Гормон роста, который выделяют из гипофиза, стимулирует рост всех тканей организма, которые способны расти. Кроме того, известно, что гормон роста оказывает следующие основные действия на метаболические процессы в организме:
1. Повышает скорость синтеза протеинов во всех клетках организма.
2. Снижает скорость утилизации углеводов в клетках организма.
3. Повышает активацию свободных жирных кислот и использование жирных кислот для выработки энергии.
Недостаточная секреция гормона роста может привести к различным заболеваниям, как например к карликовости.
Для высвобождения гормона роста известны различные пути. Так, например, такие химические соединения, как аргинин, L-3,4-дигидроксифенилаланин (L-ДОРА), глюкагон вазопрессин и инсулин вызывают гипогликемию, а также проявления таких активностей, как сонливость и физическая активность, косвенным путем вызывая выделение гормона роста из гипофиза за счет воздействия каким-либо образом на гипоталамус и, возможно, либо снижая секрецию соматостатина, либо повышая секрецию известного фактора, усиливающего высвобождение гормона роста (GPF), либо неизвестного эндогенного гормона, высвобождающего гормон роста, либо все это вместе.
В тех случаях, когда желательно повысить уровень содержания гормона роста, проблему обычно разрешают, обеспечивая экзогенный гормон роста либо вводя GPF либо пептидное соединение, которое стимулирует продуцирование и/или выделение гормона роста. В любом случае пептидная природа соединения требует, чтобы его вводили путем инъекций. Вначале источником гормона роста служили экстракты гипофиза, взятого у трупов. Это приводило к чрезвычайно высокой стоимости продукта и включало риск переноса реципиенту гормона роста заболеваний, связанных с источником гипофиза. В последнее время стал доступен гормон роста, полученный рекомбинантными способами, что, хотя и устраняет риск передачи заболевания, все еще не снижает чрезвычайно высокой стоимости продукта, который следует вводить либо за счет инъекций, либо в виде назального спрея.
Были разработаны другие соединения, которые стимулируют выделение эндогенного гормона роста, такие как аналоговые пептидильные соединения, родственные GPF, или пептиды патента США 4411890. Эти пептиды, хотя и значительно меньшего размера, нежели гормоны роста, все еще подвержены действию различных протеаз. Как и для большинства пептидов, их потенциальная биодоступность при пероральном приеме низка. Рассматриваемые соединения являются непептидными аналогами, промотирующими выделение гормона роста, которые стабильны в различных физиологических средах и которые можно вводить парентерально, через нос, либо перорально.
Настоящее изобретение охватывает некоторые спиросоединения, которые обладают способностью стимулировать выделение природного или эндогенного гормона роста. Таким образом, эти соединения можно использовать для лечения состояний, которые требуют стимуляции продукцирования или секреции гормона роста, таких, которые встречаются у людей с недостаточностью гормона роста, или для животных, которых используют для пищевых целей, где стимуляция гормона роста приводит к выращиванию более крупных, более продуктивных животных. Таким образом, целью настоящего изобретения является описание спиросоединений. Другой целью настоящего изобретения является описание способов получения таких соединений. Еще одной целью является описание применения таких соединений для усиления секреции гормона роста у людей и животных. И еще одной целью является описание композиций, содержащих спиросоединения для использования при лечении людей и животных для повышения уровня секреции гормона роста. Остальные цели станут очевидны при чтении следующего описания.
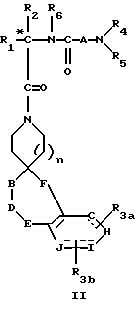
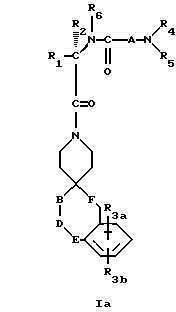
Новые спиросоединения настоящего изобретения наилучшим образом описываются следующими структурными формулами I и II: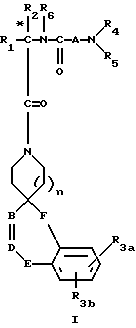
R1 представляет C1-C10алкил, арил, арил(C1-C6алкил) и C3-C7циклоалкил(C1-C6алкил) или C1-C5алкил-K-(C1-C5-алкил), арил (C0-C5алкил)-K-(C1-C5алкил), C3-C7циклоалкил- (C0-C5алкил)-K-(C1-C5алкил), где K представляет O, S(O)m, N(R2)C(O), C(O)N(R2), OC(O), C(O)O или -CR2=CR2- или 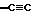 , где арильная группа определена далее, а R2 и алкильные группы могут быть замещены далее 1-9 галоидами, S(O)mR2a, 1-3 OR2a или C(O)OR2a и арильные группы могут быть замещены фенилом, фенокси, галоидфенилом, 1-3 C1-C6алкилом, 1-3 галоидами, 1-2 OR2, метилендиокcи, S(O)mR2, 1-2 CF3, OCF3, нитро, N(R2)(R2), N(R2)C(O)R2, C(O)OR2, C(O)N(R2)(R2), SO2N(R2)(R2),
, где арильная группа определена далее, а R2 и алкильные группы могут быть замещены далее 1-9 галоидами, S(O)mR2a, 1-3 OR2a или C(O)OR2a и арильные группы могут быть замещены фенилом, фенокси, галоидфенилом, 1-3 C1-C6алкилом, 1-3 галоидами, 1-2 OR2, метилендиокcи, S(O)mR2, 1-2 CF3, OCF3, нитро, N(R2)(R2), N(R2)C(O)R2, C(O)OR2, C(O)N(R2)(R2), SO2N(R2)(R2),
N(R2)S(O)2арил или N(R2)SO2R2;
R2 представляет водород, C1-C6алкил, C3-C7циклоалкил, и если две C1-C6алкильные группы присутствуют на одном атоме, они могут быть необязательно соединены с образованием C3-C8циклического кольца, необязательно содержащего кислород, серу или NR2a;
R2a представляет водород или C1-C6алкил;
R3a и R3b независимо представляют водород, галоид, C1-C6алкил, OR2, циано, OCF3, метилендиокси, нитро, S(O)mR, CF3 или C(O)OR2, и если R3a и R3b находятся в ортоположении, они могут быть соединены с образованием C5-C8 алифатического или ароматического кольца, необязательно содержащего 1 или 2 гетероатома, выбранные из кислорода, серы или азота;
R4 и R5 независимо представляют водород, C1-C6алкил, замещенный C1-C6алкил, где заместителями могут быть 1-5 галоидов, 1-3 гидрокси, 1-3 C1- C10алканоилокси, 1-3 C1-C6алкокси, фенил, фенокси, 2-фурил, C1-C6алкоксикарбонил, S(O)m (C1-C6алкил); или R4 и R5, взятые вместе, образуют -(CH2)rLa(CH2)s - где La представляет C(R2)2, O, S(O)m или N(R2), r и s независимо равны 1-3, а R2 имеет указанные ранее значения;
R6 представляет водород или C1-C6алкил;
A представляет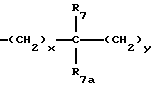
или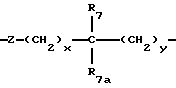
где x и y независимо представляют 0-3;
Z представляет N-R2 или O;
R7 и R7a независимо представляют водород, C1-C6алкил, OR2, трифторметил, фенил, замещенный C1-C6алкил, где заместителями являются имидазолил, фенил, индолил, p-гидроксифенил, OR2, 1-3 фтор, S(O)mR2, C(O)OR2, C3-C7циклоалкил, N(R2)(R2), C(O)N(R2)(R2); или R7 и R7a могут быть независимо соединены с одной или обоими R4 и R5 группами с образованием алкиленовых мостиков между концевым азотом и алкильной частью R7 или R7a групп, причем мостик может содержать от 1 до 5 атомов углерода, B, D, E и F независимо представляют C(R8)(R10), O, C= O, S(O)m или NR9, так что один или два из B, D, E или F могут необязательно отсутствовать, что обеспечит образование 5-, 6- или 7-членного кольца; и при условии, что B, D, E и F могут быть C(R8)(R10) или C= O, только если один из оставшихся B, D, E и F представляет одновременно O, S(O)m или NR9; B и D или D и E, взятые вместе, могут представлять CR8=CR10 при условии, что один из других B и E или F одновременно представляет O, S(O)m или NR9;
R8 и R10 независимо представляют водород, R2, OR2, (CH2)qарил, (CH2)qC(O)OR2, (CH2)qC(O)(CH2)qарил или (CH2)q (1H-тетразол-5-ил), а арил может быть необязательно замещен 1-3 галоидами, 1-2 C1-C8алкилом, 1-3 OR2 или 1-2 C(O)OR2;
R9 представляет R2, (CH2)qарил, C(O)R2, C(O)(CH2)q- арил, SO2R2, SO2(CH2)qарил, C(O)N(R2)(R2), C(O)N(R2)-(CH2)qарил, C(O)OR2, 1-H-тетразол-5-ил, SO3H, 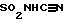 , SO2N(R2)арил, или SO2N(R2)(R2), и (CH2)q может быть необязательно замещен 1-2 С1-С4алкилом, а R2 и арил могут необязательно быть замещены еще 1-3 OR2a, O(CH2)qарилом, 1-2 C(O)OR2a, 1-2 C(O)O(CH2)qарилом, 1-2 C(O)N(R2a)(R2a), 1-2 C(O)N(R2a)(CH2)qарилом, 1-5 галоидом, 1-3 C1-C4алкилом, 1,2,4-триазолилом, 1-H-тетразол-5-илом, C(O)NHSO2R2a, S(O)mR2a,
, SO2N(R2)арил, или SO2N(R2)(R2), и (CH2)q может быть необязательно замещен 1-2 С1-С4алкилом, а R2 и арил могут необязательно быть замещены еще 1-3 OR2a, O(CH2)qарилом, 1-2 C(O)OR2a, 1-2 C(O)O(CH2)qарилом, 1-2 C(O)N(R2a)(R2a), 1-2 C(O)N(R2a)(CH2)qарилом, 1-5 галоидом, 1-3 C1-C4алкилом, 1,2,4-триазолилом, 1-H-тетразол-5-илом, C(O)NHSO2R2a, S(O)mR2a, 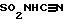 , C(O)NHSO2(CH2)qарилом, SO2NHC(O)R2a, SO2NHC(O)(CH2)qарилом, N(R2)C(O)N(R2a)-(R2a), N(R2a)C(O)N(R2a)(CH2)qарилом,
, C(O)NHSO2(CH2)qарилом, SO2NHC(O)R2a, SO2NHC(O)(CH2)qарилом, N(R2)C(O)N(R2a)-(R2a), N(R2a)C(O)N(R2a)(CH2)qарилом,
N(R2a)(R2a), N(R2a)C(O)R2a, N(R2a)C(O)(CH2)qарилом, OC(O)N(R2a)(R2a), OC(O)N(R2a)(CH2)qарилом или SO2(CH2)qCONH-(CH2)wNHC(O)- R11, где w равно 2-6, a R11 представляет биотин, арил или арил, замещенный 1 или 2 OR2, 1-2 галоидом, азидо или нитро;
m = 0,1 или 2;
n = 1 или 2;
q = 0, 1, 2, 3 или 4; и
G, H, I и J представляют атомы углерода, азота, серы или кислорода, так что по крайней мере один гетероатом и один из G, H, I или J может необязательно отсутствовать, что обеспечивает образование 5- или 6-членного гетероциклического ароматического кольца;
и его фармацевтически приемлемые соли и отдельные диастереоизомеры.
В вышеуказанных формулах и далее во всем описании термины имеют следующие значения.
Указанные ранее алкильные группы включают алкильные группы указанной длины, разветвленные или неразветвленные, которые могут необязательно содержать двойные или тройные связи. Примерами таких алкильных групп могут служить: метил, этил, пропил, этинил, изопропил, бутил, втор-бутил, третичный бутил, пентил, изопентил, гексил, изогексил, алкил, пропенил, бутенил, бутадиенил и т.п.
Указанные ранее алкоксигруппы включают алкоксигруппы указанной длины, разветвленные или неразветвленные, которые могут необязательно содержать двойные или тройные связи. Примеры таких алкоксигрупп включают метокси, этокси, пропокси, изопропокси, бутокси, изобутокси, трет-бутокси, пентокси, изопентокси, гексокси, изогексокси, аллилокси, пропинилокси, изобутенилокси, 2-гексенилокси и т.п.
Термин "галоид" включает атомы галоидов: фтор, хлор, бром и йод.
Термин "арил" включает фенил и нафтил и ароматические остатки 5- и 6-членных колец, с 1-3 гетероатомами, или конденсированные 5- или 6-членные бициклические кольца, содержащие 1-3 гетероатома, выбранные из азота, серы или кислорода. Примерами таких гетероциклических ароматических колец являются пиридин, тиофен, бензотиофен, тетразол, N-метилиндол, дигидроиндол, индазол, N-формилиндол, бензимидазол, тиазол, фуран, диримидин и тиадиазол.
Некоторые из вышеопределенных терминов могут встречаться более одного раза в вышеуказанной формуле, и в таком случае каждый термин должен быть определен независимо от других.
Предпочтительными соединениями настоящего изобретения являются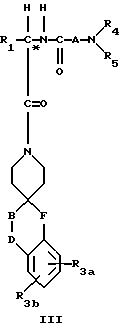
где R1 представляет C1-C10алкил, арил(C1-C4алкил), C3-C6циклоалкил(C1-C4алкил), (C1-C4алкил)-K-(C1-C4алкил), арил(C0-C5алкил)-K-(C1-C4алкил), или (C3-C7циклоалкил)(C0-C5алкил)-K-(C1- C4алкил), где K представляет O, S(O)m, -CR2=CR2-,  , или N(R2)C(O), где R2 и алкильные группы могут быть замещены далее 1-7 галоидом, S(O)m C1-C4алкилом, OR2, и арильные группы могут быть замещены далее C1-C4алкилом, 1-2 галоидом, 1-2 OR2, CF3, OCF3, метилендиокси, S(O)mR2, SON(R2)(R2), N(R2)SO2R2 или C(O)OR2; R2 представляет водород, C1-C6алкил, или C3-C7циклоалкил, и, если две C1-C6алкильные группы присутствуют на одном атоме, они могут быть необязательно соединены с образованием C4-C6-циклического кольца, которое необязательно включает 1-2 гетероатома, выбранные из кислорода, серы или NR2a;
, или N(R2)C(O), где R2 и алкильные группы могут быть замещены далее 1-7 галоидом, S(O)m C1-C4алкилом, OR2, и арильные группы могут быть замещены далее C1-C4алкилом, 1-2 галоидом, 1-2 OR2, CF3, OCF3, метилендиокси, S(O)mR2, SON(R2)(R2), N(R2)SO2R2 или C(O)OR2; R2 представляет водород, C1-C6алкил, или C3-C7циклоалкил, и, если две C1-C6алкильные группы присутствуют на одном атоме, они могут быть необязательно соединены с образованием C4-C6-циклического кольца, которое необязательно включает 1-2 гетероатома, выбранные из кислорода, серы или NR2a;
R2a представляет водород или C1-C6алкил;
R3a и R3b независимо представляют водород, галоид, C1-C4алкил, OR2, метилендиокси, нитро, S(O)m C1-C4-алкил, CF3 или C(O)OR2;
R4 и R5 независимо представляют водород, C1-C6алкил, замещенный C1-C6алкил, где заместителями могут быть 1-5 галоид, 1-2 гидрокси, 1-2 C1-C6алканоилокси, 1-2 C1-C6-алкилокси, или S(O)m (C1-C4алкил);
A представляет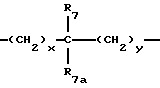
или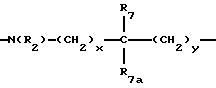
где x и y независимо равны 0, 1 или 2;
R7 и R7a независимо представляют водород, C1-C4алкил, или замещенный C1-C4алкил, где в качестве заместителей могут быть 1-3 фтор, имидазолил, фенил, индолил, или S(O)m- C1-C4алкил C(O)R2, или R7 и R7a могут быть независимо соединены с одной или обоими R4 и R5 группами с образованием алкиленовых мостиков между концевым азотом и алкильной частью R7 или R7a групп, где мостик содержит 1-3 атома углерода;
B, D и F независимо представляют C(R8)(R10), C=O, S(O)m или NR9, так что один из B, D или F может необязательно отсутствовать, что обеспечивает образование 5- или 6-членного кольца, и при условии, что один из B, D и F представляет C(R8)(R10), или C=O, только если одна из остальных B, D и F групп представляет одновременно O, S(O)m или NR9;
R8 и R10 независимо представляют водород, R2, OR2, (CH2)qарил, (CH2)qC(O)OR2, (CH2)qC(O)O(CH2)qарил, (CH2)q(1Н-тетразол-5-ил), где арил может быть необязательно замещен 1-3 галоидом, 1-2 C1-C4алкилом, 1-3 OR2 или 1-2 C(O)OR2;
R9 представляет R2, (CH2)qарил, C(O)R2, C(O)(CH2)q-арил, SO2R2, SO2(CH2)qарил, C(O)N(R2)(R2), C(O)N(R2)-(CH2)qарил, 1-H-тетразолил-5-ил,  , SO2N(R2) - арил, SO2N(R2)(R2), где (CH2)q может быть необязательно замещен 1-2 C1-C2алкилом, а R2 может быть необязательно замещен 1-2 OR2a, O(CH2)qарилом, 1-2 C(O)OR2a, C(O)N(R2a)- (R2a), S(O)mR2a, 1-H-тетразол-5-илом, C(O)NHSO2R2a, C(O)NHSO2(CH2)qарилом, (R2a)C(O)N(R2a)(R2a), или N(R2a)C(O)N(R2a)(CH2)qарилом, где арил может быть необязательно замещен 1-3 OR2a, 1-2 галоидом, 1-2 C1-C4алкилом, C(O)OR2a, или 1-H-тетразол-5-илом; SO2 (CH2)wCONH(CH2)w- NHC(O)R11, где w равно 2-6, a R11 представляет биотин, арил или арил, замещенный 1 или 2 OR2a, 1-2 галоидом, азидо или нитро;
, SO2N(R2) - арил, SO2N(R2)(R2), где (CH2)q может быть необязательно замещен 1-2 C1-C2алкилом, а R2 может быть необязательно замещен 1-2 OR2a, O(CH2)qарилом, 1-2 C(O)OR2a, C(O)N(R2a)- (R2a), S(O)mR2a, 1-H-тетразол-5-илом, C(O)NHSO2R2a, C(O)NHSO2(CH2)qарилом, (R2a)C(O)N(R2a)(R2a), или N(R2a)C(O)N(R2a)(CH2)qарилом, где арил может быть необязательно замещен 1-3 OR2a, 1-2 галоидом, 1-2 C1-C4алкилом, C(O)OR2a, или 1-H-тетразол-5-илом; SO2 (CH2)wCONH(CH2)w- NHC(O)R11, где w равно 2-6, a R11 представляет биотин, арил или арил, замещенный 1 или 2 OR2a, 1-2 галоидом, азидо или нитро;
m = 0, 1 или 2;
q = 0, 1, 2 или 3; и
арил представляет фенил, нафтил, пиридил, тиенил, индолил, тиазолил или пиримидинил;
и его фармацевтически приемлемые соли и отдельные диастереоизомеры.
Еще одна группа предпочтительных соединений реализуется, если в соединении III отсутствует F.
Другие предпочтительные соединения настоящего изобретения имеют структурную формулу IV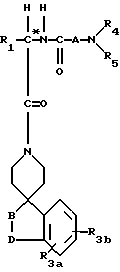
где R1 представляет C1-C10алкил, арил(C1-C4алкил), C5-C6циклоалкил(C1-C4алкил), (C1- C4алкил)-K-C1-C2алкил-, арил(C0- C2алкил)-K-(C1- C2алкил), или C3- C6циклоалкил(C0-C2алкил)-K-(C1-C2алкил),
где K представляет O или S(O)m, а арил может быть замещен далее 1-2 C1-C4-алкилом, 1-2 галоидом, OR2, C(O)OR2, CF3 или S(O)mR2;
R2 представляет водород, C1-C4алкил, цикло C3-C6алкил, и, если два C1-C4алкила присутствуют на одном атоме, они могут быть необязательно соединены с образованием C5-C6циклического кольца, необязательно содержащего гетероатомы кислорода или NR2a; R2a представляет водород или C1-C4алкил;
R3a и R3b независимо представляют водород, галоид, C1-C4алкил, C(O)OR2, гидрокси, C1- C4алкокси, S(O)m- C1-C4алкил, или CF3;
R4 и R5 независимо представляют водород, C1-C4алкил, или замещенный C1-C4алкил, где заместителями могут быть 1-2 гидрокси или S(O)m (C1-C3алкил);
A представляет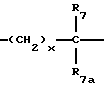
где x = 0 или 1;
R7 и R7a независимо представляют водород, или C1-C3-алкил, или R7 и R7a могут быть независимо соединены с одной или обоими R4 и R5 группами с образованием алкиленового мостика между концевым азотом и алкильной частью R7 и R7a групп с образованием 5- или 6-членных колец, содержащих концевой азот;
B и D независимо представляют C(R8)(R10), C=O, O, S(O)m или NR9 при условии, что один из B и D может быть C(R8)(R10) или C=O, только если другой из B и D представляет O, S(O)m и NR9;
R8 и R10 независимо представляют водород, R2, OR2, или (CH2)q арил, где арил может необязательно быть замещен 1-2 галоидом, 1-2 C1-C4алкилом, OR2, или 1-2 C(O)OR2;
R9 представляет C(O)R2, C(O)(CH2)qарил, SO2R2, SO(CH2)qарил, C(O)N(R2)(R2), или C(O)N(R2)(CH2)qарил, где (CH2)q может быть необязательно замещен 1-2 C1-C2-алкилом, а R2 может быть необязательно замещен 1-2 OR2a, O(CH2)qарилом, C(O)OR2a , C(O)N(R2a)(R2a), S(O)mR2a, 1-Н-тетразол-5-илом, C(O)NHSO2aR2a или N(R2a)C(O)N(R2a)-(R2a), и арил может необязательно замещен 1-2 OR2a, 1-2 галоидом, 1-2 C1-C2алкилом C(O)OR2a, 1-Н-тетразол-5-илом, S(O)mR2a; или SO2(CH2)qCONH(CH2)wNHC(O)R11, где w равно 2-6, a R11 может необязательно представлять биотин, арил, а арил может быть необязательно замещен 1-2 OR2, 1-2 галоидом, азидо, нитро;
m = 0, 1 или 2;
q = 0, 1, 2, или 3;
арил представляет фенил, нафтил, пиридил, индолил, тиенил или тетразолил; и
фармацевтически приемлемые его соли и отдельные диастереоизомеры.
Наиболее предпочтительные соединения настоящего изобретения имеет формулу V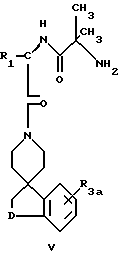
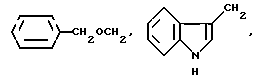
где R1 выбирают из: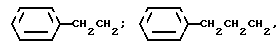
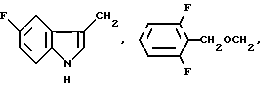
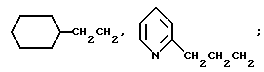
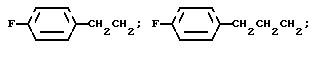
R3a представляет водород или фтор;
D представляет O, S, S(O)m, N(R2), NSO2(R2), NSO2(CH2)qOH, NSO2(CH2)tарил, NC(O)(R2), NSO2(CH2)qCOOR2, N-SO2(CH2)qC(O)N(R2)(R2),
N-SO2(CH2)qC(O)-N(R2)(CH2)wOH,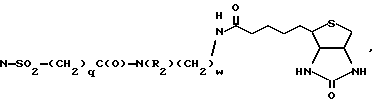
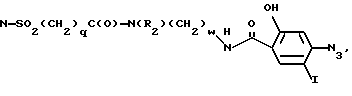
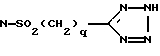
и арил представляет фенил или пиридил, а фенил может быть замещен 1-2 галоидом;
R2 представляет водород или C1-C4алкил;
m = 1 или 2;
t = 0, 1 или 2;
q = 1, 2 или 3:
w = 2 - 6;
и их фармацевтически приемлемые соли и отдельные диастереоизомеры.
Наиболее предпочтительные соединения настоящего изобретения, способствующие выделению гормона роста, включают следующие:
1. N-[1(R)-[(1,2-дигидро-1-метансульфонилспиро[3Н- индол-3,4'-пиперидин] -1'-ил)карбонил]-2-(1H-индол-3-ил)- этил]-2-амино-2-метилпропанамид;
2. N-[1(R)-[(1,2-дигидро-1-метанкарбонилспиро[3Н- индол-3,4'-пиперидин] -1'-ил)карбонил]-2-(1H-индол-3-ил)- этил]-2-амино-2-метилпропанамид;
3. N-[1(R)-[(1,2-дигидро-1-бензолсульфонилспиро[3Н- индол-3,4'-пиперидин]-1'-ил)карбонил]-2-(1H-индол-3-ил)- этил]-2-амино-2-метилпропанамид;
4. N-[1(R)-/(3,4-дигидро-спиро/2H-1-бензопиран-2,4'-пиперидин/-1'- ил)карбонил/-2-(1H-индол-3-ил)этил/-2-амино-2-метилпропанамид;
5. N-/1(R)-/(2-ацетил-1,2,3,4-тетрагидроспиро/изохинолин- 4,4'-пиперидин/-1'-ил)карбонил/-2-(индол-3-ил)этил/-2-амино-2- метилпропанамид;
6. N-/1(R)-/(1,2-дигидро-1-метансульфонилспиро/3Н- индол-3,4'-пиперидин] -1'-ил)карбонил/-2-(фенилметилокси)- этил/-2-амино-2-метилпропанамид;
7. N-/1(R)-/(1,2-дигидро-1-метансульфонилспиро/3Н- индол-3,4'-пиперидин] -1'-)карбонил/-2-(фенилметилокси)- этил/-2-амино-2-метилпропанамидмезилат, соль;
8. N-/1(R)-/(1,2-дигидро-1-метансульфонилспиро/3Н- индол-3,4'-пиперидин] -1'-ил)карбонил/-2-(2', 6'-дифторфенилметилокси) этил/-2-амино-2-метилпропанамид;
9. N-/1(R)-/(1,2-дигидро-1-метансульфонил-5-фтороспиро/3Н- индол-3,4'-пиперидин] -1'-ил)карбонил/-2-(фенилметилокси) этил/-2-амино-2-метилпропанамид;
10. N-/1(S)-/(1,2-дигидро-1-метансульфонилспиро/3Н- индол-3,4'-пиперидин]-1'-ил)карбонил/-2-(фенилметилтио)- этил/-2-амино-2-метилпропанамид;
11. N-/1(R)-/(1,2-дигидро-1-метансульфонилспиро/3Н- индол-3,4'-пиперидин]-1'-ил)карбонил/-3-(фенилпропил)-2- амино-2-метилпропанамид;
12. N-/1(R)-/(1,2-дигидро-1-метансульфонилспиро/3Н- индол-3,4'-пиперидин]-1'-ил)карбонил/-3-циклогексилпропил)-2- амино-2-метилпропанамид;
13. N-/1(R)-/(1,2-дигидро-1-метансульфонилспиро/3Н- индол-3,4'-пиперидин]-1'-ил)карбонил/-4-фенилбутил/-2- амино-2-метилпропанамид;
14. N-/1(R)-/(1,2-дигидро-1-метансульфонилспиро/3Н- индол-3,4'-пиперидин] -1'-ил)карбонил/-2-(5-фтор-1H-индол-3-ил)этил/-2- амино-2-метилпропанамид;
15. N-/1(R)-/(1,2-дигидро-1-метансульфонил-5-фтороспиро/ 3Н-индол-3,4'-пиперидин] -1'-ил)карбонил/-2-(5-фтор-1H-индол-3-ил) этил/-2-амино-2-метилпропанамид;
16. N-/1(R)-/(1,2-дигидро-1-(2-этоксикарбонил)метилсульфонилспиро/ 3Н-индол-3,4'-пиперидин] -1'-ил)карбонил/-2-(1H-индол-3-ил) этил/-2-амино-2-метилпропанамид;
17. N-/1(R)-/(1,2-дигидро-1,1-диоксоспиро/ 3Н-бензотиофен-3,4'-пиперидин]-1'-ил)карбонил/-2-(фенилметилокси)- этил/-2-амино-2-метилпропанамид;
и их фармацевтически приемлемые соли.
Представительные примеры используемой номенклатуры приведены далее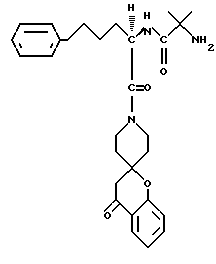
N-/1(R)-/3,4-дигидро-4-оксоспиро/2H-1-бензопиран-2,4'- пиперидин/-1'-ил)карбонил/-4-фенилбутил/-2-амино-2-метилпропанамид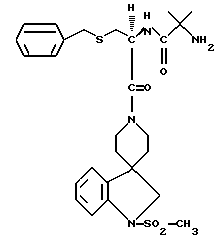
N-/1(S)-/(1,2-дигидро-1-метаносульфонилспиро/3Н-индол- 3,4'-пиперидин/-1'-ил)карбонил/-2-(фенилметилтио)этил/- 2-амино-2-метилпропанамид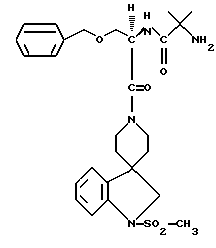
N-/1(R)-/(1,2-дигидро-1-метаносульфонилспиро/3Н-индол- 3,4'-пиперидин/-1'-ил)карбонил/-2-(фенилметилокси)этил/- 2-амино-2-метилпропанамид.
В настоящем описания сокращения имеют следущие значения:
BOC - трет-бутилоксикарбонил,
ВОР - бензотриазол-1-илокси трис/диметиламино/фосфоний- гексафторфосфат,
CBZ - бензилоксикарбонил,
DCC - дициклогексилкарбодиимид,
DMF - N,N-диметилформамид,
EDC - 1-(3-диметиламинопропил)-3-этилкарбодиимидгидрохлорид,
FAB-MS - масс-спектроскопия с бомбардировкой быстрыми атомами,
GHRP - пептид, высвобождающий гормон роста,
HOBT - гидроксибензтриазол,
LAH - литийалюминийгидрид,
HPLC - высокоэффективная жидкостная хроматография,
MHz - мегагерц = МГц,
MPLC - жидкостная хроматография среднего давления,
NMM - N-метилморфолин,
NMR - ядерный магнитный резонанс = ЯМР,
OXONE - пероксимоносульфат калия,
PLC - препаративная тонкослойная хроматография,
PCC - пиридинийхлорхромат,
Ser - серин,
TFA - трифторуксусная кислота,
THF - тетрагидрофуран,
TLC - тонкослойная хроматография = ТСХ,
TMS - тетраметилсилан = ТМС.
Соединения настоящего изобретения содержат по крайней мере один асимметричный центр, который отмечен звездочкой в структурной формуле I и II ранее. В молекуле могут быть и дополнительные асимметричные центры в зависимости от природы различных заместителей в этой молекуле. За счет каждого асимметричного центра образуются два оптических изомера, и следует считать, что все такие оптические изомеры как разделенные, чистые, или частично очищенные оптические изомеры, рацемические смеси или диастероизомерные их смеси, входят в объем настоящего изобретения. В том случае, если асимметричный центр отмечен звездочкой, было найдено, что более активный и тем самым более предпочтительный изомер представлен формулой Ia. Эта предпочтительная абсолютная конфигурация применима как к формуле I, так и II. Если R2 заместитель представляет водород, специальная конфигурация асимметричного центра соответствует конфигурация в D-аминокислоте. В большинстве случаев ее также обозначают как R-конфигурацию, хотя это будет изменяться в соответствии со значениями R1 и R2, используемыми при отнесении к стереохимической конфигурации R- или S-.
Соединения настоящего изобретения обычно выделяют в форме их фармацевтически приемлемых солей присоединения кислот, например солей, полученных с неорганическими или органическими кислотами. Примерами таких кислот могут служить соляная, азотная, серная, фосфорная, муравьиная, уксусная, трифторуксусная, пропионовая, малеиновая, янтарная, малоновая, метансульфоновая и т. п. Кроме того, некоторые соединения, содержащие кислотную функцию, такую как карбокси, можно выделять в форме их неорганических солей, в которых противоион может быть выбран из натрия, калия, лития, кальция, магния и т.п., а также из органических оснований.
Соединения I и II настоящего изобретения можно получить в результате последовательного или конвергентного синтеза. Подробные схемы последовательного синтеза соединений I и II представлены в следующих схемах реакций.
Защищенные аминокислотные производные 1 во многих случаях коммерчески доступны, если защитная группа L, является, например BOC или CBZ-группой. Другие защищенные аминокислотные производные 1 можно получить известными из литературы способами, многие из спиропиперидинов и спироазепинов (n = 2) формулы 2 и 2a известны из литературы и могут быть получены производные по арильным группам такими стандартными способами, как галоидирование, нитрация, сульфонилирование и т.д. В другом варианте, различные фенил- или гетероарилзамещенные спиропиперидины и спироазепины (n = 2) можно получить в соответствии с литературными способами, используя промежуточные фенил и гетероарилпроизводные.
В схемах, следующих после схемы 1, синтетические методы иллюстрируются только для спиропиперидинов, хотя специалисты отдают должное этому факту, что проиллюстрированные превращения могут быть выполнены с продуктами более высокого гомологического ряда, приводя к получению соединений формулы I и II, где n = 2.
Промежуточные соединения формул 3 и 3а можно синтезировать в соответствии со схемой 1, приведенной в конце описания. Сочетание спиропиперидинов формул 2 и 2а до защищенных аминокислот формулы 1, где L представляет подходящую защитную группу, обычно ведут в таком инертном растворителе, как дихлорметан, с помощью таких обеспечивающих реакцию реагентов, как DCC и EDC в присутствии HOBT. В другом варианте реакцию сочетания можно осуществить за счет таких реагентов, как BOP, в инертном растворителе, например дихлорметане. Отделение нежелательных побочных продуктов и очистку промежуточных соединений осуществляют хроматографически на силикагеле, используя флеш-хроматографию (W. C. Still, M.Kahn and A.Mitra, J.Org. Chem. 1978, 43, 2923), СДЖХ или препаративную ТСХ.
Превращение 3 и 3а в промежуточные соединения 4 и 4a можно осуществлять в соответствии со схемой 2, приведенной в конце описания. Бензилоксикарбонильные группы можно удалить различными способами, известными специалистам; например, каталитическим гидрированием водородом в присутствии палладиевого или платинового катализатора в таком протонном растворителе, как метанол. В тех случаях, когда каталитическое гидрирование противопоказано из-за присутствия других потенциально реакционноспособных групп, удалить бензилоксикарбонильные группы можно за счет обработки раствором бромистого водорода в уксусной кислоте. Удаление BOC защитных групп ведут в таком растворителе, как метиленхлорид или метанол, такой сильной кислотой, как соляная или трифторуксусная кислота.
Условия, необходимые для удаления других защитных групп, которые могут присутствовать, можно найти у Greene, N. Wuts, P.G.M. Protective Groups Organic Synthesis, John Wiley and Sons, NY, 1991.
Промежуточные соединения формул 5 и 5b, где A представляет метилен или замещенную метиленовую группу, можно получить в соответствии со схемой 3, приведенной в конце описания, за счет сочетания промежуточных 4 и 4a с аминокислотами формулы 6 и снова в инертном растворителе, таком как дихлорметан, за счет такого реагента сочетания, как EDC или DCC в присутствии HOBT. Эти аминокислоты 6 являются хорошо известными аминокислотами, или аминокислотами, которые легко синтезировать известными специалистам способами. В другом варианте реакцию сочетания можно осуществить за счет такого реагента сочетания, как ВОР, в таком инертном растворителе, как дихлорметан. Кроме того, если R4 или R5 представляют водород, тогда аминокислоту формулы 7 используют в реакции сочетания, где L представляет защитную группу, как указано ранее, до получения 5a и 5c. Удаление защиты у 5a и 5c (L = защитная группа) можно осуществить в условиях, известных специалистам.
Соединения формул I и II, где R4 и/или R5 представляет водород, можно далее превратить в новые соединения I и II (предпочтительная боковая цепь R7 = CH2-CH/OH/-CH2X, где X= H или OH), которые замещены по аминогруппе, как указано на схеме 4, приведенной в конце описания. Восстановительное аминирование I и II альдегидом осуществляют в условиях, известных специалистам; так, например, каталитическим гидрированием водородом в присутствии платинового, палладиевого или никелевого катализаторов, или за счет таких химических восстанавливающих агентов, как цианоборгидрид натрия в таком инертном растворителе, как метанол или этанол. Алкилирование до получения аминоспиртов также можно осуществить в реакции раскрытия эпоксидного кольца.
Соединения формул I и II, где A представляет N(R2)- (CH2)z-C(R7)(R7a)-(CH2)y, можно получить в соответствии со схемой 5, приведенной в конце описания, за счет взаимодействия 4 или 4a с реагентами 8, где X представляет легко отщепляемую группу, например, Cl, Br или I, имидазол. В другом варианте 4 и 4a можно подвергнуть взаимодействию с изоцианатом формулы 9 в таком инертном растворителе, как 1,2-дихлорэтан. Если R4 или R5 представляет водород в финальном продукте, реагенты 8 и 9 должны содержать удаляемую L защитную группу вместо R4 или R5.
Соединения I и II настоящего изобретения можно также получить обычным способом, как указано на реакционных схемах 6, 7 и 8, приведенных в конце описания.
Защищенные аминокислотные производные 10 являются во многих случаях коммерчески доступными, если M = метиловый, этиловый или бензиловый сложные эфиры. Другие защищенные сложным эфиром аминокислоты можно получить классическими способами, известными специалистам. Некоторые из этих способов включают реакцию защищенной аминокислоты с диазоалканом и удаление защитной группы L, реакцию аминокислоты с соответствующим спиртом в присутствии сильной кислоты, подобной соляной кислоте или пара-толуолсульфокислоте. Синтетический способ получения новых аминокислот представлен на схемах 11, 12 и 13, приведенных в конце описания.
Промежуточные соединения 11 и 11a можно получить в соответствии со схемой 6, в реакции сочетания аминов 10 с аминокислотами 6 и/или 7, где L представляет защитную группу, как представлено на схеме 3. Если мочевинная связь присутствует в 11 или 11a, ее можно ввести в соответствии со схемой 5.
Превращение сложного эфира 11 и 11a в промежуточную кислоту 12 или 12a можно осуществить рядом способов, известных специалистам, как представлено на схеме 7; так, например, метиловый и этиловый сложные эфиры можно гидролизовать гидроксидом лития в протонном растворителе, например в водном метаноле. Кроме того, удаление бензильной группы можно осуществить рядом восстановительных способов, включая гидрирование в присутствии платинового или палладиевого катализатора в таком протонном растворителе как метанол. Аллиловый сложный эфир можно расщепить тетракис-трифенилфосфинпалладиевым катализатором в присутствии 2-этоксигексановой кислоты в различных растворителях, включая этилацетат и дихлорметан (см. J.Org. Chem. 1982, 42, 587).
Кислоту 12 или 12a можно затем превратить в 5 и 5a и 5b и 5c, как представлено на схеме 8. Сочетания спиропиперидинов формулы 2 и 2a с кислотами формулы 12 и 12a, где L представляет подходящую защитную группу, обычно ведут в инертном растворителе, таком как дихлорметан, за счет такого сочетающего реагента, как дициклогексилкарбодиимид (DCC) и EDC в присутствии 1-гидроксибензтриазола (HOBT). В другом варианте сочетание можно также осуществить за счет такого сочетающего агента, как бензотриазол-1-илокситрис/диметиламино/фосфонийгексафторфосфат ("BOP") в таком инертном растворителе, как дихлорметан. Превращение 5a и 5c в I и II достигают, удаляя защитную группу L. Если R4 и/или R5 представляет H, по атому азота можно при желании добавить замещенные алкильные группы в соответствии со схемой 4.
На схеме 9, приведенной в конце описания, представлено получение окисленных спироинданилпиперидиновых промежуточных соединений, в которых R3a и R3b оба представляют водород. Гидроборирование защищенного спироиндена 13 с последующей оксидативной обработкой пиридинийхлорхроматом приводит к получению спироинданона 14.
Превращение спироинданов в промежуточные бензолактамы представлено на схеме 10, приведенной в конце описания. Обработка спироинданона азотоводородной кислотой в таком инертном растворителе, как хлороформ (реакция Шмидта), представляет один из многих подходящих литературных способов такого превращения. В этом примере получают смесь двух бензолактамов. Полученные изомеры легко разделить хроматографически на силикагеле. Затем у этих промежуточных можно удалить защиту и ввести в соединения, ускоряющие секрецию гормона роста, как представлено на схемах 1 и 8, используя общее промежуточное соединение 2.
Алкилирование 15 и 16 алкилгалоидом в таком растворителе, как DMF в присутствии NaH приводит к получению 17 и 18 (R2 = C1-C4алкил).
Если L представляет соответствующую защитную группу, например бензильную группу, амиды можно восстановить литийалюминийгидридом до аминов 19 и 21. Эти амины, в которых R2=H, можно затем алкилировать, арилировать, ацилировать или подвергнуть взаимодействию с замещенными сульфонилгалоидами или изоцианатами, используя условия, известные специалистам, до получения соединений 20 и 22. Удаление защитной группы (L) за счет гидрогенолиза с использованием палладиевого катализатора приводит к получению промежуточных соединений, которые можно ввести в соединения, ускоряющие секрецию гормона роста настоящего изобретения, используя реакции, представленные на схемах 1 и 8, в которых используют общее промежуточное соединение 2.
В другом варианте 1,2,3,4-тетрагидроспиро/изохинолин-4,4'-пиперидиновое/ кольцо можно получить в соответствии со схемой 12. Озонолиз защищенного спироиндена с последующей обработкой диметилсульфидом дает полуацеталь - промежуточное соединение 24, которое в условиях восстановительного аминирования и ацилирования дает амин 25. Аминозащитная группа (L) была определена ранее.
Циклические аналоги формулы 26, где X, Y представляют H, H; OH, H; H, OH; и = O, можно получить способами, описанными в литературе и известными специалистам. Так, например, в соответствии со схемой 13 спиро/2H-1-бензопиран-2,4'-пиперидиновый/ аналог можно получить из замещенного или незамещенного 2-гидроксиацетофенона и соответствующим образом защищенного 4-пиперидона, как указано Kabbe, H.J. Synthesis 1978, 886-887 и в приведенных там ссылках. 2-Гидроксиацетофеноны, в свою очередь, являются либо коммерчески доступными, либо их можно получить способами, известными из литературы специалистам. Такие способы описаны Chang, C.T. et al., в J.Am. Chem. Soc. 1961, 3414-3417, Elliott. J.M. et al., в J. Med. Chem. 1992, 36, 3973-3976. Удаление защитных групп описано в Protective Groups in Organic Synthesis, Creene, T. W. , Wats, P.G., John Wiley and и Olojson, R.A. et al., J.Org. Chem. 1984, 49, 2081-2082, раскрывает амины, которые можно ввести в соединения, ускоряющие выделение гормона роста в соответствии со схемами 1 и 8, в которых используют общее промежуточное соединение 2.
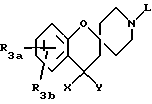
Кетоновую функциональность в соединениях общей структуры 27 можно восстановить до спирта, используя боргидрид натрия или можно полностью восстановить до метилена, также используя условия, известные специалистам. Так, например, восстановление кетона боргидридом натрия с последующей обработкой концентрированной соляной кислотой и последующим гидрированием приводит к получению соединения общей формулы 29. Амин формулы 27, 28 или 29 можно затем ввести в соединение, ускоряющее выделение гормона роста, за счет реакций, представленных на схемах 1 и 8, используя общее соединение формулы 2. В другом варианте кетон часто можно восстановить после введения в соединения формулы 1.
Получение хиральных аналогов гидроксиспиро/2H-1-бензопиран-2,4'-пиперидина/ можно осуществить, используя оптически активные восстанавливающие агенты и кристаллизацию диастереоизомерных солей.
Соединения формул I и II настоящего изобретения получают из различных замещенных природных и неприродных амино- кислот, таких как представлены формулами 30 и 6 и 7, где A представляет -(CH2)x-C(R7)(R7a)-(CH2)y. Получение многих из этих кислот описано в патенте США 5206237.
Получение этих промежуточных соединений в рацемической форме осуществляют классическими способами, знакомыми специалистам /Williams, R.M. "Synthesis of Optically Active α-Amino Acids" Pergamon Precc: Oxford, 1989, Vol. 7).
Существует несколько способов разделения (DZ)-аминокислот.
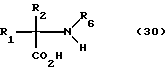
Одним из общих способов является разделение амино или карбоксизащищенных промежуточных соединений за счет кристаллизации солей, полученных из оптически активных кислот или аминов. В другом варианте аминогруппу карбоксизащищенного промежуточного можно присоединить к оптически активным кислотам, используя описанные ранее реакции. Разделение отдельных диастереоизомеров либо хроматографически, либо за счет кристаллизации с последующим гидролизом хиральных амидов приводит к получению разделенных аминокислот. Аналогично, аминозащищенные промежуточные соединения можно превратить в смесь хиральных диастереоизомерных сложных эфиров и амидов. Разделение смеси с использованием описанных ранее способов и гидролиз отдельных диастереоизомеров приводит к получению (D) и (L) аминокислот. И, наконец, энзиматические способы разделения N-ацетильных производных (DZ)-аминокислот представлены Whitesides с сотр. в J. Am. Chem. Soc., 1989, 111, 6354-6364.
Если нужно синтезировать эти промежуточные соединения в оптически чистой форме, можно использовать некоторые общепринятые способы получения, которые включают:
(1) асимметричное электрофильное аминирование хиральных енолятов (J. Am. Chem. Soc., 1986, 108, 6394-6395, 6395-6397 и 6397-6399), (2) асимметричное нуклеофильное аминирование оптических активных карбонильных производных (J. Am. Chem. Soc. , 1992, 114, 1906, Tetraheoron Lett. 1987, 28, 32), (3) диастереоселективное алкилирование хиральных глициненолятных синтонов (J. Am. Chem. Soc. , 1991, 113, 9276; J. Org. Chem., 1889, 54, 3916), (4) диастереоселективное нуклеофильное присоединение к хиральному электрофильному глицинатному синтону (J. Am. Chem. Soc., 1986, 108, 1103), (5) асимметричное гидрирование производных прохиральной дегидроаминокислоты (Asymmetric Synthesis Chiral Catalysis; Morrison, J.D. Ed; Academic Press; Orlabdo FL, 1985, Vol. 5), и (6) энзиматический синтез (Angew. Chem. Ed. Engl., 1978, 17, 176).
Так, например, алкилирование енолята дифенилоксазинона 31 (J. Am. Chem. Soc., 1991, 113, 9276) циннамилбромидом в присутствии натрийбис/триметилсилил/амида протекает спокойно до получения 32, который превращают в целевую (D)-2-амино-5-фенилпентановую кислоту 33, удаляя N-трет-бутилоксикарбонильную группу трифторуксусной кислотой, и гидрируя над PdCl2 катализатором (см. схему 14, приведенную в конце описания).
Промежуточные соединения формулы 30, которые представляют собой производные O-бензил-(D)-серина 34, удобно получать, используя известные процедуры, из соответствующим образом замещенных бензилгалоидов и N-защищенного (D)-серина 34. Удобно, чтобы защитными группами служили BOC и CBZ. Бензилирование 34 можно осуществить рядом способов хорошо известных из литературы, включая удаление защиты за счет двух эквивалентов гидрида натрия в таком инертном растворителе, как DMF с последующей обработкой одним эквивалентом какого-либо бензилгалоида (Synthesis, 1989, 36), как представлено на схеме 15, приведенной в конце описания.
O-Алкил-(D)-сериновые производные также получают, используя схему алкилирования, представленную на схеме 15. Другие способы, которые можно использовать для получения производных (D)-серина формулы 35, включают катализируемое кислотой бензилирование карбоксизащищенных промежуточных, полученных из 34, реагентами формулы ArCHOC(=NH)CCl3/O. Jonemitsu Chem. Pharm. Bull, 1988, 36, 4244. В другом варианте в результате алкилирования хиральных глициненолятов (J. Am. Chem. Soc., 1991, 113, 9276; J. Org. Chem., 1989, 54, 3916) ArCH2OCH2X, где X представляет отщепляемую группу, получают 35. Кроме того, D, L, -O-арил(алкил)серины и можно получить и разделить описанными ранее способами.
Алкилирование N-защищенного-(D)-цистеина 36 осуществляют способом, описанным для синтеза (D)-серинового производного, и проиллюстрированного далее для R1a-X, где X представляет отщепляемую группу, такую как галоиды и мезилоксигруппы, как представлено на схеме 16, приведенной в конце описания.
Окисление цистеинового производного 37 до сульфоксида 38 (n=1) и сульфона 30 (n= 2) можно осуществить, используя многие окисляющие агенты. (Обзор окисления сульфидов см. Org. Prer. Proced. Int., 1982, 14, 45). Перйодат натрия (J. Org. Chem., 1967, 32, 3191) часто используют для получения сульфоксидов, а кислый персульфат калия (OXONE) (Tetrahedron Lett., 1981, 22, 1287) используют для получения сульфонов.
Итак, различные замещенные аминокислоты можно ввести в соединение, ускоряющее выделение гормона роста, с помощью реакция, схемы которых представлены на схемах 1 и 8. Такие усиливающие секрецию соединения, которые содержат сульфоксидную или сульфоновую функциональную группу, можно получить из цистеинового ускоряющего секрецию соединения, используя перйодат натрия OXONE®. В другом варианте можно использовать перекись водорода в качестве окисляющего агента на последней стадии синтеза по схеме 17, приведенной в конце описания. Сульфоксидный 40 (n=1 ) и сульфоновый 40 (n=2) аналоги можно разделить с помощью препаративной тонкослойной хроматографии.
Удаление аминозащитных групп можно осуществить рядом способов известных специалистам; эти способы описаны в "Protective Groups in Organic Synthesis", T.W. Greene, N 4 1981.
Соединения формулы 1, где R4 и R5 каждый представляют водород, можно подвергнуть восстановительному алкилированию альдегидом в соответствии с вышеуказанными способами, или алкилированию за счет различных эпоксидов. Продукты, получаемые в виде солей соляной или трифторуксусной кислот, обычно очищают с помощью ВЭЖХ с обращенной фазой или за счет перекристаллизации.
Спиропиперидины формулы 41 можно получить несколькими способами, включая описанный далее синтез.
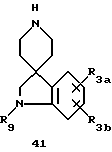
Спиропиперидины формулы 42, где L представляет определенную защитную группу, можно получить способами, известными из литературы (например, H. Ong. et al., J. Med. Chem., 1983, 23, 981-986).
Азот индолина 42, где L представляет такую защитную группу, как метил или бензил, можно подвергнуть взаимодействию с различными электрофильными соединениями до получения спиропиперидинов формулы 43, где R9 может представлять различные функциональности. Соединение 42 можно подвергнуть взаимодействию, например, с изоцианатами в таком инертном растворителе, как дихлорметан до получения производных мочевины; хлорформатами, в таком инертном растворителе, как дихлорметан до получения карбаматов; хлорангидридами ангидридами или ацилимидазолами для получения амидов; сульфонилхлоридами до поручения сульфонамидов, сульфамилхлоридами для получения сульфамидов (см. схему 18, приведенную в конце описания).
Кроме того, азот индолина 42 можно восстановительно алкилировать альдегидами в условиях, известных специалистам. Если альдегид, который используют в реакции восстановительного аминирования, представляет глиоксиловую кислоту формулы HCOCOOM, где M представляет определенную защитную группу, M можно удалить из продукта, и получать дальнейшие производные. В другом варианте, 42 можно подвергнуть взаимодействию с эпоксидами до получения 43, где R9 представляет β-гидроксизамещенную алкильную или аралкильную группы. Индолин 42 можно также превратить в соединение формулы 43, где R9 представляет фенил или замещенный фенил, гетероарил или замещенный гетероарил, осуществляя взаимодействие 42 с фторфенильным или фторгетероарильным реагентом. Эти реакции подробно описаны H. Ong. et al., J. Med. Chem., 1983, 23, 981-986.
Спиропиперидиновое промежуточное 43 (L = Me или Bn), где R9 представляет водород, или большинство из описанных ранее производных, можно деметилировать или дебензилировать до получения 44, где R9 представляет водород, или большинства описанных ранее производных, в соответствии со схемой 19, приведенной в конце описания. Для соединений формулы 43, где L = Me, деметилирование можно осуществить различными способами, известными специалистам. Так, например, деметилирование 43 можно осуществить, подвергая его взаимодействию с цианогенбромидом и карбонатом калия в таком инертном растворителе, как дихлорметан, до получения цианамида, который можно восстановить до получения 44 за счет обработки литийалюминийгидридом в кипящем с обратным холодильником тетрагидрофуране, или кипящей сильной кислоте, такой как соляная кислота, или с таким реагентом Гриньяра, как метилмагнийбромид. В другом варианте деметилирование 43 можно осуществить по способу АСЕ-Cl, как раскрыто у R. Olofson et al., Chem., 1984, 49, 2795, и в приведенных там ссылках. Для промежуточных соединений формулы 43, где L = Bn, удаление бензильной группы можно осуществить восстановительными методами, включая гидрирование в присутствии платинового или палладиевого катализатора в таком протонном растворителе, как метанол. В другом варианте дебензилирование 43, L = Bn, можно осуществить ACE-Cl способом, раскрытым у R. Olofson et al., J. Org. Chem., 1984.
Спирогетероциклические соединения 45 можно получить рядом способов, включая синтезы, представленные на схеме 20, приведенной в конце описания.
Аллильное окисление защищенного пиперидина 47 осуществляют классическими способами, известными специалистам (Rabjohn, N Org. React. 1976, 24, 261). Полученный аллильный спирт обрабатывают тионилхлоридом в таком инертном растворителе, как бензол до получения соответствующего хлорида 48. Если D = O или S, алкилирование ведут в DMF или ацетоне в качестве растворителя с карбонатом калия в качестве основания, а если D = NR9 (где R9 = H, алкил, арил, ацил, сульфонил; карбамат), реакцию ведут с гидридом натрия в качестве основания в таком инертном растворителе, как THF, до получения предшественника циклизации 49. Если L представляет определенную защитную группу, соединение 49 можно циклизовать рядом способов, известных специалистам. Так, например, диклизацию 49 можно осуществить при взаимодействии гидрида трибутилолова (Curran, D.P. Synthesis 1988, 417 и 489) в таком инертном растворителе, как бензол, до получения 46. В другом варианте, соединение 46 (D = NR9) можно получить способами, представленными на схемах 18 и 19. Если D = S, соединение 46 можно окислить до сульфоксида 47 (n = 1), и сульфоната 47 (n = 2), многими окисляющими агентами (см. схему 21, приведенную в конце описания). Так, например, перйодат натрия часто используют для синтеза сульфоксидов, а OXONE используют для синтеза сульфонов. После удаления защитных групп получают амин 45, который затем можно включить в соединение, ускоряющее выделение гормона роста в соответствии с реакциями, представленными на схемах 1 и 8, используя общее промежуточное соединение 2.
Спиропиперидины формулы 50 и 51 можно получить в реакциях, представленных на схеме 22, приведенной в конце описания.
Фталимидины формулы 53, где R11 имеет такие значения как алкил, арил (CH2)q-арил; или представляют защитную группу, либо коммерчески доступны, либо их можно синтезировать из соответствующих фталимидов способами, известными специалистам (например, Bewster et al., в J. Org. Chem., 1963, 28, 501; Mcalees et al., J. Chem. Soc., 1977, 2038). Фталимидин 53 можно алкилировать в присутствии такого основания, как гидрид калия, бис/триметилсилил/амид лития или калия, защищенным бис-2-галоидэтиламином, где L представляет такую защитную группу, как метил, бензил, трет-BOC или CBZ и т.д., а Y может быть Cl, Br, I, до получения спиропиперидина 54. Защитную группу можно удалить способами, описанными ранее, до получения соединения формулы 50. Восстановление лактама формулы 50 такими гидридами, как литийалюминийгидрид, приводит к получению соединения 51.
Следует отметить, что порядок осуществления указанных реакционных схем можно варьировать для облегчения реакции или для того, чтобы избежать образования нежелательных побочных продуктов.
Соединения, способствующие выделению гормона роста, формул I и II можно использовать in vitro как уникальные инструменты для понимания того, как происходит регулирование секреции гормона роста на уровне гипофиза. Это включает использование при оценке множества факторов, которые, как известно, или как предполагают, влияют на секрецию гормона роста (например, рост, пол, факторы питания, глюкоза, аминокислоты, жирные кислоты), а также состояние голода или сытости. Кроме того, соединения настоящего изобретения можно использовать для оценки того, как другие гормоны меняют активность выделения гормона роста. Так, например, уже было установлено, что соматостатин ингибирует выделение гормона роста. Другие гормоны, играющие важную роль, которые необходимо изучить в отношении их влияния на выделение гормона роста, включают такие гонадные гормоны, как тестостерон эстрадиол и прогестерон; такие адренальные гормоны, как кортизол и другие кортикоиды, эпинефрин и норэпинефрин; гормоны поджелудочной железы и желудочно-кишечного тракта, такие как инсулин, глюкагон, гастрин, секретин; такие вазоактивные пептиды как бомбезин, нейрокинины; и такие гормоны щитовидной железы, как тироксин и трийодотиронин. Соединения формул I и II можно также использовать для исследования возможных негативных или позитивных последствий действия некоторых гормонов гипофиза, таких как гормон роста и эндорфиновые пептиды, на гипофиз для модификации выделения гормона роста. Наибольшая научная ценность состоит в использовании этих соединений для выяснения механизма, за счет которого происходит выделение гормона роста на подклеточном уровне.
Соединения формулы I и II можно вводить животным, включая человека, для выделения гормона роста. Так, например, соединения можно вводить таким представляющим коммерческий интерес животным, как свиньи, овцы, крупный рогатый скот и т.п., для ускорения их роста и скорости привеса, для повышения эффективности корма и для увеличения производства молока у этих животных. Кроме того, эти соединения можно вводить людям in vivo в качестве диагностических инструментов для непосредственного определения того, способен ли гипофиз выделять гормон роста. Так, например, соединения формулы I и II можно вводить in vivo детям. Образцы сыворотки, отобранные до и после такого введения можно проанализировать на содержание гормона роста. Сравнение количеств гормона роста в каждом из этих образцов может служить для непосредственного определения способности гипофиза пациента выделять гормон роста.
Соответственно, настоящее изобретение включает в свой объем фармацевтические композиции, содержащие в качестве активного ингредиента по крайней мере одно соединение формулы I вместе с фармацевтическим носителем или разбавителем. Необязательно чтобы активный ингредиент фармацевтических композиций содержал анаболический агент в добавлении к по крайней мере одному из соединений формулы I, или другую композицию, обладающую другой активностью, например антибиотик, способствующий росту, или агент для лечения остеопороза, или в сочетании с кортикостероидом для минимизации катаболических побочных эффектов, или с другими фармацевтически активными материалами, если эта комбинация повышает эффективность и сводит к минимуму побочные эффекты.
Агенты, промотирущие рост, и анаболические агенты включают (но не ограничиваются ими) TRH, диэтилстилбестерол, эстрогены β-агонисты, теофиллин, анаболические стероиды, энкефалины, простагландины серии E, соединения, раскрытые в патенте США 3239345, зеранол и соединения, раскрытые в патенте США 4036979, например сулбенокс, или пептиды, раскрытые в патенте США 4411890.
Еще одним применением соединений, ускоряющих секрецию гормонов роста, настоящего изобретения является комбинация с другими соединениями, ускоряющими секрецию гормонов роста, такими как пептиды, способствующие выделению гормона роста GHRP-6, GHRP-1, как раскрыто в патенте США 4411890 и в публикациях WO 89/07110, WO 89/07111 и B-HT920, а также гексарелин и недавно открытый GHRP-2, описанный в WO 93/04081, или гормон, способствующий выделению гормона роста (GHRH, также обозначаемый как GRF), и его аналоги, или гормон роста и его аналоги, или соматомедины, включая IGF-1 и IGF-2 или α-адрегергенные агонисты, такие как клонидин или серотонин 5HTID агонисты, такие как сумитриптан, или агенты, которые ингибируют соматостатин или его выделение, такие как физоcтигмин и пиридоcтигмин.
Как хорошо известно специалистам, известное и потенциальное применения гормона роста разнообразны и многочисленны. Так, введение соединений настоящего изобретения с целью стимуляции выделения эндогенного гормона роста может оказывать такое же действие, что и сам гормон роста. Эти различные применения гормона роста можно суммировать следующим образом: стимуляция выделения гормона роста у пожилых людей; лечение дефицита гормона роста у взрослых; предотвращение катаболических побочных эффектов от применения глюкокортикостероидов, лечение остеопороза, стимуляция иммунной системы, ускорение заживления ран, ускорение сращивания костей после перелома, лечение замедленного роста, лечение острой или хронической почечной недостаточности, лечение физиологически низкого роста, включая дефицит гормона роста у детей, лечение низкого роста, связанного с хроническими заболеваниями, лечение тучности и замедления роста, связанного с тучностью, лечение замедления роста, связанного с синдромом Prader-Willi и синдромом Turuer's; ускорение выздоровления и уменьшение сроков госпитализации у пациентов с ожогами или после таких серьезных хирургических операций, как операции на желудке; лечение внутриутробного замедления роста, скелетной дисплазии, гиперкортизонизма и синдрома Кушинга; замена гормона роста у пациентов в стрессовых ситуациях; лечение остеохондродисплазии, синдрома Noonans расстройств сна, болезни Альцгеймера, задержки ранозаживления и психосоциальной депривации; лечение дисфункций легких и респираторной зависимости; ослабление протеиновой катаболической реакции после серьезных операций; лечение синдрома малабсорбции, снижение кахексии и потери протеинов из-за таких хронических заболеваний, как рак или СПИД, ускорение прироста веса и ускорение протеинов у пациентов на TPN (полное парентеральное питание); лечение гиперинсулинэмии, включая незидиобластоз; сопутствующее лечение для индуцирования овуляции и для предотвращения и лечения язвы желудка и двенадцатиперстной кишки; для стимуляции развития тимуса и предотвращения снижения функции тимуса; сопутствующая терапия для пациентов на хроническом гемодиализе; лечение пациентов с иммунодепрессией и для усиления реакции антител после вакцинации; повышения мускульной силы, мобильности, сохранения толщины кожи, метаболического гомеостаза, почечного гомеостаза у болезненных пожилых людей; стимуляция остеобластов, восстановление костной ткани и рост хрящей; лечение таких неврологических заболеваний, как периферическая и вызванная лекарствами невропатия, синдром Guillian-Barre, боковой амиотрофический склероз, цереброваскулярные нарушения и болезни, связанные с демиелинизацией; стимуляция иммунной системы у животных и лечение расстройств, связанных с возрастом у домашних животных; ускорение роста у домашнего скота и стимуляции роста шерсти у овец.
Специалистам должно быть ясно, что существует ряд соединений, которые в настоящее время используют в попытках лечения заболеваний или при терапевтических показаниях, перечисленных выше. Комбинации этих терапевтических агентов, некоторые из которых были перечислены ранее, с соединениями, ускоряющими секрецию гормона роста настоящего изобретения привносит дополнительные, комплементарные и часто синергетические свойства для усиления ускорения роста, анаболических и других желательных свойств этих различных терапевтических агентов. В этих комбинациях терапевтические агенты и соединения, ускоряющие секрецию гормона роста, настоящего изобретения могут независимо присутствовать в интервале доз от одной сотой до единицы от уровней доз, которые эффективны, если эти соединения и соединения, ускоряющие секрецию, используют отдельно.
Объединенная терапия для ингибирования костной резорбции, предотвращения остеопороза и ускорения заживления переломов костей может быть проиллюстрирована за счет сочетания бисфосфонатов и соединений, ускоряющих секрецию гормона роста, настоящего изобретения. Использование бисфосфонатов для этих целей было описано, например, у Hamdy, N.A.T., Role of Bisphosphonattes in Metabolic Bone Diseases, Treuds in Endocrinol. Metab. 1993, 4, 19-25. Бисфосфонаты для такого применения включают алендронат, тилудронат, диметил-АРД, ризедронат, этидронат, УМ-175, клодронат, памидронат и ВМ-210995. В соответствии с их эффективностью дневные уровни доз при пероральном приеме бисфосфоната составляют от 0,1 мг до 5 г, а уровни дневных доз соединений, ускоряющих секрецию гормона роста, настоящего изобретения - от 0,01 мг/кг до 20 мг/кг живого веса; которые вводят пациенту для эффективного лечения остеопороза.
Соединения настоящего изобретения можно вводить перорально, парентерально (например, внутримышечно, внутрибрюшинно, внутривенно или подкожно за счет инъекций или имплантатов), через нос, вагинально, ректально, сублингвально или поверхностно, и они могут быть приготовлены в виде дозовых форм соответствующих каждому из способов введения.
Твердые дозовые формы для перорального введения включают капсулы, таблетки, пилюли, порошки и гранулы. В таких дозовых формах активное соединение смешивают с по меньшей мере одним инертным фармацевтически приемлемым носителем, таким как сахароза, лактоза или крахмал. Такие дозовые формы могут также содержать, как это обычно практикуется, дополнительные вещества, отличные от инертных разбавителей, например скользящие, такие как стеарат магния. В случае капсул, таблеток и пилюль дозовые формы могут также содержать буферирующие агенты. Таблетки и пилюли могут также быть приготовлены с желудочными покрытиями.
Жидкие дозовые формы для перорального введения включают фармацевтически приемлемые эмульсии, растворы, суспензии, сиропы, эликсиры, содержащие инертные разбавители, которые обычно используют в таких целях, например воду. Помимо инертных разбавителей композиции могут также содержать такие адъюванты, как смачивающие агенты, эмульгаторы и суспендирующие агенты, а также подслащивающие, вкусовые агенты и отдушки.
Препараты настоящего изобретения для парентерального введения включают стерильные водные или неводные растворы, суспензии или эмульсии. Примеры неводных растворителей или носителей включают пропиленгликоль, полиэтиленгликоль, растительные масла, такие как оливковое масло и кукурузное масло, желатин и органические сложные эфиры для инъекций, такие как этилолеат. Такие дозовые формы могут также содержать такие адьюванты, как консерванты, смачивающие агенты, эмульгаторы и диспергирующие агенты. Их можно стерилизовать, например, за счет фильтрования через удерживающий бактерии фильтр, за счет введения в композиции стерилизующих агентов, за счет облучения композиций или за счет нагревания композиций. Их можно также приготовить в форме стерильных твердых композиций, которые можно растворить в стерильной воде, или в какой-либо другой стерильной среде для инъекций непосредственно перед применением.
Композиции для ректального или вагинального введения представляют собой предпочтительно суппозитории, которые могут содержать, помимо активного вещества, такие эксципиенты, как масло какао или воск для суппозиториев.
Композиции для назального или сублингвального введения также предпочтительно приготавливают со стандартными эксципиентами хорошо известными специалистам.
Дозы активного ингредиента в композициях настоящего изобретения могут меняться; однако необходимо, чтобы количество активного ингредиента было таково, чтобы получить подходящую дозовую форму. Выбираемые дозы зависят от желательного терапевтического эффекта, от способа введения, от длительности лечения. Обычно уровень доз находится в интервале от 0,0001 до 1000 мг/кг веса тела ежедневно для пациентов и животных, например млекопитающих, до получения эффективного выделения гормона роста.
Нижеследующие примеры предложены лишь с целью иллюстрации и никоим образом не ограничивают раскрытое изобретение.
Пример 1
N-/1(R)-/(2', 3'-Дигидро-2-оксоспиро/пиперидин-4,4'- (1H)-хинолин/-1'ил)карбонил/-2-(индол-3-ил)этил/-2-амино- 2-метилпропанамидгидрохлорид
Стадия А: 1'-(трет-бутилоксикарбонил)-3,4-дигидро- 3-оксоспиро/1H-инден-1,4'-пиперидин
К раствору 661 мг (2,31 ммоль) 1'-(трет- бутилоксикарбонил) спиро/1H-инден-1,4'-пиперидина/ (полученного по способу Chambers et al, J. Med. Chem. 1992, 35, 2036) в 5,0 мл THF добавляют 5,8 мл (1,0 М THF, 2,9 ммоль) 9-BBN. Реакционную смесь нагревают при 70oC до тех пор, пока по данным TCX не оказывается, что весь исходный материал израсходован. Полученный раствор концентрируют, а остаток растворяют в дихлорметане. Полученный раствор охлаждают до 0oC, и за 15 минут медленно добавляют 4,1 г (19,2 ммоль) PCC. Реакционную смесь нагревают до комнатной температуры, а затем кипятят с обратным холодильником в течение 30 минут. Полученный раствор разбавляют эфиром и фильтруют через фильтровальную лепешку, состоящую из смеси целита и флорисила. В результате очистки с помощью флеш-хроматографии (силикагель, гексан/этилацетат, 4:1) получают 326 мг (47%) указанного в заглавии соединения.
1H ЯМР (200 МГц, CDCl3): δ 7,75 - 7,60 (м, 2H), 7,50 - 7,44 (м, 2H), 4,30 - 4,15 (м, 2H), 2,85 (дт, 2H), 2,63 (с, 2H), 1,98 (дт, 2H), 1,53 - 1,40 (м, 2H), 1,49 (с, 9H).
Стадия B: Спиро/1H-инден-1,4-пиперидин/-3(2H)-он трифторацетамид
Раствор промежуточного соединения, полученного на стадии A в смеси 1:1: 0,5 трифторуксусной кислоты, дихлорметана и анизола перемешивают в течение 1 часа, а затем концентрируют и азеотропно перегоняют из толуола до получения указанного в заглавии соединения.
1H (200 МГц, CDCl3): δ 7,81 - 7,70 (м, 1H), 7,62 - 7,45 (м, 2H), 7,22 - 7,15 (м, 1H), 3,72 - 3,58 (м, 2H), 3,29 - 3,04 (м, 2H), 2,70 (с, 2H), 2,47 (дт, 2H), 1,85 - 1,75 (м, H).
Стадия C: Трифторацетамид-2,3-дигидроспиро/инден- 1,4'-пиперидин/
К раствору 1,0 г (3,21 ммоль) промежуточного соединения, полученного на стадии В, в 3,0 мл дихлорметана добавляют 0,945 мл (6,74 ммоль) триэтиламина и 50 мг DAMP, и наконец, 0,501 мл (3,63 ммоль) ангидрида трифторуксусной кислоты, реакционную смесь перемешивают в течение 3 часов, затем разбавляют 20 мл дихлорметана. Полученный раствор промывают водой, сушат над сульфатом магния и концентрируют. В результате очистки с помощью флэш-хроматографии (силикагель, гексан/этилацетат 2:1) получают 568 мг (1,91 ммоль).
1H ЯМР (200 МГц, CDCl3): δ 7,79 - 7,64 (м, 2H), 7,5 - 7,41 (м, 2H), 4,75 - 4,65 (м, 1H), 4,22 - 4,08 (м, 1H), 3,37 (дт, 1H), 2,92 (дт, 1H), 2,70 (с, 2H), 2,08 (дт, 2H), 1,71 - 1,6 (м, 2H).
Стадия D: Трифторацетамид-3', 4'-дигидро-2-оксоспиро/пиперидин-4,4'(1H)-хинолин/
К раствору 218 мг (3,36 ммоль) азида натрия в 0,285 мл воды и 1,5 мл хлороформа при 0oC добавляют 0,105 мл серной кислоты. Реакционную смесь перемешивают в течение 2,5 ч, а затем слои разделяют, и хлороформовый слой сушат над сульфатом натрия. Затем раствор гидразиновой кислоты добавляют к раствору 400 мг (1,34 ммоль) промежуточного соединения, полученного на стадии A. К этому раствору добавляют 0,400 мл и серной кислоты за 5 минут, реакционную смесь перемешивают в течение 20 минут, а затем в течение 45 минут при 45oC, и, наконец, в течение 16 часов при комнатной температуре. Слой серной кислоты добавляют ко льду, а затем подщелачивают 50% гидроксидом натрия. Водный слой экстрагируют этилацетатом, этилацетатные экстракты сушат над сульфатом натрия и концентрируют. В результате очистки порции в 100 мг сырого продукта с помощью флэш-хроматографии (силикагель, дихлорметан/этилацетат 1: 1, а затем 1:2) получают 50 мг (0,160 ммоль) материала с высоким значением RF и 16 мг (0,051 ммоль) с низким значением RF.
1H ЯМР (200 МГц, CDCl3, высокое RF): δ 8.9-8.7 (шир.с, 1H), 7.40-7.21 (м, 2H), 7.18-7.04 (м, 1H), 6.90-6.86 (м, 1H), 4.52-4.36 (м, 1H), 3.97-3.83 (м, 1H), 3.52 (дт, 1H) 3.22 (дт, 1H), 2.79 (с, 2H), 2.12-1.66 (м, 4H). 1H NMR (200 МГц, CDCl3, низкое RF): 8.12 (дд, 1H), 7.60-7.52 (м, 1H), 7.45-7.35 (м, 2H), 6.95 (шир.с, 1H), 4.56-4.43 (м, 1H), 4.03-3.96 (м, 1H), 3.64-3.62 (м, 2H), 3.49-3.35 (м, 1H), 3.11 (дт, 1H), 2.20-1.80 (м, 4H).
Стадия E: 3',4'-Дигидро-2-оксоспиро/пиперидин-4,4'- (1H)-хинолин/
Раствор 49 мг (0,157 ммоль) материала с высоким RF со стадии B в смеси метанол/вода 4: 1 с избытком гидроксида калия перемешивают в течение ночи. Полученный раствор концентрируют, и к остатку добавляют воду и этилацетат. Слои разделяют, и водный слой экстрагируют этилацетатом. Объединенные органические слои сушат над сульфатом натрия и концентрируют до получения 31 мг (0,143 ммоль) указанного в заглавии соединения.
Стадия F: N-/(R)-/(2',3'-дигидро-2-оксо,спиро/пиперидии-4,4' -(1H)-хинолин/-1'ил)карбонил/-2-(индол-3-ил)этил/-1-//1,1- диметилэтилокcикарбонил/амино/-2-метилпропанамид
К раствору 29 мг (0,134 ммоль) промежуточного соединения, полученного на стадии C, 65 мг (0,167 ммоль) 2-амино-N-/-(1R)-/2',3'-дигидро-2-оксоспиро/пиперидин-4,4'(1'H)- хинолин/-1-ил)-карбонил/-2-(1H-индол-3-ил )этил-2-метилпропанамида и 24 мг (0,174 ммоль) HOBT в дихлорметане добавляют 33 мг (0,174 ммоль) EDC. Реакционную смесь перемешивают в течение ночи, а затем обрабатывают и очищают по способу примера 1 (стадия A) за одним исключением: при хроматографической обработке используют дихлорметан/ацетон. Получают 34,8 мг (0,059 ммоль) указанного в заглавии соединения.
Стадия G: N-/1(R)-/(2', 3'-Дигидро-3-оксоспиро/пиперидин-4,4'(1H)- хинолин/-1'ил)карбонил/-2- (индол-3-ил)этил/-2-амино-2-метилпропанамидгидрохлорид
Указанное в заглавии соединение (7,2 мг, 0,013 ммоль) получают из промежуточного соединения, полученного на стадии D (14 мг, 0,023 ммоль), по способу примера 1 (стадия C) за одним исключением: гидрохлоридную соль получают из очищенного свободного амина, добавляя в этом случае 4 н. HCl в диоксане.
1Н ЯМР (400 МГц, CD3OD, 2:1 смесь ротамеров): δ 8.34 (д, 2/3H), 8.27 (д, 1/3H), 7.59 (д, 2/3H), 7.55 (д, 1/3H), 7.38 (д, 1/3H), 7.33 (д, 2/3H), 7.25 (д, 1/3H), 7.18-6.98 (м, 4H), 6.85 (д, 1/3H), 6.80 (д, 2/3H), 6.68 (д, 2/3H), 5.23-5.17 (м, 1H), 4.22-4.19 (м, 2/3H), 4.09-3.95 (м, 1/3H), 3.62-3,59 (м, 1/3H), 3.36-3.17 (м, 2 2/3H), 3.08 (дт, 1/3H), 2.75 (дт, 1/3H), 2.69 (дт, 2/3H), 2.48 (дд, 2H), 1.93-1.75 (м, 2/3H), 1.60 (с, 3H), 1.58 (с, 2H), 1.40-1.32 (м, 1H), 1.51 (с, 1H), 1.10 (м, 1/3H), 1.02 (м, 2/3H), 0.90 (м, 2/3H), 0.22 (дт, 2/3H). FAB-MS: m/e 490 (М+1).
Масс-спектр с бомбардировкой быстрыми атомами: м/е 490 (М+1).
Пример 2
N-/1(R)-/(2', 3'-Дихлор-1-оксоспиро/пиперидин-4,4'- (1H)-изохинолин/-1'ил)карбонил/-2-(индол-3-ил)этил/-2- амино-2-метилпропанамидгидрохлорид
Стадия A: /3',4'-Дигидро-1-оксоспиро/пиперидин- 4,4'(1H)-изохинолин/
Указанное в заглавии соединение (11,3 мг, 0,036 ммоль) получают из промежуточного соединения примера 1 (стадия D) с низким значением RF (16,0 мг, 0,051 ммоль) по способу примера 13 (стадия D).
1H ЯМР (200 МГц, CDCl3): δ 8,12 (дд, 1H), 7,60-7,52 (м, 1H), 7,45 - 7,35 (м, 2H), 6,95 (шир.с, 1H), 4,56 - 4,43 (м, 1H), 4,03-3,96 (м, 1H), 3,64-3,63 (м, 2H), 3,49-3,35 (м, 2H), 3,11 (дт, 1H), 2,20-1,80 (м, 4H).
Стадия B: N-/1(R)-/(2,3'-Дигидро-1-оксоспиро/пиперидин-4,4' (1H)-изoxинoлин/-1'ил)кapбoнил/-2-(индол-3-ил) этил/-2-//1,1-диметилэтилоксикарбонил/-амино/-2-метилпропанамид
Указанное в заглавии соединение (13,6 мг, 0,023 ммоль) получают из промежуточного соединения со стадии A (10,0 мг, 0,032 ммоль) и 2-амино-N-/1(R)-/2',3'-дигидро-2-оксоспиро/пиперидин-4,4' (1'Н)-хинолин/-1-)карбонил/-2-(1H-индол-3-ил)этил-2-метилпропанамида (21,6 мг, 0,055 ммоль) по способу примера 13 (стадия D).
Стадия C: N-/1(R)-/(2',3'-Дигидро-1-оксоспиро/пиперидин-4,4' (1H)-изохинолин/-1'ил)карбонил/-2-(индол-3-ил)этил/-2-амино-2- метилпропанамидгидрохлорид
Раствор 10,1 мг (0,017 ммоль) промежуточного соединения, полученного на стадии B, в 1,5 н. HCl в этилацетате перемешивают в течение ночи, а затем концентрируют и азеотропно перегоняют из метанола до получения 8,3 мг (0,015 ммоль) указанного в заглавии соединения.
1H ЯМР (400 МГц, CD3OD, 2:1 смесь ротамеров): δ 7.94 (д, 1/3H), 7.87 (д, 2/3H), 7.62-7.53 (м, 2H), 7.40-7.33 (м, 2 1/3H), 7.18-7.10 (м, 3H), 6.75 (д, 2/3H), 5.22-5.18 (м, 2/3H), 5.15 (т, 1/3H), 4.27-4.23 (м, 2/3H), 4.14-4.10 (м, 1/3H), 3.68-3.61 (м, 1H), 3.25-3.18 (м, 4H), 3.10 (дт, 2/3H), 2.87 (дт, 1/3H), 2.70 (дт, 1/3H), 2.65 (дт, 2/3H), 1.88 (дт, 1/3H), 1.75 (дт, 1/3 H), 1.62 + 1.61 +1.59+1.51 (с, 6 H total), 1.57-1.44 (м, 1H), 1.38-1.35 (м, 1/3H), 1.15-1.10 (м, 1/3H), 0.929 (дт, 2/3H), 0.19 (дт, 2/3H). FAB-MS: m/e 490 (М+1).
Масс-спектр с бомбардировкой быстрыми атомами: м/е 490 (M+1).
Пример 3
N-/1(R)-/(4H-1-Оксоспиро/3H-2-бензопиран-3,4'-пиперидин/-1'-ил) карбонил/-2-(индол-3-ил)-этил/-2-амино-2-метилпропанамидгидрохлорид
Стадия A: Спиро/3H-2-бензопиран-3,4'-пиперидин/-1(4H)-он
К суспензии 10% палладия на угле (5 мг) в 5 мл этанола добавляют 1'-бензилспиро/3H-2-бензопиран-3,4'-пиперидин/-1(4H)-он/ (20 мг, 0,059 ммоль) (Hashigaki et al., Chem. Pharm. Bull., 32, pp. 3561-3568 (1984)). Гидрирование ведут при комнатной температуре и давлении 1 атм. Реакционную смесь перемешивают в течение 2 часов при комнатной температуре до тех пор, пока по данным ТСХ реакция не заканчивается. Катализатор удаляют вакуумной фильтрацией через целит 545, и полученный фильтрат концентрируют до получения целевого продукта (12,4 мг, 98,5%).
Масс-спектр с бомбардировкой быстрыми атомами, рассчитано для C13H15NO2 217, найдено 218 (М+H, 100%).
Стадия B: N-/1(R)-/(4H-1-Оксоспиро/3H-2-бензопиран- 3,4'-пиперидин/-1'-ил )карбонил/-2-(индол-3-ил )- этил/-2-//(1,1-диметилэтилокси)карбонил/амино/ 2-метилпропанамид
Раствор промежуточного соединения со стадии A (12 мг, 0,055 ммоль) и α(R)-//2-//(1,1-диметилэтокси)карбонил/- aминo/-2,2-димeтил-1-oксoэтил/амино/-1H-индoл-3-пропанoвoй кислоты (27 мг, 0,058 ммоль) в дихлорметане охлаждают до 0oC, а затем добавляют HOBT (2 мг, 0,015 ммоль), N-метилморфолин (8,8 мг, 0,084 ммоль) и ЕДС (22 мг, 0,12 ммоль). Реакционную смесь перемешивают при комнатной температуре в течение 1 часа до тех пор, пока по данным ТСХ реакция не завершается. Полученный раствор промывают насыщенным раствором натрийхлорида и сушат над безводным сульфатом магния. Затем полученный раствор фильтруют и концентрируют. В результате очистки с помощью хроматографии на силикагеле получают указанное в заглавии соединение (15 мг, 47%). Масс-спектр с бомбардировкой быстрыми атомами, рассчитано для C33H40N4O6: 589. Найдено 589 (М+H, 39%) (489 М+H, 100, 42%, потеря трет-Boc группы).
Стадия C: N-/1(R)-/(4H-1-оксоспиро/3H-2-бензопиран- 3,4'-пиперидин/-1'-ил)карбонил/-2-(индол-3- ил)этил/-2-амино-2-метилпропанамидгидрохлодрид
Раствор промежуточного соединения со стадии В (12 мг, 0,02 ммоль) в 3 мл метанола охлаждают до 0oC. При перемешивании медленно добавляют концентрированную соляную кислоту (3 мл). Реакционную смесь перемешивают в течение 30 минут, до тех пор, пока по данным ТСХ анализа реакция не завершается. Затем полученный раствор несколько раз концентрируют из толуола. Соль соляной кислоты используют далее без дополнительной очистки (10,15 мг, 96%).
1H ЯМР (400 МГц, CD3OD): продукт существует в виде смеси двух конформеров (2:1): δ 7.977, 7.905 (2д, 2/3 H), 7.604-6.994 (м, 8H), 5.134-5.093 (м, 1 2/3 H), перекрыт 5.025-4.715 (м, 2H), 4.191-4.114 (м, 1/3H), 3.637-3.587 (м, 1H), 3.344-3.299 (м, 1H), 3.188-3.124 (м, 1H) 3.030 (с, 2/3 H), (дт 2.81 Гц 9.4 Гц, 1/3 H), 2.536 (кв, 1H), 2.301 (т, 1/3 H), 1.590, 1.571 (2с, 6H), 1.539-1.483 (м, 2/3H), 1.275 (с, 6H), 1.259-1.206 (м, 2/3H), (м, 1H), 0.633-0.545 (м, 1/3H), -0.277 -0.361 (м, 1/3H).
FAB-MS calc. for C28H32N4O4 488; found 489 (M+H, 65%).
Масс-спектр с бомбардировкой быстрыми атомами рассчитано для C28H32N4O4: 488, найдено 489 (M+H, 65%).
Пример 4
N-/(R)-/(4', 5'-Дигидpo-4'-oксocпиpo/пипepидин-4,6'- /6H/тиено[2,3-b] тиопиран/-1-ил)карбонил/-2-(индол-3-ил)- этил/-2-амино-2-пропанамидгидрохлорид
Стадия A: N-/1(R)-/(4',5'-дигидро-4'-оксоспиро/пиперидин- 4,6'/6H/тиено-2,3-b-тиопиран/-1-ил)карбонил/-2- (индол-3-ил)этил/-2-//(1,1-диметилэтилокси)карбонил/-амино/-2-пропанамид
Получают по способу примера 3, стадия В. Используют: Спиро/пиперидин-4,6'-/6H/тиено[2,3-b] тиопиран/-4'(5'H)-он- гидрохлорид (10 мг, 0,044 ммоль) (EP публикация 90313629), α(R)-//2-//(1,1-диметилэтокси)карбонил/амино/- 2,2-диметил-1-оксоэтил/амино/-1H-индол-3-пропановую кислоту (20 мг, 0,051 ммоль), HОВТ (1 экв.), N-метилморфолин (0,01 мл, 0,093 ммоль) и EDC (20 мг, 0,10 ммоль). Время реакции 5 часов, выход: 22 мг (98%).
1H ЯМР (400 МГц, CDCl3): продукт существует в виде смеси двух конформеров (2:1): δ 8.240 (с, 2/3 H), 8.063 (с, 1/3 H), 7.680 (д, 2/3 H), 7.628 (д, 1/3 H), 7.416-6.962 (м, 5 H), 5.279-5.162 (м, 1H), 4.878- 4.763 (м, 1H), 4.285 (д, 2/3H), 3.376 (д, 2/3 H), 3.342-3.196 (м, 1H), 3.129-2.973 (м, 1 2/3H), 2.715-2.662 (м, 1H), 2.285 (д, 2/3H), 2.139 (д, 2/3H), 1.683-1.567 (м, 8 1/3H), 1.503, 1.454, 1.427, 1.409 (4с, 12H), 1.278-1.217 (м, 2H), 0.708-0.628 (м, 2/3H).
Масс-спектр с бомбардировкой быстрыми атомами: рассчитано: C31H38N4O5S2 610, найдено: 611 (M+H, 32%).
Стадия B: N-/1(R)-/(4', 5'-дигидро-4'-оксоспиро/пиперидин-4,6'-/6H/тиено-2,3-b тиопиран/-1-ил)-карбонил/-2-(индол-3-ил)этил/-2-амино-2-пропанамидгидрохлорид
Получают по способу примера 15, стадия C. Промежуточное соединение с предыдущей стадии (200 мг, 0,033 ммоль) и 3 мл метанола. Время реакции 1,5 часа. Выход 12,2 мг (69%).
1H ЯМР (400 МГц, CD3OD): продукт существует в виде смеси двух конформеров (2:1): δ 7.562-7.022 (м, 6H), 5.513-5.446 (м, 6 2/3H), 5.099-5.003 (м, 1H), перекрыт 4.914-4.726 (м, 2/3H), 4.178 (д, 1H), 3.624 (д, 1H), 3.337-3.043 (м, 2 2/3H), 2.760-2.660 (м, 1H), 2.324 (д, 1H), 2.234 (д, 1H), 1.597, 1.587, 1.574, 1.510 (4с, 4H), 1.364-1.225 (м, 3H), 0.562-0.482 (м, 2/3H), -0.311 -0.391 (м, 2/3H).
Масс-спектр с бомбардировкой быстрыми атомами, рассчитано для C26H30N4O3S2 510, найдено 611 (M+H, 51%).
Пример 5
N-/1(R)-/(3-Гидроспиро/1H-изобензофуран-1,4'-пиперидин/-1'-ил) карбонил/-2-(индол-3-ил)этил/-2-амино-2-метилпропанамидгидрохлорид
Стадия A: N-/1(R)-/(3-Гидроспиро/1H-изобензофуран-1,4 -пиперидин/-1'-ил)карбонил/-2-(индол-3-ил)этил/-2- //1,1-диметилэтилоксикарбонил/амино/-2-метилпропанамид
Получают по способу примера 3, стадия В. Используют: 3-гидроспиро/1H-изобензофуран-1,4'-пиперидин/гидрохлорид (10 мг, 0,044 моля) (Bauer, et. al. , патент США 3985889), α(R)-//2-//(1,1-диметилэтокси)карбонил/аминоно/- 2,2-диметил-1-оксоэтил/амино/-1H-индол-3-пропановую кислоту (20 мг, 0,051 ммоль), HОВТ (1 экв.), N-метилморфолин (0,01 мл, 0,093 ммоль) и EDC (20 мг, 0,10 ммоль). Время реакции 5 часов. Выход 21 мг (81%). Продукт существует в виде двух конформеров (1:1):
1H ЯМР (CDCl3): δ 8.096 (с, 1H), 7.689 (т, 1H), 7.341 (д, 1H), 7.244-6.611 (м, 6H), 5.288-5.202 (м, 1/2H), 4.945 (шир.с, 1/2H), 4.161 (д, 1/2H), 4.003 (д, 1/2H) 3.338 (д, 1/2H), 3.280-3.115 (м, 2H), 3.005-2.861 (м, 1H), 2.751 (д, 1/2H), 2.416 (д, 1/2H), 1.787- 1.549 (м, 3 1/2H), 1.491, 1.461, 1.421, 1.410 (4с, 12H), 1.281-1.212 (м, 3H), 0.857 (т, 6H).
Масс-спектр с бомбардировкой быстрыми атомами, рассчитано для C32H40N4O5 560, найдено: 561 (M+H, 33%).
Стадия B: N-/1(R)-/(3-Гидроспиро/1H-изобензофуран- 1,4'-пиперидин/-1'-ил)карбонил/-2-(индол-3- ил)этил/-2-амино-2-метилпропанамидгидрохлорид
Получают до способу примера 3, стадия C. Используют промежуточное соединение с предыдущей стадии (20 мг, 0,04 ммоль) и 3 мл метанола. Время реакции 1 час. Выход 18,2 мг (93,5%).
1H ЯМР (400 МГц, CD3OD): продукт существует в виде смеси двух конформеров (1:1): δ 7.621-6.568 (м, 8H), 5.198-5.136 (м, 1H), перекрыт 4.856 (шир.с, 1H), 4.098-4.045 (м, 1H), 3.611-3.499 (м, 1H), 3.348-3.110 (м, 5 1/2H) 2.987-2.903 (м, 2 1/2H), 2.618 (д, 1/2H), 2.508 (д, 1/2 H), 1.691-1.473( м, 8H), 1.271 (шир.с, 2 1/2H), 0.081- 0.006 (м, 1/2H).
Масс-спектр с бомбардировкой быстрыми атомами, рассчитано для C27H32N4O3 461, найдено 461 (M+H, 96%).
Пример 6
N-/1(R)-/(3,4-Дигидро-6-метил-4-оксоспиро/2H-1- бензопиран-2,4'-пиперидин/-1'-ил)карбонил/-2- (индол-3-ил)- этил/-2-амино-2-метилпропанамидгидрохлорид
Стадия A: N-/1(R)-/(3,4-Дигидро-6-метил-4-оксоспиро/2H- 1-бензопиран-2,4 ' -пиперидин-1'-ил)-карбонил/-2-(индол-3-ил)этил/-2-//(1,1-диметилэтилокси) карбонил/амино/-2-метилпропанамид
Получают по способу примера 3, стадия В. Используют: 3,4-дигидро-6-метилспиро/2H-1-бензопиран-2,4'-пиперидин/- 4-онгидрохлорид (20 мг, 0,058 ммоль) (Hashigaki et. al. Chem. Pharm. Bull, 32, pp 3561-3568 (1984)), α(R)-//-2-//(1,1-диметилэтокси)карбонил/амино/-2,2-диметил-1- оксоэтил/амино/-1H-индол-3-пропановую кислоту (32 мг, 0,082 ммоль), НОВТ (1 экв.), N-метилморфолин (0,03 мл 0,28 ммоль) и EDC (40 мг, 0,21 ммоль). Время реакции 8 часов. Выход 34 мг (86%).
1H ЯМР (400 МГц, CDCl3): продукт существует в виде смеси двух конформеров (2:1): δ 8.154 (с, 2/3H), 8.088 (с, 1/3H), 7.626 (д, 2/3H), 7.591-7.060 (м, 6H), 6.725-6.688 (м, 2/3H), 5.265- 5.168 (м, 2/3H), 4.985-4.900 (м, 2/3H), 4.289-4.178 (м, 2/3H), 3.469 (с, 2/3H), 3.229-3.064 (м, 2 2/3H), 2.730 (т, 2/3H), 2.562 (с, 2 1/3H), 2.251 (д, 2 1/3H), 2.158 (д, 2/3H), 2.068 (д, 2/3H), 1.680-1.541 (м, 3H), 1.502, 1.475, 1.454, 1.427, 1.402 (5с, 15H), 1.292-1.226 (м, 3H), 0.616-0.532 (м, 1/3H), -0.495 -0.590 (м, 1/3H).
Масс-спектр с бомбардировкой быстрыми атомами, рассчитано для C34H42N4O6 602, найдено 603 (M+H, 37%).
Стадия В: N-/1(R)-/(3,4-Дигидро-6-метил-оксоспиро/2H- 1-бензопиран-2,4'-пиперидин/-1'-ил)карбонил/- 2-(индол-3-ил)этил/-2-амино-2-метилпропанамидгидрохлорид
Получают по способу примера 3, стадия C. Используют промежуточное соединение с предыдущей стадии (20 мг, 0,029 ммоль) и 3 мл метанола. Время реакции 3 часа. Выход 17,5 мг (96,5%).
1H ЯМР (400 МГц, CDCl3): продукт существует в виде смеси двух конформеров (2: 1): δ 7.550-6.768 (м, 7 1/3H), 5.089-5.016 (м, 2H), перекрыт 4.872-4.679 (м, 1H), 4.144-4.093 (м, 1H), 3.569-3.485 (м, 1H), 3.321-3.081 (м, 2 1/3H), 2.716-2.600 (м, 1 1/3H), 2.253, 2.236, 2.222, 2.196, 2.190, 2.155 (6с, 4H), 1.569, 1.541 1.475 (3с, 7H), 1.388-1.237 (м, 3 2/3H), 0.881-0.808 (м, 2H), 0.434-0.420 (м, 2/3H), 0.427-0.436 (м, 2/3H).
Масс-спектр с бомбардировкой быстрыми атомами, рассчитано для C29H34N4O4 504; найдено 503 (М+H, 97%).
Пример 7
N-/1(R)-/(3,4-Дигидро-4-оксоспиро/2H-1-бензопиран- 2,4'-пиперидин/-1'-ил)карбонил/-4-фенилбутил-2-амино-2- метилпропанамидгидрохлорид
Стадия A: α-(R)-//2-//(1,1-Диметилэтилокси)карбонил)амино/-2,2-диметил-1-оксоэтил/ амино/-4- фенилбутановой кислоты фенилметиловый сложный эфир
Дихлорметановый раствор 2(R)-амино-4-фенилбутановой кислоты фенилметилового эфира/соли толуолсульфокислоты (6,0 г, 13 ммоль) экстрагируют разбавленным раствором гидроксида натрия. Органический слой сушат над сульфатом магния и выпаривают до получения остатка. Добавляют к раствору этого остатка N-трет-бутилоксикарбонил-α -метилаланин (3,31 г, 15 ммоль), HOBT (1,7 г, 13 ммолей) в дихлорметане добавляют ЕДС (5,1 г, 26 ммоль), и полученную смесь перемешивают в течение ночи при комнатной температуре. Полученную смесь выливают в смесь рассола и 3 н. HCl и экстрагируют этилацетатом. Органический экстракт сушат, выпаривают и очищают с помощью флеш-хроматографии, элюируя 40% этилацетатом в гексане, до получения целевого продукта (5,47 г, 91%).
1H ЯМР (400 МГц, CDCl3): δ 7,34-7,07 (м, 10H), 5,15 (д, JAB=12 Гц, 1H), 5,08 (д, JBA=12 Гц, 1H), 4,86 (шир.с, 1H), 4,67-4,62 (м, 1H), 2,61-2,53 (м, 2H), 2,18-2,14 (м, 1H), 2,01-1,96 (м, 1H), 1,47 (с, 3H), 1,43 (с, 3H), 1,41 (с, 9H).
Стадия B: α(R)-//2-//(1,1-Диметилэтилокси)карбонил- /амино/-2,2-диметил-1-оксоэтил/амино/-4-фенилбутановая кислота
Промежуточное соединение с предыдущей стадии (5,37 г, 11,8 ммоль) гидрируют при комнатной температуре и давлении 1 атм, используя 10% палладий на угле в качестве катализатора (0,5 г), в течение 2 часов. Катализатор отфильтровывают через целит, раствор выпаривают до получения указанного в заглавии соединения (4,22 г, 100%).
1H ЯМР (200 МГц, CD3OD): δ 7,804-7,143 (м, 5H), 4,402-4,359 (м, 1H), 2,672 (дт, 2 Гц, 6 Гц, 2H), 2,126- 2,004 (м, 2H), 1,483-1,444 (2с, 5H), 1,423 (с, 9H), 1,422 (с, 3H).
Стадия C: N-/1(R)-/(3,4-Дигидро-4-оксоспиро/2H-1- бензопиран-2,4'-пиперидин/-1'-ил)карбонил/- 4-фенилбутил-2-//(1,1-диметилэтилокси)карбонил/амино/-2-метилпропанамид
Раствор спиро/2H-1-бензопиран-2,4'-пиперидин/-4(3H)-она (20 мг, 0,0776 ммоль) и промежуточного соединения с предыдущей стадии (31 мг, 0,085 ммоль) в дихлорметане охлаждают до 0oC, а затем добавляют НОВТ (1 экв.), N-метилморфолин (0,1 мл, 0,90 ммоль) и ЕДС (33 мг, 0,17 ммоль). Реакционную смесь перемешивают при комнатной температуре в течение 3 часов до тех пор, пока по данным ТСХ реакция не завершается. Полученный раствор промывают насыщенным раствором натрийхлорида и сушат над безводным сульфатом магния. Полученный раствор фильтруют и концентрируют. После хроматографической очистки на силикагеле получают указанное в заглавии соединение (41,6 мг, 95,5%).
1H ЯМР (400 МГц, CDCl3): продукт существует в виде смеси двух конформеров (1:1): δ 7.853-6.925 (м, 9H), 4.936-4.868 (м, 2H), 4.355-4.265 (мт, 1H), 3.605-3.565 (м, 1/2H), 3.388-3.318 (м, 1H), 3.022-2.965 (м, 1H), 2.686-2.608 (м, 3H), 2.067-1.948 (м, 2 1/2H), 1.871-1.810 (м, 1H), 1.580 (шир.с, 2H), 1.503, 1.488, 1.455, 1.411, 1.403 (5с, 15H), 1.292-1.227 (м, 2H).
Стадия D: N-/1(R)-/(3,4-Дигидро-4-оксоспиро/2H-1- бензопиран-2,4'-пиперидин/-1'-ил)карбонил/- 4-фенилбутил-2-амино-2-метилпропанамидгидрохлорид
Раствор промежуточного соединения с предыдущей стадии (40 мг, 0,071 ммоль) в этилацетате охлаждают до 0oC. Затем через раствор барботируют газообразный хлористый водород в течение 2 минут. Реакционную смесь перемешивают в течение 15 минут до тех пор, пока по данным ТСХ реакция не завершается. Полученный раствор концентрируют, и соль соляной кислоты (33,8 мг, 95,5%) используют без дополнительной очистки.
1H ЯМР (400 МГц, CD3OD): продукт существует в виде смеси двух конформеров (1:1): δ 8.271-8.229 (м, 1H), 7.799 (дд, 1 3/4 Гц, 7.84 Гц, 1H), 7.545 (кв, 1H) 7.289-7.006 (м, 7H), 4.737-4.703 (м, 1H), 4.277 (д, 1/2H), 4.186 (д, 1/2H), 3.555-3.292 (м, 1 1/2H), 3.187-3.068 (м, 1H), 2.809-2.724 (2м, 2H), 2.633-2.563 (м, 1H), 2.085-1.927 (м, 3 1/2H), 1.645, 1.639, 1.616 (3с, 6H), 1.677-1.603 (м, 3H), 1.316-1.279 (м, 2H).
Пример 8
N-/1(R)-/(3,4-Дигидро-4-оксоспиро/2H-1-бензопиран- 2,4'-пиперидин/-1'-ил)карбонил/-2-(фенилметилокси)этил/-2- амино-2-метилпропанамидгидрохлорид
Стадия A: N-/1(R)-/(3,4-Дигидро-4-оксоспиро/2H-1- бензопиран-2,4'-пиперидин/-1'-ил)карбонил/- 2-(фeнилмeтилoкси)этил/-2-//(1,1-димeтилэтилокси)карбонил/амино/-2- метилпропанамид
Раствор спиро/2H-1-бензопиран-2,4'-пиперидин/-4-(3H)она (20 мг, 0,776 ммоль) и α(R)-//2-//(1,1-диметилэтилокси)- карбонил/амино/-2,2-диметил-1-оксоэтил/амино/-3- (фенилметилокси)-3-пропановой кислоты (32 мг, 0,085 ммоль) в дихлорметане охлаждают до 0oC, а затем добавляют N-метилморфолин (0,1 мл, 0,90 ммоль) и ЕДС (33 мг, 0,171 ммоль). Реакционную смесь перемешивают при комнатной температуре в течение 4 часов до тех пор, пока по данным ТСХ реакция не завершается. Полученный раствор промывают затем насыщенным раствором натрийхлорида, и сушат над безводным сульфатом магния. Полученный раствор затем фильтруют и концентрируют. После хроматографической очистки на силикагеле получают указанное в заглавии соединение (40,8 мг, 91%). Масс-спектр с бомбардировкой быстрыми атомами, рассчитано для C32H41N3O7: 579; найдено 580 (М+H, 23%); (найдено 480 (М+H - 100, 57%, потеря трет-BOC защитной группы).
Стадия B: N-/1(R)-/(3,4-Дигидро-4-оксоспиро/2H-1- бензопиран-2,4'-пиперидин/-1'-ил)карбонил/- 2-(фенилметилокси)этил/-2-амино-2-метилпропанамидгидрохлорид
Получают по способу примера 3, стадия D. Используют промежуточное соединение с предыдущей стадии (35 мг, 0,061 ммоль) и этилацетат (10 мл). Время реакции 1 час. Выход 30,2 мг (97%).
1H ЯМР (400 МГц, CD3OD): продукт существует в виде смеси двух конформеров (1:1): δ 7.800-7.792-(м, 1H), 7.533-7.578 (м, 1H), 7.395-7.285 (м, 5H), 7.088-7.015 (м, 2H), 5.551-5.107 (м, 1H), перекрыт 4.921-4.816 (м, 1H), 4.535-4.518 (2с, 1 1/2H), 4.295 (д, 1H), 3.911-3.803 (м, 1H), 3.717-3.703 (2с, 1 1/2H), 3.499-3.400 (м, 1H), 3.309-3.291 (4с, 3 1/2H), 3.211-3.051 (м, 1H), 2.789 (кв, 1/2H), 2.633-2.513 (AB кв, 1H), 2.060 (т, 1H), 1.897 (д, 1/2H), 1.821-1.725 (м, 1/2H), 1.626-1.567 (6с, 6H), 1.564-1.410 (м, 1/2H), 1.301 (шир.с, 1 1/2H).
Масс-спектр с бомбардировкой быстрыми атомами, рассчитано для C27H33N3O5 479; найдено 480 (M+H, 100%).
Пример 9
N-/1(R)-/(6-Хлор-3H-4-оксоспиро/1H-хиназолин-2,4'- пиперидин/-1'-ил)карбонил/-2-(индол-3-ил)этил/-2-амино-2- метилпропанамидгидрохлорид
Стадия A: N-/1(R)-/(6-Хлор-3H-4-оксоспиро/1H-хиназолин-2,4' -пиперидин/-1'-ил)карбонил/-2-(индол-3-ил)этил/-2-//(1,1- диметилэтилокси)карбонил/амино/-2-метилпропанамид
Получают по способу примера 3, стадия B. Используют: 6-хлороспиро/пиперидин-4,2 (1'H)-хиназолин)-4(3H)онгидрохлорид (50 мг, 0,17 ммоль), α(R)-//2-//(1,1-диметилэтокси) карбонил/амино/-2,2-диметил-1-оксоэтил/амино/-1H-индол-3-пропановую кислоту (81 мг, 0,21 ммоль), HOBT (1 экв.), N-метилморфолин (1 экв. ) и ЕДС (80 мг, 0,42 ммоль). Время реакции 3 часа. Выход 64,5 мг (60%). Масс-спектр с бомбардировкой быстрыми электронами, рассчитано для C32H39N6O5Cl 623, найдено 624 (М+H, 29%).
Стадия B: N-/1(R)-/(6-Хлор-3H-4-оксоспиро/1H-хиназолин-2,4'-пиперидин/-1'-ил) карбонил/-2-(индол-3-ил)этил/-2-амино-2-метилпропанамидгидрохлорид
Получают по способу примера 7, стадия D. Используют промежуточное соединение с предыдущей стадии (50 мг, 0,08 ммоль). Время реакции: 1 час. Выход 40 мг (89,5%). Масс-спектр с бомбардировкой быстрыми атомами, рассчитано для C27H31N6O3Cl 523, найдено 523 (М+H, 71%).
Пример 10
N-/1(R)-/(1,4-Дигидро-4-фенил-1-оксоспиро/3H-2-бензопиран-3,4' -пиперидин/-1'-ил )карбонил/-2-(индол-3-ил)- этил/-2-амино-2-метилпропанамидгидрохлорид
Стадия A: 1,4-Дигидро-4-фенилспиро/3H-2-бензопиран- 3,4'-пиперидин/-1-он
Получают по способу примера 3, стадия A, из 1'-бензил- 1,4-дигидро-4-фенилспиро(3H-2-бензопиран-3,4'-пиперидин)-1- онгидрохлорида (8 мг, 0,019 ммоль) и 5 мл этанола. Время реакции 45 минут. Выход 5,5 мг (98,5%). Масс-спектр с бомбардировкой быстрыми атомами, рассчитано для C19H19NO2 293, найдено 294 (М+H, 93%).
Стадия B: N-/1(R)-/(1,4-Дигидро-4-фенил-1-оксоспиро/- 3H-2-бензопиран-3,4'-пиперидин/-1'-ил)карбонил/-2-(индол-3-ил) этил/-2-//(1,1-диметилэтилокси)карбонил/амино/-2-метилпропанамид
Получают по способу примера 3, стадия B. Используют промежуточное соединение с предыдущей стадии (5 мг, 0,017 ммоль), α(R)-//2-//(1,1-диметилэтокси)карбонил/амино/- 2,2-диметил-1-оксоэтил/амино/-1H-индол-3-пропановую кислоту (12 мг, 0,030 ммоль), HOBT (1 экв.), N-метилморфолин (1 экв.) и ЕДС (12 мг, 0,060 ммоль). Время реакции 5 часов. Выход 9,2 мг (86%).
1H ЯМР (400 МГц, CD3OD): продукт существует в виде смеси двух конформеров (1: 1): δ 8.185-8.072 (м, 1 1/2H), 7.885 (с, 1/2H), 7.710- 6.813 (м, 12H), 5.331-5.309 (м, 1/2H), 5.198-5.111 (м, 1/2H), 4.710-4.605 (м, 1/2H), 4.300-4.235 (м, 1/2H), 3.876 (д, 1/2H), 3.719-3.617 (м, 1H), 3.355-3.046 (м, 1 1/2H) 2.746 (кв, 1/2H), 2.006-1.960 (м, 1H), 1.678-1.574 (м, 2H), 1.438, -1.368 (м, 6H), 1.257, 1.240, 1.227, 1.208, 1.186 (5с, 5H).
Стадия C: N-/1(R)-/(1,4-Дигидро-4-фенил-1-оксоспиро- /3H-2-бензопиран-3,4'-пиперидин/-1'-ил)- карбонил/-2-(индол-3-ил)этил/-2-амино-2-метилпропанамидгидрохлорид
Получают по способу примера 7, стадия D. Используют промежуточное соединение с предыдущей стадии (9 мг, 0,015 ммоль) и 10 мл этилацетата. Время реакции 1 час, выход 8 мг (97%).
1H ЯМР (400 МГц, CDCl3): продукт существует в виде смеси двух конформеров (2: 1): δ 8.347-8.333 (м, 1H), 8.043 (т, 1/2H), 7.662-6.869 (м, 12 1/2H), 5.355-5.315 (м, 1/2H), 5.108-5.061 (м, 1/2H), перекрыт 4.897-4.768 (м, 1/2H), 4.174-4.103 (м, 1/2H), 3.717-3.526 (м, 1H), 3.387-3.237 (м, 2H), 3.179-3.067 (м, 1H), 2.660 (кв, 1/2H), 2.044-1.981 (м, 1H), 1.655-1.212 (м, 11H), 0.964-0.820 (м, 2 1/2H), 0.575-0.423 (м, 1/2H), -0.271, -0.448 (м, 1/2H).
Масс-спектр с бомбардировкой быстрыми атомами, рассчитано для C34H36N4O4 564, найдено 565 (M+H, 25%).
Пример 11
N-/2(R)-/(3,4-Дигидро-4-оксоспиро/2H-1-бензопиран- 2,4'-пиперидин/-1'-ил)карбонил/-2-(индол-3-ил)этил-2-амино- 2-метилпропанамидгидрохлорид
Стадия A: N-/1(R)-/(3,4-Дигидро-4-оксоспиро/2H-1- бeнзoпиpaн-2,4'-пипepидин/-1'-ил)кapбoнил/-2- (индол-3-ил)этил/-2-//(1,1-диметилэтилокси)- карбонил/амино/-2-метилпропанамид
Это промежуточное соединение получают из α(R)- //2-//(1,1-диметилэтокси)карбонил/амино/-2,2-диметил-1- оксоэтил/амино/-1H-индол-3-пропановой кислоты (903 мг, 2,3 ммоль) и спиро/2H-1-бензопиран-2,4'-пиперидин/-4(3H)-он- гидрохлорида (535 мг, 2,11 ммоль) (Elliot. J. et. al. J. Med. Chem. 1992, 35, 3973-3976), по способу примера 25 стадия A (1,25 г, 100%).
1H ЯМР (400 МГц, CDCl3) соединение существует в виде смеси конформеров (отношение 2: 1): δ 8.42, 8.31 (2c, 1H), 7.79, 7.75 (2дд, 1.6 Гц, 7.8 Гц, 1H), 7.66, 7.56 (2д, 8.0 Гц, 7.6 Гц, 1H), 7.47-6.78 (м, 8H), 5.37-5.15 (м, 1H), 4.98, 4.94 (2 шир.с, 1H), 4.24, 4.18 (2 шир.с 1H), 3.40, 3.32 (2 шир.д, 2H), 3.23-3.02 (м, 3H), 2.73 (дт, 3 Гц, 13 Гц, 1H), 2.47 (д, 2 Гц, 1/3H), 2.17 (д, 16.6 Гц, 2/3H), 2.08 (д, 16.7 Гц, 2/3H) 1.84 (шир.с, 2H), 1.70-1.60 (шир. дд, 1H), 1.3-1.2 (шир.дд, 1H), 0.56 (дт, 4.6, 13.8 Гц, 2/3H), -0.55 (дт, 4.6, 13.8 Гц, 2/3H). FAB-MS:
Масс-спектр с бомбардировкой быстрыми атомами, рассчитано для C33H40N4O6 588, найдено 595 (М+Li 100%).
Стадия B: N-/1(R)-/(3,4-Дигидро-4-оксоспиро/2H-1- бензопиран-2,4'-пиперидин/-1'-ил)карбонил/- 2-(индол-3-ил)этил/-2-амино-2-метилпропанамидгидрохлорид
К перемешиваемому раствору промежуточного соединения, полученного на стадии A (1,0 г, 1,7 ммоль) в 5 мл метанола добавляют 5 мл концентрированной соляной кислоты. Реакционную смесь перемешивают при комнатной температуре в течение 1 часа, и добавляют 20 мл толуола, и полученную смесь выпаривают в вакууме. Эту процедуру повторяют дважды до получения указанного в заглавии соединения (0,87 г, 98%).
1H ЯМР (400 МГц, CD3OD): соединение существует в виде смеси двух конформеров (отношение 2:1): δ 7.76-6.90 (м, 10H), 5.11 (дд, 5,11 Гц, 1H), 4.16, 4.11 (2дт, 2.0 Гц, 14 Гц, 1H), 3.60, 3.33 (2мд, 14 Гц, 1H), 3.25-3.10 (м, 2H), 2.92-2.67 (м, 2H), 2.30-2.17 (AB, с центром у w.23, 16.7 Гц, 2H), 2.85-2.80 (шир. д, 1/3H), 1.60 1.59 (2с, 6H), 1.70-1.50 (м, перекрыт), 1.40-1.30 (мд, 1H), 0.47 (дт, 5.5, 13.5 Гц, 2/3H), -0.38 (дт, 5.5, 13.5 Гц, 2/3H).
Масс-спектр с бомбардировкой быстрыми атомами, рассчитано C28H32N4O4 488, найдено 489 (M+H, 100%).
Пример 11A
N-/1(R)-/(3,4-Дигидро-4(RS)-гидроксиспиро/2H-1- бензопиран-2,4'-пиперидин/-1'-ил)карбонил/-2-(индол-3-ил)- этил-2-амино-2-метилпропанамид
К перемешиваемому раствору указанного в заглавии примера 11 соединения (55 мг, 0,09 ммоль) в 5 мл метанола при 0oC добавляют боргидрид натрия (16 мг, 0,4 ммоль) несколькими порциями. После 30 минут перемешивания при 0oC смесь выпаривают досуха и растворяют в дихлорметане, очищают с помощью флеш-хроматографии, элюируя 10% метанолом в дихлорметане до получения указанного в заглавия соединения (35 мг, 78%).
1H ЯМР (400 МГц, CD3OD): соединение существует в виде смеси двух диастереоизомеров (1: 1), и каждый изомер существует в виде двух конформеров (отношение 2: 1): δ 7.89-6.66 (м, 9H), 5.14-5.06 (м, 1H), 4.52-4.45 (2дд, 1H), 4.22-4.10 (2мд, 1H), 3.58-3.44 (2мд, 1H), 3.25-3.14 (м, 2H), 3.10- 2.59 (4дт, 1H), 2.02 (дд, 6.2, 14.7 Гц, 1/3H), 1.79-1.74 (дд, 1/3H), 1.60-1.40 (м, 3H), 1.37, 1.31, 1.28, 1.28, 1.26 (4с, 6H), 1.3-1.05 (м, перекрыт), 0.71, 0.49 (2дт, 5.6, 13.5 Гц 2/3H), -0.20, -0.47 (2дт, 4.6, 13.5 Гц, 2/3H).
Масс-спектр с бомбардировкой быстрыми атомами, рассчитано для C28H34N4O4 490, найдено 491 (М+H, 100%).
Пример 12
N-/1(R)-/(3,4-Дигидро-спиро/2H-1-бензопиран-2,4'- пиперидин/-1'-ил)карбонил/-2-(индол-3-ил)этил/-2-амино-2- метилпропанамидгидрохлорид
Стадия A: 3,4-Дигидроспиро/2H-1-бензопиран-2,4'-пиперидин/
К перемешиваемому раствору спиро/2H-1-бензопиран-2,4'- пиперидин/-4(3H)-онгидрохлорида (53 мг, 0,21 ммоль) в 5 мл метанола при 0oC добавляют боргидрид натрия (38 мг, 1 ммоль) несколькими порциями. Спустя 30 минут полученную смесь выпаривают, а затем обрабатывают концентрированной соляной кислотой (2 мл) в течение 30 минут. Полученный остаток гидрируют за счет палладия на угле (10%, 10 мг), H2 (1 атм) в этаноле в течение 2 часов. Катализатор отфильтровывают, получая чистое промежуточное соединение (89 мг), которое используют без дополнительной очистки.
1H ЯМР (400 МГц, CD3OD): δ 7,07 (проявляется как дублет, 2H), 6,84 (проявляется как триплет 7 Гц, 2H), 7,07-7,08 (м, 4H), 2,82 (т, 7 Гц, 2H), 2,02 (шир. д, 14,5 Гц), 1,90-1,85 (м, 4H), E1 - масс-спектр: рассчитано для C13H17NO 203, найдено 203 (M+, 45%).
Стадия B: N-/1(R)-/(3,4-Дигидро-спиро/2H-1-бензопиран-2,4'- пиперидин/-1'-ил)карбонил/-2- (индол-3-ил)этил/-2-//(1,1-диметилэтилокси)- карбонил/амино/-2-метилпропанамид
Это промежуточное соединение получают из продукта со стадии A и α(R)-//2//(1,1-диметилэтокси)карбонил/амино/-2,2- диметил-1-оксоэтил/амино/-1H-индол-3-пропановой кислоты в соответствии со стандартными способами сочетания пептидов.
1H ЯМР (400 МГц, CDCl3), соединение существует в виде конформеров (отношение 2:1): δ 8.04, 8.02 (2с, 1H), 7.70, 7.61 (2д, 8 Гц, 1H), 7.49-6.66 (м, 1H), 4.92 (шир.с, 1H), 4.30-4.20 (м, 1H), 3.4-3.1 (м, 4H), 2.85-2.45 (м, 3H), 1.68 (т, 7.6 Гц, 1H), 1.49, 1.45, 1.44, 1.43, 1.41 (5с, 12H), 1.30-1.21 (м, 3H), 1.11-1.07 (дд, 2.5, 14 Гц, 1/3H), 0.68 (дд, 4.5 Гц, 13 Гц, 1/3H), -0.33 - -0.43 (дт, 1/3H).
Масс-спектр с бомбардировкой быстрыми атомами, рассчитано для C33H42N4O5: 574, найдено 575 (М+H, 35%).
Стадия C: N-/1(R)-/(3,4-Дигидро-спиро/2H-1-бензопиран- 2,4'-пиперидин/-1'-ил )карбонил/-2-(индол- 3-ил)этил/-2-амино-2-метилпропанамидгидрохлорид
Указанное в заглавии соединение получают из промежуточного соединения со стадии B по способу примера 11, стадия B, (90%).
1H ЯМР (400 МГц, CD3OD): соединение существует в виде смеси конформеров (отношение 2: 1): δ 8.31, 8.21 (2д, 6.6 Гц, 2/3H), 7.58, 7.52 (2д, 7.8 Гц, 1H), 7.37 (д, 8.2 Гц, 1H), 7.15-6.60 (м, 6 1/3H), 5.17-5.13 (м, 1H), 4.14 (шир.д, 13.2 Гц, 1H), 3.35-3.10 (м, 3H), 2.90-2.45 (м, 3H), 1.70 (т, 6.9 Гц, 1H), 1.60 (с, 6H), 1.60-1.40 (м, перекрыт), 1.40- 1.20 (м, 2H), 1.11 (шир.д, 12.7 Гц, 2/3H), 0.57 (дт, 4.3, 13 Гц, 2/3H), -0.31 (дт, 4.3, 13, 2/3H).
Масс-спектр с бомбардировкой быстрыми атомами, рассчитано для C28H34N4O3 474, найдено 475 (М+H, 60%).
Пример 13
N-/1(R)-/(3,4-Дигидро-6-метансульфониламино-4-оксоспиро/2H- 1-бензопиран-2,4'-пиперидин/-1'-ил)карбонил/-2- (индол-3-ил)этил/-2-амино-2-метилпропанамидгидрохлорид
Указанное в заглавии соединение получают из α(R)- //2-//(1,1-диметилэтокси)карбонил/амино/-2,2-диметил-1- оксоэтил/амино/-1H-индол-3-пропановой кислоты и 3,4-дигидро- 6-метансульфониламино-4-оксо-спиро/2H-1-бензопиран-2,4'- пиперидина/ по способу примера 10, стадии A и B.
N-/1(R)-/(3,4-Дигидро-6-метансульфониламино-4-оксоспиро/2H- 1-бензопиран-2,4'-пиперидин/-1'-ил)карбонил/-2- (индол-3-ил)этил/2-//(1,1-диметилэтилокси)карбонил/амино/- 2-метилпропанамид
1H ЯМР (400 МГц, CDCl3): соединение существует в виде смеси конформеров (отношение 2:1): δ 8.60, 8.36 (2 шир.c, 1H), 7.63-6.81 (м, 8H), 5.20 (шир.c, 1H), 5.10-5.02 (шир.м, 1H), 3.45-3.30 (шир.м, 1H), 3.25- 3.10 (шир.м, 2H), 2.97, 2.95 (2с, 3H), 2.75-2.56 (м, 1H), 2.28 (оч.шир.с, 1H), 2.18 (д, 16.6 Гц, 1H), 2.05 (д, 16.6 Гц, 1H), 1.86-1.45 (м, перекрыт), 1.51, 1.46, 1.44, 1.43, 1.42, 1.39 (6с, 12H), 1.30-1.20 (м, 2H), 0.55-0.45 (м, 2/3H), -0.55 - -0.65 (м, 2/3H).
Масс-спектр с бомбардировкой быстрыми атомами, рассчитано для C34H43N5O8S 681, найдено 688 (М+Li, 40%).
N-/1(R)-/(3,4-Дигидро-6-метансульфониламино-4-оксо-спиро/2H-1-бензопиран- 2,4'-пиперидин/-1'-ил)карбонил/-2- (индол-3-ил)этил/-2-амино-2-метанпропанамидгидрохлорид
1H ЯМР (400 МГц, CD3OD), соединение существует в виде смеси конформеров (отношение 2: 1): δ 7.63-6.92 (м, 8H), 5.14-5.08 (м, 1H), 4.18-4.08 (2мд, 1H), 3.62-3.51 (2мд, 1H), 3.25-3.10 (м, 2H), 2.91, 2.89 (2с, 3H), 2.78-2.67 (2дд, 2 Гц, 15 Гц, 2H), 2.27 (д, 16.7 Гц, 1H), 2.19 (д, 16.6 Гц, 1H), 1.86-1.80 (м, 1/3H), 1.80-1.50 (м, перекрыт), 1.60, 1.59, 1.48 (3с, 6H), 1.40-1.30 (м, 1H), 0.47 (дт, 4.8 Гц, 13 Гц, 2/3H), -0.39 (дт, 4.8 Гц, 13 Гц, 2/3H).
Масс-спектр с бомбардировкой быстрыми атомами, рассчитано для C29H35N5O6S 581, найдено 582 (M+H, 75%).
Пример 14
N-/1(R)-/(3,4-Дигидро-4(RS)-гидрокси-6-метансульфониламиноспиро/2H-1- бензопиран-2,4'-пиперидин/-1'-ил) карбонил/-2-(индол-3-ил)этил/-2-амино-2-метилпропанамид
Указанное в заглавии соединение получают из соединения, указанного в заглавии примера 13, по способу примера 11A.
1H ЯМР (400 МГц, CD3OD), соединение существует в виде смеси 2 диастереоизомеров (1:1), и каждый изомер существует в двух конформациях: δ 7.62-7.50 (м, 1H), 7.42-7.29 (м, 2H), 7.17-6.98 (м, 4H) 6.68 (д, 8.7 Гц, 1H), 5.15-5.05 (м, 1H), 4.75-4.65 (м, 1/3H), 4.57 (1, 7 Гц, 9 Гц, 1/3H), 4.44 (дд, 6.5 Гц, 9.0 Гц, 1/3H), 4.21-4.07 (м, 1H), 3.56-3.44 (м, 1H), 3.28-3.12 (м, 3H), 3.08-3.01 (м, 2/3H), 2.89, 2.86 (2с, 3H), 12.82-2.55 (м, 1H), 2.03 (дд, 6.0 Гц, 13.8 Гц, 1/2H), 1.86 (дд, 6.0, 13.7, 1/2H), 1.70-1.35 (м, 3H), 1.33, 1.32, 1.31, 1.28, 1.24 (5с, 6H), 1.33-1.29 (м, перекрыт), 1.06 (шир.д, 13 Гц, 1/3H), 0.71 (дт, 4.6 Гц, 13 Гц, 1/3H), 0.49 (дт, 4.6 Гц, 13 Гц, 1/3H), -0.21 (дт, 4.6 Гц, 13 Гц 1/3H), -0.49 (дт, 4.6 Гц, 13 Гц, 1/3H).
Масс-спектр, с бомбардировкой быстрыми атомами, рассчитано для C29H37N5O6S 583, найдено 584 (М+H, 20%).
Пример 15
N-/1(R)-/(2-Ацетил-1,2,3,4-тетрагидроспиро/изохинолин-4,4'- пиперидин/-1'-ил)карбонил/-2-(индол-3-ил)этил/-2- амино-2-метилпропанамидгидрохлорид
Стадия A: 1,3-Дигидро-1,3-дигидроксиспиро/4H-2-бензофуран-4,4'- пиперидин/-1'-карбоновой кислоты 1,1-диметилэтиловый сложный эфир
Через перемешиваемый раствор спиро/1H-инден-1,4'-пиперидин/- 1'-карбоновой кислоты 1,1-диметилэтиловому сложному эфиру (800 мг, 1,8 ммоль) и 50 мл метанола при -78oC продувают озон до тех пор, пока раствор не становится синим. Полученную смесь оставляют при этой температуре на 20 минут, затем продувают азотом. Добавляют диметилсульфид (3 мл), и полученную смесь нагревают до комнатной температуры и перемешивают в течение двух часов. После выпаривания растворителя получают сырой продукт (940 мг), который используют без дополнительной очистки.
Стадия В: 1,2,3,4-Тетрагидроспиро/изохинолин-4,4'- пиперидин/-1'-карбоновой кислоты 1,1-диметиловый сложный эфир
Промежуточное соединение со стадии A (100 мг) перемешивают в 2 мл метанола, насыщенного аммиаком в течение одного дня, и выпаривают для удаления аммиака. Остаток снова растворяют в 3 мл метанола, и добавляют цианоборгидрид натрия (50 мг, в избытке). Полученную смесь перемешивают в течение ночи. После выпаривания и очистки получают амин.
1H ЯМР (400 МГц, CD3OD): δ 7,35-6,96 (м, 4H), 4,00 (с, 2H), 3,14 (с, 2H), 3,90 (шир. с, 2H), 2,05 (шир.с, 2H), 1,95 (шир.т, 2H), 0,69 (д, 2H), 1,49 (с, 9H).
Стадия C: 2-Ацетил-1,2,3,4-тетрагидроспиро/изохинолин- 4,4'-пиперидин/-1'-карбоновой кислоты 1,1-диметилэтиловый сложный эфир
Промежуточное соединение со стадии A (16 мг) обрабатывают 2 мл пиридина и 2 мл ангидрида уксусной кислоты в течение 2 часов, затем реакционную смесь выпаривают в вакууме до получения указанного в заглавии соединения (12 мг).
1H ЯМР (400 МГц, CDCl3), соединение существует в виде смеси 3:1 ротамеров: δ 7.36-7.05 (м, 4H), 4.72 (с, 2/4H), 4.65 (с, 6/4H), 4.10- 4.00 (шир.д, 12.8 Гц, 2H), 3.85 (шир.с, 3/4H), 3.65 (с, 1/4H), 3.11 (т, 13.1 Гц, 3/4H), 3.00 (т, 13.1 Гц, 1/4H), 2.19 (с, 3/4H), 2.18 (с, 9/4H) 2.00-1.80 (м, 2H), 1.65-1.47 (м, 2H, перекрыт), 1.47 (с, 9/4H), 1.45 (с, 27/4H).
Стадия D: 2-Ацетил-1,2,3,4-тетрагидроспиро/изохинолин- 4,4'-пиперидин/
Через раствор промежуточного соединения, полученного на стадии C (12 мг), в 5 мл этилацетата при 0oC барботируют HCl (газ) до насыщения. Спустя 30 минут реакционную смесь выпаривают в вакууме до получения целевого промежуточного соединения.
1H ЯМР (400 МГц, CD3OD): δ 7,47-7,19 (м, 4H), 4,79 (с, 2H), 3,96 (с, 2H), 3,36 (шир. д, 6,7 Гц, 2H), 2,30-2,24 (м, 1H), 2,21 (с, 3H), 1,76 (д, 13H).
Масс-спектр с бомбардировкой быстрыми атомами, рассчитано для C15H20S2O 244, найдено 245 (М+H, 100%).
Стадия E: N-/1(R)-/(2-Ацетил-1,2,3,4-тетрагидроспиро/ изохинолин-4,4'-пиперидин/-1'-ил)карбонил- /-2-(индол-3-ил)этил/-2-//(1,1-диметилэтилокси) карбонил/амино/-2-метил-пропанамид
Указанное в заглавии соединение получают из промежуточного соединения со стадии D описанным ранее способом.
Пример 16
N-/1(R)-/1,2-Дигидро-1-метансульфонилспиро/3H-индол-3,4'-пиперидин/- 1'-ил)карбонил/-2-(2', 6'-дифторфенилметилокси)этил/-2- амино-2-метилпропанамидгидрохлорид
Стадия A: Метил- α(R)-//2-//(1,1-димeтилэтoкси)кapбoнил/ амино/-2,2-димeтил-1-oксoэтил/амино/- 3-/(2',6'-дифтopфенил)мeтoкси/пpoпaнoвaя кислота
Суспензию, не содержащую масла гидрида натрия (полученного из 60% масляной дисперсии гидрида натрия в результате промывки гексанами (3 х), 1,2 г, 30,0 ммоль), в 30 мл N,N-диметилформамида добавляют к N-трет-бутоксикарбонил- (D)-серину (3,07 г, 15,0 ммоль) в 10 мл N,N- диметилформамида при комнатной температуре. Когда прекращается выделение газа, добавляют 2,6-дифторбензилбромид (2,68 г, 12,9 ммоль). После 18 часов перемешивания при комнатной температуре к реакционной смеси добавляют йодометан (1,0 мл, 16,0 ммоль). Полученную смесь перемешивают еще 1 час, а затем выливают в воду и экстрагируют этиловым эфиром. Органический слой промывают последовательно водой (5 х), рассолом и сушат над сульфатом натрия, фильтруют и концентрируют. Остаток растворяют в 20 мл хлороформа и при комнатной температуре добавляют BOC-α-метилаланин, ЕДС, HOBT и Et3N. Спустя 3 часа реакционную смесь выливают в воду и экстрагируют метиленхлоридом. Органический слой сушат над сульфатом натрия и концентрируют. Указанное в заглавии соединение получают после хроматографической очистки (гексан/этилацетат 3:1) до получения 2,37 г (35%).
1H ЯМР (300 МГц, CDCl3, смесь ротамеров): δ 7,27 (м, 1H), 7,02-6,88 (м, 2H), 4,95 (м, 1H), 4,72 (дт, 8,3 Гц, 1H), 4,58 (шир.с, 2H), 3,90 (м, 1H), 3,78 (с, 1H), 3,69 (с, 3H), 1,48 (с, 3H), 1,45 (с, 3H), 1,41 (с, 9H).
Стадия B: N-/1(R)-[(1,2-Дигидро-1-метилсульфонилспиро[3H- индол-3,4'-пиперидин] -1'-ил)карбонил] - 2-(2', 6'-дифторфенилметилокси)этил]-2-амино- 2-метилпропанамидгидрохлорид
К раствору промежуточного соединения, полученного в этом примере на стадии A (2,37 г, 5,29 ммоль) в 30 мл метанола добавляют гидроксид лития (340 мг, 8,1 ммоль) в 3 мл воды. Через 2 часа при перемешивании при комнатной температуре реакционную смесь концентрируют, а затем разбавляют водой, экстрагируют этиловым эфиром. Органический слой сливают. Водный слой подкисляют 1 н. соляной кислотой до pH = 1,5, и экстрагируют этиловым эфиром (3 х). Органический слой сушат над сульфатом натрия, фильтруют и концентрируют до получения 2,18 г (95%) кислоты. Указанное в заглавии соединение получают из кислоты (78 мг, 0,18 ммоль) и 1,2-дигидро-1-метилсульфонилспиро/3H-индол-3,4'-пиперидингидрохлорида (50 мг, 0,164 ммоль) по способу примера 20, стадия В (используя гидрохлорид в этиловом эфире вместо трифторуксусной кислоты) до получения 48 мг (44%).
1H ЯМР (400 МГц, CD3OD, смесь ротамеров): δ 7.39 (м, 2H), 7.22 (м, 1 1/2H), 7.03 (м, 1/2H), 5.14 (дд, 13,7 Гц, 1H), 4.66 (д, 16 Гц, 2H), 4.49 (м, 1H), 4.09 (м, 1H), 3.92 (шир.с, 2H), 3.76 (м, 2H), 3.25 (м, 1H), 2.97 (с, 3/2H), 2.96 (с, 3/2H), 2.87 (м, 1H), 1.95 (м, 1H), 1.76 (м, 3H), 1.61 (с, 3/2H), 1.57 (с, 3 3/2H), FAB-MS: 565 (М+1).
Масс-спектр с бомбардировкой быстрыми атомами: 565 (M+1).
Пример 17
N-[(R)-[(1,3-Дигидро-1-метилсульфонилспиро[3H-индол- 3,4'-пиперидин] -1'-ил)кapбoнил]-3-циклoгeкcилпpoпил]-2- амино-2-метилпропанамидгидрохлорид
Стадия A: трет-Бутилоксикарбонил-(D)-гексагидрогомофенилаланин
Раствор трет-бутилоксикарбонил-(D)-гомофенилаланина (100 мг, 0,358 ммоль) в 1 мл уксусной кислоты гидрируют над PtO2 при давлении 1 атм в течение 16 часов. Полученную смесь фильтруют через целит, и полученный фильтрат концентрируют и азеотропно перегоняют с толуолом.
1H ЯМР (400 МГц, CDCl3): δ 5,03 (д, 8 Гц, 1H), 4,22 (м, 1H), 1,82 (м, 1H), 1,64 (м, 6H), 1,41 (с, 9H), 1,20 (м, 6H), 0,84 (м, 2H).
Стадия В: Бензил-α(R)[[2-[[((1,1 -диметилэтилокси)- карбонил]-амино]-2,2-диметил -1-оксоэтил]амино]-4-циклогексилбутановая кислота
Раствор BOC-D-гомофенилаланина в уксусной кислоте гидрируют над PtO2 при давлении 1 атм в течение 16 часов. Полученную смесь фильтруют через целит и концентрируют. К этому остатку (44 мг) в 15 мол. DMF добавляют бензилбромид (198 мл) и K2CO3 (970 мг) при комнатной температуре. После перемешивания в течение ночи полученную смесь выливают в 200 мл эфира и промывают водой. Органическую фазу сушат над сульфатом магния, фильтруют и концентрируют. Остаток очищают с помощью флеш-хроматографии (силикагель, 7,5% этилацетат в гексанах) до получения 534 мг (95%) этого промежуточного соединения. Раствор 534 мг этого материала в 10 мл смеси 1:1 TFA/CH2Cl2 перемешивают в течение 1 часа, а затем отпаривают и азеотропно перегоняют из толуола. Остаток растворяют в 10 мл CH2Cl2 и охлаждают до 0oC. BOC-α-метилаланин (362 мг), ЕДС, HOBT и NMM добавляют, и все это перемешивают в течение ночи. Полученный раствор выливают в 250 мл этилацетата и промывают последовательно 1 н. NaHSO4 (водн.), водой и насыщенным бикарбонатом натрия. Органическую фазу сушат, фильтруют и концентрируют. В результате хроматографической очистки с помощью флеш-хроматографии (силикагель, этилацетат /гексаны) получают 638 мг указанного в заглавии соединения.
1H ЯМР (200 МГц, CDCl3): δ 0,8-0,95 (м, 3H), 1,05-1,3 (м, 7H), 1,4-1,9 (м, 19H), 2,15 (м, 2H), 4,59 (м, 1H), 4,87 (м, 1H), 5,18 (м, 2H), 6,96 (м, 1H), 7,35 (м, 5H).
Масс-спектр с бомбардировкой быстрыми атомами, рассчитано C26H40N2O5 460, найдено 461,5 (M+H).
Стадия C: N-[1(R)-[(1,2-Дигидро-1-метилсульфонилспиро[3H-индол-3,4'-пиперидин-1'- ил)карбонил] - 3-циклогексилпропил] -2-амино-2-метилпропанамидгидрохлорид
Смесь 638 мг промежуточного соединения, полученного на стадии В, и 100 мг 10% Pd на угле перемешивают в атмосфере H2 в течение 4 часов. Полученную смесь фильтруют через целит, и полученный фильтрат концентрируют. Порцию в 87 мг этого остатка растворяют в 2 мл CH2Cl2 и 49,8 мг 1,2-дигидро-1-метилсульфонилспиро/3H-индол-3,4'- -пиперидингидрохлорида, ЕДС и HOBT добавляют и перемешивают в течение 16 часов. Полученный раствор выливают в 200 мл этилацетата и промывают последовательно 1 н. NaHSO4 (водн.), водой и насыщенным водным бикарбонатом натрия. Органическую фазу сушат, фильтруют и концентрируют. В результате очистки с помощью флеш-хроматографии (силикагель, 60% этилацетат/гексаны) получают 55 мг (47%) этого промежуточного соединения. Весь полученный материал растворяют в 2 мл смеси 1:1 TFA/CH2Cl2, перемешивают в течение получаса. Раствор отпаривают, а остаток очищая с помощью флеш-хроматографии (силикагель, метанол, NH4OH (водн.), CH2Cl2). Полученное соединение растворяют в CH2Cl2, обрабатывают HCl в эфире и концентрируют до получения указанного в заглавии соединения.
1H ЯМР (400 МГц, CD3OD): δ 0.93 (м, 2H), 1.15-1.3 (м, 6H), 1.55- 1.8 (м, 1H), 2.06 (дт, 15,4 Гц, 1H), 2.88 (м, 1H), 2.97 (м, 1H) 3.35 (м, 2H), 3.8-4.1 (м, 3H), 4.51 (м, 1H), 4.83 (м, 1H), 7.06 (кв, 7 Гц, 1H), 7.22 (м, 2H), 7.37 (д, 8 Гц, 1H).
Масс-спектр с бомбардировкой быстрыми атомами, рассчитано для C27H42N4O4S 518, найдено 519,7 (М+H).
Пример 18 (способ 1)
N-/[1(R)-[(1,2- Дигидро-1-метансульфонилспиро[3H-индол-3,4'-пиперидин] -1'-ил) карбонил] -2-(фенилметилокси)- этил]-2-амино-2-метилпропанамидгидрохлорид
Стадия A: 1,2-Дигидро-1-метансульфонилспиро[3H-индол- 3,4'-пиперидин] гидрохлорид
К раствору 1,20 г (5,8 ммоль) 1'-метил-1,2-дигидроспиро/3H-индол-3,4'-пиперидина (полученного по способу H. Ong. et al., J. Med. Chem. 1983, 23, 981-986) в 20 мл сухого дихлорметана при 0oC добавляют триэтиламин (0,90 мл, 6,4 ммоль) и метансульфонилхлорид (0,49 мл, 6,35 ммоль) и перемешивают в течение 30 минут. Реакционную смесь выливают в 15 мл насыщенного водного раствора бикарбоната натрия, и экстрагируют дихлорметаном (2 х 10 мл). Объединенные органические экстракты промывают рассолом (20 мл), сушат над безводным карбонатом калия фильтруют, и растворитель удаляют при пониженном давлении до получения 1,44 г метансульфонамидного производного в виде бледно-желтого масла, которое используют без дополнительной очистки.
К раствору вышеуказанного сырого продукта в 20 мл сухого 1,2-дихлорэтана при 0oC добавляют 1,0 мл (9,3 ммоль) 1-хлорэтилхлорформата, затем перемешивают при комнатной температуре в течение 30 минут, и, наконец, при кипячении с обратным холодильником в течение 1 часа.
Реакционную смесь концентрируют до приблизительно 1/3 объема, а затем разбавляют сухим метанолом (20 мл) и кипятят с обратным холодильником в течение полутора часов. Реакционную смесь охлаждают до комнатной температуры и концентрируют до приблизительно половины объема. Осадок отфильтровывают и промывают небольшим количеством холодного метанола. В результате получают 1,0 г пиперидиновой HCl соли в виде белого твердого вещества. Полученный фильтрат концентрируют и добавляют небольшой объем метанола, а затем эфира. Осадок снова отфильтровывают, промывают холодным метанолом и сушат. В результате получают дополнительно 0,49 г целевого продукта. Полный выход 1,49 г (70%).
1H ЯМР (200 МГц, CDCl3): δ 7,43-7,20 (м, 3H), 7,10 (дд, 1H), 3,98 (шир. с, 2H), 3,55-3,40 (шир. д, 2H), 3,35-3,10 (м, 2H), 2,99 (с, 3H), 2,15 (т, 2H), 2,00 (т, 2H).
Стадия В: N-[1(R)-[(1,2-Дигидро-1-метансульфонилспиро[3H-индол-3,4'-пиперидин]-1'-ил) карбонил]-2-(фенилметилокси)этил]-2-[(1,1-диметилэтилокси) карбонил]амино-2-метилпропанамид
К 0,35 г (1,15 ммоль) (2R)-2-[(1,1-диметилэтилокси)- карбонил]амино-3-[2-(фенилметилокси)этил]-1-пропановой кислоты в 13 мл дихлорметана добавляют 1,2-дигидро-1-метансульфонилспиро[3H-индол-3,4'-пиперидин] гидрохлорида (0,325 г, 1,07 ммоль), 0,18 мл (1,63 ммоль) N-метилморфолина, (0,159 г, 1,18 ммоль) N-гидроксибензтриазола (HOBT) и перемешивают в течение 15 минут. ЕДС (0,31 г, 1,62 моль) добавляют и перемешивание продолжают в течение 1 часа. Добавляют дополнительно 60 мкл N-метилморфолина и перемешивают в течение 45 минут. Реакционную смесь выливают в 5 мл воды, и органический слой выделяют. Органический слой промывают 5 мл 0,5 н. водной соляной кислоты и 5 мл насыщенного водного раствора бикарбоната натрия. Объединенные органические слои сушат над безводным сульфатом магния и концентрируют до получения 0,627 г продукта в виде желтой пены, которую используют без дополнительной очистки.
К 0,627 г (1,07 ммоль) указанного ранее продукта в 5 мл дихлорметана добавляют 1,0 мл трифторуксусной кислоты, и перемешивают при комнатной температуре в течение 75 минут. Добавляют еще 1,00 мл трифторуксусной кислоты и перемешивают еще 10 минут, реакционную смесь концентрируют, разбавляют 5,0 мл дихлорметана и осторожно подщелачивают, выливая в 10 мл 10% водного раствора карбоната натрия. Органический слой выделяют, и водный слой далее экстрагируют (2 х 15 мл) дихлорметаном. Объединенные органически слои промывают 5 мл воды, сушат над карбонатом калия, фильтруют и концентрируют до получения 0,486 г амина в виде бледно-желтой пены, которую используют далее без дополнительной очистки.
К 0,486 г (1,01 ммоль) амина и 10 мл дихлорметана добавляют 0,26 г (1,28 ммоль) 2-[1,1-диметилэтокси)карбонил]амино- 2-метилпропановой кислоты, 0,173 г (1,28 ммоль) 1-гидроксибензтриазола (HOBT) и ЕДС (0,245 г, 1,28 моль), и перемешивают при комнатной температуре в течение ночи. Реакционную смесь выливают в 5,0 мл воды, и органический слой выделяют. Водный слой снова экстрагируют 5 мл дихлорметана. Объединенные органические слои промывают 5,0 мл 0,5 н. водной соляной кислоты, 5 мл насыщенного водного раствора бикарбоната натрия и сушат над безводным сульфатом магния, концентрируют до получения 0,751 г неочищенного продукта в виде желтой пены. Раствор этого продукта в дихлорметане обрабатывают хроматографически на 25 г силикагеля, элюируя вначале смесью гексаны/ацетон/дихлорметан (70/25/5), а затем смесью гексаны/ацетон/дихлорметан (65/30/5). В результате получают 0,63 г указанного в заглавии соединения в виде белого твердого вещества.
1H ЯМР (400 МГц, CDCl3), соединение существует в виде смеси 3:2 ротамеров: δ 7.40-7.10 (м, 6H), 7.06 (д, 1/3H), 7.02 (т, 1/3H), 6.90 (т, 1/3H), 6.55 (д, 1/3H), 5.15 (м, 1H), 4.95 (шир.с, 1H), 4.63 (шир.д, 1/3H), 4.57-4.40 (м, 2 2/3H), 4.10 (шир.д, 1/3H), 4.00 (шир.д, 1/3H), 3.82 (т, 1H), 3.78-3.62 (м, 2H), 3.60-3.50 (м, 1H), 3.04 (кв, 1H), 2.87 (с, 1H), 2.86 (с, 2H), 2.80-2.60 (м, 1H), 1.90 (шир.с, 1H), 2.85-2.75 (м, 1H), 1.82-1.60 (м, 3H), 1.55-1.45 (м, 1H), 1.45 (с, 4H), 1.42 (с, 2H), 1.39 (с, 9H).
Стадия C: N-[1()R-[(1,2-Дигидро-1-метансульфонилспиро[3H- индол-3,4'-пиперидин] -1'-ил)карбонил-2- (фенилметилокси)этил-2-амино-2-метилпропанамидгидрохлорид
К 0,637 г (0,101 ммоль) промежуточного соединения со стадии В в 5 мл дихлорметана добавляют 2,5 мл трифторуксусной кислоты и перемешивают при комнатной температуре в течение 30 минут. Реакционную смесь концентрируют до масла, помещают в 10 мл этилацетата и промывают 8 мл 10% водного раствора карбоната натрия. Водный слой экстрагируют далее 5 мл этилацетата. Объединенные органические слои промывают 10 мл воды, сушат над сульфатом магния, фильтруют и концентрируют до получения 0,512 г свободного основания в виде белой пены.
К 0,512 г свободного основания в 5 мл этилацетата при 0oC добавляют 0,2 мл насыщенной соляной кислоты в этилацетате, и перемешивают в течение 1,5 ч. Образующийся белый осадок отфильтровывают в атмосфере азота, промывают эфиром, и сушат до получения 0,50 г указанного в заглавии соединения в виде белого твердого вещества.
1H ЯМР (400 МГц, CD3OD), соединение существует в виде смеси 3:2 ротамеров: δ 7.40-7.28 (м, 4H), 7.25-7.17 (м, 2H), 7.08 (т, 1/3H), 7.00 (т, 1/3H), 6.80 (д, 1/3H), 5.16 (ддд, 1H), 4.60-4.42 (м, 3H), 4.05 (т, 1H), 3.90 (шир. с, 2H), 3.83-3.70 (м, 2H), 3.30-3.15 (м, 1H), 2.97 (с, 1H), 2.95 (с, 2H), 2.90-2.78 (м, 1H), 1.96 (т, 1/3H), 1.85-1.65 (м, 4H), 1.63 (с, 2H), 1.60 (с, 4H).
Пример 19 (способ 2)
N-[1(R)-[(1,2-Дигидро-1-метансульфонилспиро [3H-индол-3,4'-пиперидин]-1'-ил) карбонил]-2-(фенилметокси)этил]- 2-амино-2-метилпропанамидгидрохлорид
Стадия A: (2R)-[[[-2-(1,1-Диметилэтокси)карбонил]- амино]-2,2-диметил-1-оксоэтил]амино-2-(фенилметокси)этил-1-пропановой кислоты аллиловый сложный эфир
Получают из (2R)-2-[(1,1-диметилэтокси)карбонил]- амино-3-(фенилметилокси)этил-пропановой кислоты и аллилового спирта, проводя реакцию в CH3Cl2 в присутствии EDC и DMAP.
1H ЯМР (400 МГц, CDCl3): δ 7,25 (с, 5H), 5,8 (м, 1H), 5,2 (дд, 2H), 5,0 (шир. c, 1H), 4,7 (м, 1H), 4,6 (м, 2H), 4,4 (дд, 2H), 3,9 (дд, 1H), 3,6 (дд, 1H), 1,45 (Д, 6H), 1,39 (c, 9H).
Стадия В: (R)-[[[-2-(1,1-Диметилэтокси)карбонил]-амино]-2,2- диметил-1-оксиэтил]амино-2- (фенилметилокси)этил-1-пропановая кислота
К перемешиваемому раствору неочищенного промежуточного соединения, полученного на стадии A (6,7 г, 15,9 ммоль), тетракис(трифенилфосфин)-палладия (1,8 г, 0,1 экв.) и трифенилфосфина (1,25 г, 0,3 экв.) добавляют раствор калий-2- этилгексаноата (35 мл, 0,5 М раствор в EtOAc). Реакционную смесь перемешивают при комнатной температуре в атмосфере азота в течение 1 часа, а затем разбавляют эфиром (100 мл) и выливают в смесь льда и воды. Органический слой выделяют, а водную фракцию подкисляют лимонной кислотой (20%), затем экстрагируют EtOAc. EtOAc экстракты промывают рассолом, сушат над сульфатом магния, фильтруют и выпаривают до получения указанного в заглавии соединения в виде твердого вещества.
1H ЯМР (400 МГц, CD3OD): δ 7,3 (с, 5H), 4,7 (м, 1H), 4,5 (с, 2H), 4,0 (м, 1H), 3,6 (м, 1H), 1,4 (д, 6H), 1,3 (с, 9H).
Стадия C: N-[1(R)-[(1,2-Дигидро-1-метансульфонилспиро[3H-индол-3,4'- пидеридин-1'-ил)карбонил-2-(фенилметилокси)этил -2-[(1,1-диметил-этокси)карбонил]-амино-2-метилпропанамид
К раствору 1,0 г (3,44 ммоль) 1-метансульфонилспиро/- индолин-3,4'-пиперидин/гидрохлорида, 1,44 г (3,78 ммоль) (2R)-[[-2-(1,1-диметилэтокси)карбонил)амино] -2,2-диметил- 1-оксоэтил]амино-2-(фенилметилокси)этил)-1-пропановой кислоты, N-метилморфолина (0,58 мл, 5,20 ммоль) и 1-гидроксибензтриазола (HOBT) (0,58 г, 3,78 ммоль) в 50 мл дихлорметана добавляют EDC (1,03 г, 5,20 ммоль) и перемешивают при комнатной температуре в течение 16 часов, реакционную смесь разбавляют дополнительно 50 мл дихлорметана и промывая водным раствором бикарбоната натрия (50 мл), сушат над безводным сульфатом магния, фильтруют и концентрируют. После обработки флеш-хроматографией (50 г силикагеля) неочищенного маслянистого остатка получают 2,148 г (90%) целевого материала в виде бесцветной пены.
1H ЯМР (400 МГц, CDCl3), соединение существует в виде смеси 3:2 ротамеров: δ 7.40-7.10 (м, 6H), 7.06 (д, 1/3H), 7.02 (т, 1/3H), 6.90 (т, 1/3H), 6.55 (д, 1/3H), 5.15 (м, 1H), 4.95 (шир.с, 1H), 4.63 (шир.с, 1/3H), 4.57-4.40 (м, 2 2/3H), 4.10 (шир. д, 1/3H), 4.00 (шир.д 1/3H), 3.82 (т, 1H), 3.78-3.62 (м, 2H), 3.60-3.50 (м, 1H), 3.04 (кв, 1H). 2.87 (с, 1H), 2.86 (с, 2H), 2.80-2.60 (м, 1H), 1.90 (шир.с, 1H), 2.85-2.75 (м, 1H), 1.82-1.60 (м, 3H), 1.55-1.45 (м, 1H), 1.45 (с, 4H), 1.42 (с, 2H), 1.39 (с, 9H).
Стадия D: N-[1(R)-[(1,2-Дигидро-1-метансульфонилспиро[3H- индол-3,4'-пиперидин] -1'-ил)карбонил] 2-(фенилметилокси)этил]-2-амино-2-метилпропанамидгидрохлорид
К раствору 2,148 г (3,41 ммоль) промежуточного соединения со стадии C в 10 мл дихлорметана добавляют 5 мл трифторуксусной кислоты и перемешивают в течение часа. Реакционную смесь концентрируют и подщелачивают 100 мл 5% водного раствора карбоната натрия, и экстрагируют дихлорметаном (3 х 50 мл). Объединенные органические слои промывают рассолом (50 мл), сушат над безводным карбонатом калия, фильтруют и концентрируют до получения бесцветной пены. К раствору пены в 25 мл этилацетата при 0oC добавляют 4 мл 1 М раствора соляной кислоты в этилацетате. Осадок отфильтровывают и промывают вначале этилацетатом, а затем смесью этил-ацетат-эфир (1:1); сушат до получения 1,79 г (93%) указанного в заглавии соединения в виде бесцветного твердого вещества.
1H ЯМР (400 МГц, CD3OD), соединение существует в виде смеси 3:2 ротамеров: δ 7.40-7.28 (м, 4H), 7.25-7.17 (м, 2H), 7.08 (т, 1/3H), 7.00 (т, 1/3H), 6.80 (д, 1/3H), 5.16 (ддд, 1H), 4.60-4.42 (м, 3H), 4.05 (т, 1H), 3.90 (шир. с, 2H), 3.83-3.70 (м, 2H), 3.30-3.15 (м, 1H), 2.97 (с, 1H), 2.95 (с, 2H), 2.90-2.78 (м, 1H), 1.96 (т, 1/3H), 1.85-1.65 (м, 4H), 1.63 (с, 2H), 1.60 (с, 4H).
Пример 20
N-[1(R)-[1,2-Дигидро-1-метансульфонил-5-бромоспиро[- 3H-индол-3,4'-пиперидин] -1'-ил)карбонил] -2-(фенилметилокси)- этил]-2-амино-2-метилпропанамидтрифторацетат
Стадия A: N-[1(R)-[(1,2-Дигидро-1-метансульфонил-5- бромоспиро[3H-индол-3,4'-пиперидин]-1'-ил)- карбонил-2-(фенилметил-окси)этил]-2-[(1,1- диметилэтокси)карбонил]амино-2-метилпропанамид
К раствору 300 мг (1,03 ммоль) 1-метансульфонилспиро- [3H-индол-3,4'-пиперидин] гидрохлорида в 5 мл ледяной уксусной кислоты добавляют 0,28 г (2,06 ммоль) брома и перемешивают при комнатной температуре в течение 1 часа. Реакционную смесь концентрируют досуха, подщелачивают 10 мл 5% водного раствора карбоната натрия, и экстрагируют дихлорметаном (3 х 10 мл). Объединенные органические экстракты промывают рассолом (10 мл), сушат над безводным карбонатом калия, фильтруют и концентрируют до получения 0,25 г неочищенного продукта в виде масла желтого цвета, которое используют далее без дополнительной очистки.
Стадия В:
К раствору вышеуказанного сырого продукта в 10 мл дихлорметана добавляют 0,43 г (1,13 ммоль) промежуточного соединения примера 19 со стадии В (0,17 г, 31,13 ммоль) HOBT, и 0,34 г (1,70 ммоль) EDC, и перемешивают при комнатной температуре в течение 16 часов. Реакционную смесь разбавляют 15 мл эфира и промывают 10% водной лимонной кислотой (15 мл), насыщенным раствором бикарбоната натрия (15 мл), сушат над безводным сульфатом магния, фильтруют и концентрируют до получения сырого маслянистого продукта. Остаток очищают с помощью флеш-хроматографии (15 г SiO2; CH2Cl2-ацетон (10:1) в качестве элюента) до получения 0,184 г (26% за две стадии) собранного материала в виде бесцветной пены.
К 0,184 г (0,26 ммоль) вышеуказанного материала в 2 мл дихлорметана 2 мл трифторуксусной кислоты и перемешивают при комнатной температуре в течение 1 часа. Реакционную смесь выпаривают досуха до получения 0,146 г (93%) указанного в заглавии соединения в виде белого твердого вещества. Масс-спектр с бомбардировкой быстрыми атомами, рассчитано для C27H34BrN4O5S 608, найдено 609,5.
Пример 21
N-[1(R)-[(1,2-Дигидроспиро[3H-индол-3,4'-пиперидин] -1'-ил)карбонил] - 2-(индол-3-ил)этил]-2-амино-2-метилпропанамидгидрохлорид
Стадия A: Спиро/3H-индол-3,4'-пиперидин/
К раствору 1,0 г (5,0 ммоль) 1'-метилспиро/3H-индол- 3,4'-пиперидина (полученного по способу H. Ong. et al., J. Med. Chem., 1983, 23, 981-986) и 1,0 г порошка карбоната калия в 30 мл сухого дихлорметана при комнатной температуре добавляют 0,5 г цианогенбромида, и перемешивают в течение 1 часа. Реакционную смесь фильтруют через слой целита и промывают смесью хлороформ-метанол (95:5). Полученный фильтрат концентрируют, а остаток пропускают через слой силикагеля, cмывая смесью хлороформ-метанол (95:5) в качестве элюента. В результате получают примерно 1,2 г желтого масла, которое используют без дополнительной очистки.
К суспензии вышеуказанного соединения в 30 мл сухого ДМФ при 0oC добавляют 0,30 г литийалюминийгидрида, и нагревают до комнатной температуры, а затем кипятят с обратным холодильником в течение 1 часа. Реакционную смесь охлаждают до 0oC и гасят 0,30 мл воды, 0,30 мл 15% водного раствора гидроксида натрия и 0,90 мл воды. Твердую часть отфильтровывают через целит и хорошо промывают смесью хлороформ-метанол (10:1). После концентрирования фильтрата получают 0,74 г соединения в виде пены желтого цвета. Этот материал представляет собой смесь 1:1 указанного в заглавии соединения и 1'-метил-спиро/3H-индол-3,4'-пиперидина).
Стадия В: (2R)-//-2-//1,1-диметилэтокси)карбонил/- амино/-2,2-диметил-1-оксоэтил/амино/-1H-индол-3- пропановой кислоты бензиловый сложный эфир
К 5,0 г (16,5 ммоль) коммерческого N-трет-BOC-D- триптофана в 100 мл хлороформа добавляют 1,80 мл (16,5 ммоль) бензилового спирта, 0,20 г (1,65 ммоль) 4-N, N- диметиламинопиридина (DMAP) и 3,20 г EDC, и перемешивают в течение 16 часов. Реакционную смесь выливают в 100 мл воды, и органический слой выделяют. Водный слой экстрагируют дважды порциями по 100 мл хлороформа. Объединенные органические слои промывают 50 мл 10% водной лимонной кислоты, 100 мл 10% водного раствора бикарбоната натрия сушат над безводным сульфатом магния, фильтруют и концентрируют до получения густого масла.
К раствору этого масла в 10 мл дихлорметана добавляют 20 мл трифторуксусной кислоты и перемешивают в течение 1 часа. Реакционную смесь концентрируют, подщелачивают осторожно насыщенным водным раствором бикарбоната натрия и экстрагируют хлороформом (2 х 100 мл). Объединенные органические слои промывают рассолом (100 мл), сушат над безводным карбонатом калия, фильтруют и концентрируют до получения 5,46 г амина в виде масла коричневого цвета, которое используют без дополнительной очистки.
К 5,46 г вышеуказанного продукта в 100 мл хлороформа добавляют 3,40 г (22,2 ммоль) HOBT, 4,60 г (22,2 ммоль) N-BOC-α-метилаланина и 5,32 г (28,0 ммоль) EDC, и перемешивают в течение 16 часов. Реакционную смесь выливают в 100 мл воды, и органический слой выделяют. Водный слой вначале экстрагируют хлороформом (2 х 100 мл). Объединенные органические экстракты промывают 50 мл 10% водной лимонной кислоты, 100 мл 10% водного раствора бикарбоната натрия, сушат над безводным сульфатом магния, фильтруют и концентрируют до получения 6,94 г продукта в виде густого масла. Обработка с помощью флеш-хроматографии (200 г SiO2; гексан-этилацетат в качестве элюента) дает 4,75 г целевого материала в виде бесцветной пены.
1H ЯМР (200 МГц, CDCl3): δ 8,48 (шир.с, 1H), 7,54 (шир.д, 1H), 7,38-7,23 (м, 3H), 7,19 (шир.д, 2H), 7,15-7,00 (м, 1H), 6,90 (д, 1H), 6,86 (д, 1H), 5,06 (шир.с, 2H), 4,95 (ддд, 1H), 3,30 (2 дд, 2H), 1,40 (с, 15H).
Стадия C: (2R)-//-2-/(1,1-Диметилэтокси)карбонил/амино/- 2,2-диметил-1-оксоэтил/амино/-1H-индол-3- пропановая кислота
К раствору 4,75 г материала со стадии B в 100 мл этанола добавляют 1,0 г 10% Pd/C и перемешивают при комнатной температуре в атмосфере H2 в течение 18 часов. Катализатор отфильтровывают через слой целита и промывают этилацетатом. Полученный фильтрат концентрируют до получения 2,96 г кислоты в виде бесцветной пены.
1H ЯМР (200 МГц, CDCl3): δ 8,60 (шир.с, 1H), 7,55 (д, 1H), 7,26-6,90 (м, 3H), 6,88 (шир.д, 1H), 4,80 (м, 1H), 3,32 (2 дд, 2H), 1,37 (с, 3H), 1,35 (с, 12H).
Стадия D: N-/1(R)-/(1,2-Дигидро-спиро/3H-индол-3,4'- пиперидин/-1'-ил)карбонил/-2-(индол-3-ил)- этил/-2-/(1,1-диметилэтокси)карбонил/амино- 2-метилпропанамид
К раствору 0,122 г (0,542 ммоль) смеси 1:1 промежуточного соединения со стадии A и 1'-метил-спиро/3H-индол- 3,4'-пиперидина/ в 5 мл сухого хлороформа при комнатной температуре добавляют 0,105 г (0,271 ммоль) промежуточного соединения со стадии C, 41 мг (0,271 ммоль) HОВТ, и 80 мг (0,41 ммоль) EDC, и перемешивают при комнатной температуре в течение 2 часов. Реакционную смесь разбавляют 10 мл хлороформа и промывают насыщенным водным раствором бикарбоната натрия (10 мл) и 10 мл рассола, сушат над безводным карбонатом калия, фильтруют и концентрируют. Обработка с помощью флеш-хроматографии (10 г SiO2, 2% MeOH- CHCl3) остатка дает 94 мг целевого продукта в виде желтой пены.
Соединение существует в виде смеси 3:2 ротамеров.
1H ЯМР (400 МГц, CDCl3): δ 8.37 (д, 1/3H), 8.35 (д, 2/3H), 8.19 (д, 1H), 7.72 (д, 2/3H), 7.60 (д, 1/3H). 7.38 (д, 2/3H), 7.32 (д, 1/3H). 7.22-7.08 (м, 3H). 7.00 (2т, 1H), 6.93 (д, 1/3H), 6.69 (т, 1H). 6.60 (д, 1/3H), 6.56 (д, 2/3H), 6.50 (д, 2/3H), 5.30-5.15 (м, 1H), 5.00 (шир.с, 1H), 4.34 (м, 1H), 3.62-3.50 (м, 1H), 3.30-3.11 (м, 4H), 2.90 (дт, 1H), 2.40 (дт, 1/3H) 1.70-1.55 (м, 1 2/3H), 1.34 (с, 2H), 1,31 (с, 4H), 1.28 (с, 1H) 1.31 (с, 9H), 1.20-1.11 (м, 1H), 0.32 (дт, 1/3H).
Стадия E: N-[1(R)-[(1,2-Дигидро-спиро[3H-индол-3,4'- пиперидин] -1'-ил)карбонил]-2-(индол-3-ил)- этил]-2-амино-2-метилпропанамиддигидрохлорид
К 27,5 мг промежуточного соединения со стадии D добавляют 1,0 мл метанола и 1,0 мл концентрированной соляной кислоты и перемешивают при комнатной температуре в течение часа. Реакционную смесь концентрируют, подщелачивают 5 мл 10% водного раствора карбоната натрия и экстрагируют хлороформом (3 x 5 мл). Объединенные органические слои промывают рассолом (10 мл), сушат над карбонатом калия, фильтруют и концентрируют до получения густого масла. Препаративная ТСХ (0,50 мм пластина, хлороформ-метанол 96:5 + 1% NH4OH) дает 12 мг целевого продукта в виде желтого твердого вещества. Соединение существует в виде смеси 3:2 ротамеров.
1H ЯМР (400 МГц, CDCl3): δ 8.37 (д, 1/3H), 8.35 (д, 2/3H), 8.19 (д, 1H), 7.72 (д, 2/3H), 7.60 (д, 1/3H), 7.38 (д, 2/3H), 7.32 (д, 1/3H), 7.22-7.08 (м, 3H). 7.00 (2т, 1H), 6.93 (д, 1/3H), 6.69 (т, 1H), 6.60 (д, 1/3H), 6.56 (д, 2/3H), 6.50 (д, 2/3H), 5.30-5.15 (м, 1H), 4.34 (м, 1H), 3.62-3.50 (м, 1H), 3.30-3.11 (м, 4H), 2.90 (дт, 1H), 2.40 (дт, 1/3H), 1.70-1.55 (м, 1 2/3H), 1.34 (с, 2H), 1.31 (с, 4H), 1.28 (с, 1H), 1.20-1.11 (м, 1H), 0.32 (дт, 1/3H).
Пример 22
N-[1(R)-[(1,2-Дигидро-1-метилкарбонилспиро[3H-индол- 3,4'-пиперидин-1'-ил)карбонил]-2-(индол-3-ил)этил]-2-амино-2- метилпропанамидгидрохлорид
К 26 мг промежуточного соединения, полученного в примере 21 на стадии D, в 1,0 мл 1,2-дихлорметана и 55 мкл (0,14 ммоль) N-метилморфолина при 0oC добавляют 6,6 мкл (0,93 ммоль) ацетилхлорида, и перемешивают в течение 1 часа. Реакционную смесь разбавляют 5 мл эфира, промывают 5 мл 10% водной лимонной кислоты, 5 мл насыщенного раствора бикарбоната натрия, сушат над безводным сульфатом магния, фильтруют и концентрируют, в результате чего получают бледно-желтую пену, которую используют без дополнительной очистки.
К вышеуказанному материалу в 1,0 мл дихлорметана добавляют 1,0 мл трифторуксусной кислоты и перемешивают при комнатной температуре в течение 1 часа, реакционную смесь концентрируют, подщелачивают 5 мл 10% водного раствора карбоната натрия, экстрагируют хлороформом (3 х 5 мл).
Объединенные органические слои промывают рассолом (10 мл), сушат над карбонатом калия, фильтруют и концентрируют до получения густого масла. К раствору этого материала в 1,0 мл метанола добавляют 1,0 мл 4 М соляной кислоты в диоксане и концентрируют досуха до получения 16 мг указанного в заглавии соединения в виде твердого вещества желтого цвета. Соединение существует в виде смеси 3:2 ротамеров.
1H ЯМР (400 МГц, CD3OD): δ 8.43 (д, 1H), 8.35 (т, 1H), 7.72 (д, 2/3H), 7.61 (д, 1/3H), 7.40-7.25 (м, 2H), 7.20-7.08 (м, 3H), 7.05-6.95 (м, 22/3H), 6.50 (д, 1/3H), 5.25-5.10 (м, 1H), 5.00-4.84 (шир.д, 1H), 3.68-3.45 (м, 3H), 3.20 (м, 2H), 2.60-2.48 (м, 1 1/3H), 2.30 (дт, 1/3H), 2.00 (с, 1H), 1.98 (с, 2H), 1.81-1.40 (м, 4H), 1.35 (с, 2H), 1.33 (с, 2H), 1.32 (с, 1H), 1.30 (с, 1H), 1.25-1.15 (м, 1H), 1.10-1.00 (м, 1H), 0.20 (дт, 1/3H).
Пример 23
N-[1(R)-[(1,2-Дигидро-1-бензолсульфонилспиро[3H- индол-3,4'-пиперидин] -1'-ил)карбонил]-2-(индол-3-ил)этил]- 2-амино-2-метилпропанамид
К 26 мг (0,050 ммоль) промежуточного соединения примера 21 со стадии D в 1,0 мл 1,2-дихлорэтана и 5 мкл N-метилморфолина добавляют при 0oC 7,5 мкл бензолcульфонилхлорида и перемешивают в течение 1 часа. Реакционную смесь разбавляют 10 мл эфира промывают 5 мл 10% водной лимонной кислоты, 5 мл насыщенного раствора бикарбоната натрия, сушат над безводным сульфатом магния, фильтруют и концентрируют до получения 29,8 мг неочищенного продукта в виде бледно-желтой пены. К раствору этого материала в 2 мл метанола добавляют 1,0 мл концентрированной соляной кислоты и перемешивают в течение 1 часа. Этот растворитель удаляют при пониженном давлении до получения указанного в заглавии соединения в виде твердого вещества коричневого цвета.
Соединение существует в виде смеси 3:2 ротамеров.
1H ЯМР (400 МГц, CDCl3): δ 8.30 (шир.с, 1/3H), 8.20 (шир.с, 2/3H), 8.05 (шир. с, 2/3H), 7.88 (д, 1/3H), 7.72-7.45 (м, 5H), 7.43-7.30 (м, 4H), 7.20-7.05 (м, 2H), 7.00-6.90 (м, 2 2/3H), 6.35 (д, 1/3H), 5.25-5.10 (м, 1H), 4.90 (шир.с, 1H), 4.30 (дт, 1H), 4.15 (дт, 1H), 3.95 (дд, 1H), 3,60-3.40 (м, 3H), 3.25-3.20 (м, 2H), 2.90 (дт, 1H), 2.73 (дт, 2 2/3H), 2.35 (м, 1 1/3H), 1.80 (м, 1H), 1.50 (с, 1H), 1.43 (с, 2H), 1.39 (с, 3H), 1.30-1.20 (м, 2H), 1.00 (шир.д, 1/3H), 0.90-0.70 (м, 2H), 0.55 (шир.д, 1/3H), 0.48 (дд, 2/3H), -0.90 (дт, 1/3H).
Пример 24
N-[1(R)-[(1,2-Дигидро-1-сульфонилоспиро[3H-индол-3,4'-пиперидин] -1'-ил)карбонил]-2-(индол-3-ил)этил]-амино-2-метилпропанамидгидрохлорид
К раствору 0,258 г (0,50 ммоль) промежуточного соединения примера 21 стадии D в 10 мл сухого дихлорметана при 0oC добавляют 0,39 мл (1,00 ммоль) N-метилморфолина и 45 мкл (0,60 ммоль) метансульфонилхлорида, и перемешивают в течение 30 минут. Реакционную смесь разбавляют 10 мл эфира и промывают насыщенным раствором бикарбоната натрия (5 мл), рассолом (5 мл), сушат над безводным сульфатом магния, фильтруют и концентрируют до получения продукта в виде бледно-желтой пены, которую используют без дополнительной очистки. К раствору этого материала в 3,0 мл дихлорметана добавляют 1,0 мл трифторуксусной кислоты и перемешивают при комнатной температуре в течение 1 часа. Реакционную смесь концентрируют, подщелачивают 5 мл 10% водного раствора карбоната натрия и экстрагируют хлороформом (3 х 5 мл). Объединенные органические слои промывают рассолом (10 мл), сушат над карбонатом калия, фильтруют и концентрируют до получения густого масла. К раствору этого материала в 3,0 мл метанола добавляют 200 мкл 4 М соляной кислоты в диоксане и концентрируют досуха до получения 93 мг целевого материала в виде бледно-желтого твердого вещества.
Соединение существует в виде смеси 3:2 ротамеров.
1H ЯМР (400 МГц, CD3OD): δ 8.43 (д, 1H), 8.35 (т, 1H), 7.72 (д, 2/3H), 7.61 (д, 1/3H), 7.40-7.25 (м, 2H), 7.20-7.08 (м, 3H), 7.05-6.95 (м, 22/3H), 6.50 (д, 1/3H), 5.25-5.10 (м, 1H), 5.00-4.84 (шир.д, 1H), 3.68-3.45 (м, 3H), 3.20 (м, 2H), 2.82 (с, 1H), 2.80 (с, 2H), 2.60-2.48 (м, 1 1/3H), 2.30 (дт, 1/3H), 1.81-1.40 (м, 4H), 1.35 (с, 2H), 1.33 (с, 2H), 1.32 (с, 1H), 1.30 (с, 1H), 1.25-1.15 (м, 1H), 1.10-1.00 (м, 1H), 0.20 (дт, 1/3H).
Пример 25
N-1(R)-[1,2-Дигидро-1-метансульфонилспиро[3H-индол-3,4'- пиперидин] -1'-ил)карбонил]-3-фенилпропил]-2- амино-2-метилпропанамидгидрохлорид
Стадия A: N-1(R)-[1,2-дигидро-[метансульфонил- спиро[3H-индол-3,4'-пиперидин]-1'-ил)карбонил]-3-фенилпропил]-2-[1,1- диметилэтокси)-карбонил]амино-2-метилпропанамид
Указанное в заглавии соединение получают из (2R)-2- /(1,1-диметилэтокси)карбонил/-амино-4-фенил-1-бутановой кислоты и 1,2-дигидро-1-метилсульфонилспиро/3H-индол-3,4'- пиперидин/-гидрохлорида, используя способ сочетания примера 18, стадии В. Неочищенный продукт очищают на силикагеле, используя 5% ацетон в CH2Cl2.
1H ЯМР (400 МГц, CDCl3): δ 7,2 (м, 9H), 4,9 (м, 1H), 4,5 (м, 1H), 3,8 (м, 2H), 3,2 (м, 2H), 2,9 (с, 3H), 2,7 (м, 2H), 2,3 (c, 2H), 2,0 (м, 2H), 2,7 (м, 4H), 1,5 (c, 6H), 1,4 (c, 9H).
Стадия В: N-1(R)-/1,2-Дигидро-1-метансульфонил- спиро/3H-индол-3,4'-пиперидин/-1'-ил)карбонил/-3- фенилпропил/-2-амино-2-метилпропанамидгидрохлорид
Получают из промежуточного соединений, полученного на стадии A, используя способ удаления защиты, описанный в примере 18 на стадии C.
1H ЯМР (400 МГц, CD3OD): δ 7,3 (м, 9H), 4,5 (м, 1H), 3,9 (м, 2H), 3,5 (м, 2H), 3,2 (м, 2H), 2,9 (c, 3H), 2,7 (м, 4H), 2,0 (м, 4H), 1,6 (c, 6H).
Пример 26
N-/1(R)-/(1,2-Дигидро-1-трифторметилсульфонил-5- фтороспиро/3H-индол-3,4'-пиперидин/-1'-ил )карбонил/-2- (индол-3-ил)этил/-2-амино-2-метилпрапанамидтрифторацетат
Стадия A: 1,2-Дигидро-1-бензилоксикарбонил-5-фтороспиро/3H-индол-3,4'-пиперидин/
К 7,82 г 60% гидрида натрия добавляют гексан и жидкую часть декантируют. К этому добавляют раствор 11,10 мл (89 ммоль) 2,5-дифторфенилацетонитрила в 150 мл ДМСО, и перемешивают в течение 30 минут. Прикапывают раствор 15,10 г 1-хлорметилэтиламиногидрохлорида в 150 мл ДМСО, и нагревают при 75oC в течение 4 часов. Реакционную смесь выливают в 600 г льда и экстрагируют эфиром (5 х 200 мл). Объединенные органические слои промывают 2 н. соляной кислотой (3 х 100 мл). Объединенные водные экстракты подщелачивают до pH 9 50% водным раствором гидроксида натрия и экстрагируют эфиром (3 х 200 мл). Объединенные органические слои промывают рассолом (100 мл), сушат над карбонатом калия и концентрируют до получения 15,54 г густого масла.
Прикапывают 24 мл этанола к 9,90 г литийалюминийгидрида в 250 мл ДМР при 0oC, а затем нагревают до кипения с обратным холодильником. Раствор соединения в 250 мл ДМР добавляют, и полученную смесь кипятят с обратным холодильником в течение 72 часов. Реакционную смесь охлаждают до 0oC и гасят водой (10 мл), 10 мл 15 NaOH и 30 мл воды. Полученную суспензию сушат над K2CO3, фильтруют и концентрируют до получения 13,6 г густого масла. Этот неочищенный продукт тщательно растирают с гексанами, твердую часть отфильтровывают и промывают гексанами. ЯМР (200 МГц, CDCl3) твердой части (2,6 г) дает около 75% целевого спироиндолина.
К раствору 1,02 г этой смеси в 50 мл CH2Cl2 при 0oC добавляют 1,0 мл триэтиламина и 0,80 мл CBZ-C1, и полученную смесь перемешивают в течение 1 часа при комнатной температуре, реакционную смесь выливают в 50 мл 5% HCl, и водный слой выделяют. Водный слой подщелачивают 50% NaOH до pH = 10, и экстрагируют CH2Cl2 (3 х 25 мл). Объединенные органические слои промывают рассолом (50 мл), сушат над K2CO3 и концентрируют до получения 1,26 г соединения в виде густого масла.
1H ЯМР (200 МГц, CDCl3): δ 7,7-7,90 (м, 1H), 7,50-7,15 (м, 6H), 6,95-6,60 (м, 2H), 5,28 (шир.с, 2H), 3,90 (шир.с, 2H), 2,85 (шир.д, 2H), 2,30 (с, 3H), 2,20-1,80 (м, 4H), 1,65 (шир.д, 2H).
Стадия В: N-/1(R)-/(1,2-Дигидро-1-бензилоксикарбонил- 5-фтороспиро/3H-индол-3,4'-пиперидин/-1'-ил)- карбонил/-2-(индол-3-ил)этил/-//(1,1-диметилэтилокси) карбонил/амино/-2-метилпропанамид
К 1,62 г (4,62 ммоль) указанного промежуточного соединения со стадии A в 10 мл 1,2-дихлорэтана при 0oC добавляют 0,65 мл ACE-Cl и кипятят с обратным холодильником в течение часа. Реакционную смесь концентрируют до 1/3 объема и разбавляют 10 мл метанола, и нагревают до кипения с обратным холодильником в течение 1 часа. Реакционную смесь концентрируют досуха и тщательно растирают с эфиром до получения твердого вещества коричневого цвета. Этот материал растворяют в насыщенном водном растворе бикарбоната натрия (25 мл) и экстрагируют дихлорметаном (2 х 25 мл). Объединенные органические экстракты сушат над K2CO3 и концентрируют до получения 0,384 г свободного основания.
К 0,384 г этого материала в 15 мл CH2Cl2 добавляют 0,483 г кислотного промежуточного соединения со стадии C примера 21, 0,189 г HOBT и 0,34 г EDC, и перемешивают в течение 18 часов, реакционную смесь выливают в 10 мл воды и экстрагируют CH2Cl2 (2х10 мл). Объединенные органические слои промывают 20 мл 10% лимонной кислоты, 20 мл насыщенного NaHCO3, сушат над сульфатом магния и концентрируют. После обработки остатка с помощью (флеш-хроматографии на 25 г силикагеля, элюируя смесью 1:1 гексаны-ацетон, получают 0,389 г целевого материала.
1H ЯМР (200 МГц, CDCl3): δ 7,7-7,90 (м, 1H), 7,50- 7,15 (м, 6H), 6,95-6,60 (м, 2H), 5,28 (шир.с, 2H), 3,90 (шир. с, 2H), 2,85 (шир.д, 2H), 2,30 (с, 3H), 2,20-1,80 (м, 4H), 1,65 (шир.д, 2H).
Стадия C: N-/1(R)-/(1,2-Дигидро-5-фтороспиро/3H-индол-3,4'-пиперидин/-1'-ил) карбонил/-2-(индол-3-ил)этил/-//(1,1-диметилэтилокси)карбонил/- амино/-2-метилпропанамид
К раствору 0,363 г промежуточного продукта, полученного на стадии В, в 5 мл этанола добавляют 0,10 г 20% гидроксида палладия на угле, и гидрируют в атмосфере водорода в течение 1 часа. Катализатор отфильтровывают, и промывают еще метанолом. Полученный фильтрат концентрируют до получения 0,262 г целевого материала.
1H ЯМР (400 МГц, CDCl3). Этот материал существует в виде смеси 2:1 ротамеров: δ 8.85-8.60 (2 шир.с, 1H), 7.70 (д, 2/3H), 7,55 (д, 1/3H), 7.38 (д, 2/3H), 7,30 (д, 1/3H), 7.28-7.15 (м, 4H), 7.13-7.02 (м, 2H), 6.65 (дт, 2H), 6.50 (дд, 1/3H), 6.45 (дд, 2/3H), 6.14 (дд, 2/3H), 5.30-5.13 (м, 1H), 5.10 (шир. с, 1H), 4.30 (шир. д, 2/3H), 4.22 (шир.д, 1/3H), 3.50-3.30 (м, 1H), 3.30-3.00 (м, 4H), 3.00-2.80 (м, 1H), 2.73 (т, 1H), 2.53-2.40 (м, 1 1/3H), 2.20 (т, 1/3H), 1.49 (с, 3H), 1.45 (с, 3H), 1.41 (с, 9H) 1.20 (дт, 1/3H), 0.95 (шир.д, 2/3H), 0.90 (дт, 2/3H), -0.05 (дт, 1/3H).
Стадия D: N-[1(R)-[(1,2- Дигидро-1-трифторметансульфонил-6-фтороспиро[3H-индол-3,4' -пиперидин]-1'-ил)карбонил)-2-(индол-3-ил)этил]-[[(1,1- диметилэтилокси)карбонил]-амино]-2-метилпропанамид
К раствору 30 мг промежуточного соединения, полученного со стадии C, в 1 мл дихлорметана при 0oC добавляют 0,050 мл триэтиламина и 0,020 мл трифлик ангидрида и перемешивают в течение 5 минут. Катализатор отфильтровывают и промывают еще метанолом, реакционную смесь выливают в 5 мл 5% водного раствора карбоната натрия и перемешивают в течение 5 минут. Водный слой экстрагируют CH2Cl2 (2 х 5 мл), и объединенные органические экстракты сушат над сульфатом магния, фильтруют и концентрируют. В результате обработки с помощью флеш-хроматографии остатка на 3 г силикагеля, используя смесь CH2Cl2-ацетон (4:1) в качестве элюента получают 21 мг продукта.
1H ЯМР (400 МГц, CDCl3). Этот материал представляет собой смесь 2:1 ротамеров: δ 8.40 (шир.с, 2/3H), 8.25 (шир.с, 1/3H), 7.70 (д, 2/3H), 7.60 (д, 1/3H), 7.40 (д, 2/3H), 7.35-7.10 (м, 5H), 6.90-6.80 (м, 2H), 6.18 (дд, 1H), 5.30-5.13 (м, 1H), 4.95 (шир.с, 2/3H), 4.90 (с, 1/3H), 4.45 (шир.д, 2/3H), 4.35 (шир.д, 1/3H), 3.85-3.70 (м, 2H), 3.70-3.55 (м, 2H), 3.30-3.10 (м, 2H), 2.70 (т, 1H), 2.45 (т, 1/3H), 2.35 (т, 2/3H), 1.49 (с, 3H), 1.45 (с, 3H), 1.41 (с, 9H), 1.20 (дт, 1/3H), 0.95 (шир.д, 2/3H), 0.90 (дт, 2/3H), -0.05 (дт, 1/3H).
Стадия E: N-[1(R)-[(1,2-Дигидро-1-трифторметансульфонил-5- фтороспиро[3H-индол-3,4'-пиперидин] - 1'-ил)карбонил]-2-(индол-3-ил)этил]-2-метилпропанамидтрифторацетат
Раствор 21 мг промежуточного соединения, полученного на стадии D, выдерживают в 1 мл дихлорметана и 1 мл трифторуксусной кислоты при комнатной температуре в течение 30 минут. Летучие выпаривают досуха и тщательно растирают с эфиром до получения твердого вещества желтого цвета.
1H ЯМР (400 МГц, CD3OD). Этот материал существует в виде смеси ротамеров (2: 1): δ 7.65 (д, 2/3H), 7.60 (д, 1/3H), 7.42 (д, 2/3H), 7.35-7.10 (м, 5H), 6.93-6.80 (м, 2H), 6.24 (дд, 1H), 5.30-5.13 (м, 1H), 4.95 (шир.с, 2/3H), 4.90 (с, 1/3H), 4.45 (шир.д, 2/3H), 4.35 (шир.д, 1/3H), 3.85-3.70 (м, 2H), 3.70-3.55 (м, 2H), 3.30-3.10 (м, 2H), 2.70 (т, 1H), 2.45 (т, 1/3H), 2.35 (т, 2/3H), 1.49 (с, 3H), 1.45 (с, 3H), 0.93 (шир.д, 2/3H), 0.90 (дт, 2/3H), -0.05 (дт, 1/3H).
Пример 27
N-[(1(R)-[(1,2-Дигидро-1-[метоксикарбонил] метилсульфонил-5- фтороспиро[3H-индол-3,4'-пиперидин] ]-1'-ил)карбонил]- 2-(индол-3-ил)этил]-2-амино-2-метилпропанамидтрифторацетат
Стадия A: N-[1(R)-[(1,2-Дихлор -1-[метоксикарбонил]- метилсульфонил-5-фтороспиро[3H-индол-3,4'-пиперидин] ]-1'-ил) карбонил]-2-(индол-3-ил)этил]-2-метилпропанамидтрифторацетат
К раствору 77 мг промежуточного соединения, полученного на стадия C примера 26, в 1 мл дихлорметана при 0oC добавляют 0,30 мл N-метилморфолина и 0,024 мл 2-карбометоксиметансульфонилхлорида, и перемешивают в течение 1 часа. Реакционную смесь выливают в 5 мл 5% водного раствора карбоната натрия и перемешивают в течение 5 минут. Водный слой экстрагируют CH2Cl2 (2 х 5 мл), и объединенные органические экстракты промывают рассолом (5 мл), сушат над сульфатом магния, фильтруют и концентрируют. В результате обработки остатка с помощью флеш-хроматографии на 5 г силикагеля, элюируя смесью CH2Cl2/ацетон (4:1) получают 64 г продукта.
1H ЯМР (400 МГц, CDCl3). Этот материал представляет собой смесь ротамеров 2: 1: δ 8.48 (шир.с, 2/3H), 8.35 (шир.с, 1/3H), 7.70 (д, 2/3H), 7.60 (д, 1/3H), 7.40 (д, 2/3H), 7.32 (д, 1/3H), 7.25-7.00 (м, 4H), 6.90-6.78 (м, 2H), 6.18 (дд, 1H), 5.30-5.20 (м, 1H), 4.97 (шир.с, 2/3H), 4.91 (с, 1/3H), 4.50-4.35 (шир. д, 1H), 4.02 (с, 2/3H), 3.99 (с, 1/3H), 3.76 (кв, 2H), 3.58 (с, 1H), 3.56 (с, 2H), 3.08-3.07 (м, 2H), 2.72 (т, 1H), 2.50-2.30 (2т, 1H), 1.65 (т, 1/3H), 1.50 (с, 2H), 1.46 (с, 4H), 1.40 (с, 9H), 1.30 (м, 1/3H), 1.10 (шир.д, 2/3H). 0.88 (дт, 2/3H), -0.13 (дт, 1/3H).
Стадия B: N-[1(R)-[(1,2-Дигидро-1-[метоксикарбонил]- метил-сульфонил-5-фтороспиро/[3H-индол-3,4'- пиперидин]]-1'-ил)карбонил]-2-(индол-3-ил)- этил]-2-метилпропанамидтрифторацетат
Раствор 24 мг промежуточного соединения, полученного со стадии A, выдерживают в 1 мл дихлорметана и 1 мл трифторуксусной кислоты при комнатной температуре в течение 30 минут. Летучие выпаривают досуха и тщательно растирают с эфиром до получения 23 мг бесцветного твердого вещества.
1H ЯМР (400 МГц, CD3OD). Этот материал существует в виде смеси 2:1 ротамеров: δ 8,70 (шир.с, 1/3H), 8.60 (шир.с, 2/3H), 7.60 (м, 2/3H), 7.50 (д, 2/3H), 7.48 (м, 1/3H), 7.40 (д, 2/3H), 7.31 (д, 1/3H), 7.25-7.00 (м, 4H), 6.95-6.85 (м, 1H), 6.70 (дд, 1/3H), 6.15 (дд, 2/3H), 5.20-5.10 (м, 1H), 4.38 (шир.д, 1/3H), 4.28 (шир.д, 2/3H), 4.02 (с, 2/3H), 3.99 (с, 1/3H), 3.76 (кв, 2H), 3.58 (с, 1H), 3.56 (с, 2H), 3.08-3.07 (м, 2H), 2.72 (т, 1H), 2.50-2.30 (2т, 1H), 1.65 (т, 1/3H), 1.65 (с, 2H), 1.60 (с, 4H), 1.30 (м, 1/3H), 1.00 (шир.д. 2/3H), 0.88 (дт, 2/3H), -0.10 (дт, 1/3H).
Пример 28
N-[(R)-[(1,2-Дигидро-1-метансульфонил-5-фтороспиро[- 3H-индол-3,4'-пиперидин] -1'-ил)карбонил] -2- (фенилметилокси)этил]-2-амино-2-метилпропанамидгидрохлорид
Стадия A: N-[1(R)-[(1,2-Дигидро-1-бензилоксикарбонил -5-фтороспиро[3H-индол-3,4'-пиперидин]- 1'-ил)карбонил]-2-(фенилметилокси)этил]- [[(1,1-димeтилэтилoкси)кapбoнил]aмино]-2- метилпропанамид
К 0,33 г 1,2-дигидро-1-бензилоксикарбонил-5-фтороспиро- /3H-индол-3,4'-пиперидина/, полученного на стадии A примера 26, в 10 мл 1,2-дихлорметана при комнатной температуре добавляют 0,35 г N-трет-BOC-O-бензил-D-серина, 0,195 г HOBT и 0,30 г EDC и перемешивают в течение 18 часов. Реакционную смесь выливают в 10 мл воды, и экстрагируют CH2Cl2 (2 х 10 мл). Объединенные органические слои промывают 20 мл 10% лимонной кислоты, 20 мл насыщенного бикарбоната натрия, сушат над сульфатом магния и концентрируют. К раствору промежуточного соединения, полученного на стадии A, в 5 мл CH2Cl2 добавляют 5 мл трифторуксусной кислоты, и перемешивают при комнатной температуре в течение 30 минут. Реакционную смесь концентрируют, разбавляют 5,0 мл дихлорметана и осторожно подщелачивают 10 мл 10% водного раствора карбоната натрия. Органический слой выделяют, а водный слой экстрагируют далее 2 х 15 мл дихлорметана. Объединенные органические слои промывают 5 мл воды, сушат над карбонатом калия, фильтруют и концентрируют до получения 0,39 г амина в виде густого масла.
К 0,39 г вышеуказанного промежуточного соединения в 10 мл 1,2-дихлорметана при комнатной температуре добавляют 0,24 г N-трет-BOC-α-метилаланина, 0,195 г HOBT и 0,30 г EDC, и перемешивают в течение 18 часов. Реакционную смесь выливают в 10 мл воды, и экстрагируют дважды порциями CH2Cl2 по 10 мл. Объединенные органические слои промывают 20 мл 10% лимонной кислоты, 20 мл насыщенного NaHCO3, сушат над сульфатом магния и концентрируют. В результате обработки с помощью флеш-хроматографии остатка на 30 г силикагеля, используя смесь гексан-этилацетат (2:1) в качестве элюента, получают 0,33 г продукта.
1H ЯМР (200 МГц, CDCl3): δ 7.80 (шир.с, 1H), 7.50-7.15 (м, 5H), 7.10 (шир. д, 1H), 6.90-6.70 (м, 1H), 6.27 (шир.д, 1H), 7.35-7.10 (м, 5H), 5.35-5.10 (м, 3H), 4.99 (с, 1H), 4.70-4.40 (м, 3H), 3.90-3.50 (м, 4H), 3.15-2.90 (м, 2H), 2.80-2.50 (м, 2H), 1.80-1.40 (м, 2H), 1.50 (3H), 1.42 (, 6H).
Стадия В: N-[1(R)-[(1,2-Дигидро-5-фтороспиро[3H- индол-3,4'-пиперидин] -1'-ил)карбонил] -2- (фeнилмeтилoкси)этил]-[[(1,1-диметилэтилокси) карбонил] амино]-2-метилпропанамид
К раствору 0,33 г промежуточного соеденения, полученного на стадии A, в 5 мл этанола добавляют 1 каплю триэтиламина, и гидрируют из водородного баллона в течение 3 часов. Катализатор отфильтровывают через слой целита и промывают этилацетатом. Порученный фильтрат концентрируют до получения 0,269 г продукта в виде бесцветной пены.
1H ЯМР (400 МГц, CDCl3): δ 7.35-7.20 (м, 4H), 7.17-7.08 (м, 2H), 6.80-6.65 (м, 2/2/3H), 6.27 (дт, 1/3H): 5.20-5.10 (м, 1H), 4.90 (с, 1H), 4.60-4.40 (м, 3H), 4.00 (шир. т, 1H), 3.75-3.60 (м, 1H), 3.55-3.40 (м, 3H), 3.18-3.30 (м, 2H), 2.90-2.65 (м, 1H), 1.83-1.50 (м, 4H), 1.48 (с, 4H), 1.42 (с, 2H), 1.39 (с, 9H).
Стадия C: N-[1(R)-[(1,2-Дигидро-1-метансульфонил-5- фтороспиро[3H-индол-3,4'-пиперидин]-1'-ил)- карбонил]-2-(фенилметилокси)этил]-[[(1,1-диметилэтилокси) карбонил]-амино]-2-метилпропанамид
К раствору 0,134 г промежуточного соединения со стадии В, в 5 мл дихлорметана добавляют 0,080 мл N-метилморфолина и 0,022 мл метансульфонилхлорида, и перемешивают при 0oC в течение 30 минут. Реакционную смесь разбавляют дополнительно 5 мл насыщенного раствора бикарбоната натрия, рассолом (5 мл) сушат над сульфатом магния и концентрируют. В результате обработки остатка с помощью флеш-хроматографии на 20 мг силикагеля получая 0,101 г целевого продукта.
1H ЯМР (400 МГц, CDCl3): δ 7.40-7.20 (м, 5H), 7.08 (д, 1H), 6.95-6.80 (м, 2/1/3H), 6.23 (дд, 2/3H), 5.20-5.10 (м, 1H), 4.90 (шир.с, 1H), 4.60 (шир. д, 2/3H), 4.58-4.40 (м, 3/1/3H), 4.10-4.00 (м, 1H), 3.388-3.70 (м, 21/3H), 3.66-3.60 (м, 1/2H), 3.60-3.50 (м, 1H), 3.10-2.95 (м, 1H), 2.86 (с, 1H), 2.84 (с, 2H), 2.80 (т, 1/3H), 2.65 (т, 2/3H), 2.90-2.50 (м, 4H), 1.45 (с, 4H), 1.44 (с, 2H), 1.42 (с, 3H), 1.40 (с, 6H).
Стадия D: N-[1(R)-[(1,2-Дигидро-1-метансульфонил-5- фтороспиро[3H-индол-3,4'-пиперидин]-1'-ил)- карбонил]-2-(фенилметилокси)этил]-амино- 2-метилпропанамидгидрохлорид
К раствору 0,101 г промежуточного соединения со стадии C в 1 мл дихлорметана добавляют 1,0 мл трифторуксусной кислоты, и выдерживают при комнатной температуре в течение 30 минут. Реакционную смесь выпаривают досуха, подщелачивают 10% водным раствором карбоната натрия (10 мл), и экстрагируют дихлорметаном (3 х 5 мл). Объединенные органические слои промывают рассолом (5 мл), сушат над карбонатом калия и концентрируют. Этот материал растворяют в 2 мл этилацетата и при 0oC добавляют 0,10 мл 4 М HCl в EtOAc. Осадок отфильтровывают в атмосфере азота и промывают EtOAc/эфиром (1:1), сушат до получения 62 мг продукта в виде белого твердого вещества.
1H ЯМР (400 МГц, CD3OD): δ 7.40-7.20 (м, 5H), 7.08 (д, 1H), 6.95-6.80 (м, 2/1/3H), 6.23 (дд, 2/3H), 5.20-5.10 (м, 1H), 4.60 (шир.д, 2/3H), 4.58-4.40 (м, 3/1/3H), 4.10-4.00 (м, 1H), 3.388-3.70 (м, 21/3H), 3.66-3.60 (м, 1/2H), 3.60-3.50 (м, 1H), 3.10-2.95 (м, 1H), 2.86 (с, 1H), 2.84 (с, 2H), 2.80 (т, 1/3H), 2.65 (т, 2/3H), 2.90-2.50 (м, 4H), 1.45 (с, 4H), 1.44 (с, 2H).
Пример 29
Стадия A: N-[1(R)-[(1,2-Дигидро-1-бензолсульфонил- 5-фтороспиро[3H-индол-3,4'-пиперидин]-1'-ил)- карбонил]-2-фенилметилокси)этил]-2-амино-2- метилпропанамидтрифторацетат
К раствору 0,026 г промежуточного соединения со стадии В примера 27 в 2 мл дихлорметана добавляют 0,020 мл N-метилморфолина и 0,012 мл бензолсульфонилхлорида, и перемешивают при 0oC в течение 1 часа. Реакционную смесь выливают в 10 мл эфира и промывают 5 мл насыщенного раствора бикарбоната натрия, сушат над сульфатом магния и концентрируют. В результате обработки остатка с помощью флеш-хроматографии над 10 г силикагеля, используя в качестве элюента CH2Cl2-эфир (2:1), получают 0,019 г продукта.
Этот материал обрабатывают 1 мл дихлорметана и 1 мл трифторуксусной кислоты в течение 1 часа. Реакционную смесь выпаривают досуха, а остаток тщательно растирают с эфиром до получения 18 мг целевого продукта в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, CD3OD): δ 7.80 (д, 2H), 7.70-7.55 (м, 2H), 7.55-7.50 (м, 2H), 7.40-7.20 (м, 42/3H), 7.03-6.92 (м, 1H), 6.82 (дт, 2/3H), 6.47 (дт, 2/3H), 5.08 (дт, 1H), 4.60-4.48 (м, 2H), 4.33 ( шир.т, 1H), 3.94-3.85 (м, 3H), 3.75-3.65 (м, 2H), 3.10 (дт, 1H), 2.80 (дт, 1H), 1.73 (дт, 1H), 1.58 (с, 4H), 1.56 (с, 2H), 1.50 (дт, 1H), 1.38 (дт, 1H), 1.10 (дт, 2H).
Пример 30
N-[1(R)-[(1,2-Дигидро-этансульфонил-спиро[3H-индол- 3,4'-пиперидин] -1'-ил)карбонил] -2-(фенилметилокси)этил] -2- амино-2-метилпропанамидгидрохлорид
Стадия A: N-[1(R)-[(1,2-Дигидро-1-бензилоксикарбонил/- спиро/3H-индол-3,4'-пиперидин/-1'-ил)карбонил/- 2-(фенилметилокси)этил/-//(1,1-диметилэтилокси) карбонил/амино/-2-метилпропанамид
К 5 г 1,2-дигидро-1-бензилоксикарбонид-спиро/3H-индол- 3,4'-пиперидин/гидрохлорида в 100 мл дихлорметана при комнатной температуре добавляют 3,64 г N-трет-BOC-O- бензил-D-серина, 1,83 г HOBT, 2,60 мл N-метилморфолина и 3,70 г EDC, и перемешивают в течение 18 часов. Реакционную смесь выливают в 100 мл воды и экстрагируют CH2Cl2 (2 х 100 мл). Объединенные органические слои промывают 100 мл 10% лимонной кислоты, 100 мл насыщенного NaHCO3, сушат над сульфатом магния и концентрируют.
К раствору промежуточного соединения, полученного на стадии A, в 20 мл CH2Cl2 добавляют 20 мл трифторуксусной кислоты и перемешивают при комнатной температуре в течение 30 минут. Реакционную смесь концентрируют, разбавляют 50 мл дихлорметана и осторожно подкисляют 100 мл 10% водного раствора карбоната натрия. Органический слой выделяют, а водный слой экстрагируют далее 2 х 50 мл дихлорметана. Объединенные органические фракции промывают 50 мл воды, сушат над карбонатом калия, фильтруют и концентрируют до получения амина в виде густого масла.
К вышеуказанному промежуточному соединению в 50 мл дихлорметана при комнатной температуре добавляют 2,50 г N-трет-BOC-α-метилаланина, 1,83 г HOBT и 3,70 г EDC и перемешивают в течение 18 часов. Реакционную смесь выливают в 10 мл воды и экстрагируют CH2Cl2 (2 х 10 мл). Объединенные органические фракции промывают 20 мл 10% лимонной кислоты, 20 мл насыщенного NaHCO3, сушат над сульфатом магния и концентрируют. В результате обработки остатка с помощью флеш-хроматографии на 300 г силикагеля, элюируя смесью гексан-этилацетат (2:1), получают 8,1 г продукта.
1H ЯМР (400 МГц, CDCl3): δ 7.85 (шир.с, 1H), 7.45-7,20 (м, 10H), 7.20-7.05 (м, 22/3H), 6.95 (т, 1/3H), 6.88 (т, 1/3H), 6.53 (дд, 2/3H), 5.35-5.20 (м, 2H), 5.20-5.10 (м, 1H), 4.92 (шир.с, 1H), 4.65-4.20 (м, 4H), 4.05 (дд, 2/3H), 4.00-3.80 (м, 1,1/3H), 3.80-3.60 (м, 1H), 3.10 (т, 2/3H), 3.00-2.85 (м, 1/3H), 2.82-2.60 (2т, 1H), 1.90-1.55 (м, 5H), 1.49 (с, 4H), 1.42 (с, 2H), 1.40 (с, 9H).
Стадия B: N-/1(R)-/(1,2-Дигидро-спиро/3H-индол- 3,4'-пиперидин/-1'-ил)карбонил/-2-(фенилметилокси) этил/-//(1,1-диметилэтокси)карбонил/амино/-2-метилпропанамид
К раствору 8,10 г промежуточного соединения, полученного на стадии A, в 80 мл этанола добавляют 1 г 20% гидроксида палладия на угле и гидрируют водородом из баллона в течение 1 часа. Катализатор отфильтровывают через слой целита, и промывают этилацетатом. Полученный фильтрат концентрируют до получения 4,69 г продукта в виде бесцветной пены.
1H ЯМР (400 МГц, CDCl3): δ 7.35-7.20 (м, 5H), 7.18 (д, 1/2H), 7.10 (д, 1/2H), 7.04-6.98 (м, 2H), 6.75-6.60 (м, 2H), 5.20-5.10 (м, 1H), 4.97 (шир.с, 1H), 4.55-4.40 (м, 3H), 3.95 (дд, 1H), 3.73-3.61 (м, 1H), 3.60-3.50 (м, 1H), 3.50-3.33 (м, 3H), 3.10 (дт, 1H), 2.83 (дт, 1H), 1.85-1.55 (м, 5H), 1.47 (с, 4H), 1.42 (с, 2H), 139 (с, 9H).
Стадия C: N-/1(R)-/(1,2-Дигидро-1-этансульфонил- спиро/3H-индол-3,4'-пиперидин/-1'-ил)карбонил/-2- (фенилметилокси)-этил/-2-амино-2-метилпропанамид
К раствору 0,158 г промежуточного соединения, полученного на стадии В, в 5 мл дихлорметана добавляют 0,053 мл N-метилморфолина и 0,034 мл этансульфонилхлорида и перемешивают при 0oC в течение 30 минут при комнатной температуре в течение 1 часа. Реакционную смесь разбавляют дополнительно 5 мл дихлорметана и промывают 5 мл насыщенного раствора бикарбоната натрия, рассолом (5 мл), сушат над сульфатом магния и концентрируют. В результате обработки остатка с помощью флеш-хроматографии на 10 г силикагеля, элюируя смесью CH2Cl2-эфир (3:1), получают 0,057 г целевого продукта.
К раствору 0,057 г вышеуказанного промежуточного соединения в 1 мл дихлорметана добавляют 1,0 мл трифторуксусной кислоты и выдерживают при комнатной температуре в течение 30 минут. Реакционную смесь концентрируют досуха и тщательно растирают с эфиром до получения 0,034 г продукта в виде твердого вещества желтого цвета.
1H ЯМР (400 МГц, CD3OD): δ 7.40-7.25 (м, 5H), 7.25-7.13 (м, 21/2H), 7.03 (т, 1/2H), 6.95 (т, 1/2H), 6.80 (д, 1/2H), 5.18 (дт, 1H), 4.60-4.42 (м, 3H), 4.08 (т, 1H), 3.96 (с, 2H), 3.83-3.70 (м, 2H), 3.29-3.15 (м, 3H), 2.84 (дт, 1H), 1.90 (дт, 1H), 1.74-1.62 (м, 4H), 1.62 (с, 2H), 1.60 (с, 4H), 1.33 (дт, 3H).
Пример 31
Стадия A: N-/1(R)-/(1,2-Дигидро-1-/2-метил-2- пропансульфонилспиро/3H-индол-3,4'-пиперидин/-1'- ил)карбонил/-2-(фенилметилокси)этил/-//(1,1- диметилокси)карбонил/амино-2-метилпропанамид
К раствору 0,212 г промежуточного соединения со стадии B примера 29 в 2 мл 1,2-дихлорметана добавляют 0,083 мл триэтиламина и 0,054 мл изопропилсульфонилхлорида и перемешивают при 0oC в течение 30 минут и при комнатной температуре в течение 3 часов. Реакционную смесь разбавляют 5 мл дихлорметана и промывают 5 мл насыщенного раствора
бикарбоната натрия, рассолом (5 мл), сушат над сульфатом магния и концентрируют. В результате обработки остатка с помощью флеш-хроматографии на 10 г силикагеля, элюируя смесью CH2Cl2-эфир (3:1) получают целевой продукт.
К раствору 0,101 г вышеуказанного промежуточного соединения в 1 мл дихлорметана добавляют 1,0 мл трифторуксусной кислоты и выдерживают при комнатной температуре в течение 30 минут. Реакционную смесь выпаривают досуха, подщелачивают 10% водным раствором карбоната натрия (10 мл) и экстрагируют дихлорметаном (3 х 5 мл). Объединенные органические фракции промывают рассолом (5 мл), сушат над карбонатом калия и концентрируют. Этот материал растворяют в 2 мл этилацетата, и 0,10 мл 4 М HCl в EtOAc добавляют при 0oC. Осадок отфильтровывают в атмосфере азота и промывают смесью EtOAc/эфир (1:1) и сушат, в результате чего получают 88 мг продукта в виде белого твердого вещества.
1H ЯМР (400 МГц, CD3OD): δ 7.40-7.20 (м, 5H), 7.08 (д, 1H), 6.95-6.80 (м, 2/1/3H), 6.23 (дд, 2/3H), 5.20-5.10 (м, 1H), 4.60 (шир.д, 2/3H), 4.58-4.40 (м, 3/1/3H), 4.10-4.00 (м, 1H), 3.388-3.70 (м, 21/3H), 3.66-3.60 (м, 1/2H), 3.60-3.50 (м, 1H), 3.10-2.95 (м, 1H), 2.86 (с, 1H), 2.84 (с, 2H), 2.80 (т, 1/3H), 2.65 (т, 2/3H), 2.90-2.50 (м, 4H), 1.45 (с, 4H), 1.44 (с, 2H).
Пример 32
Стадия A: N-/1(R)-/(1,2-Дигидро-1-/2-карбометоксиметансульфонил-спиро/3H-индол-3,4'- пиперидин//-1'-ил)карбонил/-2-(фенилметилокси)- этил/-//(1,1-диметилэтилокси)-карбонил/- амино/-2-метилпропанамидгидрохлорид
К раствору 0,50 г промежуточного продукта со стадии В примера 29 в 10 мл дихлорметана добавляют 0,21 мл N-метилморфолина и 0,10 мл 2-карбометоксиметансульфонилхлорида и перемешивают при 0oC в течение 30 минут. Реакционную смесь разбавляют 10 мл дихлорметана и промывают 5 мл насыщенного водного раствора бикарбоната натрия, рассолом (5 мл), сушат над сульфатом магния и концентрируют. В результате обработки остатка с помощью флеш-хроматографии на 20 г силикагеля, элюируя смесью CH2Cl2-эфир (3:1) получают 0,529 г целевого продукта.
1H ЯМР (400 МГц, CDCl3): δ 7.39-7,20 (м, 5H), 7.20-7/10 (м, 21/2H), 7.08 (дт, 1H), 6.92 (т, 1/2H), 6.55 (д, 1/2H), 5.20-5.10 (м, 1H), 4.94 (шир.с, 1H), 4.60 (шир. д, 1H), 4.53-4.40 (м, 2H), 4.10 (шир.с, 2H), 4.05-3.90 (м, 2H), 3.70 (дт, 1H), 3.63 (с, 11/2H), 3.61 (с, 11/2H), 3.59-3.50 (м, 1H), 3.05 (дт, 1H), 2.70 (дт, 1H), 1.90-1.50 (м, 4H), 1.49 (с, 4H), 1.44 (с, 2H), 1.39 (с, 9H).
Стадия В: N-/1(R)-/(1,2-Дигидро-1-/2- карбометоксиметансульфонилспиро/3H-индол-3,4'-пиперидин//- -1'-ил)карбонил/-2-(фенилметилокси)этил/-2- амино-2-метилпропанамидгидрохлорид
К раствору 0,113 г вышеуказанного промежуточного соединения в 1 мл дихлорметана добавляют 1,0 мл трифторуксусной кислоты и выдерживают при комнатной температуре в течение 30 минут. Реакционную смесь выпаривают досуха, подщелачивают 10% водным раствором карбоната натрия (10 мл), экстрагируют дихлорметаном (3 х 5 мл). Объединенные органические фракции промывают рассолом (10 мл), сушат над карбонатом калия и концентрируют. Этот материал растворяют в 2 мл этилацетата, и 0,20 мл 4 М HCl в EtOAc добавляют при 0oC. Добавляют эфир, а осадок отфильтровывают в атмосфере азота, и промывают эфиром и сушат, в результате чего получают 0,108 г продукта в виде белого твердого вещества.
1H ЯМР (400 МГц, CD3OD): δ 7.40-7.20 (м, 5H), 7.08 (д, 1H), 6.95-6.80 (м, 2/1/3H), 6.23 (дд, 2H), 5.20-5.10 (м, 1H), 4.60 (шир.д, 2/3H), 4.58-4.40 (м, 3/1/3H), 4.10-4.00 (м, 1H), 3.388-3.70 (м, 21/3H), 3.66-3.60 (м, 1/2H), 3.6-3.50 (м, 1H), 3.10-2.95 (м, 1H), 2.86 (с, 1H), 2.84 (с, 2H), 2.80 (т, 1/3H), 2.65 (т, 2/3H), 2.90-2.50 (м, 4H), 1.45 (с, 4H), 1.44 (с, 2H).
Пример 33
Стадия A: N-/1(R)-/(1,2-Дигидро-1-/2-карбоксиметансульфонилспиро/3H-индол-3,4'- пиперидин//-1'-ил)карбонил/-2-/фенилметилокси/этил/-//(1,1- диметилэтилокси)карбонил/амино/-2-метилпропанамидтрифторацетат
К раствору 0,126 г промежуточного соединения со стадии A примера 32 в 3 мл метанола и 1 мл воды при 0oC добавляют 2 капли 5 н. водного гидроксида натрия и перемешивают в течение 30 минут. Реакционную смесь подкисляют до pH = 2 0,5 н. водной соляной кислотой, разбавляют рассолом (5 мл) и экстрагируют CH2Cl2 (2 х 5 мл). Объединенные органические экстракты промывают рассолом (10 мл), сушат над сульфатом магния и концентрируют до получения 0,098 г белой пены.
1H ЯМР (400 МГц, CDCl3): δ 9.80 (шир.с, 1H), 7.45 (д, 1/2H), 7.40-7.13 (м, 7H), 7.02 (т, 1/2H), 6.90 (т, 1/2H), 6.50 (д, 1/2H). 5.22-5.10 (м, 1H), 4.60-4.40 (м, 3H), 4.20-4.00 (м, 3H), 3.92 (д, 1H), 3.70-5.50 (м, 2H), 3.04 (дт, 1H), 2.70 (дт, 1H), 1.93-1.50 (м, 4H), 1.42 (с, 6H), 1.33 (с, 9H).
Стадия В: N-/1(R)-/(1,2-Дигидро-1-/2-карбоксиметансульфонил- спиро/3H-индол-3,4'-пиперидин/-1'- ил)карбонил/-2-(фенилметилокси)этил/-2-амино-2-метилпропанамид
К раствору 0,098 г промежуточного соединения со стадии A в 1 мл дихлорметана добавляют 1 мл трифторуксусной кислоты и перемешивают в течение 80 минут. Реакционную смесь выпаривают досуха и тщательно растирают с эфиром до получения 0,096 г продукта в виде белого твердого вещества.
1H ЯМР (400 МГц, CD3OD): δ 7.40-7.28 (м. 6H), 7.24-7.15 (м, 21/2H), 7.00 (дт, 1H), 6.80 (д, 1/2H), 5.17 (дт, 1H), 4.60-4.45 (м, 2H), 4.22 (д, 2H), 4.14-4.00 (м, 3H), 3.81-3.70 (м, 2H), 3.22 (дт, 1H), 2.83 (дт, 1H), 1.96 (дт, 1/2H), 1.80-1.64 (м, 41/2H), 1.62 (с, 1H), 1.60 (с, 5H).
Пример 34
Стадия A: N-/1(R)-/(1,2-Дигидро-1-/2-гидроксиэтансульфонил-спиро/ 3H-индол-3,4'-пиперидин//-1'- ил)карбонил/-2-(фенилметилокси)этил/-//(1,1- диметилэтилокси)-карбонил/-амино/-2-метилпропанамидтрифторацетат
К раствору 0,222 г промежуточного продукта со стадии A примера 32 в 2 мл безводного тетрагидрофурана при комнатной температуре добавляют 0,48 мл 2 М раствора боргидрид лития в тетрагидрофуране, и перемешивают в течение 3 часов. Реакционную смесь гасят 0,50 мл ацетона, разбавляют 15 мл воды и экстрагируют CH2Cl2 (2 х 15 мл). Объединенные органические экстракты промывают рассолом (10 мл), сушат над сульфатом магния и концентрируют до получения 0,27 г белой пены. В результате обработки остатка с помощью флеш-хроматографии на 10 г силикагеля, элюируя смесью CH2Cl2-ацетон (2:1), получают 0,129 г целевого материала в виде густого масла.
1H ЯМР (400 МГц, CDCl3): δ 7.32-7,20 (м, 6H), 7.20-7.10 (м, 2H), 7.09 (д, 1/2H), 6.98 (т, 1/2H), 6.90 (т, 1/2H), 6.54 (д, 1/2H), 5.17-5.10 (м, 1H), 5.00 (шир.с, 1H), 4.61-4.39 (м, 3H), 4.10-3.95 (м, 5H), 3.93-3.74 (м, 2H), 3.66 (ддд, 1H), 3.53 (дт, 1H), 3.27 (дт, 2H), 3.00 (дт, 1H), 2.70 (дт, 1H), 1.90-1.50 (м, 4H), 1.43 (с, 4H), 1.41 (с, 2H), 1.36 (с, 9H).
Стадия В: N-/1(R)-/(1,2-Дигидро-1-/2-гидроксиэтансульфонил-спиро/3H-индол-3, 4'-пиперидин/-1'-ил)карбонил/-2-(фенилметилокси) этил/-2-амино-2-метилпропанамидтрифторацетат
К раствору 0,129 г промежуточного соединения со стадии A в 1 мл дихлорметана добавляют 1 мл трифторуксусной кислоты и перемешивают в течение 30 минут. Реакционную смесь выпаривают досуха и тщательно растирают с эфиром до получения 0,113 г продукта в виде белого твердого вещества.
1H ЯМР (400 МГц, CD3OD): δ 7,40-7,25 (м, 6H), 7,35-7,13 (м, 21/2H), 6,98 (дт, 1H), 6,80 (д, 1/2H), 5,20- 5,10 (м, 1H), 4,60-4,43 (м, 3H), 4,10-8,90 (м, 5H), 3,81-3,70 (м, 2H), 3,40-3,33 (дт, 2H), 3,20 (дт, 1H), 3,82 (дт, 1H), 2,00-1,63 (м, 4H), 1,61 (с, 1H), 1,58 (с, 5H).
Пример 35
Стадия A: N-[1(R)-[(1,2-Дигидро-1-трифторметансульфонил-спиро[3H-индол-3,4' -пиперидин]-1'-ил)-карбонил]-2-(фенилметилокси)этил]-[[(1,1-диметилэтилокси) карбонил]-амино]-2-метилпропанамидгидрохлорид
К раствору 0,15 г промежуточного соединения со стадии B примера 29 в 5 мл дихлорметана добавляют 0,10 мл N-метилморфолина и 0,057 мл трифторметансульфонового ангидрида, и перемешивают при 0oC в течение 15 минут. Реакционную смесь разбавляют 5 мл насыщенного водного раствора бикарбоната натрия, и экстрагируют (2 х 5 мл) дихлорметаном. Объединенные экстракты промывают рассолом (5 мл), сушат над сульфатом магния и концентрируют. В результате обработки остатка с помощью флеш-хроматографии на 10 г силикагеля, элюируя смесью гексан-ацетон (3:1), получают 0,136 г целевого продукта.
1H ЯМР (400 МГц, CDCl3): δ 7.40-7.15 (м, 6H), 7.15-6.93 (м, 21/2H), 6.53 (д, 1/2H), 5.20-5.10 (м, 1H), 4.90 (шир.с, 1H), 4.70-4.60 (м, 3H), 4.15-3.90 (м, 3H), 3.70 (ддд, 1H), 3.60-3.50 (м, 1H), 3.00 (дт, 1H), 2.70 (дт, 1H), 1.93-1.55 (м, 4H), 1.46 (с, 4H), 1.43 (с, 2H), 1 40 (с, 9H).
Стадия В: N-[1(R)-[(1,2-Дигидро-1-трифторметансульфонил-спиро[3H-индол-3,4'- пиперидин-1'-ил)-карбонил] -2- (фенилметилокси)этил] -2-амино-2- метилпропанамидгидрохлорид
К раствору 0,136 г вышеуказанного промежуточного соединения в 1 мл дихлорметана добавляют 1,0 мл трифторуксусной кислоты, и выдерживают его при комнатной температуре в течение 30 минут. Реакционную смесь выпаривают досуха, подщелачивают 10% водным раствором карбоната натрия (5 мл) и экстрагируют этилацетатом (2 х 5 мл). Объединенные органические экстракты промывают рассолом (5 мл), сушат над карбонатом калия и концентрируют. Этот материал растворяют в 2 мл этилацетата и 0,20 мл 4 М HCl в EtOAc добавляют при 0oC. Добавляют эфир, и осадок отфильтровывают в атмосфере азота и промывают эфиром и сушат, в результате чего получают 0,94 г продукта в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, CD3OD): δ 7.10-7.15 (м, 6H), 7.15-6.93 (м, 21/2H), 6.53 (д, 1/2H), 5.20-5.10 (м, 1H), 4.90 (шир.с, 1H), 4.70-4.60 (м, 3H), 4.15-3.90 (м, 3H), 3.70 (ддд, 1H), 3.60-3.50 (м, 1H), 3.00 (дт, 1H), 2.70 (дт, 1H), 1.93-1.55 (м, 4H), 1.46 (с, 4H), 1.43 (с, 2H).
Пример 36
Стадия A: N-[1(R)-[1,2- Дигидро-1-бензолсульфонил-спиро[3H-индол-3,4'-пиперидин] -1'- ил)карбонил] -2-(фенилметилокси)этил]-2-амино-2-метилпропанамидгидрохлорид
К раствору 0,148 г промежуточного соединения со стадии В примера 29 в 3 мл дихлорметана добавляют 0,30 мл N-метилморфолина и 0,022 мл бензолсульфонилхлорида, и перемешивают при комнатной температуре в течение 1 часа. Реакционную смесь разбавляют 10 мл дихлорметана и промывают 10 мл насыщенного водного раствора бикарбоната натрия, сушат над сульфатом магния и концентрируют. В результате обработки остатка с помощью флеш-хроматографии на 10 г силикагеля, элюируя смесью гексан-ацетон (3:1), получают 0,190 г целевого продукта.
К раствору 0,190 г вышеуказанного промежуточного соединения в 3 мл дихлорметана добавляют 3 мл трифторуксусной кислоты и выдерживают при комнатной температуре в течение 30 минут. Реакционную смесь выпаривают досуха, подщелачивают 10% водным раствором карбоната натрия (5 мл) и экстрагируют этилацетатом (2 х 5 мл). Объединенные органические экстракты промывают рассолом (5 мл), сушат над карбонатом калия и концентрируют. Этот материал растворяют в 2 мл этилацетата, и 0,40 мл 4 М HCl в EtOAc добавляют при 0oC. Добавляют эфир, и осадок фильтруют в атмосфере азота и промывают эфиром, сушат и получают 0,136 г продукта в виде белого твердого вещества.
1H ЯМР (400 МГц, CD3OD): δ 7.82 (д, 2H), 7.67-7.58 (м, 2H), 7.52 (т, 2H), 7.40-7.20 (м, 6H), 7.10-6.90 (м, 11/2H), 6.68 (д, 1/2H), 5.10 (дт, 1H), 4.53 (ABq, 2H), 4.35 (т, 1H), 4.00-3.80 (м, 3H), 3.75-3.65 (м, 2H), 3.10 (дт, 1H), 2,73 (дт, 1H), 1.75 (дт, 1/2H), 1.48 (м, 11/2H), 1.20-1.05 (м, 2H).
Пример 37
Стадия A: N-[1(R)-[(1,2-Дигидро-[1-уреидометил- спиро[3H-индол-3,4'-пиперидин] -1'-ил)карбонил- 2-(фенилметилокси)этил]-2-амино-2-метилпропанамидтрифторацетат
К раствору 0,148 г промежуточного продукта со стадии B примера 29 в 5 мл 1,2-дихлорэтана добавляют 0,10 мл метилизоцианата и перемешивают при комнатной температуре в течение 1 часа. Реакционную смесь выпаривают досуха. В результате обработки остатка с помощью флеш-хроматографии на 15 г силикагеля, элюируя смесью CH2Cl2-ацетон (2:1), получают 0,137 г целевого продукта.
Этот материал обрабатывают 3 мл дихлорметана и 3 мл трифторуксусной кислоты в течение 30 минут при комнатной температуре. Реакционную смесь выпаривают досуха и тщательно растирают с эфиром до получения 0,126 г бледно-желтого твердого вещества.
1H ЯМР (400 МГц, CD3OD): δ 7.82 (дд, 1H), 7.42-7.35 (м, 5H). 7.30- 7.20 (м, 21/2H), 6.75 (д, 1/2H), 5.19 (дт, 1H), 4.60-4.50 (м, 3H), 4.13 (шир.д, 1H), 3.90-3.68 (м, 4H), 3.25 (т, 1H), 2.90-2.70 (2с, 4H), 1.98 (дт, 1/2H), 1.85-1.65 (м, 31/2H), 1.62 (с, 2H), 1.59 (с, 4H).
Пример 38
N-[1(R)-[(1,2-Дигидро-1-[1-метоксикарбонил-1- метилэтансульфонил-спиро[3H-индол-3,4'-пиперидин-1'-ил) карбонил]-2-(индол-3-ил)этил]-2-амино-2-метилпропанамидтрифторацетат
Стадия A: 1,2-Дигидро-1-[1-метоксикарбонил-1-метилэтансульфонил]-спиро[3H- индол-3,4'-пиперидин]
К 5,06 г 1,2-дигидро-1-бензилоксикарбонил-спиро/3H- индол-3,4'-пиперидин/гидрохлорида в 50 мл дихлорметана добавляют 3,0 мл триэтиламина и 3,40 г ди-трет-бутилкарбоната и перемешивают при комнатной температуре в течение 3 часов. Реакционную смесь выпаривают досуха и разбавляют 100 мл эфира, и промывают 50 мл 0,5 н. водной соляной кислоты, 50 мл рассола, сушат над сульфатом магния и концентрируют. К этому сырому продукту в 50 мл этанола добавляют 1 г 20% гидроксида палладия на угле и гидрируют H2 из баллона в течение ночи. К 0,506 г этого соединения в 15 мл дихлорметана при 0oC добавляют 0,74 мл триэтиламина и 0,41 мл карбометоксиметансульфонилхлорида и перемешивают в течение 1 часа. Реакционную смесь разбавляют 25 мл эфира и промывают насыщенным раствором бикарбоната натрия (20 мл), сушат над сульфатом магния и концентрируют. В результате обработки остатка с помощью флеш-хроматографии на 25 г силикагеля, элюируя смесью гексан-этилацетат (4:1) в качестве элюента, получают 1,79 г целевого материала в виде густого масла.
Гидрид натрия (0,102 г 60% в минеральном масле) промывают гексанами, а затем суспендируют в 5 мл DMF. Раствор 0,158 г вышеуказанного промежуточного соединения в 1 мл DMF добавляют и перемешивают в течение 30 минут. Добавляют метилйодид (1,85 ммоль) и перемешивают в течение 3 часов. Реакционную смесь выливают в 15 мл насыщенного водного раствора аммонийхлорида и экстрагируют эфиром (2 х 15 мл). Объединенные органические экстракты промывают водой (15 мл), рассолом (15 мл), сушат над сульфатом магния и концентрируют до получения 0,179 г целевого материала.
1H ЯМР (200 МГц, CDCl3): δ 7,32 (д, 1H), 7,20-6,90 (м, 3H), 4,13 (шир.д, 2H), 2,83 (шир.т, 2H), 1,85-1,70 (м, 4H), 1,69 (с, 6H), 1,48 (с, 9H).
Стадия В: N-[1(R)-[(1,2-Дигидро-[1-метоксикарбонил- 1-метилэтансульфонил] -спиро[3H-индол-3,4'-пиперидин] - 1'-ил)карбонил]-2-(индол-3-ил)этил]- [[(1,1-диметилэтилокси)карбонил]амино]-2- метилпропанамид
К раствору 0,179 г промежуточного соединения со стадии A добавляют 1 мл дихлорэтана и 1 мл трифторуксусной кислоты и перемешивают в течение 30 минут. Реакционную смесь выпаривают досуха, подщелачивают 10 мл 10% водного раствора карбоната натрия и экстрагируют (2 х 10 мл) дихлорметаном. Объединенные органические экстракты промывают рассолом (10 мл), сушат над карбонатом калия, фильтруют и концентрируют до 0,120 г пиперидина в виде густого масла. К раствору этого соединения в 5 мл дихлорметана добавляют 0,132 г кислотного промежуточного соединения примера 21 (стадии В), 0,055 г HOBT, 0,102 г EDC, перемешивают в течение 18 часов. Реакционную смесь разбавляют 25 мл эфира и промывают 15 мл 0,05 н. HCl, насыщенным раствором бикарбоната натрия (15 мл), сушат над сульфатом магния и концентрируют. В результате обработки остатка с помощью флеш-хроматографии на 20 г силикагеля, элюируя смесью CH2Cl2-ацетон (5:1) получают 0,094 г целевого продукта.
1H ЯМР (400 МГц, CDCl3): δ 8.60 (с, 2/3H), 8.50 (с, 1/3H), 7.70 (д, 2/3H), 7.60, (д, 1/3H), 7.35 (д, 2/3H), 7.30 (д, 1/3H), 7.26-7.00 (м, 5H), 6.90 (т, 11/3H), 6.40 (д, 2/3H), 5.28-5.16 (м, 1H), 5.05 (шир.с, 1H), 4.41 (шир.д, 2/3H), 4.32 (шир.д, 1/3H), 3.78-3.65 (м, 2H), 3.56 (с, 2H). 3.55 (с, 1H), 3.50 (шир.д, 1H), 3.20 (дт, 1H), 3.15 (ддд, 1H), 2.75 (т, 1H), 2.42 (м, 1H), 1.18 (д, 2H), 1.24 (с, 4H), 1.50 (с, 2H), 1.48 (с, 4H), 1.42 (с, 9H), 1.30-1.18 (м, 1H), 1.10-0.90 (м, 11/3H), 0.03 (дт, 2/3H).
Стадия C: N-[1(R)-[(1,2-Дигидро-[1-метоксикарбонил- 1-метилэтансульфонил] -спиро[3H-индол-3,4'- пиперидин]-1'-ил)карбонил]-2-(индол-3-ил)- этил] -2-амино-2-метилпропанамидтрифторацетат
Раствор 0,094 г промежуточного продукта со стадии C обрабатывают 1 мл дихлорметана и 1 мл трифторуксусной кислоты в течение 30 минут, выпаривают досуха и тщательно растирают с эфиром до получения 0,082 г целевого продукта.
1H ЯМР (400 МГц, CD3OD): δ 7.70 (д, 2/3H), 7.60 (д, 1/3H), 7.35 (д, 2/3H), 7.30 (д, 1/3H), 7.26-7.00 (м, 5H), 6.90 (т, 11/3H), 6.40 (д, 2/3H), 5.28-5.16 (м, 1H), 5.05 (шир.с, 1H), 4.41 (шир.д, 2/3H), 4.32 (шир.д, 1/3H), 3.78-3.65 (м, 2H), 3.56 (с, 2H), 3.55 (с, 1H), 3.50 (шир.д, 1H), 3.20 (дт, 1H), 3.15 (ддд, 1H), 2.75 (т, 1H), 2.42 (м, 1H), 1.18 (д, 2H), 1.24 (с, 4H), 1.50 (с, 2H), 1.48 (с, 4H), 1.30-1.18 (м, 1H), 1.10-0.90 (м, 11/3H), 0.03 (дт, 2/3H).
Пример 39
N-[1(R)-[(1,2-Дигидро-1-метансульфонилспиро[3H- индол-3,4'-пиперидин]-1'-ил)карбонил]-2-(индол-3-ил)этил]- 3-амино-3-метилбутанамидгидрохлорид
Стадия A: N-[1(R)-[(1,2-Дигидро-1-метансульфонилспиро [3H-индол-3,4'-пиперидин]-1'-ил)карбонил]-2-(индол-3-ил) этил/-3-амино-2-метилбутанамид
К суспензии 1,14 г 1,2-дигидро-1-метансульфонилспиро- /3H-индол-3,4'-пиперидин/гидрохлорида /полученного по способу стадии A примера 18/ в 50 мл дихлорметана добавляют 0,80 мл N-метилморфолина, 1,00 г N-трет-BOC-D-триптофана, 0,80 г HOBT и 1,20 г EDC и перемешивают при комнатной температуре в течение 18 часов, реакционную смесь разбавляют 100 мл эфира и промывают 50 мл 0,05 н. HCl, 50 мл насыщенного раствора бикарбоната натрия, сушат над сульфатом магния и концентрируют.
Раствор вышеуказанного промежуточного в 50 мл этилацетата при 0oC обрабатывают HCl (газ) в течение 2 минут, а затем перемешивают в течение 1 часа, 50 мл сухого эфира добавляют, и выпавшую в осадок твердую часть собирают фильтрованием. Выход 1,44 г.
К 0,86 г аминогидрохлорида в 30 мл дихлорметана добавляют 0,24 мл N-метилморфолина, 0,36 г HOBT, 0,56 г EDC и перемешивают в течение ночи. Реакционную смесь разбавляют 100 мл эфира и промывают 0,05 н. HCl (50 мл), 50 мл насыщенного NaHCO3, сушат над сульфатом магния и концентрируют. В результате обработки остатка с помощью флеш-хроматографии на 20 г силикагеля, элюируя смесью CH2Cl2-ацетон (5:1), получают 0,74 г целевого продукта.
Через раствор 0,74 г вышеуказанного промежуточного продукта в 5 мл этилацетата при 0oC барботируют сухой HCl газ при 0oC в течение 3 минут, и перемешивают 30 минут. Добавляют эфир для того, чтобы полностью высадить продукт. Твердую часть отфильтровывают и промывают эфиром в атмосфере азота, сушат и получают 0,57 г целевого продукта.
1H ЯМР (400 МГц, CD3OD): δ 7.69 (д, 2/3H), 7.55 (д, 1/3H), 7.37-6.90 (м, 5H), 6.82 (шир. т, 11/3H), 6.43 (д, 2/3H), 5.31-5.18 (м, 1H), 4.40 (шир.д, 2/3H), 4.30 (шир.д, 1/3H), 3.63-3.38 (м, 4H), 3.22-3.05 (м, 2H), 2.83-2.75 (м, 1H), 2.80 (с, 1H), 2.74 (с, 2H), 2.63 (дд, 1H), 2.55-2.43 (м, 2H), 2.20 (шир. д, 1H), 1.70-1.53 (м, 1H), 1.38 (2H), 1.36 (с, 2H), 1.35 (с, 1H), 1.34 (с, 1H), 1.18 (шир.д, 1H), 1.20-0.94 (м, 11/3H), 0.03 (дт, 2/3H).
Пример 40
N-[1(R)-[(1,2-Дигидро-1-метансульфонилспиро[3H-индол- 3,4'-пиперидин]-1'-ил)карбонил] -2-(индол-3-ил)этил]-[3-[2(R)- 3-гидроксилпропил]-амино]-3-метилбутанамидгидрохлорид
Стадия A: N-[1(R)-[(1,2-Дигидро-1-метансульфонилспиро- [3H-индол-3,4'-пиперидин]-1'-ил)карбонил-2- (индол-3-ил)этил]-[3-[2(R)-3-дигидроксипропил]- амино]-3-метилбутанамидгидрохлорид
К раствору 0,30 г соединения, полученного на стадии В примера 39 в 5 мл метанола добавляют 1,5 г безводного ацетата натрия, 0,30 г (R)-1,2-изопропилиденглициральдегида (Tetrahedron, 1985, 41, 3117) и перемешивают в течение 1 часа. Раствор в THF цианоборгидрида натрия (8,7 мл 1 М раствор) добавляют и полученную смесь перемешивают в течение 18 часов. Реакционную смесь разбавляют 20 мл воды и экстрагируют дихлорметаном (3 х 10 мл). Объединенные органические экстракты промывают насыщенным раствором бикарбоната натрия (10 мл), сушат над K2CO3 и концентрируют. В результате обработки остатка с помощью флеш-хроматографии на 10 г силикагеля, элюируя смесью CH2Cl2-метанол (98: 2), получают 0,146 г восстановительно аминированного соединения.
1H ЯМР (400 МГц, CDCl3): δ 8.70-8.40 (м, 2H), 7.63 (д, 2/3H), 7.55 (д, 1/3H), 7.37 (т, 1H). 7.32 (д, 1/3H), 7.28 (д, 2/3H), 7.20-6.95 (м, 41/3H), 6.52 (д, 2/3H), 5.20-5.08 (м, 1H), 4.55-4.24 (м, 3H), 4.10 (т, 2/3H), 4.05 (т, 1/3H), 3.80-3.70 (м, 1H), 3.70-3.50 (м, 4H), 3.30-3.10 (м, 2H), 2.84 (с, 1H), 2.80 (с, 2H), 2.80-2.70 (м, 2H), 2.68-2.45 (м, 1H), 2.37 (с, 2H), 1.70 (т, 2/3H), 1.52 (шир. д, 1/3H), 1.44 (с, 2H), 1.43 (с, 1H), 1.35 (с, 2H), 1.33 (с, 1H), 1.25 (с, 3H), 1.33 (с, 6H), 1.20-1.05 (м, 2H), 0.90-0.65 (м, 1/3H), 0.30 (дт, 2/3H).
Стадия В:
Раствор 0,146 г вышеуказанного промежуточного соединения перемешивают в 3 мл метанола и 0,10 мл концентрированной соляной кислоты в течение 30 минут. Реакционную смесь выпаривают досуха, и твердую часть промывают эфиром и сушат, в результате чего получают 0,109 г целевого материала.
1H ЯМР (400 МГц, CD3OD): δ 7.63 (д, 2/3H), 7.55 (д, 1/3H), 7.41 (д, 2/3H), 7.38 (д, 1/3H), 7.32 (д, 1/3H), 7.26 (д, 2/3H), 7.21-7.10 (м, 4H), 7.08-7.00 (м, 21/3H), 6.63 (д, 2/3H), 5.25 (дд, 2/3H), 5.19 (дд, 1/3H), 4.36 (шир. д, 2/3H), 4.30 (шир.д, 1/3H), 3.92-3.83 (м, 1H), 3.80-3.50 (м, 6H), 3.28-3.10 (м, 3H), 3.05-2.95 (м, 2H), 2.90 (с, 1H), 2.86 (с, 2H), 2.78-2.55 (м, 4H), 1.83-1.65 (м, 1H), 1.43 (с, 2H), 1.39 (с, 2H), 1.37 (с, 1H), 1.32 (с, 1H), 1.36-1.20 (м, 1H), 0.04-0.18 (м, 22/3H), -0.08 (дт, 2/3H).
Пример 41
N-[1(R)-[(1,2-Дигидро-1-метансульфонилспиро[3H-индол-3,4'- пиперидин]-1'-ил)карбонил] -2-(индол-3-ил)этил] - [3-[2(R)-гидроксипропил]амино]-3-метилбутанамидгидрохлорид
Стадия A: N-[1(R)-[(1,2-Дигидро-1-метансульфонилспиро- [3H-индол-3,4'-пиперидин] -1'-ил)карбонил] -2-(индол-3-ил)этил] -[3-[2- (R)-гидроксипропил] амино-3-метилбутанамидгидрохлорид
К 0,26 г промежуточного соединения со стадии B примера 39 добавляют 5 мл сухого метанола, 1,5 г безводного ацетата натрия, свежеприготовленного 0,10 г 2(R)-(тетрагидропиранил)- оксипропиональдегида, и перемешивают все при комнатной температуре в течение 1 часа. Раствор в THF цианоборгидрида натрия (8,5 мл 1 М раствор) добавляют, и все перемешивают в течение 18 часов. Реакционную смесь разбавляют 10 мл насыщенного водного раствора бикарбоната натрия и экстрагируют дихлорметаном (2 х 15 мл). Объединенные органические экстракты промывают рассолом (10 мл), сушат над K2CO3 и концентрируют. В результате обработки остатка с помощью флеш-хроматографии на 10 г силикагеля, элюируя смесью CH2Cl2-метанол (98:2), получают 0,219 г целевого продукта.
Вышеуказанный материал перемешивают в 3 мл сухого метанола с 0,10 мл концентрированной соляной кислоты, выпаривают досуха, а остаток тщательно растирают с эфиром до получения 0,174 г указанного в заглавии соединения в виде бледно-желтой пены.
1H ЯМР (400 МГц, CD3OD): δ 7.63 (д, 2/3H), 7.55 (д, 1/3H), 7.41 (д, 2/3H), 7.38 (д, 1/3H), 7.32 (д, 1/3H), 7.26 (д, 2/3H), 7.21-7.10 (м, 4H), 7.08-7.00 (м, 21/3H), 6.63 (д, 2/3H), 5.25 (дд, 2/3H), 5.19 (дд, 1/3H), 4.36 (шир. д, 2/3H), 4.30 (шир.д, 1/3H), 3.92-3.83 (м, 1H), 3.80-3.50 (м, 4H), 3.28-3.10 (м, 3H), 3.05-2.95 (м, 2H), 2.90 (с, 1H), 2.86 (с, 2H), 2.78-2.55 (м, 4H), 1.83-1.65 (м, 1H), 1.43 (с, 2H), 1.39 (с, 2H), 1.37 (с, 1H), 1.32 (с, 1H), 1.36-1.20 (м, 1H), 1.28 (д, 3H), 0.04-0.18 (м, 22/3H), -0.08 (дт, 2/3H).
Пример 42
N-[1(R)-[[3-окососпиро[изобензофуран-1(3H), 4'-пиперидин] -1' -ил]карбонил]-2-(фенилметокси)этил]-2-амино-2- метилпропанамидтрифторацетат
Стадия A:
К 0,165 г кислотного промежуточного соединения, полученного по способу стадии В примера 19, в 10 мл CH2Cl2 добавляют 0,095 г 3-оксоспиро/иэобензофуран-1(3H),4'-пиперидин/, 0,067 г HOBT и 0,110 г EDC и все перемешивают при комнатной температуре в течение 4 часов. Реакционную смесь выливают в 10 мл CH2Cl2 и промывают 20% водной лимонной кислотой (5 мл) насыщенным раствором бикарбоната натрия (5 мл), сушат над сульфатом магния и концентрируют. В результате обработки остатка с помощью флеш-хроматографии на 10 г силикагеля, элюируя смесью гексан-ацетон (3:1), получают 0,234 г продукта.
К раствору 0,024 г вышеуказанного промежуточного соединения в 1 мл CH2Cl2 добавляют 1,0 мл трифторуксусной кислоты и выдерживают при комнатной температуре в течение 30 минут. Летучие выпаривают, а остаток тщательно растирают с эфиром до получения 21 мг указанного в заглавии соединения в виде твердого вещества.
1H ЯМР (400 МГц, CD3OD): δ 7.85 (д, 1/2H), 7.80 (д, 1/2H), 7.63 (т, 1/2H), 7.54-7.40 (м, 21/2H), 7.35-7.20 (м, 51/2H), 7.06 (д, 1H), 6.58 (д, 1/2H), 5.25-5.15 (м, 1H), 4.93 (с, 1H), 4.69 (шир.с, 1H), 4.55-4.40 (м, 2H) 4.14 (шир. д, 1H), 3.70-3.40 (м, 2H), 3.18-3.10 (м, 1H), 2.13 (дт, 1H), 2.90-2.75 (м, 2H), 2.70-2.50 (м, 2H), 1.47 (с, 1.5H), 1.46 (с, 1.5H), 1.44 (с, 1.5H), 1.43 (с, 1.5H).
Пример 43
N-[1(R)-[1,2-Дигидро-1-метансульфонилспиро[3H- индол-3,4'-пиперидин] -1'-ил)карбонил]-4-фенилбутил]-2- амино-2-метилпропанамидгидрохлорид
Стадия A: N-[1(R)-[1,2-Дигидро-1-метансульфонилспиро[3H-индол-3,4'- пиперидин] -1'-ил)карбонил] -4- фенилбутил]-2-амино-2-метилпропанамидгидрохлорид
Это соединение получают из 2(R)-N-тpeт.-бутoксикapбонил- 5-фенилпентановой кислоты и 1,2-дигидро-1-метансульфонилспиро/3H-индол-3,4'-пиперидин/гидрохлорида, используя способ получения соединения примера 18.
Масс-спектр с бомбардировкой быстрыми атомами, рассчитано для C28H38N4O4S: MB = 526,2; найдено: м/е = (M+1) 527,9.
Пример 44
N-[1(R)-[1,2-Дигидро-1-метансульфонилспиро[3H-индол- 3,4'-пиперидин] -1'-ил)карбонил] -2-фенилметилтио)этил] - 2-амино-2-метилпропанамидтрифторацетат
Стадия A: N-[1(R)-[1,2-Дигидро-1-метансульфонилспиро [3H-индол-3,4'-пиперидин] -1'-ил)карбонил] -2- фенилметилтио)этил]-2-амино-2-метил- пропанамидтрифторацетат
Это соединение получают из коммерчески доступного N-трет-BOC-S-бензил-D-цистеина и 1,2-дигидро-1-метансульфонилспиро/3H-индол-3,4' -пиперидин/гидрохлорида, используя способ получения соединения примера 18.
Масс-спектр с бомбардировкой быстрыми атомами, рассчитано для C27H36N4O4S2: MB = 544,7; найдено: м/е = (M+1) 548,5.
Пример 45
N-[1(R, S)-[1,2-Дигидро-1-метансульфонилспиро[3H- индол-3,4'-пиперидин] -1'-ил)карбонил] -2-(2'-пиридометилокси) этил] -2-амино-2-метилпропанамидтрифторацетат
Стадия A: N-[1(R,S)-[1,2-Дигидро-1-метансульфонилспиро [3H-индол-3,4'-пиперидин] -1'-ил)карбонил] -2-гидрокси карбаминовой кислоты 1,1-диметилэтиловый сложный эфир
К раствору N-трет-BOC-(D)-серина (56 мг, 274 ммоль) в 2,5 мл THF при комнатной температуре добавляют 1,2-дигидро-1-метансульфонилспиро/3H-индол-3,4'-пиперидин/- гидрохлорид (полученный на стадии A примера 18, 83 мг, 0,274 ммоль), триэтиламин (45 мл, 0,33 ммоль), HOBT (44 мг, 0,33 ммоль), и EDC (63 мг, 0,33 ммоль). Спустя 3 часа полученную смесь разбавляют этилацетатом, а затем промывают последовательно водой и рассолом. Органический слой сушат над сульфатом натрия, фильтруют и концентрируют. Остаток очищают с помощью СДЖХ (силикагель, 100% этилацетат) до получения 112 мг (90%) указанного в заглавии соединения.
Стадия В: N-[1(R,S)-[1,2-Дигидро-1-метансульфонилспиро[3H-индол-3,4' -пиперидин] -1'-ил)карбонил]-2-(2'-пиридометилокси) этил]карбаминовой кислоты 1,1-диметиловый сложный эфир
К несодержащему масла гидриду натрия (полученному из 60% масляной дисперсии гидрида натрия за счет промывки гексаном (3 х), 9 мг, 0,21 ммоль) в 0,3 мл THF добавляют 2-пиколилхлорид (16 мг, 0,1 ммоль) в 0,3 мл DMF. Спустя 5 минут добавляют промежуточное соединение, полученное на стадии A (45 мг, 0,1 ммоль), и реакционную смесь перемешивают при комнатной температуре в течение 2 часов, а затем разбавляют эфиром. Эфирный слой промывают водой 5 раз, рассолом и сушат над сульфатом натрия. После очистки (препаративная TCX, силикагель, 100% этилацетат) выделяют 16 мг указанного в заглавии соединения (29).
1H ЯМР (400 МГц, CDCl3, смесь ротамеров): δ 8,53 (м, 1H), 7,77-6,83 (м, 7H); 5,75 (м, 1/3H), 4,98 (м, 2H), 4,67 (м, 2 1/2H), 4,25 (м, 1/2H), 4,10 (м, 1/2H), 3,91-3,67 (м, 4H), 3,17 (м, 1H), 2,91 (с, 3/2H), 2,89 (c, 3/2H), 2,79 (м, 1H), 1,95-1,69 (м, 4H), 1,42 (c, 9/2H), 1,41 (с, 9/2H).
Стадия C: N-[1(R,S)-[1,2-Дигидро-1-метансульфонилспиро[3H-индол-3,4' -пиперидин] -1'-ил)карбонил] - 2-(2'-пиридометилокси)этил]-2-амино-2-метилпропанамидтрифторацетат
Раствор промежуточного соединения, полученного на стадии В (16 мг, 0,029 ммоль), в 0,5 мл трифторуксусной кислоты перемешивают при комнатной температуре в течение 1/2 часа, а затем концентрируют. К раствору этого остатка в 1 мл хлороформа добавляют трет-бутилоксикарбонил-α -метилаланин (6,5 мг, 0,032 ммоль), HOBT (4,3 мг, 0,032 ммоль), триэтиламин (10 мл, 0,064 ммоль) и EDC (6 мг, 0,032 ммоль). Через 12 часов при комнатной температуре эту смесь разбавляют метиленхлоридом, а затем промывают водой и рассолом. Органический слой сушат над сульфатом натрия, фильтруют и концентрируют. Остаток очищают с помощью препаративной TCX/ силикагель, 100% этилацетат. Очищенное соединение концентрируют. К остатку добавляют трифторуксусную кислоту при комнатной температуре. Спустя 1 час полученную смесь концентрируют до получения указанного в заглавии соединения (5,3 мг).
1H ЯМР (400 МГц, CO3OD, смесь ротамеров): δ 8.70 (шир.с, 1H), 8.30 (м, 1H), 7.84 (м, 1H), 7.77 (м, 1H), 7.36 (м, 1H), 7.24 (м, 11/2H), 7.06 (м, 1 1/2H), 5.24 (т, 6 Гц, 1H), 4.86 (м, 2H), 4.56 (д, 13 Гц, 1H), 4.08 (м, 1H), 3.95 (м, 4H), 3.36 (м, 1H), 2.97 (с, 3/2H), 2.96 (с, 3/2H), 2.01-1.78 (м, 4H), 1.63 (с, 3/2H), 1.61 (с, 3/2H), 1.59 (с, 3/2H), 1.58 (с, 3/2H).
Пример 46
N-[1(R, S)-[1,2-Дигидро-1-метансульфонилспиро[3H- индол-3,4'-пиперидин] -1'-ил)карбонил] -2-[2'-пиридотиоэтил] - 2-амино-2-метилпропанамидтрифторацетат
Стадия A: N-[1(R,S)-[1,2-Дигидро-1-метансульфонилспиро[3H-индол-3,4' -пиперин] -1'ил)карбонил] -2-(2'-пиридотио)этил] карбаминовой кислоты 1,1-диметилэтиловый эфир
К несодержащему масла гидриду натрия (600 мг, 7,5 ммоль) в виде суспензии в 20 мл DMF- добавляют N-трет- BOC-D-цистеин (1,2 г, 5,4 ммоль) в 20 мл DMF при -10oC. Полученная смесь нагревается до комнатной температуры, и ее перемешивают еще один час. К реакционной смеси добавляют раствор 2-бромпиридина (0,514 мл, 5,4 ммоль) в 10 мл DMF. После нагревания в течение 20 часов при 80oC к реакционной смеси добавляют CuI (1,03 г, 5,4 ммоль) и перемешивают при той же температуре в течение еще 20 часов. Полученную смесь охлаждают до комнатной температуры и выливают в 0,5 н. соляную кислоту, экстрагируют эфиром. Эфирный слой фильтруют через целит, сушат над сульфатом натрия и концентрируют. Остаток очищают с помощью СДЖХ (силикагель, метиленхлорид/метанол = 10/1). К очищенному соединению (170 мг, 0,57 ммоль) в метиленхлориде добавляют 1,2-дигидро-1- метансульфонилспиро/3H-индол-3,4'-пиперидин/-гидрохлорид (полученный на стадии A примера 18, 172 мг, 0,57 ммоль), триэтиламин (95 мл, 0,68 ммоль), HOBT (92 мг, 0,68 ммоль) и EDC (130 мг, 0,68 ммоль) и проводят реакцию по способу примера 45 стадии A до получения указанного в заглавии соединения (310 мг, 99%).
Стадия B: N-[1(R,S)-[1,2-Дигидро-1-метансульфонилспиро[3H-индол-3,4' -пиперидин]-1'-ил)-карбонил]-2-(2'-пиридино)этил]-2-амино-2- метилпропанамидтрифторацетат
Получают из промежуточного соединения, полученного на стадии A (290 мг, 0,53 ммоль) за счет удаления защиты с помощью TFA. В результате получают указанное в заглавии соединение (102 мг).
1H ЯМР (400 МГц, CD3OD, смесь ротамеров): δ 8,45 (дд, 5,1 Гц, 1H), 7,61 (м, 1H), 7,39-7,05 (м, 6H), 5,27 (м, 1H), 4,52 (т, 12 Гц, 2H), 4,01 (м, 3H), 3,80 (м, 1H), 3,45 (м, 1H), 2,89 (c, 3H), 2,90 (м, 1H), 2,10-1,79 (м, 4H), 1,63 (c, 3/2H), 1,60 (с, 3/2H), 1,59 (с, 3/2H), 1,58 (с, 3/2H).
Масс-спектр с бомбардировкой быстрыми атомами: 532,7 (M+1).
Пример 47
N-[1(R, S)-[1,2-Дигидро-1-метансульфонилспиро[3H- индол-3,4'-пиперидин] -1'-ил)карбонил] -2-(циклогексилтио)- этил] -2-амино-2-метилпропанамидтрифторацетат
Стадия A: N-трет-BOC-циклoгeксилциcтeин
К раствору циклогексилмеркаптана (1 мл, 8,18 ммоль) и метил-2-ацетамидоакрила (1,29 г, 9 ммоль) в THF добавляют каталитическое количество гидрида натрия при комнатной температуре. Спустя 7 дней реакционную смесь концентрируют. Раствор остатка в 20 мл 6 н. соляной кислоты кипятят с обратным холодильником в течение 4 часов до комнатной температуры. Полученный раствор оставляют выстаиваться в течение 12 часов и фильтруют. Твердую часть сушат в вакууме. К смеси этой соли соляной кислоты в 1 н. растворе гидроксида натрия (15 мл) добавляют ди-трет-бутилдикарбонат (1,68 г, 7,7 ммоль) в 15 мл, 1,4 диоксана.
Через 12 часов полученную смесь выливают в 0,5 н. соляную кислоту и экстрагируют этилацетатом. Органический слой промывают водой, рассолом и сушат над сульфатом натрия. После фильтрования и концентрирования указанное в заглавии соединение выделяют с выходом 92% (2,23 г).
Стадия В: N-[1(R,S)-[1,2-Дигидро-1-метансульфонилспиро[3H-индол-3,4'-пиперидин] -1'-ил) карбонил-2-(циклогексилтио)этил] карбаминовой кислоты 1,1-диметилэтиловый сложный эфир
Получают из промежуточного соединения со стадии A (303 мг, 1,0 ммоль) по способу примера 45 (стадия A) до получения указанного в заглавии соединения (420 мг, выход 76%).
Стадия C: N-[1(R,S)-[1,2-Дигидро-1-метансульфонилспиро [3H-индол-3,4'-пиперидин] -1'-ил)карбонил] - 2-(циклогексилтио)этил]-2-[[1,1-диметилэтилокси)карбонил]амино]- 2-метилпропанамид
Раствор промежуточного соединения, полученного на стадии В (420 мг, 0,76 ммоль), в 5 мл трифторуксусной кислоты перемешивают при комнатной температуре в течение 1/2 часа, а затем концентрируют и сушат. К раствору этого остатка в 10 мл хлороформа добавляют трет-бутилоксикарбонил- α-метилаланин (170 мг, 0,84 ммоль), HOBT (113 мг, 0,84 ммоль), триэтиламин (116 мл, 0,84 ммоль) и EDC (160 мг, 0,84 ммоль). Спустя 2 часа при комнатной температуре полученную смесь разбавляют метиленхлоридом и промывают водой и рассолом. Органический слой сушат над сульфатом натрия, фильтруют и концентрируют. Остаток очищают с помощью СДЖХ (силикагель, гексаны/этилацетат = 1:1) до получения указанного в заглавии соединения (430 мг, 89%).
Стадия D: N-[1(R,S)-[1,2-Дигидро-1-метансульфонилспиро[3H-индол-3,4'-пиперидин] -1'-ил) карбонил]-2-(циклогексилтио) этил]-2-амино-2-метилпропанамидтрифторацетат
Раствор промежуточного соединения, полученного на стадии C (35 мг, 0,055 ммоль), в 0,5 мл трифторуксусной кислоты перемешивают при комнатной температуре в течение получаса, а затем концентрируют до получения указанного в заглавии соединения (33 мг).
1H ЯМР (400 МГц, CD3OD, смесь ротамеров): δ 7.38 (д, 8 Гц, 1H), 7.25-7.17 (м, 2H), 7.06 (м, 1H), 5.02 (м, 1H), 4.52 (м, 1H), 4.11 (м, 1H), 3.97 (м, 2H), 3.39 (м, 1H), 3.02 (м, 1H), 2.98 (с, 3H), 2.90-2.71 (м, 3H), 2.05-1.74 (м, 9H), 1.62 (с, 3/2H), 1.61 (с, 3/2H), 1.60 (с, 3/2H), 1.57 (с, 3/2H), 1.32 (м, 5H). FAB-MS: 537.9 (M+1).
Пример 48
N-[1(R, S)-[1,2-Дигидро-1-метансульфонилспиро[3H- индол-3,4'-пиперидин] -1'-ил)карбонил] -2- (циклогексилсульфинил)этил]-2-амино-2-метилпропанамидгидрохлорид
К раствору промежуточного соединения, полученного на стадии C примера 47 (35 мг, 0,055 ммоль), в 1 мл метанола добавляют перйодат натрия в 1 мл воды при комнатной температуре. Спустя пару часов реакционную смесь разбавляют этилацетатом и промывают водным раствором сульфита натрия.
Органический слой сушат над сульфатом натрия, фильтруют и концентрируют. Удаление защиты у остатка за счет трифторуксусной кислоты (по способу примера 47, стадии D) получают сырой продукт, который очищают с помощью препаративной TCX (силикагель, метиленхлорид/метанолгидроксид аммония = 10/1/0,1). Очищенный продукт снова подкисляют HCl, в эфире до получения указанного в заглавии соединения (21 мг).
1H ЯМР (400 МГц, CD3OD, смесь диастереоизомеров и ротамеров): δ 7,38-7,04 (м, 4H), 5,43 (м, 1H), 4,50 (м, 1H), 4,05 (м, 1H), 3,96 (м, 2H), 3,38 (м, 1H), 3,13 (м, 1H), 2,98 (с, 3H), 2,90-2,71 (м, 3H), 2,05-1,74 (м, 9H), 1,62 (м, 6H), 1,51-1,32 (м, 5H).
Масс-спектр с бомбардировкой быстрыми атомами: 553,9 (М+1).
Пример 49
N-[1(R, S)-[1,2-Дигидро-1-метансульфонилспиро[3H- индол-3,4'-пиперидин] -1'-ил)карбонил] -2-(циклогексилсульфонил) этил]-2-амино-2-метилпропанамидгидрохлорид
К раствору промежуточного соединения, полученного на стадии C примера 47 (35 мг, 0,055 ммоль), в 1 мл метанола добавляют OXONE в 1 мл воды при комнатной температуре. Спустя пару часов реакционную смесь разбавляют этилацетатом и промывают водным раствором сульфита натрия. Органический слой сушат над сульфатом натрия, фильтруют и концентрируют. К остатку добавляют трифторуксусную кислоту по способу стадии D примера 47, до получения сырого продукта, который очищают с помощью препаративной TCX (силикагель, метиленхлорид/метанол/гидроксид аммония = 10/1/0,1). Очищенный продукт снова подкисляют HCl в эфире до получения указанного в заглавии соединения (12 мг).
1H ЯМР (400 МГц, CD3OD, смесь ротамеров): δ 7.36 (дд, 7,2 Гц, 1H), 7.22 (м, 2H), 7.06 (м, 1H), 5.53 (м, 1H), 4.51 (м, 1H), 4.11 (м, 1H), 3.95 (м, 2H), 3.49 (м, 2H), 3.38 (м, 1H), 3.10 (м, 1H), 2.98 (с, 3/2H), 2.97 (с, 3/2H), 2.91 (м, 1H), 2.20-1.74 (м, 9H), 1.62 (с, 3/2H), 1.61 (с, 3/2H), 1.58 (с, 3/2H), 1.57 (с, 3/2H), 1.51-1.32 (м, 5H). FAB-MS: 569.9 (M+1).
Масс-спектр с бомбардировкой быстрыми атомами: 569,9 (M+1).
Пример 50
N-[(R)-[спиро[бензо[b] тиофен-3(2H)-4'-пиперидин] - 1'-ил]карбонил-2-индол-3-ил)этил-2-амино-2-метилпропанамидгидрохлорид
Стадия A: 1-/(1,1-Диметилэтоксикарбонил/-3-гидрокси 4-метилен-1,2,5,6-тетрагидропиридин
К суспензии/раствору метилтрифенилфосфониййодида (30 г, 74 ммоль) в 150 мл THF медленно добавляют бутиллитий (2,5 н., 25,5 мл, 63,7 ммоль) при 0oC. После перемешивания в течение часа при комнатной температуре к реакционной смеси медленно добавляют при комнатной температуре
N-трет-BOC-защищенный 4-пиперидон, полученный из 4- пиперидонмоногидратгидрохлорида (по способу, описанному в Protective Groups in Organic Synthesis, T. W.Greene, John Wiley and Sons, NY, 1981) в 50 мл THF. Эту реакционную смесь перемешивают в течение 2 часов и фильтруют. Полученный фильтрат концентрируют и очищают (СДЖХ, силикагель, гексаны/этилацетат = 10/1) до получения продукта Виттига (7,9 г, выход 82%).
К суспензии диоксида селена/силикагеля /полученной по способу, описанному в Chem. Lett. 1981, 1703/ в 30 мл метиленхлорида добавляют трет-бутилгидроперекись (1,23 мл). Через 5 минут добавляют продукт Виттига (0,72 г, 3,69 ммоль) в 5 мл метиленхлорида. Мутный раствор перемешивают с водой, рассолом и сушат над сульфатом натрия. Органический слой концентрируют и очищают с помощью флеш-хроматографии (гексаны/этилацетат = 4/1) до получения указанного в заглавии соединения с 52% выходом (0,41 г).
Стадия В: 1-/(1,1-Диметилэтокси)карбонил/-4-хлорметил- 1,2,5,6-тетрагидропиридин
Промежуточное соединение со стадии A (400 мг, 1,88 ммоль) растворяют в 10 мл бензола, и тионилхлорид (165 мл, 2,26 ммоль) добавляют и нагревают до 60oC в течение 25 минут. Полученную смесь выливают в NaHCO3 (водн.) и экстрагируют эфиром. Эфирный слой сушат над сульфатом магния и концентрируют до получения указанного в заглавии соединения (333 мг, 77%).
Стадия C: 1-/1,1-Диметилэтокси)карбонил/-4-//(2-бромфенил) тио/метил-1,2,5,6-тетрагидропиридин
Промежуточное соединение, полученное на стадии В (330 мг, 1,43 ммоль), растворяют в 10 мл ацетона и 2-бромотиофенола (172 мл, 1,43 ммоль), и карбонат калия (390 мг, 2,86 ммоль) добавляют. Реакционную смесь нагревают до 60oC в течение часа, а затем фильтруют через силикагель (100% эфир). Органический слой концентрируют и очищают с помощью флеш-хроматографии (силикагель, гексаны/этилацетат = 10/1) до получения указанного в заглавии соединения с выходом 84% (460 мг).
Стадия D: 1'-/(1,1-Диметилэтоксикарбонил/-спиро/бензо[b]тиофен- 3-(2H), y'-пиперидин
Промежуточное соединение, полученное на стадии C (450 мг, 1,17 ммоль), растворяют в 60 мл бензола и добавляют AIBN (10 мл) и гидрид трибутилолова (644 мл, 2,39 ммоль). Полученную смесь кипятят с обратным холодильником в течение 2 часов и концентрируют. Остаток растворяют в эфире и добавляют бром до тех пор, пока реакционный раствор не приобретает коричневатый цвет. К этому коричневатому раствору при комнатной температуре добавляют DBU (650 мл) по каплям. Полученный мутный раствор фильтруют через силикагель и промывают эфиром. Эфирный раствор концентрируют, а остаток очищают с помощью радиальной хроматографии (силикагель, гексаны/этилацетат = 10/1) до получения указанного в заглавии соединения (157 мг, выход 43%).
Стадия E: N-/1(1R)-/спиро/бензо[b] тиофен-3(2H), y'- пиперидин/-1'-ил)карбонил/-2-(индол-3-ил)- этил/-2-амино-2-метилпропанамидгидрохлорид
Раствор промежуточного соединения, полученного на стадии D (50 мг, 0,164 ммоль), в 0,5 мл TFA перемешивают при комнатной температуре в течение получаса, а затем концентрируют. Остаток разбавляют хлороформом и промывают NaHCO3 (водн.). Органический слой сушат над сульфатом натрия, фильтруют и концентрируют до получения свободного амина (32 мг) в 95%. К раствору свободного амина (5,1 мг, 0,025 ммоль) в 1 мл хлороформа добавляют промежуточное соединение, полученное на стадии C примера 21 (9,2 мг, 0,0246 ммоль), HOBT (4,0 мг, 0,0295 ммоль) и EDC (5,6 мг, 0,0295 ммоль) при комнатной температуре. Спустя 12 часов реакционную смесь выливают в воду и экстрагируют хлороформом. Хлороформовый слой сушат над сульфатом натрия, фильтруют и концентрируют. Остаток очищают с помощью препаративной TCX (силикагель, гексаны/этилацетат = 3/1) до получения бесцветной пены (13 мг, 94%). Указанное в заглавии соединение получают в виде бесцветной пены по способу стадии C примера 18.
1H ЯМР (400 МГц, CD3OD, смесь ротамеров): δ 7.62 (д, 8 Гц, 2/3H), 7.54 (д, 8 Гц, 1/3H), 7,39 (д, 8 Гц, 2/3H), 7.35 (д, 8 Гц, 1/3H), 7.19-7.00 (м, 6 1/3H), 2.62 (м, 1H), 1.72-1.65 (м, 2 1/3H), 1.61 (с, 4H), 1.50 (с, 2H), 0.94 (м, 1H), 0.10 (м, 2/3H).
Масс-спектр с бомбардировкой быстрыми атомами 477 (M+1).
Пример 51
Стадия A: 1',2-Диметилспиро/изоиндолин-1-он-3,4'-пиперидин/
К перемешиваемому раствору 2-метилизоиндолин-1-она (1,47 г, 10 ммоль, доступному от Aldrich Chem. Co. ) и мехлорэтамингидрохлорида (2,9 г, 15 ммоль) в 50 мл DMF при 0oC в атмосфере аргона медленно добавляют гидрид калия (35% в минеральном масле, 4,5 г, 40 ммоль). Затем реакционную смесь медленно нагревают до комнатной температуры и перемешивают еще 3 часа. По данным TCX (60 этилацетат в гексане) реакция завершается. Полученную смесь медленно выливают на лед и экстрагируют шесть раз этилацетатом. Объединенные органические экстракты сушат над сульфатом натрия и выпаривают. Остаток очищают с помощью флеш-хроматографии, элюируя градиентом растворителя 5-10% метанол в дихлорметане до получения 1,17 г продукта.
1H ЯМР (400 МГц, CDCl3): δ 7.85 (дд, J = 1.5 Гц, 6.5 Гц, 1H), 7.81 (д, J = 7.1 Гц, 1H), 7.50-7.40 (м, 2H), 3.03 (с, 3H), 2.95-2.90 (м, 2H), 2.71 (дт, J = 2.6 Гц, 11.4 Гц, 2H) 2.46 (с, 3H), 2.31 (дт, J = 4.7 Гц, 13 Гц, 2H), 1.44 (дд, J = 1.6 Гц, 13 Гц, 2H).
Масс-спектр с бомбардировкой быстрыми атомами, рассчитано для C14H18N2O: 230; найдено: 231 (М+H).
Стадия B: 2-Метилспиро/изоиндолин-1-он-3,4'-пиперидин/
Процедуру диметилирования осуществляют по Tidwell и Buchwald, J. Org. Chem. 1992, 57, 6380-6382.
К перемешиваемому раствору продукта со стадии A (1,0 г, 4,35 ммоль) в 1,2 дихлорметане (10 мл) при 0oC добавляют 1-хлорэтилхромат (0,56 мл, 5,2 ммоль), и полученную смесь перемешивают 20 минут. Добавляют 10 мл метанола, и полученную смесь кипятят с обратным холодильником в течение одного часа. После выпаривания и очистки с помощью флеш-хроматографии, элюируя 10-20% NH4OH-MeOH (1:10) в хлороформе, получают 0,63 г продукта.
1H ЯМР (400 МГц, CD3OD): δ 8.02 (д, J = 8 Гц, 1H), 7.85 (д, J = 8 Гц, 1H), 7.70 (т, J = 8 Гц, 1H), 7.60 (т, J = 8 Гц, 1H), 3.75-3.60 (м, 4H), 3.10 (с, 3H), 2.60-2.51 (м, 2H), 1.72 (шир.д, J = 14 Гц, 2H).
Масс-спектр E1, рассчитано для C13H16N2O 216; найдено: 301 (M+, 5%), 216 (M+), 185, 160.
Стадия C: 2-Метилспиро/изоиндолин-1-он-3,4'-пиперидин/-1'-карбоновой кислоты 1,1-диметилэтиловый сложный эфир
К перемешиваемому раствору 2-метилизоиндолин-1-она (100 мг) в 2 мл DMF добавляют избыток KH в минеральном масле при 0oC в атмосфере аргона. Через 5 минут добавляют биc/2-бpoмэтил/-тpeт.-бутилкарбамат (300 мг), и полученную смесь перемешивают при комнатной температуре в течение 1 часа, и нагревают при 80oC в течение ночи. Полученную смесь выливают на лед, и экстрагируют этилацетатом. Органические экстракты сушат (Na2SO4) и выпаривают. Остаток очищают с помощью препаративной TCX, элюируя 60% этилацетатом в гексане до получения 16 мг продукта.
1H ЯМР (400 МГц, CDCl3): δ 7.89 (дд, J = 1.3 Гц, 5.8 Гц, 1H), 7.75 (д, J = 6.5 Гц, 1H), 7.54-7.40 (м, 2H), 4.35-4.10 (шир.м, 2H), 3.02 (с, 3H), 2.14 (дт, J = 5.3 Гц, 13 Гц, 2H), 1.49 (с, 9H), 1.46-1.40 (м, 2H).
Масс-спектр с бомбардировкой быстрыми атомами, рассчитано для C18H24N3O3 316; найдено 317 (М+H, 100%).
Стадия D:; 2-Метилспиро/изоиндолин-1-он-3,4'-пиперидин/
Промежуточное соединение со стадии C (16 мг) обрабатывают концентрированной HCl и MeOH при комнатной температуре в течение 2 часов и выпаривают до получения целевого продукта. Все спектральные данные соответствуют данным стадии В.
Стадия E: N-/1(R)-/(2-Метилспиро/изоиндолин-1-он-3,4'- пиперидин/-1'-ил)карбонил/-2-(индол-3-ил)- этил/-2-/ (1,1-диметилэтилокси)карбонил/амино/- метилпропанамид
Это соединение получают в соответствии со стандартной методикой сочетания пептидов из α(R)-//2-//(1,1-диметилэтокси) карбонил/-амино/-2,2-диметил-1-оксоэтил/амино/-1H- индол-3-пропановой кислоты (25 мг) и продукта со стадии В.
1H ЯМР (400 МГц, CDCl3): δ 8.58, 8.44 (2шир.с, 1H), 7.80-7.15, 6.44 (м, д, J = 7.6 Гц, все 10H), 5.43-5.36, 5.22-5.15, (2м, 1H), 4.98 (шир.с, 1H), 4.60-4.50 (шир. м, 1H), 3.64-3.50 (шир.м, 1H), 3.40-3.05, 2.72-2.64, (2м, 4H), 2.88, 2.51 (2с, 3H), 2.00-1.90 (м, 2H), 1.52, 1.51 (2с, 3H), 1.49 (с, 3H), 1.45, 1.44 (2с, 9H), 1.40-0.40 (неск.м, 2H).
Масс-спектр с бомбардировкой быстрыми атомами, рассчитано для C33H41N5O5 587; найдено: 588 (М+H), 532, 488.
Стадия F: N-/1(R)-/(2-Метилспиро/изоиндолин-1-он- 3,4'-пиперидин/-1'-ил)карбонил/-2-(индол-3-ил)- этил/-2-амино-2-метилпропанамидгидрохлорид
Указанное в заглавии соединение получают, используя HCl в этилацетате, из продукта со стадии E.
1H ЯМР (400 МГц, CD3OD): δ 7.84-6.89 (м, 9H), 5.30, 5.15 (2дд, 1H), 4.50-4.40 (м, 1H), 3.85-3.77 (м, 1H), 3.65-3.50 (м), 3.40-3.15 (м), 2.95, 2.58 (2с, 3H), 2.95-2.85 (м), 2.55-2.47 (м), 2.22-2.15 (м), 2.09-2.00 (м), 1.65, 1.64, 1.60 (3с, 6H), 1.50-1.20 (м), 1.15-1.05 (м), 0.9-0.8 (м), 0.61-0.52 (м).
Масс-спектр с бомбардировкой быстрыми атомами, рассчитано для C28H33N5O3 471, найдено 472 (М+H).
Пример 52
N-/1(R)-//1-/////6-///4-Азидо-2-гидрокси-5-йодофенил/ карбонил/амино/гексил/амино/карбонил/метил/сульфонил/- 2,3-дигидроспиро/3H-индол-3,4'-пиперидин/-1'-ил/карбонил/- 2-(фенилметилокси)этил/-2-амино-2-метилпропанамидгидрохлорид
К раствору коммерчески доступного N-гидрокси-сукцинил- 4-азидо-2-гидроксибензоата в 5 мл CH2Cl2 добавляют 6- N-трет-бутоксикарбонил-н-гексиламингидрохлорид и 0,10 мл основания Ханинга и перемешивают в течение 4 часов. Реакционную смесь выпаривают досуха и обрабатывают хроматографически на 15 г силикагеля. В результате элюирования смесью гексаны-этилацетат (2:1) получают 0,229 ацилированного продукта. К 29 мг указанного ранее материала добавляют 2 мл THF и 2 мл 0,01 М водного гидроксида натрия, 25 мг йодида калия. Добавляют хлорамин-Т (15 мг) и перемешивают в течение 30 минут. Реакцию гасят 2 мл насыщенного раствора тиосульфата натрия, разбавляют 5 мл 0,05 н. HCl и экстрагируют этилацетатом (2 х 5 мл). Объединенные органические экстракты промывают рассолом (5 мл), сушат над сульфатом магния и концентрируют. В результате обработки остатка с помощью флеш-хроматографии (5 г силикагеля), элюируя смесью гексанэфир (3:1) получают 26 мг йодированного материала. Удаление защиты N-трет-BOC осуществляют 4 М HCl в этилацетате до получения 21,4 мг гидрохлорида.
К раствору этого материала в 5 мл CH2Cl2 добавляют 49 мг кислотного промежуточного соединения со стадии A примера 33, 0,016 мл NMM, 19,8 мг HOBT и 29 мг EDC и перемешивают в течение 18 часов. Реакционную смесь обрабатывают и очищают обычным способом.
Повторное удаление защиты N-трет-BOC группы осуществляют за счет 4 М HCl в этилацетате. Получают указанное в заглавии соединение в виде желто-коричневого твердого вещества. Этот материал подщелачивают, растворяя в 2 мл насыщенного NaHCO3, и экстрагируют CH2Cl2 (2 х 3 мл). Объединенные органические экстракты сушат над сульфатом натрия и концентрируют до получения указанного в заглавии соединения.
1H ЯМР (400 МГц, CDCl3): δ 8.40-8.20 (м, 1H), 7.95 (с, 2/3H), 7.90 (с, 1/3H), 7.40- 6.90 (м, 91/3H), 6.70 (с, 2/3H), 6.55 (м, 1H), 5.20-5.10 (м, 1H), 4.70-4.40 (м, 4H), 4.10-3.80 (м, 5H), 3.80-3.50 (м, 4H), 3.40-3.10 (м, 4H), 3.10-3.00 (м, 1H), 2.70 (дт, 1H), 1.90-1.20 (м, 14H), 1.30 (с, 6H).
Пример 53
N-/1(S)-/(1,2-Дигидро-1-метилсульфонилспиро/3H- индол-3,4'-пиперидин/-1'-ил)карбонил/-2-(фенилметилсульфонил) этил/-2-амино-2-метилпропанамидгидрохлорид
Стадия A: N-/1(S)-/(1,2-Дигидро-1-метилсульфонилспиро/3H-индол-3,4'- пиперидин/-1'-ил)карбонил/-2- (фенилметил)сульфонил)этил/-2-амино-2-метил-пропанамид
Образец N-/1(S)-/(1,2-Дигидро-1-метилсульфонилспиро/- 3H-индол-3,4'-пиперидин/-1'-ил)карбонил/-2-(фенилметилтио)- этил/-2-//1,1-диметилэтилокси)карбонил/амино/-2-метилпропанамида (пример 44, стадия C), 72 мг, растворяют в 0,5 мл метанола и охлаждают на ледяной бане. К этому добавляют по каплям при перемешивании раствор 101 мг OXONE (TM) в 0,5 мл воды. За ходом реакции в течение нескольких часов следят по данным TCX на пластинах силикагеля CF, проявляя смесью 2:1 EtOAc:гексан; наблюдают рост двух более полярных пятен с течением времени по мере расходования исходного материала. Когда исходный материал практически израсходован, реакционную смесь высушивают почти досуха в атмосфере азота, а остаток экстрагируют хлороформом, высушенный над сульфатом магния экстракт обрабатывают с помощью препаративной TCX на CF пластине силикагеля (10 х 10 см х 1,00 мкм), проявляя EtOAc:гексаном; выделяют две полосы. Менее полярную компоненту растворяют в 0,5 мл анизола, охлаждают на ледяной бане и обрабатывают 0,5 мл TFA. Реакцию останавливают, и реакционную смесь удаляют из бани. Спустя 30 минут основную часть TFA удаляют при пониженном давлении, а основную часть оставшегося анизола выпаривают в потоке азота. Остаток помещают в хлороформ и встряхивают с 1 М K2HPO4, к которому добавлено достаточно NaOH для создания pH более 9. Затем хлороформ удаляют, а водную фазу несколько раз экстрагируют хлороформом, объединенные органические экстракты сушат над сульфатом магния и концентрируют при пониженном давлении до смолы. В результате обработки с помощью препаративной TCX на пластине силикагеля GF 0,5:5:95 конц. NH4:MEOH:CHCl3, получают указанное в заглавии соединение в виде свободного основания, рассчитано для C27H36N4O6S2: MB 576,7, найдено: м/е = (М+1) 577,5.
Стадия В: N-/1(S)-/(1,2-Дигидро-1-метилсульфонилспиро/ 3H-индол-3,4'-пиперидин/-1'-ил)карбонил/- 2-(фенилметилсульфонил)этил/-2-амино-2-метил- пропанамидгидрохлорид
Гидрохлоридную соль соединения со стадии A получают, используя описанные ранее стандартные процедуры, и получают указанное в заглавии соединение.
Пример 54
Получение двух N-/1(S)-/(1,2-дигидро-1-метилсульфонилспиро/ 3H-индол-3,4'-пиперидин/-1'-ил)карбонил/-2-(фенилметилсульфинил)этил/-2- амино-2-метилпропанамидгидрохлоридов
Стадия A: N-/1(S)-/(1,2-Дигидро-1-метилсульфонилспиро/ 3H-индол-3,4'-пиперидин/-1'-ил)карбонил/-2- (фенилметилсульфинил)этил/-2-амино-2-метилпропанамид
Подвергая более полярную полосу со стадии A примера 53 процедуре удаления защиты TFA /анизолом с последующей обработкой с помощью препаративной TCX, выделяют две полосы, соответствующие двум ожидаемым диастереоизомерным сульфоксидам. Для менее полярного диастереоизомера: рассчитано для C27H36N4O5S2: MB 560,7, найдено м/е = (М+1) 561,7.
Для более полярного диастереоизомера рассчитано для C27H36N4O5S2: MB= 560,7; найдено м/е = (M+1) 561,7.
Стадия В: N-/1(S)-/(1,2-Дигидро-1-метилсульфонилспиро/ 3H-индол-3,4'-пиперидин/-1'-ил)карбонил/- 2-(фенилметилсульфинил)этил/-2-амино-2-метилпропанамидгидрохлорид
Указанное в заглавии соединение получают, заменяя соединение, полученное на стадии A примера 53 и на стадии В примера 53, на любое из соединений, выделенных ранее на стадии A.
Пример 55
N-/1(R)-/(1,2-Дигидро-1-метансульфонилспиро/3H-индол-3,4' -пиперидин/-1'-ил)карбонил/-2-(фенилметилокси)- этил/-2-амино-2-метилпропанамидмезилат
Это соединение получают, обрабатывая свободное основание стадий C примера 18 метансульфоновой кислотой. Указанное в заглавии соединение получают за счет перекристаллизации его из смеси этилацетат-этанол-вода. Т.плавления 166-168oC.
Пример 56
2,3,3a,4,6,6a-Гексагидро-2-оксо-1H-тиено[3,4-b]имидазол- 4(S)-пентановая кислота-6-/////1'-//(29R)-//2-амино- 2-метил-1-оксопропил/-амино/-3-(фенилметилокси)-1-оксопропил/- 2,3-дигидроспиро/3H-индол-3,4'-пиперидин/-1'-ил/ сульфонил/метил/карбонил/амино/гексилового сложного эфира трифторацетат
К раствору 0,108 г промежуточного соединения, полученного в примере 33 на стадии A, в 5 мл CH2Cl2 добавляют 20 мг 6-аминогексанола, 28 мг HOBT, и 42 мг EDC и перемешивают в течение 4 часов. Реакционную смесь разбавляют 10 мл CH2Cl2 и промывают 0,5 н. HCl (5 мл), насыщенным водным NaHCO3 (5 мл), сушат над сульфатом магния и концентрируют. Остаток очищают с помощью флеш-хроматографии (10 г силикагеля), элюируя смесью CH2Cl2-ацетон (1:1).
К 56,2 мг вышеуказанного промежуточного соединения в 2 мл CH2Cl2 и 2 мл DMF добавляют 23 мг биотина, 14 мг DMAP, 28 мг EDC и перемешивают в течение 18 часов. Реакционную смесь обрабатывают обычным способом. В результате очистки с помощью флеш-хроматографии на 5 г силикагеля, элюируя смесью CH2Cl2-ацетон (1: 1) получают 22 мг биотинового конъюгата. Удаление защиты N-трет-BOC осуществляют в CH2Cl2-TFA до получения 18,9 мг указанного в заглавии соединения в виде белого твердого вещества.
1H ЯМР (CDCl3, 400 МГц, соединение существует в виде смеси 3:2 ротамеров): δ 8.45-8.23 (м, 1H), 7.9 (с, 1H), 7.40-7.28 (м, 4H), 7.25-7.17 (м, 2H). 7.00 (дт, 2/3H), 6.80 (д. 1/3H), 5.21-5.14 (м, 1H), 4.60-4.42 (м, 4H), 4.28 (bt, 1H), 4.15-4.00 (м, 6H), 3.85-3.70 (м, 2H), 3.20-3.10 (м, 3H), 2.90 (дд, 1H), 2.83 (дт, 1H), 2.70 (д, 1H), 2.40-2.25 (м, 2H), 2.00-0.60 (м, 18H), 1.62 (с, 3H), 1.60 (с, 3H).
Данные по биологической активности заявленных соединений.
Методика анализа клеточной культуры гипофиза.
Асептически удаляют гипофиз у самцов крыс Wistar (150-200 г) и готовят культуры клеток гипофиза по методике, описанной в Cheng, K., Chan, W.W. -S., Barreto, A. , Convey, E.M. and Smith R.G (1988), Endocrinology 124, 2791-2798.
По этой же методике клетки обрабатывают различными агентами, усиливающими секрецию εH и исследуют εH секреторную активность.
Измеряют также внутриклеточные уровни с AMP и определяют EC50.
Данные испытаний представлены в таблице.
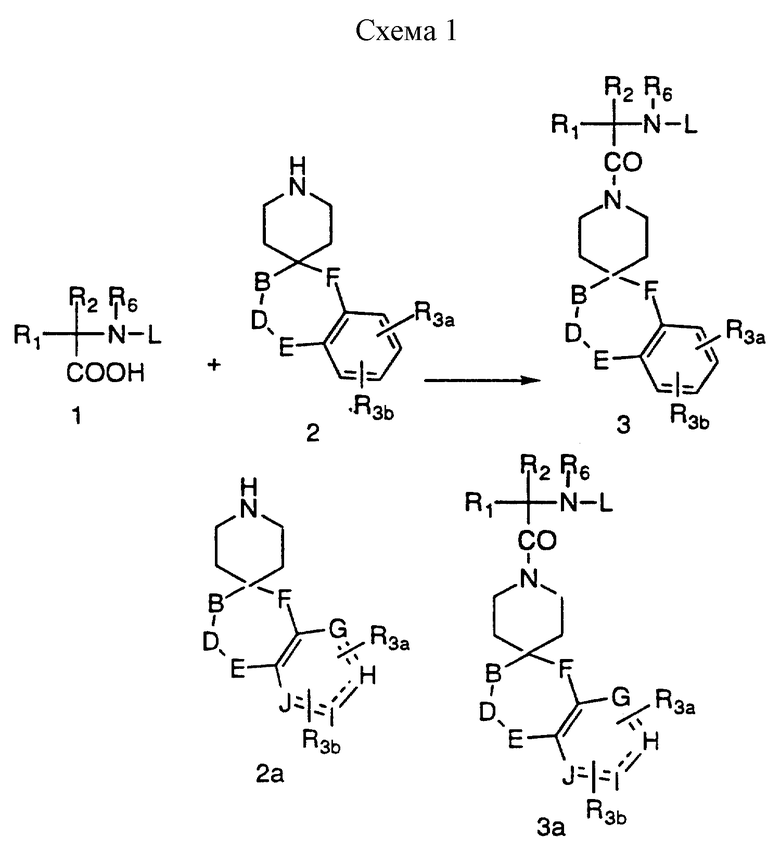
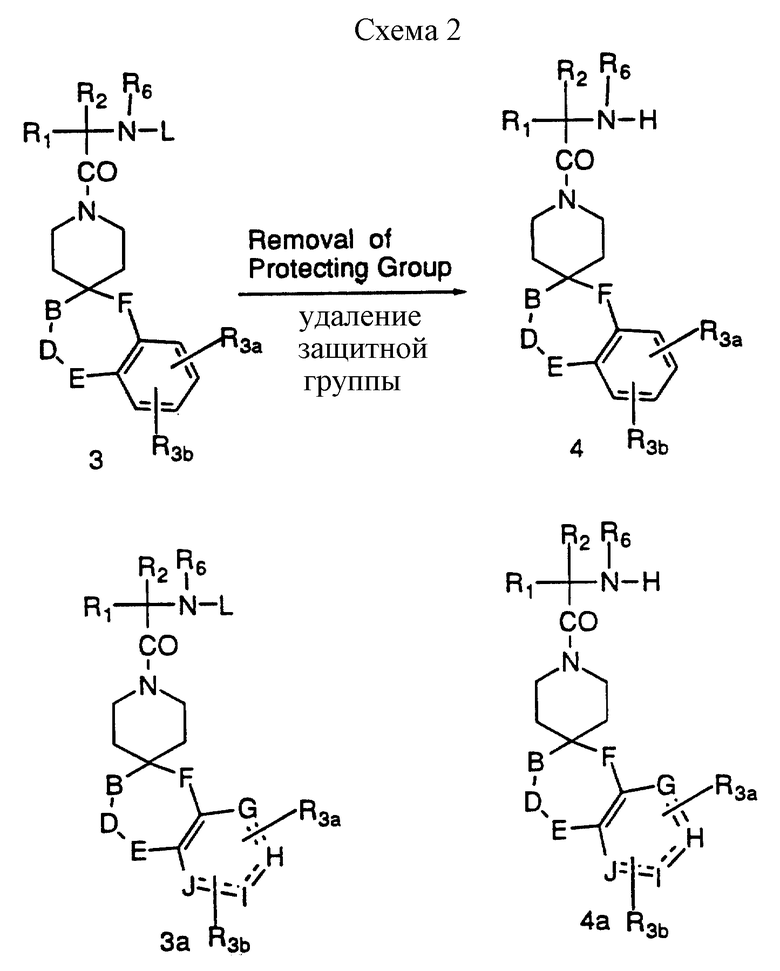
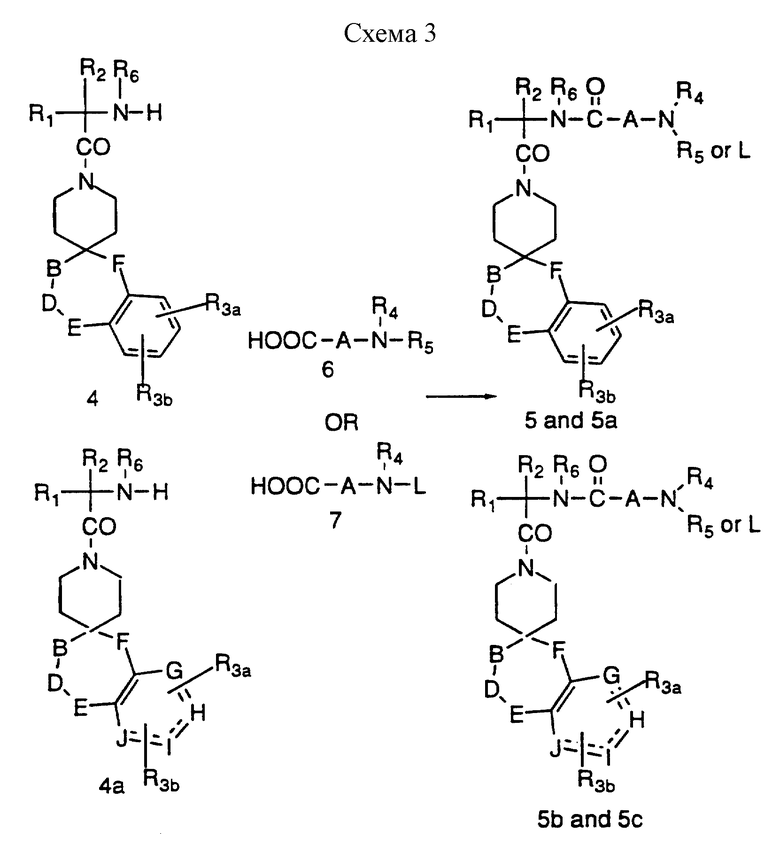
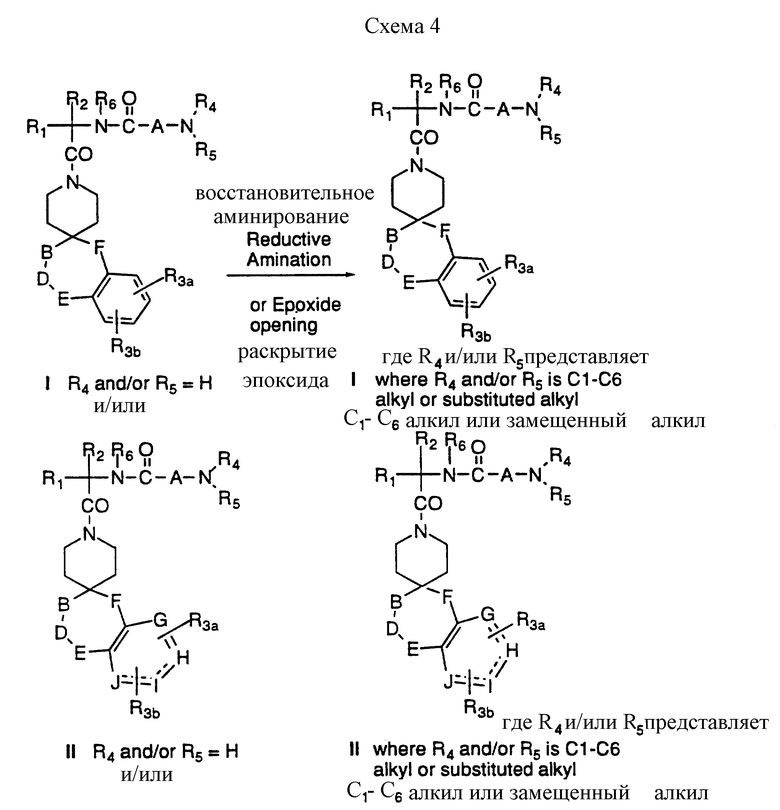
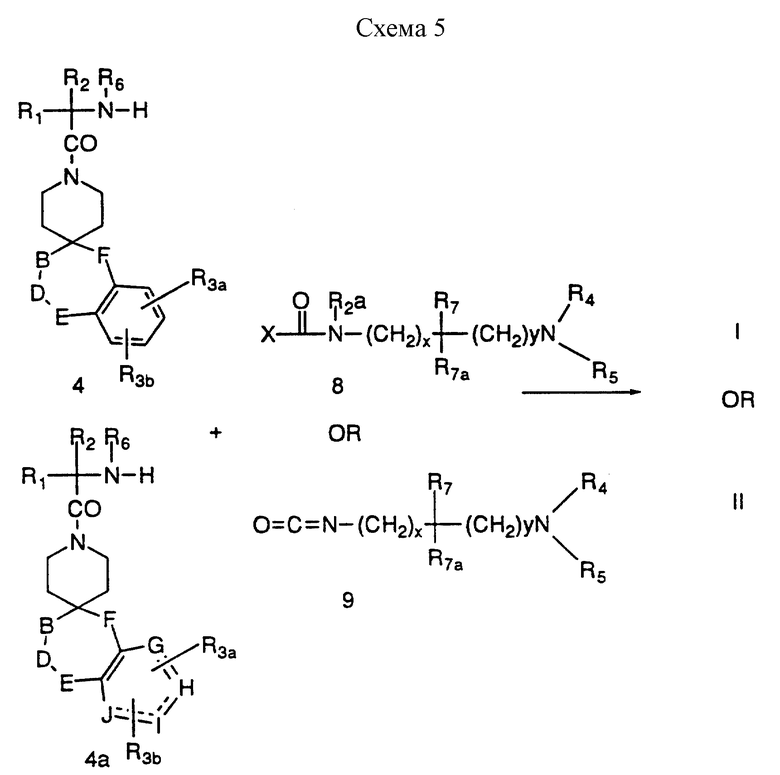
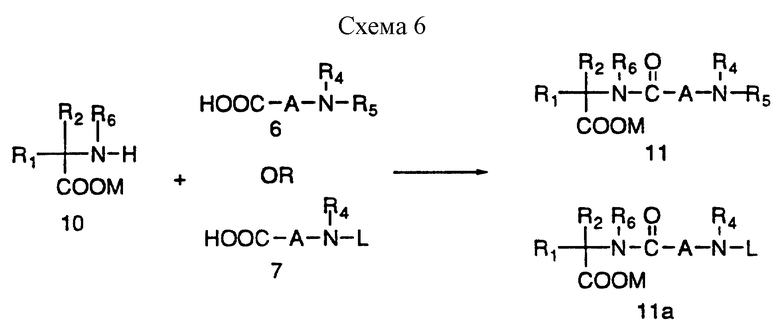
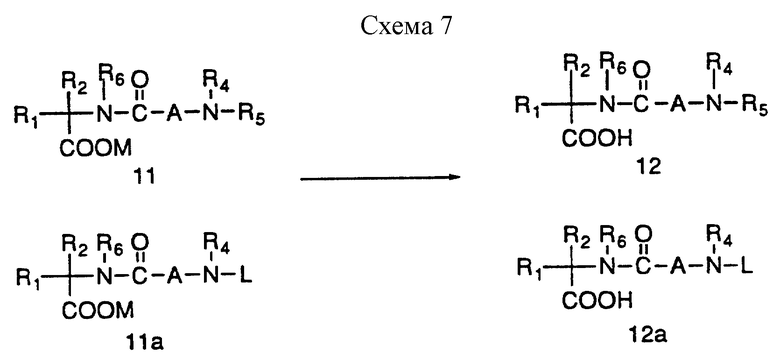
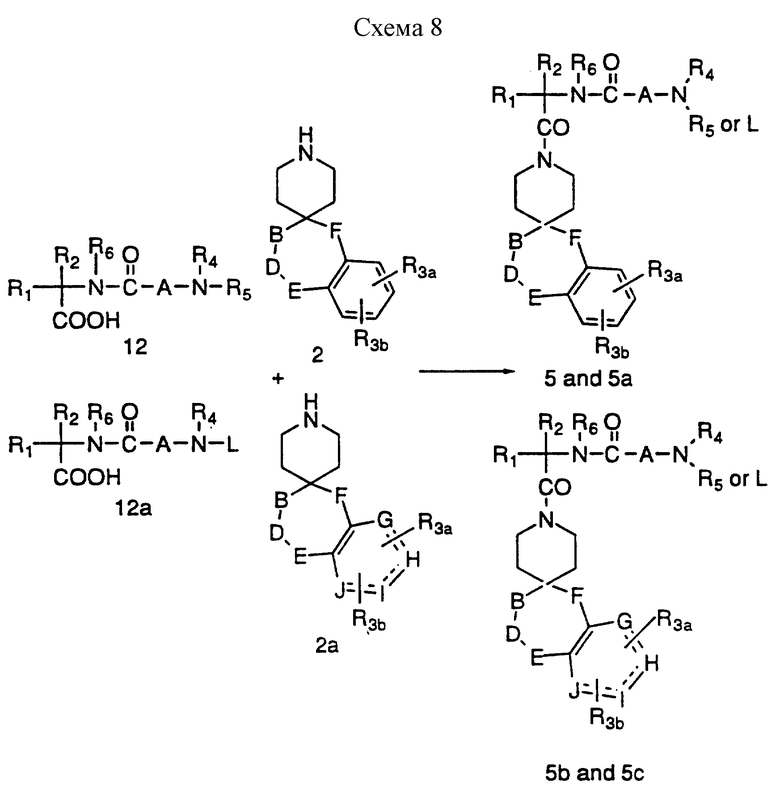
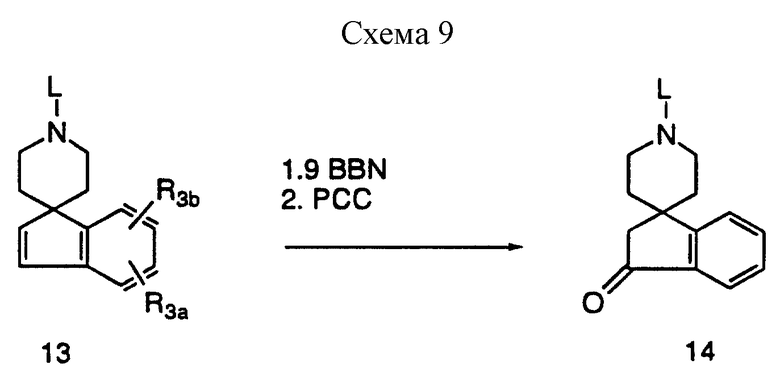
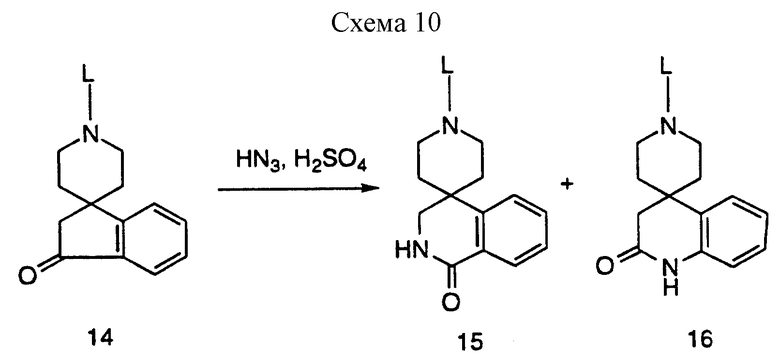
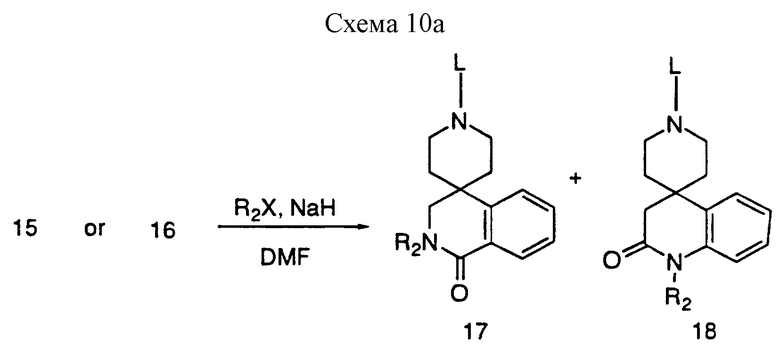
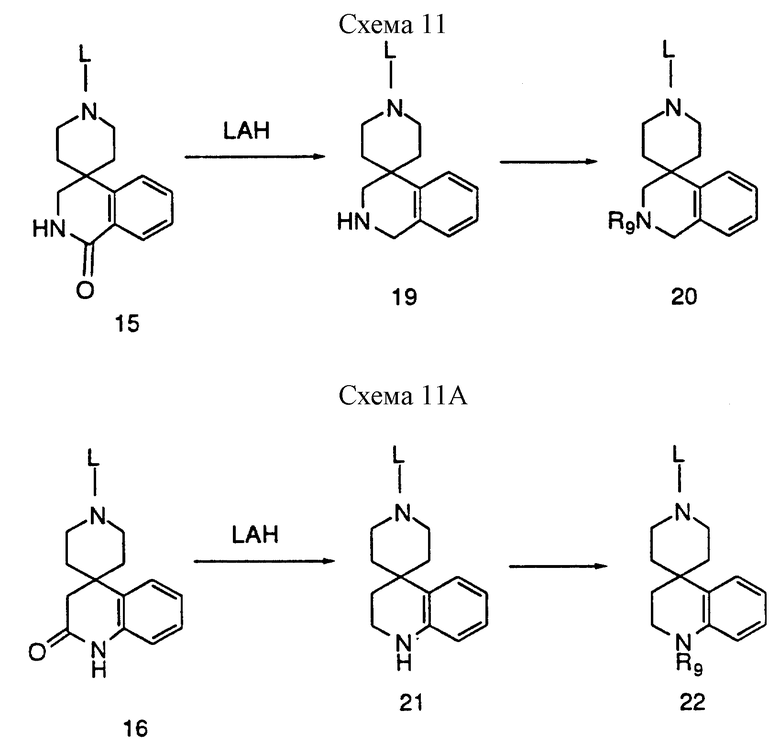
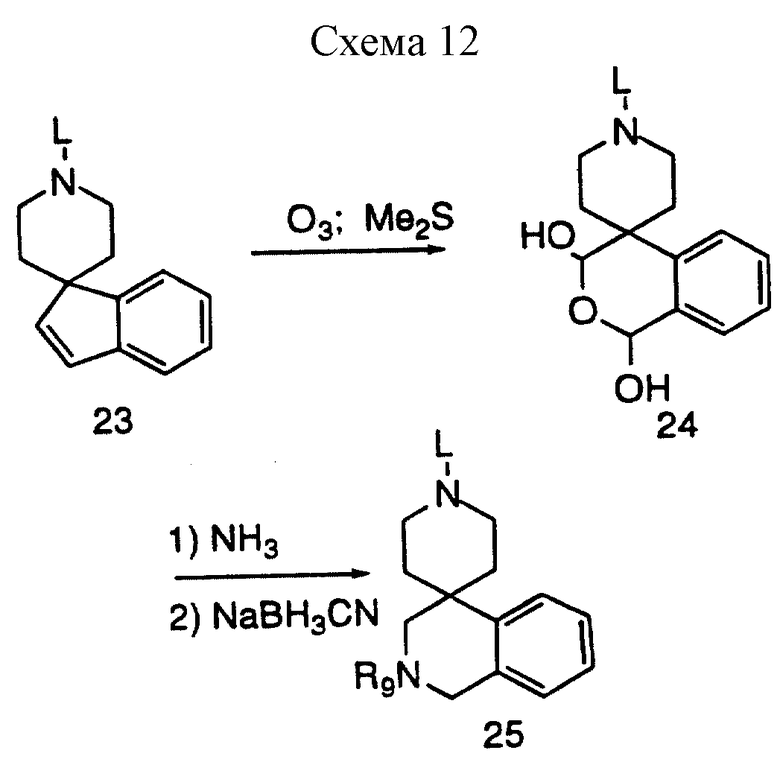
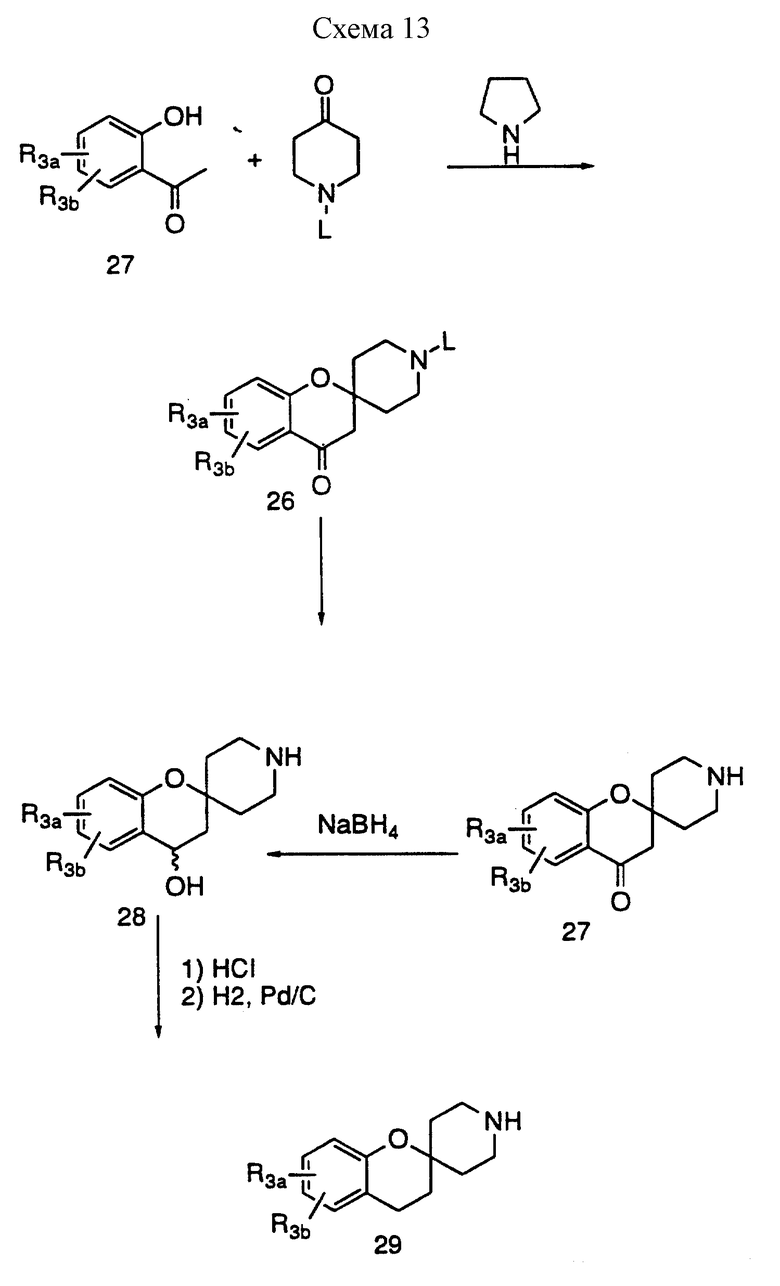
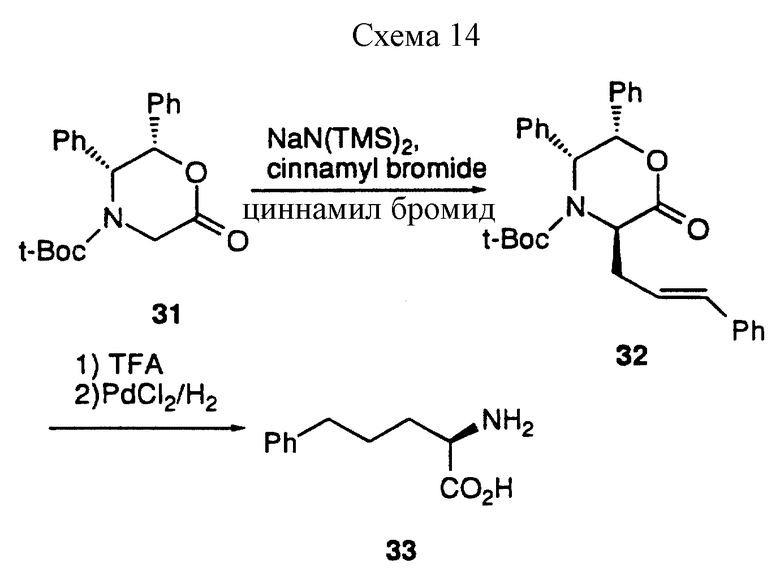
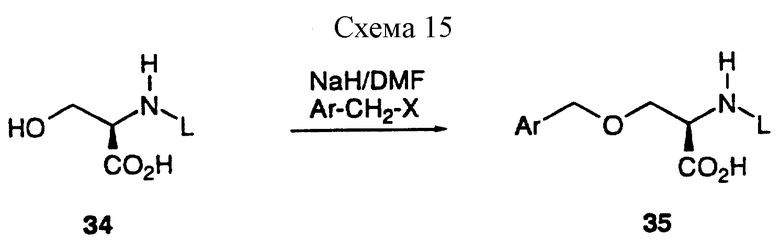
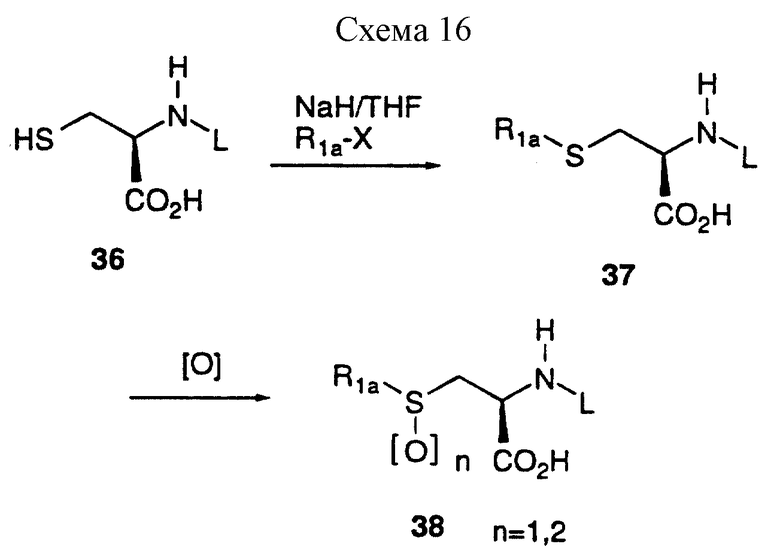
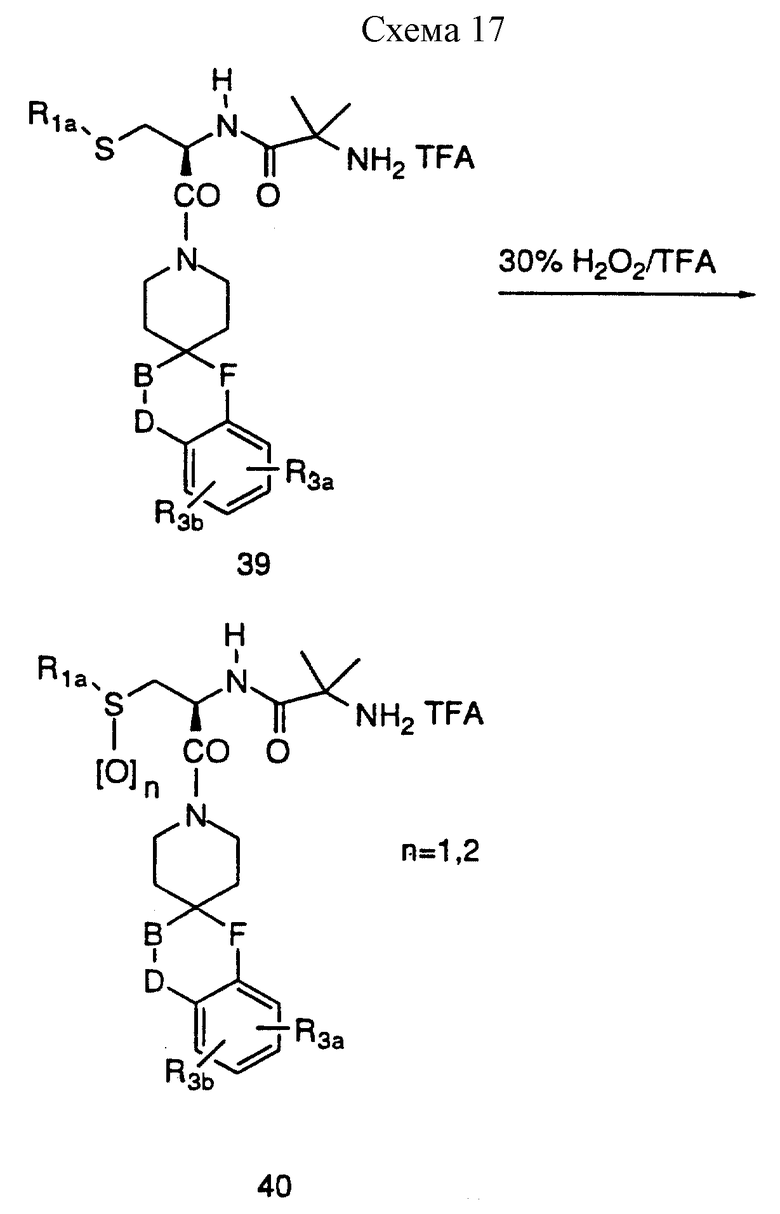
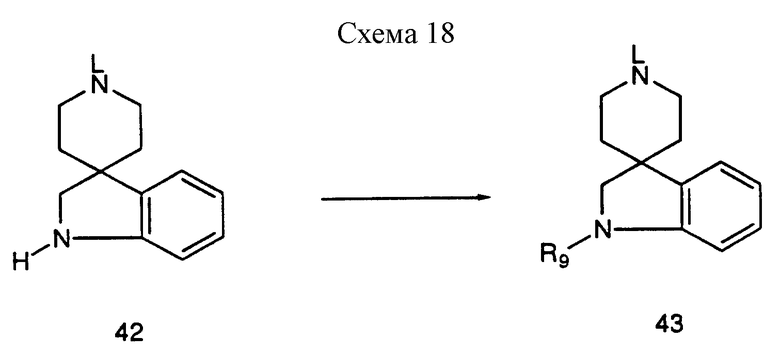
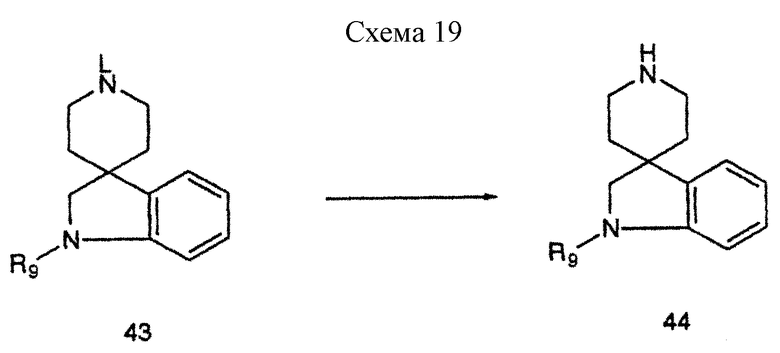
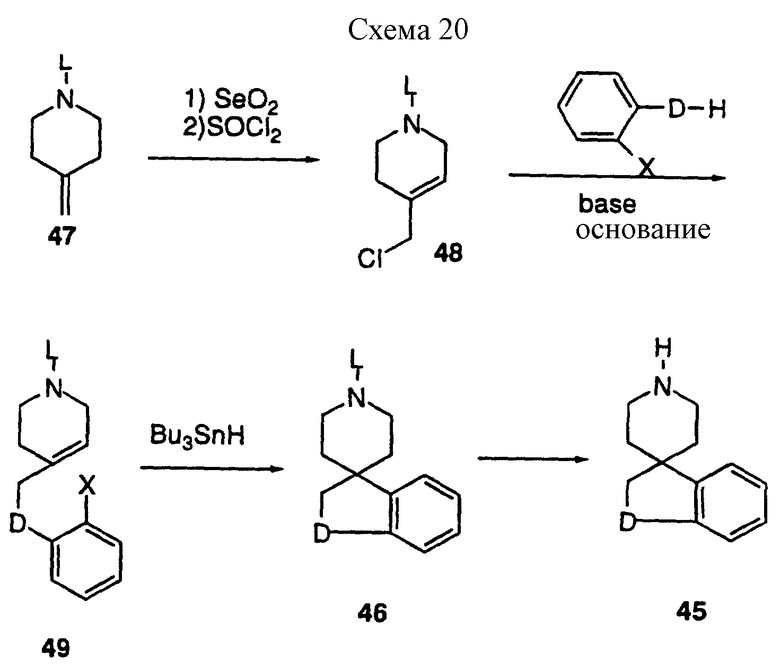
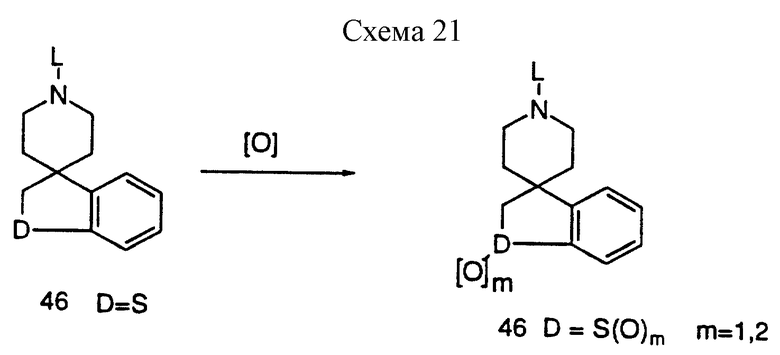
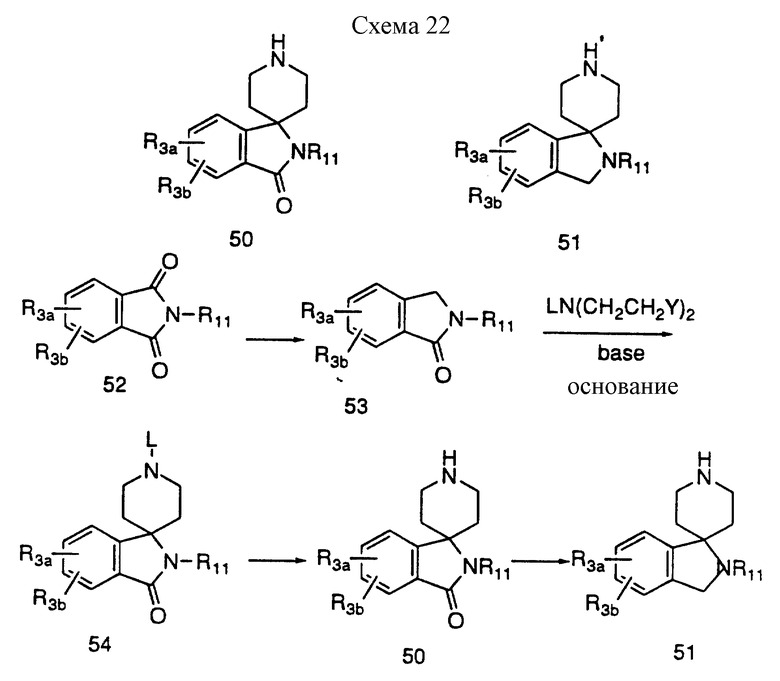
Предложены спиропиперидины формулы I или II, где R1 представляет арил-(С1-С6 алкил), С3-С7-циклоалкил-(С1-С6-алкил), арил-(С0-С5 алкил)-К-(С1-С6-алкил), С3-С7-циклоалкил-(С0-С5 алкил)-К-(С1-С5-алкил), где К представляет O, S(O)m, арил обозначает фенил, пиридил и индолил, которые могут быть замещены 1-2 атомами галогена; R2 является водородом, R3a и R3b являются водородом, атомом галогена, С1-С5 алкилом; R4 и R5 являются водородом, С1-С6-алкилом, замещенным 1-3 гидроксигруппами; R6 является водородом; А представляет группу -(СН2)x-С(R7)(R7a)-, где x = 0 или 1, R7 и R7a представляют собой С1-С6-алкил; причем один из В, D, Е, F представляет C(R8)(R10), O, С = O, S(O)m, NR9, причем В, D, Е или F может отсутствовать, обеспечивая образование 5-, 6-членного кольца, R8 и R10 представляют водород, R9 представляет R'2; SO2R'2, C(O)R'2, C(O)OR'2, SO2(CH2)q-арил, где R'2 может быть замещен галогеном, С1-С4-алкилом и др., R'2 является водородом или С1-С4-алкилом; q=0, 1; m = 0, 1 или 2; n = 1; G, Н, I и J представляют атомы углерода или серы так, что по крайней мере один является гетероатомом и один из G, Н, I и J может необязательно отсутствовать, что обеспечивает образование 5-членного гетероциклического или 6-членного ароматического кольца, и его фармацевтические соли и индивидуальные диастереоизомеры. Способ получения соединения формулы I или II включает взаимодействие соединения 4 или 4а с соединением формулы НООС-А-NR4R5 или НООС-А-N(R4)L, где L представляет защитную группу, которую затем удаляют (если она присутствует). Способ получения соединения формулы I или II путем взаимодействия соединения формулы (2) или (2а) с соединением формулы (12) или (12а), где L представляет защитную группу, которую затем удаляют. Способ повышения содержания эндогенного гормона роста у людей и животных, фармацевтическая композиция для повышения эндогенного продуцирования или выделения гормона роста у человека или животного. 5 с. и 6 з.п. ф-лы, 1 табл. 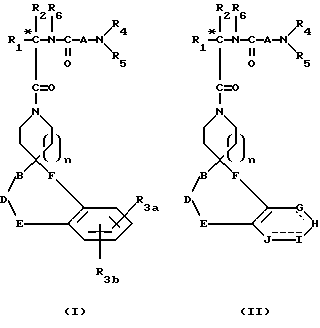
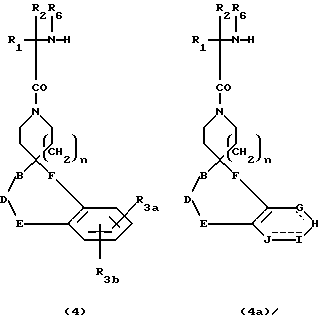
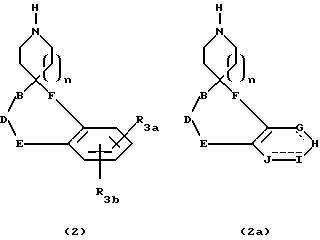
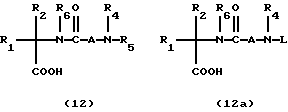

или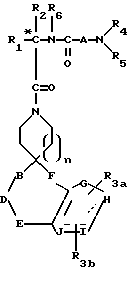
где R1 представляет арил(С1-С6-алкил), С3-С7циклоалкил(С1-С6-алкил), арил(С0-С5алкил)-К-(С1-С5-алкил), С3-С7-циклоалкил(С0-С5алкил)-К-(С1-С5-алкил), где К представляет О, S(O)m, где арильная группа представляет собой фенил, пиридил и индолил, и арильная группа может быть замещена 1-2 атомами галогена;
R2 представляет водород;
R3a и R3b независимо представляет водород, галоид, C1-С5-алкил;
R4 и R5 независимо представляют водород, замещенный C1-С6-алкил, где заместителями могут быть 1-3 гидрокси;
R6 представляет водород;
А представляет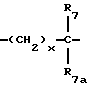
где x равно 0 или 1;
R7 и R7a независимо представляют С1-С6-алкил;
В, D, Е и F независимо представляют С(R8)(R10), О, С=O, S(O)m или NR9 так, что один или два из В, D, Е или F может необязательно отсутствовать, обеспечивая образование 5-, 6- или 7-членного кольца, и при условии, что В, D, Е или F могут представлять C(R8)(R10), или С=O только если одна из оставшихся В, D, Е или F групп представляет одновременно О, S(O)m или NR9;
R8 и R10 независимо представляют водород;
R2 представляет R'2, SO2-R'2, SO2(CH2)qарил, С(О)R'2, C(O)OR'2 и R'2 может необязательно быть дополнительно замещен OR''2, 1-5 галоидом, 1-3 С1-С4алкилом, C(O)-OR''2 и N(R''2)С(O)NR''2R''2 или SO2(CH2)qCONH(CH2)w-NHC(O)R11, w равно 2-6, а R11 представляет арил, замещенный OR''2, галогенном, азидо или нитро, R'2 и R''2 являются водородом или С1-С4алкилом,
q = 0; 1;
m = 0, 1 или 2;
n = 1;
G, H, I и J представляют атомы углерода или серы так что, по крайней мере, один является гетероатомом и один из G, H, I и J может необязательно отсутствовать, что обеспечивает образование 5-членного гетероциклического ароматического кольца или 6-членного ароматического кольца,
и его фармацевтически приемлемые соли и индивидуальные диастереоизомеры.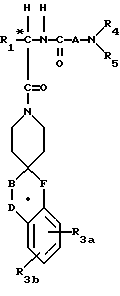
где R1 представляет арил(С1-С4-алкил), С3-С6-циклоалкил(С1-C4алкил), арил(С0-С5алкил)-К-(С1-С4-алкил) или С3-С7-циклоалкил (С0-С5алкил)-К-(С1-С4-алкил), где К представляет О, S(O)m,
R3a и R3b независимо представляет водород, галоид, C1-С4алкил;
R4 и R5 независимо представляют водород, замещенный С1-С6-алкил, где заместителями могут быть 1-2 гидрокси;
А представляет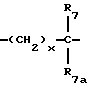
где х равно 0 или 1;
R7 и R7a независимо представляют C1-С4-алкил,
В, D и F независимо представляют C(R8)(R10), О, С=O, S(O)m или NR9 так, что один из В, D или F может необязательно отсутствовать, что обеспечивает образование 5- или 6-членного кольца, и при условии, что В, D или F представляют С(R8)(R10), или С=O, если одна из оставшихся В, D или F групп представляет одновременно О, S(O)m или NR9;
R8 и R10 независимо представляют водород;
R9 представляет R'2, SO2R'2, C(O)R'2, SO2(СН2)qарил, где R'2 может необязательно быть замещен OR''2, 1-2 атомами галогена, 1-2 C1-С4алкилом, С(O)-OR''2 и -N(R''2)C(O)NR''2R''2, SO2(CH2)wCONH(CH2)w-NHC(O)R11, w равно 1-6, а R11 представляет арил, замещенный OR''2, галогеном, азидо, R'2 и R''2 являются водородом или C1-С4-алкилом;
m равно 0, 1 или 2;
q равно 1;
арил представляет фенил, пиридил, индолил,
и его фармацевтически приемлемые соли и индивидуальные диастереоизомеры.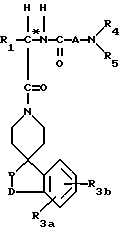
где R1 представляет арил(C1-С4-алкил), C5-С6-циклоалкил(C1-С4-алкил),
арил(С0-С5-алкил)-К-(С1-С2-алкил) или С3-C6-циклоалкил(С0-С2-алкил)-К-(С1-С2-алкил), где К представляет О или S(O)m, а арил может быть дополнительно замещен 1-2 галогеном;
R3a и R3b независимо представляет водород, галоид, С1-С4-алкил;
R4 и R5 независимо представляют водород, замещенный С1-С4-алкил, где заместителями могут быть 1-2 гидрокси;
А представляет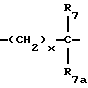
где х = 0 или 1;
R7 и R7a независимо представляют С1-С3-алкил;
В и D независимо представляют С(R8)(R10), С=0, О, S(O)m или NR9, при условии, что один из В и D может быть C(R8)(R10) или C=0, только если другой из В и D представляет О, S(O)m, или NR9;
R8 и R10 независимо представляют водород, '
R9 представляет C(O)R'2, SO2R'2, где R'2 может быть необязательно замещен OR''2, C(O)OR''2 и N(R''2)C(O)NR''2R''2, 1-2 атомами галогена, 1-2 С1-С4алкилом или SO2(CH2)qCONH(CH2)w-NHC(O)R11, q равно 2-6, а R11 может необязательно представлять арил, замещенный OR''2, галогеном, азидо;
R'2 и R''2 являются водородом или С1-С4-алкилом;
m = 0, 1 или 2;
q = 0; 1;
арил представляет фенил, пиридил, индолил,
и его фармацевтически приемлемые соли и индивидуальные диастересизомеры.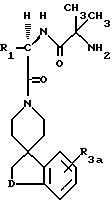
где R1 выбирают из:
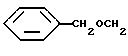
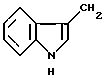
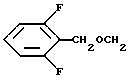
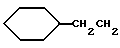
R3a представляет водород или фтор;
D представляет О, S, S(O)m, N(R''2), NSO2(R''2), NSO2(СН2)t-арил, NC(О)(R''2), NSO2(CH2)qOH, NSO2(CH2)qCOOR''2,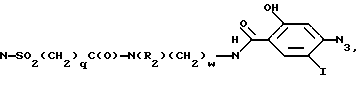
арил представляет фенил или пиридил;
R''2 представляет водород или С1-С4-алкил;
m = 1 или 2;
t = 1;
q = 1;
w = 2-6,
и его фармацевтически приемлемые соли и индивидуальные диастереоизомеры.
N-[1(R)-[(1,2-дигидро-1-метансульфонилспиро[3Н-индол-3,4'-пиперидин] -1'-ил)кapбонил]-2-(1Н-индол-3-ил)этил]-2-амино-2-метилпропанамид,
N-[1(R)-[(1,2-дигидро-1-метанкарбонилспиро[3Н-индол-3,4'-пиперидин] -1-ил)карбонил]-2-(1Н-индол-3-ил)этил]-2-амино-2-метилпропанамид,
N-[1(R)-[(1,2-дигидро-1-бензолсульфонилспиро[3Н-индол-З, 4'-пиперидин]-1'-ил)карбонил]-2-(1Н-индол-3-ил)этил-2-амино-2-метилпропанамид,
N-[1(R)-[(3,4-дигидро-спиро[2Н-1-бензопиран-2,4'-пиперидин] -1'-ил)карбонил]-2-(1Н-индол-3-ил)этил]-2-амино-2-метилпропанамид,
N-[1(R)-[(2-aцeтил-1,2,3,4-тeтparидpocпиpo[изoxинолин-4,4'-пиперидин] -1'-ил)карбонил]-2-(индол-3-ил)этил]-2-амино-2-метилпропанамид,
N-[1(R)-[(1,2-дигидро-1-метансульфонилспиро[3Н-индол-3,4'-пиперидин] -1'-ил)карбонил]-2-(фенилметилокси)-этил-2-амино-2-метилпропанамид,
N-[1(R)-[(1,2-дигидро-1-метансульфонилспиро[3H-индол-3,4'-пиперидин] -1'-ил)карбонил] -2-(фенилметилокси)-этил-2-амино-2-метилпропанамидмезилат, соль,
N-[1(R)-[(1,2-дигидро-1-метансульфонилспиро[3Н-индол-3,4'-пипepидин] -1'-ил)карбонил] -2-(2', 6'-дифторфенилметилокси)этил] -2-амино-2-метилпропанамид,
N-[1(R)-[(1,2-дигидро-1-метансульфонил-5-фтороспиро[3Н-индол-3,4'-пиперидин]-1'-ил)карбонил]-2-(фенилметилокси)этил]-2-амино-2-метилпропанамид,
N-[1(S)-[1,2-дигидро-1-метансульфонилспиро[3Н-индол-3,4'-пиперидин] -1'-ил)карбонил]-2-(фенилметилтио)этил]-2-амино-2-метилпропанамид,
N-[1(R)-[(1,2-дигидро-1-метансульфонилспиро[3Н-индол-3,4'-пиперидин] -1'-ил)карбонил]-3-фeнилпpoпил]-2-амино-2-метилпропанамид,
N-[1(R)-[(1,2-дигидpo-1-мeтaнcульфoнилcпиpo[3H-индoл-3,4'-пиперидин] -1'-ил)карбонил-3-циклогексилпропил]-2-амино-2-метилпропанамид,
N-[1(R)-[(1,2-дигидро-1-метансульфонилспиро[3Н-индол-3,4'-пиперидин] -1'-ил)карбонил]-4-фенилбутил-2-амино-2-метилпропанамид,
и их фармацевтически приемлемые соли.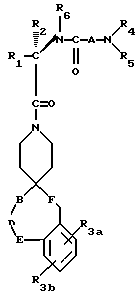
где R1, R2, R3a, R3b, R4, R5, R6, А, В, D, Е, F имеют указанные в п.1 значения.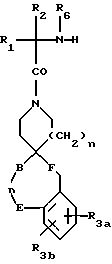
или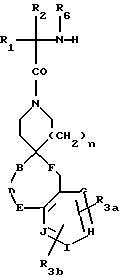
с соединением формулы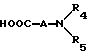
или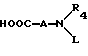
где R1, R2, R3a, R3b, R4, R5, R6, А, В, D, Е, F, G, H, I, J и n имеют указанные в п.1 значения;
L представляет защитную группу, которую затем удаляют (если она присутствует),
и, при желании, получают соли.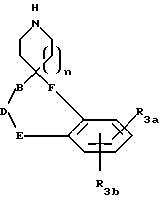
или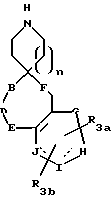
с соединением формулы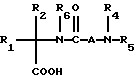
или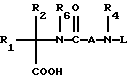
где R1, R2, R3a, R3b, R4, R5, R6, А, В, D, Е, F, G, H, I, J и n имеют указанные в п.1 значения;
L представляет защитную группу, которую затем удаляют (если она присутствует),
и, при желании, получают соли.
Приоритет по пунктам:
11.12.1992 по пп.1 - 11 (все значения радикалов, кроме NR9);
03.11.1993 по пп.1 - 11 (где NR9).
| Способ получения перхлоратов 2-стирил-4н-1,3-бензоксазин-4-ония | 1973 |
|
SU513974A1 |
| Способ преобразования малых постоянных напряжений в импульсы постоянного тока | 1960 |
|
SU144230A1 |
| Способ получения производных арилкарбоксамидов или их солей | 1976 |
|
SU694073A3 |
| Способ получения производных окса-1-диаза-3,8-спиро-(4,5)-декана | 1972 |
|
SU474150A3 |
| Справочник практического врача/Под ред | |||
| А.И.Воробьева | |||
| - М.: Медицина, 1990, с.278. | |||

