Предметом данного изобретения являются производные индола, способы их получения, содержащие их фармацевтические составы и их применение в медицине. В частности, предметом изобретения являются производные индола, которые являются сильными и специфическими антагонистами возбуждающих аминокислот.
Известно [1], что некоторые известные 2-карбоксильные производные индола являются антагонистами возбуждающих аминокислот. Известны [2] также некоторые 2-карбоксильные производные индола как соединения, терапевтически эффективные при лечении заболеваний ЦНС, являющихся результатом нейротоксического нарушения ими нейродегенеративных заболеваний. Помимо этого известны [3] замещенные по 3-му положению 2-карбоксииндольные производные, которые пригодны для лечения нейродегенеративных заболеваний, включающих цереброваскулярные расстройства.
Мы обнаружили новую группу замещенных по 3-му положению 2-карбоксииндольных производных, которые обладают специфической антагонистической активностью в нечувствительном к стрихнину глициновом сайте связывания, локализованном на N-метил-D-аспартатном (NMDA) рецепторном комплексе.
Согласно настоящему изобретению предлагают соединение формулы (I)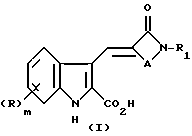
либо его соль или метаболически лабильный эфир, где R представляет собой группу, выбранную из галогено, алкил, алкоксил, амино, алкиламино, диалкиламино, гидроксил, трифторметил, трифторметокси, нитро, циано, SO2R2 или COR2 групп, где R2 является гидрокси, метокси, амино, алкиламино или диалкиламиногруппой; m равно нулю или целому числу 1 или 2;
R1 является циклоалкилом, мостиковым циклоалкилом, гетероарилом, мостиковым гетероциклическим или возможно замещенным фенилом или конденсированной бициклической карбоциклической группой, A представляет собой C1-4 цепь алкилена или цепь (CH2)pY(CH2)q, где Y является O, S(O)n или NR3, причем эти цепи могут быть замещены одной или двумя группами, выбранными из С1-6 алкила, возможно замещенного гидрокси, амино, алкиламино или диалкиламиногруппой, либо эти цепи могут быть замещены группой = O;
R3 является водородной, алкильной или азотной защитной группой;
n равно нулю или целому числу от 1 до 2;
p равно нулю или целому числу от 1 до 3;
q равно нулю или целому числу от 1 до 3 при условии, что сумма p + q равна 1, 2 или 3.
Соединения, описываемые формулой (I), могут существовать в более чем одной изомерной форме и все возможные изомеры включены в формулу (I) за исключением особо оговоренных. Так, например, в соединениях формулы (I) экзоциклическая двойная связь может существовать в цис или транс конфигурации и изобретение включает оба изомера и их смеси.
Для применения в медицине соли соединений формулы (I) должны являться физиологически приемлемыми солями. Однако для получения соединений формулы (I) или их физиологически приемлемых солей могут применяться другие соли. Поэтому за исключением оговоренных особо ссылок на соли изобретение включает как физиологически приемлемые соли, так и нефизиологически приемлемые соли соединений формулы (I).
Пригодные физиологически приемлемые соли соединений по изобретению включают соли, образованные в результате присоединения оснований, и, если целесообразно, соли, образованные в результате присоединения кислот. Пригодные физиологически приемлемые соли соединений формулы (I), образованные в результате присоединения оснований, включают соли щелочных металлов и соли щелочноземельных металлов, такие как соли натрия, калия, кальция и магния и соли аммония, образованные при взаимодействии с аминокислотами (например, лизином и аргинином) и органическими основаниями (например, прокаином, фенилбензиламином, этаноламином, диэтаноламином и N-метилглюкозамином).
Пригодные соли, образованные в результате присоединения кислот, могут быть образованы органической кислотой и неорганическими кислотами, например соляной кислотой.
Соединение формулы (I) или их соли могут образовывать сольваты (например, гидраты), и изобретение включает все подобные сольваты.
Следует отметить, что соединение формулы (I) может продуцироваться in vivo в результате метаболизма соответствующего препарата-предшественника. Такими препаратами-предшественниками могут являться, например, физиологически приемлемые метаболически лабильные сложные эфиры соединений общей формулы (I). Они могут быть образованы путем этерификации карбоксильной кислотной группы в исходном соединении общей формулы (I), у которого произведена соответствующая предварительная защита любых других реактивных групп, присутствующих в молекуле с последующим снятием защиты, если это необходимо. Примеры таких метаболически лабильных сложных эфиров включают C1-4 алкиловые сложные эфиры, например сложные метилэтиловые эфиры или t-бутиловые сложные эфиры, C3-6 алкеновые сложные эфиры, например аллиловые замещенные или незамещенные аминоалкиловые сложные эфиры (например, аминоэтиловый, 2-(N,N-диэтиламино) этиловый или 2-(4-морфолино) этиловый сложные эфиры) либо ацилоксиалкиловые сложные эфиры, такие как ацилоксиметиловый или 1-ацилоксиэтиловый, в частности пивалоилоксиметиловый, 1-пивалоилоксиэтиловый, ацетоксиметиловый, 1-ацетоксиэтиловый, 1-(1-метокси-1-метил)этилкарбонилоксиэтиловый, 1-бензоилоксиэтиловый, изопропоксикарбонилоксиметиловый, 1-изопропоксикарбонилоксиэтиловый, циклогексилоксикарбонилоксиметиловый, 1-циклогексилоксикарбонилоксиэтиловый сложный эфир, циклогексилоксикарбонилоксиметил, 1-циклогексилоксикарбонилоксиэтил, 1-(4-тетрагидропиранилокси)карбонилоксиэтил или 1-(4-тетрагидропиранил) карбонилоксиэтил.
Предпочтительные метаболически лабильные сложные эфиры соединений формулы (I) включают C1-4 алкиловые сложные эфиры, более конкретно метиловые или этиловые, аминоалкиловые сложные эфиры, более конкретно 2-(4'-морфолино)этиловые или ацилоксиалкиловые сложные эфиры, например ацетоксиметил, пивалоилоксиметил, 1-циклогексилоксикарбонилоксиэтил или 1-(4-тетрагидропиранилоксикарбонилокси) этил.
Гриппа R может располагаться в любом из четырех возможных положений на конденсированном бензольном кольце и в случае, когда m равно 2, две R группы могут быть одинаковыми или различными.
За исключением особо оговоренных случаев термин алкил, применяемый в тексте для обозначения группы или части группы, относится к алкильной группе с прямой или разветвленной цепью, содержащей от 1 до 4 атомов углерода, примеры таких групп включают метил, этил, пропил, изопропил, н-бутил, изобутил, вторичный бутил или третичный бутил.
Термин галоген обозначает атом фтора, хлора, брома или иода.
Термин циклоалкил обозначает C5-7 циклоалкильную группу, которая возможно может быть замещена 1 или 2-мя группами C1-4 алкила, например циклопентилом, циклогексилом, циклогептилом или 2-метилциклогексилом.
Термин мостиковый циклоалкил обозначает группу, содержащую от 7 до 10 углеродных атомов, которая является насыщенной или содержит одиночную двойную связь. Примеры пригодных мостиковых циклоалкильных групп включают адамантил, такой как 1-адамантил или 2-адамантил, норадамантил, бицикло(2,2,1)гептанил, например 2-норбоманил или бицикло(2,2,1)гептенил, например 5-норбоменил.
Термин гетероарил обозначает 5- или 6-членную гетероарильную группу, в которой 5-членная гетероарильная группа содержит 1 или 2 гетероатома, выбранных из кислорода, серы или азота, а 6-членная гетероарильная группа содержит 1 или 2 атома азота, причем гетероарильные группы могут быть включены в бензольное кольцо. Примеры подходящих гетероарильных групп включают фуранил, тиофенил, имидазолил, тиазолил, оксазолил, пиридинил, пиримидинил и хинолинил.
Термин мостиковый гетероцикл обозначает мостиковую гетероциклическую кольцевую систему, содержащую от 7 до 10 кольцевых членов, выбранных из углерода, кислорода или азота, причем эта мостиковая гетероциклическая система является насыщенной или содержит одну двойную связь. Предпочтительно мостиковая гетероциклическая группа содержит один гетероатом, выбранный из кислорода или азота. Примеры подходящих мостиковых гетероциклических групп включают 7-оксабицикло(2,2,1)гептанил, 7-окса-бицикло(2,2,1)гептенил, 7-аза-бицикло(2,2,1)гептанил, 7-аза-бицикло(2,2,1)гептенил или 1-аза-бицикло(2,2,2)октанил, такой как 3-хинуклидинил.
Термин сконденсированная бициклическая карбоциклическая группа обозначает 5,6/6,5 или 6,6 бициклическую карбоциклическую кольцевую систему, содержащую 9 или 10 углеродных атомов, которая может быть насыщенной или ненасыщенной. Примеры таких групп включают нафтил, тетрагидронафтил, декагидронафтил, инденил или инданил.
Если группа R1 является замещенной фенильной или сконденсированной бициклической карбоциклической группой, то это обозначает группу, которая замещена 1-3 группами, выбранными из галогена, алкил, алкоксил, амино, алкиламино, диалкиламино, C1-6 алканоиламино, уреидо, алкилсульфониламино, гидрокси, трифторметил, трифторметокси, нитро, циано, SO2R2 или COR2 групп, где R2 является гидрокси, метокси, амино, алкиламино или диалкиламиногруппой.
Если A представляет собой возможно замещенную C1-4 алкиленовую цепь, то такая цепь может являтьсяБ например, метиленом, этиленом, пропиленом, бутиленом или CH2CO. Если A представляет собой цепь (CH2)pY(CH2)q, такая цепь может являться, например, CH2OCH2, CH2NR3CH2, CH2NH, NHCO, CH2NCH3, CH2CH2NH или CH2O.
Если R3 является азотной защитной группой, то она может являться, например, возможно замещенной бензильной, алкоксикарбонильной группой, например t-бутоксикарбонилом, аралкилоксикарбонилом, триметилсилилэтоксиметилом или арилсульфонилом, в частности фенилсульфонилом.
Предпочтительным классом соединений формулы (I) являются те, у которых m равно 1 или 2, а в рамках этого класса наиболее предпочтительными являются те, у которых R находится в 4-м или 6-м положении.
Примеры подходящих R групп включают хлоро-, бромо-, иодо-, метил или этил. Наиболее предпочтительной R является хлор-группа.
Группа R3 предпочтительно является водородом или C1-4 алкилом, например метилом.
Предпочтительно цепь A является группой, выбранной из C2-3 алкиленов, возможно замещенных группой = O, например -(CH2)2-, -CH2CO- или -(CH2)3- или (CH2)pY(CH2)q, где p равно 1 или 2, q равно нулю, a Y является NH, NHCH3 или O, например -CH2NH-, -CH2NCH3-, -(CH2)2NH- или -CH2O-, либо p является нулем, Y является NH, q равно 1 или 2, а группа (CH2)q замещена группой =O, например -NHCO-.
Предпочтительный класс соединений формулы (I) включает те соединения, в которых A является цепью, выбранной из -(CH2)2, -(CH2)3-, -CH2CO-, - CH2NH-, -CH2NCH3-, -CH2O- или NHCO. В пределах этого класса наиболее предпочтительными являются те соединения, у которых A являются (CH2)2- или более конкретно -CH2NH-.
Удобно, чтобы группа R1 являлась группой, выбранной из возможно замещенных фенила, нафтила, например 1-нафтилом, пиридилом, в частности 2-пиридилом, хинолинилом, например 2-хинолинилом, циклогексилом или адамантилом, в частности 2-адамантилом.
Если R1 является возможно замещенной фенильной группой, то удобно, чтобы это был фенил или фенил, замещенный амино, ацетиламино, метансульфониламино или уреидогруппой, причем заместитель находится в мета или более предпочтительно, в пара положении.
Другим предпочтительным классом соединений формулы (I) являются соединения, у которых R1 является фенилом или фенилом, замещенным амино, ацетиламино, метансульфониламино или уреидогруппой. В рамках этого класса особенно предпочтительны те соединения, у которых R1 является фенилом.
Соединения, у которых R1 является возможно замещенным фенилом или 1-нафтилом, представляют собой уже другой предпочтительный класс соединений формулы (I).
Соединения формулы (I), где экзоциклическая двойная связь расположена в транс (E) конфигурации, представляют собой еще один предпочтительный класс соединений по изобретению.
Предпочтительной группой соединений формулы (I) являются те, у которых m равно 2, a R является хлорином в 4-м и 6-м положениях, A является цепью, выбранной из -(CH2)2-, -(CH2)3-, -CH2CO-, -CH2NH-, -CH2NCH3-, -(CH2)2NH-,
-CH2O- или -NHCO-, а R1 является группой, выбранной из возможно замещенных фенилов.
Другой предпочтительной группой соединений формулы (I) являются те, у которых m равно 2, а R является хлорином в 4-м и 6-м положениях, А является CH2NH, а R1 является возможно замещенным фенилом, 2-пиридилом, 2-хинолинилом, 1-нафтилом, циклогексилом или 2-адамантилом. В рамках этой группы наиболее предпочтительными соединениями являются их транс (E) изомеры. Более конкретно, особенно предпочтительны соединения, у которых R1 является возможно замещенным фенилом, например фенилом или фенилом, замещенным аминогруппой.
Наиболее предпочтительным соединением по изобретению является (E) 4,6-дихлор-3-(5-оксо-1-фенил-пиразолидин-4-илиденметил) -1Н-индол-2-карбоновая кислота и ее физиологически приемлемые соли, например натриевые или калиевые соли, либо ее метаболически лабильные сложные эфиры.
Другие предпочтительные соединения включают (Е)-4,6-дихлор-3- (2-оксо-1-фенил-пирролидин-3-илиденметил)-1H-индол-2-карбоновую кислоту;
(E) -4,6-дихлоро-3-(2-оксо-1-фенил-пиперидин-3-илиденметил) -1H-индол-2-карбоновую кислоту;
(Е) -4,6-дихлоро-3-[(5-оксо-1-(4-аминофенил))пиразолидин-4- илиденметил] -1H-индол-2-карбоновую кислоту;
(Z) 4,6-Дихлоро-3-(2, 5-диоксо-1-фенил-имидазолидин-4- илиденметил)-1H-индол-2-карбоновую кислоту;
(E) 4,6-Дихлоро-3-(2,5-диоксо-1-фенил-имидазолидин-4- илиденметил)-1H-индол-2-карбоновую кислоту;
4,6-Дихлоро-3-[5-оксо-1-(4-ацетиламинофенил)-пиразолидин-4- илиденметил] -1H-индол-2-карбоновую кислоту;
4,6-Дихлоро-3-[5-оксо-1-(4-уреидофенил)-пиразолидин- 4-илиденметил] -1H-индол-2-карбоновую кислоту;
4,6-Дихлоро-3-[1-(4-метилсульфамидофенил)-5-оксо-пиразолидин-4- илиденметил]-1H-индол-2-карбоновую кислоту;
4,6-Дихлоро-3-(3-оксо-2-фенил-изоксазолидин-4-илиденметил)-1H- индол-2-карбоновую кислоту;
(E) 4,6-дихлоро-3-[(5-оксо-1-(3-аминофенил))пиразолидин-4- илиденметил] -1H-индол-2-карбоновую кислоту;
(E) 4,6-дихлоро-3-(2,5-диоксо-1-фенил-пирролидин-3-илиденметил)- 1H-индол-2-карбоновую кислоту;
(E) 4,6-дихлоро-3-[(5-оксо-1-(1-нафтил)пиразолидин-4- илиденметил]-1H-индол-2-карбоновую кислоту;
и их физиологически приемлемые соли, например натриевые или калиевые соли, или их метаболически лабильные сложные эфиры.
Соединения формулы (I) и/или их физиологически приемлемые соли являются антагонистами возбуждающих аминокислот. Более конкретно они являются сильными антагонистами в невосприимчивом к стрихнину глициновом сайте связывания, ассоциированном с NMDA рецепторным комплексом. В силу этого они являются сильными антагонистами NMDA рецепторного комплекса. Кроме того, соединения по изобретению обладают хорошими показателями активности, включая хорошую биологическую доступность. Эти соединения являются поэтому перспективными для лечения или профилактики нейротоксического нарушения или нейродегенеративных заболеваний. Поэтому соединения пригодны для лечения нейротоксического повреждения, наступающего в результате церебрального удара, тромбоэмболического удара, геморрагического удара, церебральной ишемии, церебрального вазоспазма, гипогликемии, анаэзии, гипоксии, кислородной недостаточности, остановки сердца вследствие перинатальной асфиксии. Соединения перспективны для лечения хронических нейродегенеративных заболеваний, таких как: болезнь Huntingdon'a, старческое слабоумие Alzheimer'a, амиотрофический латеральный склероз, ацидоз глутарового типа, мульти-инфарктное слабоумие, эпилептическое состояние, повреждения в результате контузий (например, повреждение спинного мозга и повреждение головы), вирусные инфекции, индуцирующие нейродегенерацию (в частности, СПИД, энцефалопатия), синдром Дауна, эпилепсия, шизофрения, депрессия, тревога, боль, нейрогенный мочевой пузырь, раздраженный мочевой пузырь, лекарственная зависимость, включая симптомы абстиненции от алкоголя, кокаина, опиатов, никотина, бензодиазепина, и рвота.
Сильное и селективное действие соединения по изобретению в стрихнин-невосприимчивом глициновом сайте связывания, находящемся на NMDA рецепторном комплексе, может быть легко определено с помощью общепринятых методов анализа. Способность связываться со стрихнин-невосприимчивым глициновым сайтом связывания определяли с помощью метода [4]. Селективность действия соединений по изобретению для стрихнин-невосприимчивого глицинового сайта была подтверждена в исследованиях на других ионотропических рецепторах известных возбуждающих аминокислот. Таким образом, было установлено, что соединения по изобретению проявляют малое или вообще не проявляют сродства к рецептору дигеновой кислоты (дигенату), рецептору α-амино-3-гидрокси-5-метил-4-изоксазол-пропионовой кислоты (АМПК) или в NMDA сайте связывания.
Было также установлено, что соединения по изобретению ингибируют NMDA индуцируемые конвульсии у мышей при применении способа [5].
Нейропротективная активность соединений по изобретению может быть продемонстрирована на препарате окклюзии срединной церебральной артерии у мышей, используя способ, описанный [6].
Поэтому изобретение предлагает соединение формулы (I) и/или его физиологически приемлемую соль или метаболически лабильный эфир для применения в терапии и, в частности, применение в качестве медикамента для подавления воздействий возбуждающих аминокислот на NMDA рецепторный комплекс.
Изобретение предлагает также соединение формулы (I) и/или его физиологически приемлемую соль или метаболически лабильный эфир для изготовления лекарственного препарата для подавления воздействий возбуждающих аминокислот на NMDA рецепторный комплекс.
Согласно следующему аспекту, изобретение также предлагает способ для подавления воздействий возбуждающих аминокислот на NMDA рецепторный комплекс, включающий введение пациенту в случае необходимости антагонистического количества соединения формулы (I) и/или его физиологически приемлемой соли или метаболически лабильного эфира.
Специалисту следует иметь в виду, что ссылка на лечение распространяется как на профилактику, так и на лечение установленного заболевания или симптомов.
Далее следует иметь в виду, что количество соединения по изобретению, необходимое для применения при лечении, будет изменяться в зависимости от природы состояния, подлежащего лечению, способа введения и возраста и состояния пациента и будет в конечном итоге определяться лечащим врачом. В общем однако, дозы, применяемые для лечения взрослого человека, как правило, лежат в интервале от 2 до 800 мг в день, в зависимости от способа введения.
Так, для парентерального введения ежедневная доза, как правило, лежит в интервале 20-100 мг, предпочтительно - 60-80 мг в день. Для перорального введения ежедневная доза, как правило, лежит в интервале 200-800 мг, например 400-600 мг в день.
Желаемая доза может быть, в зависимости от удобства, скомпонована в виде отдельной дозы или в виде раздельных доз, вводимых через соответствующие интервалы, например в виде двух, трех, четырех или более под-доз в день.
Хотя возможно, чтобы для применения в терапии соединения по изобретению могло быть введено в виде исходного химического соединения, но предпочтительно представлять активный ингредиент в виде фармацевтического препарата.
Далее в изобретении предлагают фармацевтический препарат, содержащий соединение формулы (I) либо его фармацевтически приемлемую соль или метаболически лабильный эфир совместно с одним или более фармацевтически приемлемых носителей и, возможно, другими терапевтическими и/или профилактическими ингредиентами. Носитель (ли) должны быть "приемлемыми" в том смысле, что они должны быть совместимы с другими ингредиентами лекарственного препарата и не быть вредными для его реципиента.
Составы по изобретению включают составы в форме, специально изготовленной для перорального, внутриротового, парентерального введения, ингаляции или вдувания, имплантации или ректального введения.
Таблетки и капсулы для перорального введения могут содержать обычные эксципиенты, такие как связывающие агенты, например сироп, гуммиарабик, желатин, сорбит трагакант, клей из крахмала или поливинилпирролидона; наполнитель, например лактозу, сахар, микрокристаллическую целлюлозу, кукурузный крахмал, фосфат кальция или сорбит; лубриканты, например стеарат магния, стеариновую кислоту, тальк, полиэтиленгликоль или кремний; дезинтегранты, например картофельный крахмал или крахмальный гликолят натрия, либо смачивающие агенты, такие как лаурилсульфат натрия. Таблетки могут быть покрыты оболочкой согласно известным из уровня техники способам. Пероральные жидкие препараты могут быть, например, в форме водных или масляных суспензий, растворов, эмульсий, сиропов или эликсиров, либо могут быть представлены в виде сухого продукта для разведения водой или другим носителем непосредственно перед применением. Такие жидкие препараты могут содержать приемлемые добавки, такие как суспендирующие агенты, например сорбитоловый сироп, метилцеллюлозу, глюкоза/сахарный сироп; желатин, гидроксиэтилцеллюлозу, карбоксиметилцеллюлозу, гель стеарата аллюминия или пищевые гидрогенизированные жиры, эмульгирующие агенты, например лецитин, сорбитан, моно-олеат или аравийская камедь; неводные носители (которые могут включать пищевые масла), например миндальное масло, фракционированное кокосовое масло, масляные эфиры, пропиленгликоль или этилалкоголь; растворители, такие как сурфактанты, например полисорбаты или другие агенты, такие как циклодекстрины; и консерванты, например метил или пропил p-гидробензоаты или аскорбиновую кислоту. Составы могут также быть изготовлены в виде суппозиториев, например содержащих подходящие суппозиторные основания, такие как масло какао или другие глицериды.
Для внутриротового введения состав может иметь форму таблеток или лепешек, изготовленных соответствующим образом.
Состав согласно изобретению может быть приготовлен для парентерального введения путем инъекции или продолжительного вливания. Препараты для инъекции могут быть представлены в виде унифицированной дозы в ампулах, или в многодозовых контейнерах с добавлением консерванта. Составы могут иметь такие формы в виде суспензий, растворов или эмульсий в масляных или водных наполнителях и могут содержать суспендирующие, стабилизирующие и/или диспергирующие агенты. С другой стороны, активный ингредиент может находиться в порошкообразной форме для совмещения перед применением с подходящим наполнителем, например стерильной беспирогенной водой.
Для введения посредством ингаляции соединения по изобретению удобно подавать в форме аэрозоля распылением из находящейся под давлением упаковки с использованием соответствующего пропелланта, такого как дихлордифторметан, трихлорфторметан, дихлортетрафторэтан, двуокись углерода или других подходящих пропеллантов, таких как дихлордифторметан, трихлорфторметан, дихлор-тетрафторэтан, диоксид углерода или другой подходящий газ, либо из распылителя. В случае находящегося под давлением аэрозоля дозировочная единица может быть определена посредством установки клапана для выпуска отмеренного количества.
Кроме того, для введения посредством ингаляции или вдувания, соединение по изобретению может иметь форму сухого порошкообразного состава, например порошкообразной смеси соединения и подходящего носителя, такого как лактоза или крахмал. Порошковый состав может находиться в единой дозировочной форме, например в капсулах или патронах, в частности из желатина, либо в виде пузырьковых упаковок, из которых порошок может быть введен с помощью ингалятора или распылителя.
Состав согласно изобретению может быть изготовлен в виде препарата пролонгированного действия. Такие препараты пролонгированного действия могут быть введены посредством имплантации (например, подкожно или внутримышечно) или посредством внутримышечной инъекции. Так, например, соединения по изобретению могут быть скомпонованы с подходящими полимерными или гидрофобными материалами (например, в виде эмульсии в соответствующем масле), либо ионообменными смолами, либо в виде умеренно растворимых производных, например, в виде умеренно растворимой соли.
Составы согласно изобретению могут содержать от 0,1 до 99% активного ингредиента, обычно от 30 до 95% для таблеток и капсул и 3-50% для жидких препаратов.
Соединения общей формулы (I) и их соли могут быть получены общими способами, описанными ниже. В следующем описании группы R, R1 и R2, m и A те же, как определено для соединений формулы (I), за исключением особо оговоренных случаев.
Соединения формулы (I), где A имеет определенные выше значения при условии, что A не является -NHCO-, могут быть получены в результате реакции альдегида (II) (где R имеет значения, определенные выше в формуле (I), либо является его защищенным производным, R4 является карбокси защитной группой, a R5 является азот защитной группой),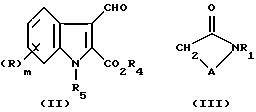
с циклическим амидом (III) (где R1 и A имеют значения, определенные выше в формуле (I), либо являются их защищенными производными, при условии, что A не является -NHCO- группой), в присутствии подходящего основания и, если необходимо или желательно, обработки полученного соединения посредством одной или более из следующих операций.
а) удаление одной или более защитных групп,
б) выделение соединения в виде его соли,
в) превращение соединения формулы (I) или его соли в метаболически лабильный эфир,
г) превращение соединения формулы (I) в его физиологически приемлемую соль.
В одном из способов реализации этого процесса альдегид (II) взаимодействует с циклическим амидом (III) в присутствии основания, такого как t-бутиллитий, диизопропиламид лития или бис(триметилсилил)амид лития, в апротонном растворителе, таком как тетрагидрофуран. Реакцию сначала проводят при температуре около -78oC, но затем ее поднимают до 0 и 30oC.
Исходный продукт этой реакции будет зависеть от природы защитных групп R4 и R5, поскольку некоторые из них могут быть отщеплены при условиях реакции. Примеры таких групп включают те группы, у которых R4 является метилом или этилом, a R5 является алкоксикарбонилом, например t-бутоксикарбонилом.
В том случае, когда реакцию проводят с применением индола формулы (II), причем карбокси защитную группу отщепляют, например R4 является этилом, а азот защитную группу R5 не отщепляют, например триметилсилил-этоксиметил, образующаяся карбоновая кислота IV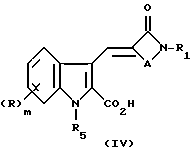
может быть превращена в соединение формулы (I) путем удаления азотной защитной группы R5. Кроме того, карбоновая кислота (IV) может быть превращена в соответствующий метиловый эфир (V) посредством реакции с диазометаном. Для этой реакции удобным источником диазометана является триметилсилилдиазометан, а реакцию можно проводить в подходящем растворителе, таком как галоуглеводород, например дихлорметан.
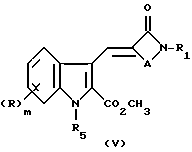
Соединение формулы (V) может быть превращено в соединение формулы (I) посредством удаления азот защитной группы R5 обычными способами с последующим гидролизом метилового эфира, если это желательно или необходимо.
Во втором способе реализации процесса альдегид (II) взаимодействует с циклическим амидом (III) в присутствии основания, такого как бутиллитий, диизопропиламин лития или бис(триметилсилил)амид лития, в апротонном растворителе, таком как тетрагидрофуран, при температуре около -78oC. В результате реакции образующегося вторичного спирта (VI)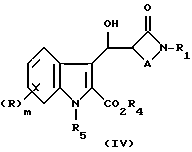
с соляной кислотой при нагревании в таком растворителе, как этанол, образуется олефин (VII)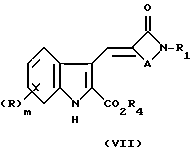
Эфир (VII) может быть превращен в соединение формулы (I) путем удаления карбокси защитной группы R4 обычными способами.
В модификации этого способа вторичный спирт (VI) может быть превращен в реактивную остаточную группу, такую как сульфонатный эфир, например п-толуолсульфонат или метансульфонат, с последующей обработкой соответствующим основанием, таким как диизопропиламид лития или этилат натрия. Полученный олефин затем может быть превращен в соединение формулы (I) путем удаления азот защитной группы R5, и если это необходимо или желательно, карбоксил защитной группы R4.
Пригодные для применения в этих реакциях карбоксил защитные группы R4 включают аллил, алкил, трихлоралкил, триалкилсилилалкил или арилметил группы, такие как бензил, нитробензил или тритил.
Пригодные азот защитные группы R5 включают алкоксикарбонил, например t-бутоксикарбонил, арилсульфонил, в частности фенилсульфонил или 2-триметилсилилэтоксиметил.
Карбоксил защитная группа может быть удалена соответствующими способами, применяемыми для удаления таких групп. У соединений, где R4 является алкильной группой, она может быть удалена посредством гидролиза с применением гидроксида щелочного металла, например гидроксида лития или гидроксида натрия, в таком растворителе, как спирт, например этанол или изопропанол, с последующим добавлением, если это желательно или необходимо, подходящей кислоты, например соляной кислоты или трифторуксусной кислоты, с образованием соответствующей свободной карбоновой кислоты.
Если R4 является аллильной группой, то она может быть удалена путем обработки рецептором аллила, таким как 5,5-диметил- 1,3-циклогександион, в присутствии тетракис(трифенилфосфин) палладия.
Кроме того, соединения, у которых R4 является алкильной или бензильной группой, могут быть превращены в соответствующую карбоновую кислоту в результате реакции с триметилсилилиодидом при нагревании в таком растворителе, как ацетонитрил.
В любой из описанных выше реакций азот защитная группа может быть удалена соответствующими способами, применяемыми для удаления таких групп, например посредством кислотного или щелочного гидролиза. Так, если R5 является алкоксикарбонилом, например t-бутоксикарбонилом, то она может быть удалена посредством щелочного гидролиза с применением, например, гидроксида лития, в соответствующем растворителе, таком как тетрагидрофуран или спирт, например изопропанол, либо посредством кислотного гидролиза, например с помощью муравьиной кислоты, трифторуксусной кислоты или хлороводорода в растворителе. Если R5 является триметилсилилэтоксиметильной группой, то она может быть удалена посредством кислотного гидролиза с помощью соляной кислоты или хлороводорода в таком растворителе, как спирт, в частности этанол.
Физиологически приемлемые соли соединений формулы (I) могут быть получены путем обработки соответствующей кислоты соответствующим основанием в подходящем растворителе. Например, соли щелочных или щелочноземельных металлов могут быть получены из гидроксидов щелочных или щелочноземельных металлов или соответствующих их карбонатных или бикарбонатных солей. Кроме того, соли щелочных или щелочноземельных металлов могут быть получены посредством прямого гидролиза карбоксил-защищенного производного соединения формулы (I) гидроксидом соответствующего щелочного или щелочноземельного металла.
Если соединение формулы (I) содержит основной центр, то его кислотно-аддитивные соли могут быть получены в результате реакции основания с соответствующей кислотой и возможно в растворителе. Кроме того, кислотно-аддитивная соль может быть получена посредством прямого гидролиза его карбоксил-защищенного и/или азото-защищенного производного в результате реакции с соответствующей кислотой.
Метаболически лабильные эфиры соединений формулы (I) могут быть получены путем этерификации карбоксильной кислотной группы или ее соли либо путем трансэтерификации соответствующими способами. Так, например, ацилоксиалкиловые эфиры могут быть получены в результате реакции свободной карбоновой кислоты или ее соли с соответствующим ацилоксиалкиловым галидом в подходящем растворителе, таком как диметилформамид. Для этерификации свободной карбоксильной группы эту реакцию предпочтительно проводить в присутствии четвертичного галида аммония, такого как тетрабутиламмонийхлорид или бензилтриэтиламмонийхлорид.
Аминоалкиловые эфиры могут быть получены посредством трансэтерификации соответствующего алкилового эфира, например метилового или этилового эфира в результате реакции соответствующего аминоалканола при повышенной температуре порядка 50-150oC.
Для взаимодействия альдегида (II) с циклическим амидом (III) может оказаться необходимым или желательным проводить реакцию с применением их защищенных производных. Например, если одно или оба соединения содержат первичную или вторичную аминогруппу, либо гидроксильную или карбоксильную группу. Эти группы могут быть защищены соответствующим способом, а защитные группы удаляют соответствующими способами, когда это необходимо.
Так, если группа R является амином или алкиламином и/или R1 содержит амин или алкиламин в качестве заместителя, а группа A содержит основную -NH- группу, то желательно, чтобы каждый такой основной атом азота был защищен, например как в его t-бутоксикарбонильном производном. Азот защитные группы могут затем быть удалены соответствующими способами; например посредством реакции с трифторуксусной кислотой в подходящем растворителе, в частности дихлорметане, или с хлороводородом в таком растворителе, как алканол.
Любая гидроксильная или карбоксильная группа может соответствующим образом быть защищена в виде ее эфира, как например t-бутоксикарбонильное производное гидроксильной группы или алкиловый или аллиловый эфир карбоксильной группы, например ее t-бутиловый или аллиловый эфир.
Соединения формулы (I), у которых A является цепью NHCO, могут быть получены в результате реакции альдегида (II) или его защищенного производного с глициновым производным (VIII)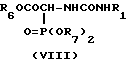
где R1 является группой, определенной в формуле (I), или ее защищенным производным, a R6 и R7 независимо являются C1-4 алкилом. Реакцию проводят в присутствии основания, такого как 1,8-диазобицикло[5,4,0]-ундец-7-ен в апротонном растворителе, таком как эфир, например тетрагидрофуран, с последующим удалением защитных групп R4 и R5 совместно с любой другой имеющейся защитной группой.
Соединения формулы (I), где A является группой CH2CO, могут быть получены в результате реакции альдегида (II) с фосфорным производным (IX), где R1 имеет значения, определенные в формуле I или является их защищенным производным.
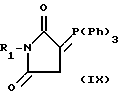
Реакцию предпочтительно проводят при нагревании в подходящем растворителе, например углеводороде, в частности толуоле, с последующим удалением защитных групп R4 и R5 соответствующим способом.
Соединения формулы (I), у которых экзоциклическая связь находится в цис конфигурации, могут быть получены в результате изомеризации соответствующего транс изомера или его защищенного производного с последующим удалением любой защитной группы. Реакцию изомеризации обычно проводят посредством облучения раствора транс изомера в соответствующем растворителе, таком как ацетонитрил, УФ светом, например посредством ртутной лампы.
Соединения формулы (II), где R4 является карбоксил защитной группой, a R5 является азот защитной группой, могут быть получены посредством обработки соответствующего индола (X)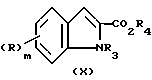
N-метилформанилидом и хлорокисью фосфора в таком растворителе, как 1,2-дихлорэтан.
Индолы формулы (X) являются либо известными соединениями, либо могут быть получены способами, аналогичными способам, описанным для известных соединений.
Циклические амиды формулы (III) являются либо известными соединениями, либо могут быть получены с применением методов, аналогичных описанным для известных соединений [7, 8].
Так, соединения формулы (III), где A является группой (CH2)pNR8, где p равно 1 или 2, a R8 является защитной группой, могут быть получены в результате реакции защищенного гидразина R1NH NHR8 с галоацилгалидом (XI),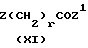
где Z и Z1 независимо являются атомами галогенов, например хлора, брома или иода, a r равно 2 или 3. Реакцию обычно проводят в присутствии такого основания, как карбонат щелочного металла, в полярном растворителе, таком как N, N-диметилформамид. Подходящей для применения в этой реакции защитной группой R8 является t-бутилоксикарбонильная группа. Если это необходимо, то эта защитная группа может быть удалена обычными способами, например в результате реакции с трифторуксусной кислотой в таком растворителе, как дихлорметан. Соединение формулы (III), где A является цепью -(CH2)pNH-, полученное таким способом, может быть затем превращено в соединение формулы (II), где A является цепью (CH2)pNR3, где R3 является алкилом, посредством обычной реакции алкилирования. Например посредством алкилирования с применением соответствующего алкилтрифторметилсульфоната в таком растворителе, как дихлорметан.
Соединения формулы (III), где A является группой (CH2)pO, где p равно 1 или 2, могут быть получены посредством реакции гидроксиламина R1NHOH с галоацилгалидом (XI) в присутствии такого основания, как карбонат калия, в полярном растворителе, таком как N,N-диметилформамид.
Гидроксиламин R1NHOH может быть получен из соответствующего нитросоединения R1NO2 обычным способом, например посредством реакции с гидразином в присутствии 5%-ного родиевого или углеродного катализатора.
Для того, чтобы изобретение стало более понятным, исключительно в качестве иллюстративного материала приведены следующие примеры.
В Промежуточных соединениях и Примерах за исключением особо оговоренных случаев определено:
Точки плавления (т. пл.) определяли с помощью Gallenkamp т.пл. аппарата либо Buchi капиллярного аппарата, и они не исправлены. Все температуры выражены в oC. Инфракрасные спектры снимали на FT-IR приборе. Спектры ядерного магнитного резонанса (1H-ЯМР) регистрировали при 300 МГц или 400 МГц, химические сдвиги приведены в ppm ниже (н) относительно Me4Si, используя внутренний стандарт, и их обозначают как синглеты (s), дуплеты (d), дуплеты дуплетов (dd), триплеты (t), квартеты (q) или мультиплеты (m). Колоночную хроматографию проводили на силикагеле (Merck AG Darmstaadt, Germany). В тексте используют следующие аббревиатуры: ЭА = этилацетат, ЦГ = циклогексан, ДХМ = дихлорметан, ДМСО = диметилсульфоксид, ДБУ = 1,8-диазобицикло[5.4.0]ундец-7-ен.
ТСХ обозначает тонкослойную хроматографию на кремниевых пластинах. Раствор сушили над безводным сульфатом натрия. Тетрагидрофуран (ТГФ) отгоняли непосредственно от K/бензофенона в атмосфере азота; химически чистые циклогексан и этилацетат применяли без дальнейшей очистки. Хроматографию проводили на силикагеле, 230-400 меш, (Merck). Выходы приведены для выделенных продуктов, которые были очищены посредством ЯМР и ТСХ.
Промежуточное соединение 1
Этил-4,6-дихлориндол-2-карбоксилат
К раствору этилпирувата (2,05 мл) в абсолютном этаноле (38 мл) при интенсивном перемешивании медленно добавляли концентрированную серную кислоту (0,5 мл). Полученную смесь перемешивали в течение 10 минут при 30oC, затем по частям добавляли гидрохлорид 3,5-дихлорфенилгидразина (4 г). Смесь прогревали с дефлегмацией в течение 4-х часов, охлаждали до 23oC, вливали в холодную воду (500 мл) и экстрагировали диэтиловым эфиром (3 х 300 мл). Органические слои отделяли и сушили. Растворитель выпаривали при пониженном давлении, получая в результате этиловый эфир 2-(3,5-дихлорфенилгидразон)пропионовой кислоты в виде вещества желтого цвета (5 г; ТСХ ДХМ, Rf= 0,79, 0,47), являвшегося смесью E и Z изомеров. Полученное вещество при перемешивании добавляли к полифосфорной кислоте (20 г), смесь прогревали при 45oC в течение 20 минут, получая в результате коричневый продукт, который кристаллизовали 95%-ным этанолом (300 мл), получая в результате соединение, указанное в заголовке, в виде вещества желто-коричневого цвета (3,3 г; т.пл. 180oC; ТСХ ДХМ, Rf= 0,54). ИК (CDCl3) Vmax (см-1) 3440 (NH), 1772-1709 (C= O).
1H-ЯМР (CDCl3) 9,00(s), 7,28(d), 4,42(q), 1,42(t).
Промежуточное соединение 2
Этил-3-формил-4,6-дихлориндол-2-карбоксилат
Раствор N-метилформанилида (5,19 г) и хлорокиси фосфора (5,53 г) перемешивали в течение 15 минут при 23oC. Добавляли 1,2 -дихлорэтан (60 мл) и промежуточный продукт 1 (6 г) и полученную суспензию перемешивали в течение 6 часов при 80oC. Реакционную смесь выливали в 50%-ный водный раствор ацетата натрия (300 мл), получая после фильтрации соединение, указанное в заголовке в виде вещества желтого цвета (4,1 г; ТСХ ЭА/ЦГ: 4/6, Rf=0,4). ИК (Nujol) Vmax(см-1) 1726 (C= O), 1663 (C=O), 1556 (C=C), 2725-2669 (CH). 1H-ЯМР (ДМСО) 13,15(s), 10,60(s), 7,54(d), 7,40(d), 4,43(q), 1,36(t).
Промежуточное соединение 3
Этил-3-формил-1-(2-триметилсилил-этоксиметил)-4,6-дихлориндол- 2-карбоксилат
К охлажденному раствору промежуточного соединения (2) (700 мг) в сухом ДМФ (20 мл) при 0oC добавляли бис-триметилсилиламидметил (3,7 мл, 1М раствор) в ТГФ. Смесь перемешивали в течение 15 минут при 0oC, затем добавляли три-метилсилилэтоксиметилхлорид (0,817 г). Спустя один час полученную смесь вливали в воду (25 мл) и экстрагировали этилацетатом (3 х 20 мл). Объединенные органические слои сушили и концентрировали под вакуумом. Остаток очищали посредством хроматографии на силикагеле с получением соединения, указанного в заголовке (950 мг) в виде бледно-желтого вещества. Rf=0,3, ЭА/ЦГ: 1,9.
Промежуточное соединение 4
Метил (Z)-4,6-дихлор-3-(2-оксо-1-фенил-пирролидин-3-илиденметил) -1-(2-триметилсилил-этоксиметил)-1H-индол-2-карбоксилат
1-Фенил-2-пирролидинон (426 мг) растворяли в ТГФ (10 мл), раствор охлаждали до -78oC и медленно добавляли трет-бутиллитий (1,80 мл, 1,6 М раствор в гексане), к полученному раствору, перемешивавшемуся при этой температуре в течение 1,5 часа, добавляли промежуточное соединение 3 (1 г), растворенное в ТГФ (10 мл), и перемешивание продолжали при -78oC в течение 3 часов. Затем смесь в течение 3 часов нагревалась до комнатной температуры и перемешивание продолжали еще 1,5 часа. Реакцию останавливали добавлением 20 мл насыщенного раствора NH4Cl, добавляли этилацетат (50 мл), органическую фазу отделяли и промывали 0,1 М соляной кислотой (2 х 20 мл), водой (20 мл), рассолом (10 мл) и сушили. Растворитель выпаривали, остаток растворяли в 20 мл смеси дихлорметан/метанол (4/1) и обрабатывали при комнатной температуре триметилсилилдиазометаном (1,70 мл, 2 М раствор в гексане) в течение 30 минут. В результате заключительной очистки посредством колоночной хроматографии (ЦГ/ЭА 8/2) получали соединение, указанное в заголовке (740 мг) в виде белого вещества. ИК (Nujol) Vmax (см-1) 1709 (C=O), 1684 (C=O).
1H-ЯМР (CDCl3) 7,85(t), 7,80(d), 7,50(d), 7,40(t), 7,20(d), 7,17 (t), 5,89(s), 3,90(s), 3,86(t), 3,53(t), 2,64(td), 0,88(t), 0,05(s).
Промежуточное соединение 5
Метил (Z)-4,6-дихлор-3-(2-оксо-1-фенил-пиперидин-3-илиденметил)- 1-(2-триметилсилил-этоксиметил)-1H-индол-2-карбоксилат
N-фенилпиперидинон (370 мг) растворяли в ТГФ (10 мл), раствор охлаждали до -78oC, добавляли трет-бутиллитий (1,30 мл, 1,6 М раствор в гексане), полученную смесь перемешивали при этой температуре в течение 1,5 часов. Добавляли промежуточное соединение 3 (800 мг), растворенное в ТГФ (10 мл), и перемешивание продолжали при -78oC в течение 3 часов. Затем реакционная смесь нагревалась до комнатной температуры в течение 3 часов и перемешивалась в течение еще 1,5 часов. Реакцию останавливали добавлением 20 мл насыщенного раствора NH4Cl, добавляли этилацетат (50 мл), органическую фазу отделяли и промывали 0,1 М соляной кислотой (2 х 20 мл), водой (20 мл), рассолом (10 мл) и сушили. Растворитель выпаривали, остаток перерастворяли в 20 мл смеси дихлорметан/метанол (4/1) и при комнатной температуре обрабатывали триметилсилилдиазометаном (1,50 мл, 2 М раствор в гексане) в течение 30 минут. В результате заключительной очистки посредством колоночной хроматографии (ЦГ/ЭА 7/3) получали соединение, указанное в заголовке, (700 мг) в виде вещества белого цвета.
ИК (Nujol) Vmax (см-1) 1713 (C=O), 1672 (C=O).
1H-ЯМР (CDCl3) 8,09(t), 7,48(d), 7,40(m), 7,26(tt), 7,18(d), 5,90(s), 3,89(s), 3,76(t), 3,52(m), 2,41(td), 1,95(m), 0,87(t), 0,06 (s).
Промежуточное соединение 6
1-трет-бутоксикарбонил-2-фенилгидразин
Дитрет-бутилдикарбонат (5,6 г) добавляли к раствору фенилгидразина (2,5 мл) в тетрагидрофуране (50 мл). Раствор перемешивали при 25oC в течение 2-х часов, а затем концентрировали под вакуумом, получая в результате соединение, указанное в заголовке (5,44 г). ТСХ ЭА/ЦГ (1/2), Rf=0,8. ИК (см-1): 1724 (C=O), 1605 (C=C).
Промежуточное соединение 7
1-трет-бутоксикарбонил-2-фенил-пиразолидин-3-он
К раствору промежуточного соединения 6 (5,4 г) в сухом диметилформамиде (50 мл) добавляли карбонат калия (6,9 г), а через 5 мин - хлорпропионилхлорид (2,4 мл). Полученную смесь перемешивали в течение 2-х часов при 25oC, разбавляли затем диэтиловым эфиром (200 мл), промывали водой (2 х 200 мл), стоили и концентрировали под вакуумом. Полученное сырое соединение кристаллизовали из смеси этилацетат/гексан, получая в результате соединение, указанное в заголовке (5,9 г). ТСХ диэтиловый эфир/петролейный эфир (1/1), Rf= 0,4. Т.пл.=150oC.
Промежуточное соединение 8
Этил-4,6-дихлор-3-формил-1-трет-битоксикарбонил-1H-индол-2- карбоксилат
К суспензии этилового эфира 4,6-дихлор-3-формил-1H-индол-2- карбоновой кислоты (8 г) в сухом тетрагидрофуране (100 мл) добавляли ди-трет-бутилдикарбонат (7,3 г) и 4-диметиламинопиридин (0,7 г). Реакционную смесь перемешивали в течение 2-х часов при 25oC, затем разбавляли этилацетатом (300 мл), промывали хлоридом аммония (нас.) (200 мл), рассолом (200 мл), сушили и концентрировали под вакуумом. Сырое соединение, указанное в заголовке, кристаллизовали из этилацетата (8,6 г). ТСХ ЭА/ЦГ (1/2), Rf=0,8. Т.пл.=141oC.
Промежуточное соединение 9
1-трет-бутоксикарбонил-2-(4-нитрофенил)гидразин
Ди-трет-бутилдикарбонат (7,8 г) добавляли к раствору фенилгидразина (2,5 мл) в тетрагидрофуране (100 мл). Раствор перемешивали при 25oC в течение 2-х часов, затем концентрировали под вакуумом, получая в результате сырой продукт, который кристаллизовали из смеси этилацетат/н-гексан (1/3) с получением соединения, указанного в заголовке (6,9 г). ТСХ ЭА/ЦГ (1/2), Rf=0,85. Т.пл.= 120oC.
Промежуточное соединение 10
1-трет-бутоксикарбонил-2-(4-аминофенил)гидразин
Раствор гидросульфита натрия (20 г) и карбоната калия (22 г) в воде (200 мл) добавляли к раствору промежуточного соединения 9 (6 г) в этаноле (350 мл). Полученную смесь перемешивали в течение 1 часа при 25oC, затем концентрировали под вакуумом и экстрагировали этилацетатом (200 мл). Органическую фазу промывали хлоридом аммония (нас.) (2 х 200 мл), сушили и концентрировали под вакуумом. Сырое соединение очищали посредством колоночной хроматографии на силикагеле с использованием ЭА/ЦГ (1/2) в качестве элюанта, получая в результате соединение, указанное в заголовке (3 г). ТСХ ЭА/ЦГ (1/1), Rf=0,2. Т.пл.=128oC.
Промежуточное соединение 11
1-трет-бутоксикарбонил-2-[4(-трет-бутоксикарбониламино)фенил] гидразин
Ди-трет-бутилдикарбонат (3,33 г) добавляли к раствору промежуточного соединения 10 (3,41 г) в тетрагидрофуране (100 мл). Раствор перемешивали в течение 15 часов при 25oC, а затем концентрировали под вакуумом, получая в результате сырое соединение, указанное в заголовке, которое затем кристаллизовали из смеси этилацетат/н-гексан (4,4 г). ТСХ ЭА/ЦГ (1/2), Rf=0,90. Т. пл.=155oC.
Промежуточное соединение 12
1-трет-бутоксикарбонил-2-[(4-трет-бутоксикарбониламино)фенил] пиразолидин-3-он
К раствору промежуточного соединения 11 (0,5 г) в сухом диметилформамиде (5 мл) добавляли карбонат калия (0,213 г), а спустя 15 мин - хлорпропионилхлорид (0,15 мл). Полученную смесь перемешивали при 25oC в течение 20 часов, затем разбавляли диэтиловым эфиром (50 мл). Ее промывали водой (50 мл) и хлоридом аммония (нас.) (50 мл). После сушки раствор концентрировали под вакуумом с получением сырого соединения, которое кристаллизовали из смеси этилацетат/н-гексан (1/4) с получением соединения, указанного в заголовке (0,252 г). ТСХ ЭА/ЦГ (1/1), Rf= 0,60. Т.пл.=179oC.
Промежуточное соединение 13
4,6-дихлориндол-3-формил-2-карбоновая кислота
К суспензии промежуточного соединения 2 (7,0 г) в этиловом спирте (250 мл) добавляли гидроксид лития (2,26 г). Желтый раствор прогревали при 50oC в течение 8 часов, затем подкисляли HCl до pH 2. Осадок собирали фильтрацией, получая в результате соединение, указанное в заголовке, в виде белого вещества (6,29 г). Т.пл.=235-240oC.
Промежуточное соединение 13а
Трет-бутиловый эфир 4,6-дихлориндол-3-формил-2-карбоновой кислоты
К суспензии промежуточного соединения 13 (1,0 г) в сухом толуоле (50 мл) в состоянии дефлегмации медленно добавляли ди-трет-бутилацеталь N,N-диметилформамида (4,38 г). Нагревание продолжали в течение 30 минут, а затем темный раствор охлаждали и промывали водой, раствором бикарбоната натрия и рассолом. Органический слой сушили, а растворитель удаляли при пониженном давлении. Остаток очищали посредством скоростной хроматографии (ЦГ/ЭА=8/2, Rf= 0,33), получая в результате соединение, указанное в заголовке, в виде белого вещества (470 мг).
ИК (Nujol) Vmax (см-1) 3335 (N-H), 1724 (C=O) и 1663 (C=O).
Промежуточное соединение 14
Трет-бутиловый эфир 4,6-дихлориндол-3-формил-1- третбутилоксикарбонил-2-карбоновой кислоты
К раствору промежуточного соединения 13a (470 мг) в сухом ТГФ (10 мл) добавляли 4-диметиламинопиридин (22 мг) и раствор ди-трет-бутилдикарбоната (392 мг) в сухом ТГФ (5 мл). Раствор перемешивали в течение 30 минут, затем растворитель удаляли при пониженном давлении. Остаток очищали посредством скоростной хроматографии (ЦГ/ЭА=9/1, Rf=0,38), получая в результате соединение, указанное в заголовке, в виде пены (468 мг).
ИК (Nujol) Vmax (см-1) 1765 (C=O), 1740 (C=O) и 1688 (C=O).
Промежуточное соединение 15
Триметиловый эфир N-(фениламинокарбонил)-α-фосфоноглицина
Раствор триметилового эфира N-(бензилоксикарбонил)-α- фосфоноглицина (3 г) в метаноле (50 мл) гидрогенизировали под давлением 1 атм в течение 5 часов в присутствии 5%-ного палладия на углероде (0,55 г). Катализатор отфильтровывали на броунмиллерите, а раствор выпаривали при пониженном давлении. Остаток растворяли в дихлорметане (15 мл), добавляли фенилизоцианат (1,1 мл) и перемешивали реакционную смесь в течение 15 часов. Растворитель выпаривали при пониженном давлении, а остаток истирали в порошок в диэтиловом эфире (50 мл), получая в результате соединение, указанное в заголовке, в виде белого порошка (264 г, т.пл. 144-146oC).
ИК (Nujol) Vmax (см-1) 1745 (C=O), 1707 (C=O).
Промежуточное соединение 16
1,2-дитретбутиловый эфир (Z)4,6-дихлориндол-3-(2,5-диоксо-1- фенил-имидазолидин-4-илиденметил)-индол-1,2-дикарбоновой кислоты
К раствору промежуточного продукта 15 (367 мг) в сухом ТГФ (8 мл) по каплям добавляли ДБА (352 мг). Раствор перемешивали при комнатной температуре в течение 10 минут, затем медленно добавляли раствор промежуточного соединения 16 (480 мг) в сухом ТГФ (10 мл). Смесь перемешивали в течение 15 минут, разбавляли этиацетатом (10 мл) и останавливали реакцию добавлением хлорида аммония. Органический слой сушили, а растворитель выпаривали при пониженном давлении. Остаток очищали посредством скоростной хроматографии (ЦГ/ЭА= 85/15, Rf=0,33), получая в результате соединение, указанное в заголовке, в виде пены (284 мг).
ИК (Nujol) Vmax (см-1) 1772 (C=O), 1726 (C=O) и 1678 (C=O).
Промежуточное соединение 17
Аллиловый эфир 4,6-дихлориндол-3-формил-2-карбоновой кислоты
К суспензии промежуточного продукта 2 (3,0 г) в аллиловом спирте (100 мл) добавляли моногидрат п-толуолсульфоновой кислоты (2,0 г). Суспензию прогревали при 90oC в течение 2,5 часов, затем растворитель удаляли при пониженном давлении. Остаток растворяли в метиленхлориде, промывали раствором Na2CO3 (10%-ный), водой и очищали посредством скоростной хроматографии (ЦГ/ЭА= 7/3, Rf= 0,34), получая в результате соединение, указанное в заголовке, в виде белого вещества (1,10 г).
ИК (Nujol) Vmax (см-1) 1720 (C=O) и 1657 (C=O).
1H-ЯМР (ДМСО) 13,20(ds), 10,61(s), 7,55(d), 7,42(d), 6,14-6,0 (m), 5,46(dd), 5,33(dd) и 4,92(d). m/z (ОБА) 298 (MH).
Промежуточное соединение 18
Аллиловый эфир 4,6-дихлориндол-1-трет-бутилоксикарбонил-3-формил- 2-карбоновой кислоты
К раствору промежуточного соединения 17 (1,10 г) в сухом ТГФ (40 мл) добавляли 4-диметиламинопиридин (49 мг) и раствор ди-трет- бутилдикарбоната (886 мг) в сухом ТГФ (10 мл). Раствор перемешивали в течение 1 часа, затем растворитель удаляли при пониженном давлении. Остаток очищали посредством скоростной хроматографии (ЦГ/ЭА=9/1, Rf=0,37), получая в результате соединение, указанное в заголовке, в виде белого вещества (853 мг; т.пл.=106,9-107,7oC).
ИК (Nujol) Vmax (см-1) 1757-1744 (C=O) и 1682 (C=O).
Промежуточное соединение 19
Смешанный 2-аллиловый и 1-третбутиловый эфир (E)4,6-дихлор- 3-(2,5-диоксо-1-фенил-имидазолидин-4-илиденметил)-индол-1,2- дикарбоновой кислоты
К раствору промежуточного соединения 15 (590 г) в сухом ТГФ (10 мл) по каплям добавляли ДБА (566 мг). Раствор перемешивали при комнатной температуре в течение 10 минут, затем медленно добавляли раствор промежуточного соединения 18 (740 мг) в сухом ТГФ (15 мл). Смесь перемешивали в течение 15 минут, разбавляли этилацетатом (10 мл) и останавливали реакцию добавлением хлорида аммония. Органический слой сушили, а растворитель выпаривали при пониженном давлении. Остаток очищали посредством тонкослойной скоростной хроматографии (ЦГ/ЭА=7/3, Rf=0,27), получая в результате соединение, указанное в заголовке, в виде пены (369 мг).
ИК (Nujol) Vmax (см-1) 3331 (N-H), 1730 (C=O), 1673 (C=O) и 1533 (C=C).
Промежуточное соединение 20
Третбутиловый эфир E-3-[(1-трет-бутоксикарбонил-3-оксо-2-фенил) пиразолидин-4-илиденметил]-4,6-дихлор-1-трет-бутоксикарбонил-1H-индол- 2-карбоновой кислоты
К раствору из Примера 15 (0,7 г) в сухом тетрагидрофуране (50 мл) добавляли ди-трет-бутил-ди-карбонат (0,33 г) и 4-диметиламинопиридин (0,03 г). Раствор перемешивали при 25oC в течение 30 минут, затем разбавляли диэтиловым эфиром (200 мл), промывали хлоридом (нас.) (200 мл), рассолом (200 мл), сушили и концентрировали в вакууме. Сырое соединение, указанное в заголовке, очищали посредством колоночной хроматографии на силикагеле с применением смеси диэтилового эфира и петролейного эфира (2/8) в качестве элюанта, получая в результате соединение, указанное в заголовке (0,54 г). ТСХ диэтиловый эфир/петролейный эфир (5/25), Rf=0,45.
1H-ЯМР (CDCl3): 1,27(s,9H), 1,67(s,9H), 4,65(d,2H), 7,18(t,1H), 7,27(d, 1H), 7,39(m,1H), 7,66 (m,2H), 8,04(d,1H).
Промежуточное соединение 21
Третбутиловый эфир (Z) 3-[(1-трет-бутоксикарбонил-3-оксо-2-фенил) пиразолидин-4-илиденметил] -4,6-дихлор-1-трет-бутоксикарбонил-1H-индол- 2-карбоновой кислоты
Раствор промежуточного соединения 20 (0,53 г) в ацетонитриле (30 мл) облучали с помощью ртутной лампы мощностью 400 Ватт в течение 45 минут. Раствор концентрировали под вакуумом, получая в результате смесь двух изомеров, которые разделяли посредством колоночной хроматографии на силикагеле с применением смеси этилацетат/циклогексан (5/95) в качестве элюанта, получая в результате соединение 6, указанное в заголовке (0,13 г). ТСХ ЭА/ЦГ (5/25), Rf=0,85.
1H-ЯМР (CDCl3): 1,27(s,9H), 1,51(s,9H), 1,65(s,9H), 4,86(m,2H), 7,07 (m, 1H), 7,09(m,1H), 7,20(d,1H), 7,28(m,2H), 7,57(m,2H), 7,97(d,1H).
Промежуточное соединение 22
1-трет-бутоксикарбонил-2-(4-метилсульфамидофенил)гидразин
К раствору промежуточного соединения 10 (0,2 г) в тетрагидрофуране (5 мл) в атмосфере азота при 0oC добавляли пиридин (0,045 мл). Через 5 минут к перемешиваемому раствору по каплям добавляли метансульфонилхлорид (0,045 мл). Затем реакционная смесь нагревалась до комнатной температуры. Через 1 час добавляли насыщенный раствор хлорида аммония (20 мл) и экстрагировали реакционную смесь этилацетатом (3 х 10 мл), а объединенные экстракты сушили. Растворитель удаляли путем дистилляции при пониженном давлении, а продукт очищали посредством колоночной скоростной хроматографии на силикагеле с ЦГ/ЭА, получая в результате соединение, указанное в заголовке, в виде желтого вещества (0,140 г).
ИК (см-1, Nujol): 3302 (NH), 1600 (C=C), 1323-1151 (SO2).
Промежуточное соединение 23
1-трет-бутоксикарбонил-2-(4-метилсульфамидофенил)-пиразолидин-3-он
К перемешиваемому раствору промежуточного соединения 22 (0,6 г) в сухом диметилформамиде (15 мл) в атмосфере азота при 0oC добавляли пиридин (0,23 мл). Затем к раствору по каплям добавляли хлорпропионилхлорид (0,2 мл). Реакционную смесь перемешивали в течение 2 часов при 25oC, разбавляли этилацетатом (50 мл), а затем промывали водой (50 мл) и насыщенным раствором хлорида аммония (50 мл). Органический раствор сушили, а затем концентрировали под вакуумом, получая в результате сырой 1-трет-бутоксикарбонил-2-(4- метилсульфамидофенил)-2-(3-хлорпропионил) гидразин в виде желтого масла. Его затем растворяли в диметилформамиде (5 мл) в атмосфере азота и при 25oC добавляли безводный карбонат калия (0,450 г). Полученную смесь перемешивали в течение 1 часа, а затем разбавляли насыщенным раствором хлорида аммония (50 мл). Водный раствор экстрагировали этилацетатом (3х50), а объединенные органические фазы сушили и концентрировали под вакуумом. Сырой остаток очищали посредством скоростной колоночной хроматографии на силикагеле с элюцией диэтиловым эфиром, получая в результате соединение, указанное в заголовке (0,25 г).
1H-ЯМР (ДМСО): 1,28(s,9H), 2,73(t,2H), 2,93(s,3H), 4,06(t,2H), 7,19(d, 2H), 7,42(d,2H), 9,66(s,1H).
ИК (см-1, Nujol): 3252-3202 (NH), 1693 (C=O), 1600 (C=C), 1310-1151 (SO2).
Промежуточное соединение 24
2-фенил-пиразолидин-3-он
Промежуточное соединение 7 (4,6 г) растворяли в сухом дихлорметане (10 мл) и трифторуксусной кислоте (10 мл). Полученный раствор перемешивали при 25oC в течение 1 часа, а затем концентрировали под вакуумом. Остаток растворяли в этилацетате (100 мл), промывали бикарбонатом натрия (нас.) (2x100 мл), рассолом (100 мл), сушили и концентрировали под вакуумом, получая в результате сырое соединение, указанное в заголовке (2,6 г). ТСХ ЦГ/ЭА (1/1), Rf=0,30.
1H-ЯМР (CDCl3): 2,78(t,2H), 3,52(q,2H), 4,75(t,1H), 7,12(tt,1H), 7636(t, 2H), 7,85 (d,2H).
ИК (см-1): 3238 (NH), 1674 (C=O).
Промежуточное соединение 25
1-метил-2-фенил-пиразолидин-3-он
К раствору промежуточного соединения 24 (1,43 г) в диметилформамиде (20 мл) по каплям добавляли при -20oC метилтрифторметансульфонат (1,4 мл). Реакционная смесь нагревалась до комнатной температуры и спустя 3 часа ее разбавляли диэтиловым эфиром (200 мл), сушили и концентрировали под вакуумом. Сырое соединение очищали посредством колоночной хроматографии на силикагеле, используя в качестве элюанта смесь этилацетат/циклогексан (3/7) и получая в результате соединение, указанное в заголовке (0,4 г). ТСХ ЦГ/ЭА (1/1), Rf=0,42.
ИК (см-1): 1697 (C=O).
Промежуточное соединение 26
N-(3-хлорпропионил)-N-фенилгидроксиламин
К 5%-ной суспензии родия на угле (0,13 г) в сухом тетрагидрофуране (23 мл) добавляли нитробензол (5 г). Смесь охлаждали до 0oC и по каплям добавляли гидрат гидразина (2 г). В процессе добавления температуру смеси поддерживали в интервале 25-30oC; затем реакционную смесь перемешивали в течение 2 часов при 25oC, фильтровали, а катализатор промывали небольшим количеством тетрагидрофурана. Раствор концентрировали под вакуумом, а остаток растворяли в диметилформамиде (30 мл) и охлаждали до -5oC, затем добавляли карбонат калия (5,6 г), а через 10 минут - хлорпропионилхлорид (3,8 мл). Реакционную смесь перемешивали в течение 1 часа при 25oC, затем разбавляли диэтиловым эфиром (150 мл), промывали водой (2x200 мл), сушили и концентрировали под вакуумом. Остаток очищали кристаллизацией, применяя смесь этилацетат/циклогексан и получая в результате соединение, указанное в заголовке, в виде твердого вещества (4 г). ТСХ ЦГ/ЭА 1/2, Rf=0,3.
1H-ЯМР (CDCl3): 2,7(bs,2H), 3,8(t,2H), 7,5(bm, 5H).
ИК (см-1): 1645 (C=O), 1626 (C=C).
Промежуточное соединение 27
3-оксо-2-фенил-изоксазолидин
К раствору промежуточного соединения 26 (3,35 г) в ацетоне (150 мл) добавляли карбонат калия (2,3 г). Реакционную смесь перемешивали в течение б часов при 25oC, а затем концентрировали под вакуумом. Остаток истирали в порошок с этилацетатом, фильтровали и концентрировали под вакуумом, получая в результате сырое соединение, указанное в заголовке, в виде масла (2,5 г). ТСХ ЦГ/ЭА 1/2, Rf=0,3.
1H-ЯМР (CDCl3): 3,0(t,2H), 4,52(t,2H), 7,14(tt,1H), 7,37(m,2H), 7,69 (m, 2H).
ИК (см-1): 1697 (C=O).
Промежуточное соединение 28
1-трет-бутоксикарбонил-2-(3-нитрофенил)гидразин
К раствору 3-нитрофенилгидразина (6 г) в диоксане (60 мл) и воды (30 мл) добавляли гидроксид натрия 1М (30 мл) и ди-трет-бутилкарбонат (7,6 г). Раствор перемешивали в течение 2 часов при 25oC, затем концентрировали под вакуумом; разбавляли этилацетатом (200 мл), промывали хлоридом аммония (нас.) (2x200 мл), рассолом (2x200 мл), сушили и концентрировали под вакуумом, получая в результате сырое промежуточное соединение, которое очищали фильтрацией со смесью этилацетат/циклогексан (19/29), получая в результате соединение, указанное в заголовке (4,3 г). ТСХ ЭА/ЦГ (1/1), Rf=0,85. Т.пл.=105oC.
Промежуточное соединение 28а
1-трет-бутоксикарбонил-2-(3-трет-бутоксикарбониламино) фенилгидразин
К раствору промежуточного соединения 28 (3,64 г) в этаноле (70 мл) добавляли порошок железа (7,0 г) и хлорид кальция (0,71 г). Реакционную смесь прогревали в течение 20 часов при 70oC, затем фильтровали через силикагель, промывая этилацетатом. Раствор сушили и концентрировали под вакуумом, получая в результате 1- трет-бутоксикарбонил-2-(3-аминофенил) гидразин (3,0 г), который растворяли в сухом тетрагидрофуране (100 мл) и добавляли ди-трет- бутил-ди-карбонат (3,68 г). Реакционную смесь очищали посредством истирания в порошок с этилацетатом, получая при этом соединение, указанное в заголовке (3,73 г). ТСХ ЭА/ЦГ (1/1); Rf=0,7.
ИК (см-1): 3329 и 3294 (NH), 1715 и 1697 (C=O), 1610 (C=C).
Промежуточное соединение 29
1-трет-бутоксикарбонил-2-[(3-трет-бутоксикарбониламино)фенил] пиразолидин-3-он
К раствору промежуточного соединения 28а (0,316 г) в сухом диметилформамиде (4,0 мл) добавляли карбонат калия (0,412 г), а через 15 минут - хлорпропионилхлорид (0,098 мл). Полученную смесь перемешивали в течение 3 часов при 25oC, затем разбавляли диэтиловым эфиром (50 мл), промывали хлоридом аммония (нас. ) (50 мл) и рассолом (50 мл). После высушивания раствор концентрировали под вакуумом, а остаток (0,4 г) растворяли в диметилформамиде (4,78 мл). Добавляли карбонат калия и перемешивали смесь в течение 3 часов при 25oC. Затем реакционную смесь разбавляли диэтиловым эфиром (50 мл), промывали хлоридом аммония (нас.) (50 мл) и рассолом (50 мл). После высушивания раствор концентрировали под вакуумом, получая в результате сырое соединение (0,4 г), которое очищали посредством колоночной хроматографии на силикагеле с применением в качестве элюанта смеси этилацетат/циклогексан (1/3), получая в результате соединение, указанное в заголовке (0,27 г). ТСХ ЭА/ЦГ (1/3), Rf=0,25. Т.пл.=129oC.
Промежуточное соединение 30 и промежуточное соединение 31
Трет-бутиловый эфир (E) 3-[(1-трет-бутоксикарбонил-2-[ (3-трет-бутоксикарбониламино)фенил] -3-оксо)пиразолидин-4-илиденметил] - 4,6-дихлор-1H-индол-2-карбоновой кислоты (30)
Трет-бутиловый эфир (E) 3-[(1-трет-бутоксикарбонил-2-[(3-трет- бутоксикарбониламино) фенил] -3-оксо)пиразолидин-4-илиденметил]-4,6-дихлор-1-трет- бутоксикарбонил-1H-индол-2-карбоновой кислоты (31)
К раствору промежуточного соединения 4 (0,196 г) в сухом тетрагидрофуране (5 мл) при -78oC по каплям добавляли раствор бис(триметисилил)амида лития 1М в тетрагидрофуране (0,564 мл). Реакционная смесь нагревалась до -20oC в течение 30 минут, затем добавляли раствор трет-бутилового эфира 4,6-дихлор-3-формил-1- [N-трет-бутоксикарбонил]-1H-индол-2-карбоновой кислоты (0,18 г) в сухом тетрагидрофуране (4 мл). Раствор выдерживали в течение 30 минут при -40oC, затем в течение 2 часов нагревали до 0oC. Раствор разбавляли этилацетатом (50 мл) и промывали хлоридом аммония (50 мл), рассолом (50 мл), сушили и концентрировали под вакуумом. Неочищенное соединение очищали посредством колоночной хроматографии на силикагеле с применением в качестве элюанта смеси этилацетат/циклогексан (3/97), получая в результате промежуточное соединение 38 [(0,05 г), ТСХ ЭА/ЦГ (1/2), Rf= 0,5] и промежуточное соединение 39 [(0,04 г), ТСХ ЭА/ЦГ].
Промежуточное соединение 30
1H-ЯМР (ДМСО): 1,23(s,9H), 1,45(s,9H), 1,51(s,9H), 4,49(d,2H), 7,17(m, 1H), 7,3-7,2(m,2H), 7,30(d,1H), 7,50(d,1H), 7,74(t,1H), 7,82(t,1H), 9,43(s, 1H), 12,4(bs, 1H). ИК (см-1): 3346 (NH), 1728 и 1684 (C=O). ms (m/z): 773, 758, 673, 561.
Промежуточное соединение 31
1H-ЯМР (ДМСО): 1,23(s,9H), 1,45(s,18H), 1,61(s,9H), 4,57(d,2H), 7,18(m, 1H), 7,25(m, 2H), 7,49(t,1H), 7,57(d,1H), 7,82(bs,1H), 7,94(d,1H), 9,44(s, 1H). ИК (см-1): 3341 (NH), 1726 (C=O). ms (m/z): 773, 242.
Промежуточное соединение 32
Ди-трет-бутиловый эфир (E)4,6-дихлор-3-(2,5-диоксо-1-фенил- пирролидин-3-илиденметил)-индол-1,2-дикарбоновой кислоты
К раствору промежуточного соединения 14 (440 мг) в сухом толуоле (20 мл) добавляли N-фенилтрифенилфосфоранилиденсукцинимид (472 мг). Раствор прогревали в течение 21 часа при 70oC, затем добавляли еще сукцинимид (236 мг) и продолжали прогревание в течение 10 часов. Растворитель удаляли при пониженном давлении, а остаток очищали посредством скоростной хроматографии (ЦГ/ЭА= 8/2, Rf=0,38), получая в результате соединение, указанное в заголовке, в виде белого вещества (309 мг).
ИК (Nujol) Vmax (см-1) 1750 (C=O).
Промежуточное соединение 33
1-трет-бутоксикарбонил-3-оксо-2-фенил-тетрагидро-пиридазин
К раствору промежуточного соединения 6 (1 г) в сухом диметилформамиде (10 мл) в атмосфере азота при 25oC добавляли пиридин (0,78 мл) и 4-бромбутирилхлорид (0,83 мл) и перемешивали реакционную смесь в течение 15 часов. Раствор разбавляли диэтиловым эфиром (80 мл) и промывали рассолом (2x80 мл). После высушивания органический раствор концентрировали под вакуумом, получая в результате желтое масло (1,7 г). К раствору этого масла (2,25 г) в диметилформамиде (15 мл) в атмосфере азота при 25oC добавляли безводный карбонат калия (1,7 г) и перемешивали полученную смесь в течение 3 часов. Раствор разбавляли диэтиловым эфиром (100 мл), промывали рассолом (2х80 мл), сушили и концентрировали под вакуумом. Сырой остаток очищали посредством скоростной колоночной хроматографии на силикагеле, применяя в качестве элюанта смесь циклогексан/этилацетат (8/2) и получая в результате соединение, указанное в заголовке (1,4 г) в виде белого порошка. ТСХ ЭА/ЦГ (1/1), Rf=0,60.
Промежуточное соединение 34
2-хинолинилгидразин
К раствору 2-хлорхинолина (15 г) в этаноле (150 мл) добавляли гидрат гидразина (40 мл). Полученный раствор прогревали с дефлегмацией в течение 6 часов, а затем разбавляли водой (400 мл) и экстрагировали диэтиловым эфиром (3x200 мл). Собранную органическую фазу промывали рассолом (300 мл), сушили и концентрировали под вакуумом, получая в результате соединение, указанное в заголовке, в виде твердого вещества (13 г).
1H-ЯМР (ДМСО): 4,29(s,2H), 6,83(d,1H), 7,15(m,1H), 7,47(m,1H), 7,52(d, 1H), 7,62(dd,1H), 7,86(d,1H), 8,05(s,1H).
Промежуточное соединение 35
1-трет-бутоксикарбонил-2-(2-хинолинил)гидразин
К раствору промежуточного соединения 34 (13 г) в тетрагидрофуране (150 мл) добавляли ди-трет-бутилдикарбонат (18 г). Раствор перемешивали в течение 1 часа при 25oC, затем концентрировали под вакуумом и истирали в порошок смесью этилацетат/н-гексан, получая в результате соединение, указанное в заголовке (19 г). ТСХ ЭА/ЦГ (1/2), Rf=0,3. Т.пл.=148-150oC.
Промежуточное соединение 36
1-трет-бутоксикарбонил-2-[3-хлорпропаноил-2-хинолинил]-гидразин
К раствору промежуточного соединения 35 (5 г) в тетрагидрофуране (60 мл) при 0oC по каплям добавляли раствор хлорпропионилхлорида (0,92 мл) в тетрагидрофуране (40 мл). Полученный гетерогенный раствор перемешивали в течение 30 минут при 0oC, фильтровали и концентрировали под вакуумом, получая в результате соединение, указанное в заголовке (2,7 г) в виде пены. ТСХ ЭА/ЦГ (1/1), Rf=0,8.
Промежуточное соединение 37
1-трет-бутоксикарбонил-2-(2-хинолинил)-пиразолидин-3-он
К раствору промежуточного соединения 36 (2,7 г), растворенного в диметилформамиде (30 мл), добавляли карбонат калия (1,3 г). Реакционную смесь перемешивали в течение 2 часов при 25oC, затем разбавляли диэтиловым эфиром (100 мл), промывали водой (100 мл), сушили и концентрировали под вакуумом. Сырой продукт очищали посредством колоночной хроматографии на силикагеле, используя в качестве элюанта смесь этилацетат/циклогексан (2/8), получая в результате соединение, указанное в заголовке (0,8 г) в виде пены. ТСХ ЭА/ЦГ (1/1); Rf=0,6. Т.пл.=112oC.
Промежуточное соединение 38
Трет-бутиловый эфир (E)3-[1-трет-бутоксикарбонил-2-(2-хинолинил)- 3-оксопиразолидин-4-илиденметил] -4,6-дихлор-1-трет-бутоксикарбонил- 1H-индол-2-карбоновой кислоты
В раствор промежуточного соединения 37 (0,17 г) в сухом тетрагидрофуране (10 мл) при -78oC по каплям добавляли раствор бис(триметилсилил)амида лития 1М в тетрагидрофуране (0,62 мл). Реакционная смесь в течение 30 минут нагревалась до -20oC, затем добавляли раствор трет-бутилового эфира 4,6-дихлор-3-формил-1- [N-трет-бутоксикарбонил]-1H-индол-2-карбоновой кислоты (0,2 г) в сухом тетрагидрофуране (10 мл). Раствор выдерживали в течение 30 минут при -20oC, затем в течение 2 часов нагревали до 25oC. Раствор разбавляли соляной кислотой (50 мл), экстрагировали диэтиловым эфиром (3 х 40 мл), а собранную органическую фазу сушили и концентрировали под вакуумом. Сырое соединение очищали посредством колоночной хроматографии на силикагеле, используя в качестве элюанта смесь ЭА/ЦГ (1/9) и получая в результате соединение, указанное в заголовке (0,125 г), ТСХ ЭА/ЦГ (1/2), Rf=0,7. Т.пл.=48-51oC.
Промежуточное соединение 39
1-трет-бутоксикарбонил-2-(2-пиридил)гидразин
2-хлорпиридин (23 г) добавляли к гидрату гидразина (110 мл). Полученный раствор прогревали при температуре дефлегмации в течение 6 часов, а затем экстрагировали диэтиловым эфиром (2 х 100 мл).
Водную фазу концентрировали под вакуумом, разбавляли водой (40 мл), затем добавляли гидроксид калия (2 г) и экстрагировали раствор диэтиловым эфиром (100 мл). Собранную органическую фазу сушили и концентрировали под вакуумом, получая в результате сырое производное 2-пиридингидразина в виде твердого вещества (13 г), которое растворяли в тетрагидрофуране (200 мл), и добавляли ди-трет-бутилдикарбонат (26 г). Раствор перемешивали в течение 1 часа при 25oC, а затем концентрировали под вакуумом и истирали в порошок из смеси этилацетат/н-гексан, получая в результате соединение, указанное в заголовке (16 г). ТСХ ЭА/ЦГ (2/1), Rf=0,54. Т.пл.=91oC.
Промежуточное соединение 40
1-трет-бутоксикарбонил-2-(3-хлорпропаноил-2-пиридил)-гидразин
К раствору промежуточного соединения 39 (4 г) в тетрагидрофуране (60 мл) при 0oC по каплям добавляли раствор хлорпропионилхлорида (0,92 мл) в тетрагидрофуране (40 мл). Полученную гетерогенную смесь перемешивали в течение 30 минут при 0oC, фильтровали и концентрировали под вакуумом, получая в результате неочищенное соединение, указанное в заголовке, в виде пены. ТСХ ЭА/ЦГ (1/1), Rf=0,8.
Промежуточное соединение 41
1-трет-бутоксикарбонил-2-(2-пиридил)-пиразолидин-3-он
К раствору промежуточного соединения 40, растворенного в диметилформамиде (30 мл), добавляли карбонат калия (1,3 г). Реакционную смесь перемешивали в течение 2 часов при 25oC, разбавляли диэтиловым эфиром (100 мл), промывали водой (100 мл), сушили и концентрировали под вакуумом. Сырой продукт очищали посредством колоночной хроматографии на силикагеле с применением в качестве элюанта смеси этилацетат/циклогексан (2/8), получая в результате соединение, указанное в заголовке (0,87 г) в виде пены. ТСХ ЭА/ЦГ (1/1): Rf= 0,65.
1H-ЯМР (CDCl3): 1,50(s,9H), 2,59(t,2H), 4,27(m,2H), 6,81(dd,1H), 6,96(m, 1H), 7,63(m,1H), 8,33(m,1H).
Промежуточное соединение 42
Трет-бутиловый эфир (E) 3-[1-трет-бутоксикарбонил-2-(2-пиридил)- 3-оксопиразолидин-4-илиденметил] -4,6-дихлор-1-трет-бутоксикарбонил -1H-индол-2-карбоновой кислоты
К раствору промежуточного соединения 41 (0,361 г) в сухом тетрагидрофуране (10 мл) при -78oC по каплям добавляли раствор бис(триметилсилил)амида лития 1М в тетрагидрофуране (1,6 мл). Реакционная смесь нагревалась до -20oC в течение 30 минут, затем добавляли раствор трет-бутилового эфира 4,6-дихлор-3-формил-1- [N-трет-бутоксикарбонил]-1H-индол-2-карбоновой кислоты (0,2 г) в сухом тетрагидрофуране (10 мл). Раствор выдерживали при -20oC в течение 30 минут, затем нагревали до 25oC в течение 2 часов. Раствор разбавляли соляной кислотой (50 мл), экстрагировали этилацетатом (2 х 50 мл) и концентрировали под вакуумом собранную органическую фазу. Сырое соединение очищали посредством колоночной хроматографии на силикагеле, используя в качестве элюента смесь этилацетат/циклогексан (2/8), получая в результате соединение, указанное в заголовке (0,06 г) в виде пены. ТСХ ЭА/ЦГ (1/1), Rf=0,85.
1H-ЯМР (CDCl3): 1,38(s, 9H), 1,53(s, 9H), 1,64(s, 9H), 4,67(d,2H), 6,19(ddd, 1H), 6,78(1H), 7,18(d,1H), 7,58(ddd,1H), 7,70(t,1H), 7,98(d,1H), 8,23(bm,1H).
Промежуточное соединение 43
1-нафтилгидразин
Раствор нитрита натрия (4,8 г) в воде (20 мл) в течение 15 минут добавляли к перемешиваемой охлаждаемой льдом суспензии 1-нафтиламина (9,58 г) в 6М соляной кислоте (80 мл). После дополнительного инкубирования в течение 30 минут на ледяной бане медленно добавляли раствор хлорида олова (44,5 г) в 6М соляной кислоте (80 мл), полученную суспензию перемешивали в течение 3 часов при 0oC. Образовавшуюся твердую фазу отфильтровывали и растворяли в смеси 40%-ного раствора гидроксида калия (100 мл) и этилацетата (150 мл). Органический слой отделяли, а водный слой экстрагировали этилацетатом (3 х 100 мл). Объединенные органические экстракты высушивали и концентрировали при пониженном давлении, получая в результате соединение, указанное в заголовке, в виде фиолетового вещества (10,58 г, Rf=0,1 в ЭА/ЦГ (1/4)). ИК (см-1): 3312-3474 (NH и NH2); 1580-1610 (C=C).
Промежуточное соединение 44
1-трет-бутоксикарбонил-2-(1-нафтил)гидразин
К раствору промежуточного соединения 43 (6,9 г) в тетра-гидрофуране (350 мл) добавляли ди-трет-бутилдикарбонат (14,3 г). Раствор перемешивали в течение 8 часов при 25oC, затем концентрировали под вакуумом, получая неочищенное соединение, указанное в заголовке, которое очищали посредством колоночной хроматографии (этилацетат/циклогексан, 1/4), получая в результате соединение, указанное в заголовке, в виде коричневого вещества (9 г). ТСХ ЭА/ЦГ (1/4), Rf=0,45. Т.пл.=109oC.
Промежуточное соединение 45
1-трет-бутоксикарбонил-2-(1-нафтил)-пиразолидин-3-он
К раствору промежуточного соединения 44 (3 г) в сухом диметилформамиде (45 мл) добавляли карбонат калия (2,2 г), а спустя 5 минут - хлорпропионилхлорид (1,5 мл). Полученную смесь перемешивали в течение 2 часов при 25oC, затем разбавляли диэтиловым эфиром (200 мл), промывали водой (2 х 100 мл), сушили и концентрировали под вакуумом. Остаток кристаллизовали из диэтилового эфира, получая в результате белое вещество (1,5 г). Его растворяли в диметилформамиде (17,9 мл) и добавляли карбонат калия (0,62 г). Реакционную смесь перемешивали в течение 3 часов при 25oC, а затем разбавляли диэтиловым эфиром (100 мл), промывали водой (100 мл), сушили и концентрировали под вакуумом, получая в результате продукт, который очищали посредством колоночной хроматографии на силикагеле, используя в качестве элюанта смесь диэтиловый эфир/циклогексан (2/1), получая в результате соединение, указанное в заголовке (1,09 г) в виде бледно-желтой пены. ТСХ диэтиловый эфир/циклогексан (2/1); Rf= 0,37.
Промежуточное соединение 46
Трет-бутиладамантилиденкарбазат
Гексановый раствор, содержавший 2-адамантон (2 г) и трет- бутилкарбазат (1,76 г), прогревали при температуре дефлегмации в течение 3 часов. После остывания раствора кристаллизовали соединение, указанное в заголовке, и отфильтровывали его (3,28 г). Т.пл. 175-177oC.
Промежуточное соединение 47
1-трет-бутоксикарбонил-2-(2-адамантил)гидразин
Раствор промежуточного соединения 46 (2,13 г), Pd/C 20%-ного в/в катализатора (0,4 г) и 150 мл абсолютного этанола помещали в колбу под давлением аппарата для гидрогенизации Paar. Поглощение водорода при 3 атм (в среднем) продолжалось 12 часов. Затем раствор фильтровали на броунмиллерите, а растворитель удаляли при пониженном давлении, получая в результате соединение, указанное в заголовке, в виде твердого вещества, т.пл. 90-92oC.
1H-ЯМР (CDCl3): 5,29(bs, 1H), 3,84(bs, 1H), 3,05(bs,1H), 2,07(m,2H), 1,9-1,4(m,12H), 1,44(s,9H).
Промежуточное соединение 48
1-трет-бутоксикарбонил-2-(2-адамантил)пиразолидин-3-он
К раствору промежуточного соединения 47 (2 г) в сухом диметилформамиде (20 мл) добавляли карбонат калия (2,07 г), а через 15 минут - хлорпропионилхлорид (0,72 мл). Полученную смесь перемешивали при комнатной температуре в течение 3 часов, затем разбавляли диэтиловым эфиром (100 мл). Затем его промывали водой (100 мл), хлоридом аммония (нас.) (100 мл) и рассолом (100 мл). После высушивания растворитель удаляли при пониженном давлении, а смесь растворяли в сухом диметилформамиде (20 мл). После добавления карбоната калия раствор перемешивали в течение 3 часов при комнатной температуре. Реакционную смесь затем разбавляли диэтиловым эфиром (100 мл) и промывали водой (100 мл), хлоридом аммония (нас.) (100 мл) и рассолом (100 мл). После высушивания растворитель удаляли при пониженном давлении, получая в результате сырое соединение, указанное в заголовке, которое затем очищали посредством колоночной хроматографии на силикагеле, используя в качестве элюанта смесь циклогексан/этилацетат (3/1) и получая в результате соединение, указанное в заголовке (1,73 г).
1H-ЯМР (CDCl3): 4,08(m, 1H), 3,91(m,2H), 2,6 (m,2H), 2,52(t,2H), 1,9-1,4(m,2H), 1,48(s,9H).
Промежуточное соединение 49
Трет-бутиловый эфир 4,6-дихлоро-3-[(5-оксо-2-(2-адамантил)-1- трет-бутоксикарбонилпиразолидин-4-ил)-гидроксиметил] -1-трет- бутоксикарбонил-1H-индол-2-карбоновой кислоты
К раствору промежуточного соединения 48 (0,386 г) в сухом тетрагидрофуране (20 мл) при -78oC по каплям добавляли раствор бис(триметисилил)амида лития 1М в тетрагидрофуране (1,44 мл). Реакционная смесь нагревалась в течение 30 минут до -20oC, затем добавляли раствор промежуточного соединения 14 (0,2 г) в сухом тетрагидрофуране (10 мл). Раствор выдерживали 30 минут при -20oC, а затем разбавляли соляной кислотой 0,1 М (50 мл) и экстрагировали этилацетатом (2 х 50 мл). Собранную органическую фазу сушили над сульфатом натрия и концентрировали под вакуумом. Сырое соединение очищали посредством колоночной хроматографии на силикагеле, используя в качестве элюанта смесь этилацетат/циклогексан (1/9), получая в результате соединение, указанное в заголовке (0,26 г), в виде пены.
1H-ЯМР (ДМСО): 1,38(s, 9H), 1,50(s, 9H), 1,63(s,9H), 1,6-1,8(m, 14H), 3,2-3,4(bs, 2H), 3,85(dt, 1H), 3,97(s,1H), 5,61(bs,1H), 5,78(d,1H), 7,53(d, 1H), 7,90(d,1H).
Пример 1
Метил (E)-4,6-дихлоро-3-(2-оксо-1-фенил-пирролидин-3-илиденметил)- 1H-индол-2-карбоксилат
Промежуточное соединение 4 (700 мг) суспендировали в этаноле (95%-ном) (10 мл), добавляли соляную кислоту (10 мл, 6М) и прогревали гетерогенную смесь в течение 10 часов с дефлегмацией. Суспензию охлаждали до комнатной температуры, осадок отфильтровывали, промывали дополнительными порциями 6М соляной кислоты и окончательно сушили под вакуумом, получая в результате соединение, указанное в заголовке (427 мг), в виде белого вещества.
ИК (Nujol) Vmax (см-1) 1692 (C=O), 1672 (C=O).
1H-ЯМР (ДМСО): 12,58(bs), 7,81(d), 7,71(t), 7,49(d), 7,42(t), 7,29(d), 7,18(t), 3,90(s), 3,88(t), 3,53(t), 2,64(dt).
Пример 2
(E)-4,6-дихлоро-3-(2-оксо-1-фенил-пирролидин-3-илиденметил)- 1H-индол-2-карбоновая кислота
Соединение из Примера 1 (136 мг) и моногидрат гидроксида лития (54 мг) растворяли в этаноле (95%-ном) (5 мл), полученный раствор прогревали с дефлегмацией в течение 1,5 часов. Растворитель выпаривали, а остаток обрабатывали при 50oC 6М соляной кислотой в течение 30 минут, затем в течение еще 30 минут при комнатной температуре. Получившееся белое вещество отфильтровывали, промывали и сушили под вакуумом, получая в результате соединение, указанное в заголовке (110 мг), в виде белого вещества.
ИК (Nujol) Vmax (см-1) 3281 (N-H), 1682 (C=O), 1630 (C=C).
1H-ЯМР (ДМСО) 13,9-13,3(bs), 12,44(s), 7,81(d), 7,72(t), 7,47(d), 7,42(t), 7,26(d), 7,18(tt), 3,90(s), 3,88(t), 3,53(t), 2,67 (td).
Пример 3
Натриевая соль (E)-4,6-дихлоро-3-(2-оксо-1-фенил-пирролидин-3- илиденметил)-1H-индол-2-карбоновой кислоты
Соединение из Примера 2 (100 мг) суспендировали при комнатной температуре в растворе гидроксида натрия (2,4 мл, 0,1 N), смесь перемешивали до тех пор, пока она не становилась лишь слегка мутной (прибл. 1 час). Затем ее лиофилизировали в течение 48 часов, получая в результате соединение, указанное в заголовке (105 мг), в виде белого вещества.
ИК (Nujol) Vmax (см-1) 3302 (N-H), 1672 (C=O), 1639 (C=C).
1H-ЯМР (ДМСО) 12,0-11,0(bs), 7,82(t), 7,78(d), 7,38(t), 7,35(d), 7,12(t), 7,04(d), 3,79(t), 2,77(td).
Пример 4
Метил (E)-4,6-дихлоро-3-(2-оксо-1-фенил-пиперидин-3-илиденметил)- 1H-индол-2-карбоксилат
Промежуточное соединение 5 (680 мг) суспендировали в этаноле (95%-ном) (10 мл), добавляли соляную кислоту (10 мл, 6М), гетерогенную смесь прогревали в течение 10 часов с дефлегмацией. Затем суспензию охлаждали до комнатной температуры, твердую фазу отфильтровывали, промывали дополнительными порциями 6М соляной кислоты и окончательно сушили под вакуумом, получая в результате соединение, указанное в заголовке (490 мг), в виде белого вещества.
ИК (Nujol) Vmax (см-1) 3285 (N-H), 1682 (C=O), 1661 (C=O).
1H-ЯМР (ДМСО) 12,50(bs), 7,88(t), 7,45(d), 7,40(m), 7,24(d), 7,24(m), 3,87(s), 3,71(t), 2,37(td), 1,84(m).
Пример 5
(E)-4,6-дихлоро-3-(2-оксо-1-фенил-пиперидин-3-илиденметил)-1H- индол-2-карбоновая кислота
Соединение из Примера 4 (490 мг) и моногидрат гидроксида лития (200 мг) растворяли в этаноле (95%-ном) (20 мл), полученный раствор прогревали с дефлегмацией в течение 1,5 часов. Растворитель выпаривали, а остаток обрабатывали при 50oC 6М соляной кислотой в течение 30 минут, затем еще в течение 30 минут при комнатной температуре. Полученное белое вещество отфильтровывали, промывали и сушили под вакуумом, получая в результате соединение, указанное в заголовке (400 мг), в виде белого вещества.
ИК (Nujol) Vmax (см-1) 3294 (N-H), 1670 (C=O), 1645 (C=O).
1H-ЯМР (ДМСО) 13,43(bs), 12,36(bs), 7,89(t), 7,43(d), 7,37(m), 7,24(m), 7,21(d), 3,71(t), 2,39(td), 1,85(m).
Пример 6
Натриевая соль (E)-4,6-дихлор-3-(2-оксо-1-фенил-пиперидин-3- илиденметил)-1H-индол-2-карбоновой кислоты
Соединение из Примера 5 (100 мг) суспендировали при комнатной температуре в растворе гидроксида натрия (2,4 мл, 0,1N), смесь перемешивали до тех пор, пока она не становилась лишь слегка мутной (прибл. 1 час). Затем ее лиофилизировали в течение 48 часов, получая в результате соединение, указанное в заголовке (105 мг), в виде белого вещества.
ИК (Nujol) Vmax (см-1) 3305 (N-H), 1670 (C=O), 1645 (C=O).
1H-ЯМР (ДМСО) 11,6(bs), 8,00(t), 7,38(m), 7,34(d), 7,22(tt), 7,03(d), 3,68(t), 2,46(td), 1,81(m).
Пример 7
(E) 3-[(1-трет-бутоксикарбонил-3-оксо-2-фенил)пиразолидин-4- илиденметил]-4,6-дихлоро-1H-индол-2-карбоновая кислота
К раствору промежуточного соединения 7 (1,6 г) в сухом тетрагидрофуране (90 мл) при -78oC по каплям добавляли раствор бис(триметилсилил)амида лития 1М в тетрагидрофуране (6,7 мл). Реакционная смесь в течение 30 минут нагревалась до -20oC, затем к ней добавляли раствор промежуточного соединения 8 (2 г) в сухом тетрагидрофуране (60 мл). Раствор выдерживали в течение 30 минут при -20oC, затем нагревали в течение 4 часов до 25oC. Раствор разбавляли диэтиловым эфиром (300 мл) и промывали 0,1 М соляной кислотой (200 мл). Водный раствор экстрагировали диэтиловым эфиром (100 мл), а собранную органическую фазу сушили и концентрировали под вакуумом. Сырое соединение кристаллизовали из смеси этилацетат/гексан, получая в результате соединение, указанное в заголовке (0,92 г). Т.пл.=182oC.
ИК (Nujol) Vmax (см-1) 1719, 1688 (C=O), 1659. ms (m/z): 502.
Пример 8
Натриевая соль (E)-4,6-дихлор-3-(5-оксо-1-фенил-пиразолидин-4- илиденметил)-1H-индол-2-карбоновой кислоты
Соединение из Примера 7 (0,255 г) растворяли в сухом дихлорметане (5 мл) и трихлоруксусной кислоте (5 мл). Полученный раствор перемешивали в течение 1 часа при 25oC, а затем концентрировали под вакуумом. В результате истирания в порошок остатка диэтиловым эфиром получали соответствующее промежуточное соединение в виде кислоты (0,177 г). Этот продукт (0,155 г) суспендировали в воде, добавляли к нему 0,1 М гидроксид натрия (3,77 мл). Гетерогенный раствор перемешивали в течение 2 часов при 25oC, а затем сушили замораживанием, получая в результате соединение, указанное в заголовке (0,160 г).
1H-ЯМР (ДМСО): 3,94(d, 2H), 6,26(t, 1H), 7.08(m+s,2H), 7,37(m+s, 3H), 7,83(bs, 1H), 7,91(m,2H). ИК (см-1): 3177 (N-H), 1674 (C=O), 1595 (C=C). ms (m/z): 494, 402.
Пример 9
(E) -3-[(1-трет-бутоксикарбонил-2-[(4-трет- бутоксикарбониламино)фенил] -3-оксо)пиразолидин-4-илиденметил]- 4,6-дихлор-1H-индол-2-карбоновая кислота
К раствору промежуточного соединения 12 (0,47 г) в сухом третрагидрофуране (15 мл) при -78oC добавляли по каплям раствор бис(триметисилил)амида 1М в тетрагидрофуране (2,6 мл). Реакционная смесь в течение 30 минут нагревалась до -20oC, а затем к ней добавляли раствор этилового эфира 4,6-дихлор-3-формил- 1-[N-трет-бутоксикарбонил] -1H-индол-2-карбоновой кислоты (0,436 г) в сухом тетрагидрофуране (10 мл). Раствор выдерживали при - 20oC в течение 30 минут, затем в течение 4 часов нагревали до 25oC. Раствор разбавляли диэтиловым эфиром (200 мл) и промывали соляной кислотой 0,1 М (100 мл). Водный раствор экстрагировали диэтиловым эфиром (100 мл), объединенную органическую фазу сушили и концентрировали под вакуумом. Неочищенное соединение кристаллизовали из смеси этилацетат/гексан (1/2), получая в результате соединение, указанное в заголовке (0,30 г). Т.пл.= 220oC.
ИК (см-1): 3418-3281 (NH, OH), 1736, 1713 (C=O), 1676, 1612 (C=C). ms (m/z): 505, 415, 242.
Пример 10
Солянокислая соль (E)-4,6-дихлор-3-[(5-оксо-1-(4-аминофенил)) пиразолидин-4-илиденметил]-1H-индол-2-карбоновой кислоты
Соединение из Примера 9 (0,830 г) растворяли в метаноле (60 мл), через раствор в течение 5 минут барботировали хлороводород. Полученный раствор перемешивали в течение 2 часов при 25oC, а затем концентрировали под вакуумом. В результате истирания в порошок остатка диэтиловым эфиром получали соединение, указанное в заголовке (0,450 г).
1H-ЯМР (ДМСО): 3,86(d,2H), 7,28(d,1H), 7,38(d,2H), 7,47(d,1H), 7,74(t, 1H), 8,00(d, 2H), 10,0(b, 3H), 12,5(bs, 1H). ИК (см-1): 3439-3325 (N-H), 1730-1676 (C=O), 1612 (C=C). ms (m/z): 417.
Пример 11
Натриевая соль (E)-4,6-дихлор-3-[(5-оксо-1-(4-аминофенил) пиразолидин-4-илиденметил)-1H-индол-2-карбоновой кислоты
Соединение из Примера 9 (0,200 г) растворяли в сухом дихлорметане (20 мл) и трифторуксусной кислоте (6 мл). Полученный раствор перемешивали в течение 2 часов при 25oC, а затем концентрировали под вакуумом. Остаток суспендировали в воде (5 мл), добавляли 1М гидроксид натрия (1 мл), а спустя 5 минут раствор с помощью 1М соляной кислоты подкисляли до pH 3. Добавляли этилацетат (3 х 100 мл), органические слои собирали и сушили. Растворитель выпаривали посредством перегонки при пониженном давлении. В результате истирания в порошок остатка диэтиловым эфиром получали соответствующее промежуточное соединение в форме кислоты (0,060 г). Этот продукт суспендировали в воде и добавляли 0,1 М гидроксид натрия (0,45 мл). Гомогенный раствор перемешивали в течение 30 минут при 5oC, затем сушили посредством замораживания, получая в результате соединение, указанное в заголовке (0,017 г).
1H-ЯМР (ДМСО): 3,82(d,2H), 6,56(d,2H), 7,21(d,1H), 7,42(d,1H), 7,54(d+s, 2H+1H), 11, 5-13,0(bm,1H). ИК (см-1): 3440-2680 (N-H), 1734 (C=O), 1587 (C= O).
Пример 12
(Z)-4,6-дихлор-3-(2,5-диоксо-1-фенил-имидазолидин-4-илиденметил)- 1H-индол-2-карбоновая кислота
Промежуточное соединение 16 (140 мг) суспендировали в муравьиной кислоте и перемешивали в течение 10 часов при комнатной температуре. Растворитель удаляли при пониженном давлении, получая в результате соединение, указанное в заголовке, в виде эмульсии (92 мг, т.пл.>250oC).
ИК (Nujol) Vmax (см-1): 3186 (N-H), 1769 (C=O), 1732 (C=O) и 1691 (C=O).
1H-ЯМР (ДМСО): 12,42(bs), 10,70(bs), 7,49(td), 7,44(d), 7,43(d), 7,40(tt), 7,25(d) и 7,04(s).
Пример 13
Аллиловый эфир (E)-4,6-дихлор-3-(2,5-диоксо-1-фенил-имидазолидин- 4-илиденметил)-1H-индол-2-карбоновой кислоты
Промежуточное соединение 19 (360 мг) суспендировали в муравьиной кислоте (20 мл) и перемешивали в течение 5 часов при комнатной температуре. Растворитель удаляли при пониженном давлении, получая в результате соединение, указанное в заголовке, в виде вещества желтого цвета (296 мг, т.пл.>250oC).
ИК (Nujol) Vmax (см-1) 3261 (N-H), 1757 (C=O), 1720 (C=O) и 1668 (C=C).
1H-ЯМР (ДМСО): 12,47(bs), 10,98(bs), 7,45(td), 7,44(d), 7,35 (tt), 7,28(dd), 7,22(d), 6,82(s), 5,94(m), 5,35(m) H4,76(d).
Пример 14
(E) -4,6-дихлор-3-(2,5-диоксо-1-фенил-имидазолидин-4-илиденметил)- 1H-индол-2-карбоновая кислота
К раствору соединения из Примера 13 (290 мг) в сухом ТГФ (10 мл) добавляли 5,5-диметил-1,3-циклогександион (98 мг) и тетракистрифенилфосфин палладия (18,4 мг). Смесь перемешивали в течение 2 часов при комнатной температуре, а затем разбавляли этилацетатом и останавливали реакцию добавлением воды. Растворитель удаляли при пониженном давлении, осадок растворяли в диэтиловом эфире и экстрагировали раствором Na2CO3 (5%-ным). Водный раствор подкисляли, в результате чего выпадал осадок, который собирали посредством фильтрации, получая в результате соединение, указанное в заголовке, в виде вещества желтого цвета (147 мг, т.пл.250oC).
ИК (Nujol) Vmax (см-1): 3335 (N-H), 1763- 1724 (C=O) и 1678-1664 (C=O).
1H-ЯМР (ДМСО): 12,35(bs), 10,95(bs), 7,50-7,28(d), 7,20(d) и 6,83 (s).
Пример 15
Трет-бутиловый эфир (E) 3-[(1-трет-бутоксикарбонил-3-оксо-2- фенил)пиразолидин-4-илиденметил]-4,6-дихлор-1H-индол-2-карбоновой кислоты
Суспензию из Примера 7 (0,7 г) в бензоле нагревали с дефлегмацией и добавляли затем по каплям ацеталь N,N- диметилформамид-ди-трет-бутила (1,5 мл). Полученный раствор нагревали при дефлегмации 30 мин, затем разбавляли этилацетатом (100 мл), промывали насыщенным бикарбонатом натрия (100 мл), рассолом (100 мл), высушивали и концентрировали под вакуумом. В результате получали неочищенное соединение, указанное в заголовке (0,7 г).
ТСХ ЭА/ЦГ (6/24), Rf= 0,8. 1H-ЯМР (CDCl3): 1,23(s,9H), 1,57 (s,9H), 4,60(d,2H), 7,14(m,1H), 7,18(d,1H), 7,35(d,1H), 7,40(m,2H), 7,70(m,2H), 7,93 (t,1H), 9,15(bs,1H).
Пример 16
3-[2-(4-аминофенил)-1-трет-бутоксикарбонил-3-оксо-пиразолидин- 4-илиденметил]-4,6-дихлор-1H-индол-2-карбоновая кислота
Соединение из Примера 9 (0,110 г) растворяли в сухом дихлорметане (11 мл) и трифторуксусной кислоте (0,55 мл). Полученный раствор перемешивали в течение 3 часов при 0oC, а затем концентрировали под вакуумом. В результате истирания в порошок остатка с диэтиловым эфиром (6 мл) получали соответствующее соединение, указанное в заголовке, в виде вещества коричневого цвета (0,070 г).
1H-ЯМР (ДМСО): 1,22(s,9H), 4,50(d,2H), 6,98(d,2H), 7,30(d,1H), 7,43(d, 2H), 7,48(d,1H), 7,76(t,1H), 12,6(s,1H).
Пример 17
3-[2-(4-ацетиламино)-фенил)-1-трет-бутоксикарбонил-3-оксо- пиразолидин-4-илиденметил]-4,6-дихлор-1H-индол-2-карбоновая кислота
К раствору соединения из Примера 16 (0,200 г) в сухом тетрагидрофуране (5 мл) в атмосфере азота добавляли триэтиламин
(0,115 мл). Спустя 5 минут при 0oC к перемешивавшемуся раствору по каплям добавляли хлорацетилхлорид (0,040 мл). Реакционная смесь нагревалась в течение 2 часов до комнатной температуры, а затем к ней добавляли раствор 5%-ного бикарбоната натрия (30 мл). Реакционную смесь экстрагировали этилацетатом (3х20 мл), а объединенные органические фазы сушили и концентрировали под вакуумом. Сырое соединение, указанное в заголовке, очищали посредством скоростной хроматографии на силикагеле (дихлорметан:метанол:уксусная кислота = 90: 5: 5), получая в результате соединение, указанное в заголовке, в виде вещества желтого цвета (0,023 г).
ИК (см-1, Nujol): 3500-2500 (ОН, NH), 1668 (C=O), 1607 (C=O, C=C).
Пример 18
4,6-Дихлор-3-[5-оксо-1-(4-ацетиламино-фенил)-пиразолидин-4- илиденметил] -1H-индол-2-карбоновая кислота
Соединение из Примера 17 (0,023 г) растворяли в сухом дихлорметане (5 мл) и трифторуксусной кислоте (5 мл). Полученный раствор перемешивали в течение 2 часов при 25oC и концентрировали под вакуумом. В результате истирания в порошок с диэтиловым эфиром (5 мл) получали соединение, указанное в заголовке, в виде вещества бледно-коричневого цвета (0,030 г).
1H-ЯМР (ДМСО): 2,02(s,3H), 3,83(m,2H), 6,40(s,1H), 7,26(d,1H), 7,45(d, 1H), 7,58(m, 2H), 7,67(t,1H), 7,82(m,2H), 9,95(s,1H), 12,44(S,1H), 13,7(s, 1H).
ИК (см-1, Nujol): 3500-2500 (OH-NH), 1678-1650 (C=O), 1601 (C=C).
Пример 19
3-[1-трет-бутоксикарбонил-3-оксо-2-(4-уреидо-фенил)-пиразолидин- 4-илиденметил]-4,6-дихлор-1H-индол-2-карбоновая кислота
К раствору соединения из Примера 16 (0,070 г) в тетрагидрофуране (4 мл) в атмосфере азота при комнатной температуре добавляли триметилсилилизоцианат (0,080 мл). Раствор прогревали с дефлегмацией в течение 4 часов, по окончании этого периода становилось видно, как из раствора выпадал осадок. Твердую фазу отфильтровывали и промывали тетрагидрофураном (10 мл), получая в результате соединение, указанное в заголовке, в виде вещества оранжевого цвета (0,042 г).
1H-ЯМР (ДМСО): 1,22(s,9H), 4,52(m,2H), 5,87(s,2H), 7,30(s,1H), 7,45(m, 4H), 7,50(m,1H), 7,80(s,1H), 8,65(s,1H), 12,5(s,1H).
ИК (см-1, Nujol): 3489-3341 (N-H).
Пример 20
4,6-Дихлор-3-[5-оксо-1-(4-уреидо-фенил)-пиразолидин-4- илиденметил] -1H-индол-2-карбоновая кислота
Соединение из Примера 19 растворяли в сухом дихлорметане (10 мл) и трифторуксусной кислоте (2 мл). Полученный раствор перемешивали в течение 2 часов при 25oC, а затем концентрировали под вакуумом. В результате истирания в порошок с диэтиловым эфиром (5 мл) получали соединение, указанное в заголовке, в виде вещества красного цвета (0,030 г).
1H-ЯМР (ДМСО): 3,81(m, 2H), 5,82(s,1H), 6,36(s,1H), 7,25(d,1H), 7,39-7,45(m,3H), 7,65(t,1H), 7,75(m,2H), 8,53(s,1H), 12,43(s, 1H), 13,71(s,1H).
ИК (см-1, Nujol): 3500- 2600 (ОН, NH).
Пример 21
3-[1-трет-бутоксикарбонил-2-(4-метилсульфамидофенил)-3-оксо- пиразолидин-4-илиденметил]-4,6-дихлор-1H-индол-2-карбоновая кислота
К раствору промежуточного соединения 23 (0,100 г) в сухом тетрагидрофуране (5 мл) при -78oC по каплям добавляли 1М раствор бис(триметилсилил)амида лития в тетрагидрофуране (0,53 мл). Реакционная смесь в течение 30 минут нагревалась до -20oC, а затем к ней добавляли раствор этилового эфира 4,6-дихлор-3-формил-1- [N-третбутоксикарбонил]-1H-индол-2-карбоновой кислоты (0,100 г) в сухом тетрагидрофуране (4 мл). Раствор выдерживали 30 минут при -20oC, а затем нагревали до 25oC в течение 4 часов. Раствор разбавляли диэтиловым эфиром (20 мл) и промывали 0,1 М соляной кислотой (10 мл). Водный раствор экстрагировали диэтиловым эфиром (15 мл), а объединенные органические фазы сушили и концентрировали под вакуумом. Сырое соединение очищали посредством скоростной хроматографии на колонке с силикагелем (дихлорметан:метанол:уксусная кислота = 90:5:5), получая в результате соединение, указанное в заголовке (0,035 г).
Пример 22
4,6-Дихлор-3-[1-(4-метилсульфамидофенил)-5-оксо-пиразолидин-4- илиденметил]-1H-индол-2-карбоновая кислота
Соединение из Примера 21 (0,035 г) растворяли в сухом дихлорметане (6 мл) и трифторуксусной кислоте (2 мл). Полученный раствор перемешивали в течение 2 часов при 25oC, а затем концентрировали под вакуумом. В результате истирания в порошок с этилацетатом (5 мл) получали соединение, указанное в заголовке, в виде вещества желтого цвета (0,010 г).
1H-ЯМР (ДМСО): 2,94(s,3H), 3,83(bm,2H), 6,40(bs,1H), 7,22(d,2H), 7,26(d, 1H), 7,45(d,1H), 7,68(d,1H), 7,86(d,2H), 9,67(s,1H), 12,48(bs,1H).
ИК (см-1, Nujol): 3186 (C=O), 1680 (C=O), 1640 (C=C).
Пример 23
(E)-3-[(2-метил-5-оксо-1-фенил)пиразолидин-4-илиденметил] - 4,6-дихлор-1H-индол-2-карбоновая кислота
К раствору промежуточного соединения 25 (0,365 г) в сухом тетрагидрофуране (10 мл) при -78oC по каплям добавляли раствор бис(триметилсилил)амида лития 1М в тетрагидрофуране (1,33 мл). Реакционная смесь в течение 30 минут нагревалась до -20oC, а затем к ней добавляли раствор промежуточного соединения 8 (0,2 г) в сухом тетрагидрофуране (10 мл). Раствор выдерживали в течение 30 минут при -20oC, а затем в течение 4 часов нагревали до 25oC. Раствор разбавляли диэтиловым эфиром (300 мл) и промывали соляной кислотой 0,1 М (200 мл). Водный раствор экстрагировали диэтиловым эфиром (100 мл), а собранную органическую фазу сушили и концентрировали под вакуумом. Сырое соединение истирали в порошок с диэтиловым эфиром, получая в результате соединение, указанное в заголовке (0,06 г).
1H-ЯМР (ДМСО): 3,31(s,3H), 3,63(bs,1H), 4,08(bs,1H), 7,16(t,1H), 7,27(d, 1H), 7,42(t,2H), 7,45(d,1H), 7,83(s,1H), 7,84(d,2H), 12,48(s,1H), 13,75(bs, 1H).
ИК (см-1): 3244 (N-H), 1676 (C=O), 1653 (C=C).
Пример 24
4,6-Дихлор-3-(3-оксо-2-фенил-изоксазолидин-4-илиденметил)-1H- индол-2-карбоновая кислота
К раствору промежуточного соединения 27 (0,475 г) в сухом тетрагидрофуране (20 мл) при -78oC по каплям добавляли раствор бис(триметилсилил)амида лития 1 М в тетрагидрофуране (3,2 мл). Реакционная смесь нагревалась в течение 30 минут до -20oC, а затем к ней добавляли раствор этил-4,6-дихлор-3-формил-1-t-бутоксикарбонил- 1H-индол-2-карбоксилата (0,94 г) в сухом тетрагидрофуране (20 мл). Раствор выдерживали в течение 30 минут при -20oC, а затем нагревали в течение 4 часов до 25oC. Раствор разбавляли диэтиловым эфиром (300 мл) и промывали соляной кислотой 0,1 М (200 мл). Водный раствор экстрагировали диэтиловым эфиром (100 мл), а собранную органическую фазу сушили и концентрировали под вакуумом. Сырое соединение очищали посредством колоночной хроматографии на силикагеле, применяя в качестве элюанта смесь этилацетат/циклогексан и получая в результате неочищенное соединение, указанное в заголовке (0,16 г) с ТСХ дихлорметан/метанол 27/3, Rf=0,3.
Пример 25
Третичный бутиловый эфир 4,6-дихлор-3-(3-оксо-2-фенил- изоксазолидин-4-илиденметил) -1H-индол-2-карбоновой кислоты
Продукт из Примера 24 обрабатывали N,N-диметилформамид-ди-трет- бутилацеталем (0,44 мл) в бензоле (15 мл). Полученный раствор прогревали в течение 30 минут с дефлегмацией, а затем концентрировали под вакуумом, получая в результате сырое соединение, которое очищали посредством колоночной хроматографии на силикагеле, применяя в качестве элюанта смесь этилацетат/циклогексан и получая в результате соединение, указанное в заголовке (0,03 г). ТСХ этилацетат/циклогексан (1/2), Rf=0,6, т.пл.=120oC.
1H-ЯМР (CDCl3): 1,59(s,9H), 5,04(d,2H), 7,17(t,1H), 7,18(d,1H), 7,34(d, 1H), 7,40(t,2H), 7,85(d,2H), 7,91(t,1H), 9,05(bs,1H).
Пример 26
4,6-Дихлор-3-(3-оксо-2-фенил-изоксазолидин-4-илиденметил)- 1H-индол-2-карбоновая кислота
Соединение из Примера 25 (0,025 г) растворяли в сухом дихлорметане (3 мл) и трифторуксусной кислоте (2 мл). Полученный раствор перемешивали в течение 1 часа при 25oC, а затем концентрировали под вакуумом. В результате истирания в порошок остатка с изопропанолом получали соединение, указанное в заголовке, в виде твердого вещества (0,014 г). Т.пл.>250oC.
1H-ЯМР (CDCl3): 1,59(s,9H), 5,04(d,2H), 7,17(t,1H), 7,18(d,1H), 7,34(d, 1H), 7,40(t,2H), 7,85(d,2H), 7,91(t,1H), 9,05(bs,1H).
ИК (см-1): 3304 (NH), 1672 (C=O), 1645 (C=C). MC (m/z): 403.
Пример 27
Хлористоводородная соль (E)-4,6-дихлор-3-[(5-оксо-1-(3- аминофенил))пиразолидин-4-илиденметил]-1H-индол-2-карбоновой кислоты
Через раствор промежуточного соединения 30 (0,03 г) и промежуточного соединения 31 (0,03 г) в метаноле (30 мл) барботировали хлороводород в течение 5 мин. при 0oC. Полученный раствор перемешивали в течение 1 часа при 25oC, а затем концентрировали под вакуумом. В результате истирания в порошок остатка с диэтиловым эфиром получали соединение, указанное в заголовке (0,01 г).
1H-ЯМР (ДМСО): 5,85(d,2H), 7,08(d,1H), 7,27(d,1H), 7,47(d,1H), 7,48(t, 1H), 7,76(t,1H), 7,83(d,1H), 8,02(s,1H), 9,5-10,5(b,3H), 12,5(bs,1H).
ИК (см-1): 3400-3200 (NH и OH), 1711 (C=O).
Пример 28
(E)-4,6-дихлор-3-(2,5-диоксо-1-фенил-пирролидин-3-илиденметил)- 1H-индол-2-карбоновая кислота
Суспензию промежуточного соединения 32 (295 мг) в муравьиной кислоте (40 мл) перемешивали в течение 6 часов при комнатной температуре, затем растворитель удаляли при пониженном давлении. Остаток истирали в порошок с этилацетатом и фильтровали, получая в результате соединение, указанное в заголовке, в виде эмульсии (148 мг, т.пл.>250oC).
ИК (Nujol) Vmax (см-1) 3204 (N-H), 1705 (C=O).
1H-ЯМР (ДМСО): 14-13(bs), 12,6(s), 8,08(t), 7,51(tt), 7,475(d), 7,43(m), 7,38(d), 7,30(d), 3,38(d).
Пример 29
(E)-3-[(1-трет-бутоксикарбонил-3-оксо-2-фенил)-тетрагидро- пиридазин-4-илиденметил]-4,6-дихлор-1H-индол-2-карбоновая кислота
К раствору промежуточного соединения 33 (0,85 г) в сухом тетрагидрофуране (8 мл) по каплям добавляли при -50oC раствор бис(триметилсилил)амида лития 1М в тетрагидрофуране (2 мл), а реакционную смесь перемешивали при -20/-40oC в течение 30 минут. Раствор охлаждали до -60oC и добавляли к нему раствор промежуточного соединения 8 (0,3 г) в сухом тетрагидрофуране (7 мл). Раствор нагревали до 25oC и перемешивали в течение 3-х часов, а затем разбавляли этилацетатом (300 мл) и промывали соляной кислотой 2М (20 мл) и рассолом (2x30 мл). Собранную органическую фазу сушили и концентрировали под вакуумом. Неочищенное соединение кристаллизовали из смеси этилацетат/н-гексан (5 мл/8 мл) при 0oC, получая в результате соединение, указанное в заголовке (0,2 г). Т.пл.>240oC.
1H-ЯМР (ДМСО): 1,28(s, 9H), 2,56(bs, 1H), 3,84(bs,2H), 7,19(tt, 1H), 7,19(d, 1H), 7,37(t, 2H), 7,46(d,1H), 7,60(dd,2H), 8,0(t, 1H), 12,8(bs,1H), 13,5(bs,1H).
Пример 30
(E)-4,6-дихлор-3-(3-оксо-2-фенил-тетрагидро-пиридазин-4- илиденметил) -1H-индол-2-карбоновая кислота
К раствору соединения из Примера 29 (0,117 г) в дихлорметане (10 мл) по каплям добавляли при 0oC трифторуксусную кислоту. Реакционную смесь нагревали до 25oC, а затем концентрировали под вакуумом. Остаток растворяли в этилацетате (30 мл), подщелачивали 5%-ным раствором гидрокарбоната натрия и подкисляли насыщенным раствором хлорида аммония. Органическую фазу сушили и концентрировали под вакуумом. Остаток порошковали в смеси этилацетат/диэтиловый эфир (2 мл/1 мл), получая в результате соединение, указанное в заголовке (0,046 г), в виде порошка бледно-желтого цвета, т.пл.>250oC.
1H-ЯМР (ДМСО): 3,10(m,2H), 3,35(m,2H), 6,02(t,1H), 7,12(t,1H), 7,22(d, 1H), 7,33(t,2H), 7,43(d,1H), 7,63(d,2H), 8,00(t,1H), 12,36(s,1H), 13,50(bs, 1H).
Пример 31
Хлористоводородная соль (E)-4,6-дихлор-3-[(5-оксо-1-(2- хинолинил)пиразолидин-4-илиденметил]-1H-индол-2-карбоновой кислоты
Через раствор промежуточного соединения 38 (0,115 г) в метаноле (15 мл) при 25oC в течение 5 мин барботировали хлороводород. Полученный раствор перемешивали в течение 4-х часов при 25oC, а затем концентрировали под вакуумом. В результате порошкования остатка с диэтиловым эфиром получали соединение, указанное в заголовке (0,035 г). Т.пл.>240oC.
Пример 32
Хлористоводородная соль (E)-4,6-дихлор-3-[(5-оксо-1-(2-пиридил)) пиразолидин-4-илиденметил]-1H-индол-2-карбоновой кислоты
Через раствор промежуточного соединения 42 (0,055 г) в метаноле (15 мл) при 25oC в течение 5 минут барботировали хлороводород. Полученный раствор перемешивали в течение 6 часов при 25oC, а затем концентрировали под вакуумом. В результате порошкования остатка с диэтиловым эфиром получали соединение, указанное в заголовке (0,02 г). Т.пл.>240oC.
Пример 33
Третичный бутиловый эфир (E)-3-[1-трет-бутоксикарбонил-2-(1- нафтил)-3-оксо] пиразолидин-4-илиденметил] -4,6-дихлор-1-трет- бутоксикарбонил-1H-индол-2-карбоновой кислоты
К раствору промежуточного соединения 45 (0,18 г) в сухом тетрагидрофуране (10 мл) при -78oC по каплям добавляли раствор бис(триметилсилил)амида лития 1М в тетрагидрофуране (0,58 мл). Реакционная смесь в течение 30 мин нагревалась до -20oC, затем реакционную смесь охлаждали до -40oC и добавляли раствор третичного бутилового эфира 4,6-дихлор-3-формил-1-[N-трет-бутоксикарбонил] -1H-индол-2-карбоновой кислоты (0,2 г) в сухом тетрагидрофуране (6 мл). Раствор выдерживали 20 мин при -40oC, а затем медленно нагревали в течение 2-х часов при 25oC. Раствор выливали в насыщенный раствор хлорида аммония и экстрагировали этилацетатом (200 мл). Органический слой промывали водой, сушили и концентрировали под вакуумом. Сырое соединение очищали посредством колоночной хроматографии на силикагеле, используя в качестве элюанта смесь диэтиловый эфир/циклогексан (3/7), получая в результате соединение, указанное в заголовке (0,030 г) в виде желтого воска, ТСХ диэтиловый эфир/циклогексан (1/2), Rf=0,55.
ИК (см-1) = 3500-2700 (NH), 1720-1690 (C=O).
Пример 34
(E)-4,6-дихлор-3-(5-оксо-1(1-нафтил)-пиразолидин-4-илиденметил)- 1H-индол-2-карбоновая кислота
Соединение из Примера 33 (0,030 г) растворяли в сухом дихлорметане (5 мл) и трифторуксусной кислоте (4 мл). Полученный раствор перемешивали в течение 1 часа при 25oC, а затем концентрировали под вакуумом. В результате истирания в порошок остатка с диэтиловым эфиром получали соединение, указанное в заголовке (0,010 г), т.пл. 200oC.
1H-ЯМР (ДМСО): 14,2-12,5 (широкий,1H), 12,46(s,1H), 8,04, 7,90 (m,3H), 7,76(t,1H), 7,66-7,56(m,4H), 7,50(d,1H), 7,30(d,1H), 6,60(s,1H), 4,04(d,2H).
ИК (см-1)= 3410-3184 (OH, NH), 1707-1686 (C=O), 1659 (C=C).
Пример 35
Трет-бутиловый эфир (E)-3-[(1-трет-бутоксикарбонил-3-оксо-2- (2-адамантил))пиразолидин-4-илиденметил]-4,6-дихлор-1H-индол-2- карбоновой кислоты
К раствору промежуточного соединения 49 (0,144 г) в тетрагидрофуране (10 мл) при 0oC по каплям добавляли раствор бис(триметилсилил)амида лития 1М в тетрагидрофуране (0,2 мл). Реакционная смесь нагревалась в течение 2-х часов до 25oC, а затем ее разбавляли соляной кислотой 0,1М (50 мл) и экстрагировали диэтиловым эфиром (2x50 мл). Собранную органическую фазу сушили над сульфатом натрия и концентрировали под вакуумом. Сырое соединение очищали посредством колоночной хроматографии с силикагелем, используя в качестве элюанта смесь этилацетат/циклогексан (1/9), получая соединение, указанное в заголовке (0,02 г) в виде пены.
1H-ЯМР (CDCl3): 0,75-2,00 (m+s+s, 30H), 2,75(m,2H), [4,22(bs)-4,34(d)3H] , [7,13(d)- 7,33(d)-7,69(t),3H], 9,15(bs,1H).
Пример 36
(E)-4,6-дихлор-3-[5-оксо-1-(2-адамантил)пиразолидин-4-илиденметил] -1H-индол-2-карбоновая кислота
Соединение из Примера 35 (0,02 г) растворяли в сухом дихлорметане (1 мл) и трифторуксусной кислоте (1 мл). Полученный раствор перемешивали в течение 1 часа при 25oC, а затем концентрировали под вакуумом. В результате истирания в порошок остатка с диэтиловым эфиром получали соединение, указанное в заголовке (0,003 г), т.пл. 190oC.
1H-ЯМР (ДМСО): 1,50-1,92(m,10H), 2,20-2,32(m,4H), 3,67(d,2H), 4,02(bs, 1H), 5,7(b, 1H), 7,26(d,1H), [7,46(d)-7,51(t),2H], 12,41 (bs,1H), 13,56(bs, 1H).
Пример 37
(Z)-4,6-дихлор-3-(5-оксо-1-фенил-пиразолидин-4-илиденметил)-1H- индол-2-карбоновая кислота
Промежуточное соединение 21 (0,125 г) растворяли в сухом дихлорметане (5 мл) и трифторуксусной кислоте (5 мл). Полученный раствор перемешивали при 25oC в течение 1 часа, а затем концентрировали под вакуумом. В результате истирания в порошок остатка с диэтиловым эфиром получали соединение, указанное в заголовке (0,041 г).
1H-ЯМР (ДМСО): 4,15(d,2H), 6,48(bm,1H), 7,05(m,1H). 7,14(d,1H), 7,18(t, 1H), 7,30(t,2H), 7,41(d,1H), 7,74(d,2H), 12,24(s,1H), 13,31(bs,1H).
Фармацевтические примеры
А. Капсулы/Таблетки
Активный ингредиент - 200,0 мг
Крахмал 1500 - 32,5 мг
Микрокристаллическая целлюлоза - 60,0 мг
Кроскармелоза натрия - 6,0 мг
Стеарат магния - 1,5 мг
Активный ингредиент смешивают с другими эксципиентами. Смесь можно применять для заполнения желатиновых капсул или прессовать для формования таблеток, применяя соответствующие штампы. На таблетки может быть нанесено покрытие с применением обычных технологий или покрытий.
Б. Таблетка
Активный ингредиент - 200,0 мг
Лактоза - 100,0 мг
Микрокристаллическая целлюлоза - 28,5 мг
Повидон - 25,0 мг
Кроскармелоза натрия - 6,0 мг
Стеарат магния - 1,5 мг
Активный ингредиент смешивают с лактозой, микрокристаллической целлюлозой и частью кроскармелозы натрия. Смесь гранулируют с повидоном после диспергирования в подходящем растворителе (например, воде). Гранулу после сушки и измельчения смешивают с оставшимися эксципиентами. Смесь может быть спрессована с применением соответствующих форм, а таблетки покрыты оболочкой, используя обычные технологии и покрытия.
В. Составы для инъецирования
Активный ингредиент - 0,1-7,00 мг/мл
Фосфат натрия - 1,0-50,00 мг/мл
NaOH с желательным pH (интервал 3-10) вода для инъекций до - 1 мл
Состав может быть упакован в стекло (ампулы) с резиновой пробкой (флаконы, шприцы) и пластиковыми/металлическими герметизаторами (только флаконы).
Г. Сухой порошок для смешивания с приемлемым наполнителем
Активный ингредиент - 0,1-100,00 мг
Маннит - До 0,02-5,00 мг
упакованный в стеклянных флаконах или шприцах с резиновой пробкой и (только флаконы) пластиковым/металлическим герметизатором.
Д. Ингаляционные патроны, мг/патрон
Активный ингредиент (микронизированный) - 5,00
Лактоза - До 25,00
Активный ингредиент микронизируют в струйной мельнице до размера мелких частиц перед смешиванием с лактозой нормального таблетного класса в высокоэнергетическом смесителе. Порошкообразной смесью заполняют подходящий контейнер для унифицированного дозирования в виде пузыря или капсулы для применения в соответствующем устройстве для ингаляций или вдуваний.
Сродство соединения по изобретению к связывающему сайту глицина, невосприимчивому к стрихнину, определяли согласно способу [4]. Значения pKi, полученные с типичными соединениями по изобретению, приведены ниже.
Пример N - pKi
3 - 7,29
6 - 7,31
8 - 8,0
10 - 7,83
12 - 7,43
14 - 7,13
18 - 7,7
20 - 7,69
22 - 7,66
23 - 7,46
26 - 7,12
27 - 7,7
28 - 7,38
34 - 7,31
Способность соединений по изобретению ингибировать NMDA индуцируемые конвульсии у мыши определяли согласно [5]. В этом тесте способность соединения ингибировать обобщенные приступы, индуцируемые внутрицеребровентрикулярной инъекцией NMDA у мышей, исследовали на нескольких дозировочных уровнях. По этим результатам вычисляли дозу, необходимую для защиты 50% животных от конвульсионного действия NMDA. Ее выражали в мг/кг и обозначали как ЕД50.
Характерные результаты, полученные для соединений по изобретению, при их внутривенном (IV) и пероральном (по) введении, представлены в табл. 1.
Соединения по изобретению существенно не токсичны при терапевтически применяемых дозах. Так например соединение из Примера 8 не показало побочных эффектов при введении анестезированным кошкам или находящимся в сознании собакам в дозах до 24 мг/кг.
ЛИТЕРАТУРА
1. U.S. Patent N. 4960786.
2. EP-A 0396124.
3. WO92/16205.
4. Kishimoto H. et al., J. Neurochem. 1981, 37, 1015-1024.
5. Chiamulera C. et al., Psychopharmacology (1990), 102, 551-552.
6. Chiamulera C. et al., European Journal of Pharmacology, 216, (1992), 335-336.
7. Manhas and Jeng, J. Org. Chem., 1967, 32, 1246-1248.
8. Hargis D.C. and Shubkin R.L. Tetrahedron Letters, Vol. 31, N. 21, pp. 2991-4, 1990.

Описываются новые производные индола формулы I или его физиологически приемлемая соль либо метаболически лабильный эфир, где m равно 2 и R является хлором в положении 4 и 6 индольного ядра; R1 представляет собой возможно замещенную фенильную группу; А представляет собой цепь, выбранную из -(СН2)2-, -(CH2)3-, -CH2CO-, -CH2NH-, -CH2NCH3-, -(CH2)2NH-, -NHCO- или -CH2O-, которые являются антагонистами возбуждающих аминокислот, а также способ их получения и другое применение в медицине. 4 с. и 6 з.п.ф-лы, 1 табл.
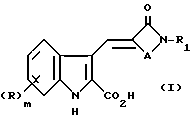
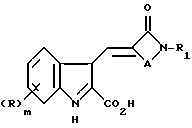
или его физиологически приемлемая соль либо метаболически лабильный эфир,
где m равно 2;
R является хлором в положении 4 и 6 индольного ядра;
R1 представляет собой возможно замещенную фенильную группу;
A представляет собой цепь, выбранную из -(CH2)2-, -(CH2)3-, -CH2CO-, -CH2NH-, -CH2NCH3-, -(CH2)2NH-, -NHCO- или -CH2O-.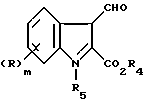
где R имеет значения, определенные в формуле I;
R4 является карбоксилзащитной группой;
R5 является азотзащитной группой,
с циклическим амидом формулы III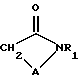
где R1 и A имеют значения, определенные в формуле I (при условии, что A не является -NHCO-), либо являются их защищенными производными,
в присутствии основания; и, если необходимо или желательно, полученное соединение подвергают одной или более чем одной из следующих операций: 1) удаление одной или более чем одной защитной группы; 2) выделение соединения в виде его соли; 3) превращение соединения формулы I или его соли в его метаболически лабильный эфир или 4) превращение соединения формулы I в его физиологически приемлемую соль.
| СПОСОБ ПОЛУЧЕНИЯ БЕЗДИСЛОКАЦИОННЫХ МОНОКРИСТАЛЛОВ КРЕМНИЯ | 0 |
|
SU396124A1 |
| Автоматический огнетушитель | 0 |
|
SU92A1 |
| Машковский М.Д | |||
| Лекарственные средства | |||
| - М.: Медицина, 1972, ч.1, с.63 - 68, 112, 119. | |||
