Изобретение касается класса новых фенилиндольных соединений, относящихся к атипичным нейролептическому сертиндолу (1-[2-[4-[5-хлор-1-(4-фторфенил)-1H-индол-3-ил] пиперидин-1-ил] этил]-2- имидазолидинон), но замещенному в 4- и/или 5-положении имидазолидинонового цикла. Указанные соединения или их соли или их пролекарства полезны как антипсихотические, антидепрессантные, антигипертензивные средства и/или при лечении экстрапирамидальных побочных эффектов, вызванных антипсихотическими лекарственными средствами, и негативных симптомов шизофрении.
В патенте США N 4710500, соответствующем ЕР 200322В, раскрыты в основном необязательно замещенные в положении 5 производные 1-арил-3-(4-пиперидил-, 1-арил-3-(1-пиперазинил)- или 1-арил-3-(1,2,3,6-тетрагидро-4-пиридил)индола, у которых заместителями при атоме азота пиперидильной, пиперазинильной или тетрагидропиридильной группы являются водород, или алкил, алкенил или гетероцикл-низший алкил. Было показано, что большинство этих соединений являются мощными антагонистами допамина in vivo с пролонгированным действием, а следовательно, полезны при лечении психозов, и было доказано, что все эти соединения являются сильнодействующими антагонистами рецепторов in vivo, что указывает на их эффективность при лечении экстрапирамидальных побочных эффектов; вызванных антипсихотическими лекарственными средствами, и негативных симптомов шизофрении. Антипсихотическая активность одного соединения, например атипичного нейролептического соединения сертиндола 1-[2-[4-[5-хлор-1-(4-фторфенил)-1H- индол-3-ил]пиперидин-1-ил]этил]-2-имидазолидинона, описана в патенте США N 5112838, соответствующем EP 392959А.
В Международной патентной заявке N WO 92/00070 было установлено, что подкласс соединений 3-(4-пиперидила), включая сертиндол, обладает анксиолитической активностью. Более того, указанные соединения 3-(4-пиперидила), как сообщается, полезны при лечении гипертензии, токсикомании и родственных нарушений (Международные патентные заявки WO 92/15301, WO 92/15302 и WO 92/15303.
Изучение метаболизма показало, что главный циркулирующий метаболит антипсихотического соединения сертиндола присутствует в организме человека. Полагают, что может быть полезным "предотвращение образования указанного главного метаболита.
Следовательно, предметом настоящего изобретение является обеспечение новых лекарственных средств с фармацевтическим профилем, аналогичным профилю сертиндола, в которых образование указанного метаболита замедляется или приостанавливается.
В настоящее время установлено, что указанный главный циркулирующий метаболит сертиндола представляет собой соединение 1-[2-[4-[5-хлор-1-(4-фторфенил)-1H-индол-3-ил] пиперидин-1- ил] этил]-1,3-дигидроимидазол-2-она, структурно идентичного сертиндолу, за исключением присутствия двойной связи в цикле 1,3- дигидроимидазол-2-она. Более того, было установлено, что класс новых фенилиндольных соединений, относящихся к сертиндолу, в которых замедляется или блокируется метаболическое превращение цикла имидазолидинона в цикл 1,3-дигидроимидазол-2-она, имеет фармакологический профиль, аналогичный профилю сертиндола.
Следовательно, настоящее изобретение касается новых соединений с общей формулой I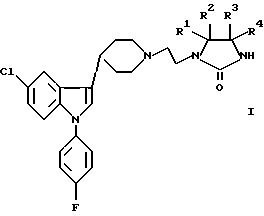
в которой R1 - R4 независимо выбирают из группы, содержащей водород, дейтерий, галоген, C1-6 алкил, циклоалкил, циклоалкил-C1-6 алкил, арил-C1-6 алкил, арил, гидрокси, C1-6 алкокси, циклоалкокси, циклоалкил-C1-6 алкокси, алкилокси, арил-C1-6 алкокси, арилокси, C1-6 алкилтио, циклоалкилтио, циклоалкил-C1-6 алкилтио, арил-C1-6 алкилтио и арилтио, при условии, что все 4 заместителя не могут быть водородом; или по крайней мере одна пара заместителей (R1, R2 или R3, R4) содержит одну оксогруппу или одну триоксогруппу и, если присутствует только одна оксо- или триоксогруппа, то остальные два заместителя выбирают из групп, перечисленных выше для R1-R4, при условии, что оба они не могут быть водородом; или R1 и R2 и/или R3 и R4 соответственно связаны друг с другом, образуя 3 - 8-членный спироцикл, необязательно содержащий в цикле один атом кислорода или серы; или пролекарство, или их кислую дополнительную соль.
Следующий аспект настоящего изобретения относится к методу получения новых соединений формулы I.
Еще один аспект настоящего изобретения касается фармацевтического состава, содержащего новое соединение формулы I и подходящий фармацевтически приемлемый носитель или разбавитель.
Еще один аспект настоящего изобретения касается использования соединений формулы 1 для получения фармацевтического состава для лечения психоза, депрессии, негативных симптомов шизофрении, гипертензии и экстрапирамидальных побочных эффектов, вызванных антипсихотическими лекарственными средствами.
Было установлено, что соединения по настоящему изобретению имеют фармакологический профиль, аналогичный профилю сертиндола, в то время как они не подлежат превращению цикла имидазолидинона в цикл 1,3-дигидроимидазол-2-она или подлежат ему в меньшей степени, причем указанное превращение имеет место относительно сертиндола. Фармакологический профиль соединений по настоящему изобретению указывает на их полезность при лечении описанных выше нарушений.
Некоторые соединения общей формулы I могут существовать в виде своих оптических изомеров, и настоящее изобретение также охватывает и эти оптические изомеры.
В общей формуле I "галоген" обозначает фтор, хлор, бром или йод.
Термин "циклоалкил" обозначает карбоцикл, включающий 3-8 атомов углерода, предпочтительно 3-6 атомов углерода, или бициклический или трициклический углеродный цикл типа адамантила.
Терминами "C1-6 алкил", "C1-6 алкокси", "C1-6 алкилтио" и т.п. обозначены такие разветвленные или неразветвленные группы, содержащие от 1 до 6 атомов углерода, предпочтительно - от 1 до 4 атомов углерода. Примерами таких групп являются метил, этил, 1-пропил, 2-пропил, 1-бутил, 2-бутил, 2-метил-2-пропил, 2-метил-1-пропил, метокси, этокси, 1-пропокси, метилтио, этилтио, 1-пропилтио, 2-пропилтио, метилсульфонил, этилсульфонил и им подобные группы.
Термин "арил" используют для обозначения карбоциклической или гетероциклической ароматической группы, необязательно содержащей один или более заместителей, независимо выбираемых из: водорода, галогена, низшего алкила, низшего алкокси, гидрокси, низшего алкилтио, низшего алкил- или диалкиламино, циано, трифторметила и трифторметилтио. Примерами таких арильных групп являются фенил, тиенил, и фуранил.
Фармацевтически приемлемыми кислыми дополнительными солями указанных соединений, используемых в настоящем изобретении, являются соли, образованные нетоксичными органическими и неорганическими кислотами. Примерами таких органических солей являются соли малеиновой, фумаровой, бензойной, аскорбиновой, янтарной, винной, щавелевой, бис-метиленсалициловой, метансульфоновой, этандисульфоновой, уксусной, пропионовой, винной, салициловой, лимонной, глюконовой, молочной, миндальной, коричной, цитраконовой, аспартиновой, стеариновой, пальмитиновой, итаконовой, гликоновой, п-аминобензойной, глютаминовой, бензолсульфоновой и теофиллинуксусной кислот, а также 8- галотеофиллинов, например, 8-бромтеофиллина. Примерами таких неорганических солей служат соли соляной, оромистоводородной, серной, сульфамидной, фосфорной и азотной кислот.
Пролекарства соединений настоящего изобретения могут быть аналогичны пролекарствам сертиндола, раскрытым в Международной патентной заявке WO 92/06089, или они могут быть эфирами C1-C18 карбоновых кислот, образованных с возможными гидроксигруппами на имидазолиноновом цикле.
В предпочтительном воплощении настоящего изобретения каждый из R1-R4 выбирают из группы, содержащей водород, дейтерий, C1-6 алкил, гидрокси, C1-6 алкокси; или одна или обе пары заместителей (R1, R2 или R3, R4) образуют одну оксогруппу или R1 и R2 и/или R3 и R4) соответственно связаны друг с другом, образуя 3-8-членный спироцикл.
Предпочтительно, чтобы оба заместителя по крайней мере из одной пары заместителей (R1, R2 или R3, R4) были отличны от водорода и образовывали вместе оксогруппу.
Согласно настоящему изобретению новые соединения формулы I получают методом, включающим:
а) взаимодействие 1-замещенного пиперидина II с соединением формулы III: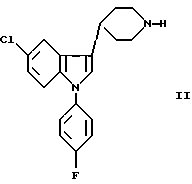
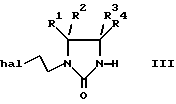
где R1-R4 были определены выше, a "hal" представляет собой хлор, бром или иод;
b) восстановление оксосоединения формулы IV или V до соответствующего гидрокси или метиленового производного: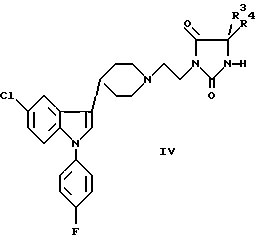
где R1-R4 были определены выше;
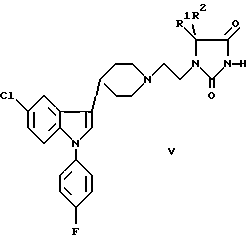
с) взаимодействие производного мочевины VI и бифункциональной группы VII, в результате которого эта группа внедряется, образуя этиленовый мостик в соединении I: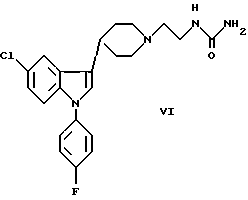
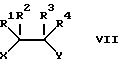
где R1-R4 были определены выше, а X и Y представляют собой хлор, бром, иод, C1-6 алкокси или гидрокси;
d) взаимодействие соединения формулы VIII со спиртом формулы R1'OH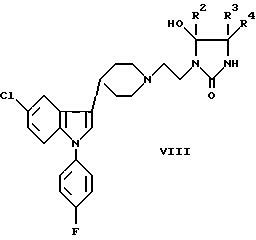
где R2-R4 были определены выше, a R1' представляет собой C1-6 алкил, циклоалкил, циклоалкил-C1-6 алкил, арил-C1-6 алкил или арил;
е) снятие защиты с соединения формулы IX, X, или XI: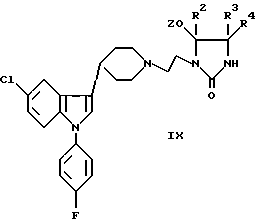
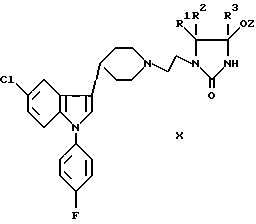
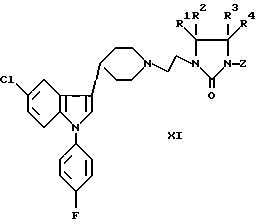
где R1-R4 были определены выше, а защитная группа Z представляет собой триалкилсилил, бензил, ацил или иную защитную группу, которую можно удалить при некислых условиях; или
f) реакцию замыкания цикла производного этилендиамина формулы XII: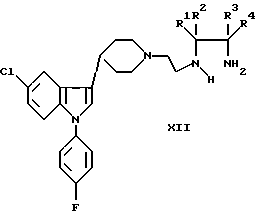
где R1-R4 были определены выше, при использовании мочевины, фосгена, диалкилкарбоната или карбаматов для включения в карбонильную группу при образовании гетероциклического цикла формулы I.
Получение промежуточного соединения II описано в работе Perregaard et al. J. Med. Chem. 1992, 35, 1092-1101, а получение промежуточного соединения III становится ясным из примеров. Алкилирование соединения II и III обычно проводят при повышенных температурах в инертном растворителе, таком как ацетон, метилизобутилкетон или N-метил-2-пирролидон в присутствии основания типа карбоната калия.
Оксосоединения IV и V легко восстановить до соответствующих гидроксисоединений, используя LiAlH4, AlH3, B2H6 или комплекс BH3 при мягких условиях, таких как охлаждение в инертных растворителях, например в диэтиловом эфире или сухом тетрагидрофуране. Получение этих оксосоединений описано в примерах.
Соединения с бифункциональными группами VII представляют собой, например, производные щавелевой кислоты, такие как оксалилхлорид или эфиры щавелевой кислоты или производные глиоксиловой кислоты. Взаимодействие этих производных с соединением VI удобно проводить в кислых или нейтральных условиях.
Взаимодействие соединения формулы VIII со спиртом R1'OH удобно проводить в инертном растворителе или при комнатной, или при повышенной температуре, используя в качестве растворителя спирт.
Защитные группы Z в формулах IХ-ХI удаляют такими методами, как расщепление защитной триалкилсилильной группы фторидом тетраалкиламмония в инертном растворителе или в условиях мягкого гидролиза. Бензильные группы удаляют каталитическим гидрированием, используя, например, в качестве катализатора Pd. Эфиры карбоновых кислот гидролизуются при мягких нейтральных или щелочных условиях, хорошо известных специалистам-химикам. Промежуточные соединения формул IX-XI можно получить общепринятыми методами.
Соединения формулы XII получают согласно методам, изложенным в Международной патентной заявке WO 92/15302, Chem. Abstr. 117 (1992) 247029 или так, как это описано в экспериментальной части. Взаимодействие соединения XII с мочевиной, диалкилкарбонатами или карбаматами обычно проводят при высоких температурах (100-200oC) или с чистыми компонентами, или в инертном апротонном растворителе, таком как N,N-диметилформамид, N-метил-2-пирролидинон или гексаметилфосфорный триамид. Фосген в качестве предшественника карбонила используют при низких температурах в инертных растворителях, таких как толуол, тетрагидрофуран, диэтиловый эфир, необязательно в присутствии основания, типа триэтиламина или карбоната калия.
Кислые аддитивные соли соединений настоящего изобретения легко получают методами, известными в этой области. Основание взаимодействует или с вычисленным количеством органической или неорганической кислоты в водном смешиваемом растворителе, таком как ацетон или этанол, при выделении указанной соли концентрированием или охлаждением, или же взаимодействует с избытком этой кислоты в водном несмешиваемом растворителе, таком как диэтиловый эфир, при этом целевую соль выделяют непосредственно. Эти соли можно также получить классическими методами двойного разложения соответствующих солей.
Указанные соединения общей формулы I, их пролекарства и их фармацевтически приемлемые кислые дополнительные соли можно применять любым подходящим способом, например орально или парентерально, и эти соединения могут быть представлены в любой удобной для такого применения форме, например в форме таблеток, капсул, порошков, сиропов или растворов или дисперсий для инъекций. Эти пролекарства могут быть легко введены в виде запасных препаратов для инъекций, растворенных в специальных маслах.
Эффективная дневная доза указанного соединения общей формулы I или его приемлемой соли составляет от 10 мкг/кг до 10 мг/кг веса тела.
Далее настоящее изобретение иллюстрируется с помощью примеров, которые никак не следует рассматривать ограничивающими данное изобретение.
Все температуры плавления определяли па оборудовании Buchi SMP-20, и они не являются точными. Спектры 1H-ЯМР регистрировались при 250 МГц на спектрофотометре Bruker АС 250. В качестве растворителей использовали дейтерированный хлороформ (99.8%D) или диметилсульфоксид (99.9%D). В качестве относительного внешнего стандарта применяли тетраметилсилан. Величины смещения химических величин выражены в ppm. Для обозначения кратности сигналов ЯМР использовали следующую аббревиатуру: s=синглет, d=дуплет, t=триплет, q=квартет, h= гептет, dd=двойной дуплет, dt=двойной триплет, dq=двойной квартет, tt=триплет триплетов, m=мультиплет.
ПРИМЕР 1 (метод b)
1-[2-[4-[5-хлор-1-(4-фторфенил)-1H-индол-3-ил] пиперидин-1-ил] этил]-5-гидроксиимидазолидин-2-он, 1
1-(2-хлорэтил)имидазолидин-2,5-дион, 1a
К суспензии глицина (49 г) в воде (750 мл) добавили гидроксид натрия (39 г) и затем полученную смесь охладили до 0oC. В течение 0.5 ч при 0-10oC каплями добавляли 2-хлор-этилизоцианат. В течение следующего часа смесь перемешивали при 10oC. pH довели до значения 1, добавив концентрированную соляную кислоту. Осажденное производное глицина отфильтровали, промыли водой и в заключение высушили в вакууме. Выход: 106 г, температура плавления 146-149oC. Все полученные таким образом производные глицина суспендировали в концентрированной соляной кислоте (520 мл) и нагревали в течение 20 мин до расплавления. Подкисленный растворитель выпарили в вакууме, оставшийся сырой продукт растворили в дихлорметане и высушили (безводный MgSO4). Дихлорметан выпарили, а кристаллический, продукт перекристаллизовывали из этилацетата, при этом получили 62 г соединения 1а, указанного в заглавии. Температура плавления 112-114oC.
1-[2-[4-[5-хлор-1-(4-фторфенил)-1H-индол-3-ил] пиперидин- 1-ил]этил]-5-гидрокси-имидазолидин-2-он, 1
Суспензию 5-хлор-1-(4-фторфенил)-3-(4-пиперидинил)-1H-индола (45 г) (полученную так, как это описано в J. Med. Chem. 1992, 35. 1092-1101), 1-(2-хлорэтил)имидазолидин-2,5-диона, 1a (45 г), карбоната калия (40 г) и иодида калия нагревали в колбе с обратным холодильником в течение 5 ч в метилизобутилкетоне (MIBK) (400 мл). Полученную смесь от фильтровали в горячем виде, а MIBK затем выпарили в вакууме. Оставшийся сырой продукт очищали колоночной хроматографией на силикагеле, используя в качестве элюента смесь этилацетат/этанол/триэтиламин (90: 10: 4). Полученное очищенное производное гидантоина перекристаллизовывали из этилацетата. Выход: 21 г, температура плавления 165-166oC. Полученное таким образом производное гидантоина (7 г) растворили в сухом тетрагидрофуране (250 мл). По каплям при 5-10oC добавляли раствор LiAlH4 (0.7 г) в сухом тетрагидрофуране (50 мл). В заключение полученную смесь в течение 5 ч перемешивали при комнатной температуре. После охлаждения до 0oC осторожно добавили водный NaOH (1.2 мл) и воду (3 мл). Неорганические соли отфильтровали, растворители выпарили в вакууме, а оставшиеся твердые частицы перекристаллизовывали из этилацетата. Выход соединения 1, указанного в заглавии, составил 5.3 г, температура плавления 174-175oC. 1H-ЯМР ( δ, CDCl3): 8.45 (широкий s, 1H), 7.60 (d, 1H), 7.40-7.30 (m, 3H), 7.25-7.10 (m, 3H), 7.05 (s, 1H), 5.25 (широкий s, 1H), 5.15 (d, 1H), 3.95 (широкий d, 1H), 3.60-3.50 (m, 1H), 3.25 (широкий d, 2H), 3.05 (t, 2H), 2.85 (tt, 1H), 2.75 (широкий t, 1H), 2.55-2.35 (m, 2H), 2.20- 2.05 (m, 3H), 1.90-1.75 (m, 2H).
ПРИМЕР 2 (метод e)
1-[2-[4-[5-хлор-1-(4-фторфенил)-1H-индол-3-ил] пиперидин-1-ил] этил]-4-гидроксиимидазолидин-2-он, 2
1-[2-[4-[5-хлор-1-(4-фторфенил)-1H-индол-3-ил] пиперидин-1-ил] этил] имидазолидин-2,4-дион, 2а
Цианат калия (30 г) суспендировали в дихлорметане (400 мл) и по каплям при 5-8oC добавляли трифторуксусную кислоту (50 мл). Раствор 5-хлор-3-[1-(N-цианометил-2-амино-этил)-4-пиперидил] -1-(4- фторфенил)-1H-индола (50 г) (Международная патентная заявка WO 92/15302, Chem.Abstr. 117 (1992) 247029) в дихлорметане (200 мл) добавляли при охлаждении, и полученную смесь перемешивали в течение всей ночи при комнатной температуре. Реакционную смесь дважды промывали холодным разбавленным раствором гидроксида натрия, а органическую фазу высушили (безводный MgSO4). Растворитель выпарили в вакууме, получив в результате 53 г сырого производного мочевины, которое перегоняли при нагревании в колбе с обратным холодильником в этаноле (700 мл) и концентрированной соляной кислоте (50 мл) в течение 1 ч. После охлаждения до комнатной температуры полученную смесь отфильтровали, получив в результате 33 г гидрохлоридной соли указанного в заглавии соединения 2а. Эту соль растворили в смеси метанола (150 мл) и триэтиламина (33 мл), и спустя несколько минут свободное основание закристаллизовалось. При этом получили 24 г соединения 2а, указанного в заглавии. Температура плавления 208-210oC.
1-[2-[4-[5-хлор-1-(4-фторфенил)-1H-индол-3-ил] пиперидин-1- ил]-этил]-3-[N-трет-бутилдиметилсилил]имидазолидин-2,4-дион, 2b
Раствор соединения 2а (8.24 г) в дихлорметане (500 мл), тетрагидрофуран (150 мл) и триэтиламин (10 мл) нагревали до расплавления. В течение 15 мин добавляли раствор хлорида трет-бутилдиметилсилила (3.35 г) в дихлорметане (100 мл). Спустя 2 ч добавили другую порцию хлорида трет-бутилдиметилсилила (3 г) в дихлорметане (50 мл) и триэтиламин (10 мл), а спустя еще 2 ч добавили такую же порцию хлорида трет-бутилдиметилсилила. Полученную смесь нагревали в колбе с обратным холодильником в течение ночи, а затем упаривали в вакууме. Остаток растворили в дихлорметане, а затем промыли водой и насыщенным соляным раствором. Органическую фазу высушили (безводный MgSO4), а затем выпарили в вакууме; было получено 12.6 г сырого указанного в заглавии продукта 2b в виде масла.
1-[2-[4-[5-хлор-1-(4-фторфеннл)-1H-индол-3-ил]пиперидин-1-ил]этил]-4-гидрокси-3-[N-трет-бутилдиметилсилил]имидазолидин-2-он, 2с
К раствору соединения 2b (16.2 г) в эфире (1 л) добавляли порциями в течение 40 мин при комнатной температуре литий алюминий гидрид (1.62 г). Эту смесь перемешивали в течение последующих 20 мин. Гашение реакции проводили водой и концентрированным гидроксидом калия, полученную смесь отфильтровали и выпарили в вакууме. После очистки колоночной хроматографией (силикагель, элюент: триэтиламин/этанол/этилацетат 4:4:100) было получено 10.1 г соединения 2с, указанного в заглавии. Температура плавления 154-155oC.
1-[2-[4-[5-хлор-1-(4-фторфенил)-1H-индол-3-ил]пиперидин-1-ил]этил]-4-гидроксиимидазолидин-2-он, 2
Раствор тригидрата фторида тетрабутиламмония (1.5 г) в тетрагидрофуране (600 мл) охладили до -53oC. Раствор соединения 2с (1.5 г) в тетрагидрофуране (300 мл) добавляли в течение 15 мин при -53oC. Полученную смесь перемешивали при -53oC в течение следующих 90 мин, а затем охладили до -70oC и при 10oC вылили на насыщенный раствор хлорида натрия. Полученную смесь экстрагировали дихлорметаном, органическую фазу промыли насыщенным соляным раствором, высушили (безводный MgSO4) и выпаривали в вакууме при комнатной температуре. Остаток повторно растворили в этилацетате и встряхивали с насыщенным соляным раствором, а затем добавили концентрированный водный аммоний до pH 12. Полученную органическую фазу высушили (безводный MgSO4) и выпаривали в вакууме при комнатной температуре. После очистки колоночной хроматографией (силикагель, элюент: триэтиламин/этанол/этилацетат 15:15:70) получили соединение, указанное в заглавии, которое кристаллизовалось из этилацетата, при этом получили 0.294 г соединения 2. Температура плавления 176-178oC. 1H-ЯМР δ, DMSO-d6): 7.70 (d, 1H), 7.62-7.58 (m, 2H), 7.47 (s, 1H), 7.47-7.34 (m, 3H), 7.16 (dd, 1H), 7.00 (s, 1H), 5.82 (d, 1H), 5.02 (t, 1H), 3.56 (dd, 1H), 3.28-3.17 (m, 2H), 3.13 (dd, 1H), 2.99 (широкий d, 2H), 2.80 (tt, 1H), 2.42 (t, 2H), 2.11 (широкий t, 2H), 1.95 (широкий d, 2H), 1,69 (q, 2H).
ПРИМЕР 3 (метод d)
1-[2-[4-[5-хлор-1-(4-фторфенил)-1H-индол-3-ил] пиперидин-1-ил] этил]-5-метоксиимидазолидин-2-он, фумарат 3
Раствор 1-[2-[4-[5-хлоро-1-(4-фторфенил)-1H-индол-3-ил] пиперидин-1-ил] этил]-5-гидроксиимидазолидин-2-она (соединение 1) (3 г) в метаноле (1000 мл) нагревали в колбе с обратным холодильником в течение 24 ч. Избыток метанола выпарили в вакууме. Оставшийся сырой продукт растворили в ацетоне и добавили фумаровую кислоту (0.75 г). При нагревании соединение 3, указанное в заглавии, кристаллизовалось в виде фумарата. Выход: 2.7 г, температура плавления 126-127oC. 1H-ЯМР ( δ, ) DMSO-d6): 7.75 (d, 1H), 7.65-7.55 (m, 2H), 7.50 (s, 1H), 7.50-7.30 (m, 3H), 7.15 (dd, 1H), 6.65 (широкий s, 1H), 6.60 (s,2H), 5.10 (d, 1H), 3.60-3.45 (m, 1H), 3.40-3.15 (m, 5H), 3.15 (s, 3H), 2.95 (широкий t, 1H), 2.80 (t, 2H), 2.55 (t, 2H), 2.05 (широкий d, 2H), 2.00-1.80 (m, 2H).
ПРИМЕР 4 (метод a)
1-[2-[4-[5-хлор-1-(4-фторфенил)-1H-индол-3-ил] пиперидин-1-ил] этил] - 4,4,5,5-тетрадейтероимидазолидин-2-он, 4
N-(2-бензилоксиэтил)оксамид, 4a
Раствор 2-бензилоксиэтиламина (25 г) и карбоната калия (50 г) в дихлорметане (500 мл) обрабатывали, по каплям метилхлороксоацетатом (25 г) при 0oC. После перемешивания в течении 3 ч при комнатной температуре эту смесь промыли водным бикарбонатом натрия и высушили над сульфатом магния. Удалив в вакууме растворитель, получили метил(2-бензилоксиэтил)аминооксоацетат в виде масла. Выход: 40 г. Это масло растворили в тетрагидрофуране, добавили концентрированный водный аммиак (500 мл), а затем в течение 0.5 ч проводили нагревание в колбе с обратным холодильником. После охлаждения фазы разделили, водную фазу экстрагировали дихлорметаном, а собранные вместе органические фазы высушивали над сульфатом магния. Удалив растворитель, получили соединение 4а в виде кристаллического вещества. Выход: 32 г.
2(2-бензилоксиэтиламино)-1,1,2,2-тетрадейтероэтиламин, 4b
Суспензию литийалюминий дейтерида (20 г) в тетрагидрофуране (600 мл) по порциям обрабатывали соединением 4а (22 г). После нагревания в колбе с обратным холодильником в течение 16 ч эту смесь охлаждали, а потом обрабатывали последовательноводой (40 мл), 15% гидроксидом натрия (20 мл) и водой (100 мл). Смесь отфильтровали, а осадок экстрагировали в дихлорметане (1000 мл). Полученный фильтрат выпарили в вакууме, а оставшееся масло смешали с дихлорметановой фазой. После высушивания над сульфатом магния и удаления в вакууме растворителя было получено соединение 4b в виде желтого масла. Выход составил 19 г.
1(2-хлорэтил)-4,4,5,5-тетрадейтероимидазолидин-2-он, 4с
Смесь соединения 4b (19 г) и мочевины (7.5 г) нагревали в течение 1 ч при 180oC. После очистки колоночной хроматографией (силикагель, элюент: метанол/этилацетат 1: 4) был получен 1-(2- бензилоксиэтил)-4,4,5,5-тетрадейтероимидазолидин-2-он в виде масла. Выход: 13.2 г. Это масло растворили в этаноле (200 мл) и добавили 5% Pd/C (3.5 г). Полученную смесь обрабатывали в течение 16 ч водородом под давлением 4 атм. После фильтрования и удаления растворителя было получено 7.8 г 1-(2-гидроксиэтил)-4,4,5,5-тетрадейтероимидазолидин-2-она в виде масла. Это масло суспендировали в сухом толуоле (50 мл). Добавили тионилхлорид (7 мл) и диметилформамид (0.5 мл), а потом в течение 2 ч нагревали до 70oC. Полученную смесь выпарили в вакууме, добавили соляной раствор, а затем провели экстракцию дихлорметаном. После высушивания над сульфатом магния, обработки углем и удаления в вакууме растворителя было получено указанное в заглавии соединение, в виде кристаллического твердого вещества. Выход: 7.4 г.
1[2-[4-[5-хлор-1-(4-фторфенил)-1H-индол-3-ил] пиперидин-1- ил] этил] -4,4,5,5-тетрадейтероимидазолидин-2-он, 4
Смесь 5-хлор-1-(4-фторфенил)-3-(4-пиперидинил)-1H-индола (12.5 г), соединения 4с (4.5 г), карбоната калия (10 г) и иодида калия (1 г) в изопропилметилкетоне выдерживали в течение 16 ч при 95oC. Смесь охладили, отфильтровали и выпарили в вакууме. После очистки колоночной хроматографией (силикагель, элюент: триэтиламин/метанол/этилацетат 1:2:17) было получено указанное в заглавии соединение в виде твердых частиц. Перекристаллизацией из этанола было получено 7.8 г кристаллического соединения 4. Температура плавления 159-162oC. 1H-ЯМР ( δ, ) CDCl3): 7.64 (d, 1H), 7.45-7.28 (m. 3H), 7.24-7.09 (m, 3H), 7.08 (s, 1H), 5.13 (s, 1H), 3.38 (t, 2H), 3.12-3.01 (m, 2H), 2.89-2.72 (m, 1H), 2.57 (t,2H), 2.28-2.13 (m, 2H), 2.11-1.98 (m, 2H), 1.90-1.69 (m, 2H).
ПРИМЕР 5 (метод a)
1[2-[4-[5-хлор-1-(4-фторфенил)-1H-индол-3-ил] пиперидин-1-ил] этил]-4,4, дидейтероимидазолидин-2-он, 5
(2-бензилоксиэтил)аминоацетонитрил, 5а
Смесь 2-бензилоксиэтиламина (40 г) и триэтиламина (50 мл) в тетрагидрофуране (500 мл) обрабатывали по каплям хлорацетонитрилом (23 г) при комнатной температуре. После нагревания в колбе с обратным холодильником в течение 2 ч эту смесь охладили и отфильтровали, добавили воду (400 мл), а полученную смесь экстрагировали дихлорметаном. Растворитель удалили в вакууме, добавили эфир (400 мл) и эфирный раствор обработали сульфатом магния и углем. После фильтрования и удаления в вакууме растворителя получили соединение 5а в виде желтого масла. Выход: 39 г.
2-(2-бензилоксиэтиламино)-1,1-дидейтероэтиламин, 5b
Суспензию литийалюминий дейтерида (15 г) в сухом тетрагидрофуране (400 мл) обрабатывали по порциям под азотом при комнатной температуре хлоридом алюминия (23 г). После перемешивания в течение 0.5 ч по каплям добавляли раствор соединения 5а (29 г) в тетрагидрофуране (50 мл), а затем в течение 1 ч проводили нагревание в колбе с обратным холодильником. После охлаждения медленно добавили 15% гидроксид натрия (80 мл), а потом воду (40 мл). Смесь отфильтровали, а осадок экстрагировали дихлорметаном (800 мл). После фильтрования фильтрат выпарили в вакууме, а оставшееся масло смешали с дихлорметановой фазой. После высушивания над сульфатом магния и удаления растворителя в вакууме было получено 22 г смеси соединения 5b и 2-бензилоксиэтилметиламина. Полученную смесь непосредственно использовали на следующей стадии.
1-(2-хлорэтил)-4,4-дидейтероимидазолидин-2-он, 5с
Смесь соединения 5b и 2-бензилоксиэтилметиламина (22 г) и мочевины (7.5 г) нагревали в течение 1 ч при 180oC. После очистки колоночной хроматографией (силикагель, элюент: триэтиламин/метанол/этилацетат 5:2:93) было получено 6.4 г 1-(2- бензилоксиэтил)-4,4-дидейтероимидазолидин-2-она в виде масла. Это масло растворили в этаноле (100 мл) и добавили 5% Pd/C (1.5 г). В течение 16 ч смесь обрабатывали водородом под давлением 4 атм. После фильтрования и удаления растворителя было получено 3.2 г 1- (2-гидроксиэтил)-4,4-дидейтероимидазолидин-2-она в виде масла. Это масло суспендировали в сухом толуоле (25 мл). Добавили тионилхлорид (3.5 мл) и диметилформамид (0.1 мл), а потом нагревали в течение 2 ч до 70oC. Полученную смесь упарили в вакууме, добавили соляной раствор, а потом экстрагировали дихлорметаном. После высушивания над сульфатом магния, обработки углем и удаления растворителя в вакууме получили указанное в заглавии соединение в виде кристаллического твердого вещества. Выход: 4.1 г.
1[2-[4-[5-хлор-1-(4-фторфенил)-1H-индол-3-ил] пиперидин- 1-ил] этил] -4,4-дидейтероимидазолидин-2-он, 5
Это соединение получали методикой, аналогичной той, которая описана для синтеза соединения 4; в качестве исходного использовали соединение 5с. Температура плавления 159-162oC. 1H-ЯМР ( δ, CDCl3): 7.64 (d, 1H), 7.45-7.28 (m, 3H), 7.24-7.09 (m, 3H), 7.08 (s, 1H), 4.45 (s, 1H), 3.56 (s, 2H), 3.38 (t, 2H), 3,12-3.01 (m, 2H), 2.89-2.72 (m, 1H), 2.57 (t, 2H), 2.28-2.13 (m, 2H), 2.11-1.98 (m, 2H), 1.90-1.69 (m, 2H).
ПРИМЕР 6 (метод a)
1[2-[4-[5-хлор-1-(4-фторфенил)-1H-индол-3-ил] пиперидин-1-ил] этил] -5,5-дидейтероимидазолидин-2-он, 6
Азидо-N-(2-бензилоксиэтил)ацетамид, 6а
Раствор 2-бензилоксиэтиламина (15 г) и карбоната калия (40 г) в ацетоне (400 мл) обрабатывали по каплям азидоацетилхлоридом при 20oC. Полученную смесь после перемешивания при комнатной температуре в течение 3 ч отфильтровали и выпарили в вакууме. Добавили дихлорметан (800 мл) и этот раствор промыли водным бикарбонатом натрия. Органическую фазу высушили над сульфатом магния. После удаления в вакууме растворителя получили желтое масло, которое отфильтровали через силикагель с этилацетатом. После удаления в вакууме растворителя получено, соединение 6а в виде бесцветного масла. Выход: 22 г.
2-(2-бензилоксиэтиламино)-2,2-дидейтероэтиламин. 6b
Суспензию литийалюминий дейтерида (10 г) в тетрагидрофуране (300 мл) обрабатывали по каплям раствором соединения 6а (14 г) в тетрагидрофуране (50 мл). После нагревания в колбе с обратным холодильником в течение 16 ч эту смесь охладили и обрабатывали последовательно водой (20 мл), 15% гидроксидом натрия (10 мл) и водой (50 мл). Потом смесь отфильтровали, а осадок экстрагировали дихлорметаном (600 мл). Фильтрат выпарили в вакууме, а оставшееся масло смешали с дихлорметановой фазой. После высушивания над сульфатом магния и удаления в вакууме растворителя получили соединение 6b в виде желтого масла. Выход: 9.4 г
1-(2-хлорэтил)-5,5-дидейтероимидазолидин-2-он, 6с
Соединение получали по методике, аналогичной той, что описана для синтеза соединения 4с; в качестве исходного применяли соединение 6b.
1[2-[4-[5-хлор-1-(4-фторфенил)-1H-индол-3-ил] пиперидин-1-ил] этил] -5,5-дидейтероимидазолидин-2-он, 6
Соединение получали по методике, аналогичной той, что описана для синтеза соединения 4; в качестве исходного применяли соединение 6с. Температура плавления 160-162oC. 1H-ЯМР ( δ, CDCl3): 7.64 (d,1H), 7.45-7.28 (m, 3H), 7.24-7.09 (m, 3H), 7.08 (s, 1H), 4.78 (s. 1H), 3.41 (s, 2H), 3.38 (t, 2H), 3.12-3.01 (m, 2H), 2.89-2.72 (m, 1H), 2.57 (t, 2H), 2.28-2.13 (m, 2H), 2.11-1.98 (m, 2H), 1.90-1.69.(m, 2H).
ПРИМЕР 7 (метод с)
1 [2-[4-[5-хлор-1-(4-фторфенил)-1H-индол-3-ил]пиперидин-1-ил] этил]-4-гидроксиимидазолидин-2,5-дион, 7а и
1[2-[4-[5-хлор-1-(4-фторфенил)-1H-индол-3-ил]пиперидин-1-ил] этил]-5-гидроксиимидазолидин-2,4-дион, 7b
5-хлоро-1-(4-фторфенил)-3-[1-(2-уреидоэтилпиперидин-4- ил]-1H-индол, 7с
Цианат калия (4.9 г) суспендировали в дихлорметане (50 мл), а потом добавляли по каплям при 0oC трифторуксусную кислоту (4.4 мл). По каплям добавляли раствор 3-[1-(2- аминоэтил)пиперидин-4-ил] -5-хлор-1-(4-фторфенил)-1H-индола (10.8 г) (Международная патентная заявка WO 9215302, Chem. Abstr. 117 (1992) 247029) в дихлорметане (100 мл), а потом перемешивали в течение 6 ч при комнатной температуре. Добавили воду (100 мл) и сделали реакционную смесь щелочной с помощью концентрированного аммиака. Получившиеся фазы разделили с последующей экстракцией дихлорметаном. Собранные вместе органические фазы высушили над сульфатом магния. После удаления в вакууме растворителя было получено густое масло, которое очищали тонкослойной хроматографией (силикагель, элюент: триэтиламин/метанол/этилацетат 5:20:75). Указанное в заглавии соединение было выделено в виде кристаллического вещества. Выход: 7.9 г, температура плавления 161-163oC.
1[2-[4-[5-хлор-1-(4-фторфенил)-1H-индол-3-ил]пиперидин- 1-ил]этил]-4-гидроксиимидазолидин-2,5-дион, 7а и
1[2-[4-[5-хлор-1-(4-фторфенил)-1H-индол-3-ил] пиперидин-1-ил] этил]- 5-гидроксиимидазолидин-2,4-дион, 7b
Смесь соединения 7с (1.6 г) и моногидрат глиоксиловой кислоты (0.5 г) в 80% уксусной кислоте (25 мл) нагревали в колбе с обратным холодильником в течение 1 ч. Эту реакционную смесь выпаривали в вакууме с последующим добавлением насыщенного раствора бикарбоната натрия (100 мл). После экстракции дихлорметаном, высушивания органической фазы над сульфатом магния и удаления в вакууме растворителя получили густое масло, которое отделяли тонкослойной хроматографией (силикагель: триэтиламин/метанол/этилацетат 5:2:93).
Фракция 1: кристаллическое вещество 7а, температура плавления 128-135oC. Выход 0.2 г. 1H-ЯМР ( δ, CDCl3): 7.59 (d, 1H), 7.45-7.28 (m, 3H), 7.24-7.08 (m, 4H), 5.35 (s, 1H), 3.71-3.56 (m, 2H), 3.40-3.22 (m, 2H), 3.00-2.54 (m, 3H), 2.32-1.71 (m, 6H).
Фракция 2: кристаллическое вещество 7b, температура плавления 172-180oC. Выход 0.35 г. 1H-ЯМР ( δ, CDCl3): 7.48-7.29 (m, 3H), 7.28- 7.12 (m, 3H), 7.09 (s, 1H), 5.09 (s, 1H), 4.20-4.06 (m, 1H), 3.37- 3.01 (m, 3H), 3.00-2.82 (m, 1H), 2.81-2.63 (m, 1H), 2.61-2.42 (m, 2H), 2.32-2.04 (m, 3H), 1.98-1.74 (m, 2H).
ПРИМЕР 8 (метод a)
1[2-[4-[5-хлор-1-(4-фторфенил)-1H-индол-3-ил] пиперидин-1-ил] этил] -4,4-диметилимидазолидин-2-он, 8
2-(2-бензилоксиэтиламино)-1,1-диметилэтиламин, 8а
К смеси 2-бром-2-метилпропионил бромида (76 г), K2CO3 (55 г) и дихлорметана (700 мл) добавляли при -10oC раствор 2-бензилоксиэтиламина (50 г) в дихлорметане (500 мл). После перемешивания в течение 1.5 ч при комнатной температуре эту смесь промыли водой (500 мл) и высушили (MgSO4). После выпаривания в вакууме растворителя получили N-(2-бензилоксиэтил)-2-бром-2-метилпропионамид в виде масла. Выход: 104 г.
Смесь сырого амида (104 г), азида натрия (23.6 г) и N-метил-2-пирролидона (500 мл) нагревали в течение 22 ч при 50oC. После охлаждения до комнатной температуры добавили воду и полученную смесь экстрагировали диметиловым эфиром (дважды по 400 мл). Собранные вместе органические фазы промыли соляным раствором (трижды по 500 мл) и высушили (Na2SO4). После выпаривания в вакууме растворителя получили 2-азидо-N-(2- бензилоксиэтил)-2-метилпропионамид в виде масла. Выход: 78 г.
Раствор 2-азидо-N-(2-бензилоксиэтил)-2-метилпропионамида (70 г) в тетрагидрофуране (750 мл) добавляли в течение 1 ч при 0oC к суспензии LiAlH4 (20 г) в тетрагидрофуране (500 мл). После нагревания в течение 4 ч в колбе с обратным холодильником смесь охладили до 0oC, добавили воду (20 мл) и водный NaOH (20 мл). Неорганические соли отфильтровали, а растворители высушили. После выпаривания в вакууме этих растворителей было получено соединение 8а в виде масла. Выход 53 г.
1-(2-хлорэтил)-4,4-диметилимидазолидин-2-он, 8b
Был получен по методике, аналогичной той, что описана для синтеза соединения 4с; в качестве исходного использовали соединение 8а.
1[2-[4-[5-хлор-1-(4-фторфенил)-1H-индол-3-ил] пиперидин-1- ил] этил]-4,4-диметилимидазолидин-2-он, 8
Был получен по методике, аналогичной той, что описана для синтеза соединения 4; в качестве исходного использовали соединение 8b. Температура плавления 142-144oC. 1H-ЯМР (CDCl3) δ: 7.65 (d, 1H), 7.45-7.30 (m, 3H), 7.25-7.10 (m, 3H), 7.05 (s, 1H), 4.40 (s, 1H), 3.35 (t, 2H), 3.25 (s, 2H), 3.25-3.00 (m, 2H), 2.90-2.70 (m, 1H), 2.60 (t, 2H), 2.30-2.15 (m, 2H), 2.15-2.00, 1.90-1.70 (m, 2H), 1.30 (s, 6H).
ПРИМЕР 9 (метод а)
1[2-[4-[5-хлор-1-(4-фторфенил)-1H-индол-3-ил] пиперидин-1-ил] этил] -5,5-диметилимидазолидин-2-он, 9
1-(2-бензилоксиэтил)-5,5-диметилимидазолидин-2,4-дион, 9а
Смесь 2-бензилоксиэтиламина (50 г), этил-2-бром-2-метилпропионата (97.5 г) и N-метил-2-пирролидона (1 л) в течение 7 ч нагревали при 100oC. Полученную смесь охладили до комнатной температуры, добавили воду, а потом эту смесь экстрагировали этилацетатом (дважды по 500 мл). Собранные вместе органические фазы промыли водой (четыре раза по 500 мл) и соляным раствором (дважды по 500 мл), высушили (Na2SO4), а растворители выпарили в вакууме. После очистки оставшегося масла колоночной хроматографией на силикагеле (этилацетат, содержащий 4% триэтиламина) был получен 2-(2-бензилоксиэтиламино)-2-метилпропионат в виде масла. Выход: 30 г.
Смесь 2-(2-бензилоксиэтиламино)-2-метилпропионата (29 г) и мочевины (8.5 г) нагревали в течение 1 ч при 180oC. После охлаждения до комнатной температуры добавили воду (250 мл) и полученную смесь экстрагировали диэтиловым эфиром (500 мл). Органические фазы промыли соляным раствором (400 мл) и высушили (Na2SO4). Выпаривание в вакууме растворителей дало соединение 9а в виде масла. Выход: 28 г.
1-(2-бензилоксиэтил)-5,5-диметилимидaзoлидин-2-oн, 9b
Раствор соединения 9а (28 г) в тетрагидрофуране (250 мл) добавили к суспензии LiAlH4 (8 г) в тетрагидрофуране (250 мл) при 20-25oC. После перемешивания в течение 1 ч при комнатной температуре смесь охладили до 0oC, добавили воду (10 мл) и водный NaOH (10 мл). Неорганические соли отфильтровали, а растворители выпарили в вакууме. Оставшееся масло очистили колоночной хроматографией на силикагеле (этилацетат), при этом получили соединение 9b в виде масла. Выход: 10 г.
1-(2-бензилоксиэтил)-5,5-диметилимидазолидин-2-он, 9с
Соединение 9b (9 г) растворили в этаноле (150 мл) и добавили 5% Pd/C (2.0 г). Эту смесь обрабатывали в течение 36 ч водородом под давлением 3 атм. После фильтрования и удаления растворителя получили 1-(2-гидроксиэтил)-5,5-диметилимидазолидин-2-он в виде масла. Выход: 5.1 г
Это масло суспендировали в сухом толуоле (50 мл). Добавили тионилхлорид (5 мл) и диметилформамид (0.5 мл) при последующем нагревании в течение 2 ч до 70oC. Смесь выпарили в вакууме, добавили соляной раствор (100 мл), а потом проводили экстракцию дихлорметаном (100 мл). Полученную органическую фазу высушили (MgSO4), и после выпаривания в вакууме растворителей получили указанное в заглавии соединение в виде масла. Выход: 5 г.
1-[2-[4-[5-хлор-1-(4-фторфенил)-1H-индол-3-ил] - пиперидин-1-ил]этил]-5,5-диметилимидазолидин-2-он, 9
Соединение было получено по методике, аналогичной той, что описана для синтеза соединения 4; в качестве исходного использовали соединение 9с. Температура плавления 146-148oC. 1H-ЯМР (CDCl3) δ: 7.65 (d, 1H), 7.45-7.30 (m, 3H), 7.25-7.10 (m, 3H), 7.05 (s, 1H), 4.70 (s, 1H), 3.25 (t, 2H), 3.15 (s, 2H), 3.20-3.05 (m, 2H), 2.90-2.70 (m, 1H), 2.60 (t, 2H), 2.35-2.20 (m, 2H), 2.15-2.00 (m, 2H), 1.95-1.70 (m, 2H), 1.30 (s, 6H).
ПРИМЕР 10 (метод f)
1-[2-[4-[5-хлор-1-(4-фторфенил)-1H-индол-3-ил] пиперидин-1-ил] этил]-5,5-диметилимидазолидин-2,4-дион, 10
Соединение было получено по методике, аналогичной той, что описана для синтеза соединения 9а; в качестве исходного использовали 2-[4-[5-хлор-1-(4-фторфенил)-1H-индол-3-ил]пиперидин-1-ил]этиламин (полученный так, как это описано для подобных соединений в J. Mod. Chem. 1992, 35, 4813-4822). Полученное соединение осаждали из этилацетата в виде триэтиламмониевой соли. Температура плавления 140-142oC. 1H-ЯМР (CDCl3) δ: 7.65 (d, 1H), 7.45-7.30 (m, 3H), 7.25-7.10 (m, 3H), 7.05 (s, 1H), 3.65 (q, 6H), 3.45 (t, 2H), 3.20-3.05 (m, 2H), 2.90- 2.80 (m, 1H), 2.70 (t, 2H), 2.40-2.20 (m, 2H), 2.15-2.00 (m, 2H), 1.95-1.70 (m, 2H), 1.50 (t, 9H), 1.45 (s, 6H).
ПРИМЕР 11 (метод b)
1-[2-[4-[5-хлор-1-(4-фторфенил)-1H-индол-3-ил] пиперидин-1-ил] этил]-4,5-дигидроксиимидазолидин-2-он, 11
Раствор соединения 7а (0.5 г) в тетрагидрофуране (25 мл) охладили до 0oC и порциями обрабатывали LiAlH4 (15 мг), строго сохраняя температуру 0oC. После перемешивания в течение 2 ч при 0oC порциями добавили еще LiAlH4 (15 мг) с последующим перемешиванием в течение 2 ч при 0oC. Провели гашение реакции водой/водным 4N NaOH, а потом в вакууме удалили растворитель. Добавили метиленхлорид (25 мл) и полученный раствор высушили над MgSO4 и выпарили в вакууме. Оставшееся масло очищали тонкослойной хроматографией (силикагель, элюент: этилацетат/метанол/триэтиламин 85:10:5). Указанное в заглавии соединение было получено в виде кристаллического материала. Температура плавления 190-192oC. Выход: 74 мг. 1H-ЯМР (DMSO-d6) δ: 7.70 (d, 1H), 7.65-7.50 (m, 2H), 7.50-7.30 (m, 4H), 7.15 (t, 1H), 4.65 (s, 1H), 4.60 (s, 1H), 3.55-3.05 (m, 3H), 3.00-2.70 (m, 2H), 2.60-2.20 (m, 3H), 2.205-1.85 (m, 3H), 1.85-1.60 (m, 2H).
ПРИМЕР 12
3-[2-[4-[5-хлор-1-(4-фторфенил)-1H-индол-3-ил] пиперидин-1-ил] этил]-1,3-диазаспиро[4,4]нонан-2-он, 12
1-азидо-N-(2-6ензилоксиэтил)циклопентан-1-карбоксамид, 12а
1-азидоциклопентан-1-карбонилхлорид (16.8 г) добавили к смеси 2-бензилоксиэтиламина (14.7 г), K2CO3 (16.6 г) и ацетона при 0oC. После перемешивания в течение 2 ч при комнатной температуре полученную смесь отфильтровали, выпарили в вакууме и после добавления воды (100 мл) экстрагировали диэтиловым эфиром (дважды по 100 мл). Собранные вместе органические фазы промыли соляным раствором и высушили (Na2SO4). После выпаривания растворителей получили сырое соединение 12а в виде масла. Выход: 22.3 г.
1-(2-бензилоксиэтиламинометил)циклопентиламин, 12b
Раствор сырого 1-азидо-N-(2-бензилоксиэтил)циклопентан-1-карбоксамида 12а (22.3 г) в тетрагидрофуране (100 мл) добавили к суспензии LiAlH4 (6 г) в тетрагидрофуране (100 мл). После нагревания в течение 2 ч в колбе с обратным холодильником полученную смесь охладили до 0oC и обрабатывали последовательно водой (6 мл), 15% водным NaOH (6 мл) и водой (6 мл). Смесь отфильтровали, а осадок экстрагировали дихлорметаном (200 мл). После высушивания собранных вместо органических фаз (MgSO4) и выпаривания в вакууме растворителей получили указанное в заглавии соединение в виде масла. Выход: 18.7 г.
3-(2-хлорэтил)-1,3-диазаспиро[4,4]нонан-2-он, 12с
Это соединение получали согласно методике, аналогичной той, что описана для синтеза соединения 4с; в качестве исходного использовали соединение 12b.
3-[2-[4-[5-хлор-1-(4-фторфенил)-1H-индол-3-ил] пиперидин-1-ил] этил]-1,3-диазаспиро[4,4]нонан-2-он, 12d
Это соединение получали согласно методике, аналогичной той, что описана для синтеза соединения 4; в качестве исходного использовали соединение 12с. Температура плавления 176-78oC. 1H-ЯМР (CDCl3) δ: 7.65 (d, 1H), 7.45-7.30 (m, 3H). 7.25-7.10 (m, 3H), 7.05 (s, 1H), 4.75 (s, 1H), 3.40-3.30 (m. 4H), 3.15-3.00 (m, 2H), 2.90-2.70 (m, 1H), 2.55 (t, 2H), 2.30-2.15 (m, 2H), 2.15-1.95 (m, 2H), 1.90-1.55 (m, 10H).
ПРИМЕР 13
1-[2-[4-[5-хлор-1-(4-фторфенил)-1H-индол-3-ил] пиперидин-1-ил] этил]-1,3-диазаспиро[4,4]нонан-2-он, 13
1-(2-бензилоксиацетамидо)циклопентил-1-кapбoкcaмид, 13а
Бензилоксиацетилхлорид (16.7 г) добавили к смеси 1-аминоциклопентан-1-карбоксамида (10.2 г), K2CO3 (13.3 г) и ацетона (150 мл) при температуре от -5 до 0oC. После перемешивания в течение 2 ч при комнатной температуре реакционную смесь отфильтровали. Осадок экстрагировали диэтиловым эфиром (50 мл), промыли водой (100 мл) и высушивали в вакууме при 50oC, в результате было получено указанное в заглавии соединение в виде кристаллического вещества. Выход: 13.8 г.
N-(1-аминометилциклопентил)-N-(2-бензилоксиэтил)амин, 13b
Это соединение получали согласно методике, аналогичной той, что описана для синтеза соединения 12b; в качестве исходного использовали соединение 13а.
1-(2-хлорэтил)-1,3-диазаспиро[4,4]нонан-2-он, 13с
Это соединение получали согласно методике, аналогичной той, что описана для синтеза соединения 4с; в качестве исходного использовали соединение 13b.
1-[2-[4-[5-хлор-1-(4-фторфенил)-1H-индол-3-ил] пиперидин- 1-ил]этил]-1,3-диазаспиро[4,4]нонан-2-он, 13
Это соединение получали согласно методике, аналогичной той, что описана для синтеза соединения 4; в качестве исходного использовали соединение 13с. Температура плавления 143-145oC. 1H-ЯМР (CDCl3) δ: 7.65 (d, 1H), 7.45-7.30 (m, 3H), 7.25-7.10 (m, 3H), 7.05 (s, 1H), 4.65 (s, 1H), 3.30-3.15 (m, 4H), 3.15-3.05 (m, 2H), 2.90-2.75 (m, 1H), 2.60 (t, 2H), 2.35-2.20 (m, 2H), 2.10-2.00 (m, 2H), 1.95-1.50 (m. 10H).
Фармакология
Соединения, используемые в настоящем изобретении, испытывались в соответствии со следующими получившими признание и надежными, методами испытаний.
Ингибирование связывания 3H-Кетастерина с рецепторами 5-HT2 в коре головного мозга крыс in vitro.
С помощью этого метода определяли in vitro ингибирование лекарственными препаратами связывания 3H-кетастерина (0.5 нМ) с рецепторами 5-HT2 в мембранах, полученных от крыс. Этот метод описан в работе Hyttel, Pharmacology & Toxicology 61, 126-129, 1987. Результаты приведены в таблице 1.
Ингибирование связывания 3H-спиперона с рецепторами допамина D2 в полосатом теле мозга крыс in vitro.
С помощью этого метода определяли in vitro ингибирование лекарственными препаратами связывания 3H-спиперона (0.5 нМ) с рецепторами допамина 02 в полосатом теле мозга крыс in vitro. Этот метод и результаты, полученные для стандартных соединений, описаны в работе Hyttel & Larsen, J. Neurochem, 44, 1615-1622, 1985). Результаты приведены в таблице 1.
Ингибирование квипазином
Квипазин представляет собой агонист 5-HT2, индуцирующий у крыс подергивание головой. Приведенное испытание представляет собой тест in vitro для влияния антагонистического воздействия 5-HT2, которым измеряют способность ослаблять квипазин, индуцирующий подергивания головы. Этот метод и результаты испытаний для некоторых стандартных веществ опубликованы Arnt et al. (Drug Development Research, 16, 59-70, 1989). Результаты приведены в таблице 2.
Каталепсия
Оценку каталепсии проводили согласно Arnt (Eur. J. Pharmacol. 90, 47-55 (1983)). Испытуемое соединение давали в разных дозах. Крысу (170-240 г) помещали на вертикальную проволочную сетку (диаметром 12 мм). Крысу считали каталептогенной, если она оставалась неподвижной в течение более чем 15 с. При каждой дозировке в группах регистрировали максимальное число крыс, демонстрирующих каталепсию в течение первых 6 ч. Эти результаты записывали по фракциям и методом логарифмического анализа вычисляли величину ED50. Результаты показаны в таблице 2.
Было установлено, что некоторые соединения настоящего изобретения также ингибируют прижигание спонтанно активных нейронов DA в покровных тканях области желудочка мозга спустя 21 день после повторяющейся оральной обработки крыс. Это испытание, проведенное по методу, описанному в работе T.Skarsfeldt, J.Perregaard, Fur. J. Pharmacol. 1990, 182. 613-614, является показателем антипсихотического воздействия на человека.
Результаты
Из таблицы 1 следует, что соединения настоящего изобретения, подобно сертиндолу, имеют сродство к рецептору серотонина 5-HT2 и к рецептору допамина D2 соответственно. Более того, из данных теста in vivo таблицы 2 видно, что эти соединения представляют собой некаталептогенные, мощные и пролонгированные антагонисты 5-HT2 in vivo.
Примеры рецептур
Фармакологические рецептуры согласно настоящему изобретению можно получить, используя методы, общепринятые в этой области. Например: таблетки можно получить, смешивая активный ингредиент с обычными адъювантами и/или разбавителями с последующим прессованием полученной смеси в подходящей таблетировочной машине. Примеры адъювантов или разбавителей включают: кукурузный крахмал, тальк, стеарат магния, желатин, лактозу, смолы и т.п. Можно использовать также любые иные адъюванты или красящие вещества, ароматизаторы, консерванты и т.д. при условии, что они совместимы с активными ингредиентами.
Растворы для инъекций могут быть получены путем растворения активного ингредиента и возможных добавок в части растворителя для инъекций, предпочтительно в стерильной воде, доведением полученного раствора до требуемого объема, стерилизацией этого раствора и заполнение им подходящих ампул или емкостей. Могут быть дополнительно введены любые подходящие добавки, которые удобно использовать в этой области, такие как тонизирующие агенты, консерванты, антиоксиданты и т.д.
Типичные примеры рецептов лекарственных средств согласно настоящему изобретению приведены ниже.
1) Таблетки, содержащие 0.5 мг соединение 6 в расчете на свободное основание:
Соединение 6 - 5.0 мг
Лактоза - 60 мг
Кукурузный крахмал - 30 мг
Гидроксипропилцеллюлоза - 2.4 мг
Микрокристаллическая целлюлоза - 19.2 мг
Кроскармеллоз натрия тип A - 2.4 мг
Стеарат магния - 0.84 мг
2) Таблетки, содержащие 1 мг соединение 8 в расчете на свободное основание:
Соединение 8 - 1.0 мг
Лактоза - 46.9 мг
Кукурузный крахмал - 23.5 мг
Повидон - 1.8 мг
Микрокристаллическая целлюлоза - 14.4 мг
Кроскармеллоз натрия тип A - 1.8 мг
Стеарат магния - 0.63 мг
3) Сироп, содержащий в 1 мл:
Соединение 4 - 5.0 мг
Сорбитол - 500 мг
Гидроксипропилцеллюлоза - 15 мг
Глицерол - 50 мг
Метил-парабен - 1 мг
Пропил-парабен - 0.1 мг
Этанол - 0.005 мл
Ароматизатор - 0.05 мг
Сахарин - 0.5 мг
Вода - До 1 мл
4) Раствор для инъекций, содержащий в 1 мл:
Соединение 6 - 0.5 мг
Сорбитол - 5.1 мг
Уксусная кислота - 0.08 мг
Вода для инъекций - До 1 млс
Описываются новые фенилиндольные соединения общей формулы I, где R1 - R4 независимо друг от друга представляют собой водород, дейтерий, C1-6 алкил, C1-6 алкокси при условии, что все четыре заместителя одновременно не могут быть водородом, или, по меньшей мере, одна пара заместителей (R1, R2 или R3, R4) представляют собой оксогруппу и, если присутствует только одна оксогруппа, два других заместителя выбирают из вышеуказанной группы, определенной для R1 - R4, при условии, что обе не могут быть одновременно водородами, или R1 - R2 и/или R3 и R4 соответственно соединяются и образуют 3-8-членный спироцикл или его кислая аддитивная соль. Соединения формулы I являются некаталептогенными антагонистами рецептора 5-НТ2 и демонстрируют в модели признаки антипсихотического воздействия. Поэтому эти соединения могут быть полезны при лечении психоза, депрессии, негативных симптомов шизофрении, гипертензии или экстрапирамидальных побочных эффектов, вызванных антипсихотическими лекарственными средствами. 2 с. и 1 з.п. ф-лы, 2 табл.
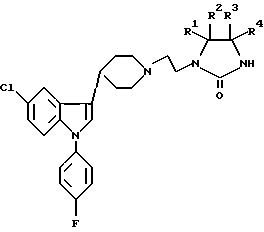
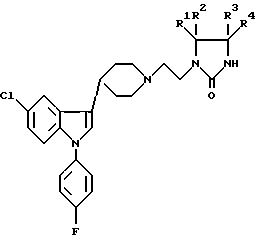
где R1-R4 независимо выбирают из группы, содержащей водород, дейтерий, С1-6 алкил, С1-6 алкокси при условии, что все четыре заместителя одновременно не могут быть водородом, или, по меньшей мере, одна пара заместителей (R1, R2 или R3, R4) представляют собой оксогруппу и, если присутствует только одна оксогруппа, два других заместителя выбирают из вышеуказанной группы, определенной для R1-R4, при условии, что обе не могут быть одновременно водородами, или R1 и R2 и/или R3, R4 соответственно, соединяются и образуют 3 - 8 членный спироцикл или его кислая аддитивная соль.
| УСТРОЙСТВО ДЛЯ ИЗМЕРЕНИЯ СКОРОСТИ ВРАЩЕНИЯ | 0 |
|
SU200322A1 |
| МЕШАЛКА ДЛЯ ВЯЗКИХ ЖИДКОСТЕЙ | 0 |
|
SU392959A1 |
| Автоматический огнетушитель | 0 |
|
SU92A1 |
| МАШКОВСКИЙ М.Д | |||
| Лекарственные средства | |||
| - М.: Медицина, 1972, ч.1, с.137 - 148. | |||

