Предлагаемое изобретение относится к аналитической химии и может быть использовано для экспресс-анализа технологических растворов, оборотных и сточных вод цветной металлургии и других отраслей промышленности.
Известен способ совместного определения сурьмы и меди методом инверсионной вольтамперометрии (см. статью: Нейман Е.Я., Суменкова М.Ф. О совместном определении сурьмы и меди методом инверсионной вольтамперометрии// Журнал аналитической химии. 1975. Т.30. Вып.8. С.1625-1628).
Сущность известного способа состоит в одновременном предварительном электроосаждении сурьмы и меди из солянокислых растворов на графитовом электроде с последующей регистрацией анодной поляризационной кривой и определении содержания сурьмы и меди в анализируемом растворе по высоте пиков электроокисления сконцентрированных на индикаторном электроде осадков. Способ осуществляют следующим образом. Проводят электроконцентрирование сурьмы (III) и меди (II) из раствора 1 М HCl на пропитанном эпоксидной смолой дисковом графитовом микроэлектроде при потенциале -0,75 В в течение 3 мин. Регистрируют поляризационную кривую электроокисления осадков со скоростью развертки потенциала 0,125 В/с в интервале напряжений (-0,75)-(0,1) В и определяют концентрацию ионов Cu(II) и Sb(III) в контролируемом растворе по высоте анодных пиков при потенциалах -0,22 и -0,07 В соответственно.
Недостатком известного способа, ограничивающим возможность его применения для контроля микропримесей сурьмы и меди в цинковом электролите, является его низкая селективность. При соотношениях CCu:CSb≥6,6; СCu:CSb≥10:1 определение ионов Sb(III) и Cu(II) становится невозможным из-за взаимного маскирования токов электроокисления сконцентрированных на индикаторном электроде осадков меди и сурьмы. Кроме того, при проведении вольтамперометрического анализа на графитовом электроде в системе сурьма-медь зависимость максимального тока ионизации контролируемого вещества от концентрации его в растворе прямо пропорциональна только для сурьмы, в то время как анодный пик меди увеличивается линейно лишь при выполнении условия CCu:СSb≤3. Это резко снижает точность вольтамперометрического контроля меди в растворах, содержащих 3-кратный избыток сурьмы.
Известен также способ определения сурьмы, меди и висмута методом амальгамной полярографии с накоплением после их экстракционного разделения (см. статью: Мальков Е.Н., Федосеева А.Г., Стромберг А.Г. Определение нанограммовых количеств сурьмы и висмута методом амальгамной полярографии с накоплением после их экстракционного разделения// Журнал аналитической химии. 1970. Т.25. Вып.9. С.1748-1751). Способ осуществляют следующим образом. Подкисленную пробу воды (после озоления водорастворимых органических веществ) обрабатывают 0,05% раствором дитизона в CCl4 до неизменной зеленой окраски. Дитизоновые экстракты промывают 4-5 мл 0,1 М HCl. Солянокислый раствор присоединяют к основной пробе воды для дальнейшего определения висмута, а дитизонат меди осторожно выпаривают на плитке досуха. Озоляют дитизон 2 мл смеси H2SO4 и KNO3 (100 мл H2SO4+5 г KNO3) в присутствии 30% Н2O2 до полного обесцвечивания осадка. Осадок смачивают 10 каплями HCl (1:1), нагревают до 70°С, приливают 10 мл 2 М HCl, 4 капли раствора HgCl2 (1 мг/мл Hg2+) (если в пробе больше 20 мкг Cu, то вводят 10-15 капель HgCl2). Медь накапливают на ртутно-графитовом электроде при потенциале -1,0 В (относительно нас. к.э.), и анодно растворяют при потенциале от -0,4 до 0,1 В. Оставшуюся водную фазу подкисляют в делительной воронке 1-2 мл соляной кислоты (пл. 1,19), прибавляют 2-3 капли бромной воды для окисления Sb(III) до Sb(V), перемешивают и экстрагируют избыток брома хлороформом в 2-3 приема по 4 мл. Приливают 4 мл 2% хлороформного раствора диэтилдитиокарбамината цинка, 5 мл 0,4 м раствора комплексона III для маскирования никеля, свинца, кадмия и других металлов и экстрагируют висмут 1 мин. Реэкстрагируют висмут 4 мл соляной кислоты (пл. 1,19). Разбавляют водой до 10 мл и фильтруют раствор через стеклянный фильтр №1 в пирексовый стакан емкостью 15-20 мл. Вводят 4 капли раствора HgCl2 (1 мг/мл Hg2+), нагревают до кипения, быстро охлаждают в холодной воде и полярографируют. Для этого поляризуют электрод 1-2 с при ϕ=-1,0 В, затем снижают потенциал до -0,6 В и ведут электролиз 1-15 мин. Потенциал анодного пика висмута на солянокислом фоне равен -0,2 В. После отделения висмута и меди раствор сливают в стакан емкостью 20 мл, при перемешивании вводят 15 капель 10% раствора сульфида натрия, быстро нагревают до энергичного кипения, охлаждают до 40-45°С и переносят в прежнюю делительную воронку. Стакан 2-3 раза промывают 3-5 мл бидистиллята, ополаскивают 2 мл 10%-ного водного раствора диэтилдитиокарбамината натрия, затем дважды 3-5 мл воды и один раз 4 мл хлороформа. Все промывные растворы, включая хлороформную фазу, сливают в ту же воронку с пробой и экстрагируют сурьму 1 мин. Хлороформный раствор диэтилдитиокарбамината сурьмы сливают в делительную воронку емк. 50 мл, а водную фазу два раза промывают хлороформом порциями по 4 мл и присоединяют к первому экстракту. Реэкстрагируют сурьму 4 мл соляной кислотой (пл. 1,19) 30-40 с. Хлороформ сливают, а солянокислый реэкстракт промывают 2 мл чистого хлороформа, вводят 5 капель раствора HgCl2 (1 мг/мл Hg2+), разбавляют водой 10 мл, фильтруют через воронку Шотта №1, быстро нагревают до кипения, охлаждают на водяной бане и полярографируют. Потенциал электролиза сурьмы равен -0,6 В, потенциал анодного пика сурьмы на солянокислом фоне составляет -0,22 В. Все полярограммы снимают со скоростью развертки потенциала 200 мВ/мин. Растворы в процессе концентрирования перемешивают газообразным азотом, который очищают от кислорода пропусканием над активированным углем марки БАУ при температуре 700-800°С и затем пропусканием газа через раствор H2SO4 и сухой NaOH. Для получения ртутно-графитового электрода в пробу перед полярографированием вводят HgCl2, поддерживая соотношение между определяемым ионом и ртутью в растворе не менее 1:400.
Способ обеспечивает определение контролируемых ионов методом амальгамной полярографии с накоплением на ртутно-графитовом электроде с чувствительностью 0,01 мг/мл и относительной ошибкой ±20%. Время накопления 15 мин. Калибровочные графики линейны в интервале концентраций 0,01-0,2 нг/мл.
Таким образом, известный способ характеризуется высокой чувствительностью и селективностью контроля ионов Cu(II), Bi(III) и Sb(III) в водных растворах. Вместе с тем, к недостаткам известного способа относятся сложность и длительность анализа на стадии пробоподготовки, необходимость использования многочисленных реактивов, проведения операций выпаривания, озоления, нагревания, кипячения, охлаждения, фильтрации, экстракции, реэкстракции, дозирования растворов реактивов. Кроме того, на стадии электрохимического концентрирования контролируемых ионных компонентов анализируемый раствор необходимо перемешивать высокочистым газообразным азотом. Известный способ не позволяет достичь высокой точности полярографического анализа (относительная ошибка ±20%), Отмеченные недостатки исключают возможность использования известного способа для оперативного контроля примесей ионов Cu(II) и Sb(III) в цинковом электролите.
Наиболее близким по технической сущности к заявляемому является способ инверсионного вольтамперометрического определения меди и сурьмы в цинковом электролите на фоне 6 М HCl (см. статью: Зарецкий Л.С., Варновский Б.И., Пидорич М.И., Кокоева Ф.Х. Анализатор микропримесей сурьмы, меди и кадмия в цинковом электролите// Заводская лаборатория. 1982. Т.48. №10. С.6-7).
Способ предусматривает одновременное определение ионов Cu(II), Cd(II) и Sb(III) в продуктах свинцово-цинкового производства методом инверсионной дифференциальной импульсной вольтамперометрии (ИДИВ) на стационарном ртутно-капельном электроде (СРКЭ) и реализуется с помощью автоматического вольтамперометрического анализатора. Известный способ осуществляется следующим образом. Анализируемый раствор смешивают в отношении 1:1 с концентрированной соляной кислотой, помещают в электрохимическую ячейку и полярографируют. Полярографирование проводят без предварительной аэрации. Электронакопление меди производят при потенциале -0,45 В в течение 50 с, а содержание ионов Cu(II) в анализируемом растворе определяют по высоте анодного пика, регистрируемого со скоростью развертки потенциала 5 мВ/с. Измерения проводят в интервале концентраций меди(II) 0,01-0,2 мг/л. Электронакопление сурьмы производят при потенциале -0,25 В в течение 50 с, а содержание ионов Sb(III) в анализируемом растворе определяют по высоте анодного пика, регистрируемого со скоростью развертки потенциала 5 мВ/с. Измерение проводят в интервале концентраций сурьмы (III) 0,01-0,1 мг/л. При совместном определении меди и сурьмы устанавливают режим измерения меди. При полярографическом определении кадмия устанавливают начальное напряжение -0,8 В и регистрируют анодный пик со скоростью развертки потенциала 5 мВ/с. Измерение производят в интервалах концентраций ионов Cd(II) 0,05-1,0 и 0,5-10,0 мг/л без предварительного электронакопления. Содержание сурьмы, меди и кадмия определяют по градуировочным графикам, построенным по эталонным растворам, приготовленным на очищенном цинковой пылью электролите. Приведенная среднеквадратичная погрешность анализа ионов Cu(II), Cd(II) и Sb(III) в промышленных пробах соответственно составляет 3, 6 и 7%.
К достоинствам известного способа относятся высокая чувствительность и оперативность измерения, а также простота пробоподготовки, позволяющие использовать его для экспресс-анализа ионов Cu(II), Sb(III), Cd(II) в продуктах свинцово-цинкового производства.
Существенным недостатком данного способа является низкая селективность. Известный способ обеспечивает полярографическое определение сурьмы при допустимом соотношении Sb3+:Cu=1:10, а полярографическое определение меди при допустимом соотношении Cu2+:Sb3+=1:10.
В зависимости от состава перерабатываемого минерального сырья, расхода реагентов (цинковой пыли и соли Шлиппе) соотношение ионных концентраций [Cu]:[Sb] в гидрометаллургических растворах цинкового производства изменяется в очень широких пределах (см. книги: Лакерник М.М., Пахомова Г.Н. Металлургия цинка и кадмия. М.: Металлургия. 1969. 488 с.; Снурников А.П. Гидрометаллургия цинка. М.: Металлургия, 1981. 384 с.). В то же время при больших расходах соли Шлиппе (ортосульфоантимоната натрия Na3Sb4·9Н2О) концентрация ионов Sb(III) в гидрометаллургических растворах существенно увеличивается, при этом соотношение ионных концентраций [Sb]:[Cu] может составлять 20:1 и больше. Соль Шлиппе выполняет роль активирующей добавки при цементационной очистке цинковых растворов от примесей. Соль Шлиппе обычно задают в процесс в виде раствора с содержанием сурьмы 10-15 г/л на первой стадии при двустадийной и на второй стадии при трехстадийной технологической схеме. Расход соли устанавливают исходя из местных заводских условий и рассчитывают по отношению к содержанию кобальта в растворе. Отношение создаваемой концентрации сурьмы к кобальту колеблется от (0,6-1):1 до (1,5-2):1, его выбирают в зависимости от исходной концентрации кобальта (см. книгу: Снурников А.П. Гидрометаллургия цинка. М.: Металлургия, 1981. С.209-211). Из-за близости потенциалов полуволн ионов Sb(III) и Cu(II) при полярографическом анализе водных растворов, включая технологические растворы цинкового производства наблюдается взаимное мешающее влияние меди и цинка (см. например, книгу: Выдра Ф., Штулик К., Юлакова Э. Инверсионная вольтамперометрия. М.: Мир, 1980. С.209-210, 226). Низкая разрешающая способность вольтамперометрического определения ионов Sb(III) и Cu(II) на СРКЭ в фоновом электролите 6 М HCl, согласно известному способу, приводит к существенному увеличению погрешности анализа и, в конечном итоге, к выдаче недостоверной информации о содержании вредных примесей сурьмы и меди в поступающем на электролиз цинковом растворе. Данные о содержании ионов Cu(II) и Sb(III) в очищенном цинковом электролите и по стадиям очистки относятся к числу важнейших режимных параметров производства цинка гидрометаллургическим методом (см. книги: Раннев Г.Г., Салин А.А. Методы и приборы автоматического контроля качества растворов. Ташкент: Изд-во «Фан», 1968. 158 с.; Раннев Г.Г. Хронопотенциографы. М.: Энергия, 1979. 136 с.). При нарушении технологического регламента процесса цементационной очистки цинковых растворов от вредных примесей меди и кадмия наблюдается резкое ухудшение технико-экономических показателей получения катодного цинка (снижение выхода по току, ухудшение качества товарной продукции, перерасход цинковой пыли и прочих реагентов).
Целью предлагаемого изобретения является повышение избирательности и точности анализа.
Поставленная цель достигается тем, что согласно способу инверсионного вольтамперометрического определения микропримесей меди (II) и сурьмы (III) в цинковом электролите на стационарном ртутно-капельном электроде, включающем измерение концентрации сурьмы (III) на фоне 6 М HCl, контроль меди (II) осуществляют непосредственно в нейтральном цинковом электролите, при этом электронакопление меди производят при потенциале (-0,15±0,01) В, анодную вольтамперную кривую регистрируют в диапазоне напряжений от (-0,15) до (+0,25) В, концентрацию меди в растворе измеряют по высоте анодного пика при потенциале (+0,095±0,001) В, сурьму (III) определяют после экстракционного отделения меди раствором диэтилдитиокарбамата свинца в хлороформе из подкисленного до рН 1-2 цинкового электролита, при этом пробу цинкового электролита смешивают в отношении 1:1,5:1,5 с 0,1 М HCl и 0,1% раствором диэтилдитиокарбамата свинца в хлороформе, экстрагируют медь в течение 2 мин, отделяют водную фазу и смешивают в отношении 1:1 с концентрированной соляной кислотой, электронакопление сурьмы производят при потенциале (-0,35±0,01) В, анодную вольтамперную кривую регистрируют в диапазоне напряжений от (-0,35) до (-0,05) В, содержание сурьмы в растворе измеряют по высоте анодного пика при потенциале -0,175±0,001 В.
В результате проведения патентных исследований не было выявлено каких-либо известных способов инверсионного вольтамперометрического определения меди и сурьмы в цинковом электролите на стационарном ртутно-капельном электроде, предусматривающих контроль ионов меди (II) непосредственно в цинковом электролите, а определение сурьмы (III) после экстракционного отделения меди раствором диэтилдитиокарбамата свинца в хлороформе из подкисленного до рН 1-2 цинкового электролита, т.е. имеющих признаки, сходные с признаками, отличающими заявляемый способ от прототипа.
Для снижения негативного воздействия ионов Cu(II) на процесс электролиза цинка очистку технологических растворов от меди, как правило, ведут до содержания 0,05-0,1 мг/л (см. книгу: Снурников А.П. Гидрометаллургия цинка. М.: Металлургия, 1981. С.239). Для экспресс-анализа низких концентраций ионов Cu(II) в цинковых электролитах применяют различные варианты вольтамперометрии. Контроль меди производят преимущественно в кислых фоновых электролитах (см., например, книги: Руководство. Методы аналитического контроля в цветной металлургии. Т.2. Производство свинца и цинка. 4.2. Методы аналитического контроля в производстве цинка. М.: Цветметинформация, 1977. С.43; Каплан Б.Я., Пац Р.Г., Салихджанова Р.М.-Ф. Вольтамперометрия переменного тока. М.: Химия, 1985. С.221, 223). Причем, для достижения приемлемой селективности полярографического анализа Cu(II) в присутствии ионов Sb(III) в качестве фона используют достаточно концентрированные растворы минеральных кислот (см. статью: Зарецкий Л.С., Варновский Б.И., Пидорич М.И., Кокоева Ф.Х. Анализатор микропримесей сурьмы, меди и кадмия в цинковом электролите// Заводская лаборатория. 1982. Т.48. №10. С.6-7).
Для избирательного вольтамперометрического контроля ионов Cu(II) в технологических растворах цинкового производства, содержащих Sb(III), могут быть использованы нейтральные фоновые электролиты. В нейтральных растворах токи электрохимического восстановления ионов Sb(III) отсутствуют, что связано с образованием полярографически неактивных форм сурьмы (см. книгу: Немодрук А.А. Аналитическая химия сурьмы. М.: 1978. С.63). Это создает возможность устранения мешающего влияния Sb(III) при полярографическом определении некоторых других ионов, включая Cu(II).
В отличие от сурьмы в некомплексообразующих нейтральных фоновых электролитах медь (II) восстанавливается на ртутном электроде в одну стадию с числом электронов, равном двум. Восстановление Cu2+→Cu(Hg) протекает обратимо. При этом регистрируется волна с ϕ1/2, близким к 0, В. При определении меди инверсионными методами обычно используют ее способность образовывать амальгаму (см. книгу: Выдра Ф., Штулик К., Юлакова Э. Инверсионная вольтамперометрия. М.: Мир, С.220-227).
Как показывает опыт (см. книгу: Крюкова Т.А., Синякова С.И., Арефьева Т.В. Полярографический анализ. М.: Госхимиздат, 1959. 772 с. и статью: Боровков Г.А., Хмаро В.В., Щербич О.В. Автоматический вольтамперометрический контроль ионов меди (II) в технологических растворах электролитического рафинирования никеля// Заводская лаборатория. 2003. Т.69. №2. С.25-28), высокая электропроводность и стабильность основных компонентов химического состава гидрометаллургических растворов позволяет в ряде случаев производить их вольтамперометрический анализ без какой-либо предварительной реагентной обработки, поскольку при полярографировании такие растворы сами выполняют роль фонового электролита.
Нами экспериментально установлено, что при определении меди непосредственно в нейтральном цинковом электролите методом дифференциальной импульсной полярографии (ДИП) на стационарном ртутно-капельном электроде (СРКЭ) при потенциале +0,090 В регистрируется четко выраженный катодный пик, пропорциональный концентрации ионов Cu(II) в анализируемом растворе. При анализе цинковых растворов методом инверсионной дифференциальной импульсной вольтамперометрии с предварительным электронакоплением ионов Cu(II) на СРКЭ при потенциале - 0,15 В и последующей съемкой вольтамперной кривой электрорастворения сконцентрированного на индикаторном электроде вещества, при потенциале +0,095 В регистрируется анодный пик, пропорциональный концентрации меди(II) в контролируемом растворе.
Нейтральный цинковый раствор может быть использован в качестве фонового электролита при полярографическом определении ионов Cd(II). Примеси кадмия также отрицательно влияют на электролиз цинка и, как правило, контролируются в цеховых экспресс-лаборатория вольтамперометрическими методами в солянокислой среде (см. например, книгу: Руководство. Методы аналитического контроля в цветной металлургии. Т2. Производство свинца и цинка. Ч2. Методы аналитического контроля в производстве цинка. М.: Цветметинформация, 1977. С.30-31). Определение ионов Cd(II) непосредственно в цинковом электролите без предварительной пробоподготовки осуществляется по высоте катодного ДИП пика, регистрируемого вторичным прибором анализатора при потенциале -0,55 В. Вольтамперную кривую снимают в интервале напряжений от -0,35 до -0,75 В.
Общепринятый допустимый предел содержания ионов Sb(III) в очищенном нейтральном цинковом электролите составляет 0,1 мг/л, но, как правило, технологи стремятся снизить концентрацию сурьмы до следовых количеств (0,01-0,02 мг/л). Оперативный контроль содержания сурьмы в технологических растворах цинкового производства позволяет сократить расход соли Шлиппе и цинковой пыли в процесс цементационной очистки, повысить выход по току цинка, улучшить качество товарного катодного цинка.
Инверсионная вольтамперометрия входит в число наиболее чувствительных физико-химических методов контроля микропримесей тяжелых металлов в продуктах цветной металлургии. Основным препятствием для широкого применения метода ИДИВ для экспресс-анализа ионов Sb(III) в цинковых электролитах является низкая селективность контроля сурьмы в присутствии избыточных концентраций меди (II) (см. статью: Нейман Е.Я., Игнатов В.И.// ЖАХ. 1976. Т.31. Вып.5. с.970-983).
В зависимости от состава перерабатываемого сырья и качества цементационной очистки соотношение ионных концентраций [Cu]:[Sb] в поступающем на электролиз раствора сульфата цинка изменяется в широких пределах, достигая в отдельных случаях значения 50:1. как показывает практика (см. книгу: Немодрук А.А. Аналитическая химия сурьмы. М., 1978. 222 с., статью: Зарецкий Л.С., Варновский Б.И., Пидорич М.И., Кокоева Ф.Х. Анализатор микропримесей сурьмы, меди и кадмия в цинковом электролите// Заводская лаборатория. 1982. Т.48. N 10. С.6-7), определение Sb(III) в цинковых электролитах методом ИДИВ на СРКЭ возможно при содержании в анализируемом растворе не более чем 10-кратного избытка ионов Cu(II).
Одним из наиболее эффективных приемов повышения селективности вольтамперометрического определения сурьмы является предварительное экстракционное отделение мешающих анализу примесей органическими растворителями. Однако разработанные для контроля слабозагрязненных и природных вод методики экстракционно полярографического определения сурьмы (см. книгу: Новиков Ю.В., Ласточкина К.О., Болдина З.Н. Методы исследования качества воды водоемов. М.: Медицина. С.222-224 и статью: Мальков Е.Н., Федосеева А.Г., Стромберг А.Г. Определение нанограммовых количеств сурьмы и висмута методом амальгамной полярографии с накоплением после их экстракционного разделения// ЖАК. 1970. Т.25. Вып.9. С.1748-1751) имеют очень сложную и длительную пробоподготовку, что существенно затрудняет их использование для экспресс-анализа Sb(III) в промышленных растворах.
Нами экспериментально установлено, что необходимая селективность и оперативность вольтамерометрического контроля сурьмы в цинковых электролитах может быть обеспечена за счет предварительного экстракционного отделения меди раствором диэтилдитиокарбамата свинца (ДЭДТК-Pb) в хлороформе.
Дитиокарбаматы (ДДТК) являются специфичными реагентами на медь, образуя с ней прочные химические соединения (см. книгу: Бырько В.М. Дитиокарбаматы. М.: Наука, 1984. 341 с.). По данным, приведенным в книге: Пятницкий И.В., Сухан В.В. Маскирование и демаскирование в аналитической химии. М.: Наука, 1990. С.30., ПРCu (ДЭДТК)2=2,8·10-30, в то время как ПРZn (ДЭДТК)2=1,2·10-17. Малорастворимый в воде комплекс ДЭДТК с медью хорошо экстрагируется хлороформом, что обеспечивает возможность ее извлечения из концентрированных цинковых растворов. Для селективного экстракционного выделения меди(II) из содержащих ионы Sb(III) цинковых электролитов в качестве экстрагента может быть использован раствор ДЭДТК-Pb в хлороформе. Диэтилдитиокарбамат свинца прочнее диэтилдитиокарбаматов многих металлов, включая сурьму, не менее прочен по сравнению с ДЭДТК-Cu (см. книгу: Немодрук А.А., Егиазарова Н.В., Боганова А.Н. и др. Фотометрические методы анализа в цветной металлургии. М.: Металлургия, 1981. С.72-73). Вследствие этого при взбалтывании раствора, содержащего ионы меди(II), с раствором ДЭДТК-Pb в хлороформе происходит замещение свинца медью с образованием в слое органического растворителя ДЭДТК-Cu. При этом в отличие от используемого в работах: Мальков Е.Н., Федосеева А.Г., Стромберга А.Г. Определение нанограммовых количеств сурьмы и висмута методом амальгамной полярографии с накоплением после их экстракционного разделения// ЖАХ. 1970. Т.25. Вып.9. С.1748-1751 и Новиков Ю.В., Ласточкина К.О., Болдина З.М. Методы исследования качества воды водоемов. М.: Медицина, 1990. С.222-224; диэтилдитиокарбамата натрия, ионы Sb(III) раствором ДЭДТК-Pb не извлекаются. Диэтилдитиокарбамат натрия реагирует с ионами Sb(III) с образованием внутрикомплексного соединения, хорошо экстрагирующегося многими органическими растворителями (см. книгу: Немодрук А.А. Аналитическая химия сурьмы. М.: Наука, 1978. С.106). Аналогичным образом ведет себя ДЭДТК-Zn, применяющийся для экстракционного выделения сурьмы из цинка и его солей. Проведенные нами экспериментальные исследования подтвердили, что использование в качестве экстрагента раствора ДЭДТК-Pb в хлороформе обеспечивает высокую селективность извлечения ионов Cu(II) из цинкового электролита в присутствии ионов Sb(III).
Таким образом, избирательный вольтамперометрический анализ технологических растворов цинкового производства на содержание ионов Cu(II) и Sb(III) согласно заявляемому способу осуществляется в две стадии. Первоначально производится определение меди (II) непосредственно в нейтральном цинковом растворе, выполняющем при этом роль фонового электролита. Затем, после экстракционного отделения меди из цинкового электролита раствором диэтилдитиокарбамата свинца в хлороформе при рН 1-2, контролируется содержание сурьмы(III) в водной фазе, смешанной в соотношении 1:1 с концентрированной HCl.
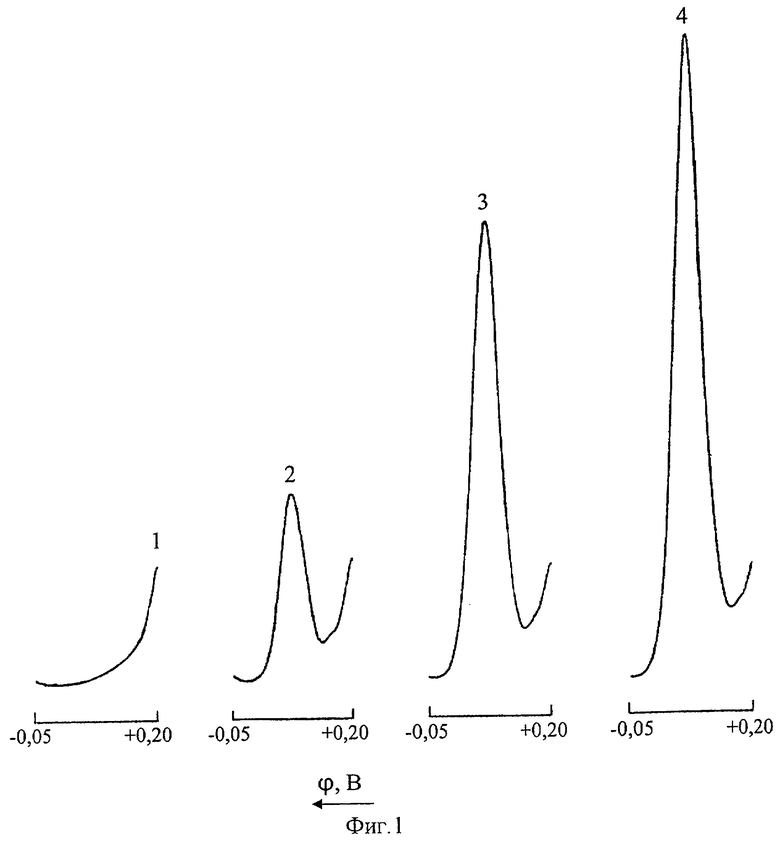
На фиг.1 представлены дифференциальные импульсные полярограммы меди в цинковом электролите: 1 - нулевой раствор (дополнительно очищенный цинковой пылью нейтральный цинковый раствор); 2 - то же после введения добавки 0,25 мг/л Cu(II); 3 - промышленная проба цинкового электролита; 4 - то же после введения добавки 0,25 мг/л Cu(II).
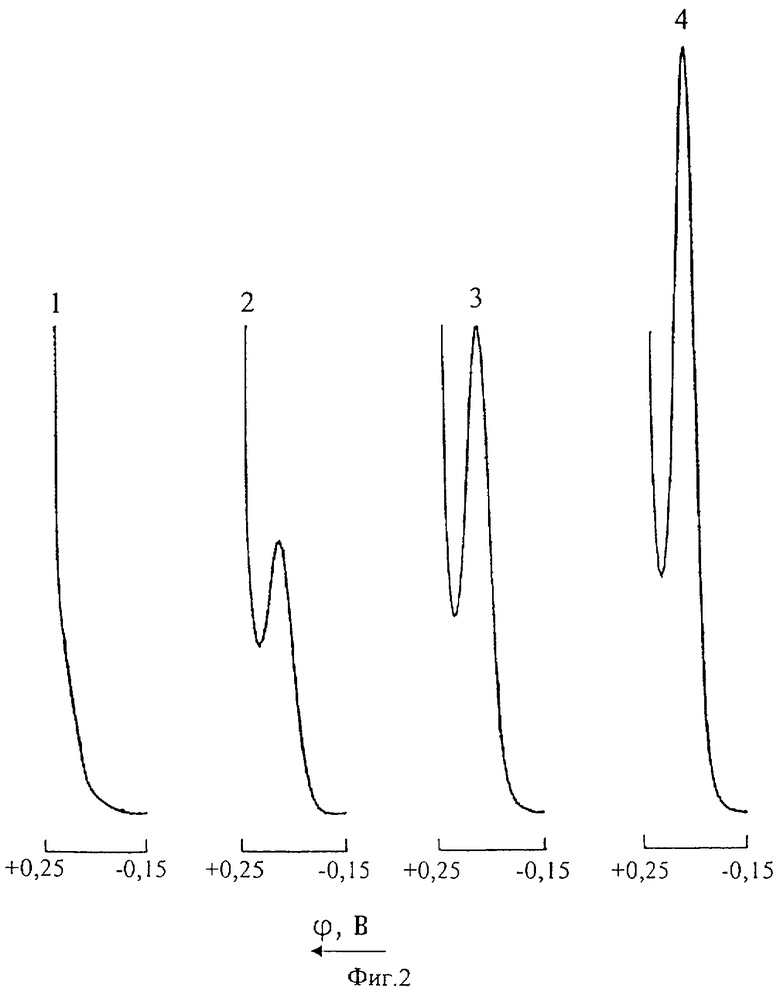
На фиг.2 представлены инверсионные вольтамперные кривые меди в цинковом электролите (Uнак=-0,15 В; τнак=30 с): 1 - нулевой раствор; 2 - то же после введения добавки 0,1 мг/л Cu(II); 3 - промышленная проба цинкового электролита; 4 - то же после введения добавки 0,1 мг/л Cu(II).
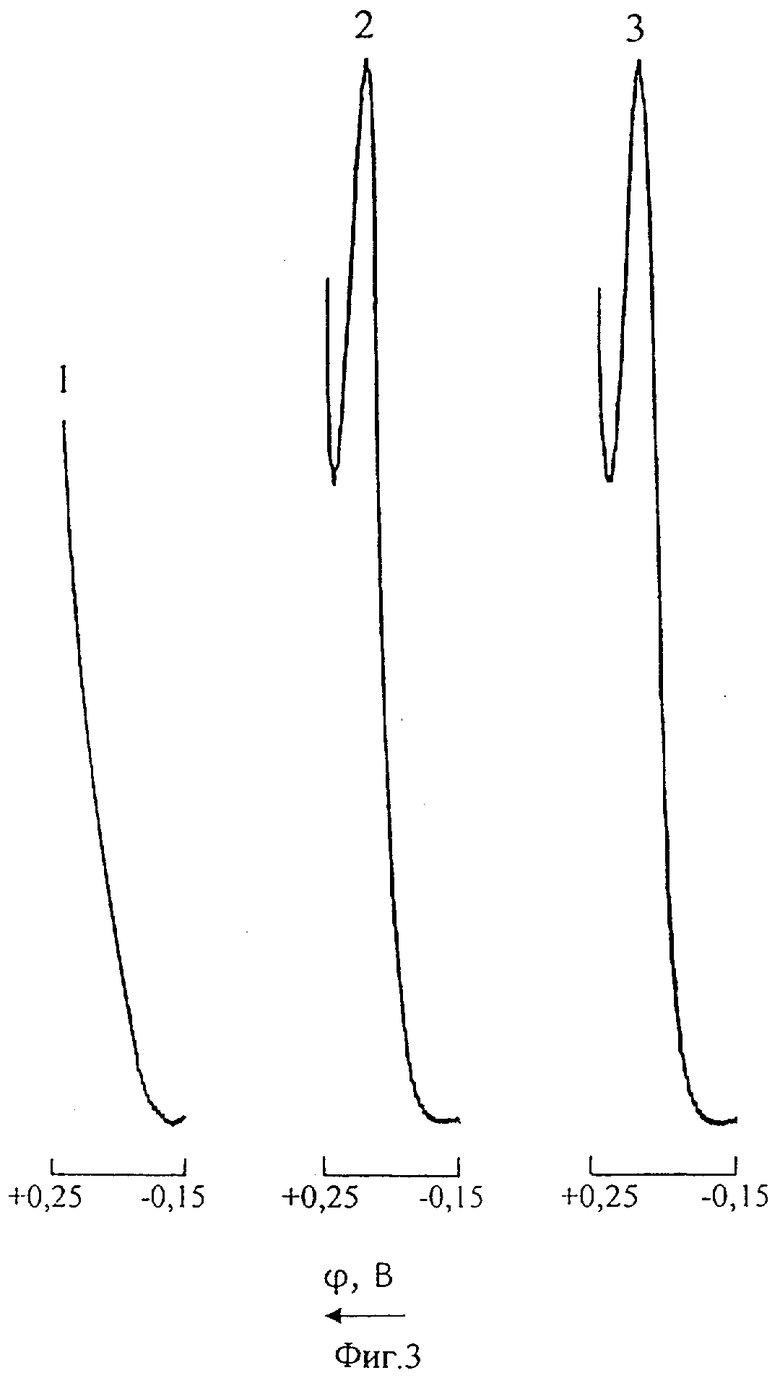
На фиг.3 представлены инверсионные вольтамперные кривые меди в цинковом электролите в присутствии избыточной концентрации сурьмы (Uнак=-0,15В; τнак=30 с): 1 - фон (нулевой раствор); 2 - CCu(II)=0,1 мг/л; CSb(III)=0; 3 - CCu(II)=0,1 мг/л; CSb(III)=20 мг/л.
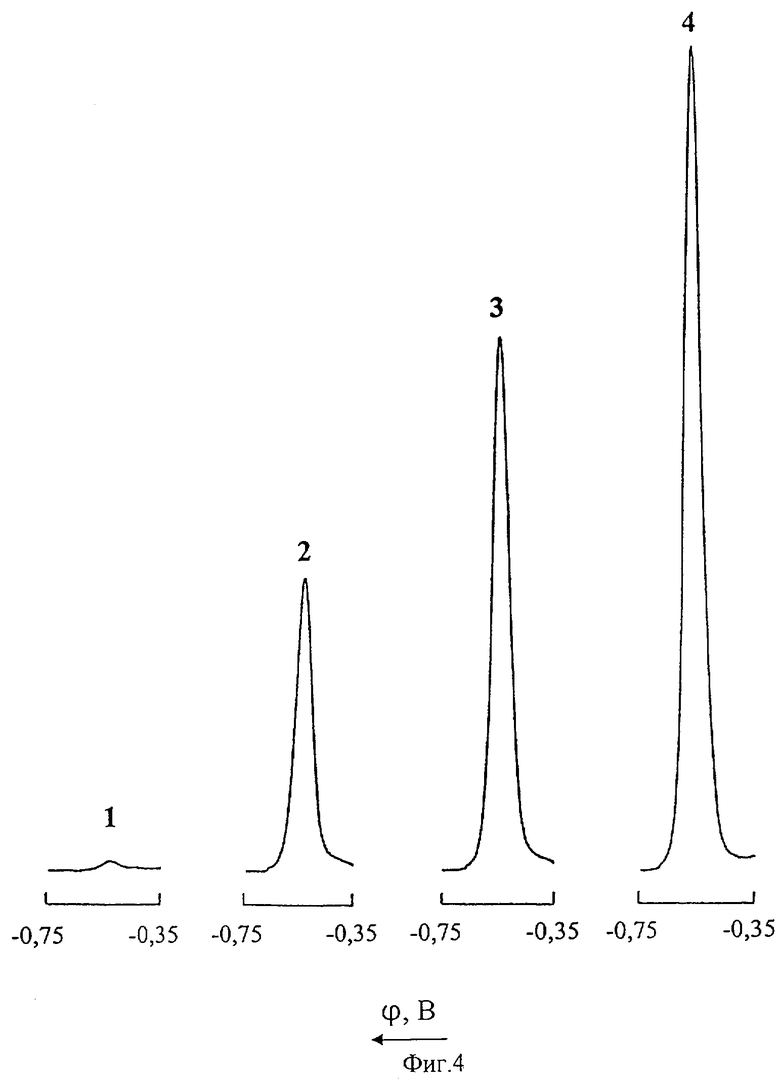
На фиг.4 представлены дифференциальные импульсные полярограммы кадмия в цинковом электролите: 1 - нулевой раствор; 2 - то же после введения добавки 1 мг/л Cd(II); 3 - промышленная проба цинкового электролита; 4 - то же после введения добавки 1 мг/л Cd(II).
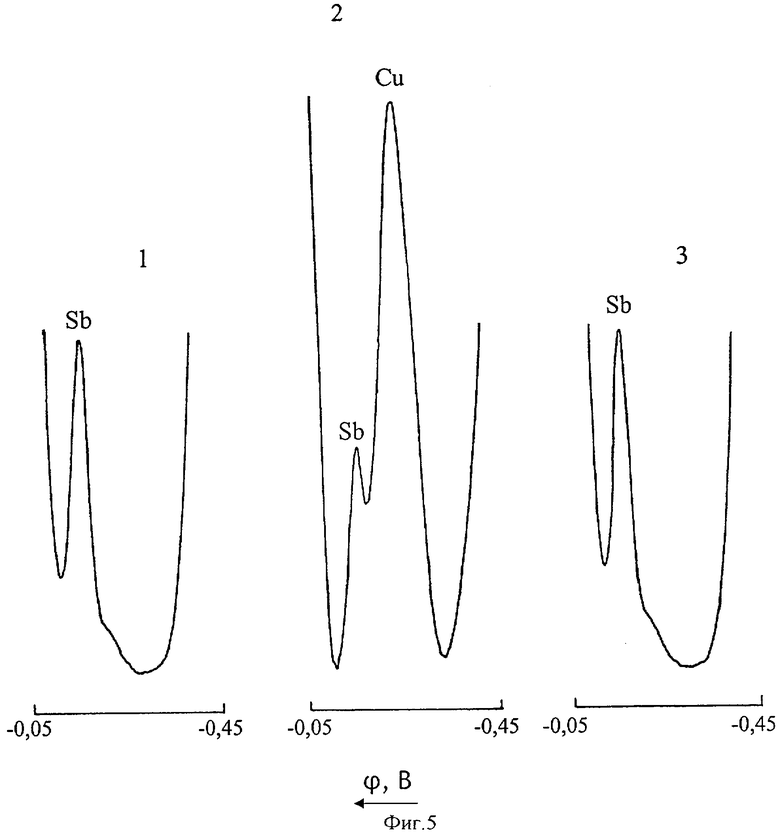
На фиг.5 представлены инверсионные вольтамперные кривые сурьмы(III) и меди(II) в цинковом электролите на фоне 6 М HCl (Uнак=-0,45 В; τнак=60 с): 1 - CSb(III)=0,05 мг/л; CCu(II)=0; 2 - CSb(III)=0,05 мг/л; CCu(II)=0,5 мг/л; 3 - то же после экстракции 0,1% раствором ДЭДТК-Pb в хлороформе.
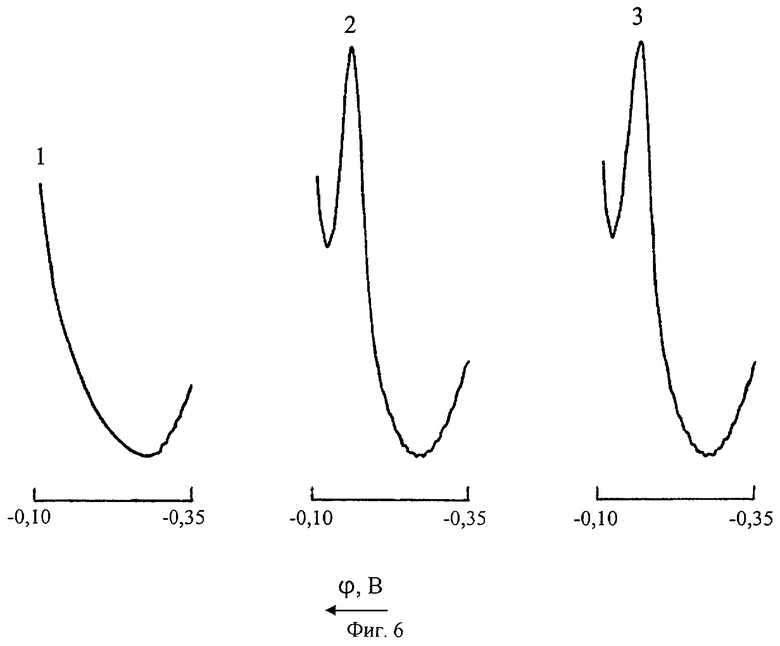
На фиг.6 представлены инверсионные вольтамперные кривые сурьмы(III) в цинковом электролите на фоне 6 М HCl (Uнак=-0,35 В; τнак=120 с): 1 - СSb(III)=CCu(II)=0; 2 - CSb(III)=0,01 мг/л; CCu(II)=0; 3 - CSb(III)=0,01 мг/л; CCu(II)=1 мг/л после экстракции 0,1% раствором ДЭДТК-Pb в хлороформе.
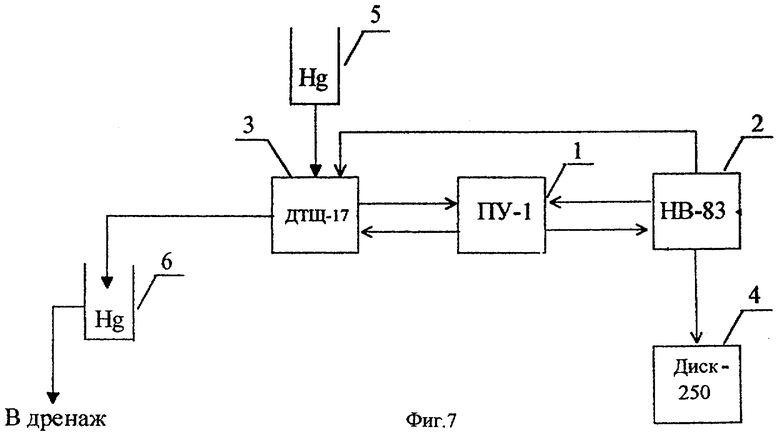
На фиг.7 представлена структурная схема вольтамперометрического анализатора тяжелых металлов в цинковом электролите.
Из фиг.1, иллюстрирующей возможность определения ионов Cu(II) в цинковом электролите методом дифференциальной импульсной полярографии (ДИП) без предварительной пробоподготовки, видно, что при полярографировании цинкового раствора в интервале напряжений от +0,20 до -0,05 В при потенциале +0,090 В регистрируется четко выраженный катодный пик меди.
Исследования проводили на промышленных пробах поступающего на электролиз нейтрального цинкового раствора, отобранных в выщелачивательном цехе завода «Электроцинк» (г.Владикавказ). Состав цинкового электролита, мг/л: (130-150)·103 Zn; (8-10)·103 Mn; 150-600 Cl; 50-150 Fe; 0,2-8 Cd; 1-2 Со; 0,05-0,4 Cu; 0,1-0,2 Ni; 0,01-0,2 Tl; 0,01-0,15 Sb.
Модельные и эталонные растворы готовили путем введения добавок меди(II) и сурьмы(III) в «нулевой» (дополнительно очищенный от примесей тяжелых металлов) цинковый электролит. Очистку проводили с помощью цинковой пыли по методике, описанной в статье: Зарецкий Л.С., Варновский Б.И., Пидорич М.И., Кокоева Ф.Х. Анализатор микропримесей сурьмы, меди и кадмия в цинковом электролите//Заводская лаборатория. 1982. Т.48. №10. С.6-7.
Измерения производили на автоматическом вольтамперометрическом анализаторе, выполненном на базе полярографа ПУ-1 (см. статью: Боровков Г.А., Хмаро В.В., Щербич О.В. Автоматический вольтамперометрический контроль ионов меди (II) в технологических растворах электролитического рафинирования никеля// Заводская лаборатория. 2003. Т.69. №2. С.25-28). Схема электрохимической ячейки трехэлектродная (рабочий электрод типа «висящая» ртутная капля, электрод сравнения - хлорсеребряный, вспомогательный электрод - стеклоуглеродный), длительность наложения прямоугольных импульсов 40 мс с периодом 80 мс. Все вольтамперные кривые регистрировали со скоростью развертки потенциала 5 мВ/с. Остальные режимные параметры (амплитуда импульсного поляризующего напряжения, амплитуда развертки потенциала, начальное напряжение или потенциал электронакопления, длительность электронакопления) выбирались в ходе экспериментальных исследований.
Метод ДИП может быть использован для определения больших концентраций меди(II) непосредственно в цинковых растворах при нарушении технологического регламента процесса цементационной очистки от примесей. Градуировочный график линеен в диапазоне содержаний 0-2 мг/л, нижний предел обнаружения составляет 0,05 мг/л Cu(II).
Из фиг.2, иллюстрирующей возможность определения ионов Cu(II) в цинковом растворе методом ИДИВ без предварительной пробоподготовки, видно, что на инверсионной вольтамперной кривой при потенциале +0,095 В проявляется четко выраженный анодный пик меди.
При анализе технологических растворов методом ИДИВ потенциал электронакопления ионов Cu(II) на СРКЭ устанавливали -0,15 В. В течение всего времени накопления (30 с) помещенную в электрохимическую ячейку пробу подвергали интенсивному перемешиванию. После окончания стадии успокоения (10 с) регистрировали анодную кривую меди в интервале напряжений от -0,15 до 0,25 В. Концентрацию ионов Cu(II) в растворе определяли по высоте ДИП пика при потенциале +0,095 В, с нижним пределом обнаружения 0,01 мг/л. Градуировачный график линеен в диапазоне содержаний 0-0,5 мг/л Cu(II).
Из фиг.3, иллюстрирующей возможность инверсионного вольтамперометрического анализа ионов Cu(II) в цинковом электролите без предварительной пробоподготовки в присутствии Sb(III), видно, что 200-кратный избыток сурьмы не мешает определению меди. Ионное соотношение [Sb]:[Cu]=200:1 не является предельным, т.к. при вольтамперометрическом измерении Cu(II) непосредственно в нейтральном цинковом растворе сурьма(III) полярографически не активна и, таким образом, не может влиять на аналитический сигнал меди.
Из фиг.4, иллюстрирующей возможность полярографического определения ионов Cd(II) непосредственно в цинковом электролите без предварительной пробоподготовки, видно, что при полярографировании цинкового раствора в интервале напряжений от -0,35 до -0,75 В при потенциале -0,55 В регистрируется катодный ДИП пик кадмия. Градуировочный график линеен в диапазонах концентраций 0-5 и 0-10 мг/л Cd(II). Нижний предел обнаружения кадмия 0,1 мг/л.
На фиг.5 представлены инверсионные вольтамперограммы 0,05 мг/л сурьмы, полученные при полярографировании нулевого цинкового раствора (кривая 1) и цинкового электролита, содержащего 0,5 мг/л Cu(II) до (кривая 2) и после (кривая 3) экстрагирования меди 0,1% раствором ДЭДТК-Pb в хлороформе. Режим вольтамперометрического анализа: потенциал электронакопления Uнак=-0,45 В, длительность электронакопления τнак=60 с. В течение всего времени электронакопления раствор подвергали интенсивному перемешиванию. Вольтамперные кривые регистрировали после 10-секундной стадии успокоения. Все стадии анализа осуществлялись в автоматическом режиме в соответствии с заданной программой. Включение и отключение приводов сброса и роста ртутной капли, мешалки и диаграммной ленты самописца производились по командам, поступающим с блока управления анализатора.
Из фиг.5 видно, что при съемке анодной вольтамперной кривой не подвергшегося экстракции цинкового электролита, содержащего ионы Cu(II) и Sb(III) (кривая 2), самописцем регистрируются пики меди (ϕп Cu=-0,25 В) и сурьмы (ϕп Sb=-0,175 В). В присутствии меди пик сурьмы искажается и несколько уменьшается по высоте. Экстракционная обработка цинкового электролита позволяет полностью нивелировать мешающее влияние меди при вольтамперометрическом определении сурьмы (кривые 1 и 3 идентичны).
Для экстракционного концентрирования ионов Cu(II), как правило, используют 0,01% раствор ДЭДТК-Pb в хлороформе (см., например, книгу: Немодрук А.А. и др. Фотометрические методы анализа в цветной металлургии М.: Металлургия, 1981. С.73). Нами экспериментально установлено, что процесс экстракции может быть значительно интенсифицирован при увеличении концентрации ДЭДТК-Pb в органической фазе до 0,1%. Ввиду того, что обменная реакция между ДЭДТК-Pb и медью происходит в кислой среде при рН 1-2 (см., например, книгу: Лурье Ю.Ю. Аналитическая химия промышленных сточных вод. М.: Химия, 1984. С.126) нейтральный цинковый электролит предварительно обрабатывали соляной кислотой. Наибольшая степень экстракционного извлечения Cu(II) достигается из смеси (1:1,5) цинкового раствора с 0,1 М HCl. Экстракция меди раствором ДЭДТК-Pb в хлороформе протекает с достаточно большой скоростью. При содержании ионов Cu(II) в цинковом электролите 0,5 мг/л уже в первые 15 с встряхивания делительной воронки из водной фазы извлекается до 95% меди, а при увеличении времени экстракции до 30 с обеспечивается 100%-ная очистка цинкового раствора. Полное удаление меди из растворов, содержащих 1 -2 мг/л Cu(II), достигается за время экстракции 90-120 с.
В результате проведенных экспериментальных исследований был выбран следующий оптимальный режим экстракционной отчистки нейтрального цинкового раствора от меди (CCu(II)≤2 мг/л) перед инверсионным вольтамперометрическим определением сурьмы (CSb(III)≥0,01 мг/л): объем пробы цинкового электролита 10 мл; объем разбавителя (0,1 М HCl) 15 мл; объем экстрагента (0,1% раствора ДЭДТК-Pb в хлороформе) 15 мл; длительность экстракции 120 с. При хранении в темной стеклянной посуде 0,1% раствор ДЭДТК-Pb устойчив в течение 5 дней.
Обработанный 0,1 М HCl водный раствор пробы цинкового электролита и экстрагента тщательно перемешивали в грушевидной делительной воронке. После разделения водной и органической фаз из очищенного от меди цинкового электролита, разбавленного 0,1 М HCl, отбирали аликвоту 10 мл и смешивали с равным объемом концентрированной соляной кислоты. Подготовленный к вольтамперометрическому анализу раствор помещали в электрохимическую ячейку анализатора и полярографировали в режиме ИДИВ.
На фиг.6 представлены вольтамперные кривые, иллюстрирующие возможность определения Sb(III) на уровне следовых концентраций (0,01 мг/л) при ионном соотношении [Cu]:[Sb] в анализируемом цинковом растворе, равном 100:1.
С учетом 5-кратного разбавления контролируемого цинкового электролита на стадии пробоподготовки растворами 0,1 М и 12 М HCl содержание ионов Sb(III) в помещенной в электрохимическую ячейку пробе фактически составляло 0,002 мг/л. Это показывает хорошие перспективы применения метода ИДИВ с предварительным экстракционным отделением меди диэтилдитиокабаматом свинца для контроля микроконцентраций сурьмы в различных объектах окружающей среды, включая почвы, атмосферный воздух, природные воды.
Градуировочные графики для определения сурьмы линейны в диапазонах концентрации Sb(III) в цинковых растворах 0-0,1 мг/л (τнак=120 с) и 0-0,5 мг/л (τнак=15 с). Для построения градуировочных графиков эталонные растворы проводили через весь ход анализа.
На подавляющем большинстве цинковых заводов России и стран СНГ цементационная очистка растворов сульфата цинка от примесей тяжелых металлов осуществляется в две стадии (см., например, книгу: Снурников А.П. Гидрометаллургия цинка М.: Металлургия, 1981, с.210). При этом возникает необходимость контроля сурьмы (III) в технологических растворах на выходе первой стадии очистки, концентрация ионов Cu(II) в которых изменяется в пределах от 30 до 60 мг/л, Sb(III) в диапазоне 2-4 мг/л. Как правило, для определения сурьмы в цинковых растворах используются фотоколориметрические методы анализа, имеющие очень сложную и трудоемкую пробоподготовку (см., например, книгу: Руководство. Методы аналитического контроля в цветной металлургии. Т.2. Производство свинца и цинка. Ч.2. Методы аналитического контроля в производстве цинка. М.: Цветметинформация, 1977. С.66). Нами изучена возможность вольтамперометрического определения сурьмы в технологических растворах после первой стадии очистки с предварительным разбавлением проб фильтрата водой для снижения концентрации ионов Cu(II) до 20 мг/л.
Исследования проводили на модельном растворе цинкового электролита, содержащем 20 мг/л Cu(II) и 0,02 мг/л Sb(III). Экспериментально установлено, что полная очистка от меди достигается в результате двукратной экстракции 0,1% раствором ДЭДТК-Pb в хлороформе, проводимой по вышеописанной методике. После первой экстракции концентрация меди снизилась до 0,45 мг/л, при этом степень извлечения достигла 98,5%. Концентрация ионов Sb(III) в анализируемом растворе после двукратной экстракции не уменьшается. Таким образом, обеспечивается возможность вольтамперометрического определения сурьмы (III) в цинковом электролите, содержащем 1000-кратный избыток меди.
Способ может быть реализован на различных отечественных и зарубежных полярографических анализаторах, обеспечивающих проведение измерений в режиме ИДИВ на СРКЭ, в частности на приборах ПУ-1, ПЛС, PAR-174, PAR-384, PA-4, АЖЭ-12 и др.
Способ осуществляется следующим образом. Из пробы нейтрального цинкового раствора отбирают аликвоту 20 мл, помещают в электрохимическую ячейку анализатора, включают мешалку и проводят электронакопление меди на СРКЭ при потенциале -0,15 В в течение 30 с. Прекращают перемешивание раствора и после стадии успокоения (в течение 10 с) снимают анодную вольтамперную кривую при линейно изменяющемся во времени потенциале в интервале напряжений (-0,15)-(+0,25) В. Развертку потенциала производят со скоростью 5 мВ/с. Регистрируют серию из 3-5 кривых и, измеряя высоту анодных пиков при потенциале +0,095 В, определяют содержание Cu(II) в растворе по градуировочному графику линейному в интервале концентраций 0-0,5 мг/л с нижним пределом обнаружения 0,01 мг/л. Сурьма (III) не мешает полярографическому анализу меди.
При концентрации меди(II) в растворе, превышающей 0,5 мг/л, анализ производят методом прямой ДИП, при этом регистрируют катодную вольтамперную кривую меди в интервале напряжений (+0,20)-(-0,05) В со скоростью развертки потенциала 5 мВ/с. Измеряют высоту пика при потенциале +0,090 В и определяют содержание Cu(II) в анализируемом растворе по градуировочному графику, линейному в интервале концентраций 0-2 мг/л. Всего снимают не менее трех ДИП пиков, при этом концентрацию меди рассчитывают по среднему арифметическому всех результатов анализа. Сурьма (III) не мешает полярографическому анализу меди.
При необходимости контроля кадмия (II) регистрируют катодную вольтамперную кривую в интервале напряжений (-0,35)-(-0,75) В со скоростью развертки 5 мВ/с. Измеряют высоту пика при потенциале -0,55 В и определяют содержание Cd(II) в анализируемом растворе по градуировочному графику, линейному в диапазонах концентраций 0-5 и 0-10 мг/л. Нижний предел обнаружения кадмия 0,1 мг/л. Всего снимают не менее трех ДИП пиков, при этом концентрацию Cd(II) рассчитывают по среднему арифметическому всех результатов анализа.
Из пробы нейтрального цинкового раствора отбирают аликвоту 10 мл и помещают в грушевидную делительную воронку, добавляют по 15 мл 0,1 М HCl и 0,1% раствора ДЭДТК-Pb в хлороформе. Встряхивают делительную воронку в течение 120 с, отделяют водную фазу и смешивают в отношении 1:1 с концентрированной соляной кислотой. Полученную смесь заливают в предварительно промытую электрохимическую ячейку, включают мешалку и проводят электронакопление сурьмы при потенциале -0,35 В. Длительность электронакопления устанавливают в зависимости от выбранного диапазона измеряемых концентраций. Прекращают перемешивание раствора, и после стадии успокоения (в течение 10 с) снимают анодную вольтамперную кривую при линейно изменяющемся во времени потенциале в интервале напряжений (-0,35)-(-0,05) В. Развертку потенциала производят со скоростью 5 мВ/с. Регистрируют серию из 3-5 кривых и, измеряя высоту анодных пиков при потенциале -0,175 В, определяют содержание Sb(III) в растворе по градуировочному графику, линейному в диапазонах концентраций 0-0,1 мг/л (τнак=120 с) и 0-0,5 мг/л (τнак=15 с). Нижний предел обнаружения сурьмы 0,005 мг/л. Определение сурьмы возможно при ионном соотношении [Cu]:[Sb] в цинковом растворе равном 100:1.
При необходимости контроля сурьмы в присутствии значительных избытков меди (при ионном соотношении [Cu]:[Sb]≤1000:1) проводят двукратную экстракцию анализируемого цинкового электролита 0,1% раствором ДЭДТК-Pb в хлороформе, после чего определяют содержание Sb(III) в водной фазе методом ИДИВ на фоне 6 М HCl. Общая длительность анализа одной пробы на содержание ионов Cu(II), Sb(III) и Cd(II) составляет 8-10 мин.
Пример. Количественное определение ионов Cu(II), Sb(III) и Cd(II) в очищенном растворе сульфата цинка выщелачивательного цеха завода «Электроцинк», содержащем: 0,2-8 мг/л Cd(II); 0,05-0,4 мг/г Cu(II); 0,01-0,15 мг/л Sb(III).
Помещают в электрохимическую ячейку вольтамперометрического анализатора 20-25 мл контролируемого раствора, включают мешалку и проводят электроконцентрирование меди на СРКЭ при потенциале -0,15 В в течение 30 с. Выключают мешалку и после 10-секундной стадии успокоения регистрируют анодную вольтамперную кривую в интервале напряжений от -0,15 до +0,25 В со скоростью развертки 5 мВ/с. Регистрируют серию из трех-пяти кривых и, измеряя высоту анодных пиков при потенциале +0,095 В, определяют содержание Cu(II) в растворе сульфата цинка по градуировочному графику в диапазоне концентраций 0-5 мг/л. Устанавливают начальное напряжение -0,35 В, отключают мешалку и регистрируют катодную вольтамперную кривую в интервале напряжений от -0,35 до -0,75 В со скоростью развертки потенциала 5 мВ/с. Измеряют высоту катодного ДИП пика при потенциале -0,55 В и определяют содержание Cd(II) по градуировочному графику в диапазоне концентраций 0-10 мг/л. Всего снимают не менее трех ДИП пиков, при этом концентрацию Cd(II) рассчитывают по среднему арифметическому всех результатов анализа. Выключают анализатор и тщательно промывают электрохимическую ячейку водой. Отбирают из пробы нейтрального цинкового раствора аликвоту 10 мл и помещают в грушевидную делительную воронку, добавляют по 15 мл 0,1 М раствора HCl и 0,1% раствора ДЭДТК-Pb в хлороформе. Проводят экстракцию в течение 2 мин, энергично встряхивая делительную воронку. Отделяют водную фазу и смешивают в отношении 1:1 с концентрированной HCl. Часть полученной пробы помещают в электрохимическую ячейку анализатора, включают мешалку и проводят электроконцентрирование сурьмы при потенциале -0,35 В в течение 90 с. Прекращают перемешивание раствора и после стадии успокоения (10 с) снимают анодную вольтамперную кривую в интервале напряжений от -0,35 до -0,05 В со скоростью развертки 5 мВ/с. Регистрируют серию из 3-5 кривых и, измеряя высоту анодных пиков при потенциале -0,175 В, определяют содержание Sb(III) в растворе по градуировочному графику в диапазоне концентраций 0-0,2 мг/л.
Заявляемый способ инверсионного вольтамперометрического определения микропримесей меди (II) и сурьмы (III) в цинковом электролите может быть реализован с помощью имеющихся технических средств, например анализаторов АЖЭ-11 и АЖЭ-12 (см. статьи: Г.А. Боровков, О.В. Щербич Автоматизация контроля ионного состава сточных вод завода «Рязцветмет»// Заводская лаборатория. 1991. Т.57. №8. С.9-12 и Г.А. Боровков, О.Г. Виноградов, В.В. Зеленский и др. Опыт разработки и внедрения автоматических электрохимических анализаторов ионного состава флотационных пульп// Цветные металлы. 1990. №9. С.108-112).
Структурная схема вольтамперометрического анализатора АЖЭ-11, предназначенного для контроля ионов Cu(II), Sb(III), Cd(II) в технологических растворах цинкового производства, представлена на фиг.7. Анализатор состоит из полярографа ПУ-1 (1), блока управления НВ-83 (2), электрохимического преобразователя (вольтамперометрического датчика) ДТЩ-17 (3) со стационарным ртутно-капельным электродом и вторичного регистрирующего прибора ДИСК-250 или А-550 (4). Электрохимический преобразователь (3) гидравлически связан с ртутным резервуаром (5) и ловушкой для отработанной ртути (6). Важнейшим узлом анализатора является электрохимический преобразователь с рабочим электродом, представляющим собой капиллярный дозатор ртути с клапаном игольчатого типа. Размеры формируемой ртутной капли определяются геометрическими параметрами капилляра и длительностью включения электромагнитного привода клапана, в момент открытия которого обеспечивается свободный исток ртути из связанного с атмосферой резервуара. Сброс капли осуществляется принудительно с помощью механической лопатки с электромагнитным приводом. Наличие в электрохимическом преобразователе электрического двигателя обеспечивает также использование лопатки в качестве мешалки анализируемого раствора.
С целью предотвращения капиллярных шумов, связанных с проникновением анализируемого раствора внутрь рабочего электрода, а также во избежание отрыва ртутной капли при механических воздействиях, например при сильных вибрациях, торец (исток) стеклянного капилляра защищен вваренной в него платиновой втулкой. Необходимые свойства капилляру такой конструкции придают предварительной электрохимической амальгамацией платиновой втулки. При этом капля удерживается на торце капилляра не только за счет сил поверхностного натяжения, но и вследствие химического взаимодействия ртути с покрывающим платину слоем амальгамы.
Схема электрохимической ячейки трехэлектродная (электрод сравнения-хлорсеребряный, электрод вспомогательный - стеклоуглеродный). При работе анализатора в режимах ДИП или ИДИВ весь измерительный процесс осуществляется на одной ртутной капле.
Блок управления программирует работу электромагнитных приводов роста и сброса ртутной капли, а также электрического привода мешалки электрохимического преобразователя, включение развертки потенциала полярографа и привода диаграммной ленты регистрирующего прибора. Входящие в состав блока управления вычислительное устройство обсчитывает показатели вольтамперной кривой (измеряет высоту ДИП пиков тяжелых металлов) и выдает электрический сигнал на вторичный прибор, отградуированный в единицах концентрации контролируемого вещества.
Контроль проб очищенного цинкового раствора на содержание ионов Cu(II) и Sb(III) методом ИДИВ осуществляется следующим образом. После заполнения ячейки электрохимического преобразователя (3) подготовленным к анализу цинковым раствором и включения анализатора в работу в соответствии с программой блока управления (2) осуществляется сброс отработанной и формирование свежей ртутной капли, включение мешалки и электрохимическое накопление меди или сурьмы на ртутном электроде при соответствующем потенциале в течение заданных промежутков времени, выключение мешалки, успокоение, регистрация и обработка инверсионных анодных пиков. При этом съемка вольтамперных кривых производится в заданном интервале напряжений, а выходные сигналы с полярографа поступают на вычислительное устройство блока управления, обрабатывающее (обсчитывающее) вольтамперную кривую и выдающее выходной сигнал на вторичный регистрирующий прибор (4), отградуированный в единицах концентрации ионов Cu(II) или Sb(III). В анализаторе также предусмотрена возможность регистрации сигналов в форме пиков, пропорциональных концентрации контролируемого вещества. Вплоть до отключения анализатор снимает в автоматическом режиме серию вольтамперных кривых, причем каждый ДИП пик регистрируется на обновленной ртутной капле.
При полярографическом определении больших концентраций Cu(II) и Cd(II) в режиме прямой ДИП программы накопления и успокоения отключаются, а регистрация катодных вольтамперных кривых производится сразу же после формирования на индикаторном электроде свежей ртутной капли.
Таким образом, заявляемый способ позволяет достичь высокой чувствительности и селективности вольтамперометрического определения меди и сурьмы в цинковых электролитах и может быть реализован на освоенных промышленностью анализаторах.
Испытания предлагаемого способа проводили на промышленных пробах очищенного цинкового раствора завода «Электроцинк». Перерабатываемое на этом предприятии сырье (цинковые концентраты) содержит примеси меди и сурьмы, оказывающие вредное влияние на процесс получения катодного цинка. Кроме того, в процесс цементационной очистки в качестве активирующей добавки дозируется соль Шлиппе, содержащая около 25% сурьмы. Для оптимизации процесса очистки растворов сульфата цинка от водорастворимых соединений меди и сурьмы необходим оперативный контроль ионов Cu(II) и Sb(III) в готовом электролите. В настоящее время для этих целей применяют методики фотоколориметрического анализа, имеющие очень сложную и трудоемкую пробоподготовку. Фотометрический контроль меди и сурьмы производится в экспресс-лаборатории выщелачивательного цеха не чаще 3-4 раз в смену, что не позволяет достичь качественной очистки цинкового электролита от вредных примесей и приводит к перерасходу дорогостоящей цинковой пыли.
В период проведения испытаний пробы отбирались из сборников очищенного раствора сульфата цинка. В качестве контрольного использовался метод стандартных добавок. Всего было проанализировано 35 проб очищенного цинкового раствора, концентрация ионов Cu(II) в которых изменялась в пределах от 0,02 до 0,33 мг/л, а содержание ионов Sb(III) - в пределах от 0,01 до 0,10 мг/л. Приведенная среднеквадратичная погрешность анализа меди (II) составила 3,54%, сурьмы (III) - 4,02%.
Заявляемый способ может быть использован для экспресс-анализа микропримесей меди и цинка в технологических растворах цинкового производства, а также в сточных и оборотных водах предприятий цветной металлургии.
Внедрение предлагаемого способа в аналитическую практику цинкового производства позволит значительно повысить оперативность и точность контроля ионов Cu(II) и Sb(III), оптимизировать технологический процесс очистки растворов сульфата цинка от примесей меди и сурьмы и, в конечном итоге, улучшить технико-экономические показатели производства катодного цинка. Использование заявляемого способа для экомониторинга сложных по составу сточных вод обеспечит снижение сброса ионов Cu(II) и Sb(III) в открытые водоемы в зоне действия предприятий цветной металлургии.
Изобретение относится к аналитической химии. Способ основан на определении ионов Cu(II) и Sb(III) методом инверсионной дифференциальной импульсной вольтамперометрии на стационарном ртутно-капельном электроде. Контроль меди (II) производится непосредственно в нейтральном цинковом растворе, выполняющем роль фонового электролита. Анализ сурьмы (III) осуществляется на фоне 6 М HCl после экстракционного отделения меди 0,1% раствором диэтилдитиокарбамата свинца в хлороформе из подкисленного до рН 1-2 цинкового электролита. Способ согласно изобретению обеспечивает повышение избирательности и точности анализа. 7 ил.
Способ инверсионного вольтамперометрического определения микропримесей меди (II) и сурьмы (III) в цинковом электролите на стационарном ртутно-капельном электроде, включающий измерение концентрации сурьмы (III) на фоне 6 М HCl, отличающийся тем, что контроль меди (II) осуществляют непосредственно в нейтральном цинковом электролите, а сурьму (III) определяют после экстракционного отделения меди раствором диэтилдитиокарбамата свинца в хлороформе из подкисленного до рН 1-2 цинкового электролита.
| Зарецкий Л.С., Варновский Б.И., Пидорич М.И., Кокоева Ф.Х | |||
| Анализатор микропримесей сурьмы, меди и кадмия в цинковом электролите// Заводская лаборатория | |||
| Устройство для видения на расстоянии | 1915 |
|
SU1982A1 |
| Приспособление для автоматической односторонней разгрузки железнодорожных платформ | 1921 |
|
SU48A1 |
| Печь-кухня, могущая работать, как самостоятельно, так и в комбинации с разного рода нагревательными приборами | 1921 |
|
SU10A1 |
| Приспособление для точного наложения листов бумаги при снятии оттисков | 1922 |
|
SU6A1 |
| Нейман Е.Я., Суменкова М.Ф | |||
| О совместном определении сурьмы и меди методом инверсионной вольтамперометрии// Журнал аналитической химии | |||
| Сплав для отливки колец для сальниковых набивок | 1922 |
|
SU1975A1 |
| Способ обработки медных солей нафтеновых кислот | 1923 |
|
SU30A1 |
| Топка с несколькими решетками для твердого топлива | 1918 |
|
SU8A1 |
| Цепной ветряный двигатель | 1923 |
|
SU1625A1 |
| SU | |||

