Изобретение относится к области промышленного производства продуктов нефтехимического синтеза, в частности, к процессу производства ацетона, широко используемого в качестве растворителя и реагента.
Традиционным способом промышленного производства ацетона является жидкофазное окисление кумола в гидропероксид кумола с последующим разложением последнего на фенол и ацетон. В продуктах окисления наряду с целевыми продуктами присутствуют побочные карбонильные соединения, в частности, альдегиды, очистка ацетона от которых представляет определенные трудности. Наличие карбонильных соединений ухудшает качественные показатели товарного ацетона, а именно резко снижает такой показатель как устойчивость ацетона к окислению марганцовокислым калием (перманганатный тест).
Известен способ очистки кетонов от примесей альдегидов с помощью реакции Тищенко, проводимой в присутствии мягких кислот Льюиса [Патент США N 4885399, от 05 декабря 1989 г.]. Недостатком способа является необходимость тщательной предварительной осушки продукта или проведения процесса в специальном растворителе, что приводит к значительному усложнению и удорожанию технологической схемы процесса.
Известен способ очистки ацетона с помощью химического связывания содержащихся в нем примесей альдегидов различными диаминами с последующей дистилляцией обработанного продукта [Патент США N 5399776, от 21 марта 1995 г.]. Способ имеет тот недостаток, что химические реагенты для проведения очистки дороги, а образующиеся в результате реакции азотистые соединения оказывают негативное воздействие на окружающую среду при их утилизации.
Наиболее распространенным способом очистки ацетона от примесей альдегидов является обработка ацетона-сырца в присутствии щелочного катализатора, основанная на реакции их конденсации с образованием альдолей [Патент США N 4626600, от 02 декабря 1986 г.; Патент США N 4722769, от 02 февраля 1988 г.; Патент США N 4340447, от 20 июля 1982 г.].
Существуют различные варианты оформления технологической схемы ректификационного выделения товарного ацетона в присутствии щелочного катализатора.
В патенте [Патент США N 4626600, от 02 декабря 1986 г.] описан вариант схемы выделения товарного ацетона, в котором реакционная масса разложения, содержащая ацетон, фенол, кумол, α -метилстирол, воду, продукты конденсации фенола и α-метилстирола и примеси, поступает в колонну разделения, основное назначение которой разделить реакционную массу разложения на ацетоновый и фенольный потоки. Полученный верхом ацетоновый поток содержит ацетон, кумол, воду и α-метилстирол и направляется на стадию выделения товарного ацетона. Выделение товарного ацетона из ацетонового потока проводится в двух колоннах, где на первой колонне отгоняются только альдегидная фракция и небольшой рецикловый поток ацетона, направляемый на стадию разложения ГПК, а весь ацетоновый поток поступает в колонну товарного ацетона, где верхом с флегмовым числом выше пяти (в высокоэффективных колоннах, заполненных насадками типа Intalox-2T, флегмовое число может быть снижено до 2,5) получают товарный ацетон, а кубовый продукт, представляющий собой гетерогенную смесь кумола с α -метилстиролом и воды, поступает в разделитель фаз. Углеводородная фракция из разделителя поступает на дальнейшую переработку, а водная фаза - на стадию нейтрализации продуктов разложения ГПК. Подача водного раствора щелочи осуществляется в обе колонны стадии выделения товарного ацетона, либо с питанием, но чаще выше точки питания (в высокоэффективных колоннах, заполненных насадками типа Intalox-2T, из-за плохой растворимости щелочи в ацетоне и, как следствие, забивки насадки подача водного раствора щелочи осуществляется предпочтительно только с питанием).
Основным недостатком используемой схемы является получение товарного ацетона с невысокой устойчивостью к окислению (перманганатный тест не выше 4 ч) из-за обратимости реакции альдольной конденсации. Проведенные нами исследования показали, что альдегиды в ацетоновом потоке, содержащем кумол и воду, распределяются между органической и водной фазами в массовом соотношении 2: 1. При обработке ацетонового потока водным раствором щелочи альдегиды, находящиеся в водной фазе, практически не вступают в реакцию альдольной конденсации. Альдегиды, находящиеся в органической фазе, легко вступают в реакцию альдольной конденсации, причем большая часть продуктов альдольной конденсации переходит в водную фазу. Таким образом, водная фаза ацетонового потока содержит свободные альдегиды и продукты альдольной конденсации. При ректификации обработанного щелочью ацетонового потока при повышенных температурах наблюдается частичный распад продуктов альдольной конденсации и, как следствие, попадание альдегидов в товарный продукт, в результате чего перманганатный тест товарного ацетона обычно не превышает 4 ч. При невысокой эффективности колонны выделения товарного ацетона продукты альдольной конденсации в незначительных количествах также попадают в товарный продукт и наряду с альдегидами снижают значение перманганатного теста.
В патенте [Патент США N 4722769, от 02 февраля 1988 г.] представлена схема выделения товарного ацетона, отличающаяся от представленной в патенте [Патент США N 4626600, от 02 декабря 1986 г.] тем, что выделение товарного ацетона проводится на одной ректификационной колонне. Так же как в патенте [Патент США N 4626600, от 02 декабря 1986 г.] реакционная масса разложения поступает в колонну разделения, в которой верхом выделяется ацетоновый поток, содержащий ацетон, кумол, воду и α-метилстирол и направляется в колонну товарного ацетона. Подача ацетонового потока в колонну осуществляется в паровой и жидкой фазах, выше точки питания подается водный раствор щелочи, товарный ацетон в жидкой фазе выводится боковым погоном, а дистиллят конденсируется и часть его подается на флегму в верхней части колонны, а часть конденсата возвращается в колонну в точке между подачей сырья и водного раствора щелочи. Авторами показано, что, так как основное взаимодействие между альдегидом и щелочью происходит на тарелках, где одновременно находятся водная и углеводородная фазы, эффективность контакта которых невелика, то для успешного проведения процесса необходимо поддерживать оптимальный режим в колонне регулированием температурного профиля. Так как боковой отбор товарного ацетона проводится в жидкой фазе, то обеспечить необходимое количество жидкости на тарелке отбора можно только за счет значительных рецикловых потоков, в данном случае за счет значительного флегмового потока. Для реализации такого процесса необходима колонна большего диаметра и соответственно за счет значительных рецикловых потоков возрастают энергетические затраты. Авторы указывают, что ограничением данного изобретения является содержание кумола в ацетоновом потоке. Как показано в патенте, для того, чтобы получать товарный ацетон требуемого качества, содержание кумола в ацетоновом потоке не должно превышать 4 мас.%. Во многих промышленных процессах содержание кумола в ацетоновом потоке значительно выше 4 мас.% и достигает 17-20 мас.%.
В патенте [Патент США N 4340447, от 20 июля 1982 г.] представлена схема выделения товарного ацетона, подобная схеме, представленной в патенте [Патент США N 4722769, от 02 февраля 1988 г.], отличающаяся тем, что ацетоновый поток в паровой фазе, содержащий ацетон, воду и примеси альдегидов, из колонны разделения через парциальный конденсатор направляется в колонну товарного ацетона. Сконденсированная в парциальном конденсаторе фаза направляется в колонну разделения на флегму. Содержание кумола в ацетоновом потоке не указывается. Ацетоновый поток поступает в колонну выделения товарного ацетона, где боковым отбором выделяется товарный ацетон, а дистиллят конденсируется и часть его подается на флегму в верхней части колонны, а часть конденсата подается на стадию разложения технического гидропероксида кумола. Подача водного раствора щелочи в колонне товарного ацетона осуществляется выше точки питания. Концентрация щелочи может варьироваться от 0,01 до 5,0 мас. %. Давление наверху колонны выделения товарного ацетона поддерживается на уровне 0,3 - 0,8 бар, при этом температура в кубе - 80 - 120oC. Массовое отношение флегмового потока к боковому потоку составляет от 4:1 до 25:1.
Представленные в патентах [Патент США N 4722769, от 02 февраля 1988 г.; Патент США N 4340447, от 20 июля 1982 г.] схемы также не позволяют получать ацетон с высокой устойчивостью к окислению по причинам, указанным выше. При выделении товарного ацетона по схемам, описанным в патентах [Патент США N 4626600, от 02 декабря 1986 г.; Патент США N 4722769, от 02 февраля 1988 г.; Патент США N 4340447, от 20 июля 1982 г.], за счет высоких флегмовых чисел расход энергии значителен.
Целью настоящего изобретения является устранение указанных недостатков и получение товарного ацетона с повышенной устойчивостью к окислению при использовании ректификационного выделения товарного ацетона в присутствии щелочного катализатора.
Проведенные нами исследования показали, что для того, чтобы получать ацетон с высокой устойчивостью к окислению (перманганатный тест выше 10 ч), необходимо получать товарный ацетон с содержанием альдегидов ниже 5 - 10 ppm и практически отсутствием в нем продуктов альдольной конденсации. Для получения товарного ацетона с такими показателями необходимо варьировать количество подаваемого щелочного катализатора, а во избежание попадания продуктов альдольной конденсации в товарный ацетон отбор ацетона проводить в паровой фазе, направляя ее в парциальный конденсатор, где за счет изменения массового соотношения парового и сконденсированного потоков удается достичь степени удаления продуктов альдольной конденсации, равной 90%.
Сущность изобретения иллюстрируется представленной на фиг. 1 и 2 схемой проведения процесса.
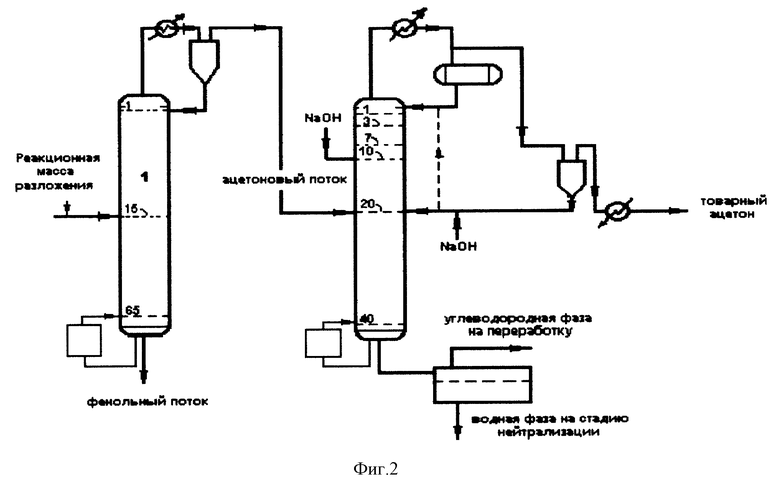
На фиг. 1 представлена трехколонная схема выделения товарного ацетона, а на фиг. 2 - двухколонная схема.
Рассмотрим схему, представленную на фиг. 1. Реакционная масса разложения поступает в колонну разделения, где верхом в паровой фазе отделяется ацетоновый поток и направляется в парциальный конденсатор. Массовое соотношение парового и сконденсированного потоков в парциальном конденсаторе поддерживается от 1:0,02 до 1:0,1. Паровая фаза парциального конденсатора, содержащая ацетон, кумол (до 40 мас.%), воду (до 20 мас.%) и α-метилстирол, направляется на стадию выделения товарного ацетона, состоящую из двух колонн ректификации, работающих либо при атмосферном давлении, либо при вакууме 400-600 мм рт.ст. Сконденсированная фаза направляется на флегму первой колонны.
Во второй колонне верхом отгоняют альдегидную фракцию, которую подают на стадию нейтрализации. При нейтрализации серной кислоты в продуктах разложения технического ГПК, осуществляемой водным раствором щелочи или водным раствором фенолята натрия, на стадии нейтрализации альдегиды превращаются в продукты альдольной конденсации и переходят в водную фазу, а ацетон с нейтрализованными продуктами разложения возвращается на питание первой колонны. Боковым погоном с 3 - 7 тарелки отбирается весь ацетон в паровой фазе с содержанием воды 1-2 мас.%, с одновременной подачей во второй колонне выше тарелки питания (на 10 тарелку) водного раствора гидроксида натрия в расчете на 100% NaOH от 0,0036 т до 0,006 т на 1 т тонну ацетона в зависимости от требуемой величины перманганатного теста от 10 до 70 ч, кубовым потоком отбираются углеводороды и вода. Боковой отбор, поступающий в паровой фазе в парциальный конденсатор, разделяется в нем на жидкую и паровую фазы при массовом соотношении потоков от 1:10 до 1:100 соответственно. Паровая фаза парциального конденсатора направляется в третью колонну, где выделяется верхом товарный ацетон колонны с флегмовым числом не выше 2,5, а кубовый продукт, представляющий собой водный раствор ацетона с содержанием воды 30-40 мас. %, возвращается на питание второй колонны. Жидкая фаза парциального конденсатора, установленного после колонны 2, обрабатывается щелочью и возвращается либо во вторую колонну, либо на стадию нейтрализации продуктов разложения гидропероксида кумола.
При осуществлении процесса получения товарного ацетона по схеме, описанной выше, варьированием количества подаваемой щелочи обеспечивается практически полное удаление альдегидов из потока выше тарелки питания, при этом, если альдегиды все-таки не будут полностью убраны, то большая их часть будет отобрана в головке колонны 2, а в боковом отборе количество альдегидов будет минимальным. Если количество альдегидов в боковом отборе будет больше 10 ppm, а также, если в данном потоке будут содержаться продукты альдольной конденсации, то содержание альдегидов и продуктов альдольной конденсации в товарном ацетоне можно обеспечить варьированием массового соотношения паровой и жидкой фаз в парциальном конденсаторе, установленном после колонны 2. При осуществлении процесса по схеме фиг. 1 назначение колонны 3 состоит только в разделении ацетона и воды. Как показывают наши исследования, расчеты и опыт промышленной эксплуатации для обеспечения требуемого содержания воды в товарном ацетоне (содержание воды в товарном ацетоне не должно быть выше 0,4 мас.%) флегмовое число на колонне 3 не должно превышать 2,5.
Обычно, согласно нашим исследованиям, путем варьирования расхода щелочного катализатора удается достичь отсутствия наличия альдегидов в ацетоне отбираемым боковым погоном. При поддержании в колонне давления ниже атмосферного содержание воды в дистилляте при постоянном флегмовом числе снижается. В таком случае для выделения товарного ацетона может быть использовано не три колонны, как это представлено на фиг. 1, а только две. На фиг. 2 представлена схема получения ацетона с двумя колоннами на стадии выделения товарного ацетона.
Ацетоновый поток в паровой фазе из парциального конденсатора первой колонны, содержащий ацетон, кумол (до 40 мас.%), воду (до 20 мас.%) и α-метилстирол, направляется во вторую колонну ректификации, где верхом в паровой фазе отбирается ацетон с содержанием воды менее 1 мас.% и направляется в парциальный конденсатор при соотношении получаемых в нем жидкой и паровой от 1:50 до 1:150, соответственно, с одновременной подачей в колонну выше тарелки питания водного раствора гидроксида натрия в расчете на 100% NaOH от 0,0036 т до 0,006 т на 1 т тонну ацетона в зависимости от требуемой величины перманганатного теста от 4 до 40 ч, конденсации паровой фазы парциального конденсатора в холодильнике с получением товарного ацетона, обработкой жидкой фазы парциального конденсатора водным раствором щелочи и последующий возврат полученного ацетона-сырца в колонну 2 либо в точку питания, либо на флегму.
Отличительными особенностями разработанного нами процесса от прототипа [Патент США N 4340447, от 20 июля 1982 г.] являются:
1) содержание кумола в ацетоновом потоке, поступающем для выделения товарного ацетона, составляет от 1 до 40 мас.%;
2) содержание воды в ацетоновом потоке, поступающем для выделения товарного ацетона, составляет от 2 до 20 мас.%;
3) изменением расхода щелочного катализатора выше точки подачи сырья (массового соотношения 100% NaOH/ацетон от 0,0036 до 0,006) обеспечивается содержание альдегидов в товарном ацетоне ниже 10 ppm;
4) cконденсированные продукты парциального конденсатора, содержащие непрореагировавшие альдегиды, альдоли и ацетон, дополнительно подвергаются обработке щелочным катализатором и либо возвращаются в колонну, либо направляются на стадию нейтрализации;
5) cтепень отделения альдолей в парциальном конденсаторе превышает 90%;
6) cодержание ацетона в кубовом потоке колонны 2 (см. фиг. 1 или 2), содержащем углеводороды и воду, не превышает 0,5 мас.%;
7) для снижения энергетических затрат отбор ацетона из колонны 2 (см. фиг. 1 или 2) проводится в паровой фазе;
8) товарный ацетон, получаемый по предлагаемым схемам, имеет высокую устойчивость к окислению (перманганатный тест от 10 до 70 ч).
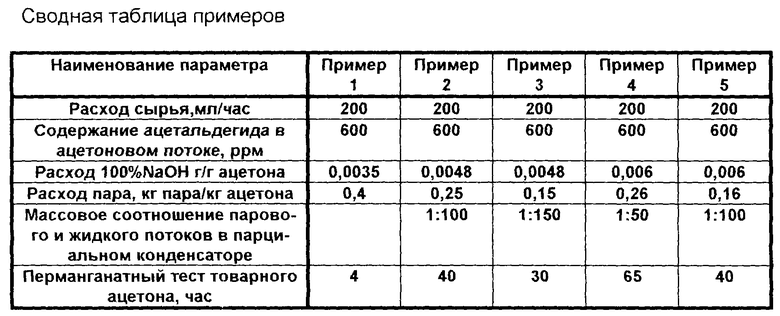
Указанные преимущества и отличия разработанной технологии демонстрируются примерами 1-5 (см. сводную таблицу примеров).
Приведенные в примерах состав ацетонового потока, концентрации щелочного катализатора, номера тарелок ввода сырья и вывода продуктов ректификации не ограничивают область применения данного изобретения.
Сопоставительное лабораторное исследование выделения товарного ацетона по схеме, представленной в патенте [Патент США N 4340447, от 20 июля 1982 г. ], и по схемам, предложенным в данном изобретении, проводилось на лабораторных насадочных ректификационных колоннах.
Пример 1 (сравнительный по прототипу).
Ацетоновый поток, содержащий ацетон, воду (3 мас.%) и такие примеси, как ацетальдегид в количестве 600 ppm, фенол в количестве 3000 ppm, поступает в колонну выделения товарного ацетона. Колонна имеет 55 реальных тарелок, диаметр колонны 38 мм. Расход сырья 200 мл/ч, подача сырья осуществлялась на 45 тарелку (нумерация тарелок производится сверху колонны). Давление на верху колонны 450 мм рт.ст. Водный раствор щелочи (15% NaOH) подавался на 35 тарелку, расход потока щелочи составлял 3,5 мл/ч. Верхом отбирался поток, содержащий ацетон и альдегиды, конденсировался и возвращался на флегму. Товарный ацетон в жидкой фазе был выделен с 10 тарелки. Товарный ацетон имел перманганатный тест, равный 4 ч. Флегмовое число (соотношение потока верха колонны и потока товарного ацетона) поддерживалось 10:1. Кубовый поток колонны содержал воду, продукты альдольной конденсации, фенол, фенолят натрия и ацетон в количестве - 1 мас.%. Расход пара - 0,4 кг пара/кг ацетона.
Пример 2.
Ацетоновый поток, содержащий ацетон (60 мас. %), кумол (20 мас.%), α-метилстирол (3 мас. %), воду (17 мас.%) и примеси ацетальдегида в количестве 600 ppm и фенола в количестве 3000 ppm, подавался в колонну 2 (процесс выделения товарного ацетона осуществляется по схеме, предложенной на фиг. 1). Колонна 2 имеет 40 реальных тарелок, диаметр колонны 28 мм. Расход сырья 200 мл/ч, подача сырья осуществлялась на 20 тарелку (нумерация тарелок производится сверху колонны). Давление наверху колонны атмосферное. Водный раствор щелочи (20% NaOH) подавался на 10 тарелку, расход щелочного катализатора составлял 2,4 мл/ч (в расчете на 100% NaOH - 0,0048 г NaOH на 1 г ацетона). Сверху отбирался поток ацетона, содержащий 25 ppm ацетальдегида в количестве 0,5 мл/ч. С пятой тарелки отбирался поток в паровой фазе в количестве 134 мл/ч, содержащий ацетон, воду - 1,5 мас.%, ацетальдегид - 15 ppm, продукты альдольной конденсации отсутствовали. Кубовый поток содержал воду, кумол, α-метилстирол, альдоли, фенол и феноляты. Содержание ацетона в кубовом потоке не превышало 0,3 мас.%. Температура в кубе поддерживалась не выше 105oC. Поток ацетона с пятой тарелки в паровой фазе направлялся в парциальный конденсатор. Массовое соотношение парового потока и сконденсированного потока в парциальном конденсаторе поддерживалось равным 1:100. Паровой поток из парциального конденсатора, содержащий ацетон, воду - 1 мас.% и ацетальдегид - 8 ppm, направлялся в колонну 3. Колонна 3 имеет 65 реальных тарелок, диаметр колонны 38 мм. Расход сырья 134 мл/ч, подача сырья осуществлялась на 50 тарелку (нумерация тарелок производится сверху колонны). Давление на верху колонны 3 - 600 мм рт.ст. Верхом колонны 3 отбирался товарный ацетон в количестве 120 мл/ч. Флегмовое число поддерживалось равным 2,5. Содержание воды в товарном ацетоне - 0,3 мас.%, содержание ацетальдегида - 10 ppm. Перманганатный тест товарного ацетона - 40 ч. Кубовый продукт колонны 3, содержащий ацетон и воду, направлялся на питание колонны 2.
Расход пара - 0,25 кг пара/кг ацетона.
Пример 3.
Ацетоновый поток, содержащий ацетон (60 мас. %), кумол (20 мас. %), α-метилстирол (3 мас. %), воду (17 мас. %) и примеси ацетальдегида в количестве 600 ppm и фенола в количестве 3000 ppm, подавался в колонну 2 (процесс выделения товарного ацетона осуществляется по схеме, предложенной на фиг. 2). Колонна 2 имеет 65 реальных тарелок, диаметр колонны 38 мм. Расход сырья 200 мл/ч, подача сырья осуществлялась на 32 тарелку (нумерация тарелок производится сверху колонны). Давление на верху колонны - 600 мм рт. ст. Водный раствор щелочи (20% NaOH) подавался на 10 тарелку, расход щелочного катализатора составлял 2,4 мл/ч (в расчете на 100% NaOH - 0,0048 г NaOH на 1 г ацетона). Сверху отбирался поток ацетона в паровой фазе в количестве 133 мл/ч, содержащий 15 ppm ацетальдегида, 0,7 мас.% воды, продукты альдольной конденсации отсутствовали. Кубовый поток содержал воду, кумол, α-метилстирол, альдоли, фенол и феноляты. Содержание ацетона в кубовом потоке не превышало 0,3 мас. %. Температура в кубе поддерживалась не выше 105oC. Поток ацетона с верха колонны направлялся в парциальный конденсатор. Массовое соотношение парового потока и сконденсированного потока в парциальном конденсаторе поддерживалось равным 1:150. Товарный ацетон получали конденсацией парового потока из парциального конденсатора. Товарный ацетон содержал воду - 0,3 мас.% и ацетальдегид - 10 ppm. Перманганатный тест товарного ацетона равнялся 30 ч. Расход пара - 0,15 кг пара/кг ацетона.
Пример 4.
Процесс выделения товарного ацетона проводится аналогично примеру 2 с тем отличием, что расход щелочи составлял 3,0 мл/ч (в расчете на 100% NaOH - 0,006 г NaOH на 1 г ацетона). Давление наверху колонны 2 поддерживалось - 600 мм рт. ст. С пятой тарелки отбирался поток в паровой фазе в количестве 133,5 мл/ч, содержащий ацетон, воду - 1,0 мас.%, ацетальдегид - 10 ppm, продукты альдольной конденсации отсутствовали. Поток ацетона с пятой тарелки в паровой фазе направлялся в парциальный конденсатор. Массовое соотношение парового потока и сконденсированного потока в парциальном конденсаторе поддерживалось равным 1: 50. Паровой поток из парциального конденсатора, содержащий ацетон, воду - 0,7 мас.% и ацетальдегид - 6 ppm, направлялся в колонну 3. Верхом колонны 3 отбирался товарный ацетон в количестве 120 мл/ч. Флегмовое число поддерживалось равным 2,0. Содержание воды в товарном ацетоне - 0,3 мас.%, содержание ацетальдегида - 7 ppm. Перманганатный тест товарного ацетона - 65 ч. Расход пара - 0,26 кг пара/кг ацетона.
Пример 5.
Процесс выделения товарного ацетона проводится аналогично примеру 3 с тем отличием, что расход щелочи составлял 3,0 мл/ч (в расчете на 100% NaOH - 0,006 г NaOH на 1 г ацетона). Давление на верху колонны - 500 мм рт.ст. Сверху отбирался поток ацетона в паровой фазе в количестве 132,7 мл/ч, содержащий 10 ppm ацетальдегида, 0,5 мас.% воды, продукты альдольной конденсации отсутствовали. Поток ацетона с верха колонны направлялся в парциальный конденсатор. Массовое соотношение парового потока и сконденсированного потока в парциальном конденсаторе поддерживалось равным 1:100. Товарный ацетон получали конденсацией парового потока из парциального конденсатора. Товарный ацетон содержал воду - 0,3 мас.% и ацетальдегид - 8 ppm. Перманганатный тест товарного ацетона равнялся 40 ч. Расход пара - 0,16 кг пара/кг ацетона.
| название | год | авторы | номер документа |
|---|---|---|---|
| БЕЗОТХОДНЫЙ ЭКОНОМИЧНЫЙ СПОСОБ ПОЛУЧЕНИЯ ФЕНОЛА И АЦЕТОНА | 1996 |
|
RU2125038C1 |
| СПОСОБ ОЧИСТКИ АЦЕТОНА | 2008 |
|
RU2403236C2 |
| СПОСОБ ОЧИСТКИ АЦЕТОНА-СЫРЦА | 2008 |
|
RU2400469C2 |
| СПОСОБ УМЕНЬШЕНИЯ И/ИЛИ УДАЛЕНИЯ ПЕРМАНГАНАТВОССТАНАВЛИВАЮЩИХ СОЕДИНЕНИЙ И С АЛКИЛИОДИДНЫХ СОЕДИНЕНИЙ | 1997 |
|
RU2181715C2 |
| УДАЛЕНИЕ ВОССТАНАВЛИВАЮЩИХ ПЕРМАНГАНАТ СОЕДИНЕНИЙ ИЗ ПОТОКА ПРОЦЕССА КАРБОНИЛИРОВАНИЯ МЕТАНОЛА | 2005 |
|
RU2372321C2 |
| СПОСОБ КОНТРОЛЯ НАД ПРОЦЕССОМ УДАЛЕНИЯ ПЕРМАНГАНАТНЫХ ВОССТАНОВЛЕННЫХ СОЕДИНЕНИЙ ПРИ ИСПОЛЬЗОВАНИИ ТЕХНОЛОГИИ КАРБОНИЛИРОВАНИЯ МЕТАНОЛА | 2005 |
|
RU2493143C2 |
| СПОСОБ ПОЛУЧЕНИЯ ГИДРОПЕРОКСИДА КУМОЛА | 1998 |
|
RU2146670C1 |
| СПОСОБ ОЧИСТКИ ФЕНОЛА ОТ АЦЕТОЛА | 2004 |
|
RU2260579C1 |
| СПОСОБ КОНТРОЛЯ НАД ПРОЦЕССОМ УДАЛЕНИЯ ПЕРМАНГАНАТНЫХ ВОССТАНОВЛЕННЫХ СОЕДИНЕНИЙ ПРИ ИСПОЛЬЗОВАНИИ ТЕХНОЛОГИИ КАРБОНИЛИРОВАНИЯ МЕТАНОЛА | 2005 |
|
RU2376276C2 |
| ВЫСОКОСЕЛЕКТИВНЫЙ СПОСОБ ПОЛУЧЕНИЯ ФЕНОЛА И АЦЕТОНА (ПРОЦЕСС ФАН-98) | 1997 |
|
RU2142932C1 |
Изобретение относится к способу получения товарного ацетона выделением его из продуктов разложения гидропероксида кумола многоступенчатой ректификацией. В первой колонне выделяют верхом ацетоновый поток, а в кубе получают фенольный поток. Ацетоновый поток направляют в парциальный конденсатор, в котором поддерживают массовое соотношение парового и сконденсированного потока от 1:0,02 до 1:0,1. Сконденсированную фазу направляют на флегму первой колонны. Несконденсированные продукты направляют во вторую колонну разделения ректификацией, в которую одновременно выше тарелки питания подают водный раствор гидроксида натрия в расчете на lOO% NaOH от 0,0036 до 0,006 т на 1 тонну ацетона в зависимости от требуемой величины перманганатного теста от 10 до 70 ч. Во второй колонне верхом отгоняют альдегидную фракцию, которую направляют на стадию нейтрализации и боковым погоном в паровой фазе отбирают поток, содержащий ацетон и воду, и его направляют в парциальный конденсатор, в котором разделяют на жидкую и паровую фазы при массовом соотношении фаз от 1: 10 до 1:100 соответственно. Паровую фазу парциального конденсатора направляют в третью колонну, где верхом выделяют товарный ацетон. Кубовый продукт третьей колонны, представляющий собой водный раствор ацетона, возвращают на питание второй колонны. Жидкую фазу парциального конденсатора обрабатывают раствором щелочи и возвращают либо во вторую колонну, либо на стадию нейтрализации продуктов разложения гидропероксида кумола. Второй вариант осуществления способа предусматривает одну колонну для выделения товарного ацетона из паровой фазы парциального конденсатора после первой колонны ректификации. При этом ацетон отбирают верхом колонны и направляют в парциальный конденсатор для получения в нем жидкой и паровой фаз в соотношении от 1:50 до 1: 150 соответственно. Паровую фазу конденсируют в холодильнике с получением товарного ацетона, а жидкую фазу обрабатывают водным раствором щелочи с последующим возвратом полученного ацетона сырца во вторую колонну либо в точку питания, либо на флегму. Технический результат - получение товарного ацетона с повышенной устойчивостью к окислению. 2 с.п. ф-лы, 1 табл., 2 ил.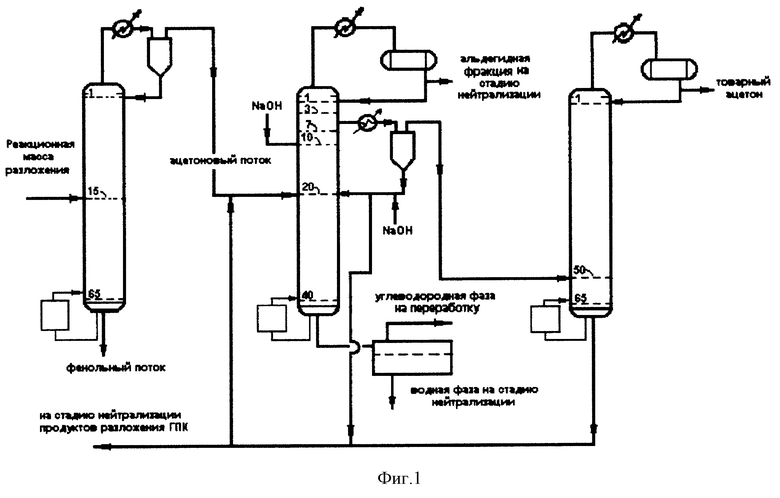
| SU 4340447 A, 20.07.1982 | |||
| US 4733769 A, 02.02.1988 | |||
| US 4626600 A, 02.12.1986 | |||
| СПОСОБ СОВМЕСТНОГО ПОЛУЧЕНИЯ ФЕНОЛА, АЦЕТОНА И АЛЬФА-МЕТИЛСТИРОЛА | 1992 |
|
RU2068404C1 |

