Изобретение относится к технологии очистки циклических лактамов, более конкретно к способу непрерывной очистки сырого капролактама, получаемого взаимодействием 6-аминокапронитрила с водой.
Известен способ непрерывной очистки сырого капролактама, получаемого взаимодействием 6-аминокапронитрила с водой, включающий отделение низкокипящих и высококипящих компонентов из сырого капролактама, гидрирование с последующей обработкой в кислой среде и перегонку в щелочной среде. При этом очищенный капролактам получают с выходом до 76% (см. патент США N 2301964, кл. 260-239, 17.11.1942 г.).
Задачей изобретения является разработка способа непрерывной очистки сырого капролактама, получаемого взаимодействием 6- аминокапронитрила с водой, который позволяет получить более чистый капролактам с более высоким выходом.
Поставленная задача решается предлагаемым способом непрерывной очистки сырого капролактама, получаемого взаимодействием 6-аминокапронитрила с водой, за счет того, что
а) сырой капролактам после отделения низкокипящих и высококипящих компонентов обрабатывают водородом при 50 - 150oC и 1,5 - 250,0 бар в присутствии катализатора гидрирования и при необходимости растворителя с получением смеси A,
б1) пропускают смесь A в растворителе при 30 - 80oC и 1 - 5 бар через ионообменник, содержащий кислые группы, с получением смеси Б1 или
б2) смесь A перегоняют в присутствии серной кислоты, причем перед добавлением серной кислоты, при необходимости, удаляют имеющийся растворитель с получением смеси Б2 и
в) смесь Б1 или Б2 перегоняют в присутствии основания с получением чистого капролактама.
Подлежащий очистке сырой капролактам, получаемый взаимодействием 6-аминокапронитрила с водой в общеизвестных условиях, представляет собой смесь, состоящую в основном из 50 - 98, предпочтительно 80 - 95 вес.% воды и от 2 до 50, предпочтительно от 5 до 20 вес.% смеси, состоящей из 50 - 90, предпочтительно 65 - 85 вес.% капролактама и из 10-50, предпочтительно 15-35 вес.% высококипящей фракции.
На стадии а) сначала отделяют низкокипящие и высококипящие компоненты путем перегонки. При этом предпочтительно из головной части колонны отделяют аммиак, имеющийся вышеуказанный растворитель, в частности алифатический спирт, избыток воды и непрореагировавший 6-аминокапронитрил, а также имеющиеся низкокипящие побочные продукты, и затем отделяют сырой капролактам перегонкой, предпочтительно из головной части колонны от высококипящих компонентов, таких как олигомеры 6-аминокапроновой кислоты. Для успеха изобретения не является решающим, отгоняют ли низкокипящие компоненты до или после высококипящих или одновременно. Предварительно очищенный сырой капролактам подвергают обработке водородом в виде расплава, предпочтительно растворенным в растворителе. В качестве растворителя предпочтительно используют такие, которые в условиях гидрирования и обработки инертны по отношению к ионообменнику; в частности, алифатические спирты с 1-3 атомами углерода, такие как метанол, этанол, н-пропанол, изо-пропанол, предпочтительно этанол, а особенно предпочтительно - воду.
Согласно предпочтительной форме выполнения изобретения используют растворители со стадии циклизации 6-аминокапронитрила, поскольку там содержится спирт или вода.
При гидрировании обычно используют от 50 до 95, предпочтительно от 70 до 95 вес. %-ные растворы сырого капролактама, считая на раствор. При передаче растворителя со стадии циклизации может быть необходимым для получения желаемой концентрации добавление или отгонка растворителя.
Обработку водородом предпочтительно проводят при температуре от 60 до 95oС, в частности от 70 до 90oC в жидкой фазе. Давление выбирают в зависимости от температуры таким, чтобы получалась жидкая фаза. Давление предпочтительно находится в области от 5 до 100, в частности от 5 до 20 бар.
Водород вводят обычно в количестве от 0,0001 до 5,0, предпочтительно от 0,001 до 0,7, в частности от 0,03 до 0,3 моль на моль капролактама.
Время реакции составляет обычно от 10 до 300, предпочтительно от 15 до 200 минут.
Нагрузку катализатора выбирают обычно от 0,1 до 15, предпочтительно от 1,5 до 10 кг капролактама на литр катализатора в час.
Гидрирование можно проводить как в суспензии, так и в неподвижном слое, причем в последнем случае предпочтительно подводить раствор капролактама вместе с водородом снизу вверх или сверху вниз через жестко закрепленный в трубчатой зоне катализатор.
В качестве катализатора гидрирования предпочтительно использовать такие, которые получены на основе металла, выбранного из группы, содержащей железо, никель, кобальт, рутений, родий, палладий, осмий, иридий и платину, особенно предпочтительно кобальт, никель и палладий, еще более предпочтительно палладий, в виде массивных катализаторов или катализаторов на носителе, предпочтительно в виде катализатора на носителе.
Согласно предпочтительной форме выполнения изобретения используют палладиевые катализаторы на носителе, которые имеют содержание палладия от 0,01 до 10, предпочтительно от 0,05 до 5, еще более предпочтительно от 0,1 до 2 вес.%, считая на катализатор. В качестве носителя используют предпочтительно активированный уголь, оксид алюминия, оксид цинка, двуокись кремния, диоксид титана, оксид лантана или диоксид циркония, или их смеси.
Согласно другой предпочтительной форме изобретения используют никелевые катализаторы на носителе, которые имеют содержание никеля от 1 до 80, предпочтительно от 5 до 50 вес.%, считая на катализатор. Кроме того, никелевый катализатор на носителе может иметь добавки на основе элементов: цирконий, марганец, медь или хром, причем эти добавки находятся в количестве от 0,1 до 20, предпочтительно от 1 до 5 вес.%, считая на количество введенного никеля, обычно в виде окисла.
В качестве носителей используют предпочтительно оксид алюминия, силикагель, глинозем, активированный уголь, силикаты магния, фосфат алюминия или фосфат бора, наиболее предпочтительно силикаты магния, фосфат алюминия, фосфат бора и оксид алюминия.
Данные для получения такого типа катализаторов, полученных методом осаждения или пропитки, можно найти, например, в Encyclopedia of Industrial Chemistry Ульмана, т. A5, стр. 348-350, 5-ое дополн. издание.
Согласно другой предпочтительной форме выполнения изобретения используют катализаторы на носителе, поверхность которых обогащена каталитически активным металлом. Такие катализаторы получают обычно известными методами путем обработки предварительно сформованного из вышеуказанных веществ носителя, в виде таблеток, шариков или прутков, водным раствором соли металла, например, нитрата, затем их сушат, прокаливают и затем активируют водородом.
Согласно предпочтительной форме выполнения изобретения палладиевый или никелевый катализатор на носителе неподвижно располагают в трубчатой зоне, например, с соотношением длины к диаметру от 10:1 до 50:1, например, в виде насыпки, и пропускают раствор сырого капролактама и водород сверху или снизу через неподвижный слой катализатора.
По имеющимся наблюдениям при обработке водородом прежде всего улучшается ультрафиолетовое число и перманганатное число, определенное титрованием, сырого капролактама.
После охлаждения и снятия давления получают смесь A, которая в основном состоит из капролактама и растворителя, если такой применялся. Если проводят гидрирование в расплаве капролактама, полученный после гидрирования продукт перед обработкой ионообменником растворяют в одном из вышеназванных для гидрирования растворителе, предпочтительно в воде.
Стадию б1) предпочтительно проводят при температуре от 50 до 60oC и при давлении от 1 до 2 бар. В качестве ионообменника используют предпочтительно сильнокислые, т. е. содержащие сульфогруппы, ионообменники в H-форме. Подходящими ионообменниками являются, например, Амберлит ® , Дауекс ® или Леватит ® (см. в Encyclopedia of Industrial Chemistry Ульмана, т. A14, стр. 451, 5-ое дополн. издание).
Нагрузку ионообменника выбирают обычно от 1 до 15, предпочтительно от 1 до 10 кг капронитрила на литр ионообменника в час.
При обработке катионообменником еще более улучшается УФ-число.
Регенерация загруженного ионообменника возможна промывкой водным раствором минеральной кислоты, например, серной или фосфорной, при этом зафиксированные на ионообменнике основные соединения могут быть выведены в виде водных растворов соответствующих солей.
Согласно изобретению вместо обработки ионообменником смесь A можно перегонять в присутствии серной кислоты (стадия б2), при этом перед добавлением серной кислоты удаляют растворитель, если он имеется.
Согласно предпочтительной форме выполнения изобретения имеющийся растворитель удаляют в перегонной колонне с двумя- четырьмя, особенно предпочтительно с двумя или тремя теоретическими тарелками при температуре куба максимально 145oC. Давление выбирают в зависимости от выбранной температуры. Обычно его выбирают от 35 до 65 мбар, в частности от 40 до 60 мбар (измерение в голове колонны), если температура куба составляет 145oC.
Освобожденный таким образом от растворителя или не имеющий растворителя сырой капролактам смешивают с серной кислотой, взятой обычно в количестве от 0,1 до 0,5, предпочтительно от 0,2 до 0,3 вес.% (считается как 100 %-ная по весу серная кислота), в расчете на капролактам.
Содержащий серную кислоту остаток после перегонки подают на расщепление серной кислоты. Согласно предпочтительной форме выполнения изобретения перегонку ведут в перегонной колонне с 12- 18, предпочтительно с 14-16 теоретическими тарелками при давлении в головной части колонны от 3 до 6, предпочтительно от 3 до 4 мбар и температуре куба максимально 145oC.
В качестве основания при перегонке смеси Б1 или Б2 используют обычно соединения щелочных или щелочноземельных металлов, такие как гидроксиды или водорастворимые карбонаты, например, гидроокись лития, гидроокись натрия, гидроокись калия, гидроокись магния, гидроокись кальция, карбонат натрия или их смеси, особенно предпочтительным является гидроокись натрия в виде натриевой щелочи.
Количество добавляемого основания выбирают обычно от 0,05 до 0,9, предпочтительно от 0,1 до 0,8 мол.%, считая на капролактам. Согласно предпочтительной форме выполнения изобретения добавляют от 0,05 до 0,25, предпочтительно от 0,1 до 0,15 вес.% натриевой щелочи (рассчитано как 100 %-ная по весу).
Перегонку проводят известным способом, при этом отделяют от капролактама растворитель, низкокипящие и высококипящие фракции.
Согласно предпочтительной форме выполнения изобретения отгоняют из смешанной с основанием смеси Б1 или Б2 сначала растворитель, в частности воду, из головной части колонны, причем температуру куба выбирают максимально 160oC и соответственно устанавливают давление. Предпочтительно работают при давлении от 35 до 65, более предпочтительно от 40 до 60 мбар (измерено в головной части колонны). Кубовый продукт подают на вторую перегоннную колонну.
Кубовый продукт из первой перегонной колонны перегоняют во второй перегонной колонне обычно при давлении от 4 до 6, предпочтительно 4 мбар (измерено в головной части колонны) и при температуре куба максимально 145oC. На этой стадии перегонки удаляют обычно низкокипящие компоненты. Затем кубовый продукт подают на третью перегонную колонну.
Кубовый продукт из второй колонны подают обычно на следующую перегонную колонну, причем работают обычно при давлении от 4 до 6, предпочтительно 4 мбар, и при температуре куба максимально 145oC. Головной продукт состоит из чистого капролактама, соответствующего спецификации.
Согласно другой форме выполнения изобретения кубовый продукт из третьей колонны можно подавать на пленочный испаритель, при этом может быть отделено дополнительное количество капролактама, которое возвращают на первую перегоннную колонну.
Далее, предпочтительным является вариант, когда в качестве основания используют натриевую щелочь. Здесь можно натрийсодержащий продукт из третьей колонны или пленочного испарителя подавать на установку для сжигания с выделением соды и пара.
Можно также операции разделения во второй и третьей колоннах объединить и использовать только одну колонну. При этом обычно отводят низкокипящие компоненты из головы колонны, высококипящие компоненты - из кубовой части и капролактам из бокового выпуска. Целесообразным является возврат частичного потока низкокипящих компонентов на стадию в) (обработка водородом). По имеющимся данным переработка перегонкой в присутствии основания еще более снижает УФ-число.
Предлагаемый способ позволяет получить чистый капролактам, который по показателям перманганатное число, полученное методом титрования (ПЧТ), перманганатное число, полученное методом фотометрии (ПЧФ), свободные основания, летучие основания (ЛО) и ультрафиолетовое число (УФ) полностью соответствует спецификации для чистого капролактама, полученного по методу Бекмана. Содержание хроматографически определяемых примесей составляет обычно от 100 до 150 ч. /млн (считая на капролактам). Так как некоторые примеси уже в количестве 10 ч. /млн и менее делают невозможным соблюдение требований к показателям и структура многочисленных примесей в концентрации 10 ч./млн и их химическое поведение во время очистки неизвестны, технический результат способа по изобретению нельзя было предсказать.
Предлагаемый способ поясняется следующим примером.
Пример 1. Проводили ряд операций по очистке сырого капролактама, полученного циклизацией 10%-ного этанольного раствора 6-аминокапронитрила (АКН) в присутствии двух молей воды на моль АКН:
В подогретый трубчатый реактор емкостью 25 мл (диаметр 6 мм, длина 800 мм), который заполнен диоксидом титана (анатаз) в виде прутков длиной 1,5 мм, пропускают при давлении 100 бар раствор 6- аминокапронитрила в воде и этаноле (10 вес. % АКН, 6,4 вес.% воды, остаток - этанол), при этом температура реакции составляет 240oC и время реакции 30 минут. Выходящий из реактора продукт анализируют с помощью газовой хроматографии и высокоэффективной жидкостной хроматографии: превращение 100%, выход 88%. Продукт реакции отделяют фракционной перегонкой от высококипящих и низкокипящих компонентов. Полученный таким образом сырой капролактам согласно газохроматографическому анализу имеет чистоту 99,5%. 1000 г сырого капролактама растворяют в 250 г воды. Водный раствор смешивают в автоклаве с 3,5 г 5 вес.%-ного палладия на активированном угле в качестве носителя и гидрируют при перемешивании 4 часа при 80oC и 5 бар. После охлаждения и снятия давления отфильтровывают катализатор. Фильтрат пропускают методом орошения при 50oC и нормальном давлении через 1 л сильнокислого ионообменника (Амберлит ® IR 120, H-форма) в течение 0,6 часов. Продукт после ионообменника смешивают с 4 г 25%-ного водного раствора натриевой щелочи. В перегонной колонне с двумя теоретическими тарелками отделяют воду при давлении в голове колонны 50 мбар и температуре куба 135oC.
Из кубового продукта первой колонны отгоняют во второй колонне с 15 теоретическими тарелками низкокипящие компоненты при давлении в голове колонны 3,5 мбар и температуре куба 140oC.
Кубовый продукт из второй колонны перегоняют в третьей колонне с 15 теоретическими тарелками. При давлении в голове колонны 4 мбар и температуре куба 145oC отгоняют из головной части колонны 990 г капролактама (99%, считая на введенный сырой капролактам).
Полученный чистый капролактам согласно газовой хроматографии содержит только 140 ч./млн примесей, соединения, которые могли бы отрицательно повлиять на полимеризацию капролактама в нейлон, не были обнаружены. Показатели капролактама следующие:
ПЧФ - 1,5; ПЧТ - 1,2; свободные основания < 0,05 мэкв/кг; летучие основания < 0,05 мэкв/кг; УФ - 2,5.
Таким образом, полученный из 6-аминокапронитрила капролактам соответствует требованиям спецификации для капролактама, полученного по методу Бекмана.
В таблице показано улучшение показателей УФ и перманганатного числа (ПЧТ) на отдельных стадиях очистки.
Перманганатное число методом титрования (ПЧТ)
Устойчивость капролактама к перманганату калия определяли методом титрования. Перманганатное число соответствует расходу 0,1 н. раствора перманганата калия в мл, в расчете на 1 кг капролактама, определенному титрованием сернокислым раствором.
Перманганатное число методом фотометрии (ПЧФ)
Устойчивость капролактама к перманганату калия определяется фотометрически (см. методику ИСО 8660).
Для этого прибавляют одинаковые количества 0,01 н. раствора перманганата калия к 3% (м/м) водному раствору капролактама и к холостой пробе (диет. вода). Через 10 минут сравнивают экстинкции E при 420 нм капролактамовой пробы и холостой пробы. Перманганатное число рассчитывается как (E-E0)420 •100:3.
Летучие основания (ЛО)
(Определение в аппарате по Парнасу, см. методику ИСО 8661 "Капролактам для промышленного применения, определение содержания летучих соединений").
При перегонке в щелочной среде из пробы отделяют летучие основания (аппарат Кьельдаля), улавливают их 0,01 н. соляной кислотой и определяют титрованием с 0,01 н. натриевой щелочью, навеска капролактама(20±0,1) г.
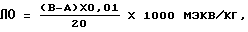
A = расход 0,01 н. натриевой щелочи;
B = расход 0,01 н. натриевой щелочи для холостого опыта.
УФ-число
Соответствующие экстинкции 50% (м/м) раствора капролактама определяют для 270, 280, 290, 300, 310, 320, 330, 340, 350 и 360 нм в 10-сантиметровой кювете. Сумма значений экстинкции умножают на два и получают УФ-число, в расчете на 100% капролактам.
Свободные основания
Для определения свободных оснований в 150 мл дистиллированной, свободной от углекислого газа, насыщенной азотом воды устанавливают с помощью 0,01 н. натриевой щелочи значение pH точно 7,0 и добавляют 50 (+/-0,1) г капролактама. Затем титруют при 25oC 0,01 н. соляной кислотой до pH 7,0. Доля свободных оснований рассчитывается по нижеприведенной формуле, где A (мл) означает расход 0,01 н. соляной кислоты:
свободные основания = 0,01 • A • 1000/50 = 0,2 • A мэкв/кг.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ОДНОВРЕМЕННОГО ПОЛУЧЕНИЯ КАПРОЛАКТАМА И ГЕКСАМЕТИЛЕНДИАМИНА | 1995 |
|
RU2153493C2 |
| СПОСОБ ПОЛУЧЕНИЯ КАПРОЛАКТАМА | 1995 |
|
RU2153492C2 |
| СПОСОБ ПОЛУЧЕНИЯ КАПРОЛАКТАМА | 1995 |
|
RU2154058C2 |
| СПОСОБ ОЧИСТКИ ЛАКТАМОВ | 1997 |
|
RU2185374C2 |
| Способ очистки капролактама | 1988 |
|
SU1709905A3 |
| СПОСОБ ОЧИСТКИ ε-КАПРОЛАКТАМА | 1997 |
|
RU2201920C2 |
| СПОСОБ ПОЛУЧЕНИЯ ε-КАПРОЛАКТОНА И 1,6-ГЕКСАНДИОЛА | 2011 |
|
RU2571082C2 |
| Способ выделения среднекипящей фракции из смеси циклогексанона и циклогексанола со средне- и высококипящими продуктами | 1990 |
|
SU1829948A3 |
| СПОСОБ ПОЛУЧЕНИЯ Н-БУТИРАЛЬДЕГИДА И/ИЛИ Н-БУТАНОЛА | 1995 |
|
RU2135456C1 |
| СПОСОБ ПОЛУЧЕНИЯ ПОЛИМЕТИЛОЛОВ | 2009 |
|
RU2530027C2 |

Изобретение относится к способу непрерывной очистки сырого капролактама, получаемого взаимодействием 6-аминокапронитрила с водой. Процесс включает отделение низко- и высококипящих компонентов из сырого капролактама, гидрирование с последующей обработкой в кислой среде и перегонку в щелочной среде. При этом после отделения низко- и высококипящих компонентов сырой капролактам обрабатывают водородом при 50 - 150°С и 1,5 - 250,0 бар в присутствии катализатора гидрирования и при необходимости растворителя. Таким образом получают смесь А, которую пропускают в растворителе при 30 - 80°С и 1 - 5 бар через ионообменник, содержащий кислые группы, с получением смеси Б1 либо перегоняют в присутствии серной кислоты, причем перед добавлением серной кислоты при необходимости удаляют имеющийся растворитель с получением смеси Б2. В качестве растворителя используют воду. Смесь Б1 или Б2 перегоняют в присутствии основания с получением чистого капролактама. В результате повышаются выход и чистота капролактама. 1 з.п. ф-лы, 1 табл.
| МЕХАНИЧЕСКАЯ ЗАБОЙКА | 2004 |
|
RU2301964C2 |
| US 3145198 A, 18.08.1949 | |||
| 0 |
|
SU150295A1 | |
| СПОСОБ ОЧИСТКИ КАПРОЛАКТАМАс>& плтс11Т1;0-<^! т7::::;1ЧЕс;:-.я Б::5л;;:;'|51^А | 0 |
|
SU172809A1 |

