Изобретение относится к способу получения промежуточных продуктов для получения имеющих антихолинергетическую активность веществ на основе производных троповой кислоты, более конкретно к способу получения чистых энантиомеров сложных эфиров (+) или (-) троповой кислоты с аминоспиртами.
Классический синтез сложных эфиров троповой кислоты разработан И. Мамлоком и Р. Вольффенштайном (см. Ber. dtsch. Chem. Ges. 41. 731 (1908 г.)), согласно которому хлорангидрид O-ацетилтроповой кислоты подвергают взаимодействию с гидрохлоридом тропина, не дает удовлетворительных результатов, возможно из-за плохой растворимости гидрохлорида тропина.
Недостатком известного способа является возможность протекания побочных реакций. Это связано с щелочными условиями, в которых имеется опасность элиминации воды. Кроме того, при применении солей аминоспиртов, которые чаще всего являются труднорастворимыми, требуется более высокая температура, приводящая к образованию сильно мешающих побочных продуктов; в этой связи следует называть дегидратизированные соединения (апосоединения) и продукты димеризации. К тому же известный способ не позволяет синтез оптически активной троповой кислоты.
Задачей изобретения является разработка синтеза чистых энантиомеров сложных эфиров (+) или (-) троповой кислоты.
Поставленная задача решается предлагаемым способом получения чистых энантиомеров сложных эфиров (+)- и (-)-троповой кислоты с аминоспиртами формулы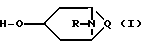
в которой Q означает группу CH2-CH2 или CH2-CH2-CH2,
R - разветвленный алкил с 1-4 атомами углерода, заключающимся в том, что
(а) соответствующую оптически активную троповую кислоту подвергают ацетилированию,
(б) полученную О-ацетилтроповую кислоту переводят в ее хлорангидрид с использованием тионилхлорида,
(в) полученный хлорангидрид подвергают взаимодействию с метансульфонатом аминоспирта общей формулы (1) при комнатной температуре в среде инертного растворителя,
(г) полученное соединение подвергают деацетилированию путем воздействия сильной кислотой и
(д) выделяют полученный оптически активный сложный эфир троповой кислоты.
Требуемую в качестве сырья (+)- и (-)-троповую кислоту можно получать из D,L-троповой кислоты путем получения известным методом соли с оптически активным основанием, которую подвергают многократной перекристаллизации. В качестве основания пригоден, например, (-)-хинин, а в качестве растворителя для перекристаллизации - этанол. Полученная таким образом (+)-троповая кислота имеет чистоту 99,8% ([α]
Ацетилирование согласно стадии (а) целесообразно осуществляют при комнатной температуре, а проводимое непосредственно после этого образование хлорангидрида согласно стадии (б) осуществляют предпочтительно без промежуточного выделения ацетилированной кислоты при комнатной температуре или при слегка повышенной температуре.
Стадию (в) осуществляют в течение нескольких дней в среде метиленхлорида в качестве предпочтительного инертного растворителя, причем для улавливания кислоты можно использовать избыточную окись алюминия. После удаления растворителя при пониженном давлении остаток можно непосредственно использовать на стадии (г), которую предпочтительно осуществляют при комнатной температуре, при этом в качестве сильной кислоты используют разбавленную водную минеральную кислоту, например 2 - 20%-ную соляную кислоту, предпочтительно 3 - 10%-ную соляную кислоту. Таким образом, например, при использовании 5%-ной соляной кислоты достигают полного деацетилирования в течение примерно двух дней.
Продукт реакции можно выделять в виде основания (стадия (д)) путем добавления кислого реакционного раствора при перемешивании к избыточному разбавленному (например, 20%-ному) натровому щелоку или к водному раствору карбоната щелочного металла, с последующим выделением получаемого кристаллического продукта путем фильтрации. При этом температура может составлять от -15oC до +50oC. Предпочтительным является использование раствора карбоната натрия при температуре примерно 20oC. Из соответствующего основания можно легко получать соли, например соответствующий гидрохлорид, а именно, путем добавления стехиометрического количества эфирной соляной кислоты к раствору основания, например, в метиленхлориде. Полученные таким образом сложные эфиры более чем на 99% состоят из чистого оптически активного соединения, при условии соответствующей чистоты исходной кислоты.
Получаемые согласно изобретению оптически активные сложные эфиры представляют собой ценные промежуточные продукты для получения соответствующих четвертичных соединений, обладающих антихолинергетической активностью, например, соответствующих метобромидов или метометансульфонатов, которые можно применять, например, в качестве лекарственного средства при астматических заболеваниях или обтурационных заболеваниях дыхательных путей.
Получение четвертичных соединений путем известных методов возможно без рацемизации.
Предлагаемый способ поясняется следующими примерами.
Пример 1
Сложный N-изопропилнортропиновый эфир (-)-троповой кислоты в виде гидрохлорида
(а) 13,3 г (-)-троповой кислоты при перемешивании при комнатной температуре добавляют к 31,4 г ацетилхлорида, после чего в течение часа образуется прозрачный раствор. По истечении дальнейшего часа согласно данным тонкослойной хроматографии достигнута практически полная конверсия. К раствору (-)-О- ацетилтроповой кислоты в течение 30 минут каплями добавляют 47,5 г тионилхлорида. Раствор перемешивают в течение ночи при комнатной температуре, а затем в течение часа при температуре 50oC. В результате упаривания при температуре 35oC при пониженном давлении получают 18,9 г коричневатой жидкости, [α]
(б) 7,26 г (0,0275 моль) метансульфоната N-изопропилнортропина и 6,20 г (0,0275 моль) полученного на стадии (а) продукта при комнатной температуре при размешивании добавляют к 45 мл метиленхлорида. По истечении семи дней реакционный раствор упаривают при температуре 30oC и при пониженном давлении. Остаток (16,9 г) используют для последующей стадии без дальнейшей переработки.
(в) 11,1 г полученного на стадии (б) продукта растворяют в 60 мл 5%-ной соляной кислоты и размешивают при комнатной температуре в течение двух дней. После этого путем тонкослойной хроматографии можно определять количественное деацетилирование.
Реакционный раствор дважды экстрагируют небольшим количеством диэтилового эфира, затем при пониженном давлении удаляют остатки простого эфира. После этого к раствору при размешивании добавляют избыточный 20%-ный водный раствор карбоната натрия, причем образуется кристаллический продукт. Последний отфильтровывают и промывают холодной водой до того, как фильтрат показывает лишь слабую щелочную реакцию, и растворяют в метиленхлориде. После сушки над сульфатом натрия растворитель упаривают при пониженном давлении и остаток перекристаллизовывают из ацетонитрила. Получают белые кристаллы, точка плавления 131oC, [α]
Для получения гидрохлорида к раствору основания в метиленхлориде добавляют стехиометрическое количество эфирной соляной кислоты. В результате перекристаллизации продукта из этанола и ацетонитрила получают гидрохлорид сложного N- изопропилнортропинового эфира (-)-троповой кислоты в виде белых кристаллов, точка плавления 214 - 218oC. [α]
Кватернизацию до метобромида можно осуществлять, например, путем взаимодействия с метилбромидом в среде метиленхлорида и ацетонитрила при комнатной температуре. Получают белые кристаллы (выход 75,2% от теории, точка плавления 238 - 242oC при разложении). [α]
Пример 2
Сложный N-изопропилнортропиновый эфир (+)-троповой кислоты в виде гидрохлорида
Исходя из (+)-троповой кислоты, полученной путем расщепления рацемата из D, L-троповой кислоты с применением (-)-хинина, [а]= +73,1 (с = 1 в этаноле; оптическая чистота 99,8%), аналогично примеру 1 получают целевое соединение в виде белых кристаллов, точка плавления 214 - 217oC при разложении, [α]
Описанным в примере 1 путем полученное соединение переводят в метобромид, точка плавления 238 - 241oC (разложение), [α]
При взаимодействии хлорангидридов O-ацетилтроповой кислоты с соединениями формулы (I) достигают выхода примерно 60 - 70%, если взаимодействие осуществляется при комнатной температуре в течение нескольких дней, причем кроме метиленхлорида в качестве среды реакции подобных результатов достигают также с применением, например, диметилформамида или ацетонитрила.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ НОРБЕНЗОМОРФАНА | 1996 |
|
RU2167868C2 |
| СЛОЖНЫЕ ЭФИРЫ ТИЕНИЛКАРБОНОВОЙ КИСЛОТЫ И АМИНОСПИРТОВ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ОБЛАДАЮЩАЯ АНТИХОЛИНЕРГИЧЕСКОЙ АКТИВНОСТЬЮ | 1990 |
|
RU2073677C1 |
| ПРОИЗВОДНЫЕ АРИЛГЛИЦИНАМИДОВ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СОДЕРЖАЩАЯ ЭТИ СОЕДИНЕНИЯ | 1996 |
|
RU2167866C2 |
| НАСАДКА И ИНГАЛЯТОР И СПОСОБ ИЗГОТОВЛЕНИЯ НАСАДКИ | 2009 |
|
RU2495726C2 |
| ПРОИЗВОДНЫЕ ОКСАДИАЗОЛА | 1997 |
|
RU2182905C2 |
| ПРОИЗВОДНЫЕ КСАНТИНА И ИХ ФАРМАКОЛОГИЧЕСКИ ПЕРЕНОСИМЫЕ СОЛИ | 1993 |
|
RU2138500C1 |
| НОВЫЕ АНТИХОЛИНЕРГИЧЕСКИЕ СРЕДСТВА, СПОСОБ ИХ ПОЛУЧЕНИЯ, А ТАКЖЕ ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ЛЕКАРСТВЕННЫХ СРЕДСТВ | 2003 |
|
RU2325388C2 |
| СПОСОБ ПОЛУЧЕНИЯ 3-(ГЕКСАДЕКАНСУЛЬФОНИЛ- ИЛИ СУЛЬФИНИЛ-2-МЕТОКСИМЕТИЛПРОПИЛ)- (2-ТРИМЕТИЛАММОНИО-ЭТИЛ)-ФОСФАТА, ИХ СТЕРЕОИЗОМЕРОВ ИЛИ ФАРМАКОЛОГИЧЕСКИ ПРИЕМЛЕМЫХ СОЛЕЙ | 1990 |
|
RU2035466C1 |
| ПРОИЗВОДНЫЕ АНЕЛЛИРОВАННОГО ДИГИДРОПИРИДИНА ИЛИ ИХ СОЛИ С ФИЗИОЛОГИЧЕСКИ ПЕРЕНОСИМЫМИ КИСЛОТАМИ И СРЕДСТВО, БЛОКИРУЮЩЕЕ НЕСЕЛЕКТИВНЫЕ КАТИОННЫЕ КАНАЛЫ | 1993 |
|
RU2127736C1 |
| ПРОИЗВОДНЫЕ ФЕНИЛАМИДИНА | 1996 |
|
RU2184726C2 |
Описывается способ получения чистых энантиомеров сложных эфиров (+) и (-)-троповой кислоты с аминоспиртами формулы 1
в которой Q означает группу СН2-СН2 или СН2-СН2-СН2, R - разветвленный алкил с 1 - 4 атомами углерода, который заключается в том, что (а) соответствующую оптически активную троповую кислоту подвергают ацетилированию, (б) полученную О-ацетилтроповую кислоту переводят в ее хлорангидрид с использованием тионилхлорида, (в) полученный хлорангидрид подвергают взаимодействию с метансульфонатом аминоспирта общей формулы (1) при комнатной температуре в среде инертного растворителя, (г) полученное соединение подвергают деацетилированию путем воздействия сильной кислотой и (д) выделяют полученный оптически активный сложный эфир троповой кислоты, с чистотой 99,8% и содержанием одного из энантиомеров более чем 99%. 2 з.п.ф-лы.
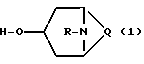
в которой Q означает группу СН2 - СН2 или СН2 - СН2 - СН2,
R - разветвленный алкил с 1 - 4 атомами углерода,
отличающийся тем, что (а) соответствующую оптически активную троповую кислоту подвергают ацетилированию, (б) полученную О-ацетилтроповую кислоту переводят в ее хлорангидрид с использованием тионилхлорида, (в) полученный хлорангидрид подвергают взаимодействию с метансульфонатом аминоспирта общей формулы (1) при комнатной температуре в среде инертного растворителя, (г) полученное соединение подвергают деацетилированию путем воздействия сильной кислотой и (д) выделяют полученный оптически активный сложный эфир троповой кислоты.
| Переднее ограждение стойл | 1984 |
|
SU1289429A1 |
| US 3505337, 1970 | |||
| ОРЕХОВ А.П | |||
| Химия алкалоидов | |||
| - М.:, 1938, с.93-98, Synthesis, 1976, Stuttgart, DE, p.311. | |||

