Изобретение касается новых классов производных (5- арилизоксазол-4-ил) замещенной 2-аминокарбоновой кислоты, которые являются рецепторами лигандов аминокислот (ЕАА), полезных при лечении церебральной ишемии, болезни Хантингтона, эпилептических нарушений, болезни Паркинсона, болезни Альцгеймера, шизофрении, боли, депрессии и тревожных состояний.
Предпосылки настоящего изобретения
В настоящее время в результате усиленного изучения возбуждающего механизма центральной нервной системы (ЦНС) на протяжении трех последних десятилетий пришли к общему мнению, что (S)-глютамат (Glu) является главным нейромедиатором ЕАА в ЦНС (Lodge D. Excitatory Amino Acids in Health and Disease. J. Wiley & Sons: Chichester, 1988; Wheal H., Thompson A. - Excitory Amino Acids and Synaptic Transmission. Academic Press: London, 1991; Meldrum B.S. Excitatory Amino Acid Antagonists. Blackwell Sci. Pubi.: Oxford, 1991; Krogs-gaard-Larsen P. , Hansen J.J. Excitory Amino Acid Receptors: Design of Agonist and Antagonist. E. Horwood: Chichester, 1992). Glu-управляемая нейротрансмиссия осуществляется большим числом рецепторов, классифицированных по меньшей мере в 5 гетерогенных семей и названных NMDA, АМРА, каиновой кислотой, метаботропным и 1-АР-4 классами рецепторов (Monaghan D.T., et al. Ann. Rev. Pharmacol. Toxicol. 1989, 29, 365-402; Watkins J.C., Krogsgaard-Larsen P. , Honore T. Trends Pharmacol. Sci. 1990, II. 25-33; Simon P.P., Excitatory Amino Acids. Thieme Med. Publ.: New York, 1992).
Существует весьма убедительное доказательство, подтверждающее точку зрения о том, что избыточное возбуждение, переносимое рецепторами ЕАА ("токсичность возбуждения") представляет собой фактор, имеющий особую важность для церебральной ишемии, вызванной ударом, повреждениями головы, асфиксией, субарахноидальным кровотечением, остановкой сердца и другими ситуациями. (Lodge D., 1988, та же ссылка; Meldrum B.S., 1991, та же ссылка). В модели на животных показано, что эти нарушения, вызванными разными ишемическими условиями, могут быть ослаблены введением Glu-антагонистов. Итак, хотя относительная важность указанных разных классов рецепторов ЕАА в феномене поддержания ишемического инсульта неясна, в целом достигнуто общее мнение, что в таких условиях антагонисты рецепторов ЕАА представляют собой мощные терапевтические средства.
Накопление доказательств, полученных от разных направлений нейрохимического и фармакологического исследования, дает возможность предположить, что нарушение механизмов рецепторов ЕАА, включая, возможно, и "токсичность возбуждения" играет роль при болезни Хантингтона (Young А.В., et al., Science, 1988, 241, 981-983), эпилептических нарушениях (Krogsgaard-Larsen P, Hansen J. J. , 1992, там же), болезни Паркинсона (Klockgether Т. Turski L. Trends Neurosci. 1989, 12, 285-286) и болезни Альцгеймера (Greenamyre J.T., Maragos W.F. Cerebrovasc. brain. Metab. Rev., 1993, 5, 61-94; Francis P.Т. et al. J. Neurochem. 1993, 60, 1589-1604).
Далее, центральные рецепторы ЕАА могут быть вовлечены в синаптический механизм, лежащий в основе шизофрении (Reynolds G.P. Trends. Pharmacol. Sci. , 1992, 13. 116-121), болей и тревожных состояний (Drejer J. In: Excitatory Amino Acid Receptors: Design of Agonists and Antagonists (Eds. Krogsgaard-Larsen P., Hansen J.J.) E. Hornwood: Chichester 1992, pp. 352-375) и депрессии (Trullas R., Skolnick P. Eur. J. Pharmacol. 1990, 185, 1-10; Trullas et al. Eur. J. Pharmacol. 1991, 203, 379-385). Итак, сниженная функция рецепторов ЕАА (гипоактивность ЕАА), как оказалось, играет роль, например, при шизофрении (Deutsch S.I, et al., Clin. Neuropharmacol. 1989, 12, 1-3) и некоторых клинических симптомах, выявляемых в болезни Альцгеймера (Greenamyre J. T. ei al. Prog. Neuro-Psychopharniacol. & Biol. Psychit. 1988, 12, 421-430). Возможно, что "токсичность возбуждения", так же, как гипоактивность ЕАА, включены в сложный механизм, связанный с болезнью Альцгеймера (Greenamyre J. T.; 1988, там же; Greenamyre J.T., Maragos W.F., 1993, там же).
Поэтому считается, что лиганды рецепторов ЕАА полезны для лечения церебральной ишемии, болезни Хантингтона, эпилептических нарушений, болезни Паркинсона, болезни Альцгеймера, тревожных состояний, шизофрении, депрессии и болей.
Большинство испытанных таким образом агонистов рецепторов ЕАА в большей или меньшей степени демонстрируют выраженную нейротоксичность в моделируемых системах, а поэтому клиническое использование таких соединений может быть ограниченным (Carlsson М., Carlsson A., Trends. Neurosci., 1990, 13, 272-276; Willetts J., Balster R.L., Leander J.D., Trends Pharmacol. Sci., 1990, II, 423-428).
С другой стороны, частичные агонисты ЕАА, демонстрирующие необходимый баланс между агонизмом и антагонизмом, представляют значительный терапевтический интерес (Greenamyre J.t., 1988, там же; Christensen 1.Т., et al.. Drug Des. Del. 1989, 5, 57-71; Francis P.Т., et al., J. Neurochem. 1993, 60, 1589-1604). Частичные агонисты за счет своего профиля антагонистов ЕАА могут демонстрировать терапевтически полезную нейрозащиту, а в некоторых случаях являются в достаточной степени агонистическими для предотвращения общей блокады нейротрансмиссии, передаваемой особыми рецепторами ЕАА.
Было обнаружено, что АТРА, представляющий собой 5-трет- бутильный аналог АМРА ((RS)-2-амино-3-(3-гидрокси-5- метилизоксазол-4-ил)пропионовой кислоты), обладает системной активностью, в то время как его нейротоксическое воздействие на животных не отмечалось (Ornstein P.L., et al., J. Med. Chem. 1993, 36, 2046-2048; Lauridsen J., Honore Т., Krogsgaard-Larsen P., J. Med. Chem., 1985, 28, 668-672).
Было обнаружено, что подобно самому АМРА, ряд его моно- и бициклических аналогов демонстрируют выборочное агонистическое влияние на рецепторы АМРА (Hansen J.J., Krogsgaard-Larsen P., Med. Res. Rev. 1990, 10, 55-94; Krogsgaard-Larsen P. , Hansen J.J., 1992, там же). Один из этих аналогов, (RS)-2-амино-3- (3-гидрокси-5-фенилизоксазол-4-ил)пропионовая кислота (АРРА), в котором метильная группа АМРА замещена фенильной группой, имеет слабый, но характерный агонистический профиль (Christensen I.T., et а1., 1989, там же).
Как видно из приведенного выше доказательства, не обладающие нейротоксичностью ЦНС-активные лиганды рецепторов ЕАА, хорошо проникающие в ЦНС, крайне желательны для лечения различных указанных выше заболеваний, и поэтому объектом настоящего изобретения является обеспечение таких новых лекарственных средств.
Резюме изобретения
В настоящее время установлено, что новый класс производных (5- арилизоксазол-4-ил) замещенных 2-карбоновых кислот представляет собой сильные лиганды рецепторов ЕАА.
Поэтому настоящее изобретение касается нового класса соединений (5-арилизоксазол-4-ил)- или (5 арилизотиазол-4- ил)замещенных 2-карбоновых кислот, имеющих общую формулу I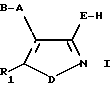
в которой A представляет собой связь или пространственную группу, выбранную из C1-6 алкилена, C2-6 алкенилена или C2-6 алкинилена, или циклоалкилена; В выбирают из группы -CH(NR'R'')-COOH, где R' и R'' независимо друг от друга представляют собой водород или C1-6 алкил, или группу формулы (II)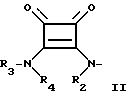
в которой R2, R3 и R4 независимо выбирают из группы, содержащей а) водород, C1-6 алкил, C2-6 алкенил, C2-6 алкинил, циклоалк(ен)ил, циклоалк(ен)ил-C1-6 алк(ен/ин)ил, фенил-C1-6 алкил, тиенил-C1-6 алкил, и b) C1-6 алкил, C2-6 алкенил и C2-6 алкинил, в которой один или более атомов углерода заменены N, О и/или S; или
R3 и R4 связаны так, что они образуют C2-C6 алкилен, C2-C6 алкенилен или C2-C6 алкинилен группу; или
R4 и R2 связаны так, что они образуют C1-C3 алкилен, C2-C3 алкенилен или C2-C3 алкинилен группу, необязательно моно- или бизамещенную гидрокси, или метилом, или CH2-O-CH2;
E представляет собой О, S, COO, (CH2)n-COO, O-(CH2)n-COO или S-(CH2)n-COO (где n - целое число от 1 до 6, 5-тетразолил, 5-тетразолил-C1-6 алкил, 3-гидроксиизоксазолил или 3-гидроксиизоксазолил-C1-6 алкил группу;
D представляет собой О или S; и
R1 представляет собой арильную или гетероарильную группу, или арильную или гетероарильную группу, замещенную одним или большим числом заместителей, выбранных из галогена, C1-6 алкила, C1-6 алкокси, гидрокси, C1-6 алкилтио, C1-6 алкилсульфонила, C1-6 алкиламино или ди-(C1-6 алкил)амино, циано, нитро, трифторметила или трифторметилтио;
при условии, что если А представляет собой метилен, B представляет собой группу -CH(NH2)-COOH, E представляет собой О, D представляет собой О, a R1 - фенил, или фенил, замещенный галогеном, или метокси, то указанное соединение должно находиться в форме чистого энантиомера.
Другой аспект настоящего изобретения относится к методу получения новых соединений формулы I.
Еще один аспект настоящего изобретения относится к фармацевтическому составу, содержащему новое соединение формулы I вместе с фармацевтически приемлемым носителем или разбавителем.
Следующий аспект настоящего изобретения касается использования соединения формулы I для лечения церебральной ишемии, болезни Хантингтона, эпилептических нарушений, болезни Паркинсона, болезни Альцгеймера, шизофрении, боли, депрессии и тревожных состояний.
Было обнаружено, что некоторые соединения настоящего изобретения являются селективными лигандами рецепторов АМРА in vitro, со сродством при низких микромолекулярных концентрациях, в то время как другие соединения, как было установлено, селективно связаны с рецепторами NMDA in vitro. Другие соединения настоящего изобретения, как было обнаружено, имеют сродство как к рецепторам АМРА, так и к рецепторам NMDA in vitro. Более того, установлено, что некоторые соединения настоящего изобретения являются агонистами, в то время как другие - антагонистами. Таким образом, соединения настоящего изобретения полезны для лечения церебральной ишемии, болезни Хантингтона, эпилептических нарушений, болезни Паркинсона, болезни Альцгеймера, шизофрении, боли, депрессии и тревожных состояний.
Детальное описание изобретения
Некоторые соединения общей формулы I могут существовать как оптические изомеры, и эти оптические изомеры также охвачены настоящим изобретением.
В общей формуле I термин "C1-6 алкил" применяют для обозначения разветвленной или неразветвленной алкил группы, содержащей от 1 до 6 атомов углерода, включая такие группы, как метил, этил, 1-пропил, 2-пропил, 1-бутил, 2-бутил, 2-метил-2- пропил и т.д. Аналогично, терминами "C2-6 алкенил" и "C2-6 алкинил" обозначены разветвленные или неразветвленные группы, содержащие от 2 до 6 атомов углерода, а термины "C1-6 алкилен", "C2-6 алкенилен" и "C2-6 алкинилен" обозначают такие разветвленные или неразветвленные бивалентные группы. Термин "циклоалкил" обозначает такую группу с 3-7 атомами углерода.
Термин "алк(ен/ин)ил" обозначает, что указанная группа может быть алкил, алкенил или алкинил группой.
Термин "связь" (используемый для А) обозначает, что B может быть присоединен непосредственно в положение 4 изоксазольного цикла.
"Галоген" обозначает фтор, хлор, бром или иод.
Термин "арил" используют для обозначения карбоциклической ароматической моноциклической или слившейся бициклической группы или бифенильной группы, а термин "гетероарил" используют для обозначения ароматической моноциклической или бициклической группы, содержащей по меньшей мере один гетероатом. Примерами таких групп являются 5-членные ароматические гетероарильные группы (имеющие 1 - 4 гетероатома, выбранных из N, О и S), такие, как тиенил, фурил, пиролил, оксазолил, изоксазолил, тиазолил, изотиазолил, пиразолил, имидазолил, триазолил, оксадиазолил, тиадиазолил и тетразолил. Кроме того, примерами их являются бензотиенил, бензофуранил, индолил, фенил, пиридил, пиримидинил, пиридазинил, пиразинил, нафтил, хинолил, хиназолинил, хиноксалинил и циннолинил.
Некоторые соединения общей формулы I могут существовать как фармацевтически приемлемые соли, которые также охватываются настоящим изобретением.
Указанными солями соединений общей формулы I являются соли, образованные нетоксичными органическими солями, например, малеиновой, фумаровой, бензойной, аскорбиновой, щавелевой, винной, молочной и яблочной кислот или неорганических кислот, например, хлористоводородной, бромистоводородной, серной, фосфорной и азотной кислот; или они могут быть солями неорганических оснований, таких, как соли щелочных металлов, например, соли натрия, калия или лития, соли щелочноземельных металлов, соли кальция или магния или соли аммония или соли органических оснований.
В формуле I A предпочтительно представляет собой связь или C1-3 алкилен.
В предпочтительно представляет собой -CH(NR'R'')-COOH группу, в которой R' и R'' - водород или группа формулы II, где R2, R3 и R4 представляют собой водород или низший алкил, или R4 и R2 связаны, образуя C1-C3 алкилен группу. Более предпочтительно, чтобы B представлял собой -CH(NH2)-COOH или группу формулы II, в которой каждый из R2, R3 и R4 - водород.
Предпочтительно, чтобы E представлял собой О, COO, -O(CH2)n- COO (n = 1, 2 или 3) или тетразолил, a D - кислород.
Особенно подходящими группами R1 являются тиенил, замещенный тиенил, оксазолил, изоксазолил, тиазолил, изотиазолил, пиразолил, имидазолил, оксадиазолил, замещенный оксазолил, тиадиазолил, тетразолил, триазолил, пиридил, фенил, бифенил и нафтил. Предпочтительны группы 2-тиенил, 3-тиенил, фенил, 2-пиридил, 4-пиридил, 2-тиенил и фенил, замещенные галогеном или метилом.
В предпочтительном воплощении настоящего изобретения А представляет собой связь или C1-C3 алкилен, В представляет собой - CH(NH2)-COOH или группу формулы II, где каждый из R2, R3 и R4 представляет, собой водород, E и D оба представляют собой кислород, а R1 - 2-пиридил, 4-пиридил, тиенил, фенил, замещенный тиенил или замещенный фенил.
Согласно настоящему изобретению новые соединения формулы I получают методом, включающим:
а) для получения соединения формулы I, в которой B представляет собой -CH(NR'R'')-COOH группу, где R' и R''' были определены выше, - снятие защиты с соединения общей формулы III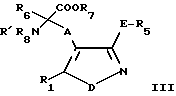
в которой R1, R', A, D и E были определены выше, R5, R7 и R8 - защитные группы, a R6 - водород или защитная группа;
b) для получения соединения формулы I, в которой B представляет собой -CH(NR'R'')-COOH группу, где R' и R'' оба представляют собой водород, - снятие защиты с соединения общей формулы IV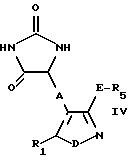
в которой R1, R5, А, D и E были определены выше;
с) для получения соединения формулы I, в которой B представляет собой группу формулы II, - реакцию присоединения- элиминирования соединения общей формулы V с соединением общей формулы VI
в этих формулах R1-R4, A, D и E были определены выше, a R'5 представляет собой водород или защитную группу;
d) для получения соединения формулы I, в которой В представляет собой группу формулы II, где R4 и R2 связаны между собой, образуя C1-3 алкилен, C2-C3 алкенилен или C2-C3 алкинилен группу, необязательно моно- или бизамещенную гидрокси или метилом, - взаимодействие соединения формулы VII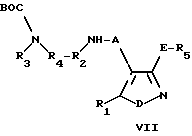
в которой R1, R3, A, D и E были определены выше, R4 и R2 связаны вместе, образуя указанную выше группу, а ВОС представляет собой трет-бутоксикарбонил; с 3,4-диэтокси-3-цикло-бутен-1,2-дионом при последующем замыкании цикла и снятии защиты;
е) для получения соединения формулы I, в которой B представляет собой группу формулы II, а один или более из R2-R4 отличен от водорода - алкилирование соединения общей формулы VIII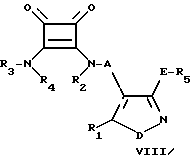
в которой R1, R2, R3, R4, A, D и E были определены выше, притом, что по крайней мере один из R2-R4 представляет собой водород, R'5 - водород или защитную группу.
В методе по настоящему изобретению предпочтительными защитными группами являются R5 и R'5 - низший алкил, бензил или бензолсульфонил группа; R6 - низший алкоксикарбонил, R7 - низший алкил и R8 - низший алкилкарбонил.
Одноступенчатое снятие защиты согласно методу а) проводят обработкой соединения формулы III подходящей водной кислотой, удобно использовать водный раствор 48% HBr, насыщенный раствор HBr в уксусной кислоте или 2-12 н. водный раствор HCl. Снятие защиты можно проводить в несколько стадий, последовательно используя водные кислоты и водные основания; удобно его осуществлять последовательно в водной кислоте, такой, как 1-12 н. HCl, водном основании, таком, как 1-8 н. NaOH и в водной кислоте, такой, как 1-12 н. HCl; или последовательно в водном основании, таком, как 1-8 н. NaOH, и в водной кислоте, такой, как 1-12 н. HCl. В случае, когда R5 представляет собой бензил, снятие защиты с E-группы завершают каталитическим гидрированием, которое удобно проводить или до или после снятия защиты с α- аминокислоты, используя в качестве катализатора палладий.
Исходные вещества для соединения общей формулы III удобно получать так, как это описано в работе Christensen I.T., et al.. Drug Design and Delivery, 1989, 5, 57-71 и в работе Christensen S.B., et al., Acta Chem. Scand., 1978, B32 27-30. Исходные вещества для соединения общей формулы III, в которой E представляет собой COO, (CH2)n-COO, O-(CH2)n-COO, S-(CH2)n-COO (при n=1-6), 5- тетразолил-C1-6 алкил группу или 3-гидроксиизоксазолил-C1-6 алкил группу, удобно получать так, как это описано в работах: Krogsga-ard-Larsen P., et al. , J. Med. Chem., 1991, 34, 123-130; Madsen U., Bio. Med. Chem. Lett., 1993, 8, 1649-1654; Madsen U., Wong E., J. Med. Chem. 1992, 35, 107-111.
Исходные вещества для соединения общей формулы III, в которой R1 представляет собой гетероарильную группу, нестойкую в кислых условиях, удобно получать из (3-алкокси-4-метилизоксазол-5-ил) карбоновой кислоты путем образования гетероарила в положении 5 бромированием 4-метилизоксазольной группы с последующим алкилированием предшественником аминокислоты, например диэтилацетамидомалонатом. Алкил, защищенный 3-гидроксиизоксазольной группой, может быть повторно защищен или может не быть защищен повторно подходящей защитной группой, подобной бензолсульфонильной группе, до, во время или после синтеза гетероарила в положении 5 изоксазола (3-алкокси-4- метилизоксазол-5-ил)карбоновую кислоту удобно получить из 3-алкокси-4,5-диметилизоксазола (синтезированного, как это описано в работе Hansen J.J., J. Chem. Soc. Perkin Trans., 1980, 1, 1826 - 1833) бромированием с последующим окислением 5-метилизоксазольной группы в соответствующее соединение 5-изоксазолкарбоновой кислоты.
В других случаях исходные вещества общей формулы III, в которой R1 представляет, например, пиридил группу, удобно получать модификацией метода, описанного в работе Tomita К., Ann. Sankyo Res. Lab., 1973, 25, 3-5. Защита 3-гидрокси группы в полученном 3-гидрокси-5-арилизоксазоле с последующим введением гидроксиметильной группы в положение 4 изоксазольного цикла приводит к промежуточным соединениям, которые затем легко превращаются в исходные вещества формулы III.
В случае b) одноступенчатое снятие защиты проводят обработкой соединения формулы IV подходящей водной кислотой или основанием, это удобно осуществлять в 2-8 н. водной соляной кислоте. Снятие защиты можно также провести последовательно по стадиям, используя водные кислоты и водные основания, как это было указано выше для метода а). Цикл гидантоина также можно расщепить с помощью водного раствора Ba(OH)2, водной 10-70% серной кислоты или использованием энзимов, таких, как гидантоинаса. Указанное расщепление цикла гидантоина может быть осуществлено или до, или после снятия защиты с E-группы.
Циклы гидантоина в соединениях общей формулы IV легко образуются согласно методам, описанным Ware Е., Chem Rev., 1950, 46, 403-470. Расщепление цикла гидантоина удобно проводить по аналогии с методами, описанными Curry К. , et а1., J. med. Chem., 1988, 31, 864-867, Farrington G.K., et al., J. Mod. Chem. , 1987, 30. 2062-2067, Grunewald G.L., et al., J. Med. Chem., 1980, 23, 754-758, Hirol К., et al., Chem. Pharm. Bull., 1968, 16. 444-447, Stark G.R., et al, J. Biol. Chem. 1963, 238, 214-226.
Исходные вещества для получения соединений формулы IV можно получить по аналогии с методами, описанными Madsen U., Eur. J. Med. Chem., 1993, 28, 791-800.
Если R5 представляет собой бензил, то снятие защиты с E-группы легко осуществить гидрированием, используя в качестве катализатора палладий.
Реакцию присоединения-элиминирования согласно методу с) удобно проводить в протоном органическом растворителе, таком, как спирт, предпочтительно в присутствии подходящего неорганического основания, такого, как NaOH, при комнатной температуре. Промежуточные соединения формулы VI можно получить методами, описанными в работах Cohen S., et al., J. Amer. Chem. Soc., 1966, 88. 1533-1536, EP-A2-0496561 или Kinney W.A. et al., J. Med. Chem., 1992, 35. 4720-4726.
Промежуточное соединение общей формулы V легко получают синтезом первичных аминов по Габриелю, как это описано Sheehan J.C. et al., J. Amer. Chem. Soc. , 1950, 72, 2786-88. Исходные для этого синтеза алкилгалогениды удобно получить, как это описано, используя в качестве исходных веществ, применяемые в методе а).
Снятие защиты удобно проводить, используя водную кислоту или водное основание, предпочтительно 0.5-8 н. HCl или 0.5-8 н. NaOH или при комнатной температуре, или при повышенных температурах. Если R'5 представляет собой бензил, то снятие защиты может быть осуществлено гидрированием с использованием в качестве катализатора палладия.
В методе d) взаимодействие и последующие замыкание цикла и снятие защиты проводят так, как это описано Kinney et al., EP-A2- 0496561.
Исходные вещества для получения соединения формулы VII могут быть получены взаимодействием, например, 4-бромметилизоксазола, полученного так, как это описано для исходных веществ в методе а), с моно-ВОС-защищенным алкилендиамином (EP-A2-0496561).
Алкилирование соединений общей формулы VIII согласно методу е) удобно осуществлять в инертном органическом растворителе, таком, как подходящий спирт, кетон или диметилформамид, предпочтительно в присутствии подходящего основания, такого, как гидрид натрия, карбонат калия или триэтиламин, как описано Kinney W.A., ЕР-А2-0496561. Исходные вещества формулы VIII получены согласно методу с).
Если β- кетоэфиры используют в качестве исходных для получения 3-гидроксиизоксазолов, которые, в свою очередь, применяют как исходные для получения соединений III-V и VII-VIII, то они или доступны в коммерческом отношении, или же их можно получить методами, описанными в работах Cason J., et al., J. Org. Chem., 1953, 18, 1594-1600 и Hannick S.M., et al., J. Org. Chem., 1983, 48. 3833-3835. Соответствующие исходные изотиазолы можно получить согласно методу, описанному в ЕР-А1-0336555.
Повторное растворение соединений общей формулы I удобно проводить при образовании солей диастереизомеров, используя активные кислоты или основания, например 1-фенилэтиламин. В некоторых случаях это повторное растворение удобно проводить при образовании соединений диастереоизомеров с последующим разделением диастереоизомеров тонкослойной хроматографией или кристаллизацией.
Соли соединений настоящего изобретения легко получить методами, хорошо известными в этой области, например взаимодействием указанного соединения или с эквивалентным количеством кислоты или основания в смешиваемом с водой растворителе (таком, как ацетон или этанол) с последующим выделении соли при выпаривании и охлаждении, или с избытком кислоты или основания в растворителе, не смешиваемом с водой (таком, как диэтиловый эфир или хлороформ) при непосредственном выделении требуемой соли. Эти соли можно также получить классическими методом двойного разложения соответствующих солей.
Соединения общей формулы I и их фармацевтически приемлемые кислые дополнительные соли могут быть введены любым подходящим способом, например орально или парентерально, а указанные соединения могут находиться в любой, пригодной для такого введения форме, например в форме таблеток, капсул, порошков, сиропов или растворов или дисперсий для инъекций.
Эффективная дневная доза соединения общей формулы I или его фармацевтически приемлемой соли составляет от 10 мкг/кг до 50 мг/кг веса тела.
Все значения температуры плавления определяли на оборудовании Buchi SMP-20, и они не являются точными. Спектры 1H-ЯМР и 13C-ЯМР регистрировались на спектрофотометре Bruker 250 МГц (250.13 МГц для первого метода и 62.90 МГц для второго). В качестве внешнего стандарта, если иное не оговорено, применяли тетраметилсилан. Для соединений 13а, 13Ь, 14с и 14d, указанных в заглавиях, спектры 1H-ЯМР и 13C-ЯМР записывали на спектрофотометре Brucker 200 MГц (200.0 МГц для 1H-ЯМР и 50.3 МГц для 13C-ЯМР), при этом в качестве внешнего стандарта, если иное не оговорено, использовали тетраметилсилан.
Масс-спектры получали на системе Quattro MS-MS от VG Biotech, Fisons instruments, Manchester, GB. Эта система MS-MS была присоединена к модулярной системе высокоэффективной жидкостной хроматографии HP 1050. 20-50 мкл образца (0.1-0.05 мг/мл), растворенного в смеси ацетонитрил/вода/ концентрированный водный аммиак (25%) при объемном соотношении компонентов 25:25:1 вводили в электрораспылитель через пробоотборник при скорости тока 30 мкл/мин. Спектры получали при стандартных рабочих условиях для получения информации о молекулярных весах (MH+). Фон вычитали.
Энантиомерный избыток энатиомерных соединений определяли хиральной высокоэффективной жидкостной хроматографией на хиральной краун-эфирной колонке. Хроматографию проводили в 150 х 4 мм колонке Daicel Crownpak CR(-) или Crownpak CR(+) при элюировании при 15-40oC водной перхлорной кислотой/ метанолом (100-85%/0-15%) со скоростью 0.4-1.0 мл/мин. Использовали один из следующих приборов:
1) Насос Jusco 880-PU, инжектор Rheodyne 7125 и детектор Waters 480 UV, установленный на 210 нм, соединенный с Merck-Hitachi D-2000 Chromato-Integratot,
2) Насос Hitachi-Merch L-6200, автоматический пробоотборник Hitachi-Merch 655А-40 и детектор Hitachi-Merch L-4000 UV, установленный на 210 нм, соединенный с Hitachi-Merch D-2500.
Чистота энантиомеров вычислялась исходя из площади пиков.
Пример 1
(RS)-2-амино-3-[3-гидрокси-5-(2-тиенил)изоксазол-4- ил] пропионовая кислота, 1а
Смесь 2-бромтиофена (250.0 г, 1.53 мол) и CuCN (157.5 г, 1.76 мол) кипятили в колбе с обратным холодильником в N-метилпирролидоне (NMP) в течение 90 мин. Полученную смесь охладили до 100oC и вылили в горячий раствор NaCN в воде (150 г NaCN в 2.5 л воды). Смесь энергично перемешивали в течение 30 мин при 80oC, а потом отфильтровали в горячем виде. После охлаждения смесь экстрагировали диэтиловым эфиром (три раза по 2 л), а потом упарили в вакууме до 1.5 л. Органическую фазу промыли водой (750 мл) и водным насыщенным раствором NACl (750 мл). Органическую фазу высушили (MgSO4) и упаривали в вакууме до получения красно-фиолетового масла. Последнее перегоняли при пониженном давлении (1 мм Hg), в результате чего получили 2-тиофенкарбонитрил (105.3 г, 63%).
Смесь 2-тиофенкарбонитрила (25.0 г), активированного цинка (22.5 г) и CuBr2 (0.2 г, 0.9 ммол) в бензоле (350 мл) нагревали до температуры расплавления (83oC). В течение 60 мин при 83oC добавляли этил-2-бромпропионат (62.2 г) в бензоле (150 мл). Полученную смесь нагревали в течение 2 ч в колбе с обратным холодильником, а потом охладили до 0oC. В течение 60 мин добавляли 15% водную H2SO4 (400 мл) при температуре ниже 10oC, и полученную смесь перемешивали при 20oC в течение 20 ч. Смесь затем отфильтровали и полученные фазы разделили. Водную фазу экстрагировали диэтиловым эфиром (два раза по 500 мл), а собранные вместе органические фазы высушили (MgSO4) и выпарили в вакууме. После колоночной хроматографии (силикагель, элюент: этилацетат/н- гептан/метанол 4:4:1) был получен этил 2-метил-3-(2-тиенил)-3- оксопропионат в виде масла (35.0 г, 72%).
Смесь этил 2-метил-3-(2-тиенил)-3-оксопропионата (25.3 г) и NaOH (5.0 г, 0.12 мол) в смеси метанол/вода (10:1, 200 мл) охладили до - 30oC. Добавили охлажденный льдом (0oC) раствор NH2OH, HCl (16.6 г) и NaOH (10.0 г) в смеси метанол/вода (10: 1, 200 мл); полученный раствор перемешивали в течение 3 ч при - 30oC. Раствор выдерживали до достижения им температуры 5oC, а затем добавляли к концентрированной HCl (280 мл) в течение 45 мин при 80oC. Полученную смесь нагревали в колбе с обратным холодильником в течение 1 ч при 80oC. Выпарили метанол и добавили воду (250 мл). Полученный раствор охладили до 5oC, а образовавшиеся кристаллы собрали при фильтровании. Эти кристаллы растворили в CH2Cl2 (500 мл). Органическую фазу высушили (MgSO4) и выпарили в вакууме, при этом получили 4-метил-5-(2-тиенил)изоксазол-3-ол (12.0 г, 55%).
Суспензию 4-метил-5-(2-тиенил)изоксазол-3-ола (12.5 г) и K2CO3 (14.4 г) в ацетоне (250 мл) нагревали до температуры расплавления. В течение 25 мин при температуре расплавления добавляли этилбромид (8.0 г) и полученную смесь нагревали в колбе с обратным холодильником в течение 3 ч. Добавили еще одну порцию этилбромида (8.0 г) и полученную смесь нагревали в колбе с обратным холодильником в течение следующих 3 ч. После фильтрования и удаления растворителя остаток обработали методом колоночной хроматографии (силикагель, элюент: этилацетат/н-гептан 1:1), в результате выделили 3-этокси-4-метил-5-(2-тиенил)изоксазол в виде масла (7.3 г, 51%).
Смесь 3-этокси-4-метил-5-(2-тиенил)изоксазола (5.1 г) и N-бромсукцинимида (5.1 г) в CCl4 (400 мл) нагревали в колбе с обратным холодильником в течение 18 ч. После фильтрования и удаления растворителя получили 4-бромметил-3-этокси-5-(2-тиенил)изоксазол (7.8 г, 100%).
К смеси диэтилацетамидомалоната (4.6 г) и трет-бутоксида калия (2.4 г) в N-метилпирролидоне (NMP) (100 мл) при 22oC добавили раствор 4-бромметил-3-этокси-5-(2-тиенил)изоксазола (3.0 г) в NMP (25 мл). Полученный раствор в течение 1 ч перемешивали при 22oC, а потом вылили на смесь льда с водой. Водную фазу экстрагировали диэтиловым эфиром, а собранные вместе органические фазы промыли водным насыщенным раствором NaCl. Органические фазы высушили (MgSO4) и упарили в вакууме. Полученный остаток обработали колоночной хроматографией (силикагель, элюент: этилацетат/н-гептан/метанол 5:5:1), в результате получили 2-ацетамидо-2-этокси-карбонил-3- [3- этокси-5- (2-тиенил)изоксазол-4-ил]пропионат (3.0 г, 68%).
Смесь 2-ацетамидо-2-этоксикарбонил-3-[3-этокси-5-(2- тиенил)изоксазол-4-ил]пропионата (2.7 г) и 48% HBr (20 мл) нагревали в колбе с обратным холодильником в течение 1 ч. Смесь упарили в вакууме, а остаток растворили в воде (50 мл) и обработали углем. После фильтрования добавили 4 н. водную NaOH и довели pH до 3. Полученные кристаллы собрали при фильтровании и высушили в вакууме, при этом получили указанное в заглавии соединение 1а (0.5 г, 30%). Температура плавления 237-239oC; CHN: вычислено: 47.23, 3.97, 11.02; найдено: 47.21, 4.02, 10.92.
1H-ЯМР (DMSO-d6): δ 9.90 (b, 1H), 7.82 (dd, 1H), 7.55 (dd, 1H), 7.24 (dd, 1H), 3.70 (dd, 1H), 2.95-2.88 (m, 2H).
13C-ЯМР (DMSO-d6): δ 171.42, 171.23, 159,62, 129.27, 128.65, 128.22, 127.15, 101.89, 52.59, 24.99.
Аналогичным способом получили соединение:
(RS)-2-амино-3-[3- гидрокси-5-(3-тиенил)изоксазол-4-ил]пропионовую кислоту, 1b, температура плавления 239-240oC
1H-ЯМР (DMSO-d6): δ 8.03-7.96 (m, 1Н), 7.76-7.70 (m, 1H), 7.48-7.43 (m, 1H), 3.70-3.63 (m, 1H), 3.00-2.76 (m, 2H).
Используя бензолсульфонильную группу как защитную для 3-гидроксиизоксазольной группы, аналогичным образом получили:
(RS)-2-амино-3-[3-гидрокси-5-(2-нафтил)изоксазол-4- -ил]пропионовую кислоту, гидрат 1с, температура плавления 235- 237oC.
1H-ЯМР (DMSO-d6): δ 2.89-3.16 (m, 2H), 3.70-3.77 (m, 1H), 7.56-7.63 (m, 2H), 7.74 (dd, 1H), 7.92-8.09 (m, 3H), 8.22 (s, 1H).
13C-ЯМР (DMSO-d6): δ 25.10; 53.03; 103.03; 124.30; 125.90; 126.89; 126,96; 127.45; 127.79; 128.70(2С); 132.74; 133.18; 164.67; 171.41; 171.54.
MS (MH+) m/z: 299.
(RS)-2-амино-3- [3-гидрокси-5-(4-трифторметилфенил) изоксазол-4-ил] пропионовую кислоту. 1d, температура плавления 237-239oC.
1H-ЯМР (DMSO-d6): δ 2.80 (dd, 1H), 2.97 (dd, 1H), 3.71 (dd, 1H), 7.87 (s, 4H).
13C-ЯМР (DMSO-d6, 5% CF3COOH) δ 23.35: 50.94; 101.72; 121.94; 126.11; 126.16; 127.77(2С); 130.34 (q, CF3); 131.74; 164.13; 170.29(2С).
MS (MH+) m/z: 317.
(RS)-2-амино-3-[3-гидрокси-5-(3-бензо[b]тиенил)изоксазол-4- ил]пропионовую кислоту, семигидрат, 1е, температура плавления 222-224oC.
1H-ЯМР (DMSO-d6): δ 2.75 (dd, 1H), 2.96 (dd, 1H), 3.70 (dd, 1H), 7.43-7.55 (m, 2H); 7.96-8.03 (m, 1H); 8.05-8.14 (m, 1H); 8.21 (s, 1H).
13C-ЯМР (DMSO-d6): δ 25.02: 52.96; 104.05; 123.17; 123.34; 123.67; 125.27(2С); 129.49; 136.80; 139.35; 161.49; 171.11; 171.31.
MS (MH+) m/z: 305.
Приведенное ниже соединение получили аналогичным способом из 2-ацетамидо-2-этоксикарбонил-3-[3-этокси-5-(2-тиенил)изоксазол-4- ил]пропионата введением оксида лития и метилированием в положение 5 тиенильной группы с последующим снятием защиты с помощью кипящей 47% водной HBr.
(RS)-2-амино-3-[3-гидрокси-5-(5-метил-2-тиенил)изоксазол-4- ил] пропионовая кислота, гидрат 1f, температура плавления 242-244oC.
1H-ЯМР (DMSO-d6): δ 2.51 (s, 3H), 2.84-2.93 (m, 2H), 3.63-3.72 (m, 1H), 6.94 (dd, 1H); 7.33 (d, 1H).
13C-ЯМР (DMSO-d6): δ 14.91; 24.95; 52.62; 101.24; 126.63; 126.88; 127.23; 142.33; 159.71; 171.20; 171.31.
MS (MH+) m/z: 269.
Пример 2
(RS)-2-амино-2-(3-гидрокси-5-фенилизоксазол-4-ил)уксусная кислота, 2a
Смесь 3-этокси-4-метил-5-фенилизоксазола (3.0 г, 15 ммол), полученного, как это описано в работе Cristensen I.T., 1989, там же, NBS (5.5 г, 31 ммол), и дибензоилпероксида (0.2 г, 0.8 ммол) в CCl4 (100 мл) в течение 20 ч нагревали в колбе с обратным холодильником. Полученную смесь охладили до температуры окружающей среды, отфильтровали и упарили в вакууме. Остаток растворили в воде (95 мл) и нагревали в колбе с обратным холодильником в течение 20 ч. После охлаждения водную фазу экстрагировали диэтиловым эфиром (три раза по 100 мл). Собранные вместе органические фазы высушили (Na2SO4) и выпарили в вакууме, в результате был получен 4-(3-этокси-5-фенилизоксазол)карбальдегид (2.4 г, 75%).
Смесь 4-(3-этокси-5-фенилизоксазол)карбальдегида (1.9 г, 8.7 ммол), KCN (2.7 г, 40.5 ммол) и (NH4)2CO3 (7.8 г, 81.1 ммол) в 50% водном метаноле (250 мл) нагревали в течение 6 ч в колбе с обратным холодильником. Метанол выпарили в вакууме, а водную фазу экстрагировали этилацетатом (три раза по 150 мл). Собранные вместе органические фазы высушили (Na2SO4), а растворитель удалили выпариванием в вакууме. Остаток обрабатывали колоночной хроматографией (силикагель, элюент: этилацетат/н-гептан/метанол 5:5:1), в результате получили 3-этокси-4-[5-(имидазолидин-2,4- дион)] -5-фенилизоксазол (1.0 г, 40%).
Суспензию 3-этокси-4-[5-(имидазолидин-2,4-дион)]-5- фенилизоксазола (800 мг, 2.8 ммол) в 6 н. водной HCl (20 мл, 120 ммол) нагревали в течение 48 ч в колбе с обратным холодильником. Полученную смесь выпарили в вакууме, а остаток растворили в воде (200 мл) и триэтиламине (870 мг, 8.5 ммол). Добавили ди-трет-бутил бикарбонат (930 мг, 4.3 ммол) в тетрагидрофуране (50 мл), и полученный раствор перемешивали в течение 20 ч при 20oC. Тетрагидрофуран выпарили в вакууме, а pH довели до 6.5 с помощью 0.1 н. водной HCl. Водную фазу экстрагировали диэтиловым эфиром, который потом вылили. pH водной фазы довели до 2 с помощью 0.1 н. водной HCl, а потом эту фазу экстрагировали диэтиловым эфиром (три раза по 100 мл). Собранные вместе органические фазы высушили (Na2SO4) и упарили в вакууме. Остаток растворили в диэтиловом эфире (40 мл) и добавили насыщенный раствор HCl в диэтиловом эфире. Полученную в результате смесь перемешивали в течение 20 ч при 20oC. Осадок собрали при фильтровании и высушили. Полученные кристаллы растворили в воде (5 мл) и pH довели до 3. Осадок собрали при фильтровании и высушили, в результате получили указанное в заглавии соединение 2а (100 мг, 15%). Температура плавления 228-230oC; CHN: вычислено 56.40, 4.31, 11.96; найдено 56.20, 4.37, 11.74.
Данные 1H-ЯМР и 13C-ЯМР для соединения 2а, HCl:
1H-ЯМР (DMSO-d6): δ 7.83-7.68 (m, 2H), 7.64- 7.40 (m, 3H), 5.12-4.95 (m, 1H), 3.90 (широкий).
13C-ЯМР (DMSO-d6): δ 169.10, 168.38, 168.31, 131.39, 129.60(2С), 127.73(2С), 126.95, 99.69, 46.06.
Способом, аналогичным методу получения 4-изоксазолкарбальдегидов из соответствующих 4-изоксазолбромметильных соединений с использованием горячих (115oC) диметилсульфоксида и кислого карбоната натрия, были получены следующие соединения:
(RS)-2-амино-2-(3-гидрокси-5-(2-тиенил)изоксазол-4- ил)уксусная кислота, 2b. Температура плавления 191-193oC.
1H-ЯМР (DMSO-d6): δ 4.50 (s, 1H), 7.26 (dd, 1H), 7.77 (d, 1H), 7.84 (d, 1H).
13C-ЯМР (соль HCl) (DMSO-d6): δ 46.16, 98.87, 127.62, 129.15, 129.83, 131.20, 163.56, 168.45, 169.39.
MS (MH+) m/z: 241.
(RS)-2-амино-2-(3-гидрокси-5-(4-трифторметилфенил)изоксазол- 4-ил)уксусная кислота, гидрат, 2с. Температура плавления 200-201oC.
1H-ЯМР (DMSO-d6): δ 4.46 (s, 1Н), 7.92 (d, 2H), 8.11 (d, 2H).
13C-ЯМР (соль HCl) (DMSO-d6): 45.84, 101.16, 121.98, 126.43, 126.50, 128.76(2С), 130.75, 131.00 (q, CF3), 166.75, 168.20, 169.21.
Пример 3
(RS)-2-амино-5-(3-гидроксифенилизоксазол-4- ил)пентановая кислота, гидрат. 3а
Соединение, указанное в заглавии, получали по аналогии с методом, описанным в работе Chiristensen I. Т., et al., Drug Design and Delivery, 1989, 5, 57-71, со следующими модификациями: 1) в качестве исходного использовали этил бензоилбензоат; 2) изоксазольный цикл в промежуточном 5-фенил-4-(2-пропенил)-3- изоксазоле получали по методу Sato К., et al., Agric. Biol. Chem. , 1986, 5(7, 1831-1837; 3) амфотерный ион указанного в заглавии соединения 3а получали при доведении pH водной фазы до 3.5 с помощью 0.1 н. водной NaOH. Температура плавления 210-212oC; CHN: вычислено: 54.62, 6.40, 9.10; найдено: 54.67, 6.36, 9.13.
1H-ЯМР (DMSO-d6): δ 7.72-7.61 (m, 2H), 7.59-7.43 (m, 3H), 3.64-3.40 (m, 2H), 3.50 (широкий), 3.31-3.19 (m, 1H), 1.82-1.53 (m, 4H).
13C-ЯМР (DMSO-d6): δ 170.75, 163.44, 161.65, 129.72, 129.23(2С), 128.58, 126.19(2С), 105.68, 53.93, 30.57, 25.13, 20.88.
Пример 4
(RS)-2-амино-4-[5-(4-фторфенил)-3-гидроксиизоксазол-4- ил]бутановая кислота, гидрат, 4а
β- кетоэфир, этил 3-оксо-3-(4-фторфенил)пропионат получали по аналогии с методом, описанным в примере 1, при этом использовали 4-фторбензонитрил (25.0 г, 0.21 мол), этил 2-бромацетат (51.7 г, 0.31 мол), активированный цинк (20.3 г, 0.31 мол), CuBr2 (0.2 г, 0.9 ммол) и бензол (500 мл). После колоночной хроматографии остатка (силикагель, элюент: этилацетат/н-гептан/метанол 5:5:1) получили этил 3-оксо-3-(4-фторфенил)пропионат в виде масла (28.0 г, 65%).
К раствору NaH (4.4 г, 0.15 мол, 80% в минеральном масле) в этаноле (500 мл) добавили 3-оксо-3-(4-фторфенил)пропионат в виде масла (28.0 г, 0.13 мол). Полученный раствор перемешивали в течение 1.5 ч при 25oC. В течение 30 мин при 25oC добавляли этил 3-хлорпропионат (20.0 г, 0.15 мол), потом полученную смесь нагревали в колбе с обратным холодильником в течение 20 ч. растворитель удалили выпариванием, а остаток растворили в воде (400 мл) и экстрагировали этилацетатом. Собранные вместе органические фазы высушили (MgSO4) и упарили в вакууме. После колоночной хроматографии остатка (силикагель, элюент: этилацетат/н-гептан/метанол 4:4:1) получили этил 4-этоксикарбонил-5-(4-фторфенил)-5-оксопентаноат (27.5 г, 67%).
Этил 3-[5-(4-фторфенил)-3-гидроксиизоксазол-4-ил] пропионат получали по аналогии с методом, описанным в примере 1, со следующими модификациями: 1) в качестве исходного использовали этил 4-этоксикарбонил-5-(4-фторфенил)-5-оксопентаноат (27.5 г, 89 ммол); 2) после выпаривания метанола из раствора метанол/вода водную фазу экстрагировали диэтиловым эфиром. Собранные вместе органические фазы высушили (MgSO4) и выпарили в вакууме. Остаток растворили в этаноле (400 мл) и добавили ацетилхлорид (40 мл). Полученную смесь нагревали в течение 20 ч в колбе с обратным холодильником, охладили, а растворитель удалили выпариванием в вакууме. После колоночной хроматографии остатка (силикагель, элюент: этилацетат/н-гептан/метанол 2:2:1) получили этил 3-[5-(4-фторфенил)-3-гидроксиизоксазол-4-ил]пропионат (9.3 г, 31%).
Этил 3-[3-этокси-5-(4-фторфенил)изоксазол-4-ил] пропионат получали так, как это описано в примере 1, со следующими модификациями: 1) в качестве исходного использовали этил 3-[5-(4- фторфенил)-3-гидроксиизоксазол-4-ил] пропионат (5.5 г, 20 ммол); 2) вместо двух эквивалентов этилбромида добавляли один эквивалент его. После колоночной хроматографии остатка (силикагель, элюент: этилацетат/н-гептан/метанол 10:10:1) получили этил 3-[3-этокси-5- (4-фторфенил)изоксазол-4-ил]пропионат (4.0 г, 65%).
3-[3-этокси-5-(4-фторфенил)изоксазол-4-ил] пропаналь получали из этил 3-[3-этокси-5-(4-фторфенил)изоксазол-4-ил] -пропионата согласно методу, описанному Rich D.H., et al., J. Org. Chem., 1978, 43. 3624-3626.
3-этокси-5-(4-фторфенил)-4-[2-[5-(имидазолидин-2,4-дион)] этил]изоксазол получали так, как это описано в примере 2, используя в качестве исходного соответствующий альдегид. Полученный сырой продукт дважды перекристаллизовывали из этанола для получения чистого вещества.
Соединение 4а, указанное в заглавии, получали из гидантоина по аналогии с методом, описанным в примере 2 при выходе 19%. Температура плавления 235-237oC; CHN: вычислено 55.71, 4.68, 10.00; найдено 54.59, 4.61, 9.97.
1H-ЯМР (DMSO-d6, 340 K): δ 7.78-7.54 (m, 2H), 7.39-7.19 (m, 2H), 5.92(b), 3.35-3.18 (m, 1H), 2.81-2.53 (m, 2H), 2.04-1.70 (m, 2H).
13C-ЯМР (DMSO-d6): δ 172.19, 170.79, 164.70 и 160.76(C-F), 162.96, 128.64, 128.50, 125.13, 116.55, 116.21, 104.52, 52.35, 31.44, 17.81.
Аналогичным образом были получены приведенные ниже соединения:
(RS)-2-амино-4-[3-гидрокси-5-фенилизоксазол-4-ил] бутановая кислота, гидрат, 4b, температура плавления 230-233oC.
1H-ЯМР (D2O): δ 7.59-7.48 (m, 2H), 7.47-7.32 (m, 3H), 3.61-3.50 (m, 1H), 2.57-2.41 (m, 2H), 2.08-1.90 (m, 2H).
13C-ЯМР (DMSO-d6): δ 179.12. 176.64. 161.12, 130.41, 128.79(2C), 128.13, 125.72(2C), 107.13, 53.31, 32.97, 18.62.
(RS)-2-амино-4-[3-гидрокси-5-(2-тиенил)изоксазол-4-ил]бутановая кислота, гидрат, 4с, температура плавления 214-216oC.
1H-ЯМР (DMSO-d6, 320 K): δ 7.84-7.73 (m, 1Н), 7.59- 7.45 (m, 1H), 7.29-7.18 (m, 1H), 3.50 (очень широкий), 3.32- 3.10 (m, 1Н), 2.93-2.60 (m, 2H), 2.00-1.75 (m, 2H).
13C-ЯМР (DMSO-d6): δ 172.11, 170.64, 159.47, 129.41, 128.41(2C), 126.35, 103.73, 52.42, 30.76, 17.93.
(RS)-2-амино-4-[3-гидрокси-5-(3-тиенил)изоксазол-4-ил]бутановая кислота, 4d, температура плавления 212-214oC.
1H-ЯМР (DMSO-d6): δ 8.02-7.95 (m, 1H), 7.78-7.72 (m, 1H), 7.48-7.43 (m, 1H), 3.28-3.16 (m, 1H), 2.93-2.77 (m, 1H), 2.75-2.54 (m, 1H), 2.00-1.75 (m, 2H).
(RS)-2 -амино-4-[3-гидрокси-5-(2-нафтил)изоксазол-4-ил]бутановая кислота, гидрат, 4е, температура плавления 209- 211oC.
1H-ЯМР (DMSO-d6): δ 1.87-2.05 (m, 2H), 2.60-2.85 (m, 2H), 2.90-3.06 (m, 1H), 7.56-7.66 (m, 2H), 7.80 (dd, 1H), 7.93-8.16 (m, 3H), 8.26 (s, 1H).
13C-ЯМР (DMSO-d6): δ 17.92, 31.50, 52.40, 105.02, 123.21, 125.66, 125.95, 127.04, 127.44, 127.76, 128.75, 128.91, 132.82, 133.09, 163.71, 170.70, 172.30.
MS (MH+) m/z: 313.
Пример 5
4-[(2-амино-3,4-диоксо-1-циклобутен-1-ил)аминометил] -3- -гидрокси-5-фенилизоксазол, полугидрат, 5а
К смеси фталимида калия (1.0 г, 5.4 ммол) в N,N-диметилформамиде (50 мл) добавили 4-бромметил-3-этокси-5- фенилизоксазол (1.4 г, 5.0 ммол) в N,N-диметилформамиде при 90oC. 4-бромметил-3-этокси-5-фенилизоксазол получали по аналогии с описанным выше методом получения 4-бромметил-3-этокси-5-(2- тиенил)изоксазола. Полученную при этом смесь перемешивали в течение 40 мин при 90oC, потом добавили CH2Cl2 (200 мл) и вылили эту смесь в воду. Провели разделение фаз, и водную фазу экстрагировали диэтиловым эфиром. Собранные вместе органические фазы промыли 0.1 н. )NaOH и водой, высушили (Na2SO4), а потом упаривали в вакууме. В результате получили N-[(3-этокси-5-фенилизоксазол-4-ил)метил]фталимид (1.5 г, 88%).
Суспензию N-[(3-этокси-5-фенилизоксазол-4-ил)метил] фталимида (1.3 г, 3.7 ммол) в водной 48% HBr (20 мл) и уксусной кислоте (20 мл) перемешивали в течение 6 ч при 110oC. Смесь упарили в вакууме, а остаток растворили в воде. Полученную водную фазу экстрагировали диэтиловым эфиром, который потом удалили. Водную фазу упарили в вакууме и потом к остатку добавили ацетон (5 мл). Осадок собрали фильтрованием и высушили, при этом получили 4-аминометил-5-фенил-3-изоксазол, гидробромид (700 мг, 70%).
К раствору 3-амино-4-этокси-3-циклобутен-1,2-диона (365 мг), полученного согласно методу, описанному Cohen S., et al., J. Amer. Chem. Soc., 1966, 88, 1533-1536, и гидробромида 4-аминометил-5- фенил-3-изоксазола (700 мг, 2.6 ммол) в этаноле (75 мл) добавили раствор NaOH (210 мг, 5.2 ммол) в воде (5 мл) при 22oC. Полученную смесь перемешивали при 22oC в течение 4 ч. Растворитель выпарили, а остаток растворили в воде (200 мл). pH водной фазы довели до 8.5. Водную фазу экстрагировали диэтиловым эфиром, который потом удалили. pH довели до 3.75, а осадок собрали при фильтровании и высушили, в результате было получено указанное в заглавии соединение 5а (675 мг, 92%). Температура плавления 259-261oC; CHN: вычислено: 57.13, 4.12, 14.28; найдено 57.70, 3.85, 14.29.
1H-ЯМР (DMSO-d6): δ 7.81-7.24 (m, 5H), 4.78-4.56 (m. 2H), 3.5 (s, очень широкий).
13C-ЯМР (DMSO-d6): δ 183.34, 183.21, 170.14, 169.48, 169.29, 165.84, 130.58, 129.32(2C), 127.51, 126.72(2C), 102.52, 35.46.
Аналогичным способом были получены следующие соединения:
4-[(2-амино-З, 4-диоксо-1-циклобутен-1-ил)аминометил] -3- -гидрокси-5-(2-тиенил)изоксазол, 5b. Температура сублимации 237-239oC.
1H-ЯМР (DMSO-d6): δ 7.88 (dd, 1H), 7.67 (dd, 1H), 7.26 (dd, 1H), 4.78-4.63 (m, 2H), 3.30 (s, очень широкий).
13C-ЯМР (DMSO-d6): δ 183.36, 183.18, 169.97, 169.51, 168.23, 161.29, 129.80, 128.59, 128.25, 127.82, 101.41, 35.20.
4-[(2-амино-3,4-диоксо-1-циклобутен-1-ил)аминометил] -3- -гидрокси-5-(3-тиенил)изоксазол, 5с. Температура плавления 276-278oC.
1H-ЯМР (DMSO-d6): δ 8.16-8.13 (m, 1H), 7.82-7.77 (m, 1H), 7.56-7.50 (m, 1H), 4.76-4.66 (m, 2H).
4-((2-амино-3,4-диоксо-1-циклобутен-1-ил)аминометил] -3- гидрокси-5-(4-трифторметилфенил)изоксазол, 5d. Температура плавления 269-272oC.
1H-ЯМР (DMSO-d6): δ 4.72 (d, 2H), 7.93 (dd, 4H).
13C-ЯМР (DMSO-d6): δ 35.29, 104.19, 121.75, 126.12, 126.17, 127.60(2C), 130.25 (q, CF3), 131.11, 164.09, 168.21, 169.51, 170.11, 183.29(2C).
MS (MH+) m/z: 354.
Пример 6
4-[2-[(2-амино-3,4-диоксо-1-циклобутен-1-ил)амино] этил]-3- гидрокси-5-(2-тиенил)изоксазол, гидрат, 6а
Смесь цианида натрия (4.0 r,81 ммол) в диметилсульфоксиде (50 мл) нагревали до 90oC. К горячей реакционной смеси по каплям добавляли 4-бромметил-3-этокси-5-(2-тиенил)изоксазол (4.9 г, 16 ммол), растворенный в диметилсульфоксиде (50 мл). Полученную смесь перемешивали еще в течение 30 мин, а потом вылили в смесь льда с водой. Водную фазу экстрагировали CH2Cl2, а собранные органические фазы промыли насыщенным раствором NaCl. Органические фазу высушили (MgSO4) и выпарили в вакууме. Остаток обработали колоночной хроматографией (силикагель, элюент: этилацетат/н-гептан 1:3), при этом получили (3-этокси-5-(2-тиенил)изоксазолацетонитрила (2.2 г, 58%).
Смесь AlCl3 (1.2 г, 9 ммол) в сухом диэтиловом эфире (50 мл) добавляли каплями к смеси LiAlH4 (0.34 г, 9 ммол) в сухом диэтиловом эфире (50 мл). Реакционную смесь перемешивали еще 10 мин при комнатной температуре. К перемешиваемому раствору добавили смесь [3-этокси-5-(2-тиенил)изоксазол] ацетонитрила (2.0 г, 9 ммол) в сухом диэтиловом эфире (50 мл) и реакционную смесь перемешивали еще 45 мин. Потом реакционную смесь охладили до 5oC и добавили воду (30 мл) и 6 М H2SO4 (5 мл). С помощью 6 М NaOH pH полученного кислого раствора повысили до 11. Полученные при этом фазы разделили, и водную фазу экстрагировали диэтиловым эфиром. Собранные вместе органические фазы высушили (MgSO4) и упарили в вакууме. Остаток обработали колоночной хроматографией (силикагель, элюент: этилацетат/н-гептан/метанол/триэтиламин 90:10:5:5. Было получено 4-(2-аминоэтил)-3-этокси-5-(2-тиенил)изоксазола (1.7 г, 76%).
Смесь 4-(2-аминоэтил)-3-этокси-5-(2-тиенил)изоксазола (1.7 г, 7 ммол) и 47% водной HBr нагревали в колбе с обратным холодильником в течение 1 ч. Реакционную смесь упарили в вакууме и добавили ацетон. Полученные кристаллы собрали при фильтровании и высушили в вакууме, был получен 4-(2-аминоэтил)-3-гидрокси-5(2- тиенил)изоксазол, гидробромид (1.9 г, 94%).
К раствору 3-амино-4-этокси-3-циклобутен-1,2-диона (480 мг, 3.4 ммол), полученному так, как это было описано ранее, и гидробромида 4-(2-аминоэтил)-3-гидрокси-5(2-тиенил)изоксазола (1.0 г, 3.4 ммол) в этаноле (30 мл) добавили раствор NaOH (270 мг, 6.8 ммол) в воде (5 мл) при 22oC. Полученный раствор перемешивали 18 ч при комнатной температуре. Реакционную смесь упарили в вакууме, и добавили воду и диэтиловый эфир. Полученные фазы разделили, и водную фазу экстрагировали диэтиловым эфиром, который потом удалили. Водную фазу подкислили до pH 2.5 2М HCl и оставили на 18 ч при 5oC. Образовавшиеся кристаллы собрали фильтрованием и промыли водой, ацетоном и диэтиловым эфиром. После высушивания кристаллов в вакууме было получено соединение 6а, указанное в заглавии (906 мг, 87%). Температура плавления 248-250oC. CHN: вычислено 50.39, 3.75, 13.57; найдено 50.66, 3.72, 13.55.
1H-ЯМР (DMSO-d6): δ] 2.77 (dd, 2H), 3.51-3.78 (m, 2H), 7.22 (dd, 2H), 7.57 (d, 1H), 7.81 (d, 1H).
13C-ЯМР (DMSO-d6): δ 24.12, 42.06, 102.06, 126.65, 127.94, 128.53, 129.00, 160.41, 168.65, 169.41, 170.05, 182.82, 183.00.
Пример 7
(RS)-2-амино-3-[3-карбоксиметокси-5-(2-тиенил) изоксазол-4-ил] пропионовая кислота, моногидрат, 7а
Суспензию 4-метил-5-(2-тиенил)изоксазол-3-ола (10.0 г, 55 ммол), полученного так, как это описано ранее, и K2CO3 (19.1 г, 138 ммол) в ацетоне (350 мл) в течение 30 мин перемешивали при комнатной температуре, а затем нагрели до температуры расплавления. В течение 45 мин добавляли этилхлорацетат (17.6 мл, 166 ммол) в ацетоне (125 мл), и полученную смесь кипятили в колбе с обратным холодильником в течение 210 мин. Потом смесь охладили до 5oC, отфильтровали и выпарили в вакууме. Остаток растворили в CH2Cl2 (500 мл), промыли водой (2 раза по 500 мл) и водным насыщенным раствором NaCl (500 мл). Органическую фазу высушили (MgSO4) и выпарили в вакууме. После тонкослойной хроматографии (силикагель, элюент: н-гептан/этилацетат/метанол 20: 10: 1) и последующего упаривания в вакууме получили этил [4-метил-5-(2-тиенил)- -3-изоксазолилокси]ацетат (8.7 г, 59%).
Смесь этил [4-метил-5-(2-тиенил)-3-изоксазолилокси]ацетата (2.5 г, 9.4 ммол) и NBS (2.0 г, 11.2 ммол) в CCl4 (125 мл) кипятили в колбе с обратным холодильником в течение 8 ч. Полученную смесь охладили, отфильтровали и упарили в вакууме, при этом был получен этил [4-бромметил-5-(2-тиенил)-3-изоксазолилокси]ацетат (2.8 г, 88%).
Смесь диэтилацетамидомалоната (3.5 г, 16.2 ммол) и трет-бутоксид калия (0.9 г, 17.0 ммол) в NMP (30 мл) перемешивали в течение 30 мин при комнатной температуре (25oC). Добавили этил [4- бромметил-5-(2-тиенил)-3-изоксазолилокси]ацетат (2.8 г, 8.1 ммол) в NMP (5 мл) (температура 25-28oC), и полученную смесь перемешивали в течение 1 ч при 28oC, а потом вылили в смесь льда и воды (250 мл). Водную фазу экстрагировали диэтиловым эфиром (три раза по 300 мл), а собранные вместе органические фазы промыли насыщенным раствором NaCl (200 мл), высушили (MgSO4) и выпарили в вакууме. После тонкослойной хроматографии (силикагель, элюент: CH2Cl2/этилацетат 7:1) получили этил 2-ацетиламино-2-этокси-карбонил-3-[3- [(этоксикарбонил)метокси] -5-(2-тиенил)изоксазол-4-ил] пропионата (2.3 г, 59%). Суспензию этил 2-ацетиламино-2-этоксикарбонил-3- [3-[(этоксикарбонил)метокси] -5-(2- -тиенил)изоксазол-4-ил] пропионата (2.0 г, 4.1 ммол) в 1 М HCl (130 мл) кипятили в колбе с обратным холодильником в течение 24 ч. Реакционную смесь охладили, экстрагировали CH2Cl2 (2 раза по 150 мл) и обработали углем. После выпаривания в вакууме получили соединение, указанное в заглавии, в виде своей хлоридной соли (1.2 г, 86%). Соединение 7а, указанное в заглавии, получили при добавлении воды (1.0 г, 71%). Температура плавления 228-230oC. CHN: вычислено 43.64, 4.27, 8.48; найдено 43.07, 4.17, 8.43.
1H-ЯМР (DMSO-d6): δ 2.86-3.28 (m, 2H), 3.79 (dd, 1H), 4.69 (s, 2H), 7.26 (dd, 1H), 7.69 (dd, 1H), 7.86 (dd, 1H).
13C-ЯМР (DMSO-d6): δ 23.50, 52.42, 67.39, 120.45, 127.91, 128.23, 128.48, 129.47, 161.44, 169.63, 170.14, 170.57.
MS (MH+) m/z: 313.
Аналогичным способом, используя в качестве алкилирующего агента вместо этилхлорацетата этил 4-бромбутират, получили приведенные ниже соединения:
(RS)-2-амино-3-[3-карбоксипропокси-5-(2-тиенил)изоксазол-4- ил]пропионовая кислота, 7b. Температура плавления 197-198oC. CHN: вычислено 49.41, 4.74, 8.23. Найдено 49.27, 4.73, 8.30.
1H-ЯМР (DMSO-d6): δ 2.01 (qui, 2H), 2.23-2.60 (m, 2H), 2.71-3.08 (m, 2H), 3.56 (dd, 1H), 4.25 (t, 2H), 7.26 (dd, 1H), 7.70 (dd, 1H), 7.85 (dd, 1H).
13C-ЯMP (DMSO-d6): δ 24.37, 25.20, 32.44, 53.03, 69.65, 101.01, 127.92, 128.69, 128.79, 129.37, 161.15, 170.39, 170.97, 175.44.
MS (MH+) m/z: 341.
Пример 8
[4-[(2-амино-3,4-диоксо-1-циклобутен-1-ил)аминометил] -5-(2- тиенил)-3-изоксазолилокси]уксусная кислота, моногидрат, 8а
Фталимид калия (0.71 г, 3.8 ммол) суспендировали в диметилформамиде (30 мл) и нагрели до 90oC. Потом в течение 20 мин добавляли этил [4-бромметил-5-(2-тиенил)-3-изоксазолилокси] ацетат (1.2 г, 3.5 ммол), синтезированного так, как это выше описано, в диметилформамиде (20 мл), и полученную при этом смесь перемешивали при 90oC в течение 40 мин. Смесь охладили и добавили CH2Cl2 (200 мл) и воду (200 мл). Полученные при этом фазы разделили, органическую фазу промывали насыщенным раствором CaCl2 (два раза по 150 мл), высушили (MgSO4) и упаривали в вакууме. В результате тонкослойной хроматографии (силикагель, элюент: н-гептан/этилацетат 2:1) был получен этил [4-(N-фталимидометил)-5-(2-тиенил)-3-изоксазолилокси] ацетат. Последний растворили в CH2Cl2 (200 мл), промыли водным насыщенным раствором CaCl2 (два раза по 200 мл), высушили (MgSO4), упарили в вакууме и потом кристаллизовали из EtOH. В результате были получены белые игольчатые кристаллы (0.9 г, 64%).
Этил [4-(N-фталимидометил)-5-(2-тиенил)-3-изоксазолил- окси]ацетат (0.60 г, 1.5 ммол) кипятили в колбе с обратным холодильником в 1 М NaOH (60 мл) в течение 45 мин. Полученный раствор охладили, экстрагировали диэтиловым эфиром (три раза по 60 мл) и подкислили до pH 1-2 концентрированной HCl (5 мл). Водную фазу экстрагировали CH2Cl2 (три раза по 80 мл) и диэтиловым эфиром (четыре раза по 80 мл). Собранные вместе органические фазы упарили в вакууме, в результате получили [4-[N-(2-карбоксибензамидо)метил]-5-(2-тиенил)-3 -изоксазолилокси]уксусную кислоту (0.6 г, 100%).
[4-[N-(2-карбоксибензамидо)метил] -5-(2-тиенил)-3- изоксазолилокси] уксусную кислоту (0.60 г, 1.5 ммол) кипятили в колбе с обратным холодильником в 1М HCl (125 мл) в течение 45 мин. Полученный раствор охладили, экстрагировали диэтиловым эфиром (четыре раза по 200 мл) и упаривали в вакууме. Был получен гидрохлорид [4-аминометил-5-(2-тиенил)-3-изоксазолилокси] уксусной кислоты (0.38 г, 100%).
К смеси гидрохлорида [4-аминометил-5-(2-тиенил)-3- изоксазолилокси уксусной кислоты (0.30 г, 1.0 ммол) и 3-амино-4- этокси-3-циклобутен-1,2-диона (0.16 г, 1.1 ммол), полученного, как было описано выше, в EtOH (50 мл) добавили NaOH (0.08 г, 2.1 ммол), растворенного в воде (3 мл). Потом смесь перемешивали в течение 18 ч при 22oC. Полученную суспензию упаривали в вакууме, растворили в воде и экстрагировали диэтиловым эфиром (два раза по 150 мл). pH водной фазы (50 мл) довели до значения 3 с помощью 1 М HCl. Получившиеся при этом кристаллы собрали фильтрованием и высушили в вакууме. Было получено соединение 8а, указанное в заглавии (0.26 г, 69%). Температура плавления 222-223oC. CHN: вычислено 45.78, 3.57, 11.44; найдено 45.13, 3.61, 11.16.
1H-ЯМР (DMSO-d6): δ 4.75 (d, 2H), 4.86 (s, 2H), 7.30 (dd, 1H), 7.70 (d, 1H), 7,94 (d, 1H).
13C-ЯМР (DMSO-d6): δ 34.88, 66.27, 100.97, 127.45, 128.52, 128.67, 130.48, 162.42, 168.19, 168.74, 169.55, 169.95, 183.29.
MS (MH+) m/z: 350.
Пример 9
(RS)-2-амино-3-[3-карбокси-5-(2-тиенил)изоксазол-4- -ил] пропионовая кислота, гидрат, 9а
К охлажденному раствору натрия (4.1 г, 0.18 мол) в EtOH (100 мл) добавили диэтилоксалат (24 мл, 0.18 мол). В течение 15 мин к этому холодному раствору добавляли 1-(2-тиенил)-1-пропанол (20 мл, 0.16 мол) в EtOH (10 мл), полученную при этом смесь перемешивали в течение 2 ч при 0oC, а потом - в течение 16 ч при 22oC. Смесь упарили в вакууме и остаток растворили в воде (400 мл). Водную фазу подкислили 1 М HCl до pH 3-4 и экстрагировали CH2Cl2 (три раза по 300 мл). Собранные вместе органические фазы высушили (MgSO4) и упарили в вакууме. После тонкослойной хроматографии (силикагель, элюент: н-гептан/этилацетат 3: 1) был получен этил 2,4-диоксо-3-метил-4-(2-тиенил)бутират (20.5 г, 53%).
К кипящему раствору хлорида гидроксиламмония (12.6 г, 0.18 мол) в EtOH (200 мл) добавили этил 2,4-диоксо-3-метил-4-(2- тиенил)бутират в EtOH (60 мл). Полученную смесь кипятили в течение 2 ч в колбе с обратным холодильником, потом охладили и упарили в вакууме. После тонкослойной хроматографии (силикагель, элюент: н-гептан/этилацетат 4:1) получили этил [4-метил-5-(2- тиенил)изоксазол)-3-ил]карбоксилат (13.2 г, 92%).
Смесь этил [4-метил-5-(2-тиенил)изоксазол)-3-ил]-карбоксилата (8.0 г, 34 ммол), NBS (6.6 г, 37 ммол) и дибензоилпероксида (0.1 г, 0.4 ммол) в CCl4 кипятили в колбе с обратным холодильником в течение 6 ч и затем оставили на 14 ч при 22oC. Полученную смесь охладили, отфильтровали и упарили в вакууме, в результате был получен этил [4-бромметил-5-(2- тиенил)изоксазол-3-ил]карбоксилат (10.7 г, 100%).
Этил 2-ацетамидо-2-этоксикарбонил-3-[3-этоксикарбонил-5-(2- тиенил)изоксазол-4-ил] пропионат получали исходя из этил [4- бромметил-5-(2-тиенил)изоксазол-3-ил]карбоксилата по аналогии с методом, описанным в примере 7.
Смесь этил 2-ацетамидо-2-этоксикарбонил-3-[3-этоксикарбонил- 5-(2-тиенил)изоксазол-ил] пропионата (1.0 г, 2.2 ммол) и 47% водной HBr (50 мл) нагревали до температуры расплавления и кипятили в течение 40 мин. Полученный раствор упаривали в вакууме, а потом добавили воду (20 мл). Добавили водную NaOH (0.1 М, 22 мл) и смесь перемешивали в течение 2 ч при 22oC. Реакционную смесь упарили в вакууме и добавили воду (20 мл). Образовавшиеся кристаллы собрали фильтрованием и высушили в вакууме. Эти кристаллы растворили в 47% HBr и полученный раствор экстрагировали диэтиловым эфиром, который потом отбросили. Водный раствор упаривали в вакууме, а потом добавили диэтиловый эфир. Полученную реакционную смесь перемешивали в течение 16 ч при 22oC, а кристаллы собрали декантацией диэтилового эфира. Кристаллы высушили в вакууме и по каплям добавляли водную NaOH (0.1 М) до pH 2.5. Образовавшиеся кристаллы собрали при фильтровании и суспендировали в воде (15 мл). Реакционную смесь перемешивали в течение 48 ч при 22oC, а полученные кристаллы собрали при фильтровании. Было получено указанное в заглавии соединение 9а (270 мг, 43%). Температура плавления 227-228oC. CHN: вычислено 46.07, 3.69, 9.77; найдено 46.15, 3.68, 9.74.
1H-ЯМР (D2O; DSS): δ 2.93-3.06 (m, 1H), 3.15-3.24 (m, 1H), 3.46 (dd, 1H), 7.26 (dd, 1H), 7.66-7.72 (m, 2H).
13C-ЯМР (D2O, pH= 12 (NaOD); диоксан): δ 29.06, 57.18, 111.38, 129.00, 129.01(2C), 129.79, 162.28, 163.26, 167.76, 182.65.
MS (MH+) m/z: 283.
Пример 10
(RS)-2-амино-3-[3-(5-тетразолил)-5-(2-тиенил)изоксазол-4- ил]пропионовая кислота, гидрат, 10а
К раствору этил [4-метил-5-(2-тиенил)изоксазол)-3-ил]- карбоксилата (3.7 г, 15.6 ммол), полученного так, как это описано выше, в тетрагидрофуране (20 мл) добавили водную HCl (6 M, 100 мл). Реакционную смесь кипятили в течение 6 ч в колбе с обратным холодильником. Охлажденную реакционную смесь экстрагировали диэтиловым эфиром (три раза по 100 мл), а собранные вместе органические фазы промыли водным насыщенным раствором NaCl, высушили (MgSO4) и упарили в вакууме. К остатку добавили CH2Cl2, и полученные кристаллы собрали при фильтровании. Фильтрат упарили в вакууме и добавили насыщенный водный раствор NaHCO3. Водную фазу промыли CH2Cl2 (два раза по 60 мл) и с помощью 6 М HCl подкислили до pH 1-2. Водную фазу экстрагировали диэтиловым эфиром (три раза по 80 мл). Собранные вместе органические фазы высушили (MgSO4) и упарили в вакууме. Выход [4-метил-5-(2-тиенил)изоксазол-3-ил] карбоновой кислоты составил 1.6 г (48%).
Смесь [4-метил-5-(2-тиенил)изоксазол-3-ил] карбоновой кислоты (4.0 г, 19.1 ммол), SOCl2 (40 мл) и N,N-диметилформамида (0.1 мл) нагревали в колбе с обратным холодильником в течение 60 мин. Полученный раствор упарили в вакууме. Остаток растворили в тетрагидрофуране (50 мл) и органическую фазу при 0-5oC вылили в водный (25%) раствор аммиака. Реакционную смесь перемешивали в течение 1 ч при 22oC и экстрагировали диэтиловым эфиром (четыре раза по 250 мл). Собранные вместе органические фазы промыли водой и насыщенным водным раствором NaCl, высушили (MgSO4) и упарили в вакууме. После этого получили [4-метил-5-(2-тиенил)изоксазол-3-ил]карбоксамид (3.7 г, 93%).
Смесь [4-метил-5- (2-тиенил)изоксазол-3-ил] карбоксамида (3.4 г, 16.3 ммол) и POCl3 (40 мл) нагревали в колбе с обратным холодильником в течение 20 мин. Реакционную смесь упарили в вакууме, а остаток растворили в диэтиловом эфире. Органические фазы вылили в смесь воды со льдом. Полученные фазы разделили и водную фазу экстрагировали диэтиловым эфиром (три раза по 100 мл). Собранные вместе органические фазы промыли водой и насыщенным водным раствором NaCl, высушили (MgSO4) и упаривали в вакууме. В результате получили [4-метил-5-(2-тиенил)изоксазол-3- ил]карбонитрил (2.9 г, 95%).
[4-бромметил-5-(2-тиенил)изоксазол-3-ил] карбонитрил получали исходя из [4-метил-5-(2-тиенил)изоксазол-3-ил] карбонитрила по аналогии с методом, описанным в примере 7.
Этил 2-ацетиламино-2-этоксикарбонил-3-[3-циано-5-(2- -тиенил)изоксазол-4-ил]пропионат получали из [4-бромметил- -5-(2-тиенил)изоксазол-3-ил] карбонитрила по аналогии с методом, описанным в примере 7.
Суспензию этил 2-ацетиламино-2-этоксикарбонил-3-[3-циано-5- (2-тиенил)изоксазол-4-ил] пропионата (2.0 г, 4.9 ммол), NaN3 (0.4 г, 6.2 ммол) и гидрохлорид триэтиламина (0.9 г, 6.2 ммол) в диметоксиэтане (80 мл) кипятили в течение 48 ч в колбе с обратным холодильником. Добавили еще NaN3 (0.4 г, 6.2 ммол) и гидрохлорида триэтиламина (0.9 г, 6.2 ммол) и реакционную смесь нагревали в колбе с обратным холодильником еще 20 ч. Охлажденную реакционную смесь упарили в вакууме, а остаток обработали тонкослойной хроматографией (силикагель, элюент: этилацетат/уксусная кислота 10:1). После выпаривания растворителя получили этил 2-ацетиламино- 2-этоксикарбонил-3-[3-(5-тетразолил)-5-(2-тиенил)изоксазол-4- ил]пропионат (0.7 г, 32%).
Смесь этил 2-ацетиламино-2-этоксикарбонил-3-[3-(5- тетразолил)-5-(2-тиенил)изоксазол-4-ил] пропионата (0.6 г, 1.3 ммол) и 4% водной HBr (20 мл) нагревали в колбе с обратным холодильником в течение 30 мин. Реакционную смесь охладили и добавили воду (50 мл). Водную фазу экстрагировали диэтиловым эфиром (три раза по 75 мл). Собранные вместе органические фазы экстрагировали водой (50 мл). Собранные вместе водные фазы упаривали в вакууме. Добавили воду (25 мл) и водный NaOH (0.1 М, 16 мл), и полученные кристаллы собрали при фильтровании. Кристаллы высушили в вакууме, в результате получили указанное в заглавии соединение 10а (0.4 г, 90%). Температура плавления 209-211oC.
1H-ЯМР (DMSO-d6): δ 3.27-3.54 (m, 2H), 4.37-4.47 (m, 1H), 7.33 (dd, 1H), 7.76 (d, 1H), 7.94 (d, 1H).
13C-ЯМР (DMSO-d6): δ 24.51, 51.82, 108.03, 127.80, 127.95, 128.57, 129.71, 151.63, 155.91, 161.96, 170.40.
MS (MH+) m/z: 307.
Пример 11
(RS)-2-амино-3-[3-гидрокси-5-(2-оксазолил)изоксазол-4- ил] пропионовая кислота, ацетат, 11а
3-гидрокси-4,5-диметилизоксазол получали методом, описанным Jacquier R., et al.. Bull., Soc. Chim. Fr., 1970, 2685-90 и модифицированным Sato К., et al., Agric. Biol. Chem., 1986, 50(7). 1831-1837.
Суспензию 3-гидрокси-4,5-диметилизоксазола (68.5 г, 0.6 мол) и K2CO3 (125.6 г, 0.9 ммол) в ацетоне (1000 мл) нагрели до температуры расплавления. Раствор этилбромида (99.1 г, 0.9 мол) в ацетоне добавляли к реакционной смеси по каплям. Реакционную смесь перемешивали еще 8 ч, а потом охладили, отфильтровали и упарили в вакууме. После тонкослойной хроматографии (силикагель, элюент: этилацетат/гептан 1:3) остатка получили 3-этокси-4,5- диметилизоксазол (52.7 г, 62%).
К охлажденной (5oC) смеси 3-этокси-4,5-диметилизокса-зола (65.6 г, 0.5 мол) в CCl4 (500 мл) добавили Br2 (150 г, 0.9 мол). Реакционную смесь в течение 96 ч перемешивали в темном месте. Добавили воду (200 мл), и при добавлении сульфита натрия реакционная смесь обесцветилась. Фазы разделили и полученную водную фазу экстрагировали CH2Cl2. Собранные вместе органические фазы промыли насыщенным водным раствором NaCl, высушили (MgSO4) и упарили в вакууме. Остаток растворили в смеси вода/NMP (15:85, 700 мл) и реакционную смесь перемешивали в течение 24 ч при 100oC. Добавили воду и полученный водный раствор экстрагировали диэтиловым эфиром.
Собранные вместе органические фазы промыли водой и насыщенным водным раствором NaCl, высушили (MgSO4) и упарили в вакууме. Остаток обработали тонкослойной хроматографией (силикагель, элюент: этилацетат/н-гептан 1:2). В результате получили 3-этокси-5- гидроксиметил-4-метилизоксазол (26,3 г, 36%).
К раствору 3-этокси-5-гидроксиметил-4-метилизоксазола (25,0 г, 0.16 мол) в смеси H2SO4/H2O/уксусная кислота (1:2:7, 200 мл) добавили смесь CrO3/H2O/уксусная кислота (1: 2: 2, 160 мл). Полученную реакционную смесь перемешивали в течение 18 ч при 22oC. Добавили воду и полученную водную фазу экстрагировали диэтиловым эфиром. Собранные вместе органические фазы промыли водой и насыщенным водным раствором NaCl, высушили (MgSO4) и упарили в вакууме. В результате этого получили (3-этокси-4-метилизоксазол-5- ил)карбоновую кислоту (22.7 г, 83%).
Раствор (3-этокси-4-метилизоксазол-5-ил)карбоновой кислоты (5.0 г, 29 ммол) в CH2Cl2 (250 мл), SOCl2 (4.3 мл) и N,N-диметилформамиде (0.2 мл) в течение 2 ч кипятили в колбе с обратным холодильником. Реакционную смесь упарили в вакууме, растворили в CH2Cl2 (100 мл) и по каплям добавляли к охлажденной (5oC) смеси аминоацетальдегиддиметилацеталя (3.5 мл, 3.2 ммол) и K2CO3 (6.0 г, 4.4 ммол) в CH2Cl2 (100 мл). Полученный раствор перемешивали в течение 4 ч при 22oC. Добавили воду и образовавшиеся фазы разделили. Органическую фазу промыли водой и насыщенным водным раствором NaCl, высушили (MgSO4) и упарили в вакууме. При этом получили 5-[N-(ацетальдегиддиметилацеталь)карбоксамид] -3-этокси-4- метилизоксазол (7.0 г, 93%).
Смесь 5-[N-(ацетальдегиддиметилацеталь)карбоксамид] -3-этокси-4-метилизоксазола (6.4 г, 25 ммол), P2O5 (7.1 г, 50 ммол) и концентрированной H2SO4 (150 мл) нагревали в колбе с обратным холодильником в течение 30 мин. Охлажденную (5oC) реакционную смесь вылили в смесь льда с водой и полученную водную фазу экстрагировали диэтиловым эфиром (два раза по 500 мл). Собранные вместе органические фазы промыли водой и насыщенным водным раствором NaCl, высушили (MgSO4) и упарили в вакууме. Остаток обработали тонкослойной хроматографией (силикагель, элюент: CH2Cl2/этилацетат/метанол 1:1:0.5) и получили 3-гидрокси-4-метил-5-(2-оксазолил)изоксазол (340 мг, 8.3%).
К охлажденному (-5oC) раствору 3-гидрокси-4-метил- -5-(2-оксазолил)изоксазола (340 мг, 2.0 ммол) в триэтанол- амине (0.33 мл) и тетрагидрофуране (40 мл) добавили раствор бензолсульфонилхлорида (0.27 мл, 2.1 ммол) в тетрагидрофуране (40 мл). Полученную реакционную смесь перемешивали в течение 18 ч при 22oC. После выпаривания в вакууме растворителя и тонкослойной хроматографии (силикагель, элюент: этилацетат/н-гептан 1:3) получили 3-бензолсульфонилокси-4-метил-5-(2-оксазолил)изоксазол (520 мг, 85%).
Смесь 3-бензолсульфонилокси-4-метил-5-(2-оксазолил) изоксазола (500 мг, 1.6 ммол), NBS (300 мг, 1.7 ммол) и пероксида дибензоила (0.1 г, 0.4 ммол) в CCl4 (100 мл) в течение 24 ч кипятили в колбе с обратным холодильником. Охлажденную реакционную смесь отфильтровали и упарили в вакууме. В результате получили 3-бензолсульфонилокси-4-бромметил-5-(2- оксазолил)изоксазол (440 мг, 70%).
Этил 2-ацетиламино-2-этоксикарбонил-3- [3-бензолсульфонилокси-5-(-2-оксазолил)изоксазол-4-ил]пропионат (120 мг, 22%) получили исходя из 3-бензолсульфонилокси-4-бромметил-5-(2- оксазолил)изоксазола (440 мг) по аналогии с методом, описанным в примере 7.
К раствору этил 2-ацетиламино-2-этоксикарбонил-3-[3- бензолсульфонилокси-5-(2-оксазолил)изоксазол-4-ил]пропионата (120 мг, 0.25 ммол) в метаноле (10 мл) добавили NaOH (20 мг, 0.5 ммол) в метаноле (10 мл). Реакционную смесь перемешивали в течение 2 ч при 22oC, а растворитель выпарили в вакууме. К остатку добавили воду, и водную фазу экстрагировали CH2Cl2. Водную фазу подкислили 0.1 М HCl до pH 2 и экстрагировали CH2Cl2. Потом водную фазу выпарили в вакууме и добавили 1 М водную HCl (10 мл). Реакционную смесь кипятили в колбе с обратным холодильником в течение 3 ч. Реакционную смесь упарили в вакууме и добавили эфир (10 мл). Кристаллы, полученные в результате этого, собрали при фильтровании и высушили в вакууме. Полученные кристаллы растворили в 1 М HCl, и водную фазу промыли диэтиловым эфиром. Водную фазу выпарили в вакууме, а остаток растворили в воде. Потом водную фазу пропустили сквозь колонку, содержащую ионообменную смолу [Amberlite IRA 400 (С1, 150 мл)] , используя в качестве элюента 1 М уксусную кислоту. После выпаривания растворителя получили указанное в заглавии соединение 11а (15 мг, 27%).
1H-ЯМР (D2O, диоксан): δ 3.26-3.36 (m, 2H), 4.11-4.21 (m, 1H), 7.40 (s, 1H), 8.04 (s,1H).
13C-ЯМР (D2O, диоксан): δ 23.41, 54.17, 105.83, 128.96, 142.16, 152.56, 171.55, 177.26, 177.74.
MS (MH+) m/z: 240.
Пример 12
(RS)-2-амино-3-[3-гидрокси-5-(2-тиазолил)изоксазол-4- -ил] пропионовая кислота, 12а
Смесь 5-[N-(ацетальдегиддиметилацетат)карбоксамид] -3- -этокси-4-метилизоксазола (6.5 г, 25.2 ммол), полученного, как это описано в примере 11, и P2S5 (5.6 г, 25.2 ммол) в толуоле нагревали в течение 2 ч в колбе с обратным холодильником. Реакционную смесь упарили в вакууме, а остаток подвергли тонкослойной хроматографии (силикагель, элюент: толуол/этилацетат 11:1), в результате получили 3-этокси-4-метил-5-(2- тиазолил)изоксазол (0.5 г, 9%).
Смесь 3-этокси-4-метил-5-(2-тиазолил)изоксазола (0.5 г, 2.4 ммол) и NBS (0.5 г, 2.6 ммол) в CCl4 (50 мл) кипятили в течение 36 ч до расплавления. Охлажденную реакционную смесь отфильтровали и упарили в вакууме, в результате получили 4-бромметил-3-этокси-5-(2- тиазолил)изоксазол (0.6 г, 87%).
Этил 2-ацетиламино-2-этоксикарбонил-3-[3-этокси-5-(2-тиазолил)изоксазол-4-ил] пропионат (320 мг, 41%) получали исходя из 4-бромметил-3-этокси-5-(2-тиазолил)изоксазола (540 мг), по аналогии с методом, описанным в примере 7.
Суспензию этил 2-ацетиламино-2-этоксикарбонил-3-[3-этокси-5-(2-тиазолил)изоксазол-4-ил] пропионата (245 мг, 0.6 ммол) в водной 47% HBr (5 мл) кипятили в колбе с обратным холодильником в течение 30 мин. Реакционную смесь выпарили в вакууме и добавили воду (30 мл). Водный раствор обработали углем, отфильтровали и упарили в вакууме. Остаток растворили в воде и полученный водный раствор пропустили сквозь колонку, содержащую ионообменную смолу [Amberlite IRA 400, (Cl, 150 мл], используя в качестве элюента 2 М уксусную кислоту. После выпаривания растворителя получили указанное в заглавии соединение 12а (66 мг, 45%).
1H-ЯМР (DMSO-d6, 10% CF3COOH): δ 3.13-3.27 (m, 1H), 3.30-3.45 (m, 1H), 4.14-4.28 (m, 1H), 8.03 (d, 1H), 8.10 (d, 1H).
13C-ЯМР (DMSO-d6, 10%, CF3COOH): δ 22.98, 51.03, 102.43, 122.64, 144.76, 154.97, 159.81, 170.31, 170.50.
MS (MH+) m/z: 256.
(RS)-2-амино-3-[3-гидрокси-5-(5-тетразолил)изоксазол-4- ил] пропионовую кислоту получали аналогичным способом при описанных ниже модификациях:
(3-этокси-4-метилизоксазол-5-ил)карбоновую кислоту, полученную так, как это было описано выше, превращали в соответствующий карбоксамид известными методами, используя SOCl2 и водный аммиак (25%). Соответствующий нитрил, (3-этокси-4- метилизоксазол-5-ил)карбонитрил, получен дегидрированием амина с помощью POCl3. Бромированием (3-этокси-4-метил- изоксазол-5-ил)карбонитрила с помощью NBS и последующим взаимодействием с диэтилацетамидомалонатом получен этил 2-ацетиламино-2-этоксикарбонил-3-(5-циано-3-этоксиизоксазол-4- -ил)пропионат. Образование соответствующего соединения 5-(5- -тетразолила) предпочтительно проводить взаимодействием этил 2-ацетиламино-2-этоксикарбонил-3-(5-циано-3-этоксиизоксазол-4- ил)пропионата с NaN3 и гидрохлоридом триэтиламина в диметоксиэтане. Снятие защиты с этил 2-ацетиламино-2-этоксикарбонил-3-[3-этокси- 5-(5-тетразолил)изоксазол-4-ил)пропионата проводили, используя водную 47% HBr, в результате получили (RS)-2-амино-3-[3-гидрокси-5-(5-тетразолил)изоксазол-4- -ил] пропионовую кислоту.
Пример 13
(RS)-2-амино-3-[3-гидрокси-5-(2-пиридил)изоксазол-4- -ил)пропионовая кислота, гидрат, 13а
Раствор 5-(2-пиридил)изоксазол-3-ола (1.14 г, 7.0 ммол) (полученного из гидробромида метил 2,3-дибром-3-(2-пиридил) пропионата с использованием модификации метода, описанного Tomita К., Ann. Sankyo Res. Lab., 1973, 25. 3-5) и NaOH (0.28 г, 7.0 ммол) в воде (10 л) и этаноле (10 мл) выпарили досуха и продолжали дальнейшее высушивание в течение 2 ч при 0.1 мм Нg. Полученный остаток суспендировали в сухом N,N-диметилформамиде (10 мл) и охладили до -10oC. По каплям добавляли диметилсульфат (0.73 мл, 7.7 ммол) и полученную смесь в течение 1 ч перемешивали при -10oC, а затем в течение 15 ч при 22oC. Полученный в результате раствор упаривали в вакууме. К остатку добавили воду и полученную смесь экстрагировали метиленхлоридом (три раза по 30 мл). Собранные вместе органические фазы высушили и выпарили в вакууме. Остаток обработали тонкослойной хроматографией (силикагель, элюент: толуол/этилацетат/ледяная уксусная кислота 1:1:1), в результате получили 3-метокси-5-(2-пиридил)изоксазол (0.73г, 59%).
Раствор 3-метокси-5-(2-пиридил)изоксазола (1.53 г, 8.68 ммол) в сухом тетрагидрофуране (40 мл) охладили до -78oC. В течение 5 мин добавляли раствор н-бутиллития в гексане (9.0 мл, 1.6 М, 14 ммол), а потом - п-формальдегид (2.48 г, 83 ммол). Полученную смесь перемешивали в течение 15 мин при -78oC, а затем в течение 2 ч при 22oC. Реакционную смесь выпарили в вакууме, добавили воду (25 мл) и метиленхлорид (40 мл), pH довели до 6 с помощью разбавленной HCl. Фазы разделили, и водную фазу экстрагировали метиленхлоридом (два раза по 30 мл). Собранные вместе органические фазы высушили и выпарили в вакууме. С помощью тонкослойной хроматографии (силикагель, элюент: толуол/этилацетат 19: 1) выделили 357 мг 4-гидроксиметил-3-метокси-5-(2-пиридил)изоксазола (выход 20%).
Смесь 4-гидроксиметил-3-метокси-5-(2-пиридил)-изоксазола (357 мг, 1.73 ммол) и тионилхлорида (10 мл) кипятили в колбе с обратным холодильником в течение 2 ч. Потом реакционную смесь выпарили в вакууме. К остатку добавили водный раствор кислого карбоната натрия (5%, 15 мл) и полученную смесь экстрагировали метиленхлоридом (два раза по 25 мл). Собранные вместе органические фазы высушили и выпарили в вакууме, в результате получили 4-хлорметил-3-метокси-5- (2-пиридил)изоксазол (388 мг, 100%).
К раствору диметилацетамидомалоната (361 мг, 1.91 ммол) в сухом N,N-диметилформамиде (4 мл) небольшими порциями добавляли гидрид натрия (84 мг, 60% в масле, 2.09 ммол). Полученную смесь перемешивали в течение 45 мин при 22oC. К реакционной смеси по каплям добавляли раствор 4-хлорметил-3-метокси-5-(2- пиридил)изоксазола (390 мг, 1.74 ммол) в сухом N,N-диметилформамиде (4 мл), и полученную смесь перемешивали еще 6 ч при 22oC. Смесь упарили в вакууме и к остатку добавили воду (10 мл). Водную реакционную смесь экстрагировали метиленхлоридом. Собранные вместе органические фазы высушили и выпарили в вакууме. В результате тонкослойной хроматографии (силикагель, элюент: толуол/этилацетат 19: 1) получили метил 2-ацетиламино-2-метоксикар6онил-3-[3- -метокси-5-(2-пиридил)изоксазол-4-ил]пропионат (500 мг, 6%).
Смесь метил 2-ацетиламино-2-метоксикарбонил-3-[3-метокси-5- (2-пиридил)изоксазол-4-ил]пропионата (500 мг, 1.3 ммол) и водной бромистоводородной кислоты (47%, 20 мл) кипятили в колбе с обратным холодильником в течение 1 ч. Смесь упарили в вакууме и к остатку добавили воду (10 мл). Полученный водный раствор обработали углем и к фильтрату осторожно до pH 3 добавили водный 2 н. )раствор карбоната натрия. Спустя 24 ч при 5oC собрали осадок, промыли его водой и высушили в вакууме. В результате получили соединение 13а, указанное в заглавии (174 мг, 54%).
CHN: вычислено 52.07, 4.57, 16.56; найдено 52.03, 4.50, 16.38.
1H-ЯМР (200 МГц, DMSO-d6): δ 8.70 (d, 1H), 7.95 (m, 1H), 7.80 (d, 1H), 7.45 (m, 1H), 3.75 (m, 1H), 3.40 (dd, 1H), 3.15 (dd, 1H).
13C-ЯМР (200 МГц, DMSO-d6): δ 171.83, 171. 53, 162.88, 149.68, 147.64, 137.82, 124.41, 121.17, 104.86, 53.30, 24.24.
Аналогичным способом получили:
(RS)-2-амино-3-[3-гидрокси-5-(4-пиридил)изоксазол-4- ил)пропионовая кислота, гидрат, 13b.
CHN: вычислено 49.44, 4.90, 15.72; найдено 49.49, 4.88, 15.83.
1H-ЯМР (200 МГц, DMSO-d6): δ 8.70 (d, 2H), 7.65 (d, 2H), 3.75 (m, 1H), 3.05 (dd, 1H), 2.90 (dd, 1H).
13C-ЯМР (200 МГц, DMSO-d6): δ 172.97, 171.20, 161.73, 150.52, 135.30, 121.01, 105.39, 52.70, 24.69.
Пример 14
(S)-(+)-2-амино-3-[3-гидрокси-5-фенилизоксазол-4- ил)пропионовая кислота, гидрат, 14а
Диастереомерную соль (RS)-2-амино-3-[3-гидрокси-5- фенилизоксазол-4-ил)пропионовой кислоты, 0.5 H2O (11.0 г, 44 ммол) (полученную так, как это описано в работе Chrictensen I.Т., 1989, там же) и (R)-(+)-1-фенилэтиламин (5.1 г, 44 ммол) осаждали из этанола (300 мл) при 0oC. Полученные кристаллы растворили в воде и получившуюся смесь подкислили до pH 2.5 с помощью 0.1 н. )соляной кислоты. Указанное в заглавии кристаллическое соединение собрали при фильтровании (320 мг, 6%). Температура плавления 251-253oC, [α]D: +35.3o (c= 0.25, 1 н. )HCl, 20oC), ee=99.0%; CHN: вычислено 58.05, 4.88, 11.29. Найдено 58.03, 5.22, 11.33.
(R)-(-)-2-амино-3-[3-гидрокси-5-фенилизоксазол-4-ил)про- пионовая кислота, гидрат, 14b, получили аналогичным способом, используя (S)-(-)-1-фенилэтиламин (5.1 г, 44 ммол). Выход соединения, указанного в заглавии, составил 1.4 г, 26%. Содержание воды, как было определено, составляет 7.7%. Температура плавления 252-254oC; [α]D: -37.8o (с=1, 1 н. )HCl, 20oC), ее=99.8%; CHN: вычислено 54.12, 5.31, 10.52; найдено: 54.10, 5.27, 10.54.
Приведенные ниже соединения были получены аналогичным способом исходя из (RS)-2-амино-3-[5-(4-фторфенил)-3-гидрок- сиизоксазол-4-ил)пропионовой кислоты по аналогии с (RS)-2-амино-3- [3-гидрокси-5-(2-тиенил)изоксазол-4-ил] пропионовой кислотой, как это описано в примере 1:
(-)-2-амино-3-[5-(4-фторфенил)-3-гидроксиизоксазол-4-ил)пропионовая кислота, 14с.
Температура плавления 247oC, ее= 99.6%; CHN: вычислено 54.12, 4.16, 10.56. Найдено 54.16, 4.12, 10.48.
1H-ЯМР (200 МГц, D2O, NaOD, диоксан): δ 2.56 (dd, 1H), 2.82 (dd, 1H), 3.40 (dd, 1H), 7.22 (dd, 2H), 7.64 (dd, 2H).
(+)-2-амино-3-[5-(4-фторфенил)-3-гидроксиизоксазол-4-ил)пропионовая кислота, 14d.
Температура плавления 246oC, [α]D: +37.9o (с=0.25, 1 н. )HCl, 20oC); ee= 99.8%. CHN: вычислено 54.12, 4.16, 10.56. Найдено 54.12, 4.06, 10.45.
1H-ЯМР (200 МГц, D2O, NaOD, диоксан): δ идентично 14с.
Пример 15
(-)-2-амино-3-[3-гидрокси-5-(2-тиенил)изоксазол-4-ил)пропионовая кислота, 15а
Соединение 1а (20.8 г, 81.8 ммол), полученное так, как это описано в примере 1, добавили к смеси этанола (350 мл) и ацетилхлорида (35 мл) при 25oC. Полученную реакционную смесь кипятили в колбе с обратным холодильником в течение 3.5 ч. Охлажденный раствор выпарили в вакууме, получив при атом (RS)-2-амино-3- [3-этокси-5-(2-тиенил)изоксазол-4-ил)пропионовую кислоту (25.0 г, 96%).
Раствор (RS)-2-амино-3-[3-этокси-5-(2-тиенил)изоксазол-4- ил)пропионовой кислоты (24.9 г, 78.1 ммол) в CH2Cl2 (1200 мл) и триэтаноламине (45 мл) нагревали до температуры расплавления. К горячей реакционной смеси в течение 40 мин добавляли (S)-(+)-2- метокси-2-фенилацетилхлорид (16.2 г, 87.9 ммол), полученный известными методами из (S)(+)-2-метокси-2-фенилуксусной кислоты в CH2Cl2 (250 мл). Полученную реакционную смесь кипятили еще 1 ч в колбе с обратным холодильником. Охлажденную реакционную смесь упарили в вакууме и подвергли тонкослойной хроматографии (силикагель, элюент: н-гептан/этилацетат/уксусная кислота 55:45:1. В результате был получен (RS)-этил 2-[N-(2-метокси-2- фенилацетамидо] -3-[3-гидрокси-5-(2-тиенил)изоксазол-4- ил]пропионат (27.2 г, 81%).
К охлажденному до 0oC раствору (RS)-этил 2-[N-(2-мeтокси- 2-фенилацетамидо] -3-[3-гидрокси-5-(2-тиенил)изоксазол- -4-ил] пропионата (27.2 г, 61.1 ммол) в сухом тетрагидрофуране (600 мл) и триэтаноламине (10 мл) в течение 160 мин добавляли бензолсульфонилхлорид (12.5 г, 70.8 ммол) в сухом тетрагидрофуране (300 мл). Полученный раствор перемешивали 18 ч при 22oC. Реакционную смесь упарили в вакууме и добавили к ней воду и CH2Cl2. Полученные фазы разделили, органическую фазу высушили (MgSO4) и выпарили в вакууме. Полученный остаток, содержащий два диастереоизомера (RS)-этил 2-[N-(2-метокси-2-фенилацетамидо] -3-[3-бензолсульфонилокси- -5-(2-тиенил)изоксазол-4-ил] пропионата подвергли тонкослойной хроматографии (силикагель, элюент: толуол/этилацетат 7: 1). Выход элюированного первым изомера (изомер 1) составил 10.4 г (60%), а выход вымываемого вторым изомера (изомер 2) составил 9.0 г (51%).
Изомер 1 (10.4 г, 18.2 ммол) растворили в метаноле (600 мл). Добавили NaOH (0.8 г, 20.0 ммол) в метаноле (20 мл), и полученную реакционную смесь в течение 10 мин перемешивали при 22oC. К реакционной смеси добавили воду (5000 мл) и с помощью концентрированной HCl pH водного раствора довели до 1. Водную фазу экстрагировали диэтиловым эфиром и CH2Cl2. Собранные вместе органические фазы высушили и упарили в вакууме. После тонкослойной хроматографии (силикагель, элюент: этилацетат/н-гептан/уксусная кислота 50:50:1) был получен изомер 1 (RS)-этил 2-[N-(2-метокси- 2-фенилацетамидо]-3-[3- -гидрокси-5-(2-тиенил)изоксазол-4-ил]пропионата (5.8 г, 74%).
Смесь полученного таким образом изомера 1 (5.8 г, 13.5 ммол) и водной 47% HBr (250 мл) кипятили в колбе с обратным холодильником в течение 1 ч. Реакционную смесь выпарили в вакууме и добавили воду. Водный раствор промыли CH2Cl2 и обработали углем. Объем этого водного раствора уменьшили до 200 мл упариванием в вакууме. Цвиттерион изомера 1 кристаллизовали из этого водного раствора добавлением к нему до pH 3 водного NaOH. Получившиеся кристаллы собрали при фильтровании, при этом получили (-)-2-амино-3-[3-гидрокси-5- -(2-тиенил)изоксазол-4-ил)пропионовую кислоту, 15а (1.0 г, 30%). Температура плавления 258-260oC; [α]D: -26.8o (с=0.25, 1 н. )HCl, 20oC); ее=96.9%; CNH: вычислено 47.23, 3.97, 11.02; найдено 46.88, 3.97, 10.89.
MS (MH+) m/z: 255.2.
Аналогичным способом при обработке изомера 1 получили:
(+)-2-амино-3-[3-гидрокси-5-(2-тиенил)изоксазол-4-ил)пропионовую кислоту, 15b.
Температура плавления 259-261oC, [α]D: +24.4o (с=0.25, 1 н. )HCl, 20oC); ee=98.9%; CNH: вычислено 47.23, 3.97, 11.02. Найдено 47.15, 4.05, 10.88.
MS (MH+) m/z: 255.2.
Соединение 4с обработали аналогично, чтобы получить:
(-)-2-амино-4-[3-гидрокси-5-(2-тиенил)изоксазол-4- ил)бутановую кислоту, 15с. Температура плавления 217-219oC; [α]D: -16.3o (с=0.25, 1 н. )HCl, 20oC); ее=85.1%; и
(+)-2-амино-4-[3-гидрокси-5-(2-тиенил)изоксазол-4-ил)бутановую кислоту, 15d. Температура плавления 217-219oC [α]D: +15.6o (с=0.25, 1 н. )HCl, 20oC); ee=81.9%.
Фармакология
Соединения настоящего изобретения испытывали следующими хорошо известными и надежными методами исследований. Результаты по испытанию связывания рецепторов приведены в таблице 1, а результаты исследований электрофизиологического кортикального препарата крыс - в таблице 2 (см. в конце описания).
Связывание [3H]АМРА,
В этом испытании сродство лекарственного средства к рецепторам АМРА определялось измерением способности замещать [3H]АМРА из рецепторов АМРА.
Испытание проводилось согласно модифицированной версии метода Honore Т. and Nielsen М. , Neurosci. Lett., 1985, 54, 27-32, в присутствии KSCN. Это означает, что были отмечены только места, обладающие сильным сродством связывания [3H] АМРА.
Использовались препараты мембран, полученные по методу Ransom R.W. and Stec, J. Neurochem., 1988, 51. 830-836.
Связывание [3H]СРР
В этом испытании сродство лекарственного средства к рецептору NMDA определялось измерением способности замещать [3H] СРР (4-(3- фосфонопропил)пиперазин-2-карбоновая кислота) из мест связывания переносчика рецептора NMDA.
Испытание проводилось согласно методу Murphy D.E., et al., J. Pharm. Exp. Ther., 1987, 240. 778-784. Использовались препараты мембран, полученные по методу, описанному выше.
Модель кортикального клина
Модель кортикального клина представляет собой испытание, в котором для количественной оценки воздействия лигандов Glu-рецепторов и оценки фармакологического профиля этих лигандов (например, свойства агониста/антагонист) исследовались тонкие срезы мозга крыс in vitro. Испытание проводили так, как это описано Harrison N.L. and Simmonds М.А., Br. J. Pharmacol., 1985, 84, 381- 391 в модификации по Wheatley P.L., Br. J. Pharmacol., 1986, 87, 159P.
Было установлено при фармакологической характеристике кортикального препарата, что некоторые из соединений являются агонистами рецепторов АМРА и NMDA, другие соединения представляют собой селективные антагонисты NMDA, а еще ряд соединений являются неселективными антагонистами рецепторов АМРА и NMDA. Все соединения демонстрируют активность в мкМ диапазоне.
Результаты
Из таблицы 1 видно, что некоторые соединения настоящего изобретения селективно замещают [3H]АМРА из рецепторов АМРА in vitro при низких микромолярных концентрациях. Другие соединения селективно замещают [3H]СРР из рецепторов NMDA in vitro. Еще ряд соединений настоящего изобретения, как было обнаружено, замещают как [3H]С]АМРА, так и [3H]СРР соответственно из рецепторов АМРА и NMDA in vitro.
В кортикальных препаратах некоторые соединения настоящего изобретения, как было найдено, являются агонистами или частичными агонистами, в то время как другие представляют собой антагонисты. Поэтому настоящее изобретение включает соединения с различными агонистическими/антагонистическими профилями при рецепторах глутаминовой кислоты, обладающие активностью при низких микромолярных концентрациях.
Примеры рецептур
Фармакологические рецептуры согласно настоящему изобретению можно получить, используя методы, общепринятые в этой области.
Например: таблетки можно получить, смешивая активный ингредиент с обычными адъювантами и/или разбавителями с последующим прессованием полученной смеси в подходящей таблетировочной машине. Примеры адъювантов или разбавителей включают: кукурузный крахмал, тальк, стеарат магния, желатин, лактозу, смолы и т.п. Можно использовать также любые иные адъюванты или красящие добавки, ароматизаторы, консерванты и т.д. при условии, что они совместимы с активными ингредиентами.
Растворы для инъекций могут быть получены растворением активного ингредиента и возможных добавок в части растворителя для инъекций, предпочтительно в стерильной воде, доведением полученного раствора до требуемого объема, стерилизацией этого раствора и заполнением им подходящих ампул или емкостей. Могут быть дополнительно введены любые подходящие добавки, которые удобно использовать в этой области, такие как тонизирующие агенты, консерванты, антиоксиданты и т.д.
Типичные примеры рецептов лекарственных средств согласно настоящему изобретению приведены ниже:
1) Таблетки, содержащие 5 мг соединения 1а:
Соединение 1а - 5.0 мг
Лактоза - 60 мг
Кукурузный крахмал - 30 мг
Гидроксипропилцеллюлоза - 2.4 мг
Микрокристаллическая целлюлоза - 19.2 мг
Кроскармеллоз натрия тип А - 2.4 мг
Стеарат магния - 0.84 мг
2) Таблетки, содержащие 1 мг соединения 4с:
Соединение 4с - 1.0 мг
Лактоза - 46.9 мг
Кукурузный крахмал - 23.5 мг
Повидон - 1.8 мг
Микрокристаллическая целлюлоза - 14.4 мг
Кроскармеллоз натрия тип А - 1.8 мг
Стеарат магния - 0.63 мг
3) Сироп, содержащий в 1 мл:
Соединение 1а - 5.0 мг
Сорбитол - 500 мг
Гидроксипропилцеллюлоза - 15 мг
Глицерол - 50 мг
Метил-парабен - 1 мг
Пропил-парабен - 0.1 мг
Этанол - 0.005 мл
Ароматизатор - 0.05 мг
Сахарин - 0.5 мг
Вода - До 1 мл
4) Раствор для инъекций, содержащий в 1 мл:
Соединение 4а - 0,5 мг
Сорбитол - 5,1 мг
Уксусная кислота - 0,08 мг
Вода для инъекций - До 1 млт
| название | год | авторы | номер документа |
|---|---|---|---|
| СОЕДИНЕНИЯ ФЕНИЛИНДОЛА | 1994 |
|
RU2156249C2 |
| КОНДЕНСИРОВАННОЕ БЕНЗОСОЕДИНЕНИЕ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1993 |
|
RU2141959C1 |
| АРИЛСУЛЬФОНИЛМЕТИЛЬНЫЕ ИЛИ АРИЛСУЛЬФОНАМИДНЫЕ ПРОИЗВОДНЫЕ АРОМАТИЧЕСКИХ СОЕДИНЕНИЙ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ И СПОСОБ ЛЕЧЕНИЯ РАССТРОЙСТВ, ВОСПРИИМЧИВЫХ К ЛЕЧЕНИЮ ЛИГАНДАМИ ДОФАМИНОВЫХ D РЕЦЕПТОРОВ, С ИХ ПОМОЩЬЮ | 2005 |
|
RU2442781C2 |
| Гибридные производные (1Н-1,2,4) триазола и серосодержащих гетероциклов: производных тиазолидин-2,4-диона, тиоморфолин-3-она и 1,4-тиазепан-3-она, обладающих антимикробной активностью | 2020 |
|
RU2771027C1 |
| ПРОИЗВОДНЫЕ [1,2,4] ТРИАЗОЛО [4,3-А]ХИНОКСАЛИНОНА ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ, СПОСОБ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБ ИНГИБИРОВАНИЯ РЕЦЕПТОРОВ АМРА | 1995 |
|
RU2149162C1 |
| СОЕДИНЕНИЕ МОЧЕВИНЫ, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ЕГО МЕДИЦИНСКОЕ ПРИМЕНЕНИЕ | 2017 |
|
RU2741596C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ 1-АЦИЛ-4-ФЕНИЛСУЛЬФОНИЛПРОЛИНАМИДА И НОВЫЕ ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ | 2012 |
|
RU2615997C2 |
| ДИСПИРО 1,2,4-ТРИОКСОЛАНЫ КАК ПРОТИВОМАЛЯРИЙНЫЕ СРЕДСТВА | 2008 |
|
RU2493159C2 |
| ФЕНИКОЛОВЫЕ ПРОТИВОБАКТЕРИАЛЬНЫЕ СРЕДСТВА | 2013 |
|
RU2593204C2 |
| ПРОИЗВОДНЫЕ ИНДАНУКСУСНОЙ КИСЛОТЫ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2002 |
|
RU2314298C2 |
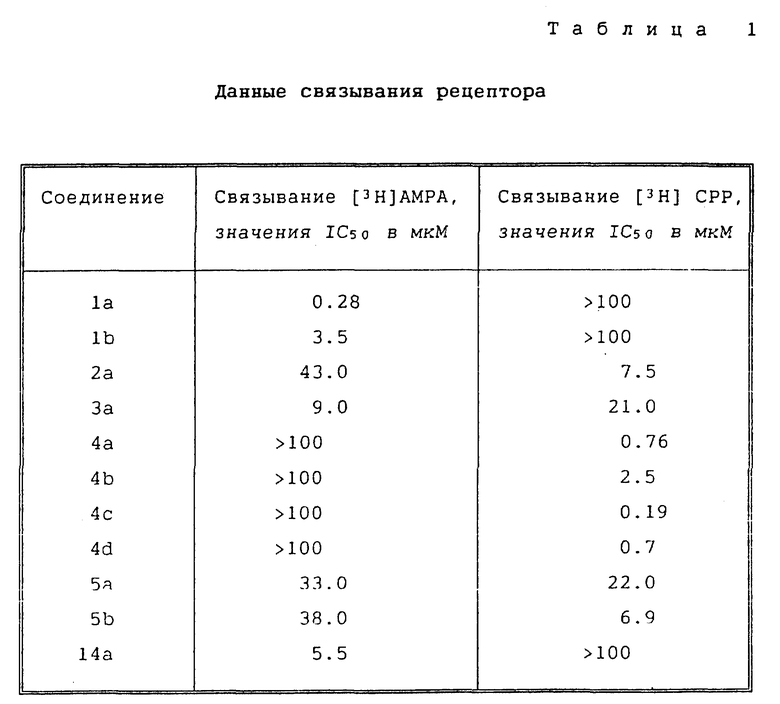
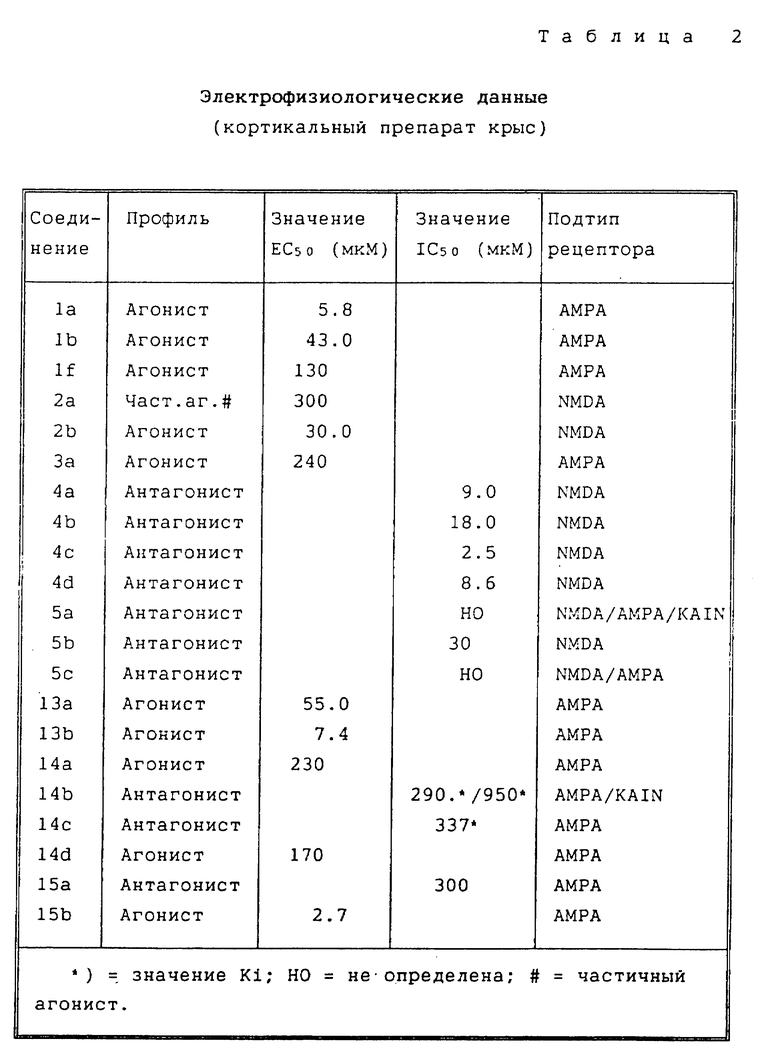
Описываются новые соединения 2-аминокарбоновой кислоты, замещенной 5-арилизоксазол-4-илом, имеющие общую формулу I где А представляет собой связь или пространственную группу С1-6 алкилен, В представляет собой -CH(NН2)-COOH или является группой формулы II; Е представляет собой O, СОО, O(CH2)n-COO (где n - целое число от 1 до 6) или 5-тетразолил; D представляет собой O; R1 представляет собой фенил, нафтил, тиенил, бензотиенил, пиридил, оксазолил, тиазолил, пиразолил, триазолил, тетразолил, хинолинил или фурил, или фенил, тиенил, пиразолил, триазолил, тетразолил, пиридил или фурил, замещенные одним или двумя заместителями, выбранными из группы, содержащей галоген, С1-6 алкил или трифторметил, при условии, что если А представляет собой метилен, В - группу-CH(NH2)-COOH, Е представляет собой O; D представляет собой O, а R1 - фенил или фенил, замещенный галогеном, то указанное соединение находится в форме чистого энантиомера; или фармацевтически приемлемая соль этих соединений. Новые соединения представляют собой сильные лиганды рецепторов ЕАА. 3 с. и 7 з.п.ф-лы, 2 табл.
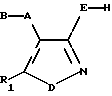
где А представляет собой связь или пространственную группу С1-6алкилен,
B представляет собой -СН(NН2)-СООН или является группой формулы II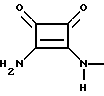
Е представляет собой О, СОО, O(СН2)n-СОО (где n - целое число от 1 до 6) или 5-тетразолил;
D представляет собой О;
R1 представляет собой фенил, нафтил, тиенил, бензотиенил, пиридил, оксазолил, тиазолил, пиразолил, триазолил, тетразолил, хинолинил или фурил, или фенил, тиенил, пиразолил, триазолил, тетразолил, пиридил или фурил, замещенные одним или двумя заместителями, выбранными из группы, содержащей галоген, С1-6алкил или трифторметил,
при условии, что если А представляет собой метилен, В -группу -СН(NН2)СООН, Е представляет собой О, D представляет собой О, а R1 - фенил или фенил, замещенный галогеном, то указанное соединение находится в форме чистого энантиомера, или фармацевтически приемлемая соль этих соединений.
| СПОСОБ ПОЛУЧЕНИЯ 3,5-ДИЗАМЕЩЕННЫХ ИЗОКСАЗОЛ-4-КАРБОНОВЫХ КИСЛОТ | 0 |
|
SU189434A1 |
| СПОСОБ ПОЛУЧЕНИЯ 5-МЕТИЛ-З-лг-АМИНОФЕНИЛИЗОКСАЗОЛ- 4-КАРБОНОВОЙ КИСЛОТЫ | 0 |
|
SU393279A1 |
| Способ получения 3,3-диамино-4,4дианилинодифенилсульфона | 1972 |
|
SU456519A1 |
| Гидропривод | 1973 |
|
SU440503A1 |
| из 4940575 А, 1990. | |||
