Настоящее изобретение касается новых производных 1,5-бензодиазепина, способов их получения, фармацевтических составов, включающих в себя указанные производные, а также их использования в медицине. Конкретнее, настоящее изобретение касается соединений, проявляющих агонистичную активность по отношению к рецепторам ХЦК-А, что позволяет указанным соединениям регулировать функционирование гормонов гастрина и холецистокинина (ХЦК) у млекопитающих.
Холецистокинины (ХЦК) и гастрин являются структурно близкими пептидами, присутствующими в ткани желудка и кишечника, а также в центральной нервной системе. К числу холецистокининов относятся ХЦК-33 (нейропептид, состоящий в своей естественной форме из 33 аминокислот), его карбокси-терминальный октапептид (который тоже представляет собой естественно встречающийся нейропептид, обозначаемый ХЦК-8), а также 39- и 12-аминокислотные формы. Гастрин встречается в 34-, 17- и 14-аминокислотных формах, причем его минимальная активная последовательность соответствует C-терминальному тетрапептиду Trp-Met-Asp-Phe-NH2 (ХЦК-4), являющемуся общим структурным элементом как для ХЦК, так и для гастрина.
ХЦК и гастрин представляют собой желудочно-кишечные гормоны и нейропередатчики в нейронной и периферической системах, выполняющие свойственные им биологические функции путем связывания с определенными рецепторами, локализованными в различных участках тела. Существует по меньшей мере два подтипа рецепторов холецистокинина, обозначаемые ХЦК-А и ХЦК-Б, причем оба указанных подтипа встречаются как в периферической, так и в центральной нервной системе.
Рецепторы ХЦК-А, обычно обозначаемые как рецепторы "периферического типа", в первую очередь обнаруживаются в поджелудочной железе, желчном пузыре, подвздошной кишке, пилорическом сфинктре, а также на афферентных тяжах блуждающего нерва. А-тип рецепторов ХЦК встречается также в определенных районах головного мозга и служит для обеспечения некоторых эффектов ЦНС. Ввиду того, что избирательные агонисты ХЦК-8 и А-типа ХЦК могут подавлять принятие пищи у некоторых видов животных, возник определенный интерес к разработке новых веществ, являющихся избирательными агонистами А-типа рецепторов ХЦК и способными служить в качестве аноректических агентов.
Рецепторы ХЦК-Б или гастрина обнаруживаются в периферических нейронах, гладкой мускулатуре желудочно-кишечной системы, а также в слизистой оболочке желудочно-кишечного тракта, особенно - в париетальных клетках, ECL-клетках, Д-клетках и главных клетках. Кроме того, рецепторы ХЦК-Б преобладают в головном мозге и вовлечены в регуляцию беспокойства, возбуждения и действия нейролептических агентов.
Описана группа производных 3-ациламино 1-алкил-5-фенил 1,5-бензодиазепина, проявляющих свойства антагонистов холецистокинина и способных обращать или блокировать действие, оказываемое соответствующими эндогенными гормонами по отношению к их рецепторам (US 4988692).
Описаны производные пептида, проявляющие агонистичную по отношению к ХЦК-А активность. Указанные соединения были предложены для использования в качестве регуляторов аппетита, а также для лечения и/или предупреждения желудочно-кишечных заболеваний, либо заболеваний центральной нервной системы у животных и, в особенности, у человека (US 4490304, WO 90/06937,WO 91/19733).
В настоящем изобретении описана новая группа 3-амино 1,5-бензодиазепиновых соединений, проявляющих агонистичную активность по отношению к рецепторам ХЦК-А, что позволяет указанным соединениям регулировать функционирование гормонов гастрина и холецистокинина (ХЦК) у млекопитающих. Некоторые из указанных соединений проявляют также антагонистичную активность по отношению к рецепторам ХЦК-Б.
Таким образом, настоящее изобретение предусматривает соединения, отвечающие общей формуле (I),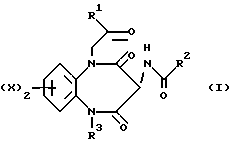
а также физиологически приемлемые соли и сольваты указанных соединений, где
X представляет собой атом водорода, трифторметил, алкил, C1-4-алкилтио, -О(C1-4-алкил), либо атом галогена;
R1 обозначает либо радикал, соответствующий формуле (II),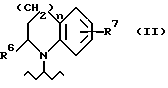
либо остаток NR4R5;
R2 представляет собой либо
(1) гетероцикл, присоединенный по своему положению 2 и выбранный из группы, включающей в себя пиррол, тетрагидропиррол, индол, бензофуран, тиофен, бензотиофен, индолин, хинолин или 4-оксобензопиран, причем указанный пиррол, тетрагидропиррол, индол или индолин могут быть замещены по атому азота, входящему с состав соответствующего кольца, группой R8 (см. ниже), а указанный индол, индолин, хинолин, бензофуран, бензотиофен или 4-оксобензопиран может быть замещен по своему бензольному кольцу группой R9 (см. ниже); либо
(2) фенил, возможно одно- или двузамещенный группами, независимо выбранными из числа следующих: атом галогена, гидрокси, циано, карбокси, -O(C1-4-алкил), -O(CH2C6H5), -COO(C1-4-алкил), амино, диметиламино, -NHR10, 1-пирролидинил или тетразолил; либо
(3) пиридин или пиридинил, одно- или двузамещенный группами, независимо выбранными из числа следующих: атом галогена, метил, гидрокси, нитро, циано, карбокси, -O(C1-4-алкил), -O(CH2C6H5), -COO(C1-4-алкил), амино или диметиламино;
либо
(4) NR11, где R11 соответствует приведенному ниже обозначению или является 7-индазолилом, содержащим в положении N-1 группу R10;
R3 представляет собой атом водорода, C1-6-алкил, C3-6-циклоалкил, фенил или фенил, одно- или двузамещенный независимыми атомами галогена;
R4 обозначает C3-6-алкил, C3-6-циклоалкил, C3-6-алкенил, фенил,-(CH2)pCN или -(CH2)pCOO(C1-4-алкил); а
R5 независимо представляет собой C3-6-алкил, C3-6-циклоалкил, C3-6-aлкенил, бензил, фенил или фенил, одно- или двузамещенный группами, независимо выбранными из числа следующих: C1-3-алкил, циано, гидрокси, диметиламино, -O(C1-4-алкил), -O(CH2C6H5), -NH(C1-4-aлкил), -COO(C1-4-алкил), -N(C1-4-алкил)2, пирролидино, морфолино или атом галогена; либо
R4обозначает C1-2-алкил, а
R5 является фенилом, замещенным по своему положению 2 или 4 атомом хлора, метилом, метокси или метоксикарбонилом;
R6 соответствует атому водорода или метилу;
R7 представляет собой атом водорода, гидрокси, атом фтора, диметиламино, -O(C1-4-алкил) или -O(CH2C6H5);
R8 является группой -(CH2)cCOOH;
R9 обозначает метил, атом хлора, нитро, гидрокси, метокси или NHR10;
R10 представляет собой атом водорода, ацетил, C1-4-алкил, -SO3H, -SO2CH3, -SO2CF3 или -SO2C6H5, C1-4-алкоксикарбонил;
R11 либо соответствует фенилу или фенилу, одно- или двузамещенному группами, независимо выбранными из числа следующих: атом фтора, трифторметокси, C1-4-алкилтио, -(CH2)CCOOH, -(CH2)CCOO(C1-4-алкил), -(CH2)CSH3, -(CH2)CSOCH3, -(CH2)CSO2CH3, -(CH2)CCONH2, -SCH2COOH, -CONH(SO2CH3) -CONH(SO2CF3), -(CH2)CN(C1-4-алкил)2, -(CH2)CNH(SO2CF3), -(CH2)CN(SO2CF3)- (C1-4-алкил), -(CH2)CSO2NHCO(C1-4-алкил), -(CH2)CSO2N(C1-4-алкил)CO- (C1-4-алкил), -(CH2)CCONHSO2(C1-4-алкил), -(CH2)CCON- (C1-4-алкил)SO2(C1-4-алкил), -(CH2)COR12, -(CH2)CNHR10 или фенил, однозамещенный -(CH2)C(тетразолилом), -(CH2)C(карбоксамидотетразолилом) или -(CH2)C(пирролидинилом);
либо обозначает пиридин или пиридинил, одно- или двузамещенный группами, независимо выбранными из числа следующих: атом галогена, метил, гидрокси, нитро, циано, карбокси, -O(C1-4-алкил), амино, диметиламино, -NHR10;
R12 представляет собой атом водорода, C1-6-алкил, C3-6-циклоалкил, -CH2C6H5, -CH2COOH, -CH2CONH2, -CH2CONH(C1-4-алкил), -CH2CON(C1-4-алкил)2 или группу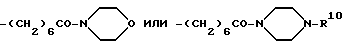
z равно 1 или 2;
n равно 1 или 2;
p равно целому числу от 1 до 4;
b равно целому числу от 0 до 3; а
c равно 0 или 1.
в том случае, если радикал R1 представляет собой группу, отвечающую формуле (II), к числу указанных групп принадлежат такие, в которых R6 является атомом водорода или, более предпочтительно, - метилом, R7 обозначает атом водорода, гидроксил или атом фтора, а
n = 1.
В том случае, если R1 представляет собой радикал NR4R5, к числу приемлемых указанных радикалов относятся такие, в которых R4 является C3-6-алкилом, таким как пропил или изопропил, циклогексил или фенил, а R5 обозначает C3-6-алкил, бензил или фенил, возможно замещенный по пара-положению гидрокси, диметиламино метокси, атомом фтора, пирролидино или морфолино-группами. Среди указанных радикалов R1 наиболее приемлемыми являются такие, в которых
R4 представляет собой пропил и, в особенности, изопропил, а
R5 обозначает фенил, возможно замещенный по пара-положению группами, выбранными из числа следующих: гидрокси, метокси, диметиламино, атом фтора или морфолино-группа.
К числу наиболее приемлемых радикалов R1 относятся такие, в которых R1 соответствует формуле (II), где R6 представляет собой метил, n = 1, а R7 обозначает атом водорода, гидрокси, атом фтора или метокси; либо
R1 является радикалом NR4R5, где R4 представляет собой пропил или изопропил, а R5 обозначает фенил, возможно замещенный по пара-положению группами, выбранными из числа следующих: гидрокси, метокси, атом фтора, диметиламино, пирролидино или морфолино-группа.
В том случае, если R2 принадлежит к числу следующих групп: индол, индолин, бензофуран, бензотиофен, хинолин или 4-оксобензопиран, возможный заместитель R9 обычно является группой, выбранной из числа следующих: атом водорода, метил, метокси, гидрокси, нитро или амино, а также, при необходимости, возможный заместитель при атоме азота (R8) представляет собой остаток -CH2CO2H.
В том случае, если R2 обозначает возможно замещенную фенильную группу, указанная группа соответствует фенилу, возможно замещенному одной, либо двумя одинаковыми или разными группами, выбранными из числа следующих: атом хлора, атом фтора, амино, гидрокси-группа или карбоксил.
В том случае, если R2 является группой NHR11, остаток R11 обычно представляет собой: фенил (возможно замещенный атомом фтора, гидрокси, амино, диметиламино, трифторметилсульфониламино, C1-4-алкоксикарбонилом, карбокси, 1Н-тетразол-5-илом, ацетиламино или остатком OR12,
где R12 обозначает атом водорода, метил, бензил, а также группы CH2CO2H, CH2ONH2, CH2CONHCH3, CH2CON(CH3)2,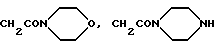
либо
7-индазолильную группу, в которой заместитель при N-1 (R10) является атомом водорода.
В том случае, если R11 представляет собой однозамещенную фенильную группу, соответствующий заместитель обычно находится в мета-положении.
К числу особо приемлемых радикалов R2 относятся индол, бензофуран, тиофен, бензотиофен, индолин, хинолин, 4-оксобензопиран, возможно замещенная фенильная группа или остаток NHR11. Обычно радикал R2 выбирают из числа следующих групп: индол, индолин или бензофуран, возможно замещенная фенильная группа или группа NHR11. В наиболее приемлемых случаях радикал R2 представляет собой индол, возможно замещенный фенил, либо остаток NHR11.
В том случае, если R3 обозначает C1-6-алкил, примерами приемлемых групп служат метил, этил, пропил, изопропил, бутил, т-бутил или изоамил.
В том случае, если R3 соответствует C3-6-циклoaлкилу, примерами приемлемых групп являются циклопропил, циклопентил или циклогексил.
В том случае, если R3 представляет собой фенил, однозамещенный или независимым образом двузамещенный атомами галогена, к числу приемлемых групп относятся такие, в которых соответствующий галогеновый заместитель является атомом фтора, например, 2-фторфенил или 4-фторфенил.
Примерами особо приемлемых групп R3 служат атом водорода, метил, циклогексил, 2-фторфенил или фенил, в наиболее приемлемом случае - фенил.
К числу особо приемлемых соединений, предусмотренных настоящим изобретением, принадлежат такие, в которых R1 обозначает:
группу, описываемую формулой (II), где R6 соответствует метилу, n = 1, а R7 представляет собой атом водорода, атом фтора, гидрокси или метокси-группу, либо в более приемлемом случае остаток NR4R5, где R4 является пропилом или изопропилом, а R5 обозначает фенил, возможно замещенный по пара-положению группами, выбранными из числа следующих: гидрокси, метокси, атом фтора, диметиламино или морфолино-группу; R2 представляет собой фенил (возможно независимым образом одно- или двузамещенный группами, выбранными из числа следующих: атом хлора, атом фтора, гидрокси-группа, амин или карбокси), остаток NHR11,
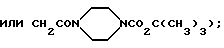
где R11 обозначает фенил (возможно замещенный амино, диметиламино, трифторметилсульфониламино, карбоксигруппой, 1Н-тетразол-5-илом, ацетиламино или группой OR12, где R12 является атомом водорода, метилом, бензилом, либо остатком CH2CO2H, CH2CONH2,
CH2CONHCH3, CH2CON(CH3)2,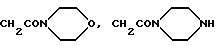
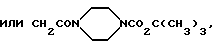
причем указанный заместитель предпочтительно находится в мета-положении); либо индол, в котором соответствующий атом азота возможно замещен остатком CH2CO2H, а соответствующее бензольное кольцо возможно замещено атомом хлора, метилом, метокси, нитро, гидрокси или амино-группой;
R3 представляет собой атом водорода, метил, циклогексил, 2-фторфенил или фенил, либо, в более предпочтительном случае, 2-фторфенил или фенил;
X соответствует атому фтора; а
z = 1; либо в более предпочтительном случае
X обозначает атом водорода.
Среди соединений, предусмотренных настоящим изобретением, особо интересный класс соединений, проявляющих высокое и избирательное сродство к рецепторам ХЦК-А, а также исключительную эффективность, получается, когда радикал R2 представляет собой индольную группу. Предпочтительная группа соединений указанного класса включает в себя соединения, в которых вышеупомянутая индольная группа замещена по соответствующему атому азота остатком CH2CO2H, либо, в более предпочтительном случае, рассматриваемый атом азота остается незамещенным, а бензольное кольцо указанного индольного радикала замещено группой, выбранной из числа следующих: атом хлора, метил, метокси, нитро, гидрокси или амино-группа.
Наиболее предпочтительным соединением, предусмотренным настоящим изобретением, является (1-[изопропил-(4-метоксифенил) карбамоилметил]-2,4-диоксо-5-фенил-2,3,4,5-тетрагидро-1Н-бензо [b] [1,4] диазепин-3-ил}-амид 1H-индол-2-карбоновой кислоты, а также его энантиомеры.
В настоящем описании под термином "алкил" обычно подразумевают алифатические изомеры как линейных, так и разветвленных цепей соответствующего алкила. Например, понятие "C1-6-алкил" охватывает метил, этил, н-пропил, изопропил, н-бутил, изобутил, трет-бутил, н-пентил и т.д.
Под термином "циклоалкил" в настоящем описании понимают алициклические изомеры соответствующего алкила. Например, понятие "C3-6-циклоалкил" предусматривает такие группы, как циклопропил, циклопентил и циклогексил.
Термин "атом галогена" обозначает атом фтора, хлора, брома или иода.
Понятие "тетразол", используемое в качестве обозначения группы или части группы, подразумевает (1Н)-тетразол-5-ильный остаток и его таутомеры.
Специалистам в данной области понятно, что в соединениях, отвечающих формуле (I), имеются стереоцентры. В соответствии с этим, настоящее изобретение охватывает все возможные стереоизомеры и геометрические изомеры, описываемые формулой (I), а также включает в себя не только рацемические соединения, но и оптически активные изомеры. В том случае, если соединение, отвечающее формуле (I), желают получить в форме чистого энантиомера, его можно выделить либо с помощью разделения конечного продукта, либо путем стереоспецифического синтеза, используя в качестве исходного вещества чистый изомер или любое приемлемое промежуточное соединение. Разделение конечного продукта, промежуточного соединения или исходного вещества можно осуществлять с помощью любого известного приемлемого способа (Eliei E.L. Etereo chem. Carb Comp, 1962, Wilen S.H., Tables Resolv. Agents). Кроме того, в тех случаях, если возможны таутомеры соединений, отвечающих формуле (I), настоящее изобретение охватывает все таутомерные формы указанных соединений.
Специалистам в данной области следует также отметить, что соединения, предусмотренные настоящим изобретением, могут быть использованы также в форме их фармацевтически приемлемых солей или сольватов. К числу физиологически приемлемых солей соединений, предусмотренных формулой (I), относятся традиционные соли, образуемые фармацевтически приемлемыми неорганическими или органическими кислотами, а также четвертичные соли аммония, получаемые в результате добавления кислоты. Более конкретными примерами приемлемых солей служат соли соляной, бромистоводородной, серной, фосфорной, азотной, перхлорной, фумаровой, уксусной, пропионовой, янтарной, гликолиевой, муравьиной, молочной, яблочной, винной, лимонной, памовой, малоновой, гидроксималеиновой, фенилуксусной, глутаминовой, бензойной, салициловой, фумаровой, толуолсульфоновой, метансульфоновой, нафталин-2-сульфоновой, бензолсульфоновой и т. п. кислот. Другие, не являющиеся фармацевтически приемлемыми кислоты, такие как щавелевая, могут быть использованы при получении солей промежуточных соединений в ходе синтеза предусмотренных настоящим изобретением соединений, а также их фармацевтически приемлемых солей. Используемое далее понятие "соединение, предусмотренное настоящим изобретением", охватывает как соединения, отвечающие формуле (I), так и их фармацевтически приемлемые соли и сольваты.
Предусмотренные настоящим изобретением соединения проявляют агонистичную активность по отношению к ХЦК-А и могут рассматриваться в качестве полных или частичных агонистов холецистокинина, поскольку указанные соединения связываются с рецепторами ХЦК-А и либо полностью, либо частично стимулируют сокращение желчного пузыря и/или снижают потребление пищи в понимании этого термина применительно к животным.
В качестве агонистов рецепторов ХЦК-А соединения, предусмотренные настоящим изобретением, являются удобными противовозбудительными агентами, пригодными для использования при лечении ожирения, а также близких заболеваний, таких как диабет или гипертензия. Кроме того, описанные в настоящем изобретении соединения предполагают новый подход для вызова чувства насыщения, используемый при регуляции аппетита и корректировке приема пищи у млекопитающих, в особенности - у человека, предназначенный для регуляции аппетита, лечения ожирения и продолжительного снижения веса.
В дополнение к этому, некоторые соединения, предусмотренные настоящим изобретением, также могут проявлять некоторую антагонистичную активность по отношению к определенным сайт-специфическим рецепторам ХЦК-Б и гастрина, что продемонстрировано по их способности подавлять вызванное ХЦК-4 сокращение изолированного продольного мускульно-миентерического сплетения подвздошной кишки морских свинок, а также вызванное пентагастрином выделение кислоты изолированной слизистой желудка крыс (J.pharmac, 1992, 106: 275-282, ibid, 1985, 86: 677-684).
Степень сродства предусмотренных настоящих изобретением соединений по отношению к рецепторам ХЦК-А и ХЦК-Б можно определить с помощью известных традиционных способов (J.pharm. Exp. Ther 1992, 261:1056-1063).
Способность предусмотренных настоящим изобретением соединений подавлять выделение желудочного сока, в частности вызванное пентагастрином выделение кислоты, можно определять у находящихся в сознании крыс с желудочными фистулами (J.Physiol, 1977, 267:191-194).
Конкретно, настоящее изобретение предусматривает соединение, отвечающее формуле (I), либо фармацевтически приемлемую соль или сольват указанного соединения для использования в терапии, в частности - в медицине.
Еще один аспект настоящего изобретения предусматривает использование соединения, отвечающего формуле (I), либо фармацевтически приемлемой соли или сольвата указанного соединения при производстве медикамента, предназначенного для лечения заболеваний, при которых модификация действия ХЦК и/или гастрина имеет благоприятный терапевтический эффект.
Еще один аспект настоящего изобретения предусматривает способ лечения млекопитающих, включая человека, особенно лечения заболеваний, при которых модификация действия ХЦК и/или гастрина имеет благоприятный терапевтический эффект, причем указанный способ предполагает применение больному терапевтически активного количества соединения, отвечающего формуле (I), либо фармацевтически приемлемой соли или сольвата указанного соединения.
Специалистам в данной области следует отметить, что в настоящем описании понятие "лечение" охватывает профилактику, а также лечение развившихся заболеваний или симптомов. Кроме того, следует обратить внимание на то, что необходимое для лечения количество предусмотренного настоящим изобретением соединения должно варьировать в зависимости от заболевания, а также от возраста и состояния пациента, и должно определяться исключительно лечащим врачом или ветеринаром. Вместе с тем, дозы, применяемые при лечении взрослого человека, обычно должны находиться в пределах 0,02 - 5000 мг в день, в том числе 1 - 1500 мг в день. Желаемая доза может применяться традиционным образом в виде одной дозы или нескольких доз, вводимых через определенные промежутки времени, например в виде двух, трех, четырех или большего числа субдоз в день.
Несмотря на то, что предусмотренные настоящим изобретением соединения можно терапевтически применять в химически чистом виде, предпочтительно использовать указанный активный ингредиент в форме фармацевтического состава. В соответствии с этим, настоящее изобретение предусматривает также фармацевтический состав, включающий в себя соединение, отвечающее формуле (I), либо фармацевтически приемлемую соль указанного соединения, вместе с одним или большим числом фармацевтически приемлемых носителей, а также, возможно, другими терапевтическими и/или профилактическими ингредиентами. Указанные носители должны быть "приемлемы" в смысле их совместимости с другими ингредиентами соответствующего состава, а также безвредности по отношению к их реципиенту.
К числу составов, предусмотренных настоящим изобретением, принадлежат такие составы, которые специально предназначены для перорального, трансбуккального, парэнтерального, имплантационного или ректального введение, вместе с тем, предпочтительным является пероральное введение. При трансбуккальном введении указанный состав может иметь форму таблеток полученных традиционным образом. Таблетки и капсулы, предназначенные для перорального введения, могут включать в себя традиционные эксципиенты, такие как связывающие агенты (например, сироп, гуммиарабик, желатин, сорбитол, трагакант, крахмальный клей или поливинилпирролидон), наполнители (например, лактозу, сахар, микрокристаллическую целлюлозу, кукурузный крахмал, фосфат кальция или сорбитол), способствующие скольжению агенты (например, стеарат магния, стеариновая кислота, тальк, полиэтилен гликоль или окись кремния), дезинтегрирующие агенты (например, картофельный крахмал или крахмальный гликоллат натрия) или смачивающие агенты, такие как лаурил сульфат натрия. Указанные таблетки могут быть покрыты оболочкой в соответствии с хорошо известными способами.
В альтернативном случае предусмотренные настоящим изобретением соединения могут быть включены в жидкие составы, предназначенные для перорального введения, такие как, например, водные или масляные суспензии, растворы, эмульсии, сиропы или эликсиры. Кроме того, составы, включающие в себя указанные соединения, могут представлять собой сухой продукт, предназначенный для смешивания с водой или другими приемлемыми носителями перед употреблением. Указанные жидкие препараты могут содержать традиционные добавки, такие как агенты, способствующие образованию суспензий (в том числе, сорбитольный сироп, метил целлюлоза, глюкозо/сахарный сироп, желатин, гидроксиэтилцеллюлоза, карбоксиметилцеллюлоза, алюминий стеаратный гель или гидрогенированные пищевые жиры); агенты, способствующие образованию эмульсий (в том числе лецитин, моно-олеат сорбитана или гуммиарабик); не водные носители, которые могут включать в себя пищевые масла (в том числе миндальное масло, фракционированное кокосовое масло, масляные эфиры, пропилен гликоль или этиловый спирт); а также консерванты (в том числе метил или пропил п-гидроксибензоаты или сорбиновая кислота). Указанные препараты могут быть составлены также в виде суппозиториев, т.е. могут содержать традиционные основы суппозиториев, такие как масло какао или другие глицериды.
Кроме того, составы, предусмотренные настоящим изобретением, могут быть предназначены для парэнтерального введения с помощью инъекции или продолжительного вливания. Составы, предназначенные для инъекций, могут иметь форму суспензий, растворов или эмульсий в масляных или водных носителях, а также могут включать в себя дополнительные агенты, такие как способствующие образованию суспензий, стабилизирующие и/или диспергирующие агенты. В альтернативном случае указанный активный ингредиент может представлять собой порошок, предназначенный для смешивания с приемлемым носителем (например, со стерильной, не содержащей пирогена водой) перед употреблением.
Предусмотренные настоящим изобретением составы могут быть созданы в форме препарата с задержанным действием. Указанные долгодействующие составы могут вводиться посредством имплантации (например, подкожно или внутримышечно), либо с помощью внутримышечной инъекции. В соответствии с этим, предусмотренные настоящим изобретением соединения могут быть присоединены к приемлемым полимерными или гидрофобными веществами (такими, как например эмульсия в приемлемом масле), ионообменными смолами, либо их слабо растворимыми производными, в том числе, например, слабо растворимыми солями.
Предусмотренные настоящим изобретением составы могут содержать от 0,1 до 99% соответствующего активного ингредиента, обычно - от 30 до 95% в случае таблеток и капсул, а также от 3 до 50% в случае жидких препаратов.
Соединения, отвечающие общей формуле (I), можно получить с помощью описанных ниже общих способов. Если это не оговорено особо, использованные в нижеследующем описании обозначения X и R1-R12 соответствуют таковым в случае соединений, предусмотренных формулой (I).
Соединения, отвечающие общей формуле (I), а также их соли могут быть получены с помощью описанных ниже общих способов. Если это не оговорено особо, использованные в нижеследующем описании обозначения X и R1-R12 соответствуют таковым в случае соединений, предусмотренных формулой (I).
В соответствии с первым общим способом А, соединения, отвечающие формуле (I), можно получить, приводя амин, описываемый формулой (III),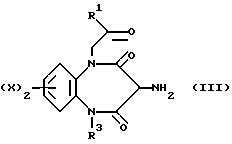
где R1, R2, R3, X и z соответствуют обозначениям в формуле (I), во взаимодействие с соединением (IV),
R11Y
в котором
Y представляет собой группу -NCO, HNCOCl или NHCORa где Ra является нитрозамещеной феноксигруппой или 1-имидазольной группой.
Указанную реакцию обычно проводят в присутствии приемлемого растворителя, такого как галогенированный углеводород (например, дихлорметан), эфир (например, тетрагидрофуран) или нитрил (например, ацетонитрил), либо в присутствии смеси указанных растворителей при температуре в пределах от 0 до 80oC.
Соединения, отвечающие формуле (IV), в которой Y обозначает группу -NCO, можно купить, либо получить, приводя амины, описываемые формулой H2N-R11, во взаимодействие с фосгеном или трифосгеном в приемлемом растворителе, таком как метиленхлорид. Соединения, отвечающие формуле (IV), где Y представляет собой группу NHCOCl, также можно получить, приводя амины, описываемые формулой H2N-R11, во взаимодействие с фосгеном или трифосгеном в приемлемом растворителе, таком как метиленхлорид. Соединения, отвечающие формуле (IV), в которой Y обозначает остаток NHCORa, а радикал Ra является 1-имидазольной группой, получают, обрабатывая амины, описываемые формулой H2N-R11, карбонил диимидазолом в приемлемом растворителе (дихлорметане, эфире, тетрагидрофуране) при температуре в пределах от 0 до 80oC (обычно - при комнатной температуре). Соединения, отвечающие формуле (IV), где Y представляет собой остаток NHCORa, a радикал Ra является нитрозамещенной феноксигруппой, получают, обрабатывая амины H2N-R11 соответствующим хлороформатом RaCOCl в присутствии основания (пиридина, триэтиламина) в приемлемом растворителе (дихлорметане) при температуре в пределах от 0 до 50oC.
В соответствии с еще одним общим способом Б, соединения, предусмотренные формулой (I), можно получить, приводя промежуточное соединение, описываемое формулой (V),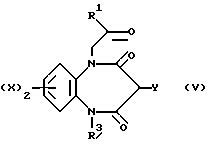
в которой Y представляет собой группу -NCO, HNCOCl или NHCORa, где Ra является нитрозамещеной феноксигруппой или 1-имидазольной группой,
во взаимодействие с амином (VI),
H2N-R11
возможно в присутствии основания, такого как третичный амин (например, триэтиламин).
Указанную реакцию обычно осуществляют в приемлемом растворителе, таком как галогенированный углеводород (например, дихлорметан), эфир (например, тетрагидрофуран) или амид (например, N, N-диметилформамид), возможно при температуре в пределах от комнатной до температуры дефлегмации соответствующего растворителя.
Соединения, отвечающие формуле (V), обычно получают in situ из амина (III).
В конкретном случае способа Б, если Y представляет собой остаток NHCORa, где Ra является 1-имидазольной группой, соответствующий имидазол (V) можно получить in situ, для чего соответствующий амин, предусмотренный формулой (VI), смешивают с соединением, отвечающим формуле (III)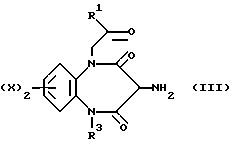
в вышеописанных условиях в присутствии карбонилдиимидазола.
В случае способа Б, если Y представляет собой остаток NHCORa, где Ra является нитрозамещенной феноксигруппой, указанную реакцию взаимодействия с первичным амином (VI) предпочтительно осуществляют в присутствии основания, такого как третичный амин, например триэтиламин.
В случае способа Б, если Y является изоцианатной группой -N=C=O, указанную реакцию взаимодействия с первичным амином (VI) предпочтительно осуществляют в присутствии апротонного растворителя, такого как галогенированый углеводород, например метилен хлорид. Указанный изоцианат обычно получают in situ перед добавлением первичного амина (VI).
Соединения, отвечающие формуле (V), в которой Ra представляет собой возможно замещенную феноксигруппу, могут быть получены из первичного амина (III) с помощью реакции с соответствующим нитрозамещенным фенил хлороформатом в присутствии основания, такого как пиридин. Указанную реакцию можно проводить в растворителе, таком как галогенированный углеводород (например дихлорметан) при температуре от 0 до 50oC.
Соединения, предусмотренные формулой (V), в которой Ra обозначает 1-имидазольную группу, можно получить, приводя соединение, описываемое формулой (III) во взаимодействие с карбодиимидазолом в присутствии приемлемого растворителя, такого как галогенированный углеводород (например дихлорметан) или эфир (например тетрагидрофуран) при температуре от 0 до 80oC (обычно - при комнатной температуре).
Соединения, отвечающие формуле (V), где радикал Y является изоцианатной группой -N= C=O или карбамоил хлоридом -NHCOCl, могут быть получены из первичного амина (III) с помощью реакции с фосгеном (COCl2) или трифосгеном в приемлемом растворителе, таком как метилен хлорид.
В соответствии с еще одним общим способом В, соединения, предусмотренные формулой (I), также можно получить, приводя соединение, описываемое формулой (VII),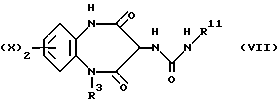
во взаимодействие с ацетилбромидом или хлоридом, отвечающим формуле (VIII),
R1COCH2hal (VIII)
где hal обозначает атом хлора или брома.
Указанную реакцию обычно осуществляют, обрабатывая соединение, описываемое формулой (VII), сильным основанием, таким как гидрид натрия, в полярном апротонном растворителе, таком как N, N-диметилформамид, после чего приводят его во взаимодействие с вышеупомянутым ацетил галогенидом (VIII).
Указанный ацетил галогенид (VIII) получают в результате реакции между амином R1-H и соответствующим галогенированным ацетил бромидом в дихлорметане при температуре 0oC в присутствии приемлемого основания, такого как триэтиламин.
Амин R1-H, где R1 представляет собой группу R4R5, можно получить с помощью восстановительного алкилирования амина H2N-R5 с приемлемым альдегидом или кетоном.
В соответствии с общим способом Г, соединения, предусмотренные общей формулой (I), также можно получить в результате реакции между промежуточным соединением, описываемым формулой (III), с кислотами, отвечающими формуле (IX), как это описано ниже.
HOOC-R2 (IX)
Указанную реакцию между промежуточными соединениями, описываемыми формулой (III), с кислотой, отвечающей формуле (IX), можно осуществить в присутствии приемлемого дегидратирующего агента, такого как дициклогексилкарбодиимид (ДЦК), 1-(3-диметиламинопропил)- 3-этилкарбодиимид гидрохлорид (ЭДХ) или бис(2-оксо-3-оксазолидинил)фосфин хлорид (БОФ).
В альтернативном случае соединения, предусмотренные общей формулой (I), можно получить в результате реакции между промежуточным соединением, описываемым формулой (III), с активированными производными кислот (IX), такими как хлорид кислоты или его ангидрид, включая смешанные ангидриды.
В число предпочтительных растворителей в случае общего способа Г входят N, N-диметилформамид или дихлорметан. Предпочтительная температура находится в пределах от 0 до 60oC. К числу предпочтительных оснований в случае указанной реакции относятся триэтиламин или N,N-диметиламинопирид (ДАМП).
В соответствии с еще одним общим способом Д, предусмотренные настоящим изобретением соединения можно превращать в другие соединения, соответствующие настоящему изобретению. Так, например, соединения, отвечающие формуле (I), в которой R8 представляет собой группу (CH2)bCO2H, можно получить в результате реакции между соединением, описываемым формулой (I), в которой R8 является атомом водорода, и соединением Br(CH2)bCOOR*, где R* обозначает C1-4-алкил, в присутствии сильного основания, такого как гидрид натрия, после чего с помощью традиционных методов (т.е. кислотного или основного гидролиза) удаляют остаток, защищающий соответствующую карбоксильную группу.
Соединения, отвечающие формуле (III), могут быть получены в результате восстановления соединений, предусмотренных формулой (X),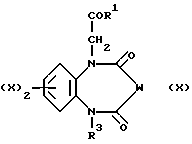
в которой W представляет собой остаток CH-N3 или C=N-NHPh.
Соединения, описываемые формулой (X), в которой W обозначает группу CH-N3, могут быть восстановлены до соединения, отвечающего формуле (III), посредством гидрогенирования в присутствии приемлемого катализатора, такого как 5-10%-ный палладий на угольной или карбонатно-кальциевой основе, либо оксид платины (IV). Указанную реакцию обычно осуществляют в присутствии растворителя, такого как спирт (например, этанол), эфир (например, этил ацетат) или уксусная кислота.
Соединения, отвечающие формуле (X), в которой W обозначает группу C=N-NHPh, могут быть восстановлены до соединения, описываемого формулой (III), в результате реакции с цинком или уксусной кислотой. Указанную реакцию можно проводить при температуре в пределах от 0 до 50oC.
Соединения, предусмотренные формулой (X), в которой W является группой CH-N3, могут быть получены из соединений, отвечающих формуле (X), в которых W представляет собой группу CH2 с помощью обработки сильным основанием, таким как гидрид натрия или трет-бутоксид калия, а затем - азидом три-изопропил бензолсульфонила или ди-третбутоксиазидодикарбоксилатом. Указанную реакцию обычно осуществляют в растворителе, таком как эфир (например, тетрагидрофуран), при температуре в пределах от -78 до 20oC.
Соединения, отвечающие формуле (X), в которой W обозначает группу C=N-NHPh или CH2, можно получить в результате реакции между орто-фенилендиамином (XI) и диацид хлоридом (XII),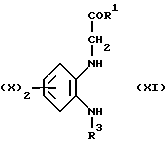
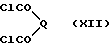
где Q представляет собой группу C=N-NHPh или CH2, в приемлемом растворителе, таком как эфир, например, тетрагидрофуран.
Соединение, предусмотренное формулой (XII), в которой Q является группой C=N-NHPh, можно получить в результате реакции между кетомалоновой кислотой и фенил гидразоном, после чего проводят обработку пентахлоридом фосфора.
Соединения, описываемые формулой (XI), либо являются известными, либо могут быть получены с помощью аналогичных способов. Так, например, соединение, отвечающее формуле (XI), можно получить путем алкилирования амина (XIII).
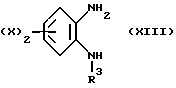
Так, амин (XIII) можно привести во взаимодействие с соединением R1COCH2hal, где hal обозначает атом хлора или брома, возможно в присутствии иодида натрия, в растворителе, таком как N,N-диметилформамид.
Альтернативный способ получения промежуточного соединения, описываемого формулой (III), как это показано ниже, состоит в обработке промежуточного соединения, отвечающего формуле (XIV),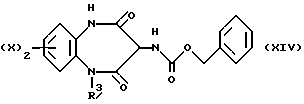
гидридом натрия, после чего добавляют ацетил галогенид (VIII) в приемлемом растворителе, таком как N,N-диметилформамид, при температуре 0oC с образованием защищенного промежуточного соединения, соответствующего формуле (XV).
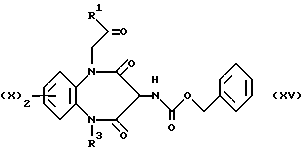
Промежуточное соединение (XV) превращают в искомый амин (III) путей каталитического гидрирования (40-60 пси) с использованием приемлемого катализатора, такого как 5-10%-ный палладиево-угольный катализатор, при комнатной температуре в приемлемом растворителе, таком как метанол, этанол, этил ацетат, хлороформ или уксусная кислота. В альтернативном случае промежуточное соединение (XVI)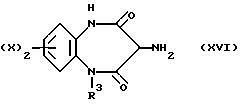
можно превратить в амин (III), обрабатывая бромистоводородной кислотой в метилен хлориде.
Промежуточное соединение (XIV) получают из промежуточного соединения, описываемого формулой (XVI), в результате реакции с бензилхлороформатом в дихлорметане, используя в качестве основания триэтиламин. Указанную реакцию обычно проводят при комнатной температуре.
Промежуточное соединение (XVI) получают из фенилен диамина (XIII) следующим образом.
Диамин (XIII) приводят во взаимодействие с п-метиоксилбензоилхлоридом, после чего образующийся при этом амид восстанавливают литий-алюминий гидридом, с получением N-защищенного диамина (XVII).
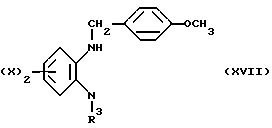
Соединение (XVII) приводят во взаимодействие с диацид хлоридом (XII, где Q обозначает группу C= N-NHPh), после чего образовавшийся продукт восстанавливают цинком или уксусной кислотой с получением амина (XVIII).
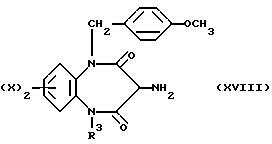
Соединение, отвечающее формуле (XVIII), может быть превращено в искомое соединение (XVI) с помощью реакции с Ce(NO2)6NH4 (церий-аммониевым нитратом).
Соединения, предусмотренные формулой (I), содержат по меньшей мере один асимметричный атом углерода, а именно - атом углерода в составе диазепинового кольца, к которому присоединен замещенный остаток мочевины. Конкретные энантиомеры соединений, отвечающих формуле (I), могут быть получены посредством разделения соответствующих рацемических соединений с помощью традиционных способов, таких как хиральная ЖХВД. В альтернативном случае искомый энантиомер можно синтезировать из соответствующего энантиомерного амина, описываемого формулой (III), с использованием любого из вышеперечисленных способов, предназначенных для получения соединений, предусмотренных формулой (I), из амина (III). Указанные энантиомеры амина (III) можно выделить из соответствующего рацемического амина (III) с помощью традиционных способов, таких как образование соли с приемлемой оптически активной кислотой, либо с помощью препаративной хиральной ЖХВД.
Примеры
Приведенные ниже примеры призваны иллюстрировать синтез некоторых конкретных соединений, предусмотренных настоящим изобретением, а также дополнительно продемонстрировать конкретные варианты реализации основных способов А - Д. В соответствии с этим, следующий ниже раздел "Примеры" не ограничивает каким бы то ни было образом область рассматриваемого изобретения.
Основные методы
Если это не оговорено особо, все исходные вещества получали из коммерческих источников и использовали без дополнительной очистки. Использованы следующие сокращенные названия растворителей и реагентов: ТГФ (тетрагидрофуран), ДМСО (диметилсульфоксид), ДХМ (дихлорметан), ТФУ (трифторуксусная кислота), ДМФ (диметилформамид), КДИ (1,1-карбодиимидазол), и БХФ (изобутилхлороформат), ГОС (гидроксисукцинимид), ГОБТ (N-гидроксибензотриазол), ЭДХ (1-(3-диметиламинопропил)-3-этилкарбодиимида гидрохлорид), БОФ (бис(2-оксо-3-оксазолидинил) фосфиновый хлорид), БОК (трет-бутилоксикарбонил), БзОК (бензилоксикарбонил).
Спектры 1H ЯМР регистрировали на аппаратуре либо Varian VXR-300, либо Varian Unity-300. Величину химических сдвигов выражали в виде частей на миллион (ppm, единицы d). Константы связывания измеряли в Герцах (Гц). Характеристики возбуждения обозначали следующим образом: s - синглет, d - дуплет, t - триплет, q - квадруплет, m - множественное, b - широкое.
Низкоразрешающие спектры масс (МС) регистрировали с помощью спектрометров JOEL JMS-AX505HA, JOEL SX-102 или SCIEX-ARIiii. Все спектры масс получали в режиме положительных ионов посредством ионизации распыленными электронами (ESI), химической ионизации (CI), электронных ударов (EI) или бомбардировки быстрыми атомами (FAB). Инфракрасные (ИК) спектры получали с помощью спектрометра Nicolet 510 FT-IR с использованием 1 мм ячеек NaCl. Вращения регистрировали с помощью поляриметра Perkin-Elmer 241. Все реакции отслеживали с помощью тонкослойной хроматографии на 0,25 мм пластинах силикагеля Е. Merck (60F-254), выявляли с помощью УФ света в 7%-ной спиртовой фосфомолибденовой кислоте или растворе п-анисальдегида. Импульсную хроматографию на колонке проводили на силикагеле (сито 230-400, Merck).
Очистку продуктов осуществляли с помощью препаративной обратно-фазной жидкостной хроматографии под высоким давлением (ОФ-ЖХВД) с использованием установки Waters Model 3000 Delta Prep, оборудованной радиальным компрессорным картриджем Deltapack (C18, 300 А, 15 м, 47 мм • 300 мм). Во всех случаях использовали линейные градиенты, скорость потока равнялась 100 мл/мин (t0 = 5,0 мин). Все растворители содержали 0,1% трифторуксусной кислоты (ТФУ). Аналитическую чистоту достигали с помощью ОФ-ЖХВД с использованием системы Waters 600E, оборудованной диодным лучевым спектрометром Waters 990 (t в пределах 200-400 нМ). Стационарная фаза представляла собой колонку Vydac C18 (5 м, 4,5 мм • 250 мм). Скорость потока составляла от 1,0 до 1,5 мл/мин (t0 = 2,8 или 3,0 мин), система растворителей соответствовала вышеописанной. Полученные данные представляли в виде tr, время удерживания в мин (% ацетонитрила в течение времени).
С помощью описанных выше основных способов А - Д были получены следующие соединения, предусмотренные настоящим изобретением.
Пример 1
2-[2,4-Диоксо-5-фенил-3-(3-фенил-уреидо)-2,3,4,5- тетрагидробензо[b] [1,4]диазепин-1-ил]-N-изопропил-N-фенил-ацетамид.
К перемешиваемому раствору 1-(2,4-диоксо-1-фенил-2,3,4,5- тетрагидро-1H-бензо[b] [1,4] диазепин-3-ил)-3-фенил мочевины (0,100 г) в N,N-диметилформамиде (2 мл), охлажденному до температуры 3oC, добавляли при перемешивании гидрид натрия (0,0104 г; 60%-ная суспензия в минеральном масле). Указанную смесь перемешивали в течение 20 мин, после чего одной порцией добавляли 2-бром-N-изопропилN-фенил ацетамид (0,0656 г). Образовавшуюся при этом смесь перемешивали при комнатной температуре в течение ночи. Полученную грубую реакционную смесь в течение 30 мин очищали с помощью препаративной ОФ-ЖХВД с градиентной элюцией 60-72%-ным ацетонитрилом в воде с 0,1%-ным трифторуксусным буфером со скоростью 100 мл/мин. Объединяли фракции, содержащие искомое вещество, замораживали их и лиофилизировали с выходом поименованного соединения (0,0653 г) в виде белого порошка.
1H ЯМР (300 МГц, DMSO-d6): d 0,95 (d, J = 7,3 Гц, 3H); 0,98 (d, J = 7,3 Гц, 3H); 4,19 (d, J = 16,6 Гц, 3H); 4,48 (d, J = 16,9 Гц, 1H); 4,79 (m, 1H); 5,04 (d, J = 7,8 Гц, 1H); 6,87-6,92 (m, 1H); 6,95 (d, J = 7,6 Гц, 1H); 7,18-7,57 (m, 17H); 9,41 (s, 1H); MC (FAB): m/z = 562 (MH+);
ТСХ (смесь CH2Cl2 и CH3OH в соотношении 19:1); Rf = 0,19;
ОФ-ЖХВД (Vydac C-18, 25 см • 4,6 мм; 60-72%-ный CH3CN в H2O с 0,1%-ным буфером ТФУ; 30 мин; 1 мл/мин): tr = 17,5 мин (t0 = 2,5 мин);
т.пл.: 230-235oC.
Энантиомеры поименованного соединения (0,014 г) разделяли на колонке Пиркла с ковалентным (L)-фенилглицином, 25 см • 10,0 мм, с использованием в качестве изократического элюента смеси метанола и воды в соотношении 80:20 при скорости 5 мл/мин. Объединяли фракции четырех введений, соответствующие первому элюированному энантиоподу, и высушивали их при пониженном давлении с получением энантиомера 1 в виде белого порошка. Аналогичным образом объединяли фракции, соответствующие второму элюированному энантиоподу, и высушивали их при пониженном давлении с получением энантиомера 2 в виде белого порошка.
Энантиомер 1: Хиральная ЖХВД (колонка Пиркла с ковалентным (L)-фенилглицином, 25 см • 4,6 мм; изократическая смесь CH3OH и H2O в соотношении 78: 22; 1,5 мл/мин): tr = 14,5 мин (t0 = 2 мин); MC (FAB): m/z = 562,1 (MH+).
Энантиомер 2: Хиральная ЖХВД (колонка Пиркла с ковалентным (L)-фенилглицином, 25 см • 4,6 мм; изократическая смесь CH3OH и H2O в соотношении 78: 22; 1,5 мл/мин): tr = 18 мин (t0 = 2 мин); MC (FAB): m/z = 562,0 (MH+).
Пример 2
[1-(Изопропил-фенил-карбомоилметил)-2,4-диоксо-5-фенил- 2,3,4,5-тетрагидро-1H-бензо[b][1,4]диазепин-3-ил]-амид-1H-индол-2- карбоновой кислоты
К интенсивно перемешиваемому раствору 2-(3-амино-2,4-диоксо-5-фенил-2,3,4,5-тетрагидро-бензо[b][1,4] диазепин-1-ил)-N-изопропил-N-фенил ацетамида (0,116 г) в N,N-диметилформамиде (5 мл) при комнатной температуре последовательно добавляли индол-2-карбоновую кислоту (0,0423 г; 0,262 мМ), N-гидроксибензотриазол (0,0354 г) и гидрохлорид 1-(3-диметиламинопропил)-3-этил-карбодиимида (0,0503 г). Для сохранения достигнутого уровня основности (pH = 9) к указанному раствору по каплям добавляли триэтиламин (8 капель). Образовавшуюся при этом смесь перемешивали при комнатной температуре в течение 5 ч. Растворитель выпаривали в вакууме с получением желтого масла, которое очищали с помощью импульсной хроматографии на силикагеле (9 г) с использованием в качестве элюента смеси этил ацетата и гексана в соотношении 2: 3 (200 мл). Объединяли фракции, содержащие искомый продукт, и выпаривали их в вакууме с выходом поименованного соединения (0,141 г) в виде белой пены.
1H ЯМР (300 МГц, CDCl3): d 1,06 (d, J = 7,3 Гц, 3H); 1,09 (d, J = 7,3 Гц, 3H); 4,22 (d, J = 16,6 Гц, 1H); 4,40 (d, J = 16,4 Гц, 1H); 5,02 (m, 1H); 5,50 (d, J = 7 Гц, 1H); 7,02 (d, J = 8,1 Гц, 1H); 7,10-7,47 (m, 16H); 7,57 (d, J = 6,8 Гц, 1H); 7,67 (d, J = 7,8 Гц, 1H); 9,29 (br s, 1H);
MC (FAB): m/z = 586,0 (MH+);
ТСХ (смесь EtOAc и гексана в соотношении 2:3); Rf = 0,16;
ОФ-ЖХВД (Vydac C-18, 25 см • 4,6 мм; 51-60%-ный CH3CN в H2O с 1%-ным буфером ТФУ; 30 мин; 1 мл/мин): tr = 19,5 мин (t0 = 3 мин).
Пример 3
(1 -[Изопропил-(4-метоксифенил)карбомоилметил] -2,4-диоксо-5-фенил- 2,3,4,5-тетрагидро-1H-бензо[b] [1,4]диазепин-3-ил]-амид 1H-индол-2- карбоновой кислоты
К раствору 2-(3-амино-2,4-диоксо-5-фенил-2,3,4,5-тетрагидро- бензо[b] [1,4] диазепин-1-ил)-N-изопропил-N-(4-метокси-фенил)-ацетамида (500 мг) в N, N-диметилформамиде (15 мл) при комнатной температуре и перемешивании последовательно добавляли индол-2-карбоновую кислоту (174 мг), N-гидроксибензотриазол (143 мг) и гидрохлорид 1-(3-диметиламинопропил)-3-этилкарбодиимида (0,203 г). Образовавшуюся при этом смесь перемешивали при комнатной температуре в течение 18 ч. Выпаривали растворитель при пониженном давлении с получением масла, которое отбирали в этил ацетат (75 мл), промывали водой (2•30 мл), высушивали над сульфатом натрия, фильтровали и концентрировали при пониженном давлении с выходом рыжевато-коричневой пены. Полученный грубый продукт очищали в течение 30 мин с помощью препаративной ЖХВД на колонке Delta-Pak С-18, элюция линейным градиентом от 50% до 60% ацетонитрила в воде с 0,1%-ным буфером трифторуксусной кислоты со скоростью 100 мл/мин. Объединяли соответствующие фракции, замораживали и лиофилизировали с выходом ТФУ соли поименованного соединения (0,550 г) в виде белого порошка.
1H ЯМР (300 МГц, CDCl3): d 1,06 (m, 6H); 3,85 (s, 3H); 4,27 (d, J = 16,6 Гц, 1H); 4,34 (d, J = 16,6 Гц, 1H) ; 4,99 (m, 1H); 5,51 (d, J = 7,4 Гц, 1H); 6,96-7,42 (m, 17H); 7,66 (m, 2H); 9,54 (br s, 1H);
ТСХ (смесь дихлорметана и метанола в соотношении 9:1); Rf = 0,64;
МС (FAB): m/z = 616,2 (МН+) (расчетн. для C36H33N5O5 = 615,2484)
Пример 4
2-[1-(Изопропил-фенил-карбомоилметил)-2,4-диоксо-5-фенил- 2,3,4,5-тетрагидро-1H-бензо[b] [1,4] диазепин-3-ил-карбамоил]-индол- 1-ил-уксусная кислота
К интенсивно перемешиваемому раствору [1-(изопропил- фенилкарбамоилметил)-2,4-диоксо-5-фенил-2,3,4,5-тетрагидро-1H-бензо- [b] [1,4] диазепин-3-ил)-амида 1H-индол-2-карбоновой кислоты (0,101 г) в N,N-диметилформамиде (3 мл), охлажденному до температуры 3oC, добавляли гидрид натрия (0,0083 г; 60%-ная суспензия в минеральном масле). Через 20 мин добавляли т-бутилбромацетат (0,0336 г). Образовавшуюся при этом смесь перемешивали в течение 90 мин при охлаждении на ледяной бане, после чего медленно нагревали до комнатной температуры и перемешивали в течение ночи. Выпаривали растворитель при пониженном давлении с получением коричневого масла, которое растворяли в дихлорметане (30 мл) последовательно промывали насыщенным водным раствором бикарбоната натрия (20 мл) и солевым раствором (20 мл). Образовавшийся при этом раствор высушивали над сульфатом натрия, фильтровали и выпаривали с выделением желтого масла (0,142 г), которое очищали с помощью импульсной хроматографии на силикагеле (9 г) с использованием в качестве элюента смеси этил ацетата и гексана в соотношении 1:2 (200 мл). Объединяли фракции, содержащие искомое вещество, и выпаривали их с выходом трет-бутилевого эфира (2-[1-(изопропил-фенил-карбомоилметил)-2,4-диоксо-5-фенил-2,3,4,5- тетрагидро-1H-бензо[b] [1,4] диазепин-3-ил-карбамоил] -индол-1-ил) уксусной кислоты (0,092 г) в виде белой пены.
1H ЯМР (300 МГц, CDCl3): d 1,07 (d, J = 4,9 Гц, 3H); 1,09 (d, J = 4,6 Гц, 3H); 1,37 (s, 9H); 4,17 (d, J = 16,6 Гц, 1H); 4,44 (d, J = 16,9 Гц, 1H); 5,01 (m, 1H); 5,18 (d, J = 17,1 Гц, 1H); 5,24 (d, J = 18 Гц, 1H); 5,47 (d, J = 7,6 Гц, 1H); 7,01 (dd, J = 1,2; 8,3 Гц, 1H); 7,13-7,51 (m, 1H); 7,57 (d, J = 7,3 Гц, 1H); 7,67 (d, J = 7,8 Гц, 1H);
MC (FAB): m/z = 700,2 (MH+);
ТСХ (смесь EtOAc и гексана в соотношении 2:3); Rf = 0,35;
ОФ-ЖХВД (Vydac C-18, 25 см • 4,6 мм; 60-70%-ный CH3CN В H2O с 1%-ным буфером ТФУ; 30 мин; 1 мл/мин): tr = 17,5 мин (t0 = 3 мин).
К раствору трет-бутилового эфира (2-[1-(изопропил-фенил- карбомоилметил)-2,4-диоксо-5-фенил-2,3,4,5-тетрагидро-1H-бензо [b] [1,4] -диазепин-3-ил-карбамоил] -индол-1-ил)-уксусной кислоты (0,072 г) в дихлорметане (4 мл) при комнатной температуре и перемешивании постепенно добавляли трифторуксусную кислоту (1,5 мл). После того, как указанную реакционную среду перемешивали в течение 30 мин, при пониженном давлении выпаривали трифторуксусную кислоту и дихлорметан с выходом чистого стеклообразного вещества. Полученное стеклообразное вещество очищали в течение 30 мин с помощью препаративной ОФ-ЖХВД на колонке C-18, градиентная элюция 45-55%-ным ацетонитрилом в воде с 0,1%-ным буфером трифторуксусной кислоты со скоростью 100 мл/мин. Объединяли фракции, содержащие искомое вещество, замораживали их и лиофилизировали с выходом поименованного соединения (0,050 г) в виде белого порошка.
1H ЯМР (300 МГц, CDCl3): d 1,07 (d, J = 4,4 Гц, 3H); 1,10 (d, J = 4,4 Гц, 3H); 4,23 (d, J = 16,6 Гц, 1H); 4,40 (d, J = 16,6 Гц, 1H); 5,01 (m, 1H); 5,06 (s, 2H); 5,44 (d, J = 7,1 Гц, 1H); 7,03 (dd, J = 1,2; 8,1 Гц, 1H); 7,17-7,52 (m, 17H); 7,67 (d, J - 8,1 ГЦ, 1H); 7,74 (d, J = 7,1 Гц, 1H);
MC (ES): m/z = 644,2 (МН+);
ТСХ (смесь CH2Cl2 и CH3OH в соотношении 19:1); Rf = 0,15;
ОФ-ЖХВД (Vydac C-18, 25 см • 4,6 мм; 45-55%-ный CH3CN в H2O с 1%-ным буфером ТФУ; 30 мин; 1мл/мин): tr = 22 мин (t0 = 3 мин).
Пример 5
2-(2,4-диоксо-5-фенил-3-(3-[3-(1H-тетразол-5-ил)-фенил] -уреидо)- 2,3,4,5-тетрагидро-1H-бензо[b] [1,4] диазепин-1-ил]-N-изопропил-N- фенилацетамид
К интенсивно перемешиваемому раствору 2-(3-амино-2,4-диоксо-5- фенил-2,3,4,5-тетрагидро-бензо[b] [1,4] диазепин-1-ил)-N-изопропил-N- фенил-ацетамида (0,070 г) в тетрагидрофуране (3 мл) при комнатной температуре добавляли 1,1-карбонилдиимидазол (0,025 г) в виде одной порции. Образовавшуюся при этом смесь перемешивали в течение 90 мин при комнатной температуре. Одной порцией добавляли гидрохлорид 3-(2H-тетразол-5-ил)-фениламина (31,3 мг) и в течение ночи выдерживали указанную смесь при температуре дефлегмации. Фильтровали образовавшуюся при этом реакционную смесь, а соответствующий фильтрат концентрировали с выделением желтого масла. Полученное масло в течение 30 мин очищали с помощью препаративной ОФ-ЖХВД на колонке C-18, градиентная элюция 43-53%-ным ацетонитрилом в воде с 0,1%-ным буфером трифторуксусной кислоты со скоростью 100 мл/мин. Объединяли фракции, содержащие искомое вещество, замораживали их и лиофилизировали с выходом поименованного соединения в виде белого порошка (50 мг).
1H ЯМР (300 МГц, DMSO-d6): d 0,96 (d, J = 7,3 Гц, 3H); 0,98 (d, J = 7,3 Гц, 3H); 4,20 (d, J = 16,8 Гц, 1H); 4,49 (d, J = 17,1 Гц, 1H); 4,79 (m, 1H); 5,06 (d, J = 7,3 Гц, 1H); 6,98 (m, 2H); 7,24-7,55 (m, 17H); 8,17 (s, 1H); 9,44 (s, 1H);
MC (FAB): m/z = 630,2 (МН+) (расчетн. для C34H31N9O4 = 629,2502)
ТСХ (смесь CH2Cl2 и CH3OH в соотношении 9:1); Rf = 0,24;
ОФ-ЖХВД (Vydac C-18, 25 см • 4,6 мм; 43-53%-ный CH3CN в H2O с 1%-ным буфером ТФУ; 30 мин; 1 мл/мин): tr = 15 мин (t0 = 3 мин).
Пример 6
Этиловый эфир 3-{ 3-[1-(изопропил-фенил-карбамоилметил)-2,4-диоксо-5-фенил-2,3,4,5-тетрагидро-1H-бензо[b] [1,4] диазепин-3-ил] -уреидо}-бензойной кислоты
К раствору 2-(3-амино-2,4-диоксо-5-фенил-2,3,4,5-тетрагидро-бензо [b] [1,4]диазепин-1-ил)-N-изопропил-N-фенил-ацетамида (288 мг) в дихлорметане (3 мл) добавляли раствор 3-этоксикарбонил фенилизоцианата (124 мг) в дихлорметане (3 мл). Образовавшуюся при этом реакционную смесь перемешивали в течение 30 мин при комнатной температуре. В вакууме выпаривали дихлорметан, а полученный остаток суспендировали в ацетонитриле и выдерживали при температуре дефлегмации в течение 1 ч при перемешивании. По мере охлаждения указанной смеси до температуры 0oC, соединение 40 выпадало в осадок. Соответствующий фильтрат промывали холодным ацетонитрилом с выходом поименованного соединения в виде белого твердого вещества (312 мг, 76%).
1H ЯМР (300 МГц, DMSO-d6): d 9,4 (S, 1H); 8,05 (s, 1H), 7,6-6,9 (m, 18H); 5,05 (d, 9 Гц, 1H); 4,8 (m, 1H); 4,48 (d, 16 Гц, 1H); 4,3 (dd, 6,8 Гц, 2H); 4,18 (d, 15,8 Гц, 1H); 1,27 (t, 7,2 Гц, 3H); 0,96 (m, 6H);
MC (FAB): m/z = 634 (МН+).
Пример 7
3-{3-[1-(изопропил-фенил-карбамоилметил)-2,4-диоксо-5-фенил- 2,3,4,5-тетрагидро-1H-бензо[b][1,4]диазепин-3-ил]-уреидо}-бензойная кислота.
Раствор этилового эфира 3-{3-[1-(изопропил-фенил-карбамоилметил)- 2,4-диоксо-5-фенил-2,3,4,5-тетрагидро-1H-бензо[b] [1,4]диазепин-3-ил]- уреидо}-бензойной кислоты (312 мг; 0,493 M) в метаноле (23 мл) и тетрагидрофуране (10 мл) нагревали до температуры дефлегмации. Добавляли 5%-ный водный раствор карбоната калия (6,5 мл) и продолжали выдерживать образовавшуюся смесь при температуре дефлегмации в течение 2,5 ч. Указанную реакционную смесь концентрировали в вакууме, нейтрализовали выделенный остаток и растирали его в 1н HCl и воде с получением грубого продукта. Выделенный грубый продукт растворяли в этил ацетате (20 мл), выдерживали при температуре дефлегмации в течение 3 ч, после чего охлаждали. Образовавшийся при этом осадок отбирали с помощью фильтрации и высушивали в вакууме с выходом поименованного соединения в виде белого твердого вещества (225 мг, 75%).
1H ЯМР (300 МГц, DMSO-d6): d 9,4 (s, 1H); 8,05 (s, 1H); 7,6-6,9 (m, 18H); 5,05 (d, 9 Гц, 1H); 4,8 (m, 1H); 4,48 (d, 16 Гц, 1H); 4,18 (d, 15,8 Гц, 1H); 0,96 (m, 6H);
MC (FAB): m/z = 606 (MH+).
Приведенные выше Примеры призваны лучше иллюстрировать приемы синтеза, используемые для получения соединений, предусмотренных настоящим изобретением. Кроме того, конкретные промежуточные соединения, которые можно использовать в рассматриваемых основных способах, могут быть получены следующим образом.
Промежуточное соединение 1
2-(Фенилгидразоно)-малоновая кислота
К интенсивно перемешиваемому раствору моногидрата кетомалоновой кислоты (29,33 г) в этаноле (140 мл) и воде (300 мл) при комнатной температуре в течение 40 мин по каплям добавляли фенилгидразин (23,3 г). Образовавшуюся при этом массу перемешивали в течение ночи при комнатной температуре. С помощью фильтрации отделяли твердое вещество, последовательно промывали его холодной водой (100 мл) и этанолом (25 мл), после чего подсушивали на воздухе. Последующее высушивание проводили в течение ночи при температуре 75oC в вакууме с выходом поименованного соединения в виде желтого твердого вещества (42,38 г).
1H ЯМР (300 МГц, DMSO-d6): d 7,12 (t, 1H); 7,35-7,48 (m, 4H); т.пл.: 155 -157oC (с разлож.).
Промежуточное соединение 2
Дихлорид 2-(фенилгидразоно)-пропандиола
К перемешиваемой массе Промежуточного соединения 1 (14,73 г) в хлороформе (90 мл) при температуре 5oC в течение 20 мин по частям добавляли пентахлорид фосфора (36,84 г). По завершении добавления указанный раствор нагревали до комнатной температуры и перемешивали в течение 1 ч, после чего выдерживали при температуре дефлегмации в течение 3 ч. Охлаждали полученный раствор на ледяной бане, с помощью фильтрации отбирали образовавшийся при этом осадок, промывали его холодным гексаном (50 мл) и высушивали в вакууме в течение ночи с выходом поименованного соединения (13,4 г) в виде ярко-желтого твердого вещества.
1H ЯМР (300 МГц, DMSO-d6): d 7,12 (t, 1H); 7,20-7,56 (m, 4H); т.пл.: 135-138oC (с разлож.).
Промежуточное соединение 3
4-Метокси-N-(2-фениламинофенил)бензамид
Интенсивно перешиваемый раствор N-фенил-1,2-фенилендиамина (20,15 г) в дихлорметане (325 мл) и триэтиламине (11,07 г) охлаждали на ледово-ацетоновой бане в атмосфере азота. В течение 20 мин по каплям добавляли п-анозоил хлорид (18,66 г), растворенный в дихлорметане (100 мл), поддерживая при этом температуру на уровне ниже 5oC. Образовавшейся реакционной смеси позволяли нагреться до комнатной температуры и перемешивали в течение 2 ч. Полученный органический раствор последовательно промывали водой (200 мл), 2н водной HCl (80 мл) и насыщенным солевым раствором (160 мл), после чего высушивали над сульфатом натрия и пропускали через фильтр из диоксида кремния (150 г). Указанный диоксид кремния элюировали этил ацетатом (1 л), а использованный элюент выпаривали в вакууме с выходом розового твердого вещества. Выделенное твердое вещество в течение ночи растирали с этиловым эфиром (350 мл), охлаждали на ледяной бане, фильтровали и высушивали в вакууме с выходом поименованного соединения в виде светло-розового твердого вещества (21,67 г).
1H ЯМР (300 МГц, CDCl3): d 3,82 (s, 3H); 5,75 (br s, 1H); 6,80- 6,91 (m, 5Н); 7,12-7,29 (d, J = 8,8 Гц, 2Н); 8,15 (dd, J = 1,7; 7,8 Гц, 1H); 8,36 (s, 1H);
ТСХ (смесь EtOAc и гексана в соотношении 1:4); Rf = 0,24; т.пл. : 148-150oC.
Промежуточное соединение 4
N-(4-Метоксибензил)-N'-фенил-бензол-1,2-диамин
К перемешиваемому раствору литий-алюминий гидрида (1,0 г) в ТГФ (40 мл), охлажденному до температуры 5oC, в течение 45 мин добавляли раствор 4-метокси-N-(2-фениламино-фенил)-бензамида (5,0 г) в ТГФ (30 мл). По завершении добавления образовавшуюся реакционную смесь выдерживали при температуре дефлегмации в течение 1,5 ч. Указанный раствор охлаждали до комнатной температуры и удаляли избыток литий-алюминий гидрида, добавляя этанол до тех пор, пока не прекратится выделение водорода. Добавляли насыщенный водный раствор кислого карбоната натрия (100 мл), а образовавшийся при этом раствор экстрагировали этил ацетатом (3х100 мл). Объединяли соответствующие органические экстракты, высушивали их над сульфатом натрия и фильтровали через окись кремния. Использованный фильтр промывали этилацетатом (500 мл) и объединяли полученные органические фракции. Указанный фильтрат концентрировали в вакууме с выделением коричневого масла, которое затвердевает при стоянии с выходом поименованного соединения (4,78 г).
1H ЯМР (300 МГц, CDCl3): d 3,79 (s, 3H); 4,27 (s, 2H); 4,52 (br s, 1H); 5,08 (s, 1H); 6,67-6,74 (m, 4H); 6,79-6,86 (m, 3H); 7,04-7,24 (m, 6H);
ТСХ (смесь EtOAc и гексана в соотношении 1:4); Rf = 0,57.
Промежуточное соединение 5
1-(4-Метоксибензил)-5-фенил-3-(фенилгидразоно)-1,5-дигидро-бензо[b][1,4] диазепин-2,4-дион
Растворы Промежуточного соединения 4 (4,86 г) в ТГФ (40 мл) и дихлорида 2-(фенилгидразоно)пропандиола (5,58 г) в ТГФ (40 мл) в течение 30 мин по каплям добавляли друг к другу при перемешивании на ледово-метанольной бане. Указанному раствору позволяли нагреться до комнатной температуры и перемешивали его в течение ночи. С помощью фильтрации отделяли образовавшийся желтый осадок, промывали его холодной ТГФ (40 мл), подсушивали на воздухе и высушивали в течение ночи в вакууме с выходом поименованного соединения (6,23 г) в виде желтого твердого вещества.
1H ЯМР (300 МГц, CDCl3): d 3,78 (s, 3H); 4,69 (d, J = 14,7 Гц, 1H); 5,76 (d, J = 14,9 Гц, 1H); 6,80-6,87 (m, 3H); 7,02-7,12 (m, 4H); 7,19-7,40 (m, 11H); 11,19 (s, 1H);
MC (FAB): m/z = 477,0 (MH+).
ТСХ (смесь EtOAc и гексана в соотношении 1:4); Rf = 0,18.
Промежуточное соединение 6
3-Амино-1-(4-метоксибензил)-5-фенил-1,5-дигидро-бензо[b] -[1,4] диазепин-2,4-дион
К интенсивно перемешиваемой массе цинковой пыли (6,49 г) в уксусной кислоте (50 мл), охлажденной до температуры 10oC, в течение 15 мин добавляли массу 1-(4-метоксибензил)-5-фенил-3- (фенилгидразоно)-1,5-дигидро-бензо[b] [1,4] диазепин-2,4-диона (5,75 г; 12,1 мМ) в уксусной кислоте (30 мл). По завершении добавления указанный раствор нагревали до комнатной температуры и перемешивали в течение 3 ч. С помощью фильтрации отделяли использованный цинк и промывали его этил ацетатом (75 мл). Соответствующий фильтрат концентрировали в вакууме и вводили в смесь воды (60 мл) и этил ацетата (100 мл). С помощью насыщенного водного раствора карбоната натрия доводили pH до 9 и разделяли полученные фазы. Выделенную водную фазу экстрагировали этил ацетатом (2х75 мл), объединяли соответствующие органические фазы, высушивали над сульфатом магния, фильтровали и концентрировали в вакууме с выходом желтого масла, которое высушивали в вакууме с выходом поименованного соединения (4,79 г).
1H ЯМР (300 МГц, CDCl3): d 3,05 (s, 2H); 3,75 (s, 3H); 4,35 (s, 1H); 4,64 (d, J = 14,7 Гц, 1H); 5,82 (d, J = 14,7 Гц, 1H); 6,59-6,85 (m, 6H); 7,06-7,29 (m, 6H); 7,51 (d, J = 7,4 Гц, 1H);
MC (FAB): m/z = 388,2 (MH+);
ТСХ (смесь CH2Cl2 и CH3OH в соотношении 9:1); Rf = 0,50.
Промежуточное соединение 7
3-Амино-1-фенил-1,5-дигидро-бензо[b][1,4]диазепин-2,4-дион
К перемешиваемому раствору 3-амино-1-(4-метоксибензил)-5-фенил- 1,5-дигидро-бензо[b][1,4]диазепин-2,4-диона (0,50 г) в смеси ацетонитрила и воды в соотношении 9:1 (12 мл) при комнатной температуре в течение 10 мин частями добавляли нитрат церрий-аммония (1,84 г). Полученный раствор перемешивали в течение ночи при комнатной температуре. Указанный раствор концентрировали в вакууме, а образовавшееся при этом твердое вещество вводили в смесь насыщенного водного раствора карбоната калия (40 мл) и этанола (60 мл). Разделяли соответствующие фазы, а водную фазу экстрагировали этанолом (4х50 мл). Объединяли полученные этанольные фракции, высушивали над сульфатом натрия и концентрировали в вакууме с выделением рыжевато-коричневого твердого вещества. Указанное вещество экстрагировали кипящим CH2Cl2 (10х60 мл) до истощения, объединяли соответствующие органические фазы, высушивали над сульфатом натрия, фильтровали и концентрировали с выходом поименованного соединения (0,30 г) в виде рыжевато-коричневого твердого вещества.
1H ЯМР (300 МГц, DMSO-d6): d 1,98 (br s, 2H); 4,08 (s, 1H); 6,86 (d, J = 8,4 Гц, 1H); 7,11-7,46 (m, 8H); 10,78 (br s, 1H);
13C (75,429 МГц, DMSO-d6): d 56,98; 123,41; 126,22; 126.51; 127,34; 128,30; 128,89; 130,15; 132,29; 134,42; 142,36; 168,13; 169,39;
MC (FAB): m/z = 268,10 (MH+).
ТСХ (смесь CH2Cl2 и CH3OH в соотношении 15:1); Rf = 0,21.
Промежуточное соединение 8
1-(2,4-Диоксо-1-фенил-2,3,4,5-тетрагидро-1Н-бензо[b] [1,4]- диазепин-3-ил)-3-фенил мочевина
К массе 3-амино-1-фенил-1,5-дигидро-бензо[b] [1,4] диазепин-2,4-диона (0,398 г) в дихлорметане (5 мл) при комнатной температуре и перемешивании постепенно добавляли фенил изоцианат (0,177 г). Указанную реакционную среду перемешивали в течение 2 ч при комнатной температуре, после чего с помощью фильтрации отбирали образовавшийся кремообразный осадок с выходом поименованного соединения (0,413 г).
1H ЯМР (300 МГц, DMSO-d6): d 4,97 (d, J = 7,5 Гц, 1H); 6,88-6,97 (m, 3H); 7,13-7,47 (m, 12H); 9,16 (s, 1H); 10,78 (br s, 1H);
ТСХ (смесь CH2Cl2 и CH3OH в соотношении 19:1); Rf = 0,21.
Промежуточное соединение 9
N-Изопропил-N-фенил-2-(2-фениламинофениламино)-ацетамид
К раствору N-фенилфенилен диамина (9,2 г) в ДМФ и 2-бром-N-изопропил-N-фенил ацетамида (12,7 г) в ДМФ (200 мл) добавляли карбонат калия (6,9 г) и перемешивали образовавшуюся смесь в течение ночи. Выпаривали ДМФ в вакууме, а полученный остаток растворяли в этил ацетате (400 мл) и промывали водной 1н HCl (4х250 мл) до истощения. Соответствующую органическую фазу промывали водой (2х200 мл), высушивали над сульфатом натрия и выпаривали с получением 17,8 г грубого алкилированного продукта. Выделенное масло очищали с помощью хроматографии на силикагеле (600 г), элюция сначала CHCl3 (8000 мл), а затем - смесью гексана и этил ацетата в соотношении 2:1 (8000 мл) с выходом поименованного соединения (10 г) в виде масла.
1H ЯМР (300 МГц, CDCl3): d 7,42-6,8 (m, 14H); 6,36 (d, 1H); 4,95 (m, 1H); 3,22 (s, 2H); 1,05 (d, 6H);
MC (FAB) = 360 (MH+);
ТСХ (CHCl3; Rf = 0,18.
Промежуточное соединение 10
2-[2,4-Диоксо-5-фенил-3-(фенилгидразоно)-2,3,4,5-тетрагидро-бензо[b] [1,4]диазепин-1-ил]-N-изoпpoпил-N-фенилaцетaмид
N-изопропил-N-фенил-2-(2-фениламинофениламино)-ацетамид (10 г) и дихлорид 2-(фенил-гидразоно)-пропандиола (6,83 г) независимо растворяли в ТГФ (100 мл), а полученные растворы одновременно добавляли в колбу, содержащую ТФУ (100 мл), при температуре 0oC, при перемешивании в атмосфере азота. Образовавшейся смеси позволяли нагреться до комнатной температуры, после чего перемешивали в течение 4 ч. Выпаривали ТГФ в вакууме, а соответствующий остаток растворяли в этил ацетате (200 мл). Полученный этил ацетатный раствор промывали 10%-ным водным раствором карбоната натрия (2х200 мл) и водой (2х200 мл), высушивали над сульфатом натрия и концентрировали в вакууме. Образующуюся при этом пену обрабатывали диэтиловым эфиром (50 мл) с выпадением осадка поименованного соединения в виде ярко-желтого твердого вещества (7,5 г). Соответствующий маточный раствор концентрировали с выходом рыжевато-коричневой пены (2,5 г).
1H ЯМР (300 МГц, CDCl3): d 11,4 и 10,85 (s, 1H); 7,6-6,8 (m, 19H); 5,05 (m, 1H); 4,4 (m, 2H); 1,05 (m, 6H);
MC (FAB) = 532 (MH+);
ТСХ (смесь гексана и этил ацетата в соотношении-2:1); Rf = 0,19
Промежуточное соединение 11
2-(3-Амино-2,4-диоксо-5-фенил-2,3,4,5 -тетрагидро-бензо[b] [1,4]диазепин-1-ил)-N-изопропил-N-фенилацетамид
К массе 2-[2,4-диоксо-5-фенил-3-(фенилгидразоно)-2,3,4,5-тетрагидро-бензо[b] [1,4]диазепин-1-ил]-N-изопропил-N-фенилацетамида (7,5 г) в ледяной уксусной кислоте, охлажденной до температуры 0oC, порциями добавляли цинковую пыль (9,1 г). Указанной реакционной смеси позволяли нагреться до комнатной температуры и перемешивали в течение 1 ч. С помощью фильтрации через целит отделяли использованный цинк, а ледяную уксусную кислоту выпаривали в вакууме. Полученный остаток растворяли в этил ацетате (200 мл), промывали 10%-ным водным раствором карбоната натрия (2х100 мл) и водой (2х100 мл), высушивали над сульфатом натрия и выпаривали с образованием рыжевато-коричневого масла. Указанное масло растирали с гексаном и этил ацетатом с выходом поименованного соединения в виде светло-бурого порошка (6,3 г).
1H ЯМР (300 МГц, CDCl3): d 7,6-6,8 (m, 14H) ; 5,05 (m, 1H); 4,3-4,0 (m, 3H); 1,05 (d, 6H);
MC (FAB): m/z = 448 (MH+);
ТСХ (смесь хлороформа и метанола в соотношении 9:1); Rf = 0,25.
Промежуточное соединение 12
2-(3-Амино-2,4-диоксо-5-фенил-2,3,4,5-тетрагидро-бензо[b] - [1,4]диазепин-1-ил)-N-изопропил-N-(4-метокси-фенил)-ацетамид
К перемешиваемому раствору 2-[2,4-диоксо-5-фенил-3-(фенилгидразоно) -2,3,4,5-тетрагидро-бензо[b] [1,4] диаpепин-1-ил]-N-изопропил-N- (4-метокси-фенил)ацетамида (4,28 г) в уксусной кислоте (50 мл) при комнатной температуре добавляли цинковую пыль (4,11 г) и перемешивали образовавшуюся смесь в течение 3 ч. Отфильтровывали использованный цинк, соответствующий фильтрат концентрировали в вакууме, а образовавшееся при этом масло вводили в смесь воды (60 мл) и этил ацетата (100 мл). С помощью 6н гидроксида натрия доводили pH до 8 и разделяли полученные фазы. Выделенную водную фазу экстрагировали этил ацетатом (2х75 мл), объединяли соответствующие органические фазы, высушивали над сульфатом магния, фильтровали и концентрировали в вакууме с выходом желтой пены. Указанный грубый продукт очищали с помощью импульсной хроматографии на силикагеле (80 г), элюция последовательно этил ацетатом (260 мл; для удаления загрязнений) и смесью метилен хлорида и метанола в соотношении 19: 1 (200 мл; для элюции продукта). Объединяли соответствующие фракции и концентрировали их с выходом поименованного соединения (2,58 г) в виде желтой пены.
1H ЯМР (300 МГц, CDCl3): d 1,08 (d, J = 6,6 Гц, 6H); 2,22 (br s, 2H); 3,85 (s, 3H); 4,12-4,35 (m, 3H); 5,01 (m, 1H); 6,91- 7,00 (m, 3, Н); 7,12 (m, 2H); 7,22-7,43 (m, 8H);
ТСХ (смесь CH2Cl2 и CH3OH в соотношении 19:1); Rf = 0,25.
Промежуточное соединение 13
2-[2,4-Диоксо-5-фенил-3-(фенилгидразоно)-2,3,4,5-тетрагидро-бензо[b] [1,4]диазепин-1-ил]-N-изопропил-N-(4-метоксифенил)-ацетамид
Растворы N-изопропил-N-(4-метоксифенил)-2-(2-фениламино- фениламино)-ацетамида (3,00 г) в ТГФ (30 мл) и дихлорида 2-(фенилгидразоно)пропандиола (1,89 г) в ТГФ (30 мл) в течение 30 мин по каплям добавляли друг к другу при перемешивании на ледово-метанольной бане. По завершении добавления указанному раствору позволяли нагреться до комнатной температуры и перемешивали его в течение ночи. Выпаривали растворитель при пониженном давлении, а образовавшееся при этом масло отбирали в этил ацетат (250 мл), промывали его насыщенным раствором бикарбоната натрия, высушивали над сульфатом натрия, фильтровали и концентрировали при пониженном давлении с выходом поименованного соединения (4,28 г) в виде желтой пены.
1H ЯМР (300 МГц, CDCl3): d 1,13 (m, 6H); 3,87 (s, 3H); 4,17- 4,55 (m, 2H); 5,05 (m, 1H); 6,88-7,60 (m, 18H); 10,68 (s, 0,5Н); 11,44 (s, 0,5H);
ТСХ (смесь EtOAc и гексана в соотношении 2:3); Rf = 0,38.
Промежуточное соединение 14
N-Изопропил-N-(4-метокси-фенил)-2-(2-фениламинофениламино)-ацетамид
К раствору N-фенилбенpол-1,2-диамина (3,08 г) в ДМФ (35 мл) добавляли карбонат калия (2,31 г) и 2-бром-N-изопропил-N-(4-метоксифенил)-ацетамид (4,79 г) и перемешивали образовавшуюся смесь в течение 18 ч при комнатной температуре. Выпаривали растворитель в вакууме, а полученное масло растворяли в этил ацетате (250 мл), промывали 1н HCl (4х100 мл), высушивали над сульфатом натрия, фильтровали и концентрировали с получением коричневого масла. Указанное масло подвергали импульсной хроматографии на силикагеле (70 г), элюция смесью этил ацетата и гексана в соотношении 1:4 (1 л). Объединяли фракции, содержащие искомый продукт и концентрировали их при пониженном давлении с выходом поименованного соединения в виде рыжевато-коричневой пены (3,95 г).
1H ЯМР (300 МГц, CDCl3): d 1,05 (d, J = 6,9 Гц, 6H); 3,44 (s, 2H); 3,87 (s, 3H); 4,97 (m, 1H); 5,37 (br s, 1H); 6,36 (d, J = 7,4 Гц, 1H); 6,69 (t, 1H); 6,71-7,21 (m, 11H);
ТСХ (смесь EtOAc и гексана в соотношении 1:4); Rf = 0,18.
Промежуточное соединение 15
2-Бром-N-Изопропил-N-(4-метоксифенил)-ацетамид
К раствору изопропил-(4-метокси-фенил)-амина (25,11 г) в дихлорметане (250 мл) при комнатной температуре и перемешивании добавляли триэтиламин (15,38 г). Образовавшийся раствор охлаждали на ледяной бане до температуры ниже 3oC и в течение 45 мин при перемешивании на ледяной бане по каплям добавляли бромид бромацетила (30,68 г), растворенный в дихлорметане (100 мл). Указанную реакционную среду перемешивали в течение ночи при комнатной температуре, промывали 0,3 н HCl (300 мл) и солевым раствором (300 мл), высушивали над сульфатом натрия, фильтровали и выпаривали при пониженном давлении с получением темно-коричневого масла. Выделенное масло фильтровали через силикагель (150 г), который затем элюировали смесью этил ацетата и гексана в соотношении 1:1 (900 мл), а соответствующий фильтрат выпаривали при пониженном давлении с выходом поименованного соединения (41,05 г) в виде коричневого масла, затвердевающего, при стоянии.
1H ЯМР (300 МГц, CDCl3): d 1,04 (d, J = 6,8 Гц, 6H); 3,53 (s, 2H); 3,84 (s, 3H); 4,93 (m, 1H); 6,93 (d, J = 9,1 Гц, 2H); 7,10 (d, J = 9,1 Гц, 3H);
ТСХ-(смесь EtOAc и гексана в соотношении 3:17); Rf = 0,18.
Промежуточное соединение 16
Изопропил-(4-метокси-фенил)-амин
К перемешиваемому раствору 4-метокси-фениламина (1,24 г) в метаноле (15 мл) при комнатной температуре последовательно добавляли ледяную уксусную кислоту (415 мг), ацетон (669 мг) и 1М цианоборогидрид натрия в ТГФ (12,7 мл). Образовавшуюся реакционную смесь перемешивали в течение ночи при комнатной температуре. С помощью 6н HCl доводили pH до 2 и перемешивали в течение 30 мин, после чего полностью удаляли избыток цианоборогидрида натрия. Затем с помощью 1н NaOH доводили pH до 8,5, а образовавшийся при этом раствор экстрагировали диэтиловым эфиром (2х50 мл) и этил ацетатом (50 мл). Объединяли соответствующие экстракты, высушивали над сульфатом натрия, фильтровали и концентрировали при пониженном давлении с выходом поименованного соединения (1,42 г) в виде желтого масла.
1H ЯМР (300 МГц, CDCl3): d 1,18 (d, J = 6,1 Гц, 6H); 2,92 (br s, 1H); 3,55 (m, 1H); 3,75 (s, 3H); 6,57 (d, J = 9,1 Гц, 2H); 6,78 (d, J = 8,8 Гц, 2H);
ТСХ (смесь EtOAc и гексана в соотношении 2:3); Rf =0,72.
Промежуточное соединение 17
3-Аминобензолацетонитрил
Раствор 3-нитробензолацетонитрила (8,0 г) в этаноле (100 мл) в течение 4 ч подвергали гидрогенизированию при давлении 1 атм и комнатной температуре на 5%-ном палладиево-угольном катализаторе (0,8 г). С помощью фильтрации через Hyflo удаляли использованный катализатор и выпаривали соответствующий фильтрат. Полученный остаток подвергали хроматографии, элюция смесью ЭА и гексана в соотношении 1:2 с выходом поименованного соединения (5,25 г) в виде оранжевого масла.
ТСХ (смесь гексана и ЭА в соотношении 2:1); Rf = 0,45.
1H ЯМР (300 МГц, CDCl3: d 3,79 (s, 2H); 3,9 (br, 2H); 6,7 (m, 3H); 7,2 (M, H).
Промежуточное соединение 18
Гидрохлорид 3-(2Н-тетразол-5-ил)-фениламина
Смесь 3-аминобензонитрила (10,0 г) и трибутилтиназида (42 г) выдерживали в атмосфере азота при температуре 160oC в течение 120 мин. Охлаждали указанную смесь, разбавляли ее эфиром (300 мл), экстрагировали 2н. водной HCl (2х200 мл), объединяли соответствующие водные экстракты и охлаждали их на ледово-метанольной бане в течение 30 мин. С помощью фильтрации отделяли образовавшийся при этом осадок, промывали его эфиром (100 мл) и высушивали с получением бледно-розового твердого вещества. Указанное вещество перекристаллизовывали из метанола (600 мл) с выходом поименованного соединения в виде беловатого твердого вещества (12,1 г).
1H ЯМР (300 МГц, DMSO-d6): d 7,32 (d, J = 7,8 Гц, 1H); 7,57 (t, 1H); 7,82 (m, 2H);
т.пл.: 256-262oC (разложение).
Используя вышеописанные общие способы А-Д, получены также следующие предусмотренные настоящим изобретением соединения, представленные для удобства в таблицах 1-8 вместе с описанием синтеза соответствующих соединений. Для удобства описания, а также для простоты идентификации соединений, примеры реализации А-3 соответствуют таблицам 1-8. Группы R13-21 представлены в таблицах 1-8 для удобства описания, а также для простоты идентификации соединений.
Тест с использованием желчных путей морских свинок
Получение ткани:
У морских свинок, умертвленных посредством сворачивания шеи, удаляли желчные пузыри. Каждый выделенный желчный пузырь очищали от прилегающей соединительной ткани и разрезали в виде двух колец 2-4 мм длиной. Указанные кольца последовательно суспендировали в камерах для инкубирования органов, содержащих солевой физиологический раствор следующего состава (мМ): NaCl (118,4); MgSO4•H2O (1,2); CaCl2•2H2O (2,5); KH2PO3 (1,2); NaHCO3 (25), декстроза (11,1). Температуру инкубационной жидкости поддерживали на уровне 37oC и аэрировали смесью 95% O2 и 5% CO2. С помощью золотых колец и передаточных соединений, выполненных из нержавеющей стали, ткани соединяли с изометрическими датчиками изменения поверхностного напряжения (Grass, Model FT03D). В дальнейшем регистрируемые ответы записывали на полиграфе (Grass, Model 7E). Одну из тканей в случае каждого животного использовали в качестве контроля времени или среды и не добавляли к ней тестируемые соединения.
Тест:
В течение 120 мин кольца ткани постепенно растягивали до достижения базового уровня напряжения в 1 gm, который поддерживали по ходу эксперимента. В течение доводки базового уровня напряжения указанные кольца ткани четырежды обрабатывали ацетилхолином (АХ, 10-6 М) для того, чтобы удостоверится в сокращаемости ткани. Затем рассматриваемые ткани подвергали действию субмаксимальной дозы сульфатированного ХЦК-8 (Sigma, 3•10-9 М). Для того, чтобы восстановить стабильный базовый уровень после получения стабильного ответа, указанные ткани быстро промывали трижды, а затем - через каждые 5-10 мин в течение 1 ч.
Соединения растворяли в диметилсульфоксиде (ДМСО), затем разбавляли водой и анализировали при помощи кривой кумулятивной зависимости "доза-эффект" для тестируемого соединения (в концентрации от 10-11 до 3•10-6 М), после чего строили кривую "доза-эффект" для сульфатированного ХЦК-8 (в концентрации от 10-10 до 10-6 М) в присутствии наивысшей дозы тестируемого соединения. В качестве финального теста добавляли АХ (10 мМ) для индукции максимального сокращения. Для каждого тестируемого соединения проводили по меньшей мере три определения его активности.
В следующей ниже таблице 9 приведены экспериментальные данные для некоторых представителей соединений, отвечающих общей формуле (I). Агонистическая активность на модели желчных пузырей морских свинок (ЖПМС) выражена в процентах максимального сокращения, вызванного ацетилхолином (АХ) при концентрации тестируемого соединения 30 мкМ.
Судя по результатам, свидетельствующим об отсутствии токсичных эффектов на крысах при применении тестируемых соединений в дозах вплоть до 12 мг/кг, специалистам следует отметить, что предусмотренные настоящим изобретением соединения могут терапевтически применяться в пределах указанных дозовых режимов в отсутствие токсичных эффектов.
Парадигма 18-часового прерывания кормления
Самцов крыс линии Long-Evans (Charles River Co., Raleigh, NC) весом 300-375 г содержали поодиночке по меньшей мере в течение недели в подвешенных клетках (размером 17,8 • 25,4 •17,8 см), выполненных из сетки из нержавеющей стали, с предоставлением свободного доступа к воде (подаваемой через автоматические устройства для питья в задней стороне клетки) и пище (Lab Blox, Purina Rodent Laboratory Chow #5001), при 12-часовом световом дне (освещение с 6 до 18 ч) и температуре приблизительно 22,8oC. Перед началом эксперимента в 16 ч изымали всю пищу, оставляя доступ к воде. В 9 ч следующего утра проводили взвешивание крыс. В 9 ч 45 мин крысам внутрибрюшинно (в. б.), перорально (п.о.) или через канюлю, введенную в двенадцатиперстную кишку, вводили тестируемое соединение или носитель (2 мл/кг), после чего указанных крыс возвращали в их клетки. В 10 ч возобновляли подачу пищи. В 10 ч 30 мин проводили взвешивание оставшейся пищи и спилажа.
Фармацевтические примеры
Таблетки
a. Активный ингредиент - 50 мг
Безводная лактоза USP - 163 мг
Микрокристаллическая целлюлоза NF - 69 мг
Предварительно желатинизированный крахмал Ph. Eur. - 15 мг
Стеарат магния USP - 3 мг
Общий вес - 300 мг
Активный ингредиент, микрокристаллическую целлюлозу, лактозу и предварительно желатинизированный крахмал просеивают через сито с размером ячеек 500 мкм и смешивают в приемлемом миксере. Стеарат магния просеивают через сито с размером ячеек 250 мкм и смешивают с вышеописанной активной смесью. Из полученной смеси с помощью приемлемых прессов изготавливают таблетки.
б. Активный ингредиент - 50 мг
Моногидрат лактозы USP - 120 мг
Предварительно желатинизированный крахмал Ph.Eur. - 20 мг
Кросповидон NF - 8 мг
Стеарат магния USP - 2 мг
Общий вес - 200 мг
Активный ингредиент, лактозу и предварительно желатинизированный крахмал смешивают друг с другом и гранулируют с водой. Полученную влажную массу высушивают и измельчают. Стеарат магния и Кросповидон просеивают через сито с размером ячеек 250 мкм и смешивают с указанными гранулами. Из образовавшейся смеси с помощью приемлемых прессов изготавливают таблетки.
Капсулы
а. Активный ингредиент - 50 мг
Предварительно желатинизированный крахмал Ph.Eur. - 148 мг
Стеарат магния USP - 2 мг
Общий вес содержимого капсулы - 200 мг
Активный ингредиент и предварительно желатинизированный крахмал просеивают через сито с размером частиц 500 мкм, смешивают друг с другом и смазывают стеаратом магния (просеянным через сито с размером ячеек 250 мкм). Полученной смесью наполняют твердые желатиновые капсулы приемлемого размера.
б. Активный ингредиент - 50 мг
Моногидрат лактозы USP - 223 мг
Повидон USP - 12 мг
Кросповидон NF - 12 мг
Стеарат магния - 3 мг
Общий вес содержимого капсулы - 300 мг
Активный ингредиент и лактозу смешивают друг с другом и гранулируют с раствором Повидона. Полученную влажную массу высушивают и измельчают. Стеарат магния и Кросповидон просеивают через сито с размером ячеек 250 мкм и смешивают с указанными гранулами. Образовавшейся смесью наполняют твердые желатиновые капсулы приемлемого размера.
Литература
1. Патент США N 4,988,692.
2. Патент США N 4,490,304.
3. Заявка PCT N WO 90/06937.
4. Заявка PCT N WO 91/19733.
5. Eliel E.L. Stereochemistry of Carbon Compounds. Mcgraw Hill, 1962.
6. Wilen S.H. Tables of Resolving Agents.
7. Patel M. and Spraggs C.F. Br. J. Pharmac., 1992, 106: 275-282.
8. Reeves J.J. and Stables R. Br. J. Pharmac., 1985, 86: 677-684.
9. Fornos et al., J. Pharmacol. Exp. Ther., 1992, 261: 1056-1063.
10. Hedges and Parsons, J. Physiol., 1977, 267: 191-194.
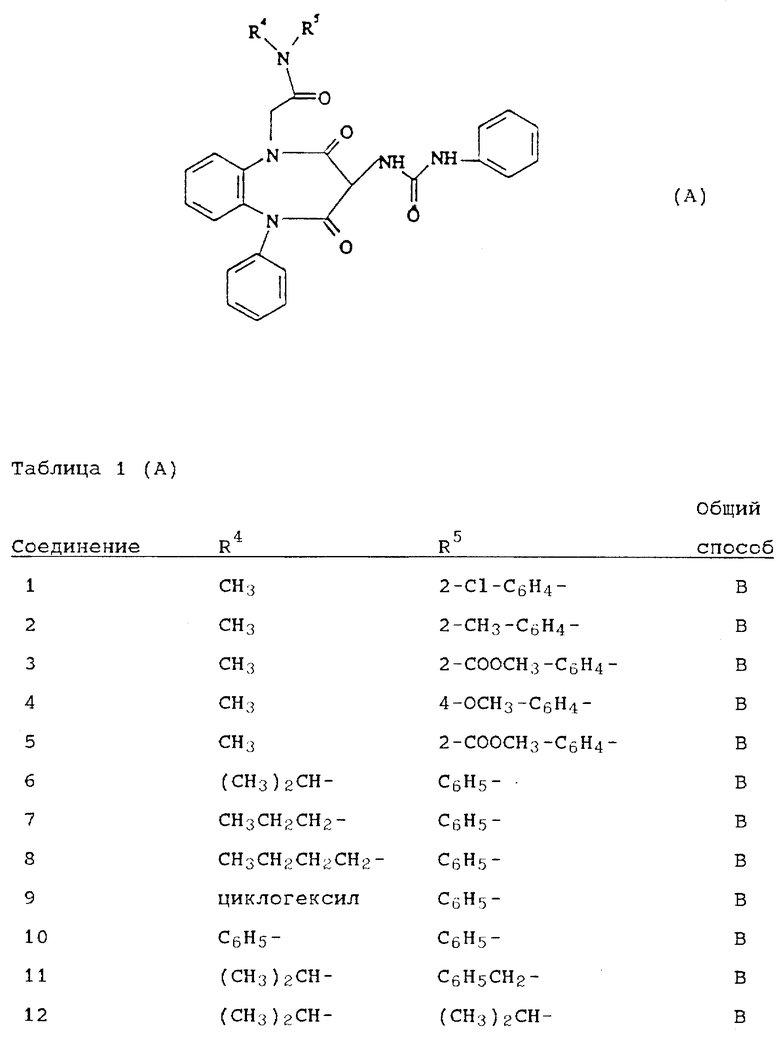
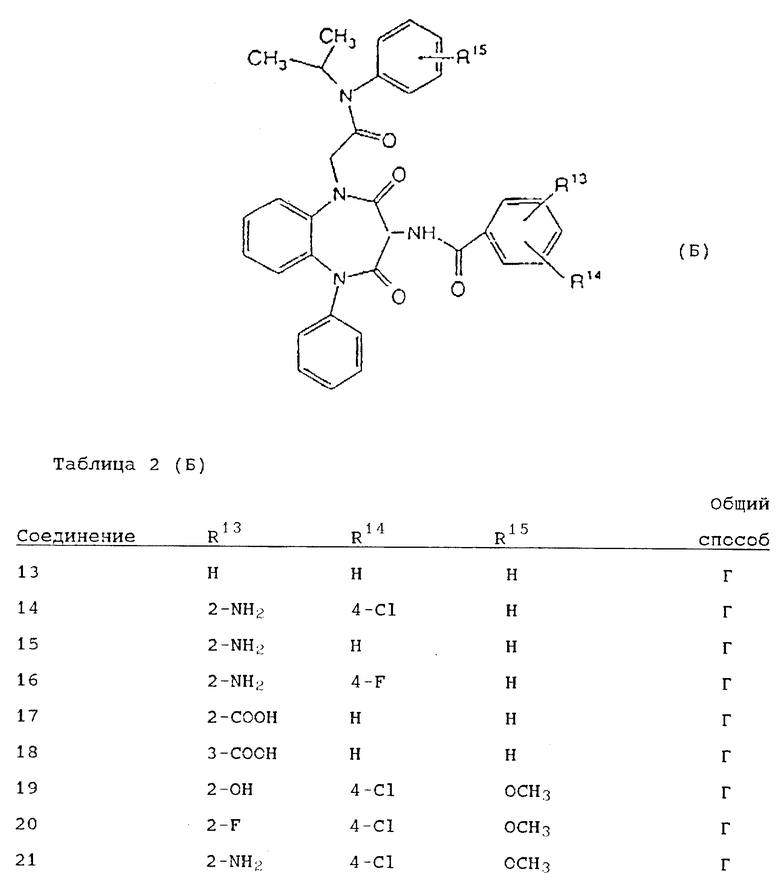
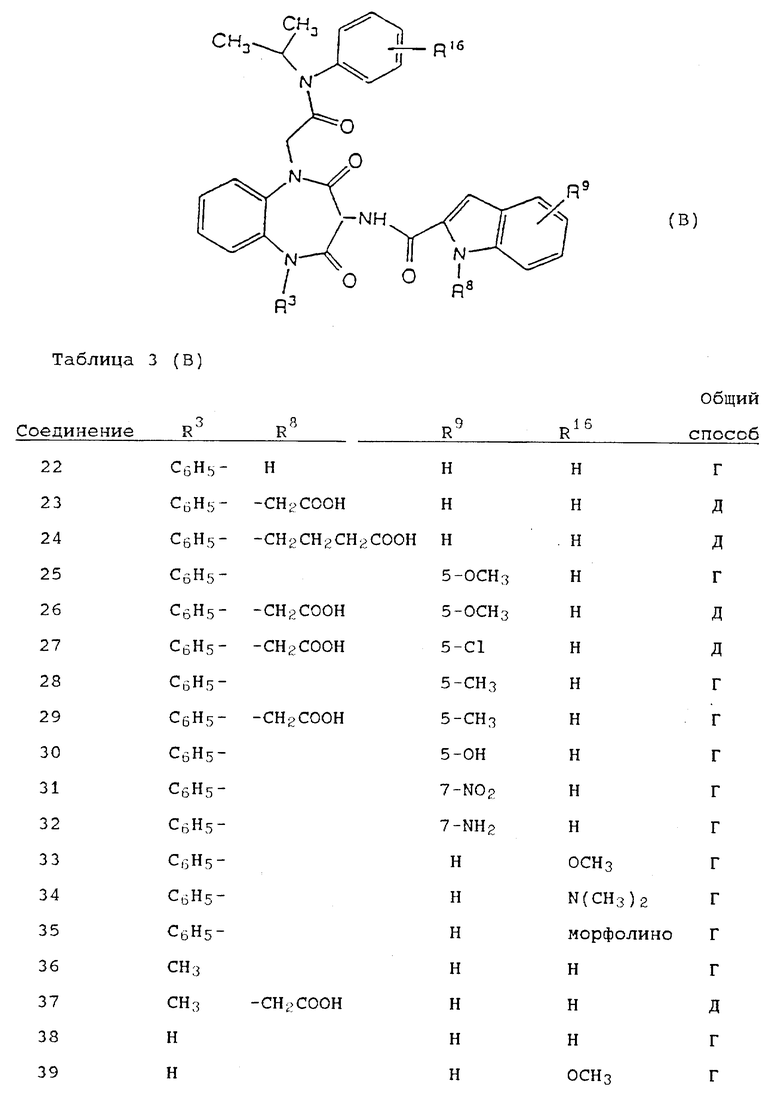
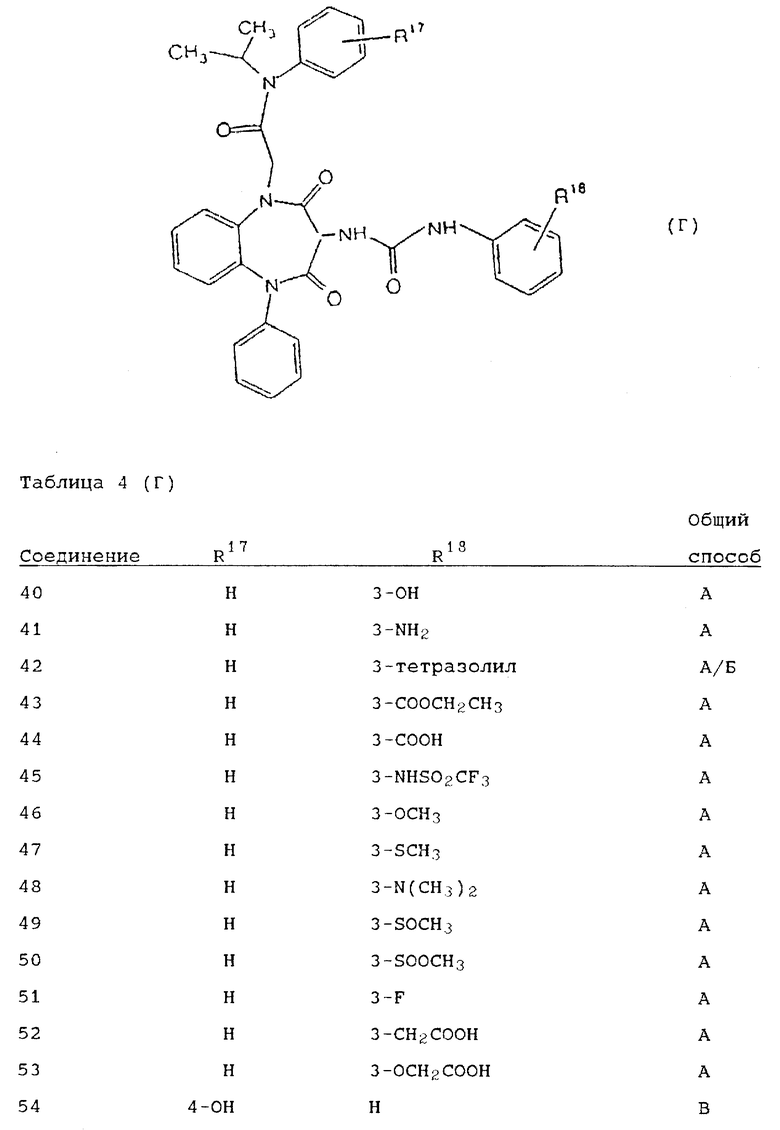

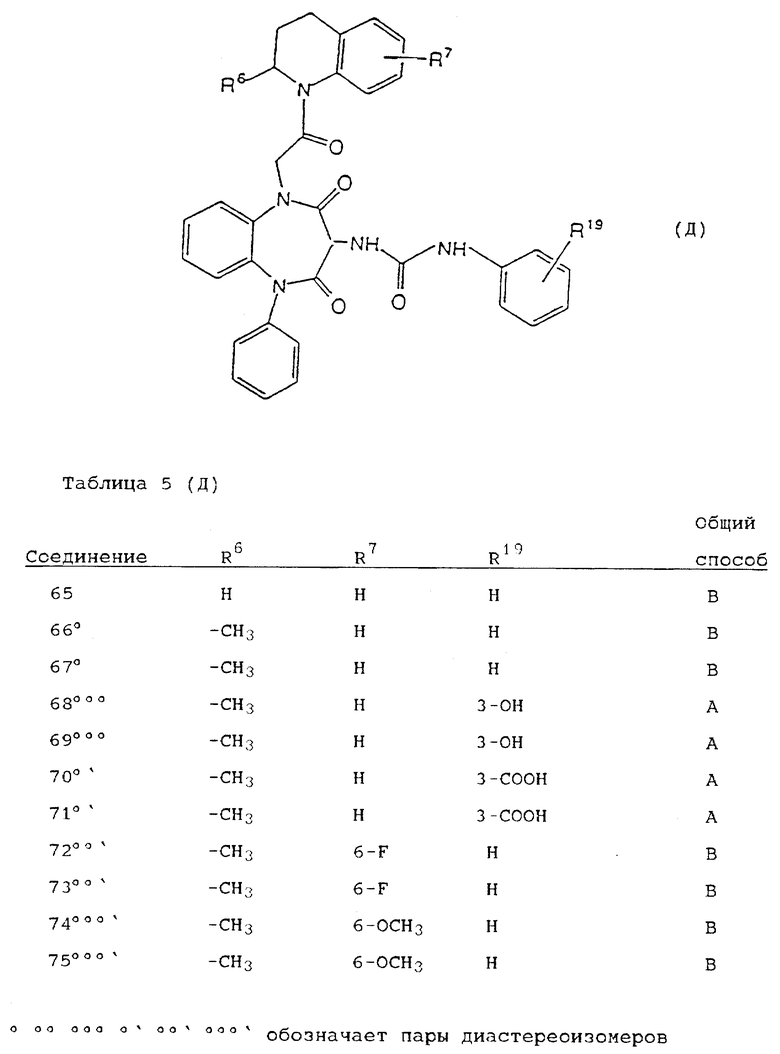
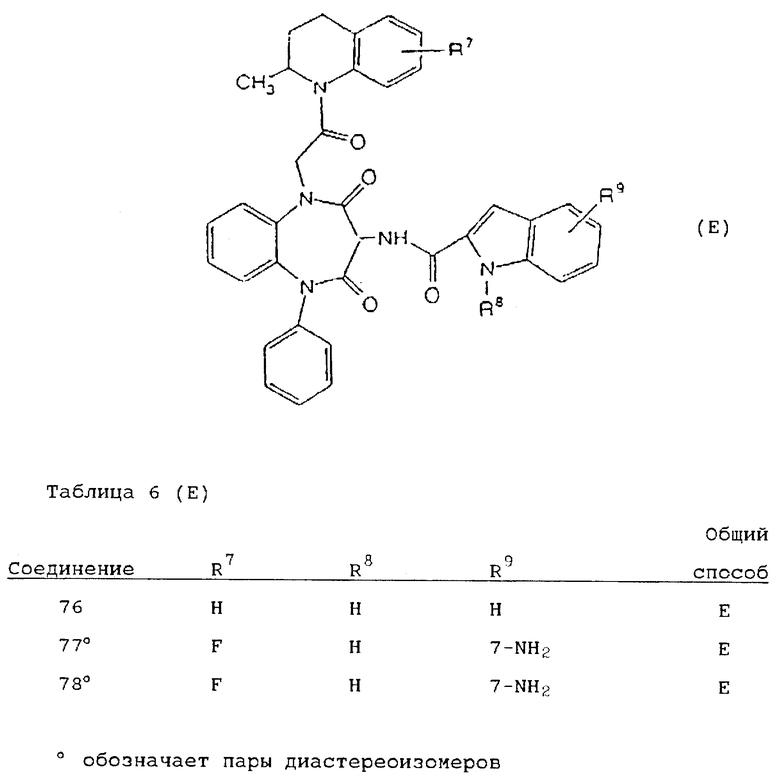
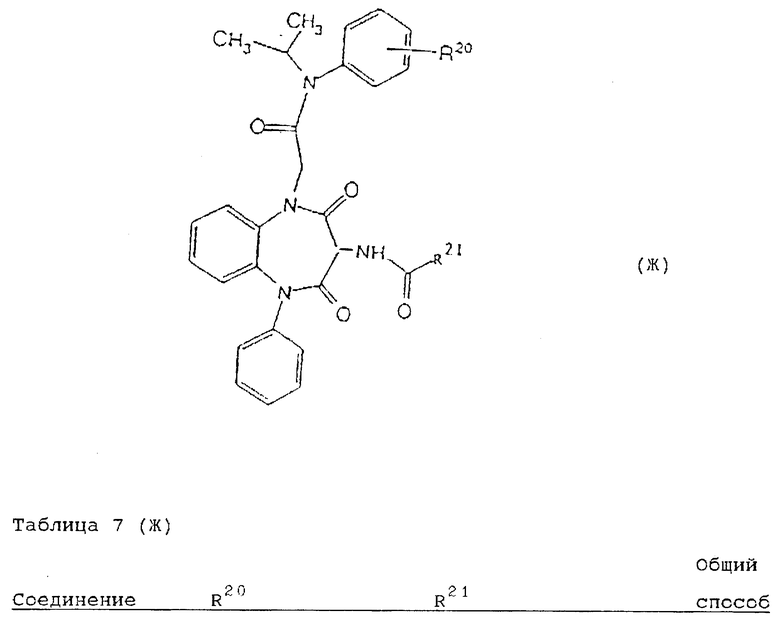

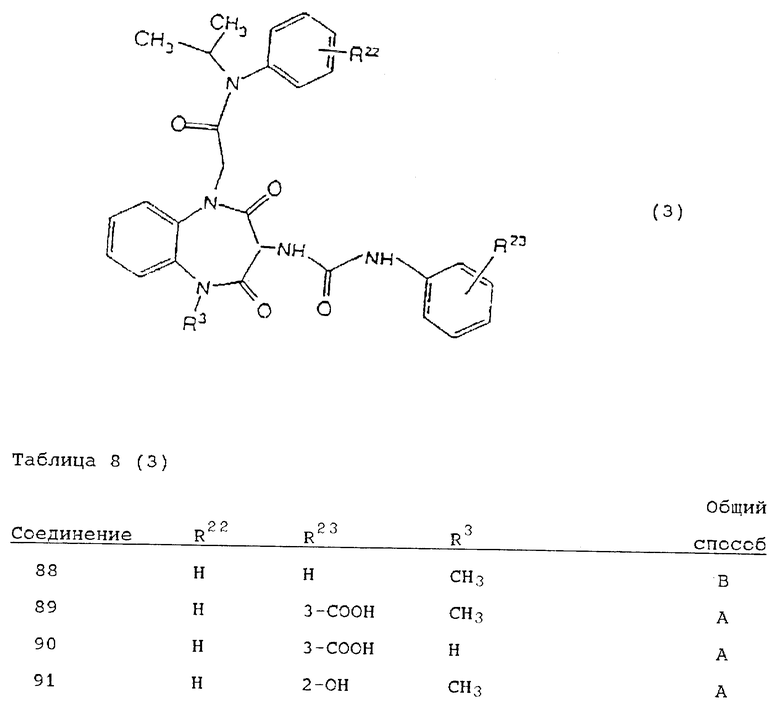


Производные 1,5-бензодиазепина формулы I, где Х=Н, R1 - радикал формулы II или NR4R5; R2 - индол, бензофуран, тиофен, бензотиофен, индолин, хинолин или 4-оксобензопиран; фенил, возможно замещенный галогеном, карбокси, амино или диметиламино, лобо NHR11, R11 - 7-индазолил; R3- Н, C1-6-алкил, С3-6-циклоалкил или фенил (значения других радикалов см. в п.1 формулы изобретения), а также их физиологически приемлемые соли и сольваты проявляют агонистическую активность по отношению к рецепторам ХЦК-А, что позволяет указанным соединениям регулировать функционирование гормонов гастрина и холецисткинина (ХЦК) у млекопитающих. 3 с. и 8 з.п. ф-лы, 9 табл.
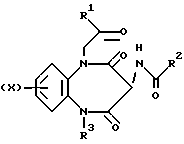
а также физиологически приемлемые соли и сольваты указанного соединения,
где Х представляет собой атом водорода;
R1 обозначает либо радикал, соответствующий формуле II,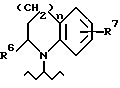
либо остаток NR4R5;
R2 представляет собой либо
(I) гетероцикл, присоединенный по своему положению 2 и выбранный из числа следующих: индол, бензофуран, тиофен, бензотиофен, индолин, хинолин или 4-оксобензопиран, причем указанный индол или индолин могут быть замещены по атому азота, входящему в состав соответствующего кольца, группой R8, как она определена ниже, а указанный индол, индолин, хинолин, бензофуран, бензотиофен или 4-оксобензопиран может быть замещен по своему бензольному кольцу группой R9, как она определена ниже;
либо
(2) фенил или фенил, одно- или двузамещенный группами, независимо выбранными из числа следующих: атом галогена, гидрокси, карбокси, амино или диметиламино;
либо
(3) NHR11, где R11 соответствует приведенному ниже обозначению или R11 является 7-индазолилом, содержащим в положении N-1 группу R10;
R3 представляет собой атом водорода, С1-6-алкил, C3-6-циклоалкил или фенил;
R4 обозначает независимо C3-6-алкил, С3-6-циклоалкил, фенил, a R5 независимо представляет собой С3-6-алкил, бензил, фенил или фенил, одно- или двузамещенный группами, независимо выбранными из числа следующих: гидрокси, диметиламино, -O(С1-4-алкил), морфолино-группа или атом галогена; либо R4 обозначает С1-2-алкил, а R5 является фенилом, замещенным по своему положению 2 или 4 атомом хлора, метилом, метоксилом или метокси-карбонилом;
R6 соответствует атому водорода или метилу;
R7 представляет собой атом водорода, гидрокси, атом фтора или -О(С1-4алкил);
R8 является группой -(СН2)bCOОН;
R9 обозначает метил, атом хлора, нитро, гидрокси, метоксигруппу или NHR10;
R10 представляет собой атом водорода;
R11 соответствует фенилу или фенилу, одно- или двузамещенному группами, независимо выбранными из числа следующих: атом фтора, С1-4-алкилтио, -(СН2)СООН, -(СH2)cСОО(С1-4-алкил), -(СН2)сSCH3, -(CH2)cSOCH3, -(CH2)cSO2CH3,
-(CH2)cN(C1-4-алкил)2, -(CH2)cNH(SO2CF3), -(CH2)cOR12, -(CH2)cNHR10 или фенилу, однозамещенному -(СН2)c(тетразолилом);
R12 представляет собой атом водорода, С1-6-алкил, -СН2СООН;
n равно 1 или 2;
p равно целому числу от 1 до 4;
b равно целому числу от 0 до 3;
с равно 0 или 1.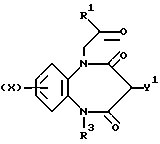
подвергают взаимодействию с соединением формулы R11Y2 (IV), в которых R1, R3 и Х соответствуют обозначениям в формуле (I), одна из групп Y1 и Y2 представляет собой аминогруппу, тогда как другая является группой NCO, NHCOCl или NHCORa, где Ra обозначает нитрозамещенную феноксигруппу или I-имидазольную группу, и R11 соответствует обозначению в формуле (1) или представляет собой группу, превращаемую в указанный остаток, после чего, при необходимости или желании, превращают полученное соединение в другое соединение по изобретению.
| Пожарный двухцилиндровый насос | 0 |
|
SU90A1 |
| Устройство для управления тяговой рамой автогрейдера | 1973 |
|
SU487207A1 |
| US 4988692, 1991 | |||
| Способ получения 2-диалкиламино-4-метил-3н-1,5-бензодиазепинов | 1986 |
|
SU1362732A1 |
| US 4517195 A, 1985 | |||
| DE 3731840 А1, 1988 | |||
| DE 3402507 A, 1985. | |||

