (0,550 ммоль) в 5 мл безводного тетрагидрофурана при -78oC в атмосфере азота обрабатывали по каплям 1,54 мл бис(триметилсилил)амидом калия (0,5 М в толуоле, 0,770 ммоль, 1,4 экв.). После перемешивания в течение 15 мин добавляли 212 мг 2,4,6-триизопропилбензолсульфонилазида (0,687 ммоль, 1,25 экв. [J.Org. Chem. 1984, 49, 1430-1434]). После перемешивания в течение 4 мин реакционную смесь охлаждали добавлением 78,6 мл ледяной уксусной кислоты (1,38 ммоль, 2,5 экв.) и оставляли нагреваться до температуры окружающей среды. Реакционную смесь упаривали под вакуумом до остатка и очищали его на флэш-кремнеземе, используя смесь 1:1 этилацетат/гексан. Соответствующие фракции объединяли, упаривали под вакуумом и сушили в глубоком вакууме, получая в результате 170 мг соединения, указанного в заголовке, в виде аморфного твердого вещества.
MC низкого разрешения (ББА) m/e 518 (МН+); ТСХ (кремнезем) 3:1 этилацетат:гексан Rf = 0,68.
Промежуточное соединение 43
2-(3-Амино-7-Фтор-2,4-диоксо-5-пиридин-3-ил-2,3,4,5- тетрагидро-бензо[b] [1,4]диазепин-1-ил)-N-изопропил-N-(4-метокси-фенил) ацетамид
Раствор 152 мг 2-(3-азидо-7-фтор-2,4-диоксо-5-пиридин-2,3,4,5- тетрагидробензо[b][1,4]диазепин-1-ил)-N-изопропил-N-(4-метоксифенил)- ацетамида в 10 мл этилацетата смешивали с 60 мг палладия на угле (10 мас.%) и гидрогенизировали под баллоном газообразного водорода в течение 16 часов. Смесь фильтровали, упаривали под вакуумом и очищали на флэш-силикагеле, используя смесь 3:7 метанол/этилацетат. Соответствующие фракции объединяли, упаривали под вакуумом и сушили в глубоком вакууме, получая в результате 82 мг соединения, указанного в заголовке, в виде пены. МС низкого разрешения (ББА) m/e 492 (МН+); ТСХ (кремний) 95:5 дихлорметан/метанол Rf = 0,30.
Фармацевтический пример
Таблетка
Активный ингредиент: - 50 мг
Безводная лактоза USP: - 163 мг
Микрокристаллическая целлюлоза NF: - 69 мг
Предварительно желатинизированный крахмал Ph.Eur. - 15 мг
Стеарат магния USP - 3 мг
Общий вес: - 300 мг
Активный ингредиент, микрокристаллическую целлюлозу, лактозу и предварительно желатинизированный крахмал просеивают через сито с размером ячейки 500 мкм и смешивают в соответствующем смесителе. Стеарат магния просеивают через сито с размером ячейки 250 мкм и смешивают с активной смесью. Смесь прессуют в таблетки с помощью соответствующих прессов, а затем покрывают ацетатфталатом целлюлозы.
ТЕСТ НА СВЯЗЫВАНИЕ С ХЦК-А РЕЦЕПТОРОМ
Приготовление ткани:
Готовили растворы 0,3 М и 0,2 М сахарозы и охлаждали их в течение ночи при 4oC. На следующий день добавляли ингибиторы таким образом, чтобы их конечные концентрации составляли 0,01% для ингибитора соевого трипсина (50 мг/500 мл сахарозы) и 100 мМ для фенилметилсульфонилфторида (8,5 мг/500 мл сахарозы).
Крыс умерщвляли посредством обезглавливания с помощью гильотины. Крысиные наружные брюшные стенки смачивали метанолом и удаляли мех и кожу. Брюшную полость вскрывали, осторожно высекали поджелудочные железы и помещали их в мензурку емкостью 50 мл, содержавшую 0,3 М сахарозу. После сбора всех поджелудочных желез удаляли избыточный жир и лимфатические узлы. Ткань поджелудочных желез разделяли на аликвоты весом примерно 4,0 г и помещали их в мензурки емкостью 30 мл, каждая из которых содержала 1,0 мл 0,3 М сахарозы.
В помещении, охлажденном до 4oC, поджелудочные железы измельчали с помощью ножниц и разбавляли 1:10 вес:объем 0,3 М сахарозой. Аликвоты гомогенизировали в охлажденном Wheaton баллоне посредством 4 ударов вверх-вниз пестиком "Б", а затем 4 ударов вверх-вниз пестиком "А". Гомогенаты фильтровали через 2 слоя фильтровальной ткани в охлажденную мензурку емкостью 500 мл, затем разбавляли при перемешивании 2,0 М сахарозой, получая в результате гомогенат с конечной концентрацией сахарозы, равной 1,3 М. Полученный 1,3 М гомогенат распределяли на льду в 18 тонкостенных полиалломерных пробирок (приблизительно 30 мл гомогената на пробирку) и в каждую пробирку последовательно наслаивали 0,3 М сахарозы до тех пор, пока жидкость не достигала приблизительно 0,5 см от края пробирки. Образцы центрифугировали в Sorvall RC70 ультрацентрифуге при скорости вращения 27500 об/мин (100000 xg) в течение 3 часов при 4oC. В охлажденный градуированный цилиндр собирали пограничный слой, разбавляли его и смешивали с холодной дистиллированной водой до общего объема 312 мл и центрифугировали при 100000 xg в течение 50 мин при 4oC. Осадки ресуспендировали в KRH буферах, переносили в Wheaton баллон и гомогенизировали посредством 4 ударов вверх-вниз подогнанного (герметичного) пестика "А". Этот гомогенат переносили в 2-мл поликарбонатные пробирки и центрифугировали при 100000 xg в течение 30 мин при 4oC. Осадок ресуспендировали (1 мл KRH буфера/гм веса исходной ткани), переносили в баллон соответствующего размера и гомогенизировали посредством 4 ударов вверх-вниз подогнанного пестика "А". Аликвоты объемом 1 мл хранили при -70oC в микроцентрифужных пробирках.
Результаты представлены в таблице.
Тест:
Тестируемые соединения разводили в буфере для тестирования связывания в базовых концентрациях в 10 раз больших, чем требуемые конечные концентрации для теста.
50 мл соединения + 400 мл буфера + 25 мл [125I] сульфатированного ХЦК-8, меченого посредством Bolton and Hunter реагента (Amersham, 2000 Cl/ммоль), + 25 мл приготовленных мембран из крысиных поджелудочных желез инкубировали в течение 30 минут при 25oC при осторожном перемешивании в процессе инкубирования.
Для определения неспецифического связывания применяли 1 мМ L-364718 (конечная концентрация).
Реакцию останавливали с помощью Brandell Cell Harvester, промывали 3 х 3 мл охлажденного на льду (4oC) буфера для тестирования связывания на промывку.
Ткани собирали на Whatman GF/B фильтровальную бумагу, предварительно смоченную буфером для тестирования, и анализировали ее с помощью счетчика гамма-излучения.
ТЕСТ НА СВЯЗЫВАНИЕ С ХЦК-В РЕЦЕПТОРОМ
Приготовление ткани:
Самцов Hartley морских свинок (250-300 г, Charles River) умерщвляли путем отсечения головы. Извлекали головной мозг и помещали его в 4oC буфер (буфер 50 мМ Трис/HCl, pH=7,4).
Кору головного мозга отсекали и помещали в 4oC буфер. Определяли общий сырой вес всех тканей коры и разбавляли их 1:10 (вес:объем) буфером.
Кору головного мозга крошили с помощью Tekmar Tissuemizer, а затем гомогенизировали в буфере посредством 5 ходов поршня вверх-вниз, используя мотор, присоединенный к гомогенизатору стекло/тефлон. Препарат хранили при 4oC (на льду).
Мембраны осаждали центрифугированием в Sorvall RC5C при 4oC с помощью SA 600 ротора, вращавшегося со скоростью 16000 об/мин (47800 X g Максимум). Осадок собирали, а супернатант отбрасывали. Осадки объединяли и ресуспендировали в буфере при 4oC, используя тот же объем, как и указанный выше, и приготовляли смесь, как описано выше, посредством 5 ходов поршня вверх-вниз в гомогенизаторе стекло/тефлон с механическим приводом, используя тот же объем, что и ранее. Полученные гомогенаты центрифугировали при 16000 об/мин (47800 X g Максимум, 36592 X g в среднем) в течение 15 минут при 4oC. Осадки собирали, а супернатанты отбрасывали. Осадки затем объединяли с буфером до конечного объема 300 мл и готовили смесь с помощью Tekmar Tissuemizer. Посредством Biorad белкового теста определяли исходное содержание белка. Объем суспензии доводили буфером таким образом, чтобы в результате доведения объема конечная концентрация составляла 4 мг/мл, что подтверждалось посредством Biorad белкового теста. Конечную суспензию переносили в виде аликвот объемом 4,0 мл в пластиковые пробирки и замораживали при -70oC.
Анализ:
Буфер = 20 мМ HEPES, 1мМ EGTA, 118 мМ NaCl, 5 мМ KCl, 5 мМ MgCl2, 0,05% БСА при pH 7,4.
Скатроновые фильтры пропитывали буфером с 0,1% бычьего сывороточного альбумина (БСА) за час до сбора.
Готовили свежие 100 мМ бестаин и 3 мМ фосфорамидон. (Конечная опытная концентрация должна была составлять точно 10 мМ).
Тестируемые соединения разводили в буфере для тестирования связывания в базовых концентрациях, в 10 раз более концентрированных по сравнению с необходимыми конечными концентрациями для анализа. Разводили [125I]-сульфатированный ХЦК-8, меченый посредством Bolton-Hunter реагента (Amersham, 200 Ки/ммоль). 25 мл 100 мМ бестаина + 25 мл фосфорамидона + 25 мл тестируемого соединения + 50 мл радиолиганда + 25 мл буфера + 100 мл мембран коры головного мозга морских свинок инкубировали в течение 150 минут при комнатной температуре.
Для определения Во буфер для тестирования связывания заменяли тестируемым соединением.
Для определения связывания с фильтром буфер для анализа заменяли тестируемым соединением и мембранами коры головного мозга морских свинок.
Для определения неспецифического связывания 1 мМ сульфатированного ХЦК-8 (Sigma) заменяли тестируемым соединением.
Реакцию останавливали фильтрованием с помощью автоматического Skatron Cell Harvester. Фильтры промывали с помощью 4oC буфера. Затем фильтры перфорировали, помещали в пробирки и анализировали посредством счетчика гамма-излучения.
АНАЛИЗ ЖЕЛЧНОГО ПУЗЫРЯ МОРСКОЙ СВИНКИ
Приготовление ткани:
У морских свинок, умерщвленных посредством сворачивания шеи, удаляли желчные пузыри. Извлеченные желчные пузыри очищали от прилипшей соединительной ткани и разрезали на два кольца от каждого животного (2-4 мм в длину). Кольца затем суспендировали в контейнерах для органов, содержавших физиологический солевой раствор следующего состава (мМ): NaCl (4,7), MgSO4•H2O (1,2); CaCl2 • 2H2O (2,5); KH2PO3 (1,2); NaHCO3 (25) и декстроза (11,1). Омывающий раствор содержали при температуре 37oC и аэрировали смесью 95% O2/5% CO2. Ткани присоединяли посредством золотых цепочек и поддерживающих спиц из нержавеющей стали к изометрическому силовому датчику замещения (Grass, Model FT03 D). Затем на полиграфе (Grass, Model 7E) регистрировали ответы. По одной ткани от каждого животного служили в качестве контроля время/растворитель и не получали тестируемого соединения.
Анализ:
Кольца постепенно растягивали (в течение 120 минут) до базового остаточного напряжения, составлявшего 1 гм, которое поддерживали в течение эксперимента. В процессе создания базового напряжения кольца четыре раза погружали в ацетилхолин (АЦХ, 10- М) для контроля сократимости. Затем ткани обрабатывали субмаксимальной дозой сульфатированного ХЦК-8 (Sigma, 3 х 10- М). После получения стабильного ответа ткани быстро промывали 3 раза по 5-10 минут каждый час для восстановления стабильной базовой линии.
Соединения растворяли в диметилсульфоксиде (ДМСО), затем разводили водой и тестировали посредством кумулятивной кривой зависимости ответа от концентрации для тестируемого соединения (от 10-11 до 3 х 10-6 М), а затем кривой зависимости ответа от концентрации для сульфатированного ХЦК-8 (от 10-11 до 10-6 М) в присутствии наибольшей дозы тестируемого соединения. В качестве заключительного анализа добавляли АЦХ (10 мМ) для индукции максимальной концентрации. Для каждого тестировавшегося соединения проводили минимум три определения активности.
Ниже приведены результаты, полученные в этом тесте с типичными соединениями по изобретению. Соединения тестировали в концентрации 1 мкМ, а результаты выражали как % максимального ответа сульфатированного ХЦК-8.
Пример N - Сокращение
1
2
3 - 91
4 - 64
5 - 83
6 - 67
7 - 51
8 - 32
9 - 77
10 - 81
11 - 83
12 - 67
13 - 96
14 - 84
15 - 93
ПРИМЕР 18-ЧАСОВОГО ИНДУЦИРОВАННОГО ДЕПРИВАЦИЕЙ ПИТАНИЯ
Самцов Long-Evans крыс (Carles River Co. , Raleighhh, NC), весивших 300-375 г, индивидуально адаптировали в течение по меньшей мере одной недели в подвешенных камерах из сетки из нержавеющей стали (17,8 х 25,4 х 17,8 см высотой) при свободном доступе к воде (вводившейся через автоматические питьевые трубки в задней части камеры) и пищи (Lab Blox, Purina Rodent Laboratory Chow # 5001) при 12-часовом цикле свет/темнота (свет от 0600-1800 часов или ч) при приблизительно 22,8oC. Перед тестированием на 16.00 ч удаляли весь корм, но не воду. На 09.00 ч следующим утром крыс взвешивали. На 09.45 ч крысам вводили внутрибрюшинно (в.б.), перорально (п.о.) или посредством введения канюли в двенадцатиперстную кишку тестировавшееся соединение либо наполнитель (2 мл/кг) и возвращали их в родные клетки. Пищу давали на 10.00 ч. На 10.30 ч оставшуюся пищу и отбросы взвешивали.
Соединения по изобретению по существу нетоксичны в терапевтически применяемых дозах. Поэтому не наблюдали никаких побочных эффектов при введении соединений крысам в терапевтически применяемых дозах.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ 1,5-БЕНЗОДИАЗЕПИНА, СПОСОБ ИХ ПОЛУЧЕНИЯ И СОДЕРЖАЩИЙ ИХ ФАРМАЦЕВТИЧЕСКИЙ СОСТАВ | 1994 |
|
RU2135486C1 |
| ПРОИЗВОДНЫЕ ИНДОЛА | 1994 |
|
RU2144535C1 |
| ПРОИЗВОДНЫЕ 1,3-ДИОКСОИНДЕНА, ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМАЯ СОЛЬ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ АНТИВИРУСНАЯ КОМПОЗИЦИЯ, СОДЕРЖАЩАЯ ИХ В КАЧЕСТВЕ АКТИВНОГО ИНГРЕДИЕНТА | 2012 |
|
RU2566761C2 |
| НОВЫЕ ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ И ИХ ПРИМЕНЕНИЕ В МЕДИЦИНЕ И КОСМЕТИКЕ | 2015 |
|
RU2712971C2 |
| ПРОИЗВОДНЫЕ АРИЛАМИДОВ В КАЧЕСТВЕ БЛОКАТОРОВ TTX-S | 2011 |
|
RU2535671C1 |
| ПРОИЗВОДНЫЕ 3-(5-ТЕТРАЗОЛИЛБЕНЗИЛ)АМИНОПИПЕРИДИНА, СПОСОБЫ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 1994 |
|
RU2136675C1 |
| ПРОИЗВОДНЫЕ 1,5-БЕНЗОДИАЗЕПИНА И СОДЕРЖАЩИЙ ИХ ФАРМАЦЕВТИЧЕСКИЙ СОСТАВ | 1994 |
|
RU2135485C1 |
| ПРОИЗВОДНЫЕ ПИПЕРИДИНА, СПОСОБЫ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 2001 |
|
RU2298550C2 |
| ПРОИЗВОДНЫЕ ДИАЗЕПИНА, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБ ЛЕЧЕНИЯ АРИТМИИ | 1994 |
|
RU2155587C2 |
| ПРОИЗВОДНЫЕ ПИРАЗОЛОПИРИДИНА В КАЧЕСТВЕ БЛОКАТОРОВ TTX-S | 2013 |
|
RU2652117C2 |
Описываются новые соединения - 5-гетероцикло-1,5-бензодиазепины общей формулы (I) и их фармацевтически приемлемые соли, где Х является водородом или галогеном; R1 является группой NR4R5, где R4 обозначает С3-6алкил, a R5 обозначает фенил, замещенный в пара-положении метоксилом; R2 является индолом, фенилом или группой NHR11, где R11 - фенил или фенил, замещенный карбоксигруппой; R3 является гетероциклической группой, присоединенной к азоту через атом углерода ее циклического кольца, выбранной из пиридила, пиримидинила или пиразолила, последний в свою очередь трижды замещен С1-4 алкилом; z равно 1. Соединения обладают агонистической активностью в отношении ХЦК-А рецептора, что дает им возможность модулировать гормоны гастрин и холецистокинин (ХЦК) у млекопитающих. Некоторые из этих соединений обладают также агонистической активностью по отношению к ХЦК-Б рецепторам. 5 з.п. ф-лы, 1 табл.
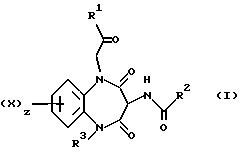
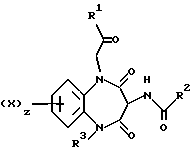
и их фармацевтически приемлемые соли,
где X является водородом или галогеном;
R1 является группой NR4R5, где R4 обозначает C3-6алкил;
R5 обозначает фенил, замещенный в пара-положении метоксилом;
R2 является индолом, фенилом или группой -NHR11, где R11 обозначает фенил или фенил, замещенный карбоксигруппой;
R3 является гетероциклической группой, присоединенной к азоту через атом углерода ее циклического кольца, выбранной из пиридила, пиримидинила или пиразолила, последний в свою очередь трижды замещен C1-4алкилом,
z равно 1.
| US, 4988692, A, 1990 | |||
| EP, 0558104, A, 1993 | |||
| SU, 94037768, A, 10.08.96 | |||
| SU, 460627, A, 1975. |
