Настоящее изобретение относится к способам и композициям для лечения и профилактики артериосклероза и/или ксантомы.
В последнее время во всем мире отмечается тенденция к повышению частоты заболеваний коронарных артерий и артериосклероза, включая атеросклероз, даже в тех странах, в которых до настоящего времени они не превалировали. Среди факторов, ответственных за это повышение, можно отметить изменение образа жизни, включая "западную" диету, содержащую много мяса, и принятие этой диеты даже в тех странах, в которых она не является традиционной, а также общее увеличение среднего возраста популяции. В результате эти заболевания, и в частности артериосклероз, широко распространились, а артериосклероз хорошо известен как возможная причина внезапной смерти, например, в результате такого последствия артериосклероза, как инфаркт миокарда.
Одним из основных факторов риска, имеющих значение при развитии этих заболеваний, является высокий уровень липидов в плазме крови, в частности высокий уровень холестерина. Предпринималось множество попыток использовать агент, снижающий уровень холестерина, для предотвращения и лечения этих заболеваний, и было найдено много соединений, в большей или меньшей степени обладающих этим эффектом. Например, одно такоe соединение, которое хорошо известно и успешно применяется, представляет правастатин, который является агентом, регулирующим липиды, и ингибитором 3-гидрокси-3-метил-глутарил-карермент А редуктазы (в настоящем документе упоминается как "ингибитор ГМГ-КоА редуктазы"), и, как предполагают, действует на этап биосинтеза холестерина, определяющий его скорость. Сообщалось о том, что у кроликов, получающих правастатин, можно предотвратить коронарный артериосклероз и ксантому, однако его эффективность остается недостаточной (Biochimica et Biophysica Acta, 960, 294-302 (1988)). Эксперименты по контролю коронарного артериосклероза и ксантомы проводили, используя комбинацию двух агентов, регулирующих липиды, правастатина и холеcтирамина, который хорошо известен как агент для снижения уровней липопротеинов, однако эффективность этой комбинации также остается недостаточной (Atherosclerosis, 83, 69-80, 1 (1990)).
В патентной заявке Японии КоKai N Hei 7-41423 предполагается, что специфический класс агентов, улучшающих резистентность к инсулину, например троглитазон, может быть эффективным при лечении и профилактике артериосклероза, в частности атеросклероза, но и в этом случае эффективность подобных соединений не вполне удовлетворительна.
В настоящее время неожиданно было обнаружено, что применение комбинации одного или более ингибиторов ГМГ-КоА редуктазы и одного или более сенсибилизирующих к инсулину агентов демонстрирует синергический эффект и значительно успешнее предотвращает и/или лечит артериосклероз и/или ксантому, чем каждый из компонентов этой комбинации по отдельности. На самом деле, применяя новую комбинацию по настоящему изобретению, эти заболевания можно медленно, но надежно излечить.
Таким образом, целью настоящего изобретения является создание комбинации одного или более ингибиторов ГМГ-КоА редуктазы и одного или более сенсибилизирующих к инсулину агентов или агентов, улучшающих резистентность к инсулину.
Еще одной и более специфической целью настоящего изобретения является создание подобной комбинации, демонстрирующей синергический эффект.
Еще одной целью настоящего изобретения является создание способов и композиций, использующих подобную комбинацию, для предотвращения и/или лечения артериосклероза и/или ксантомы.
Другие цели и преимущества настоящего изобретения станут очевидными из последующего описания.
Таким образом, в первом аспекте настоящее изобретение заключается в способе предотвращения или лечения артериосклероза или ксантомы, который включает в себя введение пациенту, страдающему или предрасположенному к артериосклерозу или ксантоме, первого агента, выбранного из группы, состоящей из ингибиторов ГМГ-КоА редуктазы, и второго агента, выбранного из группы, состоящей из сенсибилизирующих к инсулину агентов; указанные первый и второй агенты вводят совместно или в течение такого периода времени, чтобы они действовали синергически.
Настоящее изобретение также относится к упакованной фармацевтической композиции для лечения или профилактики артериосклероза или ксантомы, которая содержит первый агент, выбранный из группы, состоящей из ингибиторов ГМГ-КоА редуктазы, и второй агент, выбранный из группы, состоящей из сенсибилизирующих к инсулину агентов; указанные первый и второй агенты находятся в смеси или находятся в отдельных упаковках.
В еще одном аспекте настоящее изобретение относится к фармацевтической композиции для лечения или профилактики артериосклероза или ксантомы, которая содержит первый агент, выбранный из группы, состоящей из ингибиторов ГМГ-КоА редуктазы, и второй агент, выбранный из группы, состоящей из сенсибилизирующих к инсулину агентов.
В настоящее время экспериментальные данные предполагают, что синергический эффект вызывается взаимодействием между механизмами действия двух классов соединений, ингибиторов ГМГ-КоА редуктазы и сенсибилизирующих к инсулину агентов, и, таким образом, предполагается, что химическое строение этих соединений имеет меньшее значение, чем их активность. Соответственно в качестве первого агента можно использовать любое соединение, обладающее ингибирующей активностью в отношении ГМГ-КоА редуктазы, в то время как в качестве второго агента можно использовать любое соединение, обладающее сенсибилизирующей к инсулину активностью.
Ингибиторы ГМГ-КоА редуктазы широко применяются для лечения или профилактики гиперлипемии и могут включать встречающиеся в природе вещества, образующиеся в ходе метаболизма микроорганизмов, полусинтетические вещества, выделенные из них, и полностью синтетические вещества. Среди этих соединений предпочтительными примерами могут служить правастатин, ловастатин, симвастатин, флувастатин, ривастатин и аторвастатин. Правастатин описан в японской патентной публикации N Sho 61-13699 и в патентах США N 4346227 и 4448979; его формула (в виде натриевой соли) 1,2,6,7,8,8а-гексагидро-6,8-тетрагидрокси- 2-метил-1-нафталингептаноат натрия. Ловастатин описан в японской патентной заявке КоKai N Sho 58-16875 и европейском патенте N 22478; его формула 6-{2-[1,2,6,7,8,8а-гексагидро-8-(2-метилбутирилокси)- 2,6-диметил-1-нафтил] этил} тетрагидро-4-гидрокси-2H-пиpaн-2-oн. Симвастатин описан в японской патентной заявке KoKai N Hei 1-1476 и в европейском патенте N 33538; его формула 6-{2-[1,2,6,7,8,8а-гексагидро-8-(2,2-диметилбутилокси)-2,6-диметил-1-нафтил] этил}-тетрагидро-4-гидрокси-2Н-пиран-2-он. Флавастатин описан в японской патентной публикации N Hei 2-46031 и в патенте США N 4739073; его формула (в виде соли натрия) 7-[3-(4- фторфенил)-1-метилэтил)-1Н-индол-2-ил)] -3,5-дигидрокси-6-гептаноат натрия. Ривастатин описан в японской патентной заявке KoKai N Hei 1-216974 и в патентах США N 5006530, 5169857 и 5401746; его формула (в виде соли натрия) 7-(4-фторфенил)-2,6-диизопропил-5- метоксиметилпиридин-3-ил)-3,5-дигидрокси-6-гептаноат натрия. Аторвастатин описан в японской патентной заявке КоКai N Hei 3-58967 и в патенте США N 5273995; его формула 2-(4-фторфенил)-5- (1-метилэтил)-3-фенил-4-(фенилкарбамоил)-1Н-пиррол-1-(2,4- дигидроксигексановая) кислота.
Сенсибилизирующий к инсулину агент, другой активный ингредиент согласно настоящему изобретению, может упоминаться также как агент, улучшающий резиcтентность к инсулину, и обычно используется для предотвращения и/или лечения диабета. Этот термин включает в себя большое множество соединений, обычно тиазолидиндионовые соединения, оксазолидиндионовые соединения и оксатиадиазоловые соединения.
Эти соединения описаны, например, в японских патентных заявках KoKai N Неi 4-69383 и Hei 7-330728, WO 89/08651, WO 91/07107, WO 92/02520, WO 94/01433 и патентах США N 4287200, 4340605, 4438141, 4444779, 4461902, 4572912, 4687777, 4703052, 4725610, 4873255, 4897393, 4897405, 4918091, 4948900, 5002953, 5061717, 5120754, 5132317, 5194443, 5223522, 5232925, 5260445; в европейском патенте N 676398 и т.д. Среди этих соединений предпочтительные примеры включают в себя троглитазон, пиоглитазон, энглитазон, ВRZ-49653, 5-(4-{ 2-[l-(4,2'-пиридилфенил)этилиденаминоокcи] этокси} бензил)-тиазолидин-2,4-дион (в настоящем документе "Соединение А"), 5-{-4-(5-метокси-3-метилимидазо [5,4-в] пиридин-2-ил-метокси)бензил} тиазолидин-2,4-дион (предпочтительно в виде его гидрохлорида), 5-[4-(6-метокси-1-метилбензимидазол-2-ил-метокси)бензил -тиазолидин-2,4-дион, 5-4-(1-метилбензимидазол-2-ил-метокси)бензил -тиазолидин-2,4-дион и 5-4-(5-гидрокси-1,4,6,7- тетраметилбензимидазол-2-ил-метокси)бензил] тиазолидин-2,4-дион. Троглитазон описан в японской патентной публикации N Неi 2-31079 и в патенте США N 4572912; его формула 5-{4-[(6-гидрокси-2,5,7,8- тетраметилхроман-2-ил)-метокси] бензил} -2,4-тиазолидиндион. Пиоглитазон описан в японской патентной публикации N Sho 62-42903 и N Hei 5-66956 и в патентах США N 4287200, 4340605, 4438141, 4444779 и 4725610; его формула 5-{4-[2-(5-этилпиридин-2-ил)этокси} бензил} -2,4-тиазолидиндион. Энглитазон описан в японской патентной публикации N Hei 5-86953 и в патенте США N 4703052; его формула 5-[3,4-дигидро-2-(фенилметил)-2Н-бензопиран-6-ил-метил] -2,4-тиазолидиндион. BRZ-49653 описан в японской патентной заявке KoKai N Неi 1-131169 и в патентах США N 5002953, 5194443, 5232925 и 5260445; его формула 5-{4-[2-метил-2-(пиридин-2-ил-амино)этокси] бензил} 2,4-тиазолидиндион. Соединение А описано в европейском патенте N 708098. 5-{4-(5-метокои-3-метилимидазо[5,4-в]пиридин-2-ил- метокси)бензил}-тиазолидин-2,4-дион (и его гидрохлорид) описаны в японской патентной заявке KoKai N Hei 7-330728 и в европейском патенте N 676398. Вышеназванные соединения можно получить согласно известным способам, упомянутым выше. 5-[4-(6-метокси-1- метилбензимидазол-2-ил-метокси)бензил] тиазолидин-2,4-дион, 5-[4-(1-метилбензимидазол-2-ил-метокси)бензил] тиазолидин-2,4-дион и 5-[4-(5-гидрокси-1,4,6,7-тетраметилбензимидазол-2-ил-метокси) бензил] тиазоилидин-2,4-дион описаны в европейской патентной заявке N 96303940.9, и могут быть получены, как описано в этих документах.
Активные ингредиенты, которые используются в настоящем изобретении, включают, во-первых, один или более ингибиторов ГМГ-КоА редуктазы, и, во-вторых, один или более сенсибилизирующих к инсулину агентов или агентов, улучшающих резистентность к инсулину. Согласно настоящему изобретению комбинация ингибитора ГМГ-КоА редуктазы и сенсибилизирующего к инсулину агента демонстрирует синергический эффект по сравнению с применением каждого из ингредиентов в отдельности, как показано ниже. Интересно, что этот синергизм проявляется даже в том случае, когда соединения этих двух классов не всегда присутствуют в организме одновременно. Иными словами, синергический эффект наблюдается даже когда концентрация одного из соединений этих двух классов в крови меньше, чем требуется самому этому соединению, чтобы проявить сколько-нибудь значительный эффект. Полагают, хотя это всего лишь предположение, что когда соединение одного из двух классов вводится в организм и транспортируется к рецепторам, оно приводит в действие "переключатель" in vivo. Спустя некоторое время уровень этого соединения в крови может понизиться до значений, при которых эффект более не должен наблюдаться, однако "переключатель" все еще может действовать, поддерживая, таким образом, превентивный и/или терапевтический эффект в отношении артериосклероза и/или ксантомы, присущий соединениям этого класса. Когда пациенту в этом состоянии вводят соединение другого класса, эффект по предотвращению и/или лечению артериосклероза и/или ксантомы может комбинироваться с эффектом предыдущего введения другого соединения, и эффекты двух соединений благоприятным образом действуют синергически. Разумеется, очевидно, что в клинической практике может быть удобным также введение двух соединений одновременно. Таким образом, ингибитор ГМГ-КоА редуктазы и сенсибилизирующий к инсулину агент можно вводить одновременно в форме комбинированного препарата. Альтернативно, если смешать эти два агента трудно, из-за несовместимости или по другим причинам, например из-за трудностей технологии смешивания, эти два активных агента можно вводить отдельно в форме разовых доз. Как описано выше, поскольку соединения этих двух классов демонстрируют при совместном применении синергический эффект, их можно вводить почти одновременно или с удобными интервалами. Максимальный интервал, приемлемый для введения соединений этих двух классов для достижения синергического эффекта по настоящему изобретению, может быть установлен клинической практикой или экспериментами на животных.
Ингибиторы ГМГ-КоА редуктазы и сенсибилизирующие к инсулину агенты по настоящему изобретению могут вводиться в основном перорально. Соответственно соединения двух классов могут изготавливаться раздельно, в виде двух стандартных лекарственных форм, или могут физически смешиваться с получением разовой стандартной лекарственной формы. Примеры таких составов включают, например, порошки, гранулы, таблетки или капсулы. Эти фармацевтические препараты можно производить обычными способами, хорошо известными в фармации.
В настоящем изобретении индивидуальные дозы ингибиторов ГМГ-КоА редуктазы и сенсибилизирующих к инсулину агентов, и соотношение между количествами ингибиторов ГМГ-КоА редуктазы и сенсибилизирующих к инсулину агентов могут широко варьировать в зависимости от активности каждого соединения и других факторов, таких как состояние, возраст и вес тела пациента. Например, в случае сенсибилизирующего к инсулину агента, сила BRZ-49653 почти в 100 раз больше, чем у троглитазона in vivo у животных со смоделированным диабетом, в результате чего доза этих двух соединений может теоретически отличаться почти на два порядка по величине и на практике отличаться почти на порядок по величине.
Доза каждого из ингибиторов ГМГ-КоА редуктазы и сенсибилизирующих к инсулину агентов, если они применяются для лечения артериосклероза или ксантомы, в большинстве случае будет ниже той дозы, которая применяется при раздельном использовании этих двух соединений для обычных целей, т.е. в качестве антигиперлипидемических и антидиабетических агентов. Их дозы далее снижаются до некоторой степени благодаря синергическому эффекту комбинации соединений этих двух классов. Например, если правастатин и троглитазон применяются в соответствии с настоящим изобретением, их суточные дозы предпочтительно находятся в пределах от 1 до 40 мг и от 1 до 500 мг соответственно по сравнению с пределами от 5 до 80 мг и от 10 до 1000 мг соответственно, если эти соединения применяются для обычных целей в качестве антигиперлипидемических и антидиабетических агентов.
В большинстве случаев, несмотря на то что, как отмечалось выше, дозы ингибиторов ГМГ-КоА редуктазы и сенсибилизирующих к инсулину агентов по настоящему изобретению могут широко варьировать, суточная доза обычно находится в пределах от 0,01 до 40 мг, предпочтительно от 1 до 40 мг, и от 0,05 до 500 мг, предпочтительно от 1 до 500 мг соответственно.
Соотношения между соединениями этих двух классов также может широко варьировать, однако предпочтительно, чтобы соотношение между ингибитором ГМГ-КоА редуктазы и сенсибилизирующим к инсулину агентом находилось в пределах от 1:200 до 200:1 по весу, предпочтительно от 1:100 до 10:1 и более предпочтительно от 1:50 до 5:1 по весу.
Ингибитор ГМГ-КоА редуктазы и сенсибилизирующий к инсулину агент в соответствии с настоящим изобретением предпочтительно вводят одновременно или почти одновременно в суточных дозах, описанных выше, и могут вводиться разовой или разделенными дозами.
Соединения и композиции по настоящему изобретению могут вводиться в различных формах в зависимости от заболевания или нарушения, подвергаемого лечению, и возраста, состояния и веса тела пациента, что хорошо известно специалистам. Например, в случае, если соединения или композиции должны вводиться перорально, они должны быть изготовлены в форме таблеток, капсул, гранул, порошков или сиропов; или для парентерального введения они должны быть изготовлены в форме растворов для инъекций (внутривенных, внутримышечных или подкожных), препаратов для капельной инфузии или в форме суппозиториев. Для нанесения на слизистую оболочку глаза они должны быть изготовлены в форме глазных капель или глазных мазей. Эти препараты могут быть изготовлены обычными способами и, если это желательно, активный ингредиент можно смешивать с любым обычным вспомогательным веществом, таким как наполнитель, связывающий агент, дезинтегрирующий агент, лубрикант, корригент, солюбилизирующий агент, суспендирующий агент, эмульгатор или покрывающий агент.
Примерами наполнителей, которые можно применять, являются органические наполнители, такие как производные сахаров, включая лактозу, сахарозу, глюкозу, маннит и сорбит; производные крахмала, такие как кукурузный крахмал, картофельный крахмал, α-крахмал, декстрин и карбоксиметилкрахмал, производные целлюлозы, такие как кристаллическая целлюлоза; малозамещенная гидроксипропилцеллюлоза, гидроксипропилметилцеллюлоза, карбоксиметилцеллюлоза, карбоксиметилцеллюлоза-кальций и карбоксиметилцеллюлоза-натрий с внутренними мостиками; аравийская камедь; декстран; Pulluane и неорганические наполнители, включая производные кремния, такие как легкий ангидрид кремниевой кислоты, синтетический силикат алюминия и алюминат метасиликат магния; фосфаты, такие как фосфат кальция; карбонаты, такие как карбонат кальция, и сульфаты, такие как сульфат кальция.
Смазывающие агенты, которые можно применять, включают стеариновую кислоту, стеараты металлов, такие как стеарат кальция и стеарат магния; тальк; коллоидный диоксид кремния; воски, такие как пчелиный воск и спермацетовый воск; борную кислоту; адипиновую кислоту; сульфаты, такие как сульфат натрия; гликоль; фумаровую кислоту; бензоат натрия; DZ-лейцин; натриевые соли жирных кислот; лаурилсульфаты, такие как лаурилсульфат натрия и лаурилсульфат магния; силикаты, такие как ангидрид кремниевой кислоты и гидрат кремниевой кислоты и ранее упомянутые производные крахмала.
Связывающие агенты, которые можно применять, включают поливинилпирролидон; макрогол; и те же соединения, которые упоминались выше как наполнители.
Дезинтегрирующие агенты, которые можно применять, включают те же соединения, которые упоминались выше как наполнители; и химически модифицированные крахмалы и целлюлозы, такие как кросскармеллоза-натрий, карбоксиметил-крахмал-натрий и поливинилпирролидон с внутренними мостиками.
Стабилизаторы, которые можно применять, включают параоксибензоаты, такие как метилпарабен и пропилпарабен; спирты, такие как хлорбутанол, бензиловый спирт и фенилэтиловый спирт; хлорид бензалкония; фенолы, такие как фенол и крезол; тимерозал; дегидроуксусную кислоту и сорбиновую кислоту.
Корригенты, которые можно применять, включают подсластители, подкислители и специи.
Настоящее изобретение далее иллюстрируется следующими примерами, которые демонстрируют увеличенную активность, которая достигается синергической комбинацией по настоящему изобретению. Помимо этого последующие композиции иллюстируют фармацевтические композиции, которые можно приготовить, и приготовления, которые иллюстируют приготовление некоторых сенсибилизирующих к инсулину агентов, применяемых в настоящем изобретении.
Пример 1.
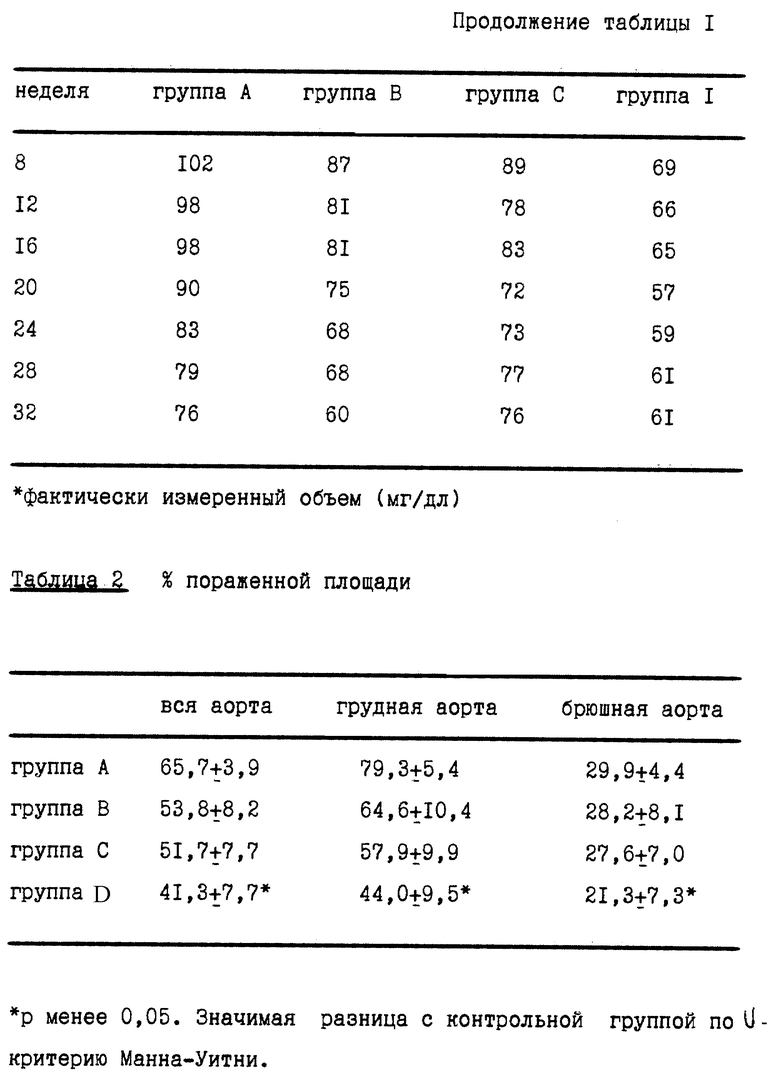
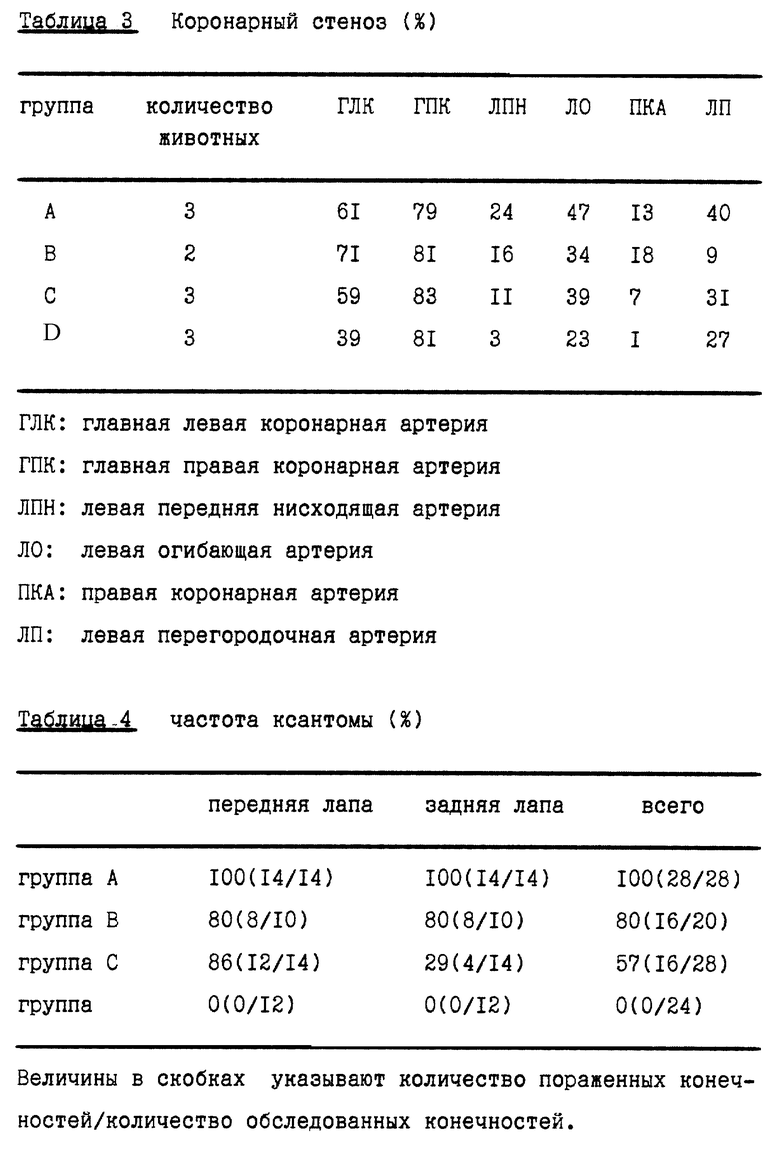
Кроликов НГЛВ (2-3-месячного возраста, кролики с наследственной гиперлипидемией Ватанабе, описанные в Biochimica et Biophysica Acta, 960, 294-302 (1988)) слепым методом отбирали в контрольную группу, (7 животных, группа A), группу, получающую только правастатин (5 животных, группа B), группу, получающую только троглитазон (7 животных, группа C), и группу, получающую комбинацию обоих активных веществ (6 животных, группа D). Правастатин вводили перорально с помощью желудочного зонда в дозе 50 мг/кг/день один раз в день, и троглитазон давали с кормом, содержащим 100 мг/кг вещества, в течение 32 недель. Потребляемое количество ограничивали до 120 г на кролика в день. У животных брали кровь непосредственно перед экспериментом и спустя 4, 8, 12, 16, 24, 28 и 32 недели после начала эксперимента, и в каждом образце определяли общий уровень холестерина (мг/дл). Эти уровни выражали в процентах (%) от уровней, определенных непосредственно перед началом эксперимента. Результаты представлены в табл. 1. Животных умерщвляли и вскрывали через 32 недели для определения (а) процента пораженных площадей (%) в целом, в грудной или брюшной части аорты; (в) стеноза (%) коронарных артерий и (с) частоты (%) ксантомы в межфаланговых суставах.
Результаты представлены в табл. 2, 3 и 4. Измеренные величины представлены как среднее значение+стандартная ошибка.
Как видно из приведенного примера, не наблюдалось значимой разницы в изменении уровней холестерина в плазме через 32 недели эксперимента между группой D (которая получала комбинацию обоих агентов) и группой B (которая получала только правастатин). Напротив, в соотношении процента площади поражений (площадь поражений/общая площадь артерии в %) наблюдался отчетливый синергизм при сравнении группы D (комбинированное лечение) и групп В и С (лечение одним агентом), как показано выше. Наблюдался синергизм в предотвращении стеноза коронарных артерий в левой передней нисходящей артерии, левой огибающей артерии и правой коронарной артерии. Развитие ксантомы в межфаланговых суставах было полностью предотвращено в группе D, что продемонстрировало, таким образом, отчетливый синергизм.
Таким образом, несмотря на то что не было обнаружено значимой разницы в уровнях холестерина плазмы между группами, получавшими комбинацию ингибитора ГМГ-КоА редуктазы и сенсибилизирующего к инсулину агента, и группами, получавшими только один активный агент, комбинация обоих активных агентов синергически предотвращала прогрессирование артериосклероза, в частности в грудной аорте. Эти результаты невозможно было представить исходя из ранее известных данных в этой области.
Пример 2.
Самцов кроликов НГЛВ (с наследственной гиперлипидемией Ватанабе), почти не имевших артериальных поражений, отбирали слепым методом в контрольную группу (7 животных, группа А), группу, получавшую только правастатин перорально (6 животных, 50 мг/кг, группа В), группу, получавшую только пиоглитазон перорально (7 животных, 20 мг/кг, группа С), группу, получавшую перорально 5-(4-{ 2-[1-(4-2'-пиридилфенил) этилиденаминоокси]этокси}бензил)тиазолидин-2,4-дион (в настоящем документе - соединение А, как описано в ЕР 708098, 7 животных, 10 мг/кг, группа D), группу, получавшую перорально комбинацию правастатина и пиоглитазона (6 животных, 50 + 20 мг/кг, группа Е), и группу, получавшую перорально комбинацию правастатина и соединения А (7 животных, 50 + 10 мг/кг, группа F).
Каждое испытуемое соединение вводили в течение восьми месяцев кроликам в форме водной суспензии (0,5%-ной добавленной карбоксиметилцеллюлозы). В контрольной группе вводили только 0,5%-ный раствор карбоксиметилцеллюлозы. Спустя один месяц после начала введения и далее было обнаружено, что уровень сывороточного холестерина в группах В, E и F поддерживался на более низком уровне, чем в контрольной группе, и в этих группах наблюдалось снижение уровней сывороточного холестерина на 22-34%. Тем не менее в группах С и D не наблюдалось снижения уровней сывороточного холестерина.
Процент площади поражений в дуге аорты и во всей аорте представлен в табл. 5. Фактически измеренные величины представлены здесь как среднее плюс или минус стандартная ошибка. Числа в скобках представляют процент площади поражений от контрольной группы; *р менее 0,05, **р менее 0,01; значимая разница по сравнению с контролем по U-критерию Манна-Уитни.
Значимая разница (р менее 0,05) наблюдалась между группами В и F, между группами С и Е и между группами D и F в дуге аорты, и между группами С и Е во всей аорте.
Измеряли среднее утолщение интимы аорты; результаты представлены в табл. 6. Фактически измеренные величины представлены здесь как среднее плюс или минус стандартная ошибка (мкм). Числа в скобках представляют процент утолщения интимы от контрольной группы. Значимая разница наблюдалась между группами D и F в дуге аорты и между группами С и Е и между группами D и F во всей аорте (p менее 0,05) по U-критерию Манна-Уитни. Среднее утолщение интимы подсчитывалось по поперечному сечению интимы аорты в дуге аорты и по двум поперечным сечениям в грудной и брюшной частях, деленным на длину средней оболочки.
Незначительное подавление утолщения интимы наблюдалось в группе В, в то время как в группах С и D не наблюдалось подавления гипертрофии. В противоположность группам С и D в группах Е и F наблюдалось подавление утолщения интимы.
Определялось содержание холестерина в аорте. Среднюю и внутреннюю оболочки дуги аорты и грудной и брюшной аорты отслаивали пинцетом и нарезали на кусочки. Эти кусочки экстрагировали смесью 2:1 по объему хлороформа и метанола.
Фазу хлороформа отделяли и выпаривали досуха, а остаток растворяли в изопропаноле. Общий холестерин и свободный холестерин определяли обычным ферментным способом. Результаты представлены в табл. 7а и 7в.
Данные представлены как среднее плюс или минус стандартная ошибка (мг/г ткани). Величины в скобках представляют процент от контроля. Значимая разница по сравнению с контрольной группой наблюдалась по непарному t-критерию Стьюдента: *p менее 0,05; **р менее 0,02.
Как следует из табл. 7, уровни общего холестерина в грудной и брюшной аорте были ниже в группах E и F, чем в группах В, С и D. Не было отчетливого тренда между уровнями свободного и этерифицированного холестерина. Эти результаты сходны с таковыми для интенсивности поражений.
Определяли частоту и степень выраженности ксантомы на четырех конечностях. Результаты представлены в табл. 8.
Как видно из табл. 8, в контрольной группе ксантома встречалась в 100% случаев для всех передних и задних конечностей. В группах B, C и D частота ксантомы была несколько меньше. В группах E и F, получавших комбинацию активных агентов, частота ксантомы была значимо ниже. Такая же тенденция и в случае массивной ксантомы, где в группах E и F не наблюдалась или была низкая частота ксантомы по сравнению с группами от A до D.
Смысл этих результатов в том, что две комбинации правастатина, ингибитора ГМГ-КоА редуктазы, и одного из тиазолидиндионовых сенсибилизирующих к инсулину агентов продемонстрировали синергические эффекты при лечении атеросклероза и наличие ксантомы.
Пример 3.
Изучали синергический эффект ингибиторов ГМГ-КоА редуктазы и тиазолидиндионовых сенсибилизирующих к инсулину агентов в отношении регрессии уже существующих атеросклеротических поражений у кроликов, которым давали холестерин.
Самцов новозеландского белого кролика (5-месячного возраста) в течение двух месяцев кормили рационом с 2%-ным содержанием холестерина, и в конце этого срока сывороточный холестерин у этих кроликов возрос до 1,100-4,100 мг/дл.
Кроликов распределяли слепым способом по группам (3-9) животных на группу и давали им испытуемые соединения перорально в течение двух месяцев, в то время как они получали нормальный по содержанию липидов рацион. Дозировки испытуемых соединений составляли: в случае одного правастатина 3 мг/кг или 5 мг/кг; в случае одного флувастатина 0,8 мг/кг или 1,5 мг/кг; в случае одного троглитазона 10 мг/кг; в случае только соединения A 2,5 мг/кг. В случае комбинаций активных агентов дозировки составляли: правастатин 3 мг/кг + троглитазон 10 мг/кг; правастатин 5 мг/кг + соединение А 2,5 мг/кг; флувастатин 0,8 мг/кг + троглитазон 10 мг/кг и флувастатин 1,5 мг/кг + соединение А 2,5 мг/кг.
Результаты представляют процент пораженной площади в грудной аорте (табл. 9).
Фактически измеренные величины представлены здесь как среднее ± стандартная ошибка. Величины в скобках представляют процент уменьшения поражений по сравнению с контрольной группой.
Как видно из табл. 9, каждый из ингибиторов ГМГ-КоА редуктазы или тиазолидиндионовых сенсибилизирующих к инсулину агентов в отдельности не вызывал или вызывал малое уменьшение поражений, в то время как все комбинации этих двух компонентов демонстрировали синергическое уменьшение поражений.
Пример 4.
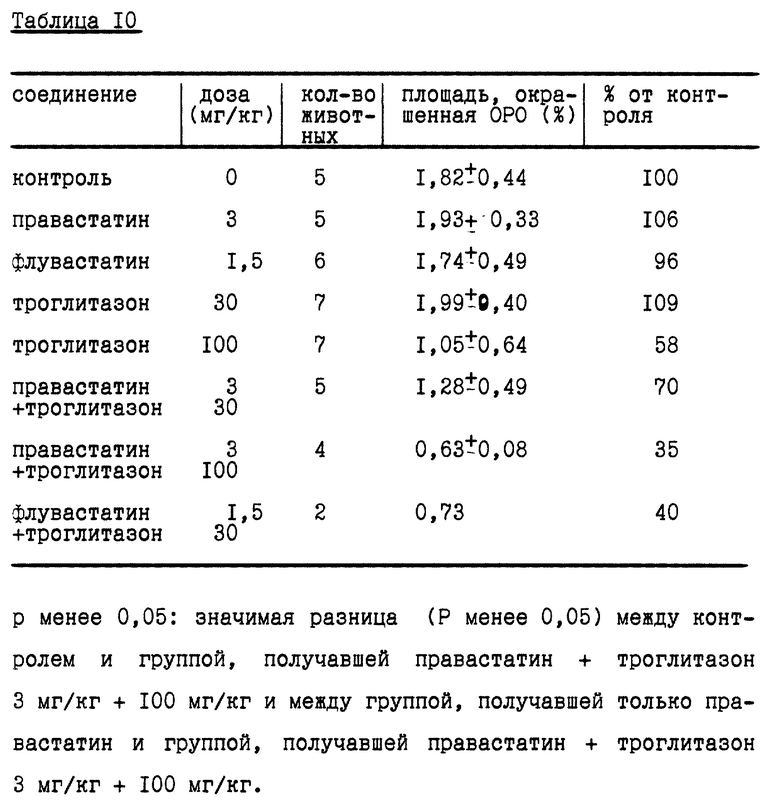
Синергизм ингибиторов ГМГ-КоА редуктазы и тиазолидиндиновых сенсибилизирующих к инсулину агентов изучали на другой модели регрессии, т.е. регрессии ранее сформированного атеросклероза у хомячков. Самцам хомяков F1b (весом около 130 г) давали рацион, содержавший 0,05% холестерина, в течение 13 недель. Из этих животных слепым способом сформировали группы (2-7 животных на группу), а затем давали испытуемые препараты в течение 4 недель, в то время как хомячки получали рацион с нормальным содержанием липидов. Правастатин и флувастатин смешивали с питьевой водой в дозе 3 и 1,5 мг/кг соответственно, в то время как троглитазон смешивали с кормом в дозе 30 или 100 мг/кг.
В случае комбинированных групп дозировка составляла 3 мг/кг + 30 мг/кг или 3 мг/кг + 100 мг/кг для группы правастатин + троглитазон и 1,5 мг/кг + 30 мг/кг для группы флувастатин + троглитазон.
Артериальные повреждения оценивали протяженностью площади, окрашивавшейся красным маслом О (ORO), как описано в Atherosclerosis, 114, 19-28 (1995). А именно, дугу аорты окрашивали ORO для приготовления образцов анфас. Вычисляли процент площади, которая окрашивалась ORO, от всей площади, что и представляло степень поражения аорты.
После лечения между группами не наблюдалось значимой разницы по уровню общего холестерина и триглицеридов сыворотки крови. Результаты представлены в табл. 10.
Как видно из табл. 10, не наблюдалось регрессии аортальных поражений в группах, получавших правастатин, флувастатин или троглитазон (30 мг/кг) по отдельности, хотя наблюдалась регрессия при применении троглитазона в отдельности в дозе 100 мг/кг.
В случае комбинации правастатина и троглитазона наблюдалась регрессия с дозозависимой в отношении троглитазона тенденцией. В случае комбинации флувастатина и троглитазона наблюдалась сходная синергическая регрессия аортальных поражений.
Суммируя, можно заключить, что комбинация ингибитора ГМГ-КоА редуктазы и тиазолидиндионового сенсибилизирующего к инсулину агента демонстрирует как класс профилактический и лечебный эффекты в отношении атеросклероза и ксантомы.
Приготовление 1
5-[4-(1-метилбензимидазол-2-ил-метокси)бензил-тиазолидин-2,4-дион
1(а) метил-4-нитрофеноксиацетат
Смесь 56 г 4-нитрофенола, 90 г метилбромацетата, 100 г карбоната калия и 50 мл диметилформамида перемешивали при комнатной температуре в течение двух дней. По окончании этого времени растворитель удаляли дистилляцией при пониженном давлении. Полученный остаток смешивали с водой, и водную смесь экстрагировали этилацетатом. Экстракт промывали водой и высушивали над безводным сульфатом натрия, после чего растворитель удаляли дистилляцией при пониженном давлении. Полученный остаток стирали с гексаном с получением 63,3 г указанного в заголовке соединения, которое плавилось при 98-99oC.
1(b) метил-4-аминофеноксиацетат
Раствор 30,8 г метил-4-нитрофеноксиацетата (приготовленного как описано в этапе (а) выше) в 50 мл метанола встряхивали в атмосфере водорода и в присутствии 5,0 г 10% в/в палладия на древесном угле в течение 6 ч. По окончании этого времени реакционную смесь фильтровали, а фильтрат концентрировали выпариванием при пониженном давлении с получением 25,8 г указанного в заглавии соединения, имеющего величину Rf = 0,79 (на тонкослойной хроматографии на силикагеле; выделяющий растворитель этилацетат).
1(с) метил-4-(2-бром-2-бутоксикарбонилэтил-1-ил)-феноксиацетат
98 г 47% в/в водного раствора бромистоводородной кислоты, а затем 33 мл водного раствора, содержавшего 12,8 г нитрита натрия, добавляли к раствору 25,8 г метил-4-амино-феноксиацетата (приготовленного как описано в этапе (b) выше) в 263 мл смеси 2:5 по объему метанола и ацетона, охлаждая во льду, и полученную смесь перемешивали, охлаждая во льду, в течение 30 мин. Затем добавляли 18,2 г бутилакрилата, и реакционную смесь перемешивали еще 30 мин, охлаждая во льду. Затем к смеси добавляли 3,2 г бромида меди (1), и смесь перемешивали в течение ночи при комнатной температуре. По окончании этого времени реакционную смесь освобождали от растворителя путем дистилляции при пониженном давлении, а остаток смешивали с водным раствором хлорида натрия. Затем его экстрагировали этилацетатом. Этот экстракт промывали водным раствором хлорида натрия и высушивали над безводным сульфатом натрия. После удаления растворителя дистилляцией получали 51,7 г указанного в заголовке соединения, имеющего величину Rf = 0,46 (на тонкослойной хроматографии на силикагеле; выделяющий растворитель смесь 5:1 по объему гексана и этилацетата) в виде сырого продукта.
1(d) 5-[4-(этоксикарбонилметокси)бензил]тиазолидин-2,4- дион
Смесь 100 г метил-4-(2-бром-2-бутоксикарбонилэтил-1- ил)феноксиацетата (приготовленного как описано в этапе (с) выше), 22 г тиомочевины и 200 мл этанола нагревали с обратным холодильником в течение 2,5 ч, после чего к реакционной смеси добавляли 2Н водный раствор хлористоводородной кислоты. Смесь затем нагревали с обратным холодильником в течение 5 ч. По окончании этого времени реакционную смесь освобождали от растворителя дистилляцией при пониженном давлении. Полученный остаток разводили водой, и водную смесь экстрагировали этилацетатом. Экстракт высушивали над безводным сульфатом магния, после чего растворитель удаляли дистилляцией при пониженном давлении. Полученный остаток очищали колоночной хроматографией на силикагеле с 2:5 смесью по объему этилацетата и гексана в качестве элюента с получением 19,4 г указанного в заглавии соединения с т.пл. 105-106oC.
1(е) 5-[4-(1-метилбензимидазол-2-ил-метокси)бензил]- тиазолидин-2,4-дион
Смесь 1,0 г N-метил-1,2-фенилендиамина, 3,8 г 5-[4-(этоксикарбонилметокси)бензил] тиазолидин-2,4-диона (приготовленного как описано в этапе (d)) выше), 20 мл концентрированного водного раствора хлористоводородной кислоты, 10 мл 1,4-диоксана и 10 мл воды нагревали с обратным холодильником в течение 5 ч. По окончании этого времени нерастворимые материалы, выпавшие в осадок из реакционной смеси, собирали фильтрованием, и полученный таким способом осадок растворяли в тетрагидрофуране. Затем к раствору добавляли воду. Полученную водную смесь нейтрализовали добавлением гидрокарбоната натрия и затем экстрагировали этилацетатом. Экстракт промывали водным раствором хлорида натрия и высушивали над безводным сульфатом натрия. Растворитель удаляли выпариванием при пониженном давлении, а полученный остаток очищали колоночной хроматографией на силикагеле, используя этилацетат, а затем этанол в качестве элюента. Этот продукт затем дважды перекристаллизовывали из смеси тетрогидрофурана и эталацетата с получением 1,3 г указанного в заглавии соединения с т. пл. 230-231oC.
Приготовление 2
5- [4-(6-метокси-1-метилбензимидазол-2-ил- метокси)-бензил]тиазолидин-2,4-дион
2(а) 5-метокси-2-нитроанилин
70 мл 28% в/о раствора метоксида натрия в метаноле добавляли при комнатной температуре к раствору 25 г 5-хлор-2- -нитроанилина в 500 мл 1,4-диоксана, и полученную смесь нагревали с обратным холодильником в течение 4 ч, после чего растворитель удаляли дистилляцией при пониженном давлении. Полученный остаток разводили водой, и полученную водную смесь экстрагировали этилацетатом. Экстракт промывали насыщенным водным раствором хлорида натрия и высушивали над безводным сульфатом натрия, после чего растворитель удаляли дистилляцией при пониженном давлении. Полученный остаток очищали колоночной хроматографией на силикагеле, используя метод градиентного элюирования, со смесями этилацетата и гексана от 1:4 до 1:2 по объему в качестве элюента с получением 16,3 г указанного в заглавии соединения с т.пл. 124-128oC.
2(b) N-т-бутоксикарбонил-5-метокси-2-нитроанилин
25 г ди-т-бутилбикарбоната, 15 мл пиридина и 0,6 г 4-диметиламинопиридина добавляли при комнатной температуре к раствору 16 г 5-метокси-2-нитроанилина (приготовленного как описано в этапе (а) выше) в 500 мл дегидратированного тетрагидрофурана, и полученную смесь перемешивали в течение 2 ч. По окончании этого времени реакционную смесь освобождали от растворителя дистилляцией при пониженном давлении, и полученный остаток разводили водой. Полученную водную смесь экстрагировали этилацетатом. Экстракт промывали насыщенным водным раствором хлорида натрия и высушивали над безводным сульфатом натрия, после чего растворитель удаляли дистилляцией при пониженном давлении. Полученный остаток очищали колоночной хроматографией на силикагеле, используя смесь 1:10 по объему этилацетата и гексана в качестве элюента, с получением 12,5 г указанного в заглавии соединения с т.пл. 112-114oC.
2(с) N-т-бутоксикарбонил-N-метил-5-метокси-2-нитроанилин
Раствор 49,6 г N-т-бутоксикарбонил-5-метокси-2-нитроаналин (приготовленного как описано в этапе (b) выше) в 300 мг дегидратированного диметилформамида добавляли, охлаждая во льду, к суспензии 12,0 г гидрида натрия (в виде 55% в/в дисперсии в минеральном масле) в 300 мл дегидратированного диметилформамида, и полученную смесь перемешивали при комнатной температуре в течение 30 мин, после чего добавляли 17,2 мл метилиодида при комнатной температуре. Реакционную смесь перемешивали в течение 1 ч, после чего ее оставляли стоять на ночь при комнатной температуре. Затем ее концентрировали приблизительно до одной пятой ее первоначального объема путем выпаривания при пониженном давлении. Этот концентрат смешивали со льдом-водой, и полученную водную смесь экстрагировали этилацетатом. Экстракт промывали водой и насыщенным водным раствором хлорида натрия в указанном порядке, после чего его высушивали над безводным сульфатом натрия. После удаления растворителя дистилляцией получали 52,1 г указанного в заглавии соединения с т.пл. 122-124oC.
2(d) N-метил-5-метокси-2-нитроаналин
750 мл 4Н раствора хлористого водорода в 1,4-диоксане добавляли к 52 г N-т-бутоксикарбонил-N-метил-5-метокси-2-нитроанилина (приготовленного как описано в этапе (с) выше) при комнатной температуре, и полученную смесь перемешивали в течение 2 ч. По окончании этого времени реакционную смесь освобождали от растворителя дистилляцией при пониженном давлении, и полученный остаток смешивали с водой и этилацетатом. Эту смесь затем нейтрализовали добавлением гидрокарбоната натрия, после чего ее экстрагировали этилацетатом. Экстракт промывали насыщенным водным раствором хлорида натрия и высушивали над безводным сульфатом натрия. После удаления растворителя дистилляцией получали 35,3 г указанного в заглавии соединения с т.пл. 107-110oC.
2(е) 5-метокси-N-метил-1,2-фенилендиамин
346 г хлорида олова (2) добавляли к смеси 35 г N-метил-5-метокси-2- нитроанилина (приготовленного как описано в этапе (d) выше), 900 мл т-бутанола и 100 мл этилацетата при комнатной температуре, и полученную смесь перемешивали при 60oC в течение 2 ч, после чего добавляли порциями 11 г боргидрида натрия при 60oC приблизительно в течение 1 ч. Реакционную смесь затем перемешивали при 60oC в течение 3 ч, после чего ее оставляли стоять при комнатной температуре в течение двух дней. Затем ее выливали в лед-воду, и водную смесь нейтрализовали добавлением гидрокарбоната натрия. Смесь экстрагировали этилацетатом, и экстракт промывали насыщенным водным раствором хлорида натрия и высушивали над безводным сульфатом натрия. Растворитель удаляли из смеси путем дистилляции при пониженном давлении, и полученный остаток очищали колоночной хроматографией на силикагеле, используя смесь 3:2 по объему этилацетата и гексана в качестве элюента, с получением 21,9 г указанного в заглавии соединения, имеющего величину Rf = 0,18 (на тонкослойной хроматографии на силикагеле; выделяющий растворитель смесь 1:1 по объему этилацетата и гексана).
2(f) 5-(4-метоксикарбонилметоксибензил)-3-трифенилметил- тиазолидин-2,4-дион
126 г карбоната цезия добавляли при комнатной температуре к раствору 120 г 5-(4-гидроксибензил)-3- трифенилметилтиазолидин-2,4-диона в 2,5 л ацетона, после чего добавляли также 36 мл метилбромацетата, и полученную смесь перемешивали в течение 1 ч. По окончании этого времени реакционную смесь освобождали от растворителя путем дистилляции при пониженном давлении, и полученный остаток смешивали с водой. Эту водную смесь затем экстрагировали этилацетатом. Экстракт промывали водой, а затем насыщенным водным раствором хлорида натрия, после чего его высушивали над безводным сульфатом магния. Растворитель удаляли путем дистилляции при пониженном давлении, после чего к маслянистому остатку добавляли 1 л диэтилового эфира. Смесь затем перемешивали ультразвуком в течение 10 мин. Выпавшее в осадок твердое вещество собирали фильтрованием с получением 126,3 г указанного в заглавии соединения с т.пл. 158-162oC.
2(g) 5-(4-метоксикарбонилметоксибензил)тиазолидин-2,4-дион
1700 мл уксусной кислоты, а затем 400 мл воды добавляли при комнатной температуре к суспензии 344 г 5-(4- метоксикарбонилметоксибензил)-3-трифенилметилтиазолидин-2,4-диона (приготовленного как описано в этапе (f) выше) в 400 мл 1,4- диоксана, и полученную смесь перемешивали в течение 5 ч при 80oC. По окончании этого времени реакционную смесь освобождали от растворителя выпариванием при пониженном давлении, и полученный остаток очищали колоночной хроматографией на силикагеле, используя смесь 1:2 по объему этилацетата и гексана, смесь 2:1 по объему этилацетата и гексана, а затем этилацетат в качестве элюентов с получением 161,7 г указанного в заглавии соединения с т.пл. 100-106oC.
2(h) 5-[4-(6-метокси-1-метилбензимидазол-2-ил-метокси)- бензил] тиазолидин-2,4-дион
Смесь 21,8 г 5-метокси-]-N-метил-1,2-фенилендиамина (приготовленного как описано в этапе (е) выше), 63,4 г 5-(4-метоксикарбонилметоксибензил)-тиазолидин-2,4-диона (приготовленного как описано в этапе (g) выше), 250 мл 1,4- диоксана и 750 мл концентрированной водной хлористоводородной кислоты нагревали с обратным холодильником в течение 60 ч. По окончании этого времени реакционную смесь охлаждали льдом, после чего твердое вещество собирали фильтрованием. К этому веществу добавляли 800 мл 5% в/о водного раствора гидрокарбоната натрия, и полученную смесь перемешивали при комнатной температуре в течение 2 ч. Затем нерастворимые материалы собирали фильтрованием и растворяли в смеси 1000 мл диметилформамида и 200 мл метанола. Полученный раствор обесцвечивали действием активизированного древесного угля, который затем удаляли фильтрованием. Фильтрат затем концентрировали выпариванием при пониженном давлении до объема приблизительно 50 мл. Полученный концентрат добавляли к 750 мл диэтилового эфира, и полученный таким способом раствор оставляли стоять в течение двух дней. По окончании этого времени полученный осадок собирали фильтрованием с получением 20,1 г указанного в заглавии соединения с т.пл. 267-271oC и величиной Rf = 0,68 (на тонкослойной хроматографии на силикагеле; выделяющий растворитель метиленхлорид с 5% о/о этанола).
Приготовление 3
5-[4-(5-гидрокси-1,4,6,7-тетраметилбензимидазол-2-ил-метокси) бензил]-тиазолидин-2,4-дион.
3(а) триметилбензохинон
Суспензию 25,6 г хлорида железа (3) в 50 мл воды добавляли при комнатной температуре к раствору 20 г триметилгидрохинона в 150 мл ацетона, и полученную смесь перемешивали в течение 1 ч, после чего ее оставляли стоять в течение двух дней. По окончании этого времени ее концентрировали до приблизительно половины первоначального объема, и концентрат смешивали с водой. Полученную водную смесь экстрагировали этилацетатом, и экстракт промывали водой и насыщенным водным раствором хлорида натрия в указанном порядке, после чего его высушивали над безводным сульфатом натрия. Растворитель удаляли дистилляцией при пониженном давлении, и полученный остаток очищали колоночной хроматографией на силикагеле с использованием смеси 1:6 по объему этилацетата и гексана в качестве элюента с получением 16,9 г указанного в заглавии соединения с величиной Rf = 0,48 (на тонкослойной хроматографии на силикагеле; выделяющий растворитель смесь 1:6 по объему этилацетатa и гексана).
3(b) 2,3,6-триметилбензохинон-4-оксим
Раствор 7,04 г гидрохлорида гидроксиламина в 30 мл воды добавляли при комнатной температуре к раствору 16,9 г триметилбензохинона (приготовленного как описано в этапе (а) выше) в 150 мл метанола, и полученную смесь перемешивали в течение 2 ч, после чего ее оставляли стоять в течение двух дней. По окончании этого времени реакционную смесь разводили 1000 мл воды. Отделившийся осадок собирали фильтрованием и перекристаллизовывали из смеси этилацетата и гексана с получением 11,2 г указанного в заглавии соединения с т.пл. 188-190oC.
3(с) 4-гидрокси-2,3,5-триметиланилин
152 г гидросульфита натрия добавляли при охлаждении льдом к смеси 36,15 г 2,3,6- триметилбензохинона-4-оксима (приготовленного как описано в этапе (b) выше) и 880 мл 1Н водного раствора гидроксида натрия, и полученную смесь перемешивали при комнатной температуре в течение 1 ч, после чего ее оставляли стоять в течение ночи. Реакционную смесь затем выливали в лед-воду, и pH водной смеси доводили до 4-5 путем добавления 5H водной хлористоводородной кислоты, после чего ее нейтрализовали гидрокарбонатом натрия. Полученную таким способом смесь экстрагировали этилацетатом. Экстракт промывали насыщенным водным раствором хлорида натрия и высушивали над безводным сульфатом натрия. Растворитель затем удаляли дистилляцией при пониженном давлении, после чего кристаллический осадок стирали с диизопропиловым эфиром и собирали фильтрованием. После промывания диизопропиловым
эфиром получали 30,1 г указанного в заглавии соединения с т.пл. 131-134oC.
3(d) N-т-бутоксикарбонил-4-гидрокси-2,3,5-триметиланилин
22,0 мл триэтиламина добавляли при комнатной температуре к раствору 20 г 4-гидрокси-2,3,5-триметиланилина (приготовленного как описано в этапе (с) выше) в 500 мл тетрагидрофурана, а затем добавляли 34,6 г ди-т-бутилбикарбоната, и полученную смесь перемешивали в течение 6 ч, после чего ее оставляли стоять в течение ночи. По окончании этого времени реакционную смесь освобождали от растворителя дистилляцией при пониженном давлении, и полученный остаток смешивали с водой. Полученную водную смесь экстрагировали этилацетатом. Экстракт промывали насыщенным водным раствором хлорида натрия и высушивали над безводным сульфатом натрия. Растворитель удаляли дистилляцией при пониженном давлении, после чего кристаллический остаток стирали с гексаном с получением 31,9 г указанного в заглавии соединения с т.пл. 158-161oC.
3(е) N-метил-т-гидрокси-2,3,5-триметиланилин
Раствор 15 г N-т-бутоксикарбонил-4-гидрокси-2,3,5-триметиланилина (приготовленного как описано в этапе (d) выше) в 200 мл дегидратированного тетрагидрофурана добавляли к суспензии 6,8 г гидрида литий-алюминия в 300 мл дегидратированного тетрагидрофурана, охлаждая льдом, и полученную смесь перемешивали при комнатной температуре в течение 3 ч, после чего ее нагревали с обратным холодильником в течение 2 ч. По окончании этого времени смесь 10 мл воды и 30 мл тетрагидрофурана добавляли к реакционной смеси, чтобы разрушить избыток гидрида литий-алюминия. Реакционную смесь затем перемешивали при комнатной температуре в течение 1,5 ч, после чего нерастворимые материалы отфильтровывали через фильтр Целит (торговое наименование). Эти материалы промывали этилацетатом, и эти промывки объединяли и высушивали над безводным сульфатом натрия. Растворитель удаляли дистилляцией при пониженном давлении, и полученный остаток очищали колоночной хроматографией на силикагеле с использованием смеси 1: 3 по объему этилацетата и гексана в качестве элюента с получением 5,1 г указанного в заглавии соединения с т.пл. 120-122oC.
3(f) N-т-бутоксикарбонил-N-метил-4-гидрокси-2,3,5- триметиланилин
5,0 мл триэтиламина и раствор 7,92 г ди-т-бутилбинарбоната в 30 мл тетрагидрофурана добавляли при комнатной температуре к раствору 5,0 г N-метил-4-гидрокси-2,3,5-триметиланилина (приготовленного как описано в этапе (е) выше) в 70 мл тетрагидрофурана, и полученную смесь перемешивали в течение 1 ч, после чего ее оставляли стоять в течение ночи. По окончании этого времени реакционную смесь освобождали от растворителя путем дистилляции при пониженном давлении, и полученный остаток смешивали с водой. Водную смесь экстрагировали этилацетатом. Экстракт промывали водой и насыщенным водным раствором хлорида натрия в указанном порядке, после чего его высушивали над безводным сульфатом магния. После удаления растворителя дистилляцией кристаллический остаток стирали с гексаном и собирали фильтрованием. Получали 7,35 г указанного в заглавии соединения с т.пл. 163-166oC.
3(g) N-т-бутоксикарбонил-N-метил-4-ацетокси-2,3,5- триметиланилин
5,64 мл дегидратированного триэтиламина и 2,9 мл ацетилхлорида добавляли при комнатной температуре к раствору 7,2 г N-т-бутоксикарбонил-N-метил-4-гидрокси-2,3,5-триметиланилина (приготовленного как описано в этапе (f) выше) в 100 мл дегидратированного тетрагидрофурана, и полученную смесь перемешивали в течение 1 ч, после чего ее оставляли стоять в течение ночи. Реакционную смесь затем разводили водой, и водную смесь экстрагировали этилацетатом. Экстракт промывали водой и насыщенным водным раствором хлорида натрия в указанном порядке, после чего его высушивали над безводным сульфатом магния. Растворитель удаляли путем дистилляции при пониженном давлении, после чего остаток стирали с охлажденным во льду гексаном, чтобы вызвать кристаллизацию. Кристаллы собирали фильтрованием и промывали охлажденным во льду гексаном с получением 6,25 г указанного в заглавии соединения с т.пл. 103-104oC.
3(h) N-метил-4-ацетокcи-2,3,5-триметиланилин гидрохлорид
Смесь, приготовленную добавлением 100 мл 4H раствора хлористого водорода в 1,4-диоксане к 5,45 г N-т-бутоксикарбонил-N-метил-4- ацетокси-2,3,5-триметиланилина (приготовленного как описано в этапе (g) выше) при комнатной температуре перемешивали в течение 3 ч. По окончании этого времени реакционную смесь освобождали от растворителя путем дистилляции при пониженном давлении, и полученный остаток стирали с диизопропиловым эфиром. Полученные таким способом кристаллы собирали фильтрованием, после чего их промывали диизопропиловым эфиром с получением 4,36 г указанного в заглавии соединения с т.пл. 172-176oC.
3(i) N-метил-4-ацетокси-2,3,5-триметил-6-нитроанилин
4,3 г N-метил-4-ацетокси-2,3,5-триметиланилин гидрохлорида (приготовленного как описано в этапе (h) выше) добавляли к охлажденной льдом концентрированной водной азотной кислоте, и полученную смесь перемешивали, охлаждая льдом, в течение 10 мин, а затем при комнатной температуре в течение 10 мин. По окончании этого времени реакционную смесь выливали в лед-воду, и водную смесь нейтрализовали добавлением гидрокарбоната натрия, после чего ее экстрагировали этилацетатом. Экстракт промывали насыщенным водным раствором хлорида натрия и высушивали над безводным сульфатом натрия. Растворитель затем удаляли путем дистилляции при пониженном давлении, после чего к остатку добавляли 50 мл диизопропилового эфира и 50 мл гексана. Смесь затем перемешивали ультразвуком в течение 5 мин. Нерастворимый осадок стирали со смесью 1:1 по объему диизопропилового эфира и гексана. Полученные кристаллы собирали фильтрованием, после чего их промывали смесью 1:1 по объему диизопропилового эфира и гексана с получением 2,76 г указанного в заглавии соединении с т.пл. 143-146oC.
3(j) 4-ацетокcи-N-метил-3,5,6-триметил-1,2-фенилендиамин
Раствор 2,65 г N-метил-4-ацетокси-2,3,5-триметил-6-нитроанилина (приготовленного как описано в этапе (i) выше) в смеси 20 мл этанола и 20 мл этилацетата встряхивали при комнатной температуре в течение 3,5 ч, а затем при 40oC в течение 3 ч в атмосфере водорода и в присутствии 0,2 г оксида платины. По окончании этого времени реакционную смесь фильтровали для удаления оксида платины, и фильтрат освобождали от растворителя путем дистилляции при пониженном давлении. Полученный остаток очищали колоночной хроматографией на силикагеле с использованием смеси 1:1 по объему этилацетата и гексана в качестве элюента с получением 1,3 г указанного в заглавии соединения в т.пл. 113-116oC.
3(k) 5-[4-(5-гидрокси-1,4,6,7-тетраметилбензимидазол-2-ил- метокси)бензил]тиазолидин-2,4-дион
Смесь 1,0 г 4-ацетокси-N-метил-3,5,6-триметил-1,2-фенилендиамина (приготовленного как описано в этапе (j) выше), 2,7 г 5-(4-метокси- карбонилметоксибензил)тиазолидин-2,4-диона (приготовленного как описано в этапе 2 (g) приготовления 2), 5 мл 1,4-диоксана и 25 мл концентрированной водной хлористоводородной кислоты нагревали с обратным холодильником в течение двух дней. По окончании этого времени реакционную смесь добавляли ко льду-воде, и полученную смесь нейтрализовали добавлением гидрокарбоната натрия. Затем ее экстрагировали этилацетатом. Экстракт промывали насыщенным водным раствором хлорида натрия и высушивали над безводным сульфатом магния. Затем растворитель удаляли путем дистилляции при пониженном давлении, после чего остаток очищали колоночной хроматографией на силикагеле с использованием этилацетата в качестве элюента. Фракции, содержавшие указанное в заглавии соединение, собирали и растворитель удаляли путем дистилляции при пониженном давлении с получением остатка в виде красного масла. К этому маслу добавляли 150 мл диэтилового эфира, и смесь перемешивали ультразвуком в течение 5 мин. Выпавший осадок собирали фильтрованием и растворяли в 300 мл тетрагидрофурана. Полученный раствор концентрировали до объема приблизительно 10-20 мл путем выпаривания при пониженном давлении. К концентрату добавляли 200 мл этилацетата, и смесь перемешивали ультразвуком в течение 20 мин. Выпавший осадок собирали фильтрованием с получением 0,52 г указанного в заглавии соединения с т. пл. 240-244oC и величиной Rf = 0,44 (на тонкослойной хроматографии на силикагеле; выделяющий растворитель этилацетат).
Пример 5.
Препаративная форма 1
Капсулы
Смешивали 0,5 г правастатина-натрия, 20 г троглитазона, 1,5 г кросповидона (дезинтегратора поливинилпирролидона) и 0,2 г лаурилсульфата натрия. Смесь разделяли между 100 пустыми капсулами (номер 1) для получения 100 капсул, каждая из которых содержала 5 мг правастатина-натрия и 200 мг троглитазона.
Пример 6
Препаративная форма 2
Таблетки
40 г 5% в/о водного раствора гидроксипропилцеллюлозы добавляли к смеси 5 г правастатина-натрия, 2 г соединения А, 24 г гидроксипропилцеллюлозы (низкой степени замещения) и 86,9 г лактозы, и полученную смесь перемешивали для получения композиции. Эту композицию пропускали через сито меш 10 (стандартный меш Tyler) и высушивали, после чего ее пропускали через сито меш 15 (стандартный меш Тyler) для получения гранул одинакового размера. Смешивали 11,9 г гранул и 0,1 г стеарата магния, и смесь превращали в таблетки с помощью машины для таблетирования, в результате чего получали таблетки диаметром 6,5 мм и весом 120 мг, каждая из которых содержала 5 мг правастатина-натрия и 2 мг соединения A.










Изобретение относится к медицинe. Комбинацию одного или более ингибиторов ГМГ-КоА редуктазы (например, правастатина, ловастатина, симвастатина, флувастатина, ривастатина или аторвастатина) и одного или более сенсибилизирующих к инсулину агентов (например, троглитазона, пиоглитазона, энглитазона, BRZ-49653, 5-(4-{ 2-[4,2'-пиридилфенил)этилиденаминоокси] этокси} бензил)-тиазолидин-2,4-диона, 5-{4-(5-метокси-3-метилимидазо[5,4-в]пиридин-2-ил-метокси)бензил} тиазолидин-2,4-диона или его гидрохлорида, 5-[4-(6-метокси-1-метилбензимидазол-2-ил-метокси)бензил]тиазолидин-2,4-диона, 5-[4-(1-метилбензимидазол-2-ил-метокси)бензил] тиазолидин-2,4-диона и 5-[4-(5-гидрокси-1,4,6,7-тетраметилбензимидазол-2-ил-метокси)бензил] тиазолидин-2,4-диона) вводят для лечения артериосклероза и/или ксантомы. Предложен также упакованный фармацевтический препарат, содержащий данную комбинацию. Способ лечения более эффективен. 3 с. и 75 з.п. ф-лы, 10 табл.
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| Способ лечения атеросклероза | 1986 |
|
SU1475667A1 |
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| US 5260305 A, 09.11.1993 | |||
| Переносная печь для варки пищи и отопления в окопах, походных помещениях и т.п. | 1921 |
|
SU3A1 |
| US 5316765 A, 31.05.1994 | |||
| Очаг для массовой варки пищи, выпечки хлеба и кипячения воды | 1921 |
|
SU4A1 |
| Огнетушитель | 0 |
|
SU91A1 |
| Кипятильник для воды | 1921 |
|
SU5A1 |
| Приспособление для точного наложения листов бумаги при снятии оттисков | 1922 |
|
SU6A1 |
| ЧАЗОВ Е.И | |||
| и др | |||
| Новое в изучении патогенеза и лечения атеросклероза: Обзор | |||
| Клиническая медицина | |||
| - М.: Медицина, 1991, N 3, с.7. | |||

