Изобретение может быть использовано в пищевой, фармакологической, микробиологической, медицинской, химической промышленности при анализе микроколичеств йода в органических соединениях, в основном в йодсодержащих белках, и относится к способам определения микроколичеств йода в йодсодержащих веществах и соединениях.
Известен титрометрический способ, основанный на восстановлении йода в результате химической реакции с последующим оттитровыванием его подходящим химическим реагентом ("метод йодометрии"). Этот метод обладает очень низкой точностью (1-10 мг) и недостаточным порогом обнаружения (примерно 10 мг/кг), хотя легко доступен и не требует затрат на приобретение дорогостоящих реактивов /1/.
Известен способ оптической эмиссии (плазменно-эмиссионная спектроскопия), основанный на возбуждении атомов йода при высокой температуре (8000 - 12000oC) с последующим изучением спектра эмиссии (электромагнитного излучения) в оптическом диапазоне. По интенсивности излучения атомов йода судят о его количестве. Этот способ имеет предел обнаружения примерно 100 мкг/кг и характеризуется высокой точностью, но требует наличия дорогостоящего оборудования (150-300 тыс.$ ) и использования дорогостоящих расходных материалов (аргон высокой чистоты, расход 12 л/мин). Этот метод могут использовать только "богатые" лаборатории с хорошим приборным парком /2/.
Известен рентген-флуоресцентный способ, основанный на возбуждении изучаемых атомов рентгеновским излучением, с последующим изучением спектра гамма-излучения. Этот способ не требует сложной пробоподготовки, удобен в использовании, но предел обнаружения составляет всего 5 г/кг, кроме того необходима защита от воздействия вредного излучения. Этот способ применяется в основном для исследования геологических образцов и в металлургии /3/.
Известен полярографический способ определения ионов, находящихся в диссоциированном состоянии в проводящем растворе. В этом способе о количестве исследуемых ионов судят по величине вольт-амперной характеристики, измеряемой при изменении потенциала на ртутном электроде электрохимической ячейки /4/. Однако известный способ не позволяет измерять количество йода в йодсодержащих органических соединениях.
В качестве прототипа был выбран способ ионной хроматографии, состоящий из пробоподготовки, заключающейся в переводе исследуемых атомов в ионную форму и очистки раствора от "мешающих" ионов, с последующим определением количества интересующих ионов в результате разделения ионов на специальной ионообменной колонке при помощи ионного хроматографа. Этот способ дает вполне удовлетворительные результаты (предел обнаружения порядка мг/кг), но требует тщательной очистки от "мешающих" ионов, а также наличия ионного хроматографа (стоимостью 30-50 тыс.$ ). Этот метод применяется в основном для анализа питьевой и природной воды /5/.
Все эти методы предназначены для решения либо своего узкого круга задач и не предназначены для определения йода в йодсодержащих органических соединениях и веществах или же, являясь универсальными, обладают недостаточной чувствительностью, или требуют наличия дорогостоящей аппаратуры.
Решаемая техническая задача состояла в создании способа определения йода в йодсодержащих органических соединениях и веществах, позволяющего определять количество йода в пределах от нескольких десятков микрограмм до нескольких десятков миллиграмм йода на килограмм исследуемого органического вещества, в частности белка. При этом способ должен быть доступен рядовым отечественным производителям йодсодержащих пищевых добавок.
Сущность изобретения состоит в том, что сначала определенным образом производят подготовку пробы из исследуемого вещества, а затем, используя приемы, присущие полярографическому методу, определяют количество йода. Отличительные особенности предлагаемого способа состоят в том, что навеску исследуемого материала смачивают раствором карбоната калия, прокаливают до полного озоления, смешивают полученную золу с водой, добавляют азотную кислоту и метанол, помещают полученную смесь в электрохимическую ячейку, осаждают ионы йода на поверхность ртутного электрода электрохимической ячейки при положительном потенциале на этом электроде, измеряют полярограмму при изменении потенциала на ртутном электроде к нулевому значению, а о количестве йода судят по результатам измерений полярограммы. При этом количество карбоната калия выбирают из расчета 2-10% от массы навески; прокаливание осуществляют при температуре от 450 до 550oC; азотную кислоту добавляют в количестве, необходимом для достижения pH раствора в пределах от 1 до 2; метанол добавляют в количестве, соответствующем 15-30% конечного объема раствора; ионы йода осаждают на ртутной поверхности электрода при потенциале от +1 до +1,5 В; изменение потенциала на электроде с ртутной поверхностью к нулевому значению проводят по линейному закону, при этом этот потенциал может быть модулирован переменным напряжением с частотой 25-35 Гц и амплитудой 20-50 мВ. Следует отметить, что анализируемый в электрохимической ячейке раствор можно отделять или не отделять от осадка, а в качестве электрода можно использовать как ртутную каплю, так и любой другой электрод, на поверхности которого можно создать слой ртути.
Другими словами, сущность изобретения состоит в том, что после соответствующей пробоподготовки сначала на рабочий ртутный электрод в течение некоторого времени подают положительный потенциал. За это время йодид-ионы оседают на ртутной поверхности электрода, образуя пленку нерастворимого йодида ртути (Hgl). Затем плавно меняют потенциал в отрицательную область (до 0 В). При этом происходит электрохимическое растворение йодида ртути по реакции:
HgI --->Hg++I-
На полярограмме получается пик йода с максимумом при потенциале 0,8 В. Площадь и высота пика прямо пропорциональны количеству ионов йода, находившихся в растворе. Для повышения чувствительности метода на прямолинейно меняющийся потенциал накладывают переменный потенциал.
Работа способа поясняется графиками на фиг. 1 - фиг.4.
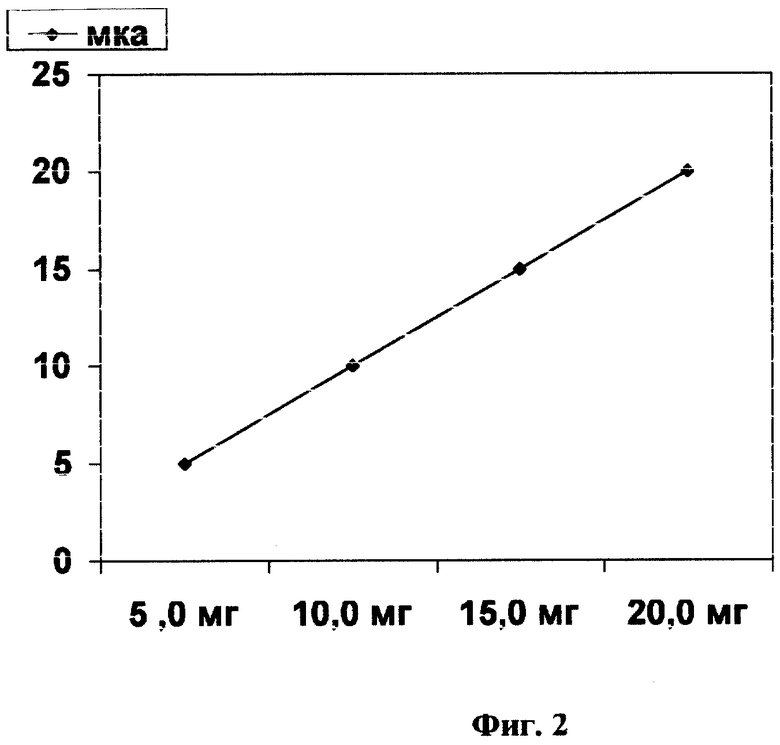
На фиг. 1 представлены результаты измерения вольтамперной зависимости, полученной при определении концентрации йода в хлебе. Кривая 1 соответствует фоновой кривой (азотная кислота 10 мл + метанол 5 мл + вода 5 мл). Кривая 2 соответствует анализируемой пробе хлеба, шкала абсцисс соответствует напряжению в мВ на электроде с ртутной поверхностью, шкала ординат соответствует току в мкА, протекающему в электрохимической ячейке.
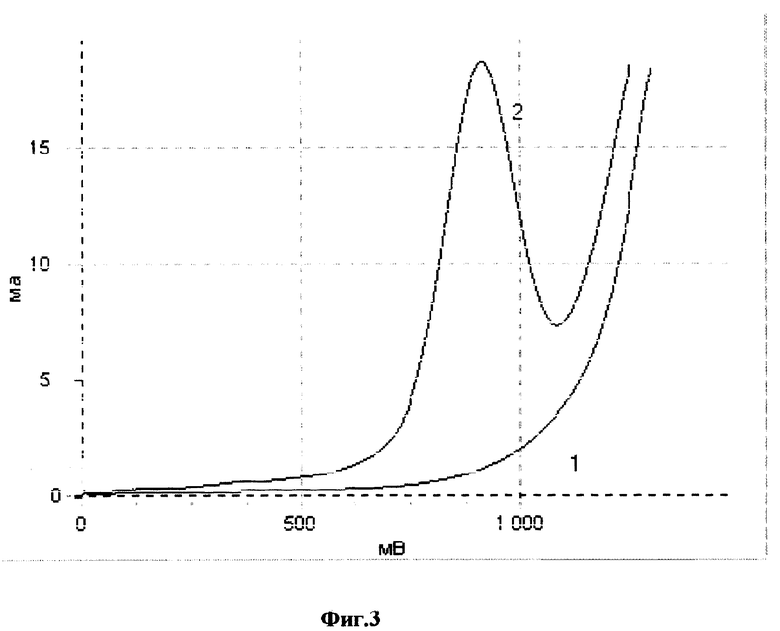
На фиг. 2 представлена колибровочная зависимость значения тока в мкА на кривой 2 фиг. 1 от концентрации йода в мг.
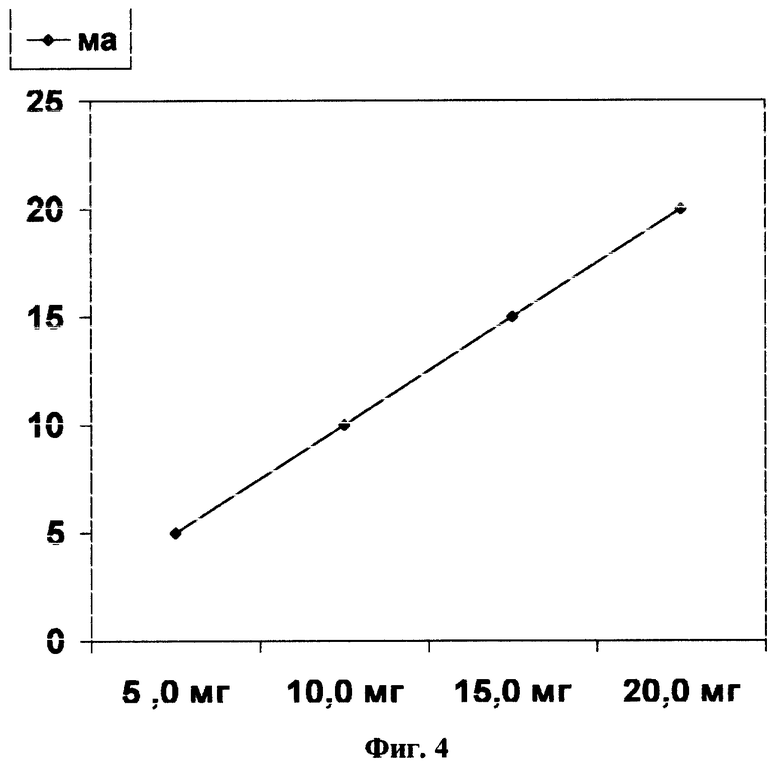
На фиг. 3 представлены результаты измерения вольтамперной зависимости, полученной при определении концентрации йода в твороге. Кривая 1 соответствует фоновой кривой (азотная кислота 10 мл + метанол 5 мл + вода 5 мл). Кривая 2 соответствует анализируемой пробе творога, шкала абсцисс соответствует напряжению в мВ на электроде с ртутной поверхностью, шкала ординат соответствует току в мА, протекающему в электрохимической ячейке.
На фиг. 4 представлена колибровочная зависимость значения тока в мА на кривой 2 фиг. 3 от концентрации йода в мг.
Способ осуществляется следующим образом: Точные навески анализируемого материала примерно по 2 г помещают в фарфоровые тигли N 3, предварительно прокаленные при температуре 600oC в течение 3-4 часов. Навески смачивают 4 мл раствора карбоната калия (0.5 н.) для связывания йода и после набухания материала (в течение 8-10 ч) высушивают в сушильном шкафу при 100oC до абсолютно сухого состояния и затем еще примерно 1 ч при 150-200oC. Тигли с навесками ставят на открытую электроплитку, где нагревают до прекращения выделения летучих веществ. Дальнейшее озоление проводят в муфельной печи при 500+50oC в течение 1-2 ч. При неполном озолении (зола должна быть серого или белого цвета) для окисления несгоревшего углерода зольный остаток смачивают примерно 0,3 мл раствора KNO3 (0.5 н.). Затем осадки в тиглях высушивают при 150oC (30 мин) и вновь прокаливают при 480oC (30 мин). Операцию смачивания, высушивания и прокаливания до полного выгорания углерода при необходимости повторяют еще 1-2 раза.
После полного озоления золу из тигля переносят в пробирку вместимостью 10 мл с делениями, смывая ее 4-5 порциями бидистиллированной воды (по 1-1,5 мл). Объем жидкости доводят до 5 мл и переносят в центрифужную пробирку. Пробы центрифугируют в течение 20 мин при 1500 об/мин. Надосадочную жидкость переливают в другую пробирку, добавляют 10 мл 0,1 н. азотной кислоты и 5 мл метанола. Затем этот раствор переносят в полярографическую ячейку и проводят измерение: при потенциале +1,0 В происходит накопление Hgl в течение 5 мин, затем при потенциале +1,2 В проводили накопление в течение 5 мин, затем успокоение раствора 15 с и после этого провели переменно-токовую полярографию с разверткой потенциала на ртутном электроде от +1,5 В до 0.0 B с модуляцией 30 мВ, частотой 30 Гц и скоростью развертки 100 мВ/с.
Результаты представлены полярограммой на фиг.1 с учетом калибровочной кривой на фиг. 2.
Пример 2. Провели анализ творога с добавлением йодированного казеина (содержанием йода до 15 мг на 1 кг творога). Результаты измерений, выполненных в условиях аналогичных примеру 1, приведены на фиг. 3 с учетом калибровочной кривой (фиг.4).
Используемые источники информации:
1. Методические указания: определение йода в соли поваренной пищевой йодированной. МУК 4.1.699-98.
2. Г.Юинг. Инструментальные методы анализа. М. "Мир", 1989, с. 203-204.
3. Chr. Reiners, H. Yanscheid, М. Labmann et al. X-ray fluorescence analysis (XFA) of thyroidal iodine content (TIC) with an improved measuring system. Exp. Clin. Endocrinol Diabetes. 106, 1998, Suppl 3. S 31-33.
4. Г.Юинг. Инструментальные методы анализа. М. "Мир", 1989, с. 331- 358.
5. М. Мархол. Ионообменники в аналитической химии. М. "Мир". 1985, т.2, с. 295-298.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ КОЛИЧЕСТВЕННОГО ОПРЕДЕЛЕНИЯ ЙОДА | 2002 |
|
RU2206086C1 |
| СПОСОБ ВОЛЬТАМПЕРОМЕТРИЧЕСКОГО ИЗМЕРЕНИЯ КОНЦЕНТРАЦИИ ЙОДА | 2003 |
|
RU2238551C1 |
| ВОЛЬТАМПЕРОМЕТРИЧЕСКИЙ СПОСОБ ОДНОВРЕМЕННОГО ОПРЕДЕЛЕНИЯ СЕЛЕНА И ЙОДА | 2009 |
|
RU2415411C1 |
| СПОСОБ ОПРЕДЕЛЕНИЯ СОДЕРЖАНИЯ НИТРОКСИЛЬНЫХ РАДИКАЛОВ В СЫРЬЕВЫХ ПОТОКАХ НЕПРЕДЕЛЬНЫХ МОНОМЕРОВ | 2017 |
|
RU2658048C1 |
| СПОСОБ ОПРЕДЕЛЕНИЯ СОДЕРЖАНИЯ ЙОДА В БИОСУБСТРАТАХ ОРГАНИЗМОВ | 2008 |
|
RU2366952C1 |
| ВОЛЬТАМПЕРОМЕТРИЧЕСКИЙ СПОСОБ КОЛИЧЕСТВЕННОГО ОПРЕДЕЛЕНИЯ L-ТИРОКСИНА | 2010 |
|
RU2428690C1 |
| СПОСОБ ИЗГОТОВЛЕНИЯ МОДИФИЦИРОВАННОГО ЭЛЕКТРОДА ДЛЯ ИНВЕРСИОННО-ВОЛЬТАМПЕРОМЕТРИЧЕСКОГО МЕТОДА ОПРЕДЕЛЕНИЯ СЛЕДОВ ТЯЖЕЛЫХ И ТОКСИЧНЫХ МЕТАЛЛОВ | 1997 |
|
RU2124720C1 |
| СПОСОБ ИНВЕРСИОННОГО ВОЛЬТАМПЕРОМЕТРИЧЕСКОГО ОПРЕДЕЛЕНИЯ МИКРОПРИМЕСЕЙ МЕДИ (II) И СУРЬМЫ (III) В ЦИНКОВОМ ЭЛЕКТРОЛИТЕ | 2004 |
|
RU2297626C2 |
| Способ определения иодид-ионов катодной вольтамперометрией | 2016 |
|
RU2645003C2 |
| СПОСОБ ПОЛЯРОГРАФИЧЕСКОГО АНАЛИЗА НЕОРГАНИЧЕСКИХ ВЕЩЕСТВ В РАСТВОРЕ | 1997 |
|
RU2135989C1 |
Использование: в пищевой, фармакологической, микробиологической, медицинской, химической промышленности при анализе микроколичеств йода в органических соединениях. Технический результат изобретения заключается в обеспечении возможности определения количества йода в пределах от нескольких десятков микрограмм до нескольких десятков миллиграмм на килограмм исследуемого органического вещества, в частности белка, и в удешевлении способа. Способ заключается в подготовке пробы из исследуемого органического вещества и в определении количества йода в ней. Подготовку пробы осуществляют следующим образом: навеску исследуемого вещества смачивают раствором карбоната калия, прокаливают ее до полного озоления, смешивают полученную золу с водой и добавляют азотную кислоту и метанол. А для определения количества йода полученную пробу помещают в электрохимическую ячейку, осаждают ионы йода на ртутную поверхность электрода электрохимической ячейки при положительном потенциале на этом электроде, измеряют вольтамперную зависимость при изменении потенциала на электроде с ртутной поверхностью к нулевому значению, а о количестве йода судят по результатам измерений вольтамперной зависимости. 7 з. п. ф-лы, 4 ил.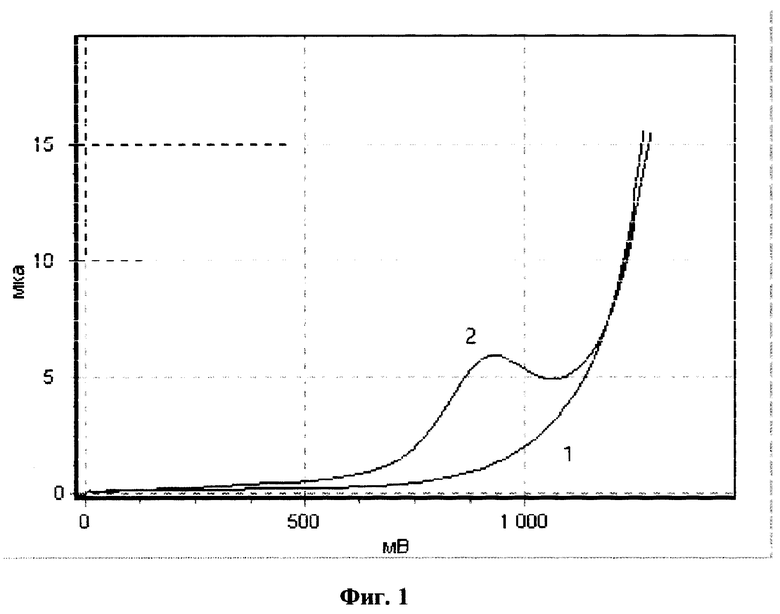
| М.Мархол | |||
| Ионообменники в аналитической химии | |||
| - М.: Мир, 1985, т.2, с | |||
| УСТРОЙСТВО ПАРОПЕРЕГРЕВАТЕЛЯ | 1920 |
|
SU295A1 |
| US 4054414, 18.10.1977 | |||
| Способ гистохимического определения йода | 1985 |
|
SU1435993A1 |
| US 5137690 A, 11.08.1992. | |||

