Изобретение относится к фармацевтически активным агентам. Эти агенты пригодны, например, для лечения рака, в частности для повышения эффективности существующей химиотерапии рака и для предотвращения резистентности к множеству лекарственных препаратов. Более конкретно, изобретение относится к ряду производных 10,11-метандибензосуберана. Изобретение также относится к фармацевтическим композициям и способам хемосенсибилизации, например, для лечения рака, включая полное преодоление резистентности к множеству лекарственных препаратов, применениям новых агентов для получения фармацевтических композиций, включающих эти агенты, и к способам их получения.
Уровень техники
Одной из проблем, с которыми сталкиваются в химиотерапии рака, является развитие резистентности к схемам лечения. У опухолей, которые в начале хорошо реагируют на определенное лекарство или лекарства, часто развивается толерантность к лекарству(ам). Это состояние болезни, называемое резистентностью к множеству лекарственых препаратов, подробно описано Kuzmich и Tew в "Detoxication Mechanisms and Tumor Cell Resistance to Anticancer Drugs", в частности, в разделе VII "The Multidrug-Resistant Phenotip (MDR)", Medical Research Reviews, т. 11, N 2, 185-217, в частности, на страницах 208-213 (1991); и Georges, Sharom и Ling в "Multidrug Resistance and Chemosensitization: Therapeutic Implications for Cancer Chemotherapy", Advances in Pharmacology, т. 21, 185-220 (1990).
Определенные активные агенты, называемые хемосенсибилизирующими агентами или потенцирующими агентами, были предложены в качестве агентов, модифицирующих резистентность для предотвращения резистентности к множеству лекарств, но они обладали различными недостатками. Они включали, например, верапамил (блокатор вхождения кальция, который понижает кровяное давление и также, как было установлено, эффективен in vitro для лечения устойчивой к лекарствам малярии), стероиды, трифторперазин (агент ЦНС), виндолин и резерпин ( α -2-блокатор некоторых функций ЦНС). Таким образом, сохраняется потребность в активных агентах для лечения, т.е. реверсии, ингибирования и/или предотвращения резистентности к множеству лекарств, предпочтительно с минимальными нежелательными побочными эффектами или с их отсутствием.
Хемосенсибилизирующие агенты взаимодействуют с P-гликопротеином, насосом для оттока лекарства, обнаруженным в клеточных мембранах, в частности в клетках опухоли с резистентностью к множеству лекарств, клетках желудочно-кишечного тракта и эндотелиальных клетках, формирующих мозговой кровяной барьер. Блокируя этот насос, хемосенсибилизирующие агенты ингибируют отток лекарств, применяемых в химиотерапии рака, от клеток опухоли и могут увеличить проникновение питательных веществ или активных агентов через желудочно-кишечный тракт и проникновение активных агентов через мозговой кровяной барьер.
В патенте США 5112817 Fukazawa и др. описывают определенные производные хинолина, пригодные в качестве потенцирующих средств противоракового лекарства, предназначенного для лечения резистентности к множеству лекарств. Одним из описанных в этом патенте первоначально перспективных активных агентов является MS-073, имеющий следующую структуру: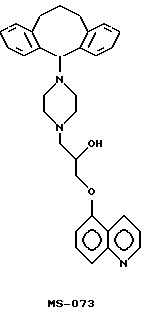
Несмотря на высокую активность, показанную в опытах in vitro, MS-073, как оказалось, имеет плохую оральную биопригодность и имеет недостатки, связанные с нестабильностью в растворе. Другие соединения этого ряда, такие как производное бифенилметилкарбонила, MS-209, как было обнаружено, имеет лучшую стабильность и оральную биопригодность, однако это достигается за счет введения более высоких эффективных доз. Таким образом, существует проблема разработки потенцирующего средства противоракового лекарства, обладающего активностью MS-073, а также хорошей оральной биопригодностью и стабильностью. Целями настоящего изобретения являются решения этих проблем.
Краткое описание изобретения
Одним из предметов настоящего изобретения являются производные 10,11-метандибензосуберана, т.е. соединения формулы I: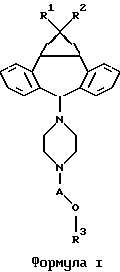
где A обозначает -CH2-CH2-, -CH2-CHRa-CH2- или -CH2-CHRa-CHRb-CH2-, где один из Ra или Rb обозначает H, OH или низший ацилокси, а другой обозначает H;
R1 обозначает H, F, Cl или Br;
R2 обозначает H, F, Cl или Br; и
R3 обозначает гетероарил или фенил, необязательно замещенный F, Cl, Br, CF3, CN, NO2 или OCHF2;
и их фармацевтически приемлемые соли.
Предпочтительный аспект изобретения относится к определенным соединениям формулы I и, в частности, к ее простым изомерам, более конкретно включающим соединение, где A обозначает -CH2-CHRa-CH2-, где Ra обозначает OH, R1 обозначает F, R2 обозначает F и R3 обозначает хинолил. Наиболее предпочтительным является (2R)-анти-изомер.
Другим предметом изобретения является фармацевтическая композиция, содержащая терапевтически эффективное количество соединения формулы I или его фармацевтически приемлемой соли. В предпочтительном варианте выполнения изобретения такая фармацевтическая композиция может также включать фармацевтически приемлемый инертный наполнитель.
Еще одним предметом изобретения является способ лечения путем введения млекопитающему, при необходимости такого лечения, соединения формулы I или его фармацевтически приемлемой соли или применение соединения формулы I или его фармацевтически приемлемой соли для получения фармацевтической композиции в количестве, терапевтически эффективном для потенцирования эффективности совместно вводимого противоракового химиотерапевтического агента. Примерами таких противораковых химиотерапевтических агентов являются антиметаболиты, такие как 6-меркаптопурин, 5-фторурацил, цитозинарабинозид и его метаболиты и производные этих агентов. Другими противораковыми химиотерапевтическими агентами являются антифолаты, такие как метотрексат, или агенты, полученные из природных продуктов, например, полученные из винкоалкалоидов, такие как винбластин, винкристин и колхицин; адриамицин, даунорубицин, доксорубицин, тенипозид или этопозид. Такие противораковые химиотерапевтические агенты также включают платинусодержащие противораковые лекарства, такие как цисплатин и карбоплатин. Кроме того, агенты по настоящему изобретению могут вводиться совместно с циклофосфамидом, бисульфоном, прокарбазином, дакарбазином, кармустином, ломустином, мехлоретамином, хлорамбуцилом, гидроксимочевиной, мелфаланом, митотоном, таксолом и спирогерманием. Наиболее часто резистентность к множеству лекарств наблюдается по отношению к винкоалкалоидам, антрациклинам даунорубицина, доксорубицина и адриамицина; а также к этопозиду и тенипозиду; реже к антиметоболитам и другим химиотерапевтическим агентам.
Еще одним предметом изобретения является способ лечения резистентности к лекарству у млекопитающего путем введения млекопитающему, при необходимости, терапевтически эффективного количества соединения формулы I или его фамацевтически приемлемой соли. Один из вариантов этого предмета включает способ лечения резистентной к лекарствам малярии. В предпочтительном варианте выполнения изобретения способ лечения резистентного к множеству лекарств рака у млекопитающего, проявляющего клиническую резистентность к противораковому химиотерапевтическому агенту, состоит в совместном введении модифицирующего резистентность количества соединения или соли формулы I с терапевтически эффективным количеством противоракового химиотерапевтического агента, к которому проявлена резистентность.
Еще одним предметом изобретения является способ хемосенсибилизации для повышения оральной биопригодности фармацевтически активного агента, включающий введение млекопитающему, при необходимости, соединения или соли формулы I в количестве, достаточном для увеличения проникновения активного агента через мозговой кровяной барьер или желудочно-кишечный тракт.
Еще одним предметом изобретения является способ получения соединений формулы I.
Подробное описание изобретения
Определения и общие параметры
Приведенные ниже определения предназначены для иллюстрации и определения значений и объема различных терминов, используемых для описания настоящего изобретения.
Термин "алкил" обозначает полностью насыщенный одновалентный радикал, содержащий только углерод и водород и который может быть циклическим, разветвленным или прямоцепочечным радикалом. Примерами для этого термина являются такие радикалы, как метил, этил, трет.-бутил, пентил, неопентил, гептил и адамантил.
Термин "низший алкил" обозначает циклический, разветвленный или прямоцепочечный одновалентный алкильный радикал, имеющий от одного до шести атомов углерода. Примерами для этого термина являются такие радикалы, как метил, этил, н-пропил, изопропил, н-бутил, трет.-бутил, изобутил (или 2-метилпропил), циклопропилметил, изоамил, н-амил и гексил.
Термин "алкилен" обозначает полностью насыщенный двухвалентный радикал, содержащий только углерод и водород, который может быть разветвленным или прямоцепочечным радикалом. Примерами для этого термина являются такие радикалы, как метилен, этилен, н-пропилен, трет.-бутилен, изопентилен и н-гептилен.
Термин "низший алкилен" обозначает двухвалентный алкильный радикал, имеющий от одного до шести атомов углерода. Примерами для этого термина являются такие радикалы, как метилен, этилен, н-пропилен, изопропилен, н-бутилен, трет. -бутилен, изобутилен (или 2-метилпропилен), изоамилен, пентилен и н-гексилен.
Термин "низший ацилокиси" обозначает группу -O-C(O)-R', где R' обозначает низший алкил.
Термин "арил" обозначает одновалентный ненасыщенный ароматический карбоциклический радикал, обычно содержащий 6-16 атомов углерода, имеющий одно кольцо (например, фенил) или два конденсированных кольца (например, нафтил), который (радикал) необязательно может быть независимо моно-, ди- или три-замещен фтором, хлором, бромом, трифторметилом, цианом, азотом и/или дифторметоксигруппой.
Термин "гетероарил" обозначает одновалентный ненасыщенный ароматический гетероциклический радикал, обычно содержащий 2-12 атомов углерода, имеющий в кольце по крайней мере один гетероатом, обычно от 1 до 3 гетероатомов, таких как N, O или S. Гетероарильный радикал обычно может иметь одно кольцо, как, например, пиридил, или два конденсированных кольца, как, например, хинолил, бензофуранил и бензофуразанил.
Термин "гало" обозначает фтор, бром, хлор или иод.
"Необязательный" или "необязательно" означают, что описываемое далее действие или обстоятельство может иметь место либо его может не быть и что описание включает примеры, где указанное действие или обстоятельство имеет место, и примеры, в которых оно отсутствует.
"Фармацевтически приемлемая соль" может быть любой солью, образованной из неорганической или органической кислоты. Термин "фармацевтически приемлемый анион" обозначает анион такой кислотно- аддитивной соли. Выбранные соль и/или анион не должны быть биологически или иным образом нежелательными.
Анионы получены из неорганических кислот, таких как, соляная кислота, бромистоводородная кислота, серная кислота (образующая сульфатную и бисульфатную соли), азотная кислота, фосфорная кислота и т.п., и из органических кислот, таких как уксусная кислота, пропионовая кислота, гликолевая кислота, пировиноградная кислота, щавелевая кислота, яблочная кислота, малоновая кислота, янтарная кислота, малеиновая кислота, фумаровая кислота, винная кислота, лимонная кислота, бензойная кислота, коричная кислота, миндальная кислота, метансульфоновая кислота, этансульфоновая кислота, салициловая кислота, п-толуолсульфокислота, гексановая кислота, гептановая кислота, циклопентанпропионовая кислота, молочная кислота, о-(4-гидроксибензоил)бензойная кислота, 1,2-этандисульфоновая кислота, 2-гидроксиэтансульфоновая кислота, бензолсульфоновая кислота, п-хлорбензолсульфоновая кислота, 2-нафталинсульфокислота, камфорсульфоновая кислота, 4-метилбицикло[2.2.2]окт-2-ен-1-карбоновая кислота, глюкогептоновая кислота, 4,4'-метиленбис(3-гидрокси-2-нафтойная) кислота, 3-фенилпропионовая кислота, триметилуксусная кислота, трет. -бутилуксусная кислота, лаурилсерная кислота, глюкуроновая кислота, глутаминовая кислота, 3-гидрокси-2-нафтойная кислота, стеариновая кислота, муконовая кислота и т.п.
Термин "лечение" или "лечить" означает любое лечение болезни у млекопитающего, включая:
(1) предупреждение болезни, т.е. приводящее к тому, чтобы клинические симптомы болезни не развивались;
(2) ингибирование болезни, т.е. прекращение развития клинических симптомов; и/или
(3) облегчение болезни, т.е. приводящее к регрессии клинических симптомов.
Термин "эффективное количество" означает дозу, достаточную для обеспечения лечения такого состояния болезни, которое поддается лечению. Оно может варьироваться в зависимости от пациента, болезни и осуществляемого лечения.
Термин "совместное введение" означает введение более одного активного агента как части одной лечебной схемы вне зависимости от того, вводятся ли они одновременно или в различное время.
"Структура формулы I" обозначает общую структуру соединений по изобретению. Химические связи, обозначенные волнистой линией, например, в формуле II, показывают неспецифическую стереохимию, например, в положении 5 дибензосуберана, т.е. углерод, к которому присоединена группа пиперазина.
"Изомерия" относится к соединениям, имеющим одинаковую атомную массу и число атомов, но отличающимся одним или несколькими физическими или химическими свойствами.
"Стереоизомер" относится к одному из двух химических соединений, имеющих одинаковый молекулярный вес, химический состав и строение, но атомы которых сгруппированы по-разному. Это означает, что определенные идентичные химические части имеют различную ориентацию в пространстве и, следовательно, в чистом виде обладают способностью отклонять плоскость поляризации света. Однако некоторые чистые стереоизомеры могут иметь настолько малое оптическое отклонение, что оно не может быть обнаружено современными приборами.
"Оптическая изомерия" описывает один тип стереоизомерии, который проявляется в том, что изомер, чистый или в растворе, отклоняет плоскость поляризации света. Ее причиной во многих случаях является присоединение четырех различных химических атомов или групп по крайней мере к одному атому углерода в молекуле. Эти изомеры могут быть обозначены как d-, l- либо d, l-пара или D-, L- либо D,L-пара; или (R)-, (S)- либо (R,S)-пара в зависимости от того, какая номенклатурная система используется.
Соединения формулы I существуют в двух изомерных конфигурациях, определяемых взаимосвязью 10,11-метан- и 5-пиперазинил-заместителей в дибензосуберане (см., например, структуру, представленную в формуле II, в прилагаемом ниже Номенклатурном описании). Когда 10,11-метан- и 5-пиперазинил-заместители оба ориентированы в одном и том же направлении по отношению к дибензосуберану (например, оба вверх или оба вниз), изомерная форма называется "син". Когда 10,11-метан- и 5-пиперазинил-заместители ориентированы в противоположных направлениях по отношению к дибензосуберану (например, один вверх, а другой вниз), изомерная форма называется "анти".
Определенные соединения формулы I могут иметь асимметричный центр в группе, обозначаемой "A", где Ra или Rb не являются водородом. Эти соединения могут существовать в двух стереохимических формах, называемых (+) и (-) или называемых (R)- и (S)- или в виде смеси двух указанных стереоизомеров. В описании используются обозначения (R)- и (S)-.
Несмотря на то, что описаны и указаны конкретные стереоизомеры, объем настоящего изобретения включает как индивидуальные стереоизомеры, так и их смеси, рацематы и др.
Номенклатура
Соединения формулы I названы и перечислены как описано ниже со ссылкой на формулу II.
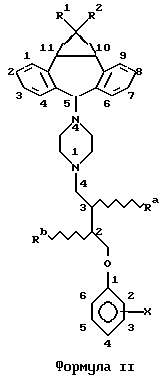
Например, соединение, где R1 и R2 обозначают хлор, Ra обозначает гидроксил и фенильная группа (из R3 в формуле I) замещена NO2 в положении 3, называется (3R, S)-анти, син-1-{ 4-[4-(10,11-дихлорметандибензосубер-5-ил)пиперазин-1-ил]-3-гидроксибутокси}-3-нитробензолом.
Соединение, где R1 и R2 обозначают водород, Rb обозначает ацетокси (в изомерной форме, расположенной как бы под плоскостью страницы), фенильная группа (из R3 в формуле I) замещена трифторметилом в положении 5, и связь, соединяющая положение 4 пиперазина с положением 5 бензосуберана, находится в изомерной форме, расположенной как бы над плоскостью страницы, называется (2S)-син-1-{ 4-[4-(10,11-метандибензосубер-5-ил)пиперазин-1- ил] -2-ацетоксибутокси}-5-трифторметилбензолом.
Предпочтительное соединение данного изобретения, изображенное ниже в виде формулы III: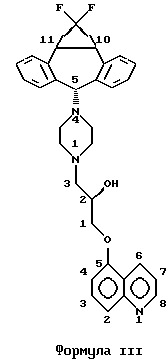
которое является соединением формулы I, где R1 и R2 обозначают F, A обозначает (2R)-тидроксипропил и R3 обозначает хинолил, присоединенный в положении 5 к кислороду, называется (2R)-анти-5-{3-[4-(10,11-дифторметандибензосубер-5- ил)пиперазин-1-ил] -2-гидроксипропокси} хинолином. В противоположность этому по номенклатуре Chemical Abstracts соединение формулы III называется 1-(4-анти(1,1-дифтор-1a,10b-дигидродибензо[a,e]циклопропа [c]циклогептен-6-ил)пиперазин-1-ил)-(2R)-3-(5-хинолилокси)-2-пропанолом (нумерация, представленная в формуле III, не применяется в номенклатурной системе Chemical Abstracts). Поскольку любая номенклатурная система адекватно описывает соединения настоящего изобретения, первая из двух названных систем будет применяться для настоящей спецификации.
Параметры реакции синтеза
Термины "растворитель", "инертный органический растворитель" или "инертный растворитель" означают растворитель, инертный в условиях описываемой при этом реакции [они включают, например, бензол, толуол, ацетонитрил, тетрагидрофуран (ТГФ), диметилформамид (ДМФ), хлороформ, метиленхлорид (или дихлорметан), диэтиловый эфир, метанол и т.п.]. Если не указано иное, то растворители, используемые в реакциях по настоящему изобретению, являются инертными органическими растворителями.
Термин "д.к." означает добавление количества, достаточного для достижения заданного назначения, например, доведение раствора до желаемого объема (т.е. до 100%).
Если не указано иное, приведенные в описании реакции происходят при атмосферном давлении в диапазоне температур от 5oC до 100oC (предпочтительно от 10oC до 50oC; наиболее предпочтительно при "комнатной" температуре или температуре "окружающей среды", т.е. при 20oC). Однако ясно, что некоторые реакции, для которых используется температурный диапазон химической реакции, могут протекать при температурах выше или ниже указанных температурных диапазонов. Кроме того, если не указано иное, время и условия реакции рассматриваются как приблизительные, например, как протекающие примерно при атмосферном давлении в пределах температурного диапазона от примерно 5oC до примерно 100oC (предпочтительно от примерно 10oC до примерно 50oC; наиболее предпочтительно при примерно 20oC) в течение периода времени от примерно 1 часа до примерно 10 часов (предпочтительно примерно 5 часов). Параметры, указанные в примерах, рассматриваются как конкретные, а не приблизительные.
Выделение и очистка соединений и промежуточных продуктов, описанных ниже, могут быть выполнены, если требуется, с помощью любого подходящего метода разделения или очистки, как, например, фильтрацией, экстрагированием, кристаллизацией, колоночной хроматографией, тонкослойной хроматографией, толстослойной хроматографией или комбинацией этих методов. Конкретные иллюстрации пригодных методов разделения и выделения описаны в примерах, приведенных ниже. Однако, разумеется, могут быть использованы другие равноценные методы разделения и выделения.
Синтез соединений формулы I
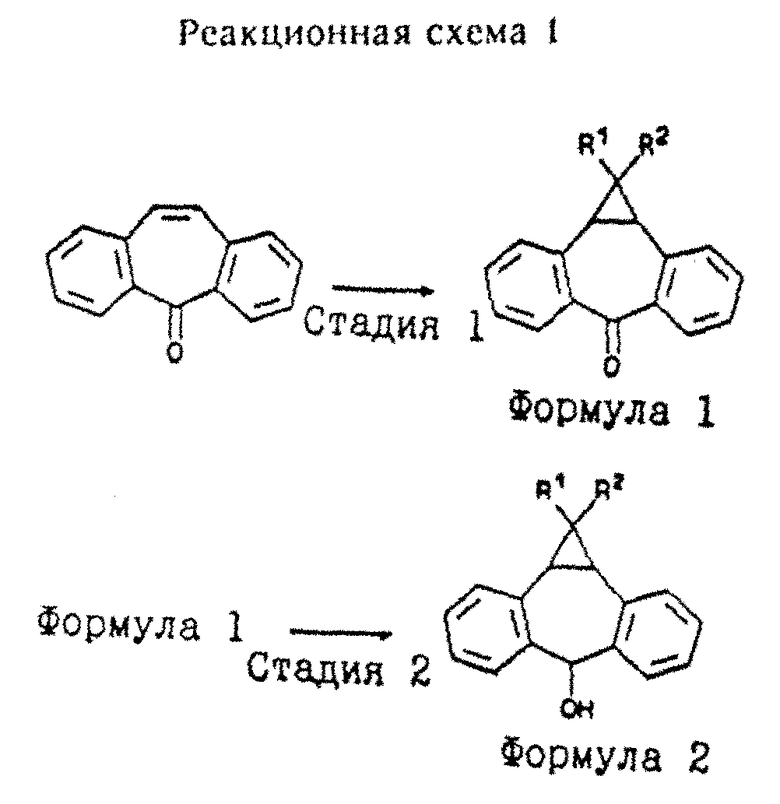
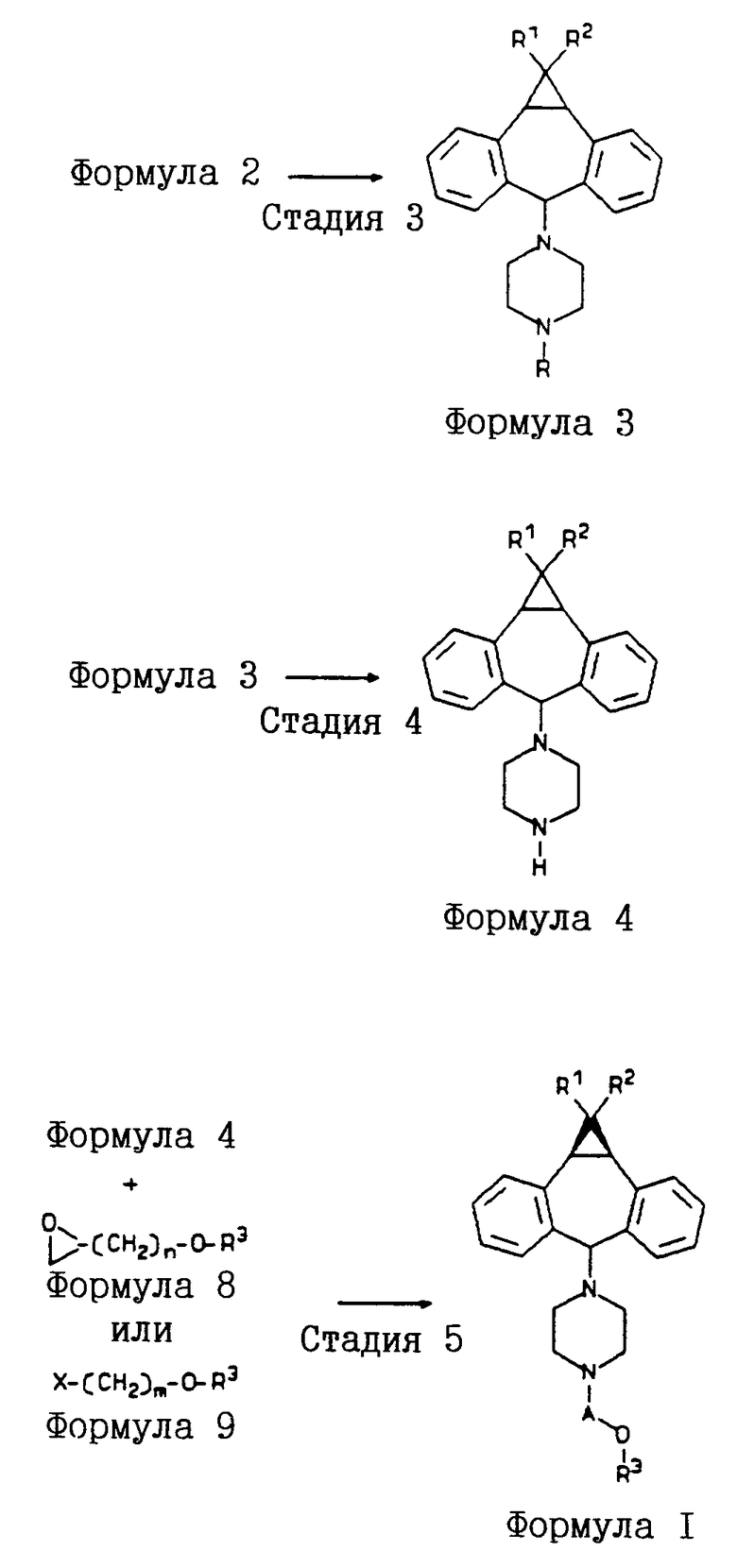
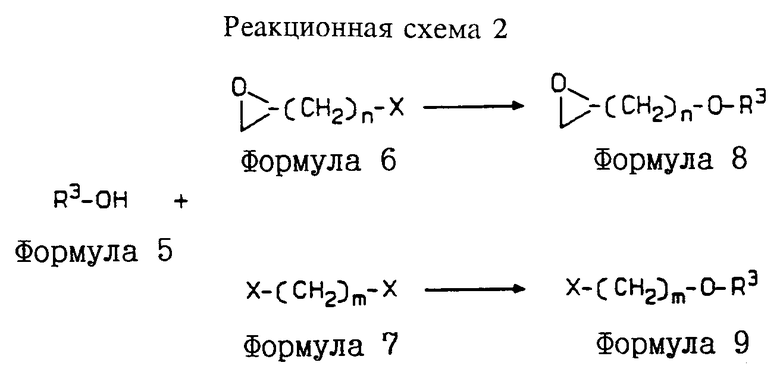
Соединения формулы I могут быть получены в соответствии со способами, описанными в патенте США 5112817, включенном в настоящее описание в качестве ссылки, путем замещения дибензосуберона необязательно замещенным 10,11-метандибензосубероном, полученным, например, как описано Ciganek и др. в "Imine Analogues of Tricyclic Antidepressants", J. Med. Chem., 1981, 24, 336-41; или Coyne и Cusic в "Aminoalkyldibenzo[a,e]cyclopropa[c]cycloheptene Derivatives. A Series of Potent Antidepressants", J. Med. Chem., 1974, т. 17, N 1, 72-75, которые включены в настоящее описание в качестве ссылки. Различные синтезы соединений формулы I описаны ниже со ссылкой на реакционные схемы 1, 2 и 3.
Ключевую роль в синтезе соединений формулы I играет 1-[10,11-метандибензосубер-5-ил] пиперазин приведенной ниже формулы 4, который необязательно может быть замещен в положениях 10 и 11. Этот ключевой промежуточный продукт соединяют или конденсируют с реагентом, который содержит или превращается в группу A-O-R3 для включения в соединения формулы I; или с реагентом, который содержит по существу группу A-эпоксида или ее производное, группу A'-O-R3, или превращается в нее, для включения в соединения формулы I. Такие превращения включают, например, ацилирование гидроксигруппы, присутствующей в группе A', для получения ацилоксигруппы, или удаление ацилоксигруппы в группе A', или превращение группы A-эпоксида с R3-OH в соединение формулы I. При таком превращении, когда R3-OH добавляется к группе A-эпоксида, эпоксидное кольцо открывается, причем образуется гидроксигруппа, соседняя с атомом углерода, к которому присоединяется -OR3.
Краткое описание реакционных схем
Реакционная схема 1 иллюстрирует синтез соединений формулы I, где Ra обозначает H или OH.
Реакционная схема 2 иллюстрирует синтез соединений формул 8 и 9; которые используются как реагенты на стадии 5 реакционной схемы 1 в качестве предшественников при синтезе соединений формулы I.
Реакционная схема 3 иллюстрирует синтез соединений формулы I, где Rb обозначает OH.
Используемые в реакционных схемах заместители A, R1, R2, R3, Ra и Rb имеют те же самые значения, которые приведены в "Кратком описании изобретения". Заместитель "X" обозначает галогруппу; n равно 1 или 2; и m равно 1, 2, 3 или 4.
Исходные материалы
Соединение 5H-дибензо[a,d]циклогептен-5-он (называемое также дибензо[a, d] -5H-циклогептен-5-оном или дибензосубероном) является коммерчески доступным, например, производится Aldrich Chemical Company, Милуоки, WI. Другие реагенты, такие как эпибромгидрин и 1-бром-3,4-эпоксибутан, также являются коммерчески доступными или могут быть легко получены специалистами в данной области техники с использованием обычно применяемой методологии синтеза. (Реакционную схему 1 см. в конце текста).
Получение соединений формулы I
Раствор ацетата (такого, как хлордифторацетат натрия, метилтрихлорацетат, этилтрихлорацетат; в зависимости от желаемых заместителей для R1 и R2) в растворителе (таком, как диглим, бензол или петролейный эфир) добавляют в течение 4-8 часов (предпочтительно 6 часов) к раствору дибензосуберенона (например, в диглиме) при перемешивании в атмосфере азота, поддерживая температуру реакции на уровне 160-165oC. (Могут быть использованы другие температурные режимы реакции в зависимости от применяемых реагентов, как описано Ciganek и др. и Coyne и Cusic). Реакционную смесь доводят до комнатной температуры, затем сливают в воду и экстрагируют (например, простым эфиром). Желаемый 10,11-замещенный-метандибензосуберон выделяют и очищают обычными способами, например промывают водой органическую (например, бензол) фазу, высушивают (например, над Na2SO4), упаривают и остаток перекристаллизовывают (например, из этанола и необязательно повторно, например, из ацетона/гексана).
В противоположность этому соединения формулы 2, где R1 и R2 различные, такие как H и Cl соответственно, могут быть получены аналогично описанному в журнале J. Med. Chem., т. 17, 72 (1974), который включен в настоящее описание в качестве ссылки. Соединение формулы 2, где R1 и R2 оба обозначают водород, может быть получено аналогично описанному у Coyne и Cusic в статье "Aminoalkyldibenzo[a, e] cyclopropa[c] cyclogepten Derivatives. A Series of Potent Antidepressants", J. Med. Chem., 1974, т. 17, N 1, 72-75, которая ранее упоминалась в качестве ссылки.
Получение соединений формулы 2
Раствор 10,11-(необязательно замещенного)-метандибензосуберона в растворителе (например, ТГФ/метаноле) охлаждают (например, в ледяной бане) и добавляют по частям восстанавливающий агент (например, борогидрид натрия). Реакционной смеси дают дойти до комнатной температуры и перемешивают в течение 1-5 часов (предпочтительно 2 часа), затем сливают в воду. Продукт выделяют (например, с помощью фильтрации) и очищают обычными способами (например, промывают водой и высушивают) до получения соответствующего 10,11-(необязательно замещенного)-метандибензосуберола.
Получение соединений формулы 3, где R обозначает формил
Раствор 10,11-(необязательно замещенного)-метандибензосуберола в растворителе (например, диоксане) охлаждают (например, в ледяной бане), далее галогенируют [например, добавляя по каплям тионилхлорид, поддерживая повышенную температуру (от 40 до 70oC, предпочтительно 50oC) на протяжении 2-5 часов (предпочтительно 4 часа)]. Реакционную смесь упаривают досуха, получая смесь син- и анти-изомеров соответствующего 5-гало-10,11-(необязательно замещенного)-метандибензосуберана. Этот галогенированный суберан растворяют без дальнейшей очистки (например, в ацетонитриле) и вводят пиперазин путем нуклеофильного замещения галогенида [например, добавляя при перемешивании 1-пиперазинкарбоксальдегид, предпочтительно в атмосфере сухого N2 при повышенной температуре (например, 100oC) в течение 10-30 часов (предпочтительно 20 часов)] . Реакционную смесь упаривают досуха и выделяют желаемый продукт 1-[10,11-(необязательно замещенный)-метандибензосубер-5-ил] -4-формилпиперазин и очищают его обычными способами [например, осадок разделяют между водным NaHCO3 и этилацетатом, органическую фазу промывают водой, высушивают (например, над K2CO3) и упаривают]. Индивидуальные син- и анти-изомеры разделяют, например, с помощью быстрой хроматографии остатка на силикагеле (30% ацетон/гексан).
Получение соединений формулы 4
Раствор 1-[10,11-(необязательно замещенный)метандибензосубер-5-ил]-4-формилпиперазина и гидроксида калия в растворителе (например, 9:1 этанол/H2O) нагревают с обратным холодильником в течение 0,5-2 часов (предпочтительно 1 час), затем охлаждают. Охлажденную реакционную смесь концентрируют, растворяют в воде, экстрагируют (например, этилацетатом), высушивают (например, над K2CO3) и упаривают органическую фазу, получая соответствующий 1-[10,11-(необязательно замещенный)метандибензосубер-5-ил] пиперазина.
Получение соединений формулы I
Раствор соединения формулы 8 [например, 1-(арилокси- или гетероарилокси)-2,3-эпоксипропан или 1-(арилокси- или гетероарилокси)-3,4-эпоксибутан] или арилокси- или гетероарилоксиалкилгалогенид формулы 9 и 1-[10,11-(необязательно замещенный)метандибензосубер-5-ил] пиперазин нагревают с обратным холодильником в растворителе (например, изопропаноле) в течение 10-30 часов (предпочтительно 20 часов). Желаемый продукт, соответствующее производное 10,11-метандибензосуберана формулы I, выделяют и очищают обычными способами [например, упаривая досуха и хроматографируя на силикагеле (например, с использованием смеси 70:30:1 этилацетат/гексан/триэтиламин)]. (Реакционную схему 2 см. в конце текста).
Получение соединений формулы 8
Ариловый или гетероариловый спирт (такой, как бензофуразан-4-ол, хинолин-5-ол или 2-нитрофенол) растворяют в растворителе (например, ацетонитриле, ТГФ или диметилформамиде), обрабатывают небольшим избытком сильного основания (например, гидрида натрия или трет.-бутоксида калия). Смесь нагревают (например, при 50oC) в течение 10 минут - 2 часов (предпочтительно 30 минут). Добавляют соединение формулы 6 (такое, как 1-хлор-2,3-эпоксибутан, 1-бром-2,3-эпоксибутан, эпибромгидрин, эпихлоргидрин или их тозил- или мезил-производное) и смесь нагревают (например, при 60oC в течение 1-5 часов; предпочтительно 2 часа). Реакционную смесь сливают в воду и экстрагируют (например, этилацетатом). Органическую фазу промывают водой, высушивают над Na2SO4 и упаривают, получая соответствующий 1-(арилокси- или гетероарилокси)-2,3-эпоксипропан или 1-(арилокси- или гетероарилокси)-3,4-эпоксибутан, который затем выделяют и очищают обычными способами [например, хроматографией на силикагеле (50% этилацетат/гексан)].
Соединения формулы 8, такие как 1-(5-хинолилокси)-2,3-эпоксипропан, могут быть синтезированы аналогично описанному в публикации Drug Design and Discovery, т. 9, 69, (1992), которая включена в настоящее описание в качестве ссылки.
Получение соединений формулы 9
Как показано на реакционной схеме 3 (см. в конце текста), анион арилового или гетероарилового спирта формулы 5 подвергают взаимодействию с дигалоалкильным соединением формулы 7, таким как 1-бром-2-хлорэтан, 1-бром-3-хлорпропан или 1-бром-4-хлорбутан, в растворителе (таком, как ацетон, ТГФ или ДМФ) при температуре в диапазоне от комнатной температуры до температуры кипения используемого растворителя, получая соответствующее галоалкилоксиарильное (или гетероарильное) соединение формулы 9. Этот синтез описан в патенте США 5112817, ранее указанном в настоящем описании в качестве ссылки.
Получение соединений формулы 10
Как показано на реакционной схеме 3, стадия 1, 1-[10,11-(необязательно замещенный)-метандибензосубер-5-ил] пиперазин формулы 4 подвергают взаимодействию с соединением формулы 6 (например, с соединением, где n равно 2, как показано на реакционной схеме 3; другие соединения формулы 6 дадут соответствующие продукты) в условиях, описанных выше для получения формулы 8 (на реакционной схеме 2), получая соединение формулы 10 1-[10,11-(необязательно замещенный)-метандибензосубер-5-ил]-4-(3,4-эпоксибутил)пиперазин.
Получение соединений формулы I, где Rb обозначает OH
Как показано на реакционной схеме 3, стадия 2, соединение формулы 10 1-[10,11-(необязательно замещенный)-метандибензосубер-5-ил] -4-(3,4-эпоксибутил)пиперазин подвергают взаимодействию с ариловым или гетероариловым спиртом (таким, как бензофуразан-4-ол, хинолин-5-ол или 2-нитрофенол) в условиях, описанных выше для получения формулы 1 (на реакционной схеме 1), получая соответствующее соединение формулы I, где Rb обозначает OH.
Получение соединений формулы I, где Ra или Rb обозначает низший ацилокси
Соединения формулы I, где Ra или Rb обозначает низший ацилокси, получают аналогично описанному в патенте США 5112817, указанном ранее в качестве ссылки, начиная с соответствующего соединения формулы I, где Ra или Rb обозначает OH (полученного в соответствии с описанным выше). Например, соединение формулы I, где Ra или Rb обозначает OH, подвергают взаимодействию с ацилхлоридом для образования соответствующего ацилокси-соединения.
Получение солей соединения формулы I
Соединения формулы I могут быть превращены в соответствующие кислотно-аддитивные соли. Превращение осуществляется при помощи обработки стехиометрическим количеством соответствующей кислоты, такой как соляная кислота (например, 3 молярных эквивалента для образования тригидрохлоридной соли в случае использования соединения формулы I, включающего три основных атома азота). Если R3 обозначает фенил, соединение формулы I имеет только два основных атома азота и может абсорбировать только два эквивалента кислоты для получения кислотно-аддитивной соли. Если заместитель R3 включает два основных атома азота, основание формулы I может абсорбировать четыре эквивалента кислоты. Предпочтительные кислотно-аддитивные соли по изобретению могут включать два или три эквивалента кислоты. Более предпочтительными являются кислотно-аддитивные соли с тремя эквивалентами кислоты. На стадии образования соли по настоящему изобретению обычно свободное основание растворяют в полярном органическом растворителе, таком как метанол или этанол, и кислоту добавляют в воду, метанол или этанол. Температуру поддерживают в диапазоне от 0oC до 50oC. Соответствующая соль осаждается самопроизвольно, или может быть выведена из раствора с помощью менее полярного растворителя, или путем выпаривания растворителя, или путем охлаждения раствора.
На стадии высвобождения свободного основания формулы I в соответствии с настоящим изобретением кислотно-аддитивные соли соединений формулы I могут быть разложены до соответствующих свободных оснований путем обработки избытком подходящего основания, таким как аммоний или бикарбонат натрия, обычно в присутствии водного растворителя и при температуре между 0oC и 50oC. Свободное основание выделяют обычными способами, таким как экстрагирование органическим растворителем. Очевидно, что стехиометрический избыток должен быть взят с учетом числа эквивалентов кислоты, связанных основанием формулы I.
Предпочтительные свободные основания формулы I включают 2 или 3 основных атомов азота. Основания с 3 основными атомами азота являются наиболее предпочтительными.
Предпочтительные способы и заключительные стадии
Для получения соответствующего производного 10,11-метандибензосуберана формулы I объединяют 1-(арилокси- или гетероарилокси)-2,3-эпоксипропан или 1-(арилокси- или гетероарилокси)-3,4-эпоксибутан, или арилокси- или гетероарилоксиалкилгалогенид и 1-[10,11-(необязательно замещенный)-метандибензосубер-5-ил]пиперазин.
Соединение формулы I, где Ra или Rb обозначает OH, подвергают взаимодействию с ацилхлоридом для образования соответствующего ацилокси-соединения.
Соединение формулы 10 подвергают взаимодействию со спиртом R3-OH для получения соответствующего производного 10,11-метандибензосуберана формулы I, где Rb обозначает OH.
Соединение формулы I подвергают взаимодействию с фармацевтически приемлемой кислотой для образования соответствующей кислотно-аддитивной соли.
Фармацевтически приемлемую кислотно-аддитивную соль формулы I подвергают взаимодействию с основанием для образования соответствующего свободного основания формулы I.
Предпочтительные соединения
Предпочтительными являются соединения формулы I, где R1 и R2 обозначают фтор. Также предпочтительными являются те соединения, где A обозначает 2-гидроксипропилен. Также предпочтительными являются те соединения, где R3 обозначает 5-хинолил или 4-бензофуразил. Кроме того, предпочтительными являются те соединения, в которых объединены отмеченные выше характеристики. Также предпочтительны простые изомеры.
Наиболее предпочтительным является соединение 5-{3-[4-(10,11-дифторметандибензосубер-5-ил)пиперазин-1-ил] -2- гидроксипропокси}-хинолин; в частности, его (2R)-анти-изомер.
Полезность, испытание и введение
Общая полезность
Соединения по настоящему изобретению являются хемосенсибилизирующими или потенцирующими агентами, и они также пригодны в качестве агентов, модифицирующих резистентность. Они также пригодны для лечения резистентности к множеству лекарств (т.е. после проявления клинической резистентности) и могут также вводиться во время начальной химиотерапии (т.е. до проявления любой клинической резистентности) для повышения активности противораковых агентов, вводимых впервые. Соединения по настоящему изобретению пригодны также для лечения резистентной к лекарствам малярии.
Испытание
Активность in vitro хемосенсибилизирующих или потенцирующих агентов, в частности, для лечения резистентности к множеству лекарств определяют с помощью количественного анализа пролиферации МТТ, например, с помощью модифицированного количественного анализа, описанного T.Mosmann в "Rapid Colorimetric Assay for Cellular Growth and Survival: Application to proliferation and cytotoxicity assays", J. Immunol. Meth., т. 65, 55-63 (1983). Другой количественный анализ пролиферации МТТ описан Alleu и др. в "Feasibility of Drug Screening with Panels of Human Tumor Cell Lines Using a Microculture Tetrasolium Assay", Cancer Research, т. 48, 589-601 (1988).
Активность in vivo хемосенсибилизирующих или потенцирующих агентов, в частности, для лечения резистентности к множеству лекарств, определяют, например, аналогично описанному Slate и Mchelson в "Drug Resistance Reversal Strategies: A Comparison of Experimental Data with Model Prediction", J. Natl. Cancer Inst., т. 83, 1574-1580 (1991). Другие методы испытаний in vivo описаны Sato и др. в "Circumvention of Multidrug Resistance by a Newly Synthesized Quinoline Derivative, MS-073, "Cancer Research, т. 51, 2420-2424 (1991); Tsuruo и др. в "Circumvention of Vincristine and Adriamycin Resistance in Vitro and in Vivo by Calcium Influx Blockers", Cancer Research, т. 43, 2905-2910, (1983); и Tsuruo и др. в "Overcoming of Vincristine Resistance in P388 Leukemia in Vivo and in Vitro through Enhanced Cytotoxicity of Vincristine and Vinblastine by Verapamil", Cancer Research, т. 41, 1967-1972 (1981).
Стабильность соединений в воде определяют обычными методами, например, путем измерения количества соединения, сохранившегося в растворе при различных значениях pH и температурах.
Введение
Соединения формулы I вводят в терапевтически эффективной дозе, например в дозе, достаточной для обеспечения лечения ранее описанных состояний болезни, обычно путем совместного назначения со вторым активным агентом, предпочтительно противораковым химиотерапевтическим агентом, в частности, выбранным из группы агентов, перечисленных выше, и наиболее предпочтительно с противораковым химиотерапевтическим агентом, к которому проявилась клиническая резистентность у млекопитающего, подлежащего лечению. Введение соединений по изобретению или их фармацевтически приемлемых солей может быть осуществлено посредством любых принятых форм введения для агентов, служащих для подобных целей.
Хотя уровни дозировки соединений по настоящему изобретению для человека еще должны быть оптимизированы, обычно суточная доза составляет приблизительно от 0,01 до 4,0 мг/кг веса тела, предпочтительно приблизительно от 0,1 до 2,0 мг/кг веса тела и наиболее предпочтительно приблизительно от 0,3 до 1,0 мг/кг веса тела. Таким образом, при введении человеку весом 70 кг диапазон дозировки должен составлять приблизительно от 0,7 до 280 мг в сутки, предпочтительно приблизительно от 7,0 до 140 мг в сутки и наиболее предпочтительно приблизительно от 21 до 70 мг в сутки. Количество вводимого активного соединения должно, конечно, зависеть от субъекта и состояния болезни, подлежащей лечению, тяжести поражения, способа и режима введения (например, оральное введение за день до противораковой химиотерапии и внутривенное введение во время противораковой химиотерапии) и мнения лечащего врача.
При использовании соединений по настоящему изобретению для лечения указанных выше состояний может быть использована любая фармацевтически приемлемая форма введения. Соединения формулы I могут вводиться либо по отдельности, либо в сочетании с другими фармацевтически приемлемыми инертными наполнителями, включая твердые, полутвердые, жидкие или аэрозольные формы дозировки, такие как, например, таблетки, капсулы, порошки, жидкости, суспензии, суппозитории, аэрозоли или т.п. Для пролонгированного введения соединения с заранее определенной нормой расхода, предпочтительно в форме стандартных доз, пригодных для однократного введения точных доз, соединения формулы I могут также вводиться в виде лекарственных форм с непрерывным или контролируемым высвобождением, включая инъекцию веществ замедленного всасывания, осмотические насосы, пилюли, трансдермальные (включая электротранспортирующие) повязки и т.п. Композиции могут обычно включать типичный фармацевтический носитель или инертный наполнитель и соединение формулы I или его фармацевтически приемлемую соль. Кроме того, эти композиции могут включать другие медицинские агенты, фармацевтические агенты, носители, адъюванты и т. д., такие как перечисленные выше противораковые химиотерапевтические агенты.
В целом, в зависимости от предполагаемой формы введения, фармацевтически приемлемая композиция может содержать приблизительно от 0,1 вес.% до 90 вес. %, предпочтительно приблизительно от 0,5 вес.% до 50 вес.% соединения или соли формулы I, остальное составляют фармацевтически пригодные инертные наполнители, носители и т.п.
Одним из предпочтительных способов введения для состояний, детально описанных выше, является оральный с использованием обычного режима суточной дозы, которая определяется в зависимости от степени поражения. Для такого орального введения формируют фармацевтически приемлемую нетоксичную композицию путем включения любых обычно применяемых инертных наполнителей, таких как, например, маннитол, лактоза, крахмал, стеарат магния, сахарин натрия, тальк, целлюлоза, кросскармелоза натрия, глюкоза, желатин, сахароза, карбонат магния и т.п. Такие композиции включают растворы, суспензии, таблетки, диспергируемые таблетки, пилюли, капсулы, порошки, формы с непрерывным высвобождением и т.п.
Предпочтительно композиция должна иметь форму пилюли или таблетки. Таким образом, наряду с активным ингредиентом, композиция может содержать: разбавитель, такой как лактоза, сахароза, дикальцийфосфат или т.п.; смазывающее вещество, такое как стеарат магния или т.п.; и связывающее вещество, такое как крахмал, аравийская камедь, желатин, поливинилпирролидон, целлюлоза и их производные и т.п.
Жидкие вводимые фармацевтические композиции могут, например, быть приготовлены путем растворения, диспергирования и т.д. активного соединения, как показано выше, и необязательных фармацевтических адъювантов в носителе, таком как, например, вода, физиологический раствор, водная декстроза, глицерин, гликоль, этанол и т.п., для образования таким образом раствора или суспензии. Если требуется, вводимая фармацевтическая композиция может также содержать меньшие количества нетоксичных вспомогательных веществ, таких как смачиватели, эмульгаторы или солюбилизаторы, забуферивающие pH агенты и т.п. , например, ацетат, цитрат натрия, производные циклодекстрина, сорбитанмонолаурат, натрийацетат триэтаноламина, триэтаноламинолеат и т.д. Современные способы получения таких лекарственных форм известны или могут быть очевидными для специалистов в этой области техники; например, см. Remington's Pharmaceutical Sciences, Mack Publishing Company, Easton, Pennsylvania, 15-е изд. , 1975. Вводимая композиция или форма в любом случае должна содержать активное вещество в количестве, достаточном для облегчения симптомов субъекта, подлежащего лечению.
Могут быть приготовлены лекарственные формы или композиции, содержащие активный ингредиент в диапазоне от 0,005% до 95% с дополнением недостающего до баланса количества за счет нетоксичного носителя.
Для орального введения фармацевтически приемлемую нетоксичную композицию готовят путем включения любого, обычно применяемого инертного наполнителя, такого как, например, фармацевтически чистые маннитол, лактоза, крахмал, стеарат магния, тальк, производные целлюлозы, кросскармалоза натрия, глюкоза, сахароза, карбонат магния, сахарин натрия и т.п. Такие композиции могут быть в виде растворов, суспензий, таблеток, капсул, порошков, форм с непрерывным высвобождением и т.п. Такие композиции могут содержать 0,01%-95% активного ингредиента, предпочтительно 0,1-50%.
Для получения твердой лекарственной формы раствор или суспензию, например, в пропилене или этиленкарбонате или их смеси, в растительных маслах или триглицеридах, предпочтительно включают в желатиновую капсулу. Такие растворы и способы их получения и включение в желатиновые капсулы описаны в патентах США 4328245; 4409239 и 4410545. Для получения жидкой лекарственной формы раствор, например, в полиэтиленгликоле, может быть разбавлен достаточным количеством фармацевтически приемлемого жидкого носителя, например водой, для облегчения нормирования при введении.
В противоположность этому жидкие или полутвердые оральные формы могут быть получены путем растворения или диспергирования активного вещества или соли в растительных маслах, гликоле, триглицеридах, эфирах пропиленгликоля (например, пропиленкарбонате), этиленкарбонате и их смесях и т.п. и включения этих растворов или суспензий в твердые или мягкие желатиновые капсульные оболочки.
Другие пригодные формы включают такой набор, который описан в патентах США Re. 28819 и 4358603.
Другим преимуществом настоящего изобретения является пригодность для орального и парэнтерального введения вследствие очень высоких характеристик стабильности, которые были обнаружены для соединений формулы I, что являлось проблемой для MS-073.
Под парэнтеральным назначением обычно подразумевают инъекцию - подкожную, внутримышечную либо внутривенную. Инъецируемые препараты могут быть приготовлены в обычных формах в виде жидких растворов или суспензий, в твердых формах, пригодных для получения раствора или суспензии в жидкости до инъекции, или в виде эмульсий. Пригодными инертными наполнителями являются, например, вода, физиологический раствор, декстроза, глицерин, этанол и т.п. Кроме того, если требуется, фармацевтические композиции для введения могут также содержать меньшие количества нетоксичных добавочных веществ, таких как смачивающие или эмульгирующие агенты, забуферивающие pH агенты, повышающие растворимость агенты и т.п., такие как, например, ацетат натрия, сорбитанмонолаурат, триэтаноламинолеат, циклодекстрины и т.д.
В наиболее современном подходе для парэнтерального введения применяется имплантация системы с медленным высвобождением или с непрерывным высвобождением, что позволяет поддерживать постоянный уровень дозировки. См., например, патент США 3710795.
Процентная концентрация активного вещества, входящего в такие парэнтеральные композиции, в большой степени зависит от их специфической природы, а также от активности вещества и от нужд субъекта. Однако при этом обычно принятые процентные концентрации активного ингредиента в растворах составляют от 0,01% до 10%, и они должны быть выше, если композиция твердая, которую, в свою очередь, необходимо разбавлять до вышеуказанных процентных концентраций. Предпочтительно, чтобы композиция включала 0,2-2% активного агента в растворе.
Также могут вводиться назальные растворы активного вещества, индивидуально или в комбинации с другими фармацевтически приемлемыми инертными наполнителями.
Формы активного вещества или его соли могут также вводиться через дыхательную систему в виде аэрозоля или раствора, применяемого с помощью распылителя, или в виде ультратонкого порошка для вдувания, индивидуально или в комбинации с инертным носителем, таким как лактоза. В этом случае частицы композиции имеют диаметры менее 50 микрон, предпочтительно менее 10 микрон.
Примеры
Следующие препараты и примеры приведены для того, чтобы предоставить возможность специалистам в данной области техники более ясно понять и осуществить настоящее изобретение. Они не должны рассматриваться как ограничивающие объем данного изобретения, а только как иллюстрирующие и поясняющие его.
Пример 1
10.11-дифторметандибензосуберон
1А. Формула 1, где R1 и R2 обозначают F
Раствор хлордифторацетата натрия (350 г) в диглиме (1400 мл) по каплям при перемешивании верхнего слоя и в атмосфере азота в течение 6 часов добавляли к раствору дибензосуберинона (25 г) в диглиме (500 мл), поддерживая температуру реакции на уровне 160-165oC. Охлажденную реакционную смесь сливали в воду (1,8 л) и экстрагировали эфиром (1,8 л). Органическую фазу промывали водой, сушили над Na2SO4 и упаривали. Остаток перекристаллизовали из этанола, затем из смеси ацетон/гексан, получая 14 г 10,11-дифторметандибензосуберона, tпл 149,6oC. С помощью быстрой хроматографии объединенных маточных растворов на силикагеле и элюирования в смеси 20% ацетон/гексан дополнительно получали 6,5 г целевого материала.
1Б. Формула 1 с различными R1 и R2
В соответствии с процедурой, описанной в части А, и заменяя хлордифторацетат натрия следующими веществами:
а. метилтрихлорацетатом,
б. метилтрибромацетатом, и
в. дихлорфторацетатом натрия,
получали соответственно следующие соединения:
а. 10,11-дихлорметандибензосуберон,
б. 10,11-дибромметандибензосуберон, и
в. 10,11-хлорфторметандибензосуберон.
Пример 2
10,11-дифторметандибензосуберол
2А. Формула 3, где R1 и R2 обозначают F
Раствор 10,11-дифторметандибензосуберона (20,4 г) в ТГФ/MeOH (1:2, 900 мл) охлаждали в ледяной бане. По частям добавляли борогидрид натрия (12 г). Охлаждающую баню убирали, реакционную смесь перемешивали при температуре окружающей среды в течение 2 часов и сливали в воду. Продукт отфильтровывали, промывали водой и сушили, получая 20 г 10,11-дифторметандибензосуберола, tпл 230,1-230,6oC.
2Б. Формула 2 с различными R1 и R2
В соответствии с процедурой, описанной в части А, и заменяя 10,11-дифторметандибензосуберон следующими веществами:
а. 10,11-дихлорметандибензосубероном,
б. 10,11-дибромметандибензосубероном,
в. 10,11-метандибензосубероном, и
г. 10,11-хлорфторметандибензосубероном;
получали соответственно следующие соединения:
а. 10,11-дихлорметандибензосуберол,
б. 10,11-дибромметандибензосуберол,
в. 10,11-метандибензосуберол, и
г. 10,11-хлорфторметандибензосуберол.
Пример 3
Син- и анти-1-(10,11-дифторметандибензосубер-5-ил)-4-формилпиперазин
3А. Формула 3, где R1 и R2 обозначают F и R обозначает формил
К раствору 10,11-дифторметандибензосуберола (5,2 г) в диоксане (70 мл), охлажденному в ледяной бане, по каплям добавляли тионилхлорид (4,5 мл). Температуру повышали до 50oC и поддерживали в течение 4 часов. Реакционную смесь упаривали досуха, получая смесь син- и анти-5-хлор-10,11-дифторметандибензосуберана (5,7 г), которую растворяли в ацетонитриле (200 мл), и добавляли 1-пиперазинкарбоксальдегид (10 мл). Смесь перемешивали в атмосфере сухого азота при 100oC (температура бани) в течение 20 часов и затем упаривали досуха. Остаток разделяли между водным NaHCO3 и этилацетатом. Органическую фазу промывали водой, сушили над K2CO3 и упаривали. С помощью быстрой хроматографии остатка на силикагеле (30% ацетон/гексан) получали син-1-(10,11-дифторметандибензосубер-5-ил)-4-формилпиперазин (2,4 г), tпл 213oC и анти-1-(10,11-дифторметандибензосубер-5-ил)-4-формилпиперазин (2,6 г), tпл 238oC.
3Б. Формула 3 с различными R1 и R2
В соответствии с процедурой, описанной в части А, и заменяя 10,11-дифторметандибензосуберол следующими веществами:
а. 10,11-дихлорметандибензосуберолом,
б. 10,11-дибромметандибензосуберолом,
в. 10,11-метандибензосуберолом, и
г. 10,11-хлорфторметандибензосуберолом,
получали соответственно следующие соединения:
а1. анти-1-(10,11-дихлорметандибензосубер-5-ил)-4-формилпиперазин, tпл 205oC,
а2. син-1-(10,11-дихлорметандибензосубер-5-ил)-4-формилпиперазин,
б1. анти-1-(10,11-дибромметандибензосубер-5-ил)-4-формилпиперазин,
б2. син-1-(10,11-дибромметандибензосубер-5-ил)-4-формилпиперазин,
в1. анти-1-(10,11-метандибензосубер-5-ил)-4-формилпиперазин, tпл 195oC,
в2. син-1-(10,11-метандибензосубер-5-ил)-4-формилпиперазин,
г1. анти-1-(10,11-хлорфторметандибензосубер-5-ил)-4-формилпиперазин, и
г2. син-1-(10,11-хлорфторметандибензосубер-5-ил)-4-формилпиперазин.
Пример 4
Анти-1-(10,11-дифторметандибензосубер-5-ил)пиперазин
4А. Формула 4, где R1 и R2 обозначают F
Раствор анти-1-(10,11-дифторметандибензосубер-5-ил)-4-формилпиперазина (2,55 г) и гидроксида калия (3,0 г) в этаноле/воде (9:1, 100 мл) нагревали с обратным холодильником в течение 1 часа, затем охлаждали. Охлажденную реакционную смесь концентрировали, разбавляли водой, экстрагировали этилацетатом и сушили над K2CO3. Высушенную органическую фазу упаривали и получали анти-1-(10,11-дифторметандибензосубер-5-ил)пиперазин (2,35 г), tпл 131oC.
4Б. Формула 4 с различными R1 и R2
В соответствии с процедурой, описанной в части А, и заменяя анти-1-(10,11-дифторметандибензосубер-5-ил)-4-формилпиперазин следующими веществами:
а. син-1-(10,11-дифторметандибензосубер-5-ил)-4-формилпиперазином,
б. анти-1-(10,11-дихлорметандибензосубер-5-ил)-4-формилпиперазином,
в. син-1-(10,11-дихлорметандибензосубер-5-ил)-4-формилпиперазином,
г. анти-1-(10,11-дибромметандибензосубер-5-ил)-4-формилпиперазином,
д. син-1-(10,11-дибромметандибензосубер-5-ил)-4-формилпиперазином,
е. анти-1-(10,11-метандибензосубер-5-ил)-4-формилпиперазином,
ж. син-1-(10,11-метандибензосубер-5-ил)-4-формилпиперазином,
з. анти-1-(10,11-хлорфторметандибензосубер-5-ил)-4-формилпиперазином, и
и. син-1-(10,11-хлорфторметандибензосубер-5-ил)-4-формилпиперазином;
получали соответственно следующие соединения:
а. син-1-(10,11-дифторметандибензосубер-5-ил)пиперазин, tпл 225,5oC,
б. анти-1-(10,11-дихлорметандибензосубер-5-ил)пиперазин, tпл 199oC,
в. син-1-(10,11-дихлорметандибензосубер-5-ил)пиперазин,
г. анти-1-(10,11-дибромметандибензосубер-5-ил)пиперазин,
д. син-1-(10,11-дибромметандибензосубер-5-ил)пиперазин,
е. анти-1-(10,11-метандибензосубер-5-ил)пиперазин, tпл 103oC,
ж. син-1-(10,11-метандибензосубер-5-ил)пиперазин,
з. анти-1-(10,11-хлорфторметандибензосубер-5-ил)пиперазин, и
и. син-1-(10,11-хлорфторметандибензосубер-5-ил)пиперазин.
Пример 5
1-(4-бензофуразанилокси)-2,3-эпоксипропан
5А. Формула 8, где R3 обозначает бензофуразанил и n равно 1
Гидрид натрия (620 мг; 60% дисперсия в масле) добавляли по частям к бензофуразан-4-олу (1,74 г) в диметилформамиде (30 мл). Смесь выдерживали при 50oC в течение 30 минут. Добавляли эпибромгидрин (1,6 мл) и смесь выдерживали при 60oC в течение 2 часов. Реакционную смесь сливали в воду и экстрагировали этилацетатом. Органическую фазу промывали водой, сушили над Na2SO4 и упаривали. Остаток хроматографировали на силикагеле (50% этилацетат/гексан) и получали 1-(4-бензофуразанилокси)-2,3-эпоксипропан (1,6 г), tпл 75oC.
5Б. Формула 8 с различными R3 и n
В соответствии с процедурой, описанной в части А, и заменяя бензофуразан-4-ол и эпибромгидрин следующими веществами:
а. хинолин-5-олом и 1-хлор-3,4-эпоксибутаном,
б. 2-нитрофенолом и эпибромгидрином,
в. 2-хлорфенолом и эпибромгидрином,
г. 2-дифторметоксифенолом и эпибромгидрином,
д. пиридин-3-олом и эпибромгидрином, и
е. хинолин-5-олом и эпибромгидрином;
получали соответственно следующие соединения:
а. 1-(5-хинолилокси)-3,4-эпоксибутан,
б. 1-(2-нитрофенокси)-2,3-эпоксипропан,
в. 1-(2-хлорфенокси)-2,3-эпоксипропан,
г. 1-(2-дифторметоксифенокси)-2,3-эпоксипропан,
д. 1-(3-пиридилокси)-2,3-эпоксипропан, и
е. 1-(5-хинолилокси)-2,3-эпоксипропан.
5В. Формула 9 с различными R3 и n
В соответствии с процедурой, описанной в части А, и заменяя бензофуразан-4-ол и эпибромгидрин следующими веществами:
а. хинолин-5-олом и 1-бром-3-хлорпропаном,
б. хинолин-5-олом и 1-бром-4-хлорбутаном, и
в. 2-нитрофенолом и 1-бром-3-хлорпропаном,
получали соответственно следующие соединения:
а. 1-(5-хинолилокси)-3-хлорпропан,
б. 1-(5-хинолилокси)-4-хлорбутан, и
в. 1-(2-нитрофенокси)-3-хлорпропан.
Пример 6
(2R, S)-анти-5-{ 3-[4-(10,11-дихлорметандибензосубер-5-ил)-пиперазин-1- ил]-2-гидроксипропокси}хинолин
6А. Формула 1, где R1 и R2 обозначают F, A обозначает (2R,S)-гидроксипропил и R3 обозначает 5-хинолил
Раствор 1-(5-хинолилокси)-2,3-эпоксипропана (586 мг) и анти-1-(10,11-дифторметандибензосубер-5-ил)пиперазина (950 мг) в изопропаноле (20 мл) нагревали с обратным холодильником в течение 20 часов. Реакционную смесь упаривали досуха и остаток хроматографировали на силикагеле (70:30:1 этилацетат/гексан/триэтиламин), получая (2R, S)-анти-5-{3-[4-(10,11-дихлорметандибензосубер-5-ил)-пиперазин-1- ил]-2-гидроксипропокси}хинолин (1,33 г), который превращали в тригидрохлоридную соль, tпл 193,5oC, путем взаимодействия с 3 молярными эквивалентными HCl.
6Б. Изомеры формулы I с различными R1, R2, R3 и A
В соответствии с процедурой, описанной в части А, и заменяя анти-1-(10,11-дифторметандибензосубер-5-ил)пиперазин и 1-(5-хинолилокси)-2,3-эпоксипропан следующими веществами:
а. син-1-(10,11-дифторметандибензосубер-5-ил)пиперазином и 1-(5-хинолилокси)-2,3-эпоксипропаном,
б. анти-1-(10,11-дифторметандибензосубер-5-ил)пиперазином и (2R)-1-(5-хинолилокси)-2,3-эпоксипропаном,
в. анти-1-(10,11-дифторметандибензосубер-5-ил)пиперазином и (2S)-1-(5-хинолилокси)-2,3-эпоксипропаном,
г. син-1-(10,11-дифторметандибензосубер-5-ил)пиперазином и (2R)-1-(5-хинолилокси)-2,3-эпоксипропаном,
д. син-1-(10,11-дифторметандибензосубер-5-ил)пиперазином и (2S)-1-(5-хинолилокси)-2,3-эпоксипропаном,
е. анти-1-(10,11-дихлорметандибензосубер-5-ил)пиперазином и 1-(5-хинолилокси)-2,3-эпоксипропаном,
ж. анти-1-(10,11-дихлорметандибензосубер-5-ил)пиперазином и (2R)-1-(5-хинолилокси)-2,3-эпоксипропаном,
з. анти-1-(10,11-дихлорметандибензосубер-5-ил)пиперазином и (2S)-1-(5-хинолилокси)-2,3-эпоксипропаном,
и. анти-1-(10,11-метандибензосубер-5-ил)пиперазином и 1-(5-хинолилокси)-2,3-эпоксипропаном,
к. анти-1-(10,11-дифторметандибензосубер-5-ил)пиперазином и 1-(4-бензофуразанилокси)-2,3-эпоксипропаном,
л. син-1-(10,11-дифторметандибензосубер-5-ил)пиперазином и 1-(4-бензофуразанилокси)-2,3-эпоксипропаном,
м. анти-1-(10,11-дихлорметандибензосубер-5-ил)пиперазином и 1-(4-бензофуразанилокси)-2,3-эпоксипропаном,
н. анти-1-(10,11-дифторметандибензосубер-5-ил)пиперазином и 1-(2-нитрофенокси)-2,3-эпоксипропаном,
о. анти-1-(10,11-дифторметандибензосубер-5-ил)пиперазином и 1-(2-хлорфенокси)-2,3-эпоксипропаном,
п. анти-1-(10,11-дифторметандибензосубер-5-ил)пиперазином и 1-(2-дифторметоксифенокси)-2,3-эпоксипропаном,
р. анти-1-(10,11-дифторметандибензосубер-5-ил)пиперазином и 1-(3-пиридилокси)-2,3-эпоксипропаном,
с. анти-1-(10,11-дифторметандибензосубер-5-ил)пиперазином и 1-(5-хинолилокси)-3-хлорпропаном,
т. анти-1-(10,11-дифторметандибензосубер-5-ил)пиперазином и 1-(5-хинолилокси)-4-хлорбутаном,
у. анти-1-(10,11-дифторметандибензосубер-5-ил)пиперазином и 1-(2-нитрофенокси)-3-хлорпропаном, и
ф. анти-1-(10,11-дифторметандибензосубер-5-ил)пиперазином и 1-(5-хинолилокси)-3,4-эпоксибутаном,
получали соответственно следующие соединения:
а. (2R, S)-син-5-{3-[4-(10,11-дифторметандибензосубер-5- ил)пиперазин-1-ил]-2-гидроксипропокси}хинолин, tпл 208oC (тригидрохлорид),
б. (2R)-анти-5-{3-[4-(10,11-дифторметандибензосубер-5- ил)пиперазин-1-ил]-2-гидроксипропокси}хинолин, tпл 190oC (тригидрохлорид),
в. (2S)-анти-5-{3-[4-(10,11-дифторметандибензосубер-5- ил)пиперазин-1-ил]-2-гидроксипропокси}хинолин, tпл 195oC (тригидрохлорид),
г. (2R)-син-5-{3-[4-(10,11-дифторметандибензосубер-5- ил)пиперазин-1-ил] -2-гидроксипропокси}хинолин, tпл 193oC (тригидрохлорид),
д. (2S)-син-5-{3-[4-(10,11-дифторметандибензосубер-5- ил)пиперазин-1-ил] -2-гидроксипропокси}хинолин, tпл 188,5oC (тригидрохлорид),
е. (2R,S)-анти-5-{3-[4-(10,11-дихлорметандибензосубер-5- ил)пиперазин-1-ил]-2-гидроксипропокси}хинолин, tпл 195oC (тригидрохлорид),
ж. (2R)-анти-5-{3-[4-(10,11-дихлорметандибензосубер-5- ил)пиперазин-1-ил]-2-гидроксипропокси}хинолин, tпл 218oC (тригидрохлорид),
з. (2S)-анти-5-{3-[4-(10,11-дихлорметандибензосубер-5- ил)пиперазин-1-ил]-2-гидроксипропокси}хинолин, tпл 215oC (тригидрохлорид),
и. (2R,S)-анти-5-{3-[4-(10,11-метандибензосубер-5-ил)пиперазин- 1-ил]-2-гидроксипропокси} хинолин, tпл в диапазоне 87,4-99,3oC; МС: молекулярный ион = 491,
к. (2R,S)-анти-4-{3-[4-(10,11-дифторметандибензосубер-5- ил)пиперазин-1-ил]-2-гидроксипропокси}бензофуразан, tпл 186oC (дигидрохлорид),
л. (2R, S)-син-4-{3-[4-(10,11-дифторметандибензосубер-5- ил)пиперазин-1-ил]-2-гидроксипропокси}бензофуразан, tпл 188oC (дигидрохлорид),
м. (2R,S)-анти-4-{3-[4-(10,11-дихлорметандибензосубер-5- ил)пиперазин-1-ил]-2-гидроксипропокси}бензофуразан,
н. (2R,S)-анти-1-{3-[4-(10,11-дифторметандибензосубер-5- ил)пиперазин-1-ил]-2-гидроксипропокси}-2-нитробензол,
о. (2R,S)-анти-1-{3-[4-(10,11-дифторметандибензосубер-5- ил)пиперазин-1-ил]-2-гидроксипропокси}-2-хлорбензол,
п. (2R,S)-анти-1-{3-[4-(10,11-дифторметандибензосубер-5- ил)пиперазин-1-ил]-2-гидрокспропокси}-2-дифторметоксибензол,
р. (2R,S)-анти-3-{3-[4-(10,11-дифторметандибензосубер-5- ил)пиперазин-1-ил]-2-гидроксипропокси}пиридин,
с. (2R,S)-анти-5-{3-[4-(10,11-дифторметандибензосубер-5- ил)пиперазин-1-ил]пропокси}хинолин,
т. (2R,S)-анти-5-{4-[4-(10,11-дифторметандибензосубер-5- ил)пиперазин-1-ил]бутокси}хинолин,
у. (2R,S)-анти-5-{3-[4-(10,11-дифторметандибензосубер-5- ил)пиперазин-1-ил]пропокси}-2-нитробензол, и
ф. (2R,S)-анти-5-{3-[4-(10,11-дифторметандибензосубер-5- ил)пиперазин-1-ил]-3-гидроксибутокси}хинолин.
Пример 7
В этом примере проиллюстрировано получение характерной фармацевтической композиции для орального введения, содержащей активное вещество формулы I, например, (2R)-анти-5-{3-[4-(10,11-дифторметандибензосубер-5-ил)пиперазин-1- ил]-2-гидроксипропокси}хинолин.
Ингредиенты - Количество на таблетку, мг
Активное вещество - 200
Лактоза, высушенная распылением - 148
Стеарат магния - 2
Вышеперечисленные ингредиенты смешивают и вводят в желатиновую капсулу с твердой оболочкой. Если используется тригидрохлоридная соль, то требуется 243 мг соли.
Другие соединения формулы I, такие как полученные в соответствии со способами, описанными в примерах 1-6, могут использоваться в качестве активного вещества для получения композиций этого примера для орального введения.
Пример 8
В этом примере проиллюстрировано получение другой характерной фармацевтической композиции для орального введения, содержащей активное вещество формулы I, например, (2R)-анти-5-{ 3-[4-(10,11-дифторметандибензосубер-5-ил)пиперазин-1- ил]-2-гидроксипропокси}хинолин.
Ингредиенты - Количество на таблетку, мг
Активное вещество - 400
Кукурузный крахмал - 50
Лактоза - 145
Стеарат магния - 5
Вышеперечисленные ингредиенты смешивают до однородности и прессуют в виде набора отдельных таблеток. Если используется тригидрохлоридная соль, то требуется 486 мг соли.
Другие соединения формулы I, такие как полученные в соответствии со способами, описанными в примерах 1-6, могут использоваться в качестве активного вещества для получения композиций этого примера для орального введения.
Пример 9
В этом примере проиллюстрировано получение характерной фармацевтической композиции, содержащей активное вещество формулы I, например, (2R)-анти-5-{ 3-[4-(10,11-дифторметандибензосубер-5- ил)пиперазин-1-ил] -2-гидроксипропокси}хинолин.
Суспензию для орального введения готовят на основе следующей композиции:
Ингредиенты - Количество
Активное вещество - 1,0 г
Фумаровая кислота - 0,5 г
Хлорид натрия - 2,0 г
Метилпарабен - 0,1 г
Гранулированный сахар - 25,5 г
Сорбитол (70% раствор) - 12,85 г
Veegum K (Vanderbilt Co.) - 1,0 г
Корригент - 0,035 мл
Красители - 0,5 г
Дистиллированная вода - д.к. до 100 мл
Если для получения суспензии используется тригидрохлорид, то требуется 1,215 г соли. Другие соединения формулы I, такие как полученные в соответствии со способами, описанными в примерах 1-6, могут использоваться в качестве активного вещества для получения композиций этого примера для орального введения.
Пример 10
В этом примере проиллюстрировано получение характерной фармацевтической композиции, содержащей активное вещество формулы I, например, (2R)-анти-5-{ 3-[4-(10,11-дифторметандибензосубер-5- ил)пиперазин-1-ил] -2-гидроксипропокси}хинолин.
Инъецируемый препарат, забуференный до pH 4, готовят на основе следующей композиции:
Ингредиенты - Количество
Активное вещество - 0,2 г
Буферный раствор ацетата натрия (0,4 М) - 2,0 мл
HCl (1Н) - д.к. до pH 4
Вода (дистиллированная, стерильная) - д.к. до 20 мл
Если для получения инъецируемого препарата используется тригидрохлоридная соль, то требуется 0,243 г соли. Другие соединения формулы I, такие как полученные в соответствии со способами, описанными в примерах 1-6, могут использоваться в качестве активного вещества для получения инъецируемых композиций этого примера.
Пример 11
В этом примере проиллюстрировано получение характерной фармацевтической композиции, содержащей активное вещество формулы I, например, (2R)-анти-5-{ 3-[4-(10,11-дифторметандибензосубер-5- ил)пиперазин-1-ил] -2-гидроксипропокси}хинолин.
Суппозиторий общим весом 2,5 г готовят на основе следующей композиции:
Активное вещество - 500 мг
Витепсол H-15* - баланс
(* триглицериды насыщенных жирных кислот растительного происхождения; продукт фирмы Riches-Nelson, Inc., Нью-Йорк, N.Y.).
Если используется тригидрохлоридная соль, то требуется 607 мг соли. Другие соединения формулы I, такие как полученные в соответствии со способами, описанными в примерах 1-6, могут использоваться в качестве активного вещества для получения суппозиторных композиций этого примера.
Пример 12
Определение стабильности при кислом pH
Испытываемые соединения (15 мкг) растворяли в 3 мл 0,01н. HCl (pH 2) и инкубировали при 37oC. Через различные промежутки времени отбирали аликвотные пробы и впрыскивали в 3 мкм картриджную колонку Pecosphera C-18 (3,3 х 0,46 см) для анализа с помощью ЖХВД (подвижная фаза: 35% ацетонитрил 18% тетрагидрофуран/47% одноосновный фосфат калия, содержащая 4 мМ N,N-диметилоктиламина; скорость потока 1,0 мл/мин). Испытываемые соединения и продукты их распада контролировали с помощью УФ-адсорбции при 240 нм. Уменьшение количества исходного соединения выражали в процентах по отношению к высоте пика в нулевой момент времени, на основе чего графически определили следующие значения t1/2:
MS-073 (патент США 5112817), t1/2 = 15 мин,
(2R, S)-анти-5-{ 3-[4-(10,11-метандибензосубер-5-ил)пиперазин-1- ил]-2-гидроксипропокси}хинолин, t1/2 = 2,5 ч,
(2R, S)-5-{3-[4-(10,11-дифторметандибензосубер-5-ил)пиперазин-1- ил]-2-гидроксипропокси}хинолин, t1/2 ≥ 72 ч, и
(2R, S)-анти-5-{ 3-[4-(10,11-хлорметандибензосубер-5-ил)пиперазин-1- ил] -2-гидроксипропокси}хинолин, t1/2 ≥ 72 ч.
При тестировании указанным методом соединения по настоящему изобретению проявляют более значительную стабильность при кислом pH по сравнению с MS-073.
Пример 13
Определение активности in vitro с использованием количественного анализа МТТ
Этот метод является модификацией анализа, описанного Mosmann, T. в "Rapid Colorimetric Assay For Cellular Growth And Survival: Application to proliferation and cytotoxity assays", J. Immunol. Meth., т. 65, 55-63, (1983).
Штамм клеток (0,3 мл), содержащий резистентные к множеству лекарств клетки CHRC5 китайского хомяка (приблизительно 2 • 105 клеток/мл) для образования суспензии вводят в среду (2,7 мл), содержащую испытываемое соединение или контроль с носителем, в присутствии или при отсутствии адриамицина (1 мкг/мл). Аликвоты (0,1 мл) клеточной суспензии затем помещают в восемь лунок на каждой из трех 96-луночных планшетов для микротитрования. После инкубации в тканевом, культуральном инкубаторе при 37oC берут по одному планшету в каждый из нижеуказанных моментов времени: 24 часа, 48 часов и 72 часа. После удаления из инкубатора в каждую лунку планшета, который затем возвращают в инкубатор на 3 часа, добавляют 3-[4,5-диметилтиазол-2-ил]-2,5-дифенилтетразолийбромид, МТТ (10 мкл, из раствора штамма с концентрацией 5 мг/мл в фосфатном забуференном физиологическом растворе).
Кристаллы формазана, образованные в результате активности митохондриальных ферментов в живых клетках, солюбилизируют путем удаления среды и добавления ДМСО (150 мкл/лунку) при перемешивании, осуществляемом на орбитальном вибраторе. A570 (соответствующая длина волны 650 нм) считывают на считывающем устройстве Molecular Devices microplate и результаты выражают в процентах к контролю с носителем или к контролю с адриамицином ежедневно или в виде графика по времени A570.
Соединения по настоящему изобретению проявляют активность при тестировании указанным методом. Более конкретно, соединение (2R)-анти-5-{3-[4-(10,11-дифторметандибензосубер-5-ил)пиперазин-1- ил] -2-гидроксипропокси} хинолин более чем в 3 раза активнее MS-73; соответствующий (2S)-анти-изомер приблизительно в 1,7 раза более активен, чем MS-73, и (2R,S)-анти-смесь приблизительно в 1,6 раз более активна, чем MS-73.
Пример 14
Определение активности in vivo с использованием количественного анализа MDR (резистентности к множеству лекарств)
Этот метод является модификацией анализа, описанного Slate и Michelson в "Drug Resistance Reversal Strategies: A Comparison of Experimintal Data With Model Predictions", J. Natl. Cancer Inst., т. 83, 1574-1580 (1991).
Мышей (B6D2F1, 7-8 недельных самок, весом приблизительно 20 г, лаборатории Jackson) выбирали случайным образом, взвешивали и разделяли на группы по 6-7 особей в каждой. В момент начала эксперимента (день 0) каждой мыши впрыскивали внутрибрюшинно резистентные к множеству лекарств мышиные лейкозные клетки P388/ADR, 2,4 • 107 клеток/мл. Спустя два часа каждой мыши имплантируют внутрибрюшинно 7-дневный мининасос Alzet (модель 2001, Alza Corporation, Пало Альта, СА), содержащий носитель (ДМСО/ЗФР) плюс адриамицин (3 мг/кг/день), одно испытываемое соединение (30 мг/кг/день) или адриамицин плюс испытываемое соединение (по 0,3, 3, 10 и 30 мг/кг/день). Состояние мышей контролировали ежедневно и регистрировали смертность, начиная с 7 дня. Более продолжительное время выживания по сравнению с группами, которым ввели только контроль с носителем или адриамицин, свидетельствует об активности.
Соединения по настоящему изобретению проявляют активность при испытывании указанным методом. Более конкретно, соединение (2R)-анти-5-{3-[4-(10,11-дифторметандибензосубер-5-ил)пиперазин-1- ил] -2 гидроксипропокси} хинолин более чем в 4 раза активнее MS-73; и соответствующий (2S)-анти-изомер приблизительно в 1,3 раза более активен, чем MS-73.
Кроме того, в анализе в отношении маточной саркомы человека соединение (2R)-анти-5-{3-[4-(10,11-дифторметандибензосубер-5- ил)пиперазин-1-ил]-2-гидроксипропокси}хинолин в 3 раза более активно, чем MS-73.
Пример 15
Токсичность соединений формулы I
Мыши получали (2R)-анти-5-{ 3-[4-(10,11-дифторметандибензосубер-5- ил)пиперазин-1-ил] -2-гидроксипропокси}хинолин внутривенно инъекцией ударной дозы вещества один раз в день в течение двух недель дозами по 0 (носитель: 5 вес. % по объему маннитола в очищенной воде), 10, 30 и 100 мг/кг. Все мыши в группах 0, 10 и 30 мг/кг выжили. У всех мышей, получавших 100 мг/кг, проявились мышечные конвульсии, и они умерли после однократного применения дозы. Причина смерти не была определена.
| название | год | авторы | номер документа |
|---|---|---|---|
| КАРБОКСАМИДЫ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1995 |
|
RU2163232C2 |
| МОСТИКОВЫЕ ИНДОЛЫ В КАЧЕСТВЕ ИНГИБИТОРОВ МАТРИЧНЫХ МЕТАЛЛОПРОТЕАЗ | 1996 |
|
RU2191779C2 |
| 1-ФЕНИЛАЛКАНОНЫ - НОВЫЕ ЛИГАНДЫ 5-НТ-РЕЦЕПТОРА | 1994 |
|
RU2170228C2 |
| ПРОИЗВОДНЫЕ БЕНЗОЦИКЛОАЛКИЛАЗОЛОТИОНА | 1995 |
|
RU2145321C1 |
| ПРОИЗВОДНЫЕ АМИДОВ, ЯВЛЯЮЩИЕСЯ ИНГИБИТОРАМИ МАТРИЧНЫХ МЕТАЛЛОПРОТЕАЗ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1994 |
|
RU2132851C1 |
| ЗАМЕЩЕННЫЕ КАРБОКСАМИДЫ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 1994 |
|
RU2132327C1 |
| СПОСОБ ПОЛУЧЕНИЯ АРИЛОКСИОКСИПРОПИЛЕН-ПИПЕРАЗИНИЛАЦЕТАНИЛИДОВ, ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫХ СЛОЖНЫХ ЭФИРОВ, ИЛИ КИСЛОТНОАДДИТИВНЫХ СОЛЕЙ | 1984 |
|
RU2071471C1 |
| Способ получения (4,2,0)бициклооктановых производных, или их фармацевтически приемлемых нетоксичных солей, или фармацевтически приемлемых нетоксичных сложных эфиров | 1986 |
|
SU1500153A3 |
| ПРОИЗВОДНЫЕ ПИПЕРАЗИНА И ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СЛОЖНЫЕ ЭФИРЫ И КИСЛОТНО-АДДИТИВНЫЕ СОЛИ | 1991 |
|
RU2083570C1 |
| Способ получения (4,2,0)-бициклооктановых производных или их фармацевтически приемлемых нетоксичных солей | 1987 |
|
SU1588275A3 |
Изобретение относится к фармацевтически активным агентам. Описываются производные 10,11-метандибензосуберана общей формулы I, где обозначает -СН2-СН2; -СН2-CHRa-CH2, где Ra обозначает Н, ОН или низший ацилокси; или -СН2-СНRa-CHRb-СН2, где один из Ra или Rb обозначает Н,ОН или низший ацилокси, а другой обозначает Н, R' обозначает Н, F, Сl или Br, R2 обозначает Н, F, Cl или Вг; R3 обозначает гетероарил, состоящий из двух конденсированных колец, содержащих от 1 до 3 гетероатомов, выбранных из N, О или S, или их фармацевтически приемлемые соли. Эти соединения проявляют противоопухолевую активность и пригодны для лечения рака, в частности для повышения эффективности существующей химиотерапии рака и для предотвращения резистентности к множеству лекарственных препаратов. Описывается также способ преодоления резистивности к противоопухолевым лекарствам и способ лечения опухолевых заболеваний. 4 с. и 13 з.п. ф-лы.
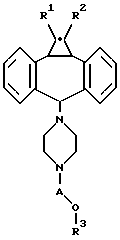
где А обозначает -СН2-СН2-, -СН2-СНRа-СН2-, где Rа обозначает Н, ОН или низший ацилокси, или -СН2-СНRа-СН2Rb-СН2-, где один из Rа или Rb обозначает Н, ОН или низший ацилокси, а другой обозначает Н;
R1 обозначает Н, F, Cl или Br;
R2 обозначает Н, F, Cl или Br;
R3 обозначает гетероарил, состоящий из двух конденсированных колец, содержащих от 1 до 3 гетероатомов, выбранных из N, О или S,
или их фармацевтически приемлемые соли.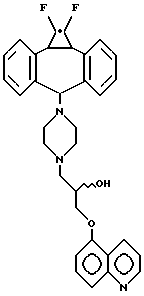
или его фармацевтически приемлемая соль.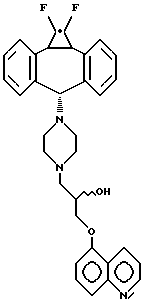
или его фармацевтически приемлемая соль.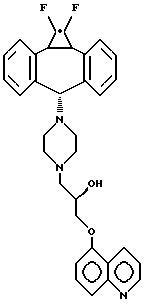
или его фармацевтически приемлемая соль.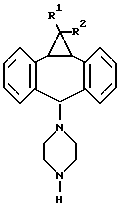
с соединением формулы 8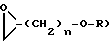
где R1, R2, R3 имеют значения, указанные в п.1, и n = 1 или 2, с получением соединения формулы I, в которой А обозначает -СН2-СНRа-СН2, где Rа обозначает ОН,
с последующим превращением при необходимости в их фармацевтически приемлемые соли, или конденсацией соединения формулы 4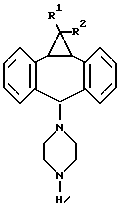
с соединением формулы 9
Х-(СН2)mОR3,
где R1, R2, R3 имеют вышеуказанные значения, m = 2, 3 или 4, а Х обозначает галоген,
с получением соединения формулы I, в которой А обозначает -СН2-СН2-, -СН2-СН2-СН2 или -СН2-СН2-СН2-СН2-, или их фармацевтически приемлемых солей; (b) конденсацией эпоксида формулы 10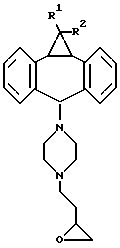
где R1 и R2 имеют вышеуказанные значения,
с соединением формулы R3ОН, где R3 имеет указанное выше значение, с получением соединения формулы I, в которой R1, R2 и R3 имеют вышеуказанные значения, а А означает радикал -СН2-СНRа-СНRb-СН2-, где Rа означает водород, а Rb означает ОН; (в) реакцией соединения формулы I, где Rа или Rb обозначает ОН, с низшим ацилгалогенидом формулы R'-С(О)-Х, где R' обозначает низший алкил и Х обозначает галоген, для получения соединения формулы I, где R1, R2 и R3 имеют вышеуказанные значения, а Rа или Rb обозначает низший ацилокси; (г) взаимодействием соединения формулы I с фармацевтически приемлемой кислотой для образования соответствующей кислотно-аддитивной соли или (д) взаимодействием кислотно-аддитивной соли соединения формулы I с основанием для образования соответствующего свободного основания формулы I.
| ИНВЕРТОР | 0 |
|
SU363212A1 |
| СТАТОР КОМПРЕССОРА | 2014 |
|
RU2567885C1 |
| US 5112817 А, 1992 | |||
| Кешликова Э.Д | |||
| Биохимические механизмы лекарственной устойчивости к N-нитрозомочевинам | |||
| Автореферат диссертации на соискание ученой степени кандидата медицинских наук | |||
| - М., 1986 | |||
| Авербух Л.А | |||
| Развитие устойчивости опухолевых клеток в эксперименте к некоторым антибластомным средствам при их изолированном и совместном применении | |||
| Автореферат на соискание ученой степени кандидата биологических наук | |||
| - М., 1979 | |||
| Онкология/Под ред | |||
| Трапезникова Н.Н., Экхардта Г.И | |||
| - М.: Медицина, 1981, с.148 - 165. | |||
